Данное изобретение принадлежит к области фармацевтической химии и синтетической органической химии, и предоставляет асимметрический процесс синтеза ключевого промежуточного соединения при получении дулоксетина, соли соляной кислоты (+)-N-метил-3-(1-нафталинилокси)-3- (2-тиенил)-пропанамина.
Дулоксетин представляет собой фармацевтическое средство, находящееся на стадии исследования и разработки в качестве антидепрессантного средства. Он ингибирует поглощение, как норэпинефрина, пинефрина, так и серотонина, и в настоящее время проходит клиническую оценку. Данное соединение описано в патенте США NN 5023269 и 4956388 на имя Робертсона и др., а синтез его обсуждался более подробно Дитером и др., в Tetrahedron Letters, 31/49/, 7101-04 (1990). Процесс получения дулоксетина представлен следующим образом.
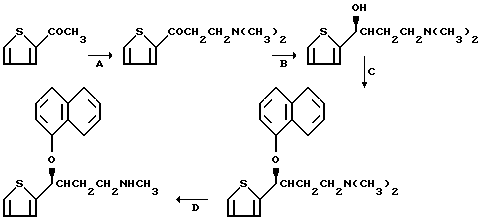
Продукт стадии Д, представленной выше, когда он находится в форме хлоргидратной соли, представляет собой дулоксетин.
Настоящее изобретение предоставляет улучшенные условия осуществления стадии С в представленной выше схеме, с помощью которых продукт стадии С получается более быстро с лучшей чистотой и выходом, чем это было возможно ранее.
Предоставляется также особенно полезная соль для выделения промежуточного продукта.
Настоящее изобретение предоставляет процесс получения (S)-(+)-N, N-диметил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамида, включающий реакцию (S)-(-)-N, N-диметил-3-(2-тиенил)-3- гидроксипропанамина с гидридом натрия, бензоатом калия или ацетатом калия, и 1-фторнафталином в органическом растворителе; и, если необходимо, выделение продукта в виде фосфорнокислой соли. Соль фосфорной кислоты также является одним из аспектов и воплощений данного изобретения.
Настоящее изобретение предоставляет асимметрический процесс получения конкретного энантиомера, показанного выше, в качестве продукта стадии С на представленной выше схеме. Он называется (S)-(+) энантиомером, и данная номенклатура будет использоваться на протяжении данного документа. Исходный материал для процесса данного изобретения, гидроксипропанамин, называется (S)-(-)-энантиомером.
Предпочтительным растворителем для осуществления настоящего изобретение является диметилсульфоксид. Могут также использоваться другие растворители, такие как диметилформамид, пиридин и аналогичные. Данный процесс может эффективно осуществляться при необычно высоких концентрациях, таких как примерно от 0.5 до 1.0 молярной, предпочтительно при 0.6 - 0.9 молярной. Однако при этом концентрация не оказывает серьезного отрицательного влияния на скорость и выход данного процесса, если только конечно не превышается предел растворимости.
Порядок и способ сочетания реагентов не являются существенными и могут варьироваться. Реагенты могут добавляться к реакционной смеси в виде твердых веществ, или могут растворяться по отдельности и соединяться в виде растворов. Далее, любые из реагентов могут растворяться вместе в виде подгрупп, и эти растворы могут сочетаться или объединяться в любом порядке.
Предпочтительным способом осуществления изобретения является растворение исходного материала и гидрида натрия вместе, добавление соединения калия, а затем добавление 1-фторнафталина, и далее подробно будет описано данное воплощение процесса.
Когда исходный материал растворится, предпочтительно при температуре окружающей среды, такой как примерно от 10o до 35oC, добавляется порция гидрида натрия. Количество гидрида натрия является эквимолярным с исходным материалом; при использовании избытка гидрида натрия не обнаружено никаких особых преимуществ. Реакционная смесь затем перемешивается в течение некоторого периода, такого как примерно от 5 до 60 минут, а затем соединяется с бензоатом калия или ацетатом калия и 1-фторнафталином.
Необходимо лишь небольшое количество соединения калия, примерно от 0.05 до 1 эквивалента. Обычно преимущества настоящего изобретения наилучшим образом достигаются, когда используется лишь примерно от 0.1 до 0.3 эквивалента калиевого соединения. При использовании большего количества нет никаких неудобств за исключением явного неудобства, связанного со стоимостью.
После добавления соединения калия дается дополнительное время на перемешивание, а затем добавляется 1-фторнафталин. Для гарантии потребления более дорогого тиофенового исходного материала может успешно использоваться небольшой избыток 1-фторнафталина. После того, как фторнафталин добавлен, реакционная смесь предпочтительно подогревается до температуры примерно от 40o до 75oC, предпочтительно примерно от 45o до 70oC, и наиболее предпочтительно при температуре примерно от 60o до 65oC, и смесь перемешивается в течение короткого периода времени, такого как примерно от 1 до 5 часов, наиболее предпочтительно примерно от 1.5 до 4 часов. Желаемый продукт затем выделяется с помощью общепринятых приемов экстракции и фильтрования, и, если это необходимо, продукт может преимущественно превращаться в соль фосфорной кислоты с помощью реакции с фосфорной кислотой в органическом растворителе, таком как этилацетат.
Преимущество настоящего изобретения найдено в его способности давать целевой продукт с выходами в пределах 95%, с весьма незначительной, если она и есть, рацемизацией, и в короткие периоды времени, описанные выше. Прежние процедуры занимали более чем сутки, и давали продукт худшей чистоты.
Гидроксипропанамин, который является исходным материалом для настоящего процесса, может получаться с помощью известных в технике приемов, а для сведения читателей настоящего документа ниже дается описание получения 1.
Продукт настоящего процесса, диметильное соединение, превращается в дулоксетин с помощью деметилирования с получением желаемого монометильного фармацевтического агента, и превращения в гидрохлоридную иди хлоридгидратную соль. Получение 2 ниже иллюстрирует данное превращение, которое, как и получение гидроксильного исходного материала, известно из предшествующего уровня техники и не составляет части настоящего изобретения.
Получение 1
(S)-(-)-N,N-диметил-3-гидрокси-3-(2-тиенил)пропанамин
Смесь 8.18 г 2-ацетилтиофена, 6.66 г хлоргидрата диметиламина, 2.9 г параформальдегида и 0.31 г концентрированной соляной кислоты в 20 мл изопропанола нагревалась до кипения с обратным холодильником и перемешивалась в течение 6 часов. Смесь затем охлаждалась до 0oC и перемешивалась в течение еще одного часа. Суспензия затем фильтровалась, и твердое вещество промывалось холодным этанолом. Промытое твердое вещество сушилось в течение 16 часов при 50oC, давая 12.5 г хлоргидрата 2-тиенил-2-диметиламиноэтилкетона в виде белого твердого вещества. 12.0 г порция данного промежуточного продукта перемешивалась в 40 мл этанола при температуре окружающей среды, и величина pH раствора поднималась до 11 - 12 медленным добавлением гидроокиси натрия. Добавлялась порция боргидрида натрия в 1.03 г, и смесь перемешивалась при температуре окружающей среды в течение 4 часов. Затем добавлялось 7.5 мл ацетона, и смесь перемешивалась еще в течение 20 минут. Смесь затем концентрировалась выпариванием до белой суспензии, и добавлялось 120 мл метил-трет-бутилового эфира. Смесь подкислялась до pH 1-1.5 добавлением концентрированной соляной кислоты, и раствор перемешивался в течение 10 минут. Смесь затем подщелачивалась до pH 12 медленным добавлением гидроокиси натрия.
Слои затем разделялись, водная фаза экстрагировалась 30 мл метил-трет-бутилового эфира, а органические фазы объединялись и промывались один раз 50 мл воды. Органическая фаза концентрировалась с помощью выпаривания до 118 мл, и нагревалась до 50oC.
В отдельной емкости, (S)-(+)-миндальная кислота (4.18 г) растворялась в 12 мл этанола при 50oC, и раствор миндальной кислоты медленно добавлялся к первому раствору. Получающаяся суспензия затем нагревалась до дефлегмации и перемешивалась в течение 45 минут. Затем она охлаждалась до температуры окружающей среды, перемешивалась в течение одного часа и фильтровалась, и твердое вещество промывалось метил-трет-бутиловым эфиром. Твердое вещество затем сушилось в вакууме при 50oC, давая 7.29 г соли желаемого продукта и миндальной кислоты, и продукт выделялся в виде свободного амина с помощью растворения в воде, подщелачивания раствором гидроокиси натрия, экстрагирования органическим растворителем и выпаривания для удаления растворителя.
Пример 1
Фосфорнокислая соль (S)-(+)-N,N-диметил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина
13.5 г порция (S)-(-)-N,N-диметил-3-гидрокси-3-(2-тиенил)-пропанамина растворялась в 80 мл диметилсульфоксида при 25oC. К раствору медленно добавлялось 3 г гидрида натрия в виде 60% дисперсии в минеральном масле при энергичном перемешивании. После 15-минутного перемешивания добавлялось 1.17 г бензоата калия, и перемешивание продолжалось приблизительно при постоянной температуре в течение еще 15 минут. Затем к реакционной смеси медленно добавлялось 12.8 г 1-фторнафталина, и после завершения добавления смесь нагревалась и перемешивалась в течение 2.5 часов при 60 - 65oC. Смесь затем медленно выливалась в 190 мл холодной воды, и величина pH доводилась до 4.8 добавлением уксусной кислоты. Температура смеси доводилась до 25oC, и добавлялось 75 мл гексана, и перемешивание продолжалось в течение 10 минут. Слои затем разделялись, и водная фаза перемешивалась снова с 75 мл гексана, и фазы разделялись. Величина pH водной фазы доводилась до 10.2 с помощью добавления водной гидроокиси натрия, и добавлялось 75 мл этилацетата. Данная смесь перемешивалась в течение 15 минут при 25oC, и 2-фазная смесь фильтровалась в вакууме через слой фильтровального средства. Фазы фильтрата оставлялись разделяться, и водная фаза экстрагировалась 75 мл. этилацетата. Экстракт объединялся с предыдущим этилацетатным слоем, и данная смесь промывалась 100 мл воды. Органический слой перемешивался при 25oC, и к нему добавлялось по каплям 7 г 85% фосфорной кислоты. После завершения добавления смесь перемешивалась еще в течение 20 минут, а затем охлаждалась до 0oC и перемешивалась в течение 1 часа при этой температуре. Суспензия затем фильтровалась, и твердые вещества промывались три раза 20 мл порциями холодного этилацетата. Твердое вещество сушилось при 60oC, давая 24.19 г целевого соединения в виде белого твердого вещества, tпл = 135 - 139oC 98.1% крепости, выход 79.6%, 91% ЕЕ. Методика анализа.
Продукт данного примера анализировался с помощью жидкостной хроматографии высокой разрешающей способности, с использованием прибора Спектра Физикс SР 8800, снабженного SР 4400 интегратором, и детектора Спектрофлоу 757, установленного на 230 нм, при чувствительности 0.5 единиц абсорбции, время поднятия на фильтре 1 секунда. Колонка представляла Дюпон Цорбакс Rx С8, 4,6 мм•25 см. Элюентом были 70% ацетонитрила, 30% 0,01 М фосфатного с рН 6, скорость потока 1.0 мл/минуту, инжекционный объем 20 микролитров. Образцы приготавливались путем разбавления 0.1 - 0.3 г реакционной смеси или экстракта до 50 мл смесью 1:1 ацетонитрил:вода. Пик продукта элюируется в пределах 13 - 17 минут, исходный материал - 6 - 8 минут; фторнафталин - через 5 - 6 минут; диметилсульфоксид - через 2 - 3 минуты; и бензоат калия - через 2 - 2,5 минуты.
Когда необходимо было проводить хиральный анализ, тот же прибор устанавливался на 280 нм, с чувствительностью 0.1 единицы абсорбции, и использовалась колонка Хиралцел ОД. Элюент для хирального анализа представлял 2% изопропанола, 0.2% диэтиламина, и 97.8% гексана. Использовались те же показатели инжекции и потока. Образцы приготавливались разбавлением 0.1 - 0.3 г реакционной смеси или экстракта до 5 мл дихлорметаном, промыванием смеси примерно 5 мл воды, и сушкой органической фазы над сульфатом натрия. Получающийся раствор фильтровался и разбавлялся до 25 мл элюентом. Желаемый энантиомер элюируется через 5 - 5.5 минут, нежелательный энантиомер - через 6 - 6.5 минут, и фторнафталин - через 3 - 4 минуты. Пример 2
(S)-(+)-N,N-диметил-3-(1-нафталинилокси)-3-(2-тиенил) пропанамин
Порция в 1.60 г (S)-(-)-N, N-диметил-3-гидрокси-3-(2- тиенил)-пропанамина растворялась в 8 мл диметилсульфоксида при температуре окружающей среды, к раствору добавлялось 0.35 г гидрида натрия в виде 60% дисперсии в минеральном масле при энергичном перемешивании. После 30-минутного перемешивания добавлялось 0.28 г бензоата калия, и перемешивание продолжалось еще в течение 10 минут. Затем добавлялось 1.52 г 1-фторнафталина, и смесь затем перемешивалась при 50oC в течение 8 часов. Реакционная смесь медленно выливалась в 30 мл холодной воды, и величина pH доводилась до 4.8 добавлением уксусной кислоты. Добавлялось 15 мл гексана, смесь перемешивалась в течение 10 минут, и слои разделялись. Водная фаза перемешивалась снова с 15 мл гексана, и фазы разделялись. Величина pH водной фазы доводилась до 12.5 добавлением водной гидроокиси натрия, и добавлялось 15 мл этилацетата. Основная смесь перемешивалась при температуре окружающей среды в течение 10 минут, и слои разделялись. Водная фаза экстрагировалась еще одной 15 мл порцией этилацетата, и органические экстракты объединялись, промывались один раз 30 мл воды, и сушились над сульфатом магния. Растворитель удалялся в вакууме, давая красно-коричневое масло, которое растворялось в минимальном количестве смеси 1: 1 этилацетат: гексан. Раствор пропускался через слой силикагеля с использованием в качестве элюента смеси этилацетат: гексан: метанол: гидроокись аммония, 47:47:5.8:0.2. Фракция продукта выпаривалась под вакуумом, давая 2.3 г желаемого продукта в виде янтарного масла.
Получение 2
Гидрохлорид (S)-(+)-N-метил-3-(1-нафталинилокси)-3- (2-тиенил)-пропанамина
Пять грамм продукта примера 1 перемешивались в смеси 40 мл толуола и 40 мл воды при 40oC, и добавлялось 2.5 мл 30% раствора гидроокиси аммония. Смесь перемешивалась в течение 10 минут при постоянной температуре, и слои разделялись. Органическая фаза промывалась водой, сушилась сульфатом магния и фильтровалась. Фильтрат концентрировался до половины объема под вакуумом и нагревался до 55oC. Затем добавлялось 0.16 г диизопропилэтиламина с последующим добавлением по каплям 2.39 г фенилхлорформиата. Смесь перемешивалась при 55oC в течение 1.25 часа, и добавлялось 50 мл 1% раствора бикарбоната натрия. Смесь перемешивалась в течение 10 минут при 40 - 50oC, и фазы разделялись. Органическая фаза промывалась дважды 0.5 норм. соляной кислотой, и затем промывалась 1% раствором бикарбоната натрия. Промытая органическая фаза разделялась пополам, и одна аликвота выпаривалась под вакуумом, а к остатку добавлялось 26 мл диметилсульфоксида. Смесь нагревалась до 45oC, и по каплям добавлялись 1 г гидроокиси натрия и 6 мл воды. Основная смесь перемешивалась в течение 18 часов при 50oC; разбавлялась 17 мл воды, и подкислялась до pH 5.0 - 5.5 добавлением уксусной кислоты. Затем добавлялось 20 мл гексана, и смесь перемешивалась в течение 10 минут, и фазы разделялись. Водная фаза подщелачивалась до pH 10.5 добавлением 50% водной гидроокиси натрия, и добавлялось 17 мл этилацетата. После перемешивания в течение 10 минут, фазы разделялись, и водный слой экстрагировался еще 17 мл этилацетата. Объединенные органические экстракты промывались водой и концентрировались до 10 мл под вакуумом. К остатку добавлялось 0.46 г концентрированной соляной кислоты, а затем добавлялись затравочный кристалл и дополнительные 10 мл этилацетата. Смесь перемешивалась в течение 30 минут еще, и раствор концентрировался до 10 мл под вакуумом. Остаток перемешивался в течение 1 часа при температуре окружающей среды; и в течение 1 часа при 0oC, давая суспензию, которая фильтровалась. Твердое вещество промывалось охлажденным этилацетатом, давая 1.32 г желаемого продукта, который представлял дулоксетин в виде белого твердого вещества крепости 99.8%.
Приведенный выше пример иллюстрирует удобство и превосходные результаты настоящего процесса.
Изобретение относится к области фармацевтической химии и синтетической органической химии и представляет асимметрический процесс синтеза ключевого промежуточного соединения при получении дулосектина - антидепрессантного средства. Способ получения вышеуказанного соединения взаимодействием (S)-(-)-N, N-ди-метил-3-(2-тиенил)-3-гидроксипропанамина с гидридом натрия и 1- фторнафталином в органическом растворителе отличается тем, что процесс проводят с использованием бензоата калия или ацетата калия и целевой продукт выделяют в свободном виде или, при необходимости, в виде соли фосфорной кислоты. Изобретение позволяет ускорить процесс, улучшить чистоту и выход целевого продукта. 2с. и 4 з.п.ф-лы.
| Способ получения производных 2-(тиенил-2)- или 2-(тиенил-3) этиламина | 1982 |
|
SU1148563A3 |
| АВТОМАТИЗИРОВАННАЯ СИСТЕМА НАЛИВА СВЕТЛЫХ НЕФТЕПРОДУКТОВ | 0 |
|
SU363869A1 |
| СПОСОБ ПОЛУЧЕНИЯ РУТЕНИЕВЫХ ПОКРЫТИЙ | 0 |
|
SU298703A1 |

