Настоящее изобретение относится к производному тиоформамида, полезному в качестве лекарства.
Гипертензия известна как болезнь, вызывающая осложнения в виде удара или заболеваний сердца, которые занимают высокое место в списке причин смерти в Японии, вследствие чего проведены разнообразные исследования по ее предупреждению и лечению. Система регулирования кровяного давления сложна и поэтому применяют различные типы гипотензивных средств.
Такие противогипертензивные средства включают диуретики, β- блокаторы и ACE-ингибиторы. Однако диуретики вызывают нежелательно действие в виде гиперурикемии, обострения сахарного диабета, приступы подагры и т.п.; β- блокаторы - сердечную недостаточность, бронхоспазм и т. п.; ACE-ингибиторы - одышку, замедление деятельности сердца и т.п. Таким образом, гипотензивные средства известного уровня техники соответственно вызывают нежелательные действия, в частности сердечные заболевания, что затрудняет их применение или делает его нежелательным.
В Японии число пациентов с нарушениями коронарных сосудов сердца достигло того же уровня, что и в странах Европы и Америки, по мере перехода на западный образ жизни, и предполагается, что имеется весьма значительное число пациентов с ишемическими болезнями сердца (такими как ангина пекторис (стенокардия) или инфаркт миокарда), включающими болезни, причиной которых является гипертензия.
Хотя нитритные препараты и β- блокаторы и применяются в качестве профилактических и лечебных средств от стенокардии, нитритные препараты имеют тот недостаток, что они вызывают толерантность в пациентах и сопровождаются нежелательным действием, таким как метемоглобинемия или глазная гипертензия, в то время как β- блокаторы являются причиной, как упомянуто выше, нежелательного действия, такого как сердечная недостаточность или бронхоспазм. Поэтому требуется осторожность в применении этих лекарств.
Между тем, число пациентов, страдающих астмой, за последнее время увеличилось, предположительно вследствие изменения пищевых привычек или ухудшения среды обитания, вызванного загрязнением воздуха, воды и т.д.
До последнего времени в качестве профилактических и лечебных средств от астмы применялись β- стимуляторы, метилксантины, стабилизаторы тучных клеток и стероиды. Однако β- стимуляторы вызывают в качестве нежелательного действия аритмию, гипертензию, головную боль и т.п.; метилксантины - желудочно-кишечные расстройства или нейропатию; стероиды - сахарный диабет или остеопороз. Итак, эти лекарства иногда вызывают серьезные нежелательные действия и поэтому требуют осторожности при их назначении. Хотя стабилизаторы тучных клеток не являются причиной каких-либо нежелательных действия, они имеют те недостатки, что их применение для детей и пожилых представляет трудности, так как они могут применяться только как лекарственная форма для ингаляции, что они не эффективны против серьезной астмы и что они могут использоваться только как профилактическое средство.
При таких обстоятельствах возникает потребность в разработке гипотензивного средства или средства профилактики и лечения сердечных болезней или астмы, которое имеет различные механизмы действия и может легко применяться благодаря его безопасности и качества, превосходящим качества препаратов известного уровня техники.
Авторы настоящего изобретения провели интенсивные исследования, изучая соединения, полезные в качестве таких лекарств. В результате исследований они направили свое внимание на активность по открытию ATP-чувствительного калиевого канала и провели исследования с целью найти соединение, имеющее такую активность.
В результате этих исследований авторы настоящего изобретения нашли, что производные тиоформамида, которые будут описаны ниже, могут достичь вышеуказанной цели. Настоящее изобретение сделано на основе этого открытия.
Хотя производные тиоформамида, имеющие активность в качестве лекарств, раскрыты, например, в японской патентной публикации N 59150/1990 и в японских выложенных патентных заявках NN 63260/1991, 289543/1990, 286659/1990, 211566/1989, 273/1990, 308275/1989 и 258760/1990, соединения по настоящему изобретению отличаются от них структурой.
Сущность изобретения.
Настоящее изобретение относится к производному тиоформамида, представленному следующей общей формулой (I), или его фармакологически приемлемой соли: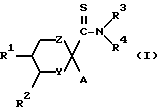
в которой
Y обозначает группу, представленную формулой -O-, группу, представленную формулой  , (где n - нуль или целое число от 1 до 2), группу, представленную формулой
, (где n - нуль или целое число от 1 до 2), группу, представленную формулой 
группу, представленную формулой 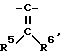 (где R5 и R6 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, низший алкоксил, циано, цианоалкил, возможно защищенный карбоксил, возможно (необязательно) защищенный карбоксиалкил или ацил), группу, представленную формулой
(где R5 и R6 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, низший алкоксил, циано, цианоалкил, возможно защищенный карбоксил, возможно (необязательно) защищенный карбоксиалкил или ацил), группу, представленную формулой 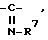 (где R7 обозначает группу, представленную формулой -OR8, где R8 обозначает водород, циано, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил или возможно защищенный карбоксиалкил), группу, представленную формулой
(где R7 обозначает группу, представленную формулой -OR8, где R8 обозначает водород, циано, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил или возможно защищенный карбоксиалкил), группу, представленную формулой  (где R9 и R10 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил, или возможно защищенный карбоксиалкил, или альтернативно R9 и R10 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу), водород, циано, цианоалкил, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил, или возможно защищенный карбоксиалкил), или группу, представленную формулой
(где R9 и R10 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил, или возможно защищенный карбоксиалкил, или альтернативно R9 и R10 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу), водород, циано, цианоалкил, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, возможно защищенный карбоксил, или возможно защищенный карбоксиалкил), или группу, представленную формулой  {где R11 и R12 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, арил, арилалкил, цианоалкил, ацилалкил, возможно защищенный карбоксиалкил, группу, представленную формулой -(CH2)s-O-R13, [где R13 обозначает водород, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, низший алкоксиалкил, ацил, карбамоил, алкилкарбамоил, возможно замещенный арилкарбамоил, возможно замещенный арилалкилкарбамоил, возможно замещенный арилалкилсульфинил, возможно замещенный гетероарилалкилсульфинил, возможно замещенный арилсульфонил, возможно замещенный гетероарилсульфонил, алкилсульфонил, возможно защищенный карбоксиалкил, аминоалкил, в котором амино- часть может быть замещена, или группу, представленную формулой
{где R11 и R12 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, арил, арилалкил, цианоалкил, ацилалкил, возможно защищенный карбоксиалкил, группу, представленную формулой -(CH2)s-O-R13, [где R13 обозначает водород, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, низший алкоксиалкил, ацил, карбамоил, алкилкарбамоил, возможно замещенный арилкарбамоил, возможно замещенный арилалкилкарбамоил, возможно замещенный арилалкилсульфинил, возможно замещенный гетероарилалкилсульфинил, возможно замещенный арилсульфонил, возможно замещенный гетероарилсульфонил, алкилсульфонил, возможно защищенный карбоксиалкил, аминоалкил, в котором амино- часть может быть замещена, или группу, представленную формулой  (где B обозначает серу, кислород или группу, представленную формулой =N-CN; и R16 и R17 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный арилалкил, или возможно замещенный гетероарилалкил, или альтернативно R16 и R17 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу); и s - нуль или целое число от 1 до 10], или группу, представленную формулой
(где B обозначает серу, кислород или группу, представленную формулой =N-CN; и R16 и R17 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный арилалкил, или возможно замещенный гетероарилалкил, или альтернативно R16 и R17 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу); и s - нуль или целое число от 1 до 10], или группу, представленную формулой  [где R14 и R15 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, низший алкоксиалкил, ацил, карбамоил, алкилкарбамоил, возможно замещенный арилкарбамоил, возможно замещенный арилалкилкарбамоил, возможно замещенный арилсульфонил, возможно замещенный гетероарилсульфонил, алкилсульфонил, возможно замещенный арилалкилсульфинил, возможно замещенный гетероарилалкилсульфинил, группу, представленную формулой
[где R14 и R15 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, возможно замещенный арилалкил, возможно замещенный гетероарилалкил, низший алкоксиалкил, ацил, карбамоил, алкилкарбамоил, возможно замещенный арилкарбамоил, возможно замещенный арилалкилкарбамоил, возможно замещенный арилсульфонил, возможно замещенный гетероарилсульфонил, алкилсульфонил, возможно замещенный арилалкилсульфинил, возможно замещенный гетероарилалкилсульфинил, группу, представленную формулой  (где D обозначает серу, кислород или группу, представленную формулой = N-CN; и R18 и R19 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный арилалкил или возможно замещенный гетероарилалкил, или альтернативно R18 и R19 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу), возможно защищенный карбоксиалкил, или аминоалкил, в котором аминная часть может быть замещена, или, альтернативно R14 и R15 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу; и t - нуль или целое число от 1 до 10]};
(где D обозначает серу, кислород или группу, представленную формулой = N-CN; и R18 и R19 могут быть одинаковыми или различными и каждый обозначает водород, циано, низший алкил, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный арилалкил или возможно замещенный гетероарилалкил, или альтернативно R18 и R19 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу), возможно защищенный карбоксиалкил, или аминоалкил, в котором аминная часть может быть замещена, или, альтернативно R14 и R15 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу; и t - нуль или целое число от 1 до 10]};
Z обозначает группу, представленную формулой -O-, группу, представленную формулой 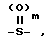 (где m - нуль или целое число от 1 до 2) или группу, представленную формулой -(CH2)p - (где p - нуль или целое число от 1 до 2);
(где m - нуль или целое число от 1 до 2) или группу, представленную формулой -(CH2)p - (где p - нуль или целое число от 1 до 2);
A обозначает арил, тиенил, фурил, бензофуразанил, пирролил, имидазолил, триазолил, тетразолил, пиридонил, пиразолил, изоксазолил, изотиазолил, оксазолил, бензимидазолил, имидазопиридил, имидазопиразинил, имидазопиримидинил, имидазопиридазинил, имидазоксазинил или имидазотиазинил, при условии, что каждая гетероарильная группа может иметь один или два заместителя, и незамещенный имидазолил исключается, когда Y обозначает группу, представленную формулой -O- или -S- или когда Z обозначает группу, представленную формулой -O- или -S-;
R1 и R2 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, возможно замещенный арилалкил, или возможно замещенный гетероарилалкил, или альтернативно R1 и R2 могут вместе обозначать бензольное кольцо;
R3 и R4 могут быть одинаковыми или различными и каждый обозначает водород, низший алкил, циклоалкил, низший алкокси, гидроксил, арил, арилалкил, гетероарил, или гетероарилалкил или альтернативно R3 и R4 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу, которая может содержать атом кислорода, азота или серы.
Низший алкил, указанный в отношении R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18 и R19, представляет собой линейный или разветвленный алкил, имеющий от 1 до 8 атомов углерода, примерами его являются метил, этил, пропил, изопропил, бутил, изобутил, втор.-бутил, трет. -бутил, пентил (амил), изопентил, неопентил, трет.-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил, гексил, изогексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этил-1-метилпропил, 1-этил-2-метилпропил, гептил и октил. Среди этих групп предпочтительны метил, этил, пропил и изопропил, а наиболее предпочтительны метил и этил.
Низший алкокси, указанный в отношении R3 и R4, представляет собой линейный или разветвленный алкокси, имеющий от 1 до 8 атомов углерода, и его примерами являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор. -бутокси, трет. -бутокси, 1-метилбутокси, 2-метилбутокси, 1,2-диметилпропокси и гексилокси. Среди этих групп предпочтительны метокси и этокси, причем наиболее предпочтителен метокси.
Циклоалкил, указанный в отношении R3 и R4, представляет собой циклоалкил, имеющий от 3 до 8 атомов углерода, предпочтительно от 3 до 7 атомов углерода и наиболее предпочтительно 5 или 6 атомов углерода.
Арил, указанный в отношении R3, R4, R11, R12, R14, R15, R16, R17, R18, R19 и A, включает фенил, нафтил и толил.
Арильная группа, входящая в арилалкил, указанный в отношении R1, R2, R3, R4, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18 и R19, является той же самой, что и группы, указанные выше. Алкильная группа (то есть алкиленовая группа), входящая в арилалкил, является группой, имеющей от 1 до 6 атомов углерода.
Гетероарил, указанный в отношении R3, R4, R16, R17, R18 и R19, является гетероарилом, происходящим из 5- или 6-членного кольца, содержащего один или два гетероатома, выбранных из атомов азота, серы и кислорода.
Гетероарильная группа, входящая в гетероарилалкил, указанный в отношении R1, R2, R3, R4, R7, R8, R9, R10, R13, R14 и R15, является той же самой, что и группы, указанные выше. Алкильная группа (то есть алкиленовая цепь), входящая в гетероарилалкил, является группой, имеющей от 1 до 6 атомов углерода.
Арилалкилсульфинил, указанный в отношении R13, R14 и R15, является арилалкилсульфинилом, производным от любой из вышеприведенных арилалкильных групп.
Гетероарилалкилсульфинил, указанный в отношении R13, R14 и R15, является гетероарилалкилсульфинилом, производным от любой из вышеприведенных гетероарилалкильных групп.
Арилсульфонил, указанный в отношении R13, R14 и R15, является арилсульфонилом, производным от любой из вышеприведенных арильных групп.
Гетероарилсульфонил, указанный в отношении R13, R14 и R15, является гетероарилсульфонилом, производным от любой из вышеприведенных гетероарильных групп.
Алкилсульфонил, указанный в отношении R13, R14 и R15, является алкилсульфонилом, производным от любой из вышеприведенных низших алкильных групп.
Как указано выше, R14 и R15 могут обозначать вместе с атомом азота, к которому они присоединены, циклическую группу. Циклическая группа включает в себя группы, производные от пиперидинового и пиразолинового колец. Циклическая группа может далее содержать атом кислорода, азота или серы и примерами такой группы являются группы, производные от морфолинового и пиперазинового колец. Циклическая группа не ограничена этими группами. Альтернативно R14 и R15 могут совместно представлять ацильную группу, производную от дикарбоновой кислоты, т. е. они могут образовывать имидную группу вместе с атомом азота.
Предпочтительные примеры заместителя для вышеприведенных арила и гетероарила включают водород, низший алкил, галоген, нитро, низший алкокси, алкилтио, алкилсульфинил, алкилсульфонил, галоидзамещенный низший алкил, циано, карбоксил, возможно защищенный, арил, возможно замещенный, и гетероарил, возможно замещенный.
Как указано выше, R5, R6, R8, R9 и R10 могут каждый обозначать возможно защищенный карбоксил, и защитная группа, подходящая для этого случая, включает низшие алкильные группы, такие как метил, этил и т-бутил; фенилзамещенные низшие алкильные группы, в которых фенильная группа может быть замещена, такие как п-метоксибензил, п-нитробензил, 3,4-диметоксибензил, дифенилметил, тритил и фенетил; галоидированные низшие алкильные группы, такие как 2,2,2-трихлорэтил и 2-иодоэтил; низшие алканоилокси-низшие алкильные группы, такие как пивалоилоксиметил, ацетоксиметил, пропионилоксиметил, бутиролоксиметил, валерилоксиметил, 1-ацетоксиэтил, 2-ацетоксиэтил, 1-пивалоилоксиэтил и 2-пивалоилоксиэтил; высшие алканоилокси-низшие алкильные группы, такие как пальмитоилоксиэтил, гептадеканоилоксиметил и 1-пальмитоилоксиэтил; низшие алкоксикарбонилокси-низшие алкильные группы, такие как метоксикарбонилоксиметил, 1-(бутоксикарбонилокси)этил и 1-(изопропоксикарбонилокси)этил; карбокси-низшие алкильные группы, такие как карбоксиметил и 2-карбоксиэтил; гетероциклические группы, такие как 3-фталидил; возможно замещенные бензоилокси-низшие алкильные группы, такие как 4-глицилоксибензоилоксиметил; (замещенный диоксилен)-низшие алкильные группы, такие как (5-метил-2-оксо-1,3- диоксолен-4-ил)метил; циклоалкилзамещенные низшие алканоилокси-низшие алкильные группы, такие как 1-циклогексилацетилоксиэтил; и циклоалкилоксикарбонилокси-низшие алкильные группы, такие как 1-циклогексилоксикарбонилоксиэтил. Далее, карбоксильная группа может быть защищена в форме различных амидов кислот. Короче говоря, защитная группа может быть любой группой, которая может быть удалена некоторыми средствами in vivo с получением свободной карбоксильной группы.
Как указано выше, R8, R9, R10, R11, R12, R13, R14 и R15 могут каждый представлять возможно защищенный карбоксиалкил, а защитная группа, подходящая для этого случая, является той же самой, что и описанная выше в отношении возможно защищенного карбоксила. Далее, алкиленовая группа, входящая в возможно защищенный карбоксиалкил, является группой, производной от любой из вышеприведенных алкильных групп.
Хотя имидазолил, указанный выше в отношении A, может быть любым, предпочтителен имидазолил, представленный формулой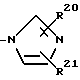
(где R20 и R21 могут быть одинаковыми или различными и каждый обозначает водород, циано, нитро, низший алкил, галоген, низший алкокси, алкилтио, алкилсульфинил, алкилсульфонил, галоидзамещенный низший алкил, возможно замещенный арил, возможно замещенный гетероарил или возможно защищенный карбоксил).
Имидазопиридил, указанный в отношении A, может быть имидазопиридилом, производным от любого имидазопиридина, а его примеры включают
(1) группы, производные от имидазо/1,2-a/пиридина, такие как имидазо/1,2-a/пиридин-2-ил, имидазо/1,2-a/пиридин-3-ил, имидазо/1,2-a/пиридин-5-ил, имидазо/1,2-a/пиридин-6-ил, имидазо/1,2-a/пиридин-7-ил и имидазо/1,2-a/пиридин-8-ил;
(2) группы, производные от имидазо/1,5-a/пиридина, такие как имидазо/1,5-a/пиридин-1-ил, имидазо/1,5-a/пиридин-3-ил, имидазо/1,5-a/пиридин-5-ил, имидазо/1,5-a/пиридин-6-ил, имидазо/1,5-a/пиридин-7-ил и имидазо/1,5-a/пиридин-8-ил;
(3) группы, производные от имидазо/4,5-b/пиридина, такие как имидазо/4,5-b/пиридин-1-ил, имидазо/4,5-b/пиридин-2-ил, имидазо/4,5-b/пиридин-3-ил, имидазо/4,5-b/пиридин-5-ил, имидазо/4,5-b/пиридин-6-ил и имидазо/4,5-b/пиридин-7-ил;
(4) группы, производные от имидазо/4,5-c/пиридина, такие как имидазо/4,5-c/пиридин-1-ил, имидазо/4,5-c/пиридин-2-ил, имидазо/4,5-c/пиридин-3-ил, имидазо/4,5-c/пиридин-4-ил, имидазо/4,5-c/пиридин-6-ил и имидазо/4,5-c/пиридин-7-ил.
Предпочтительные примеры заместителя для имидазопиридила включают циано, нитро, низший алкил, галоген, низший алкокси, алкилтио, алкилсульфинил, алкилсульфонил, галоидзамещенный низший алкил, возможно замещенный арил, возможно замещенный гетероарил и возможно защищенный карбоксил.
Ацил, указанный выше в отношении R5, R6, R13, R14 и R15, может быть ацилом, производным от любой из алифатических насыщенных монокарбоновых, алифатических насыщенных дикарбоновых, алифатических ненасыщенных карбоновых, насыщенных и ненасыщенных карбоциклических карбоновых, гетероциклических карбоновых, гидроксикарбоновых, алкоксикарбоновых и любых других карбоновых кислот. Примеры ацила включают низшие алканоильные группы, такие как формил, ацетил, пропионионил, бутирил, валерил, изовалерил и пивалоил; ароильные группы, такие как бензоил, толуол и нафтоил; и гетероароильные группы, такие как фуроил, никотиноил и изоникотиноил.
Цианоалкил, указанный выше в отношении R11 и R12, является алкильной группой, описанной выше, в которой один из атомов водорода, входящий в алкильную группу, заменен на цианогруппу.
Ацилалкил, указанный выше в отношении R11 и R12, является ацилалкилом, производным от любой из вышеприведенных низших алкильных групп путем замены одного из водородных атомов ацильной группой.
Фармакологически приемлемая соль в соответствии с настоящим изобретением включает соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат и фосфат; соли органических кислот, такие как ацетат, малеат, тартрат, метансульфонат, бензолсульфонат и толуолсульфонат; и соли аминокислот, такие как аргининат, аспартат и глутамат.
Далее, производное согласно настоящему изобретению может образовывать соль металла, такую как соль натрия, калия, кальция или магния. Фармакологически приемлемая соль в соответствии с настоящим изобретением включает эти соли металлов.
Далее, производные в соответствии с настоящим изобретением могут быть представлены в виде геометрических или оптических изомеров в зависимости от заместителя. Настоящее изобретение включает эти изомеры.
Ниже описываются типичные способы получения соединений в соответствии с настоящим изобретением.
Способ получения 1.
Соединение, представленное общей формулой (I), в которой R3 есть водород, a Y есть группа, представленная формулой  может быть получено согласно следующему способу:
может быть получено согласно следующему способу:
где
R1, R2, R4, A и Z имеют каждый вышеуказанное значение.
Конкретнее, целевое соединение (III) может быть получено путем проведения реакции соединения, представленного общей формулой (II), с соединением, представленным общей формулой (IV), а присутствии основания.
Основание включает алкоксиды щелочного металла, такие как т-бутоксид калия; гидриды щелочного металла, такие как гидрид натрия; и литийорганические соединения, такие как н-бутиллитий.
Растворитель, используемый в вышеприведенной реакции, представляет собой предпочтительно эфир, такой как тетрагидрофуран, полярный апротонный растворитель, такой как N,N-диметилформамид, или их смесь, хотя им может быть любой органический растворитель, инертный в условиях данной реакции.
Температура реакции может быть в пределах от -78oC до температуры дефлегмации применяемого растворителя.
Способ получения 2.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой  , может быть получено согласно следующему способу:
, может быть получено согласно следующему способу: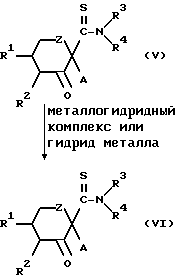
где
R1, R2, R3, R4, A и Z каждый имеет вышеприведенные значения.
Конкретнее, целевое соединение (VI) может быть получено путем восстановления соединения, представленного общей формулой (V) металлогидридным комплексом или гидридом металла.
Металлдогидридный комплекс включает боргидрид натрия и алюминийлитийгидрид, а в качестве гидрида металла предпочтительно используется диизобутилалюминийгидрид.
Растворитель, используемый в вышеприведенной реакции, представляет собой предпочтительно спирт, такой как метанол, эфир, такой как тетрагидрофуран, или углеводород, такой как толуол, хотя им может быть любой органический растворитель, инертный в условиях данной реакции.
Температура реакции может быть в пределах от -78oC до приблизительно 50oC.
Альтернативно соединение, представленное общей формулой (VI), может быть получено также восстановлением соединения, представленного общей формулой (V) алкоксидом алюминия в спиртовом растворителе. В этой реакции предпочтительно применять изопропиловый спирт в качестве растворителя и алкоксид алюминия в качестве восстановителя.
Температура реакции может быть в пределах от комнатной температуры до температуры дефлегмации применяемого растворителя.
Способ получения 3.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой  (где R
(где R
где
R1, R2, R3, R4, A, Z и R
Конкретнее, целевое соединение (VII) может быть получено конденсацией соединения, представленного общей формулой (VI), с соединением, представленным общей формулой (VIII) или реакционноспособным производным его, обычным способом.
Альтернативно можно использовать соль соединения (VIII) и превращать его в соединение (VIII) в реакционной системе.
Реакционноспособное производное соединение (VIII) включает его галоидангидриды и симметричные ангидриты кислот. Когда соединение (VIII) используется в форме свободного спирта, обычно вместе с ним используется конденсирующее средство.
Предпочтительные примеры конденсирующего средства включают дифонилфосфорамид, N-гидроксибензотриазол, N-гидроксисукцинамид, этилхлорформиат, метансульфонилхлорид, 1,3-дициклогексилкарбониимид, 1,1'-карбонилдиимидазол, диэтилазодикарбоксилат и дипиридилдисульфид.
Далее, вышеуказанная конденсация может проводиться в присутствии основания в некоторых случаях. Основание, применяемое в этом случае, является предпочтительно органическим основанием, таким как диизопропилэтиламин, триэтиламин, пиридин, пиколин, лутидин, N,N-диметиланилин или 4-диметиламинопиридин, или неорганическим основанием, таким как карбонат калия или гидроксид натрия, хотя им может быть любое основание.
Растворитель, применяемый в этой реакции, представляет собой предпочтительно спирт, такой как этанол, эфир, такой как тетрагидрофуран, углеводород, такой как толуол, галоидный растворитель, такой как дихлорметан, полярный апротонный растворитель, такой как этилацетат, N,N-диметилформамид или ацетонитрил или пиридин.
Температура реакции может быть в пределах от -20oC до температуры дефлегмации применяемого растворителя.
Способ получения 4.
Соединение, представленное общей формулой (I), в которой Y есть группа, представленная формулой  , может быть также получено следующим способом:
, может быть также получено следующим способом:
(Стадия 1)
где
R1, R2, A и Z каждый имеет вышеуказанные значения, R22 обозначает низший алкил или бензил: и Q обозначает уходящую или удаляемую группу.
Конкретнее, целевое соединение (X) может быть получено путем реакции соединения, представленного общей формулой (II), сначала с дисульфидом углерода, а затем с соединением, представленным общей формулой (IX), в присутствии основания.
Уходящая группа, указанная в отношении Q, представляет предпочтительно галоген или метансульфонилокси.
Предпочтительные примеры основания включают алкоксиды щелочного металла, такие как т-бутоксид калия; гидриды щелочных металлов, такие как гидрид натрия; и литийорганические соединения, такие как н-бутиллитий.
Растворитель, используемый в реакции, предпочтительно представляет собой эфир, такой как тетрагидрофуран, полярный апротонный растворитель, такой как N,N-диметилформамид или их смесь, хотя он может быть любым органическим растворителем, инертным в условиях реакции.
(Стадия 2)
где
R1, R2, R3, R4, R22, A и Z каждый имеет вышеуказанные значения.
Целевое соединение (XI) может быть получено проведением реакции соединения, представленного общей формулой (X), с соединением, представленным общей формулой (XII), обычно в избытке, в присутствии или в отсутствии растворителя и, если необходимо, под давлением.
Когда в вышеуказанной реакции имеется в виду использование растворителя, то растворителем предпочтительно является спирт, такой как этанол, эфир, такой как тетрагидрофуран, или углеводород, такой как толуол, хотя им может быть любой органический растворитель, инертный в условиях реакции.
Температура реакции может быть в пределах от комнатной температуры до температуры дефлегмации растворителя.
Полезно, чтобы R22-SH, образованный в ходе реакции, захватывался в виде тяжелого металла путем добавления, например, хлорида ртути в реакционную систему.
Способ получения 5.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой  может быть получено следующим способом
может быть получено следующим способом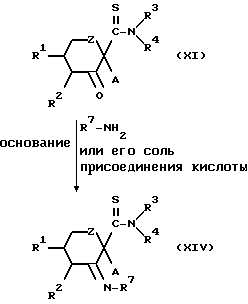
где
R1, R2, R3, R4, R7, A и Z каждый имеет вышеуказанные значения.
Конкретнее, целевое соединение (XIV) может быть получено конденсацией производимого тиоформамида, представленного общей формулой (XI), с соединением, представленным общей формулой (XIII) или его солью присоединения кислоты (кислотно-аддитивной солью) в присутствии основания.
Основание представляет собой предпочтительно пиридин или неорганическое основание, такое как ацетат натрия.
Растворитель, используемый в реакции, является предпочтительно спиртом, таким как метанол, пиридином или водой, хотя им может быть любой органический растворитель, инертный в условиях реакции.
Температура реакции может быть в пределах от приблизительно 0oC до температуры дефлегмации применяемого растворителя.
Способ поучения 6.
Соединение, представленное общей формулой (I), в которой Y есть группа, представленная формулой  может быть получено также следующим способом
может быть получено также следующим способом
где
R1, R2, R3, R4, R7, A и Z каждый имеет вышеуказанные значения.
Конкретнее, целевое соединение (XIV) может быть получено конденсацией производного тиоформамида, представленного общей формулой (XI), с соединением, представленным общей формулой (XIII), или его солью присоединения кислоты в присутствии кислоты Льюиса.
Кислотой Льюиса является предпочтительно тетрахлорид титана.
Растворитель, используемый в реакции, предпочтительно представляет собой галоидный органический растворитель, такой как дихлорметан, хотя им может быть любой органический растворитель, инертный в условиях реакции.
Температура реакции может быть в пределах от приблизительно 0oC до температуры дефлегмации применяемого растворителя.
Способ получения 7.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой  (где R15 имеет вышеуказанное значение), может быть получено следующим способом:
(где R15 имеет вышеуказанное значение), может быть получено следующим способом: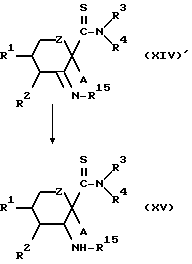
где
R1, R2, R3, R4, R15, A и Z каждый имеет вышеуказанные значения.
Конкретнее, целевое соединение (XV) может быть получено восстановлением производного тиоформамида (XIV)', полученного по способу 6 в обычных условиях.
Это восстановление может проводиться обычным способом, например способом с использованием металлогидридного комплекса или каталитического гидрирования.
Металлогидридный комплекс включает цианогидроборат натрия и боргидрид натрия.
Растворитель, используемый в этом случае, предпочтительно представляет собой эфир, такой как тетрагидрофуран, или спирт, такой как метанол. Предпочтительно, чтобы температура реакции была между приблизительно -20oC и приблизительно 50oC.
Каталитическое гидрирование может быть проведено путем использования обычного катализатора, такого как платина/углерод, оксид платины, никель Ренея или родий/глинозем.
Растворитель, используемый при каталитическом гидрировании, предпочтительно представляет собой спирт, такой как метанол, углеводород, такой как толуол, эфир, такой как тетрагидрофуран, N,N-диметилформамид или этилацетат. Предпочтительно, чтобы температура реакции была между 0oC и температурой дефлегмации применяемого растворителя.
Способ получения 8.
Соединение, представленной общей формулой (I), в которой Y - группа, представленная формулой 
(где R5 и R6 каждый имеет вышеуказанные значения), может быть получено следующим способом:
где
R1 , R2, R3, R4, R5, R6, A и Z каждый имеет вышеуказанные значения; R23 представляет низший алкил; R24 представляет фенил; и Hal представляет галоген.
Конкретнее, целевое соединение (XVI) может быть получено проведением реакции соединения, представленного общей формулой (XI), с соединением, представленным общей формулой (XVII) или (XVIII) в соответствии с реакцией Виттига в обычных условиях.
Предпочтительные примеры основания, применяемого в этой реакции, включают гидриды щелочных металлов, такие как гидрид натрия; литийорганические соединения, такие как н-бутиллитий; и алкоксиды щелочных металлов, такие как т-бутоксид калия.
Растворитель, применяемый в реакции, предпочтительно представляет собой эфир, такой как тетрагидрофуран, или полярный апротонный растворитель, такой как N, N-диметилформамид, хотя им может быть любой растворитель, инертный в условиях реакции.
Температура реакции может быть в пределах от приблизительно -78oC до температуры дефлегмации применяемого растворителя.
Когда по меньшей мере один из R3 и R4 - водород, иногда желательно, чтобы тиоформамидная группа соединения (XI) была защищена соединением, представленным общей формулой: R22-Q (где R22 и Q каждый имеет вышеуказанные значения) перед реакцией с соединением (XVII) или (XVIII).
Способ получения 9.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой 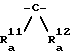 /где R
/где R (где R14 и R15 каждый имеет вышеуказанные значения) и -OR13 (где R13 имеет вышеуказанные значения) исключены/, может быть получено следующим способом:
(где R14 и R15 каждый имеет вышеуказанные значения) и -OR13 (где R13 имеет вышеуказанные значения) исключены/, может быть получено следующим способом: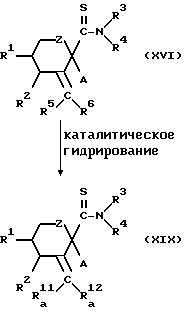
где
R1, R2, R3, R4, R5, R6, R
Конкретнее, целевое соединение (XIX) может быть получено каталитическим гидрированием соединения, представленного общей формулой (XVI), в обычных условиях.
Катализатор, применяемый в процессе гидрирования, включает палладий/углерод, оксид платины, никель Ренея и родий/глинозем.
Предпочтительные примеры растворителя, применяемого в реакции, включают спирты, такие как метанол, углеводороды, такие как толуол, эфиры, такие как тетрагидрофуран, N,N-диметилформамид и этилацетат.
Температура реакции может быть в пределах от приблизительно 0oC до температуры дефлегмации применяемого растворителя.
Способ получения 10.
Соединение, представленное общей формулой (I), в которой Y - группа, представленная формулой  (где n есть целое число от 0 до 2), может быть получено следующим способом:
(где n есть целое число от 0 до 2), может быть получено следующим способом: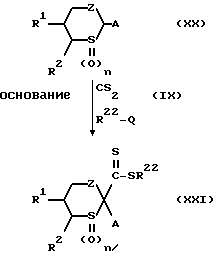
где
R1, R2, R22, A, Z, Q и n каждый имеет вышеуказанные значения.
Конкретнее, целевое соединение (XXI) может быть получено путем реакции соединения, представленного общей формулой (XX), сначала с дисульфидом углерода, а затем с соединением, представленным общей формулой (IX), в присутствии основания.
Уходящая группа, указанная в отношении Q, представляет собой предпочтительно галоген или метансульфонилокси.
Предпочтительные примеры основания включают алкоксиды щелочного металла, такие как т-бутоксид калия, и диалкиламид щелочного металла, такой как диизопропиламид лития.
Растворитель, применяемый в реакции, представляет собой предпочтительно эфир, такой как тетрагидрофуран, который может быть использован вместе с гексаметилфосфорамидом в качестве сорастворителя в некоторых случаях.
Предпочтительно, чтобы температура реакции была между приблизительно -78oC и температурой дефлегмации применяемого растворителя.

где
R1, R2, R3, R4, R22, A, Z и n каждый имеет вышеуказанные значения.
Конкретнее, целевое соединение (XXII) может быть получено путем реакции соединения (XXI), полученного на стадии 1, с соединением, представленным общей формулой (XII).
Растворитель, применяемый в реакции, предпочтительно представляет собой спирт, такой как этанол, эфир, такой как тетрагидрофуран, или углеводород, такой как толуол, хотя им может быть любой органический растворитель, инертный в условиях реакции.
Предпочтительно, чтобы температура реакции была между комнатной температурой и температурой дефлегмации применяемого растворителя.
Полезно, чтобы R22-SH, образованный в ходе реакции, удавливался бы в виде соли тяжелого металла путем добавления в реакционную систему, например, хлорида ртути.
Способ получения 11.
Соединение, представленное общей формулой (I), имеющее несимметричный атом углерода, может быть получено в виде оптически активного вещества следующим способом:
где
R1, R2, R3, R4, A, Y и Z каждый имеет вышеуказанные значения.
Когда производное тиоформамида, представленное общей формулой (I), имеет основные свойства, оптически активное вещество (I') может быть получено путем реакции производного с оптически активной кислотой, такой как дибензоилвинная кислота, с образованием диастереоизомерной кислотной смеси, подвержения смеси фракционной кристаллизации с использованием подходящего растворителя с получением чистой диастереоизомерной соли, и нейтрализации этой диастереизомерной соли.
Далее, оптически активное вещество (I') может быть получено также путем реакции производного тиоформамида, представленного общей формулой (I), с оптически активным реагентом оптического разделения (таким как оптически активный гидразин) с образованием диастереоизомерной смеси, разделения диастереоизомерной смеси с помощью колоночной хроматографии или фракционной кристаллизации с получением чистого диастереизомера от оптически активного реагента.
Далее, оптически активное вещество (I') может быть получено также непосредственным разделением производного тиоформамида, представленного общей формулой (I), с помощью хроматографии с использованием хиральной колонки.
Далее, оптически активное вещество (I') можно также получать, подвергая производное тиоформамида, представленное общей формулой (I), кинетическому разделению с использованием асимметричного восстановителя и окислению полученного диастереизомерного продукта обычным способом.
Эффект изобретения.
Ниже описываются примеры фармакологических экспериментов с целью иллюстрации действия изобретения.
(1) Вазогипотоническая активность на образце крысиной аорты.
Соединения согласно настоящему изобретению исследовались на вазогипотоническую активность с помощью аорты, удаленной из крысы.
Экспериментальный метод.
Грудную аорту быстро удаляли из самца крысы SD, имеющего вес 250 - 400 г, и из аорты готовили спиральный образец. Этот спиральный образец вертикально суспендировали в Органванне, заполненной раствором Кребса-Хенслейта при 30oC, через которую пропускали газовую смесь, состоящую из 95% кислорода и из 5% двуокиси углерода. Изменение в напряжении образца измеряли изотонически под нагрузкой 1 г.
После того как натяжение образца стабилизировалось, в ванну добавляли 20 мМ KCl. После того как возникшее натяжение стабилизировалось, в Органванну кумулятивно добавляли исследуемое соединение для определения релаксации.
Величину релаксации, т.е. активность по замедлению возникновения натяжения, определяли, принимая натяжение, возникающее при добавлении KCl, до 100% и подсчитывая значение IC50. Значения IC50, определенные таким образом, приведены в таблице.
(2) Активность в отношении потенциальной (скрытой) продолжительности действия на правой вентрикулярной папиллярной мышце морской свинки.
Соединения согласно изобретению исследовались на эффект активации калиевого канала с использованием правой вентрикулярной папиллярной мышцы морской свинки.
Экспериментальный метод.
Потенциальная длительность действия при 90%-ной реполяризации (ниже обозначаемая сокращенно как APD90) отражает выходящий ток калия. Соединение, имеющее эффект активации калийного канала, может активировать выходящий калийный канал, сокращая APD90 (см. Proc. Natl. Acad. Sci., USA, 85, 8360 - 8364 (1988)).
Использовали самцов морских свинок Хартли весом от 300 до 500 г. Морскую свинку умерщвляли кровопусканием, папиллярную мышцу полностью удаляли из ее правого желудочка и фиксировали в Органванне. Поверхность папиллярной мышцы промывали раствором Tyrode при 36 ± 0,5oC, через который пропускали смесь кислорода/двуокиси углерода. Полученный в результате образец стимулировали приложением к нему прямоугольного волнового импульса 1 Гц для записи потенциала действия методом стеклянного микроэлектрода.
После того как потенциал действия стабилизировался, образец промывали растворами Tyrode, содержащими каждое из исследуемых соединений, в течение 30 минут для определения кривой доза-отклик.
Отношение (процент) APD90, обнаруживаемое при каждой концентрации исследуемого соединения в растворе Tyrode к начальному APD90, обнаруживаемому до введения соединения, подсчитывалось для определения концентрации соединения (-log M), при котором APD90 сокращалось на 50% от первоначального значения. Определенные таким образом концентрации соединения представлены в таблице как значения IC50.
(3) Гипотензивная активность и активность по повышению коронарного кровотока у анестезированной собаки с вскрытой грудной клеткой.
Соединения согласно настоящему изобретению исследовалась на гипотензивную активность и активность по повышению коронарного кровотока путем использования анестезированной собаки с вскрытой грудной клеткой.
Экспериментальный метод.
Использовали взрослую гибридную собаку. У собаки вскрывали грудную клетку под ингаляционным наркозом с применением энфлурана/веселящего газа. Устанавливали датчик электромагнитного расходомера на огибающей ветви (ramus circumflexes) левой коронарной артерии для определения коронарного кровотока. Артериальное давление определялось путем помещения наконечника датчика давления с катетером в дугу аорты (arcus aortae).
Каждое испытуемое соединение вводили внутривенно в дозе 3 мкг/кг через катетер, вставленный в бедренную вену.
Гипотензивная активность и активность по повышению коронарного кровотока для каждого соединения приведены в таблице как изменение кровяного давления и коронарного кровотока по сравнению с таковыми перед введением соединения.
(4) Гипотензивное действие на спонтанно гипертензивную крысу в состоянии пробуждения.
Соединения согласно настоящему изобретению исследовались на гипотензивную активность путем орального введения их в спонтанно гипертензивные крысы (SHR).
Экспериментальный метод.
Систолическое давление спонтанно самцов гипертензивных крыс (вес: 260 - 360 г), которых не кормили в течение 12 часов, определяли бескровно методом хвостовой манжеты. Каждое лекарство суспендировали в 0,5%-ном водном растворе метилцеллюлозы и вводили орально в дозе 1 мг/кг. Контрольным крысам давали только растворитель.
Кровяное давление у крыс через 2 часа после введения лекарства показано в таблице, причем кровяное давление до введения принято за 100.
Экспериментальные результаты.
Результаты вышеупомянутых экспериментальных примеров от (1) до (13) приведены в таблице. Соединения 1 - 13 представляют следующие:
соединение 1
(-)-1-(имидазо/1,2-a/пиридин-6-ил)-N-метил-2-оксоциклогексанкарботиоамид
соединение 2
(-)-N-этил-1-(имидазо/1,2-a/пиридин-6-ил)-2-оксоциклогексанкарботиоамид
соединение 3
(-)-N-метил-1-(2-метилимидазо/1,2-a/пиридин-6-ил)-2- оксоциклогексанкарботиоамид
соединение 4
(-)-2-бензоилокси-1-(имидазо/1,2-a/пиридин-6-ил)-2-N- метилциклогексанкарботиоамид
соединение 5
2-бензилокси-1-(имидазо/1,2-a/пиридин-6-ил)-N-метил- циклогексанкарботиоамид
соединение 6
анти-2-бензилоксиимино-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид
соединение 7
2-бензиламино-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид
соединение 8
2-(имидазо/1,2-a/пиридин-6-ил)-N-метилтетрагидротиопиран-2- карботиоамид-1-оксид
соединение 9
2-бензоилокси-N-метил-1-(4-нитроимидазол-1-ил)циклогексанкарботиоамид
соединение 10
2-бензилокси-N-метил-1-(4-нитроимидазол-1-ил)циклогексанкарботиоамид
соединение 11
анти-2-бензилоксиимино-N-метил-1-(4-нитроимидазол-1- ил)циклогексанкарботиоамид
соединение 12
(-)-1-(2-трифторметилимидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамид
соединение 13
(-)-2-(2-метилимидазо/1,2-a/пиридин-6-ил)-N-метилтетрагидротиопиран-2- карботиоамид-1-оксид
Как видно из результатов фармакологических экспериментов, соединения согласно настоящему изобретению могут активировать калиевый канал гладкой мышцы, расслабляя тем самым кровяную гладкую мышцу и проявляя таким образом активность в повышении коронарного кровотока и гипотензивную активность. В соответствии с этим соединения согласно настоящему изобретению эффективны в качестве средства открытия калиевого канала и благодаря этой активности они могут быть использованы в качестве лекарства.
Так, соединения согласно настоящему изобретению эффективны в профилактике и лечении ишемических болезней сердца, таких как ангина пекторис и гипертензия.
Далее, соединения согласно настоящему изобретению менее токсичны и имеют большой запас безопасности, что делает их ценными также и в этом смысле.
Соединение согласно настоящему изобретению вводятся как терапевтическое и профилактическое средство при вышеупомянутых болезнях в форме таблетки, порошка, гранулы, капсулы, сиропа или ингаляции. Хотя доза соединения колеблется в зависимости от серьезности симптома, возраста и вида болезни, ежедневная доза для взрослого составляет от приблизительно 0,1 до 1000 мг, предпочтительно от 1 до 500 мг, и ее можно вводить в один или несколько приемов в день.
Когда соединение согласно настоящему изобретению вводится в виде инъекции, доза обычно составляет от 1 до 3000 мкг/кг, предпочтительно приблизительно от 3 до 1000 мкг/кг.
Фармацевтические препараты в соответствии с настоящим изобретением готовятся с использованием обычных носителей обычным способом.
Конкретнее, твердый препарат для орального введения в соответствии с настоящим изобретением готовится путем добавления к активному ингредиенту наполнителя и, если необходимо, связующего, дизинтегратора, смазывающего вещества, красящих и/или модифицирующих веществ и формования полученной смеси в таблетки, покрытые таблетки, гранулы, порошок или капсулы обычным способом.
Примеры наполнителей включают лактозу, зерновой крахмал, сахарозу, глюкозу, сорбит, кристаллическую целлюлозу и двуокись кремния; примеры связующего включают поливиниловый спирт, поливиниловый эфир, этилцеллюлозу, метилцеллюлозу, акацию, трагакант, желатину, шеллак, гидроксипропилцеллюлозу, гидроксиметилцеллюлозу, цитрат кальция, декстрин и пектин; примеры смазывающего вещества включают стеарат магния, тальк, полиэтиленгликоль, кремнезем и гидрогенизированное растительное масло; примеры красящего вещества включают разрешенные фармацевтические добавки, а примеры модифицирующих веществ включают порошок какао, мяту, ароматический порошок, масло мяты, борнеол и измельченную в порошок кору коричневого дерева. Конечно, таблетки, гранулы могут быть подходящим образом покрыты сахаром, желатиной или тому подобным веществом, если это необходимо.
Инъекционный раствор в соответствии с настоящим изобретением готовится путем добавления при необходимости к активному ингредиенту регулятора pH, буфера, стабилизатора и/или солюбилизирующего агента и формулирования смеси в инъекцию для подкожного, внутримышечного или внутривенного введения обычным способом.
Пример.
Ниже будут описаны примеры в соответствии с настоящим изобретением, хотя нет необходимости говорить, что настоящее изобретение не ограничивается ими.
Далее будут даны препаративные примеры, описывающие приготовление препаратов исходных материалов, используемых в получении целевых соединений согласно настоящему изобретению.
В нижеследующих препаративных примерах и примерах Me представляет метил, а Et - этил.
Препаративный пример 1.
2-(Имидазо/1,2-a/пиридин-6-ил)циклогексанон
49,35 г магния добавляли к 500 мл тетрагидрофурана, и к полученной смеси прикапывали 11,4 мл бромэтана при комнатной температуре при перемешивании в атмосфере азота для приготовления реактива Гриньяра. Полученную реакционную смесь охлаждали водой и в эту реакционную смесь в течение приблизительно одного часа по каплям прибавляли раствор 100 г 6-бромимидазо/1,2-a/пиридина и 102 мл бромэтана в 1 л тетрагидрофурана. После окончания прикапывания полученную смесь нагревали с обратным холодильником в течение 30 минут.
Полученную реакционную смесь охлаждали льдом с последующим добавлением по каплям раствора 263 г 2-метоксициклогексанона в 200 мл тетрагидрофурана. После окончания прикапывания полученную смесь перемешивали при комнатной температуре в течение 3 часов.
Полученную реакционную смесь снова охлаждали льдом и закаливали добавлением насыщенного водного раствора хлорида аммония. Полученную смесь подкисляли 6 н. хлористоводорнодной кислотой. Водную фазу промывали этилацетатом, подщелачивали концентрированным водным аммиаком и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя, получая 122,5 г коричневого масла.
К этому маслу порциями прибавляли 300 мл концентрированной серной кислоты. Полученную смесь встряхивали при комнатной температуре достаточно энергично, получая коричневый раствор, что занимало приблизительно 2 часа. Полученный коричневый раствор выливали на лед и полученную смесь подщелачивали концентрированным водным аммиаком и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученный остаток очищали хроматографически в колонках с силикагелем /растворитель: дихлорметан/метанол (50 : 1 - 30 : 1)/ и полученное твердое тело тщательно промывали эфиром, получая 63,46 г главного соединения в виде порошка цвета светлой охры.
Далее, промывные воды концентрировали и очищали хроматографически в колонке с силикагелем /растворитель: дихлорметан/метанол (30 : 1)/. Полученное твердое вещество промывали эфиром, получая 6,83 г главного соединения в виде порошка цвета светлой охры. Таким образом, всего было получено 70,29 г заглавного соединения (выход: 65%).
Т.пл. (oC): 120 - 121,5.
1H-ЯМР (400 МГц, CDCl3) δ : 1,76 - 1,12 (4H, m), 2,16 - 2,28 (1H, m), 2,29 - 2,38 (1H, m), 2,44 - 2,62 (2H, m), 3,59 (1H, dd, J = 5,5, 12,4 Гц), 6,95 (1H, dd, J = 1,6, 9,3 Гц), 7,52 (1H, dd, J = 0,5, 1,3 Гц), 7,57 (1H, d, J = 9,3 Гц), 7,60 (1H, d, J = 1,3 Гц), 7,94 (1H, dd, J = 0,5, 1,3 Гц).
Препаративный пример 2.
2-(2-Метилимидазо/1,2-a/пиридин-6-ил)циклогексанон
7,70 мл (0,103 моль) этилбромида прикапывали к суспензии 32,08 г (1,32 моль) металлической магниевой стружки в 350 мл тетрагидрофурана в атмосфере азота. Как только было замечено образование этилмагнийбромида, к полученной смеси по каплям добавляли раствор 69,52 г (0,329 моль) 6-бром-2-метилимидазо-/1,2-a/пиридина и 66,0 мл (0,880 моль) этилбромида в 700 мл тетрагидрофурана в течение часа с такой скоростью, чтобы продолжалось спонтанное мягкое дефлегмирование. После окончания прикапывания полученную смесь нагревали с обратным холодильником в течение 30 минут и охлаждали льдом. К реакционной смеси добавляли раствор 171,1 г (1,34 моль) 2-метоксициклогексанона в 150 мл тетрагидрофурана в течение 30 минут с такой скоростью, чтобы температура основной массы смеси не превышала 30oC. Полученную смесь встряхивали при комнатной температуре в течение 3 часов, охлаждали льдом и подкисляли добавлением насыщенного водного раствора хлорида аммония и 6 н. хлористоводородной кислоты в указанном порядке. Полученную кислую водную фазу промывали этилацетатом, подщелачивали концентрированным водным аммиаком и экстрагировали хлороформом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме.
К полученному сырому 2-метокси-2-(2-метилимидазо-/1,2-a/-пиридин- 6-ил)циклогексанону (73,7 г) добавляли порциями в течение 30 минут 200 мл концентрированной серной кислоты, охлаждая получающуюся смесь подходящим образом на водяной бане. Полученную красновато-пурпурную жидкость перемешивали при комнатной температуре в течение 16 часов.
Полученную реакционную смесь выливали на лед, охлаждая льдом. Полученную смесь подщелачивали концентрированным водным аммиаком и экстрагировали дихлорметаном. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток (62,3 г) очищали хроматографически на колонках с силикагелем /растворитель: дихлорметан/метанол (50 : 1)/, получая 51 г желтого кристалла. Кристаллы перекристаллизовывали из дихлорметана/этилацетата, получая 18,85 г главного соединения в виде бесцветных кристаллов. Далее, маточный раствор (26,7 г) порошковали эфиром, получая 16,89 г указанного в заголовке соединения в виде светло-коричневого порошка. Таким образом всего получено 35,74 г указанного в заголовке соединения (выход: 48%).
Т. пл. (oC): 147 - 148.
1H-ЯМР (400 МГц, CDCl3) δ : 1,76 - 2,08 (4H, м), 2,16 - 2,24 (1H, м), 2,31 (1H, м), 2,44 (3H, д, J = 0,9 Гц), 2,46 - 2,60 (2H, м), 3,57 (1H, дд, J = 12,4, 5,4 Гц), 6,90 (1H, дд, J = 9,3, 1,6 Гц), 7,27 (1H, шир. с.), 7,45 (1H, д, J = 9,2 Гц), 7,84 (1H, дд, J = 1,6, 0,8 Гц).
Препаративный пример 3.
2-(Имидазо/1,2-a/пиридин-6-ил)-2-((4-метоксибензилтио)(метилимино) метил)циклогексанол
2,50 г 2-Гидрокси-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамида (M-форма) суспендировали в 45 мл N,N-диметилформамида с последующим добавлением 1,32 г безводного карбоната калия и 1,29 мл 4-метоксибензилхлорида. Полученную смесь перемешивали при комнатной температуре в течение 10 часов.
Полученную реакционную смесь концентрировали, затем добавляли воду. Полученную смесь фильтровали, получая нерастворимый материал. Этот материал промывали 50%-ным водным этанолом и эфиром последовательно, получая 3,19 г указанного в заголовке соединения в виде слегка желтого порошка (выход: 90%).
Т. пл. (oC): 203 - 206 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ : 1,36 - 1,70 (5H, м), 1,83 - 1,90 (1H, м), 2,59 - 2,68 (1H, м), 3,33 (1H, д, J = 12,3 Гц), 3,55 (3H, м), 3,72 (3H, с), 3,75 (1H, д, J = 12,3 Гц), 4,37 (1H, ддд, J = 2,7, 2,9, 10,1 Гц), 6,53 - 6,57 (2H, м), 6,68 - 6,73 (2H, м), 7,49 - 7,56 (3H, м), 7,61 (1H, м), 8,74 (1H, шир. с.).
Препаративный пример 4.
2-Бенхзилкокси-1-(имидазо/1,2-a/пиридин-6-ил)-1-((4-метоксибензилтио) (метилимино)метил)циклогексан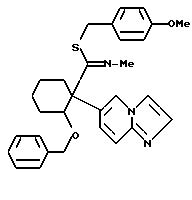
1,04 г (2-Имидазо/1,2-a/пиридин-6-ил)-2-((4-метоксибензилтио) (метилимино))метил)циклогексанола суспендировали в 25 мл тетрагидрофурана. Полученную суспензию охлаждали до -60oC с последующим добавлением сразу 300 мг т-бутоксида калия. Полученную смесь перемешивали при -60oC в течение 2,5 часов с последующим добавлением 0,32 мл бензилбромида. Температуру полученной смеси постепенно поднимали до 0oC в течение приблизительно одного часа, и полученную смесь перемешивали при 0oC в течение 2 часов, затем добавляли воду. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографически на колонках с силикагелем /растворитель: дихлорметан/метанол (50:1)/, получая 500 мг указанного в заголовке соединения в виде бледно-желтого масла (выход: 39%).
1H-ЯМР (400 МГц, CDCl3) δ : 1,14 - 1,28 (1H, м), 1,45 - 1,76 (3H, м), 1,88 - 2,02 (2H, м), 2,19 (1H, дт, J = 3,5, 12,7 Гц), 3,35 (1H, д, J = 12,3 Гц), 3,60 (3H, с), 3,70 (3H, с), 3,72 (1H, д, J = 12,3 Гц), 4,07 (1H, д, J = 11,7 Гц), 4,35 (1H, шир. с), 4,41 (1H, д, J = 11,7 Гц), 6,51 - 6,56 (2H, м), 6,64 - 6,68 (2H, м), 6,83 (2H, дд, J = 1,5, 8,4 Гц), 7,04 - 7,17 (4H, м), 7,46 (2H, т, J = 4,4 Гц), 7,61 (1H, д, J = 1,3 Гц), 7,98 (1H, м).
Препаративный пример 5.
6-Имидазо/1,2-a/пиридинкарбальдегид
1,97 г 6-бромидазо/1,2-a/пиридина суспендировали в 40 мл безводного эфира. Полученную суспензию охлаждали до -70oC и перемешивали в атмосфере азота с последующим добавлением 6,9 мл 1,6 М раствора н-бутиллития в гексане. Полученную смесь перемешивали в течение одного часа, затем добавляли сразу 1,5 мл N,N-диметилформамида. Через час температуру смеси повышали до комнатной температуры, и полученную смесь перемешивали в течение 30 минут.
Добавляли ледяную воду к смеси, и полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли в вакууме для удаления растворителя. Добавляли эфир к полученному остатку, и полученную смесь фильтровали, получая нерастворимый материал. Таким образом, было получено 0,43 г указанного в заголовке соединения в виде светло-коричневого порошка. Далее, фильтрат концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (50 : 1)/, получая 0,47 г указанного в заготовке соединения в виде бледно-желтого твердого вещества. Таким образом всего было получено 0,90 г целевого соединения (выход: 62%).
Т.пл. (oC): 151 - 153,5.
1H-ЯМР (400 МГц, CDCl3) δ : 7,66 (1H, дд, J = 1,6, 9,3 Гц), 7,71 (1H, д, J = 9,3 Гц), 7,74 (1H, дд, J = 0,9, 1,3 Гц), 7,77 (1H, д, J = 1,3 Гц), 8,70 (1H, ддд, J = 0,5, 0,9, 1,6 Гц), 9,96 (1H, д, J = 0,5 Гц).
Препаративный пример 6.
(Имидазо/1,2-a/пиридин-6-ил)метанол
0,90 г 6-имидазо/1,2-a/пиридинкарбальдегида, полученного в препаративном примере 5, растворяли в 20 мл метанола. Полученный раствор перемешивали при охлаждении льдом, затем к нему добавляли 90 мг боргидрида натрия. Полученную смесь перемешивали в течение 50 минут с последующим добавлением воды. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученный остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 30 : 1 до 10 : 1)/, получая 0,52 г указанного в заготовке соединения в виде слабо оранжево-желтого твердого тела (выход: 57%).
Т.пл. (oC): 106 - 107.
1H-ЯМР (400 МГц, CDCl3) δ : 4,69 (2H, д, J = 0,9 Гц), 7,12 (1H, дд, J = 1,6, 9,3 Гц), 7,50 (1H, д, J = 9,3 Гц), 7,52 (1H, дд, J = 0,5, 1,3 Гц), 7,58 (1H, д, J = 1,3 Гц), 8,10 (1H, ддд, J = 0,5 0,9, 1,6 Гц).
Препаративный пример 7.
6-((4-Хлорбутил)тиометил)имидазо/1,2-a/пиридин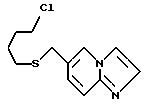
11,23 г (имидазо/1,2-a/пиридин-6-ил)метанола, полученного в препаративном примере 6, растворяли в 170 мл хлороформа, а затем добавляли по каплям 11,1 мл тионилхлорида при комнатной температуре при перемешивании. Спустя час, отгоняли растворитель. Таким образом получали сырой 6-хлорметилимидазо/1,2-a/пиридин-гидрохлорид в виде светло-коричневого порошка.
Порошок добавляли к смеси, содержащей 40 мл метанола и 40 мл этанола, с последующим добавлением 6,92 г тиомочевины. Полученную смесь нагревали с обратным холодильником в течение 4 часов, охлаждали, давая ей постоять, и перегоняли для удаления растворителя. Таким образом, получали сырой S-((имидазо/1,2-a/пиридин- 6-ил)метилизотиомочевину-дигидрохлорид в виде светло-коричневого порошка.
Данный порошок растворяли в 40 мл воды с последующим добавлением 15,2 мл 10 н. водного раствора гидроксида натрия. Полученную смесь нагревали с обратным холодильником в течение 2 часов и охлаждали льдом. К смеси последовательно добавляли 9,1 мл 10 н. водного раствора гидроксида натрия, 11,4 мл 1-бром-4-хлорбутана и 40 мл этанола. Полученную смесь перемешивали при комнатной температуре в течение 2 часов.
К смеси добавляли воду, а затем экстрагировали этилацетатом. Органическую фазу экстрагировали разбавленной хлористоводородной кислотой. Водную фазу подщелачивали карбонатом калия и экстрагировали снова этилацетатом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученный остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (50 : 1)/, получая 11,79 г указанного в заголовке соединения в виде слегка коричневого масла (общий выход: 61%).
1H-ЯМР (400 МГц, CDCl3) δ : 1,70 - 1,79 (2H, м), 1,82 - 1,90 (2H, м), 2,46 (2H, т, J = 7,1 Гц), 3,53 (2H, т, J = 6,2 Гц), 3,69 (2H, д, J = 0,4 Гц), 7,20 (1H, дд., J = 1,6, 9,3 Гц), 7,54 (1H, дд., J = 0,5, 1,1 Гц), 7,59 (1H, д. , J = 9,3 Гц), 7,62 (1H, д., J = 1,1 Гц), 8,05 (1H, ддд., J = 0,4, 0,5, 1,6 Гц).
Препаративный пример 8.
6-((-Хлорбутил)сульфинилметил)имидазо/1,2-a/пиридин
11,79 г 6-((4-хлорбутил)тиометил)имидазо/1,2-a/пиридина, полученного в препаративном примере 7, растворяли в 160 мл дихлорметана. Полученный раствор охлаждали льдом с последующим добавлением 7,99 г м-хлорпербензойной кислоты в течение приблизительно 40 минут. Полученную смесь перемешивали. Через 30 минут к ней добавляли еще 0,4 г м-хлорпербензойной кислоты. Полученную смесь дополнительно перемешивали в течение одного часа, а затем к ней добавляли водный раствор тиосульфата натрия. Полученную смесь энергично перемешивали при комнатной температуре в течение 30 минут и экстрагировали хлороформом. Органическую фазу промывали водным раствором карбоната калия, сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученный остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 30 : 1 до 10 : 1)/, получая 9,51 г указанного в заголовке соединения в виде темно-кремового масла (выход: 76%).
1H-ЯМР (400 МГц, CDCl3) δ : 1,85 - 2,04 (4H, м), 2,64 (2H, дд., J = 6,8, 7,7 Гц), 3,58 (2H т, J = 6,0 Гц), 3,87 (1H, д., J = 13,4 Гц), 3,98 (1H, д, J = 13,4 Гц), 7,07 (1H, дд, J = 1,6, 9,2 Гц), 7,60 (1H, дд, J = 0,7, 1,2 Гц), 7,64 (1H, д, J = 9,2 Гц), 7,66 (1H, д, J = 1,2 Гц), 8,19 (1H, дд, J = 0,7, 1,6 Гц).
Препаративный пример 9.
2-(Имидазо/1,2-a/пиридин-6-ил)тетрагидротиопиран 1-оксид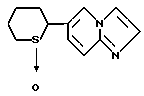
8,68 г т-бутоксида калия добавляли к 80 мл смеси /4:1 (об./об.)/ тетрагидрофурана и гексаметилфосфорного триамида с последующим охлаждением льдом. В полученную смесь по каплям добавляли раствор 9,51 г 6-((4-хлорбутил)-сульфинилметил)имидазо/1,2-a/пиридина, полученного в препаративном примере 8, в 30 мл смеси /4:1 (об./об.)/ тетрагидрофурана и гексаметилфосфорного триамида. Полученную смесь перемешивали в течение одного часа, после чего к ней добавляли ледяную воду. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученное масло отверждали, давая ему постоять, с последующим добавлением эфира. Полученную смесь фильтровали, получая 5,84 г указанного в заголовке соединения в виде темно-кремового порошка (выход: 71%). ЯМР-спектроскопический анализ этого порошка показал, что порошок представляет собой смесь, содержащую два диастереометра в соотношении приблизительно 2:1.
1H-ЯМР (400 МГц, CDCl3) δ : 1,53 - 2,45 (5H, м), 2,56 - 2,81 (2H, м), 3,16 - 3,61 (2H, м), 7,09 - 7,17 (полн. 1H, д, J = 1,8, 9,3 Гц), 7,56 - 7,67 (3H, м), 8,17 - 8,19 (1H, м).
Препаративный пример 10.
2-(4-Нитроимидазол-1-ил)циклогексанон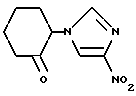
84,0 мл (0,736 моль) 2-хлорциклогексанона и 92,54 г (0,670 моль) карбоната калия добавляли к суспензии 75,16 г (0.665 моль) 4-нитроимидазола в 1 л ацетонитрила. Полученную смесь нагревали с обратным холодильником в течение 18 часов и охлаждали, давая ей постоять, после чего к ней добавляли воду. Полученную смесь экстрагировали этилацетатом. Полученную органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток порошковали этилацетатом, получая 86,46 г указанного в заголовке соединения в виде светло-коричневого порошка (выход: 62%).
Т. пл. (oC): 122 - 123.
1H-ЯМР (400 МГц, CDCl3) δ/ : 1,81 (1H, кв. т., J = 13,4, 3,8 Гц), 1,94 (1H, м), 2,12 (1H, кв.д. J = 12,8, 3,4 Гц), 2,18 (1H, м), 2,27 (1H, м), 2,53 (1H, тдд. , J = 13,8, 6,2, 0,7 Гц), 2,60 (1H, м), 2,70 (1H, д.квин., J = 13,9, 2,2 Гц), 4,90 (1H, дд., J = 13,0, 5,7 Гц), 7,42 (1H, д, J = 1,6 Гц), 7,77 (1H, д. J = 1,6 Гц).
Препаративный пример 11.
2-(4-Нитроимидазол-1-ил)циклогексанол
40,0 мл (395 ммоль) циклогексеноксида и 86,37 г (625 ммоль) карбоната калия добавляли к суспензии 50,08 г (443 ммоль) 4-нитроимидазола в 800 мл N, N-диметилформамида. Полученную смесь перемешивали при нагревании при 100oC в течение 3 дней и 20 часов, получая желтовато-коричневую суспензию. Эту суспензию охлаждали, давая ей постоять, и фильтровали для удаления нерастворимых веществ. Фильтрат концентрировали в вакууме. Добавляли воду к полученному остатку для образования кристаллического осадка. Кристаллический осадок удаляли фильтрованием и промывали водой. Полученный сырой кристалл растворяли в хлороформе, и полученный раствор промывали насыщенным водным раствором поваренной соли. Органическую фазу сушили над безводным сульфатом магния и концентрировали в вакууме. В ходе концентрирования начинали осаждаться кристаллы. Кристаллы удаляли фильтрованием, получая 53,26 г указанного в заголовке соединения в виде бесцветных кристаллов. Далее, фильтрат перекристаллизовывали из дихлорметан/изопропилового эфира, получая 5,14 г целевого соединения в виде светло-коричневых кристаллов. Таким образом, всего получено 58,30 г целевого соединения (выход: 70%).
Т. пл. (oC): 150 - 151.
1H-ЯМР (400 МГц, CDCl3) δ : 1,36 - 1,54 (3H, м), 1,76 (1H, м), 1,84 - 1,98 (2H, м), 2,12 - 2,26 (2H, м), 3,18 (1H, д, J = 4,2 Гц), исчезал при добавлении D2O, 3,71 (1H, м, когда добавляли D2O, тд. J = 9,9, 4,5 Гц), 3,79 (1H, ддд, J = 12,3, 9,5, 3,8 Гц), 4,47 (1H, д, J = 1,6 Гц), 7,79 (1H, д, J = 1,6 Гц).
Препаративный пример 12.
2-(4-Нитроимидазол-1-ил)циклогексанон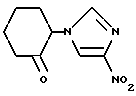
Раствор 19,0 мл (218 ммоль) оксалилхлорида в 250 мл дихлорметана охлаждали в бане из сухого льда и метанола. В вышеуказанный раствор в течение 10 минут по каплям добавляли раствор 27,0 мл (380 ммоль) диметилсульфоксида в 90 мл дихлорметана с такой скоростью, чтобы температура основной массы раствора поддерживалась в пределах от -60 до -50oC. Полученную смесь далее перемешивали в течение 4 минут, получая бесцветный раствор. В бесцветный раствор по каплям добавляли раствор 31,92 г (151 ммоль) 2-(4-нитроимидазол-1-ил)циклогексанола, полученного в препаративном примере 11, в смеси, содержащей 240 мл дихлорметана и 30 мл диметилсульфоксида, в течение 7 минут с такой скоростью, чтобы температура основной массы не превышала -50oC. Полученную смесь далее перемешивали в течение 15 минут. В полученную смесь в течение 7 минут прикапывали 80,0 мл (570 ммоль) триэтиламина с такой скоростью, чтобы температура основной массы не превышала -50oC. Полученную смесь далее перемешивали в течение 7 минут и после этого убирали баню из сухого льда и метанола. Через 30 минут температура основной массы достигала +10oC. К полученной в результате смеси добавляли насыщенный водный раствор кислого карбоната натрия, и полученную смесь оставляли стоять для расслоения жидкость-жидкость. Органическую фазу концентрировали в вакууме, полученный остаток растворяли в этилацетате. Далее, водную фазу экстрагировали этилацетатом, и этилацетатную фазу объединяли с этилацетатным раствором, полученным ранее. Полученную смесь промывали насыщенным водным раствором поваренной соли. Органическую фазу сушили над безводным сульфатом магния и концентрировали в вакууме, получая 33,9 г сырых кристаллов. Сырые кристаллы перекристаллизовывали из дихлорметана/этилацетата, получая 26,53 г целевого соединения в виде бесцветного кристалла. Далее, маточный раствор перекристаллизовывали из дихлорметана/диизопропилового эфира, получая 2,52 г целевого соединения в виде светлых желтовато-коричневых кристаллов. Таким образом, всего получали 29,05 г целевого соединения (выход: 92%).
Препаративный пример 13.
2-((4-Метоксибензилтио)(метилимино)метил)-2-(4-нитроимидазол- 1-ил)циклогексанол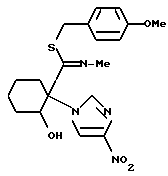
21,7 мл (160 ммоль) 4-метоксибензилхлорида и 32,94 г (238 ммоль) карбоната калия добавляли к раствору 45,23 г (159 ммоль) 2-гидрокси-N-метил-1-(4-нитроимидазол-1-ил)циклогексанкарботиоамида (L-форма), полученного в примере 17, в 300 мл N,N-диметилформамида. Полученную смесь перемешивали при комнатной температуре в течение 17 часов, получая желтовато-коричневую суспензию. К этой суспензии добавляли воду и поваренную соль, и полученную смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток (65,5 г) перекристаллизовывали из дихлорметана/этилацетата, получая 42,78 г указанного в заголовке соединения в виде светлых желтовато-коричневых кристаллов (выход: 66%).
Т. пл. (oC): 135 - 136.
1H-ЯМР (400 Мгц, CDCl3) δ : 1,28 - 1,48 (3H, м), 1,56 - 1,76 (3H, м), 1,90 (1H, м), 2,63 (1H, м), 3,44 (1H, д, J = 12,5 Гц), 3,58 (3H, с), 3,74 (1H, д, J = 12,5 Гц), 3,77 (3H, с), 4,38 (1H, дт, J = 11,0, 3,1 Гц при добавлении D2O, дд, J = 11,3, 3,6 Гц), 4,78 (1H, шир.с. исчезала при добавлении D2O), 6,74 (2H, д, J = 8,6 Гц), 6,85 (2H, д, J = 8,6 Гц), 7,99 (1H, д, J = 1,6 Гц), 8,27 (1H, д, D = 1,6 Гц).
Препаративный пример 14.
2-Бензилокси-1-((4-метоксибензилтио)(метилимино)метил)- 1-(4-нитроимидазол-1-ил)циклогексан
Раствор 42,76 г (106 ммоль) 2-((4-метоксибензилтио)(метилмино)метил- 2-(4-нитроимидазол-1-ил)циклогексанола, полученного в препаративном примере 13, в 300 мл тетрагидрофурана по каплям добавляли к суспензии 5,13 г (60%-ная дисперсия в минеральном масле, 128 ммоль) гидрида натрия в 60 мл тетрагидрофурана в атмосфере азота при комнатной температуре в течение 20 минут. Полученную смесь далее перемешивали при 50oC в течение 10 минут с последующим добавлением 13,2 мл (111 ммоль) бензилбромида при 50oC. Полученную смесь перемешивали при 50oC в течение одного часа и затем в условиях дефлегмации при нагревании в течение 20 часов. Полученную коричневую суспензию оставляли охлаждаться и наливали в нее ледяную воду. Полученную смесь экстрагировали этилацетатом. Полученную органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток (55,8 г) очищали хроматографией на колонке с силикагелем /растворитель: бензол/ацетон (от 25 : 1 до 5 : 1)/, получая 6,15 г указанного в заголовке соединения в виде коричневого масла (выход: 12%).
1H-ЯМР (400 МГц, CDCl3) δ : 1,28 - 1,49 (2H, м), 1,49 - 1,60 (1H, м), 1,60 - 1,78 (2H, м), 1,91 (1H, м), 2,19 (1H, ддд, J = 12,8, 8,8, 2,9 Гц), 2,39 (1H, м), 3,58 (3H, с), 3,59 (1H, д, J = 19,4 Гц), 3,76 (1H, д, J = 19,4 Гц), 3,77 (3H, с), 4,24 (1H, д, J = 11,4 Гц), 4,29 (1H, дд, J = 6,9, 2,5 Гц), 4,53 (1H, д, J = 11,4 Гц), 6,75 (2H, д, J = 8,8 Гц), 6,91 (2H, д, J = 8,8 Гц), 7,08 (2H, дд, J = 7,5, 1,8 Гц), 7,22 - 7,32 (3H, м), 7,61 (1H, д, J = 1,1 Гц), 7,69 (1H, д, J = 1,1 Гц).
Препаративный пример 15.
2-(2-(Трифторметилимидазо/1,2-a/-пиридин-6-ил)-циклогексанон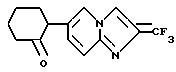
6,9 г магния добавляли к 120 мл тетрагидрофурана, и полученную смесь перемешивали в атмосфере азота. К образовавшейся смеси добавляли 1,6 мл бромэтана по каплям при комнатной температуре с получением реактива Гриньяра. К вышеуказанной реакционной смеси прикапывали раствор 18,9 г 6-бром-2-трифторметилимидазо/1,2-a/пиридина и 14,4 мл бромэтана в 120 мл тетрагидрофурана. По окончании прикапывания полученную смесь нагревали с обратным холодильником в течение 30 минут и затем охлаждали льдом. К полученной смеси по каплям добавляли 36,2 мл 2-метоксициклогексана. Полученную смесь перемешивали при комнатной температуре в течение 3 часов и охлаждали снова льдом с последующим добавлением насыщенного водного раствора хлорида аммония. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя с получением коричневого масла.
К этому маслу порциями добавляли 70 мл концентрированной серной кислоты. Полученную смесь перемешивали при комнатной температуре в течение 2 часов и выливали на лед. Полученную смесь подщелачивали концентрированным водным аммиаком и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: н-гексан/этилацетат (от 2 : 1 до 1 : 1)/ и полученное твердое вещество промывали эфиром, получая 10,18 г указанного в заголовке соединения в виде слегка желтого порошка (выход: 51%).
Т. пл. (oC): 166,5 - 169.
1H-ЯМР (400 МГц, CDCl3) δ : 1,76 - 2,03 (3H, м), 2,07 (1H, м), 2,24 (1H, м), 2,35 (1H, м), 2,46 - 2,62 (2H, м), 3,62 (1H, дд, J = 5,3, 12,4 Гц), 7,09 (1H, дд, J = 1,6 Гц, 9,3 Гц), 7,62 (1H, д, J = 9,3 Гц), 7,82 (1H, м), 7,98 (1H, м).
Препаративный пример 16.
2-Метилмидазо/1,2-a/пиридин-6-метанол
В атмосфере азота 78,56 г магния добавляли к 2 л тетрагидрофурана и 0,3 г иода, и 18 мл бромэтана добавляли к полученной смеси для получения реактива Гриньяра. К вышеуказанной реакционной смеси медленно добавляли по каплям раствор 170,5 г 6-бром-2-метилимидазо/1,2-a/пиридина и 162 мл бромэтана в 500 мл тетрагидрофурана. По окончания прикапывания полученную смесь нагревали с обратным холодильником в течение 30 минут и затем охлаждали льдом. Формальдегид генерировался с помощью нагревания контейнера, содержащего 242 г параформальдегида, до 200oC и вводился в реакционную смесь с использованием струи азота. Через 30 минут к образовавшейся смеси добавляли разбавленную хлористоводородную кислоту, и полученную смесь фильтровали для удаления нерастворимых веществ. Фильтрат подщелачивали концентрировали водным аммиаком и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 30:1 до 10:1)/, получая 120 г целевого соединения в виде бледно-желтого твердого вещества (выход: 91,9%).
Т. пл. (oC): 136 - 137
1H-ЯМР (400 МГц, CDCl3) δ : 1,8 (1H, шир.), 2,43 (3H, д, J = 0,9 Гц), 4,66 (2H, с), 7,06 (1H, дд, J = 1,6 Гц, 9,2 Гц), 7,27 (1H, с), 7,39 (1H, д, J = 9,2 Гц), 7,99 (1H, м).
Препаративный пример 17.
6-((4-Хлорбетил)тиометил)-2-метилимидазо/1,2-a/-пиридин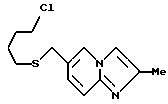
6,80 г 2-метилимидазо/1,2-a/пиридин-6-метанола, полученного в препаративном примере 16, растворяли в 90 мл хлороформа, и затем по каплям добавляли к полученному раствору 6,1 мл тионилхлорида при перемешивании при комнатной температуре. Через два часа растворитель отгоняли. Получали сырой 6-хорметил-2-метилимидазо/1,2-a/пиридин-гидрохлорид в виде коричневого масла.
Масло растворяли в смеси, содержащей 40 мл метанола и 40 мл этанола, с последующим добавлением 3,8 г тиомочевины. Полученную смесь нагревали с обратным холодильником в течение 3,5 часов и затем охлаждали, давая ей постоять. Полученную смесь перегоняли для удаления растворителя, получая сырой S-((2-метилимидазо/1,2-a/пиридин- 6-ил)метил)изотиомочевину-дигидрохлорид в виде светлого оранжево-желтого аморфного вещества.
К этому веществу добавляли 40 мл воды и 8,4 мл 10 н. водного раствора гидроксида натрия. Полученную смесь нагревали с обратным холодильником в течение одного часа и охлаждали льдом с последующим добавлением 5,8 мл 10 н. водного раствора гидроксида натрия, 5,0 мл 1-бром-4-холорбутана и 2,2 мл этанола в указанном порядке. Полученную смесь перемешивали при комнатной температуре в течение 2 часов, затем к ней добавляли воду. Полученную смесь экстрагировали этилацетатом, и органическую фазу экстрагировали разбавленной хлористоводородной кислотой. Водную фазу подщелачивали карбонатом калия и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 100 : 1 до 50 : 1)/, получая 4,95 г указанного в заготовке соединения в виде желтого масла (выход: 44%).
1H-ЯМР (400 МГц, CDCl3) δ/ : 1,69 - 1,79 (2H, м), 1,81 - 1,89 (2H, м), 2,45 (3H, д, J = 0,9 Гц), 2,50 (2H, т, J = 7,1 Гц), 3,53 (2H, т, J = 7,1 Гц), 3,67 (2H, с), 7,14 (1H, дд, J = 1,8 Гц, 9,3 Гц), 7,29 (1H, с), 7,47 (1H, д, J = 9,3 Гц), 7,95 (1H, м).
Препаративный пример 18.
6-((4-Хлорбутил)сульфинилметил)-2-метилимидазо/1,2-a/-пиридин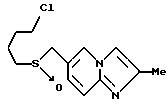
4,95 г 6-((4-хлорбутил)тиометил)-2-метилимидазо/1,2-a/-пиридина, полученного в препаративном примере 17, растворяли в 50 мл дихлорметана с последующим охлаждением полученного раствора льдом. В образовавшийся раствор по каплям добавляли в течение 20 минут раствор 3,18 г м-хлорпербензойной кислоты в 20 мл дихлорметана. Через один час в полученную смесь еще добавляли 0,16 г м-хлорпербензойной кислоты. Полученную смесь дополнительно перемешивали в течение 30 минут, затем к ней добавляли водный раствор тиосульфата натрия. Полученную смесь энергично перемешивали при комнатной температуре в течение 15 минут, и затем к ней добавляли водный раствор карбоната калия. Полученную смесь экстрагировали дихлорметаном. Органическую фазу сушили над безводным карбонатом калия и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихрометан/ метанол (от 50 : 1 до 30 : 1)/, получая 4,18 г указанного в заголовке соединения в виде бледно-розового твердого тела (выход: 79 %).
1H-ЯМР (400 МГц, CDCl3) δ : 1,83 - 2,03 (4H, м), 2,46 (3H, д, J = 0,9 Гц), 2,66 (2H, м), 3,57 (2H, м), 3,86 (1H, д, J = 13,6 Гц), 3,96 (1H, д, J = 13,6 Гц), 7,02 (1H, дд, J = 1,8 Гц, 9,3 Гц), 7,35 (1H, м), 7,52 (1H, д, J = 9,3 Гц), 8,08 (1H, м).
Пример 1.
1-(Имидазо/1,2-a/пиридин-6-ил)-N-метил-2-оксо-циклогексанкарботиоамид
13,00 г 2-(имидазо/1,2-a/пиридин-6-ил)циклогексанона, полученного в препаративном примере 1, суспендировали в 200 мл безводного тетрагидрофурана с последующим добавлением 7,50 г т-бутоксида калия при охлаждении льдом. Полученную смесь перемешивали один час. К смеси добавляли раствор 4,88 г метилизоцианата в 10 мл тетрагидрофурана с последующим добавлением 20 мл N, N-диметилформамида. Полученную смесь перемешивали при комнатной температуре в течение 45 минут и выливали в насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя.
Полученное твердое вещество промывали смесью хлороформа и этилацетата, получая 12,53 г указанного в заголовке соединения в виде слегка желтого порошка (выход: 72%).
Т. пл. (oC): 241 - 243 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ : 1,74 - 1,86 (1H, м), 1,88 - 1,96 (2H, м), 1,98 - 2,09 (1H, м), 2,30 - 2,64 (2H, м), 2,79 (2H, т, J = 6,2 Гц), 3,18 (3H, д, J = 4,8 Гц), 7,01 (1H, дд, J = 2,0, 9,7 Гц), 7,52 - 7,57 (2H, м), 7,62 (1H, д, J = 1,1 Гц), 8,05 (1H, дд, J = 0,9, 2,0 Гц), 8,96 (1H, шир.).
Пример 2.
(-)-1-(Имидазо/1,2-a/пиридин-6-ил)-N-метил-2-оксо- циклогексанкарботиоамид
5,60 г 1-(имидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамида, полученного в примере 1, суспендировали в 130 мл смеси ацетона и воды /3 : 1 (об./об.)/ с последующим добавлением раствора 7,34 г моногидрата (+)-дибензоил-D-винной кислоты в 20 мл смеси воды с ацетоном /3 : 1 (об./об. )/. Полученную смесь перемешивали при комнатной температуре в течение 30 минут с получением осадка. Осадок удаляли фильтрованием и промывали ацетоном, получая 5,85 г белого порошка. Порошок растворяли в 100 мл смеси этанола и воды /5 : 1 (об./об.)/ при нагревании. К полученному раствору добавляли воду до тех пор, пока раствор не становился мутным. Полученную смесь оставляли стоять при комнатной температуре с образованием осадка. Этот осадок удаляли фильтрованием. Получали 3,33 г белых игловидных кристаллов. Кристаллы перекристаллизовывали из 300 мл смеси этанола и воды /3 : 5 (об./об.)/, получая еще 2,52 г белых игловидных кристаллов, которые перекристаллизовывали снова из 150 мл смеси этанола с водой /3 : 5 (об./об.)/, получая 2,15 г белых игольчатых кристаллов. Кристаллы добавляли к разбавленному водному аммиаку, и полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя, получая кристаллический остаток. Этот кристаллический остаток перекристаллизовывали из ацетонитрила, получая 0,32 г указанного в заголовке соединения в виде пластинчатых кристаллов (выход: 6%).
Оптическая чистота этого продукта, определенная высокоэффективной жидкостной хроматографией с использованием хиральной колонки, была 100%.
Характеристика высокоэффективной жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка) (продукт фирмы Дайцел Кемикал Индастриз, Лтд) (250 мм х 4,6 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол /3 : 2 (об./об.)/.
Скорость потока: 1,0 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 10,2 мин (время удержания рацемической модификации было 7,8 и 10,2 минут)
Т. пл. (oC): 225 - 227 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ : 1,74 - 1,86 (1H, м), 1,87 - 1,97 (2H, м), 1,98 - 2,09 (1H, м), 2,30 - 2,64 (2H, м), 2,79 (2H, т, J = 6,0 Гц), 3,18 (3H, д, J = 4,8 Гц), 7,01 (1H, дд, J = 2,0, 9,5 Гц), 7,54 (1H, д, J = 9,5 Гц), 7,54 (1H, д, J = 0,9, 1,3 Гц), 7,62 (1H, д, J = 1,3 Гц), 8,04 (1H, д, J = 0,9, 2,0 Гц), 8,94 - 9,03 (1H, щир.).
Удельное вращение:  -236o (C = 1,0, метанол).
-236o (C = 1,0, метанол).
Пример 3.
-Этил-1-(имидазо/1,2-a/пиридин-6-ил)-2-оксоциклогексанкарботиоамид
5,00 г (23,4 ммоль) 2-(имидазо/1,2-a/пиридин-6-ил)циклогексанона, полученного в препаративном примере 1, растворяли в 50 мл сухого тетрагидрофурана. Полученный раствор охлаждали до 0oC, а затем добавляли к нему т-бутоксид калия. Полученную смесь перемешивали в течение 30 минут с последующим медленным добавлением по каплям раствора 2,45 г N-этилизоцианата в 5 мл полученной смеси. Температуру полученной смеси постепенно повышали до комнатной, и образовавшуюся в результате смесь оставляли на ночь перемешиваться с получением кристаллического осадка. Этот кристаллический осадок отделяли фильтрованием и промывали этилацетатом, получая 4,8 г целевого соединения в виде белых кристаллов (выход: 72%).
Т. пл. (oC): 176 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ : 1,24 (3H, т, J = 7,3 Гц), 1,74 - 2,10 (4H, м), 2,50 - 2,62 (2H, м), 2,70 - 2,84 (2H, м), 3,59 - 3,76 (2H, м), 7,01 (1H, дд, J = 1,9 Гц, 9,6 Гц), 7,50 - 7,58 (2H, м), 7,62 (1H, д, J = 1,3 Гц), 8,05 - 8,08 (1H, м), 8,75 (1H, шир.с.).
Пример 4.
(-)-N-Этил-1-(имидазо/1,2-a/пиридин-6-ил)-2-оксоциклогексанкарботиоамид и (+)-N-этил-1-(имидозо-/1,2-a/пиридин-6-ил)-2-оксоциклогексанкарботиоамид
1,43 г рацемической модификации, приготовленной в примере 3, и 2,06 г (+)-дибензоил-D-винной кислоты-моногидрата растворяли в 10 мл смеси воды и ацетона (3:1) при нагревании. Полученный раствор медленно охлаждали, получая 1,5 г кристаллического осадка. Этот осадок извлекали фильтрованием, снова растворяли в 7 мл смеси воды и ацетона (3:1) и перекристаллизовывали из нее с получением 1,0 г кристаллов. Эти кристаллы растворяли в водном растворе бикарбоната натрия, и полученный раствор экстрагировали хлороформом, получая свободное соединение. Данное соединение перекристаллизовывали из этилацетата, получая 210 мг целевого соединения.
Оптическая чистота этого продукта, определенная с помощью высокоэффективной жидкостной хроматографии, используя хиральную колонку, была 100%.
Характеристика высокоэффективной жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка) (продукт фирмы Дайцел Кемикал Индастриз. Лтд.) (250 мм х 4,6 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол/диэтиламин (60:300:1).
Скорость потока: 0,5 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 16,7 мин (время удержания рацемической модификации было 14,2 и 16,7 минут).
Т. пл. (oC): 208 (разл.).
Удельное вращение:  - 262,6o (C = 1,0, этанол).
- 262,6o (C = 1,0, этанол).
Пример 5.
N-Метил-1-(2-метилимидазо/1,2-a/пиридин-6-ил)-2- оксоциклогексанкарботиоамид
В атмосфере азота раствор 35,79 г (0,157 моль) 2-(2-метилимидазо/1,2-a/пиридин-6-ил)циклогексанона, приготовленного в препаративном примере 2, в 550 мл тетрагидрофурана охлаждали льдом, а затем к нему добавляли 19,55 г (0,174 моль) т-бутоксида калия. Полученный желтый раствор перемешивали при охлаждении льдом в течение одного часа, а затем к нему добавляли раствор 11,8 мл (0,173 моль) метилизоцианата в 50 мл тетрагидрофурана и 60 мл N,N-диметилформамида в этом порядке. Полученную реакционную смесь перемешивали при охлаждении льдом один час с последующим добавлением насыщенного водного раствора хлорида аммония. Полученную смесь экстрагировали хлороформом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. В ходе концентрирования начинали осаждаться кристаллы. Кристаллы выделяли фильтрованием. Было получено 37,09 г целевого соединения в виде бесцветных кристаллов. Далее, маточную жидкость (14,5 г) очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (100:3)/ и перекристаллизовывали из дихлорметана/этилацетата с получением 3,21 г целевого соединения в виде бледно-розового кристалла. Таким образом, всего было получено 40,3 г целевого соединения (выход: 85%).
Т. пл. (oC): 217 - 220 (разл.).
1H-ЯМР (400 МГц, диметилсульфоксид-d6) δ/ : 1,58 - 1,82 (3H, м), 1,88 - 2,02 (1H, м), 2,22 (1H, м), 2,31 (3H, д, J = 0,7 Гц), 2,40 (1H, дт, J = 13,6, 3,5 Гц), 3,00 (1H, м), 3,01 (3H, д, J = 4,4 Гц), 3,13 (1H, ддд, J = 13,6, 11,5, 6,4 Гц), 7,16 (1H, дд, J = 9,5, 1,8 Гц), 7,32 (1H, д, J = 9,5 Гц), 9,67 (1H, шир.), 8,24 (1H, дд, J = 2,0, 0,9 Гц), 9,80 (1H, шир.д, J = 4,4 Гц).
Пример 6.
(-)-N-Метил-1-(2-метилимидазо/1,2-a/пиридин-6-ил)-2- оксоциклогексанкарботиоамид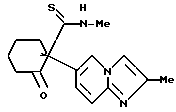
Раствор 20,26 г (53,8 ммоль) (+)-дибензоил-D-винной кислоты-моногидрата в 110 мл смеси ацетона и воды (4:1) добавляли к раствору 16,21 г (53,8 ммоль) (±)-N-метил-1-(2-метилимидазо/1,2-a/ пиридин-6-ил)-2-оксоциклогексанкарботиоамида, приготовленного в примере 5, в 900 мл смеси ацетона и воды (4:1). Полученную смесь оставляли стоять при комнатой температуре на 2 часа, получая (+)-N-метил-1-(2- метилимидазо/1,2-a/пиридин-6-ил)-2-оксоциклогексанкарботиоамид (+)-дибензоил-D-винной кислоты в виде бесцветных призматических кристаллов. Кристаллы (14,6 г, 95% или выше) отфильтровывали.
Фильтрат концентрировали в вакууме, и полученный остаток растворяли в 900 мл смеси этанола и воды (9:1) в условиях дефлегмации. Полученный раствор оставляли стоять на 12 часов, получая бесцветные игольчатые кристаллы (20,4 г). Кристалл выделяли фильтрованием и растворяли снова в 900 мл смеси этанола и воды (9:1) в условиях дефлегмации. Полученный раствор охлаждали, давая ему стоять 2 часа, и получали бесцветные игольчатые кристаллы. Эти кристаллы выделяли фильтрованием. Таким образом, получали 14,17 г (98%) (-)-N-метил-1-(2-метилимидазо/1,2-a/пиридин-6-ил)-2- оксоциклогексанкарботиоамида (+)-дибензоил-D-винной кислоты.
К суспензии 14,17 г (21,5 ммоль) соли, приготовленной выше, в 200 мл воды добавляли 100 мл 1 н. водного раствора гидроксида натрия. Полученную смесь перемешивали при комнатной температуре в течение 5 минут, получая бесцветный раствор, который экстрагировали хлороформом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. В ходе концентрирования начиналось выпадение осадка кристаллов. Образовавшиеся кристаллы извлекали фильтрованием. Таким образом, было получено 5,22 г целевого соединения в виде бесцветных кристаллов (выход: 32%, оптическая чистота: 99%).
Оптический выход определялся с помощью высокоэффективной жидкостной хроматографии с использованием хиральной колонки.
Характеристика жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка) (продукт фирмы Дайцел Кемикал Индастриз, Лтд) (250 мм х 4,6 мм внутр. диам.).
Элюент: н-гексан/2-пропанол /4,1 (об./об.)/.
Температура колонки: комнатная температура.
Скорость потока: 1 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удерживания: 16 мин (времена удержания рацемической модификации были 14 и 16 минут).
Т.пл. (oC): 219 - 222 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ: 1,79 (1H, м), 1,90 (2H, квинт, J = 6,4 Гц), 2,00 (1H, м), 2,43 (3H, д, J = 0,9 Гц), 2,55 (2H, м), 2,80 (2H, м), 3,16 (3H, д, J = 4,8 Гц), 6,96 (1H, дд, J = 9,5, 2,0 Гц), 7,30 (1H, шир.с), 7,43 (1H, шир.д, J = 9,5 Гц), 7,96 (1H, дд, J = 2,0, 0,9 Гц), 8,98 (1H, шир.с).
Удельное вращение:  -218o (C = 1,09, метанол).
-218o (C = 1,09, метанол).
Пример 7.
(+)-N-Метил-1-(2-метилимидазо/1,2-a/пиридин-6-ил)-2- оксоциклокарботиоамид
Суспензию 14,6 г (22,1 ммоль) (+)-N-метил-1-(2-метилимидазо/1,2-a/ пиридин-6-ил)-2-оксоциклогексанкарботиоамида (+)-дибензоил-D-винной кислоты, отфильтрованного в примере 6, в 1 л смеси метанола и воды (9:1) нагревали с обратным холодильником в течение 30 минут и охлаждали, давая постоять 90 минут. Полученную в результате суспензию фильтровали, получая кристаллы.
К суспензии 12,02 г (18,2 ммоль) полученной соли в 200 мл воды добавляли 100 мл 1 н. водного раствора гидроксида натрия. Полученную смесь перемешивали при комнатной температуре в течение 5 минут, получая бесцветный раствор, который экстрагировали хлороформом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. В ходе концентрирования начинали выпадать в осадок кристаллы. Образовавшиеся таким образом кристаллы извлекали фильтрованием. Было получено 4,30 г целевого соединения в виде бесцветных кристаллов (выход: 27%, оптическая чистота: 99%).
Оптическую чистоту определяли с помощью высокоэффективной жидкостной хроматографии с использованием хиральной колонки.
Характеристика жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка) (продукт фирмы Дайцел Кемикал Индастриз, Лтд.) (250 мм х 4,6 мм внутр. диам.).
Элюент: н-гексан/2-пропанол /4:1 (об./об.)/.
Температура колонки: комнатная температура.
Скорость потока: 1 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 14 мин (времена удержания рацемической модификации были 14 и 16 минут).
Т. пл. (oC): 219 - 222 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ: 1,78 (1H, м), 1,91 (2H, квинт, J = 6,3 Гц), 2,00 (1H, м), 2,43 (3H, д, J = 0,7 Гц), 2,56 (2H, м), 2,80 (2H, м), 3,16 (3H, д, J = 4,6 Гц), 6,96 (1H, дд, J = 9,4, 1,9 Гц), 7,30 (1H, шир.с), 7,42 (1H, шир.д, J = 9,5 Гц), 7,96 (1H, дд, J = 2,0, 0,9 Гц), 9,00 (1H, шир.с).
Удельное вращение:  -227o (C = 1,03, метанол).
-227o (C = 1,03, метанол).
Пример 8.
2-Гидрокси-1-(имидазо/1,2-a/пиридин-6-ил)-N-метил- циклогексанкарботиоамид (M-форма)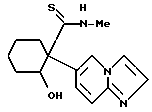
24,9 г 1-имидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиомида, полученного в примере 1, суспендировали в 280 мл метанола. Полученную суспензию охлаждали льдом с последующим добавлением 1,27 г боргидрата натрия. Полученную смесь перемешивали в течение 30 минут.
Реакционную смесь анализировали с помощью тонкослойной хроматографии на силикагеле /проявитель: бензол/этилацетат/метанол (10 : 10 : 1)/, при этом было установлено, что реакционный продукт состоит из низкополярного диастереомера (L-форма) в качестве второстепенного компонента и высокополярного диастереомера (M-форма) в качестве основного компонента.
К реакционной смеси добавляли воду, и полученную смесь фильтровали для выделения нерастворимого материала. Таким образом, было получено 15,12 г целевого соединения в форме чистого диастереомера в виде бледно-желтого порошка (выход: 62%).
Т. пл. (oC): 231 - 233 (разл.).
1H-ЯМР (400 МГц, диметилсульфоксид-d6) δ: 1,27 - 1,41 (3H, м), 1,52 - 1,71 (2H, м), 1,92 - 2,01 (1H, м), 2,16 - 2,26 (1H, м), 2,47 - 2,56 (1H, м), 2,92 (3H, д, J = 4,4 Гц), 4,59 - 4,68 (1H, м), 4,88 - 4,97 (1H, м), 7,32 (1H, дд, J = 1,8, 9,5 Гц), 7,44 (1H, д, J = 9,5 Гц), 7,51 (1H, д, J = 1,3 Гц), 7,95 (1H, м), 8,63 (1H, м), 9,21 - 9,28 (1H, м).
Пример 9.
2-Бензоилокси-1-(имидазо/1,2-a/пиридин-6-ил)-N-метил- циклогексанкарботиоамид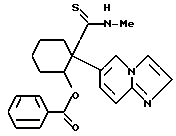
500 мг 2-гидрокси-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиамида (M-форма), приготовленного в примере 8, суспендировали в 5 мл N, N-диметилформамида с последующей добавкой 470 мг бензойного ангидрида, 0,29 мл триэтиламина и каталитического количества 4-диметиламинопиридина. Полученную смесь перемешивали при комнатной температуре в течение 2 часов, затем к ней добавляли воду. Образовавшуюся смесь экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали и перегоняли для удаления растворителя. Остаток очищали хроматографически в колонке с силикагелем /растворитель: дихлорметан/метанол (30 : 1)/, и полученное твердое вещество перекристаллизовывали из смеси хлороформ/эфир, получая 40 мг целевого соединения в виде белого порошка (выход: 6%).
Т. пл. (oC): 273 - 274 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ: 1,38 - 1,76 (4H, м), 1,90 - 2,02 (1H, м), 2,35 - 2,51 (2H, м), 2,69 - 2,78 (1H, м), 3,04 (3H, д, J = 4,8 Гц), 6,19 (1H, дд, J = 3,5, 8,8 Гц), 7,31 (1H, дд, J = 1,8, 9,5 Гц), 7,35 - 7,45 (3H, м), 7,50 - 7,56 (2H, м), 7,60 (1H, м), 7,46 (1H, д, J = 1,1 Гц), 7,81 - 7,86 (2H, м), 8,59 (1H, м).
Пример 10.
(-)-2-Гидрокси-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид (M-форма)
Суспензию 1,00 г (-)-1-имидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамида (97%), приготовленного в примере 2, в 20 мл метанола охлаждали льдом с последующим добавлением 53 мг боргидрида натрия. Полученную смесь перемешивали в течение 30 минут.
Реакционную смесь анализировали с помощью тонкослойной хроматографии на силикагеле /проявитель: бензол/этилацетат/метанол (10 : 10 : 1)/, при этом было установлено, что продукт реакции состоял из низкополярного диастереомера (L-форма) в качестве второстепенного компонента и высокополярного диастереомера (M-форма) в качестве главного компонента.
В реакционную смесь добавляли воду и получающуюся смесь фильтровали для извлечения нерастворимого материала. Таким образом, было получено 0,61 г целевого соединения в форме чистого диастереомера в виде белого порошка (выход: 60%).
Т. пл. (oC): 244 - 246 (разл.).
1H-ЯМР (400 МГц, диметилсульфоксид-d6) δ: 1,26 - 1,40 (3H, м), 1,52 - 1,72 (2H, м), 1,93 - 2,02 (1H, м), 2,15 - 2,25 (1H, м), 2,47 - 2,55 (1H, м), 2,92 (3H, д. J = 4,4 Гц), 4,60 - 4,66 (1H, м), 4,88 - 4,94 (1H, м), 7,32 (1H, дд, J = 1,8, 9,5 Гц), 7,44 (1H, д, J = 9,5 Гц), 7,51 (1H, д, J = 1,3 Гц), 7,96 (1H, м), 8,63 (1H, м), 9,20 - 9,30 (1H, м).
Удельное вращение:  -44o (C = 0,68, N,N-диметилформамид).
-44o (C = 0,68, N,N-диметилформамид).
Пример 11.
(-)-2-Бензоилокси-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид
400 мг (-)-2-Гидрокси-1-(имидазо/1,2-a/пиридин-6-ил)- метилциклогексанкарботиоамида (M-форма), полученного в примере 10, суспендировали в 10 мл N, N-диметилформамида с последующим добавлением 0,24 мл триэтиламина, 380 мг бензойного ангидрида и каталитического количества 4-пирролидинпиридина. Полученную смесь перемешивали при комнатной температуре в течение 2 часов, затем к ней добавляли насыщенный раствор бикарбоната натрия. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали последовательно водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 50 : 1 до 30 : 1)/ и полученное твердое вещество перекристаллизовывали из дихлорметана/эфира, получая 380 мг указанного в заголовке соединения в виде игольчатых кристаллов (выход: 69%).
Оптическая чистота вышеупомянутого продукта была 98% и определялась высокоэффективной жидкостной хроматографией с использованием хиральной колонки.
Характеристика высокоэффективной жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка) (продукт фирмы Дайцел Кемикал Индастриз, Лтд.) (250 мм х 4,6 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол /4 : 1 (об./об.)/.
Скорость потока: 0,5 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 11,2 мин (времена удержания рацемической модификации были 8,8 и 11,2 минуты).
Т. пл. (oC): 231,5 - 233 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ: 1,39 - 1,51 (1H, м), 1,56 - 1,76 (3H, м), 1,91 - 2,01 (1H, м), 2,35 - 2,51 (2H, м), 2,70 - 2,78 (1H, м), 3,05 (3H, д, J = 4,6 Гц), 6,19 (1H, дд, J = 3,5, 8,6 Гц), 7,34 (1H, дд, J = 1,8, 9,5 Гц), 7,36 - 7,41 (2H, м), 7,42 - 7,48 (1H, м), 7,50 - 7,56 (2H, м), 7,58 (1H, м), 7,63 (1H, д, J = 1,3 Гц), 7,82 - 7,86 (2H, м), 8,55 (1H, м).
Удельное вращение:  -238o (C = 0,5, метанол).
-238o (C = 0,5, метанол).
Пример 12.
2-Бензилокси-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид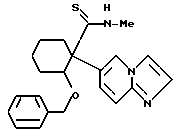
500 мг 2-бензилокси-1-(имидазо/1,2-a/пиридин-6-ил)-1-((4- метоксибензилтио)(метилимино)метил)циклогексан, приготовленного в препаративном примере 4, растворяли в 5 мл дихлорметана с последующим добавлением 1 мл анизола. Полученную смесь охлаждали льдом с последующим постепенным добавлением 5 мл трифторуксусной кислоты. Полученную смесь перемешивали один час, затем к ней добавляли насыщенный водный раствор бикарбоната натрия. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом натрия и перегоняли для удаления растворителя. Полученный остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (50: 1)/. Полученное масло кристаллизовали из эфира, и полученные кристаллы извлекали фильтрованием. Таким образом, было получено 300 мг целевого соединения в виде белого порошка (выход: 79%).
Т. пл. (oC): 196 - 198.
1H-ЯМР (400 МГц, CDCl3) δ: 0,98 - 1,11 (1H, м), 1,43 - 1,60 (2H, м), 1,71 - 1,83 (2H, м), 2,14 - 2,22 (1H, м), 2,38 - 2,46 (1H, м), 2,63 (1H, дт, J = 3,3, 13,5 Гц), 3,03 (3H, д, J = 4,8 Гц), 4,32 (1H, дд, J = 4,4, 11,7 Гц), 4,43 (1H, д, J = 11,2 Гц), 4,71 (1H, д, J = 11,2 Гц), 7,21 (1H, дд, J = 1,8, 9,5 Гц), 7,24 - 7,36 (5H, м), 7,54 (1H, шир.), 7,55 (1H, дд, J = 0,9, 1,3 Гц), 7,58 (1H, д, J = 9,5 Гц), 7,63 (1H, д, J = 1,3 Гц), 8,80 (1H, дд, J = 0,9, 1,8 Гц).
Пример 13.
Анти-2-бензилоксиимино-1-(имидазо/1,2-a/пиридин-6-ил)-N- метилциклогексанкарботиоамид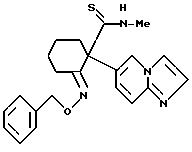
600 мг 1-(имидазо/1,2-a/пиридин-6-ил)-N-метил-2-оксогексанкарботиоамида, приготовленного в примере 1, растворяли в 5 мл пиридина с последующим добавлением 730 мг O-бензилгидроксиламина-гидрохлорида. Полученную смесь перемешивали при 80oC в атмосфере азота в течение 13 часов, затем к ней добавляли водный раствор карбоната калия. Полученную смесь экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Полученный остаток очищали хроматографически на колонке с силикагелем /растворитель: дихлорметан/метанол (от 100:1 до 50:1)/, и полученное масло кристаллизовали из эфира. Образовавшиеся кристаллы извлекали фильтрованием. Таким образом получали 610 мг целевого соединения в виде белого похожего на ленты кристаллов (выход: 74%).
Т. пл. (oC): 159 - 161,5.
1H-ЯМР (400 МГц, CDCL3) δ: 1,50 - 1,79 (3H, м), 1,83 - 1,94 (1H, м), 2,15 - 2,27 (2H, м), 2,88 - 3,01 (2H, м), 3,05 (3H, д, J=4,9 Гц), 5,06 (2H, с), 6,91 (1H, дд, J = 1,8, 9,5 Гц), 7,25 - 7,30 (2H, м), 7,32 - 7,44 (5H, м), 7,57 (1H, д, J = 1,3 Гц), 7,80 (1H, дд, J = 0,9, 1,8 Гц), 8,24 (1H, шир. ).
Пример 14.
2-Бензиламино-1-(имидазо/1,2-a/пиридин-6-ил)-N-метил- циклогексанкарботиоамид
1,50 г 1-(имидао/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамида, приготовленного в примере 1, и 2,35 г бензиламина растворяли в 20 мл дихлорметана. Полученный раствор охлаждали до 0oC.
В вышеприготовленный раствор медленно прикапывали 5,5 мл раствора (1 ммоль/мл) TiCl4 в дихлорметане, и температуру полученной смеси повышали до комнатной температуры. Образовавшуюся смесь перемешивали в течение 2 часов с последующим добавлением водного раствора бикарбоната натрия. Полученную смесь фильтровали через целит для удаления нерастворимых частиц. Фильтрат экстрагировали трижды хлороформом. Объединенные органические фазы сушили над сульфатом натрия и перегоняли в вакууме для удаления растворителя с получением масла. Масло как таковое (без очистки) растворяли в 10 мл смеси дихлорметана с метанолом (1:1) с последующим добавлением 350 мг NaBH3CN при комнатной температуре. К полученному раствору добавляли уксусную кислоту для доведения pH до 3. Образовавшийся в результате раствор перемешивали один час с последующим добавлением раствора бикарбоната натрия. Полученную смесь экстрагировали трижды хлороформом. Объединенные органические фазы сушили над сульфатом натрия и перегоняли в вакууме для удаления растворителя, получая масло. Это масло очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (50:1)/, получая 950 мг целевого соединения (выход: 45%).
1H-ЯМР (400 МГц, CDCl3) δ: 1,19 - 1,33 (1H, м), 1,43 - 1,78 (4H, м), 2,05 - 2,13 (1H, м), 2,37 - 2,56 (2H, м), 3,70 (3H, д, J = 4 Гц), 3,55 (1H, дд, J = 4 Гц, 12 Гц), 3,64 (1H, д, J = 12 Гц), 3,90 (1H, д, J = 12 Гц), 7,22 - 7,31 (4H, м), 7,31 - 7,38 (2H, м), 7,52 - 7,57 (2H, м), 7,61 (1H, д, J = 1,61 Гц), 8,70 (1H, шир.с.), 8,98 (1H, с).
Пример 15.
2-(Имидазо/1,2-a/пиридин-6-ил)-N-метилтетрагидротиопиран-2- карботиоамид 1-оксид
3,00 г 2-(имидазо/1,2-a/пиридин-6-ил)-тетрагидротиопирана 1-оксида, приготовленного в препаративном примере 9, суспендировали в 40 мл смеси тетрагидрофурана с гексаметилфосфорным триамидом /4:1 (об./об.)/. Полученную суспензию охлаждали льдом с последующим добавлением 1,58 г т-бутоксида калия. Полученную смесь перемешивали один час, затем к ней прикапывали 1,5 мл дисульфид углерода. Полученную смесь перемешивали 30 минут и затем к ней прикапывали 2,4 мл метилиодида. Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов с последующим добавлением ледяной воды. Полученную смесь экстрагировали этилацетатом, органическую фазу промывали водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и перегоняли для удаления растворителя.
Полученное красное масло растворяли в 10 мл метанола, затем добавляли к раствору 10 мл 40%-ного раствора метиламина в метаноле. Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов и концентрировали. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (от 20 : 1 до 10 : 1)/ и кристаллизовали из этилацетата, получая 160 мг целевого соединения в виде слегка желтого порошка (выход: 4%).
Т. пл. (oC): около 235 (разл.).
1H-ЯМР (400 МГц, CDCl3) δ: 1,56 - 1,84 (3H, м), 2,09 - 2,25 (2H, м), 2,65 - 2,75 (1H, м), 3,01 - 3,08 (1H, м), 3,28 (3H, д, J = 4,8 Гц), 3,92 - 4,03 (1H, м), 6,94 - 7,00 (1H, м), 7,17 - 7,22 (1H, м), 7,42 (1H, м), 7,54 (1H, д, J = 1,3 Гц), 8,08 (1H, м), 9,74 - 9,83 (1H, шир).
Пример 16.
N-Метил-1-(4-нитроимидазол-1-ил)-2-оксоциклогенсанкарботиоамид
В атмосфере азота суспензию 76,40 г (0,365 моль) 2-(4-нитроимидазол-1-ил)-циклогексанона, приготовленного в препаративном примере 10, в 1,2 л тетрагидрофурана охлаждали льдом с последующим добавлением 45,16 г (0,402 моль) т-бутоксида калия. Полученную красновато-пурпурную реакционную смесь перемешивали при охлаждении льдом в течение одного часа с последующим добавлением раствора 27,5 мл (0,402 моль) метилизоцианата в 100 мл тетрагидрофурана и 300 мл диметилформамида в этом порядке. Реакционную смесь перемешивали один раз, при этом ее температура повышалась до комнатной. К полученной смеси последовательно добавляли насыщенный водный раствор хлорида аммония и воду с последующей экстракцией хлороформом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Остаток порошковали хлороформом, получая 46,93 г указанного в заголовке соединения в виде светло-коричневого порошка. Маточную жидкость концентрировали в вакууме и остаток порошковали хлороформом снова, получая 9,97 г целевого соединения в виде светло-коричневого порошка. Далее, маточную жидкость концентрировали в вакууме и остаток порошковали этилацетатом, получая 3,24 г целевого соединения в виде коричневого порошка. Таким образом, было получено всего 60,14 г целевого соединения (выход: 58%).
Т. пл. (oC): 205 - 208 (разл.).
1H-ЯМР (400 МГц, диметилсульфоксид-d6) δ:/ 1,58 - 1,78 (2H, м), 1,86 (1H, м), 1,98 (1H, м), 2,50 - 2,62 (2H, м), 3,07 (3H, д, J = 3,5 Гц), 3,08 - 3,18 (2H, м), 7,79 (1H, д, J = 1,6 Гц), 8,25 (1H, д, J = 1,6 Гц), 10,48 (1H, шир.с).
Пример 17.
2-Гидрокси-N-метил-1-(4-нитроимидазол-1-ил)-циклогексанкарботиоамид (L-форма)
Суспензию 56,89 г (202 ммоль) N-метил-1-(4-нитроимидазол-1-ил)- 2-оксоциклогексанкарботиоамида, приготовленного в примере 16, в 160 мл метанола охлаждали льдом. К полученной суспензии порциями в течение 3 минут добавляли 2,68 г (70,8 ммоль) боргидрида натрия таким образом, что температура основной массы не превышала 10oC. Полученную смесь перемешивали при охлаждении льдом в течение 30 минут с последующим добавлением насыщенного водного раствора хлорида аммония и воды в указанном порядке. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором поваренной соли и сушили над безводным сульфатом магния. Образовавшуюся органическую фазу анализировали хроматографией на колонке с силикагелем /проявитель: бензол/этилацетат (1:2)/, при этом было установлено, что продукт этой стадии состоит из низкополярного диастереомера (L-форма, Rf : 0,41) и высокополярного диастереомера (M-форма, Rf : 0,29) в соотношении 2: 1. Органическую фазу концентрировали. В ходе концентрирования начиналось выпадение кристаллов. Образовавшиеся таким образом кристаллы извлекали фильтрованием. Таким образом было получено целевое соединение в форме диастереомера в виде бледно-желтого кристалла (выход: 50%).
Т. пл. (oC): 198 - 201 (разл.).
1H-ЯМР (400 МГц, диметилсульфоксид-d6) δ: 1,06 (1H, м), 1,28 - 1,48 (2H, м), 1,52 (1H, м), 1,60 (1H, м), 1,83 (1H, м), 2,45 (1H, м), 2,55 (1H, м), 2,99 (3H, с), 4,64 (1H, м, при добавлении D2O, дд, J = 10,3, 3,5 Гц), 5,87 (1H, шир., исчезали при добавлении D2O), 8,12 (1H, д, J = 1,1 Гц), 8,54 (1H, д, J = 1,1 Гц), 9,48 (1H, шир.с., исчезала при добавлении D2O).
Пример 18.
2-Бензоилокси-N-метил-1-(4-нитроимидазол-1-ил)-циклогексанкарботиоамид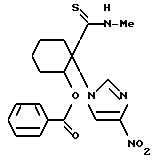
К суспензии 4,99 г (17,5 ммоль) 2-гидрокси-N-метил-1-(4- нитроимидазол-1-ил)циклогексанкарботиоамида (L-форма), приготовленного в примере 17, в 50 мл дихлорметана добавляли 2,7 мл (19,4 ммоль) триэтиламина, 4,38 г (19,4 ммоль) бензойного ангидрида и 215 мг (1,8 ммоль) N,N-диметиламинопиридина. Полученную смесь перемешивали при комнатной температуре в течение 14 часов, получая оранжевый раствор, затем к нему добавляли насыщенный раствор бикарбоната натрия. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали последовательно водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Остаток (7,5 г) очищали хроматографически на колонке с силикагелем /растворитель: дихлорметан/ацетон (от 20:1 до 5:1)/ и перекристаллизовывали из дихлорметана/диизопропилового эфира, получая 3,15 г целевого соединения в виде бледно-желтых кристаллов (выход: 46%).
Т. пл. (oC): 209 - 210.
1H-ЯМР (400 МГц, CDCl3) δ: 1,32 (1H, м), 1,56 - 1,88 (4H, м), 2,34 (1H, шир.д.кв., J = 13,2, 4,2 Гц), 2,56 (1H, шир.д., J = 15,2 Гц), 3,00 (1H, ддд, J = 15,3, 12,5, 3,1 Гц), 3,11 (3H, д, J = 4,8 Гц), 6,27 (1H, дд, J = 10,8, 3,8 Гц), 7,46 (2H, т, J = 7,7 Гц), 7,59 (1H, т, J = 7,5 Гц), 7,75 (1H, шир. с), 7,89 (2H, д, J = 7,9 Гц), 7,95 (1H, д, J = 1,3 Гц), 8,21 (1H, д, J = 1,3 Гц).
Пример 19.
2-Бензилокси-N-метил-1-(4-нитроимидазол-1-ил)-циклогексанкарботиоимид
6,15 г (12,4 ммоль) 2-бензилокси-1-((4-метоксибензилтио)(метил)- 1-(4-нитроимидазол-1-ил)циклогексан, приготовленного в препаративном примере 14, растворяли в 25 мл трифторуксусной кислоты при охлаждении льдом с последующим добавлением 5 мл анизола. Полученную смесь перемешивали при 0oC в течение 10 минут, получая темно-коричневый раствор, который выливали на смесь, содержащую водный раствор бикарбоната натрия и лед. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Остаток (8,02 г) очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/ацетон (40:1)/ и перекристаллизовывали из дихлорметана/диизопропилового эфира, получая 3,14 г целевого соединения в виде бесцветных кристаллов (выход: 67%).
Т. пл. (oC): 165 - 166.
1H-ЯМР (400 МГц, CDCl3) δ: 0,98 (1H, кв.т., J = 13,5, 3,4 Гц), 1,38 (1H, кв. д. , J = 12,6, 3,7 Гц), 1,50 (1H, кв.т. J = 12,9, 3,7 Гц), 1,70 (1H, м), 1,76 (1H, м), 2,14 (1H, м), 2,37 (1H, д.кв. J = 15,4, 2,7 Гц), 2,97 (1H, ддд, J = 15,5, 13,8, 3,8 Гц), 3,14 (3H, д, J = 4,8 Гц), 4,50 (1H, д, J = 10,8 Гц), 4,65 (1H, дд, J = 12,0, 4,3 Гц), 4,67 (1H, д, J = 10,8 Гц), 7,22 (2H, дд, J = 7,6, 1,7 Гц), 7,28 - 7,36 (3H, м), 7,65 (1H, шир.с.), 7,88 (1H, д, J = 1,6 Гц), 8,10 (1H, д, J = 1,6 Гц).
Пример 20.
Анти-2-бензилоксиимино-N-метил-1-(4-нитроимидазол-1- ил)циклогексанкарботиоамид
К суспензии 6,06 г (21,5 ммоль) N-метил-1-(4-нитроимидазол-1-ил)- 2-оксоциклогексанкарботиоамида, приготовленного в примере 16, в 50 мл пиридина добавляли 10,01 г (62,7 ммоль) O-бензилгидроксиламина- гидрохлорида. Полученную смесь перемешивали при нагревании при 100oC в течение 19 часов и охлаждали, давая ей постоять. К образовавшейся в результате смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органическую фазу промывали насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и концентрировали в вакууме. Остаток (12,9 г) очищали хроматографически на колонке с силикагелем /растворитель: дихлорметан/ацетон (от 30:1 до 5:1)/ и перекристаллизовывали из дихлорметана/этилацетата, получая 2,27 г целевого соединения в виде бесцветных кристаллов (выход: 275 г).
Т. пл. (oC): 175 - 177.
1H-ЯМР (400 МГц, CDCl2) δ: 1,58 - 1,76 (3H, м), 1,84 (1H, м), 2,39 (1H, ддд, J = 14,1, 8,4, 3,5 Гц), 2,49 (1H, ддд, J = 15,6, 8,3, 5,0 Гц), 2,73 (1H, м), 2,92 (1H, м), 3,02 (3H, д, J = 4,8 Гц), 5,10 (2H, AB кв. J = 12,3 Гц), 7,29 (2H, м), 7,36 - 7,41 (3H, м), 7,39 (1H, д, J = 1,6 Гц), 7,70 (1H, д, J = 1,6 Гц), 8,08 (1H, шир.с).
Пример 21.
1-(2-Трифторметилимидазо/1,2-a/пиридин-6-ил)-R-метил-2- оксоциклогексанкарботиоамид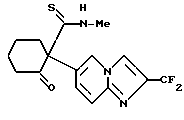
К суспензии 6,0 г 2-(2-трифторметилимидазо/1,2-a/пиридин- 6-ил)циклогексанона, приготовленного в препаративном примере 15, в 70 мл тетрагидрофурана добавляли при охлаждении льдом 2,63 г т-бутоксида калия. Полученную смесь перемешивали один час с последующим добавлением раствора 1,7 г метилизотиоцианата в 5 мл тетрагидрофурана. Затем к полученной смеси добавляли 7 мл N,N-диметилформамида. Через час реакционную смесь выливали в насыщенный водный раствор хлорида аммиака и затем экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом магния и перегоняли для удаления растворителя. Остаток очищали хроматографией на колонке с силикагелем /растворитель: дихлорметан/метанол (50 : 1)/, и полученное твердое вещество промывали эфиром, получая 5,99 г целевого соединения в виде белого порошка (выход: 79%).
Т. пл. (0oC): 116 - 120.
1H-ЯМР (400 МГц, CDCl2) δ:/ 1,76 - 2,12 (4H, м), 2,51 - 2,63 (2H, м), 2,69 (1H, м), 2,81 (1H, м), 3,18 (3H, д, J = 4,8 Гц), 7,15 (1H, дд, J = 2,0, 9,7 Гц), 7,61 (1H, д, J = 9,7 Гц), 7,86 (1H, с), 8,08 (1H, м), 8,79 (1H, шир.).
Пример 22.
(-)-1-(2-Трифторметилимидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамид
1-(2-Трифторметилимидазо/1,2-a/пиридин-6-ил)-N-метил-2- оксоциклогексанкарботиоамид, приготовленный в примере 21, подвергали препаративной хроматографии с использованием оптически активной колонки для проведения оптического разделения.
Характеристика препаративной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка), (продукт фирмы Дайцел Кемикал Индастриз, Лтд.), (250 мм х 20 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол (3 : 1 (об./об.)).
Скорость потока: 6 мл/мин.
Детектирование: УФ-детектор (254 нм).
Пик для препаративных целей: более поздний пик между двумя пиками.
Твердое вещество, полученное препаративной хроматографией, перекристаллизовывали из н-гексана/этилацетата, получая целевое соединение в виде белых кристаллов.
Оптическая чистота этого продукта, определенная с помощью высокоэффективной жидкостной хроматографии с использованием хиральной колонки, была 99%.
Характеристика высокоэффективной жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка), (продукт фирмы Дайцел Кемикал Индастриз, Лтд.), (250 мм х 4,6 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол (3 : 1 (об./об.)).
Скорость потока: 1,0 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 20,0 мин (времена удержания рацемической модификации были 12,8 и 20,0 минут).
Т. пл. (oC): 184 - 186.
Удельное вращение: 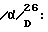 -229o (C = 1,02, метанол).
-229o (C = 1,02, метанол).
Пример 23.
2-(2-Метилимидазо/1,2-a/пиридин-6-ил)-N- метилтетрагидротиопиран-2-карботиоамид 1-оксид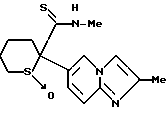
3,86 г т-бутоксида калия растворяли в 30 мл смеси (1 : 1 (об./об.)) тетрагидрофурана и гексаметилфосфорного триамида. Полученный раствор охлаждали льдом с последующим добавлением по каплям раствора 3,68 г 6-((4-хлорбутил)сульфинилметил)-2- метилимидазо/1,2-a/пиридина, приготовленного в перепаративном примере 18, в 30 мл смеси (1 : 1 (об./об.)) тетрагидрофурана и гексаметилфосфорного триамида. Через 1,5 часа в полученную смесь прикапывали 3,45 мл дисульфида углерода с последующим перемешиванием образовавшейся смеси при +10oC. Через 1,5 часа в смесь прикапывали 3,57 мл метилиодида, и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов и выливали в ледяную воду. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным водным раствором поваренной соли, сушили над безводным сульфатом магния и перегоняли для удаления растворителя, получая коричневое твердое вещество.
Это твердое вещество растворяли в 10 мл метанола, добавляли в раствор 10 мл 40%-ного раствора метиламина в метаноле. Полученную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли для удаления растворителя. Остаток очищали хроматографически на колонке с силикагелем /растворитель: дихлорметан/метанол (20 : 1)/, и полученное твердое вещество промывали эфиром, получая 160 мг целевого соединения в виде белого порошка (выход: 3%).
Т. пл. (oC): 227 - 231.
1H-ЯМР (400 МГц, CDCl2) δ: 1,55 - 1,81 (3H, м), 2,08 - 2,24 (2H, м), 2,42 (3H, д, J = 0,7 Гц), 2,71 (1H, м), 3,04 (1H, м), 3,23 (3H, д, J = 4,8 Гц), 3,88 (1H, м), 6,91 (1H, д, J = 9,5 Гц), 7,03 (1H, дд, J = 2,0 Гц, 9,5 Гц), 7,24 (1H, с), 8,29 (1H, с), 9,39 (1H, шир.).
Пример 24.
(-)-2-(2-Метилимидазо/1,2-a/пиридин-6-ил)-N-метилтетрагидротиопиран- 2-карботиоамид 1-оксид
2-(2-Метилимидазо/1,2-a/пиридин-6-ил)-N-метилтетрагидротиопиран- 2-карботиоамид 1-оксид, приготовленный в примере 20, подвергали препаративной хроматографии с использованием оптически активной колонки для проведения оптического разделения.
Характеристика препаративной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка), (продукт фирмы Дайцел Кемикал Индастриз, Лтд.), (750 мм х 50 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол/диэтиламин (750 : 250 : 1).
Скорость потока: 100 мл/мин.
Детектирование: УФ-детектор (254 нм).
Пик для препаративных целей: более поздний пик между двумя пиками.
Твердое вещество, полученное с помощью препаративной хроматографии, очищали хроматографически на колонке с силикагелем /растворитель: дихлорметан/метанол (10 : 1)/ и перекристаллизовывали из водного этанола, получая целевое соединение в виде белых кристаллов.
Оптическая чистота этого продукта, определенная с помощью высокоэффективной жидкостной хроматографии с использованием хиральной колонки, была 100%.
Характеристика высокоэффективной жидкостной хроматографии.
Колонка: Хирацел (зарегистрированная торговая марка), (продукт фирмы Дайцел Кемикал Индастриз, Лтд.), (250 мм х 4,6 мм внутр. диам.).
Растворитель: н-гексан/2-пропанол/диэтиламин (750 : 250 : 1).
Скорость потока: 0,7 мл/мин.
Детектирование: УФ-детектор (254 нм).
Время удержания: 19,4 мин (времена удержания рацемической модификации были 14,5 и 19,4 минуты).
Т. пл. (oC): 219 - 223 (разл.).
Удельное вращение: 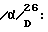 -352o (C = 1,02, метанол).
-352o (C = 1,02, метанол).

Производные тиоформамида общей формулы I, где Y - группа формулы -C(O)-, -CH(OH)-, 
 Z представляет -CH2-; A - замещенный или незамещенный имидазопиридил; R1, R2, R3 и R4 - водород или низший алкил; или их фармацевтически приемлемые соли представляют собой превосходные гипотензивные средства.
Z представляет -CH2-; A - замещенный или незамещенный имидазопиридил; R1, R2, R3 и R4 - водород или низший алкил; или их фармацевтически приемлемые соли представляют собой превосходные гипотензивные средства.

5 з.п. ф-лы, 1 табл.
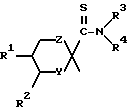
в которой y представляет группу формул: CH(OH),
CH(OH),

Z представляет -CH2-;
A представляет незамещенный или замещенный имидазопиридил;
R1, R2, R3 и R4 независимо представляют водород или низший алкил,
или их фармакологически приемлемые соли.
имидазо [1,2-a] пиридильную группу, выбранную из группы, состоящей из имидазо [1,2-a] пиридин-2-ила, имидазо [1,2-a] -пиридин-3-ила, имидазо [1,2-a] пиридин-5-ила, имидазо [1,2-a]-пиридин-6-ила, имидазо [1,2-a] пиридин-7-ила, имидазо [1,2-a] пиридин-8-ила;
имидазо-[1,5-a] пиридильную группу, выбранную из группы, состоящей из имидазо [1,5-a] пиридин-1-ила, имидазо [1,5-a] -пиридин-3-ила, имидазо [1,5-a] пиридин-5-ила, имидазо [1,5-a]-пиридин-6-ила, имидазо [1,5-a] пиридин-7-ила, и имидазо [1,2-a] пиридин-8-ила;
имидазо-[4,5-b] пиридильную группу, выбранную из группы, состоящей из имидазо [4,5-b] пиридин-1-ила, имидазо [4,5-b] пиридин-2-ила, имидазо [4,5-b] пиридин-3-ила, имидазо [4,5-b] пиридин-5-ила, имидазо [4,5-b] пиридин-6-ила и имидазо-[4,5-b] пиридин-7-ила; или
имидазо-[4,5-c] пиридильную группу, выбранную из группы, состоящей из имидазо [4,5-c] пиридин-1-ила, имидазо [4,5-c] -пиридин-2-ила, имидазо [4,5-c] пиридин-3-ила, имидазо [4,5-c]-пиридин-4-ила, имидазо [4,5-c] пиридин-6-ила, и имидазо-[4,5-c] пиридин-7-ила,
или их фармакологически приемлемые соли.
| JP, заявка вылож | |||
| Машина для прокладки снежноледяных дорог | 1941 |
|
SU63260A1 |
