Эта заявка является частичным продолжением заявки на патент США с регистрационным N 07/810661, поданной 19 декабря 1991, которая в настоящее время аннулирована.
Предметом настоящего изобретения являются гетероциклические углеродные соединения, обладающие лекарственными свойствами и биологической активностью, а также способы их получения и применения. В частности, настоящее изобретение относится к 1,4-бизамещенным производным пиперазина, в которых одним заместителем является индол-3-ил-алкильная группа, а другим заместителем является алкокси-замещенное пиримидин-4-ильное или пиридин-4-ильное кольцо. Эти соединения обладают уникальным серотонергическим профилем, что делает их полезными для лечения васкулярных головных болей, таких как мигрень.
В патенте США N 3188313 Арчер рассматривает целый ряд индолил-алкилпиперазинов, являющихся депрессантами центральной нервной системы. Среди большого числа возможных заместителей в положении у 4-го атома азота пиперазинового кольца указывались пиримидиновое и пиридиновое кольца (оба незамещенные). В патенте США N 3562278 Арчер описывает ряд 1-индолилэтил-4-замещенных пиперазинов. В качестве возможных заместителей в положении у 4-го атома называются пиридинил и 2-пиримидинил, которые также являются незамещенными. Фармакологическое действие этих прототипных соединений связано с общей депрессией центральной нервной системы и психомоторной депрессией, что не имеет никакого отношения к лечению мигрени. Таким образом Арчером не предлагаются алкоксипиридиновые или алкоксипиримидиновые соединения, предназначенные для лечения мигрени по настоящему изобретению.
Довай и др. в опубликованной заявке на патент Великобритании N 2124210 рассмотрели ряд производных 3-алкиламиноиндола в качестве полезных лекарственных средств для лечения мигрени. Одно из этих соединений специально рассматривается в последующей заявке на патент Великобритании N 2162522, поданной Оксфордом и опубликованной 5 февраля 1986 г. Данное соединение известно в литературе как суматриптан /1/.

Маноури и др. описали ряд новых производных индолина в заявке на Европейский патент N 354094, поданной 7 февраля 1990. В этой заявке указывается, что эти соединения являются полезными для лечения различных нарушений центральной нервной системы, включая депрессию, беспокойство и мигрень. Эти прототипные соединения имеют формулу /2/.
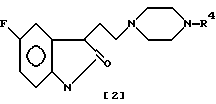
в которой
R4 представляет арильную, пиридиновую или хинолиновую части.
Ни одно из этих прототипных соединений не имеет отношения к новым алкоксипиридин-4-ильным и алкоксипиримидин-4-ильным производным индол-3-ил-алкилпиперазинов, которые предназначены для лечения васкулярной головной боли, такой как мигрень.
Мигрень входит в более широкий класс головных болей, который также включает "гистаминовые" головные боли и другие головные боли, которые по своей этиологии имеют сосудистый характер. Такие головные боли классифицируются как васкулярные головные боли. Для ознакомления с текущим кратким обзором головных болей и с методами их лечения см.: главу 13. Лекарственные средства, применяемые для лечения мигрени и других головных болей. Drag Evaluation, 6-е издание, 1986, с. 239-253, Американская медицинская ассоциация, У.В. Сондерс компани, Филадельфия, шт. Пенсильвания.
Нерегулярные часто возникающие головные боли присущи большому числу людей, но обычно они бывают резкими и непродолжительными. Головную боль этого типа, как правило, облегчают слабые анальгетики, такие как аспирин или ацетаминофен. Такие головные боли являются довольно обычными, и, хотя они являются болезненными и неприятными, очень редко вызывают нетрудоспособность и ухудшение общего самочувствия. Однако хронически повторяющиеся головные боли сосудистого характера обычно заставляют больного обратиться к врачу из-за тяжелой боли, которая часто вызывает нетрудоспособность.
Хотя не существует общепринятой системы классификации головной боли, васкулярная головная боль в соответствии с целями настоящего изобретения определяется главным образом как мигрень или "гистаминовая" головная боль. Определение мигрени включает обычный или классический тип, а также разновидности мигрени, которые хорошо известны специалистам в этой области. Другие подтипы, такие как васкулярные головные боли и головные боли, вызываемые токсичными веществами и гипертонией, хроническая пароксизмальная гемикрания, а также некоторые головные боли, вызываемые сокращением мышц, или имеющие смешанный сосудисто-мышечный характер, также входят в категорию васкулярных головных болей и поддаются лечению в соответствии с настоящим изобретением. Специалистам в этой области известно, что не существует одного эффективного метода лечения для всех больных, которым поставлен один и тот же диагноз, в результате чего возникает большая неопределенность в отношении классификации головных болей.
Лекарственные средства, чаще всего используемые для лечения головной боли, делятся на следующие группы: эрготалкалоиды, бетаблокирующие средства, средства, блокирующие кальциевый канал, антидепрессанты и их смеси.
Лечение повторяющейся васкулярной головной боли осложняется отсутствием единого метода лечения, который был бы эффективен для всех больных, страдающих головной болю одного и того же типа, и необходимостью выбора абортивного или профилактического метода лечения этих головных болей. Другую сложность создают лекарственные средства, которые вызывают зависимость при длительном применении, например, эрготамин. Другим важным соображением, которое учитывалось при разработке настоящего изобретения, было то, что применяемые в настоящее время для лечения мигрени более эффективные лекарственные средства, например, эрготы, метисергид, вызывают серьезные побочные эффекты, ограничивающие их применение в течение длительного времени.
Таким образом существует потребность в безопасном и эффективном лекарственном средстве, предназначенном для лечения мигрени и связанных с нею расстройств, которое можно использовать в профилактических целях или для облегчения приступа головной боли.
Целью изобретения является использование новых алкоксипиридин-4-ильных и алкоксипиримидин-4-ильных производных индол-3-ил-алкилпиперазинов для лечения васкулярных головных болей, в частности мигрени; способы их получения; фармацевтические составы, включающие эти соединения, и их применение в лечебных целях.
Способ по настоящему изобретению предназначен для облегчения васкулярных или подобных головных болей, наиболее известными примерами которых являются мигрень и "гистаминовая" головная боль. Этот способ включает введение алкоксипиридин-4-ильного или алкоксипиримидин-5-ильного производного индол-3-ил-алкилпиперазина либо его фармацевтически приемлемой соли человеку, нуждающемуся в таком лечении. При применении способа по настоящему изобретению предпочтение отдается пероральному или назальному введению фармацевтических составов, содержащих новые лекарственные средства против мигрени.
В широком смысле настоящее изобретение относится к лечению выскулярных головных болей при помощи алкоксипиридиновых и пиримидиновых производных индол-3-ил-алкилпиперазинов. Определенный аспект настоящего изобретения относится к новым соединениям, обладающим полезными серотонергическими свойствами против мигрени и имеющим формулу I.
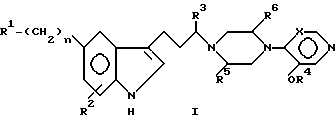
В формуле I R1 представляет заместитель, выбираемый из группы заместителей, включающих амино, циано, нитро, -OCH2CN; -OCH2CONR7R8; -O2CR9; -O2CNR7R8; -SO2NR7R8; -SO2R9; -COR8; -CO2R8; -CONR7R8; -NR7CO2R9; -NR7COR8 и 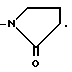
R2 выбирают из группы, включающей водород, галоген, низший алкил, низший алкокси и -CO2R9. Описательный термин "низший" используется в этой заявке для определения органического радикала, содержащего от 1 до 4 атомов углерода.
R3 и R7 независимо друг от друга выбирают из водорода и низшего алкила. Однако R3 не может быть водородом, если часть молекулы R1-(CH2)n - представляет -CONH2.
R4 представляет низший алкил, при этом предпочтение отдается метилу.
R5 и R6 независимо друг от друга выбирают из водорода и низшего алкила либо R5 и R6 вместе образуют метиленовый или этиленовый мостик.
R8 выбирают из группы, включающей водород, низший алкил. R7-замещенный фенил-низший алкил и трифторметил.
R9 выбирают из группы, включающей низший алкил и R7-замещенный фенил-низший алкил.
X может представлять -CH-, в результате чего образуется пиридиновое кольцо, или -N-, в результате чего образуется пиримидиновое кольцо.
Символ n означает нуль или целые числа 1 и 2.
Соединения формулы I можно подразделить на две структурные категории в зависимости от значения X. Если X представляет N-/-формула IA/,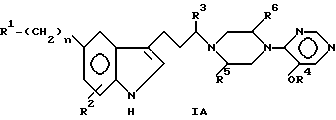
соединения относятся к категории пиримидина, а R1, R6 и n имеют указанные выше значения.
Если X представляет -CH- /формула IB/.

соединения относятся к категории пиридина, причем объем этой категории может быть несколько расширен за счет того, что R1 может дополнительно включать водород, галоген, низший алкокси, R7-замещенный фенил-низший алкокси и NR7SO2R9.
Поэтому расширенный ряд соединений определяется формулой XXI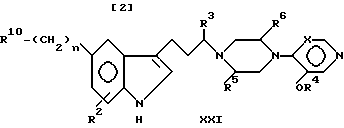
в которой R10 ограничивается значениями R1, при которых X представляет -N-, при этом R1 представляет водород, галоген, низший алкил, низший алкокси, R7-замещенный фенил-низший алкокси, гидрокси и -NR7SO2R9, если X представляет -CH-. Элементы R2-R9, n и X имеют указанные выше значения.
Кроме того, соединения формулы XXI также включает все фармацевтически приемлемые соли присоединения кислоты и/или их сольваты. Предпочтительными сольватами являются гидраты. Настоящее изобретение также включает стереоизомеры, а также оптические изомеры, например, смеси энантиомеров, отдельные энантиомеры и диастереоизомеры, которые образуются в результате структурной асимметрии, характерной для определенных соединений настоящего ряда. Выделение отдельных изомеров осуществляется с помощью различных методов, которые хорошо известны специалистам в этой области. Термин "низший алкил" относится к углеродным радикалам с прямой и разветвленной цепью, содержащим от 1 до 4 атомов углерода включительно. Примерами этих радикалов могут служить углеродные цепи, которыми являются метил, этил, пропил, изопропил, 1-бутил, 1-метилпропил, 2-метилпропил. Галогеном может быть фтор, хлор, бром и йод, при этом предпочтение отдается фтору и хлору и наиболее предпочтительным является фтор.
Фармацевтически приемлемыми солями присоединения кислоты по настоящему изобретению являются такие соли, в которых противоион не оказывает значительного влияния на токсичность или фармакологическую активность соли, и, будучи таковыми, они являются фармакологическими эквивалентами оснований формулы XXI. Обычно им отдается предпочтение для применения в медицинских целях. В некоторых случаях они обладают физическими свойствами, которые делают их более желательными для изготовления фармацевтических составов, такими как растворимость, отсутствие гигроскопичности, прессуемость при создании таблеток и совместимость с другими ингредиентами, вместе с которыми данное вещество может использоваться в фармацевтических целях. Соли обычно получают путем смешивания основания формулы XXI с выбранной кислотой, предпочтительно посредством контактирования в растворе с применением избыточного количества обычно используемых инертных растворителей, таких как вода, простой эфир, бензол, метанол, этанол, этилацетат и ацетонитрил. Их также можно получить с помощью реакции обмена или в результате обработки ионообменной смолой в таких условиях, при которых анион одной соли вещества формулы XXI замещается другим анионом, и при этом создаются возможности для отделения желаемого соединения, например, путем осаждения из раствора или экстрагирования введением в растворитель либо элюирования из ионообменной смолы, или удерживания в ней. Фармацевтически приемлемые кислоты, используемые для образования солей веществ формулы XXI, включают серную, фосфорную, хлористоводородную, бромистоводородную, иодистоводородную, лимонную, уксусную, бензойную, коричную, фумаровую, миндальную, щавелевую, фосфорную, азотную, слизевую, изэтионовую, пальмитиновую, энантовую и другие кислоты.
Соединения формулы XXI можно получать в результате применения общего способа, изображенного на схеме 1, приведенной в конце текста. Способы синтеза промежуточных соединений представлены на схеме 2, приведенной в конце текста. Кроме того, определенные соединения и способы их синтеза более детально рассматриваются в приводимом ниже разделе, посвященном конкретным вариантам осуществления настоящего изобретения.
В способе, изображенном на схеме 1, элементы с R2 по R6, R10 и n имеют указанные выше значения. Реагент Y-X является реагентом с органической отщепляемой группой, в котором X представляет отщепляемую группу, такую как тозил, мезил, галогенид, сульфат, фосфат и так далее; а Y является или протоном, или противоином: например, Y-X может представлять HBr, мезил- или тозилхлорид и подобное соединение. Реакции, представленные на схеме 1, и их применение известны специалистам в области органического синтеза, поэтому легко могут быть поняты изменения, вносимые в условия реакций и реагенты.
Индолилалкиловый спирт формулы VI превращают в активированное промежуточное соединение формулы V, в котором спиртовая часть молекулы превращается в органическую отщепляемую группу. В результате взаимодействия промежуточного соединения V с промежуточным соединением пиперазина формулы III образуется продукт формулы XXI.
Схема 2. Способы синтеза промежуточных соединений - приведена в конце текста.
На схеме 2 изображено несколько способов синтеза промежуточных соединений формулы VI. Специалист в области синтеза легко поймет, каким образом следует использовать эти способы для получения конкретных соединений формулы VI. Способ N 1 представляет общие способы восстановления с целью превращения карбонильной части соединения II в спирт. Выбор условий реакции определяется реакционной способностью элемента R10. Символ Z обозначает низший алкил, низший алкоксил или гидроксил. Если Z является низшим алкилом, R3 в промежуточном продукте VI также будет низшим алкилом.
В соответствии со схемой 1 может осуществляться модифицированный вариант способа 1, при котором взаимодействие соответствующего соединения формулы II с промежуточным соединением пиперазина формулы III с последующей обработкой цианоборогидридом натрия позволяет получить целевой продукт формулы XXI. Этот вариант является желательным для синтеза продуктов, в которых R3 представляет низший алкил, а R10означает часть молекулы, которая является химически инертной по отношению к цианоборогидриду натрия.
Способ N 2 представляет синтез индолилалкилового спирта с использованием промежуточных соединений гидразона VII и VIII и последующим замыканием кольца в индольную систему.
Способ N 3 представляет особенно полезный метод получения промежуточных соединений формулы VI, в которой R1 является -SO2NR7R8. Конкретные примеры, включающие применение способа N 3, детально рассматриваются в приводимом ниже экспериментальном разделе.
Дополнительные продукты формулы XXI также можно получить в результате химического превращения R10-индолил-заместителя.
Примеры включают превращение цианогруппы в аминокарбонильный заместитель под воздействием сильных гидроксидов, таких как КОН; превращение аминогрупп в амиды, лактамы и сульфонамиды, а также различные превращения гидроксильного заместителя R10 в другие функциональные группы показано на схеме 4, приведенной в конце текста.
Реагенты, растворители и условия реакций, применяемые в описанных выше стадиях этих способов, хорошо известны специалистам в области органического синтеза, так как эти способы включают стандартные реакции органического синтеза, детальное описание которых приводится в химической литературе. В эти способы могут быть внесены изменения с целью получения других соединений, входящих в объем настоящего изобретения, но не рассматриваемых специально. Специалист в этой области легко поймет изменения, вносимые в эти методы с целью несколько иного получения тех же соединений.
Для более детального описания настоящего изобретения в разделе "Описание конкретных вариантов осуществления настоящего изобретения" приводятся типичные примеры синтеза.
Была установлена связь серотонина с патофизиологией мигрени на основании собранных данных, свидетельствующих о повышенном выделении метаболитов серотонина после приступа мигрени и об уменьшении содержания серотонина в тромбоцитах во время мигрени. Последний признак, по-видимому, является характерным для мигрени и не возникает в результате боли или напряжения (Энтони и др. Содержание серотонина в плазме в случае мигрени и стресса. - Arch. Neurol. 1967, 16: 544 - 552). Более важным свидетельством является то, что внутримышечная инъекция резерпина уменьшает содержание серотонина в плазме и вызывает характерную для мигрени головную боль у людей, страдающих мигренью. Такую искусственно вызванную головную боль можно ослабить посредством медленного внутривенного вливания креатининсульфата серотонина (Кимбалл и др. Влияние серотонина на больных, страдающих мигренью. - Neurology, Нью-Йорк, 1960, 10: 107 - 111).
Хотя было продемонстрировано, что серотонин оказывает эффективное действие при лечении приступов мигрени, его применению препятствуют вызываемые им побочные эффекты, такие как беспокойство, тошнота, слабость, гипервентиляция легких, покраснение лица и увеличение щитовидной железы (Ланс и др. Регулирование краниальных артерий с помощью гуморальных механизмов и его связь с синдромом мигрени. - Headache, 1967, 7: 93-102). По этой причине более конкретные средства на основе серотонина, которые позволяли бы лечить мигрень без побочных воздействий, являются потенциально полезными лекарственными средствами против мигрени. Накопление данных привело к осознанию того, что соединения с избирательным воздействием на подтип 5-HTID рецепторов серотонина должны быть весьма эффективными при лечении мигрени. В этой связи следует отметить, что соединения по настоящему изобретению демонстрируют сильное сродство и агонистическое действие на участок 5-HTID. Соединения формулы XXI обладают эффективностью, которая обеспечивает достижение концентрации 50%-ного ингибирования (IC50) при использовании этих соединений в количестве менее 100 нмоль. Предпочтительные соединения обеспечивают достижение IC50 при их содержании менее 10 нмоль.
Определение характеристик связывания 5-HTID производилось по методике, описанной Герингом и Пероутка. J. Neurosci, 7/3/, 1987, 894 - 903; при внесении незначительных изменений. Значения IC50(nM) определяли при испытании в лабораторных условиях для соединений по настоящему изобретению с использованием меченого тритием серотонина.
Другим аспектом настоящего изобретения предусматривается способ лечения больных, страдающих васкулярной головной болью, который включает системное введение такому больному терапевтически эффектного количества соединения формулы XXI или его фармацевтически приемлемой соли и/или сольвата.
Способ введения и схема приема лекарственного средства на основе соединений формулы XXI аналогичны таким же параметрам, указанным для эталонного соединения, которым является суматриптан (Оксфорд, заявка на патент Великобритании N 2162522). Хотя доза и схема приема должны тщательно определяться в каждом случае исходя из профессионального опыта врача с учетом возраста, веса и состояния больного, способа введения и сложности заболевания, суточная доза обычно составляет от 0,05 до 10 мг/кг, предпочтительно от 0,1 до 2 мг/кг при парентеральном введении и от 1 до 50 мг/кг, предпочтительно от 5 до 20 мг/кг при пероральном введении. В некоторых случаях достаточное лечебное воздействие может быть достигнуто при более низких дозах, а в других случаях могут потребоваться более высокие дозы. Системное введение относится к пероральному, назальному, ректальному и парентеральному введению (то есть внутримышечно, внутривенно и подкожно). Следует отметить, что при пероральном введении соединения по настоящему изобретению необходимо большее количество активного средства для достижения такого же эффекта, который обеспечивается меньшим количеством соединения, вводимого назально или парентерально. В соответствии с лечебной практикой предпочтение отдается введению соединений по настоящему изобретению при концентрации, которая обеспечивает эффективное воздействие на мигрень без возникновения каких-либо вредных или нежелательных побочных эффектов.
Соединения по настоящему изобретению можно вводить в целях лечения мигрени в виде отдельных лекарственных средств или в виде смесей с другими лекарственными средствами. В лечебных целях эти соединения обычно применяют в виде фармацевтических составов, включающих необходимое для устранения мигрени количество соединения формулы XXI или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. Предпочтение отдается фармацевтическим составам, содержащим от 1 до 500 мг активного ингредиента в унифицированной дозе, которые обычно готовят в виде таблеток, лепешек, капсул, порошков, водных или масляных суспензий, сиропов, эликсиров и водных растворов.
Характер фармацевтического состава, несомненно, зависит от желаемого способа введения. Например, пероральные составы могут иметь форму таблеток или капсул и могут содержать обычные наполнители, такие как связывающие вещества (например, крахмал) и смачиватели (например, лаурилсульфат натрия). Растворы или суспензии соединения формулы XXI с известными фармацевтическими носителями применяются для изготовления назальных и парентеральных составов, таких как водный раствор для внутривенного вливания или масляная суспензия для внутримышечного вливания.
Описание конкретных вариантов осуществления настоящего изобретения
Соединения, входящие в объем этого изобретения, способы их получения и их биологическое действие станут более понятными при рассмотрении следующих соединений, которые приводятся только в иллюстративных целях и не ограничивают область и объем настоящего изобретения. В следующих примерах, используемых для иллюстрации представленных выше способов синтеза, температуры выражены в градусах по шкале Цельсия, а температуры плавления не скорректированы. Характеристики спектра ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам (δ), выраженным в виде частей на миллион (ppm) по сравнению с тетраметилсиланом (TMS), используемым в качестве эталона. Относительная площадь, указанная для различных сдвигов в данных спектра 1H ЯМР, соответствует числу атомов водорода определенного функционального типа в молекуле. Характер сдвигов с точки зрения мультиплетности выражается в виде широкого синглета (ш.с.), синглета (с.), мультиплета (м.), триплета (т.) или дублета (д.). В примерах применяются такие сокращения, как DMSO-d6 (дейтеродиметилсульфоксид), CDCl3 (дейтерохлороформ), которые являются общепринятыми. Описания инфракрасного спектра (IP) включают только величины абсорбционных волн (см-1), имеющие показатель идентификации функциональной группы. Определения инфракрасного спектра производились с использованием бромида калия (KBr) в качестве разбавителя. Величины элементного анализа указаны в массовых процентах.
В следующих примерах детально описываются способы получения соединений формулы XXI, а также синтез промежуточных продуктов в каждом способе. Специалисту в этой области должно быть ясно, что изменения, вносимые в используемые материалы и методы, позволяют получить другие соединения, рассматриваемые в этой заявке. Описание изобретения и нижеследующие примеры позволяют сделать вывод о том, что специалист в этой области может в полной мере использовать настоящее изобретение.
А. Получение промежуточных соединений
Ниже приведены некоторые типичные процедуры получения синтетических промежуточных соединений, используемых в трех способах, изображенных на схеме 2. Большинство исходных материалов и определенные промежуточные соединения (например, соединения формул II и V) выпускаются промышленностью либо способы их синтеза легко найти в химической литературе, что делает возможным их применение специалистом в области химии органического синтеза.
Соединения формулы VI
Соединения типа: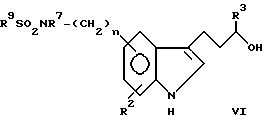
Пример 1 3-/5-Этансульфониламино-1H-индол-3-ил/пропанол
5-Этансульфониламино-1H-индол
К раствору 5-амино-1H-индола (10,0 г, 76 ммоль) и триэтиламина (15,8 мл, 114 ммоль) в 100 мл CH2Cl2 при температуре 0oC по каплям добавляли раствор этансульфонилхлорида (7,9 мл, 83 ммоль) в 25 мл CH2Cl2. Полученный раствор оставляли на 20 ч для медленного нагревания до 23oC. Реакционную смесь концентрировали в условиях вакуума, а остаток растворяли в 400 мл этилацетата. Органический слой промывали 100 мл воды, 50 мл 0,1 М раствора HCl, 50 мл насыщенного раствора NaHCO3 и 50 мл насыщенного раствора NaCl. Органический слой сушили над безводным K2CO3, фильтровали и концентрировали в условиях вакуума с образованием 5-этансульфониламино-1H-индола (16,9 г, > 99%), который использовали без дальнейшей очистки.
5-Метил/этансульфонил/амино-1H-индол
К раствору 5-этансульфонамино-1H-индола (8,96 г, 40 ммоль) в 200 мл безводного тетрагидрофурана при 0oC по каплям добавляли 2,5 М раствор н-BuLi в гексане (17,6 мл, 44 ммоль). После перемешивания в течение 30 мин при 0oC по каплям добавляли метилиодид (6,25 г, 44 ммоль). Реакционную смесь оставляли на 66 ч для нагревания до температуры 23oC, производили перемешивание в течение всего этого периода. Эту смесь выливали в этилацетат, промывали пятью 100 мл порциями 1 н. раствора NaOH и, наконец, насыщенным раствором NaCl. Органический слой сушили над безводным K2CO3, фильтровали и концентрировали в условиях вакуума с образованием 5-метил/этансульфонил/амино-1H-индола (9,52 г, > 99%).
3-/5-Этансульфониламино-1H-индол-3-ил/пропановая кислота
Раствор 5-этансульфониламино-1H-индола (8,0 г, 36 ммоль), акриловой кислоты (5,15 г, 71 ммоль) и уксусного ангидрида (7,3 г, 71 ммоль) в 35 мл уксусной кислоты нагревали при температуре 90oC в течение 20 ч. Летучие вещества удаляли в высоком вакууме при температуре 90oC. Образовавшийся остаток растворяли в 1 н. растворе NaOH, а затем подкисляли до pH 1 концентрированной хлористоводородной кислотой (HCl). После этого водный раствор экстрагировали пятью порциями этилацетата. Органические экстракты соединяли, сушили над безводным MgSO4, фильтровали и концентрировали в условиях вакуума с образованием 3-/5-этансульфониламино-1H-индол-3-ил/пропановой кислоты (10,6 г неочищенного материала). Спектр ЯМР и масс-спектр показали наличие целевого продукта. Полученное вещество не очищали, а сразу же подвергали восстановлению в соответствии с процедурой, описанной ниже.
3-/5-Этансульфониламино-1H-индол-3-ил/пропанол
К раствору неочищенной 3-/5-этансульфониламино-1H-индол-3-ил/пропановой кислоты (10,6 г, 36 ммоль) в 50 мл тетрагидрофурана при температуре 0oC добавляли 1,0 М раствор борана в тетрагидрофуране (161 мл, 161 ммоль). Реакционную смесь оставляли на два часа для нагревания до 23oC. Реакционную смесь охлаждали до 0oC и медленно добавляли 100 мл 5 н. раствора KOH. После выстаивания в течение 16 ч органический слой отделяли, а водную фазу экстрагировали четырьмя порциями тетрагидрофурана. Водную фазу нейтрализовали концентрированной HCl и экстрагировали этилацетатом. Соединенные органические слои сушили над безводным K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография остатка на силикагеле (95 : 5 : 0,5 смеси CH2Cl2, метанола и NH4OH) позволила получить 3-/5-этансульфониламино-1H-индол-3-ил/пропанола (1,76 г, 18%).
Соединения типа: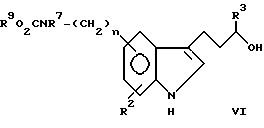
Пример 2. 3-/5-/Фенилметоксикарбонил/амин/-1H-индол-3-ил/пропанол
Фенилметил-/1H-индол-5-ил/карбамат
К раствору 5-аминоиндола (10,0 г, 76 ммоль) и триэтиламина (7,67 г, 76 ммоль) в ацетонитриле (400 мл) при температуре 0oC по каплям добавляли раствор карбоксибензилоксихлорида (12,93 г, 75,8 ммоль) в ацетонитриле (80 мл). После окончания добавления реакционную смесь оставляли для нагревания до 23oC и выстаивали в течение 60 ч. Растворитель удаляли в условиях вакуума, а остаток обрабатывали водой, содержащей Na2CO3 (76 ммоль). Эту смесь экстрагировали четырьмя порциями CH2Cl2. Соединенные органические экстракты промывали насыщенным раствором NaCl, сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь гексана и этилацетата с градиентом от 4:1 до 1:1) концентрата позволила получить фенилметил-/1H-индол-5-ил/карбамат (5,11 г, 25%).
Фенилметил-[3-[/диметиламино/метил]-1H-индол-5-ил]карбамат
К раствору фенилметил-/1H-индол-5-ил/карбамата (3,76 г, 14,1 ммоль) в этаноле (6 мл) добавляли диметиламин (1,75 мл, 40%-ный водный раствор) и формальдегид (1,25 мл, 40%-ный водный раствор). Реакционную смесь нагревали с обратным холодильником в течение 16 ч. Растворитель удаляли в условиях вакуума, а остаток обрабатывали 10%-ным раствором Na2CO3 и экстрагировали четырьмя порциями этилацетата. Соединенные органические экстракты промывали насыщенным раствором NaCl, сушили с помощью Na2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь CH2Cl2, метанола и NH4OH с градиентом от 93:7:0,7 до 90:10:1) указанного концентрата позволила получить фенилметил-[3-[/диметиламино/метил] -1H-индол-5-ил]карбамат (2,30 г, 51%).
Иодид N, N,N-триметил-1-[5-[/фенилметоксикарбонил/амин]-1H- индол-3-ил] метанаминия
К раствору фенилметил[3-[/диметиламино/метил]-1H-индол-5-ил]-карбамата (2,30 г, 7,1 ммоль) в тетрагидрофуране (75 мл) при температуре 0oC по каплям добавляли метилиодид (1,52 г, 10,7 ммоль). Реакционную смесь оставляли для нагревания до 23oC и выстаивали в течение 2 ч. Осадок фильтровали и сушили в условиях вакуума с образованием йодида N,N,N-триметил-1-[5-[/фенилметоксикарбонил/амин]-1H- индол-3-ил]метанаминия (2,94 г, 89%).
Метил-2-метоксикарбонил-3-[5-[/фенилметоксикарбонил/амин] -1H- индол-3-ил]пропионат
К раствору диметилмалоната натрия (1,12 г, 7,3 ммоль) в метаноле (15 мл) добавляли раствор метанола (50 мл), содержащий иодид N,N,N- триметил-1-[5-[/фенилметоксикарбонил/амин] -1H-индол-3-ил] метанаминия. Полученный раствор нагревали с обратным холодильником в течение 24 ч. Растворитель удаляли в условиях вакуума. Концентрат обрабатывали насыщенным раствором NaHCO3 и экстрагировали тремя порциями этилацетата. Соединенные органические экстракты промывали насыщенным раствором NaCl, сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь гексана и этилацетата в отношении 60:40) полученного концентрата привела в образованию метил-2-метоксикарбонил-3[5-[/фенилметоксикарбонил/-амин] -1Н-индол-3-ил] пропионата (1,17 г, 59%), структура которого была подтверждена спектром ЯМР и масс-спектром.
Метил-3-[5-[/фенилметоксикарбонил/амин]-1Н-индол-3-ил]пропионат
К раствору метил-2-метоксикарбонил-3-[5-[/фенилметоксикарбонил/амин]-1Н-индол-3-ил] пропионата (1,17 г, 2,8 ммоль) в пиридине (15 мл) добавляли йодид натрия (0,83 г, 5:5 ммоль), после чего полученный раствор нагревали с обратным холодильником в течение 24 ч. Растворитель удаляли в условиях вакуума. Концентрат обрабатывали насыщенным раствором NaHCO3 и экстрагировали четырьмя порциями этилацетата. Соединенные органические экстракты промывали насыщенным раствором NaCl, сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь гексана и этилацетат в отношении 70:30) указанного концентрата позволила получить метил-3-[5[/фенилметоксикарбонил/амин] -1Н-индол-3-ил]-пропионат (0,47 г, 48%), структура которого была подтверждена спектром ЯМР и масс-спектром.
3-[5-[/фенилметоксикарбонил/амин]-1Н-индол-3-ил]пропанол
К суспензии LiAlH4 (0,08 г, 2 ммоль) в тетрагидрофуране при температуре - 20oC по каплям добавляли раствор метил-3-[5-[/фенилметоксикарбонил/амин] -1Н-индол-3-ил] пропионата (0,47 г, 1,3 ммоль) в тетрагидрофуране (3 мл). После окончания добавления реакционную смесь оставляли для выстаивания при температуре - 20oC в течение 5 ч. Избыток восстановителя разрушали путем добавления по каплям воды (0,1 мл), затем 15%-ного раствора NaOH (0,1 мл) и, наконец, 0,3 мл воды. Полученную смесь оставляли для нагревания до 23oC и выстаивания в течение 16 ч. Реакционную смесь фильтровали через слой целита, а фильтровальный осадок промывали простым этиловым эфиром. Соединенные органические фазы сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь гексана и этилацетата в отношении 1:1) указанного концентрата позволила получить 3-[5-[/фенилметоксикарбонил/амин]-1Н-индол-3-ил]пропанол (0,31 г, 72%), структура которого была подтверждена спектром ЯМР и масс-спектром.
Соединения типа: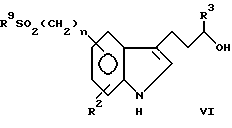
Пример 3. 3-[5-[/Метилсульфонил/метил]-1Н-индол-3-ил]пропанол
1-[2-/Метилсульфонил/метил]-4-нитробензол
Раствор 4-нитробензилбромида (2,16 г, 10 ммоль) и метансульфината натрия (1,12 г, 11 ммоль) в диметилформамиде (25 мл) нагревали с обратным холодильником в течение 0,5 ч. Растворитель удаляли в условиях вакуума, а остаток экстрагировали CH2Cl2 и водой. Соединенные органические фазы промывали насыщенным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали в условиях вакуума с образованием 1-[2-/метилсульфонил/метил]-4-нитробензола (1,54 г, 71,6%), который использовали без дальнейшей очистки
1-[2-/Метилсульфонил/этил]-4-нитробензол
Это соединение получали аналогичным образом из 1-/2-бромэтил/-4-нитробензола и метансульфината натрия.
4-амино-1-[2-/метилсульфонил/метил]бензол
Суспензию 1-[2-/метилсульфонил/метил]-4-нитробензола (27 г, 126 ммоль) и концентрированной HCl (5 мл) в 300 мл 66% этанола (водный раствор) гидрировали (под давлением 50 фунтов на кв. дюйм) в присутствии катализатора на основе 10% палладированного угля (4 г), выполняя эту операцию при температуре 23oC в течение 16 ч. Катализатор удаляли фильтрованием, а фильтрат концентрировали в условиях вакуума с целью удаления этанола. Оставшуюся водную фазу подщелачивали до реакции на лакмусе путем добавления 50%-ного раствора NaOH, в результате чего происходило осаждение продукта, представляющего 4-амсино-1-[2-/метилсульфонил/метил]бензол (21,54 г, 93%).
1-[2-/5-Гидроксипентилиден/гидразинил]-4-[/метилсульфонил/-метил]бензол.
К концентрированному раствору 4-амино-1-[/метилсульфонил/метил]бензола (8,0 г, 43 ммоль) в хлористоводородной кислоте (40 мл) добавляли воду (40 мл). Реакционную смесь охлаждали до 0oC, после чего по каплям добавляли раствор, содержащий NaNO3 (3,58 г, 51 ммоль) в воде (10 мл). Реакционную смесь в течение 1 ч перемешивали при температуре 0oC, а затем выливали в раствор SnCl2 (48,78 г, 216 ммоль) в 6 н. растворе HCl (160 мл) при температуре -40oC. Реакционную смесь оставляли для нагрева до 23oC и с помощью 50%-ного раствора NaOH доводили показатель pH до 2. Добавляли этанол (300 мл) и дигидропиран (4,73 г, 56 ммоль), после чего реакционную смесь перемешивали в течение 16 ч. Эту смесь подщелачивали 50%-ным раствором NaOH (pH 10), а затем фильтровали через целит. Полученный раствор концентрировали в условиях вакуума, и оставшуюся водную фазу экстрагировали этилацетатом. Соединенные органические слои промывали водой, сушили над MgSO4, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь CH2Cl2, метанола и NH4OH в отношении 95:5:0,5) указанного концентрата позволила получить целевой гидразон (4,34 г, 38%).
3-[5-[/Метилсульфонил/метил]-1Н-индол-3-ил]пропанол
Раствор 1,2-диметоксиэтана, содержащий 1-[2-/5-гидроксипентилиден/гидразинил] -4-[/метилсульфонил/]бензол (4,74 г, 17 ммоль) и ZnCl2 (11,4 г, 84 ммоль) нагревали с обратным холодильником в течение 24 ч. Реакционную смесь концентрировали в условиях вакуума, а остаток растворяли в воде и экстрагировали этилацетатом. Соединенные органические слои промывали водой, сушили над MgSO4, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (смесь CH2Cl2, метанола и NH4OH с соотношением 95:5: 0,5) указанного концентрата позволила получить 3-[5-[/метилсульфонил/метил] -1Н-индол-3-ил]пропанол (1,23 г, 28%).
Соединения типа: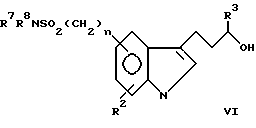
Пример 4. 3-/3-Гидроксипропил/-N-метил-1Н-индол-5-метансульфонамид
4-[2-/5-Гидроксипентилиден/гидразинил]-N-метилбензолметансульфонамид
К суспензии 14,0 г (69,9 ммоль, 1,0 экв.) 4-//N-метил/-метансульфонамидо/метил/анилина в 140 мл концентрированного водного раствора хлористоводородной кислоты и 70 мл воды при температуре 0oC добавляли 4,82 г нитрита натрия в 70 мл воды. Эту смесь перемешивали 15 мин при температуре 0oC. Тем временем готовили раствор 78,9 г хлористого олова в 140 мл концентрированного водного раствора хлористоводородной кислоты и охлаждали его до температуры -45oC. Раствор соли диазония фильтровали в раствор хлористого олова. Реакционную смесь оставляли на один час для нагревания до 0oC, а затем осторожно доводили показатель pH до 3,5 путем добавления твердого гидроксида калия. Реакционную смесь разбавляли 420 мл этанола и добавляли 7,65 мл (83,9 ммоль, 1,2 экв.) 3,4-дигидро-2Н-пирана. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Показатель pH далее увеличивали до 10 путем добавления твердого гидроксида калия. Смесь фильтровали и удаляли этанол в условиях вакуума. Оставшийся водный слой трижды экстрагировали 500 мл порциями этилацетата. Соединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали с образованием масла. Это масло хроматографировали на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 7,85 г (38%) 4-[2/5-гидроксипентилиден/гидразинил]-N-метил-бензолметансульфонамида.
3-/3-Гидроксипропил/-N-метил-1H-индол-5-метансульфонамид
К раствору 23,6 г (173 ммоль, 5,0 экв.) хлористого цинка в 1000 мл безводного 1,2-диметоксиэтана в атмосфере азота добавляли раствор 10,37 (34,65 ммоль, 1,0 экв.) 4-[2-/5-гидроксипентилиден/гидразинил]-N-метилбензолметансульфонамида в 200 мл безводного 1,2-диметоксиэтана. Этот раствор нагревали до температуры кипения с обратным холодильником в течение 30 ч. Реакционную смесь охлаждали до комнатной температуры, и полученный объем концентрировали до 500 мл в условиях вакуума, остаток разбавляли 1000 мл этилацетата и промывали 200 мл порциями воды и насыщенного водного раствора хлорида натрия. Органический слой сушили над сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло очищали с помощью хроматографии на силикагеле при использовании 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 5,16 г (53%) 3-/3-гидроксипропил/-N-метил-1H-индол-5-метансульфонамида.
Пример 5. Альтернативный способ синтеза (по способу N 3 схемы 2)
4-Амино-3-иод-N-метилбензолметансульфонамид
К суспензии 1,06 г (5,31 ммоль, 1,0 экв.) 4-амино-N-метилбензолметансульфонамида в 20 мл ацетонитрила добавляли 0,862 г (5,31 ммоль, 1,0 экв.) однохлористого йода. Реакционную смесь 15 мин перемешивали при комнатной температуре. Эту смесь распределяли между 25 мл этилацетата и 15 мл 20%-ного водного раствора тиосульфата натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло фильтровали через набивку из силикагеля с использованием смеси 40% этилацетата в гексане с образованием 1,13 г (65%) 4-амино-3-иод-N-метилбензолметансульфонамида.
1-трет-Бутилдиметилсилилокси-4-пентин
К суспензии 18,0 г (749 ммоль, 1,05 экв.) гидрида натрия в 500 мл тетрагидрофурана при температуре 0oC добавляли раствор 60,0 г (713 ммоль, 1,00 экв. ) 4-пентин-1-ола в 150 мл тетрагидрофурана. После окончания добавления реакционную смесь оставляли на один час для нагревания до комнатной температуры. К этой смеси добавляли 113 г (749 ммоль, 1,05 экв.) трет-бутилдиметилсилилхлорида в 150 мл тетрагидрофурана. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем разбавляли 1200 мл гексана. Органический слой промывали 500 мл насыщенного водного раствора бикарбоната натрия, сушили над безводным сульфатом натрия и концентрировали в условиях вакуума. Полученную жидкость перегоняли при температуре 39-42oC и давлении 0,05 мм рт.ст., в результате чего образовалось 135 г (96%) 1-трет-бутилдиметилсилилокси-4-пентина.
1-трет-Бутилдиметилсилилокси-5-триметилсилил-4-пентин
К раствору 135 г (682 ммоль, 1,0 экв.) 1-трет-бутилдиметилсилилокси-4-пентина в 700 мл тетрагидрофурана при температуре -78oC добавляли 311 мл (716 ммоль, 1,05 экв.) 2,3 М раствора н-бутиллития в гексане. Удаляли охлажденную ванну, поддерживающую температуру на уровне -78oC, и контролировали температуру реакционной смеси по мере ее повышения до 0oC. Реакционную смесь вновь охлаждали до -78oC и по каплям добавляли 90,8 мл (716 ммоль, 1,05 экв. ) триметилсилилхлорида. Реакционную смесь оставляли на 16 ч для медленного нагревания в холодной ванне. Реакционную смесь разбавляли 2000 мл гексана, промывали 500 мл насыщенного водного раствора бикарбоната натрия, сушили над безводным сульфатом натрия и концентрировали в условиях вакуума. Полученную жидкость перегоняли при температуре 71 - 74oC и давлении 0,05 мм рт. ст. с образованием 168 г (91%) 1-трет-бутилдиметилсилилокси-5-триметилсилил-4-пентина.
3-/3-трет-Бутилдиметилсилилоксипропил/-2-триметилсилил-N-метил-1Н- индол-5-метансульфонамид
К раствору 0,326 г (1,0 ммоль, 1,0 экв.) 4-амино-3-йод-N-метилбензолметансульфонамида в 20 мл диметилформамида добавляли 0,541 г (2,0 ммоль, 2,0 экв. ) 1-трет-бутилдиметилсилилокси-5-триметилсилил-4-пентина, 0,278 г (1,0 ммоль, 1,0 экв. ) хлорида тетрабутиламмония, 0,530 г (5,0 ммоль, 5,0 экв.) карбоната натрия, 0,0131 г (0,05 ммоль, 0,05 экв.) трифенилфосфина и 0,0112 г (0,05 ммоль, 0,05 экв.) ацетата палладия (II). Эту смесь нагревали в масляной бане с температурой 100oC в течение 16 ч. Полученную смесь охлаждали до комнатной температуры и в условиях вакуума удаляли диметилформамид. Остаток разбавляли 75 мл этилацетата и промывали 25 мл насыщенного водного раствора бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло фильтровали через набивку из силикагеля с использованием смеси 25% этилацетата в гексане, в результате чего было получено 0,411 г (88%) 3-/3-трет-бутилдиметилсилилоксипропил/-2-триметилсилил-N-метил-1H-индол- 5-метансульфонамида.
3-/3-Гидроксипропил/-2-триметилсилил-N-метил-1Н-индол-5-метансульфонамид
К раствору 0,0738 г (0,157 ммоль, 1,0 экв.) 3-/3-трет-бутилдиметилсилилоксипропил/-2-триметилсилил-N-метил-1H-индол-5-метансульфонамида в 2 мл пиридина при температуре 0oC добавляли 1 мл 48%-ного водного раствора фтористоводородной кислоты. Полученный раствор 20 мин перемешивали при температуре 0oC. Этот раствор разбавляли 25 мл этилацетата и промывали 10 мл 10%-ного водного раствора карбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума, в результате чего было получено 0,0551 г (99%) 3-/3-гидроксипропил/-2-триметилсилил-N-метил-1H-индол-5-метансульфонамида, однородность которого была подтверждена спектром 1H ЯМР и тонкослойной хроматографией.
3-/3-Гидроксипропил/-N-метил-1H-индол-5-метансульфонамид
К раствору 0,0551 г (0,155 ммоль, 1,0 экв.) /3-/3-гидроксипропил/-2-триметилсилил-N-метил-1H-индол-5 -метансульфонамида в 2 мл метиленхлорида при температуре 0oC добавляли 0,01 мл (0,155 ммоль, 1,0 экв.) трифторуксусной кислоты. Через 15 мин реакционную смесь оставляли для нагревания до комнатной температуры. По истечении в общей сложности тридцати минут реакционную смесь выливали в 15 мл этилацетата и промывали 5 мл насыщенного водного раствора бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием 0,0413 г (95%) 3-/3-/гидроксипропил/-N-метил-1Н-индол-5- метансульфонамида, однородность которого была подтверждена спектром 1H ЯМР и тонкослойной хроматографией.
Пример 6. 3-/3-Гидроксипропил/-N-N-диметил-1H-индол-5-метансульфонамид
4-Нитробензилсульфонат натрия
Суспензию 100 г (463 ммоль, 1,0 экв.) 4-нитробензилбромида и 64,2 г (509 ммоль, 1,1 экв.) сульфита натрия в 500 мл метанола и 500 мл воды нагревали до температуры кипения с обратным холодильником. Ход реакции контролировали посредством тонкослойной хроматографии. После того как бромид был израсходован, реакционную смесь оставляли для охлаждения до комнатной температуры. Осажденный продукт собирали фильтрованием. Его тщательно сушили в течение 16 ч при температуре 65oC и давлении 0,05 мм рт. ст., в результате чего было получено 100 г (90%) 4-нитробензилсульфоната натрия.
4-Нитробензилсульфонилхлорид
К суспензии 28,3 г (118 ммоль, 1,0 экв.) тонко измельченного 4-нитробензилсульфоната натрия в 250 мл толуола порциями добавляли 24,6 г (118 ммоль, 1,0 экв.) тонко измельченного пентахлорида фосфора в виде твердого вещества. После окончания добавления эту смесь нагревали до температуры кипения с обратным холодильником в течение одного часа. Эту смесь охлаждали, а летучие вещества удаляли в условиях вакуума. Остаток растворяли в 500 мл хлороформа и 250 мл воды. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с образованием 27,8 г (100%) 4-нитробензилсульфонилхлорида, чистота которого была подтверждена спектром 1H ЯМР.
4-Нитро-N-N-диметилбензолметансульфонамид
Безводный диметиламин барботировали через 250 мл хлороформа при температуре 0oC в течение 30 мин. При температуре 0oC по каплям добавляли раствор 27,8 г (118 ммоль, 1,0 экв.) 4-нитробензилсульфонилхлорида. Происходила экзотермическая реакция, после чего эту смесь оставляли для нагревания до комнатной температуры. Полученную смесь концентрировали в условиях вакуума с образованием 32,2 г 4-нитро-N-N-диметилбензолметансульфонамида, загрязненного гидрохлоридом диметиламина. Эту смесь использовали на последующей стадии процесса без очистки.
4-Амино-N-N-диметилбензолметансульфонамид
К раствору 22,3 г неочищенного 4-нитро-N,N-диметилбензолметансульфонамида (загрязненного гидрохлоридом диметиламина, как это указывалось выше), в 45 мл 2 н. раствора хлористоводородной кислоты, 100 мл этанола и 200 мл воды добавляли 5,0 г 10% палладированного угля. Полученную смесь гидрировали в гидрогенизаторе Парра при давлении от 60 до 50 фунтов на кв. дюйм в течение трех часов. Эту смесь фильтровали и удаляли этанол в условиях вакуума. Полученный водный раствор обрабатывали 15%-ным водным раствором гидроксида калия. При показателе pH, равном 4, начинал формироваться осадок. Добавление гидроксида калия продолжали до тех пор, пока показатель pH не достигал 9. Образовавшийся продукт отделяли фильтрованием и сушили в течение 16 ч при температуре 65oC и давлении 0,05 мм рт. ст., в результате чего было получено 21,0 г (83%, две стадии) 4-амино-N,N-диметилбензолметансульфонамида.
Гидрохлорид 4-гидразинил-N-N-диметилбензолметансульфонамида
К суспензии 4,30 г (20,1 ммоль, 1,0 экв.) 4-амино-N,N-диметилбензолметансульфонамида в 40 мл концентрированного водного раствора хлористоводородной кислоты и 20 мл воды, охлажденной до 0oC, по каплям добавляли раствор 1,38 г (20,1 ммоль, 1,0 экв.) нитрита натрия в 20 мл воды. Во время добавления реакционная смесь становилась однородной. Реакционную смесь перемешивали 15 мин при температуре 0oC после окончания добавления. Тем временем готовили раствор 22,6 г (100 ммоль, 5,0 экв.) дигидрата хлористого олова в 40 мл концентрированного водного раствора хлористоводородной кислоты и охлаждали его до -40oC. Раствор соли диазония фильтровали в раствор хлористого олова. Реакционную смесь оставляли на один час для нагревания до комнатной температуры. Затем реакционную смесь вновь охлаждали до 0oC и собирали осажденный продукт фильтрованием. Твердое вещество сушили при температуре 65oC и давлении 0,05 мм рт.ст. в течение 45 мин с образованием 5,02 г (94%) гидрохлорида 4-гидразинил-N,N-диметилбензолметансульфонамида.
4-[2-/5-Гидроксипентилиден/гидразинил] -N,N- диметилбензолметансульфонамид
К раствору 5,02 г (18,9 ммоль, 1,0 экв.) 4-гидразинил- N,N-диметилбензолметансульфонамида в 20 мл воды и 20 мл этанола добавляли ацетат натрия в количестве, достаточном для повышения показателя pH до 4. К полученной суспензии добавляли 1,90 мл (20,8 ммоль, 1,1 экв.) 3,4-дигидро-2H-пирана. Полученную реакционную смесь перемешивали в течение шестнадцати часов, а затем разбавляли 200 мл этилацетата. Органический слой промывали 10%-ным водным раствором карбоната калия, сушили над безводным сульфатом натрия и концентрировали в условиях вакуума с образованием масла. Хроматография на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, позволила получить 1,67 г (28%) чистого 4-[2-/5-гидроксипентилиден/гидразинил]-N,N- диметилбензолметансульфонамида в виде масла.
3-/3-Гидроксипропил/-N,N-диметил-1H-индол-5-метансульфонамид
К раствору 3,63 г (26,6 ммоль, 5,0 экв.) хлористого цинка в 150 мл безводного 1,2-диметоксиэтана в атмосфере азота добавляли раствор 1,67 г (5,33 ммоль, 1,0 экв.) 4-[2-/5-гидроксипентилиден/гидразинил]- N,N-диметилбензолметансульфонамида в 50 мл безводного 1,2-диметоксиэтана. Полученный раствор нагревали до температуры кипения с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали ее объем до 25 мл в условиях вакуума. Остаток разбавляли 100 мл этилацетата и промывали 20 мл порциями воды и насыщенного водного раствора хлорида натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием масла. Полученное масло очищали посредством хроматографии на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 0,409 г (26%) 3-/3-гидроксипропил/-N,N-диметил-1H-индол-5-метансульфонамида.
Соединения типа: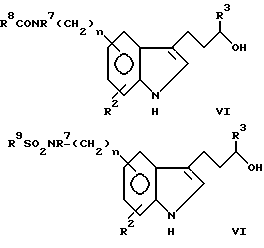
Пример 7. N-[[3-/3-Гидроксипропил/-1H-индол-5-ил]метил]метансульфонамид
N-[/4-Нитрофенил/метил]метансульфонамид
К раствору 25,0 г (133 ммоль, 1,0 экв.) гидрохлорида 4-нитробензиламина в 500 мл метиленхлорида при температуре 0oC добавляли 46,4 мл (331 ммоль, 2,5 экв. ) триэтиламина, после чего по каплям добавляли 11,3 мл (146 ммоль, 1,1 экв.) метансульфонилхлорида. Реакционную смесь тридцать минут перемешивали при температуре 0oC, а затем разбавляли 500 мл хлороформа. Органический слой промывали 150 мл порциями 1 н. раствора HCl и насыщенного хлорида натрия. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с образованием 25,8 г (85%) N-[/4-нитрофенил/-метил]метансульфонамида.
N-[/4-Аминофенил/метил]метансульфонамид
К суспензии 25,9 г (113 ммоль, 1,0 экв.) N-[/4-нитрофенил/метил]метансульфонамида в 113 мл 1 н. раствора HCl (113 ммоль, 1,0 экв.), 113 мл воды и 225 мл этанола добавляли 5,18 г 10% палладированного угля. Полученную смесь гидрировали в гидрогенизаторе Парра при давлении 60 фунтов на кв. дюйм в течение 16 ч. Эту смесь фильтровали через целит. Этанол удаляли в условиях вакуума и доводили показатель pH до 9, когда начиналось осаждение продукта. Осадок удаляли фильтрованием и сушили в условиях вакуума с образованием 18,0 г (80%) N-[/4-аминофенил/метил]метансульфонамида.
Гидрохлорид N-[/4-гидразинилфенил/метил]метансульфонамида
К суспензии 18,0 г (89,9 ммоль, 1,0 экв.) N-[/4-аминофенил/ метил]метансульфонамида в 85 мл воды и 175 мл концентрированного водного раствора хлористоводородной кислоты при температуре 0oC добавляли раствор 6,21 г (89,9 ммоль, 1,0 экв.) нитрита натрия в 85 мл воды. Реакционную смесь перемешивали в течение 15 мин. Тем временем готовили раствор 101,6 г дигидрата хлористого олова в 175 мл концентрированного водного раствора хлористоводородной кислоты и охлаждали его до -55oC. Раствор соли диазония фильтровали в раствор хлористого олова. Реакционную смесь в течение одного часа выдерживали при температуре от -55oC до -35oC. Кристаллическое вещество собирали фильтрованием и сушили в условиях высокого вакуума при температуре 65oC в течение 16 ч, в результате чего было получено 23,9 г (> 100%) неочищенного гидрохлорида N-[/4- гидразинилфенил/метил]метансульфонамида.
N-[[4-[2-/5-Гидроксипентилиден/гидразинил]фенил]метил]- метансульфонамид
К раствору 23,9 г неочищенного гидрохлорида N-[/4-гидразинил/ фенил/метил] метансульфонамида в 175 мл воды и 300 мл этанола добавляли ацетат натрия в количестве, достаточном для доведения показателя pH до 4. К полученной суспензии добавляли 10,4 мл (114 ммоль, 1,3 экв.) 3,4-дигидро-2H-пирана. Затем эту смесь перемешивали при комнатной температуре в течение 20 ч. Этанол удаляли в условиях вакуума. Показатель pH повышали до 10 путем добавления твердого карбоната калия и трижды экстрагировали эту смесь 50 мл порциями метиленхлорида. Соединенные органические слои промывали 25 мл насыщенного водного раствора хлорида натрия, сушили над сульфатом магния и фильтровали с образованием масла. Полученное масло очищали посредством хроматографии на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 11,1 г (39%, две стадии) чистого N-[[4-[2-/5-гидроксипентилиден/гидразинил]фенил]метил]метансульфонамида.
N-[[3-/3-Гидроксипропил/-1H-индол-5-ил]метил]метансульфонамид
К раствору 25,2 г (185 ммоль, 5,0 экв.) хлористого цинка в 900 мл безводного 1,2-диметоксиэтана в атмосфере азота добавляли раствор 11,1 г (37,0 ммоль, 1,0 экв.) N-[[4-[2-/5-гидроксипентилиден/гидразинил]фенил]метил] метансульфонамида. Раствор нагревали с обратным холодильником в течение 48 ч, а затем охлаждали до комнатной температуры. Реакционную смесь фильтровали и концентрировали в условиях вакуума. Остаток растворяли в 500 мл этилацетата и промывали 100 мл порциями воды и насыщенным водным раствором хлорида натрия. Органический слой сушили над сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием 11,0 г масла. Это масло очищали посредством хроматографии на силикагеле с применением градиентного элюирования при использовании от 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, до 10% метанола в метиленхлориде, содержащем 1% концентрированного водного раствора гидроксида аммония, в результате чего было получено 6,97 г (67%) чистого N-[[3-/3-гидроксипропил/-1H-индол-5-ил]метил]метансульфонамида.
Замена алкилсульфонилхлорида анилгалогенидом на стадии 1 вышеуказанной последовательности операций позволяет получить ацильный аналог сульфонильного продукта.
Соединения типа: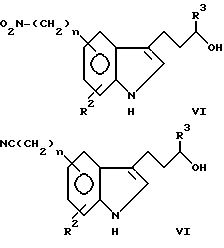
Пример 8. /5-Циано-1H-индол-3-ил/пропанол
5-Циано-3-/диметиламино/метил-1H-индол
К раствору 14,0 г (98,5 ммоль, 1,0 экв.) 5-цианоиндола в 35 мл этанола добавляли 8,7 мл (108 ммоль, 1,1 экв.) 40%-ного водного раствора формальдегида и 12,2 мл (108 ммоль, 1,1 экв.) 40%-ного водного раствора диметиламина. Колбу оснащали обратным холодильником, поверх которого устанавливали конденсатор холодного орошения, действующий при температуре -78oC. Раствор нагревали до температуры кипения с обратным холодильником в течение 8 ч, после чего тонконслойная хроматография показала отсутствие исходного материала. Полученный раствор выливали в 350 мл хлороформа и промывали один раз 50 мл 10%-ного водного раствора карбоната натрия. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с образованием 19,6 г (100%) 5-циано-3-/диметиламино/метил-1H-индола, чистота которого была подтверждена спектром 1H ЯМР.
Иодид N,N,N-триметил-1-/5-циано-1H-индол-3-ил/метанаминия
К раствору 2,7 г (13,6 ммоль, 1,0 экв.) 5-циано-3-/диметиламино/метил-1H-индола в 125 мл тетрагидрофурана при температуре 0oC по каплям добавляли 1,3 мл (20,3 ммоль, 1,5 экв.) метил-иодида. Через пять минут полученный раствор оставляли для нагревания до комнатной температуры, производя перемешивание в течение одного часа. Полученный осадок фильтровали и сушили в условиях вакуума с образованием 4,4 г (96%) йодида N,N,N-триметил-1-/5-циано-1H-индол-3-ил/метанаминия, чистота которого была подтверждена спектром 1H ЯМР.
Метил-2-метоксикарбонил-3-/5-циано-1H-индол-3-ил/пропаноат
К раствору 21,5 г (63,0 ммоль, 1,0 экв.) иодида N,N,N-триметил-1-/5-циано-1H-индол-3-ил/метанаминия в 300 мл метанола добавляли 14,6 г (94,5 ммоль, 1,5 экв. ) диметилнатрмалоната. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч. Раствор охлаждали до комнатной температуры и разбавляли 1000 мл хлороформа. Органический слой один раз промывали 250 мл насыщенного водного раствора бикарбоната натрия. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток фильтровали через набивку из силикагеля с использованием смеси этилацетата и гексана в отношении 1:1 с целью удаления непрореагировавшего диметилмалоната. Это позволило получить 18,95 г (100%) метил-2-метоксикарбонил-3-/5-циано-1H-индол-3-ил/пропаноата.
Метил-3-/5-циано-1H-индол-3-ил/пропаноат
К раствору 1,14 г (3,98 ммоль, 1,0 экв.) метил-2-метоксикарбонил-3-/5-циано-1H-индол-3-ил/пропаноата в 20 мл пиридина добавляли 1,19 г (7,96 ммоль, 2,0 экв. ) иодида натрия. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч. Раствор охлаждали и разбавляли 150 мл хлороформа. Органический слой один раз промывали 25 мл насыщенного водного раствора бикарбоната натрия. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток фильтровали через набивку из силикагеля с использованием смеси этилацетата и гексана в отношении 1:1 с целью удаления окрашенных примесей, в результате чего было получено 0,734 г (81%) метил-3-/5-циано-1H-индол-3-ил/пропаноата.
/5-Циано-1H-индол-3-ил/пропанол
К суспензии 0,184 г (4,86 ммоль, 2,0 экв.) алюмогидрида лития в 10 мл тетрагидрофурана при температуре 0oC по каплям добавляли раствор 0,555 г метил-3-/5-циано-1H-индол-3-ил/пропаноата. Полученную суспензию перемешивали тридцать минут при температуре 0oC, а затем резко охлаждали 0,18 мл воды, 0,18 мл 15%-ного водного раствора гидроксида натрия и 0,54 мл воды. Образовавшуюся суспензию фильтровали через целит. Раствор концентрировали, а остаток фильтровали через набивку из силикагеля с использованием смеси этилацетата и гексана в отношении 1:1, что позволило получить 0,470 г (97%) /5-циано-1H-индол-3-ил/пропанола.
Пример 9. 5-Нитро-3-/3-гидроксипропил/индол
5-/5-Нитроиндол-3-ил-метил/-2,2-диметил-1,3-диоксан-4,6-дион
Применяли вариант процедуры, описанный Флау1. Таким образом, раствор 5-нитроиндола (50,0 г, 0,31 моль), кислоты Мелдрума (46,0 г, 0,32 моль), 37%-ного водного раствора формальдегида (26,0 мл, 0,32 моль) и пролина (1,8 г, 0,016 моль) в 200 мл ацетонитрила перемешивали при комнатной температуре в течение 18 ч. Образовавшуюся густую суспензию желтого цвета фильтровали, после чего фильтровальный осадок промывали ацетонитрилом, затем ацетоном и, наконец, простым эфиром. Полученное вещество сушили в условиях вакуума с образованием указанного в заголовке соединения (80,0 г, 81%) в виде ярко-желтого твердого вещества, температура плавления 182oC (разложение). Маточный раствор концентрировали, а затем разбавляли H2O, образовавшееся твердое вещество собирали, промывали и сушили так, как указывалось выше. Это позволило получить сбор продукта (7,0 г) в виде более темного желтого твердого вещества. Общий выход 87,0 г (89%).
Этил-5-нитро-3-индолпропионат
К раствору 5-/5-нитроиндол-3-ил-метил/2,2-диметил-1,3-диоксан-4,6-диона (10,0 г, 0,031 моль) в смеси пиридина (80 мл) и абсолютного этанола (20 мл) добавляли 0,1 г медного порошка, после чего эту смесь нагревали с обратным холодильником в атмосфере Ar в течение 2 ч. После перемешивания при комнатной температуре в течение 66 ч эту смесь нагревали с обратным холодильником еще в течение 1 ч. Охлажденную смесь фильтровали, а фильтрат выпаривали. Полученный остаток истирали в порошок с простым эфиром и небольшим количеством CH2Cl2, в результате чего было получено указанное в заголовке соединение (7,3 г, 89%) в виде твердого вещества, температура плавления 118-121oC.
5-Нитро-3-/3-гидроксипропил/индол
К суспензии 95% LiAlH4 (2,20 г, 0,058 моль) в 60 мл сухого тетрагидрофурана при температуре 0oC в атмосфере Ar добавляли раствор этил-5-нитро-3-индолпропионата (7,30 г, 0,028 моль) в 100 мл сухого тетрагидрофурана. После перемешивания в течение 20 мин эту смесь резко охлаждали путем осторожного добавления 3 мл H2O. Образовавшуюся суспензию перемешивали в течение 10 мин, после чего ее фильтровали, а фильтровальный осадок промывали дополнительным количеством тетрагидрофурана. Фильтрат выпаривали, остаток растворяли в простом эфире, сушили (Na2SO4) и выпаривали. Образовавшееся твердое вещество истирали в порошок с гексаном, что позволило получить указанное в заголовке соединение (4,30 г, 70%) в виде твердого вещества желтого цвета, температура плавления 107-110oC.
Пример 10. 1-/5-Ацетил-1H-индол-3-ил/-3-пропанол
4-Амино-3-йод-ацетофенон
К раствору, содержащему 4-аминоацетофенон (4,05 г, 30 ммоль) в ледяной уксусной кислоте (60 мл) и воду (10 мл), по каплям добавляли раствор однохлористого йода (4,97 г, 31 ммоль) в уксусной кислоте (15 мл). После окончания добавления реакционную смесь пять минут нагревали при температуре 90oC, а затем охлаждали до 23oC и оставляли для выстаивания на один час. Избыток ICl устраняли путем добавления насыщенного бисульфита натрия (15 мл). Растворитель удаляли в условиях вакуума, а остаток растворяли в CH2Cl2, промывали насыщенным раствором NaCl и, наконец, водой. Органическую фазу сушили с помощью Na2SO4, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (градиент смеси гексана и этилацетата; 10-50% этилацетата) позволила получить 4-амино-3-иод-ацетофенон (6,15 г, 82,3%).
5-Ацетил-3-[3-/трет-бутилдиметилсилокси/пропил] -2-триметилсилил- -1H-индол
К раствору 4-амино-3-иод-ацетофенона (6,15 г, 25 ммоль) в 1,2-диметоксиэтане (250 мл) добавляли насыщенный раствор Na2CO3 (20 мл), 1-триметилсилил-5-трет-бутилдиметилсилоксипент-1-ин (13,36 г, 49 ммоль) и Pd(PPh3)4 (2,85 г, 2 ммоль). После нагревания полученной смеси с обратным холодильником в течение 48 ч удаляли растворитель в условиях вакуума. Остаток растворяли в этилацетате и промывали насыщенным раствором NaCl, а затем водой. Органическую фазу сушили над MgSO4, фильтровали и концентрировали в условиях вакуума. Хроматография на силикагеле (градиент смеси гексана и этилацетата; 10-50% этилацетата) и кристаллизация из смеси CH2Cl2 и гексана позволили получить 5-ацетил-3-[3-/трет-бутилдиметилсилокси/пропил] -2-триметилсилил-1H-индол (5,76 г, 58%).
1-[5-Ацетил-1H-индол-3-ил]-3-пропанол
К раствору 5-ацетил-3-[3-/трет-бутилдиметилсилокси/пропил]-2- -триметилсилил-1H-индола (1,8 г, 5 ммоль) в ацетонитриле (100 мл) добавляли 50%-ный раствор фтороводорода (4 мл). После перемешивания в течение 24 ч при температуре 23oC реакционную смесь подщелачивали (pH 10) путем добавления 50%-ного раствора NaOH и концентрировали в условиях вакуума. Остаток растворяли в этилацетате и промывали насыщенным раствором NaCl, а затем водой. Органическую фазу сушили над MgSO4, фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь CH2Cl2, метанола и NH4OH с отношением 95:5:0,5) позволила получить 1-[5-ацетил-1H-индол-3-ил]-3-пропанол (0,91 г, 94%).
Пример 11. 5-Фтор-3-/3-гидроксипропил/индол
5-Фториндол-3-пропионовая кислота
Применяли модифицированный вариант процедуры, описанной Джонсоном (Г.И. Джонсон и Д.Дж. Кросби, J, Org. Chem., 25, 569, 1969) для получения индол-3-пропионовой кислоты.
Так, раствор 5-фториндола (1,35 г, 0,010 моль) в 10 мл уксусной кислоты, содержащей акриловую кислоту (1,5 мл, 0,022 моль) и уксусный ангидрид (1,9 мл, 0,02 моль), нагревали (масляная баня) в атмосфере аргона при 90oC в течение 5 дней. Летучие вещества удаляли в условиях вакуума, а остаток растворяли в 3 н. растворе NaOH. Нерастворимое вещество удаляли фильтрованием, фильтрат подкисляли концентрированной HCl, а затем экстрагировали CH2Cl2. Органический экстракт сушили (Na2SO4) и выпаривали с образованием продукта (1,191 г, 57%) в виде твердого вещества, которое использовали без дальнейшей очистки: инфракрасный спектр (в чистом виде) 3420, 1710 см-1;
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 7,94 (широкий синглет, 1H); 7,28-7,18 (м., 3H); 7,05 (д., J = 2,5 Гц, 1H); 6,93 (двойной триплет, J = 9,0, 2,6 Гц, 1H); 3,05 (т., J = 7,5 Гц, 2H); 2,73 (т., J = 7,7 Гц, 2H).
Путем внесения соответствующих изменений в общий метод можно легко получить другие соединения формулы II.
5-Фтор-3-/3-гидроксипропил/индол
К суспензии LiAlH4 (433 мг, 11,4 ммоль) в 20 мл сухого тетрагидрофурана в атмосфере аргона при температуре 5-10oC добавляли раствор 5-фториндол-3-пропионовой кислоты (1,179 г, 5,7 ммоль) в 5 мл тетрагидрофурана. Через 10 мин удаляли охлаждающую ванну, смесь перемешивали при комнатной температуре в течение 30 мин, а затем 30 мин нагревали до температуры кипения с обратным холодильником. Образовавшуюся смолистую смесь оставляли для охлаждения до комнатной температуры, после чего реакционную смесь резко охлаждали в результате последовательного добавления 0,5 мл H2O, 0,5 мл 15%-ного раствора NaOH и, наконец, 1,5 мл H2O. Затем эту смесь разбавляли этилацетатом, сушили (MgSO4) и выпаривали с образованием желто-зеленого масла. Испарительная хроматография (силикагель, смесь CH2Cl2 и этилацетата с отношением 2:1) позволила получить целевой продукт (918 мг, 83%) в виде масла: инфракрасный спектр (в чистом виде) 3420, 1583 см-1;
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 7,94 (широкий синглет, 1H); 7,28 - 7,20 (м., 2H); 7,03 (д., J = 2,4 Гц, 1H); 6,92 (двойной триплет, J = 9,1 2,5 Гц, 1H); 3,71 (т., J = 6,4 Гц, 2H); 2,80 (т., J = 7,5 Гц, 2H); 2,02 - 1,88 (м., 2H); 1,33 (широкий синглет, 1H).
Пример 12. Этил-3-[3-гидроксипроп-1-ил]-1H-индол-5-карбоксилат
Этил-3-[3-[[/1,1-диметилэтил/диметилсилил] окси] проп-1-ил] -2-триметилсилил -1H-индол-5-карбоксилат
Смесь этил-4-амино-3-иод-бензоата (Гиршфелд и др., J. Med. Chtm, 1992, 35, 2231 - 2238) (7,80 г, 26,8 ммоль), [5-[[/1, 1-диметил-этил/диметилсилил] окси] -1-пентинил] триметилсилана (9,41 г, 34,8 ммоль), хлорида тетра-н-бутиламмония (7,44 г, 26,8 ммоль), карбоната натрия (14,20 г, 0,134 ммоль), трифенилфосфина (0,35 г, 1,3 ммоль) и ацетата палладия (II) (0,30 г, 1,3 ммоль) в 200 мл диметилформамида нагревали в атмосфере N2 при температуре 98oC в течение 2 ч. Полученный раствор охлаждали и удаляли основную часть диметилформамида в условиях вакуума. Эту смесь разбавляли этилацетатом, органический раствор промывали водным раствором NaHCO3, сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь гексанов и этилацетата в отношении 10:1) позволила получить указанное в заголовке соединение (7,49 г, 65%) в виде кристаллического твердого вещества. Перекристаллизация из гексанов дала пробу для анализа в виде кристаллического твердого вещества белого цвета: температура плавления 115 - 116oC.
Элементный анализ:
Высчитано для C23H39N1O3Si2: C 63,69; H 9,06; N 3,23.
Обнаружено: C 63,54; H 9,11; N 3,17.
Этил-3-[3-гидрокси-проп-1-ил]-1H-индол-5-карбоксилат
Раствор этил-3-[3[/1,1-диметилэтил/диметилсилилокси/проп-1-ил] - 2-триметилсилил-1H-индол-5-карбоксилата (5,0 г, 11,5 ммоль) и 48%-ного водного раствора фтороводорода (1,9 г, 46,9 ммоль) в 140 мл MeCN перемешивали при 23oC в течение 3,5 ч. Полученный раствор резко охлаждали с помощью 10% Na2CO3 и экстрагировали органическое вещество введением в этилацетат. Органический раствор сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (градиент смеси 50 - 100% этилацетата и гексанов) позволила получить указанное в заголовке соединение (2,79 г, 98%) в виде кристаллического твердого вещества белого цвета: температура плавления 111 - 112oC.
Элементный анализ:
Высчитано для C14H17N1O3 C 68,00; H 6,93; N 5,66.
Обнаружено: C 67,94; H 6,78; N 5,65.
Пример 13. 3-/3-Гидроксипропил/-1H-индол-5-карбоновая кислота
К раствору метил-3-/3-гидроксипропил/-1H-индол-5-карбоксилата (5,0 г, 21,46 ммоль) в 50 мл этанола добавляли 30 мл 15%-ного раствора гидроксида калия в этаноле. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 1 ч. Смесь охлаждали до 0oC и подкисляли концентрированной хлористоводородной кислотой до pH 6. Этанол удаляли в условиях вакуума. Остаток разбавляли 250 мл этилацетата и промывали 50 мл воды. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием 3-/3-гидроксипропил/-1H-индол-5-карбоновой кислоты (4,30 г, 92%).
Пример 14. 3-/3-Гидроксипропил/-N-/фенилметил/-1H-индол-5-карбоксамид
К смеси 3-/3-гидроксипропил/-1H-индол-5-карбоновой кислоты (4,30 г, 19,63 ммоль), триэтиламина (3,97 г, 39,26 ммоль) и бензиламина (2,10 г, 19,63 ммоль) в 150 мл ацетонитрила добавляли иодид 2-хлор-1-метилпиридиния (6,04 г, 23,68 ммоль). Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч. Раствор охлаждали до комнатной температуры, добавляли 70 мл 0,5 н. раствора хлористоводородной кислоты и удаляли ацетонитрил в условиях вакуума. Остаток экстрагировали 250 мл этилацетата. Органический слой отделяли, а водный слой вновь экстрагировали тремя 50 мл порциями этилацетата. Соединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь CH2Cl2 метанола и NH4OH в отношении 95:5:0,5) позволила получить 3-/3-гидроксипропил/-N-/фенилметил/-1H-индол-5-карбоксамид (2,26 г, 37%).
Соединения формулы V
Превращение соединений формулы VI в соединения формулы V производится в соответствии со стандартными реакциями синтеза, при осуществлении которых алканольная часть молекулы превращается в часть алкил-(отщепляемая группа). Следующие примеры служат для демонстрации этого процесса, но не ограничивают объем изобретения.
Пример 15. 3-/5-Этансульфониламино-1H-индол-3-ил/пропилметансульфонат
К раствору 3-/5-этансульфониламино-1H-индол-3-ил/пропанола (0,59 г, 2,1 ммоль) в 20 мл тетрагидрофурана при 0oC добавляли триэтиламин (0,58 мл, 4,2 ммоль), после чего по каплям вводили метансульфонилхлорид (0,24 мл, 3,1 ммоль). Реакционную смесь перемешивали 30 мин при температуре 0oC. Реакционную смесь выливали в смесь насыщенного раствора NaHCO3 и этилацетата. Органическую фазу отделяли, а водную фазу вновь экстрагировали тремя порциями этилацетата. Соединенные органические слои промывали насыщенным раствором NaCl, сушили над безводным K2CO3, фильтровали и концентрировали в условиях вакуума с образованием 3-/5-этансульфониламино-1H-индол-3-ил/пропилметансульфоната (0,75 г, > 99%), который использовали без дальнейшей очистки.
Пример 16. 3-[5-[/Фенилметоксикарбонил/амин]-1H-индол-3-ил]пропил-4-метилбензолсульфонат
К раствору спирта (0,31 г, 0,96 ммоль), триэтиламина и 4-диметиламинопиридина (DMAP) (0,01 г) в CH2Cl2 (10 мл) при температуре 0oC добавляли TsCl (0,27 г, 0,96 ммоль). Реакционную смесь оставляли для нагревания до 23oC и выстаивали в течение 16 ч. В конце этого периода вводили дополнительное количество TsCl (0,068 г) и триэтиламин (0,038 г) и продолжали взаимодействие в течение 3 ч. Реакционную смесь обрабатывали смесью воды со льдом, органическую фазу отделяли, экстрагировали насыщенным раствором NaCl, сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь гексана и этилацетата в отношении 70:30) позволила получить 3-[5-[/фенилметоксикарбонил/амин]-1H-индол-3-ил]пропил-4-метилбензолсульфонат (0,39 г, 85%), структура которого была подтверждена спектром ЯМР и масс-спектром.
Пример 17. 3-/5-///Метиламино/сульфонил/метил/-1H-индол-3-ил/пропилметансульфонат
К раствору 5,16 г (18,3 ммоль, 1,0 экв.) 3-/3-гидроксипропил/-N-метил-1H-индол-5-метансульфонамида и 3,84 мл (27,4 ммоль, 1,5 экв.) триэтиламина в 100 мл безводного ацетонитрила и 100 мл метиленхлорида при температуре 0oC по каплям добавляли 1,77 мл (22,8 ммоль, 1,25 экв.) метансульфонилхлорида. Реакционную смесь тридцать минут перемешивали при 0oC, а затем разбавляли 250 мл этилацетата. Органический слой промывали 50 мл порциями насыщенного водного раствора бикарбоната натрия и насыщенного водного раствора хлорида натрия. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием масла, которое без очистки использовали на следующей стадии.
Пример 18. 5-Нитро-3-/3-бромпропил/индол
К раствору трифенилфосфина (6,70 г, 0,025 моль) в 80 мл ацетонитрила добавляли раствор 5-нитро-3-/3-гидроксипропил/индола (4,30 г, 0,020 моль) в 75 мл ацетонитрила, а затем раствор CBr4 (9,00 г, 0,027 моль) в 25 мл ацетонитрила, производят эту операцию в атмосфере аргона при температуре 0oC. Полученную смесь перемешивали при комнатной температуре в течение 3 ч, а затем выпаривали и хроматографировали остаток (силикагель, смесь этилацетата и гексана сначала в отношении 1:9, а затем 1:4), что позволило получить указанное в заголовке соединение (4,60 г, 84%) в виде твердого вещества, температура плавления 92 - 95oC.
Пример 19. 5-Фтор-3-/паратолуолсульфонилоксипропил/индол
К раствору 5-фтор-3-/3-гидроксипропил/индола (917 мг, 4,75 ммоль) в 20 мл CH2Cl2 в атмосфере аргона при температуре 0oC добавляли триэтиламин (728 мкл, 5,23 ммоль), затем раствор паратолуолсульфонилхлорида (994 мг, 5,23 ммоль) в 5 мл CH2Cl2 и, наконец, каталитическое количество 4-диметиламинопиридина (59 мг, 0,48 ммоль). Реакционную смесь перемешивали 30 мин при температуре 0oC, а затем при комнатной температуре в течение 1,5 ч. Выпаривание смеси с последующей хроматографией (силикагель/CH2Cl2) остатка позволило получить смолу. Эту смолу растворяли в простом эфире и разбавляли этот раствор гексаном до отделения масла. Добавление небольшого количества CH2Cl2 привело к растворению масла и кристаллизации продукта. В результате хранения при температуре -20oC, фильтрования и сушки остатка в условиях вакуума был получен продукт (1,378 г, 84%) в виде рыхлых белых игл: температура плавления 99oC.
Инфракрасный спектр (CH2Cl2) 3470, 1360, 1178 см-1.
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 7,90 (широкий синглет, 1H); 7,61 (д., J = 8,4 Гц, 2H); 7,30 (д., J = 8,4 Гц, 2H); 7,27 - 7,20 (м., 1H); 7,08 (двойной дублет, J = 9,6, 2,6 Гц, 1H); 6,96 - 6,94 (м., 1H); 6,88 (двойной дублет, J = 9,0, 2,5 Гц, 1H); 4,06 (т., J = 6,2 Гц, 2H); 2,74 (т., J = 7,2 Гц, 2H); 2,42 (с., 3H); 1,99 (двойной квартет, J = 7,2, 6,2 Гц, 2H).
Пример 20. Этил-3-[3-иодопроп-1-ил]-1H-индол-5-карбоксилат
Смесь этил-3-[3-гидрокси-проп-1-ил] -1H-индол-5-карбоксилата (2,64 г, 10,7 ммоль), триэтиламина (1,52 г, 15 ммоль) и метансульфонилхлорида (1,83 г, 16 ммоль) в 50 мл ацетонитрила перемешивали при температуре 0oC в течение 1 ч. Ацетонитрил удаляли в условиях вакуума, а органическое вещество разбавляли этилацетатом. Органический раствор промывали водным раствором Na2CO3, сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума. Концентрат растворяли в 100 мл ацетонитрила и добавляли порошкообразный KI (3,55 г, 21,4 ммоль). Этот раствор нагревали с обратным холодильником в течение 1,5 ч, затем охлаждали, выливали в воду и экстрагировали введение в этилацетат. Соединенные органические экстракты сушили (рассол, MgCO4), фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь гексанов и этилацетата в отношении 5:1) позволила получить указанное в заголовке соединение (3,50 г, 92%) в виде кристаллического твердого вещества белого цвета: температура плавления 81oC.
Элементный анализ:
Высчитано для C14H16I1N1O2: C 47 08; H 4,51; N 3,92.
Обнаружено: C 47,12; H 4,44; N 3,89.
Соединения формулы III
Пример 21. 1-/5-Метокси-4-пиримидинил/пиперазин - Метод 1
К раствору пиперазина (38,40 г, 0,45 моль) в CH3CN (225 мл) по каплям добавляли раствор CH2CN (100 мл), содержащий 4-хлор-5-метоксипиримидина (6,45 г, 0,04 моль), производя эту операцию в атмосфере азота. После окончания добавления реакционную смесь нагревали при 60oC в течение 0,75 ч. Реакционную смесь концентрировали при пониженном давлении, остаток растворяли в CH2Cl2 и экстрагировали 5%-ным раствором NaHCO3 и H2O. Органическую фазу сушили с помощью K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография концентрата на силикагеле (смесь CH2Cl2, метанола и NH4OH в отношении 92: 8: 0,8) позволила получить соединение формулы II (7,63 г, 88,1%). Обработка основания (1,0 г) раствором HCl в этаноле и кристаллизация из смеси этанола и изо-PrOH привела к образованию хлористоводородной соли соединения формулы II (0,50 г, 39,1%), (температура плавления 207 - 211oC).
1-/5-Метокси-4-пиримидинил/пиперазин - Метод 2
4,6-Дигидрокси-5-метоксипиримидин
Применяли модифицированный вариант процедуры, описанной Бретшнейдером, Рихтером и Клотцером, Monatsh. Chem. 96/6/, 1661 - 76 (1965). Абсолютный метанол (1,0 л) добавляли при охлаждении в бане со льдом к метоксиду натрия (175 г, 3,24 моль), производя эту операцию в круглодонной колбе емкостью 3 л. Когда эта смесь охлаждалась до температуры ниже 20oC, добавляли диметилметоксималонат (162,14 г, 1,00 моль), а затем добавляли твердый формамидинацетат (104,11 г, 1,00 моль). Эту смесь перемешивали в бане со льдом в течение 30 мин, после чего нагревали с обратным холодильником в течение 1 ч. Смесь охлаждали в ванне с холодной водой и добавляли концентрированную HCl (около 350 мл) до тех пор, пока смесь становилась сильно кислой при определении с помощью индикаторной бумаги. Осадок фильтровали, суспендировали в холодной воде (около 400 мл), а затем снова фильтровали. Белый порошок сушили в условиях вакуума (125,84 г, 88,7%) и использовали без дальнейшей очистки.
4,6-Дихлор-5-метоксипиримидин
Применяли модифицированный вариант процедуры, описанной Бретшнейдером, Рихтером и Клотцером, Monatsh. Chem. /96/6/, 1661-76 (1965). Смесь 4,6-дигидрокси-5-метоксипиримидина (125,84 г, 0,887 моль), POCl3 (700 мл) и N,N-диэтиланилина (50 мл) нагревали с обратным холодильником в течение 3 ч с образованием коричневого раствора. Полученный раствор охлаждали и удаляли избыток POCl3 в условиях вакуума. К остатку добавляли гексан (около 300 мл), после чего смесь нагревали с обратным холодильником при одновременном перемешивании. Слой горячего гексана сливали в химический стакан, и остаток еще два раза обрабатывали горячим гексаном. Гексановые экстракты (общий объем около 1 л) концентрировали в условиях вакуума с образованием неочищенного продукта в виде белого твердого вещества (116,5 г, 73,3%). Это вещество перекристаллизовывали из петролейного эфира, что позволило получить бесцветные иглы (92,0 г + 16,51 г второго сбора, общий выход 93,1%).
6-Хлор-5-метокси-/1-пиперазинил/пиримидин
Пиперазин (30 г) растворяли в воде (150 мл), а затем добавляли твердый 4,6-дихлор-5-метоксипиримидин (10,00 г, 55,8 ммоль). Полученную смесь интенсивно перемешивали в течение 2 ч при комнатной температуре, причем в течение этого периода происходило растворение 4,6-дихлор-5-метоксипиримидина. Продукт экстрагировали из водного раствора реакционной смеси метиленхлоридом (выход 12,67 г, 99,2%). Пробу (5 г) неочищенного продукта хроматографировали на силикагеле с применением градиента смеси 20-40% метанола и этилацетата в качестве элюента. Полученный продукт растворяли в ацетонитриле и добавляли концентрированную HCl с образованием соли в виде белого порошка, которую сушили в условиях вакуума, что позволило получить пробу для анализа (4,0 г, температура плавления 169-173oC с кипением).
Элементный анализ:
Высчитано для C9H13N4OCl • 1,5 HCl • 0,2 H2O: C 37,67; H 5,24; N 19,53; H2O 1,26.
Обнаружено: C 37,63; H 4,99; N 19,46; H2O 1,47.
1-/5-Метокси-4-пиримидинил/пиперазин
Пиперазин (20 г) растворяли в воде (100 мл) в сосуде Парра, а затем добавляли твердый 4,6-дихлор-5-метоксипиримидин (5,00 г, 27,9 ммоль). Полученную смесь энергично перемешивали в течение 2 ч при комнатной температуре, причем во время этого периода происходило растворение 4,6-дихлор-5-метоксипиримидина. Удаляли перемешивающий стержень, в мутный раствор вводили катализатор (10% палладированный уголь, 1,0 г) и гидрировали смесь при комнатной температуре (давление 60 фунтов на кв. дюйм, 3 ч). Катализатор отфильтровывали, а фильтрат трижды экстрагировали CH2Cl2. Экстракты из CH2Cl2 сушили над Na2SO4 и концентрировали в условиях вакуума с образованием светлого масла, которое затвердевало при выстаивании (3,34 г, 61,7%). Этот неочищенный продукт перегоняли по методу Кюгельроха (выход 3,24 г), растворяли в ацетонитриле и добавляли концентрированную HCl с целью осаждения продукта в виде белого порошка, который сушили в условиях вакуума (4,32 г, 94,0% из неочищенного продукта, температура плавления 219-221,5oC).
Пример 22. 1-/5-Метокси-4-пиримидинил/-2-метилпиперазин - Метод 1
1-/трет-Бутоксикарбонил/-3-метилпиперазин
К холодному (-5oC) раствору 2-метилпиперазина (5,00 г, 0,05 моль) в 200 мл CH2Cl2 в атмосфере аргона в течение 1 ч добавляли раствор ди-трет-бутилдикарбоната (10,9 г, 0,05 моль) в 100 мл CH2Cl2. Образовавшуюся смесь перемешивали при температуре -5oC в течение 1 ч, а затем при комнатной температуре в течение 2 ч. Раствор промывали (H2O), сушили (Na2SO4) и выпаривали с образованием масла, которое хроматографировали на силикагеле этилацетатом, а затем смесью этилацетата, метанола и NH4OH в отношении 10:1:0,1, что позволило получить целевой продукт (4,30 г, 43%) в виде масла. Это вещество использовали без дальнейшей очистки.
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 4,15-3,75 (широкий синглет, 2H); 3,0-2,6 (м. , 4H); 2,47-2,35 (м., 1H); 1,48 (с., 9H); 1,08 (д., J = 6,7 Гц, 3H).
1-/трет-Бутоксикарбонил/-4-/5-метокси-4-пиримидил/-3-метил- пиперазин
Смесь 1-/трет-бутоксикарбонил/-3-метилпиперазина (2,0 г, 0,01 моль), 4-хлор-5-метоксипиримидина (1,5 г, 0,01 моль) и диизопропилэтиламина (2,6 мл, 0,015 моль) в 25 мл сухого ацетонитрила нагревали до температуры кипения с обратным холодильником в атмосфере аргона в течение 60 ч. Полученный раствор разбавляли простым эфиром, промывали (H2O, рассол), сушили (Na2SO4) и выпаривали с образованием смолы. Эту смолу растирали в порошок с гексаном (x3), а всплывающий слой выпаривали, получая при этом смолу. Испарительная хроматография (силикагель, смесь этилацетата и гексана в отношении 1:1, затем этилацетата) этого вещества привела к образованию сначала 4-хлор-5-метоксипиримидина (0,4 г, 27%), а затем целевого продукта (1,2 г, 30%) в виде светло-розового твердого вещества: температура плавления 70-72oC.
Инфракрасный спектр (KBr) 1690, 1575 см-1.
Спектр 1H-ЯМР (200 МГц, CDCl3) δ: 8,33 (с., 1H); 7,90 (с., 1H); 4,79 (широкий синглет, 1H); 4,4-3,8 (м., 3H); 3,86 (с., 3H); 3,35-2,90 (м., 3H); 1,48 (с., 9H); 1,21 (д., J = 6,7 Гц, 3H).
1-/5-Метокси-4-пиримидинил/-2-метилпиперазин
Раствор 1-/трет-бутоксикарбонил/-4-/5-метокси-4-пиримидинил/- 3-метилпиперазина (1,70 г, 4,2 ммоль) и трифторуксусной кислоты (5 мл) в 50 мл CH2Cl2 перемешивали при комнатной температуре в атмосфере аргона 18 ч. Полученный раствор выпаривали, остаток растворяли в воде и подщелачивали смесь (pH 8) 15%-ным раствором NaOH. Полученную (pH 8) смесь экстрагировали этилацетатом, органическую фазу промывали (H2O, рассол), сушили (Na2SO4) и выпаривали с образованием полутвердого вещества. Это вещество растворяли в простом эфире, фильтровали с целью удаления нерастворимого материала, а раствор выпаривали, что позволило получить целевой продукт в виде масла. Его использовали без дальнейшей очистки.
Инфракрасный спектр (в чистом виде) 3300, 1580, 1540 см-1.
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 8,32 (с., 1H); 7,87 (с., 1H); 4,83 - 4,68 (м. , 1H); 4,26 - 4,19 (м., 1H); 3,85 (с., 3H); 3,26 - 3,17 (м., 1H); 3,12 - 3,01 (м., 2H); 2,94 - 2,80 (м., 2H); 1,29 (д., J = 6,8 Гц, 3H).
4-/5-Метокси-4-пиримидинил/2-метилпиперазин - Метод 2
Раствор 2-метилпиперазина (20 г) в воде (100 мл) подвергали взаимодействию с твердым 4,6-дихлор-5-метоксипиримидином (5,00 г, 27,9 ммоль) в соответствии с процедурой, аналогичной той, которая описывалась для метода 2 в примере 14. После гидрогенизации и фильтрования катализатора продукт экстрагировали из фильтра с помощью CH2Cl2. Экстракты концентрировали в условиях вакуума, а остаток перегоняли по методу Кюгельроха с образованием светлого масла (5,46 г 99,8%). Это масло растворяли в ацетонитриле и добавляли концентрированную HCl, что приводило к образованию соли, которую перекристаллизовывали из изо-PrOH и сушили в условиях ваккума, что позволило получить целевой продукт в виде белого порошка (4,02 г, температура плавления 185 - 188oC).
Пример 23. 1-/5-Этокси-4-пиримидинил/пиперазин
5-Этоксипиримидин
Натрий (2,89 г, 125,8 ммоль) растворяли в этаноле (110 мл) и добавляли 5-бромпиримидин (10,0 г 62,9 ммоль). Реакционную смесь нагревали в автоклаве при 120oC в течение 17 ч, а затем оставляли для выстаивания при температуре 23oC на 60 ч. Этанол удаляли при пониженном давлении и добавляли к концентрату воду (5 мл). Водную фазу экстрагировали CH2Cl2 (4•100 мл). Соединенные органические экстракты промывали насыщенным раствором NaCl, сушили над безводным K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография концентрата на силикагеле (смесь гексана и этилацетата в отношении 70:30) позволила получить 5-этоксипиримидин (2,00 г, 25,6%).
5-Этоксипиримидин-N-оксид
К раствору 5-этоксипиримидина (2,00 г, 16,1 ммоль) в CH2Cl2 (100 мл) добавляли 3-хлорпероксибензойную кислоту, 50 - 60% технический сорт (6,13 г, 17,7 ммоль), после чего реакционную смесь перемешивали при 23oC в течение 18 ч. Реакционную смесь экстрагировали водой (мл), содержащей Na2CO3 (1,71 г, 16,1 ммоль). Органическую фазу сушили с помощью безводного K2CO3, фильтровали и концентрировали при пониженном давлении с образованием N-оксида (1,75 г, 78%).
4-Хлор-5-этоксипиримидин
К раствору триэтиламина (1,90 г, 18,6 ммоль) и оксихлорида фосфора (2,87 г, 18,6 ммоль) с CHCl3 (60 мл) порциями добавляли 5-этоксипиримидин-N-оксид (1,75 г, 12,5 ммоль). После окончания добавления реакционную смесь нагревали с обратным холодильником в течение 3 ч. После охлаждения этой смеси до 0oC добавляли CHCl3 (60 мл) и воду (10 мл), после чего порциями добавляли NaHCO3 (3,15 г, 37,5 ммоль). После прекращения вспенивания органическую фазу отделяли, сушили с помощью MgSO4, фильтровали и концентрировали при пониженном давлении с образованием хлорсодержащего продукта (1,98 г), который использовали без дальнейшей очистки.
4-/Этоксикарбонил/-1-/5-этокси-4-пиримидинил/пиперазин
Смесь 4-хлор-5-этоксипиримидина (1,98 г, 12,5 ммоль), этил-1-пиперазинкарбоксилата (5,93 г, 37,5 ммоль) и микроизмельченного K2CO3 (5,18 г, 37,5 ммоль) нагревали с обратным холодильником в CH3CN (75 мл) в течение 4 ч. После охлаждения до 23oC растворитель удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 и промывали водой (5 мл). Органическую фазу сушили с помощью безводного K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография концентрата на силикагеле (смесь CH2Cl2 и метанола в отношении 98:2) позволила получить целевой продукт (2,29 г, 65%).
1-/5-Этокси-4-пиримидинил/пиперазин
К раствору КОН (10 н., 20 мл) добавляли 4-/этоксикарбонил/1-/5-этокси-4-пиримидинил/пиперазин (2,29 г, 8,18 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 24 ч, после чего при пониженном давлении удаляли воду. Остаток растворяли в CH2Cl2, промывали насыщенным раствором NaCl, сушили с помощью безводного K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография концентрата на силикагеле (смесь CH2Cl2, метанола и NH4OH в отношении 95:5:0,5) позволила получить соединение формулы III (0,71 г, 42%).
Пример 24. 4-/5-Метокси-4-пиримидинил/-2-метилпиперазин - Метод 1
Смесь 2-метилпиперазина (27,74 г, 0,28 ммоль) и 4-хлор-5-метоксипиримидина (8,0 г, 0,06 ммоль) нагревали в сосуд высокого давления Парра при температуре 100oC в течение 1,5 ч. Реакционную смесь растворяли в CH2Cl2 и экстрагировали 5%-ным раствором NaHCO3 и H2O. Органическую фазу сушили с помощью K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография концентрата на силикагеле (смесь CH2Cl2, метанола и NH4OH в отношении 93:7:0,7) позволила получить продукт (9,02 г, 78,2%). Обработка основания (1,0 г) раствором HCl в этаноле и кристаллизация из смеси изо-PrOH и этанола привела к образованию хлористоводородной соли (0,45 г, 32,1%, температура плавления 191 - 193oC).
4-/5-Метокси-4-пиримидинил/2-метилпиперазин - Метод 2
Раствор 2-метилпиперазина (20 г) в воде (100 мл) подвергали взаимодействию с твердым 4,6-дихлор-5-метоксипиримидином (5,00 г, 27,9 ммоль) в соответствии с процедурой, аналогичной той, которая описывалась для метода 2 в примере 21. После гидрогенизации и фильтрования катализатора полученный продукт экстрагировали из фильтра с помощью CH2Cl2. Экстракты концентрировали в условиях вакуума, а остаток перегоняли по методу Кюгельроха с образованием светлого масла (5,46 г, 99,8%). Это масло растворяли в ацетонитриле и добавляли концентрированную HCl с целью образования соли, которую перекристаллизовывали из изо-PrOH и сушили в условиях вакуума, что позволило получить продукт в виде белого порошка (4,02 г, температура плавления 185 - 188oC).
Пример 25. 1-/3-Метокси-4-пиридинил/пиперазин
Этил-4-/3-нитро-4-пиридинил/-1-пиперазинкарбоксилат
Раствор 3-нитропиридона технического сорта (25 г, 177 ммоль) (содержит 20 мас. % 3,5-динитропиридона) в 150 мл оксихлорида фосфора нагревали при 90oC в течение 2 ч.
Избыток оксихлорида фосфора удаляли перегонкой, а оставшееся масло выливали в смесь воды со льдом. Водный раствор экстрагировали пятью 300 мл порциями CH2Cl2. Экстракты из CH2Cl2 сушили (MgSO4), фильтровали и концентрировали в условиях вакуума с образованием 26,7 г неочищенного 4-хлор-3-нитропиридина. К раствору неочищенного 4-хлор-3-нитропиридина (26,7 г) в 200 мл MeCN добавляли карбонат калия (25,7 г, 186 ммоль) и этил-1-пиперазинкарбоксилат (27,3 мл, 186 ммоль). Реакционную смесь перемешивали 16 ч при 23oC, а затем концентрировали в условиях вакуума. Эту смесь разбавляли атилацетатом (1 л), органический раствор промывали водой, сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума с образованием твердой массы. Хроматография этой смеси на силикагеле позволила получить этил-4-/3-нитро-4-пиридинил-/1-пиперазинкарбоксилат (34,73 г, 73%): температура плавления 101 - 103oC, и этил-4-//3,5-динитро-4-пиридинил/-1-пиперазинкарбоксилат (7,07 г, 22%): температура плавления 165 - 167oC.
Элементный анализ:
Высчитано для C12H16N4O4: C 51,42; H 5,75; N 19,99.
Обнаружено: C 51,26; H 5,69; N 19,91.
Элементный анализ:
Высчитано для C12H15N5O6: C 44,31; H 4,65; N 21,53.
Обнаружено: C 44,51; H 4,65; N 21,36.
Этил-4-/3-Амино-4-пиридинил/-1-пиперазинкарбоксилат
Механически перемешиваемый раствор этил-4-/3-нитро-4-пиридинил/-1-пиперазинкарбоксилата (10 г, 35,7 ммоль) и железных опилок (40 меш) (21,7 г, 389 ммоль) в смеси 50 мл этанола, 7 мл воды и 0,36 мл концентрированной HCl нагревали до температуры кипения с обратным холодильником в течение 4 ч. Горячий раствор фильтровали и концентрировали в условиях вакуума с образованием этил-4-/3-амино-4-пиридинил/-1-пиперазинкарбоксилата (8,5 г, 95%). Пробу для анализа получали путем перекристаллизации из этилацетата в гексане: температура плавления 141 - 142oC.
Элементный анализ:
Высчитано для C12H18N4O2: C 57,58; H 7,25; N 22,38.
Обнаружено: C 57,43; H 7,19; N 22,24.
Этил-4-/3-йод-4-пиридинил/-1-пиперазинкарбоксилат
К раствору этил-4-/3-амино-4-пиридинил/-1-пиперазинкарбоксилата (2,0 г, 8,0 ммоль) в 4 мл смеси воды со льдом, содержащей 0,51 мл концентрированной H2SO4, добавляли нитрат натрия (0,55 г, 8,0 ммоль) в 1,2 мл воды в течение 1 мин. Полученную смесь 20 мин перемешивали при 0oC, а затем добавляли 0,16 мл концентрированной H2SO4, после чего эту смесь выливали в раствор иодида калия (1,6 г, 9,6 ммоль) в 2 мл воды, имеющей температуру 0oC. Через несколько минут добавляли 0,1 г Cu (Бронза), нагревали смесь до 23oC и перемешивали в течение 2 ч. Водный слой экстрагировали с помощью CHCl3 (3 • 50 мл). Органические слои соединяли, сушили (рассол, MgSO4) и концентрировали в условиях вакуума. Остаток очищали посредством хроматографии на колонках из силикагеля (смесь этилацетата и гексана в отношении 1:1), в результате чего было получено промежуточное соединение, представляющее этилкарбоксилат (1,35 г, 48%).
Элементный анализ:
Высчитано для C12H16N3O2I: C 39,91; H 4,46; N 11,63.
Обнаружено: C 40,11; H 4,46; N 11,77.
Метил-4-/3-метокси-4-пиридинил/-1-пиперазинкабоксилат
Раствор этил-4-/3-иод-4-пиридинил/-1-пиперазинкабоксилата (3,85 г, 10,7 ммоль), метилата натрия (6,4 г, 118,7 ммоль) и хлорида меди (0,071 г, 0,535 ммоль) в 38 мл диметилфомамида и 19,3 мл метанола нагревали при 90oC в течение 1 ч. Реакционную смесь охлаждали, фильтровали и выливали в 150 мл воды. Водный слой экстрагировали этилацетатом, органические экстракты сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума. Полученное твердое вещество белого цвета перекристаллизовывали из смеси этилацетата и гексанов с образованием промежуточного соединения, представляющего метилкарбоксилат (2,18 г, 80%): температура плавления 117-118oC.
Элементный анализ:
Высчитано для C12H17N3O3: C 57,36; H 6,82; N 16,72.
Обнаружено: C 57,34; H 6,74; N 16,69.
1-/3-Метокси-4-пиридинил//пиперазин
Раствор метил-4-/3-метокси-4-пиридинил/-1-пиперазинкарбоксилата (1,2 г, 4,7 ммоль) в 50 мл этанола, содержащего 1 мл 85% гидразингидрата и гидроксида калия (12 г, 210 моль), нагревали до температуры кипения с обратным холодильником в течение 12 ч. Реакционную смесь охлаждали, концентрировали в условиях вакуума, а остаток растворяли в этилацетате. Органические экстракты сушили (рассол, MgSO4), фильтровали и концентрировали в условиях вакуума. Хроматография остатка на силикагеле (смесь CH2Cl2, метанола и этилнитрида в отношении 3:1:0,01) позволяла получить целевое соединение (0,42 г, 46%).
Соединения формулы XXI
Пример 26. 1-[3-/5-Этансульфониламино-1H-индол-3-ил/пропил] -4-/5-метокси-4-пиримидинил/-3-метилпиперазин
К раствору 3-/5-этансульфониламино-1H-индол-3-ил/пропил-метансульфоната (0,75 г, 2,1 ммоль) в 15 мл ацетонитрила добавляли диизопропилэтиламин (0,54 г, 4,2 ммоль), иодид калия (0,05 г, 0,3 ммоль), бисульфат трет-бутиламмония (0,04 г, 0,1 ммоль) и 4-/5-метокси-4-пиримидинил/-3-метилпиперазин (0,87 г, 4,2 ммоль). Реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 20 ч. Реакционную смесь концентрировали в условиях вакуума, остаток обрабатывали 5%-ным раствором NaHCO3 и экстрагировали тремя порциями этилацетата. Соединенные органические слои промывали насыщенным раствором NaCl, сушили над безводным K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография остатка на силикагеле (смесь CH2Cl2 и метанола в отношении 98:2) позволила получить 0,32 г (32%) целевого продукта. Хлористоводородную соль получали путем добавления раствора HCl в этаноле. Эту соль перекристаллизовывали из этанола с образованием гидрата 1-[3-/5-этансульфониламино-1H-индол-3-ил/-пропил] -4-/5- метокси-4-пиримидинил/-3-метилпиперазингидрохлорида (0,15 г, 39%), температура плавления 212-214oC.
Элементный анализ:
Высчитано для C23H32N6O3S • 2HCl • 0,4 H2O • 0,3 EtOH: C 50,04; H 6,52; N 14,84.
Обнаружено: C 50,12; H 6,27; N 14,90.
Пример 27. 1-[3-[5-[Метил/трифторметансульфонил/амино]-1H-индол- 3-ил] -пропил]-4-/5-метокси-4-пиримидинил/пиперазин
Целевой продукт с температурой плавления > 230oC получили аналогичным образом с использованием в качестве исходного вещества 5-трифторметансульфониламино-1H-индола.
Элементный анализ:
Высчитано для C22H27F3N6O3S • 2HCl: C 45,14; H 5,00; N 14,36.
Обнаружено: C 44,75; H 4,90; N 14,42.
Пример 28. 1-[3-[5-[[/Фенилметокси/карбонил]амино]-1H-индол-3-ил]-пропил]-4-/5-метокси-4-пиримидинил/пиперазин
Раствор 3-[5-[/фенилметоксикарбонил//амин] -1H-индол-3-ил] пропил-4-метилбензолсульфоната (0,54 г, 1,13 ммоль), 4-/5-меткси-4-пиримидинил/пиперазина (0,44 г, 2,26 ммоль), диизопропилэтиламина (0,29 г, 2,26 ммоль), иодида калия (0,19 г, 1,13 ммоль) и бисульфата трет-бутиламмония (0,02 г, 1,13 ммоль) в CH3CN (10 мл) нагревали с обратным холодильником в течение 20 ч. После концентрирования реакционной смеси в условиях вакуума остаток обрабатывали 5%-ным раствором NaHCO3 и экстрагировали четырьмя порциями CH2Cl2. Соединенные органические фазы промывали насыщенным раствором NaCl, сушили с помощью K2CO3, фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь CH2Cl2 и метанола в отношении 96:4) позволила получить 1-[3-[5-[[/фенилметокси/карбонил]-амино] -1H-индол-3-ил] пропил]-4-/5- метокси-4-пиримидинил/пиперазин (0,44 г, 77%), температура плавления 174-175oC.
Элементный анализ:
Высчитано для C28H32N6O: C 67,19; H 6,45; N 16,79.
Обнаружено: C 67,05; H 6,47; N 16,88.
Пример 29. 4-/5-Метокси-4-пиримидинил/-1-[3-[5-[[/метиламино/ сульфонил] -метил]-1H-индол-3-ил]пропил]пиперазин
К раствору неочищенного 3-]5-[[/метиламино/сульфонил]метил]-1H- индол-3-ил] пропил] метансульфоната (18,3 ммоль, 1,0 экв.) в 225 мл безводного ацетонитрила добавляли 3,03 г (18,3 ммоль, 1,0 экв.) иодида калия, 3,81 мл (21,9 ммоль, 1,2 экв) N,N-диизопропилэтиламина и 3,89 г (20,1 ммоль, 1,1 экв. ) 1-/5-метокси-4-пиримидинил/пиперазина. Эту смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч. Реакционную смесь концентрировали в условиях вакуума и растворяли остаток в 500 мл хлороформа. Органический слой промывали 100 мл порциями 10% карбоната калия и насыщенного водного раствора хлорида натрия. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло очищали посредством хроматографии на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, после чего производили хроматографию на силикагеле с использованием смеси 20% метанола в этилацетате, что позволило получить 4,83 г (58%) 4-/5-метокси-4-пиримиднил/-1-[3-[5-[[/метиламино/сульфонил]метил]-1H- индол-3-ил]пропил]пиперазина. Хлористоводородную соль получали путем добавления 3 н. раствора HCl в этаноле. Две перекристаллизации из горячего метанола привели к образованию 4,05 г (68%) 4-/5-метокси-4-пиримидинил/-1-[3-[5-[[/метиламино/сульфонил]метил]-1H- индол-3-ил]-пропил]пиперазингидрохлорида, температура плавления 170oC (разложение).
Элементный анализ:
Высчитано для C22H30N6O3S (3 HCl): C 46,52; H 5,86; N 14,80.
Обнаружено: C 46,14; H 6,22; N 14,51.
Пример 30. Гидрат 4-/5-метокси-4-пиримидинил/-1-[3-[5- [[/метиламино/-сульфонил]метил]-1H-индол-3-ил]пропил]-3- метилпиперазингидрохлорида
Производные 4-/5-метокси-4-пиримидинил/-3-метилпиперазина синтезировали аналогичным образом путем замены 1-/5-метокси-4-пиримидинил/пиперазина 1-/5-метокси-4-пиримидинил//-2-метилпиперазином на стадии алкилирования, температура плавления 148oC (разложение).
Элементный анализ:
Высчитано для C23H33N6O3S (2,8 HCl) 0,8 C3H8O: C 48,99; H 6,67; N 13,49.
Обнаружено: C 48,98; H 6,77; N 13,27.
Пример 31. 1-[3-[5-[[/Диметиламино/сульфонил]метил]-1H-индол-3-ил]-пропил]-4-/5-метокси-4-пиримидинил/пиперазин
К раствору неочищенного 3-[5-[[/диметиламино/сульфонил] метил-1H- индол-3-ил] -пропил] метансульфоната (1,4 ммоль, 1,0 экв.) в 10 мл безводного ацетонитрила добавляли 0,225 г (1,50 ммоль, 1,1 экв.) йодида натрия, 0,26 мл (1,50 ммоль, 1,1 экв.) N,N-диизопропилэтиламина и 0,291 г (1,50 ммоль, 1,1 экв. ) 1-/5-метокси-4-пиримидинил/пиперазина. Смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч. Реакционную смесь концентрировали в условиях вакуума и растворяли остаток в 50 мл этилацетата. Органический слой промывали 10 мл порцией 10%-ного раствора карбоната калия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло очищали посредством хроматографии на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 0,403 г (62%) 1-[3-[5-[[/диметиламино/-сульфонил]метил]-1H-индол-3-ил]пропил] -4-/5-метокси-4-пиримидинил/-пиперазина. Хлористоводородную соль получали путем добавления 3 н. раствора HCl в этаноле. Две перекристаллизации из метанола привели к образованию 0,323 г (64%) гидрата 1-[3-[5-[[/диметиламино/сульфонил]метил]-1H-индол-3-ил] пропил] -4-/5-метокси-4-пиримидинил/пиперазингидрохлорида, температура плавления 210oC (разложение).
Элементный анализ:
Высчитано для C23H32N6O3S (3 HCl) 0,5 H2O: C 46,74; H 6,14; N 14,22.
Обнаружено: C 46,82; H 6,42; N 14,09.
4-/5-метокси-4-пиримидинил/-1-[[5[[/метилсульфонил/амино] -метил] -1H-индол-3-ил]пропил]пиперазин
К раствору неочищенного 3-[5-[[/метилсульфонил/амино] -метил] -1H-индол-3-ил] пропилметансульфоната (15,1 ммоль, 1,0 экв.) в 200 мл безводного ацетонитрила добавляли 2,51 г (15,1 ммоль, 1,0 экв.) иодида калия, 3,15 мл N, N-диизопропилэтиламина и 3,23 г 1-/5-метокси-4-пиримидинил/пиперазина. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток растворяли в 2540 мл хлороформа, промывали 50 мл порциями 10%-ного водного раствора карбоната калия и насыщенного водного раствора хлорида натрия. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием масла. Хроматография на силикагеле с использованием смеси 7,5% метанола в метиленхлориде, содержащем 0,75% концентрированного водного раствора гидроксида аммония, позволяла получить 3,93 г (57%) 4-/5-метокси-4-пиримидинил/-1-[[5-[[/метилсульфонил/амино] метил] -1H- индол-3-ил] пропил] пиперазина. Гидрохлорид получали путем добавления 3 н. раствора HCl в этаноле, что привело к образованию раствора основания в этаноле. Перекристаллизация из этанола с последующей сушкой в условиях вакуума при температуре 65oC позволила получить 0,268 г (53%) этанолят гидрохлорида, температура плавления 204-206oC.
Элементный анализ:
Высчитано для C22H30N6O3S (2,4 HCl) 0,4 C2H6O: C 48,51; H 6,21; N 14,89.
Обнаружено: C 48,59; H 6,21; N 14,95.
Пример 33. 4-/5-Метокси-4-пиримидинил-1-[[5-[[метил/ метилсульфонил/-амино]метил]-1H-индол-3-ил]пропил]пиперазин
К раствору 1,93 г (4,21 ммоль, 1,0 экв.) 4-/5-метокси-4-пиримидинил/1-[[5-[[/метилсульфонил/амино]метил]-1H- индол-3-ил]-пропил]пиперазина в 84 мл безводного тетрагидрофурана при температуре -78oC по каплям добавляли 1,9 мл (4,41 ммоль, 1,05 экв.) 2,32 М раствора н-BuLi в гексане. Во время добавления происходило образование осадка. После окончания добавления реакционную смесь перемешивали сорок минут при температуре -78oC. Затем реакционную смесь помещали в баню со льдом и в чистом виде по каплям добавляли 0,275 мл (4,41 ммоль, 1,05 экв.) метилиодида. Реакционную смесь медленно нагревали в бане со льдом до комнатной температуры в течение 16 ч. Реакционную смесь разбавляли 250 мл этилацетата и промывали 50 мл порциями 10%-ного водного раствора карбоната калия и насыщенного водного раствора хлорида натрия. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях вакуума с образованием масла. Это масло хроматографировали с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, что позволило получить 1,1 г твердого вещества. Это твердое вещество перекристаллизовывали с образованием 0,760 г (38%) чистого 4-/5-метокси-4-пиримидинил/-1-[[5-[[метил-/метилсульфонил/амино] метил] -1H-индол-3-ил] пропил] пиперазина, температура плавления 164-165oC, C22H30N6O3S.
Элементный анализ:
Высчитано для C22H30N6O3S: C 58,45; H 6,82; N 17,78.
Обнаружено: C 58,12; H 6,75; N 17,56.
Пример 34. 1-[[5-[[Этил/метилсульфонил/амино] метил] -1H-индол- 3-ил] пропил]-4-/5-метокси-4-пиримидинил/пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 1-[[5-[[этил/метилсульфонил/амино] метил] -1H-индол- 3-ил]пропил]-4-/5-метокси-4-пиримидинил/пиперазин с использованием этилиодида на стадии алкилирования: температура плавления 197oC (разложение).
Элементный анализ:
Высчитано для C23H32N6O3S (3,7 HCl): C 45,47; H 5,92; N 13,83.
Обнаружено: C 45,32; H 6,10; N 13,73.
Пример 35. 4-/5-Метокси-4-пиримидинил/-1-[[5-[[фенилметил/метилсульфонил/-амино]метил]-1H- индол-3-ил]пропил]пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 4-/5-метокси-4-пиримидинил/-1-[[5-[[фенилметил/метилсульфонил/амино] метил] -1H-индол-3-ил] пропил] пиперазин с использованием бензилбромида на стадии алкилирования: температура плавления 203-204oC.
Элементный анализ:
Вычислено для C29H36N6O3S (1,1 HCl): C 59,16; H 6,35; N 14,27.
Обнаружено: C 59,04; H 6,38; N 14,11.
Пример 36. 1-[[5-[[/Этилсульфонил/амино]метил]-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 1-[[5-[[/этилсульфонил/амино] метил] -1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/пиперазин с использованием этансульфонилхлорида на стадии сульфонилирования: температура плавления 136oC (разложение).
Элементный анализ:
Высчитано для C23H32N6O3S (2,1 C4H4O4) 0,6 H2O: С 51,70; H, 5,73; N 11,38.
Обнаружено: C 51,32; H 5,70; N 11,54.
Пример 37. 1-[[5-[[/Метилсульфонил/амино]метил]-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/-3-метилпиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 1-[[5-[[/метилсульфонил/амино] метил] -1H-индол-3-ил] пропил] -4-/5-метокси-4- пиримидинил/-3-метилпиперазин с использованием 1-/5-метокси-4-пиримидинил/-2-метилпиперазина на стадии сочетания: температура плавления 210-214oC.
Элементный анализ:
Высчитано для C23H32N6O3S (2,1 HCl) 0,4 C2H6O: C 50,36; H 6,48; N 14,81.
Обнаружено: C 50,34; H 6,47; N 14,74.
Пример 38. 1-[[5-[[/Этилсульфонил/амино]метил]-1H-индол-3-ил]пропил]-4-/5-метокси-4 -пиримидинил/-3-метилпиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 1-[[5-[[/этилсульфонил/амино] метил] -1H-индол-3-ил] пропил]-4-/5-метокси-4 -пиримидинил/-3-метилпиперазин с использованием 1-/5-метокси-4-пиримидинил/-2-метилпиперазина на стадии сочетания и этансульфонилхлорида на стадии сульфонилирования: температура плавления 202-210oC.
Элементный анализ:
Высчитано для C24H34N6O3S (2,9 HCl): C 48,66; H 6,28; N 14,19.
Обнаружено: C 48,41; H 6,35; N 13,93.
Пример 39.
4-/5-Метокси-4-пиримидинил/-1-[[5-[[метил/этилсульфонил/амино] -метил] -1H-индол-3-ил]пропил]пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 4-/5-метокси-4-пиримидинил/-1-[[5-[[метил/этилсульфонил/амино] метил]-1H-индол -3-ил] пропил]пиперазин с использованием метилиодида на стадии алкилирования и этансульфонилхлорида на стадии сульфонилирования: температура плавления 113-115oC.
Элементный анализ:
Высчитано для C24H34N6O3S (C4H6O4): C 55,61; H 6,67; N 13,90.
Обнаружено: C 55,43; H 6,67; N 13,74.
Пример 40. 4-/5-Метокси-4-пиримидинил/-1-[[5-[[фенилметил/этилсульфонил/-амино]-метил]-1H -индол-3-ил]пропил]пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 4-/5-метокси-4-пиримидинил/-1-[[5-[[фенилметил/этилсульфонил/амино] метил] -1H-индол-3-ил] пропил] пиперазин с использованием бензилбромида на стадии алкилирования и этансульфонилхлорида на стадии сульфонилирования: температура плавления 73-75oC.
Элементный анализ:
Высчитано для C30H38N6O3S (1,2 C4H6O4): C 59,34; H 6,47; N 11,93.
Обнаружено: C 59,02; H 6,17; N 11,89.
Пример 41. 1-[[5-[[/Ацетил/амино]метил]-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/ пиперазин
В соответствии с процедурой, описанной в примере 32, синтезировали 1-[[5-[[/ацетил/амино]метил]-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/ пиперазин с использованием уксусного ангидрида для ацилирования вместо применения сульфонилхлорида для сульфонилирования: температура плавления 169-174oC.
Элементный анализ:
Высчитано для C23H30N6O2 (2,8 HCl) 1,2 H2O: С 50,57; H 6,50; N 15,39.
Обнаружено: C 50,22; H 6,87; N 15,25.
Пример 42. 1-[3-/5-Циано-1H-индол-3-ил/пропил]-4-/5-метокси-4-пиримидинил/пиперазин
К раствору 3-/5-циано-1H-индол-3-ил/пропилметансульфоната (2,35 ммоль, 1,0 экв.) в 25 мл безводного ацетонитрила добавляли 0,874 г (4,5 ммоль, 1,5 экв. ) (5-метокси-4-пиримидинил)-пиперазина и 1,0 мл N,N-диизопропилэтиламина. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 13 ч. Раствор охлаждали, разбавляли 100 мл хлороформа и промывали один раз 20 мл 10%-ного водного раствора карбоната натрия. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле с использованием смеси 5% метанола в метиленхлориде, содержащем 0,5% концентрированного водного раствора гидроксида аммония, в результате чего было получено 0,375 г (42%, две стадии) (1-[3-/5-циано-1H-индол-3-ил/-пропил]-4-/5-метокси-4-пиримидинил/пиперазина, чистота которого была подтверждена спектром 1H ЯМР.
Пример 43. 1-[3-/5-Аминокарбонил-1H-индол-3-ил/пропил]-4-/5-метокси-4-пиримидинил/ пиперазин
К раствору 1,19 г (3,17 ммоль, 1,0 экв.) 1-[3-/5-циано-1H-индол-3-ил/пропил] -4-/5-метокси-4-пиримидинил/пиперазина в 16 мл этанола добавляли раствор 1,85 г (33,0 ммоль, 10,4 экв.) гидроксида калия в 16 мл этанола. Полученный раствор нагревали до температуры кипения с обратным холодильником в течение 16 ч. Тонкослойная хроматография показала, что реакция была завершена менее, чем на 50%. Реакционную смесь разбавляли 150 мл этилацетата и один раз промывали 25 мл насыщенного водного раствора бикарбоната натрия. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с образованием 1,22 г масла. Это масло хроматографировали на силикагеле с использованием смеси 10% метанола в метиленхлориде, содержащем 1% концентрированного водного раствора гидроксида аммония, что позволило получить 0,293 г (23%) 1-[3-/5-аминокарбонил-1H-индол-3-ил/пропил] -4-/5-метокси-4-пиримидинил/ пиперазина. Превращение в хлористоводородную соль привело к образованию C21H24N6O (2,8 HCl) • 0,7 H2O, температура плавления 220-225oC (разложение).
Элементный анализ:
Высчитано: C 51,35; H 5,79; N 17,11.
Обнаружено: C 51,61; H 6,08; N 16,82.
Пример 44. 3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил]пропил]-5-аминоиндол
5-Нитро-3-/3-бромпропил/индол
К раствору трифенилфосфина (6,70 г, 0,025 ммоль) в 80 мл ацетонитрила добавляли раствор 5-нитро-3-/3-гидроксипропил/индола (4,30 г, 0,020 ммоль) в 75 мл ацетонитрила, а затем раствор CBr4 (9,00 г, 0,027 ммоль) в 25 мл ацетонитрила, производя эту операцию в атмосфере аргона при температуре 0oC. Полученную смесь перемешивали при комнатной температуре в течение 3 ч, после чего ее выпаривали, а остаток хроматографировали (силикагель, смесь этилацетата и гексана с отношением 1:9, затем 1:4), в результате чего было получено бромсодержащее соединение (4,60 г, 84%) в виде твердого вещества, температура плавления 92-95oC.
3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил]пропил]-5-нитроиндол
Смесь 5-нитро-3-/3-бромпропил/индола (0,57 г, 2,0 ммоль), 1-/5-метокси-4-пиримидил/пиперазина (0,47 г, 2,4 ммоль), иодида калия (0,40 г, 2,4 ммоль) и диизопропилэтиламина (1,75 мл, 10,0 ммоль) в 20 мл ацетонитрила нагревали с обратным холодильником в атмосфере аргона в течение 6 ч. Охлажденную реакционную смесь разбавляли этилацетатом и промывали (H2O, рассол). Промывочные воды вновь экстрагировали с помощью CH2Cl2 и промывали органическую фазу (H2O, рассол). Соединенные органические фазы сушили (Na2SO4) и выпаривали, а остаток хроматографировали (силикагель, смесь CH2Cl2 и метанола в отношении 95:5) с образованием твердого вещества. Это вещество истирали в порошок со смесью CH2Cl2 и гексана, в результате чего было получено указанное в заголовке соединение (0,55 г, 70%) в виде твердого вещества желтого цвета, температура плавления 163-166oC.
3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил]пропил]-5-аминоиндол
К раствору 3-[3-[4-/5-метокси-4-пиримидил/-1-пиперазинил]пропил]-5-нитроиндола (550 г, 1,39 ммоль) в смеси этанола (120 мл) и тетрагидрофурана (40 мл) добавляли 10% палладированный уголь (0,30 г), после чего смесь гидрировали в аппарате для встряхивания Парра под давлением 40 фунтов на кв. дюйм в течение 18 ч. Затем эту смесь фильтровали через целит и промывали фильтровальный осадок дополнительным количеством смеси этанола и тетрагидрофурана. Выпаривание фильтрата позволило получить в основном чистое указанное в заголовке соединение (0,557 г, 100%) в виде пены коричневого цвета. Пробу этого вещества (0,143 г) обрабатывали избытком раствора HCl в метаноле, после чего полученный раствор разбавляли ацетоном с образованием осадка. Этот осадок фильтровали, а затем кристаллизовали из этанола, что позволило получить 0,100 г твердого вещества с фиолетовым оттенком, температура плавления 192oC (разложение).
Инфракрасный спектр (KBr): 3410, 3200, 1630, 1540 см-1.
Спектр 1H ЯМР (DMCO-d6, 200 МГц) δ: 11,22 (широкий синглет, 1H); 10,20 (широкий синглет, 2H); 8,60 (д., 1H); 8,20 (с., 1H); 7,55 (д., J = 1,6 Гц, 1H); 7,55 (д., J = 1,6 Гц, 1H); 7,45 (д., J = 8,6 Гц, 1H); 7,35 (д., J = 2,1 Гц, 1H); 7,07 (двойной дублет, J = 8,6, 1,9 Гц, 1H); 4,89-4,82 (м., 2H); 3,91 (с. , 3H); 3,8-3,0 (широкий мультиплет, 8H); 2,76 (м., 2H); 2,12 (широкий мультиплет, 2H).
Элементный анализ:
Высчитано для C20H26N6O•4HCl•H2O: C 45,29; H 6,08; N 15,85.
Обнаружено: C 45,32; H 5,97; N 15,59.
Пример 45. 3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил] пропил]-5- /метилкарбамоил/оксииндол
К раствору 3-[3-[4-/5-метокси-4-пиримидинил]-1-пиперазинил] пропил]-5-гидроксииндола (0,170 г, 0,46 ммоль) в 6 мл CH2Cl2 добавляли метилизоцианат (60 мкл, 1,0 ммоль), после чего эту смесь перемешивали при комнатной температуре в течение 4 дней. Полученную смесь выпаривали, и образовавшуюся пену розовато-серого цвета истирали в порошок со смесью изопропанола и простого эфира, что позволило получить твердое вещество светло-серого цвета. Это вещество хроматографировали (силикагель, смесь этилацетата и метанола в отношении 95: 5, затем 90:10) с образованием указанного в заголовке соединения (0,120 г, 60%) в виде твердого вещества светло-серого цвета, температура плавления 120-122oC. Инфракрасный спектр (KBr):3400, 3220, 1730 см-1.
Спектр 1H ЯМР (CDCl3, 200 МГц) δ: 8,33 (с., 1H); 8,06 (широкий синглет, 1H); 7,88 (с., 1H); 7,34-7,26 (м, 2H); 6,99-6,91 (м., 2H); 5,00 (широкий мультиплет, 1H); 3,85 (с., 3H); 3,79 (т., J = 4,7 Гц, 4H); 2,91 (д., J = 4,8 Гц, 3H); 2,75 (т., J = 7,4 Гц, 2H); 2,54 (т., J = 4,8 Гц, 4H); 2,45 (т., J = 7,6 Гц, 2H); 2,04-1,83 (м., 2H).
Элементный анализ:
Высчитано для C22H28N6O3•0,8H2O: C 60,20; H 6,80; N 19,15.
Обнаружено: C 60,13; H 6,40; N 18,75.
Пример 46. 3-[3-[4-/5-Метокси-4-пиримидинил]-1-пиперазинил] пропил]-5-/цианометил/оксииндол.
К суспензии NaH (60% в масле, 0,020 г, 0,5 ммоль) в 10 мл сухого тетрагидрофурана добавляли суспензию 3-[3-[4-/5-метокси-4-пиримидил/- 1-пиперазинил]пропил]-5-гидроксииндола (0,183 г, 0,5 ммоль) в 20 мл тетрагидрофурана в атмосфере аргона при комнатной температуре. Через 30 мин выделение газа превращалось и образовался светлый раствор. К этому раствору добавляли раствор хлорацетонитрила (35 мкл, 0,55 ммоль) в 5 мл сухого тетрагидрофурана, эту смесь перемешивали при комнатной температуре в течение 2 ч, а затем нагревали с обратным холодильником в течение 1,5 ч. Охлажденную смесь разбавляли CH2Cl2, промывали (вода, рассол), сушили (Na2SO4) и выпаривали с образованием пенообразного продукта. Хроматография на колонках (силикагель, смесь CH2Cl2 и метанола в отношении 95:5, а затем смесь CH2Cl2, метанола и NH4OH в отношении 95:4,5:0,5) позволила получить указанное в заголовке соединение (0,150 г, 75%) в виде пены. Этот материал обрабатывали избытком раствора HCl в метаноле, после чего раствор выпаривали с образованием стеклообразного продукта. Истирание в порошок со смесью метанола и этанола (1:9) позволило получить гидрохлорид (0,110 г) в виде твердого вещества нестандартного белого цвета, температура плавления 198-200oC (разложение):
Инфракрасный спектр (KBr) 3400, 3220, 1630 см-1.
Спектр 1H ЯМР (DMCO-d6, 200 Мгц) δ: 11,39 (широкий синглет, IH); 10,87 (широкий синглет, IH); 8,61 (c., IH); 8,20 (c., IH); 7,30 (д., J = 8,8 Гц, IH); 7,24 (д. , J = 2,4 Гц, IH); 7,22 (д., J = 2,0 Гц, IH); 6,83 (двойной дублет, J = 8,7, 2,4 Гц, IH); 5,16 (c., 2H); 4,90-4,83 (м., 2H); 3,90 (с., 3H); 3,70-3,36 (м., 4H); 3,13 (широкий синглет, 4H); 2,75 (м., 2H); 2,12 (м. , 2H).
Элементный анализ:
Высчитано для C22H26N6O2 • 2,5 HCl • 0,3 C2H6O : C 53,07; H 5,97; N 16,43.
Обнаружено C 53,26; H 5,70; N 16,11.
Пример 47. 3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил]пропил]-5-/карбоксамидометил/оксииндол
К суспензии NaH (60% в масле, 0,020 г, 0,5 ммоль) в 5 мл сухого тетрагидрофурана добавляли суспензию 3-[3-[4-/5-метокси-4-пиримидил/-1-пиперазинил] пропил] -5-гидроксииндола (0,184 г, 0,5 ммоль) в 35 мл сухого тетрагидрофурана, после чего эту смесь перемешивали при комнатной температуре в атмосфере аргона в течение 30 мин. К образовавшемуся светлому раствору добавляли раствор 2-хлорацетамида (0,047 г, 0,5 ммоль) в 5 мл тетрагидрофурана, выдерживали эту смесь при комнатной температуре в течение 2 ч, а затем нагревали ее с обратным холодильником в течение 2 ч. Охлажденную смесь разбавляли этилацетатом, промывали (вода, рассол), сушили (Na2SO4) и выпаривали. Остаток хроматографировали (силикагель/ тетрагидрофуран) с образованием загрязненного примесями исходного вещества (0,117 г). Дальнейшее элюирование привело к образованию указанного в заголовке соединения (0,100 г, 47%) в виде смолы. Это вещество обрабатывали избытком раствора HCl в метаноле, после чего раствор выпаривали, а остаток кристаллизовали из смеси метанола и простого эфира, что позволило получить гидрохлорид (0,095 г) в виде сероватого твердого вещества, температура плавления 90oC.
Инфракрасный спектр (KBr): 3400, 3250, 1680, 1630 см-1.
Спектр 1H ЯМР (DMCO-d6, 200 МГц) δ: 11,55 (широкий синглет, 1H); 10,76, (широкий синглет, 1H); 8,69 (c., 1H); 8,21 (с., 1H); 7,49-7,40 (широкий мультиплет, 2H); 7,25 (д., J = 8,6 Гц, IH); 7,16 (д., J= 2,1 Гц, IH); 7,05 (д., J = 2,2 Гц, IH); 6,81 (двойной дублет, J = 8,7, 2,3 Гц, 1H); 4,99-4,92 (широкий мультиплет, 2H); 4,41 (c., 2H); 3,91 (с., 3H); 4,3-3,5 (м., 4H); 3,11 (широкий синглет, 4H); 2,72 (м., 2H); 2,10 (широкий синглет, 2H).
Элементный анализ:
Высчитано C22H28N6O3 • 3HCl • H2O: C 47,87; H 6,03; N 15,23.
Обнаружено: C 48,24; H 5,89; N 14,83.
Пример 48. 1-[3-[5-ацетил-1H-индол-3-ил]пропил]-4-/5метокси-4 -пиримидинил/пиперазингидрохлорид
Это соединение получали обычным способом из 1-/5-ацетил-1H-индол-3-ил/-3-пропанола, температура плавления 215-217oC (разложение).
Элементный анализ:
Высчитано для C22H27N5O2 (2,7 HCl): C 53,72; H 6,09; N 14,24.
Обнаружено: C 53,52; H 6,23; N 14,28.
Пример 49. 4-/5-Метокси-4-пиримидинил/-1-[3-[5-[2-/метилсульфонил/ метил]-IH-индол-3-ил]пропил]пиперазин
Температура плавления 190-195oC.
Элементный анализ:
Высчитано для C22H29N5O3S • 3,0 HCl: C 47,79; H 5,84; N 12,67.
Обнаружено: C 47,58; H 5,96; N 12,32.
Пример 50. 4-/5-Метокси-4-пиримидинил/-1-[3-[5-[2-/метилсульфонил/ метил]-IH-индол-3-ил] пропил]-3-метилпиперазин
Температура плавления 208-210oC (разложение).
Элементный анализ:
Высчитано для C23H32N5O3S • 2,5 HCl: C 50,25; H 6,33; N 12,47.
Обнаружено: C 50,17; H 6,04; N 12,58.
Пример 51. 4-/5-Метокси-4-пиримидинил/-1-[3-[5-[2-/метилсульфонил/ этил] -1H-индол-3-ил]пропил]пиперазин
Температура плавления 153-154oC.
Элементный анализ:
Высчитано для C23H31N5O3S: C 60,37; H 6,83; N 15,30.
Обнаружено: C 60,24; H 6,79; N 15,42.
Пример 52. 4-/5-Метокси-4-пиримидинил/-1-[3-[5-[2-/метилсульфонил/ этил] -1H-индол-3-ил]пропил]-3-метилпиперазин
Температура плавления 220-223oC.
Элементный анализ:
Вычислено для C24H33N5O3S • 2,5 HCl: C 51,22; H 6,36; N 12,44.
Обнаружено: C 51,20; H 6,26; N 12,36.
Пример 53. Гидрат 4-/5-этокси-4-пиримидинил//-1-[3-/1H-индол-3-ил/пропил]- пиперазингидрохлорида
Смесь 3-/1H-индол-3-ил/пропил-4-метилбензолсульфоната (0,87 г, 2,65 ммоль), 1-/5-этокси-4-пиримидинил/пиперазина (1,10 г, 5,29 ммоль), микроизмельченного K2CO3 (0,73 г, 5,29 ммоль) и бисульфата тетрабутиламмония (0,04 г, 0,13 ммоль) в CH3CN (15 мл) нагревали с обратным холодильником в атмосфере азота в течение 2,5 ч. Реакционную смесь оставляли для охлаждения до 23oC и выставляли в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, а остаток растворяли в CH2Cl2 и экстрагировали водой. Органическую фазу сушили безводным K2CO3, фильтровали и концентрировали при пониженном давлении. Хроматография остатка на силикагеле (смесь CH2Cl2 и метанола в отношении 98:2) позволила получить свободное основание (0,70 г, 72%), которое обрабатывали раствором HCl в этаноле, при этом целевой продукт (0,85 г, 99%, температура плавления > 230oC) получали после кристаллизации из смеси этанола и метанола.
Элементный анализ:
Высчитано для C21H27N5O • 2HCl • 7H2O: C 55,93; H 6,80; N 15,53; H2O 2,80.
Обнаружено: C 55,67; H 6,66; N 15,40; H2O 2,50.
Пример 54. 1-[3-/1H-индол-3-ил/бутил]-4-/5-метокси-4-пиримидинил/-пиперазин
К смеси 1-/5-метокси-4-пиримидил/пиперазина (1,55 г, 8,0 ммоль) гидрохлорида триэтиламина (1,10 г, 8,0 ммоль) и NaCNBH3 (1,76 г, 28 ммоль) в 20 мл сухого тетрагидрофурана добавляли раствор 3-/3-оксобутил/индола (Дж. Жмускович и др. , J. Am. Chem. Soc. 79, 2819 (1957) (0,75 г, 4,0 ммоль) в 5 мл тетрагидрофурана, после чего эту смесь перемешивали при комнатной температуре в атмосфере аргона в течение 20 ч. Затем реакционную смесь выливали в 10%-ный насыщенный раствор NaHCO3 и экстрагировали этилацетатом (x 2). Органический экстракт промывали H2O (x 2), а затем 25 мл 0,1 н. раствора HCl. Полученный органический раствор экстрагировали 1 н. раствором HCl (x 2), водяную фазу промывали CH2Cl2 (x 2), после чего ее охлаждали при 0oC и подщелачивали 50%-ным раствором NaOH. Это позволило получить смолистый осадок, который экстрагировали введением в этилацетат (x 4), а соединенный органический экстракт промывали (рассол), сушили (Na2SO4) и выпаривали с образованием смолы. Испарительная хроматография (силикагель, смесь ацетонитрила и метанола в отношении 95:5, затем 80:20) этого вещества позволила получить чистый продукт (968 мг, 66%) в виде пены белого цвета.
Эту пену растворяли в CH2Cl2 и обрабатывали избытком раствора HCl в этаноле. Полученный раствор выпаривали, а остаток снова растворяли в избытке раствора HCl в этаноле. Выпаривание этого раствора привело к образованию пены светло-коричневого цвета, которую кристаллизовали из горячей смеси метанола и ацетона с образованием гидрохлорида (916 мг) в виде белого порошка: температура плавления 169-172oC (разложение).
Инфракрасный спектр (KBr) : 3400, 1632, 1550, 1275 см-1.
Спектр 1H ЯМР (200 МГц, CDCl3) δ: 11,68 (широкий синглет, 1H); 10,89 (с. , 1H); 8,66 (с., 1H); 8,20 (с., 1H); 7,56 (д., J = 7,4 Гц, 1H); 7,33 (д., J = 7,7 Гц, 1H); 7,21 (д., J = 2,3 Гц); 7,10-6,93 (м., 2H); 5,03-4,96 (м., 2H); 3,91 (с., 3H); 3,85-3,75 (м., 2H); 3,54-3,25 (м., 5H); 2,88-2,60 (м., 2H); 2,51-2,35 (м., 2H); 1,88-1,81 (м., 1H); 1,38 (д., J = 6,5 Гц, 3H).
Элементный анализ:
Высчитано для C21H27N5O • 2HCl • 1,4H2O : C 54,50; H 6,91; N 15,11.
Обнаружено: C 54,39; H 6,93; N 15,37.
Пример 55. 3-[3-[4-/5-Метокси-4-пиримидинил/-1-пиперазинил] пентил]индол
Смесь 3-//3-бромпентил/индола (1,33 г, 5,0 ммоль), 1-/5-метокси-4-пиримидил/пиперазина (2,00 г, 10 ммоль), тонко измельченного K2CO3 (0,70 г, 5,1 ммоль) и тонко измельченного иодида калия (0,90 г, 5,1 ммоль) в 20 мл ацетонитрила нагревали до температуры кипения с обратным холодильником в атмосфере аргона в течение 5 ч. Охлажденную реакционную смесь разбавляли этилацетатом, промывали (H2O, рассол), сушили (Na2SO4) и выпаривали с образованием смолы. Хроматография этого вещества (силикагель, сначала этилацетат, а затем смесь 10% метанола и этилацетата) позволила получить продукт (0,65 г, 34%) в виде смолы. Эту смолу растворяли в простом эфире, после чего добавляли раствор HCl в метаноле. Образовавшееся твердое вещество фильтровали, промывали простым этиловым эфиром и сушили в условиях вакуума, что позволило получить твердое вещество нестандартного белого цвета (0,50 г), температура плавления 145oC (разложение).
Инфракрасный спектр (KBr): 3400, 1633, 1550 см-1.
Элементный анализ:
Высчитано для C22H29N5O • 3HCl • 0,75 H2O: C 52,59; H 6,72; N 13,94.
Обнаружено: C 52,70; H 6,34; N 13,91.
Пример 56. 1-[3-/5-Гидрокси-1H-индол-3-ил/пропил]-4-/5-метокси- 4-пиримидинил//пиперазин
Смесь 1-[3-/5-бензилокси-1H-индол-3-ил/пропил]-4-/5-метокси- 4-пиримидинил//пиперазина (пример 71; 1,40 г, 3,06 ммоль) и 10% Pd (OH2 ) (C (0,85 г)) в 25 мл этанола гидрировали в аппарате для встряхивания Парра под давлением 30-40 фунтов на кв. дюйм в течение 3,5 ч. Эту смесь фильтровали, после чего к фильтрату добавляли свежий катализатор (0,75 г) и возобновляли гидрогенизацию под давлением около 45 фунтов на кв. дюйм, продолжая ее в течение 22 ч. Полученную смесь фильтровали через целит, а фильтрат выпаривали с образованием пены (0,995 г, 88%). Эту пену растворяли в растворе HCl в метаноле, в результате чего происходило осаждение твердого вещества белого цвета. Твердое вещество фильтровали, промывали холодным метанолом, а затем простым эфиром и сушили в условиях вакуума, что позволило получить гидрохлорид в виде твердого вещества слегка розоватого цвета (0,59 г), температура плавления 215oC.
Инфракрасный спектр (KBr): 3320 (Br), 1630, 1550 см-1.
Спектр 1H ЯМР: 7,07 (с., 1H); 6,80 ((д., J = 2,1 Гц, 1H); 6,59 (двойной дублет, J = 2,2, 8,6 Гц, 1H); 4,91-4,84 (м., 2H); 3,90 (с., 3H); 3,8-3,4 (м. , 7H); 3,2-3,0 (м., 4H); 2,69-2,62 (м., 2H); 2,2-2,0 (м., 2H).
Элементный анализ:
Высчитано для C20H25N5O2 • 2,25 HCl: C 53,44; H 6,11; N 15,58.
Обнаружено: C 53,30; H 5,90; N 15,40.
Пример 57. Гидрохлорид N-бутил-3-[3-[4-/5-метокси-4-пиримидинил/ -пиперазин-1-ил]проп-1-ил]-1H-индол-5-карбоксамида
Металлический натрий (0,84 г, 36,67 ммоль) растворяли в 15 мл безводного метанола и добавляли н-бутиламин (2,68 г, 36,67 ммоль) при 0oC. Затем добавляли раствор метил-3-[3-[4-[/5-метокси-4-пиримидинил/пиперазин-1-ил] проп-1-ил]-1H- индол-5-карбоксилата (1,5 г, 3,67 ммоль) в 5 мл метанола. Реакционную смесь нагревали с обратным холодильником в течение 24 ч. Реакционную смесь охлаждали, а затем резко охлаждали 5 мл воды. Метанол удаляли в условиях вакуума. Остаток экстрагировали тремя 30 мл порциями этилацетата. Соединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях вакуума. Хроматография концентрата на силикагеле (смесь CH2Cl2, метанола и NH4OH в отношении 94:6:0,5) привела к образованию N-бутил-3-[3-[4-/5-метокси-4-пиримидинил/пиперазин-1-ил] проп-1-ил] - 1H-индол-5-карбоксамида (0,59 г, 36%). Хлористоводородную соль получали из 0,59 г N-бутил-3-[3-[4-/5-метокси-4-пиримидинил/пиперазин-1-ил] проп-1-ил]- 1H-индол-5-карбоксамида, в результате чего достигался выход указанного в заголовке соединения (0,23 г, 36%): температура плавления 180-185oC.
Элементный анализ:
Высчитано для C25H34N6O2 • 2,0 HCl: C 57,36; H 6,93; N 16,05.
Обнаружено: C 57,01; H 6,95; N 15,98.
Пример 58. 1-[3-[5-[/Трифторметил/карбонил]амино-1H-индол-3-ил] пропил] -4-/5-метокси-4-пиримидинил/пиперазингидрохлорид
К раствору 1-[3-[5-амино-1-H-индол-3-ил] пропил]-4-/5-метокси-4- пиримидинил/пиперазина (0,5 г, 1,36 ммоль) в метиленхлориде (8 мл) при температуре 0oC добавляли трифторуксусный ангидрид (0,34 г, 1,63 ммоль). Реакционную смесь оставляли на 2 ч для нагревания до комнатной температуры. Полученный раствор разбавляли водой, органический слой сушили (MgSO4), фильтровали и концентрировали в условиях вакуума. Хроматография на колонках из силикагеля (смесь этилацетата и метанола в отношении 95:5) полученного концентрата привела к образованию 1-[3-[5-[/трифторметил/карбонил]амино-1-H-индол-3-ил] пропил] -4-/5- метокси-4-пиримидинил/пиперазина (0,2 г, 32%). Свободное основание (0,2 г, 0,43 ммоль) растворяли в минимальном объеме этанола и превращали в его хлористоводородную соль с использованием 2 мл 3 н. раствора HCl в этаноле. Летучие вещества удаляли в условиях вакуума с образованием указанного в заголовке соединения (0,19 г, 0,28 ммоль, 67%), температура плавления 295 - 300oC.
Элементный анализ:
Высчитано для C22H25F3N6O2 • 5,5 HCl: C 39,86; H 4,64; N 12,68.
Обнаружено: C 40,05; H 5,03; N 12,33.
Пример 59. 1-[3-[5-[[/4-Метилфенил/сульфонил] амино]-1H-индол-3-ил]-пропил]-4-/5-метокси-4-пиримидинил/пиперазиноксалат
Паратолуолсульфонилхлорид (0,30 г, 1,56 ммоль) растворяли в 2 мл тетрагидрофурана и охлаждали полученный раствор до 0oC. К раствору паратолуолсульфонилхлорида добавляли смесь 1-[3-[5-амино-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/ пиперазина (0,5 г, 1,42 ммоль) и триэтиламина (0,16 г, 1,56 ммоль) в 3 мл тетрагидрофурана. Этот раствор перемешивали при 0oC в течение 30 мин, нагревали до 23oC (1 ч) и концентрировали в условиях вакуума. Хроматография на колонках из силикагеля (смесь CH2Cl2, метанола и 30% NH4OH) полученного концентрата привела к образованию 1-[3-[5-[[/4-метилфенил/сульфонил] амино-1H-индол-3-ил] пропил] -4-/5-метокси-4-пиримидинил/-пиперазина (0,42 г, 0,8 ммоль, 57%). Свободное основание (0,42 г, 0,80 ммоль) растворяли в минимальном объеме ацетонитрила. При перемешивании добавляли концентрированный раствор щавелевой кислоты (0,072 г, 0,8 ммоль) в CH3CN. Осадок отделяли фильтрованием, умеренно промывали CH3CN и сушили при 70oC в условиях вакуума в течение ночи, что позволило получить указанное в заголовке соединение (0,40 г, 0,66 ммоль) 82%): температура плавления 125 - 127oC.
Элементный анализ:
Высчитано для C27H32N6O3S1 • 1,0 C2H2O4: C 55,40; H 5,77; N 13,36.
Обнаружено: C 55,29; H 5,41; N 13,15.
Пример 60. 1-[3-[5-[2-Пирролидинон-1-ил]-1H-индол-3-ил]пропил]-4-/5-метокси-4-пиримидинил/пиперазиноксалат
К смеси 1-[3-[5-амино-1H-индол-3-ил]пропил]-4-/5-метокси-4- пиримидинил/пиперазина (2,3 г, 6,3 ммоль) и карбоната натрия (0,67 г, 6,3 ммоль) в 25 мл ацетона, охлажденного до 0oC, добавляли по каплям 4-хлорбутирилхлорид (0,89 г, 6,3 ммоль). Вводили дополнительное количество ацетона (10 мл) и перемешивали суспензию в течение 19 ч при 23oC. Нерастворимое вещество отделяли фильтрованием и удаляли ацетон в условиях вакуума. Неочищенное вещество растворяли в этилацетате, полученный раствор промывали водой, сушили (рассол, Mg SO4), фильтровали и концентрировали в условиях вакуума с образованием неочищенного 1-[3-[5-[4-хлорбутириламино]-1H-индол-3-ил]пропил]-4-/5-метокси- 4-пиримидинил/пиперазина (1,98 г, 4,2 ммоль, 67%) в виде смолы. К раствору неочищенного 1-[3-[5-[4-хлорбутирил-амино]-1H-индол-3-ил] пропил] -4-/5-метокси-4-пиримидинил/пиперазина (1,0 г, 2,1 ммоль) в этаноле (5 мл) по каплям добавляли этилат натрия (0,67 мл, 2,1 ммоль, 21% в этаноле). После окончания добавления реакционную смесь нагревали при 78oC в течение 90 мин, а затем охлаждали до комнатной температуры. Нерастворимое твердое вещество отделяли и концентрировали фильтрат в условиях вакуума. Хроматография на колонках из силикагеля (смесь CH2Cl2, метанола и 30% NH4OH в отношении 95:5: 0,5) позволила получить 1-[3-[5-[2-пирролидинон-1-ил]-1-H-индол-3-ил]пропил] -4-/5-метокси -4-пиримидинил/пиперазин (0,33 г, 0,76 ммоль, 36%). Свободное основание (0,25 г, 0,58 ммоль) растворяли в минимальном объеме ацетонитрила. При перемешивании добавляли концентрированный раствор щавелевой кислоты (0,052 г, 0,58 ммоль) в CH3CN. Осадок отделяли фильтрованием, умеренно промывали CH3CN и сушили при 70oC в условиях вакуума в течение ночи, что позволило получить указанное в заголовке соединение (0,17 г, 56%); температура плавления 100 - 101oC.
Элементный анализ:
Высчитано для C24H30N6O2 • 1,0 C2H2O4 • 1,15 H2O: C 57,26; H 6,34; N 15,41.
Обнаружено: C 56,90; H 5,96; N 15,51.
Пример 61. 4-/5-Метокси-4-пиридинил/-1-[3-[5-[[/метиламино/сульфонил]-метил] -1H-индол-3-ил]пропил]пиперазингидрохлорид
Раствор 3-/3-индопропил/-N-метил-1H-индол-5-метансульфонамида (0,556 г, 1,26 ммоль), 1-/3-метокси-4-пиридинил/пиперазина (0,4 г, 2,02 ммоль) и K2CO3 (0,5 г, 3,6 ммоль) в 30 мл MeCN нагревали до температуры кипения с обратным холодильником в течение 12 ч. Реакционную смесь охлаждали, концентрировали в условиях вакуума и растворяли остаток в этилацетате. Этилацетатный раствор промывали водой, сушили (MgSO4), фильтровали и концентрировали в условиях вакуума. Хроматография остатка на силикагеле (смесь CH2Cl2, метанола и этилнитрида в отношении 100:3:1) позволила получить 4-/5-метокси-4-пиридинил/-1-[3-[5-[[/метиламино//сульфонил] метил]-1H-индол-3-ил]пропил]пиперазин (300 мг, 52%). Хлористоводородную соль получали путем растворения свободного основания в 3 экв. 1,5 М раствора HCl в этаноле. Этот раствор концентрировали в условиях вакуума, а остаток перекристаллизовывали из метанола с образованием указанного в заголовке соединения (380 мг, 50%): температура плавления 100 - 103oC.
Элементный анализ:
Высчитано для C23H31N5O3S • 3,8 CHl: C 46,34; H 5,88; N 11,78.
Обнаружено: C 46,32; H 6,18; N 11,67.
Пример 62. 1-[3-/1H-Индол-3-ил/пропил]-4-/3-метокси-4-пиридинил/пиперазиндигидрохлорид
В соответствии с процедурой, описанной в примере 61, получали указанный в заголовке пиридинильный продукт 1B, температура плавления 225oC (разложение).
Элементный анализ:
Высчитано для C12H26N4O•2,0 HCl•0,15 H2O: C 59,20; H 6,69; N 13,15.
Обнаружено: C 58,96; H 6,91; N 12,85.
Пример 63. 1-[3-/1H-Индол-3-ил/пропил]-4-/3-метокси-4-пиридинил/- 2-метилпиперазингидрохлорид
В соответствии с процедурой, описанной с примере 61, получали указанный в заголовке пиридинильный продукт, температура плавления 209 - 211oC (разложение).
Элементный анализ:
Высчитано для C22H28N4O • 1,85 HCl • 0,13 C3H8O: C 60,23; H 7,12; N 12,55.
Обнаружено: C 60,57; H 7,22; N 12,48.
В результате соответствующего изменения процедур, описанных в представленных выше примерах синтеза, можно получить дополнительные соединения формулы XII. Выбранные соединения, полученные в соответствии с дополнительными примерами, приведены в табл. 1
Сродство с участками связывания 5-HT1D, характерное для типичных соединений, приведено в табл. 2.
Результаты исследований и состав соединения примера 2 приведены в табл. 3.
Соединение с нейтральными компонентами измельчают в порошок, перемешивают и помещают в сухие желатиновые капсулы.
Состав ингредиентов приведен в табл. 4.
Соединение тонко измельчают и смешивают с также измельченными инертными ингредиентами согласно рецептуре, а затем прессуют в таблетки на стандартном оборудовании.
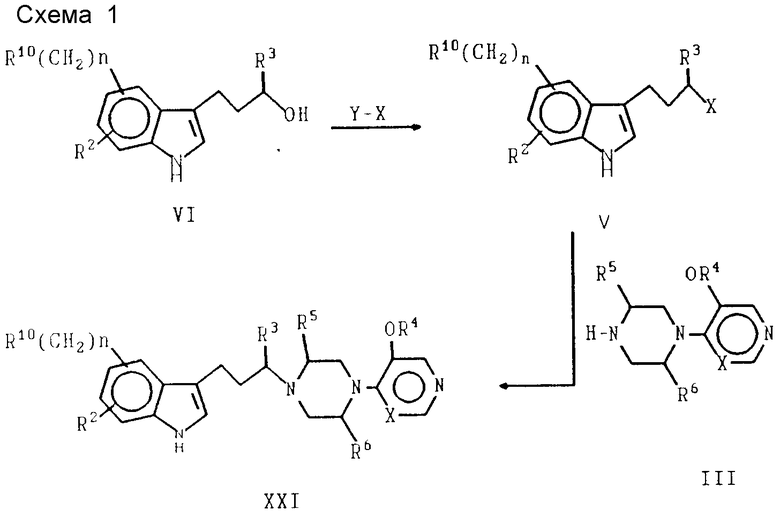
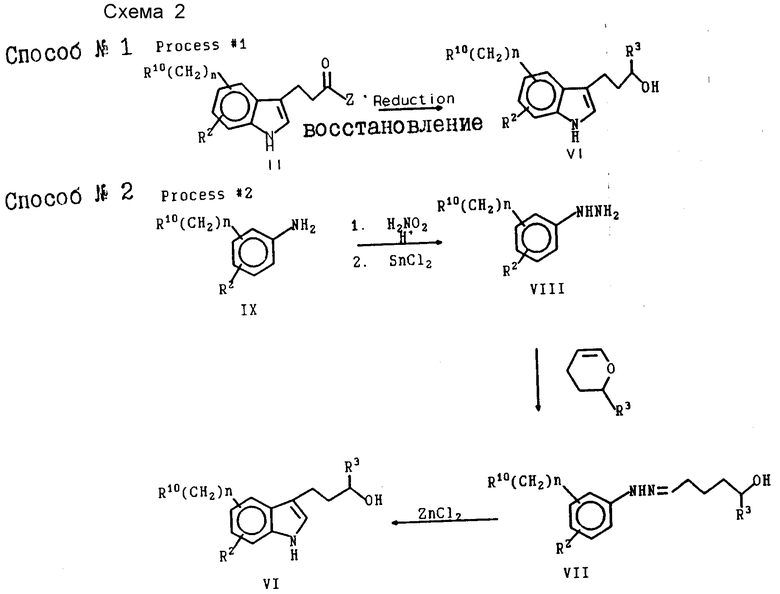
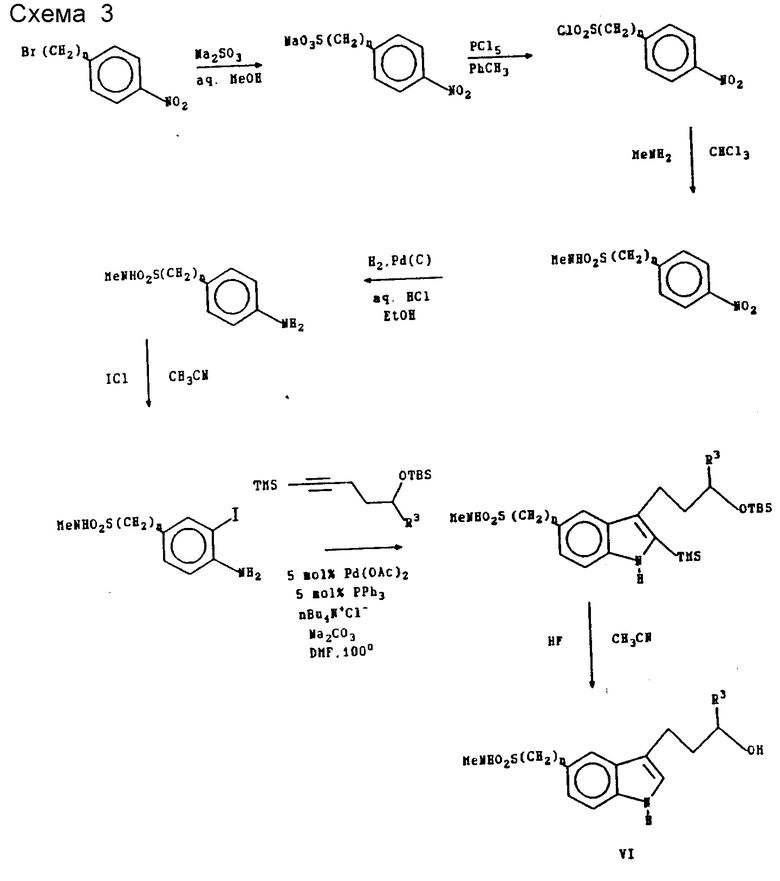
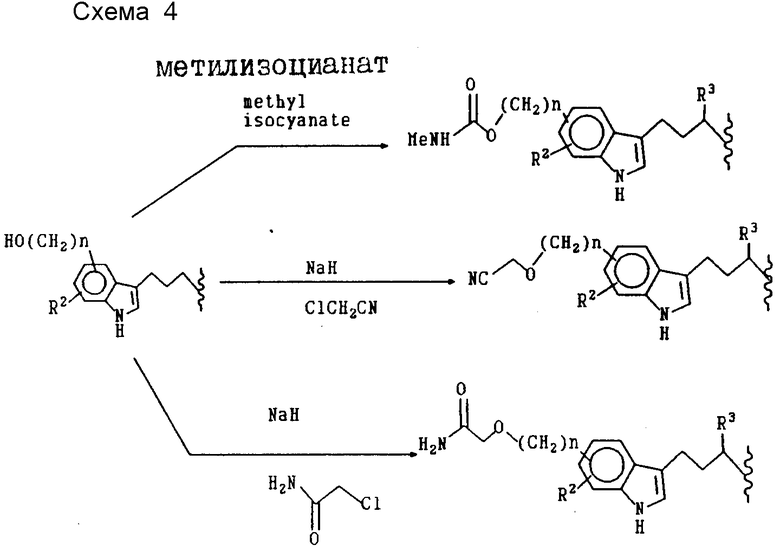

Изобретение относится к 4-пиримидин- или пиридинильным производством индол-3-ил-алкилпиперазинов формулы 1, где X - = N - или -CH=; R1 - водород, галоген, низшая алкоксигруппа, аминогруппа, цианогруппа, гидрокси, нитрогруппа, группа формулы: -OCH2CN, -OCH2CONH2, -SO2NH2, -O2CR9, NR7SO2R9, где R7 - водород, низший алкил; R9 - низший алкил; или группа формулы: 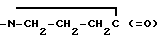 R2 - водород, низшая алкоксигруппа; группа формулы: CO2R9, где R9 - низший алкил, галоген; R3 - водород, низший алкил; R4 - низший алкил; R5 - водород; R6 - водород, низший алкил; при условии, что часть молекулы R1 - (CH2) не может быть водородом, низшим алкилом, низшей алкоксигруппой, -N- R7SO2R9, если X == N-. Соединения формулы I используют в качестве активного начала в количестве 1 - 500 в фармацевтической композиции агонистического действия в отношении рецепторов серотонина 5 - НТ1Д, которая содержит и фармацевтически приемлемые целевые добавки.
R2 - водород, низшая алкоксигруппа; группа формулы: CO2R9, где R9 - низший алкил, галоген; R3 - водород, низший алкил; R4 - низший алкил; R5 - водород; R6 - водород, низший алкил; при условии, что часть молекулы R1 - (CH2) не может быть водородом, низшим алкилом, низшей алкоксигруппой, -N- R7SO2R9, если X == N-. Соединения формулы I используют в качестве активного начала в количестве 1 - 500 в фармацевтической композиции агонистического действия в отношении рецепторов серотонина 5 - НТ1Д, которая содержит и фармацевтически приемлемые целевые добавки.
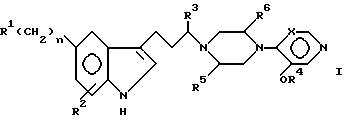
2 с. и 8 з.п.ф-лы, 4 табл.
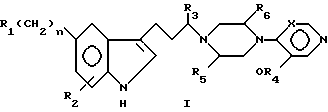
где X - =N- или -CH=;
R1 - водород, галоген, низшая алкоксигруппа, аминогруппа, цианогруппа, гидроксигруппа, нитрогруппа, группа формулы -OCH2CN, -OCH2CONH2, -SO2NH2, -O2CR9, NR7SO2R9, где R7 - водород, низший алкил, R9 - низший алкил, или группа формулы
R2 - водород, низшая алкоксигруппа, группа формулы CO2R9, где R9 - низший алкил, галоген;
R3 - водород, низший алкил;
R4 - низший алкил;
R5 - водород;
R6 - водород, низший алкил;
n равно 0 или целому числу 1 или 2,
при условии, что часть молекулы R1-(CH2) не может быть водородом, низшим алкилом, низшей алкоксигруппой, -NR7SO2R9, если X является =N-,
или их фармацевтически приемлемые соли присоединения кислоты. .
.
Приоритеты по признакам:
19.12.91 - относится к пиримидинам, где X - N;
13.10.92 - относится к пиридинам, где X - CH.
| EP, 0354094, A 61 K 31/40, 1990. |
