Настоящее изобретение относится к получению класса терапевтически активных соединений. Более конкретно настоящее изобретение относится к усовершенствованному способу получения триптаминовых производных и родственных им соединений, молекулы которых содержат 1,2,4-триазол-1-иловый остаток.
В описании к заявке на патент EP-A-0497512, опубликованном 5 августа 1992 г, представлен, помимо прочего, класс 1,2,4-триазол-1-иловых производных, к молекулам которых присоединен заместитель, который вводит триптаминовый или аналогичный остаток. Было установлено, что эти соединения являются селективными агонистами так называемых "5-HT1-подобных" рецепторов и, следовательно, могут найти конкретное применение при лечении мигрени и таких связанных с этим состоянием, как сильная приступообразная головная боль с периодическими рецедивами, хроническая пароксизмальная гемикрания, головная боль, связанная с сосудистыми расстройствами, гипертоническая головная боль и детская мигрень.
Авторами настоящего изобретения теперь разработан эффективный и экономически выгодный способ получения соединения вышеуказанного класса.
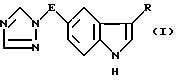
Таким образом, в соответствии с настоящим изобретением предлагается способ получения соединения формулы I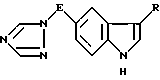
где E обозначает прямую или разветвленную алкиленовую цепь, содержащую от 1 до 4 углеродных атомов:
R - группа -CH2CHR1-NR2R3, где R1 - водород, R2 и R3 одинаковые и означают алкил C1-C6,
причем при осуществлении этого способа предусмотрены нижеследующие стадии: i) реакция 4-амино-1,2,4-триазола формулы II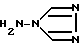
с соединением формулы III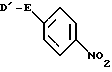
где значения E определены выше;
D1 обозначает легко отщепляемую группу,
в результате чего образуется соединение формулы IV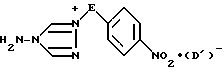
где значения E и D1 определены выше;
ii) деаминирование полученной таким образом аминотриазолиевой соли формулы IV путем обработки азотистой кислотой с последующей нейтрализацией, в результате чего получают соединение формулы V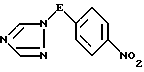
где значения E определены выше;
iii) восстановление полученного таким образом нитросоединения формулы V трансферной гидрогенизацией с использованием катализатора гидрирования и в присутствии донора водорода, в результате чего образуется соединение формулы VI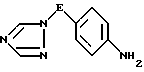
где значения E определены выше;
и iv) обработку полученного таким образом анилинового производного формулы VI азотистой кислотой, а затем сульфитом щелочного металла с последующим подкислением, в результате чего образуется гидразиновое производное формулы VII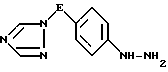
где значения E определены выше,
после чего проводят реакцию этого соединения по месту его получения с соединением формулы VIII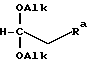
где Ra соответствует группе R, определенной выше.
Алкиленовая цепь E может, например, представлять собой метилен, этилен, 1-метилэтилен, пропилен или 2-метилпропилен.
Приемлемой легко отщепляемой группой D1 в соединениях формулы III является атом галогена, предпочтительно бром.
На стадиях (i) и (ii) вышеописанного способа предусмотрены алкилирование 4-амино-1,2,4-триазола в 1-м положении с последующим деаминированием полученной аминотриазолиевой соли обработкой азотистой кислотой и затем нейтрализацией. Все превращение удобно проводить с созданием реакционных условиях, аналогичных тем, что описаны в журнале J. Org. Chem. 1989 г, 54, 731. Таким образом, стадию (i) удобно осуществлять совместной выдержкой реагентов при температуре кипения с обратным холодильником в подходящем растворителе, предпочтительнее в таком полярном растворителе, как ацетонитрил, или низшем алканоле, например изопропиловый спирт, в течение промежутка времени от 1 до 10 ч, оптимально в течение около 7,5 ч. Азотистую кислоту, используемую на стадии (ii), выгодно получать по месту использования смешиванием нитрата натрия с такой минеральной кислотой, как концентрированная соляная кислота, поддерживая в идеальном варианте температуру в интервале от -5 до +5oC и избегая слишком большого избытка азотистой кислоты. По истечении соответствующего периода, обычно приблизительно через 15 мин, перемешивая при температуре ниже 5oC реакционной смеси дают нагреться до комнатной температуры, после чего нейтрализуют с использованием в идеальном варианте водного раствора аммиака.
Аминотриазолиевую соль формулы IV можно выделить как таковую по завершении стадии (i) способа в соответствии с настоящим изобретением, причем в этом случае стадии (i) и (ii) можно осуществлять независимо друг от друга или же процедуру на стадии (ii) можно проводить по месту реакционной смеси, полученной на стадии (i), без выделения аминотриазолиевой соли IV. Другими словами, стадии (i) и (ii) можно с успехом совмещать осуществлением "одночановой" процедуры.
В варианте, который описан в сопроводительном примере 1, стадии (i) и (ii) осуществляют раздельно. Тем не менее в примере 1 описана "одночановая" процедура получения нитросоединения формулы V.
Было установлено, что аминотриазолиевая соль может быть получена на стадии (i) с выходом продукта обычно в интервале 95 - 96%. Было установлено также, что на стадии (ii) нитросоединение V обычно получают с выходом продукта приблизительно 92 - 97%. Таким образом, если в общем рассматривать стадии (i) и (ii) совместно с условиями, эквивалентными тем, что описаны в сопроводительных примерах, то можно ожидать совокупного выхода нитросоединения V в диапазоне примерно 83 - 93%.
В ранее описанной процедуре (см., в частности, пример 5 заявки на патент EР-A-0497512, стадия 1) нитросоединение формулы V, которая описана выше, готовят непосредственно алкилированием с использованием реагента, соответствующего соединению формулы III, которая представлена выше, натриевой соли, 1,2,4-триазола. Однако выход такой реакции, согласно сообщению, оказывается умеренным - 52%. Это можно рассчитать по тому факту, что при попытке адаптировать эту процедуру для применения в промышленном масштабе полученный продукт, как оказалось, загрязнен большими количествами стильбенового производного формулы IX.
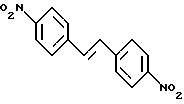
Последующие попытки устранить эту проблему и действительно усовершенствовать всю процедуру в общем с использованием 1,2,4-триазолового свободного основания в присутствии безводного карбоната калия оказали, тем не менее, слабое влияние на эффективность процесса, в результате чего описанное нитросоединение V было получено с достижением выхода всего примерно 45%. Таким образом, из вышеизложенного обсуждения совершенно очевидно, что процедура, описанная на стадиях (i) и (ii) способа в соответствии с настоящим изобретением, исключительно выгодна в отношении эффективности, поскольку ожидаемым ее результатом является эффективное удвоение выхода продукта.
Использование 4-амино-1,2,4-триазола II в качестве исходного материала при получении соединений формулы V выгодно также и с других точек зрения. Во-первых, он является недорогим реагентом, что позволяет улучшить весь процесс в целом с экономической точки зрения. Кроме того, функциональная аминогруппа в молекуле реагента II направляет ход реакции таким образом, что воздействие происходит исключительно через азотный атом в 1-м положении. Было установлено, что продукт, полученный в результате реакции с использованием натриевой соли 1,2,4-триазола, загрязнен заметным количеством нежелательного региоизомера, что пагубно отражается на степени чистоты полученного из него продукта. Тот факт, что со стадий (i) и (ii) способа в соответствии с настоящим изобретением получают более чистый продукт с эффективным достижением его удвоенного выхода в сравнении с тем, который достижим при осуществлении ранее известной процедуры, означает также, что при этом гарантируются значительно упрощенные рекуперация и возможность возврата в процесс используемого при этом растворителя.
Стадии (i) и (ii), которые описаны выше, и которые осуществляют для получения нитросоединений формулы V, где стадию (i) осуществляют в присутствии изопропилового спирта, по праву можно считать новыми и составляющими дополнительные отличительные признаки настоящего изобретения.
На стадии (iii) способа в соответствии с настоящим изобретением предусмотрено восстановление нитросоединения V до анилинового производного VI посредством трансферной гидрогенизации. При осуществлении этой процедуры применяют такой катализатор гидрогенизации, как палладий на угле, в идеальном варианте 10%-ный палладий на угле, в присутствии водородного донора, в частности аммонийформиата, гипофосфита натрия, триэтиламмонийформиата или формиата калия, предпочтительнее аммонийформиата. В том случае, когда в качестве водородного донора используют формиат аммония, реакцию удобно проводить в таком растворителе, как метанол или этанол, или же в водном метаноле или этаноле. В случае, если в качестве растворителя используют метанол или водный метанол, реакцию с успехом проводят при температуре в диапазоне 35 - 45oC. Когда применяют этанол или водный этанол, реакцию удобнее проводить при температуре в интервале 60 - 75oC. Время, которое отнимает реакция до ее завершения, варьируется в зависимости, помимо прочего, от температуры реакционной смеси и количества гидрогенизационного катализатора в реакционной смеси. В случае применения в качестве катализатора 10%-ного палладия на угле этот катализатор обычно содержится в количестве приблизительно от 1 до 4 мас.% в идеальном варианте от 2 до 3,5 мас.% от массы нитросоединения формулы V, причем в этом варианте при условии использования сочетаний растворителя и реакционной температуры, которые указаны выше, требуемая продолжительность реакции обычно составляет приблизительно 2 ч. С целью обеспечить полноту реакционного процесса при необходимости можно периодически добавлять в реакционную смесь дополнительные аликвоты гидрогенизационного катализатора. В таких условиях можно ожидать достижения практически количественного выхода анилинового производного VI.
В ранее описанных процедурах (см., в частности, пример 5 заявки на патент EP-A- 0497512, стадия 2) процесс восстановления нитросоединения формулы V, определенной по вышеизложенному, до анилинового производного формулы VI, которая определена выше, проводят путем обычной каталитической гидрогенизации. Хотя, как сообщается, при этом достигают практически количественного выхода целевого продукта, эта процедура страдает, тем не менее, практическим недостатком, состоящим в том, что ее необходимо осуществлять в автоклаве, а это означает, что ее нелегко осуществлять в более крупном масштабе при попытке применения в промышленных условиях. Осуществление методологии трансферной гидрогенизации не требует между тем специальных предметов лабораторного оборудования, поскольку она может быть легко приспособлена для осуществления в любых стандартных лабораторных сосудах неограниченного размера, поэтому она оказывается значительно более гибкой процедурой, чем обычная каталитическая гидрогенизация с производственной точки зрения.
Процедура, описанная для стадии (iii) способа в соответствии с настоящим изобретением также обладает достоинством, состоящим в возможности выделения амина VI в форме свободного основания. Это соединение является стойким. В противоположность сказанному в результате осуществления ранее описанной процедуры получают гидрохлоридную соль. Следствием этого является не только непостоянство продукта, поскольку на практике получают смеси моно- и дигидрохлоридных солей и тому подобного, но, по наблюдениям, эти гидрохлоридные соли характеризуются, также светочувствительностью, практическим эффектом этого свойства является то, что окраска полученного продукта, как это было отмечено, со временем становится темнее, что считается совершенно нежелательным явлением.
На стадии (iv) способа в соответствии с настоящим изобретением вначале проводят конверсию анилинового производного VI в гидразиновое производное формулы VII путем обработки азотистой кислотой, а затем обрабатывают сульфитом щелочного металла с последующим подкислением. Не выделяя это гидразиновое соединение VII, далее проводят его реакцию по месту получения с альдегидом VIII или его карбонилблокированной формой, обычно с диметилацеталевым производным, в результате чего получают целевой триазолилзамещенный индол формулы I. Как и на вышеописанной стадии (ii), азотистую кислоту, используемую на стадии (IV), выгоднее получать по месту использования путем смешения нитрита натрия с такой минеральной кислотой, как концентрированная соляная кислота, причем в идеальном варианте необходимо температуру в это время поддерживать в пределах от 0 до 5oC и избегать слишком большого избытка азотистой кислоты.
Затем полученную таким образом диазониевую соль восстанавливают в той же среде до гидразинового производного VII обработкой сульфитом щелочного металла, что удобно производить постепенным нагреванием реакционной смеси с последующим подкислением, которое в идеальном варианте осуществляют серной кислотой. Приемлемым используемым сульфитом щелочного металла может служить сульфит натрия и сульфит калия, предпочтительнее сульфит натрия.
В ранее описанной процедуре (см, в частности, пример 5 заявки на патент EP-A- 0497512, стадия 3) предусмотрено восстановлением диазониевой соли, полученной обработкой анилинового производного формулы VI, которая представлена выше, азотистой кислотой, до гидразина VII реакцией с дигидратом дихлорида олова в концентрированной соляной кислоте. Однако соли олова известны своей токсичностью, из-за чего возникают значительные проблемы, связанные с избавлением от них, поэтому замена дихлорида олова сульфитом натрия является очевидным усовершенствованием с экологической точки зрения, в особенности в том случае, когда такой процесс приспосабливают к производству в промышленном масштабе. Более того, соли олова обладают стойкостью, их следы часто наблюдают до самых конечных стадий процесса синтеза, если не предусмотрена тщательная хроматографическая обработка, поэтому совершенно очевидно, что замена дихлорида олова сульфитом натрия позволяет устранить этот недостаток.
Реакция между соединениями VII и VIII является примером хорошо известного индолового синтеза фишера. Эту реакцию проводят посредством начального нециклизованного промежуточного продукта формулы X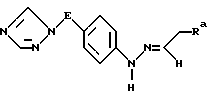
где значения A и Ra определены выше.
Однако при правильном подборе реакционных условий, обычно в случае проведения реакции при достаточно высокой температуре можно избежать операции выделения промежуточного продукта X. Эту реакцию выгоднее проводить перемешиванием реагентов в присутствии 4% серной кислоты при повышенной температуре, обычно при приблизительно 90oC.
Существенное достоинство, связанное с тем фактом, что стадию (iv) способа в соответствии с настоящим изобретением осуществляют в один этап, без выделения каких-либо промежуточных продуктов, обусловлено затруднениями технологического порядка при выделении гидразиновых производных формул VII. Обычно гидразин VII обладает исключительной водорастворимостью, вследствие чего для его получения с практическим выходом требуется тщательная экстракционная обработка. Это может частично объяснить оцениваемый как умеренный выход (56%) гидразинового производного формулы VII, которая описана выше, достигаемый в случае выделения и очистки этого соединения в соответствии с ранее описанным способом (см. пример 5 заявки на патент EP-A- 0497512, стадия 3)
Было установлено, что как и в случае с амином VI, гидрохлоридная соль гидразинового производного VII светочувствительна и также существует в форме смесей переменного состава монодигидрохлоридных и тому подобных солей, что обуславливает целесообразность устранения операции выделения гидразинового производного VII.
В соответствии с ранее описанным способом (сравни примеры 5 и 17 заявки на патент EP-A-0497512), при осуществлении которого соединение, отвечающее формуле I, представленной выше, получают из амина формулы VI, описанной выше, в ходе проведения многостадийной процедуры, общий выход из амина VI может составлять согласно расчетам, примерно 10 - 20%. Теперь, как установлено, с использованием на стадии (iv) вышеописанного метода при осуществлении способа в соответствии с настоящим изобретением, благодаря чему проводят конверсию аминопроизводного VI в той же реакционной среде непосредственно в целевой продукт формулы I, общий выход из амина VI обычно составляет примерно 40 - 45%. Это вновь демонстрирует превосходство способа в соответствии с настоящим изобретением перед ранее описанным. Более того, поскольку все реакционные стадии от амина VI до конечного продукта 1 осуществляют в одном и том же сосуде, можно использовать, как было установлено, более крупные партии материалов, чем это было до сих пор.
Существо настоящего изобретения проиллюстрировано далее с помощью нижеследующих пояснительных примеров.
Пример 1. Стадия (i): 4-амино-1-(4-нитробензил)-4H-1,2,4- триазолийбромид (IV, E-CH32, D1 - атом брома).
Смесь 250 г (2,976 моль) 4-амино-1,2,4-триазола с 99%-ным 4-нитробензилбромидом (ex Janssen, 2,83 моль) довели до температуры кипения с перемешиванием. Непосредственно после того, как смесь превратилась в раствор, при температуре кипения с обратным холодильником кристаллизовалась целевая триазолиевая соль. Эту смесь перемешивали, выдерживая при температуре кипения с обратным холодильником в течение 7,5 ч, а затем ей дали остыть до комнатной температуре в течение ночи. На следующий день смесь охладили до температуры 0 - 5oC, выдержали в течение 1 ч и образовавшийся продукт отфильтровывали, промыли небольшим количеством изопропилового спирта, а затем высушили в вакууме при 50oC, получив в виде белого твердого вещества 808 г указанной в заголовке триазолиевой соли (95%-ный выход) с температурой плавления 199oC) (с разложением)
Стадия (ii): 1-(4-нитробензил)-1,2,4-триазол (V, E-метилен).
Раствор 206 г (2,98 моль) нитрита натрия в 840 мл воды добавили при температуре 0 - 5oC в течение 70 мин под поверхность жидкости в суспензию 808 г (2,69 моль) вышеуказанной триазолиевой соли в 5,6 л воды и 505 мл концентрированной соляной кислоты. Бледно-желтый шлам перемешивали при температуре менее 5oC в течение 15 мин и затем ему дали нагреться до температуры 25oC в течение 1 ч. Добавлением 380 мл 18 н. водного раствора аммиака величину pH бесцветного раствора довели до 9, поддерживая температуру на уровне ниже 30oC. Эту смесь охладили до 0 - 5oC и перемешивали в течение 1 ч. Твердый материал собрали фильтрованием, промыли 400 мл воды, содержащей 20 мл 18 н. водного раствора аммиака, и высушили под пониженным давлением при 50oC, получив 535 г (97%-ный выход) указанного в заголовке нитросоединения с температурой плавления 102 - 103oC.
Стадия (iii): 1-(4-аминобензил)-1,2,4-триазол (VI: E-метилен)
803 г (3,9 моль) вышеуказанного нитросоединения, 1,16 кг (18,4 моль) формиата аммония и 28 г 10%-ного палладия на угле в 8 л метанола перемешивали в токе азота и нагрели до 30oC. Нагревание прекратили и начали охлаждение с целью контролировать экзотермическую реакцию, поддерживая температур 35 - 45oC в течение 2 ч. Реакционную смесь охладили до 20oC и фильтрованием с помощью вспомогательного фильтровального вещества Hyflo удалили катализатор. Фильтрованный тампон промыли 2 л метанола. Фильтрат сконцентрировали и остаток разбавили 12 л этилацетата и 1,57 л воды. Нижний, водный слой обработали 10 мл 18 н. водного раствора гидрата окиси аммония, доведя величину pH до 9. Водный слой отделили и подвергли экстракционной обработке 2 порциями (6 и 3 л) этилацетата. Объединенный экстракт промыли 1,57 л насыщенного водного раствора бикарбоната натрия, высушили и выпарили под пониженным давлением, получив 679 г 99%-ный выход) указанного в заголовке амина с температурой плавления 127 - 128oC.
Стадия (iv): N,N-диметил-2[5-(1,2,4-триазол-1-илметил)-1H- индол-3]-этиламин (I, E - метилен, R - группа - CH2 - CH2 - NMe2).
Раствор 16,7 г (0,24 моль) нитрита натрия в 22,7 мл воды ввели под поверхность раствора 40 г (0,23 моль) вышеуказанного амина в 65,3 мл соляной кислоты и 162 мл воды, поддерживая температуру менее 5oC. Этот раствор перемешивали при 0 - 5oC в течение 1 ч. Затем раствор в токе азота добавили в суспензию 72,4 г (0,57 моль) сульфита натрия в 227 мл воды, охлажденной до температуры 5 - 10oC. Красный раствор перемешивали при 5 - 10oC в течение 10 мин, дали ему нагреться до 20oC в течение 20 мин, после чего выдержали при 70oC в течение 45 мин. Этот раствор перемешивали при 70oC в течение 2,5 ч и охладили до 65oC. Далее в раствор в течение 15 мин добавили 45,8 мл концентрированной серной кислоты, поддерживая температуру на уровне 70 - 80oC. Раствор перемешивали при 70oC в токе азота в течение 2 ч, а затем ему дали остыть до 20oC в течение ночи. Раствор полученного гидразина (VII, E - метилен) нагрели до 25oC и в течение 15 мин добавили в него 44,3 г (0,28 моль) 4-(N,N-диметиламино)-1,1-диметоксибутана, поддерживая температуру ниже 35oC. Раствор перемешивали при 30 - 35oC в течение 30 мин. Далее смесь выдерживали при 90oC в течение 30 мин, а затем при 90 - 93oC в течение 15 мин. Смесь охладили до 15oC и добавили в нее 68 г вспомогательного фильтровального вещества Hyflo, а затем 200 мл 18 н. водного раствора гидрата окиси аммония, доведя величину pH до 11 - 12. Смесь профильтровали и фильтрат, а также вещество Hyflo подвергали экстракционной обработке 5 порциями по 300 мл этилацетата. Экстракт высушили над сульфатом натрия и выпарили под пониженным давлением. Остаток подвергли хроматографической обработке на 550 г двуокиси кремния, элюируя смесью ацетата с метанолом, меняя соотношение от 80 : 20 до 50 : 50. Содержащие продукт фракции выпарили под пониженным давлением, получив в форме свободного основания 27,8 г (45%-ный выход) указанного в заголовке продукта.
Пример 2. Стадия (i): "Одночановое" получение 1-(4-нитробензил)-1,2,4-триазола (V, E - метилен).
64,22 г 4-нитробензилбромида, 26 г 4-амино-1,2,4-азола и 586 мл изопропилового спирта совместно перемешивали при температуре кипения с обратным холодильником в течение 7 ч. Изопропаноловый растворитель заменили водой, используя процедуру азеотропной вакуумной перегонки в роторном испарителе Бучи. Объем конечного водного шлама составлял 750 мл 675 мл этого шлама охладили до температуры -2oC и в течение нескольких минут в него добавили 50,8 мл 12 М соляной кислоты. Далее по каплям в течение 40 мин добавили раствор 21,7 г нитрита натрия в 86 мл воды его введением под поверхность жидкости. В течение всей операции добавления температуру массы поддерживали в интервале от -2 до -1oC, а затем ей дали нагреться в течение 30 мин до 18oC. После этого температуру повысили до 28oC и выдержали при ней в течение 1 ч с последующей обработкой раствора 4,5 г угля (Fisons) в течение 15 мин. Уголь удалили и 750 мл фильтрата разделили поровну на две части. Одну половину раствора подщелачивали добавлением 22 мл раствора аммиака, а выпавшее в осадок основание собрали, промыли 2 порциями по 30 мл воды и высушили в вакууме в течение 16 ч при 35oC. В результате получили 22,73 г (выход - 83,3%) 1-(4-нитробензил)-1,2,4-триазола.
Стадия (ii): 1-(аминобензил)-1,2,4-триазол (VI, E - метилен)
В сосуде из нержавеющей стали током азота создали инертную атмосферу и его подключили к мобильной скрубберной установке. В скруббер загрузили очень разбавленную соляную кислоту. В реакционный сосуд загрузили 40 кг 96%-ного этанола, а затем 9,62 кг 1-(4-нитробензил)-1,2,4-триазола. Током азота вновь создали в нем инертную атмосферу и в перемешиваемую массу с помощью капельной воронки добавили в качестве катализатора 192 г шлама 10%-ного палладия на угле (типа Englhardt 4505), в воде. Сосуд, воронку и трубку промыли водой. Общее количество использованной воды составляло 16 л. Массу хорошенько перемешали и нагрели до 60oC. В реактор в течение 1 ч добавили раствор 13,95 кг формиата аммония в 30 мл воды. Температуру массы поддерживали на уровне 65oC. По завершении этой операции добавления массу выдержали в течение последующего 1 ч при 70 - 75oC. Тонкослойная хроматография показала завершение реакции и образование единственного пятна. Массу охладили до 60oC и пропустили через фильтр, работающий под давлением, покрытый 1,5 вещества Hyflo, направив в другой сосуд. Реакционный сосуд, фильтр и трубки промыли 10 л воды. После этого аналогичным образом восстановили другую порцию, 9,585 кг 1-(4-нитробензил)-1,2,4-триазола. Эту порцию профильтровали и добавили к ранее профильтрованной массе совместно с 10 л воды после промывки ею сосуда, фильтра и трубок. 215 л объединенной профильтрованной массы перегнали под атмосферным давлением до удаления из нее 70 л дистиллята (на этом этапе температура паров составляла 83oC). Добавили 30 л воды и перегонку продолжили до тех пор, пока температура паров не достигла 95oC) в общей стоимости 106 л дистиллята). Далее давление в перегонном сосуде понизили с целью обеспечить возможность перегонки при температуре кипения не выше 70oC. В результате дополнительно выделили 70 л дистиллята, а остаточный объем составил 69 л. На этом этапе масса кристаллизовалась и шлам охладили в течение 1 ч до температуры не выше 20oC. Затем массу охладили до 0oC, выдержали для "старения" в течение 1 ч с последующим фильтрованием. Твердый материал промыли 10 л воды при 0 - 5oC. После этого массу высушили в течение ночи при 50oC в вакууме с подачей слабого тока азота, получив 1-(4-аминобензил)-1,2,4-триазола (96%-ный выход).
Изобретение относится к способу получения производных индола общей формулы I, где F означает прямую или разветвленную С1-С4-алкиленовую цепь; R - группа формулы -CH2-CHR1-NR2R3, где R1 водород; R2 и R3 одинаковые и означают C1-C6-алкил. Способ заключается во взаимодействии 4-амино-1,2,4-триазола с нитробензоловым производным, содержащим легко замещаемую группу, деаминировании полученной таким образом аминотриазолиевой соли обработкой азотистой кислотой с последующей нейтрализацией, восстановлении полученного таким образом триазолинилнитробензолового производного трансферной гидрогенизацией, обработке полученного таким образом триазолиланилинового производного азотистой кислотой и затем сульфатом щелочного металла с последующим подкислением и после этого реакцию полученного таким образом триазолилгидразинового производного в реакционной среде подходящим карбонильным соединением формулы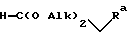
где Alk - метил. Целевой продукт получают с выходом 96%, без выделения промежуточных соединений.
9 з.п. ф-лы.
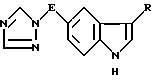
E означает прямую или разветвленную C1 - C4-алкиленовую цепь;
R означает группу формулы -CH2-CHR1 - NR2R3, где R1 - водород;
R2 и R3 одинаковые и означают C1 - C6-алкил,
отличающийся тем, что осуществляют нижеследующие стадии: i) реакцию 4-амино-1,2,4-триазола формулы II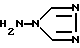
с соединением формулы III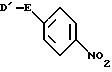
где значение E определено выше;
D1 означает легко отщепляемую группу,
в результате чего образуется аминотриазолиевая соль формулы IV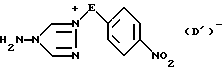
где значения E и D' определены выше;
ii) деаминирование полученной соли азотистой кислотой, с последующей нейтрализацией, в результате чего получают нитросоединение формулы V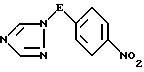
где значение E определено выше,
iii) восстановление полученного нитросоединения трансферной гидрогенизацией с использованием катализатора гидрирования и в присутствии донора водорода с образованием производного анилина формулы VI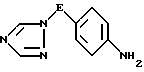
где значение E определено выше и
iv) обработку полученного соединения азотистой кислотой, а затем сульфитом щелочного металла с последующим подкислением, в результате чего образуется производное гидразина формулы VII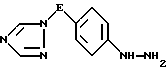
где значение E определено выше,
которое подвергают взаимодействию с соединением формулы VIII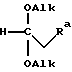
где Ra означает R;
каждый Alk - метил.
| EP 0313397, 1989 | |||
| Способ получения замещенных 3-(2-4фенил-1-пиперазинил-этил)-индолинов или их солей, или их четвертичных аммониевых солей | 1972 |
|
SU488408A3 |

