Настоящее изобретение относится к усовершенствованному способу синтеза антибактериального агента и к новому промежуточному веществу, предназначенному для использования в этом способе.
Известен [публикация заявки на Европейский патент N 0416953A2] новый класс трициклических антибактериальных агентов и способы их получения. Наиболее предпочтительным соединением, описанным в этом источнике, является (4S, 8S, 9R, 10S, 12R)-4-метокси-10-(1-гидроксиэтил)-11-оксо-1-азатрицикло [7.2.0.03,8] -ундека-2-ен карбоновая кислота и ее физиологически приемлемая соль. Также в описании показывается, что данное соединение может быть получено следующим способом.
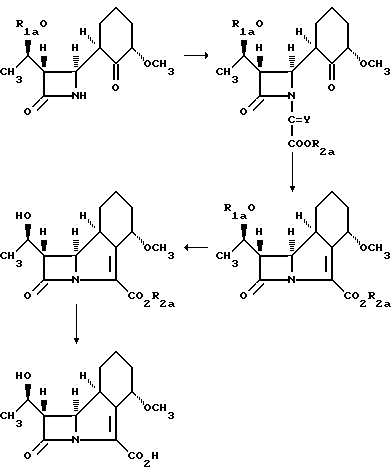
В приведенных выше формулах R1a представляет собой группу, защищающую гидроксил, например трет-бутилдиметилсилил, Y представляет собой кислород или фосфиновую группу, a R2a представляет собой защищающую карбоксил группу, и включает арилметил, например бензил или аллил. В частности, описание содержит примеры синтеза соединения с использованием промежуточных веществ, где R1a представляет собой третбутилдиметилсилильную группу, Y - кислород, а R2a - аллил, или где R1a - третбутилдиметилсилильная группа, Y - трифенилфосфин, a R2a - бензил.
В этих примерах различные промежуточные соединения получают в виде масел, что нежелательно при многостадийном синтезе, используемом в производственных процессах. Неожиданно было установлено, что пригодность процессов, описанных выше, для заводского использования, может быть значительно повышена, если использовать в данных синтезах в качестве группы, защищающей карбоксил, третбутилбензильную группу. Приведенное ниже промежуточное соединение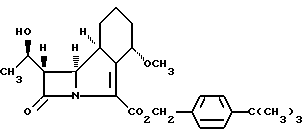
в окончательном виде является твердым веществом, при этом оно обладает такими свойствами, которые позволяют осуществлять сравнительно простую стадию очистки, например путем рекристаллизации, если это требуется перед проведением конечной стадии синтеза.
Таким образом, настоящее изобретение относится к способу получения соединения формулы I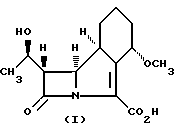
или его соли, который включает гидрогенолиз соединений формулы II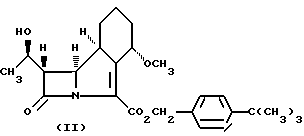
и, если это желательно или необходимо, превращение полученной карбоновой кислоты в ее соль. Реакцию гидрогенолиза целесообразно проводить с использованием водорода и металлического катализатора, такого как палладий, в растворителе, таком как алканол (например, этанол, изопропанол), эфиры (этилацетат) или кетон (ацетон). Эту реакцию предпочтительно проводят в присутствии основания, при этом подходящие для использования в реакции основания включают четвертичные органические основания, такие как триалкиламины, например триэтиламин. Карбоновая кислота (I) или ее соль могут быть превращены в физиологически приемлемые соли без выделения продукта гидрогенолиза. Т. о. , например, натриевая соль может быть получена путем добавления ацетона и этилгексаноата натрия в реакционные раствор, с последующим добавлением осадителя, например эфира, в частности диизопропилового эфира. В этом способе может быть полезным добавление в качестве зародышей кристаллов требуемой натриевой соли.
Соли соединения формулы (I) включают физиологически приемлемые соли, а также физиологически неприемлемые соли.
Подходящие физиологически приемлемые соли соединения формулы (I) включают соли щелочных металлов, например натрия или калия, щелочноземельных металлов, например кальция, аминокислот, например лизина или аргинина, и органических оснований, например прокаина, фенилбензиламина, этаноламина, диэтаноламина и N-метилглюкозамина. Неприемлемые физиологически соли соединения формулы (I) могут быть использованы в качестве промежуточных соединений для получения и/или выделения соединения формулы (I) или его физиологически приемлемой соли.
В изобретении предложено также новое соединение формулы (II).
Соединение формулы (II) может быть получено циклизацией соединения формулы (III)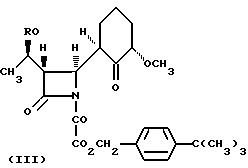
где R - группа, защищающая гидроксил, с последующим удалением защищающей гидроксил группы R.
В качестве подходящих групп, защищающих гидроксил, используют такие традиционные защищающие гидроксил группы, которые можно удалить гидролизом в условиях буферного раствора или в условиях безводной среды. Примерами таких групп являются гидрокарбосилильные группы, такие как три(C1-C4алкил)силильные группы, например трет-бутилдиметилсилил или триметилсилил.
Реакцию циклизации проводят путем обработки соединения формулы (III) соединением фосфора формулы (IV)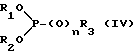
где R1, R2 и R3 независимо представляют собой C1-C4алкил, бензил или фенил, и n = 0 или 1, в подходящем растворителе, при температуре от 50 до 200oC.
Подходящими растворителями являются углеводороды, такие как н-октан, нонан, толуол, ксилол, этилбензол, галогенсодержащие углеводороды, такие как хлорбензол, дихлорэтан, дихлорбензол, трихлорметан или 1,1,2-трихлорэтан, простые эфиры, такие как тетрагидрофуран или сложные эфиры, такие как этилацетат или бутилацетат.
Предпочтительные соединения формулы (III) для использования в процессе циклизации включают такие соединения, в которых R1, R2 и R3 представляют собой алкильные группы, например этил, а n = 1, или R1 и R2 представляют собой этил, n = 0, а R3 - метил. Последние соединения могут быть получены in situ известными методами.
Защищающая гидроксил группа R может быть удалена хорошо известными стандартными методами [Protective groups in Organic Chemistry pages 46-119, Edited by JFW McOmie (Plenum Press 1973]. Таким образом, например, когда R-третбутилдиметилсилильная группа, она может быть удалена путем реакции с фторидом тетрабутиламмония в уксусной кислоте, или путем взаимодействия с фторидионами в присутствии соответствующего катализатора фазового переноса, такого как бромид тетрабутиламмония, в присутствии уксусной кислоты. В качестве наиболее подходящего источника фторидионов можно использовать фторид калия или фторид цезия.
Соединения формулы (II) могут быть получены из соединений формулы (V)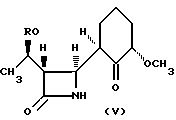
где R соответствует формуле (III) по реакции с n-третбутилбензилоксалилхлоридом в присутствии соответствующего основания, такого как пиридин, триэтиламин и/или карбонат калия, в растворителе, таком как углеводород, например, ксилол или циклогексан, или галогенсодержащий углеводород, например, хлорбензол, или дихлорметан или их смесь.
В приведенной выше формуле сплошные клиновидные связи показывают, что связь находится выше плоскости листа, а штриховые клинообразные связи показывают, что они находятся ниже плоскости листа.
Следующие примеры приведены только с целью иллюстрации.
Для всех промежуточных соединений и для всех примеров все температуры даны в oC, а под осушенными растворами понимают растворы, высушенные над безводным сульфатом натрия.
Промежуточное соединение 1. (3S,4R)-1-[4-третбутилбензилокси)оксалил]-3-(R)-1-(третбутилдиметилсилилокси) этил[-4-[(2'R, 6'S)-6'-метокси-1'-оксоциклогексил]азетидин-2-он.
К раствору триэтиламина (42,4 мл), карбоната натрия (2,32 г) и 4-[(3S, 4R)-3[(R)-1-(третбутилдиметилсилилокси)этил] -4-[(2 'R, 6'S)-6'-метокси-1'-оксациклогексил]азетидин-2-она (42 г) в циклогексане (400 мл), прибавляли по каплям чистый n-третбутилбензилоксалилхлорид (64 г) в течение приблизительно 10 мин при комнатной температуре в атмосфере азота. Через 10 мин реакционную смесь промывали водой (3 порции по 400 мл) и концентрировали органический слой (до объема приблизительно 125 мл). Полученный раствор разбавлялся изопропанолом (1040 мл), остатки циклогексана отгоняли азеотропной перегонкой (до объема приблизительно 700 мл) и затем по каплям добавляли воду (230 мл) в течение приблизительно 10 мин. В помутневшую реакционную смесь вносили затравку и затем перемешивали в течение часа при комнатной температуре. Далее в течение 30 мин по каплям прибавляли воду (450 мл) и перемешивали полученную суспензию в течение часа. Твердое вещество отфильтровывали, промывали смесью вода: изопропанол в соотношении 4:1 (200 мл), затем после сушки под вакуумом при 40oC получали белое кристаллическое целевое соединение (57.3 г) (Т. пл. 68-69oC)•(1H-ЯМР (CDCl3):7.36(dd), 5.28(dd), 4.35-4.25(m), 3.76(m), 3.52(t), 3.33(t), 3.22(s), 2.2(m), 2.1-2.0(m), 1.68(m), 1.46(m), 1.31(s), 1.19(d), 0.78(s), 0.04(s), -0.05(s) ppm.
Промежуточное соединение 2. 4-Третбутилбензил-(4S,8S,9R,10S,12R) -4-метокси-10[1-(третбутилметилсилилокси)этил-11-оксо-1-азатрицикло- [7.2.0.0. 3,8]-ундека-2-ен-2-карбоксилат
Раствор промежуточного соединения 1 (5.1 г), триэтилфосфита (17.8 мл) и гидрохинона (0.1 г) в этилбензоле (330 мл) кипятили с обратным холодильником в атмосфере азота в течение 15 ч, охлаждали до комнаткой температуры и концентрировали приблизительно до 178 мл. Полученный раствор обрабатывали 5% перекисью водорода (66 мл) и перемешивали смесь в течение 40 мин. Органический слой после отделения промывали водным 5% раствором карбоната натрия (51 мл), водой (51 мл) и затем концентрировали до масла, которое растворяли в петролейном эфире (153 мл). Полученный раствор промывали водой (3 порции по 153 мл) и выпаривали до получения прозрачного масла, которое растворяли в смеси изопропанол:воды 4:1 (26 мл); остатки органических растворителей отгонялись азеотропной перегонкой с получением неочищенного целевого соединения. Полученное неочищенное целевое соединение растворяли в изопропаноле (51 мл) и по каплям добавляли воду (26 мл) в течение 10 мин. В мутную реакционную смесь добавляли затравку, и затем перемешивали в течение часа при комнатной температуре. Далее в течение 15 мин по каплям прибавляли воду (35 мл) и перемешивали полученную взвесь в течение полутора часов. Отфильтрованный твердый осадок промывали смесью вода:изопропанол 4:1 (10 мл), затем после сушки под вакуумом при 45oC получали белое кристаллическое целевое соединение (4.2 г) (T. пл. 71-72oC). 1H-ЯМР(CDCl3): 7.38 (dd), 5.25 (dd), 4.96 (t), 4.2 (m), 4.13 (dd), 3.21 (s), 3.18 (dd), 3.2-3.12 (m), 2.05 (m), 1.9-1.7 (m),1.7-1.5 (m), 1.5-1.35 (m), 1.32 (s), 1.23 (d), 0.86 (s), 0.8 (s) ppm.
Промежуточное соединение 3. 4-третбутилбензил-окси-оксалилхлорид.
К перемешиваемому раствору оксалилхлорида (100 мл) в диэтиловом эфире (500 мл) при -10oC в атмосфере азота по каплям прибавляли раствор 4-третбутилбензилового спирта (200 мл) в диэтиловом эфире (1000 мл) при охлаждении так, чтобы температура поддерживалась в пределах от -10 до -5oC, растворитель выпаривали в вакууме с получением целевого соединения (289 г) в виде прозрачного масла (T. кип. 120-122oC при 15 мм рт.ст.).
Пример 1. 4-Трeтбутилбензил-[4S, 8S, 9R, 10S, 12R)-4-метокси-10 [1-(гидpокси)этил-11-оксо-1-азатрицикло-[7.2.0.0. 3,8] -ундека-2-ен-2-карбоксилат
Способ А. К раствору промежуточного соединения 2 (15.5 г) и уксусной кислоты (9.5 мл) в тетрагидрофуране (53 мл) по очереди добавляли тетрабутиламмонийбромид (40 г) и измельченный фторид калия (7.3 г ). Реакционную смесь перемешивали в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры смесь разбавляли этилацетатом (295 мл) и промывали водным 10% раствором гидрокарбоната натрия (217 мл) и водой (2 порции по 295 мл). Органический слой упаривали до 40 мл и медленно (в течение приблизительно 5 минут) по каплям при интенсивном перемешивании добавили к н-гексану (295 мл). Образовавшуюся взвесь белого цвета в течение часа перемешивали при комнатной температуре и еще час на ледяной бане. Отфильтрованный твердый осадок промывали смесью н-гексан: циклогексан 1:1, затем после сушки под вакуумом получали белое кристаллическое целевое соединение (9.6 г) (Т. пл. 112-113oC).
1H-ЯМР (CDCl3): 7.4(dd), 5.35(d), 5.2(d), 4.95(t), 4,25(m), 4.2(dd), 3.3-3.25(dd+m), 3.2(s), 2.05(111), 1.9-1.1(m) ppm.
Способ Б. К раствору промежуточного соединения 2 (2.77 г) в тетрагидрофуране (13.6 мл) по очереди добавляли уксусную кислоту (3 мл) и кристаллогидрат тетрабутиламмонийфторида (14.3 г), после чего реакционную смесь перемешивали 3 часа при 40oC в атмосфере азота. Далее смесь разбавляли этилацетатом (480 мл) и промывали насыщенным водным раствором гидрокарбоната натрия (480 мл), водой (2 порции по 350 мл) и рассолом (350 мл). Органический слой после осушки выпаривали до получения светлой пены, после чего добавляли смесь петролейного и диэтилового эфиров 10:1 (70 мл) и перемешивали полтора часа при комнатной температуре. Отфильтрованный твердый осадок целевого соединения (1.31 г) сушили под вакуумом.
Пример 2. (4S,8S,9R,10S,12R)-4-метокси-10-(1-гидроксиэтил)- 11-оксо-1-азатрицикло-[7.2.0.0.3,8-ундека-2-ен-2-карбосилат натрия.
10% палладий на угле (0.42 г) и триэтиламин (0.48 мл) добавляли к раствору из примера 1 (1.22 г) в н-пропаноле (10.6 мл) в атмосфере азота. Гидрировали 30 мин, отфильтрованный катализатор промывали ацетоном (4.2 мл) и добавляли кристаллический этилгексаноат натрия (0.615 г) к фильтрату. Далее к раствору по каплям прибавляли диизопропиловый эфир (432 мл) в течение более 20 мин и смесь перемешивали 2 часа при комнатной температуре. Отфильтрованный в атмосфере азота твердый осадок промывали смесью диизопропиловый эфир: н-пропанол: ацетон 10:2.5:1 (2.1 мл), затем после сушки под вакуумом получали белое кристаллическое целевое соединение (0.38 г).
Пример 3. (4S,8S,9R,10S,12R)-4-метокси-10-(1-гидроксиэтил)-11-оксо- -1-азатрицикло-[7.2.0.0.3,8]-ундека-2-ен-2-карбоксилат натрия
10% палладий на угле (300 мг) и триэтиламин (11 мл) добавляли к раствору целевого соединения из примера 1 (3.2 г) в н-пропаноле (21 мл) в атмосфере азота. Гидрировали 45 мин при комнатной температуре, отфильтрованный катализатор промывали ацетоном (6 мл) и добавляли кристаллический этилгексаноат натрия (1.34 г) к объединенному фильтрату. Далее к раствору по каплям прибавляли диизопропиловый эфир (126 мл) в течение более 45 мин и смеси давали отстояться и перемешивали 2 часа при комнатной температуре. Отфильтрованный в атмосфере азота твердый осадок промывали диизопропиловым эфиром (15 мл), затем после сушки под вакуумом получали белое кристаллическое целевое соединение (1.78 г) 1H-ЯМР (300 МГц, D2O): 4.74(1H,m), 4.08(1H,m), 4.03(1H,dd), 3.28(1H, dd), 3.08(3H,s), 2.99(1H,m), 1.85(1H,m), 1.72(1H,m), 1.6-1.3(3H,m), 1.20(1H,m), 1.11(3H,d) ppm.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭФИРЫ 10(1- ГИДРОКСИЭТИЛ) -11-ОКСО-1-АЗАТРИЦИКЛО [7,2,0,0]УНДЕЦ-2-ЕН-2-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2043354C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 1992 |
|
RU2054006C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА | 1994 |
|
RU2144535C1 |
| ПРОИЗВОДНЫЕ СОРДАРИНА И ОБЛАДАЮЩИЙ ПРОТИВОГРИБКОВОЙ АКТИВНОСТЬЮ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 1995 |
|
RU2152398C1 |
| 5-ГЕТЕРОЦИКЛО-1,5-БЕНЗОДИАЗЕПИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1995 |
|
RU2152939C1 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| ПРОИЗВОДНЫЕ 5-АМИНО-2-(1-ГИДРОКСИЭТИЛ)ТЕТРАГИДРОПИРАНА | 2009 |
|
RU2525541C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-4-ФЕНИЛ-4-ОКСОМАСЛЯНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139850C1 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИ-1,2,3,4-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ-МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ, СРЕДИ ПРОЧЕГО, ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНОГО ТРАКТА | 2011 |
|
RU2586333C1 |
Способ получения антибактериального соединения формулы I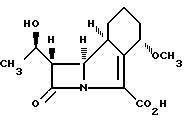
или его соли, который включает гидрогенолиз нового соединения формулы II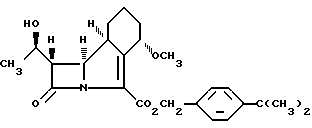
и в случае, когда это необходимо или желательно, выделение полученной карбоновой кислоты в виде ее соли. Предложено также соединение формулы II, использующееся в качестве промежуточного в синтезе соединения формулы I. Использование соединения формулы II упрощает стадию очистки, если это требуется перед проведением конечной стадии синтеза. 2 с. и 7 з.п. ф-лы.
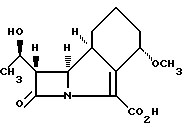
или его соли, отличающийся тем, что проводят гидрогенолиз соединения формулы II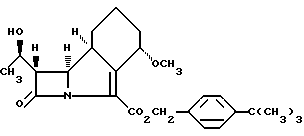
и включающий, если это необходимо, превращение полученной карбоновой кислоты в ее соль.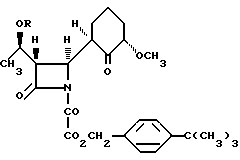
где R - группа, защищающая гидроксил, с последующим удалением группы R.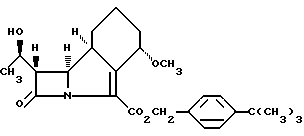
| 1972 |
|
SU416953A3 | |
| Protective Groups in Organic | |||
| Chemistry, Ed | |||
| by JFW McOnie, p.p | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
