Изобретение относится к классу ароматических соединений, которые используются в качестве антагонистов тахикининов. Конкретнее, соединения настоящего изобретения содержат аминозамещенную азогетероциклическую группу.
Тахикинины представляют собой группу встречающихся к природе пептидов, широко распространенных, как обнаружено, в тканях млекопитающих, как в центральной нервной системе, так и в периферической нервной системе и в сердечно-сосудистой системе.
Тахикинины отличаются сохраняющейся карбоксил-концевой последовательностью Phe-X-Gly-Leu-Met-NH2.
В настоящее время известны три тахикинина млекопитающих, упоминаемых как вещество P, нейрокин A (NKA, вещество K, нейромедин L) и нейрокин B (NKB, нейромедин K) (см. обзор J.E.Meggio, Peptides (1985) 6 (Suppl. 3), 237-242). Общепринятая номенклатура определяет три тахикининовых рецептора, опосредующих биологическое действие вещества P, NKA и NKB как NK1-, NK2- и NK3- рецепторы, соответственно.
Доказательства полезности антагонистов тахикининовых рецепторов при боли, головной боли, особенно - при мигрени, болезни Альцгеймера, рассеянном склерозе, синдроме отмены морфина, сердечно-сосудистых изменениях, отеках, таких как отеки, вызываемые термическим ожогом, хронических воспалительных заболеваниях, таких как ревматоидный артрит, астме бронхиальной повышенной реактивности и других респираторных заболеваниях, включая аллергический ринит, воспалительных заболеваниях кишечника, включая неспецифический язвенный колит и болезнь Крона, глазной травме и глазных воспалительных заболеваниях, пролиферативной витреоретинопатии, синдроме раздражения толстой кишки и нарушениях функции мочевого пузыря, включая цистит и гиперрефлексию мышц мочевого пузыря, рассматриваются в "Tachykinin Receptors and Tachykinina Receptor Antagonicts", C. A.Maggi, R. Patacchini, R. Rovero and A. Giachetti, J. Auton. Pharmacol (1993) 13, 23-93.
Например, полагают, что вещество P, среди прочих, вовлекается в нейротрансмиссию болевых ощущений (Otsuka et al, "Role of Substance Pas c Sensory Transmitter in Spinal Cord and Sympathetic Ganglia" b 1982 Substance P in Nervous System, Ciba Foundation Symposium 91, 13-34 (published by Pitman), u Otsuka and Yanagisawa, "Does Substance P Actas a Pain (1987), 8, 506-510), особенно - в Ttansmitter?" TIPS передачу боли при мигрени (B.E.B. Sandberg et al. , J. Med. Chem, (1982), 25, 1009) в артрите (Levine et al., Science (1984) 226, 547-549). Тахикинины также вовлекаются в желудочно-кишечные (G1) расстройства и болезни G1-тракта, такие как воспалительные заболевания кишечника [(Mantyh et al. , Neuroscience (1988) 25(3), 817-37, u D. Regoli, "Trends in Cluster Headache" Ed. Sicuteri et al., Elsevier Scientific Publishers, Amsterdam (1987) page 85)] и рвота [E.D. Tattersall et al., Eur. J. Pharmacol. , (1993), 250, R5-R6). Предполагается также, что существует нейрогенный механизм для артрита, при котором некую роль может играть вещество P (Kidd et al., "Neurogenic Mechanism for Symmetrical Arthiritis" в The Lancet, 11 November 1989, u Gronblad et al., "Neuropeptides in Synovium of Patients with Pheu matoid Arthritis and Osteoartritis" в Rhtumatol, (1988), 15 (12), 1807-10). Поэтому полагают, что вещество P включается в воспалительную реакцию при таких болезных, как ревматоидный артрит и остеоартрит, и фиброзит (O'Byrne et al., Arthritis and Rheumatism (1990), 33, 1023-8). Другими связанными с болезнями областями, при которых, как полагают, полезны антагонисты тахикинина, являются аллергические состояния (Hamelet et al, Can. J. Pharmacol Physiol (1986) 66, 1361-7) иммунорегуляция (Lotz et al, Science (1988) 241, 1218-21, и Kimball et al, J.Immunol (1988) 141 (10), 3564-9), вазодилатация, спазм бронхов, рефлекторная и нейронная регуляция внутренних органов (Mantyh et al, PNAS (1988), 85, 3235-9), и, возможно, посредством купирования или ослабления β -амилоидопосредованных нейродегенеративных изменений (Yankner et al, Science (1990) 250, 279-82), при сенильном слабоумии типа Альцгеймера, болезни Альцгеймера и синдроме Дауна.
Антагонисты тахикининов также могут быть полезны при лечении мелкоклеточного рака, в частности, мелкоклеточного рака легких (SCLC) (Landgon et al, Cancer Research (1992) 52, 4554-7).
Вещество P также может играть некоторую роль при демиелинизирующих болезнях, таких как рассеянный склероз и боковой амиотрофический склероз (J.Luber-Narod et al, poster C.I.N.P. XVIIIth Congress, 28th June-2nd July 1992), и при расстройствах функции мочевого пузыря, таких как гиперрефлексия мышц мочевого пузыря (Lancet, 16th May 1992, 1239).
Кроме того, предполагается, что тахикинины имеют вспомогательное значение при следующих нарушениях: депрессия, дистимические нарушения, хроническая непроходимость дыхательных путей, аллергические расстройства, такие как под действием сумаха, вазопастические болезни, такие как стенокардия и болезнь Рейна (Reynauld), фиброзные и коллагеновые болезни, такие как склеродермия и эозинофильный фасцит (fascioliasis), рефлекторная симпатическая дистрофия, такая как плечевой синдром, болезни вследствие вредных привычек, такие как алкоголизм, связанные со стрессом соматические расстройства, невропатия, невралгия, нарушения, связанные с иммунным усилением или угнетением, такие как системная красная волчанка (описание европейского патента N 0436334), глазные болезни, такие как конъюнктивит, весенний конъюнктивит и т.п., и кожные болезни, такие как контактный дерматит, атопический дерматит, крапивница и другие экзематозные дерматиты (Европейский патент N 0394989).
В Европейском патенте N 0577394 (опубликован 5 января 1994) описаны морфолиновые и тиоморфолиновые антагонисты тахикининовых рецепторов общей формулы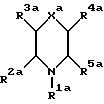
где R1a представляет собой самые разные заместители;
R2a и R3a, среди прочего, представляют собой водород;
R4a, среди прочего, представляет собой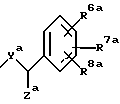
R5a представляет собой, среди прочего, необязательно замещенный фенил;
R6a, R7a и R8a представляет собой самые разные заместители;
Xa представляет собой O, S, SO или SO2;
Ya представляет собой, среди прочего, O; и
Za представляет собой водород или C1-4-алкил.
В Европейском патенте N 0528495 (опубликован 24 февраля 1993) описаны ациклические производные, пригодные в качестве антагонистов тахикининов, которые имеют общую формулу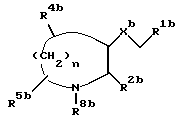
где n равен 1, 2 или 3;
Xb представляет собой O или S;
R1b представляет собой необязательно замещенный фенил,
R2b представляет собой арил, гетероарил, бензгидрил или бензил;
R4b и R5b представляет собой, независимо, H, галоген, CH2OR9b, C1-6- алкил, оксогруппу, CO2R10b или CONR10bR11b;
R8b представляет собой H, COR9b, CO2R10b или необязательно замещенный C1-6- алкил,
R9b представляет собой H, C1-6-алкил или фенил, и
R10b и R11b, независимо, представляет собой H или C1-6-алкил.
Заявители обнаружили еще один класс непептидных соединений, которые являются сильными антагонистами тахикининов, особенно, вещества P.
Желательно, чтобы соединения можно было вводить перорально и посредством инъекций. В настоящем изобретении раскрываются соединения, которые действуют как сильные непептидные антагонисты тахикининов, и которые, благодаря их преимущественной растворимости в воде, особенно легко включаются в состав для введения как пероральным путем, так и посредством инъекций, например, в водной среде.
Кроме того, соединения настоящего изобретения обладают особенно выгодным профилем активности, обладая сильной активностью антагониста к NK1-рецептору, и имеют длительный период действия. Соединения настоящего изобретения, и, в особенности, их фармацевтически приемлемые соли присоединения кислот, являются также весьма подходящими для широкого ряда фармацевтических составов благодаря своей стабильности.
Настоящее изобретение относится к соединениям формулы (1)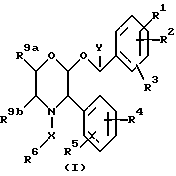
где R1 представляет собой водород, галоген, C1-6 -алкил, C1-6-алкокси, CF3, NO2, CN, SRa, SORa, SO2Ra, CO2Ra, CONRaRb, C2-6-алкенил, С2-6-алкинил или C1-4-алкил, замещенные C1-4-алкокси, где Ra и Rb, каждый, независимо, представляет собой водород или C1-4 алкил,
R2 представляет собой водород, галоген, C1-6-алкил, C1-6-алкокси, замещенную C1-4-алкокси, или CF3,
R3 представляет собой водород, галоген или CF3,
R4 представляет собой водород, галоген, C1-6-алкил, C1-6-алкокси, CF3, NO2, CN, SRa, SORa, SO2Ra, CO2Ra, CONRaRb, C2-6-алкенил, C2-6-алкинил или C1-4-алкил, замещенные C1-4-алкокси-группой, где Ra и Rb, каждый независимо, представляет собой водород или C1-4-алкил,
R5 представляет собой водород, галоген, C1-6 алкил, C1-6-алкокси, замещенную C1-4 -алкокси, или CF3,
R6 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O, =S или C1-4-алкильной группой, и необязательно замещенное группой формулы ZNR7R8, где
Z представляет собой C1-6-алкилен или C3-6- циклоалкилен,
R7 представляет собой водород, C1-4-алкил, C3-7-циклоалкил или C3-7-циклоалкил-C1-4 алкил, или C2-4-алкил, замещенный C1-4-алкокси-группой или гидроксилом,
R8 представляет собой водород, C1-4-алкил, C3-7-циклоалкил или C3-7-циклоалкил-C1-4-алкил, или C2-4-алкил, замещенный одним или двумя заместителями, выбираемыми среди C1-4-алкоксигруппы, гидроксила или 4-, 5-, или 6-членного гетероалифатического кольца, содержащего один или два гетероатома, выбираемых среди N, O и S.
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, необязательно замещенное одной или двумя группами, выбираемыми среди гидроксила или C1-4-алкила, необязательно замещенного C1-4-алкоксигруппой или гидроксильной группой, и необязательно содержащее двойную связь, и это кольцо может, необязательно, содержать кольцевой атом кислорода или серы, группу S(O) или S(O)2 или второй атом азота, который будет являться частью группы NH или NRc, где Rc представляет собой C1-4-алкил, необязательно замещенный гидроксильной группой или C1-4-алкоксигруппой;
или R7, R8 и атом азота, к которому они присоединены, образуют неароматическую азабициклическую кольцевую систему с 6-12 кольцевыми атомами;
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо с 4-7 атомами в кольце, которое, необязательно, может содержать кольцевой атом кислорода;
R9a и R9b, каждый и независимо, представляет собой водород или C1-4-алкил, или R9a и R9b соединяются, вместе с атомами углерода, к которым они присоединены, таким образом, что образуется кольцо C5-7,
X представляет собой алкиленовую цепь и 1-4 атомов углерода, необязательно замещенную оксогруппой, и
Y представляет собой C1-4-алкильную группу, необязательно замещенную гидроксильной группой,
при условии, что если Y представляет собой C1-4-алкил, R6 является замещенным по крайней мере группой формулы ZNR7R8, определение которой дается выше;
и их фармацевтических приемлемым солям и пролекарствам.
Некоторые особенно подходящие соединения настоящего изобретения включают соединения, в которых R1 представляет собой водород, C1-4-алкил, C1-4-алкокси, галоген или CF3.
Наиболее подходяще, когда R2 представляет собой водород, C1-4-алкил, C1-4-алкокси, галоген или CF3.
Наиболее подходяще, когда R3 представляет собой водород, фтор, хлор или CF3.
Благоприятно, когда R1 представляет собой фтор, хлор или CF3.
Благоприятно, когда R2 представляет собой водород, фтор, хлор или CF3.
Благоприятно, когда R3 представляет собой водород, фтор, хлор или CF3.
Предпочтительно, когда R1 и R2 находятся в положениях 3 и 5 фенильного кольца.
Более предпочтительно, когда R1 представляет собой 3-фтор или 3-CF3.
Более предпочтительно, когда R2 представляет собой 5-фтор или 5-CF3.
Более предпочтительно, когда R3 представляет собой водород.
Более предпочтительно когда R1 представляет собой 3-F или 3-CF3, R2 представляет собой 5-CF3, и R3 представляет собой водород.
Наиболее подходяще, когда R4 представляет собой водород.
Наиболее подходяще, когда R5 представляет собой водород, фтор, хлор или CF3.
Предпочтительно, чтобы R4 представлял собой водород, и R5 представлял собой водород или 4-фтор.
Наиболее подходяще, когда R9a и R9b, каждый, независимо, представляет собой водород или метил.
R9a, предпочтительно, представляет собой водород, R9b, предпочтительно, представляет собой водород. Наиболее предпочтительно, когда R9a и R9b - оба представляют собой водороды.
Из вышеизложенного понятно, что особенно подходящей подгруппой соединений настоящего изобретения являются соединения формулы (Ia) и их фармацевтически приемлемые соли и пролекарства.
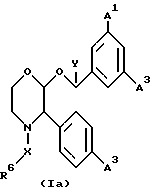
где A1 представляет собой фтор или CF3;
A2 представляет собой фтор или CF3;
A3 представляет собой фтор или водород;
X, Y и R6 имеют значения, установленные при определении формулы (I).
В соответствии с другим аспектом настоящего изобретения, предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых Y представляет собой C1-4-алкильную группу, замещенную гидроксильной группой; или их фармацевтически приемлемые соли или пролекарства.
В соответствии с еще одним, или альтернативным аспектом настоящего изобретения, другим предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых Y представляет собой C1-4-алкильную группу, при условии, что R6 является замещенным по крайней мере группой формулы ZNR7R8, определение которой дается выше, или их фармацевтически приемлемые соли или пролекарства.
В соответствии с еще одним аспектом настоящего изобретения, другим предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых
Y представляет собой C1-4-алкильную группу, и
R6 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O или =S, и замещенное группой формулы ZNR7R8, в которой
Z представляет собой C1-6-алкилен или C3-6-циклоалкилен,
R7 представляет собой водород, C1-4-алкил, C3-7-циклоалкил или C3-7-циклоалкил-C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой или гидроксилом,
R8 представляет собой водород, C1-4-алкил, C3-7-циклоалкил или C3-7-циклоалкил-C1-4-алкил, или C2-4-алкил, замещенный одним или двумя заместителями, выбираемыми среди C1-4-алкоксигруппы, гидроксила или 4-, 5- или 6-членного гетероалифатического кольца, содержащего один или два гетероатома, выбираемых среди N, O и S,
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, необязательно замещенное гидроксильной группой, и содержащее, необязательно, двойную связь, и это кольцо может содержать, необязательно, кольцевой атом кислорода или атом серы, группу S(O) или S(O)2, или второй атом азота, который будет являться частью группы NH или NRc, или Rc представляет собой C1-4-алкил, необязательно замещенный гидроксильной группой или C1-4-алкоксигруппой,
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое, необязательно, может содержать кольцевой атом кислорода;
или их фармацевтически приемлемые соли или пролекарства.
В соответствии с еще одним аспектом настоящего изобретения, другим предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых
Y представляет собой C1-4-алкильную группу; и
R6 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O или =S, и замещенное группой формулы ZNR7R8, где
Z представляет собой C1-6-алкилен или C3-6-циклоалкилен,
R7 представляет собой водород или C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой или гидроксильной группой, R8 представляет собой водород или C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой, гидроксилом или 5- или 6-членным гетероалифатическим кольцом, содержащим один или два гетероатома, выбираемых среди N, O и S,
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, необязательно замещенное гидроксильной группой, и это кольцо может содержать, необязательно, кольцевой атом кислорода или атом серы, группу S(O) или S(O)2, или второй атом азота, который будет являться частью группы NH или NRC, где RC представляет собой C1-4-алкил, необязательно замещенный гидроксильной группой или C1-4-алкоксигруппой,
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо и 4-7 атомов, которое, необязательно, может содержать кольцевой атом кислорода,
или их фармацевтически приемлемые соли или пролекарства.
В соответствии с еще одним аспектом настоящего изобретения, другим предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых
Y представляет собой C1-4-алкильную группу, и
R6 представляет собой 5- или 6-членное гетероциклические кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O или =S, и необязательно замещенное группой формулы ZNR7R8, где
Z представляет собой C1-6-алкилен или C3-6-циклоалкилен,
R7 представляет собой водород или C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой или гидроксильной группой, R8 представляет собой водород или C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой или гидроксилом,
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое может содержать, необязательно, кольцевой атом кислорода или второй атом азота, который будет являться частью группы NH или NRc, где Rc представляет собой C1-4-алкил, необязательно замещенный гидроксильной группой или C1-4-алкоксигруппой,
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое, необязательно, может содержать кольцевой атом кислорода,
или их фармацевтически приемлемые соли или пролекарства.
В соответствии с еще одним аспектом настоящего изобретения, другим предпочтительным классом соединений формулы (I) или (Ia) являются соединения, в которых R6 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O или =S, и необязательно замещенное группой формулы ZNR7R8, где
Z представляет собой C1-6-алкилен или C3-6-циклоалкил,
R7 представляет собой водород или C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкоксигруппой или гидроксильной группой, R8 представляет собой водород или C1-4-алкил, или C2-4-алкоксигруппой или гидроксилом,
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое может содержать, необязательно, кольцевой атом кислорода или второй атом азота, который будет являться частью группы NH или NRc, где Rc представляет собой C1-4-алкил, необязательно замещенный гидроксильной группой или C1-4-алкоксигруппой,
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое, необязательно, может содержать кольцевой атом кислорода,
или их фармацевтически приемлемые соли или пролекарства.
Предпочтительной группой Y для соединений формул (I) или (Ia) является группа CH2OH.
Другой предпочтительной для соединений формул (I) или (Ia) группой Y является группа CH3.
Особенно подходящие значения X для соединений формул (I) или (Ia) включают группы CH2, CH(CH3) и CH2CH2, из которых предпочтительной является группа CH2.
Благоприятно, когда R6 представляет собой 5-членное кольцо.
В частности, R6 может представлять собой, имея в виду условие, выставленное при определении формулы (I), гетероциклическое кольцо, выбираемое среди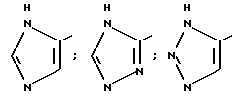
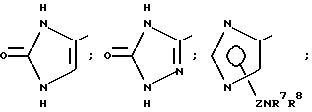
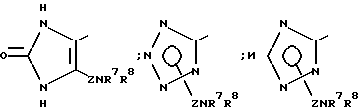
Особенно предпочтительным гетероциклическими кольцами, которые представляет R6, могут выбираться среди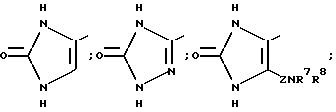
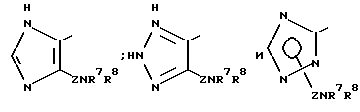
R6 может представлять собой гетероциклическое кольцо, выбираемое, главным образом, среди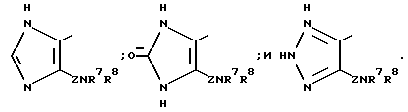
Особенно предпочтительным гетероциклическим кольцом, которое изображается R6, является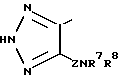
Одной из предпочтительных групп соединений настоящего изобретения являются соединения формулы (Ib) и их фармацевтически приемлемые соли и пролекарства.

где A1, A2 и A3 имеют значения, указанные при определении формулы (Ia),
Z, R7 и R8 имеют значения, указанные для формулы (I).
Другой предпочтительной группой соединений настоящего изобретения являются соединения формулы (Ic) и их фармацевтически приемлемые соли и пролекарства.
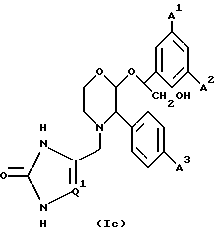
где A1, A2 и A3 имеют значения, указанные для формулы (1a), и
Q1 представляет собой CH или N, или C-ZNR7R8, где R7 и R8 имеют значения, установленные для формулы (1).
Еще одной предпочтительной группой соединений настоящего изобретения являются соединения формулы (1d) и их фармацевтически приемлемые соли и пролекарства.
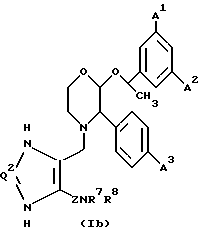
где A1, A2 и A3 имеют значения, установленные для формулы (1a), Q2 представляет собой CH или N, и Z, R7 и R8 имеют значения, установленные для формулы (1).
Что касается соединений формул (1), (1a), (1b), (1c) и (1d), в них Z может представлять собой линейную, разветвленную или циклическую группу. Предпочтительно, Z содержит 1 - 4 атома углерода, и наиболее предпочтительно - 1 или 2 атома углерода. Особенно предпочтительной группой Z является группа CH2.
Что касается соединений формулы (1), (1a), (1b), (1c) и (1d), группа R7 может, подходящим образом, представлять собой C1-4-алкильную группу или C2-4-алкильную группу, замещенную гидроксилом или C1-2-алкоксигруппой, R8 может представлять собой C1-4-алкильную группу или C1-4-алкильную группу, замещенную гидроксилом или C1-2-алкоксигруппой, или R7 и R8 могут быть соединены таким образом, что вместе с атомом азота, к которому они присоединены, они образуют азетидинильную группу, пирролидильную группу, пиперидильную группу, морфолиногруппу, тиоморфолиногруппу, пиперазиногруппу или пиперазиногруппу, замещенную у атома азота C1-4-алкильной группой, замещенной гидроксильной группой или C1-2-алкоксигруппой.
Когда группа NR7R8 представляет собой гетероалифатическое кольцо с 4-7 атомами в кольце и упомянутое кольцо содержит двойную связь, особенно предпочтительной группой является 3-пирролиновая группа.
Когда группа NR7R8 представляет собой неароматическую азабициклическую кольцевую систему, такая система может содержать в себе от 6 до 12 атомов, предпочтительно - от 7 до 10 атомов. К таким подходящим кольцевым системам относятся
5-азабицикло[2.1.1] гексил, 5-азабицикло[2.2.1] гептил, 6-азабицикло[3.2.1]октил, 2-азабицикло[2.2.2]октил, 6-азабицикло[3.2.2]нонил, 6-азабицикло[2.3.1] нонил, 6-азабицикло[3.2.2]децил, 7-азабицикло[4.3.1]децил, 7-азабицикло[4.4.1] ундецил и 8-азабицикло[5.4.1]додецил, в особенности, 5-азабицикло[2.2.1]гептил и 6-азабицикло[3.2.1]октил.
Когда R8 представляет собой C2-4-алкильную группу, замещенную 5- или 6-членным гетероалифатическим кольцом, содержащим один или два гетероатома, выбираемых среди N, O и S, подходящие кольца представляют собой пирролидиногруппу, пиперидиногруппу, пиперазиногруппу, морфолиногруппу, или тиоморфолиногруппу. Особенно предпочтительными являются азотсодержащие гетероалифатические кольца, в особенности кольца пирролидиногруппы и морфолиногруппы.
Особенно подходящие группы ZNR7R8 включают группы, в которых Z представляет собой CH2 или CH2CH2, и NR6R8 представляет собой аминогруппу, метиламиногруппу, диметиламиногруппу, диэтиламиногруппу, азетидильную группу, пирролидиногруппу и морфолиногруппу.
Другими предпочтительными группами, изображаемыми ZNR7R8, являются группы, в которых Z представляет собой CH2 или CH2CH2, R7 представляет собой водород, C1-4-алкил или C3-6-циклоалкил, и R8 представляет собой C2-4-алкил, замещенный одним или двумя заместителями, выбираемыми среди гидроксильной группы, C1-2-алкоксигруппы, азетидинила, пирролидиногруппы, пиперидиногруппы, морфолиногруппы или тиоморфолиногруппы.
В частности, Z представляет собой, предпочтительно, CH2, или NR7R8 представляет собой, предпочтительно, диметиламиногруппу, азетидинил или пирролидиногруппу, в особенности - диметиламиногруппу.
При рассмотрении соединений формул (1a), (1b), (1c) и (1d) A1, предпочтительно, представляет собой фтор или CF3, A2, предпочтительно, представляет собой CF3 и A3, предпочтительно, представляет собой фтор.
Используемые здесь термины "алкил" или "алкокси" в отношении группы или части группы означает, что такая группа является линейной или разветвленной. Примерами подходящих алкильных групп являются метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил и трет-бутил. Примерами подходящих алкоксигрупп являются метоксигруппа, этоксигруппа, н-пропоксигруппа, изо-пропоксигруппа, н-бутоксигруппа, втор-бутоксигруппа и трет-бутоксигруппа.
Циклоалкильные группы, которые здесь упоминаются, могут представлять собой, например, циклопропил, циклобутил, циклопентил или циклогексил. Подходящая циклоалкилалкильная группа может представлять собой, например, циклопропилметил.
Используемые здесь термины "алкенил" или "алкинил" для обозначения группы или части группы, означают, что такая группа является линейной или разветвленной. Примерами подходящих алкенильных групп являются винильная группа и аллильная группа. Подходящей алкинильной группой является пропаргил.
Когда здесь используется термин "галоген", он означает фтор, хлор, бром и иод. Наиболее подходящими галогенами являются фтор и хлор, из которых предпочтительным является фтор.
К конкретным соединениям, входящим в объем настоящего изобретения, относятся
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(2,3- дигидро-5-(N, N-диметиламино)метил-2-оксо-1,3-имидазол-4-ил)метил- 3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-5-(N, N-диметиламино)метил-2-оксо-1,3- имидазол-4-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)- (3-фтор-5-(трифторметил)фенил)этокси)морфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)- фенил)этокси)-4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол- 4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол- 4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-5-(4-гидроксипиперидино)метил-2-оксо- 1,3-имидазол-4-ил)метилморфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)- фенил)этокси)-4-(2,3-дигидро-5-морфолинометил-2-оксо-1,3-имидазол- 4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-5-морфолинометил-2-оксо-1,3-имидазол- 4-ил)метилморфолин,
4-(5-азетидинилметил-2,3-дигидро-2-оксо-1,3-имидазол-4-ил)- метил-2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3- (4-фторфенил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-5-(N-метилпиперазинил)метил-2-оксо-1,3- имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)(фенил)этокси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-5-(N-(2-морфолиноэтил)аминометил)-2- оксо-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3(S)-(4-фторфенил)-4-(2,3- дигидро-2-оксо-5(N-(2-пирролидиноэтил)аминометил)-1,3-имидазол-4-ил)- метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-диметиламино)-метил- 1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)-морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси-3-(S)-(4-фторфенил)-4-(N- (N'-метиламиноэтил)-1,2,4-триазол-3-ил)метилморфолин,
и их фармацевтически приемлемые соли или пролекарства.
Другими предпочтительными соединениями, входящими в объем настоящего изобретения, являются
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4- (5-(N-метиламинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(4-аминометил)-1,2,3-триазол-4-ил)метил-2-(R)-(1-(R)-(3,5-бис(трифторметил) фенил)этокси)-3-(S)-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4- (5-пирролидинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-(азатидинилметил)-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)-2- (R)-(1-(R)-(4-фтор-5-(трифторметил)фенил)этокси)-морфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)-фенил) этокси)-4-(5-(пирролидинометил)-1,2,3-триазол-4-ил)метилморфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)- фенил)этокси)-4-(5-(морфолинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-(N, N-диметиламинометил)-1,2,3-триазол-4-ил)метил-3- (S)-(4-фторфенил)-(R)-(1(R)-(3-трифторметил)фенил)этокси)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4- (5-(N'-метилпиперазинометил)-1,2,3-триазол-4-ил)-метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4- (1-(2-пирролидиноэтил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-фенил-4- (2-(2-пирролидиноэтил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S) (4-фторфенил)-4-(5-(морфолиометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-азетидинилметил)-1,2,3-триазол-4-ил)-метил- 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-4-фторфенил) морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S) (4-фторфенил)-4-(5-(пирролинометил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5- (бис(метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)- 4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(2- хлор-5-морфолинометил-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)- морфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- N,N-диметиламинометил)-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)- морфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- N,N-диметиламинометил)-1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)- морфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- N-(2,2-диметоксиэтил)-N-метиламинометил)-1,2,3-триазол-4-ил)метил-3- (S)-фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- (2-метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- (N-(2-метоксиэтил)-N-метил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)- фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- (N-изопропил-N-(2-метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3- (S)-фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- (N-циклопропил-N-(2-меткосиэтил)аминометил)-1,2,3-триазол-4-ил)-метил-3- (S)-фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- N, N-дибутиламинометил-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R))-(3,5-бис(трифторметил)фенил)этокси)-4-(5- N, N-диизопропиламинометил-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
и их фармацевтически приемлемые соли или пролекарства.
Другими предпочтительными соединениями, входящими в объем настоящего изобретения, являются следующие соединения:
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)- 3-(S)-(4-фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)- метилморфолин,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)- 3-(S)-4-(фторфенил)-4-(1,2,4-триазол-3-ил)метилморфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-(4-фторфенил)- 2-(R)-(1-(S)-(3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)- морфолин,
4-(2,3-дигидро-2-оксо-1,3-имидазол-4-ил)метил- 2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)- 3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол-4-ил)метил- 2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)- 3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)-3-(S)-фенил- 2-(R)-(1-(S)-(3-(трифторметил)фенил)-2-гидроксиэтокси)морфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)- 2-(R)-(1-(S)-(3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)- фенилморфолин,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси-4- (2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)-3-(S)-фенилметил-морфолин,
3-(S)-фенил-4-(1,2,4-триазол-3-ил)- 2-(R)-(1-(S)-(3-(трифторметил)фенил)-2-гидроксиэтокси)морфолин,
и их фармацевтически приемлемые соли или пролекарства.
Другие предпочтительные соединения, входящие в объем настоящего изобретения, описываются в приведенных здесь примерах.
В соответствии с еще одним аспектом настоящего изобретения, будет предлагаться получать соединения формулы (1) в форме фармацевтически приемлемых солей, в особенности, в форме солей присоединения кислот.
Соли соединений формулы (1) для применения в медицине будут представлять собой нетоксичные фармацевтически приемлемые соли. Однако, другие соли могут быть пригодными при получении соединений по настоящему изобретению или их нетоксичных фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений настоящего изобретения включают соли присоединения кислот, которые могут быть образованы, например, путем смешения раствора соединения по настоящему изобретению с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, фумаровая кислота, п-толуолсульфоновая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота, угольная кислота, фосфорная кислота или серная кислота. Соли аминогрупп также могут включать четвертичные аммониевые соли, в которых атом азота аминогруппы несет подходящую органическую группу, такую как алкильная, алкенильная, алкинильная или аралкильная группа. Кроме того, соединения настоящего изобретения несут кислотную группу, подходящие фармацевтически приемлемые соли которой могут включать соли металлов, такие как соли щелочных металлов, например, натриевые или калиевые соли; и соли щелочноземельных металлов, например, кальциевые или магниевые соли.
Настоящее изобретение включает в себя объем пролекарства соединений формулы (1), приведенной выше. Вообще, такие пролекарства будут представлять собой функциональные производные соединений формулы (1), которые являются легко превращаемыми в требуемые соединения формулы (1) in vivo. Обычные процедуры для выбора и получения пролекарственных производных описываются, например, в "Desing of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
Пролекарство может представлять собой фармакологически неактивное производное биологически активного вещества ("материнского лекарственного вещества" или "материнской молекулы"), которое требует трансформации в организме, чтобы выделить активное лекарственное средство, и которое обладает улучшенными свойствами доставки относительно молекулы материнского лекарственного вещества. Трансформация in vivо может являться, например, результатом некоторого метаболического процесса, такого как химический или ферментный гидролиз эфира карбоновой, фосфорной или серной кислоты, или восстановление или окисление поддающейся этому процессу функциональной группы.
Так, например, некоторые предлагаемые пролекарства могут не быть антагонистами тахикинина, в частности, вещества P, и не быть достаточно активными (или вовсе не обладать активностью). Однако такие соединения еще являются полезными при лечении различных описанных здесь состояний, особенно, когда предпочтительная формулировка, которую можно вводить инъекцией.
Преимущества пролекарства могут заключаться в его физических свойствах, таких как повышенная растворимость в воде по сравнению с материнским лекарственным веществом, что удобно при парентеральном введении, или оно может увеличивать абсорбцию из желудочно-кишечного тракта, или может повышать устойчивость лекарственного препарата при длительном времени хранения. В идеале, пролекарство будет улучшать общую эффективность материнского лекарственного вещества, например, за счет снижения токсичности и побочного действия лекарственных препаратов путем регулирования их поглощения, содержания в крови, метаболизма, распределения и усвоения клеткой.
Особенно предпочтительным классом пролекарства соединений настоящего изобретения являются те пролекарства, в которых гидроксильная группа в группе Y в формуле (1) (когда Y представляет собой C1-4-алкил, замещенный гидроксилом), модифицирована.
Необходимо учесть, что другим классом пролекарства соединений настоящего изобретения являются соединения, в которых преобразована гетероциклическая группа, изображаемая R6 в формуле (1), или, с другой стороны, когда преобразованы как гидроксильная группа в группе Y (когда Y представляет собой C1-4-алкил, замещенный гидроксилом), так и гетероциклическая группа, обозначенная R6 в формуле (1).
Подходящие пролекарственные производные включают группы
(a) -(CHR10)n-PO(OH)O- • M+,
(b) -(CHR10)n-PO(O-)2 • 2M+,
(c) -(CHR10)n-PO(O-)2 • D2+,
(d) -(CHR10)n-SO3 -) • M+,
(e) -COCH2CH3CO2- • M+,
(f) -COH,
(g) -CO(CH2)nN(R10)2, и
(h) -(CH(R10)O)n-COR11,
при этом n равен нулю или 1,
M+ представляет собой фармацевтически приемлемый одновалентный контр-ион,
D2+ представляет собой фармацевтически приемлемый двухвалентный контр-ион,
R10 представляет собой водород или C1-3-алкил, и
R11 представляет собой группу, выбираемую среди O(CH2)2 NH3 - • M-, -O(CH2)2NH2(R12)+ • M-, -OCH2CO2- • M+, -OCH(CO2 - • M+) CH2CO2- • M+, -OCH2CH(NH3 -)CO2-, -OC(CO2- • M+) (CH2CO2 - • M+)2, и
где M- представляет собой фармацевтически приемлемый одновалентный контр-ион, и R12 представляет собой водород, C1-4-алкил или C1-4-алкил, замещенный гидроксилом или C1-4-алкоксигруппой.
Особенно предпочтительными пролекарственными производными являются производные, содержащие группы
(a) -(CHR10)n-PO(OH)O- • M+,
(b) -(CHR10)n-PO(O-)2 • 2M+,
(c) -(CHR10)n-PO(O-)2 • D2+,
в особенности, когда n равен нулю.
Термины "материнская молекула", "материнское соединение" или "материнское лекарственное вещество" относятся к биологически активному продукту, который высвобождается путем ферментативного действия метаболического или катаболического процесса, или при химическом процессе, следующим за введением пролекарства. Материнское соединение также может быть исходным веществом для получения своего соответствующего пролекарства.
Хотя все обычные способы введения подходят для вышеупомянутых пролекарств, предпочтительными способами введения являются пероральный способ и внутривенный. После всасывания в желудочно-кишечном тракте или внутривенного введения пролекарства гидролизуются, или, иначе, расщепляются, in vivo до соответствующих материнских соединений формулы (1) или их фармацевтически приемлемых солей. Поскольку материнские соединения могут иметь растворимость менее оптимальной, вышеупомянутые пролекарства имеют определенное преимущество благодаря своей относительно повышенной растворимости в воде.
Примеры отрицательных одновалентных контр-ионов, определяемых здесь как "M-", включают ацетат, адипат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфаросульфонат, цитрат, этансульфонат, фумарат, гемисульфат, 2-оксиэтилсульфонат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, лактат, малат, малеат, метансульфонат, 2-нафталинсульфонт, оксалат, памоат, персульфат, пикрат, пивалат, пропионат, салицилат, стеарат, сукцинат, сульфат тартрат, тозилат(п-толуолсульфонат) и ундеканоат.
Основные соли (которые содержат фармацевтически приемлемые одновалентные катионы, обозначаемые здесь как "M+", или фармацевтически приемлемые двухвалентные катионы, обозначаемые здесь как "D2+", если подходит) включают соли аммония, соли щелочных металлов, таких как натрий, литий и калий, соли щелочноземельных металлов, таких как алюминий, кальций и магний, соли с органическими основаниями, такими как дициклогексиламин, N-метил-D-глюкамин, и соли с аминокислотами, такими как аргинин, лизин, орнитин и т.п. Если M+ представляет собой одновалентный катион, считается, что если определено, что присутствуют 2M+, каждый из M+ может быть одинаковым или разным. Кроме того, также считается, что если присутствуют 2M+ вместо них может присутствовать двухвалентный катион D2+. Также основные азотсодержащие группы могут быть кватернизованы такими агентами как низшие галоидалкилы, такие как метил, этил, пропил и бутилхлориды, -бромиды и -иодиды; диалкилсульфаты, подобные диметил-, диэтил- и дибутилсульфатам; длинноцепные галогениды, такие как децил-, лаурил-, мезитил- и стеарил-хлориды, -бромиды и -иодиды; аралкилгалогениды, подобные бензилбромиду, и другими агентами. Предпочтительными являются нетоксичные физиологически приемлемые соли, хотя другие соли также являются пригодными, например, при выделении или очистке продукта.
Соли могут быть образованы обычными способами, такими как взаимодействие продукта в форме свободного основания с одним или несколькими эквивалентами соответствующей кислоты в растворителе или в среде, в которой соль является нерастворимой, или в растворителе, таком как вода, который удаляется в вакууме или сушкой вымораживанием, или путем замены анионов, имеющихся в соли, на другие, подходящие анионы ионно-обменной смолы.
Особенно предпочтительной подгруппой пролекарств соединений настоящего изобретения являются пролекарства, определяемые формулой (1e), и их фармацевтически приемлемые соли.

где R1, R2, R3, R4, R5, R6, R9a, R9b и X имеют значения, указанные при определении формулы (1), и P в кружочке представляет собой PO(OH(O- • M+'PO(O-)2 • 2M+ или PO(O-)2 • D2+.
Другой предпочтительной подгруппой пролекарств соединений настоящего изобретения являются пролекарства, определяемые формулой (1f), и их фармацевтически приемлемые соли.
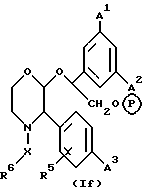
где A1, A2 и A3 имеют значения, установленные для формулы (1a), X и R6 имеют значения, установленные для формулы (1), и P в кружочке представляет собой PO(OH)O2 • M+, PO(O-)2 • 2M+ или PO(O-)2 • D2+.
Особенно предпочтительной подгруппой пролекарств соединений настоящего изобретения являются пролекарства, определяемые формулой (1g), и их фармацевтически приемлемые соли.
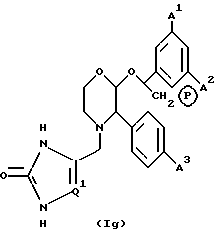
где A1, A2 и A3 имеют значения, указанные для формулы (1a), Q1 имеет значения, установленные для формулы (1c), и P в кружочке представляет собой PO(OH)O- • M+, PO(O-)2 • 2M+ или PO(O-)2 • D2+.
Еще одной предпочтительной подгруппой пролекарств соединений настоящего изобретения являются пролекарства, определяемые формулой (1h), и их фармацевтически приемлемые соли.
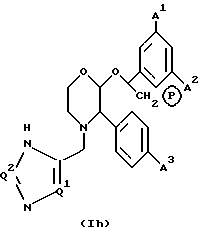
где A1, A2 и A3 имеют значения, указанные для формулы (1a), Q1 и Q2 имеют значения, указанные для формулы (1c) и (1d), соответственно, и P в кружочке представляет собой PO(OH)O-, M+, PO(O-)2•2M+ или PO(O-)2•D2+. Конкретными пролекарственными производными, входящими в объем настоящего изобретения, являются
2-(R)-1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S) -(4-фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метилморфолин,
2-(R)-1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S) -(4-фторфенил)-4-(1,2,4-триазол-3-ил)метилморфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-2-(R)-(1-(S)-3-фтор-5 -(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S)-фенилморфолин,
2-(R)-1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-4- (2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-фенилморфолин,
и их фармацевтически приемлемые соли.
Что касается соединений формул (1f), и (1g) и (1h), в них A1, предпочтительно, представляет собой фтор или CF3, A2, предпочтительно, представляет собой CF3, и A3, предпочтительно, представляет собой фтор.
Настоящее изобретение включает в свой объем сольваты соединений формулы (1) и их солей, например, гидраты.
Соединения, соответствующие настоящему изобретению, имеют, по крайней мере, три асимметрических центра, и могут, соответственно, существовать как в виде энантиомеров, так и в виде диастереоизомеров. Необходимо понимать, что все такие изомеры и их смеси заключаются в объеме настоящего изобретения.
Предпочтительные соединения формул (1), (1a), (1b), (1c), (1d), (1e, (1f) и (1h) будут иметь цис-конфигурацию заместителей 2- и 3-, и предпочтительной стереохимией в положении 2 является стереохимия, которой обладает соединение примера 1 (т.е. 2-(R)-), и предпочтительная стереохимия в положении 3 та, которой обладает соединение примера 1 (т.е., 3-(S)-), и предпочтительная стереохимия атома углерода, к которому присоединяется группа Y, является либо (R), когда Y представляет собой C1-4-алкил (например, метил), либо (S), когда Y представляет собой C1-4-алкил, замещенный гидроксильной группой (например, CH2OH). Это выглядит, например, так, как показано в формуле (1i).
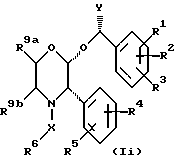
Настоящее изобретение также относится к фармацевтическим композициям, содержащим одно или несколько соединений формулы (1) в сочетании с фармацевтически приемлемым носителем.
Предпочтительно, композициями по настоящему изобретению являются единичные дозированные формы, как таблетки, пилюли, капсулы, порошки, гранулы, растворы или суспензии, или суппозитории, для перорального, парентерального или ректального введения, или для введения путем ингаляции или инсуффляции.
Для приготовления твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим носителем, например, с обычными ингредиентами для приготовления таблеток, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, дикальцийфосфат, или камеди, и другими фармацевтическими разбавителями, например, с водой, чтобы образовать твердую предпрепаративную композицию, содержащую однородную смесь соединения настоящего изобретения или его нетоксичной фармацевтически приемлемой соли. Когда такие предпрепаративные композиции упоминаются как однородные, это означает, что активный ингредиент равномерно диспергирован в композиции, таким образом, что композицию можно легко разделить на равно эффективные единицы лекарственной формы, такие как таблетки, пилюли или капсулы. Такую твердую предпрепаративную композицию затем разделяют на единичные лекарственные формы описанного выше типа, содержащие от 0,1 до 500 мг активного ингредиента по настоящему изобретению. На таблетки или пилюли из новой композиции затем может быть нанесено покрытие, или они могут быть составлены иным способом, для того, чтобы получить лекарственную форму, имеющую преимущество пролонгированного действия. Например, таблетки или пилюли могут содержать внутренний и наружный компонент лекарственной формы, причем последний является формой оболочки для первого. Оба компонента могут быть разделены энтеросолюбильным слоем, который служит для сопротивления разрушению в желудке, и дает возможность внутреннему компоненту проходить незатронутым в двенадцатиперстную кишку или задерживаться при выделении. Ряд материалов может быть использован для таких энтеросолюбильных слоев или покрытий, причем такие материалы включают некоторые полимерные кислоты и смеси полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в состав которых входят композиции по настоящему изобретению для введения их перорально или путем инъекции, включают водные растворы, сиропы с соответствующим вспомогательным лекарством, водные или масляные суспензии, и эмульсии со вспомогательным лекарством с пищевыми маслами, такими как хлопковое масло, сезамное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические носители. Подходящие для водных суспензий диспергирующие или суспендирующие агенты включают синтетические и природные смолы, такие как трагакант, аравийскую камедь, альгинат, декстран, натрийкарбоксиметилцеллюлозу, метилцеллюлозу, поливинилпирролидон или желатин.
Предпочтительные композиции для введения путем инъекции включают композиции, содержащие соединение формулы (1) в качестве активного ингредиента в сочетании с поверхностно-активным веществом (или смачивателеи или поверхностно-активной добавкой), или в форме эмульсии (такой как эмульсия "вода" в масле" или "масло в воде").
Подходящие поверхностно-активные вещества включают анионогенные материалы, такие как бис-(2-этилгексил)сульфосукцинат натрия (докузат натрия), катионогенные вещества, такие как бромиды алкилтриметиламмония (например, бромид цетилтриметиламмония (центримид), и особенно - неионогенные вещества, такие как полиоксиэтиленсорбитаны (например, твинтм 20, 40, 60, 80 или 85) и другие сорбитаны (например, спантм 20, 40, 60, 80 или 85). Композиции с поверхностно-активным веществом обычно будут содержать от 0,05 до 5% поверхностно-активного вещества, предпочтительно - от 0,1 до 2,5%. Следует принять во внимание, что, при необходимости, могут добавляться другие ингредиенты, например маннит, или другие фармацевтически приемлемые носители.
Подходящие эмульсии могут быть приготовлены при использовании коммерчески доступных жировых эмульсий, такие как IntralipidТМ, LyposynТМ, InfonutrolTM, LipofundinTM и LipiphysanTM. Активный ингредиент может быть либо растворен в предварительно смешанной эмульсионной композиции, либо, с другой стороны, он может быть растворен в масле (например, в соевом масле, подсолнечном масле, хлопковом масле, сезамном масле, кукурузном масле или миндальном масле), и эмульсия образуется при смешении с фосфолипидами (например, с фосфолипидами яиц, соевыми фосфолипидами или соевым лецитином) и водой. Следует принять во внимание, что могут добавляться другие ингредиенты, например, глицерин или глюкоза, чтобы установить изотоничность эмульсии. Подходящие эмульсии, как правило, будут содержать до 20% масла, например, от 5 до 20%. Жировая эмульсия будет содержать, предпочтительно, жировые капли размером от 0,1 до 1,0 мкм, особенно - от 0,1 до 0,5 мкм, и иметь pH в интервале от 5,5 до 8,0.
Особенно предпочтительными эмульсионными композициями являются композиции, полученные путем смешения соединения формулы (1) с IntralipidTMили его компонентами (соевое масло, яичные фосфолипиды, глицерин и вода).
Композиции для ингаляции или инсуффуляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях, или в их смесях, и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые добавки, упомянутые выше. Предпочтительно, композиции вводят респираторным путем через рот или нос для местного или системного действия. Композиции в стерильных, предпочтительно, фармацевтически приемлемых растворителях могут быть распылены с помощью инертных газов. Распыляемые растворы могут вдыхаться непосредственно из распыляющего устройства, или распыляющее устройство может быть присоединено к маске для лица, палатке или дыхательному аппарату с периодическим избыточным давлением. Композиции в виде растворов, суспензий или порошков могут вводиться, предпочтительно, через рот или нос, из устройств, которые доставляют препарат соответствующим способом.
Настоящее изобретение также относится к способу получения фармацевтической композиции, содержащей соединение формулы (1), причем упомянутый способ включает сочетание соединения формулы (1) с фармацевтически приемлемым носителем или добавкой.
Соединения формулы (1) являются ценными при лечении широкого ряда клинических состояний, которые характеризуются избыточной активностью тахикинина, в частности, вещества P. Такие состояния могут включать расстройства центральной нервной системы, такие как страх, депрессия, психоз и шизофрения; эпилепсию; нейродегенеративные нарушения, такие как слабоумие, включая старческое слабоумие типа Альцгеймера, болезнь Альцгеймера и синдром Дауна; демиелинизирующие болезни, такие как рассеянный склероз (MS) и боковой аминотрофический склероз (ALS), и другие невропатологические нарушения, такие как периферическая невропатия, например, диабетическая и вызванная химиотерапией невропатия, и постгерпетическая и другие невралгии; мелкоклеточный рак, такой как мелкоклеточный рак легких; респираторные заболевания, в частности, заболевания, связанные с избыточными слизистыми выделениями, такие как хроническая непроходимость дыхательных путей, бронхопневмония, хронический бронхит, мусковисцидоз и астма, и бронхоспазм; воспалительные заболевания, такие как воспалительные заболевания кишечника, псориаз, фиброз, остеоартрит, ревматоидный артрит, пруриго и солнечная эритема; аллергии, такие как экзема и ринит; аллергические расстройства, такие как вызванные сумахом; глазные болезни, такие как конъюнктивит, весенний конъюнктивит и т.п.; глазные заболевания, ассоциированные с клеточной пролиферацией, такие как пролиферативная витреоретинопатия; кожные болезни, такие как контактный дерматит, атопический дерматит, крапивница и другие экзематозные дерматиты; заболевания вследствие вредных привычек, такие как алкоголизм; связанных с стрессом соматические расстройства; рефлекторная симпатическая дистрофия, такая как плечевой синдром; дистимические нарушения; побочные иммунологические реакции, такие как отторжение трансплантированных тканей, и нарушения, связанные с иммунным усилением или угнетением, такие как системная красная волчанка; желудочно-кишечные расстройства и болезни желудочно-кишечного тракта, такие как болезни, связанные с нейронной регуляцией внутренних органов, неспецифический язвенный колит, болезнь Крона, синдром раздражения толстой кишки и рвота, включая острую, позднюю или опережающую рвоту, такую как вызванную химиотерапией, облучением, токсинами, вирусными или бактериальными инфекциями, при беременности, вестибулярных расстройствах, нарушениях движения, при операциях, мигрени и колебаниях внутричерепного давления, в частности, например, вызванную лекарственными препаратами или облучением рвоту, или послеоперационная тошнота и рвота; нарушения функций мочевого пузыря, такие как цистит, гиперрефлексия мышц мочевого пузыря и недержание; фиброзные и коллагеновые болезни, такие как склеродермия и эозинофильный фасцит; нарушения кровотока, вызванные вазодилатационными и вазоспастическими болезнями, такими как стенокардия, мигрень и болезнь Рейно; и боль или ноцицептия, например, свойственные или связанные с любым из вышеупомянутых состояний, особенно, передач боли при мигрени.
Соединения формулы (1) также имеют ценность при лечении сочетаний вышеупомянутых состояний, в частности, при лечении сочетания послеоперационной боли и послеоперационной тошноты и рвоты.
Соединения формулы (1) особенно пригодны при лечении рвоты, включая острую, позднюю и опережающую рвоту, такую как рвота, вызванная химиотерапией, облучением, токсинами, при беременности, вестибулярных нарушениях, нарушениях движения, при операциях, мигрени и колебаниях внутричерепного давления. Соединения формулы (1) в наибольшей мере применимы при лечении рвоты, вызванной противоопухолевыми (цитотоксическими) средствами, включая те средства, которые обычно применяются в химиотерапии рака.
Примеры таких химиотерапевтических средств включают алкилирующие агенты, например, азотистые иприты, соединения этиленимина, алкилсульфонаты и другие соединения с алкилирующим действием, такие как нитромочевины, цисплатин и дакарбазин; антиметаболиты, например, фолиевую кислоту, пуриновые или пиримидиновые антагонисты; митотические ингибиторы, например, винкаалкалоиды и производные подофиллотоксина; и цитотоксические антибиотики.
Конкретные примеры химиотерапевтических средств описываются, например, в D. J. Stewart, Nausea and Vomiting: Recent Recearch and Clinical Advаnces, Eds. J. Kucharczyk et al, CRC Press.Inc., Boca Raton. Florida, USA (1991), pages 177-203, в особенности, на с.188. Обычно применяемыми химиотерапевтическими средствами являются цисплатин, дакарбазин (DTIC), лактиномицин, мехлорэтамин (азотистый иприт), стрептозоцин, циклофосфамид, кармустин (BCNU), ломустин (CCNU), доксорубицин (адриамицин), даунорубицин, прокарбазин, митомицин, цитарабин, этопозид, метотрексат, 5-флуороурацил, винбластин, винкристин, беомицин и хлорамбуцил (P.J.Gralla et al, Cancer Treatment Reports (1984) 68 (1), 163-172).
Соединения формулы (1) также полезны при лечении рвоты, вызванной облучением, в том числе, лучевой терапией, такой как при лечении рака, или лучевой болезнью; и при лечении послеоперационной тошноты и рвоты.
Следует принять во внимание, что соединения формулы (1) могут даваться вместе с другим лечебным средством в виде комбинированного препарата для одновременного, раздельного или последовательного применения для успокоения рвоты. Такие комбинированные препараты могут существовать, например, в форме сдвоенной упаковки.
Другой аспект настоящего изобретения включает соединения формулы (1) в сочетании с 5-HT3-антагонистом, таким как ондансетрон, гранизетрон или тропизетрон, или с другими противорвотными лекарственными средствами, например, с антагонистом допамина, таким как метохлорпрамид. Кроме того, соединение формулы (1) может вводиться в сочетании с противовоспалительным кортикостероидом, таким как дексаметазон. Кроме того, соединение формулы (1) может вводиться в сочетании с химиотерапевтическим средством, таким как алкилирующий агент, антиметаболит, митотический ингибитор или цитотоксический антибиотик, описанные выше. Вообще, для использования в таких сочетаниях подходящими будут доступные на сегодня лекарственные формы известных лечебных препаратов.
При испытаниях на хорьковой модели с вызываемой цисплатином рвотой, описанной F.D. Tattersall et al, Eur. J. Pharmacol., (1993) 250, R5-R6, обнаруживается, что соединение настоящего изобретения смягчают позывы к рвоте и рвоту, вызываемые цисплантином.
Соединения формулы (1) особенно пригодны также при лечении боли или ноцицепции и/или воспаления и расстройств, связанных, например, с невропатией, такой как диабетическая и вызываемая химиотерапией невропатия, постгерпетической и другой невралгией, астмой, остеоартритом, ревматоидным артритом и, особенно, мигренью.
Настоящее изобретение также относится к соединению формулы (1) для применения в терапии.
В соответствии с еще одним аспектом, настоящее изобретение относится к соединению формулы (1) для применения при производстве лекарственного средства для лечения физиологических расстройств, связанных с избытком тахикининов, особенно, вещества P.
Настоящее изобретение также относится к способу лечения или предупреждения физиологических расстройств, связываемых с избытком тазикининов, особенно, вещества P, и упомянутый способ включает введение пациенту, нуждающемуся в таком лечении, снижающего тахикинины количества соединения формулы (1) или композиции, содержащей соединение формулы (1).
При лечении некоторых состояний может быть желательно использовать соединение по настоящему изобретению в сочетании с другим фармакологически активным агентом. Например, при лечении респираторных заболеваний, таких как астма, соединение формулы (1) может применяться в сочетании с бронхолитическим средством, таким как антагонист β2- адренергического рецептора, или антагонистом тахикинина, который воздействует на NK-2-рецепторы. Соединение формулы (1) и бронхолитическое средство могут вводиться пациенту одновременно, последовательно или в сочетании.
Настоящее изобретение относится, соответственно, к способу лечения респираторного заболевания, такого как астма, и упомянутый способ включает введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы (1) и эффективного количества бронхолитического средства.
Настоящее изобретение также относится к композиции, содержащей соединение формулы (1), бронхолитическое средство и фармацевтически приемлемый носитель.
Отличный фармакологический профиль соединений настоящего изобретения дает благоприятную возможность для их применения при лечении в низких дозах, за счет чего снижается риск возникновения нежелательных побочных эффектов.
При лечении состояний, ассоциируемых с избытком тахикининов, подходящий уровень дозировки составляет от 0,001 до 50 мг/кг сутки, в частности - от 0,01 до 25 мг/кг, и такой как от 0,05 до 10 мг/кг в сутки.
Например, при лечении состояний, включающих нейротрансмиссию болевых ощущений, подходящий уровень дозировки составляет от 0,001 до 25 мг/кг в сутки, предпочтительно - от 0,005 до 10 мг/кг в сутки, и особенно - от 0,005 до 5 мг/кг в сутки. Композиции могут вводиться в режиме 1-4 раза в сутки, предпочтительно - один или два раза в сутки.
При лечении рвоты с применением составов для инъекций подходящий уровень дозировки составляет от 0,001 до 10 мг/кг в сутки, предпочтительно - от 0,005 до 5 мг/кг в сутки, и особенно предпочтительно - от 0,01 до 2 мг/кг в сутки. Соединения могут вводиться в режиме 1-4 раза в сутки, предпочтительно - один или два раза в сутки.
Следует принять во внимание, что количество соединения формулы (I), требуемое для применения при каком-либо лечении, будет изменяться не только с изменением выбранного конкретного соединения или выбранной композиции, но также в зависимости от способа введения, природы состояния, подвергаемого лечению, и возраста и состояния пациента и будет определяться, в конечном счете, лечащим врачом.
В соответствии с общим способом (A), соединения по настоящему изобретению могут быть получены из соединений формулы (II)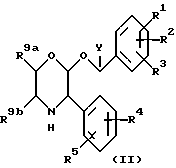
где R1, R2, R3, R4, R5 и Y имеют значения, указанные для формулы (I), путем взаимодействия с соединением формулы (III)
X1-X-R6a
где X имеет значения, указанные для формулы (I); R6a представляет собой группы формулы R6a, определение которой дается при определении формулы (Ia), или ее предшественника, и X1 представляет собой отщепляющуюся группу, такую как бром или хлор, и, если R6a представляет собой группу-предшественник, превращение ее в группу R6 (в процессе, при котором любая реакционноспособная группа может быть защищена, и затем, если желательно, защитная группа может быть удалена).
Это взаимодействие может быть осуществлено обычным способом, например, в органическом растворителе, таком как диметилформамид, в присутствии акцептора кислоты, такого как карбонат калия.
В соответствии с другим способом (B), соединения формулы (I), где R6 представляет собой 1,2,3-триазол-4-ил, замещенный CH2NR7R8, и X представляет собой -CH2-, могут быть получены при взаимодействии соединения формулы (IV)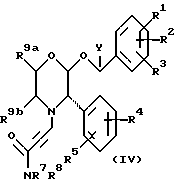
с азидом, например, азидом натрия, в подходящем растворителе, таком как диметилсульфоксид, при температуре от 40oC до 100oC, с последующим восстановлением карбонильной группы, соседней с -R7R8, при использовании подходящего восстановителя, такого как алюмогидрид лития, при температуре от -10oC до комнатной температуры, обычно - при комнатной температуре.
С другой стороны, в соответствии со способом (c), соединения формулы (I), где R6 представляет собой 1,2,3-триазол-4-ил, замещенный CH2NR7R8, и X представляет собой -CH2-, могут быть получены путем взаимодействия соединения формулы (V)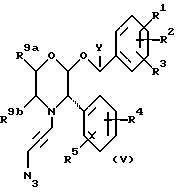
с амином формулы NHR7R8 в подходящем растворителе, таком как простой эфир, например, диоксин, при повышенной температуре, например, от 50oC до 100oC, в запаянной трубке или подобным образом. Эта реакция основывается на взаимодействии, описанном в Chemische Berichte (1989) 122, p. 1963.
В соответствии с еще одним способом (D), соединения формулы (I), где R6 представляет собой замещенный или незамещенный 1,3,5-триазин, могут быть получены путем взаимодействия промежуточного соединения формулы (VI)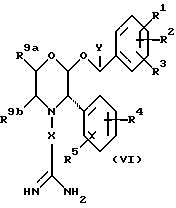
с замещенным или незамещенным 1,3,5-триазином.
Реакцию удобно осуществлять в подходящем органическом растворителе, таком как ацетонитрил, при повышенной температуре, такой как 80-90oC, предпочтительно - при 82oC.
В соответствии с еще одним способом (E), соединения формулы (I), где R6 представляет собой замещенный или незамещенный 1,2,4-триазин, могут быть получены путем взаимодействия промежуточного соединения формулы (VII) с дикарбонильным соединением формулы (VIII)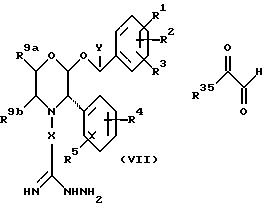
где R35 представляет собой H или подходящий заместитель, такой как ZNR7R8.
Реакцию удобно осуществлять в подходящем органическом растворителе, таком как простой эфир, например, в тетрагидрофуране, при температуре окружающей среды.
В соответствии с другим способом (F), соединения формулы (I), где R6 представляет собой замещенную 1,2,4-триазольную группу, могут быть получены путем взаимодействия промежуточного соединения формулы (I) с соединением формулы (IX)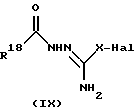
где X имеет значения, установленные для формулы (I), Hal представляет собой атом галогена, например, брома, хлора или иода, и R18 представляет собой H, CONH2 или OCH3 (который превращается в оксозаместитель при условиях реакции),
в присутствии основания, с последующим, при необходимости, преобразованием соединения формулы (I), например, посредством восстановления группы CONH2 до CH2NH2.
Подходящие основания для применения в этой реакции включают карбонаты щелочных металлов, такие как, например, карбонат калия. Реакцию удобно осуществлять в безводном органическом растворителе, таком как, например, безводный диметилформамид, предпочтительно, при повышенной температуре, такой как 140oC.
Подходящим восстановителем для группы CONH2 является алюмогидрид лития, который применяют при температуре от - 10oC до комнатной температуры.
В соответствии с еще одним способом (G), соединения формулы (I), в которых R6 представляет собой тиоксотриазолил, могут быть получены из промежуточных соединений формулы (X)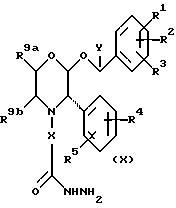
путем взаимодействия с соединением формулы HNCS в присутствии основания.
Подходящими основаниями для применения в этой реакции являются органические основания, такие как, например, 1,8-диазабицикло[5.4.1]ундек-7-ен (ДБУ). Реакцию удобно осуществлять в подходящем органическом растворителе, таком как спирт, например, в бутаноле.
Другие детали соответствующих процедур можно найти в прилагаемых примерах.
Соединения формулы (I) также можно получать из других соединений формулы (I), используя соответствующие методы взаимопревращения. Например, соединения формулы (I), в которых X представляет собой C1-4-алкил, могут быть получены из соединений формулы (I), в которых X представляет собой C1-4-алкил, замещенный оксогруппой, посредством восстановления, например, при применении борана или алюмогидрида лития. Подходящие процедуры взаимопревращения будет очевидны для специалистов в этой области техники.
Промежуточные соединения формулы (IV) могут быть получены из промежуточных соединений формулы (II) путем взаимодействия с ацетиновым соединением формулы HC= C-CH2-Hal в присутствии основания, такого как карбонат калия, в подходящем растворителе, таком как диметилформамид, обычно, при комнатной температуре, с последующим взаимодействием получающегося в результате ацетиленового промежуточного соединения с амидом формулы Hal-CO-NR7R8 в присутствии подходящих катализаторов, включая бис(трифенилфосфин)палладий(II)хлорид, иодид меди (I) и трифенилфосфин, в подходящем растворителе, таком как триэтиламин, предпочтительно, при кипячении с обратным холодильником.
Промежуточные соединения формулы (V) можно получить из соединений формулы (ХI)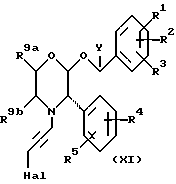
где Hal представляет собой атом галогена, например, хлора, брома или иода, особенно - хлора, путем взаимодействия с азидом, например, с азидом натрия, в подходящем растворителе, таком как диметилсульфоксид, при комнатной температуре или при температуре ниже комнатной,
Соединения формулы (ХI) могут быть получены посредством добавления по каплям промежуточного соединения формулы (II) к дигалогенацетилену формулы Hal-CH2-C ≡ C-CH2-Hal, где каждый Hal представляет собой, независимо, хлор, бром или иод, особенно - хлор. Реакцию удобно осуществлять в подходящем растворителе, таком как диметилформамид, в присутствии основания, такого как карбонат калия.
Промежуточные соединения формулы (VI) можно получать из промежуточных соединений формулы (II) посредством взаимодействия с соединением формулы Hal-X-C(NH)NH2, где Hal и X имеют ранее установленные значения.
Промежуточные соединения формулы (VII) могут быть получены из промежуточных соединений формулы (II) путем взаимодействия с соединением формулы Hal-X-C(NH)NHNH-Boc, где Hal и X имеют ранее установленные значения, и Boc обозначает трет-бутокси-карбонил, с последующим отщеплением группы в условиях кислой среды.
Соединения формулы (VIII) являются коммерчески доступными, или их можно получить известными способами из коммерчески доступных соединений.
Соединения формулы (IX) можно получить так, как описано в J.Med. Chem. (1984), 27, 849.
Промежуточные соединения формулы (X) могут быть получены из соответствующего сложного эфира путем обработки гидразином. Реакцию удобно осуществлять в подходящем органическом растворителе, таком как спирт, например, в этаноле, при повышенной температуре.
В случае соединений, где R6 представляет собой гетероцикл, замещенный группой ZNR7R8, где • представляет собой CH2, некоторые преимущественные соединения формулы (I) могут быть получены из соответствующих соединений с атомом водорода вместо группы ZNR7R8. Так, например, соединение формулы (I), где R6 представляет собой имидазолиноновую группу, несущую группу CH2NR7R8, можно получить из соответствующего соединения, не содержащего группу CH2NR7R8, путем взаимодействия с формальдегидом и амином NHR7R8 в условиях, принятых для реакции Манниха, например, в метаноле при нагревании. При желании можно использовать предварительно образованный реагент, такой как R7R8N+= CH2•1-, и третичный амин, такой как триэтиламин - в качестве акцептора кислоты.
С другой стороны, соединение формулы (I), где R6 представляет собой имидазолиноновую группу, лишенную CH2NR7R8, может быть введено во взаимодействие с параформальдегидом и амином, например, со вторичным амином, таким как пирролидин, что дает соединение, в котором имидазолиноновое кольцо замещено группой CH2NR7R8, где R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4-7 атомов, которое может содержать, необязательно, кольцевой атом кислорода или второй атом азота, который будет являться частью группы NH или NRc, где Rc имеет значения, установленные ранее.
Эта реакция может быть выполнена обычным способом, например, в подходящем растворителе, таком как спирт, например, в метаноле, при повышенной температуре до температуры кипения растворителя.
Другой альтернативный способ получения некоторых соединений формулы (I) включает в себя взаимодействие промежуточного соединения формулы (II), определение которой дается выше, с одним из соединений формулы (XII)
где все LG, которые могут быть одинаковыми или разными, представляют собой отщепляющиеся группы, такие как алкил- или арилсульфонилоксигруппа (например, мезилат или тозилат), или, в частности, атом галогена (например, атом брома, хлора или иода), и X и Z имеют значения, установленные для формулы (I), с последующим взаимодействием образовавшегося в результате соединения с амином NHR7R8 для завершения образования группы ZR7R8.
Эту реакцию удобно осуществлять в органическом растворителе, таком как диметилформамид, в присутствии акцептора кислоты, такого как карбонат калия.
Следует принять во внимание, что, при необходимости, реакционноспособные группы можно защитить, так, например, NH-группы имидазолинона формулы (XIIa) можно защитить любой подходящей защитной для аминогруппы группой, такой как ацетильная группа.
Предпочтительные фосфатные пролекарства соединений настоящего изобретения могут быть получены ступенчатым способом из соединения формулы (I), в котором Y представляет собой, например, -CH2OH-.
Так, оксисоединение сначала обрабатывают дибензилоксидиэтиламинофосфином в подходящем растворителе, таком как тетрагидрофуран, предпочтительно, в присутствии кислотного катализатора, такого как тетразол. Полученное в результате соединение (Y=CH2OP-(OCH2Ph)2) затем окисляют, используя, например, 4-метилморфолин-N-оксид, и получают фосфат с защитной дибензильной группой. Отщепление защитной группы путем каталитического гидрирования или гидрирования с переносом (катализатор палладий-на-угле и формиат аммония) в подходящем растворителе, таком как метанол, при кипячении с обратным холодильником дает нужное фосфатное пролекарство, которое можно превратить в любую нужную соль обычными способами.
При другом двухстадийном способе оксисоединение формулы (I) может быть введено во взаимодействие с подходящим основанием, таким как гидрид натрия, в тетрагидрофуране, и добавленным тетрабензилпирофосфатом, что приводит к образованию защищенного дибензилом фосфата, от которого можно отщепить защитную группу, как описано выше.
Соединения формулы (II) могут быть получены так, как показано на схеме, на которой Ar1 представляет собой R1, R2, R3, замещенные фенильной группой, Ar2 представляет собой R4, R5, замещенные фенильной группой, и Ph означает фенил.
L-Селектрид представляет собой три-втор-бутилборогидрид.
В перечисленных далее ссылках описываются способы, которые может применить специалист для химического синтеза вышеупомянутых соединений, которые приведены в настоящем описании.
(i) D.A.Evans et al., J. Am. Chem. Soc., (1990), 112, 4011
(ii) I.Yanagisawa et al., J.Med. Chem., (1984) 27, 849
(iii) R. Duschinsky et al., J. Am. Chem. Soc., (1948), 70, 657
(iv) F.N. Tebbe et al., J. Am. Chem. Soc., (1978) 100, 3611
(v) N.A. Petasis et al., J. Am. Chem. Soc., (1990) 112, 6532
(vi) K. Takai et al., J. Org. Chem., (1987) 52, 4412.
Приведенные здесь примеры раскрывают, главным образом, получение предпочтительных изомеров. Изомеры, не являющиеся предпочтительными, также получаются в виде второстепенных компонентов. Если желательно, их можно выделить обычными способами, например, хроматографией с использованием соответствующей колонки, и использовать для получения различных стереоизомеров. Однако специалисту следует принять во внимание, что хотя примеры оптимизованы для получения предпочтительных изомеров, изменение растворителя, реагентов, способа хроматографии и т.п. можно использовать, чтобы получить на выходе другие изомеры.
Следует принять во внимание, что соединения формулы (I), В которых R6 содержит заместитель =O или =S, могут существовать в таутомерных формах. Все такие таутомерные формы и их смеси включаются в пределы настоящего изобретения. Наиболее подходящим заместителем из числа =O или =S в R6 является заместитель =O.
Когда вышеупомянутые промежуточные соединения формулы (III) не являются коммерчески доступными, их можно получить способами, описанными в прилагаемых примерах, или иными способами, которые будут очевидными для специалиста в этой области техники.
При выполнении какой-либо, из числа вышеупомянутых, последовательности реакций синтеза может оказаться необходимым и/или желательным защитить чувствительные или реакционноспособные группы любой из молекул, имеющих отношение к этому процессу. Это можно достичь с помощью обычных защитных групп, таких, какие описаны в Protective Groups in Organic Chemistry, ed, J.E.W. McOmie, Plenum Press, 1973; T.W. Greene and P.G.M.Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991. Защитные группы могут быть удалены на подходящей стадии последовательности с применением методов, известных в технике.
Приведенные в качестве примеров соединения настоящего изобретения испытывают методами, изложенными на с. 36-39 описания международного патента ВОИС 93/01165. Обнаружено, что соединения или, в случае пролекарства, материнские соединения являются активными с величиной IC50 при NK1-рецепторе менее 10 нМ - при испытаниях по упомянутым методам.
Пример-описание 1
(S) -4-(фторфенил)глицин
Путем хирального синтеза
Стадия A. 3-(4-фторфенил)ацетил-4-(S)-бензил-2-оксазолидинон
Высушенную в печи 1-литровую 3-горлую колбу, снабженную мембраной, вводом для азота, термометром и магнитной мешалкой, продувают азотом и загружают в нее раствор 5,09 г (33,0 ммоль) 4-фторфенилуксусной кислоты в 100 мл безводного эфира. Раствор охлаждают до -10oC и обрабатывают 5,60 мл (40,0 ммоль) триэтиламина, а затем 4,30 мл (35,0 ммоль) триметилацетилхлорида. Сразу образуется белый осадок. Получающуюся в результате смесь перемешивают при -10oC в течение 40 минут, а затем охлаждают до -78oC.
Высушенную в печи 250-миллилитровую круглодонную колбу, снабженную мембранной и магнитной мешалкой, продувают азотом и загружают в нее раствор 5,31 г (30,0 ммоль) 4-(S)-бензил-2-оксазолидинона в 40 мл сухого ТГФ. Раствор перемешивают в течение 10 минут на бане из сухого льда с ацетоном, и затем постепенно добавляют 18,8 мл 1,6 М раствора н-бутиллития в гексане. Через 10 минут к вышеупомянутой смеси в 3-горловой колбе через трубочку добавляют раствор литиированного оксазолидинона. Охлаждающую баню убирают от получающейся в результате смеси и дают возможность температуре подняться до 0oC. Реакцию гасят 100 мл насыщенного водного раствора хлорида аммония, переносят смесь в 1-литровую колбу, и эфир и ТГФ удаляют в вакууме. Концентрированную смесь распределяют между 300 мл метиленхлорида и 50 мл воды и слои разделяют. Органический слой промывают 100 мл 2н. водной соляной кислотой, 300 мл насыщенного водного раствора бикарбоната натрия, сушат над сульфатом магния и концентрируют в вакууме. Флэш-хроматография на 400 г силикагеле с использованием в качестве элюента смеси гексана с эфиром (3:2 по объему) дает 8,95 г масла, которое медленно отверждается при стоянии. Перекристаллизация из смеси гексана с эфиром (10:1) дает 7,89 г (83%) указанного в заголовке соединения в виде белого твердого вещества. Т.пл. 64-66oC. М.-с. (FAB): m/z 314 (M+ + H, 100%), 177 (M-ArCH2CO+H, 85%). 3H ЯМР (400 МГц, CDCl3 δ 2,76 (1H, д. д., J = 13,2, 9,2), 3,26 (дд., J = 13,2, 3,2), 4,16 - 4,34 (4H, м), 4,65 (1H, м), 7,02 - 7,33 (9H, м). Элем. анализ:
вычислено для C18H16FNO3: C 69,00, H 4,15, N 4,47, F 6,06,
найдено: C 68,86, H 5,14, N 4,48, F 6,08.
Стадия B. 3-((S)-Азидо-(4-фторфенил))ацетил-4-(S)-бензил-2-оксазолидинон
Высушенную в печи 1-литровую 3-горлую колбу, снабженную мембраной, вводом для азота, термометром и магнитной мешалкой, продувают азотом и загружают в нее раствор 58,0 ил 1M раствора бис(триметилсилил)амида калия в толуоле и 85 мл ТГФ, и охлаждают до -78oC. Высушенную в печи 250-мл круглодонную колбу, снабженную мембраной и магнитной мешалкой, продувают азотом и загружают в нее раствор 7,20 г (23,0 ммоль) 3-(4-фторфенил)ацетил-4-(S)-бензил-2-оксазолидинона (со стадии A) в 40 мл ТГФ. Раствор ацилоксазолидинона перемешивают на бане со смесью сухого льда с ацетоном в течение 10 минут, затем переносят, через трубочку, в раствор бис(триметилсилил)амида калия с такой скоростью, чтобы температура внутри смеси оставалась ниже -70oC. Колбу из-под ацилоксазолидинова ополаскивают 15 мл ТГФ, и эту промывную жидкость добавляют, через трубочку, к реакционной смеси, и получающуюся в результате смесь перемешивают при -78oC в течение 30 минут. Высушенную в печи 250-мл круглодонную колбу, снабженную мембраной и магнитными стержнями для перемешивания, продувают азотом и загружают в нее раствор 10,89 г (35,0 ммоль) 2,4,6-триизопропилфенилсульфонилазида в 40 мл ТГФ. Раствор азида перемешивают на бане со смесью сухого льда с ацетоном в течение 10 минут, затем переносят, через трубочку, в реакционную смесь с такой скоростью, чтобы температура внутри смеси оставалась ниже -70oC. Через 2 минуты реакцию гасят 6,0 мл ледяной уксусной кислоты, охлаждающую баню удаляют и смесь перемешивают при комнатной температуре в течение 18 часов. Погашенную реакционную смесь распределяют между 300 мл этилацетата и 300 мл 50% водного насыщенного раствора бикарбоната натрия. Органический слой отделяют, сушат над сульфатом магния и концентрируют в вакууме. Флэш-хроматография на 500 г силикагеля с использованием в качестве элюента смеси гексана с метиленхлоридом, сначала 2: 1, по объему, затем - 1:1, по объему, дает 5,45 г (67%) указанного в заголовке соединения в виде масла. ИК-спектр (чистый, см-1): 2104, 1781, 1702. 3H ЯМР (400 МГц, CDCl3) δ 2,86 (1H, д.д., J = 13,3, 9,6), 3,40 (1H, д.д., J 13,2, 3,2), 4,09 - 4,19 (2H, м), 4,62 - 4,68 (1H, м), 6,14 (1H, с), 7,07 - 7,47 (9H, м). Элем. анализ: вычислено для C18H15FN4O3: C 61,01, H 4,27, N 15,81, F 5,36,
найдено: C 60,99, H 4,19, N 15,80, F 5,34.
Стадия C. (S)-Азидо-(4-фторфенил)уксусная кислота
Раствор 5,40 г (15,2 ммоль) 3-((S)-азидо-(4-фторфенил))-ацетил-4-(S)-бензил-2-оксазолидинона (со стадии B) в 200 мл смеси ТГФ с водой (3:1 по объему) перемешивают на ледяной бане в течение 10 минут. Добавляют в один прием 1,28 г (30,4 ммоль) моногидрата гидроксида лития и получающуюся в результате смесь перемешивают при охлаждении в течение 30 минут. Реакционную смесь распределяют между 100 мл метиленхлорида и 100 мл 25% насыщенного водного раствора бикарбоната натрия и слои разделяют. Водный слой промывают 2 х 100 мл метиленхлорида и подкисляют до pH 2 2н. раствором соляной кислоты. Получающуюся в результате смесь экстрагируют 2 х 100 мл этилацетата; экстракты объединяют, промывают 50 мл насыщенного водного раствора хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме, получают 2,30 г (77%) указанного в заголовке соединения в виде масла, которое используют на следующей стадии без дополнительной очистки. ИК-спектр (чистый, C-1): 2111, 1724. 1H ЯМР (400 МГц, CDCl3) δ 5,06 (1H, с), 7,08 - 7,45 (4H, м), 8,75 (1H, ш.с.).
Стадия (D). (S)-(4-фторфенил)глицин
Смесь 2,30 г (11,8 ммоль) (S)-азидо-(4-фторфенил)уксусной кислоты (со стадии C), 250 мг 10% палладия-на-угле как катализатора и 160 мл смеси воды с уксусной кислотой (3:1 по объему) перемешивают в атмосфере водорода в течение 18 часов. Реакционную смесь фильтруют через целит, и колбу и остаток на фильтре хорошо ополаскивают ~ 1 л смеси воды с уксусной кислотой (3:1 по объему). Фильтра концентрируют в вакууме до объема около 50 мл. Добавляют 300 мл толуола, и смесь концентрируют до получения твердого вещества. Твердое вещество суспендируют в смеси метанола с эфиром (1:1 по объему), фильтруют и сушат, получают 1,99 г (100%) названного в заголовке соединения. 3H ЯМР (400 МГц, D2O + NaOD) δ 3,97 (1H, с), 6,77 (2H, прибл.т., J = 8,8), 7,01 (2H, прибл.т. J = 5,6).
Через расщепление
Стадия A'. (4-фторфенил)ацетилхлорид
Раствор 150 г (0,974 ммоль) 4-(фторфенил)уксусной кислоты и 1 мл N,N-диметилформамида в 500 мл толуола при 40oC обрабатывают 20 мл тионилхлорида и нагревают до 40oC. Добавляют еще 61,2 мл тионилхлорида по каплям в течение 1,5 часов. После добавления раствор нагревают при 50oC в течение 1 часа, растворитель удаляют под вакуумом, и оставшееся масло перегоняют при пониженном давлении (1,5 мм рт.ст.), и получают 150,4 г (89,5%) указанного в заголовке соединения, т.кип. 68-70oC.
Стадия B'. Метил-2-бром-3-(4-фторфенил)ацетат
Смесь 150,4 г (0,872 моль) 4-(фторфенил)ацетилхлорида (со стадии A') и 174,5 г (1,09 моль) брома облучают при 40-50oC кварцевой лампой в течение 5 часов. Реакционную смесь добавляют по каплям к 400 мл метанола, и раствор перемешивают в течение 16 часов. Растворитель удаляют под вакуумом, и оставшееся масло перегоняют при пониженном давлении (1,5 мм рт.ст.), и получают 198,5 г (92%) указанного в заголовке соединения, т.кип. 106-110oC.
Стадия C'. Метил (±)-(4-фторфенил)глицин
Раствор 24,7 г (0,1 моль) метил-2-бром-2-(4-фторфенил)-ацетата (со стадии B') и 2,28 г (0,01 моль) хлорида бензилтриэтиламмония в 25 мл метанола обрабатывают 6,8 г (0,105 моль) азида натрия, и получающуюся в результате смесь перемешивают в течение 20 часов при комнатной температуре. Реакционную смесь фильтруют, фильтрат разбавляют 50 мл метанола и гидрируют в присутствии 0,5 г 10% Pd/C при 345 кПа (50 ф/д2) в течение 1 часа. Раствор фильтруют и растворитель удаляют в вакууме. Остаток распределяют между 10% водным раствором карбоната натрия и этилацетатом. Органическую фазу промывают водой, насыщенным водным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме, получают 9,8 г указанного в заголовке соединения в виде масла.
Стадия (D'). Метил-(S)-(4-фторфенил)глицинат
Раствор 58,4 г метил-(±)-4(фторфенил)глицината (со стадии C') в 110 мл смеси этанола с водой (7:1 по объему) смешивают с раствором 28,6 г (0,0799 моль) O,O'-(+)-дибензолилвинной кислоты ((+)-DBT) (28,6 г), (0,799 ммоль) в 110 мл смеси этанола с водой (7:1 по объему) и получающийся в результате раствор оставляют стоять при комнатной температуре. После завершения кристаллизации добавляют этилацетат (220 мл) и получающуюся в результате смесь охлаждают до -20oC и фильтруют, получают 32,4 г соли (+)-DBT (S)-(4-фторфенил)глицината. Маточные жидкости концентрируют в вакууме и выделяют свободное основание посредством распределения между этилацетатом и водным раствором карбоната натрия. Раствор полученного таким образом свободного основания в 110 мл смеси этанола с водой (7:1 по объему) смешивают с раствором 28,6 г (0,0799 моль) O,O'-(-)-дибензоилвинной кислоты ((-)-DBT) (28,6 г, 0,0799 моль) в 110 мл смеси этанола с водой (7:1 по объему) и получающийся в результате раствор оставляют стоять при комнатной температуре. После завершения кристаллизации добавляют этилацетат (220 мл) и получающуюся в результате смесь охлаждают до -20oC и фильтруют, получают 47,0 г соли (-)-DBT метил-(R)-(4-фторфенил)глицината (ee= 75,8%). Повторный цикл с маточной жидкостью и добавление (+)-DBT дают второй сбор из 7,4 г соли (+)-DBT (S)-(4-фторфенил)глицината (ee = 96,4%). Оба сбора (S)-аминоэфира (39,8 г) объединяют в 200 мл смеси этанола с водой (7:1 по объему), нагревают в течение 30 минут и охлаждают до комнатной температуры. Добавление этилацетата, охлаждение и фильтрация дают 31,7 г соли (+)-DBT (S)-(4-фторфенил)глицината (ee > 98%). Энантиомерный избыток определяют хиральной ЖХВР (Crowпрак (CR(+), 5% MeOH в водн. HClO4, pH 2, 1,5 мл/мин, 40oC, 200 нм).
Смесь 17,5 г соли (+)-DBT (S)-(4-фторфенил)глицината и 32 мл 5,5 н. HCl кипятят с обратным холодильником в течение 1,5 часов. Реакционную смесь концентрируют под вакуумом и остаток растворяют в 40 мл воды. Водный раствор промывают (3 х 30 мл этилацетат) и разделяют слои. Доводят pH водного слоя до 7, используя гидроксид аммония, и выпавшее в осадок твердое вещество отфильтровывают, и получают 7,4 г указанного в заголовке соединения (ee = 98,8%).
Пример-описание 2
4-Бензил-3-(S)-(4-фторфенил)-2-морфолинон
Стадия A. N-Бензил-(S)-(4-фторфенил)глицин
Раствор 1,87 г (11,05 ммоль) (S)-(4-фторфенил)глицина (из примера-описания 1) и 1,12 мл (11,1 ммоль) бензальдегида в 11,1 мл 1н. водного раствора гидроксида натрия и 11 мл метанола при 0oC обрабатывают 165 мг (4,4 ммоль) борогидрида натрия. Охлаждающую баню удаляют и получающуюся в результате смесь перемешивают при комнатной температуре в течение 30 минут. Добавляют к реакционной смеси вторые порции бензальдегида (1,12 мл, 11,1 ммоль) и борогидрида натрия (165 мг, 4,4 ммоль) и перемешивание продолжают еще в течение 1,5 часов. Реакционную смесь распределяют между 100 мл эфира и 50 мл воды и слои разделяют. Отделяют водный слой и фильтруют для удаления небольшого количества нерастворимых веществ. Фильтрат подкисляют до pH 5 2н. водной хлористоводородной кислотой, и твердое вещество, которое выпало в осадок, отфильтровывают, тщательно промывают водой, затем эфиром, и сушат, получают 1,95 г указанного в заголовке соединения. 1H ЯМР (400 МГц, D2O+NaOD) δ 3,33 (2H, AB, к., J = 8,4) 3,85 (1H, c), 6,79 - 7,16 (4H, м).
Стадия B. 4-Бензил-3-(S)-(4-фторфенил)-2-морфолинон
Смесь 1,95 г (7,5 ммоль) N-бензил-(S)-(4-фторфенил)глицина, 3,90 мл (22,5 ммоль) N,N-диизопропилэтиламина, 6,50 мл (75,0 ммоль) 1,2-дибромэтана и 40 мл N, N-диметилформамида перемешивают при 100oC в течение 20 часов (растворение всех твердых веществ происходит при нагревании). Реакционную смесь охлаждают и концентрируют в вакууме. Остаток распределяют между 250 мл эфира и 100 мл 0,5 н. раствора гидросульфата калия и слои разделяют. Органический слой промывают 100 мл насыщенного водного раствора бикарбоната натрия, 3х150 мл воды, сушат над сульфатом магния и концентрируют под вакуумом. Флэш-хроматография на 125 г силикагеля со смесью гексана с эфиром (3:1 по объему) в качестве элюента дает 1,58 г (74%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 2,65 (1H, дт, J = 3,2, 12,8), 3,00 (1H, дт, J = 12,8, 2,8), 3,16 (1H, дт, J = 13,6), 3,76 (1H, д, J = 13,6), 4,24 (1H, с), 4,37 (1H, д, J = 13,2, 3,2), 4,54 (1H, дт, J = 2,8, 13,2), 7,07 - 7,56 (9H, м).
Пример-описание 3
4-Бензил-2-(R)-(3,5-бис(трифторметил)бензоилокси)-3-(S)-(4-фторфенил) морфолин.
Раствор 2,67 г (10,0 ммоль) 4-бензил-3-(S)-(4-фторфенил)-2-морфлинона (пример-описание 2) в 40 мл сухого ТГФ охлаждают до -78oC. Холодный раствор обрабатывают 12,5 мл 1,0 М L-селектрида® в ТГФ, поддерживая температуру в смеси ниже - 70oC. Получающийся в результате раствор перемешивают в холодном состоянии в течение 45 минут и в реакционную смесь добавляют 3,60 мл (20,0 ммоль) 3,5-бис(трифторметил)бензоилхлорида. Получающуюся в результате желтую смесь перемешивают в холодном состоянии в течение 30 минут и реакцию гасят 50 мл насыщенного водного раствора бикарбоната натрия. Погашенную смесь распределяют между 300 мл эфира и 50 мл воды и слои разделяют. Органический слой сушат над сульфатом магния. Водный слой экстрагируют 300 мл эфира, экстракт сушат и объединяют с первоначальным органическим слоем. Объединенные органические растворы концентрируют в вакууме. Флэш-хроматография на 150 г силикагеля с применением смеси гексана с эфиром (37:3 по объему) в качестве элюента дает 4,06 г (8)%) указанного в заголовке соединения в виде твердого вещества. 1H ЯМР (200 МГц, CDCl3) δ 2,50 (1H, дт, J = 3,4, 12,0), 2,97 (1H, прибл. д, J = 12,0), 2,99 (1H, д, J = 13,6), 3,72 - 3,79 (1H, м), 3,82 (1H, д, J = 2,6), 4,00 (1H, д, J = 13,6), 4,20 (дт, J = 2,4, 11,6), 6,22 (1H, д, J = 2,6), 7,22 - 7,37 (7H, м), 7,57 (2H, прибл. д, J = 6,8), 8,07 (1H, c), 8,47 (2H, c), M.-c. (FAB) m/z 528 (M+H), 25%), 270 (100%). Элем. анализ: вычислено для C26H20F7NO3: C 59,21, H 3,82, N 2,66, F 25,21. Найдено: C 59,06, H 4,05, N 2,50, F 25.18.
Пример-описание 4
4-Бензил-2-(R)-(1-(3,5-бис(трифторметил)фенил)-этенилокси)-3-(S)-(4-фторфенил) морфолин
Стадия A. Диметилтитаноцен.
Раствор 2,49 г (10,0 ммоль) дихлорида титаноцена в 50 мл эфира в темноте при 0oC обрабатывают 17,5 мл 1,4 М раствора метиллития в эфире, поддерживая температуру в пределах смеси ниже 5oC. Получающуюся в результате желтую или оранжевую смесь перемешивают при комнатной температуре в течение 30 минут и реакцию гасят, добавляя постепенно 25 г льда. Погашенную реакционную смесь разбавляют 50 мл эфира и 25 мл воды и разделяют слои. Органический слой сушат над сульфатом магния и концентрируют в вакууме, получают 2,03 г (98%) названного в заголовке соединения в виде чувствительного к свету твердого вещества. Диметилтитаноцен можно хранить в виде раствора в толуоле при 0oC, по крайней мере, 2 недели без заметных признаков химического разложения. 1H ЯМР (200 МГц, CDCl3) δ 0,15 (6H, c), 6,06 (10H, c).
Стадия B. 4-Бензил-2-(R)-(1-(3,5-бис(трифторметил)-фенил) этенилокси)-3-(S)-(4-фторфенил)морфолин
Раствор соединения примера-описания 3 (2,50 г, 4,9 ммоль) и 2,50 г (12,0 ммоль) диметилтитаноцена (со стадии A) в 35 мл смеси ТГФ с толуолом (1:1 по объему) перемешивают на масляной бане при 80oC в течение 16 часов. Реакционную смесь охлаждают и концентрируют в вакууме. Флэш-хроматографии на 150 г силикагеля с применением смеси гексана с метиленхлоридом (3:1 по объему) в качестве элюента дает 1,71 г (69%) указанного в заголовке соединения в виде твердого вещества. Образец для анализа получают перекристаллизацией из изопропанола. 1H ЯМР (400 МГц, CDCl3) δ 2,42 (1H, дт, J = 3,6, 12,0), 2,90 (1H, прибл. д., J = 12,0), 2,91 (1H, д, J = 13,6), 3,62 - 3,66 (1H, м), 3,72 (1H, д, J = 2,6), 3,94 (1H, д, J = 13,6), 4,09 (1H, дт, J = 2,4, 12,0), 4,75 (1H, д, J = 3,2), 4,82 (1H, д, J = 3,2), 5,32 (1H, д, J = 2,6), 7,09 (2H, т, J = 8,8), 7,24 - 7,33 (5H, м), 7,58 - 7,62 (2H, м), 7,80 (1H, c), M.c-c. (FAB) 526 (M+H, 75%), 270 (100%). Элем. анализ: вычислено для C27H22F7NO2: C 61,72, H 4,22, N 2,67, F 25,31. Найдено: C 61,79, H 4,10, N 2,65, F 25,27%.
Пример-описание 5
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил) морфолин
Соединение примера-описания 4 (4,0 г) растворяют в этилацетате (50 мл) и изопропаноле (16 мл). К этому раствору добавляют палладий-на-угле (1,5 г) и смесь гидрируют при давлении 275 кПа (40 ф/д2) в течение 36 часов. Катализатор удаляют фильтрацией через цеолит и растворители удаляют в вакууме. Остаток очищают флэш-хроматографией на силикагеле, применяя 100% этилацетата, и затем 1-10% метанола в этилацетате. Такая процедура дает 500 мг изомера A (15%) и 2,6 г изомера B (80%) в виде прозрачных масел - изомер B кристаллизуется при стоянии. Для указанного в заголовке соединения 1H ЯМР (400 МГц, CDCl3) δ 1,16 (3H, д, J = 6,8 МГц), 1,80 (1H, ушир.с), 3,13 (1H, слитно, J = 3,2, 12,4 Гц), 3,23 (1H, дт, J = 3,6, 12,4 Гц), 3,63 (1H, дд, J = 2.4, 11,2 Гц), 4,01 (1H, д, J = 2,4 Гц), 4,13 (1H, дт, J = 3,2, 12,0 Гц), 4,42 (1H, д, J = 2,4 Гц), 4,19 (1H, кв, J = 6,8 Гц), 7,04 - 7,09 (2H, м), 7,27 - 7,40 (4H, м), 7,73 (1H, c), M.c-c. (FAB) 438 (M + H, 75%), 180 (100%).
Получение HCl соли. К раствору свободного основания (0,77 г) в диэтиловом эфире (10 мл) добавляют 1 М HCl в метаноле (1,75 мл). Раствор упаривают досуха, и при добавлении эфира образуются кристаллы. Раствор фильтруют, и остаток промывают диэтиловым эфиром, и получают гидрохлорид указанного в заголовке соединения, т.пл. 248-250oC. Найдено: C 50,46, H 3,85, N 3,01, Cl 7,31, C20H18F7NO2 • HCl, вычислено C 50,70, H 4,04, N 2,96, Cl 7,48%.
Пример-описание 6
4-Бензил-3-(S)-(4-фторфенил)-2-(R)-(3-фтор-5-(трифторметил)бензоилокси) морфлин
Указанное в заголовке соединение получают при взаимодействии соединения примера-основания 2 с 3-фтор-5-(трифторметил)бензоилхлоридом в соответствии с процедурой, изложенной в примере-описании 3. 1H ЯМР (360 МГц, CDCl3) δ 2,50 (1H, дт, J = 3,3, 12,0), 2,96 (1H, д, J = 12,0), 2,98 (1H, д, J = 13,6), 3,75 (1H, дд, J = 1,7, 11,5), 3,80 (1H, д, J = 2,5), 3,92 (1H, д, J = 13,6), 4,19 (1H, дт, J = 2,1, 12,0), 6,20 (1H, д, J = 2,5), 6,99 (2H, т, J = 8,7), 7,2 - 7,37 (5H, м), 7,51 - 7,55 (3H, м), 7,89 (1H, д, J = 8,4), 8,09 (1H, c), M. -c. (Cl+) m/z 478 (M+ + 1, 100%). Элем. анализ: вычислено для C25H20F5NO3: C 62,88, H 4,23, N 2,93. Найдено: C 62,59, H 4,03, N 3,07%.
Пример-описание 7
4-Бензил-3-(S)-(4-фторфенил)-2-(R)-(1-3-фтор-5-(трифторметил)фенил) этенилокси)морфолин
Указанное в заголовке соединения получают с выходом 85% из соединения примера-описания 6 в соответствии с процедурой, приведенной в примере-описание 4. 1H ЯМР (360 МГц, CDCl3) δ 2,42 (1H, дт, J=3,6, 12,0), 2,90 (1H, д, J= 12,0), 2,91 (1H, д, J=13,6), 3,60-3,62 (1H, м), 3,72 (1H, д, J=2,6), 3,92 (1H, д, J=13,6), 4,09 (1H, дт, J=2,4, 12,0), 4,67 (1H, л, J=2,9), 5,28 (1H, д, J= 2,6), 7,07 (2H, т, J=8,7), 7,2-7,37 (7H, м), 7,53 (1H, c), 7,57-7,61 (2H, м). М.-с. (Cl+) 476 (M+1, 100%).
Пример-описание 8
3-(S)-4-(фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил) этокси)формофлин
Гидрируют соединение примера-описания 7 в соответствии со способом, описанным в примере-описании 5. Получают смесь 2 эпимерных продуктов - изомера A и изомера B (главный продукт) - в виде прозрачных масел. Для соединения, указанного в заголовке: 1H ЯМР (360 МГц, CDCl3) δ 1,42 (3H, д, J=6,6 Гц), 1,91 (1H, c), 3,11 (1H, дд, J=3,2, 12,4 Гц), 3,22 (1H, дт, J=3,6, 12,4 Гц), 3,58-3,62 (1H, м), 4,01 (1H, д, J=2,3 Гц), 4,11 (1H, дт, J=3,2, 12,0 Гц), 4,41 (1H, д, J=2,3 Гц), 4,80 (1H, кв, J=6,6 Гц), 6,41 (1H, д, J= 9,2 Гц), 6,86 (1H, c), 7,02 (2H, т, J=8,7 Гц), 7,08 (2H, д, J=9,2 Гц), 7,21-7,26 (2H, м). М. -с. (Cl+) m/z 387 (M+1, 100%). Элем. анализ: вычислено для C19H18F5NO2: C 58,91, H 4,69, N 3,62. Найдено: C 58,88, H 4,81, N 3,76%.
Пример-описание 9
2-(R)-(1-(R)-(3,5-Бис(трифторметил(фенил)этокси)-3-(S)-(4- фторфенил)-4-(2,3-дигидро-2-оксо-1,3-имидазол-4-ил)метилморфолин
Смесь соединения примера-описания 5 (1 г), N,N-диацетил-4-брометил-2-имидазолинона (0,62 г) (полученного в соответствии со способом Dolan and Dushinsky, JACS 1948, 70, 657) и карбоната калия (0,63 г) в 10 мл диметилформамида перемешивают при комнатной температуре в течение 15 минут. Реакционную смесь разбавляют этилацетатом (100 мл) и промывают водой и соляным раствором. Этилацетатный слой сушат (MgSO4) и упаривают в вакууме. Получающееся в результате масло растворяют в этаноле (10 мл), добавляют 33% этанольный раствор метиламина (1 мл) и смесь перемешивают при комнатной температуре в течение 10 минут. Смесь концентрируют в вакууме и получают твердое вещество. Перекристаллизация из этилацетата/метанола дает указанное в заголовке соединение (0,63 г). Т.пл. 192-194oC. 1H ЯМР (360 МГц, ДМСО-d6) 1,35 (3H, д, J=6,5 Гц), 2,25 (1H, дт, J=8,6 Гц), 2,60 (1H, д, J=13,8 Гц), 2,89 (1H, д, J= 11,6 Гц), 3,28-3,36 (2H, м), 3,62 (1H, д, J=10,2 Гц), 4,1 (1H, д, J=10,0 Гц), 4,31 (1H, д, J=2,7 Гц), 4,92 (1H, кв, J=6,5 Гц), 5,97 (1H, c), 7,06 (2H, т, J=8,8 Гц), 7,36 (2H, c), 7,65-7,85 (2H, м), 7,84 (1H, c), 9,58 (1H, c), 9,8 (1H, c).
Пример-описание 10
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил) этокси)-4-(2,3-дигидро-2-оксо-1,3-имидазол-4-ил)метилморфолин
Указанное в заголовке соединение получают из соединения примера-описания 8, используя процедуру, аналогичную процедуре примера-описания 9. Т.пл. 209-210oC. ( α )D = +92,8 (c=1,0, метанол). 1H ЯМР (360 МГц, ДМСО-d6) δ 1,31 (3H, д, J= 6,5 Гц), 2,24 (1H, дт, J=3,0, 11,9 Гц), 2,6 (1H, д, J=13,9 Гц), 3,61 (1H, д, J=11,2 Гц), 4,1 (1H, т, J=11,0 Гц), 4,29 (1H, д, J=2,3 Гц), 4,8 (1H, кв, J=6,5 Гц), 6,00 (1H, c), 6,55 (1H, д, J=9,3 Гц), 6,94 (1H, c), 7,11 (2H, т, J=8,7 Гц), 7,398 (1H, д, J=8,4 Гц), 7,51 (2H, c), 9,59 (1H, c), 9,84 (1H, c).
Пример-описание 11
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4- фторфенил)-4-(1,2,4-триазол-3-ил)-метилморфолин
Раствор соединения примера-описания 5 (3,77 г) и карбоната калия (3,59 г) в сухом диметилформамиде (7 мл)) перемешивают при комнатной температуре в течение 10 минут. Добавляют N-формил-2-хлорацетамидразон (полученный по I. Yanagisawa, J. Med. Chem. (1984) 27, 849) и реакционную смесь нагревают при 60oC в течение 1 часа. Затем температуру поднимают до 140oC в течение 2 часов. Смесь охлаждают и распределяют между этилацетатом и водой, и органическую фазу промывают водой, соляным раствором, сушат (MgSO4) и упаривают, получают коричневое масло. Остаток очищают хроматографией на силикагеле, используя 1-5% метанола в дихлорметане. 1H ЯМР (360 МГц, ДМСО) δ 8,25 (1H, c), 7,85 (1H, c), 7,50 (2H, т), 7,11 (2H, т, J=9,0 Гц), 4,93 (1H, кв, J=6,6 Гц), 4,32 (1H, д, J=2,8 Гц), 4,09 (1H, дт, J=11,5 Гц), 3,63 (1H, д, J=14,1 Гц), 3,59 (1H, д, J=3,0 Гц), 3,17 (1H, д, J=14,0 Гц), 2,49 (1H, дт, J=15,7 Гц), 1,36 (3H, J= 6,6 Гц). М. -с. (Cl+) m/z 519. Элем. анализ: вычислено для C23H19F7NO2: C 53,29, H 4,08, N 10,81. Найдено: C 52,92, H 3,94, N 10,33.
Пример-описание 12
4-Бензил-3-(S)-(4-фторфенил)-2-(R)-(3-трифторметил)бензоилокси)морфолин
Указанное в заголовке соединение получают при взаимодействии соединения примера-описания 2 с 3-(трифторметил)бензоилхлоридом в соответствии с процедурой, описанной в примере-описании 3. 1H ЯМР (360 МГц, CDCl3) δ 2,48 (1H, дт, J= 12,0, 3,5), 2,94 (1H, д, J=13,6), 3,73 (1H, прибл.д, J=11,4), 3,78 (1H, д, J=2,7), 3,91 (1H, д, J=13,6), 4,21 (1H, дт, J=11,7, 2,4), 6,20 (1H, д, J= 2,8), 6,97 (2H, т, J=8,7), 7,25-7,37 (5H, м), 7,53 (2H, м), 7,61 (1H, т, J= 7,8), 7,84 (1H, д, J=8,0), 8:21 (1H, д, J=7,8), 8,30 (1H, c). М.-с. (Cl+) m/z 460 (M+1, 100%).
Пример-описание 13
4-Бензил-3-(S)-(4-фторфенил)-2-(R)-(1-(3-(трифторметил)фенил) этенилокси)морфолин
Указанное в заголовке соединение получают из соединения примера-описания 12 в соответствии с процедурой, описанной в примере-описании 4. 1H ЯМР (360 МГц, CDCl3) δ 2,40 (1H, дт, J=11,9, 3,6 Гц), 2,87 (1H, прибл.д, J=11,8 Гц), 2,89 (1H, д, J=13,5 Гц), 3,62 (1H, прибл.д, J=11,5 Гц), 3,70 (1H, д, J=2,7 Гц), 3,91 (1H, д, J=13,5 Гц), 4,12 (1H, дт, J=11,7, 2,4 Гц), 4,62 (1H, д, J= 2,7 Гц), 4,74 (1H, д, J=2,7 Гц), 5,30 (1H, д, J=2,7 Гц), 7,07 (2H, т, J=8,7 Гц), 7,21-7,32 (5H, м), 7,40 (1H, т, J=7,8 Гц), 7,53-7,63 (4H, м), 7,74 (1H, c). М.-с. (Cl+) m/z 458 (M+1, 100%).
Пример-описание 14
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-(трифторфенил)фенил)этокси)морфолин
Соединение примера-описания 13 гидрируют по способу, описанному в примере-описании 5. Получают смесь 2 эпимерных продуктов - изомер A и изомер B, приблизительно в равных количествах, в виде желтых масел. Для указанного в заголовке соединения (изомер B): 1H ЯМР (360 МГц, CDCl3) δ 1,43 (3H, д, J= 6,6), 3,11 (1H, дд, J=12,6, 2,9), 3,22 (1H, дт, J=12,4, 3,7), 3,60 (1H, дд, J= 11,1, 2,8), 3,99 (1H, д, J=2,2), 4,13 (1H, дт, J=11,6, 3,2), 4,42 (1H, д, J= 2,2), 4,81 (1H, кв. , J=6,6), 6,84 (1H, д, J=7,8), 6,96-7,03 (3H, м), 7,16-7,27 (3H, м), 7,38 (1H, д, J=7,5), M.-c. (Cl+) m-z 370 (M+1, 100%). Элем. анализ: вычислено для C19H19F4NO2: C 61,77, H 5,20, N 3,79. Найдено: C 61,60, H 5,16, N 3,95%.
Пример-описание 15
4-Бензил-3-(S)-фенил-2-морфолинон
Cnflbz A. N-Бензио-(S)-фенилглицин
Раствор 1,51 г (10,0 ммоль) (S)-фенилглицина в 5 мл 2н. водного раствора гидроксида натрия обрабатывают 1,0 мл (10,0 ммоль) бензальдегида и перемешивают смесь при комнатной температуре в течение 20 минут. Раствор разбавляют 5 мл метанола, охлаждают до 0oC и осторожно обрабатывают 200 мг (5,3 ммоль) борогидрида натрия. Охлаждающую таню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Реакционную смесь разбавляют 20 мл воды и экстрагируют 2х25 мл метиленхлорида. Водный слой подкисляют концентрированной соляной кислотой до pH 6 и твердое вещество, которое выпало в осадок, отфильтровывают, промывают 50 мл воды, 50 мл смеси метанола с этиловым эфиром (1:1 по объему), и сушат. Получают 1,83 г (76%) продукта, т.пл. 230-232oC. Элем.анализ: вычислено для C15H15NO2: C 74,66, H 6,72, N 5,81. Найдено: C 74,17, H 6,19, N 5,86.
Стадия B. 4-Бензил-3-(S)-фенил-2-морфолинон
Смесь 4,00 г (16,6 ммоль) N-бензил-(S)-фенилглицина (со стадии A), 5,0 г (36,0 ммоль) карбоната калия, 10,0 мл 1,2-дибромэтана и 25 мл N,N-диметилформамида перемешивают при 100oC в течение 20 часов. Смесь охлаждают и распределяют между 200 мл этилового эфира и 100 мл воды. Слои разделяют и органический слой промывают 3х50 мл воды, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают флэш-хроматографией на 125 г силикагеля, элюируя сначала смесью гексана с этиловым эфиром 9:1 (по объему), а затем 4: 1, и получают 2,41 (54%) продукта в виде твердого вещества, т.пл. 98-100oC. 1H ЯМР (250 МГц, CDCl3) δ 2,54-2,68 (1H, м), 2,96 (1H, дт, J=12,8, 2,8), 3,14 (1H, д, J=13,3), 3,75 (1H, д, J=13,3), 4,23 (1H, с), 4,29-4,37 (1H, м), 4,53 (дт, J=3,2, 11,0), 7,20-7,56 (10 нм), М.-с. (FAB): m/z 268 (M+H, 100%). Элем.анализ: вычислено для C17H17NO2: C 76,38, H 6,41, N 5,24. Найдено: C 76,06, H 6,40, N 5,78.
Пример-описание 16
4-Бензил-2-(R)-(3,5-бис(трифторметил)бензилокси)-3-(S)- фенилморфолин
Раствор 2,67 г (10,0 ммоль) соединения примера-описания 15 в 40 мл сухого ТГФ охлаждают до -78oC. Холодный раствор обрабатывают 12,5 мл 1, ОМ L-селектрида® в ТГФ, сохраняя температуру внутри смеси ниже -70oC. Получающийся в результате раствор перемешивают в течение 45 минут в холодном состоянии и в реакционную смесь загружают 3,60 мл (20,0 ммоль) 3,5-бис(трифторметил)бензолилхлорида. Получающуюся в результате желтую смесь перемешивают в холодном состоянии в течение 30 минут и гасят реакцию 50 мл насыщенного водного раствора бикарбоната натрия. Погашенную смесь распределяют между 300 мл эфира и 50 мл воды, и разделяют слои. Органический слой сушат над сульфатом магния. Водный слой экстрагируют 300 мл эфира, экстракт сушат и соединяют с первоначальным органическим слоем. Объединенные органические растворы концентрируют в вакууме. Флеш-хроматография на 150 г силикагеля с применением смеси 4,06 г (80%) указанного в заголовке соединения в виде твердого вещества. 1H ЯМР (200 МГц, ppm, CDCl3) δ 2,50 (1H, дт, J=3,4, 12,0), 2,97 (1H, прибл. д., J=12,0), 2,99 (1H, д, J=13,6), 3,72-3,79 (1H, м), 3,82 (1H, д, J=2,6), 4,00 (1H, д, J=13,6), 4,20 (ДТ, J=2,4, 11,6), 6,22 (1H, д, J=2,6), 7,22-7,37 (2H, прибл.д., J=6,8), 8,07 (1H, с), 8,78 (2Н, с). Элем. анализ: вычислено для C26H21F6NO3: C 61,29, H 4,16, N 2,75, F 22,38. Найдено: C 61,18, H 4,14, N 2,70, F 22,13.
Пример-описание 17
4-Бензил-2-(R)-(1-(3,5-бис(трифторметил)фенил)-этенилокси)- 3-(S)-фенилморфолин
Раствор 2,50 г (4,9 ммоль) соединения примера-описания 16 и 2,50 г (12,0 ммоль) диметилтитанорцена (пример-описание 4a) в 35 мл смеси ТГФ с толуолом (1: 1 по объему) перемешивают на масляной бане при 80oC в течение 16 часов. Реакционную смесь охлаждают и концентрируют в вакууме. Флеш-хроматографией на 150 г силикагеля с применением в качестве элюента смеси гексана с метиленхлоридом (3:1 по объему) получают 1,71 г (69%) указанного в заголовке соединения в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 2,42 (1H, дт, J= 3,6, 12,0), 2,89 (прибл.д, J=11,6), 2,92 (1H, д, J=13,6), 3,61-3,66 (1H, м), 3,73 (1H, д, J=2,8), 4,00 (1H, д, J=13,6), 4,09 (1H, дт, J=2,4, 11,6), 4,75 (1H, д, J= 2,8), 4,79 (1H, д, J=2,8), 5,36 (1H, д, J=2,4), 7,34-7,41 (7H, м), 7,63 (1H, прибл.д, J=7,2), 7,79 (1H, с), 7,91 (2H, с). М.-с. (FAB) m/z 508 (M+1, 25%). Элем.анализ: вычислено для C27H23F6NO2: C 63,90, H 4,57, N 2,76, F 22,46. Найдено: C 63,71, H 4,53, N 2,68, F 22,66.
Пример-описание 18
2(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)этокси)- -3-(S)-фенилморфолин
Смесь соединения примера-описания 17 (1,5 г) и 10% палладия-на-угле (750 мг) в смеси изопропанола с этилацетатом (25 мл, 3:2 по объему) перемешивают в атмосфере водорода в течение 48 часов. Катализатор удаляют фильтрацией через целит и реакционную колбу и прокладку фильтра промывают этилацетатом (500 мл). Фильтрат концентрируют под вакуумом. Флэш-хроматография дает эпимер A (106 г) и эпимер B (899 мг) в виде прозрачных масел. Названное в заголовке соединение - эпимер B - имеет следующие характеристики.
1H ЯМР (CDCl3, 400 МГц) δ 1,46 (3H, д, J=6,8 Гц), 1,92 (1H, ушир.с), 3,13 (1H, дд, J=3,0, 12,6 Гц), 3,24 (1H, дт, J=3,6, 12,6 Гц), 3,62 (1H, дд, J= 3,6, 11,2 Гц), 4,04 (1H, д, J=2,4 Гц), 4,14 (1H дт, J=3,0, 11,2 Гц), 4,48 (1H, д, J=2,4 Гц), 4,90 (1H, кв, J=6,8 Гц), 7,21-7,32 (7H, м), 7,64 (1H, с). М. -с. (Cl+) m/z 420 (M++1, 20%), 178 (100%). Элем.анализ: вычислено для C20H19F6NO2: C 57,28, H 4,57, N 3,84, F 27,18. Найдено: C 57,41, H 4,61, N 3,29, F 27,23.
Пример-описание 19
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)- 3-(S)-фенил-4-(1,2,4-триазол-3-ил)метилморфолин
Это соединение получают из соединения примера-описания 18 в соответствии с процедурой, описанной в примере-описании 11. М.-. (Cl+) m/z 501 (M++1, 100%).
Пример-описание 20
4-Бензил-2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2- гидроксиэтокси)-3-(S)-(4-фторфенил)морфолин
Соединение примера-описания 4 (12,8 г) растворяют в тетрагидрофуране (50 мл) и смесь охлаждают на льду. Добавляют по каплям боран (49 мл 1,0 М раствора в тетрагидрофуране) и реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Раствор охлаждают на ледяной бане и добавляют по каплям, осторожно, раствор гидроксида натрия (120 мл, 1М) и пероксид водорода (36 мл, 30 мас.%). Получающуюся в результате смесь перемешивают в течение 1 часа, затем разбавляют ее водой (200 мл) и экстрагируют этилацетатом (3х50 мл). Органические экстракты промывают раствором сульфита натрия и затем соляным раствором. Органическую фазу сушат (MgSO4) и упаривают, получают прозрачное масло. ТСХ (этилацетат/гексан, 50:50) показывает два главных продукта, которые разделяют флэш-хроматографией на силикагеле, используя градиент элюирования - 1-30% этилацетата в гексане. Сначала элюируют побочный продукт (2,3 г), а потом элюируется главным продукт (8 г). Главный продукт выделяют в виде белой пены. 1H ЯМР (360 МГц, ДМСО-d6) δ 2,23-2,29 (1H, м), 2,73 (1H, д), 2,80 (1H, д, J=13,0 Гц), 3,48 (1H, д, J=3,5 Гц), 3,45-3,52 (2H, м), 3,56 - 3,65 (2H, м), 4,00 - 4,06 (1H, м), 4,37 (1H, д, J = 3,0 гц), 4,81 (1H, тн, J = 6,0 гц), 4,92 (1H, т, J = 5,5 Гц), 7,14 (2H, т, J = 9,0 Гц), 7,23 - 7,33 (5H, м), 7,35 (2H, т, ArH), 7,85 (1H, c, ArH). М. -с. (Cl+) m/z 544 (M+ + 1, 100%).
Пример-описание 21
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-гидроксиэтокси)-3- (S)-(4-фторфенил)морфолин
Соединение примера-описания 20 (8 г) растворяют в этилацетате (100 мл) в изопропаноле (50 мл) и добавляют к раствору палладий-на-угле (1,5 г). Смесь гидрируют при 275 кПа (40 ф/д2) в течение ночи. Катализатор удаляют фильтрацией и растворители удаляют в вакууме. Остаток очищают флэш-хроматографией, используя в качестве элюента 1 - 10% метанола в дихлорметане. Целевой продукт получают в виде белого порошка (5,7 г, 90%).
1H ЯМР (360 МГц, CDCl3) δ 2,68 - 2,73 (1H, м), 3,03 - 3,15 (1H, м), 3,43 - 3,65 (3H, м), 3,95 (1H, д, J = 3,0 Гц), 4,12 - 4,22 (1H, м), 4,40 (1H, д, J = 3,0 Гц), 4,89 (1H, т, J = 7,0 Гц), 6,99 (т, J = 9,0 Гц, ArH), 7,15 (2H, с), 7,26 - 7,31 (1H, м, ArH), 7,62 (1H, с, ArH). М.-с. (Cl+) m/z 454 (M+ + 1, 100%).
Пример-описание 22
3-(S)-(4-Фторфенил)-2-(R)-(1-(S)-(3-фтор-5- (трифторфенил)фенил)-2-гидроксиэтокси)морфолин
Стадия A. 4-Бензил-3-(S)-(4-фторфенил)-2-(R)-(1-(S)- (3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)морфолин
Соединение примера-описания 7 (0,8 г) растворяют в тетрагидрофуране (5 мл) при комнатной температуре и добавляют боран (5 мл, 1,0 М раствор в тетрагидрофуране). Раствор перемешивают в атмосфере азота в течение 30 минут, до полного взаимодействия всех исходных продуктов. К охлаждаемому (0oC) раствору, который сильно вскипает, добавляют по каплям перекись водорода (5 мл, 29%, водн.) и гидроксид натрия (10 мл, 4н). Получающуюся в результате смесь экстрагируют этилацетатом, органическую фазу промывают раствором бисульфита натрия и насыщенным раствором соли сушат (MgSO4) и упаривают, получают бесцветное масло (1 г). Это вещество далее не очищают, а подвергают взаимодействию, как описано на следующей стадии.
Стадия B. 3-(S)-(4-Фторфенил)-2-(R)-(1-(S)-(3-фтор-5- (трифторметил)фенил)-2-гидрокси)морфолин
Соединение с вышеописанной стадии (A) (1 г) растворяют в смеси этилацетата с 2-пропанолом (20 мл, 3:1) и обрабатывают раствор Pd-на-угле (100 мг). Смесь гидрируют при 414 кПа (60 ф/д2) в течение 12 часов. Катализатор удаляют фильтрацией и растворитель удаляют в вакууме. Остаток очищают хроматографией среднего давления на диоксиде кремния (Lobar), используя в качестве элюента 5% метанола в дихлорметане. Продукт перекристаллизовывают из эфира. 1H ЯМР (360 МГц, ДМСО-d6) δ 2,77 - 3,04 (3H, м), 3,36 - 3,51 (2H, м), 3,93 (1H, ушир.с), 4,05 - 4,13 (1H, м), 4,36 (1H, д, J = 2,0 Гц), 4,72 (1H, т, J = 5,0 Гц), 4,98 (т, J = 7,0 Гц), 6,66 (1H, д, J = 9,2 Гц), 6,89 (1H, с), 7,10 (2H, т, J = 9,0 Гц), 7,33 - 7,37 (2H, м), 7,41 (1H, д, J = 9,0 Гц). М.- с. (Cl+) m/z 404 (M+1, 100%).
Пример-описание 23
N-Карбометокси-2-хлорацетамидразон (ClCH2C(=NH)- NHNHCOOCH3)
К раствору хлорацетонитрила (54,1 г) в безводном метаноле (100 мл) при 0oC добавляют метоксид натрия (20 мл, 1М). Смесь перемешивают при комнатной температуре в течение 30 минут и затем нейтрализуют уксусной кислотой (1,2 мл). Метилгидразинокарбоксилат (64,5 г, предварительно перегнанный под вакуумом) растворяют в теплом диметилформамиде (35 мл) и метаноле (300 мл) и добавляют к реакционной смеси при 0oC. Смесь перемешивают в течение 30 минут, кристаллическое твердое вещество, которое образовалось, удаляют фильтрацией, промывают его этилацетатом и получают указанное в заголовке соединение, т.пл. 138 - 140oC.
Пример-описание 24
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-гидроксиэтокси)-2- (S)-фенилморфолин
Стадия A. 4-Бензил-2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)- 2-гидроксиэтокси)-3-(S)-фенилморфолин
Соединение примера-описания 17 вводят во взаимодействие с бораном и затем с щелочной перекисью водорода в соответствии со способом, показанном в примере-описании 20. Это промежуточное соединение не очищают, и вводят его сырым во взаимодействие на следующей стадии.
Стадия B. 2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)- 2-гидроксиэтокси)-3-(S)-фенилморфолин
Соединение с вышеупомянутой стадии (A) подвергают снятию защитной группы путем гидролиза, как описано в примере-описании 21, и получают указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (360 МГц, CDCl3) δ 2,85 (1H, прибл. д, J = 11,0 Гц), 3,15 (1H, дт, J = 12,0, 3,5 Гц), 3,58 (1H, дд, J = 11,0, 3,0 Гц), 3,63 - 3,71 (2H, м), 4,02 (1H, д, J = 3,0 Гц), 4,25 (дт, J = 12,0, 3,0 Гц), 4,53 (1H, д, J = 3,0 Гц), 4,93 (1H, т, J = 5,0 Гц), 7,22 (2H, с), 7,35 (5H, ушир. с), 7,67 (1H, с). М.-с. (Cl+) m/z 436 (M + 1, 100%).
Пример-описание 25
4-Бензил-2-(R)-(3-фтор-5-(трифторметил)бензилокси)-3-(S)- фенилморфолин
Соединение примера-описания 15 вводят во взаимодействие с L-селектридом, а затем с 3-фтор-5-(трифторметил)бензоилхлоридом, в соответствии со способом, показанном в примере-описании 3, и получают указанное в заголовке соединение в виде прозрачного масла. 1H ЯМР (250 МГц, CDCl3) δ 2,47 (1H, дт, J = 8,5, 2,5 Гц), 2,93 - 2,97 (2H, м), 3,72 - 3,76 (1H, м), 3,79 (2H, д, J = 3,0 Гц), 3,97 (1H, д, J = 9,5 Гц), 4,17 (1H, дт, J = 8,5, 2,5 Гц), 6,22 (1H, д, J = 3,0 Гц), 7,19 - 7,35 (8H, м), 7,45 - 7,56 (3H, м), 7,88 (1H, ушир. д), 8,09 (1H, с). М.-с. (Cl+) m/z 460 (M + 1, 100%).
Пример-описание 26
4-Бензил-2-(R)-(3-фтор-5-(трифторметил)фенил) этенилокси)-3-(S)-фенилморфолин
Соединение примера-описания 25 вводят во взаимодействие с диметилтитаноценом в соответствии с процедурой, описанной в примере-описании 4. Получают указанное в заголовке соединение в виде прозрачного масла (66%). 1H ЯМР (250 МГц, CDCl3) δ 2,29 - 2,39 (1H, м), 2,79 - 2,86 (2H, м), 3,53 - 3,64 (2H, м), 3,92 (1H, д, J = 13,5 Гц), 4,00 - 4,09 (1H, м), 4,61 (1H, д, J = 3,0 Гц), 4,66 (1H, д, J = 3,0 Гц), 5,25 (1H, д, J = 3,0 Гц), 7,14 - 7,35 (10H, м), 7,47 (1H, с), 7,56 (2H, ушир. М.-с. (Cl+) m/z 458 (M + 1, 100%).
Пример-описание 27
2-(R)-(1-(S)-(3-Фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)- 3-(S)-фенилморфолин
Стадия A. 4-Бензил-2-(R)-(1-(S)-(3-фтор-5- (трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-фенилморфолин
Соединение примера-описания 26 вводят во взаимодействие с бораном с последующей обработкой перекисью натрия с основанием в соответствии с процедурой, показанной в примере-описании 20, и получают прозрачное масло. М.-с. (Cl+) m/z 476 (M + 1, 100%).
Стадия B. 2-(R)-(1-(S)-(3-Фтор-5-(трифторметил)фенил)-2- гидроксиэтокси)-3-(S)-фенилморфолин
Удаляют защитные группы у соединения с вышеупомянутой стадии (A), следуя методу, описанному в примере-описании 21. Получают указанное в заголовке соединение в виде белого твердого вещества. Элем. анализ: вычислено для C19H19F4NO3: C 59,22, H 4,97, N 3,63. Найдено: C 59,18, H 5,12, N 3,62. М. -с. (Cl+) m/z 386 (M + 1, 100%).
Пример-описание 28
4-Бензил-3-(S)-фенил-2-(R)-(3-(трифторметил)бензилокси)морфолин
Получают из соединения примера-описания 15 в соответствии со способом, изложенным в примере-описании 3. 1H ЯМР (250 МГц, CDCl3) δ 2,47 (1H, дт), 2,89 - 2,99 (2H, м), 3,69 - 3,82 (2H, м), 3,98 (1H, д), 4,23 (1H, дт), 6,22 (1H, д), 7,22 - 7,40 (8H, м), 7,54 - 7,66 (3H, м), 7,83 (1H, д), 8,22 (1H, д), 8,31 (1H, с).
Пример-описание 29
4-Бензил-3-(S)-фенил-2-(R)-(3-(трифторметил)фенил)- этенилокси)морфолин
Получают из соединения примера-описания 28 в соответствии со способом, изложенным в примере-описании 4.
1H ЯМР (250 МГц, CDCl3) δ 2,41 (1H, дт), 2,84 - 2,96 (2H, м), 3,58 - 3,66 (1H, д), 3,72 (1H, д), 3,99 (1H, д), 4,13 (1H, д), 4,63 (1H, д), 4,72 (1H, д), 5,34 (1H, д), 7,21 - 7,43 (9H, м), 7,50 - 7,68 (4H, м), 7,75 (1H, с).
Пример-описание 30
3-(S)-Фенил-2-(R)-(1-(S)-(3-(трифторметил)фенил)-2- гидроксиэтокси)морфолин
Стадия A. 4-Бензил-3-(S)-фенил-2-(R)-(1-(S)-(3- (трифторметил)фенил)-2-гидроксиэтокси)морфолин
Получают из соединения примера-описания 29 в соответствии со способом, изложенным в примере-описании 20.
Стадия B. 3-(S)-Фенил-2-(R)-(1(S)-(3-(трифторметил)фенил)-2- гидроксиэтокси)морфолин
Вещество со стадии (A) без очистки до указанного в заголовке соединения по способу, показанному в примере- описании 21. 1H ЯМР (250 МГц, CDCl3) δ 2,81 - 2,90 (1H, ушир.д.), 3,16 (1H, дт), 3,54 - 3,68 (3H, м), 4,02 (1H, дт), 4,28 (1H, дт), 4,53 (1H, д), 4,85 - 4,92 (1H, м), 6,85 (1H, д), 6,99 (1H, c), 7,15 - 7,24 (1H, м), 7,34 - 7,45 (6H, м).
Пример 1
2-(R)-1-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(2,3- дигидро-5-(N, N-диметиламинометил)-2-оксо-1,3-имидазол-4-ил)метио-2-(S)- (4-фторфенил)морфолин.
Соединение примера-описания 9 (0,35 г) обрабатывают N,N- диметилметиленаммонийидиодом (0,48 г) и триэтиламином (111 мкл) в тетрагидрофуране 910 мл), и смесь кипятят с обратным холодильником в течение 4 часов. Растворитель удаляют в вакууме, и остаток очищают хроматографией на силикагеле, используя в качестве элюента 1 - 10% метанола в дихлорметане, и получают указанное в заголовке соединение (0,2 г). 1H ЯМР (250 МГц, CDCl3) δ 9,72 (1H, с), 9,68 (1H, с), 7,86 (1Н, с), 7,50 - 7,60 (2H, м), 7,36 (2H, с), 7,07 (2H, т, J=8,8 Гц), 4,96 - 4,89 (1H, кв. J=6,5 Гц), 4,31 (1H, д, J=2,7 Гц), 4,08 (1H, т, J= 10,1 Гц), 3,62 (1H, д, J=10,1 Гц), 3,34 (2H, с), 3,24 (1H, д, J=13,6 Гц), 3,00 (1H, д, J=13,4 Гц), 2,85 (1H, д, J=11,1 Гц), 2,62 (1H, д, J=13,6 Гц), 2,2 (1H, т, J=11 Гц), 2,01 (6H, с) и 1,35 (3H, д, J=6,5 Гц), М.-с (Cl+) m/z 591 (M+1).
4-(2,3-Дигидро-5-(N, N-диметиламинометил)-2-оксо-1,3-имидазол -4-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-( трифторметил)фенил)этокси)морфолин.
Получают из соединения примера-описания 10 по способу, аналогичному способу примера 1. 1H ЯМР (250 МГц, CDCl3) δ 1,38 (3H, д, J=6,2 Гц), 2,22 (6H, с), 2,78 (1H, д, J=14 Гц), 2,92 (1H, д, J=11,2 Гц), 3,14 (2H, прибл.кв. J=14 Гц), 3,34 (1H, д, J=2,8 Гц), 3,46 (1H, д, J=11,2 Гц), 3,60 (1H, д, J=10 Гц), 4,22 (2H, м), 4,26 (1H, д, J=2,8 Гц), 4,74 (1H, кв, J=6,2 Гц), 6,32 (1H, д, J=8,4 Гц), 6,72 (1H, с), 7,06 (3H, т, J=8,4 Гц), 7,36 (2H, ушир, с), 8,70 (1H,ушир.с), 9,20 (1H, ушир. с).
Пример 3
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил) токси)-4-(2,3-дигидро-2-оксо-5-пирролидинометил)1,3-имидазол-4-ил) метилформолин
Смесь соединения из примера-описания 10 (0,1 г), параформальдегида (0,012 г) и пирролидина (0,04 мл) в метаноле (2 мл) нагревают при 90oC в течение 1 часа. К смеси добавляют дополнительную аликвоту параформальдегида (12 мг) и продолжают нагревание еще в течение 30 минут. Смесь охлаждают и растворитель удаляют в вакууме. Остаток очищают хроматографией на силикагеле используют, используют 0,5% водного аммиака и 5% метанола в дихлорметане. Получают продукт в виде пены. Далее продукт очищают в виде гидрохлорида. Т. пл. 157 - 150oC. 1H ЯМР (250 МГц, (свободное основание), CDCl3) δ 1,40 (3H, т, J=6,2 Гц), 1,72 (4H, ушир.с), 2,41 (4H, ушир.с), 2,76 (1H, д, J=12,9 Гц), 2,92 (1H, д, J=11,2 Гц), 3,14 - 3,50 (5H, м), 3,62 (1H, д, J=11,2 Гц), 4,16 (1H, д, J= 12,9 Гц), 4,26 (1H, д, J=2,8 Гц), 4,71 (1H, кв, J=6,2 Гц), 6,30 (1H, д, J= 8,4 Гц), 6,75 (1H, с), 7,0 (3H, т, J=8,4 Гц), 7,34 (2H, ушир.с), 8,86 (1H, ушир.с), 9,14 (1H, ушир.с). М.-м. (Cl+) m/z 567 )M++H).
Пример 4
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)токси)-3-(S)- (4-фторфенил)-4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3- имидазол-4-ил)метилморфолин
Раствор соединения примера-описания 5 (1,5 г) в безводном диметилформамиде (15 мл) при охлаждении льдом и при перемешивании добавляют по каплям, в течение 5 минут, к раствору 4,5-бис-(бромметил)-1,3-диацетил-2-имидазолинона (1,8 г) (полученного по способу Dolan and Duchinsky JACS (1984) 70, 657) в диметилформамиде (10 мл), содержащему карбонат калия (1,4 г). Реакционную смесь перемешивают в течение 10 минут и добавляют в один прием пирролидин (1,1 г), и перемешивание продолжают еще в течение 20 минут. Реакционную смесь разбавляют водой (250 мл) и экстрагируют этилацетатом (3х50 мл). Объединенные органические экстракты промывают водой (2х50 мл) и соляным раствором (1х50 мл) и затем сушат (K2CO3) и концентрируют под вакуумом. Остаток очищают хроматографией на силикагеле, используя градиент элюирования - от 100% дихлорметана до смеси дихлорметан/метанол/водный аммиак (85:15: 0,5), и получают указанное заголовке соединение в виде пены. 1H ЯМР (360 МГц, ДМСО-d6) δ 9,63 (2H, ушир.с), 7,84 (1H, с), 7,53 (2H, ушир.с), 7,36 (2H, с), 7,06 (2H, т, J=8,7 Гц), 4,94 - 4,90 (1H, кв, J=6,5 Гц), 4,31 (1H, д, J=2,68 Гц), 4,07 (1H, т, J=11,4 Гц), 3,61 (1H, д, J=11,20 Гц), 3,34 (1H, J=2,7 Гц), 3,27 (1H, д, J=13,7 Гц), 3,17 (16, д, J=13,4 Гц), 3,00 (1H, д, J= 13,4 Гц), 2,86 (1H, д, J=11,6 Гц), 2,62 (1H, д, J=13,6 Гц), 2,40 - 2,20 (5H, м), 1,64 - 1,58 (2H, м), 1,35 (3H, д, J=6,5 Гц), М.-с. (Cl+) m/z 615 (M+1H).
Соединение примеров 5 - 11 в таблице 1 получают таким же способом, какой описан в примере 4, из соответствующего морфолина, 4,5-бис(бромметил)-1,3-диацетил-2-имидазолинона и соответствующего амина.
Пример 12
2-(R)-(1-(R)-(3,5-Бис(трифторметил(фенил)этокси)-4-(5- диметиламинометил)01,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин
Способ A
a) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси-3-(S)- (4-фторфенил)-4-пропаргилморфолин
К смеси соединения примера-описания 5 (5 г) и карбоната калия (4,76 г) в сухом диметилформамиде при 23oC при перемешивании добавляют пропаргилбромид (1,9 мл). Через 15 минут реакционную смесь разбавляют водой (250 мл) и экстрагируют этилацетатом (3х100 мл). Объединенные органические фазы промывают соляным раствором (1х100 мл), затем сушат (K2CO3) и концентрируют, остается масло. Это масло очищают хроматографией на силикагеле, используя в качестве элюента этилацетат с гексаном (сначала 1:9, затем 1:4), и получают указанное в заголовке соединение в виде масла. 1H ЯМР (250 МГц, CDCl3) δ 1,50 (3H, д, J= 6,6 Гц), 2,21 (1H, с), 2,84 (1H, д, J=11,1 Гц), 2,97 (1H, тд, J=3,2, 11,7 Гц), 3,26 (2H, д, J=1,8 Гц), 3,62 (1H, д, J=2,2 Гц), 3,71 (1H, дд, J=2,3, 11,1 Гц), 4,33 (2H, м), 4,89 (1H, кв, J=6,6 Гц), 7,03 (2H, т, J=8,6 Гц), 7,18 (2H, с), 7,38 (2H, ш.с), 7,63 (1H, с). M.-c (Cl+) m/z 476 (MH, 100).
b) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4- (4-диметиламино-4-оксо-бут-2-инил)-3-(S)-(4-фторфенил)морфолин
Смесь N, N-диметилкарбамоилхлорида (0,195 мл), иодида меди (1) (2 г), бис(трифенилфосфин(палладий(11)хлорида (2 мг), трифенилфосфина (3 мг) и соединения, описанного выше на стадии (a) (1 г) в триэтиламине (4 мл) нагревают при 90oC в течение 5 часов в инертной атмосфере. Смесь охлаждают до 23oC, добавляют метанол (1 мл), удаляют растворитель в вакууме. Остаток распределяют между водой и этилацетатом и слои разделяют. Водную фазу экстрагируют этилацетатом (2х20 мл). Объединенные органические фазы промывают водой, солевым раствором, сушат (MgSO4) и концентрируют, остается масло. Остаток очищают хроматографией на силикагеле, используя в качестве элюента этилацетат в гексане (1:1) и затем этилацетат, и получают указанное в заголовке соединение в виде масла. 1H ЯМР (250 МГц, CDCl3) δ 1,49 (3H, д, J=6,6 Гц), 2,84 - 3,06 (2H, м), 3,00 (3H, с), 3,17 (3H, с), 3,44 (2H, с), 3,64 (1H, ушир. с), 3,73 (1H, д, J=2,0, 11,1 Гц), 4,33 (2H, м), 4,88 (1H, кв, J= 6,6 Гц), 7,03 (2H, т, J=8,7 Гц), 7,17 (2H, с), 7,38 (2H, ушир.с), 7,63 (1H, с), M.-c. (Cl+) m/z 547 (NH, 100%).
c) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(5-N, N- диметилкарбоксамидо-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин
Смесь соединения, описанного выше на стадии (b) (1,1 г), и азида натрия (0,65 г) в диметилсульфоксиде (7,5 мл) нагревают при 70oC в течение 17 часов. Смесь охлаждают до 23oC и избыток диметилсульфоксида удаляют под вакуумом. Остаток распределяют между соляным раствором и этилацетатом. Слои разделяют, и органический слой промывают соляным раствором (2х20 мл), затем сушат (MgSO4) и концентрируют, получают масло. Это масло очищают хроматографией на силикагеле, используя в качестве элюента сначала этилацетат в гексане (1:2, затем 1:1), и затем этилацетат, и получают названное в заголовке соединение в виде бледно-желтой пены. 1H ЯМР (360 МГц, CDCl3) δ 1,47 (3H, д, J= 6,6 Гц), 2,64 (1H, м), 2,90 (1H, д, J=11,6 Гц), 3,09 (3H, c), 3,34 (3H, c), 3,65 (3H, м), 3,92 (1H, д, J=15,5 Гц), 4,27 (1H, тд, J=2,1, 9,5 Гц), 4,35 (1H, д, J= 2,6 Гц), 4,89 (1H, кв, J=6,6 Гц), 7,01 (Т, J=8,7 Гц), 7,16 (2H, ушир.c), 7,64 (1H, c). m/z 590 (MH, 100%).
d) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)- 4-(5-(диметиламинометил)-1,2,3-триазол-4-ил)метил-3-(S)-(4- фторфенил)морфолин
К раствору соединения, описанного выше на стадии (c), (0,11 г) в сухом тетрагидрофуране (1 мл) при 23oC и в инертной атмосфере добавляют по каплям алюмогидрид лития (0,47 мл, 1М раствор в тетрагидрофуране). Через 30 минут добавляют гидроксид натрия (10 капель, 1М), а затем воду (5 капель). Затем добавляют этилацетат (50 мл) и получившуюся в результате смесь фильтруют через фильтр Hyflo. Фильтрат концентрируют под вакуумом, и остаток очищают хроматографией на силикагеле, используя в качестве элюента этилацетат в метаноле (9: 1, затем 4:1), и получают названное в заголовке соединение в виде пены. 1H ЯМР (360 МГц, CDCl3) δ 1,44 (3H, д, J=6,6 Гц), 2,25 (6H, c), 2,57 (1H, тд, J=3,4, 8,55 Гц), 2,90 (1H, д, J=11,7 Гц), 3,25 (1H, д, J=14,0 Гц), 3,43 (1H, д, J=13,6 Гц), 3,45 (1H, д, J=2,2 Гц), 3,53 (1H, д, J=13,6 Гц), 3,61 (1H, д, J=11,2 Гц), 3,78 (1H, д, J=14,0 Гц), 4,22 (1H, т, J=9,3 Гц), 4,32 (1H, д, J=2,2 Гц), 4,86 (1H, кв, J=6,6 Гц), 7,06 (2H, т, J=8,7 Гц), 7,16 (2H, c), 7,48 (2H, ушир.c), 7,63 (1H, c). m/z 576 (MH).
Способ B
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4- фторфенил)-4-(4-хлорбут-2-инил)формолин
a) Раствор продукта из примера-описания 5 (свободное основание, 5 г) в N, N-диметилформамиде (20 мл) постепенно добавляют к нагретому (50oC) раствору 1,4-дихлорбут-2-ина (2,2 мл) и карбоната калия (4,8 г) в N,N-диметилформамиде (20 мл). Раствор нагревают еще в течение 5 часов при 50oC и затем удаляют растворитель под вакуумом. К остатку добавляют воду (400 мл) и продукт экстрагируют этилацетатом (3х150 мл). Объединенные органические фазы промывают водой, насыщенным соляным раствором и сушат (MgSO4). Растворитель удаляют под вакуумом, и остаток хроматографируют на силикагеле (элюирование с 10% этилацетата в петролейном эфире, т.кип. 60-80oC), и получают указанное в заголовке соединение. 1H ЯМР (250 МГц, CDCl3) δ 1,41 (3H, д, J=6,6 Гц), 2,80 (1H, прибл.т, J=10,8 Гц), 2,87 (1H, тд, J=3,5, 11,7 Гц), 3,22 (2H, т, J= 1,9 Гц), 3,52 (1H, д, J=2,8 Гц), 3,68 (1H, д, J=1,4 Гц, 11,1 Гц), 4,00 (2H, т, J=1,9 Гц), 4,22-4,32 (2H, м), 4,81 (1H, к, J=6,6 Гц), 6,96 (2H, т, J= 8,7 Гц), 7,10 (2H, c), 7,31 (2H, ш.c), 7,56 (1H, c). m/z (Cl+) 524 (M+H, 100%).
b) N-(4-Азидобут-2-инил)-2-(R)-(1-(R)-(3,5-бис(трифторметил) фенил)этокси)-3-(S)-(4-фторфенил)морфолин
К раствору 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)- 3-(S)-(4-фторфенил)-4-(4-хлорбут-2-инил)морфолина (4 г) в диметилсульфоксиде (17 мл) добавляют азид натрия (0,562 г). Раствор перемешивают в течение 20 часов и добавляют хлорид аммония и этилацетат. Органическую фазу промывают водой (2 раза),а насыщенным соляным раствором и сушат (MgSO4). Растворитель удаляют в вакууме, и остаток хроматографируют на силикагеле (элюирование с 20% этилацетата в петролейном эфире, т.кип. 60-80oC), и получают указанное в заголовке соединение. 1H ЯМР (360 МГц, CDCl3) δ 1,48 (3H, с, J=6,6 Гц), 2,87 (1H, прибл.т, J=10,2 Гц), 2,98 (1H, тд, J=3,6, 11,7 Гц), 3,35 (2H, т, J=1,9 Гц), 3,61 (1H, д, J=2,8 Гц), 3,72 (1H, де, J=1,4 Гц, 10,0 Гц), 3,92 (2H, т, J= 1,9 Гц), 4,30-4,40 (2H, м), 4,89 (1H, кв, J=6,6 Гц), 7,03 (2H, т, J=8,7 Гц), 7,17 (2H, c), 7,27 (2H, ушир.c), 7,63 (1H, c).
c) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(5- диметиламиноэтил)-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин
Диметиламин (приблизительно 10 мл) конденсируют при 80oC в трубке для высокого давления и добавляют к нему раствор N-(4-азидобут-2-инил)-2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)- 3-(S)-(4-фторфенил)морфолина (3,2 г) в диоксане (15 мл). Трубку запаивают и раствор нагревают при 90oC в течение 16 часов. Раствор упаривают досуха, и остаток хроматографируют на силикагеле (элюирование 5% метанола в дихлорметане, содержащем 0,25% аммиака (уд. вес 0,88), и фракции, содержащие нужный продукт, упаривают в вакууме, и получают указанное в заголовке соединение. К раствору этого остатка в диэтиловом эфире добавляют 1М HCl в метаноле. Раствор упаривают досуха и снова растворяют в диэтиловом эфире, получают кристаллы дигидрохлорида указанного в заголовке соединения, т.пл. 194-198oC, (α)
Пример 13
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)- 4-(N-(2-метиламиноэтил)-1,2,4-триазол-3-ил)метилморфолин, региоизомер B
a) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(N- карбоксиметоксиметил-1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)морфолин
Соединение примера-описания 11 (2,94 г), карбонат калия (2,03 г) и метилбромацетат (0,74 мл) нагревают в диметилформамиде в течение 45 минут. Реакционную смесь обрабатывают водой и этилацетатом, органическую фазу промывают (солевой раствор), сушат (MgSO4) и очищают на диоксиде кремния, используя смеси бензина с этилацетатом. Получают два продукта - изомер A и изомер B - в виде белой пены.
Изомер A. 1H ЯМР (360 МГц, ДМСО) δ 7,89 (1H, c), 7,84 (1H, c), 7,48 (3H, c), 7,33-7,30 (3H, м, J=10,1 Гц), 5,26 (1H, д, J=17,8 Гц), 5,07 (1H, д, J= 17,8 Гц), 4,96 (1H, кв, J=6,5 Гц), 4,39 (1H, д, J=2,8 Гц), 4,04 (1H, ушир.т, J= 10,1 Гц), 3,72 (3H, c), 3,58 (2H, д, J=14,0), 3,51 (1H, д, J=2,8), 3,20 (1H, д, J= 14,0), 2,55 (1H, д, J=11,5), 2,37 (1H, ушир.т, J=3,5 Гц), 1,40 (3H, д, J=6,6).
Изомер B. 1H ЯМР (360 МГц, ДМСО) δ 8,43 (1H, c), 7,82 (1H, c), 7,44 (2H, д, J=1,4 Гц), 7,37 (2H, c), 7,31-7,25 (3H, м, J=3,2 Гц), 5,16 (2H, c), 4,91 (1H, кв, J= 6,5 Гц), 4,35 (1H, д, J=2,8 Гц), 4,08 (1H, ушир.с, J=10,1 Гц), 3,69 (3H, c), 3,60 (1H, д, J=8,8 Гц), 3,55 (1H, д, J=2,7 Гц), 3,30 (1H, д, J= 8,7 Гц), 3,08 (1H, д, J= 13,7 Гц), 2,95 (1H, д, J=11,5 Гц), 2,47 (1H, ушир.с, J=3,4 Гц), 1,35 (3H, д, J=6,5 Гц). М.-с.. (Cl+) m/z 573 (M+1).
b) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4- фторфенил)-4-(N-(N'-метилкарбоксамидо)метил-1,2,4-триазол-3-ил)метилморфолин
Газообразный монометиламин пропускают через раствор соединения вышеупомянутой стадии (b) (375 мг, изомер B) в метаноле (25 мл) в течение 10 минут, и затем систему герметизируют 16 часов. Реакционную смесь упаривают, снова растворяют в этилацетате и концентрируют в вакууме, получают белое твердое вещество (374 мг). 1H ЯМР (250 МГц, CDCl3) δ 8,09 (1H, c), 7,61 (1H, c), 7,45 (2H, ушир.c), 7,33 (2H, c), 7,31 (1H, ушир.c), 7,13 (2H, ушир.c), 4,85 (1H, кв, J=6,5 Гц), 4,76 (2H, ушир.c), 4,37 (1H, ушир.c), 4,36 (1H, ушир.c), 3,85 (1H, д, 3,66 (1H, ушир.c), 3,63 (1H, ушир.c), 3,49 (1H, д), 3,03 (1H, ушир.c), 2,82 (3H, д), 2,80 (1H, ушир.c), 1,46 (3H, д). М.-с. (Cl+) 573 (M++1).
c) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)- (4-фторфенил)-4-(N-(2-метиламиноэтил)-1,2,4-триазол-3-ил) метилморфолин
Охлажденный раствор вышеупомянутого соединения (b) (302 мг) в тетрагидрофуране (5 мл) и комплекс боран-тетрагидрофуран (1,59 мл, 1M) перемешивают в течение 60 минут, затем нагревают (60oC) еще 60 минут. Реакционную смесь упаривают, и остаток снова растворяют в CH3OH c K2CO3, и затем кипятят с обратным холодильником в течение 30 минут. Реакционную смесь выливают в этилацетат, промывают (вода х 2, соляной раствор), и сушат (MgSO4). После очистки на силикагеле с применением смесей CH3OH с дихлорметаном получают указанное в заголовке соединение в виде бесцветного масла (54 мг). 1H ЯМР (250 МГц, CDCl3) δ 7,97 (1H, с), 7,53 (1H<с), 7,39 (2H, ушир. с), 7,29 - 7,23 (3H, м, J = 2,61), 7,06 (2H, с), 4,77 (1H, кв. J = 6,6), 4,29 (1H, д, J = 2,9), 4,25 (1H, ушир. т, J = 2,6), 4,13 (2H, т, J = 5,7), 3,76 (1H, д, J = 14,2), 3,57 (1H, т, J = 3,5), 3,53 (1H, д, J = 2,8), 3,31 (1H, д, J = 14,1), 2,95 (1H, т, J = 9,3), 2,92 (2H, т, J = 5,9), 2,56 (1H, ушир.т, J = 3,5), 2,36 (3H, с), 2,16 (1H, ушир, с), 1,37 (3H, дд, = 6,6). М.-с. (Cl+) m/z 558 (M+ + 1).
Соединения примеров 14-21 в таблице 2 получают способом, подобным способу, описанному в примере 12, способ B, через соответствующие N-(4-азидобут-2-инил)морфолины и соответствующие амины.
Пример 22
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-фенил-4- (1-(2-пирролидиноэтил)-1,2,4-триазол-3-ил)метилморфолин
a) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(1-(2-оксо-2- пирролидиноэтил)-1,2,4-триазол-3-ил)метил-3-(S)-фенилморфолин
Раствор соединения примера-описания 19 (2,86 г), карбоната калия (2,37 г) и 1-бромацетилпирролидина (1,21 г) нагревают в диметилформамиде (15 мл) при 60oC. Смесь охлаждают и распределяют между водой и этилацетатом. Органическую фазу промывают водой, солевым раствором и сушат (MgSO4). Растворитель удаляют в вакууме и остаток очищают на диоксиде кремния, используя 1,5% метанола в дихлорметане в качестве элюента. Получают 2 продукта - изомер A и изомер B.
Изомер A (алкилирование в положение 2 в 1,2,4-триазоле). 1H ЯМР (250 МГц, CDCl3) δ 7,83 (1H, с), 7,61 (1H, с), 7,39 - 7,30 (5H, м), 7,16 (2H, c), 5,00 (2H, д, J = 16,4 Гц), 4,88 (1H, кв., J = 6,6 Гц), 4,67 (1H, д, J = 16,4 гц), 4,35 (1H, д, J = 2,8 Гц), 4,20 (1H, ушир. т, J = 11,6 Гц), 3,77 (1H, д, J = 14,4 Гц), 3,62 (1H, дд, J = 11,3 Гц), 3,51 - 3,44 (4H, м), 3,39 (1H, с), 3,33 (1H, д, J = 14,4 Гц), 2,90 (1H, д, J = 11,4 Гц), 2,74 (1H, ушир. т, J = 11,8 гц), 2,12 - 2,02 (2H, м), 1,97 - 1,86 (2H, м), 1,45 (3H, д, J = 6,6 Гц).
Изомер B (алкилирование в положении 1,1,2,4-триазола). 1H ЯМР (250 МГц, CDCl3) δ 8,19 (1H, с), 7,60 (1H, с), 7,47 (2H, ушир, с), 7,36 - 7,27 (3H, м), 7,14 (2H, с), 4,89 (1H, кв, J = 6,6 Гц), 4,36 (1H, д, J = 2,8 Гц), 4,31 (1H,ушир. т, J = 11,4 Гц), 3,86 (1H, д, J = 14,0 Гц), 3,60 (1H, дд, J = 11,3 Гц), 3,59 (1H, д, J = 2,7 Гц), 3,53 - 3,48 (4H, м), 3,35 (1H, д, J = 14,1 Гц), 3,03 (1H, д, J = 11,8 Гц), 2.60 (1H, ушир. т, J = 11,9 Гц), 2,08 - 2,00 (2H, м), 1,94 -1,84 (2H, м), 1,44 (3H, д, J = 6,6 гц).
b) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)- фенил-4-(1-(2-пирролидиноэтил)-1,2,4-триазол-3-ил)метилморфолин
К раствору соединения, описанного выше в пункте (a) (изомер B), в тетрагидрофуране (5 мл) при 0oC добавляют алюмогидрид лития (1,0 M раствор в тетрагидрофуране). Смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. Реакцию гасят (гидроксид натрия и вода) и реакционную смесь фильтруют через цеолит для удаления неорганических веществ. Фильтрат упаривают и остаток очищают на диоксиде кремния, используя в качестве элюента 10% метанола в дихлорметане. Получают продукт в виде желтого масла. 1H ЯМР (250 МГц, CDCl3) δ 8,08 (1H, с), 7,60 (1H, с), 7,49 (2H, ушир.с), 7,37 - 7,31 (3H, м), 7,13 (2H, с), 4,85 (1H, кв, J = 6,6 Гц), 4,36 (1H, д, J = 2,8 Гц), 4,33 - 4,24 (1H, м), 4,22 (2H, т, J = 6,5 гц), 3,86 (1H, дд, J = 14,1 Гц), 3,63 (1H, д, J = 9,2 Гц), 3,60 (1H, д, J = 2,9 Гц), 3,38 (1H, дд, J = 14,0 Гц), 3,00 (1H, д, J = 11,7 Гц), 2,89 (2H, т, J = 6,6 Гц), 2,59 (1H, ушир. т, J = 11,9 Гц), 2,59 - 2,49 (4H, м), 1,79 (4H, м), 1,43 (3H, д, J = 6,5 Гц).
Пример 23
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси-3- (S)-фенил-4-(2-(2-пирролидиноэтил)-1,2,4-триазол-3-ил) метилморфолин
Соединение, описанное в примере 22a (изомер A), вводят во взаимодействие в соответствии с процедурой, описанной в примере 22b, и получают указанное в заголовке соединение в виде желтого масла. 1H ЯМР (250 МГц, CDCl3) δ 7,80 (1H, с, CH), 7,61 (1H, с, ArH), 7,53 - 7,48 (2H, м, PhH), 7,38 - 7,34 (3H, м), 7,17 (2H, с), 4,88 (1H, кв, J = 6,5 Гц), 4,36 (1H, д, J = 2,9 Гц), 4,34 - 4,20 (1H, м), 4,23 - 4,07 (3H, м), 3,83 (1H, д, J = 14,0 Гц), 3,66 (1H, м), 3,42 (1H, д., J = 2,8 Гц), 3,27 (1H, д, J = 14,1 Гц), 2,88 - 2,73 (1H, м), 2,88 - 2,73 (2H, м), 2,88 - 2,73 (1H, м), 2,50 (3H, ушир.с), 1,73 (4H, ушир.с) 1,4 (4H, д, J = 6,6 Гц).
Пример 24
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4-фторметил)- 4-(5-морфолинометил)-1,2,3-триазол-4-ил)метилморфолин
Это соединение получают способом, описанным в примере 12 (способ A), и очищают хроматографией на диоксиде кремния, используя этилацетат, петролейный эфир (60-80oC) и метанол (3:10:0, затем 1:0:0, и затем 9:0:1) в качестве элюента, и получают указанное в заголовке соединение в виде белой пены. 1H ЯМР (360 МГц, CDCl3) δ 1,44 (3H, д, J = 6,6 Гц), 2,43 (4H, м), 2,57 (1H, д, J = 11,9, 3,4 Гц), 2,90 (1H, д, J = 11,6 Гц), 3,27 (1H, д, J = 14,1 Гц), 3,4 - 3,67 (8H, м), 3,82 (1H, д, J = 14,1 Гц), 4,23 (1H, м), 4,32 (1H, д, J = 2,8 Гц), 4,87 (1H, м), 7,06 (2H, т, J = 8,7 Гц), 7,16 (2H, с), 7,48 (2H, ушир.с), 7,64 (1H, с), M.-c. (ES+) m/z 618 (MH1, 54%).
Соединения примеров 25-27 в таблице 2 получают способом, подобным способу, описанному в примере 12, способ B, через соответствующие N-(4-азидобут-2-инил)морфолины и соответствующие амины.
Пример 28
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси-4- (2-хлор-5-морфолинометил-1,3-имидазол-4-ил)-(S)-(4-фторфенил) морфолин
Продукт примера 7 (0,2 г) и оксихлорид фосфора кипятят с обратным холодильником в течение 20 часов. Смесь охлаждают и обрабатывают ее дихлорметаном и водным раствором карбоната калия. Органический слой промывают (H2O), сушат (MgSO4) и упаривают под вакуумом. Продукт очищают хроматографией на диоксиде кремния, используя сначала 100% этилацетата, затем 5% метанола и 95% этилацетата, и получают указанное в заголовке соединение в виде масла. M.c-c. (ES+) m/z 651 (MH+, 100%).
Пример 29
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(5-(N, N-диметиламиномтил) имидазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин
Гидрохлорид 4,5-бис(хлорметил)имидазола (описание патента Великобритании GB-2068362-A) вводят во взаимодействие с соединением примера-описания 5 в соответствии со способом, описанным в примере 4, и получают указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (250 МГц, CDCl3) δ 1,44 (3H, д, J = 6 Гц), 2,19 (3H, c), 2,46-2,62 (2H, м), 2,92 - 3,07 (2H, м), 3,25 - 3,44 (3H, м), 3,56 - 3,70 (2H, м), 4,16 - 4,33 (2H, м), 4,85 (1H, кв, J = 6 Гц), 7,01 - 7,17 (4H, м), 7,38 - 7,67 (4H, м), M.-c. (ES) m/z 575 (M + 1+, 100%).
Пример 30
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(5-(N, N-диметилламинометил)- 1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)-морфолин
3,5-Бис(хлорметил)триазол (J. Het. Chem. (1986) 23, 361-368) вводят во взаимодействие с соединением примера-описания 5 в соответствии со способом, проиллюстрированным в примере 4, и получают названное в заголовке соединение в виде твердого вещества. 1H ЯМР (250 МГц, CDCl3) δ 1,27 (3H, д, J = 6,6 Гц), 2,15 (6H, c, CH3), 2,43 (1H, д, J = 11,7 Гц), 2,79 - 2,83 (1H, м), 3,16 (1H, д, J = 15,4 Гц), 3,38 (1H, д, J = 2,8 Гц), 3,43-3,48 (1H, м), 3,48 (2H, с, CH2), 3,63 (1H, д, J = 14,5 Гц), 4,12 (1H, дт, J = 11,7, 3,2 Гц), 4,15 (1H, д, J = 2,8 Гц), 4,69 (1H, кв. J = 6,6 Гц), 6,85 (2H, т, J = 8,75 Гц), 6,97 (2H, c), 7,27 (2H, ушир. т), 7,45 (1H, c). M.-c. (ES) m/z 576 (M+1, 100%).
Соединение примеров 31-37 в таблице 2 получают по способу, подобному способу, описанному в примере 12, способ B, через соответствующие N-(4-азидобут-2-инил)морфолины и соответствующие амины.
Соединения примеров 38-41 в таблице 1 получают по способу, подобному способу, описанному в примере 4, из соответствующих морфолинов, 4,5-бис(бромметил)-1,3-диацетил-2-имидазолинона и соответствующих аминов.
Пример 42
5-Оксид-2-(R)-(1-(R)-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)- 4-(2,4-дигидро-2-оксо-5-тиомофролинометил-1,3-имидазол-4-ил)метилморфолина
Соединение примера 38 (67 мг, 1 экв.) растворяют в CF3CO2H (0,3 мл) в атмосфере N2, затем раствор охлаждают до 0oC и добавляют раствор CF3CO3H (2 М в CF3CO2H, 47 мкл, 1,1 экв.). После перемешивания при 0oC в течение 1 часа растворитель удаляют в вакууме, и остаток растворяют в EtOAc и промывают насыщенным водным раствором NaHCO3, сушат (K2CO3) и концентрируют, получают в остатке желтую пену. Эту пену очищают колоночной хроматографией, используя в качестве элюента MeOH/CH2Cl2/NH3 (3:97:0,25), и получают указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (250 МГц, CDCl3) δ 9,48 (1H, c), 8,66 (1H, c), 7,64 (1H, c), 7,40 (2H, м), 7,14 (2H, c), 7,06 (2H, т, J = 8,6 Гц), 4,87 (1H, кв, J = 6,5 Гц), 4,30 (1H, д, J = 2,7 Гц), 4,23 (1H, т, J = 10,0 Гц), 3,65 (1H, д, J = 9,6 Гц), 3,45 (1H, м), 3,75 (1H, м), 3,36 (1H, д, J = 2,7 Гц), 3,30 (1H, д, J = 14 Гц), 3,20 (1H, д, J = 14 Гц), 3,05 - 2,60 (9H, м), 2,36 (1H, м), 1,46 (3H, д, J = 6,5 Гц).
Соединение примеров 43-62 в таблице 2 получают по способу, подобному способу, описанному в примере 12, способ B, через соответствующие N-(4-азидобут-2-инил)морфолины и соответствующие амины.
Пример 63
2-(R)-(2-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(1-(2-(N, N- диизопропиламино)этил)-1,2,4-триазол-3-ил)метил-3-(S)-фенилморфолин.
a) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(1-(2-гидроксиэтил)- 1,2,4-триазол-3-ил)метил-3-(S)-фенилморфолин
Соединение примера-описания 19 (3,90 г, 7,8 мМ) нагревают (60oC) в диметилформамиде (20 мл), содержащем 2-бромэтанол (1,66 мл, 23,4 мМ) и карбонат калия (3,23 г, 23,4 мМ) в течение 2 часов. Реакционную смесь выливают в этилацетат и промывают водой и солевым раствором, сушат (MgSO4) и упаривают. Два изомера очищают и разделяют на диоксиде кремния, элюируя смесями метанола с дихлорметаном (3,06 г), M.-c. (ES)+ m/z 545.
b) 2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)-3-(S)-фенил-4-(1-(2- тозилоксиэтил)-1,2,4-триазол-3-ил)метилморфолин
Спирт с вышеупомянутой стадии (a) (1,81 г, 3,22 мМ) растворяют в дихлорметане (20 мл), добавляют тозилхлорид (1,84 г, 9,66 мМ) и триэтиламин (1,34 мл, 9,66 мМ) и реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют, остаток снова растворяют в этилацетате и раствор промывают водой и солевым раствором, сушат (MgSO4) и упаривают. Продукт очищают на диоксиде кремния, элюируя смесям метанола с дихлорметаном (1.87 г).
c) 2-(R)-(2-(R)-(3,5-Бис(трифторметил)фенил)этокси)-4-(1-(2-(N, N-диизопропиламино) этил)-1,2,4-триазол-3-ил)метил-3-(S)-фенилморфолин
Тозилат с вышеупомянутой стадии (b) (0,29 г, 0,41 мМ) растворяют в диметилформамиде (5 мл), добавляют дипропиламин (0,18 мл, 1,24 мМ) и триэтиламин (0,18 мл, 1,24 мМ) и реакционную смесь нагревают в запаянной трубке в течение 18 часов. Остаток растворяют в этилацетате, промывают раствор водой и солевым раствором, сушат (MgSO4) и упаривают. После очистки на диоксиде кремния с элюированием смесями метанола с дихлорметаном получают указанное в заголовке соединение (0,095 г). 1H ЯМР (360 МГц, ДМСО-d6) δ 8,31 (1H, c), 7,82 (1H, c), 7,46 - 7,42 (2H, м), 7,36 (2H, с), 7,32 - 7,22 (3H, м), 4,89 - 4,92 (1H, кв. J = 6,5 Гц), 4,34 (1H, д, J = 2,8 Гц), 4,18 - 4,04 (3H, м), 3,60 - 3,56 (3H, м), 3,09 (1H, д, J = 13,6 Гц), 3,94 (1H, д, J = 11,5 Гц), 2,71 (2H, т, J = 5,8 Гц), 2,44 - 2,40 (1H, м), 2,30 (4H, т, J = 7,0 Гц), 1,34 (3H, д, J = 6,5 Гц), 1,32 - 1,20 (4H, м) и 0,73 (6H, т, J = 7,4 Гц). M/S + 628.
Соединения примеров 64-74 в таблице 3 получают по способу, подобному способу, описанному в примере 63, из соответствующих 1,2,4-триазол-3-илметилморфолинов и соответствующих аминов.
Пример 75
2-(R)-(1-(R)-3,5-Бис(трифторметил)фенил)этокси)-4-(5-N, N- диметиламинометил)-1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)морфолин
a) 2-(R)-(1-(R)-3,5-Бис(трифторметил)фенил)этокси)-3-(S)-(4- фторфенил)-4-(1-(тетрагидро-2-пиранил)-5-(N, N-диметиламино-метил)- 1Н-1,2,4-триазол-3-ил)метилморфолин
Соединение примера-описания 5 (1 г, 2,28 мМ) растворяют в изопропаноле (20 мл), добавляют 3,5-бис(хлорметил)-1-(тетрагидро-2-пиранил)-1Н-1,2,4-триазол (1,14 г, 4,58 мМ) полученный по способу Bradshaw, H.Het.Chem. (1986), 23, 361) и карбонат калия (0,95 г, 6,84 мМ), и реакционную смесь нагревают при 60oC в течение 18 часов. Затем добавляют диметиламин (3 экв.), и реагенты переносят в трубку, которую запаивают, и нагревают еще в течение 18 часов. Затем удаляют растворители и остаток очищают на диоксиде кремния, элюируя смесями метанола с метиленхлоридом, и получают указанное в заголовке соединение (0,62 г).
b) 2-(R)-(1-(R)-3,5-Бис(трифторметил)фенил)этокси)-4-(5-N,N- диметиламинометил)-1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)морфолин
Соединение с вышеуказанной стадии (a) с защищенной аминогруппой (0,62 г, 0,94 мМ) растворяют в метаноле (15 мл) и обрабатывают HCl в метаноле (1н, 25 мл), и перемешивают реакционную смесь в течение 1 часа при комнатной температуре. Затем растворитель удаляют, и остаток очищают на диоксиде кремния, элюируя смесями метанола с дихлорметаном и аммиаком, и получают названное в заголовке соединение (0,48 г). 1H ЯМР (250 МГц, CDCl3) δ 7,45 (1H, c), 7,30-7,22 (2H, м), 6,97 (2H, c), 6,85 (2H, т, J=8,7 Гц), 4,72-4,66 (1H, кв, J= 6,5 Гц), 4,15 (1H, д, J=2,8 Гц), 4,15-4,07 (1H, м), 3,63 (1H, д, J=14,4 Гц), 3,48 (4H, c), 3,44-3,41 (1H, м), 3,38 (1H, д, J=2,8 Гц), 3,16 (1H, д, J=14,5 Гц), 2,81 (1H, д, J=11,1 Гц), 2,50-2,39 (1H, м), 2,15 (6H, c), и 1,27 (3H, д, J=6,6 Гц). M/S ES+ 576.
Пример 76
4-(5-N,N-Диметиламинометил)-1,2,3-триазол-4-ил)метил-3-(S)-(4- фторфенил)-2-(R)-(1-(R)-(3-метилтио-5-(трифторметил)фенил)этокси)морфолин
Соединение примера 57 (270 мг, 0,51 ммоль) нагревают при 120oC с тиометоксидом натрия (178 мг, 2,55 ммоль) в безводном ДМФА (10 мл) в течение 2-5 часов. После охлаждения раствор разбавляют водой (150 мл), экстрагируют этилацетатом (4х40 мл), экстракты сушат (MgSO4) и упаривают под вакуумом. Получают сырое масло (372 мг), которое очищают флэш-хроматографией на силикагеле в 5-10% метанола в дихлорметане, и получают указанное в заголовке соединение в виде вязкого смолообразного/стеклообразного вещества.. (179 мг, 60%). 1H ЯМР (360 МГц, CDCl3) δ 1,31 (3H, д, J=6,6 Гц), 2,17 (6H, c), 2,28 (3H, c), 2,47 (1H, д, J=12,1, 3,4 Гц), 2,82 (1H, д, J=11,6 Гц), 3,14 (1H, д, J=13,9 Гц), 3,35 (2H, м), 3,46 (1H, д, J=13,5 Гц), 3,52 (1H, дд, J=11,2, 1,9 Гц), 3,70 (1H, д, J=13,9 Гц), 4,14 (1H, дт, J=11,6 Гц), 4,26 (1H, д, J=2,7 Гц), 4,66 (1H, кв, J=6,5 Гц), 6,64 (2H, c), H, c), 6,99 (2H, т, J=8,6 Гц), 7,11 (1H, c), 7,41 (2H, ушир.c), 10,0-10,8 (1H, очень широкий c). М.-с. (ES+) m/z 554 M+1, 100%).
Пример 77
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-метилтио-5-(трифторметил) фенил)этокси)-4-(5-пирролидинометил-1,2,3-триазол-4-ил)метилморфолин
Названное в заголовке соединение получают из соединения примера 18 по способу, описанному в примере 76, в виде пены (620 мг, 81%). 1H ЯМР (360 МГц, CDCl3) δ 1,40 (3H, д, J=6,6 Гц), 1,79 (4H, ушир.c), 2,36 (3H, c), 2,5-2,6 (5H, м), 2,87 (1H, д, J=11,7 Гц), 3,23 (1H, д, J=13,9 Гц), 3,43 (1H, д, J= 13,9 Гц), 3,57-3,64 (2H, м), 3,71 (1H, д, J=13,7 Гц), 3,78 (1H, д, J= 14,0 Гц), 4,21 (1H, м), 4,33 (1H, д, J=2,8 Гц), 4,74 (1H, кв, J=6,5 Гц), 6,71 (2H, c), 7,06 (2H, т, J=8,7 Гц), 7,19 (1H, c), 7,47 (2H, ушир.c). М.-с. (ES+) m/z 580 (M+1, 100%).
Пример 78
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-метилтио-5-(трифторметил) фенил)этокси)-4-(5-морфолинометил)-1,2,3-триазол-4-ил)метилморфолин
Указанное в заголовке соединение получают в виде пены из соединения примера 19 по способу, описанному в примере 76 (126 мг, 66%). 1H ЯМР (360 МГц, CDCl3) δ 1,40 (3H, с, J=6,6 Гц), 2,37 (3H, c), 2,32-2,49 (4H, м), 2,54 (1H, дт, J=11,9, 3,4 Гц), 2,90 (1H, д, J=11,7 Гц), 3,25 (1H, д, J=13,9 Гц), 3,48 (1H, д, J= 13,5 Гц), 3,57-3,68 (7H, м), 3,82 (1H, д, J=14,1 Гц), 4,23 (1H, м), 4,35 (1H, д, J=2,8 Гц), 4,75 (1H, кв, J=6,5 Гц), 6,71 (2H, c), 7,06 (2H, т, J= 8,7 Гц), 7,19 (1H, c), 7,49 (2H, ушир.c). М.-с. (ES+) m/z 596 (M+1, 55%), 203 (100%).
Пример 79
4-(5-(N, N-Диметиламинометил)-1,2,3-триазол-4-ил)-метил- 2-(R)-(1-(R)-(3-метилтио-5-(трифторметил)фенил)этокси)-3-(S)-фенилморфолин
Указанное в заголовке соединение получают в виде пены из триазола примера 102 по способу, описанному в примере 76 (116 мг, 36%). 1H ЯМР (250 МГц, CDCl3) δ 1,39 (3H, д, J=6,5 Гц), 2,24 (6H, c), 2,32 (3H, c), 2,59 (1H, дт, J=11,8, 3,3 Гц), 3,25 (1H, д, J=13,8 Гц), 3,38-3,44 (2H, м), 3,52 (1H, д, J= 13,6 Гц), 3,62 (1H, дд, J=11,2, 1,8 Гц), 3,81 (1H, д, J=13,9 Гц), 4,23 (1H, м), 4,39 (1H, д, J=2,6 Гц), 4,75 (1H, кв, J=6,5 Гц), 6,71 (2H, c), 7,17 (1H, c), 7,34-7,41 (3H, м), 7,49 (2H, ушир.c). М.-с. (ES+) m/z 536 (M+1, 100%).
Пример 80
4-(5-(N, N-Диметиламинометил)-1,2,3-триазол-4-ил)-метил-3-(S)- (4-фторфенил)-2-(R)-(1-(R)-(3-трет-бутилтио-5-(трифторметил)фенил) этокси)морфолин
Указанное в заголовке соединение получают из соединения примера 57 в виде пены, по способу, описанному в примере 76 (117 мг, 68%). 1H ЯМР (360 МГц, CDCl3) δ 1,19 (9H, c), 1,42 (3H, д, J=6,6 Гц), 2,23 (6H, c), 2,57 (1H, дт, J= 12,0, 3,5 Гц), 2,92 (1H, д, J=11,6 Гц), 3,24 (1H, д, J=13,9 Гц), 3,39-3,44 (2H, м), 3,51 (1H, д, J=14,8 Гц), 3,62 (1H, м), 3,80 (1H, д, J= 13,9 Гц), 4,23 (1H, м), 4,41 (1H, д, J=2,7 Гц), 4,77 (1H, кв, J=6,5 Гц), 6,89 (1H, c), 7,14 (1H, c), 7,31-7,35 (3H, м), 7,46 (2H, ушир.c), 7,51 (1H, с). М.-.с (ES+) m/z 578 (M+1, 100%).
Пример 81
4-(5-(N, N-Диметиламинометил)-1,2,3-триазол-4-ил)- метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-метилсульфонил- 5-(трифторметил)фенил)этокси)морфолин
Тиоэфир из примера 76 (155 мг, 0,28 ммоль) растворяют в трифторуксусной кислоте (800 мкл), охлаждают до 0oC и обрабатывают 2,0 М раствором трифторнадуксусной кислоты в трифторуксусной кислоте (153 мкл, 0,308 ммоль) и перемешивают в течение 30 минут. Реакционную смесь выливают в 0,5М раствор бикарбоната натрия (50 мл), экстрагируют смесь дихлорметаном (3х15 мл), сушат экстракты (MgSO4) и концентрируют в вакууме. Образовавшееся в результате сырое твердое вещество очищают флэш-хроматографией на силикагеле в метаноле (8%) с дихлорметаном и получают указанное в заголовке соединение в виде нерасщепленных стереоизомеров в виде белой пены (81 мг, 51%). 1H ЯМР (360 МГц, CDCl3) δ 1,44 и 1,46 (3H, всего 2 х д, J=6,6 Гц), 2,24 (6H, с), 2,56 (1H, м), 2,59 и 2,62 (3H всего 2 х с), 2,88 (1H, д, J=11,9 Гц), 3,23 и 3,26 (1H, всего 2 х д, J=13,9 Гц), 3,42-3,55 (3H, м), 3,62 (1H, ушир.д, J= 11,3 Гц), 3,75 и 3,79 (1H, всего 2 х д, J=14,4 Гц), 4,22 (1H, м), 4,32 и 4,35 (1H, всего 2 х д, J=2,7 Гц), 4,89 (1H, м), 6,85 (1/2H, с), 7,04-7,13 (3H, м), 7,24 (1/2H, с), 7,50 (2H, ушир.с), 7,73 и 7,75 (1H, всего 2 х с). М.-с. (ES+) m/z 570 (M+1, 100%).
Пример 82
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-метилсульфинил- 5-(трифторметил)фенил)этокси)-4-(5-пирролидинометил- 1,2,3-триазол-4-ил)метилморфолин
Указанное в заголовке соединение в виде нерасщепленных стереоизомеров и в виде пены получают из соединения примера 77 в соответствии со способом, описанным в примере 81 (90 мг, 63%). 1H ЯМР (360 МГц, CDCl3) δ 1,44 и 1,46 (3H, всего 2 х д, J=6,6), 1,86 (4H, ушир.с), 2,50-2,60 (1H, м), 2,59 и 2,62 (3H, всего 2 х с), 2,70-2,90 (5H, м), 3,24 и 3,26 (1H, 2 х д, J=14,0), 3,46 (1H, д, J<2), 3,62 (1H, ушир.д, J=11,2), 3,71-3,86 (3H, м), 4,20 (1H, м), 4,32 и 4,35 (1H, всего 2 х д, J=2,7), 4,89 (1H, м), 6,89 (1/2, с), 7,03-7,13 (3H, м), 7,25 (1/2H, с), 7,49 (2H, ушир.с(, 7,73 и 7,75 (1H, всего, 2 х с). М.-с. (ES+) m/z 596 (M+1, 100%).
Пример 83
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-метилсульфинил- 5-(трифторметил)фенил)этокси)-4-(5-морфолинометил- 1,2,3-триазол-4-ил)метилморфолин
Указанное в заголовке соединение получают в виде нерасщепленной смеси стереоизомеров и в виде пены, из соединения примера 78 по способу, описанному в примере 81 (113 мг, 92%). 1H ЯМР (360 МГц, CDCl3) δ 1,3 и 1,41 (3H, всего 2 х д, J= 6,6), 2,54 и 2,57 (3H, всего, 2 х с), 2,54-2,65 (1H, м), 2,82-2,89 (1H, м), 3,05-3,25 (4H, очень широкий с.), 3,35 (1H, м), 3,50 (1H, м), 3,61 (1H, м), 3,72 и 3,74 (1H, 2 д, J=14,5), 3,85-4,22 (6H, м), 4,29 и 4,33 (1H, 2 х д, J=2,5), 4,81 (1H, м), 6,99- 7,09 (3 1/2, м), 7,30 (1/2H, с), 7,42 (2H, ушир.с), 7,64 и 7,66 (1H, всего, 2 х с). М.-с. (ES+) m/z 612 (M+1, 100%).
Пример 84
3-(S)-(4-Фторфенил)-2-(R)-(1-(R)-(3-метилсульфонил- 5-(трифторфенил)фенил)этокси)-4-(5-морфолинометил- 1,2,3-триазол-4-ил)метилморфолин
Сульфоксид примера 83 (78 мг, 0,128 ммоль) растворяют в трифторуксусной кислоте (500 мкл), раствор охлаждают до 0oC и обрабатывают 2,0М раствором трифторуксусной кислоты в трифторуксусной кислоте (70 мкл, 0,140 ммоль) при перемешивании в течение 2,5 часов. После этого добавляют еще эквивалент трифторуксусной кислоты (70 мкл, 0,140 ммоль) и через 3 часа продукт очищают по способу, описанному в примере 81, получая указанное в заголовке соединение в виде пены (27 мг, 34%). 1H ЯМР (360 МГц, CDCl3) δ 1,47 (3H, д, J=6,6 Гц), 2,38-2,45 (4H, м), 2,57 (1H, дт, J=11,9, 3,5), 2,90 (1H, д, J=11,7 Гц), 2,96 (3H, с), 3,26 (1H, д, J=14, 0 Гц), 3,46-3,51 (2H, м), 3,56-3,68 (6H, м), 3,79 (1H, д, J=14,1 Гц), 4,22 (1H, м), 4,35 (1H, д, J=2,8 Гц), 4,90 (1H, кв, J=6,8 Гц), 7,07 (2H, т, J=8,6 Гц), 7,17 (1H, с), 7,50 (2H, ушир.д), 7,67 (1H, с), 7,97 (1H, с). М.-с (ES+) m/z 628 (М+1, 100%).
Пример 85
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси)- -(S)-(4-фторфенил)-4-(2-(5-((S)-(+)-2-метоксиметилпирролидинометил) -1,2,3-триазол-4-ил)этил)-морфолин
Стадия A. 2-(R)-1-(R)-(3,5-Бис(трифторметил)фенил)этокси- 4-(бут-3-инил)-3-(S)-(4-фторфенил)морфолин
Раствор соединения примера-описания 5 (1,24 г, 1 экв.), 3-бутил-1-ол-тозилата (1,43 г, 2,5 экв.) K2CO3 (1,32 г, 3,7 экв.) и Nal (кат.) в сухом ДМФА (7 мл) нагревают при 100oC в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь обрабатывают H2O и EtOAc. Слои разделяют и водную фазу экстрагируют EtOAc (2x). Объединенные органические фазы сушат (Mg SO4) и упаривают, и остаток очищают хроматографией (гексан/EtOAc 9: 1 ---> 4:1), и получают указанное в заголовке соединение в виде прозрачного бесцветного масла. М.-с m/z 490 (MH+).
Стадия B. 2-(R)-(1-(R)-(3,5-Бис-трифторметил)фенил)- этокси)-3-(S)-(4-фторфенил)-4-(4-гидроксибут-3-инил)морфолин
Ацетиленовое соединение со стадии A (1,2 г, 1,0 экв.) растворяют в сухом ТГФ, затем раствор охлаждают до -78oC и добавляют к нему н-BuLi (2,5М в гексане, 1 мл, 1,05 экв.). Реакционную смесь перемешивают при -78oC в течение 1 часа, затем через раствор пропускают газообразный HCHO до насыщения. Реакционную смесь нагревают до комнатной температуры, перемешивают в течение 1 часа. Обработкой (NH4Cl/EtOAc) с последующей очисткой на силикагеле (гексан-EtOAc) 9:1 ---> 4:1) получают указанное в заголовке соединение в виде прозрачного вязкого масла. М.-с. m/z 520 (MH+).
Стадия C. 2-((R)-(1-(R)-(3,5-Бис(трифторметил)фенил)- этокси)-4-(4-хлорбут-3-инил)-3-(S)-(4-фторфенил)морфолин
Спирт со стадии B (0,42 г, 1 экв.) растворяют в сухом ТГФ (5 мл) в атмосфере N2 и добавляют трифосген (84 мг, 0,35 экв.), а затем пиридин (128 мкл, 2,0 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 0,5 часа, затем разбавляют EtOAc и промывают H2O и солевым раствором, сушат (MgSO4) и концентрируют, остается желтое масло. Это масло очищают хроматографией (гексан/EtOAc 9:1 ---> 4:1) и получают названное в заголовке соединение в виде прозрачного вязкого масла. М.-с. m/z 538, 540 (MH6+)
Стадия D. N-(4-Азидобут-3-инил)-2-(R)-(1-(R)-(3,5-бис(три-фторметил) фенил)этокси-3-(S)-(4-фторфенил)морфолин
Хлорид со стадии C (0,23 г, 1 экв.) и NaN3 (31 мг, 1 экв.) в ДМСО (0,8 мл) перемешивают при комнатной температуре в течение 14 часов. Обработка (NH4Cl/EtOAc) дает указанное в заголовке соединение в виде масла, которое используют без дополнительной очистки.
Стадия E. 2-(R)-(1-(R)-(3,5-Бис-(трифторметил)фенил)этокси-3- (S)-(4-фторфенил)-4-(2-(5-((S)-(+)-2-метоксиметил-пирролидинометил)- 1,2,3-триазол-4-ил)этил)формолин
Раствор азида со стадии D (0,205 г, 1 экв.) и (S)-(+)-2-метоксиметилпирролидин (114 мкл, 3 экв. ) нагревают при 80oC в атмосфере N2, удаляют растворитель под вакуумом, и остаток очищают хроматографией, используя в качестве элюента CH2Cl2/MeOH/NH3 (98: 2: 0,1, затем 97:3:0,1), и получают указанное в заголовке соединение в виде белой пены. 1H ЯМР (250 МГц, CDCl3) δ 7,62 (1H, с), 7,24 (2H, м), 7,14 (2H, с), 6,95 (2H, т, J = 8,7 Гц), 4,87 (1H, кв, J = 6,5 Гц), 4,30 (2H, м), 3,95 (1H, д, J = 14 Гц), 3,70 (1H, дд, J = 2, 11,3 Гц), 3,53 - 3,34 (7H, м), 3,19 (1H, д, J = 11,6 Гц), 2,86 - 2,56 (6H, м), 2,29 (1H, м), 2,09 (1H, м), 1,88 (1H, м), 1,70 (3H, м), 1,45 (3H, д, J = 6,5 Гц), М.-с. m/z = 660.
Пример 86
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-гидрокси-этокси) -4-(5-(N, N-диметиламинометил)-1,2,3-триазол-4-ил)метил-3-(S)-(4- фторфенил)морфолин
Стадия A. 2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-трет- бутилдиметилсилилоксиэтокси)-3-(S)-(4-фторфенил)морфолин
Продукт примера-описания 21 (2 г) растворяют в безводном дихлорметане (16 мл) в атмосфере азота и раствор охлаждают до 0oC. Затем добавляют 2,6-лутидин (0,5 мл) и трет-бутилдиметилтрифторметансульфонат (1,0 мл) и смесь перемешивают в течение 15 минут. Реакционную смесь промывают (H2O, солевой раствор), сушат (MgSO4) и упаривают под вакуумом. Очистка на самотечной колонке с диоксидом кремния с применением в качестве элюента 20 - 50% этилацетата в петролейном эфире дает названное в заголовке соединение в виде бесцветного масла. 1H ЯМР (250 Мгц, CDCl3) δ 0,04 (3H, с), 0,00 (3H, с), 0,87 (9H, с), 3,15 - 3,36 (2H, м), 3,64 - 3,70 (2H, м), 3,90 - 3,96 (1H, м), 4,10 (1H, д, J = 2,2 Гц), 4,22 - 4,53 (1H, м), 4,53 (1H, д, J = 2,2 Гц), 4,91 (1H, т, J = 5,9 Гц), 7,04 - 7,14 (2H, м), 7,29 - 7,36 (4H, м), 7,74 (1H, ушир. с). М.-с. (ES+) m/z = 567.
Стадия B. 2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-трет- бутилметилсилилоксиэтокси)-3-(S)-(4-фторфенил)-4-(4-хлорбут-2-инил)морфолин
Получают способом, аналогичным способу стадии (a) примера 12, способ B, используя продукт с вышеупомянутой стадии A. Получают указанное в заголовке соединение в виде бесцветного масла. 1H ЯМР (360 МГц, CDCl3) δ 0,00 (3H, с), 0,04 (3H, с), 0,91 (9H, с), 2,95 - 3,09 (2H, м), 3,40 (2H, ушир.с), 3,72 - 3,83 (3H, м), 4,01 (1H, дд, J = 10,2, 5,5 Гц), 4,25 (2H, м), 4,50 (2H, м), 4,9 (1H, т, J = 5,9 Гц), 7,15 (2H, т, J = 8,7 Гц), 7,29 (2H, с), 7,52 (2H, ушир.с), 7,76 (1H, с).
Стадия C. 2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-трет- бутилдиметилсилилоксиэтокси)-4-(5-(N, N-диметиламинометил)-1,2,3- триазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин
По аналогии со стадиями (b) и (c) примера 12, способ B, используя продукт с вышеупомянутой стадии B, получают указанное в заголовке соединение. 1H ЯМР (250 МГц, CDCl3) δ 0,02 (3H, с), 0,00 (3H, с), 0,88 (9H, с), 2,30 (6H, с), 2,60 - 2,70 (1H, м), 2,93 - 2,98 (1H, ушир. д, J = 11,6 Гц), 3,30 (1H, д, J = 13,8 Гц), 3,48 - 3,63 (3H, м), 3,68 - 3,74 (2H, м), 3,84 - 3,97 (2H, м), 4,33 - 4,41 (1H, м), 4,46 (1H, д, J = 2,8 Гц), 4,90 (1H, т, J = 5,6 Гц), 7,16 (2H, т, J = 8,7 Гц), 7,25 (2H, ушир. с), 7,59 (2H, очень широкий м), 7,74 (1H, ушир. с).
Стадия D. 2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2- гидроксиэтокси)-4-(5-(N, N-диметиламинометил)-1,2,3-триазол-4-ил) метил-3-(S)-(4-фторфенил)морфолин
Продукт с вышеупомянутой сталии C (0,2 г) в безводном тетрагидрофуране (2 мл) перемешивают с тетрабутиламмонийфторидом (1,0 М) в тетрагидрофуране 0,42 мл) в течение 30 минут. Смесь обрабатывают раствором хлорида аммония и этилацетатом, и органический слой промывают (H2O, соляной раствор), сушат (MgSO4) и упаривают под вакуумом. Очистка с помощью самотечной колонки с диоксилом кремния с элюированием 4 - 10% MeOH/0,1% NH4OH/дихлорметан дает названное в заголовке соединение. 1H ЯМР (250 МГц, CDCl3) δ 2,26 (6H, с), 2,51 (1H, м), 3,09 (2H, м), 3,35 (2H, м), 3,51 - 3,63 (4H, м), 3,78 (2H, д, J = 13,8 Гц), 4,30 - 4,36 (2H, м), 4,88 (1H, м), 7,01 - 7,10 (4H, м), 7,50 (2H, очень широкий C), 7,59 (1H, ушир. с).
Пример 87
2-(R)-(1-(R)-(3,5-Бис(трифторметил)фенил)этокси-4-(5-N-этил-N- изопропиламинометил)-1 (или 2, или 3) -метил-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин
Продукт примера 101 (2 г) растворяют в N,N-диметилформамиде (4 мл) при комнатной температуре в атмосфере азота. Добавляют иодметан, а затем гидрид натрия (60%) (14 мг) и смесь перемешивают в течение 16 часов. Реакционную смесь обрабатывают этилацетатом и водой и органический слой промывают (H2O • 2, соляной раствор), сушат (MgSO4) и упаривают под вакуумом. Очистка самотечной хроматографией на диоксиде кремния с элюированием 100% этилацетата, а затем смесью 10% метанола, 0,1% NH4OH и дихлорметана дает указанное в заголовке соединение. 1H ЯМР (250 МГц, CDCl3) δ 0,85 - 1,02 (9H, м), 1,44 (3H, д, J = 6,6 Гц), 2,25 - 2,40 (2H, м), 2,57 - 2,68 (1H, м), 2,75 - 2,85 (1H, м), 2,96 (1H, ушир. д, J = 13,5 Гц), 3,15 (1H, д, J = 13,5 Гц), 3,38 (1H, д, J = 2,7 Гц), 3,44 (2H, с), 3,60 - 3,73 (2H, м), 4,07 (3H, с), 4,18 (1H, м), 4,35 (1H, д, J = 2,8 Гц), 4,83 (1H, м), 7,15 (2H, ушир. с), 7,33 (3H, м), 7,48 (2H, очень широкий с), 7,61 (1H, ушир.с). М.-с. (ES+) m/z = 613 (MH+, 100%).
Пример 88
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил-2-гидроксиэтокси)-3-(S)-(4- фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метилморфолин
Соединение примера-описания 6 (0,5 г), N-карбометокси-2-хлорацетамидазон (пример-описание 23) (182 мг) и карбонат калия (0,3 г) суспендируют в диметилформамиде (3,6 мл) и смесь нагревают при 60oC в течение 2 часов. Затем смесь греют при 140oC еще в течение 2 часов. Смесь охлаждают и затем фильтрацией через целит удаляют неорганическое вещество. Растворитель удаляют в вакууме азеотропной перегонкой с ксилолом. Остаток очищают флэш-хроматографией на диоксиде кремния, используя 1-10% метанола в дихлорметане. Получают указанное в заголовке соединение в виде белого порошка (300 мг). 1H-ЯМР (360 МГц, ДМСО-d6) δ 2,38 - 2,41 (1H, м), 2,78 (1H, д, J = 14,0 Гц), 2,81 - 2,84 (1H, м), 3,36 (1H, д, J = 14,0 Гц), 3,45 - 3,48 (1H, м), 3,52 (1H, д, J = 3,0 Гц), 3,58 - 3,61 (2H, м), 4,81 (1H, т, J = 6,0 Гц), 4,88 (1H, ушир. т.), 7,09 (2H, т, J = 9,0 Гц), 7,33 (2H, с), 7,50 (2H, ушир.т.), 7,85 (1H, c), 11,26 (1H, с), 11,30 (1H, с. M.-с. (Cl+) m/z 551 (M+1, 10%), 454 (M+-CH2 -триазолон, 20).
Пример 89
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил-2-гидроксиэтокси)-3-(S)-(4- фторфенил)-4-(1,2,4-триазол-3-ил)морфолин
Соединение примера-описания 6 (270 г), безводный карбонат калия (250 мг) и N-формил-2-хлорацетамидгидразон (92 мг) (полученный по I.Yanagisawa, J. Med. Chem. (1984), 27, 849) нагревают при 60oC в безводном диметилформамиде в течение 1 часа, а затем при 140oC в течение 2 часов. Реакционную смесь охлаждают и разбавляют водой (100 мл). Продукт экстрагируют этилацетатом (3 х 50 мл) и органический слой промывают солевым раствором, сушат (MgSO4), и упаривают в вакууме. Остаток очищают хроматографией на диоксиде кремния, используя в качестве элюента 7% метанола в дихлорметане. Получают указанное в заголовке соединение (200 мг, 60%) в виде белого твердого вещества. 1H ЯМР (360 МГц, ДМСО-d6) σ 2,47 (1H, т, J = 9,0 Гц), 2,89 (1H, д, J = 11,0 Гц), 3,18 (1H, д, J = 14,0 Гц), 3,44 - 3,49 (1H, м), 3,55 - 3,61 (4H, м), 3,64 (1H, д, J = 6 Гц), 4,25 (1H, т, J = 11,0 Гц), 4,34 (1H, д, J = 3,0 Гц), 4,81 (1H, т, J = 5,0 Гц), 7,11 (2H, т, J = 9,0 Гц), 7,34 (2H, c), 7,52 (2H, м), 7,85 (1H, с), 8,19 (1H, ушир.с.), М.-С. (Cl) m/z 535 (M+1, 10%).
Пример 90
4-(2,3-Дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(S) -(1-(S)-(3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)морфолин
Соединение примера-описания 22 (350 мг), N-карбометокси-2-хлорацетамидразон (150 мг) (пример-описание 23) и карбонат калия (150 мг) в диметилформамиде греют при 60oC до тех пор, пока не израсходуется все исходное вещество. Затем смесь нагревают при 140oC в течение 3 часов. Смесь охлаждают и фильтруют через целит для удаления неорганических веществ. Остаток упаривают, используя ксилол для азеотропного удаления остатков диметилформамида. Остаток очищают хроматографией на диоксиде кремния, используя 1-10% метанола и дихлорметане в качестве элюента. Получают указанное в заголовке соединение в виде пены, которую перекристаллизовывают из эфира. 1H ЯМР (360 МГц, ДМСО-d6) δ 2,34 - 2,46 (1H, м), 2,74 - 2,84 (2H, м), 3,34 - 3,43 (3H, м), 3,50 - 3,60 (2H, м), 4,21 - 4,31 (2H, м), 4,68 (1H, т, J = 5,0 Гц), 4,90 (1H, т, J = 7,0 Гц), 6,54 (1H, д, J = 9,0 Гц), 6,88 (1H, с), 7,14 (т, J = 9,0 Гц), 7,42 (1H, т, J = 9,0 Гц), 7,44 (2H, м).
Пример 91
4-(2,3-Дигидро-2-оксо-1,3-имидазол-4-ил)метил-2-(R)-(1-(S)-(3,5-бис (трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)морфолин
Смесь соединения примера-описания 6 (2 г), 4-бромметил-1,3-диацетил-2-имидазолона (1,38 г) (полученного по способу Dolan and Dushinsky, JASC (1948) 70, 657) и карбоната калия (1,2 г) в диметилформамиде (14 мл) перемешивают при комнатной температуре в течение 30 минут, пока не прореагирует весь исходный морфолин. Смесь разбавляют водой (150 мл) и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают солевым раствором и органический растворитель удаляют в вакууме. Оставшееся масло растворяют в этаноле (20 мл) и добавляют метиламин (2 мл 8М р-ра в этаноле). Этот раствор перемешивают в течение 1 часа, затем растворитель удаляют в вакууме. Оставшееся масло очищают на диоксиде кремния, используя 1-10% метанола в дихлорметане в качестве элюента. Получают продукт (2 г, 83%) в виде белой пены. Затем его обрабатывают хлористым водородом в метаноле и получают более твердое вещество 1H ЯМР (360 МГц, ДМСО-d6) σ 2,22-2,34 (1H, м), 2,62 (1H, д, J = 14,0 Гц), 2,89 (1H, прибл. д, J = 11,0 Гц), 3,26 (1H, д, J = 14,0 Гц), 3,38 (1H, д, J = 3,0 Гц), 3,43 - 3,50 (1H, м), 3,57 - 3,62 (2H, м), 4,19 - 4,28 (1H, м), 4,32 (1H, д, J = 3,0 Гц), 4,81 (1H, т, = 5,5 Гц), 4,93 (1H, т, = 6,0 Гц), 6,00 (1H, с), 7,09 (1H, т, = 9,0 Гц), 7,33 (2H, c), 7,54 (2H, ушир. т. ), 7,86 (1H, с), 9,63 (1H, с). М.-с. (Cl) m/z 550 (M+1, 20%), 454 (80) 116 (100).
Пример 92
4-(2,3-Дигидро-2-оксо-5-пирролидинометил-1,3-имидазол-4-ил)метил-2-(R)-1-(S)- (3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)-морфолин
Смесь соединения примера-описания 6 (1,8 г) 4,5-бис(бромметил)-1,3-диацетил-2-имидазолона (полученного по способу Dolan and Dushinsky, JACS (1948) 70, 657) (2,2 г) и карбоната калия в диметилформамиде (13 мл) перемешивают при комнатной температуре в течение 10 минут, пока не прореагирует все исходное вещество. К образующейся в результате коричневой смеси добавляют по каплям пирролидин (1,65 мл, избыток), что приводит к экзотермической реакции. Растворитель удаляют в вакууме, остаток экстрагируют этилацетатом (3 х 50 мл) и промывают экстракты солевым раствором. Органическую фазу сушат (MgSO4) и удаляют растворитель в вакууме. Коричневый остаток очищают хроматографией со средним давлением на силикагеле с обращенной фазой используя в качестве элюента 30% ацетонитрила в 0,1% водной трифторуксусной кислоте. Получают указанные в заголовке продукт в виде темно-желтого твердого вещества (1 г). 1H ЯМР (360 МГц, ДМСО-d6) δ 1,61 (4H, ушир.с), 2,26 - 2,30 (5H, м), 2,66 (1H, д, J = 14,0 Гц), 2,83 - 2,87 (1H, ушир.д), 3,02 (1H, д, J = 13,5 Гц), 3,15 (1H, д, J = 13,5 Гц), 3,23 (1H, д, J = 14,0 Гц), 3,37 (1H, д, J = 3,0 Гц), 3,42 - 3,47 (1H, м), 3,57 - 3,60 (2H, м), 4,17 - 4,24 (1H, м), 4,32 (1H, д, J = 3,0 Гц), 4,79 (1H, т, J = 5,5 Гц), 4,89 (1H, т, J = 5,5 Гц), 7,08 (2H, т, J = 9,0 Гц), 7,32 (2H, с), 7,56 (2H, м.ц.), 7,85 (1H, с), 9,61 (1H, с), 9,65 (1H, с). М.-с. (Cl+) m/z 633 (M+ + 1), 454 (50%).
Пример 93
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил-2-фосфорилоксиэтокси)- 3-(S)-(4-фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол- 5-ил)метилморфолин
Соединение примера 88 (200 мг) в сухом тетрагидрофуране (1 мл) обрабатывают дибензилоксидиэтиламинофосфином (200 мг) и тетразолом (100 мг). Реакционную смесь перемешивают в течение 2 часов, и затем обрабатывают ее еще 100 мг дибензилоксиэтиламинофосфина, и спустя 1 час - тетразолом (100 мг). Реакционную смесь перемешивают еще в течение 1 часа, прежде чем добавить 4-метилморфолин-N-оксид (1,0 г), после чего ее перемешивают в течение 16 часов. Реакционную смесь выливают в раствор карбоната калия и экстрагируют этилацетатом. Органический слой сушат (MgSO4), фильтруют, упаривают и очищают хроматографией на силикагеле, используя в качестве элюента смесь метанола с дихлорметаном (4 : 96), и получают масло. Это масло растворяют в метаноле (2 мл) и добавляют формиат аммония (100 мг) и гидроксид палладия (20% на угле). Реакционную смесь кипятят с обратным холодильником в течение одного часа и затем фильтруют, упаривают и сушат вымораживанием из ацетонитрила с водой, получают аммониевую соль названного в заголовке соединения (93 мг). 1H ЯМР (360 МГц, D6-ДМСО) σ 11,29 (1H, с), 7,85 (1H, с), 7,53 (2H, с), 7,36 (2H, м), 7,06 (2H, т, J = 7,2 Гц), 4,96 (1H, т, J = 5,4 Гц), 4,34 (1H, д, J = 3,6 Гц), 4,29 (1H, т, J = 11,2 Гц), 3,92 - 3,85 (1H, м), 3,68 - 3,63 (1H, м), 3,62 - 3,55 (1H, м), 3,49 (1H, д, J = 3,6 Гц), 3,38 (1H, д, J = 14,4 Гц), 2,82 - 2,79 (1H, м), 2,77 (1H, д, J = 14,4 Гц), 2,41 - 2,35 (1H, м). М. -с. (ES+) 631 (M + H).
Пример 94
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-фосфорилоксиэтокси)- 3-(S)-(4-фторфенил)-4-(1,2,4-триазол-3-ил)метилморфолин
Получают аммониевую соль названного в заголовке соединения из соединения примера 89 по способу, описанному в примере 93. 1H ЯМР (250 МГц, D6-ДМСО + 0,1% ТФК) σ 8,74 (1H, с), 7,95 (1H, с), 7,68 (2H, ушир. с), 7,54 (2H, с), 7,30 (2H, т, J = 8,7 Гц), 5,16 (1H, дд, J = 7 Гц и 5 Гц), 4,72 (1H, д, J = 1 Гц), 4,66 (1H, д, J = 1 Гц), 4,42 (1H, т, J = 11 Гц), 3,95 - 4,27 (3H, м), 3,72 (1H, д, J = 11 Гц) и 3,41 - 3,55 (1H, м).
Пример 95
4-(2,3-Дигидро-3-оксо-1,2,4-триазол-5-ил)-3-(S)-фенил-2- (R)-(1-(S)-(3-трифторметилэфенил)-2-гидроксиэтокси)морфолин
Получают из соединения примера-описания 30, следуя способу, описанному в примере 88. М.-с. (C++) m/z 465 (M + 1)+, 71%).
Пример 96
4-(2,3-Дигидро-3-оксо-1,2,4-триазол-5-ил)метил-2-(R)- (1-(S)-(3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)-3- (S)-фенилморфолин
Соединение примера-описания 27 (600 мг), N-карбометокси-2-хлораметамидразон (271 мг) и карбонат калия (258 мг) вводят во взаимодействие в диметилформамиде в соответствии с процедурой, описанной в примере 88. Получают продукт в виде белого твердого вещества, которое перекристаллизовывают из эфира с гексаном (220 мг, 30%). 1H ЯМР (360 МГц, ДМСО-d6) σ 2,38 (1H, м), 2,78 (1H, д, J = 14,0 Гц), 2,84 (1H, с), 3,38 - 3,39 (2H, м), 3,45 (1H, д, J = 14,0 Гц), 3,50 (1H, д, J = 3,0 Гц), 3,56 (1H, д, J = 11,0 Гц), 4,26 (1H, т, J = 11,0 Гц), 4,34 (1H, д, J = 3,0 Гц), 4,68 (1H, т, J = 6,0 Гц), 4,85 (1H, т, J = 6,0 Гц), 6,40 (1H, д, J = 9,0 Гц), 6,96 (1H, с), 7,33 (3H, м), 7,36 (1H, д, J = 9,0 Гц), 7,49 (2H, м). М.-с. (Cl+) m/z 483 (M + 1, 20%).
Пример 97
4-(2,3-Дигидро-3-оксо-1,2,4-триазол-5-ил)метил-2-(R)- (1-(S)-(3-фтор-5-(трифторметил)фенил)-2-фосфорилоксиэтокси)-3- (S)-фенилморфолин
Из соединения примера 96 получают аммониевую соль названного в заголовке соединения, применяя способ примера 93. 1H ЯМР (360 МГц, ДМСО-d6) σ 11,29 (1H, с), 7,49 - 7,29 (5H, м), 7,38 (1H, д, J = 10,8 Гц), 6,96 (1H, с), 6,45 (1H, д, J = 10,8 Гц), 4,84 (1H, д, J = 7,2 Гц), 4,34 (1H, д, J = 3,6 Гц), 4,28 (1H, т, J = 10,8 Гц), 3,80 - 3,76 (1H, м), 3,57 (1H, д, J = 3,6 Гц), 3,57 - 3,49 (2H, м), 3,47 (1H, д, J = 14,4 Гц), 2,83 - 2,76 (1H, м), 2,78 (1H, д, J = 14,4 Гц), 2,46 - 2,36 (1H, м).
Пример 98
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-гидроксиэтокси)- 4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-фенилморфолин
Соединение примера-описания 17 вводят во взаимодействие с N-карбоксиметокси-2-хлорацетамидразоном (пример-описание 23) и карбонатом калия по способу, описанному в примере 88. Получают продукт в виде белого твердого вещества. 1H ЯМР (360 МГц, ДМСО-d6) δ 2,42 (1H, дт, J = 12,0, 3,5 Гц), 2,76 (1H, д, J = 14,0 Гц), 2,83 (1H, д, J = 12,0 Гц), 3,39 (1H, д, J = 14,0 Гц), 3,44 - 3,47 (1H, м), 3,50 (1H, д, J = 3,0 Гц), 3,60 (2H, м), 4,22 - 4,28 (1H, м), 4,40 (1H, д, J = 3,0 Гц), 4,77 - 4,83 (2H, м), 7,25 - 7,34 (3H, м), 7,41 (2H, с), 7,48 - 7,50 (2H, м), 7,82 (1H, с), 11,20 (1H, с), 11,25 (1H, с). М.-с. (Cl) m/z 533 (M + 1, 30%) 434 (20), 117 (100).
Пример 99
2-(R)-(1-(S)-(3,5-Бис(трифторметил)фенил)-2-фосфорилоксиэтокси)- 4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-фенилморфолин
Получают аммониевую соль указанного в заголовке соединения из соединения примера 98 по способу, описанному в примере 93. 1H ЯМР (360 МГц, d6-ДМСО) δ 11,26 (1H, с), 7,83 (1H, с), 7,48 - 7,24 (7H, м), 4,95 (1H, т, J = 5,4 Гц), 4,39 (1H, д, J = 3,6 Гц), 4,29 (1H, т, J = 11,2 Гц), 3,92 - 3,89 (1H, м), 3,60 - 3,64 (1H, м), 3,5 - 3,59 (1H, м), 3,48 (1H, д, J = 3,6 Гц), 3,42 (1H, д, J = 14,4 Гц), 2,84 - 2,79 (1H, м), 2,78 (1H, д, J = 14,4 Гц), 2,42 (1H, м). ЖХВР на колонке с Zorbax Z-Ph (250 x 4,6 мм, вн.д. 5 мкМ), элюирование 40% ацетонитрилом в 25 мм KH2PO4 с 0,2% триэтиламином (pH 3), скорость потока 1 мл/мин, УФ-детектор 210 нм. Время удерживания 4,68 мин.
Пример 100
3-(S)-Фенил-4-(1,2,4-триазол-3-ил)-2-(R)-(1-(S)-3-(трифторметил)фенил)- 2-гидрокси-этокси)морфолин
Получают в виде соли - гидрохлорида - из соединения примера-описания 30, следуя способу, описанному в примере 89. M.-c. (ES+) m/z 449 (M+1)+ 100%)
Соединения примеров 101 и 201 в таблице 2 получают по способу, подобному способу, описанному в примере 12, способ B, исходя из соответствующих N-(4-азидобут-2-инил)морфолины и соответствующие амины.
В приведенных далее таблицах 1-3 (см. в конце описания) и примерах использованы следующие обозначения:
MS - м.-с. - масс-спектрометрия
Aral (analysis) - элем.анализ - элементный анализ Calcd.For - вычислено для
Found - найдено
Hz - Гц - герц
MHz - МГц - мегагерц
DMSO - ДМСО - диметилсульфоксид
NMR - ЯМР - ядерный магнитный резонанс
S - с - синглет, d - д - дублет, t - т - триплет, q - кв - квартет, qn - квин - квинтет, m - м - мультиплет, brs - ушир. с. - уширенный синглет, br d - ушир. д. - уширенный дублет, br t - ушир. т. - уширенный триплет, br qn - ушир. квин. - уширенный квинтет, br m - ушир. м. - уширенный мультиплет, vbr s - оч. ш.с. - очень широкий синглет, dd - дд - двойной дублет, dt - дт - двойной триплет, td - тд - тройной дублет, br dt - ушир. дт. - уширенный двойной триплет, HRMS - м.-с. - высокого разрешения.
Следующие далее примеры иллюстрируют композицию по настоящему изобретению.
Пример 103A.
Таблетки, содержащие 1-25 мг соединения - Количество, мг
Соединение формулы (1) - 1,0; 2,0; 25,0
Микрокристаллическая целлюлоза - 20,0; 20,0; 25,0
Модифицированный пищевой кукурузный крахмал - 20,0; 20,0; 20,0
Лактоза - 58,5; 57,5; 34,5
Стеарат магния - 0,5; 0,5; 0,5
Пример 103B.
Таблетки, содержащие 26-10 мг соединения - Количество, мг
Соединение формулы (1) - 26,0; 50,0; 100,0
Микрокристаллическая целлюлоза - 80,0; 80,0; 80,0
Модифицированный пищевой кукурузный крахмал - 80,0; 80,0; 80,0
Лактоза - 213,5; 189,5; 139,5
Стеарат магния - 0,5; 0,5; 0,5
Соединение формулы (1), целлюлозу, лактозу и часть кукурузного крахмала смешивают и гранулируют с 10% пастой кукурузного крахмала. Получающийся в результате гранулированный продукт просеивают, сушат и смешивают с оставшейся частью кукурузного крахмала и стеаратом магния. Получающийся в результате гранулированный продукт, затем прессуют в таблетки, содержащие 1,0 мг, 2,0 мг, 25,0 мг, 26,0 мг, 50,0 мг и 100 мг активного соединения на таблетку.
Пример 104.
Парентеральная инъекция - Количество
Соединение формулы (1) - 1-100 мг
Лимонная кислота, моногидрат - 0,75 мг
Фосфат натрия - 4,5 мг
Хлорид натрия - 9 мг
Вода для инъекций - до 10 мл
Фосфат натрия, моногидрат лимонной кислоты и хлорид натрия растворяют в части воды. Растворяют или суспендируют в этом растворе соединение формулы (1) и доводят до нужного объема.
Пример 105.
Наружный состав - Количество
Соединение формулы (1) - 1-10 г
Воск-эмульгатор - 30 г
Вазелиновое масло - 20 г
Бесцветный мягкий парафин - до 100 г
Бесцветный мягкий парафин нагревают до расплавления. Вводят в него вазелиновое масло и воск-эмульгатор и перемешивают до растворения. Добавляют соединение формулы (1) и продолжают перемешивание до образования дисперсии. Затем смесь охлаждают до твердого состояния.
Пример 106A.
Состав для инъекций (поверхностно-активное вещество)
Соединение формулы (1) - до 10 мг/кг
Твин 80TM - до 2,5%
(в 5% водном манните (изотонический р-р))
Соединение формулы (1) растворяют прямо в растворе коммерчески доступного твинаTM (моноалеат полиоксиэтиленсорбитана) и 5% водном растворе маннита (изотонический р-р).
Пример 106B
Состав для инъекцией (эмульсия)
Соединение формулы (1) - до 30 мг/мл
IntralipidTM - 10 - 20%
Соединение формулы (1) растворяют прямо в коммерчески доступном IntralipidTM (10 или 20%) с образованием эмульсии.
Пример 106C
Альтернативный состав, который можно вводить инъекцией (эмульсия) - Количество
Соединение формулы (1) - 0,1 - 10 мг
Соевое масло - 100 мг
Яичный фосфолипид - 6 мг
Глицерин - 22 мг
Вода для инъекций - до 1 мл
Все материалы стерилизуют и очищают от пирогенов. Соединение формулы (1) растворяют в соевом масле. Затем получают эмульсию, смешивая этот раствор с яичным фосфолипидом, глицерином и водой. Затем эмульсию помещают в стерильные ампулы, которые запаивают.
Биологические данные
Полученные в примерах соединения по настоящему изобретению испытывали способом, описанными ниже, для установления аффинности к человеческому NK1 рецептору. Было установлено, что соединения или, в случае пролекарственных препаратов, предшествующих им исходные соединения, проявляют активность к NK1 рецептору человека со значением IC50 менее 10 нМ. Следующие характерные данные приведены для иллюстрации.
Пример N - IC50 к NK1 человека (нМ)
1 - 0.07
2 - 0.05
3 - 0.07
4 - 0.05
12 - 0.1
16 - 0.2
17 - 0.2
18 - 0.2
21 - 0.2
22 - 0.3
23 - 0.1
32 - 0.5
33 - 0.35
64 - 0.14
65 - 0.45
67 - 0.3
72 - 0.1
78 - 0.3
83 - 0.9
84 - 0.3
88 - 0.05
90 - 0.35
91 - 0.06
92 - 0.06
Анализ антагонизма вещества P
A. Экспрессия рецептора в клеточную линию почки обезьяны (COS)
Чтобы временно экспрессировать клонированный человеческий нейрокинин-1-рецептор (NK1R) в COS, кДНК для человеческого NK1R клонировали в вектор экспрессии pCDM9, являющийся производным от pCDM8 (INVITRO-ГЕН), путем введения гена, устойчивого к ампициллину (нуклеотиды от 1973 до 2964 от BLUESCRIPT SK+ (торговая марка, STRATAGENE, La Jolla, CA, США), в сайт Sac II. Трансфекцию 20 уг плазмидной ДНК в 10 миллионов клеток COS осуществляли путем электропорации в 800 мкл трансфекционного буфера (135 мМ NaCl, 1.2 мМ CaCl2, 1.2 мМ MgCl2, 2.4 мМ K2HPO4, 0.6 мМ KH2PO4, 10 мМ глюкозы, 10 мМ N-2-гидроксиэтил-пиперазин-N'-2-этансульфоной кислоты (HEPES) с pH 7.4) при 260 B и 950 мкФ с использованием IBI GENEZAPPER (торговая марка IBI, New Haven, CT, США). Клетки инкубировали в 10% фетальной телячьей сыворотки, 2 мМ глутамина, 100 Ед./мл пенициллин-стрептомицина и 90% среды DMEM (модифицированная по способу Дульбекко среда Игла, GIBCO, Grand Islаnd, NY, США) в 5% CO2 при 37oC в течение трех дней перед проведением связывающего анализа.
B. Стабильная экспрессия в клеточную линию яичника китайского хомячка (CHO)
Для основания стабильной клеточной линии, экспрессирующей клонированный NK1R человека, кДНК субклонировали в вектор pRcCMV (INVITRO-ГЕН). Трансфекцию 20 уг плазмидной ДНК в клетки яичника китайского хомячка (CHO) осуществляли путем электропорации в 800 мкл трансфекционного буфера, дополненного 0,625 мг/мл ДНК спермы Херринга при 300 B и 950 мкФ с использованием IBI GENEZAPPER (IBI). Трансфекцированные клетки инкубировали в CHO среде (10% фетальной телячьей сыворотки, 100 ед. /мл пенициллин-стрептомицина, 2 мМ глутамина, 1/500 гипоксантин-тимидина (АТСС), 90% среды IMDM (среда Дульбекко, модифицированная по способу Исков, JRH BIOSCIENCES, Lenexa, KS, США), 0,7 мг/мл G418 (GIBCO)] в 5% CO2 при 37oC до тех пор, пока колония не становилась видимой. Каждую колонию отделяли и культивировали. Клеточные клоны с наиболее высоким значением NK1R человека отбирали для последующего применения, такого как лекарственный скрининг.
C. Протокол анализа с использованием COS или CHO
Анализ связывания человеческого NK1R, экспрессированного или COS, или CHO клетками, основан на использовании меченого 125I-вещества P (125I-SP от DU PONT, Boston, MA) в качестве радиоактивно меченного лиганда, который конкурирует с немеченым веществом P или любым другим лигандом за связывание человеческого NK1R.
Монослойные клеточные культуры COS или CHO диссоциировали в неферментативном растворе (SPECIALTY MEDIA, Lavellette, NJ) и ресуспендировали в подходящем объеме связывающего буфера (50 мМ Tris pH 7,5, 5 мМ MnCl2, 150 мМ NaCl, 0,04 мг/мл бацитрацина, 0,04 мг/мл лейпептина, 0,2 мг/мл альбумина бычьей сыворотки, 0,01 мМ фосфорамидона) таким образом, чтобы 200 мкл клеточной суспензии приводили к примерно 10,000 импульсам в минуту специфического 125I-SP связывания (приблизительно от 50,000 до 200,000 клеток). В связывающем анализе, 200 ул клеток добавляют в пробирку, содержащую 20 ул от 1,5 до 2,5 нМ 125I-SP и 20 мкл немеченого вещества P или любого другого тестируемого соединения. Пробирки инкубировали при 4oC или при комнатной температуре в течение 1 часа при осторожном встряхивании. Связанную радиоактивность отделяли от несвязанной радиоактивности с использованием GF/C фильтра (BRANDEL, Gaithersburg, MD), который предварительно был смочен 0,1% полиэтиленимином. Фильтр трижды промывали 3 мл промывочного буфера (50 мМ Tris pH 7.5, 5 мМ MnCl2, 150 мМ NaCl) и его радиоактивность определяли счетчиком гамма-излучения.
Активация фосфолипазы C посредством NK1R может также быть измерена в клетках CHO, экспрессирующих NK1R человека, путем определения аккумуляции монофосфата инозитола, являющегося продуктом разложения IP3. Клетки CHO высеивали в 12-луночный планшет из расчета 250,000 клеток на лунку. После инкубирования в среде CHO в течение 4 дней, в клетки вводили 5 мккюри 3H-миоинозитола в 1 мл среды на лунку путем инкубации в течение ночи. Межклеточную радиоактивность удаляли путем промывания солевым фосфатным буфером. В лунки добавляли LiCl в конечной концентрации 10 мМ вместе с или без тестируемого соединения и продолжали инкубирование при 37oC в течение 15 минут. Вещество P добавляли в лунку в конечной концентрации 0,3 нМ, чтобы активировать человеческий NK1R. После 30 минут инкубирования при 37oC среду удаляли и добавляли 0,1 н. HCl. Каждую лунку разрушали ультразвуком при 4oC и экстрагировали CHCl3/метанолом (1:1). Водную фазу наносили на 1 мл ионообменной смолы Dowex AG 1X8 на колонке. Колонку промывали 0,1 н муравьиной кислотой, а затем 0,025М формиатом аммония - 0,1 н. муравьиной кислотой.
Монофосфат инозитола элюировали 0,2 М формиатом аммония 0,1 н. муравьиной кислотой и оценивали количественно с использованием счетчика бета-излучения.
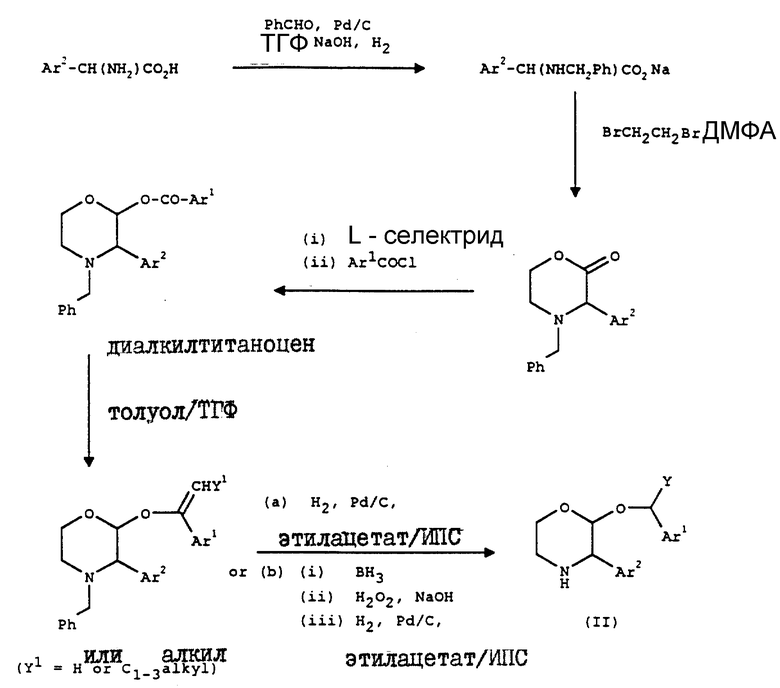
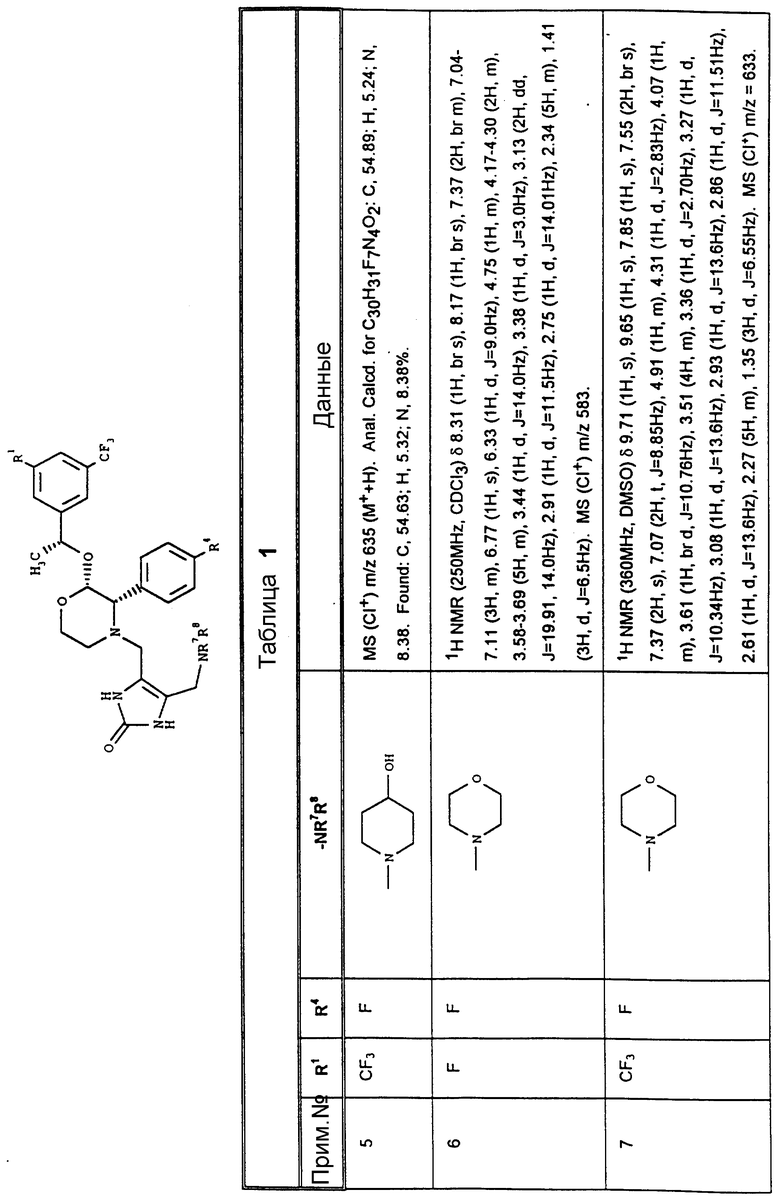
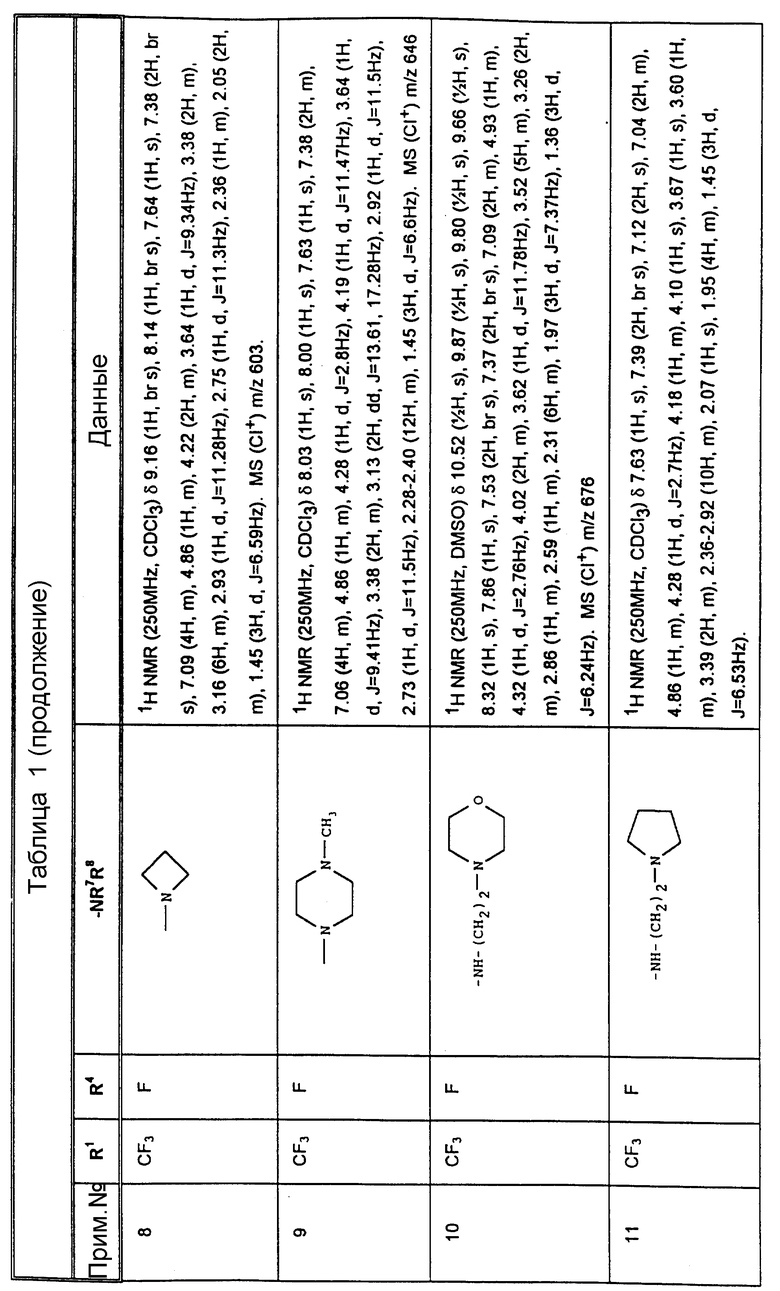
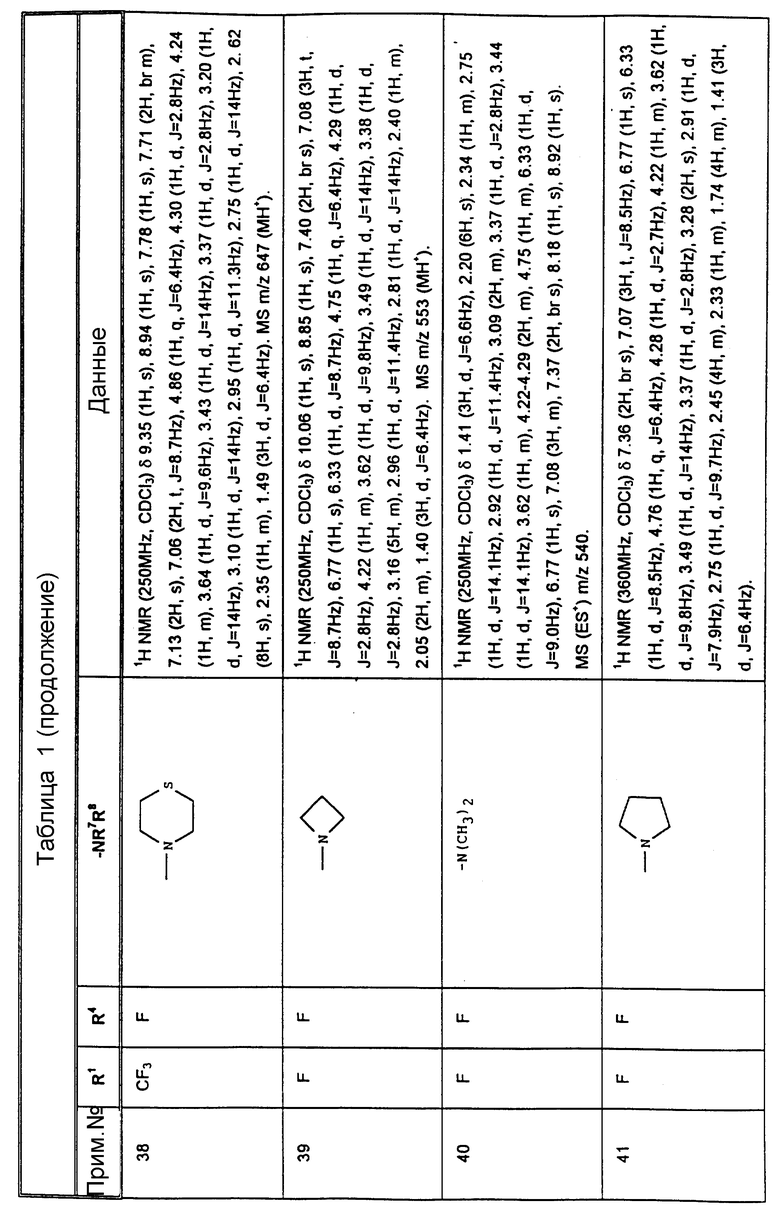


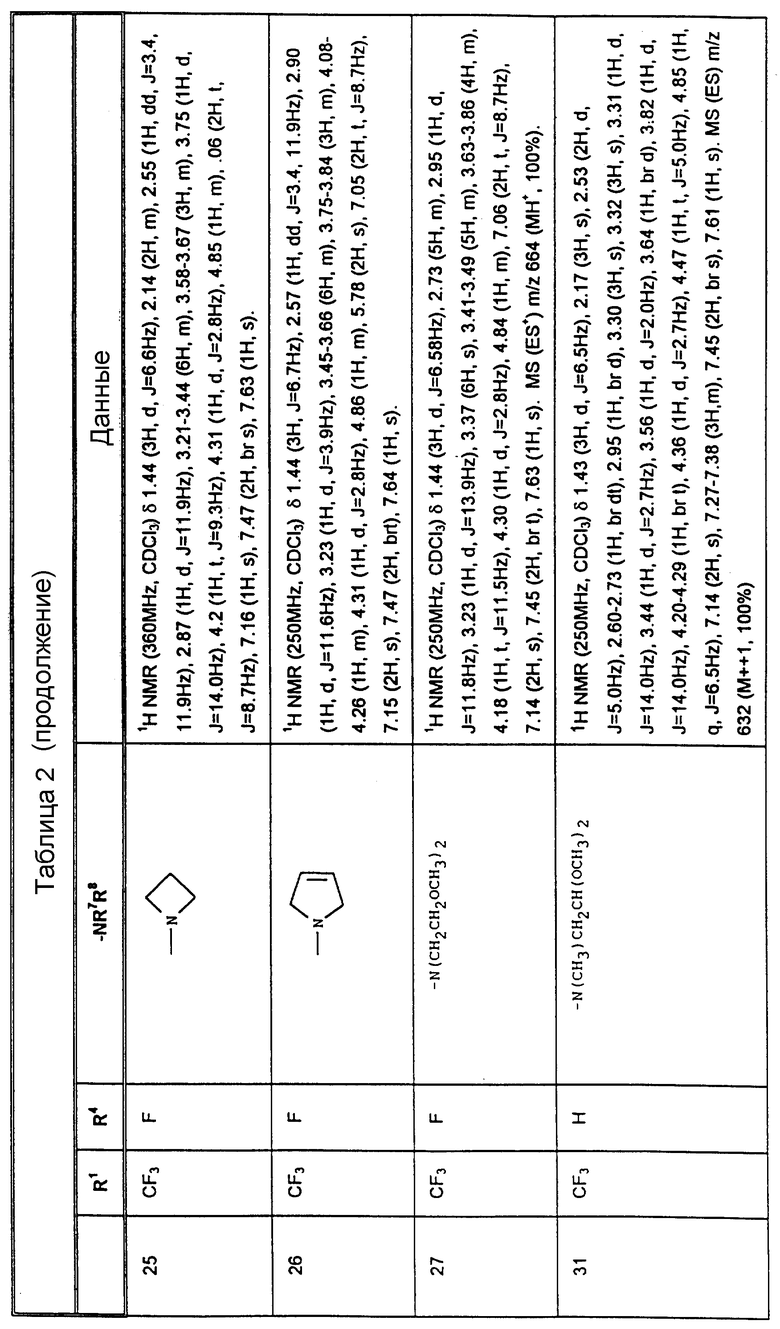

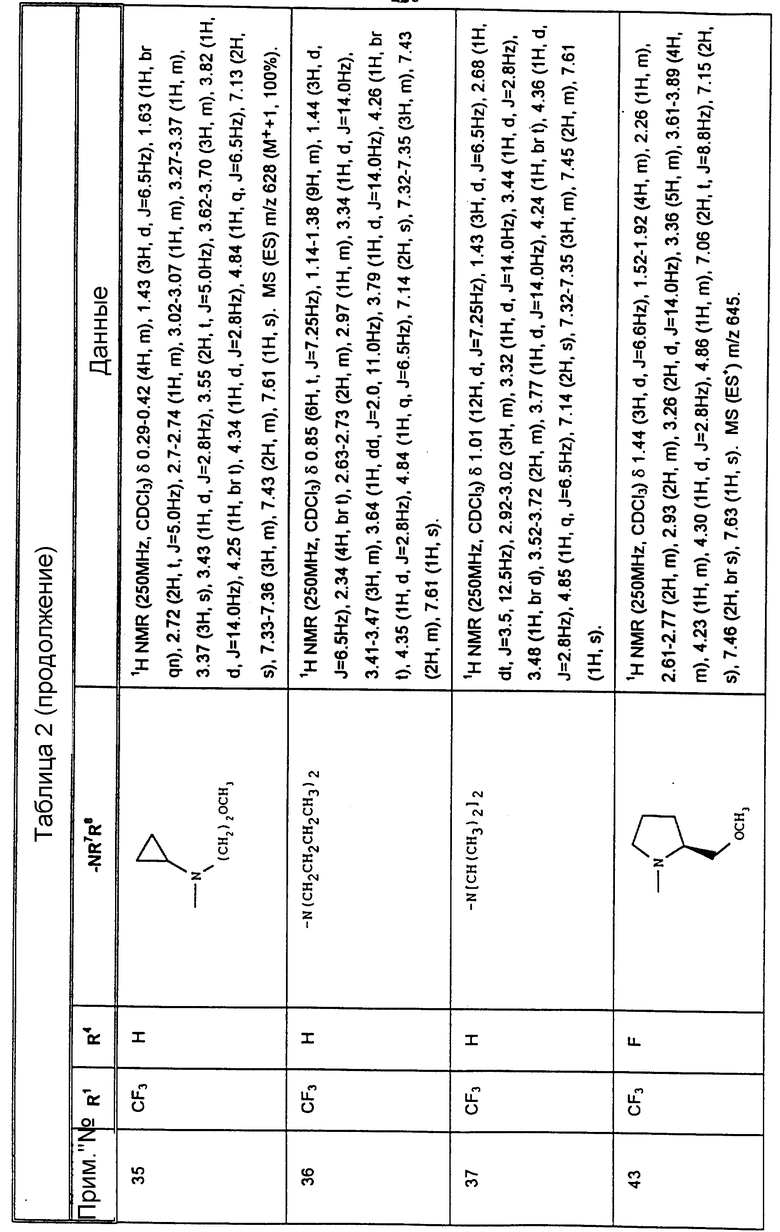
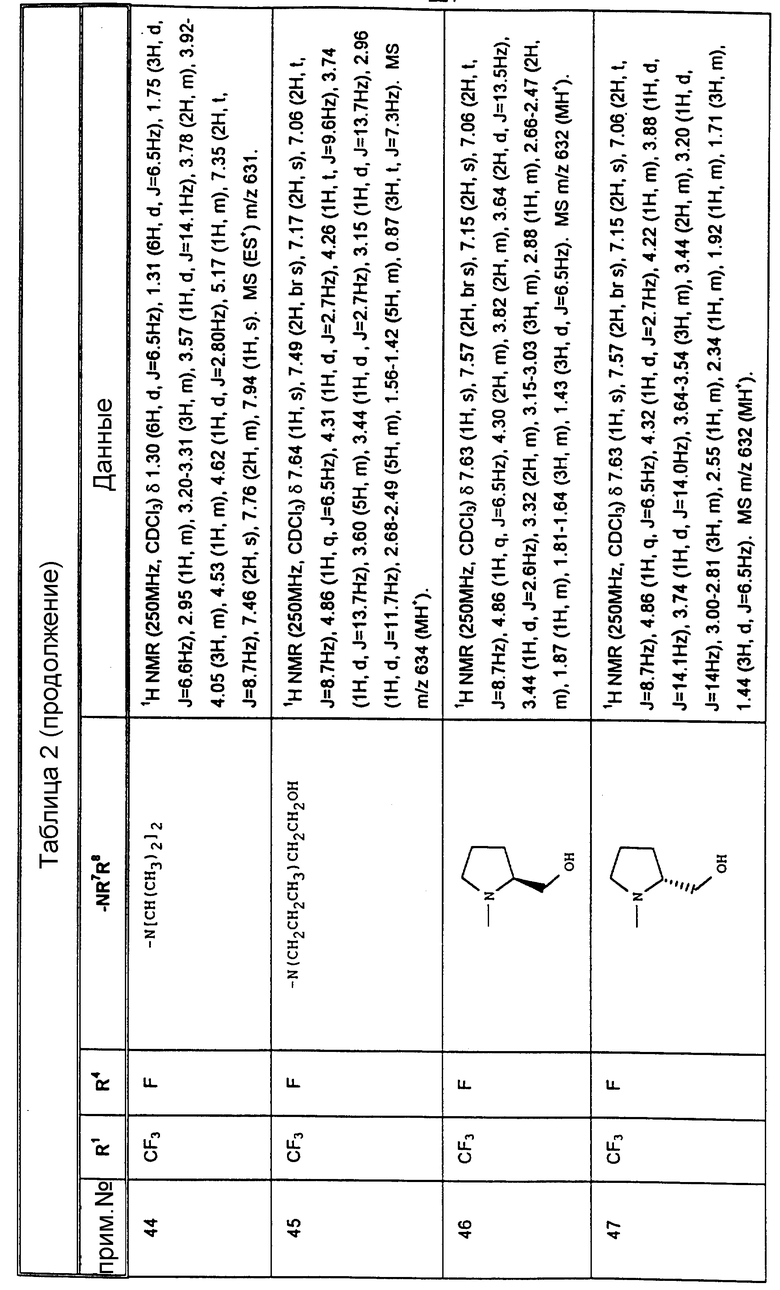

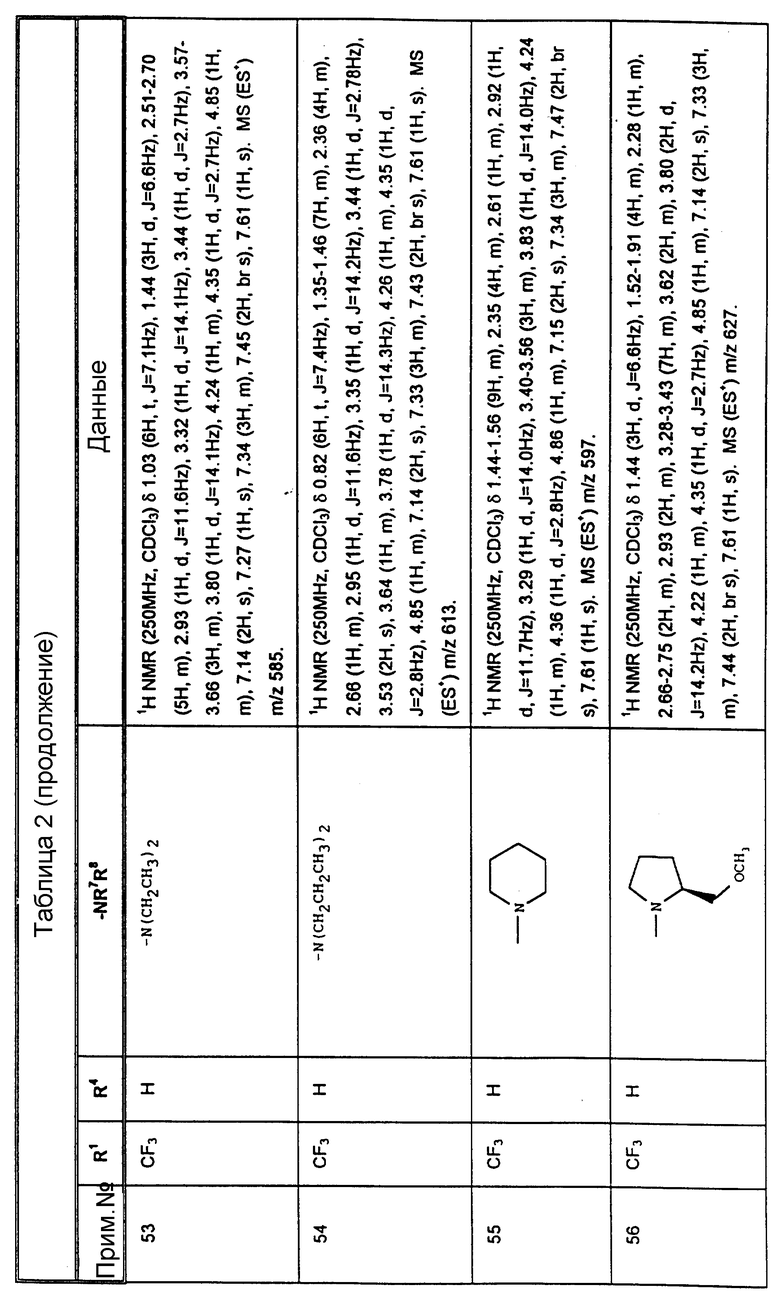
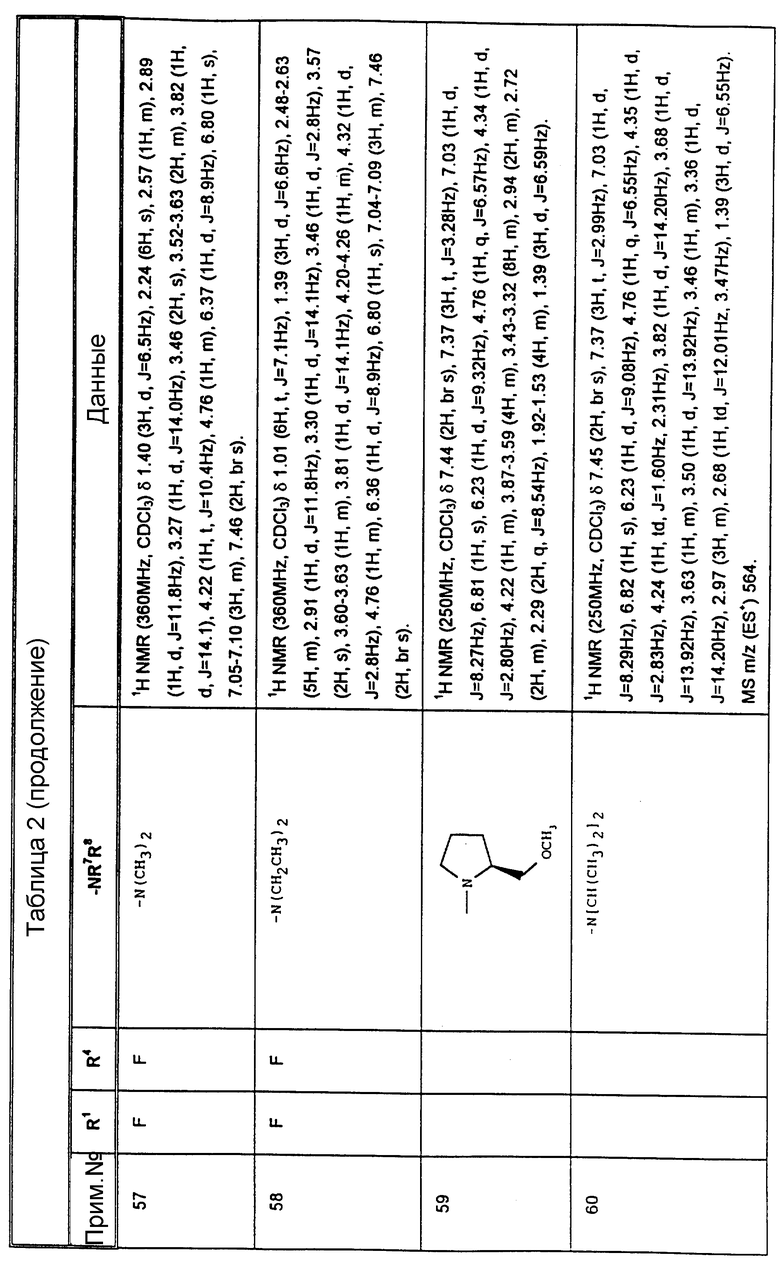

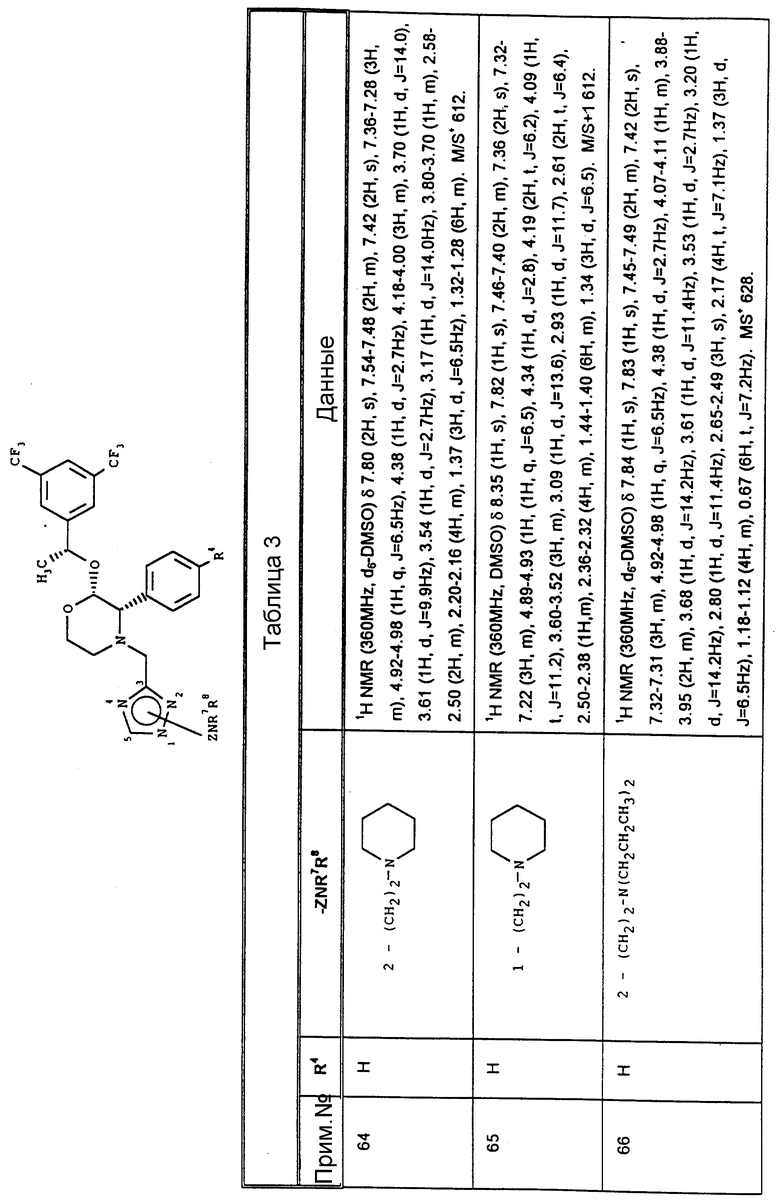

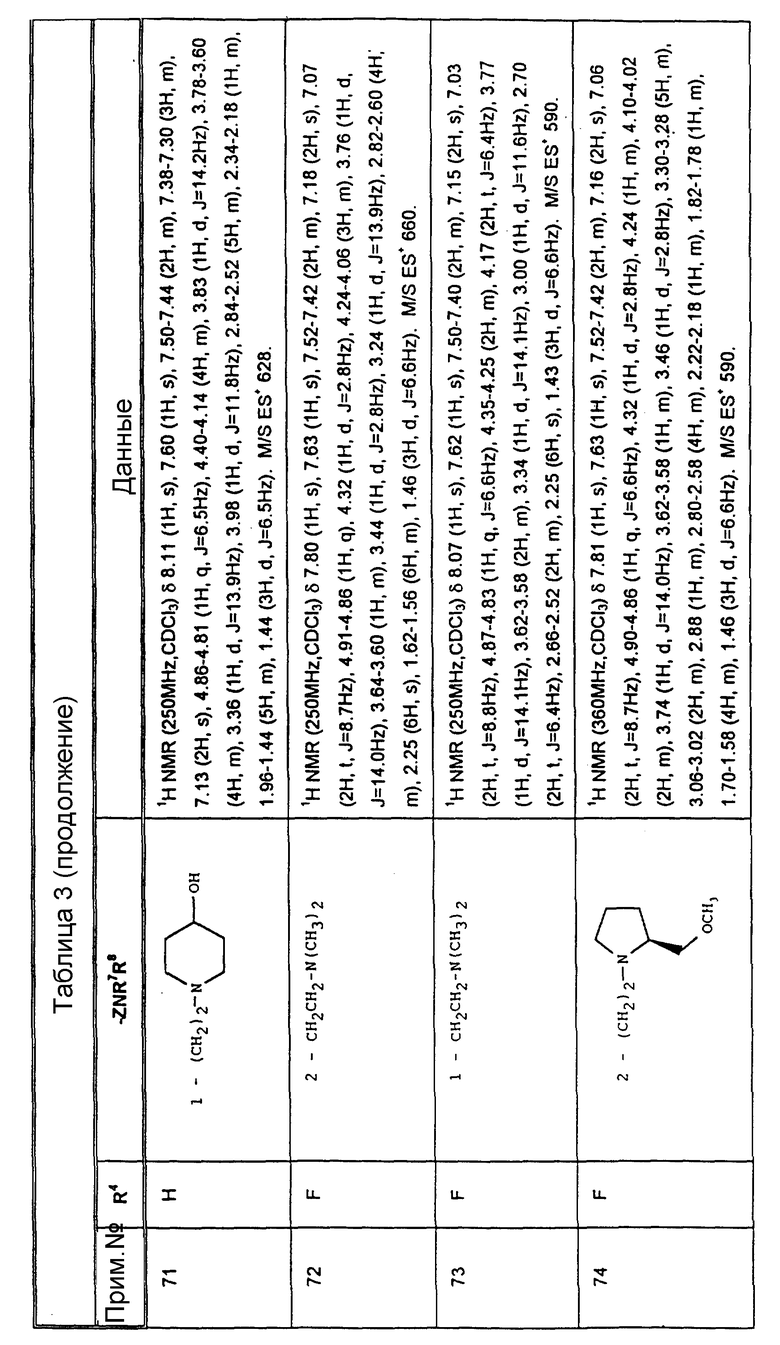
Замещенные производные морфолина формулы 1, где R1, R2 - водород, галоген, алкил, алкокси или СF3; R3 - водород, галоген или СF3; R4 - водород; R5 - водород или 4-фтор; R6 - 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, возможно замещенное =0, =S, алкилом или ZNR7R8, Z - C1-4-алкилен, R7 - водород, алкил, C3-7-циклоалкил, или C3-7-циклоалкил-C1-4-алкил или C2-4алкил, замещенный алкокси или гидроксилом (значения других радикалов см. в п.1 ф-лы), или его фармацевтически приемлемые соли, а также фармацевтическая композиция на их основе, обладает свойствами антагонистов тахикининов. 4 c.п. и 24 з.п. ф-лы, 3 табл.
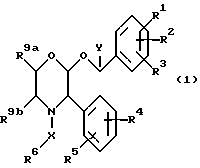
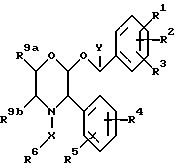
где R1 представляет собой водород, галоген, C1-4-алкил, C1-4-алкокси или CF3;
R2 представляет собой водород, галоген, C1-4-алкил, C1-4-алкокси или CF3;
R3 представляет собой водород, галоген или CF3;
R4 представляет собой водород;
R5 представляет собой водород или 4-фтор;
R6 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее 2 или 3 атома азота, необязательно замещенное =O, =S или C1-4-алкильной группой, и необязательно, замещенное группой формулы ZNR7R8,
где Z представляет собой C1-4-алкилен;
R7 представляет собой водород, C1-4-алкил, C3-7-циклоалкил, или C3-7-циклоалкил-C1-4-алкил, или C2-4-алкил, замещенный C1-4-алкокси или гидроксилом;
R8 представляет собой водород, C1-4-алкил, C3-7-циклоалкил, или C3-7-циклоалкил-C1-4-алкил, или C2-4-алкил, замещенный одним или двумя заместителями, выбранными из C1-4-алкокси, гидроксила или 4-, 5- или 6-членного гетероалифатического кольца, содержащего один или два гетероатома, выбранных из N, O и S;
или R7, R8 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4 - 7 кольцевых атомов, необязательно замещенное одной или двумя группами, выбранными из гидрокси или C1-4-алкила, необязательно замещенного C1-4-алкокси или гидроксильной группой, и содержащее, необязательно, двойную связь, и это кольцо может содержать, необязательно, кольцевой атом кислорода или серы, группы S(O) или S(O)2 или второй атом азота, который будет являться частью группы NH или NRc, где Rc представляет собой C1-4-алкил, необязательно замещенный гидрокси или C1-4-алкокси;
или R7, R8 и атом азота, к которому они присоединены, образуют неароматическую азабициклическую кольцевую систему из 6 - 12 кольцевых атомов;
или Z, R7 и атом азота, к которому они присоединены, образуют гетероалифатическое кольцо из 4 - 7 кольцевых атомов, которое, необязательно, может содержать кольцевой атом кислорода;
R9a и R9b, каждый, независимо, представляет собой водород;
X представляет собой алкиленовую цепь из 1 - 2 атомов углерода;
Y представляет собой C1-4-алкильную группу, необязательно замещенную гидроксильной группой
при условии, что, если Y представляет собой C1-4-алкил, R6 является замещенным по крайней мере группой формулы ZNR7R8, указанной выше, или его фармацевтически приемлемая соль или функциональное производное, где Y обозначает 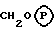 , когда
, когда  представляет PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+, где M+ обозначает фармацевтически приемлемый моновалентный противоион, а D2+ обозначает фармацевтически приемлемый дивалентный противоион.
представляет PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+, где M+ обозначает фармацевтически приемлемый моновалентный противоион, а D2+ обозначает фармацевтически приемлемый дивалентный противоион.
где A1 представляет собой фтор или CF3;
A2 представляет собой фтор или CF3;
A3 представляет собой фтор или водород;
X, Y и R6 имеют значения, указанные в п.1,
или его фармацевтически приелемая соль или функциональное производное, указанное в п.1.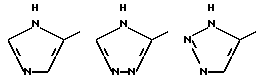

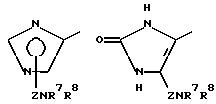
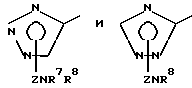
или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.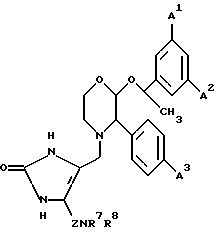
где A1, A2 и A3 имеют значения, указанные в п.2;
Z, R7 и R8 имеют значения, указанные в п.1,
или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.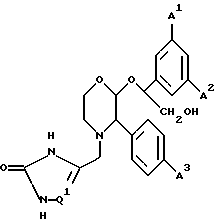
где A1, A2 и A3 имеют значения, указанные в п.2;
Q1 представляет собой CH, N или C-ZNR7R8, где Z, R7 и R8 имеют значения, указанные в п.1,
или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.
где A1, A2 и A3 имеют значения, указанные в п.2;
Q2 представляет собой CH или N;
Z, R7 и R8 имеют значения, указанные в п.1,
или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(2,3-дигидро-5(N, N-диметиламино)метил-2-оксо-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-5(N, N-диметиламино)метил-2-оксо-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил)этокси)морфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-трифторметил)фенил)этокси)-4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-5-(4-гидроксипиперидино)-метил-2-оксо-1,3-имидазол-4-ил)метилморфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил)этокси)-4-(2,3-дигидро-5-морфолинометил-2-оксо-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-5-морфолинометил-2-оксо-1,3-имидазол-4-ил)метилморфолин,
4-(5-азетидинилметил-2,3-дигидро-2-оксо-1,3-имидазол-4-ил)метил-2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-5-(N-метилпиперазинил)метил-2-оксо-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-5-(N-(2-морфолиноэтил)аминометил)-2-оксо-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-2-оксо-5-(N-(2-пирролидиноэтил)аминометил)-1,3-имидазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-диметиламино)метил-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(N-(N'-метиламиноэтил)-1,2,4-триазол-3-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(5-(N-метиламинометил)-1,2.3-триазол-4-ил)метилморфолин,
4-(5-аминометил)-1,2,3-триазол-4-ил)метил-2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)морфолин,
2(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(5-пирролидинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-(азетидинилметил-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил)этокси)морфолин),
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил)этокси)-4-(5-(пирролидинометил)-1,2,3-триазол-4-ил)метилморфолин,
3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-фтор-5-(трифторметил)фенил)этокси)-4-(5-морфолинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-(N, N-диметиламинометил)-1,2,3-триазол-4-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(R)-(3-трифторметил)фенил)-этокси)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(5-(N'-метилпиперазинометил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-(1-(2-пирролидиноэтил)-1,2-3,-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-фенил-4-(2-(2-пирролидиноэтил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(5-(морфолинометил)-1,2,3-триазол-4-ил)метилморфолин,
4-(5-азетидинилметил)-1,2.3-триазол-4-ил)метил-2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(5-(пирролинометил)-1,2,3-триазол-4-ил)метилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(бис(метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)-4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(2-хлор-5-морфолинометил-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N, N-диметиламинометил)-1,3-имидазол-4-ил)метил-3-(S)-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N, N-диметиламинометил)-1,2,4-триазол-3-ил)метил-3-(S)-(4-фторфенил)морфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-N-(2,2-диметоксиэтил)-N-метиламинометил)-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(2-метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N-(2-метоксиэтил)-N-метил)аминометил)-1,2,3-триазол-4-ил)-метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N-изопропил-N-(2-метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N-циклопропил-N-(2-метоксиэтил)аминометил)-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N, N-дибутиламинометил-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-4-(5-(N, N-диизопропиламинометил-1,2,3-триазол-4-ил)метил-3-(S)-фенилморфолин,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метилморфолин,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)-4-(1,2,4-триазол-3-ил)метилморфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-(4-фторфенил)-2-(R)-(1-(S)-(3-фтор-5-(трифторметил)фенил-2-гидроксиэтокси)морфолин,
4-(2,3-дигидро-2-оксо-1,3-имидазол-4-ил)метил-2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-2-оксо-5-пирролидинометил-1,3-имидазол-4-ил)метил-2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-(4-фторфенил)морфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)-3-(S)-фенил-2-(R)-(1-(S)-(3-(трифторметил)фенил)-2-гидроксиэтокси)морфолин,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-2-(R)-(1-(S)-(3-фтор-5-(трифторметил)фенил)-2-гидроксиэтокси)-3-(S)-фенилморфолин,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-гидроксиэтокси)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)-3-(S)-фенилметилморфолин,
3-(S)-фенил-4-(1,2,4-триазол-3-ил)-2-(R)-(1-(S)-3-(трифторметил)фенил)-2-гидроксиэтокси)морфолин, или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.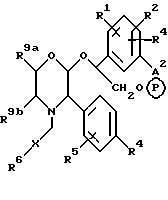
где R1, R2, R3, R4, R5, R6, R9a, R9b и X имеют значения, указанные в п. 1, и P в кружочке представляет собой PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+,
где M+ представляет собой фармацевтически приемлемый моновалентный противоион,
D2+ представляет собой фармацевтически приемлемый дивалентный противоион.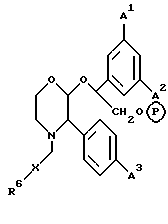
где A1, A2 и A3 имеют значения, указанные в п.2;
X и R6 имеют значения, указанные в п.1;
P в кружочке представляет собой PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+,
где M+ представляет собой фармацевтически приемлемый моновалентный противоион,
D2+ представляет собой фармацевтически приемлемый дивалентный противоион.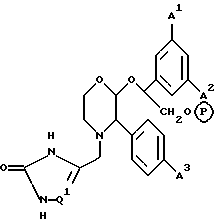
где A1, A2 и A3 имеют значения, указанные в п.2;
Q1 представляет собой CH, N или C-ZNR7R8, где Z, R7 и R8 имеют значения, указанные в п.1,
P в кружочке представляет собой PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+,
где M+ представляет собой фармацевтически приемлемый моновалентный противоион,
D2+ представляет собой фармацевтически приемлемый дивалентный противоион.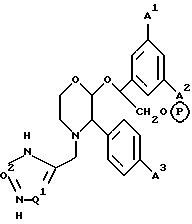
где A1, A2 и A3 имеют значения, указанные в п.2;
Q1 представляет собой CH, N или C-ZNR7R8, где Z, R7 и R8 имеют значения, указанные в п.1,
Q2 представляет собой CH или N;
P в кружочке представляет собой PO(OH)O-.M+, PO(O-)2.2M+ или PO(O-)2. D2+,
где M+ представляет собой фармацевтически приемлемый моновалентный противоион,
D2+ представляет собой фармацевтически приемлемый дивалентный противоион.
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S)-(4-фторфенил)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метилморфолина,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S)-(4-фторфенил)-4-(1,2,4-триазол-3-ил)метилморфолина,
4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-2-(R)-(1-(S)-3-фтор-5-(трифторметил)фенил)-2-фосфорилоксиэтокси)-3-(S)-фенилморфолина,
2-(R)-(1-(S)-(3,5-бис(трифторметил)фенил)-2-фосфорилоксиэтокси)-4-(2,3-дигидро-3-оксо-1,2,4-триазол-5-ил)метил-3-(S)-фенилморфолина, или его фармацевтически приемлемая соль.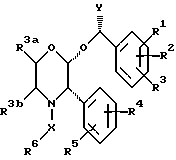
где R1, R2, R3, R4, R5, R6, R9a, R9b, X и Y имеют значения, указанные в п.1,
или его фармацевтически приемлемая соль или функциональное производное, указанное в п.1.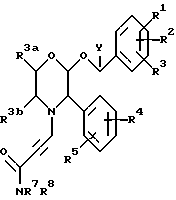
с азидом с последующим восстановлением карбонильной группы, смежной с -NR7R8; с последующим, при необходимости, удалением любой защитной группы, которая присутствует, и, когда соединение формулы I получают в виде смеси энантиомеров или диастереоизомеров, необязательном разделении смеси с получением желаемого энантиомера, и/или, если желательно, превращении полученного соединения формулы 1 или его соли в его фармацевтически приемлемую соль или функциональное производное, указанное в п.1.
Приоритет по признакам:
29.12.93 - по п. 2 подкласс (Ia); п.3 Y - гидроксиC1-4алкил; п.4 Y - C1-4-алкил; п.6 подкласс (Ib); п.7 подкласс (Ic);
22.04.94 - по п.9 разновидности; п.10 подкласс (Ie); п.11 подкласс (If); п. 12 подкласс (Ig); п.13 подкласс (Ih); п.14 разновидности (пролекарства); п. 26 способ (B); п. 28 пример 89 - 12.04.94; по п.8 подкласс (Id); п.27 пример 12;
15.08.94 - по п.9 разновидности;
23.12.94 - по п.1; п.5 R6 предпочтения; п.15 подкласс (Ij); пп.16 - 25.
| Способ получения диметилтерефталата | 1981 |
|
SU980613A3 |
| Вихретоковый накладной преобразователь для контроля цилиндрических электропроводных изделий | 1974 |
|
SU528495A1 |
| КРЕПЛЕНИЕ НАСОСА В АППАРАТЕ ДЛЯ ПРИГОТОВЛЕНИЯ НАПИТКА | 2010 |
|
RU2534915C2 |
| ТЕРМОРЕГУЛИРУЕМОЕ КРИОСТАТИОЕ УСТРОЙСТВО | 1970 |
|
SU436334A1 |
| EP 394989 A2, 1990. | |||

