Настоящее изобретение относится к трициклическим производным, представленным следующей формулой 1, или к их фармацевтически приемлемым солям, к способам их получения и к содержащим их фармацевтическим композициям.
Формула 1
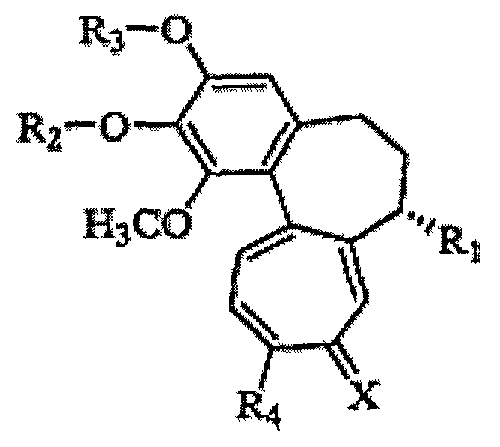
(где R1, R2, R3, R4 и X имеют указанные в описании значения).
Одно из псевдоалкалоидных соединений, колхицин, обладает противовоспалительным действием, что делает его терапевтическим агентом для лечения ревматоидных артритов [Internal Medicine, 86, No. 2, 342-345, 2000]. Производные колхицина и тиоколхицина оказывают расслабляющее действие на мышцы и обладают противовоспалительным действием (USP 5973204, EP 0870761 Al). Тиоколхикозид использовали для лечения контрактур и воспалений скелетных мышц. Кроме того, колхицин ингибирует инфильтрацию моноцитов и Т-клеток в трансплантированные органы в экспериментах на животных и в то же самое время ограничивает продуцирование TNF-α, IL-1 и IL-6, воспалительных цитокинов, что предполагает ингибирующее воздействие на иммунную реакцию [J. Am. Soc. Nephrol., 4(6), 1294-1299, 1993; Transplantation Proceedings, 32, 2091-2092, 2002]. Таким образом, колхицин является весьма привлекательным кандидатом для создания ингибитора иммунной реакции (WO 02/100824).
Колхицин ингибирует образование микротрубочек за счет взаимодействия с тубулином, что приводит к подавлению деления клеток. [The Alkaloids, 1991, 41, 125-176; USP 4533675]. Так, колхицин использовали для лечения подагры и других воспалительных заболеваний, связанных с подагрой. Однако применение колхицина при острых воспалительных заболеваниях ограничено из-за ограничений в связи с терапевтическим индексом и токсичностью в отношении желудочно-кишечного тракта [Pharmacotherapy, 11, 3, 196-211, 1991].
Все попытки получить производные колхицина в качестве противораковых лекарств до сих пор оказывались безуспешными [USP 3222253; USP 00/6080739; WO 97/01570], и лишь демеколцин использовали для лечения лейкемии. Однако токсичность в отношении желудочно-кишечного тракта и ограничения из-за терапевтического индекса до сих пор остаются проблемой при использовании демеколцина.
Авторы настоящего изобретения осуществили настоящее изобретение, создав производные колхицина, обладающие превосходными активностями в качестве противораковых и противопролиферативных агентов и ингибиторов ангиогенеза, которые теперь характеризуются стабильным терапевтическим индексом, являющимся результатом пониженной токсичности.
Задачей настоящего изобретения является создание трициклических производных или их фармацевтически приемлемых солей, обладающих превосходными активностями в качестве противораковых и противопролиферативных агентов и ингибиторов ангиогенеза со стабильным терапевтическим индексом благодаря пониженной токсичности.
Кроме того, задачей настоящего изобретения является создание способов получения трициклических производных или их фармацевтически приемлемых солей.
Дальнейшей задачей настоящего изобретения является создание фармацевтических композиций, содержащих в качестве эффективных ингредиентов трициклические производные или их фармацевтически приемлемые соли.
КРАТКОЕ ОПИСАНИЕ ФИГУР ЧЕРТЕЖЕЙ
ФИГ.1 представляет график, демонстрирующий изменения объема опухоли у BALB/c голых мышей с трансплантированной клеточной линией рака легких человека NCI-H460 после введения трициклических производных настоящего изобретения (пример 8).
ФИГ.2 представляет график, демонстрирующий изменения веса тела BALB/c голых мышей с трансплантированной клеточной линией рака легких человека NCI-H460 после введения трициклических производных настоящего изобретения (пример 8).
ФИГ.3 представляет график, демонстрирующий изменения объема опухоли у BALB/c голых мышей с трансплантированной клеточной линией рака легких человека NCI-H460 после введения трициклических производных настоящего изобретения (пример 12) в различных концентрациях (1, 3, 10 мг/кг).
ФИГ.4 представляет график, демонстрирующий изменения веса тела BALB/c голых мышей с трансплантированной клеточной линией рака легких человека NCI-H460 после введения трициклических производных настоящего изобретения (пример 12) в различных концентрациях (1, 3, 10 мг/кг).
ФИГ.5 представляет ряд фотографий, демонстрирующих объем опухолей, растущих у BALB/c голых мышей с трансплантированной клеточной линией рака легких человека NCI-H460, которые были вырезаны на 14 день после введения трициклических производных настоящего изобретения.
ФИГ.6 представляет ряд фотографий, демонстрирующих активности трициклических производных настоящего изобретения в отношении ингибирования ангиогенеза в HUVEC клетках.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Настоящее изобретение относится к трициклическим производным, представленным следующей формулой 1, или к их фармацевтически приемлемым солям.
Формула 1
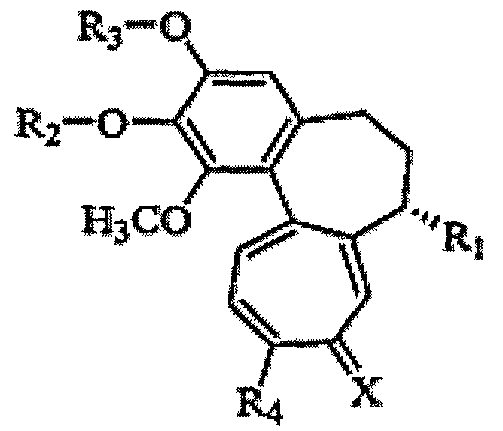
(где
(1) R1 представляет -Т1-B1;
где T1 представляет -X1-, -X1-C(X2)-, -N(R5)-, -N(R5)C(X2)-,
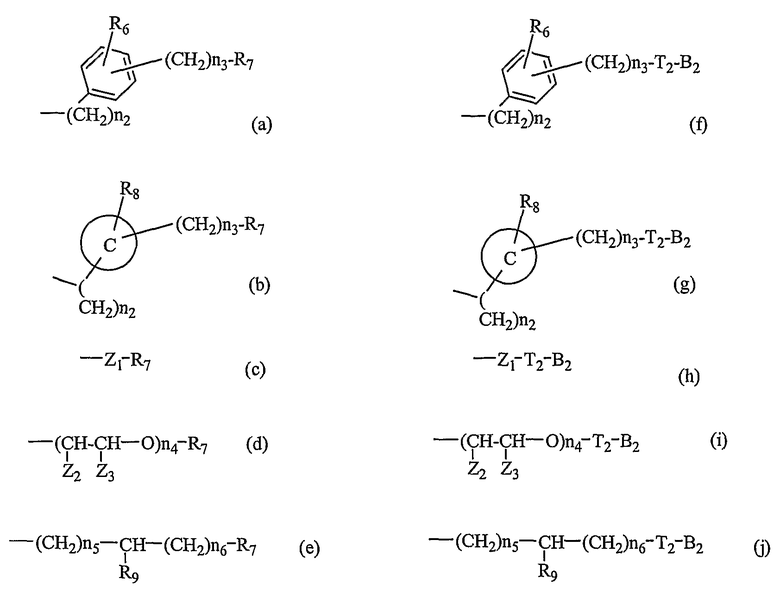
-N(R5)S(O)n1-, -N(R5)C(O)-X1- или -N(R5)C(X1)NH-, где X1 и X2, каждый, представляет O или S, каждый R5 представляет H или C1-C5алкильную группу, n1 представляет целое число 1-2; и B1 выбирают из группы, состоящей из следующих значений(a)-(j):
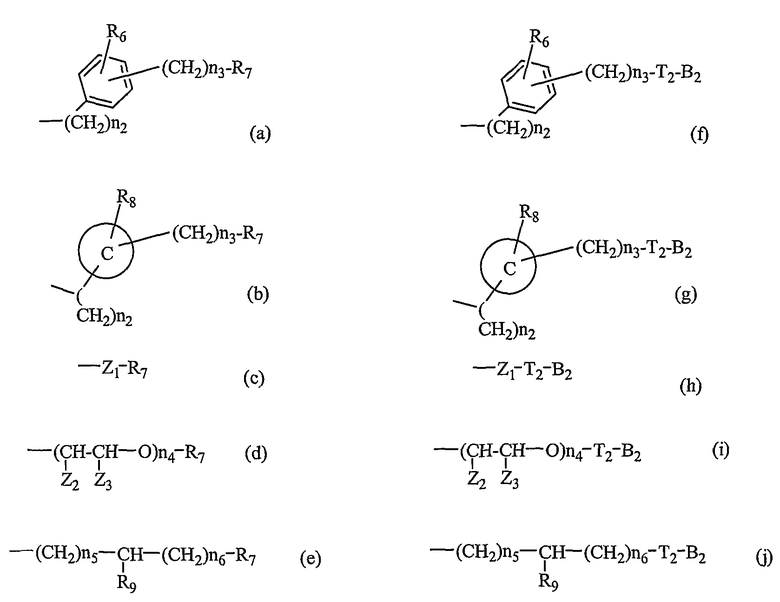
где R6 и R8, каждый, представляет H, галоген, гидрокси, С1-C3алкокси, амино, нитро, циано или C1-C3низшую алкильную группу; R7 и R9, каждый, независимо представляет галоген, гидрокси, меркапто, -ONO, -ONO2 или SNO, где R7 и R9 одинаковы или различны;
 представляет C5-C6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1-2 гетероатома, где гетероатом выбирают из группы, состоящей из O, S и N, предпочтительно
представляет C5-C6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1-2 гетероатома, где гетероатом выбирают из группы, состоящей из O, S и N, предпочтительно

более предпочтительно представляет C1 (пиридильную группу), замещенную в положениях 2 и 6 или в положениях 2 и 5, C7 (пирролильную группу), замещенную в положениях 2 и 5 или в положениях 2 и 4, C11 (тиофенильную группу) или C12 (фуранильную группу);
Z1 представляет C1-C10 разветвленную или неразветвленную алкильную группу, предпочтительно C2-C5 разветвленную или неразветвленную алкильную группу или циклоалкильную группу, содержащую заместитель; Z2 и Z3, каждый, независимо представляет H или метильную группу, где Z3 представляет H, если Z2 представляет метильную группу, Z2 представляет H, если Z3 представляет метильную группу; T2 представляет -X1- или -X1-C(X2)-, где X1 и X2, каждый, независимо представляет O или S; B2 выбирают из группы, состоящей из указанных (a), (b), (c), (d) или (e); n2 представляет целое число 0-3, n3 представляет целое число 0-5, n4 представляет целое число 1-5, n5 и n6, каждый, независимо представляет целое число 1-6;
(2) R2 и R3, каждый, независимо представляет H, -PO3H2, фосфонат, сульфат, C3-C7 циклоалкил, C2-C7 алкенил, C2-C7 алкинил, C1-C7 алканоил, C1-C7 неразветвленный или разветвленный алкил или сахар, где сахаром является моносахарид, такой как глюкуронил, глюкозил или галактозил;
(3) R4 представляет OCH3, SCH3 или NR10R11, где R10 и R11, каждый, независимо представляет H или C1-5 алкил;
(4) X представляет О или S.
Предпочтительно в соединении формулы 1
(1) R1 представляет -T1-B1;
где Ti представляет -N(R5)C(X2)-, -N(R5)C(О)-X1- или -N(R5)C(X1)NH-, где X1 и X2, каждый, представляет O, каждый R5 представляет H или C1-C5 алкильную группу; и B1 выбирают из группы, состоящей из следующих значений (a)-(j):
где R6 и R8, каждый, представляет H, галоген, гидрокси, C1-C3 алкокси, амино, нитро, циано или C1-C3 низшую алкильную группу; R7 и R9, каждый, независимо представляет галоген, гидрокси, меркапто(тиол), -ONO, -ONO2 или SNO, где R7 и R9 одинаковы или различны;
 представляет C5-C6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1-2 гетероатома, где гетероатом выбирают из группы, состоящей из О, S и N, предпочтительно состоящей из
представляет C5-C6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1-2 гетероатома, где гетероатом выбирают из группы, состоящей из О, S и N, предпочтительно состоящей из
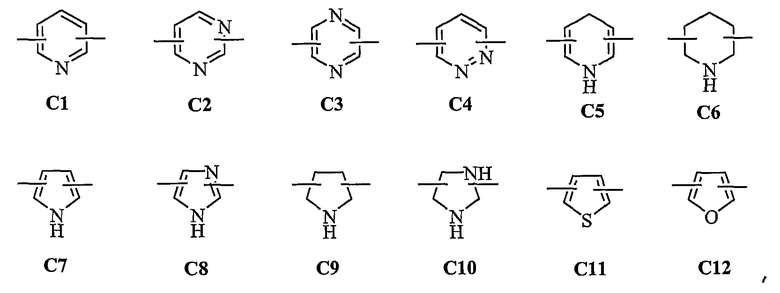
более предпочтительно C1 (пиридильной группы), замещенной в положениях 2 и 6 или положениях 2 и 5, C7 (пирролильной группы), замещенной в положениях 2 и 5 или положениях 2 и 4, C11 (тиофенильной группы) или C12 (фуранильной группы), причем связь заместителей может быть в симметричном или асимметричном положениях; Z1 представляет C1-C10 неразветвленную или разветвленную алкильную группу, предпочтительно C2-C5 неразветвленную или разветвленную алкильную группу или циклоалкильную группу, содержащую заместитель; Z2 и Z3, каждый, независимо представляет H или метильную группу, где Z3 представляет H, если Z2 представляет метильную группу, Z2 представляет H, если Z3 представляет метильную группу; T2 представляет -X1- или -X1-C(X2)-, где X1 и X2, каждый, представляет O или S; B2 выбирают из группы, состоящей из указанных (a), (b), (c), (d) или (e); n2 представляет целое число 0-3, n3 представляет целое число 0-5, n4 представляет целое число 1-3, n5 и n6, каждый, независимо представляет целое число 1-3;
(2) R2 и R3, каждый, независимо представляет C3-C7 циклоалкил или C1-C7 алкил;
(3) R4 представляет SCH3 или OCH3;
(4) X представляет О или S.
Предпочтительно, чтобы соединения формулы 1 включали
1) 6-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]никотинамид;
2) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 5-нитрооксиметилфуран-2-карбоновой кислоты;
3) N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид;
4) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид;
5) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 6-нитрооксиметилпиридин-2-карбоновой кислоты;
6) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид
5-нитрооксиметилтиофен-2-карбоновой кислоты;
7) N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид;
8) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-2-фтор-3-нитрооксиметилбензамид;
9) 2-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид;
10) 2-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
11) N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]2-фтор-3-нитрооксиметилбензамид;
12) 3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
13) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамид;
14) 3-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]- 5-нитрооксиметилбензамид;
15) N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамид;
16) 4-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
17) 2-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
18) 3-гидрокси-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
19) 3,5-бис-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
20) 2-гидрокси-4-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
21) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 4-нитрооксиметилтиофен-2-карбоновой кислоты;
22) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 3-нитрооксиметилтиофен-2-карбоновой кислоты;
23) 2-(3-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамид;
24) 3-(2-нитрооксиэтил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
25) 5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 3-нитрооксибензойной кислоты;
26) 5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 4-нитрооксимасляной кислоты;
27) 6-(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 3-нитрооксиметилбензойной кислоты;
28) 6-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 4-нитрооксимасляной кислоты;
29) 2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 3-нитрооксиметилбензойной кислоты;
30) 2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-S,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 4-нитрооксимасляной кислоты;
31) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 3-нитрооксиметилбензойной кислоты;
32) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 4-нитрооксимасляной кислоты;
33) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]бензиловый эфир 3-нитрооксиметилбензойной кислоты;
34) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]бензиловый эфир 4-нитрооксимасляной кислоты;
35) 2-нитрозотио-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
36) 3-нитрозооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
37) 3-фтор-5-нитрозооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
38) 3-нитрозотиометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
39) 3-фтор-5-нитрозотиометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
40) 3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3,10-тетраметокси-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
41) 3-нитрооксиметил-N-метил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
42) 3-фтор-N-метил-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид;
43) 2-(3-фтор-5-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамид; или
44) 2-(2-фтор-5-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамид.
В настоящем изобретении предложены также фармацевтически приемлемые соли соединения, представленного формулой 1. Фармацевтически приемлемые соли настоящего изобретения могут включать соли присоединения кислот соединения настоящего изобретения, если это соединение является полностью основным. Такие соли присоединения кислот включают соли, содержащие фармацевтически приемлемый анион неорганической кислоты, такой как галогенводород, или анион органической кислоты, или соли серной кислоты или фосфорной кислоты или соли трифторуксусной кислоты, лимонной кислоты или малеиновой кислоты. И они включают, например, гидрохлориды, гидробромиды, фосфонаты, сульфаты, алкилсульфонаты, арилсульфонаты, ацетаты, бензоаты, цитраты, малеаты, фумараты, сукцинаты, лактаты и тартраты. Если соединение настоящего изобретения является полностью кислотным, фармацевтически приемлемые соли могут включать неорганические соли или органические соли, образующие фармацевтически приемлемые катионы. Указанные неорганические соли включают соли натрия, соли калия, соли кальция или соли магния и т.д., указанные органические соли включают соли метиламина, соли диметиламина, соли триметиламина, соли пиперидина или соли морфолина и т.д.
В настоящем изобретении предложен также способ получения трициклических производных, представленных формулой 1. Способ получения трициклических производных настоящего изобретения раскрыт в приводимых далее схемах 1-8. Более конкретно, для формулы 1, если R1 представляет -Т1-B1 и B1 представляет одно из указанных значений (a), (b), (c), (d) и (e), производные получают в соответствии со способами, представленными на схемах 1-6. С другой стороны, для формулы 1, если R1 представляет -Т1-B1 и B1 представляет одно из указанных значений (f), (g), (h), (i) и (j), производные получают способами, представленными на схемах 7 и 8. И конкретное соединение формулы 1 представлено общими формулами (IIa), (IIb), (IIc), (IId), (IIe), (IIf), (IIg), (IIh), (IIi), (IIj), (IIk), (IIl), (IIm), (IIn), (IIo) и (IIp) на схемах 1-8.
Схема 1
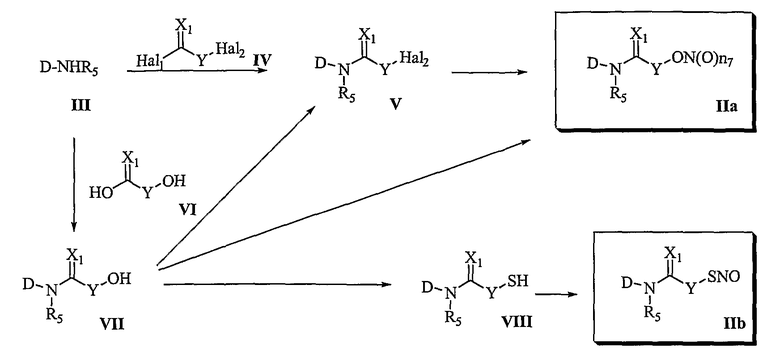
Схема 2
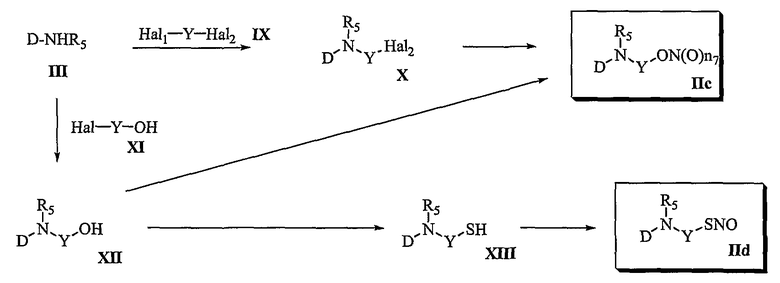
Схема 3

Схема 4
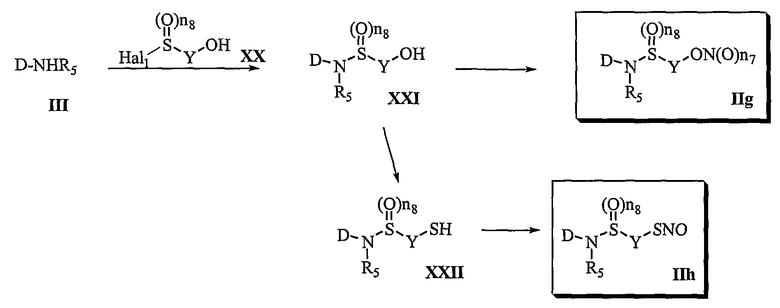
Схема 5
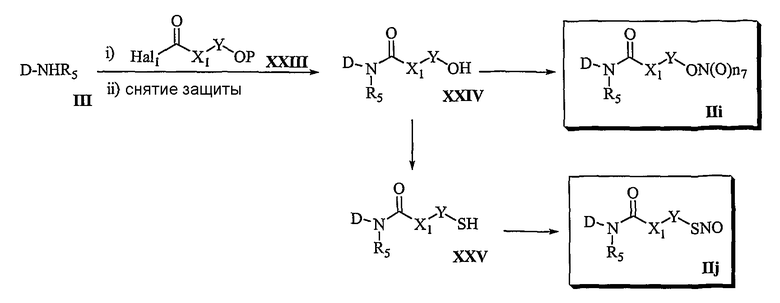
Схема 6
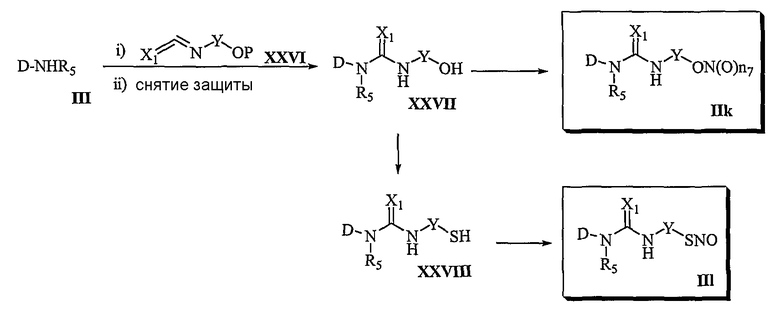
Схема 7
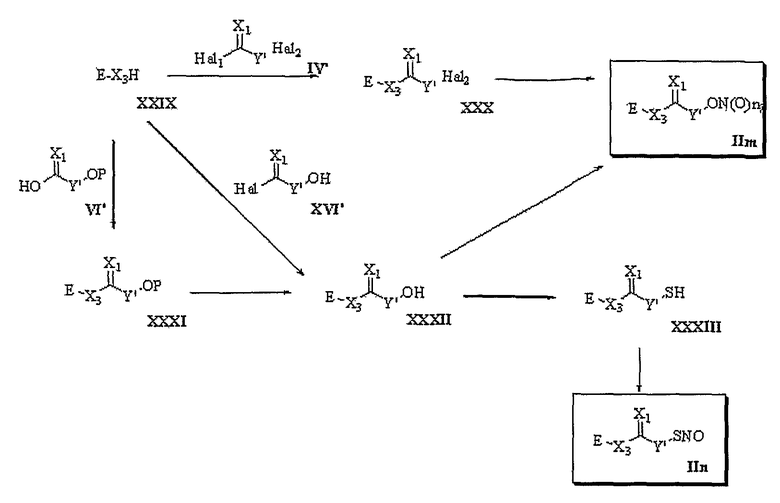
Схема 8
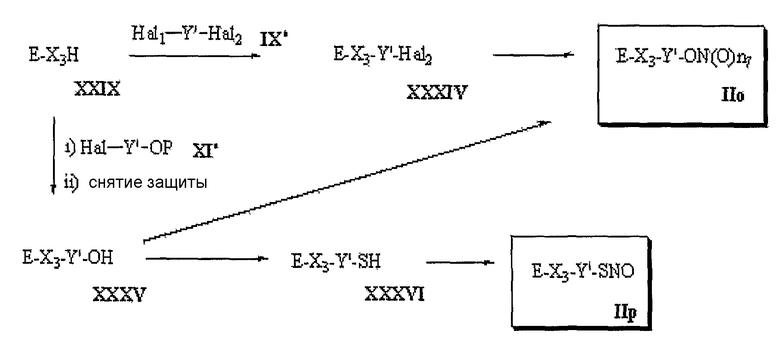
В вышеприведенных схемах Е имеет соответственно следующие значения: Е1-Е6;
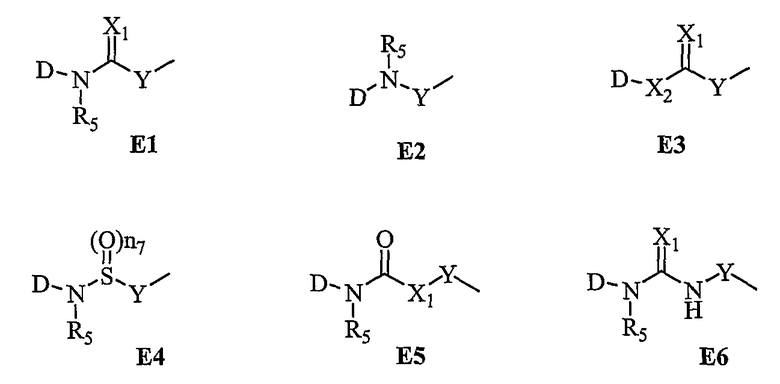
где X1, X2 и X3, каждый, представляет О или S.
В вышеуказанной схеме D представляет 
и R2, R3, R4 и X имеют указанные для формулы 1 значения;
R5 представляет H или низший алкил; X1, X2 и X3, каждый, независимо представляет O или S; Hal1 и Hal2 представляют галогены; Hal1 и Hal2 в общих формулах (IV) и (IX), каждый, представляет одинаковые или различные галогены, например F, Cl, Br или I; P представляет обычную защитную группу для гидрокси, такую как метоксиметил, трет-бутилдиметилсилил или бензил; Y и Y' одинаковы или различны и представляют следующие общие формулы (a'), (b'), (c'), (d') и (e') соответственно:

где  , R6, R8, R9, Z1, Z2, Z3, n2, n3, n4, n5 и n6 имеют указанные для формулы 1 значения; n7 и n8 представляют целое число 1-2.
, R6, R8, R9, Z1, Z2, Z3, n2, n3, n4, n5 и n6 имеют указанные для формулы 1 значения; n7 и n8 представляют целое число 1-2.
Способ получения трициклических производных настоящего изобретения проиллюстрирован далее более подробно.
Способ 1
В соответствии со способом 1 получения соединений формул (IIa) и (IIb) настоящего изобретения соединение, представленное формулой (V), получают, используя реакцию амидирования с получением амина формулы (III), который подвергают взаимодействию с галогеном формулы (IV), что составляет стадию 1. На стадии 1, основание можно исключить, но реакцию обычно ведут в растворителе, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, толуол или диметилформамид и т.д., которые не оказывают никакого влияния на ход реакции амидирования, в присутствии пиридина, триэтиламина, диэтилизопропиламина или N-метилморфолина и т.д., оснований, которые обычно можно использовать для реакций амидирования. Температура реакции конкретно не ограничена, но обычно реакцию ведут при охлаждении или при повышенной температуре, предпочтительно ее осуществляют при комнатной температуре.
На стадии 2, превращение соединения формулы (V), полученного ранее на стадии 1, в нитрооксисоединение (n7=2) формулы (IIa) и в нитрозооксисоединение (n7=1) формулы (IIa) осуществляют, используя реакцию нитрования и реакцию нитрозирования соответственно. Для реакции нитрования необходимо соединение, которое способно превращать галоген в нитрат, и реакцию осуществляют, используя нитрат серебра (AgNO3), трет-бутиламмонийнитрат (BU4NNO3) и т.д., в присутствии хлороформа, ацетонитрила, смеси ацетонитрила и водного раствора, или дихлорметана, причем все они являются растворителями, которые не оказывают влияния на ход реакции. В реакции нитрозирования можно использовать соединения, которые способны превращать галоген в нитрозат, и реакцию осуществляют, предпочтительно используя нитрит серебра (AgNO2) или нитрит натрия (NaNO2) в присутствии хлороформа, ацетонитрила, смеси ацетонитрила и водного раствора, водного раствора или дихлорметана, которые также являются растворителями, которые не оказывают влияния на ход реакции. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре.
Другой способ получения соединения формулы (IIa) состоит в следующем: осуществляют взаимодействие соединения формулы (III) с соединением формулы (VI), получая соединение формулы (VII), и затем осуществляют превращение соединения формулы (VII) в соединение формулы (IIa). Реакцию соединения формулы (III) и соединения формулы (VI) осуществляют в присутствии связующего агента, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDCI), гидрата 1-гидроксибензотриазола (HOBt) или 1,3-дициклогексилкарбодиимида (DCC). Указанную реакцию можно осуществить, не используя основание, но обычно используют основания, такие как 4-диметиламинопиридин, пиридин, триэтиламин, диэтилизопропиламин, N-метилморфолин или диметилфениламин и т.д., которые можно использовать в реакции амидирования, в растворителе, который не оказывает вредного воздействия на ход реакции, например в ацетонитриле, диметилформамиде, дихлорметане, и т.д.
Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре. Непосредственное превращение соединения формулы (VII) в соединение формулы (IIa) осуществляют при взаимодействии спирта с трифенилфосфином (PPh3), N-бромсукцинимидом (NBS) и нитратом серебра или нитритом серебра. Реакцию осуществляют в растворителе, который не влияет на ход реакции, таком как хлороформ, ацетонитрил, дихлорметан, смесь ацетонитрила и дихлорметана, и т.д. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при комнатной температуре. Другим способом превращения соединения формулы (VII) в соединение формулы (IIa) является следующий: вначале осуществляют превращение соединения формулы (VII) в галогенсодержащее соединение формулы (V) и затем снова осуществляют его превращение в соединение формулы (IIa). На этом этапе превращение в галогенсодержащее соединение осуществляют, используя реагент, который обычно используют для превращения гидроксигруппы в галогенсодержащее соединение, например, трибромфосфин, тетрабромметан и т.д., в присутствии хлороформа, ацетонитрила, дихлорметана и т.д., которые являются растворителями, которые не оказывают вредного воздействия на ход реакции. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при комнатной температуре.
Способы получения соединений формулы (IIb) в способе 1 настоящего изобретения являются следующими: водород в спирте формулы (VII) превращают в отщепляемую группу, такую как мезилат, тозилат или трифлат, используя реакцию с тиоацетатом калия, получая сложный эфир тиоацетата. В результате гидролиза соединения в присутствии основания получают соединение формулы (VIII). На этом этапе основание выбирают из традиционных оснований, которые способны осуществить гидролиз сложноэфирного соединения, например, из гидроксида натрия, гидроксида калия или тиометоксида натрия. В качестве растворителя в указанной реакции предпочтительны спиртовые растворы, такие как растворы метанола или этанола. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре. Взаимодействие соединения формулы (VIII) с нитритом натрия в кислотных условиях приводит к превращению соединения в нитрозотиосоединение формулы (IIb). Растворитель для реакции выбирают из группы, состоящей из метанола, этанола, ацетонитрила, смеси ацетонитрила и водного раствора или дихлорметана и т.д., которые не влияют на ход реакции. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре.
Способ 2
В соответствии со способом 2 настоящего изобретения получают соединения формул (IIc) и (IId). В частности, на стадии 1 осуществляют взаимодействие соединения формулы (III) с соединением формулы (IX), получая соединение формулы (X). Указанную реакцию осуществляют по аналогии со способом, раскрытым для способа 1, где превращение соединения формулы (III) в соединение формулы (V) осуществляют, используя реакцию амидирования.
На стадии 2 превращение соединения формулы (X), полученного на стадии 1, в соединение формулы (IIc) осуществляют, используя реакцию нитрования наряду с реакцией нитрозирования. Указанную реакцию осуществляют по аналогии со способом, раскрытым для способа 1, где осуществляют превращение соединения формулы (V) в соединение формулы (IIa).
Другим способом получения соединения формулы (IIc) является следующий: осуществляют взаимодействие соединения формулы (III) с соединением формулы (XI), получая соединение формулы (XII), и затем осуществляют превращение соединения формулы (XII) в соединение формулы (IIc). Реакцию соединения формулы (XI) с соединением формулы (III) осуществляют по аналогии со способом, раскрытым для способа 1, где превращение соединения формулы (III) в соединение формулы (V) осуществляют, используя реакцию амидирования. Превращение соединения формулы (XII) в соединение формулы (IIc) осуществляют в тех же условиях, что указаны для превращения соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XII) в соединение формулы (IId) осуществляют в тех же условиях, которые указаны для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Способ 3
В соответствии со способом 3 настоящего изобретения получают соединения формул (IIe) и (IIf). В частности, на стадии 1 осуществляют реакцию соединения формулы (XIV) с соединением формулы (IV), получая соединение формулы (XV). Реакция в указанном способе является реакцией превращения в сложный эфир спирта (X2=О) или тиоспирта (X2=S) с помощью ацил- или тиоацилгалогенида, которую осуществляют в присутствии оснований, которые обычно используют для реакций этерификации. Предпочтительными основаниями являются пиридин, 4-диметиламинопиридин, триэтиламин, диэтилизопропиламин, 2,6-лутидин, гидрид натрия (NaH), карбонат цезия или гидроксид натрия, и их можно использовать вместе с катализатором переноса заряда, таким как бензилтриэтиламмонийхлорид. Кроме того, вышеуказанную реакцию предпочтительно вести в растворителе, который не оказывает вредного воздействия на ход реакции, например, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, толуол, диметилформамид, ацетонитрил или водный раствор. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре.
На стадии 2 превращение соединения формулы (XV), полученного на стадии 1, в соединение формулы (IIe) осуществляют, используя реакцию нитрования наряду с реакцией нитрозирования. Указанную реакцию осуществляют по аналогии со способом, раскрытым в способе 1, где осуществляют превращение соединения формулы (V) в соединение формулы (IIa).
Другим способом получения соединений формулы (IIe) является следующий: осуществляют взаимодействие соединения формулы (XIV) с соединением формулы (VI'), у которого спиртовая группа защищена, в результате чего получают соединение формулы (XVII) с последующей реакцией удаления защитных групп, получая соединение формулы (XVIII). Осуществляют превращение полученного соединения в соединение формулы (IIe). Реакцию соединения формулы (XIV) с соединением формулы (VI') осуществляют, используя реакцию этерификации спирта (X2=О) или тиоспирта (X2=S) и карбоновой кислоты или тиокарбоновой кислоты. Реакцию осуществляют или в водном растворе, подкисленном кислотой, такой как хлористоводородная кислота, серная кислота, додецилбензолсульфоновая кислота или п-толуолсульфоновая кислота, при комнатной температуре или при повышенной температуре, или в таких же условиях, что и условия, используемые для превращения соединения формулы (III) в соединение формулы (VII) в способе 1. Другую реакцию этерификации осуществляют, используя реакцию Misunobu, в которой используют трифенилфосфин и диэтилазодикарбоксилат в растворителе, который не влияет на ход реакции, и растворитель предпочтительно выбирают из группы, состоящей из дихлорметана, хлороформа, тетрагидрофурана, диэтилового эфира, толуола или ацетонитрила. Температура реакции конкретно не ограничена, но обычно реакцию осуществляют при охлаждении или при комнатной температуре. Реакции введения и удаления групп, защищающих спиртовую группу, осуществляют обычным способом, известным в органическом синтезе.
Реакцию соединения формулы (XIV) с соединением формулы (XVI) осуществляют, получая соединение формулы (XVIII) по аналогии со способом, раскрытым в способе 3, где осуществляют превращение соединения формулы (XIV) в соединение формулы (XV).
Превращение соединения формулы (XVIII) в соединение формулы (IIe) осуществляют в таких же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XVIII) в соединение формулы (IIf) осуществляют в таких же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Способ 4
В соответствии со способом 4 настоящего изобретения получают соединения формул (IIg) и (IIh). В частности, на стадии 1 осуществляют реакцию соединения формулы (III) с соединением формулы (XX), получая соединение формулы (XXI).
Если соединение формулы (IIg) представлено сульфиниламидом (n8=1), реакцию соединения формулы (III) с сульфинилгалогенидом формулы (XX) осуществляют в отсутствие основания или в присутствии основания, которое можно использовать в реакции амидирования, например, такого как пиридин, триэтиламин, диэтилизопропиламин, N-метилморфолин или диметилфениламин, в растворителе, который не оказывает вредного воздействия на ход реакции, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, толуол или диметилформамид. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре.
Если соединение формулы (IIg) представлено сульфониламидом, (n8=2), реакцию соединения формулы (III) с сульфонилгалогенидом формулы (XX) осуществляют или в отсутствие основания или в присутствии основания, которое обычно используют в реакции амидирования, например, в присутствии пиридина, триэтиламина, диэтилизопропиламина, N-метилморфолина, гидроксида натрия, карбоната натрия или карбоната калия, в растворителе, который не оказывает вредного воздействия на ход реакции, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, толуол или диметилформамид. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при комнатной температуре.
На стадии 2 превращение соединения формулы (XXI), полученного на стадии 1, в соединение формулы (IIg) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XXI) в соединение формулы (IIh) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Способ 5
В соответствии со способом 5 настоящего изобретения соединения формулы (IIi) и формулы (IIj) получают следующим образом. На стадии 1 осуществляют взаимодействие соединения формулы (III) с соединением формулы (XXIII), в котором спиртовая группа защищена, с последующей реакцией для удаления защитной группы. Осуществляют превращение соединения формулы (XXIV), полученного в вышеуказанной реакции, в соединение формулы (IIi). На этом этапе реакцию соединения формулы (III) с соединением формулы (XXIII) осуществляют, используя реагент сочетания, такой как карбонилдихлорид, трифосген, ди-трет-бутил дикарбонат или 1,1'-карбонилдиимидазол и т.д. Указанную реакцию можно вести в отсутствие основания или в присутствии основания, которое обычно используют в реакции амидирования, например, в присутствии пиридина, триэтиламина, диэтилизопропиламина, N-метилморфолина или диметилфениламина, в таком растворителе, который не оказывает вредного воздействия на ход реакции, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, этанол или диметилформамид. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при комнатной температуре. Реакцию удаления защитных групп осуществляют обычными способами, известными в органическом синтезе.
На стадии 2 превращение соединения формулы (XXIV), полученного на стадии 1, в соединение формулы (IIi) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XXIV) в соединение формулы (IIj) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Способ 6
В соответствии со способом 6 настоящего изобретения соединения формулы (IIk) и формулы (IIl) получают следующим образом. На стадии 1 осуществляют взаимодействие соединения формулы (III) с соединением формулы (XXVI), спиртовые группы которого защищены, а затем осуществляют реакцию для удаления защитных групп. Затем осуществляют превращение полученного соединения формулы (XXVII) в соединение формулы (IIk). На этом этапе реакцию соединения формулы (III) с соединением формулы (XXVI) осуществляют или в отсутствие основания или в присутствии основания, которое приемлемо для осуществления реакции амидирования, например, в присутствии пиридина, триэтиламина, диэтилизопропиламина или N-метилморфолина и т.д., в растворителе, который не оказывает вредного воздействия на ход реакции, таком как дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, бензол, ацетонитрил и т.д. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при комнатной температуре. Реакции введения и удаления защитных групп у спиртовой группы осуществляют, используя обычные способы органического синтеза.
На стадии 2 превращение соединения формулы (XXVII), полученного на стадии 1, в соединение формулы (IIk) осуществляют в тех же условиях, которые соответствуют превращению соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XXVII) в соединение формулы (IIl) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Способ 7
В соответствии со способом 7 настоящего изобретения соединения формулы (IIm) и формулы (IIn) получают из соединения формулы (XXIX) по аналогии со способом, раскрытым в способе 3.
Способ 8
В соответствии со способом 8 настоящего изобретения соединения формулы (IIo) и формулы (IIp) получают следующим образом. На стадии 1 осуществляют взаимодействие соединения формулы (XXIX) с соединением формулы (IX'), получая соединение формулы (XXXIV). Указанную реакцию осуществляют путем реакции этерификации спирта (X2=O) или тиоспирта (X2=S) алкилгалогенидом в присутствии основания, которое можно использовать в реакции этерификации. В качестве оснований для этой цели пригодны гидрид натрия (NaH), трет-бутоксид калия (t-BuOK), н-BuLi, гидроксид натрия, гидроксид калия и предпочтительны катализаторы переноса фазы, такие как бензилтриэтиламмонийхлорид и т.д., или краун эфир. Реакцию предпочтительно ведут в растворителе, который не оказывает вредного воздействия на ход реакции, например, в дихлорметане, хлороформе, тетрагидрофуране, диэтиловом эфире, толуоле, диметилформамиде, водном растворе, диметилсульфоксиде или бензоле и т.д. Температура реакции конкретно не ограничена, но обычно реакцию можно вести при охлаждении или при повышенной температуре, и предпочтительно реакцию ведут при охлаждении или при комнатной температуре.
На стадии 2 превращение соединения формулы (XXXIV), полученного на стадии 1, в соединение формулы (IIo) осуществляют, используя реакцию нитрования или реакцию нитрозирования. Указанную реакцию осуществляют по аналогии со способом, раскрытым в способе 1, где осуществляют превращение соединения формулы (V) в соединение формулы (IIa).
Другим способом получения соединения формулы (IIo) является следующий. Осуществляют реакцию взаимодействия соединения формулы (XXIX) с соединением формулы (XI'), у которого спиртовая группа защищена защитной группой, с последующим удалением защитной группы, получая соединение формулы (XXXV). Осуществляют превращение соединения формулы (XXXV) в соединение формулы (IIo). Реакцию соединения формулы (XXIX) с соединением формулы (XI') ведут в тех же условиях, которые используют для превращения соединения формулы (XXIX) в соединение формулы (XXXIV), используя реакцию этерификации в способе 8.
Превращение соединения формулы (XXXV) в соединение формулы (IIo) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIa) в способе 1.
Превращение соединения формулы (XXXV) в соединение формулы (IIp) осуществляют в тех же условиях, которые используют для превращения соединения формулы (VII) в соединение формулы (IIb) в способе 1.
Целевые соединения, получаемые с помощью раскрытых выше реакций, можно выделить и очистить обычными способами, такими как хроматография на колонке с силикагелем, перекристаллизация и т.д.
В настоящем изобретении предложены также фармацевтические композиции, содержащие в качестве эффективного ингредиента трициклические производные, представленные формулой 1, или их фармацевтически приемлемые соли.
Трициклические производные настоящего изобретения или их фармацевтически приемлемые соли демонстрируют очень высокую цитотоксичность в отношении раковых клеточных линий, но обладают гораздо меньшей токсичностью в отношении тестируемых животных, нежели инъекции колхицина или таксола.
Если трициклические производные настоящего изобретения вводят BALB/c голым мышам, которым трансплантирована клеточная линия рака легких человека NCI-H460, размер и вес опухоли заметно снижается пропорционально введенной дозе.
Трициклические производные настоящего изобретения обладают также высокой активностью ингибирования ангиогенеза в HUVEC клетках.
Поэтому трициклические производные настоящего изобретения или их фармацевтически приемлемые соли можно эффективно использовать в качестве противораковых агентов, противопролиферативных агентов и в качестве ингибиторов ангиогенеза.
Композиция настоящего изобретения может дополнительно включать помимо трициклических производных или их фармацевтически приемлемых солей, по меньшей мере, один из активных ингредиентов, обладающих такими же или аналогичными функциями, что и указанные трициклические производные или их фармацевтически приемлемые соли.
Указанные трициклические производные или их фармацевтически приемлемые соли можно вводить перорально или парентерально, и их можно приготовить в форме обычных фармацевтических препаратов. Трициклические производные настоящего изобретения или их фармацевтически приемлемые соли можно приготовить для перорального или парентерального введения, смешивая их с традиционно используемыми наполнителями, сухими разбавителями, связующими, смачивающими агентами, разрыхлителями, разбавителями или эксципиентами. Твердыми формами для перорального введения служат таблетки, пилюли, порошки, гранулы и капсулы. Указанные твердые формы готовят, смешивая с одним или более из подходящих эксципиентов, таких как крахмал, карбонат кальция, сахароза, лактоза и желатин, и т.д. Помимо простых эксципиентов можно использовать смазывающие агенты, например, стеарат магния, тальк, и т.д. Жидкими формами для перорального введения служат суспензии, растворы, эмульсии и сиропы, и вышеуказанные формы могут содержать различные эксципиенты, такие как смачивающие агенты, подсластители, ароматизаторы и консерванты в дополнение к обычно используемым простым разбавителям, таким как вода и жидкий парафин. Формами для парентерального введения являются стерилизованные водные растворы, водорастворимые эксципиенты, суспензии, эмульсии, лиофилизированные агенты и суппозитории. Нерастворимые в воде эксципиенты и суспензии могут содержать в дополнение к активному соединению или соединениям пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, сложные эфиры для инъекций, такие как этилолеат, и т.д. Суппозитории могут содержать witepsol, макрогол, tween 61, масло какао, лауриновое масло, глицерин и желатин.
Композиции настоящего изобретения можно приготовить в формах либо для перорального, либо для парентерального введения (например, для внутривенного, подкожного, внутрибрюшинного введения или для локальных инъекций) и дозу определяют, исходя из веса, возраста, пола, состояния здоровья и диеты пациента и способа введения, скорости выведения лекарства и тяжести заболевания. Предпочтительная эффективная доза трициклических производных настоящего изобретения составляет 3-300 мг/кг (веса тела), и препарат вводят один раз или несколько раз в день.
ПРИМЕРЫ
Практические и предпочтительные варианты настоящего изобретения являются иллюстративными, как представлено в следующих примерах.
Однако настоящее изобретение следующими примерами не ограничивается.
7-Деацетилколхицин, который используют в приводимых далее примерах, получают способом, раскрытым в [EP 0493064; Synthetic Communications 1997, 27(2), 293-296].
7-Амино-1,2,3-триметокси-10-метилсульфанил-6,7-дигидро-5H-бензо[a]гептален-9-он получают способом, раскрытым в (WO 9421598; Bioorganic & Medicinal Chemistry, Vol 5, No. 12, pp 2277-2282, 1997).
Тиодемеколцин получают способом, раскрытым в (J. Med. Chem, 1985, 28, 1204-1208).
(7S)-7-амино-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-6,7-дигидро-5H-бензо[a]гептален-9-он,
(7S)-7-амино-3-изопропокси-1,2-диметокси-10-метилсульфанил-6,7-дигидро-5H-бензо[a]гептален-9-он,
(7S)-7-амино-3-этокси-1,2-диметокси-10-метилсульфанил-6,7-дигидро-5H-бензо[a]гептален-9-он получают способом, раскрытым в (WO 9611184).
Пример 1:
Получение 6-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]никотинамида
<Стадия 1>
Получение 6-гидроксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]никотинамида
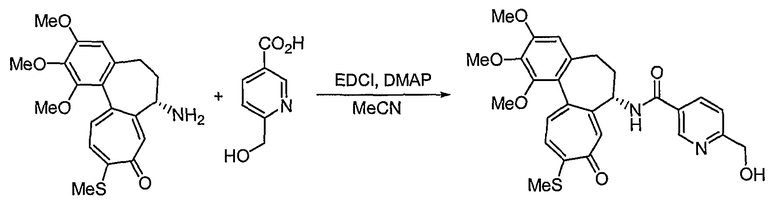
6-Гидроксиметилникотиновую кислоту синтезируют способом, раскрытым в {Bioorg. Med. Chem. Lett, 1996, 6, 3025-3028).
К раствору 7-амино-1,2,3-триметокси-10-метилсульфанил-6,7-дигидро-5H-бензо[a]гептален-9-она (300 мг, 0,80 ммоль), 6-гидроксиметилникотиновой кислоты (135 мг, 0,88 ммоль) и DMAP (60 мг, 0,48 ммоль) в 10 мл ацетонитрила добавляют EDCI (308 мг, 1,60 ммоль) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 2 час. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:метанол = 8:1), получая 244 мг (выход: 60%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,07-2,15 (м, 1H), 2,31-2,44 (м, 2H), 2,45 (с, 3H), 2,56-2,59 (м, 1H), 3,75 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,66 (кв, J=10,2 Гц, 2H), 4,90-4,93 (м, 1H), 6,56 (с, 1H), 7,13 (т, J=9,1 Гц, 2H), 7,40 (д, J=10,2 Гц, 1H), 7,52 (с, 1H), 8,15 (дд, J=2,2, 5,8 Гц, 1H), 8,80 (д, J=6,9 Гц, 1H), 8,96 (с, 1H).
<Стадия 2>
Получение 6-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]никотинамида
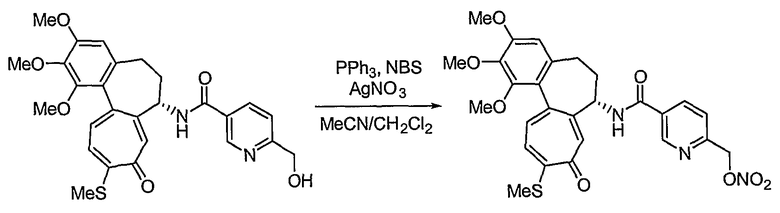
Соединение (100 мг, 0,19 ммоль), полученное на стадии 1 примера 1, и трифенилфосфин (57 мг, 0,21 ммоль) растворяют в смеси ацетонитрил/дихлорметан (1,25 мл/0,5 мл), к этому добавляют NBS (42 мг, 0,23 ммоль) при -35°C. Реакционную смесь перемешивают в течение 20 мин. Затем медленно добавляют по каплям нитрат серебра (40 мг, 0,23 ммоль) при комнатной температуре, и реакционную смесь перемешивают при комнатной температуре в течение 18 час. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:метанол = 10:1), с получением 18 мг (выход: 35%, в виде твердого вещества желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,09-2,13 (м, 1H), 2,31-2,43 (м, 2H), 2,46 (с, 3H), 2,55-2,64 (м, 1H), 3,75 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,93-4,98 (м, 1H), 5,50 (с, 2H), 6,56 (с, 1H), 7,16 (т, J=10,9 Гц, 1H), 7,26 (д, J=8,8 Гц, 1H), 7,40 (д, J=10,6 Гц, 1H), 7,59 (с, 1H), 8,27 (дд, J=2,2, 5,8 Гц, 1H), 8,77 (д, J=7,3 Гц, 1H), 9,08 (с, 1H).
Примеры 2-4
Соединения примеров 2-4 синтезируют по аналогии со способом, раскрытым в примере 1, и промежуточные соединения получают способом, раскрытым далее.
<Промежуточное соединение 1>
Получение 5-гидроксиметилфуран-2-карбоновой кислоты
5-гидроксиметилфуран-2-карбоновую кислоту синтезируют способом, раскрытым в (Helv. Chim. Acta, 1926, 9, 1068).
<Промежуточное соединение 2>
Получение 3-гидроксиметилбензойной кислоты
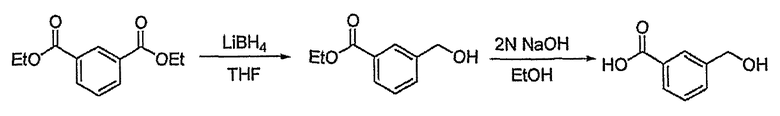
Диэтиловый эфир изофталевой кислоты (9,100 г, 40,95 ммоль) растворяют в тетрагидрофуране (20 мл). Затем медленно добавляют по каплям боргидрид лития (11,26 мл, 22,52 ммоль, 2M раствор в тетрагидрофуране) и реакционную смесь кипятят с обратным холодильником в течение 3 час. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (гексан:этилацетат = 2:1), получая 5,76 г (выход: 77,1%, бесцветная жидкость) этилового эфира 3-гидроксиметилбензойной кислоты.
Сложноэфирное соединение (1,317 г, 7,311 ммоль), полученное выше, растворяют в этаноле (6 мл). Затем медленно добавляют по каплям 2 н. водный раствор NaOH (11,0 мл, 21,93 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализуют 1% водным раствором HCl, экстрагируют этилацетатом и промывают насыщенным раствором NaCl. Объединенный органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:метиловый спирт = 5:1), получая 1,03 г (выход: 99,2%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 4,66 (с, 2H), 7,44 (т, J=7,7 Гц, 1H), 7,58 (д, J=7,7 Гц, 1H), 7,92 (д, J=7,7 Гц, 1H), 8,04 (с, 1H).
Пример 2:
[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 5-нитрооксиметилфуран-2-карбоновой кислоты
1H ЯМР (400 МГц, CDCl3): δ 2,37-2,51 (м, 3H), 2,46 (с, 3H), 2,61-2,92 (м, 1H), 3,74 (с, 3H), 3,93 (с, 3H), 3,98 (с, 3H), 4,82-4,85 (м, 1H), 4,85 (д, J=13,2 Гц, 1H), 4,90 (д, J=13,2 Гц, 1H), 6,39 (с, 1H), 6,58 (с, 1H), 6,64 (с, 1H), 7,16 (д, J=10,6 Гц, 1H), 7,42 (д, J=10,2 Гц, 1H), 7,72 (с, 1H), 8,99 (с, 1H).
Пример 3:
N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид
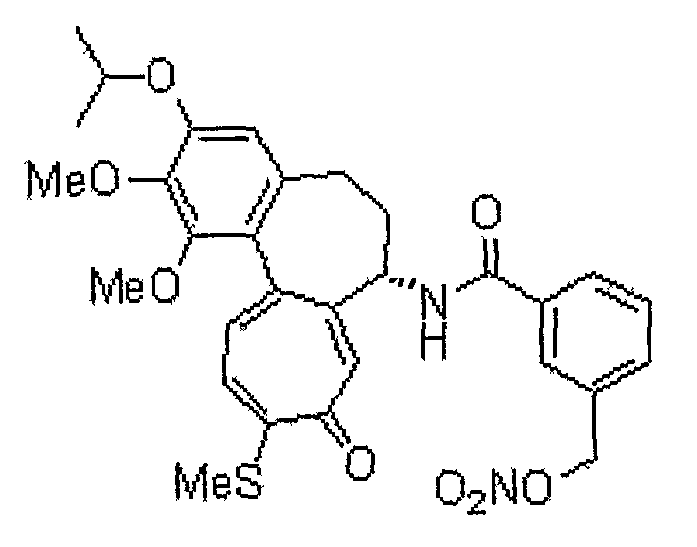
1H ЯМР (400 МГц, CDCl3): δ 1,42 (т, J=6,6 Гц, 6H), 2,22-2,27 (м, 1H), 2,34-2,49 (с, 2H), 2,54-2,57 (м, 1H), 3,75 (с, 3H), 3,96 (с, 3H), 4,59-4,63 (м, 1H), 4,93-4,97 (м, 1H), 5,23 (кв, J=13,0 Гц, 2H), 6,56 (с, 1H), 7,15 (д, J=10,6 Гц, 1H), 7,18-7,25 (м, 1H), 7,33 (д, J=8,0 Гц, 2H), 7,46 (д, J=10,2 Гц, 1H), 7,64 (с, lH), 7,71 (д, J=8,0 Гц, 1H), 7,88 (с, 1H), 8,40 (с, 1H).
Пример 4:
N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид
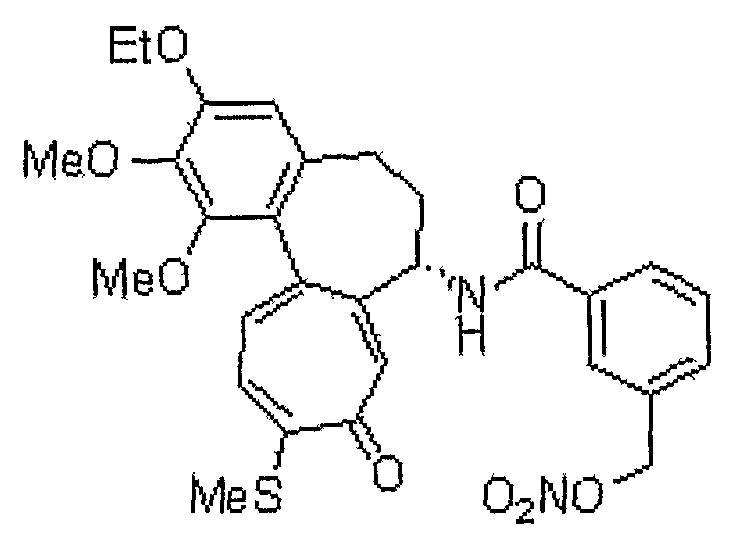
1H ЯМР (400 МГц, CDCl3): δ 1,49 (т, J=6,9 Гц, 3H), 2,17-2,24 (м, 1H), 2,34-2,47 (м, 2H), 2,45 (с, 3H), 2,49-2,58 (м, 1H), 3,75 (с, 3H), 3,97 (с, 3H), 4,12-4,15 (м, 2H), 4,89-4,96 (м, 1H), 5,24 (кв, J=12,0 Гц, 2H), 6,56 (с, 1H), 7,15 (д, J=10,2 Гц, 1H), 7,21-7,25 (м, 1H), 7,31-7,35 (м, 1H), 7,41 (д, J=10,6 Гц, 1H), 7,60 (с, 1H), 7,71 (д, J=7,3 Гц, 1H), 7,78 (с, 1H), 8,23 (с, 1H).
Пример 5:
Получение [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амида 6-нитрооксиметилпиридин-2-карбоновой кислоты
<Стадия 1>
Получение [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амида 6-гидроксиметилпиридин-2-карбоновой кислоты
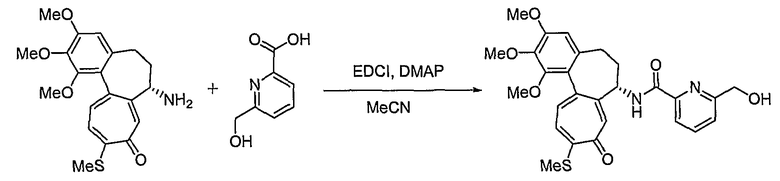
В соответствии со способом, представленным для стадии 1 примера 1, используя 6-гидроксиметилпиридин-2-карбоновую кислоту (23 мг, 0,17 ммоль), получают 35 мг (выход: 53%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ2,45 (с, 3H), 2,33-2,50 (м, 3H) 2,52-2,73 (м, 1H), 3,75 (с, 3H), 3,92 (с, 3H), 3,97 (С, 3H), 4,28 (д, J=14 Гц, 1H), 4,44 (д, J=14 Гц, 1H) 4,86-4,92 (м, 1H), 6,57 (с, 1H), 7,13 (д, J=10,4 Гц, 1H), 7,37-7,48 (м, 4H), 7,73 (с, 1H), 9,80 (д, J=8 Гц, 1H).
<Стадия 2>
Получение [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амида 6-бромометилпиридин-2-карбоновой кислоты
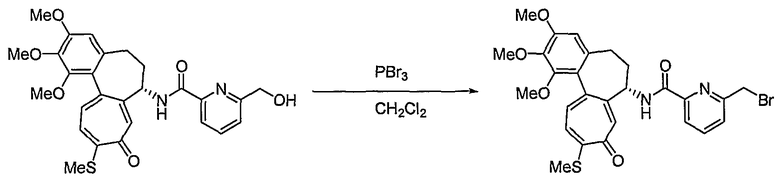
Соединение (50 мг, 0,098 ммоль), полученное на стадии 1, растворяют в дихлорметане (6 мл). Затем медленно добавляют по каплям трибромфосфин (PBr3, 0,005 мл, 0,05 ммоль) при 0°C и реакционную смесь перемешивают при комнатной температуре в течение 3 час. Добавляют метанол, чтобы погасить реакцию. Объединенный органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:метанол = 99:1), получая 40 мг (выход: 71%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,06-2,14 (м, 1H), 2,33-2,41 (м, 1H), 2,41 (с, 3H), 2,46-2,54 (м, 1H) 2,58-2,63 (м, 1H), 3,72 (с, 3H), 3,90 (с, 3H), 3,95 (C, 3H), 4,55 (3, J=2,8 Гц, 2H), 4,77-4,83 (м, 1H), 6,57 (с, 1H), 7,04 (д, J=10,4 Гц, 1H), 7,29 (с, 1H), 7,31 (д, J=10,4 Гц, 1H), 7,58 (д, J=7,6 Гц, 1H), 7,78 (т, J=7,6 Гц, 1H), 7,90 (д, J=8,0 Гц, 1H), 8,53 (д, J=7,6 Гц, 1H).
<Стадия 3>
Получение [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амида 6-нитрооксиметилпиридин-2-карбоновой кислоты
Соединение (40 мг, 0,070 ммоль), полученное на стадии 2, растворяют в ацетонитриле (3 мл). Затем медленно добавляют по каплям нитрат серебра (23 мг, 0,14 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакционную смесь промывают водой и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:метанол = 99:1), получая 17 мг (выход: 44,7%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,03-2,10 (м, 1H), 2,33-2,41 (м, 1H), 2,43 (с, 3H), 2,46-2,54 (м, 1H) 2,58-2,65 (м, 1H), 3,73 (с, 3H), 3,92 (с, 3H), 3,95 (с, 3H), 4,77-4,83 (м, 1H), 5,63 (д, J=3,2 Гц, 2H), 6,58 (с, 1H), 7,06 (д, J=10,4 Гц, 1H), 7,27 (с, 1H), 7,31 (д, J=10,4 Гц, 1H), 7,53 (д, J=7,6 Гц, 1H), 7,88 (т, J=8,0 Гц, 1H), 8,02 (д, J=7,2 Гц, 1H), 8,39 (д, J=7,2 Гц, 1H).
Примеры 6-24
Соединения примеров 6-24 синтезируют по аналогии со способом, раскрытым в примере 5, и промежуточные соединения получают раскрытым далее способом.
<Промежуточное соединение 3>
Получение 6-гидроксиметилпиридин-2-карбоновой кислоты

Этиловый эфир 6-гидроксиметилпиридин-2-карбоновой кислоты (200 мг, 1,1 ммоль) (J. Amer. Chem. Soc, 1982, 104, 2251-2257) растворяют в метаноле (1 мл). Затем медленно добавляют по каплям 2 н. водный раствор NaOH (1 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь подкисляют (pH 3), используя 2н. HCl. Растворитель концентрируют при пониженном давлении, затем растворяют в метаноле и фильтруют. Фильтрат концентрируют при пониженном давлении, получая 150 мг (выход: 89%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 5,05 (с, 2H), 8,34 (д, J=8,0 Гц, 1H), 8,47 (д, J=8,0 Гц, 1H) 8,73 (д, J=8,0 Гц, 1H).
<Промежуточное соединение 4>
Получение 5-гидроксиметилтиофен-2-карбоновой кислоты
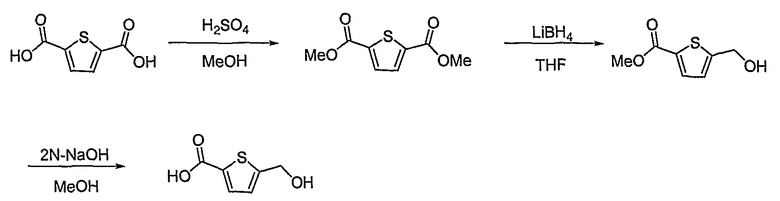
Тиофен-2,5-дикарбоновую кислоту (4 г, 23,3 ммоль) растворяют в метаноле (300 мл). К этому медленно добавляют каталитическое количество серной кислоты. Реакционную смесь кипятят с обратным холодильником, получая 3,8 г (выход: 81,7%, твердое вещество белого цвета) диметилового эфира тиофен-2,5-дикарбоновой кислоты. Диметиловый эфир тиофен-2,5-дикарбоновой кислоты (3,7 г, 18,84 ммоль) растворяют в безводном тетрагидрофуране (50 мл) при комнатной температуре в атмосфере азота. Медленно добавляют 2,0 M раствор боргидрида лития в тетрагидрофуране (5,5 мл, 11 ммоль) при 0°C. Реакционную смесь кипятят с обратным холодильником в течение 3 час, получая 2,1 г (выход: 64,7%, твердое вещество белого цвета) метилового эфира 5-гидроксиметилтиофен-2-карбоновой кислоты. Метиловый эфир 5-гидроксиметилтиофен-2-карбоновой кислоты (2,1 г, 12,2 ммоль) растворяют в метаноле (20 мл). Медленно добавляют 2 н. водный раствор NaOH (15 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, получая 1,75 г (выход: 89%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,90 (ушир., 1H), 4,79 (д, J=0,8 Гц, 2H), 6,97 (д, J=4 Гц, 1H), 7,66 (д, J=4 Гц, 1H).
<Промежуточное соединение 5>
Получение 2-фтор-3-гидроксиметилбензойной кислоты

2-фторизофталевую кислоту (3 г, 16,3 ммоль) (J. Amer. Chem. Soc., 1943, 65, 2308) растворяют в метаноле (150 мл). К этому медленно добавляют каталитическое количество серной кислоты. Реакционную смесь кипятят с обратным холодильником. Полученную смесь концентрируют при пониженном давлении для удаления растворителя и растворяют в этилацетате. Объединенный органический слой промывают насыщенным раствором карбоната натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:гексан = 1:2), получая 3,1 г (выход: 88%, твердое вещество белого цвета) диметилового эфира 2-фторизофталевой кислоты. Диметиловый эфир тиофен-2,5-дикарбоновой кислоты (3,1 г, 14,6 ммоль) растворяют в безводном тетрагидрофуране (50 мл) при комнатной температуре в атмосфере азота. Медленно добавляют 2,0 M раствор боргидрида лития в тетрагидрофуране (4,4 мл, 8,7 ммоль) при 0°C. Реакционную смесь кипятят с обратным холодильником в течение 3 час. Реакционную смесь подкисляют водным раствором HCl, концентрируют при пониженном давлении для удаления растворителя и экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и растворитель концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (дихлорметан:метанол = 99:1), получая 1,5 г (выход: 58%, твердое вещество белого цвета) метилового эфира 2-фтор-3-гидроксиметилбензойной кислоты. Метиловый эфир 2-фтор-3-гидроксиметилбензойной кислоты (1,3 г, 7,6 ммоль) растворяют в метаноле (20 мл). Медленно добавляют 2 н. водный раствор NaOH (14 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь подкисляют, используя (pH 3) 2 н. HCl. Раствор концентрируют при пониженном давлении, затем растворяют в метаноле и фильтруют. Фильтрат концентрируют при пониженном давлении, получая 1,15 г (выход: 88%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, ДМСО-d6): δ 4,58 (д, J=5,2 Гц, 2H),5,37 (т, J=5,2 Гц, 1H) 7,28 (т, J=8 Гц, 1H), 7,68 (т, J=8 Гц, 1H), 7,74 (т, J=8 Гц, 1H).
<Промежуточное соединение 6>
Получение 3-фтор-5-гидроксиметилбензойной кислоты
<Стадия 1>
Получение метилового эфира 3-фтор-5-гидроксиметилбензойной кислоты

Диметиловый эфир 5-фторизофталевой кислоты (1,6 г, 7,54 ммоль) (J. Org. Chem; 1969, 34, 1960-1961) растворяют в тетрагидрофуране (15 мл). Затем медленно добавляют по каплям 2,0 M раствор боргидрида лития в тетрагидрофуране (2,6 мл, 5,27 ммоль) при 0°C и реакционную смесь кипятят с обратным холодильником в течение 3 час. Реакционную смесь подкисляют 1 н. водным раствором HCl, концентрируют при пониженном давлении, для удаления растворителя, и экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (дихлорметан:метанол = 99:1), получая 800 мг (выход: 57%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,92 (д, J=1,6 Гц, 3H), 4,75 (д, J=4 Гц, 2H), 7,29-7,32 (м, 1H), 7,61 (дд, J=9,2, 1,6 Гц, 1H), 7,8 (д, J=0,8 Гц, 1H).
<Стадия 2>
Получение 3-фтор-5-гидроксиметилбензойной кислоты
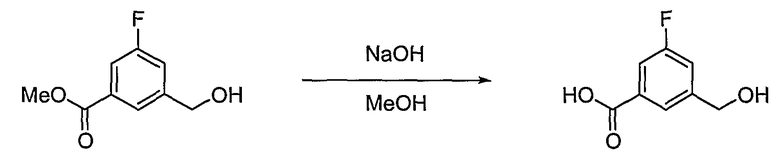
В соответствии со способом, представленным для получения промежуточного соединения 3, используя соединение, полученное на стадии 1, получают 1,6 г (выход: 94%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 4,66 (д, J=0,8 Гц, 2H), 7,33-7,36 (м, 1H), 7,56-7,59 (м, 1H), 7,83-7,84 (м, 1H).
<Промежуточное соединение 7>
Получение 4-фтор-3-гидроксиметилбензойной кислоты
<Стадия 1>
Получение 4-фторизофталевой кислоты
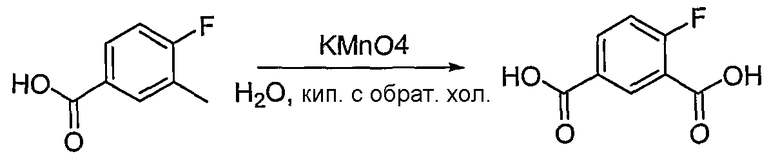
4-фтор-3-метилбензойную кислоту (2,52 г, 16,346 ммоль) и перманганат калия (10,33 г, 65,382 ммоль) растворяют, получая водный раствор (300 мл), и полученную смесь кипятят с обратным холодильником в течение 1 дня. Реакционную смесь фильтруют и полученный раствор охлаждают до комнатной температуры, затем к нему добавляют концентрированный раствор HCl. Полученный твердый продукт нагревают до тех пор, пока он полностью не расплавится. Температуру снова снижают до комнатной температуры и затем твердое вещество отфильтровывают, получая 2,08 г (выход: 69,1%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 7,32 (дд, J=10,4, 8,6 Гц, 1H), 8,21-8,25 (м, 1H), 8,59 (дд, J=7,0, 2,4 Гц, 1H).
<Стадия 2>
Получение диметилового эфира 4-фторизофталевой кислоты
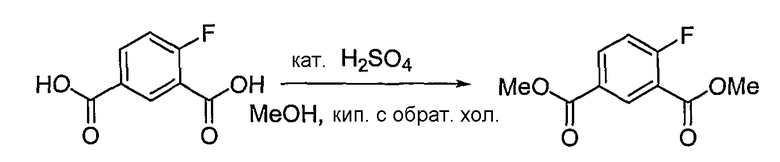
Соединение, полученное на стадии 1 (2,08 г, 11,29 ммоль), растворяют в метаноле (30 мл). Затем добавляют 10 капель концентрированной серной кислоты. Реакционную смесь кипятят с обратным холодильником в течение 1 дня, нейтрализуют насыщенным водным раствором бикарбоната натрия и экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 2,21 г (выход: 92,2%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,96 (с, 3H), 3,97 (с, 3H), 7,22 (дд, J=10,3, 8,8 Гц, 1H), 8,20-8,23 (м, 1H), 8,64 (дд, J=7,0, 2,2 Гц, 1H).
<Стадия 3>
Получение метилового эфира 4-фтор-3-гидроксиметилбензойной кислоты и метилового эфира 2-фтор-5-гидроксиметилбензойной кислоты
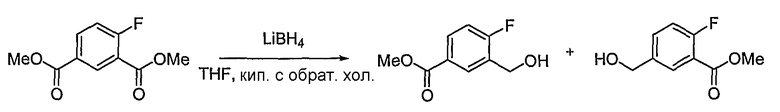
Соединение, полученное на стадии 2 (104,5 мг, 0,493 ммоль), растворяют в тетрагидрофуране (4 мл). Затем медленно добавляют 2 M раствор боргидрида лития в тетрагидрофуране (0,123 мл, 0,246 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 1 дня. Реакцию гасят водой. Затем pH доводят до 5, используя 1 M раствор HCl при 0°C. Осуществляют экстрагирование этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (гексан:этилацетат = 2:1), получая метиловый эфир 4-фтор-3-гидроксиметилбензойной кислоты (45,4 мг, выход: 50,1%, бесцветная жидкость) и метиловый эфир 2-фтор-5-гидроксиметилбензойной кислоты (15,4 мг, выход: 17,0%, бесцветная жидкость).
Метиловый эфир 4-фтор-3-гидроксиметилбензойной кислоты:
1H ЯМР (400 МГц, CDCl3): δ 2,73 (т, J=5,1 Гц, 1H), 3,90 (c, 3H), 4,78 (д, J=5,1 Гц, 2H), 7,07 (дд, J=9,2, 9,2 Гц, 1H), 7,93-7,97 (м, 1H), 8,14 (дд, J=7,1, 2,2 Гц, 1H).
Метиловый эфир 2-фтор-5-гидроксиметилбензойной кислоты:
1H ЯМР (400 МГц, CDCl3): δ 1,96 (т, J=4,4 Гц, 1H), 3,94 (с, 3H), 4,70 (д, J=4,4 Гц, 2H), 7,13 (дд, J=10,6, 8,4 Гц, 1H), 7,52-7,56 (м, 1H), 7,92 (дд, J=7,0, 2,2 Гц, 1H).
<Стадия 4>
Получение 4-фтор-3-гидроксиметилбензойной кислоты
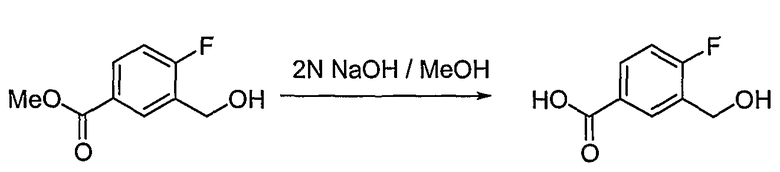
Аналогично способу получения промежуточного соединения 3, используя соединение, полученное на стадии 3 (1,074 г, 5,380 ммоль), получают 0,906 г (выход: 91,3%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 4,69 (с, 2H), 7,13 (дд, J=9,9, 8,8 Гц, 1H), 7,90-7,97 (м, 1H), 8,16 (дд, J=7,3, 2,2 Гц, 1H).
<Промежуточное соединение 8>
Получение 2-фтор-5-гидроксиметилбензойной кислоты
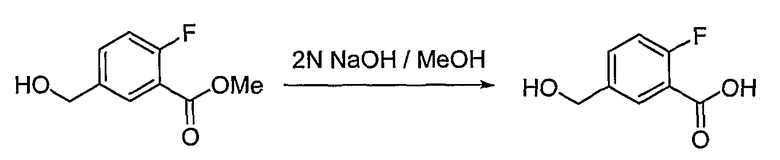
Аналогично способу получения промежуточного соединения 3, используя метиловый эфир 2-фтор-5-гидроксиметилбензойной кислоты, полученный на стадии 3 получения промежуточного соединения 7, получают целевое соединение.
1H ЯМР (400 МГц, CD3OD): δ 4,61 (с, 2H), 7,17 (дд, J=11,0, 8,4 Гц, 1H), 7,55-7,59 (м, 1H), 7,93 (дд, J=7,1, 2,4 Гц, 1H).
<Промежуточное соединение 9>
Получение 3-гидрокси-5-гидроксиметилбензойной кислоты

Метиловый эфир 5-гидроксиизофталевой кислоты (300 мг, 1,42 ммоль) растворяют в тетрагидрофуране (20 мл) при 0°C в атмосфере азота. Добавляют литийалюминийгидрид (30 мг, 0,7 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 час. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (дихлорметан:метанол = 20:1). Полученный выше метиловый эфир 3-гидрокси-5-гидроксиметилбензойной кислоты (170 мг, 0,93 ммоль) растворяют в метаноле (1 мл). Добавляют 1 н. водный раствор NaOH (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь подкисляют (pH 3), используя 2 н. HCl. Растворитель концентрируют при пониженном давлении и затем растворяют в метаноле и фильтруют. Фильтрат концентрируют при пониженном давлении, получая 150 мг (выход: 96%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 3,87 (с, 3H), 4,57 (с, 2H), 7,02 (с, 1H), 7,31 (с, 1H), 7,48 (с, 1H).
<Промежуточное соединение 10>
Получение 3,5-бис-гидроксиметилбензойной кислоты
<Стадия 1>
Получение метилового эфира 3,5-бис-гидроксиметилбензойной кислоты
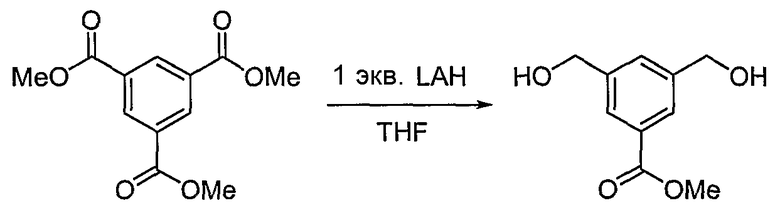
Триметиловый эфир бензол-1,3,5-трикарбоновой кислоты (1,010 г, 4,003 ммоль) растворяют в тетрагидрофуране (15 мл). Затем температуру снижают до 0°C, медленно добавляют литийалюминийгидрид (0,160 г, 4,003 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Медленно добавляют воду (0,15 мл) и 15% водный раствор NaOH (0,15 мл), чтобы погасить реакцию. Снова добавляют водный раствор (0,45 мл). Растворитель концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (гексан:этилацетат = 1:2), получая 0,34 г (выход: 43,1%, бесцветная жидкость) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 3,91 (с, 3H), 4,66 (с, 4H), 7,59 (с, 1H), 7,93 (с, 2H).
<Стадия 2>
Получение 3,5-бис-гидроксиметилбензойной кислоты
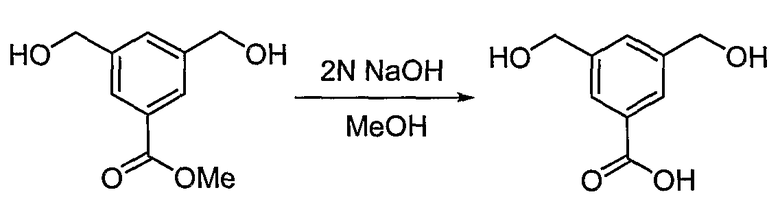
Способом, аналогичным способу получения промежуточного соединения 3, используя соединение, полученное на стадии 1 (1,50 г, 7,65 ммоль), получают 0,617 г (выход: 44,3%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 5,21 (с, 4H), 7,55 (с, 1H), 7,92 (с, 2H).
<Промежуточное соединение 11>
Получение 2-гидрокси-4-гидроксиметилбензойной кислоты
<Стадия 1>
Получение метилового эфира 2-гидрокси-4-гидроксиметилбензойной кислоты
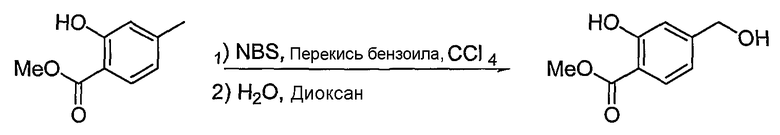
Метиловый эфир 2-гидрокси-4-метилбензойной кислоты (166 мг, 1 ммоль) растворяют в CCl4 (1,5 мл). Добавляют NBS (177 мг, 1 ммоль) и перекись бензоила (5 мг, 0,02 ммоль). Реакционную смесь перемешивают при 70°C в течение 12 час. Затем реакционную смесь промывают водой, сушат над безводным сульфатом натрия и фильтруют. Растворитель концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (дихлорметан:гексан = 1:4), получая метиловый эфир 4-бромметил-2-гидроксибензойной кислоты (130 мг, выход: 53%, твердое вещество белого цвета). Полученное соединение (130 мг, 0,53 ммоль) растворяют в водном растворе (1,5 мл) и 1,4-диоксане (1,5 мл) и смесь перемешивают при 90°C в течение 12 час. Реакционную смесь экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:гексан = 1:4), получая 55 мг (выход: 57%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,95 (с, 3H), 4,71 (д, J=6 Гц, 2H), 6,88 (д, J=8 Гц, 1H), 6,99 (с, 1H), 7,82 (д, J=8 Гц, 1H), 10,79 (с, 1H).
<Стадия 2>
Получение 2-гидрокси-4-гидроксиметилбензойной кислоты
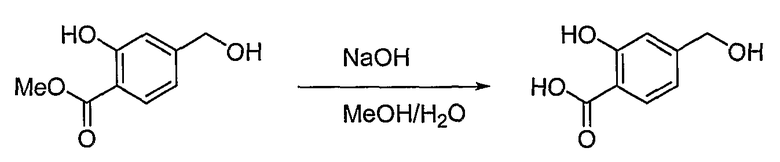
Способом, аналогичным способу получения промежуточного соединения 3, используя соединение, полученное на стадии 1, получают 45 мг (выход: 90%, твердое вещество белого цвета) целевого соединения.
<Промежуточное соединение 12>
Получение 4-гидроксиметилтиофен-2-карбоновой кислоты
<Стадия 1> A: 4-метилтиофен-2-карбоновая кислота,
B: Получение 3-метилтиофен-2-карбоновой кислоты
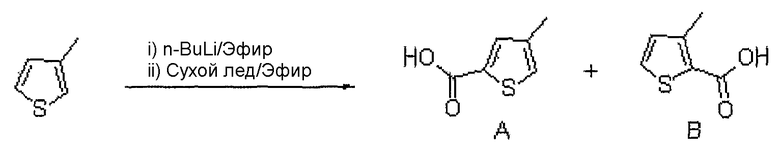
2,5 M н-бутиллитий (12,2 мл, 30,55 ммоль) растворяют в диэтиловом эфире (1 мл) при комнатной температуре в атмосфере азота. Медленно добавляют 3-метилтиофен (3 г, 30,55 ммоль), растворенный в диэтиловом эфире. Полученную смесь кипятят с обратным холодильником в течение 2 часов. Реактор охлаждают до 0°C и медленно добавляют сухой лед. Реакцию гасят, используя 45 мл воды. Слой диэтилового эфира экстрагируют и удаляют. Водный слой подкисляют, используя 1 н. раствор HCl и экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (гексан:этилацетат = 9:1), получая целевое соединение A (900 мг, твердое вещество белого цвета) и B (650 мг, твердое вещество белого цвета).
<Стадия 2>
Получение метилового эфира 4-бромметилтиофен-2-карбоновой кислоты

4-метилтиофен-2-карбоновую кислоту (900 мг, 6,33 ммоль), полученную на стадии 1, растворяют в метаноле (15 мл). Медленно добавляют каталитическое количество серной кислоты. Реакционную смесь кипятят с обратным холодильником, получая метиловый эфир 4-метилтиофен-2-карбоновой кислоты (890 мг, выход: 90%, твердое вещество белого цвета). Метиловый эфир 4-метилтиофен-2-карбоновой кислоты (200 мг, 1,28 ммоль), NBS (215 мг, 1,216 ммоль) и каталитическое количество перекиси бензоила растворяют в тетрахлорметане (5 мл). Реакционную смесь кипятят с обратным холодильником (70°C) в течение 3 часов, получая 165 мг (выход: 55%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,89 (с, 3H), 4,46 (с, 2H), 7,49 (с, 1H), 7,80 (с, 1H).
<Стадия 3>
Получение 4-гидроксиметилтиофен-2-карбоновой кислоты
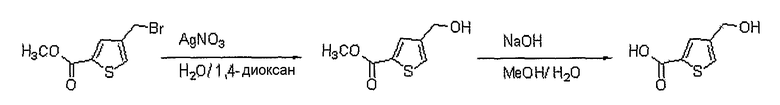
Соединение (150 мг, 0,638 ммоль), полученное на стадии 2, растворяют в 1,4-диоксане (1,5 мл) и воде (1,5 мл). Медленно добавляют нитрат серебра (130 мг, 0,765 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов, получая метиловый эфир 4-гидроксиметилтиофен-2-карбоновой кислоты (60 мг, выход: 55%, твердое вещество белого цвета). Указанное соединение (60 мг, 0,348 ммоль) растворяют в метаноле (1 мл) при комнатной температуре. Медленно добавляют 1 н. водный раствор NaOH (1 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1 часа, получая 50 мг (выход: 95%, твердое вещество белого цвета) целевого соединения.
<Промежуточное соединение 13>
Получение 3-гидроксиметилтиофен-2-карбоновой кислоты
<Стадия 1>
Получение метилового эфира 3-бромметилтиофен-2-карбоновой кислоты
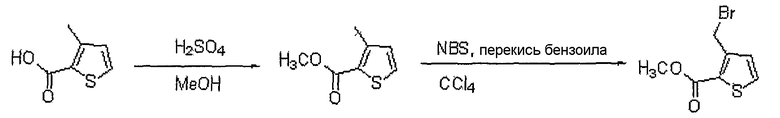
Способом, аналогичным способу стадии 2 получения промежуточного соединения 12, используя 3-метилтиофен-2-карбоновую кислоту (650 мг, 4,57 ммоль), полученную на стадии 1 получения промежуточного соединения 12, получают 750 мг (выход: 79%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ3,90 (с, 3H), 4,91 (с, 2H), 7,18 (д, J=5,2 Гц, 1H), 7,46 (д, J=5,2 Гц, 1H).
<Стадия 2>
Получение 3-гидроксиметилтиофен-2-карбоновой кислоты
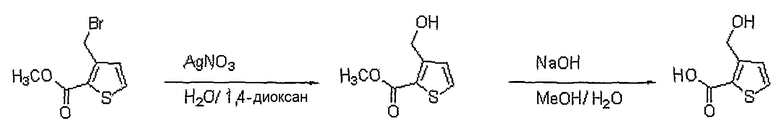
Способом, аналогичным способу стадии 3 получения промежуточного соединения 12, используя соединение, полученное на стадии 1 (750 мг, 3,19 ммоль), получают 210 мг (выход: 92%, твердое вещество белого цвета) целевого соединения.
<Промежуточное соединение 14>
Получение (3-гидроксиметилфенил)уксусной кислоты
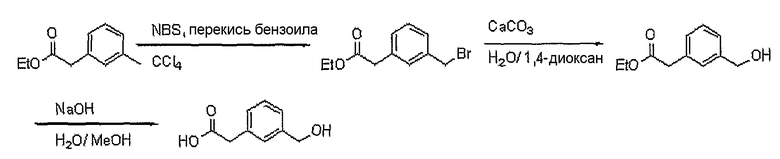
Этиловый эфир м-толилуксусной кислоты (1 г, 5,6 ммоль), NBS (948 мг, 5,33 ммоль) и каталитическое количество перекиси бензоила растворяют в тетрахлорметане (15 мл). Реакционную смесь кипятят с обратным холодильником (70°C) в течение 3 часов, получая этиловый эфир (3-бромметилфенил)уксусной кислоты (600 мг, выход: 42%, твердое вещество белого цвета). Этиловый эфир (3-бромметилфенил)уксусной кислоты (130 мг, 0,50 ммоль) и карбонат кальция (300 мг, 3 ммоль) растворяют в воде (2 мл) и 1,4-диоксане (2 мл). Реакционную смесь кипятят с обратным холодильником, получая этиловый эфир (3-гидроксиметилфенил)уксусной кислоты (85 мг, выход: 87%, твердое вещество белого цвета). Этиловый эфир (3-гидроксиметилфенил)уксусной кислоты (85 мг, 0,43 ммоль) растворяют в метаноле (1 мл) при комнатной температуре. К этому добавляют 1 н. водный раствор NaOH (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, получая 65 мг (выход: 91%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ3,66 (с, 2H), 4,57 (с, 2H), 7,21-7,32 (м, 4H).
<Промежуточное соединение 15>
Получение 3-(2-гидроксиэтил)бензойной кислоты

Изофталевую кислоту (5 г, 30 ммоль) растворяют в метаноле (50 мл). Добавляют каталитическое количество серной кислоты. Реакционную смесь перемешивают при кипении с обратным холодильником в течение 12 час, получая диметиловый эфир изофталевой кислоты (5,2 г, выход: 90%, твердое вещество белого цвета). Диметиловый эфир изофталевой кислоты (5,2 г, 26,7 ммоль) растворяют в тетрагидрофуране (30 мл). Добавляют 2 M тетрагидрофурановый раствор боргидрида лития (13 мл, 26,7 ммоль). Реакционную смесь кипятят с обратным холодильником, получая метиловый эфир 3-гидроксиметилбензойной кислоты (2,7 г, выход: 63%, бесцветная жидкость). Метиловый эфир 3-гидроксиметилбензойной кислоты (200 мг, 1,20 ммоль) растворяют в дихлорметане (5 мл). Добавляют PCC (388 мг, 1,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, получая метиловый эфир 3-формилбензойной кислоты (140 мг, выход: 72%, твердое вещество белого цвета). (Метоксиметил)трифенилфосфонийхлорид (770 мг, 2,24 ммоль) растворяют в тетрагидрофуране (5 мл) в атмосфере азота. Медленно добавляют 1 M тетрагидрофурановый раствор NaHMDS (2 мл, 2,04 ммоль) при -78°C, и реакционную смесь перемешивают в течение 1 часа. Медленно добавляют метиловый эфир 3-формилбензойной кислоты (160 мг, 0,975 ммоль), растворенный в тетрагидрофуране (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 час, получая метиловый эфир 3-(2-метоксивинил)бензойной кислоты (121 мг, выход: 65%, твердое вещество белого цвета). Метиловый эфир 3-(2-метоксивинил)бензойной кислоты (120 мг, 0,63 ммоль) растворяют в тетрагидрофуране (3 мл). Добавляют 4 M HCl (2 мл) и смесь перемешивают при комнатной температуре в течение 24 час, получая метиловый эфир 3-(2-оксоэтил)бензойной кислоты (60 мг, выход: 54%, твердое вещество белого цвета). Метиловый эфир 3-(2-оксоэтил)бензойной кислоты (60 мг, 0,036 ммоль) растворяют в этаноле (2 мл). Добавляют NaBH4 (25 мг, 0,67 ммоль) при 0°C, и смесь перемешивают в течение 1 часа, получая метиловый эфир 3-(2-гидроксиэтил)бензойной кислоты (50 мг, выход: 84%, твердое вещество белого цвета). Метиловый эфир 3-(2-гидроксиэтил)бензойной кислоты (50 мг, 0,28 ммоль) растворяют в метаноле (1 мл). Добавляют 1 н. водный раствор NaOH (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, получая 43 мг (выход: 95%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,32 (ушир., 1H), 2,89 (т, J=6,4 Гц, 2H), 3,85 (т, J=6,4 Гц, 2H), 7,36 (т, J=7,6 Гц, 1H), 7,42 (д, J=5,6 Гц, 1H), 7,88 (д, J=8,8 Гц, 1H), 7,89 (с, 1H).
Пример 6:
[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 5-нитроксиметилтиофен-2 карбоновой кислоты
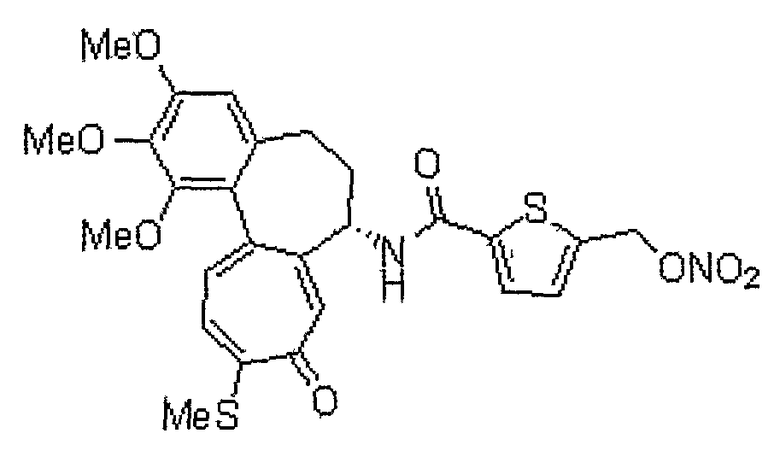
1H ЯМР (400 МГц, CDCl3): δ 2,07-2,11 (м, 1H), 2,32-2,45 (м, 2H), 2,45 (с, 3H), 2,53-2,56 (м, 1H), 3,71 (с, 3H), 3,91 (с, 3H), 3,96 (С, 3H), 4,84-4,91 (м, 1H), 5,46 (с, 2H), 6,56 (с, 1H), 6,95 (д, J=3,6 Гц, 1H), 7,13 (д, J=10,8 Гц, 1H), 7,37 (д, J=10,8 Гц, 1H), 7,58 (д, J=4,0 Гц, 1H), 7,61 (с, 1H), 8,50 (д, J=6,8 Гц, 1H).
Пример 7:
N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид
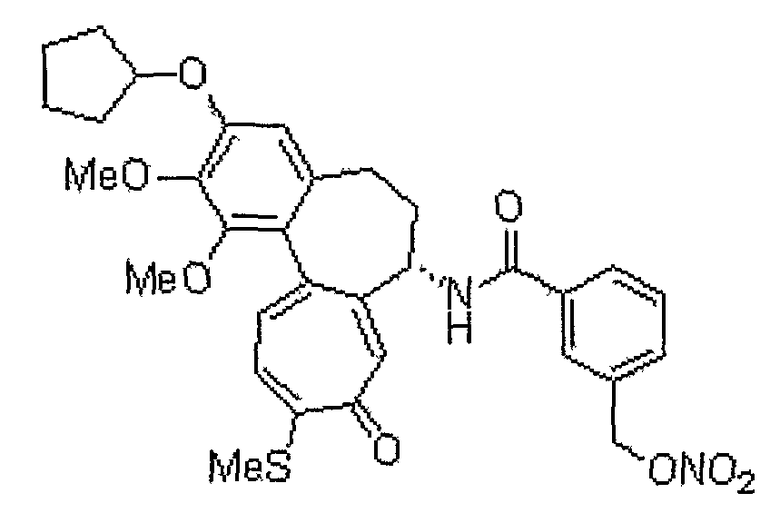
1H ЯМР (400 МГц, CDCl3): δ1,61-1,73 (м, 2H), 1,85-2,00 (м, 6H), 2,21-2,65 (м, 4H), 2,44 (с, 3H), 3,75 (с, 3H), 3,93 (с, 3H), 4,82-4,88 (м, 1H), 4,92-5,00 (м, 1H), 5,16 (дд, J=12, 31,2 Гц, 2H), 6,55 (с, 1H), 7,13-7,17 (м, 2H), 7,28 (д, J=6,4 Гц, 1H), 7,41 (д, J=10,8 Гц, 1H), 7,68-7,70 (м, 1H), 7,72 (с, 1H), 7,78 (с, 1H), 8,78 (д, J=7,2 Гц, 1H).
Пример 8:
N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-2-фтор-3-нитрооксиметилбензамид
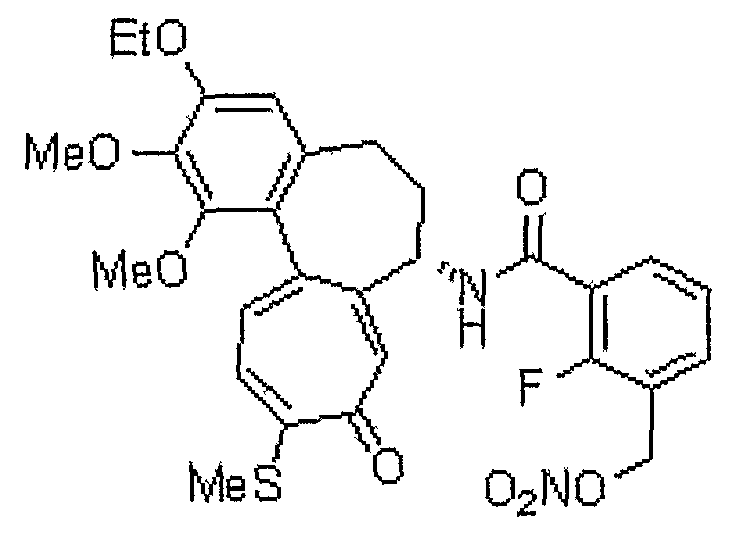
1H ЯМР (400 МГц, CDCl3): δ 1,49 (т, J=6,9 Гц, 3H), 1,93-1,98 (м, 1H), 2,31-2,39 (м, 1H), 2,43 (с, 3H), 2,46-2,49 (м, 1H), 2,51-2,59 (м, 1H), 3,73 (с, 3H), 3,96 (с, 3H), 4,11-4,14 (м, 2H), 4,83-4,86 (м, 1H), 5,53 (д, J=12,8 Гц, 1H), 5,59 (д, J=12,4 Гц, 1H), 6,56 (с, 1H), 7,07 (д, J=10,2 Гц, 1H), 7,10-7,15 (м, 1H), 7,20-7,28 (м, 1H), 7,31 (д, J=10,2 Гц, 1H), 7,57 (т, J=6,4 Гц, 1H), 7,97 (т, J=6,9 Гц, 1H).
Пример 9:
2-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-нитрооксиметилбензамид
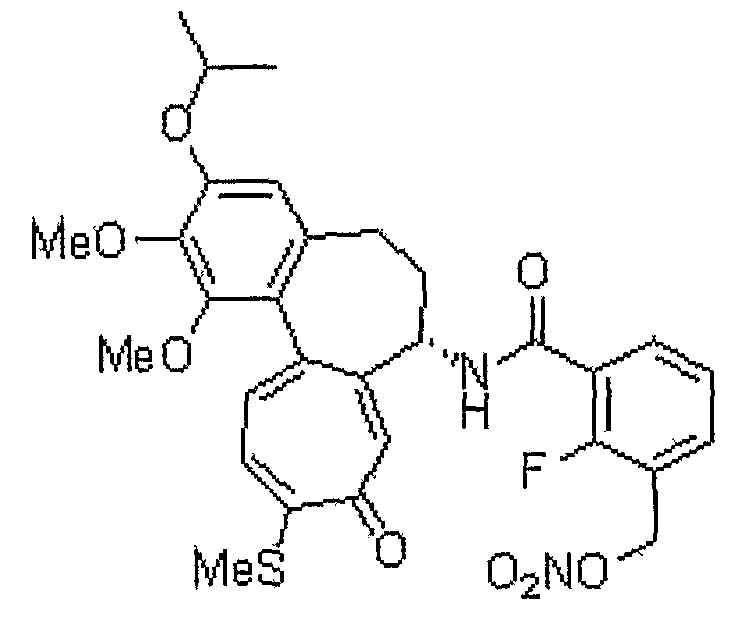
1H ЯМР (400 МГц, CDCl3): δ 1,41 (кв, J=6,0 Гц, 6H), 1,92-1,99 (м/ 1H), 2,31-2,40 (м, 1H), 2,43 (с, 3H), 2,46-2,52 (м, 1H), 2,55-2,60 (м, 1H), 3,72 (с, 3H), 3,94 (с, 3H), 4,57-4,63 (м, 1H), 4,83-4,89 (м, 1H), 5,53 (д, J=12,8 Гц, 1H), 5,59 (д, J=12,4 Гц, 1H), 6,57 (с, 1H), 7,07 (д, J=10,6 Гц, 1H), 7,15-7,19 (м, 1H), 7,23-7,27 (м, 1H), 7,34 (д, J=10,2 Гц, 1H), 7,56 (т, J=7,1 Гц, 1H), 7,7 (т, J=7,5 Гц, 1H).
Пример 10:
2-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
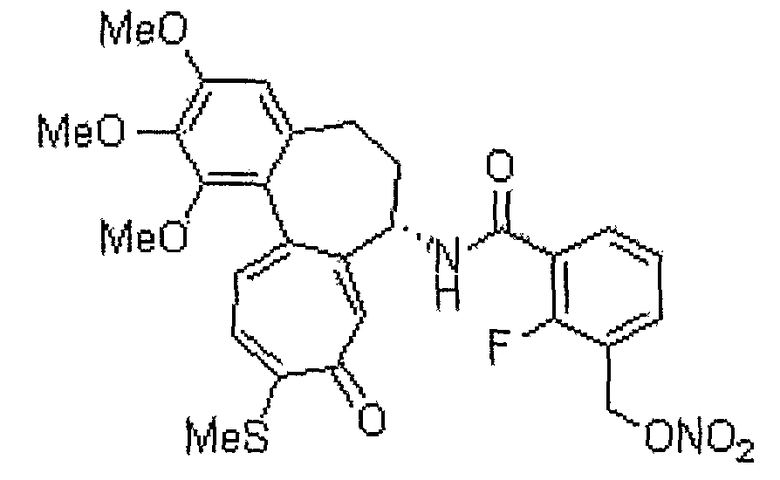
1H ЯМР (400 МГц, CDCl3): δ 1,95-2,02 (м, 1H), 2,31-2,54 (м, 2H), 2,43 (с, 3H), 2,59-2,63 (м, 1H), 3,73 (с, 3H), 3,87 (с, 3H), 3,92 (с, 3H), 4,82-4,88 (м, 1H), 5,51 (д, J=12,8 Гц, 1H), 5,60 (д, J=12,8 Гц, 1H), 6,58 (с, 1H), 7,06 (д, J=10,6 Гц, 1H), 7,22-7,27 (м, 2H), 7,28 (с, 1H), 7,32 (д, J=10,6 Гц, 1H), 7,54-7,58 (м, 1H), 7,93-7,98 (м, 1H).
Пример 11:
N-[(7S)-3-циклопентилокси-1,2-диметокси-10-
метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-2-фтор-3-нитрооксиметилбензамид

1H ЯМР (400 МГц, CDCl3): δ 1,61-1,73 (м, 2H), 1,85-2,00 (м, 6H), 2,35-2,52 (м, 4H), 2,42 (с, 3H), 3,72 (с, 3H), 3,93 (с, 3H), 4,82-4,88 (м, 2H), 5,57 (дд, J=12,8, 37,6 Гц, 2H), 6,55 (с, 1H), 7,05 (д, J=10,4 Гц, 1H), 7,09-7,13 (м, 1H), 7,26 (д, J=7,2 Гц, 2H), 7,32 (д, J=10,4 Гц, 1H), 7,55-7,59 (м, 1H), 7,96-8,00 (м, 1H).
Пример 12:
3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
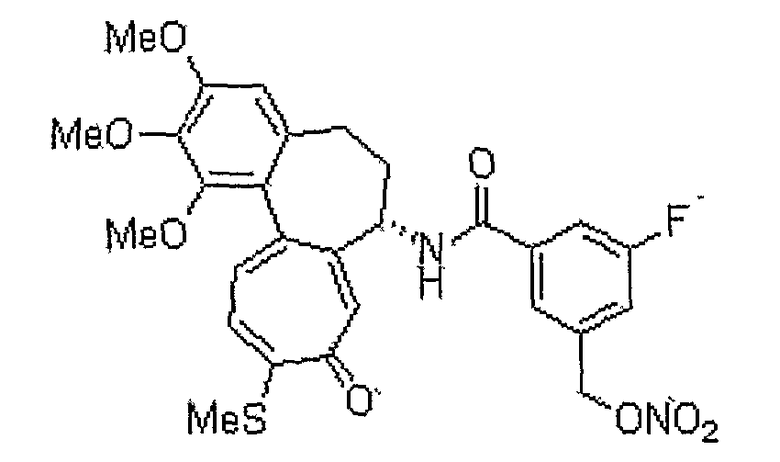
1H ЯМР (400 МГц, CDCl3): δ 2,31-2,45 (м, 3H), 2,46 (с, 3H), 2,59-2,63 (м, 1H), 3,75 (с, 3H), 3,93 (с, 3H), 3,98 (с, 3H), 4,90-4,95 (м, 1H), 5,11 (дд, J=12,8, 45,6 Гц, 2H), 6,57 (с, 1H), 7,03 (д, J=7,6 Гц, 1H), 7,19 (д, J=8,8 Гц, 1H), 7,30-7,33 (м, 1H), 7,42 (д, J=10,8 Гц, 1H), 7,55 (с, 1H), 7,68 (с, 1H), 8,76 (д, J=7,2 Гц, 1H).
Пример 13:
N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамид
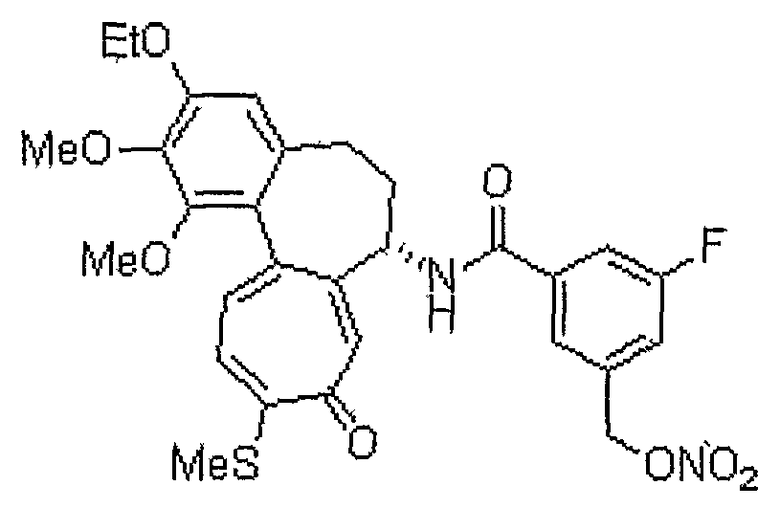
1H ЯМР (400 МГц, CDCl3): δ 1,50 (т, J=6,9 Гц, 3H), 2,33-2,42 (м, 3H), 2,46 (с, 3H), 2,51-2,59 (м, 1H), 3,75 (с, 3H), 3,98 (с, 3H), 4,11-4,16 (м, 2H), 4,90˜4,93 (м, 1H), 5,05 (д, J=12,4 Гц, 1H), 5,17 (д, J=12,4 Гц, 1H), 6,56 (с, 1H), 7,03 (д, J=7,3 Гц, 1H), 7,20 (д, J=10,6 Гц, 1H), 7,31 (д, J=9,1 Гц, 1H), 7,45 (д, J=10,6 Гц, 1H), 7,55 (с, 1H), 7,69 (с, 1H), 8,88 (д, J=7,3 Гц, 1H).
Пример 14:
3-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-5-нитрооксиметилбензамид
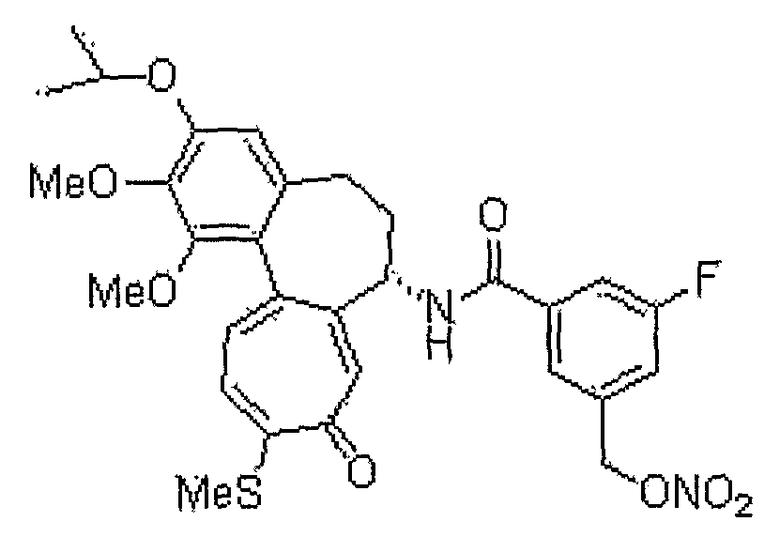
1H ЯМР (400 МГц, CDCl3): δ 1,37-1,44 (м, 6H), 2,34˜2,41 (м, 3H), 2,46 (с, 3H), 2,51-2,58 (м, 1H), 3,75 (с, 3H), 3,96 (с, 3H), 4,57-4,62 (м, 1H), 4,91˜4,95 (м, 1H), 5,05 (д, J=12,4 Гц, 1H), 5,17 (д, J=12,4 Гц, 1H), 6,56 (с, 1H), 7,03 (д, J=8,4 Гц, 1H), 7,20 (д/ J=10,6 Гц, 1H), 7,31 (д, J=7,3 Гц, 1H), 7,45 (д, J=10,6 Гц, 1H), 7,55 (с, 1H), 7,70 (с, 1H), 8,91 (д, J=7,3 Гц, 1H).
Пример 15:
N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамид
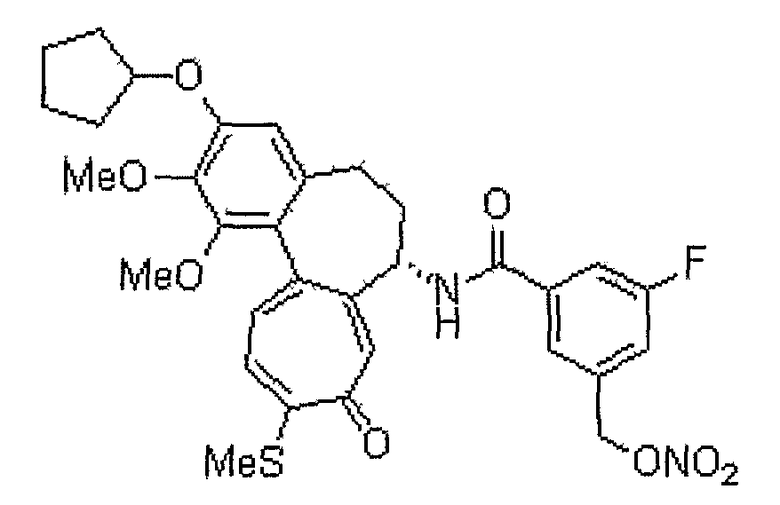
1H ЯМР (400 МГц, CDCl3): δ 1,64-1,69 (м, 2H), 1,83-1,98 (м, 6H), 2,35-2,43 (м, 3H), 2,46 (с, 3H), 2,51-2,58 (м, 1H), 3,74 (с, 3H), 3,94 (c, 3H), 4,82-4,84 (м, 1H), 4,91˜4,94 (м, 1H), 5,04 (д, J=12,4 Гц, 1H), 5,16 (д, J=12,4 Гц, 1H), 6,55 (с, 1H), 7,02 (д, J=8,4 Гц, 1H), 7,20 (д, J=10,6 Гц, 1H), 7,31 (д, J=7,3 Гц, 1H), 7,45 (д, J=10,2 Гц, 1H), 7,55 (с, 1H), 7,70 (с, 1H), 8,93 (д, J=6,9 Гц, 1H).
Пример 16:
4-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
1H ЯМР (400 МГц, CDCl3): δ 2,22-2,51 (м, 3H), 2,46 (с, 3H), 2,55-2,63 (м, 1H), 3,76 (с, 3H), 3,92 (с, 3H), 3,98 (с, 3H), 4,90-4,97 (м, 1H), 5,10 (д, J=12,1 Гц, 1H), 5,35 (д, J=12,1 Гц, 1H), 6,57 (с, 1H), 6,86 (дд, J=9,2, 8,8 Гц, 1H), 7,18 (д, J=10,3 Гц, 1H), 7,43 (д, J=10,3 Гц, 1H), 7,72 (с, 1H), 7,76-7,80 (м, 1H), 7,91 (дд, J=6,80, 2,2 Гц, 1H), 8,93 (д, J=7,2 Гц, 1H).
Пример 17:
2-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
1H ЯМР (400 МГц, CDCl3): δ 1,94-2,01 (м, 1H), 2,31-2,54 (м, 2H), 2,43 (с, 3H), 2,58-2,63 (м, 1H), 3,73 (с, 3H), 3,92 (с, 3H), 3,97 (с, 3H), 4,81-4,87 (м, 1H), 5,36 (с, 2H), 6,57 (с, 1H), 7,06 (д, J=10,3 Гц, 1H), 7,16-7,28 (м, 2H), 7,26 (с, 1H), 7,33 (д, J=10,3 Гц, 1H), 7,51-7,55 (м, 1H), 7,99 (дд, J=7,3, 2,6 Гц, 1H).
Пример 18:
3-гидрокси-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
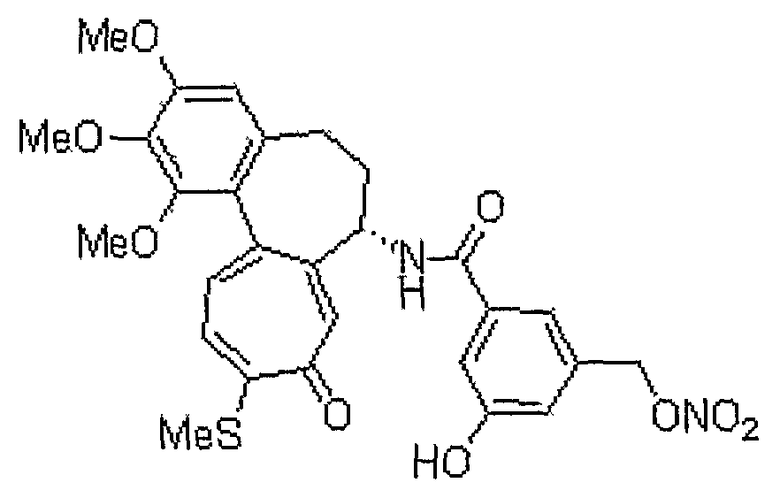
1H ЯМР (400 МГц, CDCl3): δ 2,30-2,43 (м, 6H), 2,56-2,57 (м, 1H), 3,61 (с, 3H), 3,90 (с, 3H), 3,92 (с, 3H), 4,82-4,85 (м, 1H), 5,00 (дд, J=30,8 Гц, 12,8 Гц, 2H), 6,56 (с, 1H), 6,62 (с, 1H), 7,11 (с, 2H), 7,16-7,19 (м, 2H), 7,41 (д, J=10,8 Гц, 1H), 7,66 (с, 1H), 8,57 (ушир.с, 1H, NH), 8,78 (ушир.с, 1H, OH).
Пример 19:
3,5-бис-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
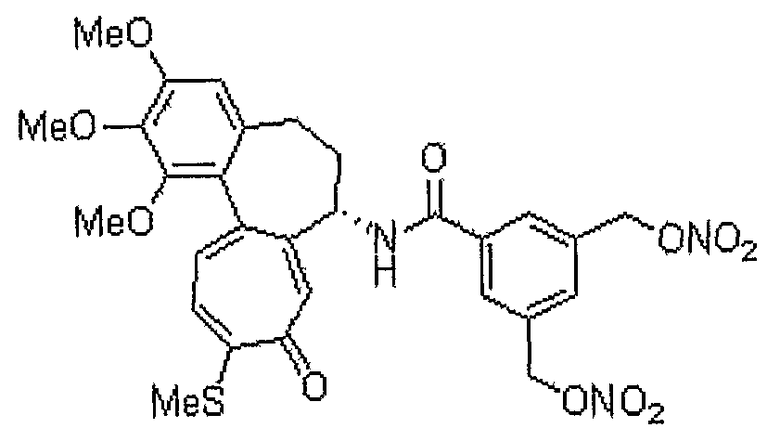
1H ЯМР (400 МГц, CDCl3): δ 2,29-2,51 (м, 3H), 2,46 (с, 3H), 2,60-2,62 (м, 1H), 3,77 (с, 3H), 3,93 (с, 3H), 3,99 (с, 3H), 4,94-4,98 (м, 1H), 5,18 (с, 4H), 6,58 (с, 1H), 7,20 (д, J=10,6 Гц, 1H), 7,26 (с, 1H), 7,45 (д, J=10,6 Гц, 1H), 7,77 (с, 2H), 7,81 (с, 1H), 9,12 (д, J=7,0 Гц, 1H).
Пример 20:
2-гидрокси-4-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
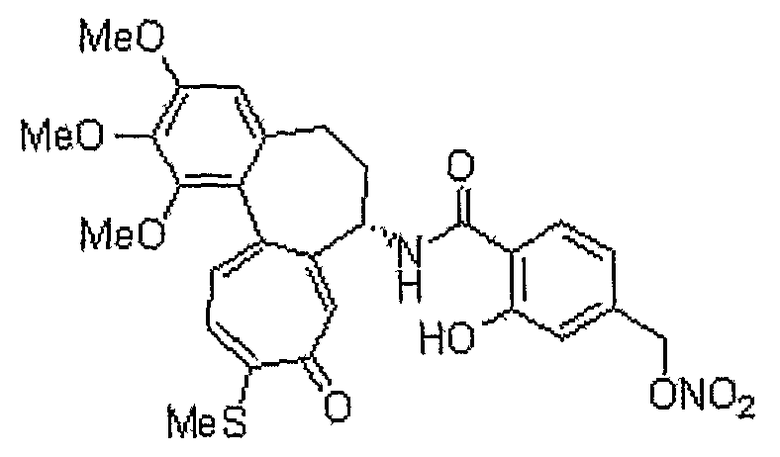
1H ЯМР (400 МГц, CDCl3): δ 2,25-2,63 (м, 4H), 2,47 (с, 3H), 3,74 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,85-4,92 (м, 1H), 5,27 (д, J=4,8 Гц, 2H), 6,53 (д, J=8,4 Гц, 1H), 6,57 (с, 1H), 6,61 (с, 1H), 7,20 (д, J=10,4 Гц, 1H), 7,42 (д, J=10 Гц, 1H), 7,63 (с, 1H), 7,72 (д, J=8 Гц, 1H), 9,13 (д, J=6,8 Гц, 1H), 12,29 (с, 1H).
Пример 21:
[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 4-нитрооксиметилтиофен-2-карбоновой кислоты
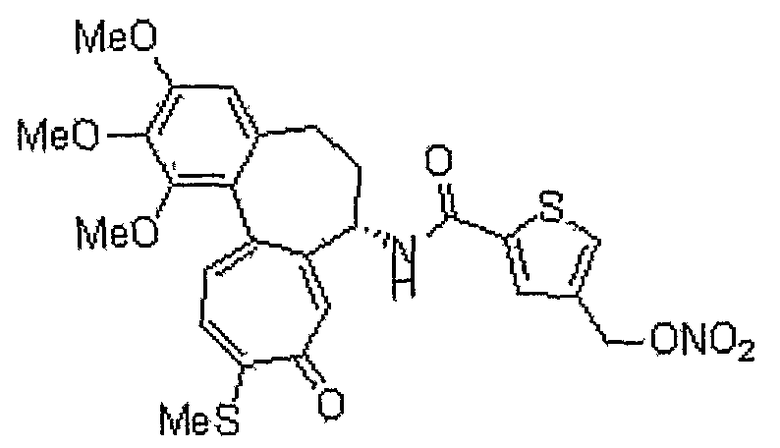
1H ЯМР (400 МГц, CDCl3): δ 2,21-2,29 (м, 1H), 2,34-2,46 (м, 5H), 2,51-2,58 (м, 1H), 3,71 (с, 3H), 3,91 (с, 3H), 3,96 (с, 3H), 4,88-4,95 (м, 1H), 5,18 (дд, J=12,8, 20 Гц, 2H), 6,56 (с, 1H), 7,17 (д, J=10,4 Гц, 1H), 7,30 (с, 1H), 7,41 (д, J=10,4 Гц, 1H), 7,61 (с, 1H), 7,74 (с, 1H), 8,81 (д, J=6,4 Гц, 1H).
Пример 22:
[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]амид 3-нитрооксиметилтиофен-2-карбоновой кислоты
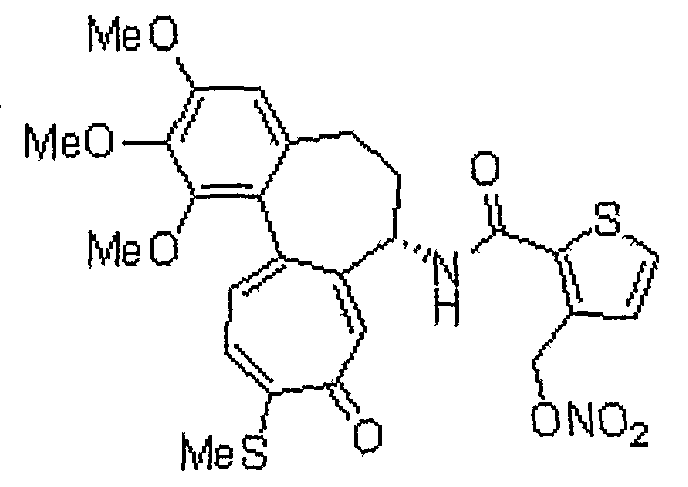
1H ЯМР (400 МГц, CDCl3): δ 2,04-2,17 (м, 1H), 2,30-2,45 (м, 5H), 2,57-2,64 (м, 1H), 3,73 (с, 3H), 3,91 (с, 3H), 3,96 (с, 3H), 4,67-4,88 (м, 1H), 5,66 (д, J=13,6 Гц, 1H), 5,80 (д, J=13,6 Гц, 1H), 6,56 (с, 1H), 7,02 (д, J=4,4 Гц, 1H), 7,08 (д, J=10,4 Гц, 1H), 7,26 (д, J=4,4 Гц, 1H), 7,29 (д, J=10,4 Гц, 1H), 7,43 (с, 1H), 7,45 (д, J=7,2 Гц, 1H).
Пример 23:
2-(3-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамид
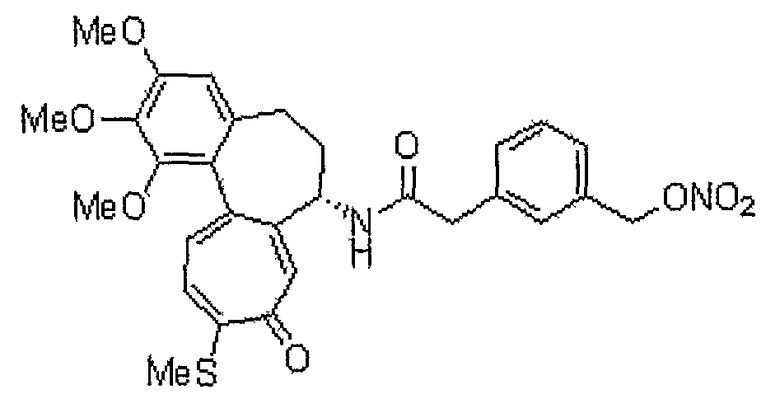
1H ЯМР (400 МГц, CDCl3): δ 1,82-1,95 (м, 1H), 2,21-2,28 (м, 1H), 2,31-2,42 (м, 1H), 2,45 (с, 3H), 2,47-2,53 (м, 1H), 3,51 (д, J=14,0 Гц, 1H), 3,64 (с, 3H), 3,66 (д, J=14,0 Гц, 1H), 3,88 (с, 3H), 3,93 (с, 3H), 4,66-4,72 (м, 1H), 5,40 (с, 2H), 6,51 (с, 1H), 7,11 (д, J=10,4 Гц, 1H), 7,24-7,38 (м, 5H), 7,48 (с, 1H), 7,90 (д, J=7,2 Гц, 1H).
Пример 24:
3-(2-нитрооксиэтил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
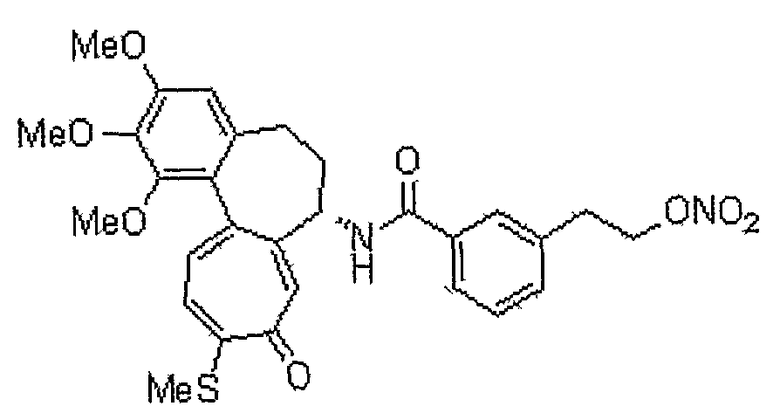
1H ЯМР (400 МГц, CDCl3): δ 2,05-2,11 (м, 1H), 2,17-2,56 (м, 2H), 2,46 (с, 3H), 2,56-2,60 (м, 1H), 2,81-2,91 (м, 2H), 3,69 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,45 (т, J=6,8, Гц, 2H), 4,89-4,95 (м, 1H), 6,56 (с, 1H), 7,12-7,24 (м, 3H), 7,39 (д, J=10,4 Гц, 1H), 7,61-7,64 (м, 2H), 7,69 (С, 1H), 8,22 (д, J=6,8 Гц, 1H).
Пример 25:
Получение 5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметилового эфира 3-нитрооксибензойной кислоты
<Стадия 1>
Получение {5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-ил}метилового эфира 3-хлорметилбензойной кислоты
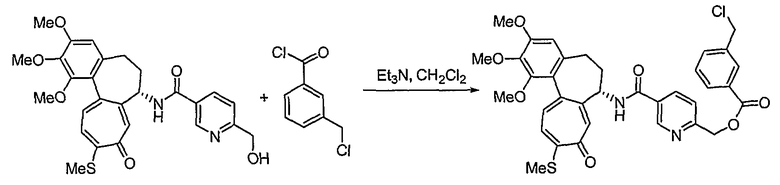
Соединение, полученное на стадии 1 примера 1 (100 мг, 0,19 ммоль), растворяют в 3 мл дихлорметана. Медленно добавляют 3-(хлорметил)бензоилхлорид (0,030 мл, 0,21 ммоль) и триэтиламин (0,082 мл, 0,59 ммоль) и смесь оставляют реагировать при комнатной температуре в течение 10 мин. Добавляют воду, чтобы погасить реакцию и водный слой экстрагируют дихлорметаном. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат), получая 99 мг (выход: 79%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,14-2,16 (м, 1H), 2,30-2,39 (м, 1H), 2,43-2,45 (м, 1H), 2,48 (с, 3H), 2,68-2,96 (м,1H), 3,67 (с, 3H), 3,90 (с, 3H), 3,91 (с, 3H), 4,83-4,93 (м, 1H), 4,88 (с, 2H), 5,51 (с, 2H), 6,77 (с, 1H), 7,27 (с, 1H), 7,52 (т, J=7,7 Гц, 1H), 7,65-7,70 (м, 2H), 8,05 (д, J=7,7 Гц, 1H), 8,14 (с, 1H), 8,28 (д, J=10,6 Гц, 1H), 9,02 (с, 1H).
<Стадия 2>
Получение 5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-ил}метилового эфира 3-нитрооксибензойной кислоты
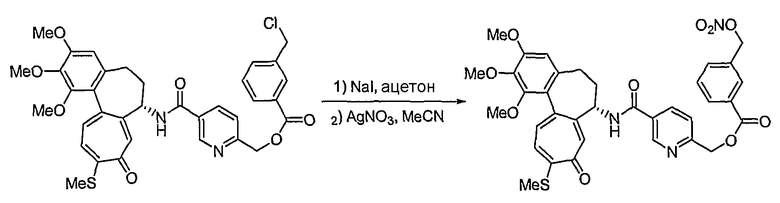
Соединение, полученное на стадии 1 (90 мг, 0,13 ммоль), и йодид натрия (31 мг, 0,20 ммоль) растворяют в 3 мл ацетона и смесь оставляют реагировать при комнатной температуре в течение 1 дня. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Раствор концентрируют при пониженном давлении. Концентрированный раствор и нитрат серебра (30 мг, 0,045 ммоль) растворяют в 5 мл ацетонитрила и смесь оставляют реагировать при комнатной температуре в течение 1 часа. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Раствор концентрируют при пониженном давлении. Остаток очищают на короткой хроматографической колонке (этилацетат) и, используя жидкостную хроматографию PLC, получают 26 мг (выход: 29%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,09-2,17 (м, 1H), 2,35-2,41 (м, 2H), 2,44 (с, 3H), 2,55-2,58 (м, 1H), 3,74 (с, 3H), 3,91 (с, 3H), 3,96 (с, 3H), 4,92-4,95 (м, 1H), 5,45 (с, 2H), 5,47 (с, 2H), 6,56 (с, 1H), 7,13 (д, J=10,2 Гц, 1H), 7,35 (т, J=9,1 Гц, 2H), 7,50 (т, J=8,5 Гц, 1H), 7,54 (с, 1H), 7,62 (д, J=7,3 Гц, 1H), 8,14 (д, J=8,3 Гц, 2H), 8,24 (дд, J=2,2, 6,2 Гц, 1H, ArH), 8,36 (д, J=6,9 Гц, 1H), 9,09 (с, 1H).
Примеры 26-34
Соединения примеров 26-34 синтезируют по аналогии со способом, раскрытым в примере 25, и промежуточные соединения получают раскрытым далее способом.
<Промежуточное соединение 16>
Получение 2-гидрокси-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида

2-гидроксибензойную кислоту (203 мг, 1,47 ммоль), EDCI (385 мг, 2,01 ммоль) и HOBt (271 мг, 2,01 ммоль) растворяют в диметилформамиде (10 мл). Добавляют триэтиламин (0,37 мл, 2,67 ммоль) и смесь перемешивают при комнатной температуре в течение 1 дня. Добавляют 7-амино-1,2,3-триметокси-10-метилсульфонил-6,7-дигидро-5Н-бензо[a]гептален-9-он (500 мг, 1,34 ммоль) и смесь перемешивают при комнатной температуре в течение 1 дня. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют диэтиловым эфиром. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (гексан:этилацетат = 1:5), и перекристаллизовывают из метанола, получая 516 мг (выход: 78%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,14-2,17 (м, 1H), 2,31-2,37 (м, 1H), 2,40-2,44 (с, 1H), 2,45 (с, 3H), 2,56-2,60 (м, 1H), 3,74 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,85-4,92 (м, 1H), 6,56 (с, 1H), 6,64 (д, J=7,7 Гц, 1H), 6,77 (д, J=8,4 Гц, 1H), 7,15 (д, J=10,6 Гц, 1H), 7,22 (д, J=7,3 Гц, 1H), 7,38 (д, J=10,6 Гц, 1H), 7,57 (с, 1H), 7,71 (д, J=8,0 Гц, 1H).
<Промежуточное соединение 17>
Получение 3-гидрокси-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида

Способом, аналогичным способу получения промежуточного соединения 16, используя 3-гидроксибензойную кислоту (497 мг, 1,33 ммоль), получают 558 мг (выход: 85%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, ДМСО-d6): δ 2,07-2,15 (м, 1H), 2,18-2,35 (м, 6H), 3,55 (с, 3H), 3,80 (с, 3H), 3,84 (с, 3H), 4,53-4,56 (м, 1H), 6,81 (с, 1H), 6,92 (д, J=6,8 Гц, 1H), 7,10 (с, 1H), 7,15-7,31 (м, 5H), 8,98 (д, J=7,6 Гц, 1H, -NH), 9,70 (с, 1H, -OH).
Пример 26:
5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 4-нитрооксимасляной кислоты
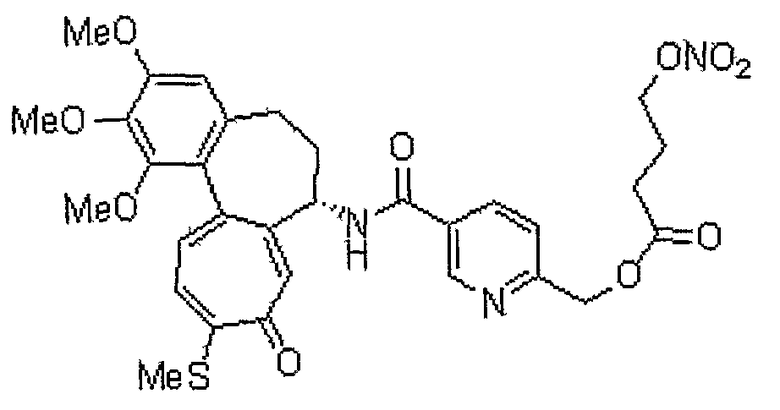
1H ЯМР (400 МГц, CDCl3): δ 2,06-2,16 (м, 1H), 2,08-2,11 (м, 2H), 2,31-2,49 (м, 1H), 2,45 (с, 3H), 2,57 (т, J=7,1 Гц, 2H), 3,75 (с, 3H), 3,91 (с, 3H), 3,96 (с, 3H), 4,52 (т, J=6,4 Гц, 2H), 4,91-4,94 (м, 1H), 5,21 (с, 2H), 6,56 (с, 1H), 7,13 (д, J=10,9 Гц, 1H), 7,21-7,25 (м, 1H), 7,38 (д, J=10,6 Гц, 1H), 7,52 (с, 1H), 8,22 (д, J=8,0 Гц, 1H), 9,05 (с, 1H).
Пример 27:
6-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 3-нитрооксиметилбензойной кислоты
1H ЯМР (400 МГц, CDCl3): δ 1,84-1,92 (м, 1H), 2,33-2,60 (м, 1H), 2,43 (с, 3H), 2,46-2,54 (м, 1H) 2,55-2,60 (м, 1H), 3,73 (с, 3H), 3,92 (с, 3H), 3,96 (с, 3H), 4,74-4,81 (м, 1H), 5,51 (с, 2H) 5,57 (д, J=4,0 Гц, 1H), 6,56 (с, 1H), 7,04 (д, J=10,4 Гц, 1H), 7,25 (д, J=10,4 Гц, 1H), 7,54-7,60 (м, 2H), 7,65 (д, J=7,6 Гц, 1H), 7,87 (т, J=8,0 Гц, 1H), 7,99 (д, J=7,6 Гц, 1H), 8,21 (с, 2H), 8,22 (с, 1H), 8,41 (д, J=7,2 Гц, 1H).
Пример 28:
6-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 4-нитрооксимасляной кислоты
1H ЯМР (400 МГц, CDCl3): δ 2,02-2,09 (м, 1H), 2,11-2,18 (м, 2H), 2,21-2,40 (м, 2H), 2,43 (с, 3H), 2,47-2,56 (м, 1H), 2,63 (т, J=7,2 Гц, 2H), 3,73 (с, 3H), 3,92 (с, 3H), 3,96 (с, 3H), 4,57 (т, J=6,4 Гц, 2H), 4,77-4,83 (м, 1H), 5,30 (д, J=4,8 Гц, 2H), 6,58 (с, 1H), 7,05 (д, J=10,4 Гц, 1H), 7,25 (с, 1H), 7,32 (д, J=10,4 Гц, 1H), 7,52 (д, J=8,0 Гц, 1H), 7,84 (т, J=8,0 Гц, 1H), 7,98 (д, J=8,0 Гц, 1H), 8,45 (д, J=6,8 Гц, 1H).
Пример 29:
2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-
тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 3-нитрооксиметилбензойной кислоты
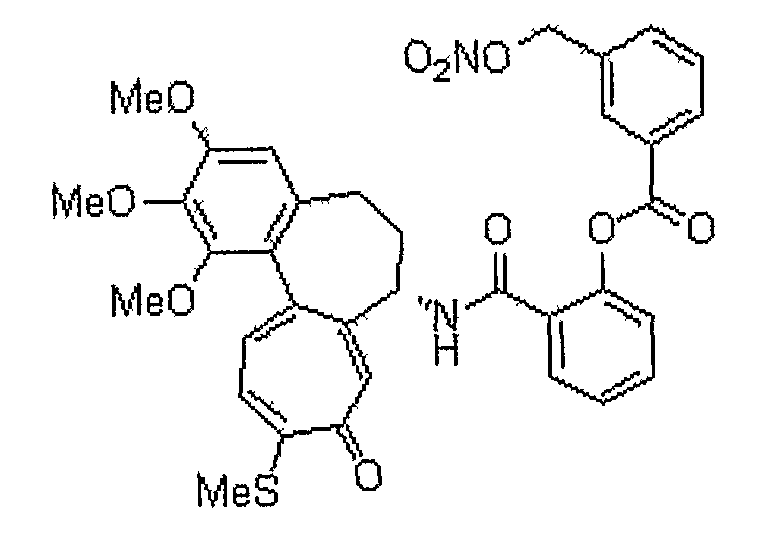
1H ЯМР (400 МГц, CDCl3): δ 1,48-1,55 (м, 1H), 1,83-1,89 (м, 1H), 2,22-2,39 (м, 2H), 2,42 (с, 3H), 3,62 (с, 3H), 3,87 (с, 3H), 3,92 (с, 3H), 4,63-4,70 (м, 1H), 5,54 (с, 2H), 6,47 (с, 1H), 6,89 (д, J=7,4 Гц, 1H), 7,03 (д, J=10,2 Гц, 1H), 7,17 (с, 1H), 7,23 (д, J=10,1 Гц, 1H), 7,35 (т, J=7,3 Гц, 1H), 7,54 (т, J=7,4 Гц, 1H), 7,58 (т, J=7,6 Гц, 1H), 7,72 (д, J=8,0 Гц, 1H), 7,83 (д, J=7,7 Гц, 1H), 8,26 (м, 2H).
Пример 30:
2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 4-нитрооксимасляной кислоты

1H ЯМР (400 МГц, CDCl3): δ 1,60-1,90 (м, 1H), 2,10-2,14 (м, 2H), 2,26-2,39 (м, 1H), 2,44 (с, 3H), 2,46-2,51 (м, 1H), 2,56-2,61 (м, 1H), 2,80 (т, J=7,1 Гц, 2H), 3,70 (с, 3H), 3,92 (с, 3H), 3,96 (с, 3H), 4,55 (т, J=6,2 Гц, 2H), 4,74-4,81 (м, 1H), 6,57 (с, 1H), 6,82 (д, J=6,9 Гц, 1H), 7,08 (д, J=10,6 Гц, 1H), 7,13 (д, J=9,1 Гц, 1H), 7,23 (с, 1H), 7,28-7,36 (м, 1H), 7,47 (т, J=9,9 Гц, 1H), 7,68 (д, J=9,5 Гц, 1H).
Пример 31:
3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-
тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 3-нитрооксиметилбензойной кислоты
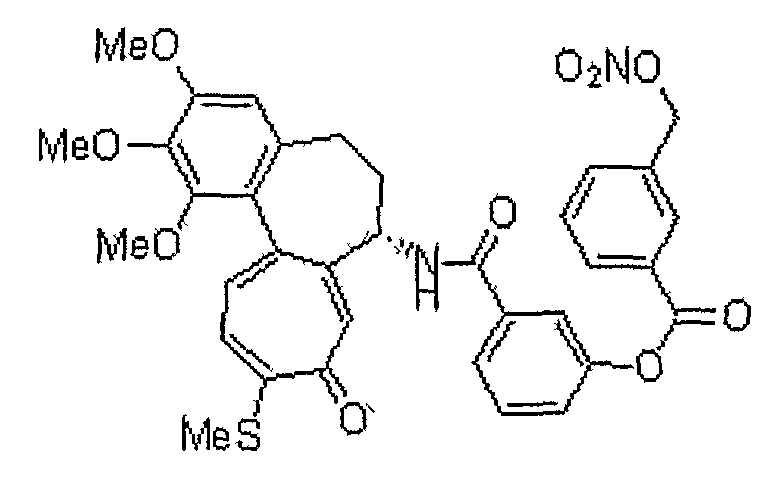
1H ЯМР (400 МГц, CDCl3): δ 2,05-2,15 (м, 1H), 2,23-2,59 (м, 6H), 3,75 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,90-5,00 (м, 1H), 5,49 (с, 2H), 6,56 (с, 1H), 7,03 (д, J=10,4 Гц, 1H), 7,27 (с, 2H), 7,34 (д, J=8,8 Гц, 2H), 7,53-7,65 (м, 2H), 7,76 (с, 1H), 7,77 (д, J=6,4 Гц, 1H), 8,13 (м, 3H).
Пример 32:
3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]фениловый эфир 4-нитрооксимасляной кислоты
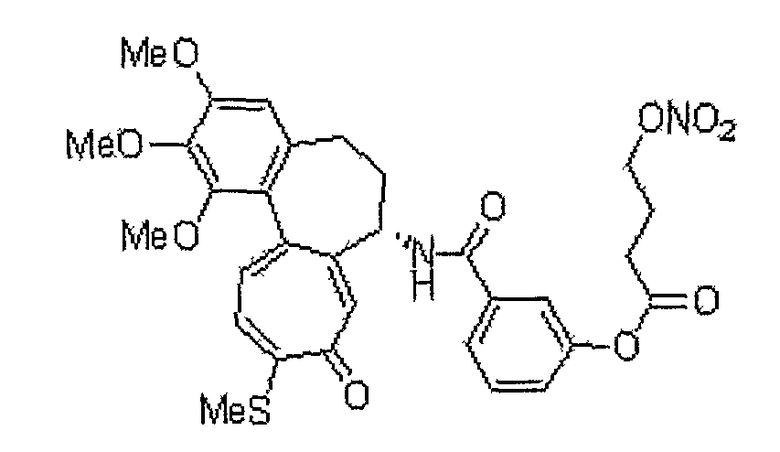
1H ЯМР (400 МГц, CDCl3): δ 2,04-2,18 (м, 3H), 2,25-2,60 (м, 6H), 2,65 (т, J=6,8 Гц, 2H), 3,74 (с, 3H), 3,9l (с, 3H), 3,96 (с, 3H), 4,56 (т, J=6,4 Гц, 2H), 4,87-4,94 (м, 1H), 6,56 (с, 1H), 7,08-7,12 (м, 2H), 7,26-7,35 (м, 2H), 7,48 (с, 1H), 7,59 (с, 1H), 7,71 (д, J=8,0 Гц, 1H), 7,75 (д, J=7,6 Гц, 1H, -NH).
Пример 33:
3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-
тетрагидробензо[a]гептален-7-илкарбамоил]бензиловый эфир 3-нитрооксиметилбензойной кислоты
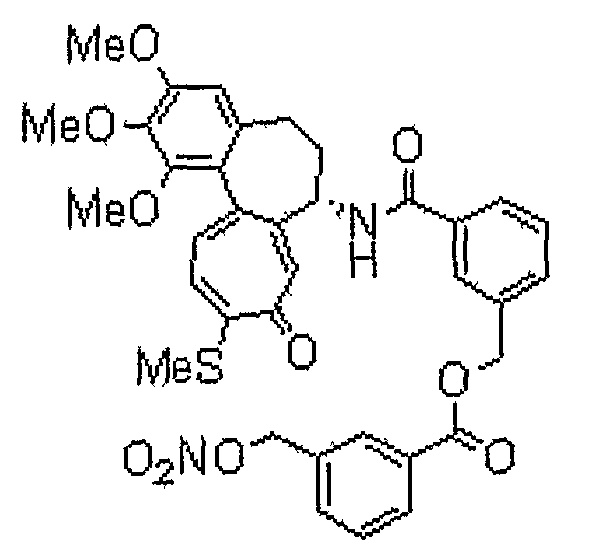
1H ЯМР (400 МГц, CDCl3): δ 2,05-2,18 (м, 1H), 2,31-2,47 (м, 2H), 2,40 (с, 3H), 2,55-2,59 (м, 1H), 3,75 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,90-4,96 (м, 1H), 5,24 (с, 2H), 5,44 (с, 2H), 6,56 (с, 1H), 7,09 (д, J=10,6 Гц, 1H), 7,27 (дд, J=7,6, 7,6 Гц, 1H), 7,36 (д, J=10,6 Гц, 1H), 7,44 (д, J=7,6 Гц, 1H), 7,45 (дд, J=8,0, 7,6 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,59 (с, 1H), 7,79 (д, J=7,6 Гц, 1H), 7,91 (с, 1H), 8,05 (д, J=7,6 Гц, 1H), 8,06 (с, 1H), 8,18 (д, J=7,2 Гц, 1H).
Пример 34:
3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]бензиловый эфир 4-нитрооксимасляной кислоты
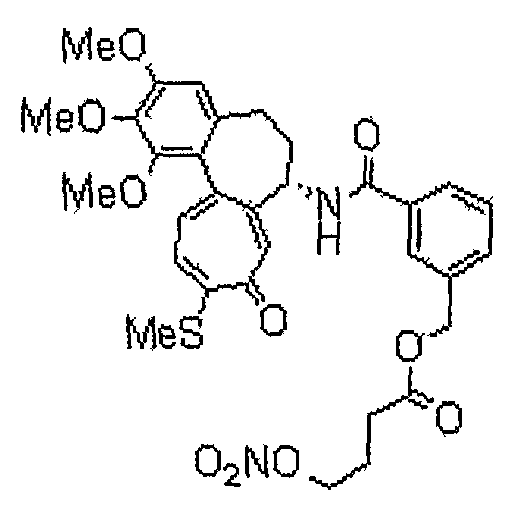
1H ЯМР (400 МГц, CDCl3): δ 2,00-2,14 (м, 3H), 2,31-2,51 (м, 4H), 2,44 (с, 3H), 2,56-2,60 (м, 1H), 3,75 (с, 3H), 3,92 (с, 3H), 3,97 (с, 3H), 4,49 (т, J=6,2 Гц, 2H), 4,89-4,95 (м, 1H), 4,99 (д, J=12,4 Гц, 1H), 5,04 (д, J=12,4 Гц, 1H), 6,56 (с, 1H), 7,11 (д, J=10,6 Гц, 1H), 7,26 (дд, J=7,6, 7,6 Гц, 1H), 7,35-7,37 (м, 2H), 7,54 (с, 1H), 7,76 (д, J=8,0 Гц, 1H), 7,81 (с, 1H), 8,00 (д, J=7,6 Гц, 1H).
Пример 35:
Получение 2-нитрозотио-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
<Стадия 1>
Получение 2-меркапто-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
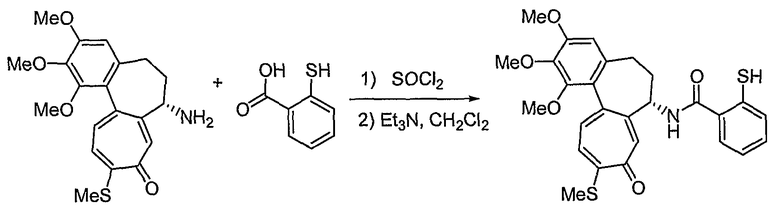
Избыток тионилхлорида добавляют в тиосалициловую кислоту (115 мг, 0,75 ммоль) и смесь перемешивают при нагревании в течение 1 дня. Реакционную смесь концентрируют при пониженном давлении для удаления тионилхлорида, получая соединение хлорида. 7-амино-1,2,3-триметокси-10-метилсульфонил-6,7-дигидро-5Н-бензо[a]гептален-9-он (243 мг, 0,62 ммоль) растворяют в очищенном дихлорметане. Медленно добавляют триэтиламин (0,26 мл, 1,86 ммоль). Добавляют также тиосалицилхлорид, растворенный в дихлорметане при 0°C, и смесь перемешивают в течение 30 мин. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:метанол = 12:1), получая 210 мг (выход: 66%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,06-2,17 (м, 1H), 2,33-2,49 (м, 2H), 2,42 (с, 3H), 2,54-2,59 (м, 1H), 3,72 (с, 3H), 3,92 (с, 3H), 3,96 (с, 3H), 4,86-4,93 (м, 1H), 6,56 (с, 1H), 7,06 (д, J=10,6 Гц, 1H), 7,14 (т, J=7,5 Гц, 1H), 7,24 (д, J=7,7 Гц, 1H), 7,31 (д, J=10,6 Гц, 1H), 7,51 (с, 1H), 7,58 (д, J=7,3 Гц, 1H), 7,63 (т, J=8,6 Гц, 2H).
<Стадия 2>
Получение 2-нитрозотио-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
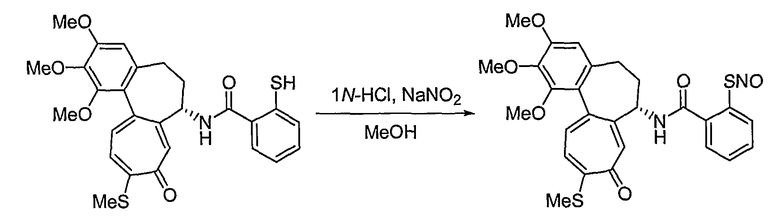
Соединение, полученное на стадии 1 (40 мг, 0,078 ммоль), растворяют в метаноле. Добавляют 1 н. водный раствор HCl (3 мл). Добавляют также нитрит натрия (NaNO2, 6,5 мг, 0,094 ммоль), растворенный в воде (1,5 мл), и смесь перемешивают при комнатной температуре в течение 1 час. Добавляют бикарбонат натрия, чтобы погасить реакцию, и водный слой экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ: метанол = 12:1) и перекристаллизовывают из метанола, получая 34,1 мг (выход: 81%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,07-2,14 (м, 1H), 2,31-2,37 (м, 1H), 2,42 (с, 3H), 2,45-2,48 (м, 1H), 2,54-2,59 (м, 1H), 3,72 (с, 3H), 3,91 (с, 3H), 3,96 (с, 3H), 4,88-4,94 (м, 1H), 6,56 (с, 1H), 7,06 (д, J=10,6 Гц, 1H), 7,10 (т, J=7,5 Гц, 1H), 7,21 (т, J=6,9 Гц, 1H), 7,31 (д, J=10,6 Гц, 1H), 7,54 (с, 1H), 7,64 (т, J=8,6 Гц, 2H), 7,72 (д, J=7,3 Гц, 1H).
Пример 36:
Получение 3-нитрозооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
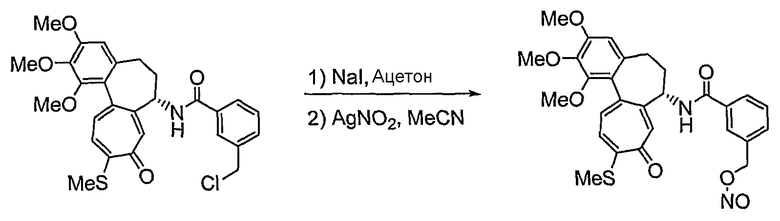
3-хлорметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид (119,8 мг, 0,228 ммоль) и йодид натрия (136,5 мг, 0,911 ммоль) растворяют в ацетоне (15 мл) и смесь перемешивают при 55°C в течение 1 дня. Реакционную смесь экстрагируют хлороформом, промывают насыщенным водным раствором хлорида натрия. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток и нитрит серебра (125,5 мг, 0,812 ммоль) растворяют в ацетонитриле (5 мл) и смесь перемешивают при комнатной температуре в течение 1 дня. Реакционную смесь экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:хлороформ = 4:1), получая 35,4 мг (выход: 32,4%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,35-2,55 (м, 3H), 2,48 (с, 3H), 2,62-2,66 (м, 1H), 3,76 (с, 3H), 3,84 (д, J=12,8 Гц, 1H), 3,92 (с, 3H), 3,98 (с, 3H), 4,26 (д, J=12,8 Гц, 1H), 4,91-4,97 (м, 1H), 6,58 (с, 1H), 6,96 (дд, J=7,7, 7,7 Гц, 1H), 7,22 (д, J=10,6 Гц, 1H), 7,26-7,30 (м, 2H), 7,44-7,47 (м, 2H), 7,86 (с, 1H), 9,30 (д, J=6,8 Гц, 1H).
Пример 37:
3-фтор-5-нитрозооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
Указанное соединение синтезируют по аналогии со способом, раскрытым в примере 36.
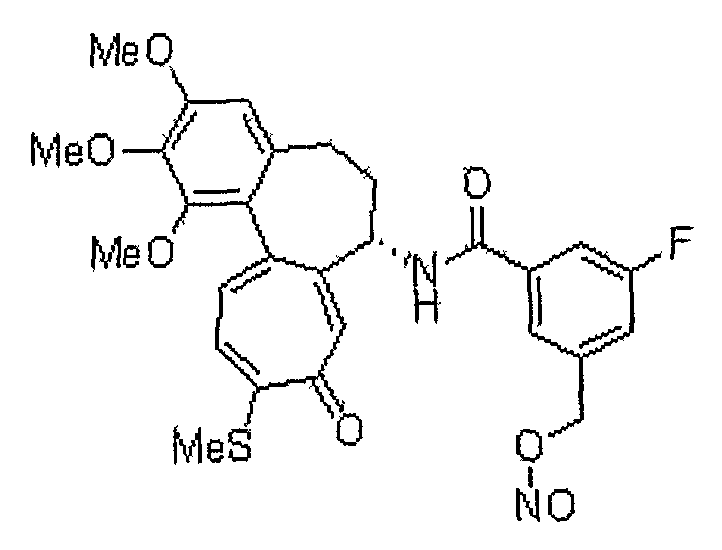
1H ЯМР (400 МГц, CDCl3): δ 2,04-2,20 (м, 1H), 2,47 (с, 3H), 2,52-2,62 (м, 2H), 2,67-2,72 (м, 1H), 3,81 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,7l (с, 2H), 4,93-4,96 (м, 1H), 6,42 (д, J=6,2 Гц, 1H), 6,58 (с, 1H), 7,14-7,21 (м, 2H), 7,30 (д, J=8,4 Гц, 1H), 7,42 (с, 1H), 7,53 (д, J=10,6 Гц, 1H).
Пример 38:
Получение 3-нитрозотиометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
<Стадия 1>
Получение 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]бензилового эфира метансульфоновой кислоты
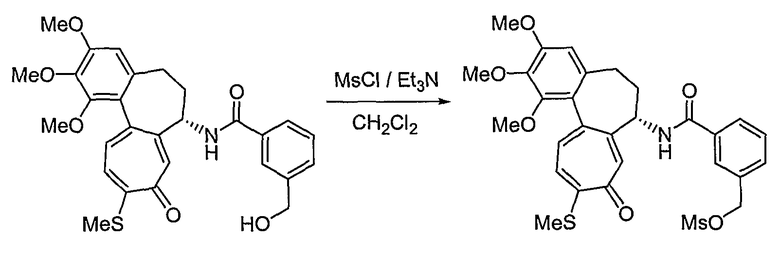
3-гидроксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид (252,5 мг, 0,497 ммоль) растворяют в дихлорметане (10 мл) и температуру снижают до 0°C. Добавляют метансульфонилхлорид (42,4 мл, 0,547 ммоль) и триэтиламин (0,104 мл, 0,745 ммоль) и смесь перемешивают при комнатной температуре в течение 2 час. Реакционную смесь экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:хлороформ = 3:2), получая 182,3 мг (выход: 62,6%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,14-2,21 (м, 1H), 2,31-2,50 (м, 2H), 2,46 (с, 3H), 2,57-2,62 (м, 1H), 2,85 (с, 3H), 3,76 (с, 3H), 3,92 (с, 3H), 3,97 (с, 3H), 4,87-4,94 (м, 1H), 5,06 (д, J=12,7 Гц, 1H), 5,10 (д, J=12,7 Гц, 1H), 6,57 (с, 1H), 7,14 (д, J=10,6 Гц, 1H), 7,28 (дд, J=7,7, 7,7 Гц, 1H), 7,38-7,42 (м, 2H), 7,56 (с, 1H), 7,74 (д, J=8,0 Гц, 1H), 8,24 (д, J=7,7 Гц, 1H).
<Стадия 2>
Получение S-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил]бензилового эфира тиоуксусной кислоты
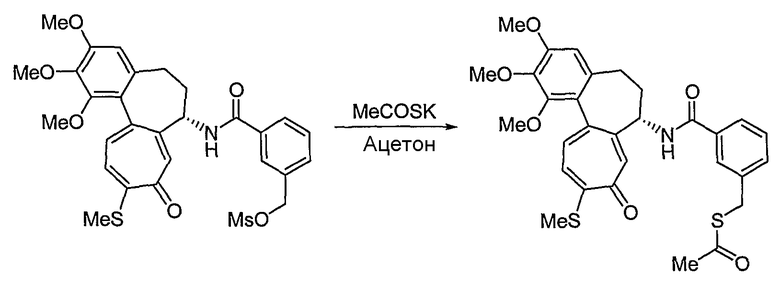
Соединение, полученное на стадии 1 (182,3 мг, 0,311 моль), растворяют в ацетоне (6 мл) и температуру снижают до 0°C. Медленно добавляют тиоацетат калия (53,2 мг, 0,467 ммоль) при 0°C и смесь перемешивают в течение 1 часа. Реакционную смесь экстрагируют хлороформом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 173,6 мг (выход: 98,6%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,05-2,14 (м, 1H), 2,30 (с, 2H), 2,32-2,49 (м, 2H), 2,44 (с, 3H), 2,52-2,61 (м, 1H), 3,74 (с, 3H), 3,92 (с, 3H), 3,97 (с, 3H), 4,01 (д, J=13,9 Гц,1H), 4,05 (д, J=13,9 Гц, 1H), 4,87-4,93 (м, 1H), 6,56 (с, 1H), 7,10 (д, J=10,3 Гц, 1H), 7,21 (дд, J=7,7, 7,7 Гц, 1H), 7,32-7,36 (м, 2H), 7,49 (с, 1H), 7,64-7,71 (м, 3H).
<Стадия 3>
Получение 3-меркаптометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
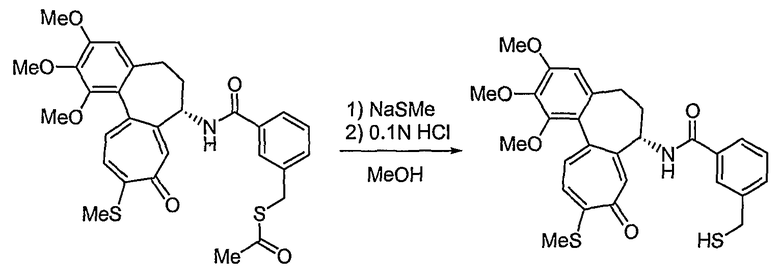
Соединение, полученное на стадии 2 (165,2 мг, 0,292 моль), растворяют в метаноле (6 мл) и температуру снижают до 0°C. Медленно добавляют тиометоксид натрия (21,5 мг, 0,307 ммоль) при 0°C и смесь перемешивают при комнатной температуре в течение 30 мин. Реакцию гасят, добавляя 0,1 н. водный раствор HCl. Реакционную смесь экстрагируют хлороформом и промывают насыщенным водным раствором хлорида натрия. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:хлороформ = 1:1), получая 182,3 мг (выход: 62,6%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,70 (т, J=7,3 Гц, 1H), 2,12-2,19 (м, 1H), 2,31-2,50 (м, 2H), 2,18 (с, 3H), 2,57-2,61 (м, 1H), 3,58 (д, J=7,3 Гц, 1H), 3,74 (с, 3H), 3,92 (с, 3H), 3,97 (с, 3H), 4,89-4,95 (м, 1H), 6,57 (с, 1H), 7,12 (д, J=10,3 Гц, 1H), 7,19 (дд, J=7,7, 7,7 Гц, 1H), 7,34 (д, J=7,7 Гц, 1H), 7,37 (д, J=10,3 Гц, 1H), 7,55 (с, 1H), 7,59 (д, J=7,7 Гц, 1H), 7,70 (с, 1H), 7,93 (д, J=7,3 Гц, 1H).
<Стадия 4>
Получение 3-нитрозотиометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
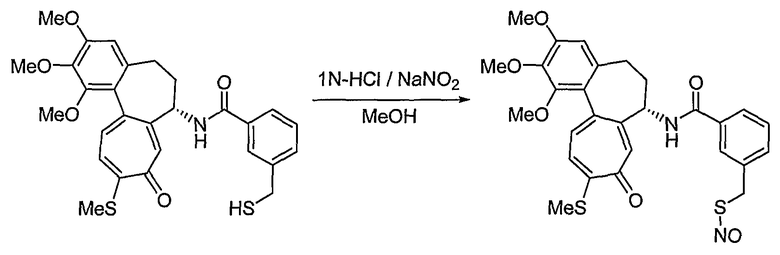
Соединение, полученное на стадии 3 (101,2 мг, 0,193 моль), растворяют в метаноле (3 мл) и диметилформамиде (3 мл) и температуру снижают до 0°C. Медленно добавляют 0,1 н. водный раствор HCl (3 мл) и нитрит натрия (16,0 мг) при 0°C и смесь перемешивают при комнатной температуре в течение 2 час. Реакционную смесь экстрагируют хлороформом и промывают насыщенным раствором карбоната натрия. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (этилацетат:хлороформ:гексан = 3:2:1,5), получая 19,5 мг (выход: 18,3%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,08-2,16 (м, 1H), 2,31-2,51 (м, 2H), 2,45 (с, 3H), 2,57-2,61 (м, 1H), 3,57 (с, 3H), 3,80 (с, 2H), 3,92 (с, 3H), 3,97 (с, 3H), 4,89-4,96 (м, 1H), 6,57 (с, 1H), 7,10 (д, J=10,3 Гц, 1H), 7,25 (дд, J=7,7, 7,7 Гц, 1H), 7,36 (д, J=10,3 Гц, 1H), 7,38 (д, J=7,7 Гц, 1H), 7,53 (с, 1H), 7,68 (д, J=7,7 Гц, 1H), 7,77 (с, 1H), 7,79 (д, J=7,0 Гц, 1H).
Пример 39:
3-фтор-5-нитрозотиометил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамид
Указанное в заголовке соединение примера 39 синтезируют по аналогии со способом, раскрытым в примере 38.
1H ЯМР (400 МГц, CDCl3): δ 2,30-2,48 (м, 3H), 2,44 (с, 3H), 2,56-2,59 (м, 1H), 3,57 (кв, J=13,9, 16,5 Гц, 2H), 3,73 (с, 3H), 3,91 (с, 3H), 3,97 (с, 3H), 4,97-5,03 (м, 1H), 6,57 (с, 1H), 7,02 (д, J=8,4 Гц, 1H), 7,15 (д, J=10,6 Гц, 1H), 7,39 (д, J=10,2 Гц, 1H), 7,43 (с, 1H), 7,64 (д, J=9,1 Гц, 1H), 7,73 (с, 1H), 8,96 (д, J=7,3 Гц, 1H).
Пример 40:
Получение 3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3,10-тетраметокси-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
<Стадия 1>
Получение 3-фтор-5-гидроксиметил-N-[(7S)-1,2,3,10-тетраметокси-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
Деацетилколхицин (150 мг, 0,42 ммоль), 3-фтор-5-гидроксиметилбензойную кислоту (85 мг, 0,50 ммоль) и HOBt (67 мг, 0,50 ммоль) растворяют в диметилформамиде (2 мл) и температуру снижают до 0°C. Медленно добавляют EDCI (95 мг, 0,50 ммоль) при 0°C и смесь перемешивают при комнатной температуре. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:этилацетат = 2:1), получая 110 мг (выход: 52%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,40-2,58 (м, 3H), 2,62-2,69 (м, 1H), 3,64-3,78 (м, 1H), 3,74 (с, 3H), 3,92 (с, 3H), 3,98 (с, 3H), 4,07 (с, 3H), 4,23-4,28 (м, 1H), 4,81-4,88 (м, 1H), 6,58 (с, 1H), 6,87 (д, J=9,2 Гц, 1H), 6,97 (д, J=9,2 Гц, 1H), 7,03 (д, J=10,4 Гц, 1H), 7,38 (с, 1H), 7,50 (д, J=10,4 Гц, 1H), 7,96 (с, 1H), 9,64 (д, J=6,0 Гц, 1H).
<Стадия 2>
Получение 3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3,10-тетраметокси-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
Аналогично способам на стадиях 2 и 3 примера 5, используя соединение, полученное на стадии 1 (90 мг, 0,18 ммоль), получают 30 мг (выход: 30%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,38-2,49 (м, 3H), 2,58-2,59 (м, 1H), 3,75 (с, 3H), 3,92 (с, 3H), 3,98 (с, 3H), 4,05 (с, 3H), 4,89-4,93 (м, 1H), 4,94 (д, J=12,8 Гц, 1H), 5,10 (д, J=12,8 Гц, 1H), 6,58 (с, 1H), 6,96 (д, J=8,4 Гц, 1H), 7,01 (д, J=10,4 Гц, 1H), 7,17 (д, J=8,4 Гц, 1H), 7,45 (с, 1H), 7,48 (д, J=10,4 Гц, 1H), 7,89 (с, 1H), 9,25 (д, J=6,4 Гц, 1H).
Пример 41:
Получение 3-нитрооксиметил-N-метил-N-[(7S)-l,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
<Стадия 1>
Получение 3-хлорметил-N-метил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
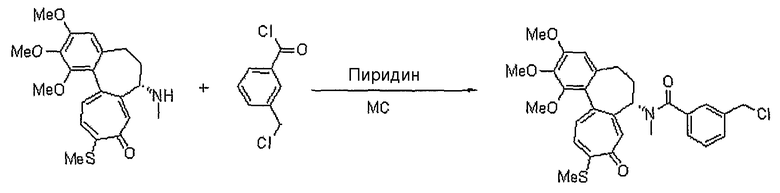
Тиодемеколцин (50 мг, 0,129 ммоль) и пиридин (0,012 мл, 0,154 ммоль) растворяют в дихлорметане и температуру снижают до 0°C. Медленно добавляют 3-(хлорметил)бензоилхлорид (0,022 мл, 0,154 ммоль) при 0°C и смесь перемешивают при комнатной температуре. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:этилацетат = 2:1), получая 45 мг (выход: 67%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,24-2,38 (м, 2H), 2,44 (с, 3H), 2,45-2,58 (м, 1H), 2,61-2,74 (м, 1H), 3,25 (с, 3H), 3,72 (с, 3H), 3,91 (с, 3H), 3,92 (с, 3H), 4,54 (с, 2H), 5,05 (ушир., 1H), 6,51 (с, 1H), 7,06 (д, J=10,0 Гц, 1H), 7,09 (с, 1H), 7,22-7,41 (м, 5H).
<Стадия 2>
Получение 3-нитрооксиметил-N-метил-N-[(7S)-l,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
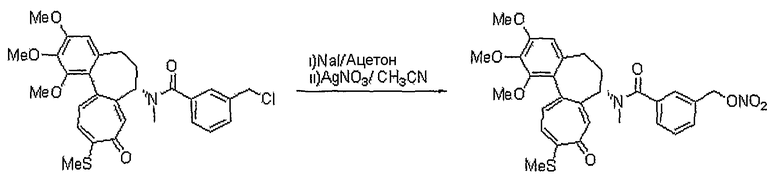
Аналогично способу стадии 2 примера 25, используя соединение, полученное на стадии 1 (45 мг, 0,085 ммоль), получают 40 мг (выход: 83%, твердое вещество желтого цвета) целевого соединения
1H ЯМР (400 МГц, CDCl3): δ 2,24-2,38 (м, 2H), 2,44 (с, 3H), 2,45-2,58 (м, 1H), 2,61-2,74 (м, 1H), 3,24 (с, 3H), 3,71 (с, 3H), 3,91 (с, 3H), 3,92 (с, 3H), 4,77 (ушир., 1H), 5,58 (с, 2H), 6,85 (с, 2H), 7,21 (д, J=10,0 Гц, 1H), 7,29 (д, J=10,4 Гц, 1H), 7,40-7,53 (м, 4H).
Пример 42:
Получение 3-фтор-N-метил-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
<Стадия 1>
Получение 3-фтор-5-гидроксиметил-N-метил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
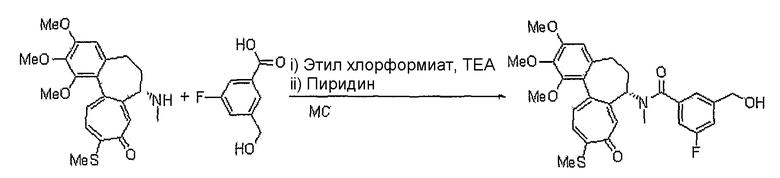
3-фтор-5-гидроксиметилбензойную кислоту (52 мг, 0,309 ммоль) растворяют в дихлорметане (3 мл) в атмосфере азота. Медленно добавляют этилхлорформиат (0,022 мл, 0,231 ммоль) и TEA (0,042 мл, 0,309 ммоль) при 0°C и смесь перемешивают при 0°C в течение 30 мин. Кроме того, добавляют пиридин (0,012 мл, 0,154 ммоль) и тиодемеколцин (60 мг, 0,154 ммоль) и смесь перемешивают при комнатной температуре в течение 3 час. Добавляют воду, чтобы погасить реакцию, и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают, используя хроматографическую колонку (хлороформ:этилацетат = 2:1), получая 30 мг (выход: 21%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,76 (ушир., 1H), 2,24-2,38 (м, 2H), 2,44 (с, 3H), 2,45-2,58 (м, 1H), 2,61-2,74 (м, 1H), 3,23 (с, 3H), 3,72 (с, 3H), 3,93 (с, 3H), 3,97 (с, 3H), 4,65 (с, 2H), 5,02 (ушир., 1H), 6,57 (с, 1H), 6,92 (с, 1H), 6,99-7,24 (м, 4H), 7,34 (с, 1H).
<Стадия 2>
Получение 3-фтор-N-метил-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]бензамида
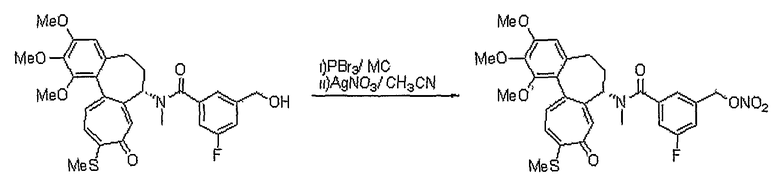
Аналогично способам стадий 2 и 3 примера 5, используя соединение, полученное на стадии 1 (55 мг, 0,101 ммоль), получают 20 мг (выход: 37%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 2,22-2,41 (м, 2H), 2,45 (с, 3H), 2,45-2,58 (м, 1H), 2,61-2,74 (м, 1H), 3,24 (с, 3H), 3,75 (с, 3H), 3,91 (с, 3H), 3,95 (с, 3H), 5,02 (ушир., 1H), 5,39 (с, 2H), 6,57 (с, 1H), 7,05-7,24 (м, 5H), 7,34 (с, 1H).
Пример 43:
Получение 2-(3-фтор-5-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамида
<Стадия 1>
Получение (3-фтор-5-гидроксиметилфенил)уксусной кислоты
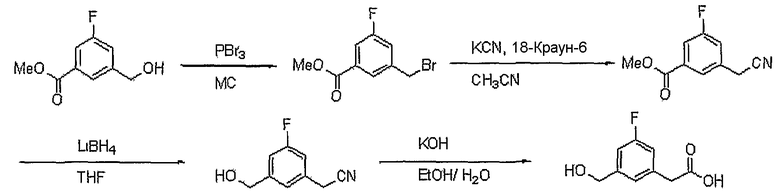
Соединение, полученное на стадии 1 получения промежуточного соединения 6 (2,5 г, 13,57 ммоль), растворяют в дихлорметане (30 мл). Медленно добавляют PBr3 (1,15 мл, 12,21 ммоль) и смесь перемешивают при комнатной температуре в течение 3 часов, получая метиловый эфир 3-бромметил-5-фторбензойной кислоты (1,6 г, выход: 50%, твердое вещество белого цвета). Метиловый эфир 3-бромметил-5-фторбензойной кислоты (1,5 г, 6,1 ммоль), KCN (1,6 г, 24,4 ммоль) и 18-краун-6 (820 мг, 3,1 ммоль) растворяют в ацетонитриле (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов, получая метиловый эфир 3-цианометил-5-фторбензойной кислоты (900 мг, выход: 77%, твердое вещество белого цвета). Метиловый эфир 3-цианометил-5-фторбензойной кислоты (900 мг, 4,65 ммоль) растворяют в тетрагидрофуране (10 мл). Медленно добавляют 2 M тетрагидрофурановый раствор боргидрида лития (2,3 мл, 4,6 ммоль) и смесь кипятят с обратным холодильником, получая (3-фтор-5-гидроксиметилфенил)ацетонитрил (430 мг, выход: 56%, твердое вещество белого цвета). (3-фтор-5-гидроксиметилфенил)ацетонитрил (400 мг, 2,42 ммоль) и гидроксид калия (1,34 г, 23,8 ммоль) растворяют в этаноле (10 мл) и воде (5 мл). Реакционную смесь кипятят с обратным холодильником в течение 24 час, получая 356 мг (выход: 80%, твердое вещество белого цвета) целевого соединения.
1H ЯМР (400 МГц, CD3OD): δ 3,62 (с, 2H), 4,59 (с, 2H), 6,94 (д, J=9,6 Гц, 1H), 7,04 (д, J=9,6 Гц, 1H), 7,08 (с, 1H).
<Стадия 2>
Получение 2-(3-фтор-5-гидроксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамида
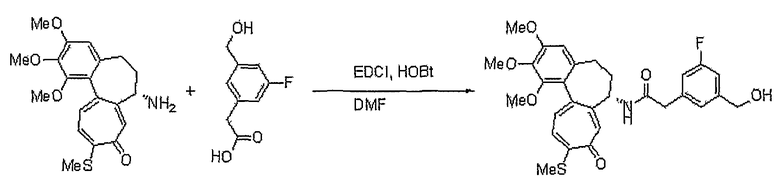
Аналогично способу стадии 1 примера 40, используя соединение, полученное на стадии 1 (30 мг, 0,16 ммоль), получают 58 мг (выход: 70%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,92-2,01 (м, 1H), 2,19-2,40 (м, 2H), 2,41 (с, 3H), 2,46-2,52 (м, 1H), 3,46 (д, J=14,0 Гц, 1H), 3,52 (д, J=14,0 Гц, 1H), 3,63 (с, 3H), 3,89 (с, 3H), 3,91 (с, 3H), 4,64-4,74 (м, 1H), 4,75 (с, 2H), 6,53 (с, 1H), 6,86-6,94 (м, 3H), 7,07 (д, J=10,8 Гц, 1H), 7,13 (с, 1H), 7,30 (д, J=10,8 Гц, 1H), 7,39 (с, 1H).
<Стадия 3>
Получение 3-фтор-5-[(7S)-(1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-илкарбамоил)метил]бензилового эфира метансульфоновой кислоты
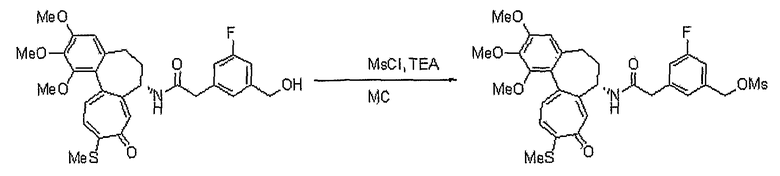
Аналогично способу получения на стадии 1 примера 38, используя соединение, полученное на стадии 2 (58 мг, 0,107 ммоль), получают 60 мг (выход: 90%, твердое вещество желтого цвета) целевого соединения
<Стадия 4>
Получение 2-(3-фтор-5-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамида

Аналогично способу получения на стадии 2 примера 25, используя соединение, полученное на стадии 3 (60 мг, 0,097 ммоль), получают 30 мг (выход: 83%, твердое вещество желтого цвета) целевого соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,92-1,98 (м, 1H), 2,22-2,42 (м, 2H), 2,46 (с, 3H), 2,49-2,52 (м, 1H), 3,50 (д, J=14,0 Гц, 1H), 3,65 (с, 3H), 3,66 (д, J=14,0 Гц, 1H), 3,89 (с, 3H), 3,93 (с, 3H), 4,64-4,74 (м, 1H), 5,37 (с, 2H), 6,52 (с, 1H), 6,97 (д, J=8,8 Гц, 1H), 7,11-7,17 (м, 3H), 7,35 (д, J=10,8 Гц, 1H), 7,50 (с, 1H), 8,04 (д, J=7,2 Гц, 1H).
Пример 44:
2-(2-фтор-5-нитрооксиметилфенил)-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил]ацетамид
Соединение примера 44 синтезируют по аналогии со способом, раскрытым в примере 43, и промежуточное соединение получают в соответствии с раскрытым далее способом.
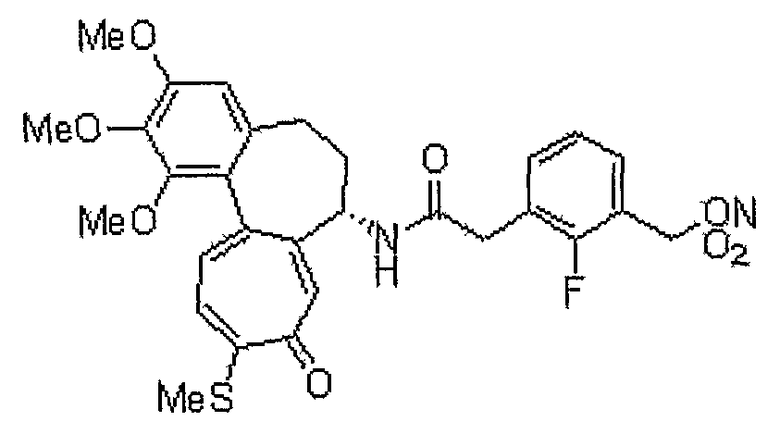
1H ЯМР (400 МГц, CDCl3): δ 1,89-1,93 (м, 1H), 2,26-2,29 (м, 1H), 2,34-2,40 (м, 1H), 2,43 (с, 3H), 2,49-2,54 (м, 1H), 3,61 (с, 3H), 3,62 (д, J=15,2 Гц, 1H), 3,72 (д, J=15,2 Гц, 1H), 3,89 (с, 3H), 3,92 (с, 3H), 4,70-4,75 (м, 1H), 5,49 (с, 2H), 6,53 (с, 1H), 7,06-7,11 (м, 2H), 7,25-7,31 (м, 2H), 7,35 (т, J=7,6 Гц, 1H), 7,44 (с, 1H), 7,57 (д, J=7,2 Гц, 1H).
<Промежуточное соединение 18>
Получение (2-фтор-5-гидроксиметилфенил)уксусной кислоты
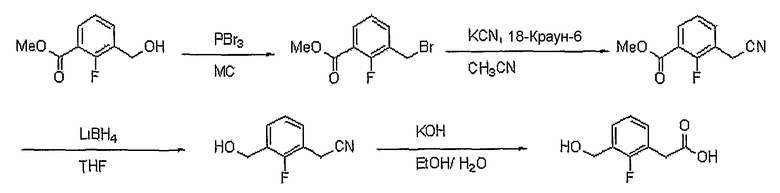
Аналогично способу получения стадии 1 примера 43, используя соединение, полученное в способе получения промежуточного соединения 5 (2,2 г, 11,94 ммоль), получают 400 мг (выход: 90%, твердое вещество белого цвета) целевого соединения.
Экспериментальный пример 1:
Тест на цитотоксичность в отношении раковых клеточных линий
Цитотоксичность в отношении A549 (Korea Research Institute of Chemical Technology), SK-OV-3 (Korea Research Institute of Chemical Technology), SK-MEL-2 (Korea Research Institute of Chemical Technology), HCT-15 (Korea Research Institute of Chemical Technology) и MCF7 (Korean Cell Line Bank, Seoul National University School of medicine) клеток определяют с помощью способа с использованием сульфородамина-В (Sulforhodamin-B (SRB)) (1989, National Cancer Institute (NCI)), который был разработан для измерения in vitro противораковой активности лекарственного средства. Клетки выделяют, используя раствор 0,25% трипсин-EDTA, в результате чего получают клеточную суспензию около 5×103-2×104 клеток/лунку. Затем суспензию переносят в 96-луночный планшет в количестве 100 мкл/лунку, культивируют в инкубаторе при 37°C, 5% CO2 в течение 24 часов. Соединения, полученные в примерах настоящего изобретения, используют в качестве образцов. А именно, соединения растворяют в диметилсульфоксиде и разбавляют средой RPMI 1640, перед тем как использовать их в качестве образцов. Конечная используемая концентрация образцов меняется в интервале 1 мкМ - 0,00001 мкМ. Среду удаляют из 96-луночного планшета, и затем разбавленный раствор образца добавляют в количестве 100 мкл/лунку, и затем продолжают культивирование в инкубаторе при 37°C, 5% CO2 в течение 48 часов. Для планшета Tz (время ноль) устанавливают с момента добавления образца. После завершения культивирования среду удаляют из каждой из лунки, также как и для планшета на время Tz, затем добавляют 10% трихлоруксусную кислоту (TCA) по 100 мкл/лунку. Планшет оставляют при 4°C на 1 час, чтобы обеспечить фиксацию клеток на дне планшета. После того как клетки фиксированы полностью, планшет промывают водой 5-6 раз для полного удаления остатков раствора трихлоруксусной кислоты и влагу полностью высушивают при комнатной температуре. 0,4% сульфородамин-B растворяют в 1% растворе уксусной кислоты для получения красящего раствора. Клетки окрашивают в течение 30 минут красителем, который добавляют по 100 мкл в каждую из лунок полностью сухого планшета. Затем планшет промывают 1% раствором уксусной кислоты 5-6 раз для удаления сульфородамина-B, который не присоединился к клеткам. Затем планшет сушат при комнатной температуре. Добавляют 10 мМ раствор Tris по 100 мкл/лунку, чтобы растворить краситель, и измеряют оптическую плотность (OD) на длине волны 520 нм, используя считывающее устройство для микропланшетов.
ED50 (концентрация, которая ингибирует рост раковых клеток на 50%, 50% эффективная доза, нМ/мл) образца в отношении раковых клеток рассчитывают следующим образом. Значение OD в момент начала культивирования с образцом определяют как величину Tz (время ноль). OD значение для лунки, которую культивируют без образца, определяют как контрольную величину (C). OD значение для лунки, предварительно обработанной образцом, определяют как экспериментальную величину (T). После расчета Tz, C и T цитотоксичность образца определяют, используя следующую математическую формулу <1>:
<Математическая формула 1>
T≤Tz, (T-Tz)/(C-Tz) × 100
T>Tz, (T-Tz)/Tz × 100
ED50, концентрацию, которая способна ингибировать рост раковых клеток на 50%, рассчитывают, используя регрессионный анализ - лотос программы, на основании степени цитотоксичности, полученной с использованием математической формулы 1.
Каждое значение ED50 для паклитакселя, доксорубицина и колхицина, которые были использованы в качестве контролей, также рассчитывают указанным выше способом.
Результаты представлены в таблице 1
Как представлено в таблице 1, трициклические производные настоящего изобретения демонстрируют очень высокую цитотоксичность в отношении раковых клеточных линий.
Экспериментальный пример 2:
Ингибирующий эффект трициклических производных настоящего изобретения на рост опухолей
Для исследования ингибирующего эффекта трициклических производных настоящего изобретения на рост опухолей проводят следующие эксперименты.
Образцы для эксперимента все время хранят в холодильнике. Тестируемым животным вводят различные дозы соединения; доза соединения, полученного в примере 8, составляет 10 мг/кг, доза соединения, полученного в примере 12, составляет 1, 3, 10 мг/кг соответственно и доза позитивного контроля составляет 2,5 мг/кг (таксол) и 2 мг/кг (адриамицин). Соединение примера 12 растворяют в 4% tween 80 и позитивный контроль таксол растворяют в смешанном растворителе, состоящем из 5% этанола + 25% кремофора + 75% PBS. Подготовленный образец осаждают таким образом, чтобы обработку ультразвуком осуществить непосредственно перед введением для получения равномерной дисперсии.
Семинедельных самок S.P.F. BALB/c голых мышей, поставляемых Charles River Co., Japan, используют в качестве тестируемых животных. Тестируемых животных адаптируют в Hepa-filter помещении в течение недели до проведения теста. Температура составляет 21±2°C, влажность 55±5%, и 12-часовой цикл свет/темнота автоматически повторяется в указанной лаборатории. Твердый корм (CheilJedang) стерилизуют, используя радиоактивное облучение, и питьевую воду также стерилизуют в автоклаве. Корм и воду поставляют животным без ограничений. В указанном эксперименте используют раковую клеточную линию NCI-H460 (клеточная линия рака легких человека), предоставленную исследовательским институтом Биологии и Биотехнологии Кореи.
Опухолевую клеточную линию, хранящуюся в жидком азоте, оттаивают и культивируют в инкубаторе при 37°C, 5% СО2 в течение нужного промежутка времени. После завершения культивирования все клетки извлекают и клеточную концентрацию культуральной жидкости доводят до значения 3×107 клеток/мл, используя PBS. Подготовленный культуральный раствор клеток вводят подкожно в подмышечную ямку между пояском правого плеча и стенкой грудной клетки в количестве 0,3 мл на мышь. Начиная со следующего дня после прививки, NCI-H460 ксенотрансплантированным голым мышам каждый день внутрибрюшинно вводят раствор образца в количестве 0,2 мл/20 г веса, один раз в день.
После трансплантации опухолевых клеток у каждого из животных проводят трехмерные измерения объема опухоли, используя штангенциркуль с нониусом, и рассчитывают по представленной далее <Математической формуле 2>.
<Математическая формула 2>
Объем опухоли = (длина × ширина × высота)/2
Изменения веса животных измеряют три раза в неделю. Каждую из ксенотрансплантированных голых мышей умерщвляют для извлечения опухоли, которую затем взвешивают.
Все результаты тестов для экспериментальной группы сравнивают с результатами для контрольной группы, используя t-Test, чтобы определить, существует ли какое-либо значительное различие между двумя группами.
Изменения объема и веса опухоли представлены в таблице 2 и отражены на ФИГ.1, 3 и 5, а изменения веса мышей представлены в таблице 3 и отражены на ФИГ.2 и ФИГ.4.
V.C-1: 4% tween 80,
V.C-2: 5% этанол + 25% кремофор + 75% PBS
(n)
(День 9-2, день 11, день 14-1)
V.C-1: 4% tween 80,
V.C-2: 5% этанол + 25% кремофор + 75% PBS
Как представлено в таблице 2 и на ФИГ.1, 3 и 5, размер и вес опухолей у NCI-Н460 ксенотрансплантированных BALB/c голых мышей заметно снижается, если им вводят трициклические производные настоящего изобретения (полученные в примере 8 и в примере 12), по сравнению с мышами, которым вводили только растворитель (V.C-1, V.C-2) или позитивный контроль (таксол, адриамицин). В частности, в случае соединения примера 12 объем и вес опухоли гораздо значительнее снижается при дозе 10 мг/кг, нежели при дозе 1 мг/кг. И в случае соединения примера 8 степень ингибирования объема опухоли соответствует 85% при введении соединения в дозе 10 мг/кг. Поэтому степени уменьшения объема и снижения веса опухоли можно принять пропорциональными вводимой дозе трициклических производных настоящего изобретения.
Как представлено в таблице 3 и отражено на ФИГ.2 и 4, вес NCI-H460 ксенотрансплантированных BALB/c голых мышей снижается примерно на 10% при введении трициклических производных настоящего изобретения (полученных в примере 8 и примере 12) по сравнению со случаем введения одного только растворителя (V.C-1, V.C-2) или позитивного контроля (таксол, адриамицин).
Поэтому трициклические производные настоящего изобретения уменьшают объем и вес опухоли дозозависимым образом, и кроме того, демонстрируют превосходный противораковый эффект. Таким образом, трициклические производные настоящего изобретения можно эффективно использовать в качестве противораковых агентов и в качестве противопролиферативных агентов.
Экспериментальный пример 3:
Ингибирующий эффект трициклических производных настоящего изобретения в отношении образования капилляроподобных трубочек в HUVEC клетках (анализ образования капилляроподобных трубочек)
В качестве матригеля, использованного в этом эксперименте, был использован продукт BioCoat. Матригель оттаивают в холодильнике в течение 24 часов перед использованием. Оттаявший матригель, 96-луночный планшет и желтую крышку помещают на лед. Затем матригель распределяют на планшете в каждую лунку по 40 мкл. Полимеризацию содержимого планшета ведут в инкубаторе при 37°C в течение 30 мин. Каждую лунку планшета инокулируют 180 мкл раствора HUVEC клеток (2×104 клеток/мл) вместе с 20 мкл соединения примера 12 в не содержащей сыворотки среде (0,3, 1, 3, 10 и 30 мкг/мкл) и затем культивируют в течение 24 часов.
Образование трубочек наблюдают в микроскоп для исследования активности ингибирования ангиогенеза.
Фумагилин и доксорубицин используют в качестве позитивного контроля.
Полученные результаты представлены на ФИГ.6.
Как видно на ФИГ.6, трициклические производные настоящего изобретения (полученные в примере 12) обладают активностью ингибирования ангиогенеза при дозах выше 0,3 мкг/мл, что является хорошим показателем по сравнению с результатами для фумагилина, позитивного контроля, при дозе 10 мкг/мл. Кроме того, трициклические производные настоящего изобретения (полученные в примере 12) полностью ингибируют ангиогенез при дозах выше 10 мкг/мл.
Таким образом, трициклические производные настоящего изобретения можно эффективно использовать в качестве ингибиторов ангиогенеза.
Экспериментальный пример 4:
Тест на острую токсичность
Пятинедельных мышей штамма ICR весом 25-35 г (SPF, SLC Co. Japan) используют в качестве тестируемых животных. Для тестирования каждого из соединений используют пару мышей: самку и самца. Создают группы T2, T3, T4 и T5 (по 10 животных в группе) для проведения теста на острую токсичность соединения, полученного в примере 12. Соединение примера 12 растворяют в растворителе [5% ДМСО; 20% tween 80; 75% PBS(-)], и вводят в брюшную полость мышей с помощью инъекций (конкретные дозы представлены в таблице 4), и затем наблюдают за животными в течение 7 дней. Контрольной группе (T1) вводят только растворитель без соединения примера 12. Полученные результаты представлены в таблице 4.
Для сравнения тестируемым животным вводят колхицин указанным выше способом. Группа, которую обрабатывают колхицином, состоит из 6 мышей. Результаты тестирования приведены в таблице 5.
Для сравнения используют также таксол, продукт Bristol Myers Sqibb Co., и инъекции и тестирование на острую токсичность осуществляют также указанным выше способом. Полученные результаты представлены в таблице 6.
Тест на острую токсичность для соединения, полученного в примере 12
Тест на острую токсичность для колхицина
Тест на острую токсичность для таксола
Как представлено в таблицах 4, 5 и 6, показано, что токсичность природного колхицина для мышей составляет LD50=2 мг/кг, что очень близко к представленному ранее значению LD50=1,6 мг/кг [Medicinal Research Reviews, Vol. 8, No.1, 77-94 (1988)]. Токсичность инъекций таксола, паклитакселя соответствует значению LD50=9-13 мг/кг (при внутривенном введении). Но токсичность соединения примера 12 настоящего изобретения соответствует LD50=60 мг/кг, что в 30 раз меньше токсичности колхицина и также меньше токсичность инъекции таксола. Таким образом доказано, что соединение настоящего изобретения обладает меньшей токсичностью в отношении нормальных клеток, нежели инъекции колхицина или таксола.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Трициклические производные настоящего изобретения обладают очень высокой цитотоксичностью в отношении раковых клеточных линий, но меньшей токсичностью для самих животных, нежели инъекции колхицина или таксола. Кроме того, трициклические производные настоящего изобретения уменьшают объем и снижают вес опухоли и эффективно ингибируют ангиогенез в HUVEC клетках. Поэтому указанные производные можно эффективно использовать в качестве противораковых агентов, противопролиферативных агентов и также в качестве ингибиторов ангиогенеза. Кроме того, трициклические производные настоящего изобретения можно легко получать и легко приготавливать в виде препаратов для перорального введения или для инъекций благодаря их растворимости в воде.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производное (S)-4-оксо-N-(1,2,3,10-тетраметокси-9-оксо-5,6,7,9-тетрагидробензо[a]гептален-7-ил)пентанамида и его применение | 2022 |
|
RU2809516C1 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ ТРИЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ИЛИ ИХ ГИДРАТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2572820C1 |
| НОВЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2011 |
|
RU2536688C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| ПРОИЗВОДНОЕ ИНДАНОНА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ ИЛИ ЭНАНТИОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ | 2012 |
|
RU2574591C2 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
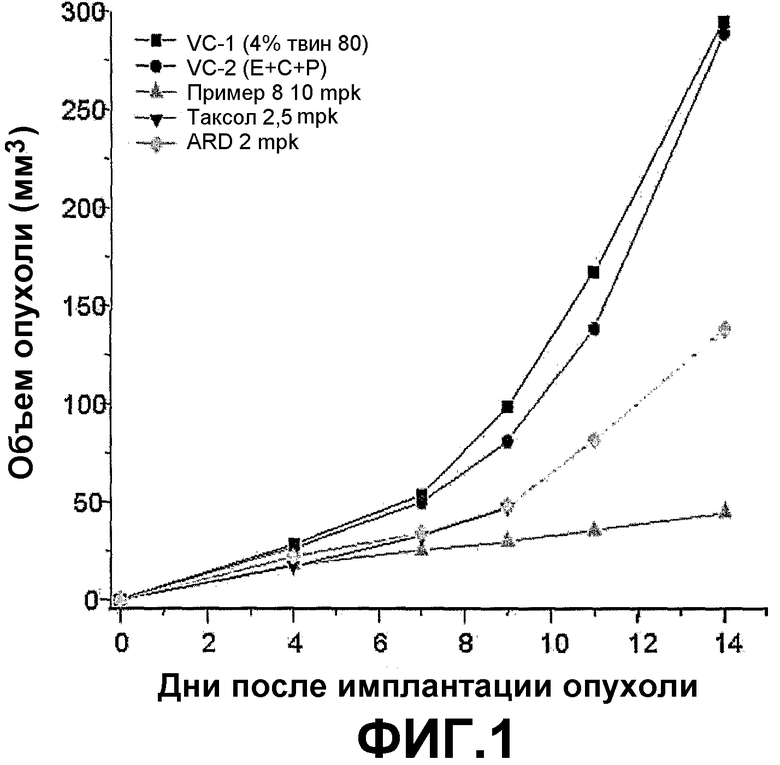
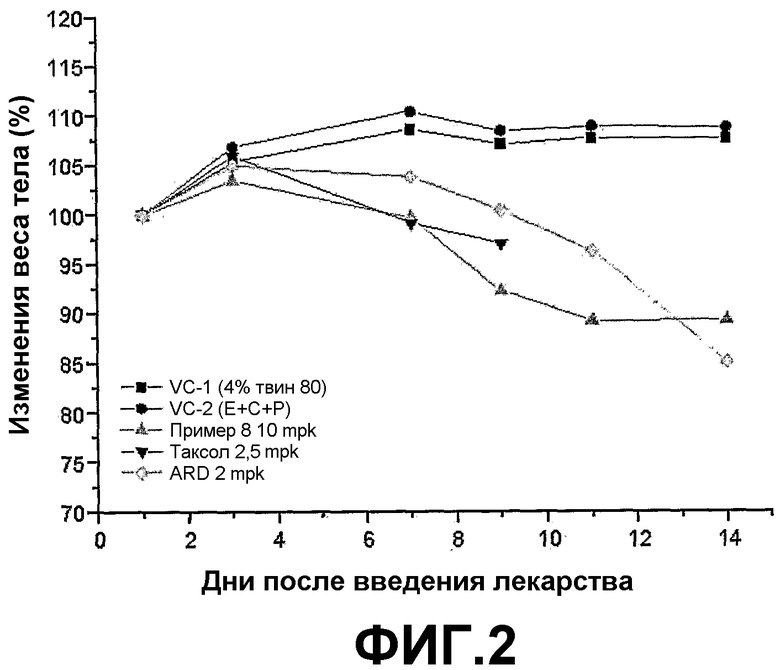
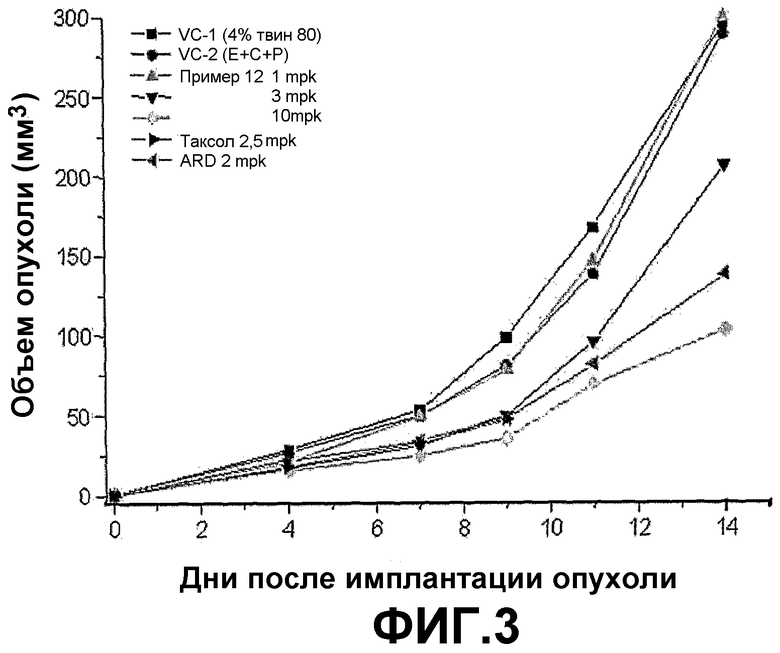
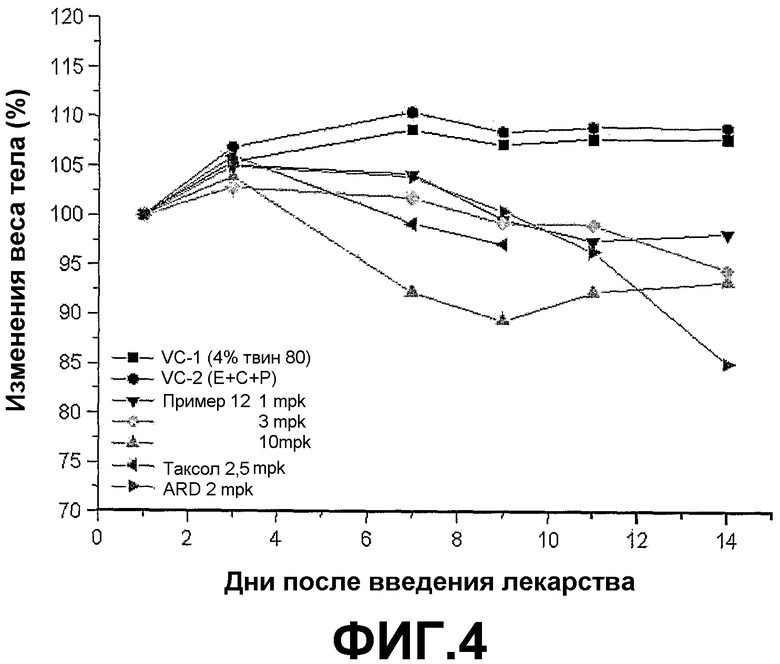
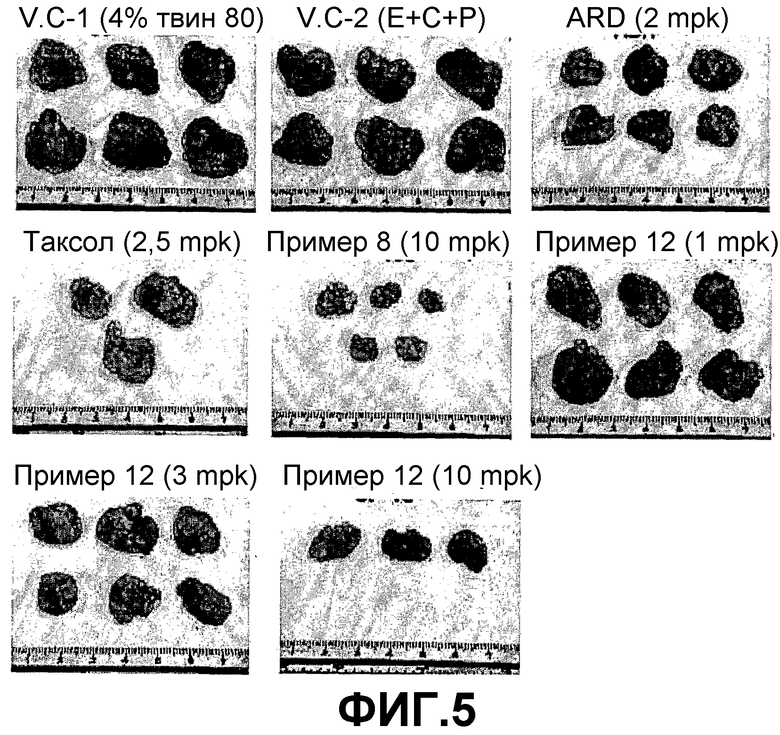
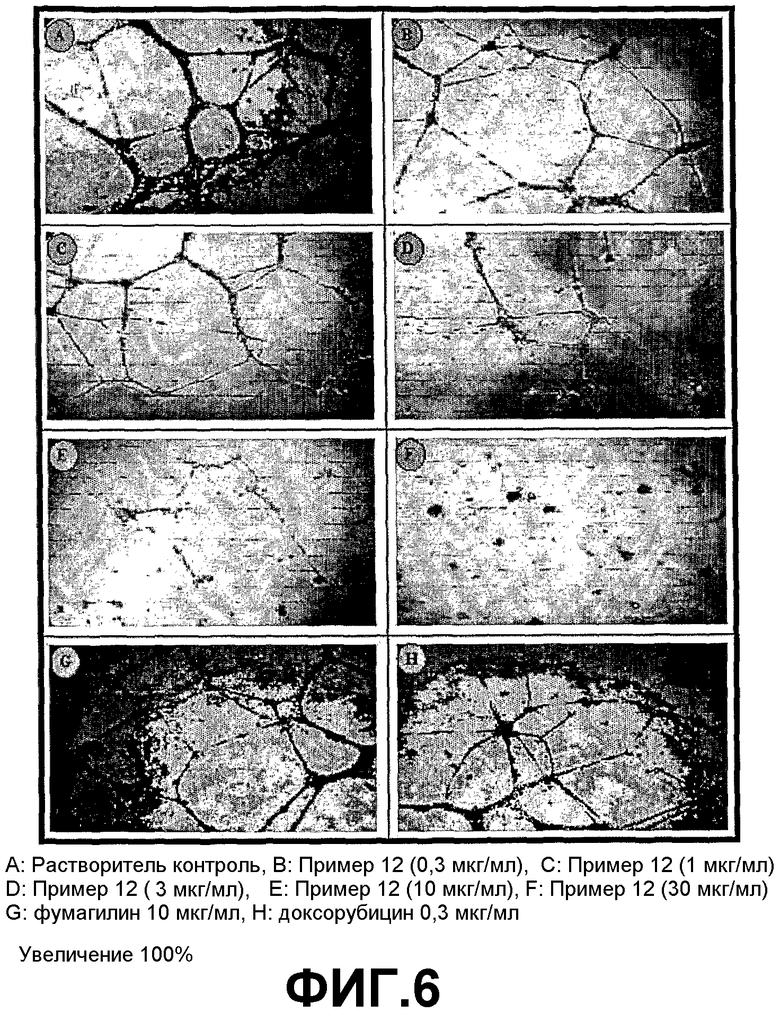
Изобретение относится к трициклическим производным, представленным формулой (I), или к их фармацевтически приемлемым солям, обладающим пролиферативной активностью и способностью ингибировать ангиогенез, к способу их получения (варианты), к пролиферативному агенту и ингибитору ангиогенеза на их основе.
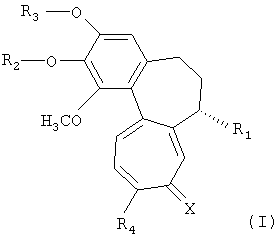 ,
,
где R1 представляет -T1-B1; T1 представляет -N(R5)C(O)-, где R5 представляет Н или C1-C5алкильную группу; и B1 выбирают из группы, состоящей из следующих значений (a), (b), (f), (g):
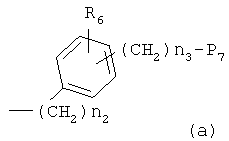
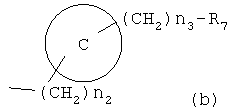
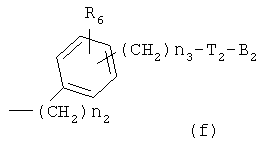
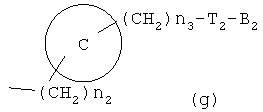
где
 представляет
представляет 
 или
или 
R6 представляет Н, галоген или гидроксигруппу; R7 представляет гидрокси или -ONO2-группу, при условии, что, если R6 представляет Н, R7 отличен от гидроксила; Т2 представляет -О-С(О)-; В2 представляет указанную группу (а) или -Z1-R7, где Z1 представляет прямолинейную С2-С5алкильную группу; n2 представляет целое число 0-3; и n3 представляет целое число 0-5; R2 представляет СН3; R3 представляет C1-C4 неразветвленный или разветвленный алкил, или С3-С7циклоалкил при условии, что, если B1 представляет (а), R6 представляет Н, R7 представляет -ONO2-группу и R3 не является СН3; R4 представляет SCH3 или ОСН3; Х представляет О. 6 н. и 1 з.п. ф-лы, 6 табл., 6 ил.
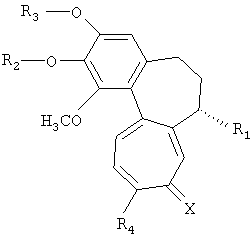
где (1) R1 представляет -T1-B1;
где T1 представляет -N(R5)C(O)-, где R5 представляет Н или C1-C5 алкильную группу; и
B1 выбирают из группы, состоящей из следующих значений (a), (в), (f), (g):
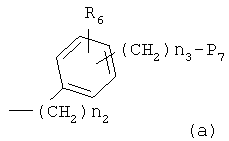
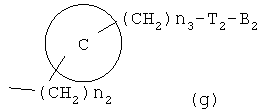
где
 представляет
представляет 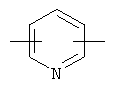 ,
, 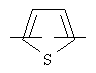 или
или 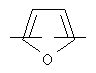 ;
;
R6 представляет Н, галоген или гидроксигруппу;
R7 представляет гидрокси или -ONO2 группу, при условии, что если R6 представляет Н, R7 отличен от гидроксила;
Т2 представляет -О-С(О)-;
В2 представляет указанную группу (а) или -Z1-R7, где Z1 представляет прямолинейную С2-С5алкильную группу;
n2 представляет целое число 0-3; и n3 представляет целое число 0-5;
(2) R2 представляет СН3;
(3) R3 представляет C1-C4 неразветвленный или разветвленный алкил, или С3-С7циклоалкил, при условии, что если B1 представляет (а), R6 представляет Н, R7 представляет -ONO2 группу и R3 не является СН3;
(4) R4 представляет SCH3 или ОСН3;
(5) Х представляет О.
1) 6-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]никотинамида;
4) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-3-нитрооксиметилбензамида;
5) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]амида 6-нитрооксиметилпиридин-2-карбоновой кислоты;
6) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидро-бензо[а]гептален-7-ил]амида 5-нитрооксиметилтиофен-2-карбоновой кислоты;
8) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-2-фтор-3-нитрооксиметилбензамида;
9) 2-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-3-нитрооксиметилбензамида;
10) 2-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфа-нил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамида;
12) 3-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфа-нил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамида;
13) N-[(7S)-3-этокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамида;
14) 3-фтор-N-[(7S)-3-изопропокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-5-нитрооксиметилбензамида;
15) N-[(7S)-3-циклопентилокси-1,2-диметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]-3-фтор-5-нитрооксиметилбензамида;
17) 2-фтор-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфа-нил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамида;
21) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]амида 4-нитрооксиметилтиофен-2-карбоновой кислоты;
22) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]амида 3-нитрооксиметилтиофен-2-карбоновой кислоты;
29) 2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]фенилового эфира 3-нитрооксиметил-бензойной кислоты;
30) 2-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]фенилового эфира 4-нитро-оксимасляной кислоты;
31) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]фенилового эфира 3-нитро-оксиметилбензойной кислоты;
32) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]фенилового эфира 4-нитро-оксимасляной кислоты;
33) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]бензилового эфира 3-нитрооксиметилбензойной кислоты;
34) 3-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]бензилового эфира 4-нитрооксимасляной кислоты;
2) [(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидро-бензо[а]гептален-7-ил]амид5-нитрооксиметилфуран-2-карбоновой кислоты;
16) 4-фтор-3-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамид;
18) 3-гидрокси-5-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метил-сульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамид;
20) 2-гидрокси-4-нитрооксиметил-N-[(7S)-1,2,3-триметокси-10-метил-сульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-ил]бензамид;
25) 5-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]пиридин-2-илметилового эфира 3-нитрооксибензойной кислоты;
27) 6-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетра-гидробензо[а]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 3-нитрооксиметилбензойной кислоты;
28) 6-[(7S)-1,2,3-триметокси-10-метилсульфанил-9-оксо-5,6,7,9-тетрагидробензо[а]гептален-7-илкарбамоил]пиридин-2-илметиловый эфир 4-нитрооксиметилбензойной кислоты.
(1) взаимодействие соединения формулы (III) с соединением формулы (IV) с получением соединения формулы (V) (стадия 1); и
(2) нитрование или нитрозирование полученного соединения формулы (V) с получением соединения формулы (IIa) (стадия 2)
Схема
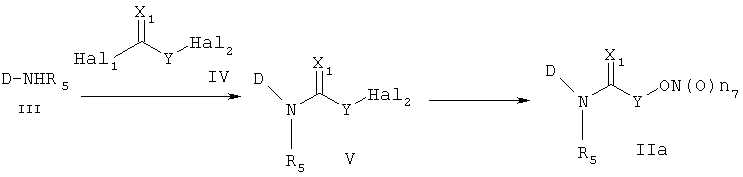
где D представляет 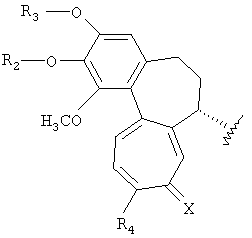 и R2, R3, R4 и Х имеют указанные для формулы I значения;
и R2, R3, R4 и Х имеют указанные для формулы I значения;
R5 представляет Н или C1-C5алкильную группу;
X1 представляет О;
Hal1 и Hal2 представляют галогены;
Hal1 и Hal2 в формуле (IV), каждый, одинаковы или различны и представляют галогены, например F, Cl, Br или I;
Y выбирают из группы, состоящей из соединений формул (а') и (b')

где , R6, n2 и n3 имеют указанные для формулы 1 значения, n7 формулы (IIa) представляет целое число 1-2.
,
где R6, R7, n2 и n3 имеют значения, указанные в п.1.
(1) взаимодействие соединения формулы (III) с соединением формулы (VI) с получением соединения формулы (VII) (стадия 1); и
(2) нитрование или нитрозирование полученного соединения формулы (VII) с получением соединения формулы (IIa) (стадия 2)
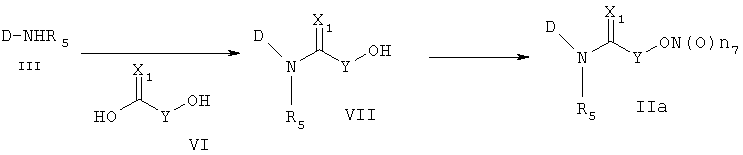
где D, R5, X1, Y, n7 имеют значения, определенные в п.3.
(1) взаимодействие соединения формулы (III) с соединением формулы (VI) с получением соединения формулы (VII), с последующим его взаимодействием с галогенсодержащим соединением, с получением соединения формулы (V) (стадия 1); и
(2) нитрование или нитрозирование полученного соединения формулы (V) с получением соединения формулы (IIa) (стадия 2)
схема
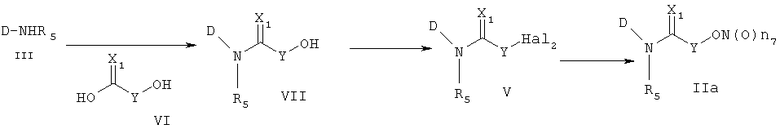
где D, R5, X1, Y, Hal2, n7 имеют значения, определенные в п.3.
| ПРОИЗВОДНЫЕ КОЛХИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2145598C1 |
| ПРОИЗВОДНЫЕ ТИОКОЛХИЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2190598C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| J.GUAN ET AL, Antitumor Agents | |||
| Способ укрепления под покрышкой пневматической шины предохранительного слоя или манжеты | 1917 |
|
SU185A1 |
| Synthesis and Biological Evaluation of Tridemethythiocolchicine Analogues as Novel Topoisomerase II Imhinhibitorslnhibitors | |||
| J | |||
| Med | |||
| Chem | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| L.Sun ET AL, Antitumor Agents | |||
| Топливник с глухим подом | 1918 |
|
SU141A1 |
| Synthesis and | |||

