Изобретение касается новых арилциклоалкильных производных, фармацевтических композиций на их основе.
Известны халконы общей формулы 1a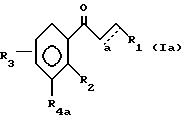
в которой R1 - замещенный фенил;
R2 - OH;
a - одинарная или двойная связь,
R3 - OH,
R4a - H, изопренил или изопентил,
и которые являются эффективными для лечения заболеваний, вызываемых гиперсекрецией андрогенов, например, гипертрофия предстательной железы, алопеция у мужчин, обыкновенные угри или себорея /Патент Японии N 281022/.
Известны также соединения указанной выше формулы 1a, в которой
R1 - замещенный фенил;
R2 - H, OH, ацетокси, карбоксиметокси или метоксикарбонилметокси;
R3 - OH, метокси, бензилокси, H;
R4a - H, изопренил, изопентил,
данные соединения обладают активностью, направленной против гиалуронидазы /Патент Японии N 026775/.
Кроме того известны соединения формулы 1a, в которой
R1 - замещенный фенил,
R2 - OH, ацетокси, карбоксиметокси, метоксикарбоксиметокси,
R3 - OH, метокси, H,
a - одинарная или двойная связь,
R4a - изопренил, изопентил, н-пропил или H, и являются полезными ингибиторами альдозоредуктазы, используемыми для лечения осложнений, вызываемых диабетом, таких как катаракты, ретиниты, неврологические нарушения или заболевания почек /Патент Японии 142166/.
А также соединения формулы 1a, в которой
R1 - замещенный фенил,
R2 - OH,
R3 - OH,
a - двойная связь,
R4a - H,
являются полезными ингибиторами альдозоредуктазы, используемыми для лечения воспалительных осложнений, называемых диабетом /Патент Японии 248389/.
Соединения формулы 1a, в которой
R1 - замещенный фенил,
R2 - H или OH,
R3 - H или OH,
a - двойная связь,
R4a - H или OH,
являются полезными ингибиторами C-киназы и противоопухолевыми средствами /Патент Японии N 144717/.
Соединения формулы 1a, в которой
R1 - замещенный фенил,
R2 - H, галоген, низший алкил, низший алкокси, CN, карбокси, нитро,
R3 - H, галоген, низший алкил, низший алкокси, CN, карбокси, нитро, гидрокси, замещенное производное уксусной кислоты,
R4a - имеет значения, указанные для R3,
обладает ингибирующим действием в отношении гидроксипростагландин-дегидрогеназы. Они могут обладать потенциальной местной активностью в отношении нарушений желудочно-кишечного тракта, таких как язва желудка и язвенные колиты. Другие возможные области применения включают лечение ревматического артрита, нарушений кровообращения, рака, отсутствия фертильности и клеточной регуляции /EP N 150166/.
Соединения формулы 1a, в которой
R1 - замещенный фенил,
R2 - H,
R3 - OH,
a - одинарная связь,
R4a - OH,
являются избирательными ингибиторами 5-липоксигеназы и обладают великолепным противоаллергическим действием, представляя безопасное противоаллергическое лекарственное средство, такое как противоастматическое, противовоспалительное и иммуноактивирующее лекарственное средство /Патент Японии N 167288/.
Настоящее изобретение относится к соединениям формулы I, в которой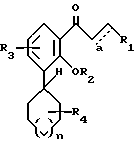
где R1 - остаток, выбираемый из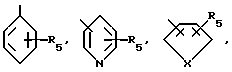
где R5 представляет один, два, три или четыре остатка, которые независимо друг от друга включают H, C1-C6-алкил, замещенный C1-C6-алкил, гидрокси, C1-C6-алкокси, C1-C4-алкил-O-C1-C4-алкил, карбокси, циано, -NHC(O)C1-3-алкил, -OC1-3 алкилфенил, -OCH2O-, -C/O/-O-C1-C4-алкил, галогены, амино, нитро, -NH- C1-C4-алкил, -n-N/C1-C4-алкил/2;
X = O, S, N-H, N-C1-C6-алкил;
R2 - H, C1-C6-алкил, -C/O/-C1-C6-алкил;
R3 - один, два или три остатка, которые независимо друг от друга представляют H, C1-C6-алкил, -C/O/-C1- C6-алкил, -C/O/-O-C1-C6-алкил, OH, -O-C1-C6-алкил, -O-C/O/-C1-C6-алкил, галоген;
R4 - H, -OH, -O-C1-C6-алкил, -O-C/O/-C1-C6-алкил, -C/O/-OH, -C/O/-O-C1-C6-алкил,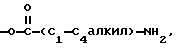
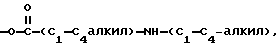
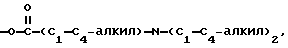
n = 0, 1 или 2;
a представляет необязательную дополнительную одинарную связь.
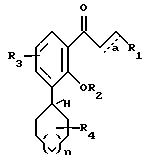
Предпочтительными соединениями являются соединения формулы II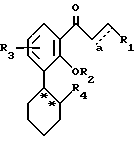
в которой R1, R2, R3, R4 и a имеют указанные выше значения.
Из этой группы соединений предпочтение отдается таким соединениям, в которых R1 представляет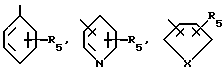
где R5 означает H, C1-C6-алкил, замещенный C1-C6-алкил, гидрокси, C1-C3-алкокси, галоген;
R4 означает H, OH или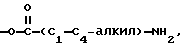
X представляет O, NH, S, N-C1-C6-алкил и
a означает необязательную дополнительную связь.
Особенно предпочтительным являются соединения формулы III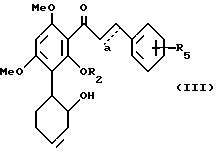
в которой R2 представляет H или C1-C3-алкил;
R5 означает один или два галогена либо одну или две C1-C6-алкильные или C1-C3-алкоксильные группы, и a означает необязательную дополнительную одинарную связь.
Приведенный выше термин "замещенный алкил" означает алкил, предпочтительно C1-3 алкил, замещенный предпочтительно одним галогеном, гидрокси, C1-C3-алкокси, амино, C1-C4-алкиламино, ди-/C1-C4-алкил/-амино, карбонилом или карбокси-C1-C4-алкилом.
Соединения по настоящему изобретению содержат два асимметричных центра, обозначенных звездочками в формуле II, в точках присоединения R4 /например, соединение формулы II, в которой R4 - H/ и арильной группы в карбоциклическом кольце; поэтому возможно получение четырех изомеров, обозначаемых в отдельности как цис-/+/, цис-/-/, транс-/+/ и транс-/-/ формы.
Настоящее изобретение включает все четыре изомера как в отдельности, так и в виде смеси двух или большего числа из четырех изомеров.
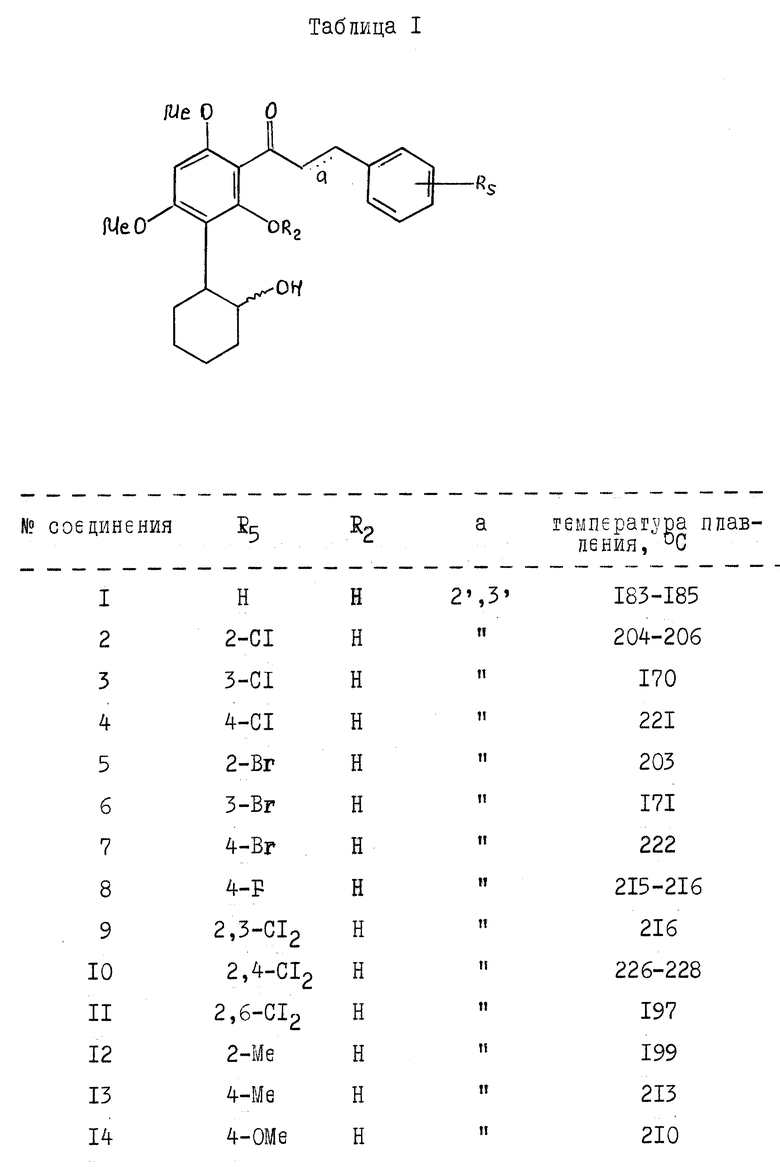
Примеры особенно предпочтительных соединений включают:
1. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-хлор-фенил// проп-2-еноил]фенилциклогексанол;
2. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2- хлорфенил// -проп-2-еноил]фенилциклогексанол;
3. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/3-хлорфенил// проп-2-еноил]фенилциклогексанол;
4. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2-бромфенил// проп-2-еноил]фенилциклогексанол;
5. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/3-бромфенил// проп-2-еноил]фенилциклогексанол;
6. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-бромфенил// проп-2-еноил]фенилциклогексанол;
7. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-фторфенил// проп-2-еноил]фенилциклогексанол;
8. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2-метилфенил// проп-2-еноил]фенилциклогексанол;
9. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-метилфенил// проп-2-еноил]фенилциклогексанол;
10. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2,3- дихлорфенил//проп-2-еноил]фенилциклогексанол;
11. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2,6- дихлорфенил//проп-2-еноил]фенилциклогексанол;
12. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/2,6- дихлорфенил// проп-2-еноил]фенилциклогексанол.
13. транс-/+/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-хлорфенил// проп-2-еноил]-фенилциклогексанол;
14. транс-/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-хлорфенил// проп-2-еноил]фенилциклогексанол;
15. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-метоксифенил// проп-2-еноил]фенилциклогексанол;
16. транс-/-/-2-[4,6-диметокси-2-гидрокси-3/3-/3-метоксифенил// проп-2-еноил]фенилциклогексанол;
17. транс-/+/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-хлор-3- нитрофенил// проп-2-еноил]фенилциклогексанол;
18. транс-/-/-2-[4,6-диметокси-2-гидрокси-3-/3-/4-хлор-3- нитрофенил// проп-2-еноил]фенилциклогексанол;
19. транс-/+/-/-1-[4,6-диметокси-2-гидрокси-3-/2-/ β -амино /ацетокси/-циклогексил]фенил-1-/3-/3,4-диметокси/фенил/пропанол гидрохлорид.
Также настоящее изобретение касается способа получения соединений формулы I, описанных выше, в соответствии с которым соединение формулы V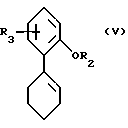
/A/ превращают в соединение формулы VI, в которой R4 означает OH,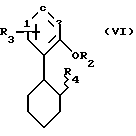
путем обработки комплексом борана с растворителем с последующим окислением, /B/ для получения соединения формулы VI соединение формулы V обрабатывают перкислотой и полученный таким образом эпоксид обрабатывают гидридом, /C/ соединение формулы VI получают путем конденсации соответствующего арена с циклогексеноксидом в присутствии кислотного катализатора, /D/ затем соединение формулы VI обрабатывают уксусным ангидридом и минеральной кислотой с образованием соединения формулы VII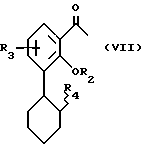
в которой R2 представляет метил, а R4 представляет O-C/O/-Me, и /E/ соединение формулы VII, описанное в пункте /D/, деметилируют путем обработки кислотой Льюиса или деметилирующим агентом с образованием соединения формулы VII, в которой R2 означает H и R4 означает OC/O/Me, /F/ соединение формулы VII, в которой R2 представляет H, а R4 означает OH, получают путем обработки соединения, полученного в пункте /E/, разбавленной щелочью, /G/ соединение формулы VII превращают в соединение формулы I /a - дополнительная связь/ путем обработки соответствующим альдегидом в присутствии основания, а соединение формулы I /a - отсутствие дополнительной связи/ получают путем гидрогенизации соединения формулы I /a - дополнительная связь/, в которой R1, R2 и R3, если нет специального указания, имеют приведенные выше значения.
Соединения формулы V получают в соответствии со способами, хорошо известными специалистам в этой области. Обычно их получают путем добавления ариллития формулы IV к циклогексанону с последующей дегидратацией, катализируемой кислотой, с образованием соединения формулы /IV/, в которой R2 и R3 имеют указанные выше значения.
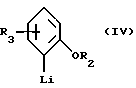
Приемлемым комплексом борана с растворителем, используемым на стадии A вышеуказанной последовательности операций, является, например, комплекс борана с тетрагидрофураном или борана с диметилсульфидом. Окисление можно производить с использованием щелочной перекиси водорода. Приемлемой перкислотой для стадии B является, например, хлорпербензойная кислота. Примером приемлемого гидрида является алюмогидрид лития.
Стадия C может осуществляться с использованием в качестве арена 1,3,5-триметоксибензола, при этом кислотным катализатором может быть хлорид алюминия.
Минеральной кислотой, необходимой для осуществления стадии, может быть, например, фосфорная кислота,
Стадия E осуществляется с использованием в качестве кислоты Льюиса трехбромистого бора, а в качестве деметилирующего агента тиолатов металла. Предпочтительной разбавленной щелочью, используемой на стадии /F/, является 2 н. раствор гидроксида натрия.
Основанием, в присутствии которого осуществляется стадия G, может быть, например, гидроксид натрия.
Продукты, полученные в соответствии с вышеуказанными стадиями реакций, можно использовать в последующих реакциях с целью получения соединений по настоящему изобретению. Большую часть указанных реакций можно выполнять в соответствии с процедурами, описанными в заявке на Европейский патент N 0241003. Дополнительную информацию об исходных продуктах, промежуточных соединениях и реакциях получения производных соединений можно получить из патентной литературы, приведенной в вводной части описания изобретения.
Физические параметры некоторых предпочтительных соединений по настоящему изобретению приведены в таблице 1.
Новые соединения по настоящему изобретению демонстрируют интересное фармакологическое действие при испытании в фармакологических моделях; соединение 4, приведенное в представленной выше таблице, будет использоваться в примерах в качестве типичного соединения.
Как показано в примерах, соединения по настоящему изобретению обладают противовоспалительными свойствами. Эти соединения являются особенно полезными для ингибирования или антагонистического воздействия на реакции, вызываемые эндогенными молекулами, такими как липоксигеназы и/или лейкотриены, интерлейкины и протеинкиназа C. Таким образом соединения по настоящему изобретению, в отдельности или в виде приемлемого состава, являются полезными лекарственными средствами для лечения воспалительных заболеваний, в частности хронических воспалительных заболеваний, таких как ревматический артрит, остеоартрит, астма и злокачественные опухоли.
Также настоящее изобретение касается способов использования вышеуказанных соединений для лечения и профилактики описанных воспалительных заболеваний путем введения активного количества одного или нескольких соединений по настоящему изобретению. Кроме того, объектом настоящего изобретения являются фармацевтические составы, содержащие одно или несколько соединений, описанных выше. Указанные фармацевтические составы можно получать и вводить в соответствии с методами, известными в этой области.
Настоящее изобретение далее иллюстрируется следующими примерами и формулой изобретения.
Пример I
Ингибирование вызываемого лейкотриеном сокращения подвздошной кишки, удаленной у морских свинок
Морских свинок обоего пола весом 300-350 г сенсибилизировали суспензией гелеобразного гидроксида алюминия и яичного альбумина. Через 21 день животных подвергали воздействию аэрозоля, содержащего 0,5% яичного альбумина, в воздухонепроницаемой камере из плексигласа, и для дальнейшего эксперимента отбирали только тех животных, у которых развивался аллергический бронхостеноз.
Животных исследователи в течение одной недели после антигенного воздействия, а затем умерщвляли ударом в голову и вскрытия сонной артерии. Быстро удаляли легкое и помещали его в аэрированный раствор Тироде, температура которого равнялась 37oC. Легкое разрезали на одинаковые полоски и каждую полоску помещали в ванну для органов, содержащую удаленную у морской свинки подвздошную кишку, соединенную с потенциометрическим самописцем через изотонический преобразователь в присутствии раствора Тироде с температурой 37oC. После стабилизации в течение 30 минут проверяли реакционную способность подвздошной кишки на гистамин путем стимуляции ее 100-200 нг/мл гистамина. Затем перфузивную жидкость заменяли раствором Тироде, содержащим атропин /10-7 М/, мепираминмалеат /10-7 М/ и метилсергид /10-7 М/. Через три минуты полоску легкого стимулировали яичным альбумином /25 мкг/мл/ и контролировали высвобождение лейкотриенов в виде медленного сокращения подвздошной кишки. Сокращение подвздошной кишки продолжалось в течение 10 - 15 минут, после чего достигалось постоянное состояние. Вслед за этим вводили испытуемое соединение /соединение 4 из таблицы 1/ с целью наблюдения релаксации. Специфичность антагонистического действия в отношении лейкотриенов определяли путем вызывания сокращения подвздошной кишки морских свинок такими агонистами, как гистамин, ацетилхолин и KCl. Соединения, оказывающие воздействие на сокращение, вызываемое липоксигеназой, обычно не ингибируют сокращение, вызываемое гистамином, ацетилхолином и KCl. Полученные данные представлены в таблице 2.
Никакого воздействия на сокращение, вызванное гистамином и KCl, не оказывалось до концентрации, равной 7,11 • 10-5 M.
Соединение 4, являющееся типичным представителем новых соединений по настоящему изобретению, ингибирует сокращение, вызываемое лейкотриенами.
Пример II
Ингибирование гранулемы, вызванной ватными шариками, у крыс
Эта модель позволяет определить потенциальную активность соединения в отношении ингибирования искусственно вызванной гранулемы. Имплантация ватных шариков, пропитанных каррагинином, вызывает образование большой хорошо выраженной гранулемы, которая легко иссекается. Эффективность соединений оценивают путем измерения величины уменьшения образования гранулематозной ткани.
Приготовление солевого раствора и ватных шариков, пропитанных каррагинином.
Для стерилизации использовали ватные тампоны весом 40 мг. Половину всех ватных шариков погружали в солевой раствор, а остальные шарики погружали в 1% водный раствор /вискарин® тип 402, фирма "Марин коллоидз инк", Спрингфилд/, где они находились до полной пропитки, после чего их слегка отжимали для удаления избытка солевого раствора или каррагинина.
Ватные шарики сушили в течение ночи под лампой. Отбирали шарики, вес которых составлял 42-44 мг.
Подготовка животных:
Крыс /группы по 6 животных, самцы или самки, Черльз ривер, Вистар, вес 140-150 г/ антестезировали простым эфиром. Шерсть на спине сбривали, а кожу очищали, протирали спиртом и в нижней области средней части спины делали разрез длиной один сантиметр. С помощью тупого пинцета создавали билатеральный канал, куда помещали один ватный шарик. Из разреза удаляли воздух и на рану накладывали швы. Испытуемое соединение готовили в 0,5% карбоксиметилцеллюлозе и вводили перорально при суточной дозе 10, 20 и 30 мг/кг в течение семи дней. Через три часа после введения последней дозы на седьмой день животных умерщвляли. Ватные шарики удаляли путем надреза кожи вдоль средней линии спины и отделения кожи от тела в обоих боковых направлениях. Ватные шарики взвешивали, а затем на ночь помещали в сушильный шкаф при температуре 140oC. Значения массы в сухом состоянии регистрировали и определяли величину гранулемы путем вычитывания первоначального веса ватных шариков из их массы во влажном и сухом состоянии. Полученные данные оценивали исходя из различий между массами в левой и правой колонках /см. таблицу 3/.
Соединение 4, являющееся типичным представителем соединений по настоящему изобретению, ингибирует образование гранулемы, вызванной каррагинином.
Пример III
Ингибирование микроанафилактического шока у морских свинок
Морских свинок обоего пола весом 300-350 г сенсибилизировали яичным альбумином, абсорбированным над Al/OH/3 гелем. Через 21 день после сенсибилизации животных помещали в воздухонепроницаемую камеру из плексигласа и подвергали воздействию аэрозоля, содержащего 0,5% яичного альбумина, с помощью распылителя EEL. Распылитель EEL приводился в действие путем подсоединения к источнику сжатого воздуха через водоотделитель и сфигмоманометр с круглой шкалой при создании постоянного давления воздуха, равного 180 мм рт. ст. Отмечали время возникновения астмы в секундах и период восстановления в минутах.
Всех животных подвергали воздействию аэрозоля на основе яичного альбумина с интервалом в 15 дней с целью сохранения у животных сопоставимой реакции на антиген. После 3 таких контрольных воздействий животными начинали вводить лекарственный препарат. В день эксперимента одну группу морских свинок, включающую 10 животных, определяли как контрольную и подвергали ее воздействию аэрозоля, содержащего 0,5% яичного альбумина, без введения лекарственного препарата. Другой группе, включающей 10 морских свинок, за 30 минут до воздействия антигеном внутрибрюшинно вводили индометацин в количестве 10 мг/кг. Другой группе, состоящей из 10 морских свинок, внутрибрюшинно вводили индометацин в количестве 10 мг/кг до начала эксперимента и через 30 минут после введения индометацина вводили испытуемое соединение /20 мг/внутрибрюшинно/. Через пятнадцать минут после введения испытуемого соединения животных подвергали воздействию аэрозоля, содержащего 0,5% яичного альбумина. В каждой группе отмечали время возникновения приступа и время восстановления /см.таблицу 4/.
Соединение 4, являющееся типичным представителем новых соединений по настоящему изобретению, защищает животных от бронхостеноза, вызываемого лейкотриенами, после воздействия аэрозолем на основе яичного альбумина.
Пример IV
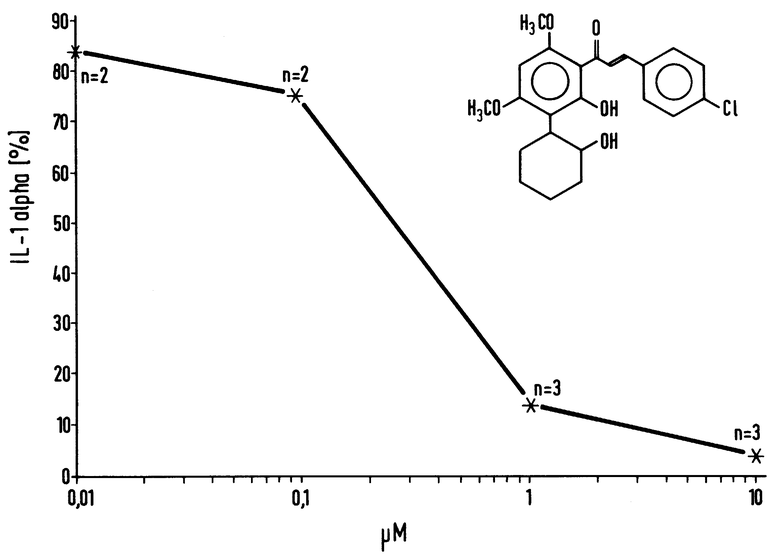
Ингибирование синтеза IL-1 мононуклеарными клетками человека
Очистка мононуклеарных клеток, получаемых из человеческой крови
Из вены, расположенной кпереди от локтевого сустава, с помощью шприца, содержащего 1 мл раствора 3,8% цитрата натрия, осторожно отбирали 10 мл человеческой крови. После разбавления 10 мл РМ 16 /Серва, Хайдельберг, ФРГ/ и подслаивания 15 мл лимфопрепа® /Молтер GmbH/ пробу центрифугировали с градиентом 400 х г в течение 40 минут при температуре 20oC. Мононуклеарные клетки, образующие белое кольцо между лимфопрепом и плазмой, осторожно отсасывали шприцем, разбавляли РМ 16 в отношении 1:1 и снова центрифугировали с градиентом 400 х г в течение 10 минут. Надосадочную жидкость промывали 10 мл RPM1 1640 /Гибко, Берлин, ФРГ/, дополнительно содержащего 300 мг /л L-глутамина, 25 ммоль/л HEPES, 100 мкг/мл стрептомицина и 100 мкг/мл пенициллина. И, наконец, с помощью устройства для подсчета клеток IT доводили суспензию до 5 • 106 клеток/мл. Эти клетки состояли примерно из 90% лимфоцитов и 10% моноцитов.
Стимуляция синтеза интерлейкина 1 мононуклеарными клетками человека в лабораторных условиях
10 мкл смеси диметилсульфоксида и воды /в объемном отношении 1:10/, содержащей испытуемое соединение, добавляли к 480 мкл суспензии, содержащей 5 • 106 мононуклеарных клеток. Синтез IL-1 стимулировали путем добавления 10 мкл смеси диметилсульфоксида и воды /в объемном отношении 1:10/, содержащей 0,5 мкг липополисахарида /Salmonella abortus equi, Сигма/. После инкубации при температуре 37oC в течение 18 часов пробы охлаждали до 0oC и центрифугировали 1 минуту в настольной центрифуге. 25 мкл аликвоты надосадочной жидкости оценивали на активность в отношении IL-1 альфа с помощью выпускаемого промышленностью набора для радиоиммуноанализа 125-J-IL-1-альфа /Амершам, Великобритания/, а также в отношении IL-1 бета с помощью типичного лабораторного набора. Контрольные эксперименты выполняли так, как описывалось выше, без испытуемого соединения или с использованием циклогексимида в качестве испытуемого соединения.
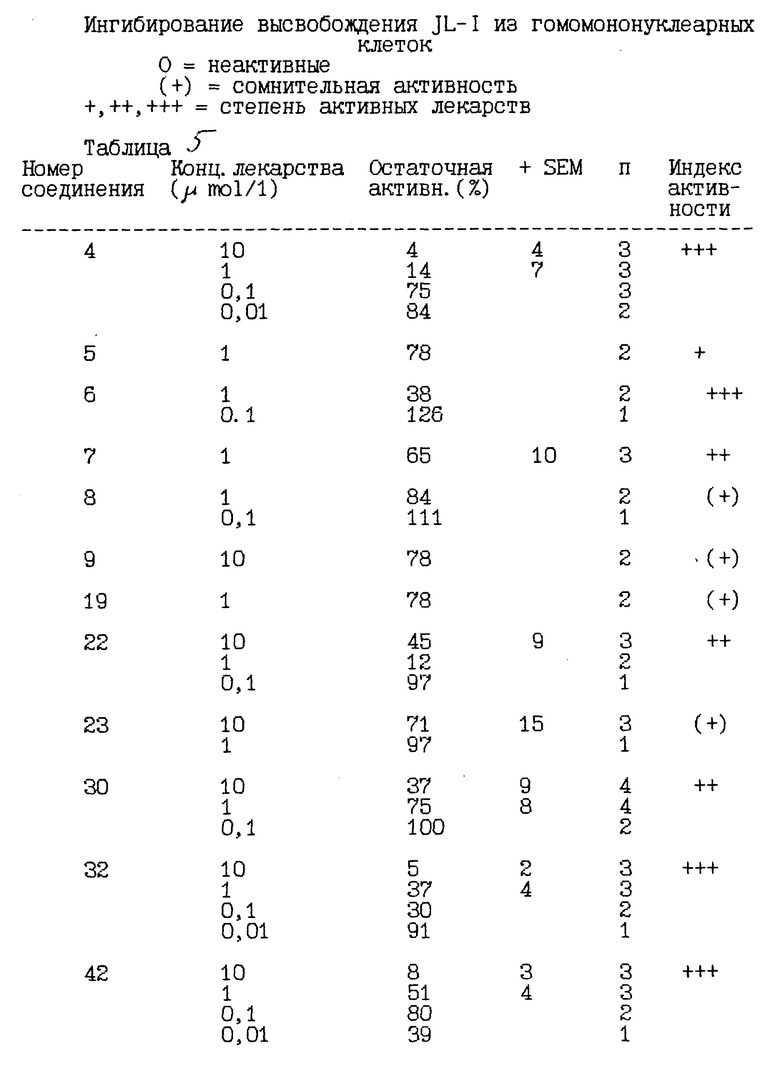
Воздействие соединения 4 в качестве ингибитора синтеза IL-1 альфа, стимулируемого липополисахаридом /LPS/ /IC50 = 200 - 300 нмоль/л/, показано на чертеже.
Соединение 4, являющееся типичным представителем соединений по настоящему изобретению, ингибирует высвобождение IL-1 альфа, стимулируемое липополисахаридом, из мононуклеарных клеток человека в лабораторных условиях.
Соединения данной заявки получены по методикам, представленным ниже.
Пример V
Получение 1-(2,4,6-триметоксифенил)циклогексена. Пример соединения формулы V, где R3 = 4,6-диметокси, R2 = CH3.
2,4,6-Триметоксибромбензол (1 экв.) помещают в высушенную в пламени 3-х горлую колбу в атмосфере азота. Добавляют сухой тетрагидрофуран (ТГФ) (988 мл) и охлаждают реакционную смесь до -30oC. По каплям добавляют раствор р-BuLi (1,3 экв.) в гексане (коммерческий), а затем реакционную смесь перемешивают в течение 30 мин. С помощью тонкослойной хроматографии определяют окончание реакции металлирования. При -30oC к смеси добавляют циклогексанон (1,1 экв.), разбавленный равным объемом сухого ТГФ, и перемешивают еще один час при -30oC. При комнатной температуре к реакционной смеси добавляют 150 мл воды и экстрагируют этилацетатом. Этилацетатный экстракт сушат безводным сульфатом натрия и упаривают. Остаток растворяют в дихлорметане и перемешивают в течение 30 мин в присутствии каталитического количества 4-толуолсульфокислоты (9 г). Дихлорметановую фазу промывают раствором бикарбоната натрия, а затем водой и сушат. Остаток кристаллизуют из диизопропилового эфира и получают указанное соединение, т.пл. 127oC, выход 64,7%.
Пример VI
Получение транс-(±)-2-(2,4,6-триметоксифенил)циклогексанола. Пример соединения формулы VI, где R2 = CH3, R3 = 4,6-диметокси и R4 = OH.
К соединению формулы V (из примера 1) (1 экв.) добавляют боргидрид натрия (4 экв.) и сухой ТГФ (2200 мл). Полученную смесь охлаждают до 0oC в атмосфере азота и по каплям добавляют трифторэфират бора (5,1 экв.). По окончании добавления температуру смеси повышают до 50oC и перемешивают в течение 30 мин. Затем реакционную смесь охлаждают до комнатной температуры и по каплям добавляют воду для разрушения избытка диборана. Для окисления органоборана добавляют 30%-ный раствор H2O2 (248 мл) и 3 М раствор NaOH (248 мл). По окончании добавления смесь нагревают до 50oC и перемешивают в течение 3 ч. После завершения окисления реакционную массу разбавляют водой и экстрагируют этилацетатом. Экстракт сушат и упаривают. Сырой продукт очищают с помощью экспресс-хроматографии на силикагеле, элюент петролейный эфир-этилацетат 9:1, т.пл. 123oC, выход 52%.
Пример VII
Получениетранс-(±)-1-/3-(2-ацетокси)циклогексил-2,4,6-триметокси/фенил-1-этанона. Пример соединения формулы VII, где R3 = 4,6-диметокси, R2 = CH3 и R4 = OCOCH3.
Продукт примера II (1 экв.) растворяют в сухом метиленхлориде (1520 мл). К раствору добавляют уксусный ангидрид (25 экв.) и фосфорную кислоту (152 мл) и перемешивают смесь при комнатной температуре в течение часа. Реакционную смесь нейтрализуют раствором карбоната натрия до щелочной реакции и экстрагируют дихлорметаном. Органический слой тщательно промывают водой и сушат, Сырой продукт после упаривания растворителя кристаллизуют из петролейного эфира, т.пл. 87oC, выход 84%.
Пример VIII
Получение транс-(±)-1-/3-(2-ацетокси)циклогексил-4,6-диметокси-2- гидрокси/фенил-1-этанона. Пример соединения формулы VII, где R2 = H, R3 = 4,6-диметокси и R4 = OCOCH3.
Продукт примера III (1 экв.) растворяют в сухом дихлорметане (5450 мл), охлаждают до 0oC, добавляют трибромид бора (1,1 экв.) с помощью шприца и перемешивают при 0oC в течение часа. Затем осторожно добавляют воду и продукт экстрагируют этилацетатом. Экстракт промывают водой, сушат безводным сульфатом натрия. Сырой продукт кристаллизуют из этилацетата, получают указанное соединение, т.пл. 151oC, выход 70 - 71%.
Пример IX
Получение транс-(±)-2-/3-ацетил-4,6-диметокси-2-гидрокси/- фенилциклогексанола. Пример соединения формулы VII, где R2 = H, R3 = 4,6-диметокси, R4 = OH.
Продукт примера IV (1 экв.) перемешивают в атмосфере азота с раствором гидроксида калия в метаноле (20 экв., MeOH : вода = 3:1) в течение 6 ч. Реакционную массу подкисляют разбавленной HCl, отфильтровывают осадок, промывают, сушат и кристаллизуют из этилацетата, т.пл. 161oC, выход 88 - 89%.
Пример X
Получение транс-(±)-2-[4,6-диметокси-2-гидрокси-3-(3-(4-хлорфенил)проп-2- (E)-еноил)] фенилциклогексанола. Пример соединения формулы II, где R1 = 4-хлорфенил, a = другая связь, R2= H, R3 = 4,6-диметокси, R4 = OH.
Смесь продукта примера V (1 экв.) 4-хлорбензальдегида (3 экв.) и 10%-ного спиртового раствора гидроксида натрия (30 экв.) перемешивают при комнатной температуре в течение 24 ч. Затем при температуре 0oC реакционную массу подкисляют разбавленной HCl до pH 5 и отфильтровывают оранжевый осадок, который перекристаллизовывают из этанола, т.пл. 221oC, выход 68%.
Пример XI
Получение транс-(±)-2-[4,6-диметокси-2-гидрокси-3-(3- (4-хлорфенил)пропаноил)] фенилциклогексанола. Пример соединения формулы II, где R1 = 4-хлорфенил, a = нет связи, R2 = H, R3 = 4,6-диметокси и R4 = OH.
Продукт из примера VI перемешивают с 10% Pd/C (5 мол.%) в этиловом спирте в атмосфере водорода в течение ночи. Катализатор отфильтровывают, фильтрат упаривают. Получают названное соединение, т.пл. 190oC, выход 90%.
Пример XII
Альтернативный метод получения транс-(±)-2-(2,4,6-триметокси)фенилциклогексанола. Пример соединения формулы VI, где R2 = CH3, R3 = 4,6-диметокси, R4 = OH.
2,4,6-Триметоксибензол (1 экв.), окись циклогексена (1,5 экв.) и сухой дихлорметан (840 мл) загружают в трехгорлую колбу, снабженную мешалкой. Реакционную массу охлаждают до -78oC и небольшими порциями в течение 1 ч добавляют хлористый алюминий (1,5 экв.). Перемешивание продолжают еще в течение 3 ч, затем добавляют воду и экстрагируют этилацетатом. Сырой продукт кристаллизуют из петролейного эфира, т.пл. 123oC, выход 63 - 64%.
Пример XIII
Расщепление (±)-транс-2-(2,4,6-триметокси)фенилциклогексанола. Соединение формулы VI, где R2 = CH3, R3 = 4,6-диметокси, R4 = OH.
Смесь (±)-транс-2-(2,4,6-триметокси)фенилциклогексанола (50,0 г, 0,18797 моль), 3-нитрофталевого ангидрида (26,299 г, 0,18797 моль) и пиридина (42,18 мл, 2,78 • 0,18797 моль) нагревают при 100oC в атмосфере азота в течение 3 ч. Реакционную смесь охлаждают до 0oC, нейтрализуют 2 н. HCl и полученный раствор экстрагируют хлороформом. Остаток после упаривания растворителя кристаллизуют из метанола (400 мл) и получают кристаллическое соединение формулы VI, где R4 = 3-нитрофталоилокси (59,0 г, т.пл. 198 - 200oC). Полученное соединение (0,1285 моль) обрабатывают (+)-цинхонином (37,85 г, 0,1285 моль) в метаноле (250 мл) на паровой бане в течение 30 мин. Растворитель упаривают под вакуумом и оставшуюся соль /96,5 г, OR (+) 84,75o (Hg, 578)/ кристаллизуют из смеси этилацетат-петройленый эфир 1:1 (1400 мл) и получают кристаллы /45,0 г, OR (+) 75,11o (Hg, 578)/ и маточный раствор 50,0 г, OR (+) 97,30o (Hg, 578)/.
Кристаллы (45,0 г) дополнительно перекристаллизовывают (трижды) из смеси этилацетат-петролейный эфир и получают обогащенную соль цинхонина /31,0 г, OR (+) 71,08o (Hg, 578)/. Обогащенную соль при 0oC обрабатывают 2 н. HCl и получают (-)-соединение формулы VI, где R4 представляет собой 3-нитрофталоилокси /16,1 г, OR (-) 37,15o (Hg, 578)/. Полученное соединение гидролизуют 7,5%-ным раствором КОН в смеси метанол-вода (1:2, 587-8 мл) при температуре кипения, а затем кристаллизуют полученный продукт из смеси этилацетат-петролейный эфир (24: 160 мл); получают (-)-транс-2-(2,4,6-триметокси)фенилциклогексанол /7,0 г, OR (-) 43,43o (Hg, 578)/.
Маточный раствор (50,0 г) обрабатывают 2 н. HCl при 0oC, продукт затем кристаллизуют (трижды) из смеси этилацетат-петролейный эфир и получают кристаллы (+)-соединения формулы VI, где R4 представляет собой 3-нитрофталоилокси /15,1 г, OR (+) 35,65o (Hg, 578)/. Полученное соединение при гидролизе 7,5%-ным раствором КОН в смеси этанол-вода (1:2, 548,5 мл) при кипячении в течение 60 ч с последующей кристаллизацией продукта из смеси этилацетат-петролейный эфир (25: 150 мл) дает (+)-транс-2-(2,4,6-триметокси)фенилциклогексанол /7,24 г, OR (+) 42,30o (Hg, 578)/.
Пример получения композиции в форме орального раствора
Пример 1
300 мг активного ингредиента, полученного в примере 10, растворяют в около 100 мл 0,5% раствора карбоксиметилцеллюлозы, получая раствор, содержащий 3 мг/мл активного ингредиента. Полученным раствором заполняют подходящие контейнеры (емкости). Этот раствор готов для орального введения пациенту в дозе 10, 20, 30 мг/кг (в расчете 1 мл раствора на мышь весом 130 - 150 г).
Пример 2
200 мг активного ингредиента, полученного в примере 11, растворяют в около 100 мл 0,5% раствора карбоксиметилцеллюлозы, получая раствор с содержанием активного ингредиента 2 мг/мл. Полученным раствором заполняют подходящие контейнеры. Раствор готов для орального введения пациенту.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИНОФЕНИЛСУЛЬФОНИЛМОЧЕВИН (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1996 |
|
RU2177003C2 |
| СРЕДСТВО ДЛЯ ЗАЩИТЫ КУЛЬТУРНЫХ РАСТЕНИЙ ОТ ФИТОТОКСИЧЕСКОГО ПОБОЧНОГО ДЕЙСТВИЯ ГЕРБИЦИДОВ, N-АЦИЛСУЛЬФОНАМИДЫ | 1997 |
|
RU2182423C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ИНДАНОНОВ | 1993 |
|
RU2110510C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ СОВМЕСТИМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО ДЛЯ СНИЖЕНИЯ ВЫСОКОГО ДАВЛЕНИЯ КРОВИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ СНИЖЕНИЯ КРОВЯНОГО ДАВЛЕНИЯ | 1993 |
|
RU2116300C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1992 |
|
RU2111211C1 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
| НОВЫЕ ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЗОРБЦИИ КОСТИ И АНТАГОНИСТОВ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2180331C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 1993 |
|
RU2126378C1 |
| НОВЫЕ ИНГИБИТОРЫ РАССАСЫВАНИЯ КОСТЕЙ И АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2195460C2 |
| НОВЫЕ ИМИНОПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЗОРБЦИИ КОСТИ И АНТАГОНИСТОВ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2197476C2 |





Производные арилциклоалкила формулы I, где R - остаток формулы (а), (б), (в), где R5 представляет 1 - 4 заместителя, выбранных из H, алкила, гидрокси, алкокси, алкилалкокси, карбокси, циано, -NHC(O)C1-3-алкила, -OC1-3-алкилфенила, -OCH2O-, -C(O)-O-C1-4-алкила, галогена, амино, нитро, -NH-C1-4-алкила, -N-(C1-4-алкил)2; X = O, S, N-H, N-C1-6-алкил; R2 = H, C1-6-алкил, -C/O/-C1-6-алкил; R2 = H, алкил, -CO-C1-6-алкил; R3 = 1 - 3 заместителя, выбранных из H, алкила, COC1-6-алкила, C/O/-O-C1-6-алкила, OH, OC1-6-алкила, O-C/O/-C1-6-алкила, галогена; R3 = H, OH, O-C1-6-алкил, OCO-алкил, COOH, COO-алкил, OCO-алкил - NH2, O-CO-алкил-NH-алкил, OCO-алкил-N-(алкил)2; n = 0, 1 или 2 и а представляет необязательно дополнительно одинарную связь. Соединения I проявляют противовоспалительные свойства и могут быть полезны для лечения для лечения воспалительных заболеваний, таких как ревматический артрит, остеоартрит, астма и злокачественные опухоли. 2 с. и 5 з.п. ф-лы, 1 ил., 5 табл.
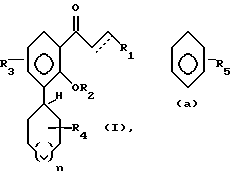
в которой R1 представляет остаток, выбираемый из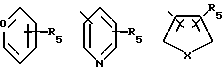
где R5 представляет один, два, три или четыре остатка, которые независимо друг от друга включают Н, С1-С6-алкил, замещенный С1-С6-алкил, гидрокси С1-С6-алкокси, С1-С4-алкил-О-С1-С1-алкил, карбокси, циано, -NHC-(O)C1-3алкил, -ОС1-3 алкилфенил, -ОСН2О-, -С(О)-О-С1-С4-алкил, галоген, амино, нитро, -NH-C1-C4-алкил, N-(C1-C4-алкил)2;
Х представляет O, S, N-H, -N-С1 - С6-алкил;
R2 представляет Н, С1-С6-алкил, -С(О)-С1 - С6-алкил;
R3 представляет один, два или три остатка, которые независимо друг от друга включают Н, С1-С6-алкил, -С(О)-С1-С6-алкил, -С(О)-О-С1-С6-алкил, ОН, О-С1-С6-алкил, -О-С(О)-С1-С6-алкил, галоген;
R4 представляет Н, -ОН, -О-С1-С6-алкил, -О-С(О)- С1-С6-алкил, -С(О)-ОН, -С(О)-О-С1-С6-алкил;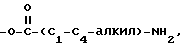
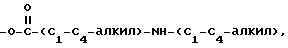
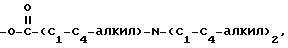
n = 0,1 или 2,
а представляет необязательную дополнительную одинарную связь.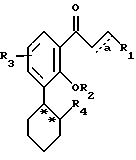
в которой R1, R2, R3, R4 и а имеют значения, указанные выше.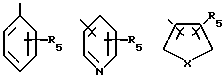
где R5 означает Н, С1-С6-алкил, замещенный С1-С6-алкил, гидрокси, С1-С3-алкокси, галоген;
R4 означает Н, ОН или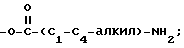
Х представляет O, NH, S, N-С1-С6-алкил;
а означает необязательную дополнительную связь.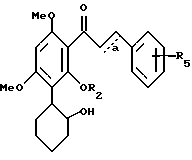
в которой R2 представляет Н и С1-С3-алкил;
R5 означает один или два галогена либо одну или две С1-С6-алкильные или С1-С3-алкоксильные группы;
а означает необязательную дополнительную одинарную связь.
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, ч.1, с.109 - 110 | |||
| JP, 2-48389, 1990. |
