Изобретение относится к гетероциклическим производным, имеющим антибактериальную активность, к способам их получения, к содержащим их композициям и к их использованию в медицине.
Таким образом, представленное изобретение дает соединения общей формулы I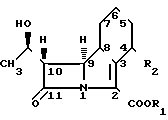 в которой R1 представляет собой группу
в которой R1 представляет собой группу O
O (O)pR5 где R4 является атомом водорода или С1-С4 алкильной группой; p= 0 или 1; R5 представляет собой группу, выбранную из С1-С6 алкила, С5-С8циклоалкила, необязательно замещенного С1-С3 алкильной группой, фенила или С1-С4 алкила, замещенного С1-С3 алкоксигруппой; R2 является группой OR3, в которой R3 представляет собой С1-С5 алкильную группу.
(O)pR5 где R4 является атомом водорода или С1-С4 алкильной группой; p= 0 или 1; R5 представляет собой группу, выбранную из С1-С6 алкила, С5-С8циклоалкила, необязательно замещенного С1-С3 алкильной группой, фенила или С1-С4 алкила, замещенного С1-С3 алкоксигруппой; R2 является группой OR3, в которой R3 представляет собой С1-С5 алкильную группу.
Кроме того, как определено в формуле I с точки зрения стереохимического расположения, молекула содержит два различных асимметричных атома углерода в положениях 4 и 8. Группа R1 также содержит по меньшей мере один асимметричный атом углерода, когда R4имеет значение, отличное от водорода. Является очевидным, что все стереоизомеры, включающие и их смеси, являющиеся результатом таких дополнительных асимметрических центров, охватываются формулой I.
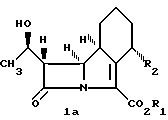
Общая формула I включает по меньшей мере 4 стереоизомера и их смеси, и эти соединения могут быть представлены формулами 1а, 1b, 1c и 1d.
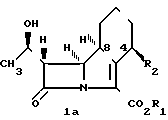

CH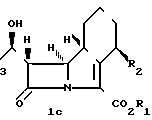

Связь, обозначенная в форме клина  обозначает, что связь располагается выше плоскости рисунка. Связь, изображенная прерывисто обозначает связь, располагающуюся ниже плоскости рисунка.
обозначает, что связь располагается выше плоскости рисунка. Связь, изображенная прерывисто обозначает связь, располагающуюся ниже плоскости рисунка.
Конфигурация, изображенная для углеродного атома в положении 8 в формулах 1а и 1b, является конфигурацией, относящейся в дальнейшем к β-конфигурации, а в формулах 1с и 1d к d-конфигурации.
Конфигурация, изображенная для углеродного атома в положении 4 в формулах 1b и 1d является конфигурацией, относящейся в дальнейшем к α-конфигурации, а в формулах 1а и 1с к β-конфигурации.
Вообще, в индивидуальных соединениях, названных ниже, β-конфигурация в положении 8 соответствует S-изомеру, а β-конфигурация в положении 4-R-изомеру. α-конфигурация в положении 8 соответствует R-изомеру, α-конфигурация в положении 4 соответствует S-изомеру. Расположение R или S конфигурации в положениях 4 и 8 соответствует правилам Сahn. Ingold and PrelogExperienta, 1956, 12, 81.
Использованный в работе термин алкил относится к алкильной группе с прямой или разветвленной цепью. Когда R4 представляет собой С1-С4алкильную группу, он может быть, например, метилом, этилом, пропилом, изопропилом или бутилом.
В том случае, когда R5 является алкильной группой, он обычно представляет собой С1-С4 алкильную группу такую, как метил, этил, изопропил или т-бутил.
В том случае, когда R5 является С1-С4 алкильной группой, замещенной С1-С3 алкокси, он может быть, например, метилом, этилом, пропилом или изобутилом, замещенным метоксигруппой.
В том случае, когда R5 представляет собой циклоалкил, необязательно замещенный С1-С3 алкилом, он может быть, например, циклопентильной, циклогексильной, циклогептильной или циклооктильной группой, замещенной метильной или этильной группой.
Предпочтительным классом соединений формулы I являются соединения, представляющие собой β-конфигурацию в положении 8. Внутри этого класса особенно предпочтительными являются соединения, имеющие α-конфигурацию в положении 4.
Другим предпочтительным классом соединений формулы I являются соединения, в которых R4 является водородом, метилом, пропилом или изопропилом, в особенности водородом или метилом.
Еще одним предпочтительным классом соединений формулы I являются такие соединения, в которых R5 представляет собой С1-С4 алкильную группу, такую, как метил, этил, изопропил или т-бутил; C1-C4 алкильную группу, замещенную метокси, такую, как 1-метокси-1-метилэтил; фенильную; или С5-С6 циклоалкильную группу, такую, как циклопентил или циклогексил, необязательно замещенную метильной или этильной группой, например как этилциклогексил.
Другим предпочтительным классом соединений, соответствующих изобретению, являются также соединения формулы I, в которых R2 является этокси или метокси группой.
Особенно предпочтительной группой эфиров, соответствующих изобретению, являются такие эфиры, в которых R4 является атомом водорода или метильной группой, p=0 или 1, а R5 представляет собой метильную, этильную, изопропильную, т-бутильную, 1-метокси-1-метилэтильную, фенильную, циклогексильную или 4-этилциклогексильную группу.
Особенно предпочтительной группой соединений изобретения являются такие, в которых углеродный атом в положении 8 характеризуется β-конфигурацией, а углеродный атом в положении 4 α-конфигурацией; R4является атомом водорода или метильной группой; R5 представляет собой С1-С4 алкил, С5-С6 циклоалкил, необязательно замещенный С1-С2 алкильной группой, фенил или C1-С4 алкил, замещененый метоксилом; р равно 0 или 1, а R2 является метоксилом.
Конкретными предпочтительными соединениями являются эфиры (4S, 8S, 9R, 10S, 12R)-4-метокси-10-(1-гидроксиэтил)-11-ок- со-1-азатрицикло [7.2.0.03.8] ундец-2-ен-2-карбоновой кислоты такие, как пивалоилоксиметиловый, 1-пивалоилоксиэтиловый, ацетоксиметиловый, 1-ацетоксиэтиловый, 1-метокси-1-метилэтилкарбонилоксимети- ловый, 1-(1-метокси-метилэтилкарбонилоксиэтиловый, 1-бензоилоксиэтиловый-1-изопропоксикарбонилоксиэтиловый, циклогексилоксикарбонилоксиметиловый, 1-(4-этил-циклогексилоксикарбонилокси) этиловый или более предпочтительно-1-циклогексилоксикарбонилоксиэтиловый эфир.
Соединения, соответствующие изобретению, при оральном приеме выявляют высокий уровень антибактериальной активности по отношению к большому разнообразию патогенных микроорганизмов и обладают очень высокой устойчивостью ко всем β-лактамазам. Соединения изобретения являются также относительно стабильными в отношении почечной дегидропептидазы.
Было установлено, что соединения изобретения выявляют значительный уровень активности против штаммов Staphylococcus aureus, Streptococcus faecalis, Streptococcus pneumonial, Escherichia coli, Klebsiella pneumonial, Proteus mirabiles Citrobacter freundii, Preudomonas aeruginosa, Cloctridium perfingens, Bacteriodes fragilis and Motganella morganii.
Соединения изобретения, таким образом, могут использоваться для лечения ряда болезней, вызванных патогенными бактериями, существующими у человека и у животных.
Следовательно, в соответствии с другим аспектом представленного изобретения, мы представляем соединение формулы I для использования в лечении или профилактике общих бактериальных инфекций у человека или животных.
В соответствии с другим аспектом изобретения мы представляем использование соединения формулы I для производства терапевтических агентов с целью лечения общих бактериальных инфекций, существующих у человека и животных.
В соответствии с еще одним аспектом изобретения мы представляем метод лечения человека или нечеловекообразных животных, включающих борьбу с бактериальными инфекциями, который состоит в приеме эффективного количества соединения формулы I.
Для специалиста в данной области понятно, что имеющиеся при этом ссылки на воздействие, распространяются на профилактику, а также и на лечение установленных инфекций и симптомов.
Существенно, что количество соединения изобретения, требуемое для использования его при лечении, будет варьироваться в зависимости от условий воздействия, возраста и условий пациента и, в конечном счете, будет раздельно выбрано соответствующим врачом или ветеринаром. Обычно применяемые дозы для лечения взрослого человека лежат в интервале 200-2000 мг в день, например, 1000 мг в день.
Необходимая доза обычно принимается в виде единичной дозы или в виде раздельных доз, принимаемых через соответствующие интервалы, например в виде двух, трех, четырех или большего числа суб-доз в день.
Хотя для использования в терапии возможен прием соединения изобретения в виде непереработанного химического препарата, предпочтительным является прием активного ингредиента в виде фармацевтического состава.
Изобретение, таким образом, далее, дает фармацевтическую композицию для орального приема, включающую соединение формулы I вместе с одним или большим числом фармацевтически приемлемых носителей его и, необязательно, другими терапевтическими и/или профилактическими ингредиентами. Носители должны быть "приемлемы" с точки зрения их совместимости с другими ингредиентами композиции и невредными для того, кто их принимает.
В соответствии с изобретением, фармацевтические композиции могут, например, быть в виде таблеток или капсул, приготовленных стандартными методами с фармацевтически приемлемыми наполнителями такими, как связующие агенты (например, предварительно желатинированный маисовый крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза), наполнители (например, крахмал, лактоза, микрокристаллическая целлюлоза или фосфат кальция), смазывающие вещества (например, стеарат магния, гидрогенизированное растительное масло, тальк, двуокись кремния, полиэтиленгликоли), дизинтегранты (например, картофельный крахмал или натриевый крахмальный гликолят) или увлажняющие агенты (например, натрий лаурилсульфат). При необходимости могут использоваться также вещества, помогающие течению, например, двуокись кремния. Таблетки могут приготавливаться с покрытием с использованием для этого хорошо известных в данной области приемов.
Жидкие составы для орального приема могут приготавливаться, например, в виде растворов, сиропов или суспензий, или они могут быть приготовлены в виде сухого продукта или для разведения водой или другим подходящим разбавителем перед использованием для приема, или в виде жидкости для непосредственного приема и последующего смывания водой или другой подходящей жидкостью. Такие жидкие составы могут быть изготовлены с использованием стандартных методов с применением приемлемых добавок, таких, как суспензирующие агенты (например, сорбитоловый сироп, метилцеллюлоза или гидрогенизированные пищевые жиры и масла, такие, как касторовое масло), эмульгирующие или загущающие агенты (например, лецитин, стеарат алюминия или акация), неводных растворителей (например, миндальное масло, кокосовое масло, синтетические сложноэфирные масла или этиловый спирт), консервирующие вещества (например, метил- или бутил-п-гидроксибензоаты или сорбиновая кислота) и подходящие ароматизирующие и подслащивающие агенты.
Соединения формулы I могут быть получены этерификацией карбоновой кислоты II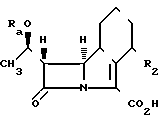 в которой Rа является водородом или гидроксил-защищающей группой, а R2имеет значение, определенное для формулы I, или соли, или химически активной производной этого соединения и, если требуется или желательно, превращение получающегося предшествующего соединения последующее или разделение его на стереохимические изомеры, удаление любой защищающей группы Rа. В том случае, когда Rа представляет собой гидроксильную защищающую группу, она, например, может быть гидроксикарбилсилильной группой, такой как триалкилсилил, например, триметилсилил или т-бутилдиметилсилил.
в которой Rа является водородом или гидроксил-защищающей группой, а R2имеет значение, определенное для формулы I, или соли, или химически активной производной этого соединения и, если требуется или желательно, превращение получающегося предшествующего соединения последующее или разделение его на стереохимические изомеры, удаление любой защищающей группы Rа. В том случае, когда Rа представляет собой гидроксильную защищающую группу, она, например, может быть гидроксикарбилсилильной группой, такой как триалкилсилил, например, триметилсилил или т-бутилдиметилсилил.
Этерификация соединения формулы II или его соли может проводиться взаимодействием с соединением R1Х, в котором R1 имеет значение, определенной в формуле I, а Х является уходящей группой, такой, как галоген-атом, например, хлор, бром или йод, или алкил или арил сульфонатом, таким, как мезилат или тозилат, в присутствии основания. Реакция проводится предпочтительно в присутствии растворителя, природа которого не является решающей, при условии, что он не имеет обратного воздействия на реакцию. Подходящие растворители включают диметилформамид, диметилацетамид или диметилсульфоксид.
В одном из воплощений данного способа реакция обычно осуществляется с использованием соли, такой, как соль щелочного металла, например, калиевой или натриевой соли карбоновой кислоты (II) в присутствии соответствующей четвертичной аммониевой соли такой, как триэтилбензиламмоний хлорид или тетрабутиламмоний бромид и предпочтительно в присутствии полярного апротонного растворителя, такого, как диметилформамид, диметилацетамид или N-метилпирролидон.
Реакция этерификации обычно проводится при использовании соединения формулы II, в которой Rа представляет собой водородный атом. Если реакция этерификации проводится на соединении формулы II, в которой Rапредставляет собой гидроксилзащищающую группу, то тогда эта группа может быть удалена с использованием стандартных методик. Например, когда Rапредставляет собой трет-бутилдиметил- силильную группу, она может быть удалена действием тетрабутиламмоний фторида и уксусной кислоты.
Соединения формулы II могут быть получены известными методами.
Соединения формулы I могут быть получены также циклизацией соединения формулы III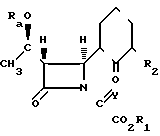 в которой группы R1 и R2 имеют значения, определенные для формулы (I), Rа является гидроксил-защищающей группой, а У является кислородным атомом или фосфиновой группой, и если требуется или желательно, превращение получающегося предшествующего соединения или в последующие для разделения на его стехиометрические изомеры с удалением защищающей группы Ra.
в которой группы R1 и R2 имеют значения, определенные для формулы (I), Rа является гидроксил-защищающей группой, а У является кислородным атомом или фосфиновой группой, и если требуется или желательно, превращение получающегося предшествующего соединения или в последующие для разделения на его стехиометрические изомеры с удалением защищающей группы Ra.
Циклизация соединения формулы III, в которой У является кислородом, обычно осуществляется нагреванием в присутствии органического фосфита. Реакция проводится предпочтительно в интервале 60-120оС. Подходящими растворителями являются углеводороды с соответствующей температурой кипения, например ароматические углеводороды, такие, как толуол или ксилол.
Подходящие органические фосфиты включают ациклические и циклические триалкилфосфиты, триарилфосфиты и смешанные алкиларилфосфиты. Особенно подходящими органическими фосфитами являются триалкилфосфиты, например, триэтилфосфиты или триметилфосфит.
Циклизация соединения формулы III, в которой У является фосфиновой группой, проводится преимущественно в растворителе при температуре в интервале 40-200оС. Подходящими растворителями являются углеводороды, такие, как ароматические углеводороды, например, ксилол или толуол, алифатические углеводороды и галогенированные углеводороды, такие, как дихлорметан, хлороформ и трихлорэтан. Примерами подходящих фосфиновых групп являются триарилфосфины, например, трифенилфосфин или триалкилфосфины, например, три-т-бутилфосфин.
Гидроксилзащищающая группа может быть удалена хорошо известными стандартными методами, такими, как описано в работе "Защитные группы в органической химии, стр. 46-119, под ред. IFW McOmil (Печатные труды пленума, 1973). Например, когда Ra является трет-бутилдиметилсилильной группой, она может быть удалена действием тетрабутиламмоний фторида и уксусной кислоты. Этот процесс обычно проводится в растворителе, таком, как тетрагидрофуран.
Аналогично, когда Rа является трихлорэтоксикарбонильной группой, она может быть удалена цинком и уксусной кислотой.
Соединения формулы III могут быть получены с использованием известных методов для получения родственных по строению соединений.
Для того, чтобы можно было более глубоко понять изобретение даются следующие примеры только в качестве иллюстрации.
В приготовлениях и примерах, если не указано особо: т.пл. определены на приборе для определения точки плавления Gallenkamp и не корректируются. Все температуры выражаются в оС.
Инфракрасные спектра были определены в растворах хлороформа -dl на приборе FT-IK. Протоновые магнитные резонансные спектры (1Н-ЯМР) записывались при 300 МГЦ в виде растворов в хлороформе -d1. Химические смещения выражаются в частях на миллион (δ) по отношению к тетраметилсилану, использованному в качестве внутреннего стандарта, и обозначаются в виде синглетов (с), дублетов (д), двойных дублетов (дд) или мультиплетов (м).
Хроматографирование проводилось через силикагель (Merk AG, Дармштадт, Германия).
Растворы сушились над безводным сульфатом натрия. "Бензин" обозначает петролейный эфир, т.кип. 40-60оС.
Метиленхлорид был дважды перегнан над гидридом кальция; тетрагидрофуран был дважды перегнан над натрием; этиловый эфир был дважды перегнан над натрием; ксилол был дважды перегнан над пятиокисью фосфора и этилацетат был высушен над активированными молекулярными ситами.
Промежуточное соединение 1.
Виниловый эфир 2-метокси-2-метил) пропионовой кислоты.
К 2-метокси-2-метилпропионовой кислоте (1,5 г) в атмосфере азота добавлялись ацетат ртути (II) (0,162 г), ацетат палладия (0,0285 г), гидроокись калия (0,067 г) и винилацетат (1,2 г). Получающийся раствор нагревался в течение 4 часов при 50оС. Затем к реакционной смеси добавлялся дополнительно винилацетат (2,4 г) и смесь нагревалась в течение 16 часов при 50оС.
После охлаждения до 20оС добавлялся этиловый эфир (15 мл) и смесь фильтровалась через тампон, а затем экстрагировалась диэтиловым эфиром (2х70 мл). Органическая фаза промывалась раствором хлористого кальция (150 мл) и высушивалась над безводным сульфатом натрия с образованием сырого озаглавленного соединения в виде "палевого" желтого масла (0,7 г); ТСХ циклогексан/этилацетат 8:2Rf=0,7; ИК (CDCl3) νmax (см-1): 1749 (C=O эфир) 1640 (С=С; 1Н-ЯМР (300 МГЦ; CDCl3) (м.ч.) 7,30 (м), 4,983 (дд), 3,297 (с), 1,464 (с).
Промежуточное соединение 2.
1-Хлорэтиловый эфир (2-метокси-2метил) пропионовой кислоты.
В растворе промежуточного соединения I (2,7 г) в этилацетате (50 мл) в течение часа при 0оС барботировали безводный хлористый водород, затем, в течение 10 мин продували азот. Растворитель выпаривали, а остаток очищали перегонкой (90оС/ 15 мм Hg) с получением озаглавленного соединения в виде бесцветного масла (2,1 г) ТСХ циклогексан/этилацетат 9:1. Rf 0,9; ИК (CDCl3) νmax (см-1): 1755 (С= 0 эфир); 1Н-ЯМР (CDCl3; 300 МГц) (мил.ч.) 6,58 (кв) 3,296 (с), 1,837 (д), 1,442 (с).
Промежуточное соединение 3.
(1-Хлор-2-метил) пропилметил карбонат.
Раствор 1-хлор-2-метилпропил хлорформиат (1,71 г) в сухом дихлорметане (5 мл) добавлялся по каплям к раствору метанола (0,83 мл) в сухом дихлорметане (5 мл) при 0оС в атмосфере азота, при перемешивании.
Затем добавлялся раствор пиридина (0,80 мл) в сухом дихлорметане (10 мл) и реакционная смесь перемешивалась в течение 18 ч при 20оС.
Смесь разбавлялась дихлорметаном (950 мл), промывалась раствором хлористого кальция (3х40 мл), сушилась над безводным сульфатом натрия и концентрировалась в токе азота при низкой температуре, образуя сырьевое озаглавленное соединение в виде бесцветного масла с количественным выходом. Н-ЯМР (300 МГц, CDCl3): 6,18 (д), 3,86 (с), 2,28-2,12 (м), 1,08 (д), 1,06 (д) мил.д.
Промежуточное соединение 4.
1-Хлорэтил-4-этилциклогексил карбонат.
Раствор 1-хлорэтил хлорформиата (5,46 г) в сухом дихлорметане (20 мл) при 0оС в атмосфере азота по каплям добавлялся к перемешиваемому раствору 4-этилциклогексанола (5 г) в сухом дихлорметане (20 мл) в присутствии 3А молекулярных сит. К реакционной смеси по каплям добавлялся раствор пиридина (3 г) в сухом дихлорметане (20 мл) в течение 20 мин при 0оС, после чего реакционная смесь нагревалась до 20оС, перемешивалась в течение 20 ч, промывалась раствором хлористого кальция (2x50 мл) и сушилась. Растворитель удалялся под вакуумом, а остаток очищался, давая озаглавленное соединение в виде бесцветного масла (7.9 г; т.кип. 130о/мбар; т.с.х. циклогексан/этилацетат 9/1Rf= 0,88; ИК (CDCl3), νmax (см-1): 1757 (С=0); 1Н-ЯМР (300 МГЦ, CDCl3): 6,43 (кв), 6,42 (кв), 4,93 (уш.с), 4,59 (Т.Т.), 2,00-1,88 (уш.с), 1,88-1,78 (м), 1,83 (д), 1,82 (д), 1,60-1,50 (м), 1,50-1,32 (м), 1,30-1,15 (м), 1,28-1,18 (м), 1,05-0,95 (м), 0,95-0,85 (м).
Промежуточное соединение 5.
1-Хлор-2метилпропил-2,2-диметилпро- пионат.
2-метилпропиональдегид (5,98 г) в течение 10 мин по каплям добавлялся к перемешиваемой смеси хлорида цинка (0,11 г) и пивалоилхлорида (10 г) в атмосфере азота при -20oС. Затем, реакционная смесь нагревалась до 23оС и перемешивали далее в течение 2 ч. Твердое вещество отделялось центрифугированием, а маслянистый остаток подвергался перегонке, образуя озаглавленное соединение в виде бесцветного масла (11,55 г; т.кип. 70оС/мбар; ИК (CDCl3), νmax (см-1): 1749 (С=0); 1Н-ЯМР (300 МГЦ, CDCl3): 6,28 (д), 2,16 (м), 1,22 (с), 1,05 (д) мил.д.).
Промежуточное соединение 6.
Циклогексилхлорметилкарбонат.
Ток хлора медленно пропускали в холодный (-10/+5оС) метилхлорформиат (6 мл) в рассеянном свете. Реакция контролировалась снятием 1-Н-ЯМР спектров и прекращалась при достижении концентрации дихлорметил хлорформиата >5 молярн. Через раствор пропускали азот до тех пор, пока он не становился прозрачным, а остаток перегоняли, получая две основные функции, содержащие требующийся промежуточный хлорметил хлорформиат; фракция а (2,48 г; молярные соотношения: метилхлорформиат/хлорметил хлорформиат/дихлорметил хлорформиат 19/77/4), фракция b (1,76 г; молярные соотношения: метил хлорформиат/хлорметил хлорформиат/ дихлорметил хлорформиат 4/90/6). К охлажденному льдом раствору циклогексанола (1,37 мл) в сухом дихлорметане (5 мл) в присутствии 3А молекулярных сит и в атмосфере азота добавлялся раствор (фракция а) (1,7 г) в течение 5 мин. Затем, в реакционную смесь в течение 30 мин при 0оС добавляется раствор пиридина (1,06 мл) в сухом дихлорметане (5 мл), и смесь медленно нагревали до 20-25оС. Спустя 5 ч раствор отфильтровывали, разбавляли дихлорметаном (30 мл), промывали водой (20 мл), солевым раствором (3х30 мл) и сушили. Растворитель отгоняли, а остаток очищали на хроматографической колонке с использованием силикагеля и смеси циклогексан /этилацетат/ в соотношении 9:1 в качестве элюента с получением озаглавленного соединения в виде белого воска (1,98 г; т.с.х. циклогексан/этилацетат 9/1Rf= 0,44; ИК (CDCl3)νmax (см-1): 1759 (С=0); 1Н-ЯМР (3000 МГц, CDCl3): 5,73 (c), 4,78-4,66 (м), 2,00-1,90 (м), 1,80-1,70 (м), 1,60-1,20 (м) мил.д.
П р и м е р 1. 1-(Циклогексилоксикарбонилокси)этил (4S, 8S, 9R, 10S, 12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатри- цикло- [7,2,7,03,8] ундец-2-ен-карбоксилат.
К раствору калий (4S,8S,9R,10S,12R)-4-метокси-10-(1-гидроксиэтил)-11-оксо-1- аза- трицикло[7,2,0,03,8]ундец-2-ен-2-карбокси- лата (называемого в дальнейшем как "Промежуточное соединение А") (0,5 г) в диметилформамиде (8 мл) добавлялись тетрабутиламмоний бромид (0,5 г) и (1-хлорэтил)-циклогексилкарбонат (0,65 г) при 22оС. Получающаяся смесь перемешивалась в течение 15 ч при 22оС затем выливалась в диэтиловый эфир (60 мл), промывалась 10%-ным водным раствором НСl (40 мл), 5%-ным водным раствором NaHCO3 (2х50 мл) и солевым раствором (2х50 мл), сушилась и концентрировалась. Остаток (1 г) растворялся в диэтиловом эфире (2 мл) и при энергичном перемешивании добавлялся петролейный эфир (20 мл). Осадок (0,1 г) отфильтровывался, а маточные растворы концентрировались, образуя остаток, который растворяли в диэтиловом эфире (1 мл) и петролейном эфире (20 мл), добавленных при тщательном перемешивании для получения большего осадка (0,14 г). Осадки объединялись (0,24 г) и подвергались дальнейшей очистке растворением в диэтиловом эфире (3 мл) и высаждением из петролейного эфира (30 мл) при тщательном перемешивании с образованием озаглавленного соединения в виде белого порошка (0,160 г; т.с.х. диэтиловый эфир Rf 0,44, т.кип. 90-100оС.
ИК (CDCl3) νmax(см-1) 1771, 1632; 1Н-ЯМР (3000 МГц, CDCl3) 6,93-6,85 (кв+кв), 4,92 (т), 4,64 (м), 4,25-4,05 (м), 3,30-3,15 (м), 3,25 (с), 3,24 (с), 2,08 (м), 2,0-1,2 (м), 1,61 (д), 1,59 (д), 1,31 (д), 1,30 (д).
В соответствии с экспериментальными методиками, описанными в примере 1, из промежуточного соединения А были получены следующие эфиры.
П р и м е р 2. 1-(Этилоксикарбонилокси)этил (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатрицикло- [7,2,7,03,8]ундец-2-ен-карбоксилат были получены в виде масла (т.с.х. диэтиловый эфир Rf 0,42).
ИК (CDCl3) νmax (см-1) 1765, 1738, 1600; 1Н-ЯМР (300 МГц, CDCl3) 6,93-6,87 (кв+кв), 4,933 (м), 4,3-1,8 (м), 3,26-3,24 (с+с), 3,32-3,20 (м), 2,08 (м), 1,94-1,3 (м), 1,62 (д), 1,60 (д), 1,36-1,28 (м) из промежуточного соединения А и (1-хлорэтил)-этилкарбоната.
П р и м е р 3. 1-(Изопропилоксикарбонилокси) этил (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатри- цикло-[7,2,0,03,8]ундец-2-ен-карбоксилат. ИК (CDCl3)νmax (см-1) 3614, 1767, 1632, 1Н-ЯМР (300 МГц, CDCl3) 6,90 (кв), 6,89 (кв), 4,95-4,83 (м), 4,3-4,2 (м), 4,19 (дд), 3,35-3,20 (м), 3,257 (с), 3,243 (с), 2,07 (м), 1,93-17,5 (м), 1,7-1,3 (м), 1,613 (д), 1,33-1,29 (д+д+д), т.с.х. циклогексан, этилацетат 4:6Rf0,4 был получен из промежуточного соединения А и (1-хлорэтил)-изопропил-карбоната.
П р и м е р 4. 1-(Ацетокси)этил (4S, 8S,9R,10S,12R)-4-метокси-10-(1'-гидрокси- этил)-11-оксо-1-азатрицикло-[7,2,0,03,8] ун- дец- 2-ен-2-карбоксилат.
Был получен в виде масла (0,160 г; циклогексан, этилацетат 4:6Rf0,4. ИК (CDCl3)νmax (см-1) 3605, 1769, 1700, 1Н-ЯМР (300 МГц, СDCl36,99 (кв), 6,98 (кв), 4,93 (т), 4,25 (м), 4,19 (дд), 3,3-3,2 (м), 3,25 (с), 3,24 (с), 2,10 (с), 2,07 (с), 2,08 (м), 1,95-1,3 (м), 1,56 (д), 1,55 (д), 1,31 (д), 1,30 (д) из промежуточного соединения А и (1-хлорэтил)ацетата.
П р и м е р 5. 1-(Циклогексилкарбонилокси)этил (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатри цикло- [7,2,0,03,8]ундец-2-ен-карбоксилат.
ИК (CDCl3) νmax (см-1) 1774, 1750, 1630; 1Н-ЯМР (300 МГц, CDCl3) 6,997 (кв), 6,977 (кв), 4,931 (т), 4,913 (т), 4,24 (м), 4,193 (дд), 3,3-3,2 (м), 3,25 (с), 3,245 (с), 2,38-2,24 (м), 2,05 (м), 1,95-1,2 (м), 1,65 (дд), 1,566 (д), 1,326 (д), 1,314 (д) был получен из промежуточного соединения А и (1-хлорэтил) циклогесанкарбоксилата.
П р и м е р 6. 1-(Бензоилокси)этил (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидрок- сиэтил)-11-оксо-1-азатрицикло [7,2,0,03,0] ундец-2-ен-2карбоксилат был получен в виде масла (0,045 г; т.с.х. циклогексан, этилацетат 1:Rf0,25).
ИК (CDCl3) νmax (см-1) 1776, 1738, 1640, 1603, 1Н-ЯМР (300 МГц, CDCl3) 8,1-8,02 (м), 7,58 (тт), 7,48-7,4 (м), 7,27 (м), 4,948 (т), 4,914 (т), 4,3-4,2 (м), 4,20 (дд), 3,3-3,2 (м), 3,23 (с), 3,21 (с), 2,05 (м), 1,9-1,3 (м), 1,725 (д), 1,326 (д), 1,302 (д), из промежуточного соединения А и (1-хлорэтил)-бензоата.
П р и м е р 7. 1-[(1,1-Диметилэтил)карбонилокси]этил (4S,8S,9R,10S, 12R)-4-метокси-10-(1'-гидроэксил)-11-оксо-1-азатри-цикло- [7,2,0,03,8]ундец-2-ен-карбоксилат был получен в виде масла (0,160 г; т.с.х. циклогексан, этилацетат 4:6 Rf 0,37); ИК (CDCl3) νmax (см-1) 3666, 1776, 1744, 1632; 1Н-ЯМР (300 МГц, CDCl3) 6,982 (кв), 6,941 (кв), 4,94-4,88 (м), 4,3-4,16 (м), 3,3-3,18 (м), 3,238 (с), 3,20 (с), 2,05 (м), 1,9-1,2 (м), 1,565 (д), 1,554 (д), 1,317 (д), 1,306 (д), 1,207 (с), 1,179 (с) из промежуточного соединения А и 1-[(1,1-диметилэтил)карбонилокси этил]хлорида.
П р и м е р 8. 1-[2-Метокси-2-метил-пропаноилокси)]этил (4S,8S,9R,10S, 12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-аза- трицикло[7,2,0,03,8[-ундец-2-ен-2-карбокси- лат был получен в виде масла (0,130 г; т.с.х. диэтиловый эфир Rf0,35);
ИК( CDCl3)Vmax (см-1) 1772, 1603, 1Н-ЯМР (300 МГц, CDCl3) 7,028 (кв), 6,984 (кв, ), 4,914 (м), 4,3-4,2 (м), 4,190 (дд), 3,3-3,2 (м), 3,260 (с), 3,248 (с), 3,290 (с), 3,226 (с), 2,06 (м), 1,9-1,35 (м), 1,604 (м), 1,437 (с), 1,429 (с), 1,403 (с) 1,400 (с), 1,315 (д) из промежуточного соединения А и хлорэтилового эфира 2-метокси-2-метиолпропановой кислоты.
П р и м е р 9. Ацетоксиметил (4S,8S,9R, 10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатрицикло[7,2,0,03,8] ундец-2-ен-2карбоксилат был получен в виде масла (0,240 г; т.с.х. циклогексан, этилацетат 4:6 Rf 0,24);
ИК (CDCl3) νmax (см-1) 1769, 1730, 1640; 1Н-ЯМР (300 МГц, CDCl3) 4,996 (т), 4,802 (с), 4,3-4,2 (м), 4,23 (дд), 3,774 (с), 3,36-3,24 (м+дд), 3,28 (с), 2,1 (м), 1,94-1,30 (м), 1,769 (д), 1,327 (д) из промежуточного соединения А и хлорметилацетата.
П р и м е р 10. [1,1-Диметил-этил)карбонилокси]метил (4S,8S,9R,10S, 12R)-4-метокси-10-(1'-гидроксилэтил-11-оксо-1-аза-три- цикло[7,2,0,03,8] ундец-2-ен-2-карбоксилат был получен в виде масла (0,260 г; т.с.х. циклогексан, этилацетат 1:1 Rf 0,26;
ИК (CDCl3) νmax (см-1) 3568, 1772, 1751, 1600; 1Н-ЯМР (300 МГц, CDCl3) 5,95 (д), 5,85 (д), 4,88 (т), 4,24 (м), 4,20 (дд), 3,27 (м), 3,25 (дд), 3,23 (с), 2,1 (м), 2,0 (уш. с), 1,95-1,6 (м), 1,5-1,20 (м), 1,31 (д), 1,21 (с), из промежуточного соединения А и [(1,1-диметилэтил)карбонилокси]метил йодида.
П р и м е р 11. 1-(2-Метокси-2-метил-пропаноилокси)метил (4S,8S,9R,10S, 12R) -4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-аза- трицикло[7,2,0,03,8]ундец-2-ен-2-карбокси- лат был получен в виде масла (0,110 г; т.с.х. диэтиловый эфир Rf0,33;
ИК (CDCl3)Vmax (см-1); 3600, 1742, 1740, 1640; 1-Н-ЯМР (300 МГц, CDCl3) 5,964 (д+д), 4,904 (м), 4,242 (м), 4,203 (дд), 3,984 (дд), 3,33-3,22 (м+дд), 3,292 (с), 3,240 (с), 2,1 (м), 1,95-1,2 (м), 1,441 (с), 1,429 (с), 1,315 (с) из промежуточного соединения А и хлорметилового эфира (2-метокси-2-метил) пропионовой кислоты.
П р и м е р 12. 1-(Метилоксикарбонилокси)-2-метилопропил (4S,8S,9R,10S, 12R)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-аза- трицикло[7,2,0,03,8]ундец-2-ен-2-карбокси- лат был получен в виде масла (0,040 г); т.с.х. циклогексан, этилацета 4:6 Rf 0,36;
ИК (CDCl3) νmax (см-1) 1767, 1734, 1680, 1Н-ЯМР (300 МГц, CDCl3), 6,661 (д), 60636 (д), 4,974 (м), 4,936 (м), 4,3-4,15 (м), 3,824 (с), 3,805 (с), 3,262 (с), 3,242 (с), 3,32-3,18 (м), 1,327 (д), 1,306 (д), 1,15-0,95 (м), 2,4-1,2 (м), из промежуточного соединения А и (1-хлор-2-метил) пропилметилкарбоната.
П р и м е р 13. 1-(Ацетилокси)бутил- (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидрок- сиэтил)-11-окcо-1-азатрицикло [7,2,0,03,8] ундец-2-ен-карбоксилат был получен в виде масла (0,050 г; т.с.х. циклогексан, этилацетат 1:1 Rf0,33);
ИК (CDCl3) νmax (см-1) 1769, 1732, 1632, 1Н-ЯМР, (300 МГц, CDCl3) 6,925 (кв), 4,948 (м), 4,28-4,16 (м), 3,3-3,2 (м), 3,251 (с), 3,243 (с) 2,105 (с), 2,069 (с), 2,12-2,04 (м), 1,94-1,74 (м), 1,74-1,58 (м), 1,54-1,349 (м), 1,318 (д), 1,307 (д), 0,962 (т), 0,957 (т) из промежуточного соединения А и 1-бромбутил ацетата.
П р и м е р 14. 1-[(4-Этилциклогексилокси)карбонилокси]этил (4S,8S,9R, 10S, 12CR)-4-метокси-10-(1'-гидроэтил)-11- оксо-1-аза- трицикло[7,2,0,03,8] ундец-2-ен-2-карбокси- лат.
К раствору промежуточного соединения А (0,3 г) в N,N-диметилформамиде (5 мл) добавлялись тетра-н-бутиламмоний бромид (0,3 г) и промежуточное соединение 4 (0,47 г) в атмосфере азота, и перемешивание продолжалось при 22оС в течение 16 ч. Реакционная смесь разбавлялась диэтиловым эфиром (15 мл), промывалась насыщенным хлоридом аммония (2х10 мл), солевым раствором (2х20 мл), сушилась и выпаривалась в вакууме. Маслянистый остаток хроматографировался через силикагель с использованием в качестве элюента смеси циклогексан/этилацетат 7/3 с получением озаглавленного соединения в виде пены (0,169 г; т. с.х. циклогексан/этилацетат 1/1Rf=0,41; ИК (CDCl3), νmax (см-1): 3640 (ОН), 1761 (С=0), 1634, (С=С); 1Н-ЯМР (300 МГц, CDCl3): 6,88 (м), 4,92 (м), 4,91 (м), 4,95-4,85 (м), 4,54 (м), 4,28-4,18 (м), 4,18 (дд), 3,30-3,20 (м), 3,24 (с), 3,23 (с), 2,05 (м), 2,00-1,75 (м), 1,70-1,50 (м), 1,50-1,09 (м), 1,31 (д), 1,29 (д), 1,25-1,15 (м), 0,86 (м), мил.д.
П р и м е р 15. (Циклогексилоксикарбонилокси) метил (4S,8S,9R,10S, 12R)-4-метокcи-10-(1'-гидроксиэтил)-11-оксо-1- азатрицикло [7,2,0,03,8]ундец-2-ен-2-карбоксилат.
К раствору промежуточного соединения А (0,22 г) в N,N-диметилформамиде (6,5 мл), в присутствии ЗА молекулярных сит добавлялись тетра-н-бутиламмоний бромид (0,222 г) и промежуточное соединение 6 (0,191 г). Образующаяся смесь перемешивалась в течение 5 ч при 22оС, разбавлялась диэтиловым эфиром, хлоридом аммония (2х30 мл), 5%-ным водным раствором гидрокаpбоната натрия (30 мл), солевым раствором (2х30 мл), водой (30 мл), и сушились. Растворитель удалялся в вакууме, а остаток подвергался хроматографированию через силикагель с использованием циклогексан/этилацетат 100/0 до 65/35 с получением озаглавленного соединения в виде белой пены (0,1 г; т.с.х. циклогексан/этилацетат 1/1Rf= 0,18; ИК (CDCl3) νmax (см-1): 3614 (ОН), 1772 (С=0 β-лактам), (С=0 эфир), 1640 (С=С); 1Н-ЯМР (300 МГц, CDCl3): 5,90 (дд), 4,93 (т), 4,67 (м), 4,30-4,20 (м), 4,20 (дд), 3,35-3,25 (м), 3,25 (с), 2,08 (м), 2,00-1,8 (м), 1,80-1,30 (м), 1,32 (д, мил.д.).
П р и м е р 16. 1-[(1,1-диметилэтил)карбонилокси]-2-метилпропил (4S,8S, 9R, 10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11- окси-1-азатрицикло [7,2,0,03,8]ундец-2-ен-2-карбоксилат.
К раствору промежуточного соединения А (0,3 г) в N,N-диметилформамиде (5 мл) добавлялись тетра-н-бутиламмоний бромид (0,3 г) и пpомежуточное соединение 5 (0,398 г) в атмосфере азота и перемешивание продолжалось при 22оС в течение 16 ч. Реакционная смесь разбавлялась диэтиловым эфиром (15 мл), промывалась насыщенным хлористым аммонием (2х30 мл), солевым раствором (2х30 мл), сушилась и выпаривалась в вакууме. Маслянистый остаток подвергался хроматографированию через селикагель с использованием в качестве элюента смеси циклогексан/этилацетат 7/3 с получением озаглавленного соединения в виде бесцветного масла (0,057 г, т.с.х. циклогексан/этилацетат 1/1Rf=0,45; ИК (CDCl3), νmax (см-1): 3611 (NH), 1774 (С=0 β-лактам), 1747 (С=0 эфир, 1632 (С=С), 1Н-ЯМР (300 МГц, CDCl3): 6,76 (д), 4,95 (т), 4,92 (т), 4,30-4,16 (м), 3,32-3,19 (м), 3,24 (с), 3,23 (с), 2,10 (м), 2,06 (м), 1,94-1,80 (м), 1,75-1,60 (м), 1,50-1,20 (м), 1,32 (м), 1,22 (с), 1,19 (с), 1,06-0,98 (д) мил.д.
П р и м е р 17. 1-(Циклогексилоксикарбонилокси)этил (4S,8S,9R,10S, 12S)-4-метокси-10-(1'-гидроксиэтил)-11-оксо-1-азатрицик- ло[7,2,0,03,8]ундец-2-ен-2-карбоксилат.
К раствору натрий (4S,8S,9R,10S,12R)-4-метокси-10-(1'-гидроксиэтил)-11- оксо-1-азатрицикло[7,2,0,03,8] ундец-2-ен-2-карб- оксилата (194 мг) в диметилформамиде (8 мл) при комнатной температуре добавлялись триэтилбензиламмоний хлорид (146 мг) им (1-хлорэтил)-циклогексилкарбонат (0,142 мл). Образующаяся смесь перемешивалась при 60оС в течение 97 мин, разбавлялась диэтиловым эфиром (30 мл) и промывалась холодной водой (60 мл). Водный раствор повторно экстрагировался диэтиловым эфиром (30 мл), а объединенные органические экстракты пpомывались соляным раствором (30 мл) и высушивались над сульфатом натрия. Органический слой концентрировался при пониженном давлении, а образовавшаяся белая пена (288 мг) перекристаллизовывалась из смеси диэтиловый эфир/петролейный эфир с образованием озаглавленного соединения (220 мг) в виде белого твердого вещества.
Фармацевтические примеры.
Таблетки
П р и м е р А мг/табл. Соединение примера 1 320 Лактоза 150 Этилцеллюлоза 20 Лаурилсульфат натрия 7 Стеарат магния 3 Масса таблетки 500 мг
Активный ингредиент и лактоза смешиваются вместе и затем гранулируются с использованием воды в качестве гранулирующей жидкости. Высушенные гранулы смешиваются с этилцеллюлозой, лаурилсульфатом натрия и стеаратом магния и таблетная масса формируется с использованием соответствующего штампа. Затем таблетки могут быть покрыты оболочкой с использованием стандартных методик и покрытий. П р и м е р В мг/табл Соединение примера 1 320 Прессуемый сахар 170 Лаурилсульфат натрия 7 Стеарат магния 3 Масса таблетки 500 мг
Активный ингредиент и наполнители смешиваются вместе и затем прессуются с использованием соответствующего штампа. При необходимости сформированные таким образом таблетки могут быть покрыты оболочкой с использованием стандартных способов.
Гранулы П р и м е р С мг/стандарт. доза Соединение примера 1 320 Крахмал 100 Целлюлоза 40 Полиметакрилат 30 Лаурилсульфат натрия 7 Стеарат магния 3 Ароматический агент Сколько нужно П р и м е р D мг/стандарт. доза Соединение примера 1 320 Этилцеллюлоза 140 Полиметакрилат 30 Лаурилсульфат натрия 7 Стеарат магния 3 Ароматизирующий агент Сколько нужно П р и м е р Е. мг/стандарт. доза Соединение примера 1 320 Прессуемый сахар 140 Полиметакрилат 30 Лаурилсульфат натрия 7 Стеарат магния 3 Ароматизирующий агент Сколько
нужно
Раствор активного ингредиента в этаноле напыляется на соответствующее жидкостное основание гранулятора, смешанного с основными наполнителями. Сформованные таким образом гранулы сушатся и просеиваются. При желании гранулы затем могут быть покрыты энтеросолюбильной оболочкой и высушены. Высушенные гранулы затем смешиваются с остающимися наполнителями, включая ароматизирующий агент и покрываются, например, энтеросолюбильной оболочкой. Полученные таким образом гранулы могут быть изготовлены в виде капсул и т.п. для единичной дозы приема или помещены в сосуды для изготовления соответствующих препаратов, предназначенных для многократного орального приема в виде жидких доз.
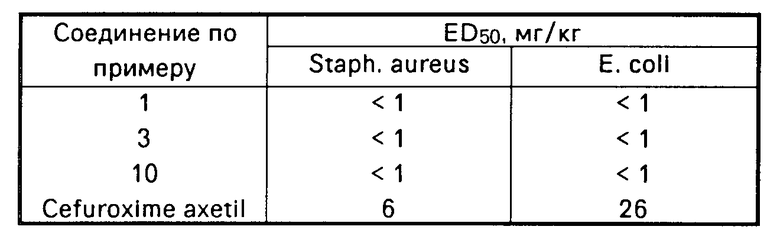
Данные по активности.
Проведенные в стандартных условиях испытания, с использованием мышей, выявили очень высокую активность соединений изобретения при их оральном приеме в защите от патогенных бактерий, как это проиллюстрировано в следующей ниже таблице, в которой эти соединения сравниваются с известным антибиотиком широкого спектра действия Cefuroxime axetil, принимаемым орально. Данные приведены в таблице.
Соединения изобретения также являются нетоксичными при уровнях доз, используемых в терапевтическом лечении. Например, не наблюдалось неблагоприятных эффектов при оральном приеме мышами соединения примера 1 в дозах, достигающих 1000 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 1992 |
|
RU2054006C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-МЕТОКСИ-10-(1-ГИДРОКСИЭТИЛ)-11-ОКСО-1-АЗАТРИЦИКЛО- (7.2.0.0)-УНДЕКА-2-ЕН КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ СОЛЕЙ, 4-ТРЕТБУТИЛБЕНЗИЛ- (4S, 8S, 9R, 10S, 12R)-4-МЕТОКСИ-10-(1-ГИДРОКСИЭТИЛ)-11-ОКСО-1-АЗАТРИЦИКЛО- (7.2.0.0)-УНДЕКА-2-ЕН-2-КАРБОКСИЛАТ | 1994 |
|
RU2127735C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| С-21 МОДИФИЦИРОВАННЫЕ ЭПОТИЛОНЫ | 2000 |
|
RU2253652C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2014 |
|
RU2666356C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТОКСИМЕТИЛПЕНЕМОВ | 1990 |
|
RU2049786C1 |
| ПРОИЗВОДНЫЕ 1,1-ДИОКСОЦЕФЕМ-4-КАРБОТИОЛОВОЙ КИСЛОТЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2091383C1 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| 2-ТИОЗАМЕЩЕННЫЕ КАРБАПЕНЕМЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2130457C1 |
Использование: в медицине в качестве антибактериальных средств. Сущность изобретения: продукты эфиры 10(гидроксиэтил)-11-оксо-1-азатрицикло 03,8 ундец-2-ен-2 карбоновой кислоты ф-лы I, где 03,8 R1-CH(R4)-O-C(O)-(O)p-R5 где R4- Н, C1-C4 -алкил; р-0 или 1, R5-C1-C6 -алкил, C5-C8 циклоалкил, фенил, R2 метоксигруппа. Реагент 1: соединение ф-лы II, где R2 -метоксигруппа, Ra-H или гидроксилзащитная группа. Реагент 2: этерифицирующий агент, служащий для введения группы R1 Условия реакции: в среде органического растворителя. 4 с. и 4 з.п.ф-лы. Структура соединений I и II: 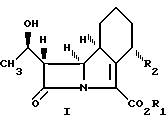

где R1 группа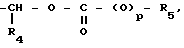
где R4 водород или С1 С4-алкильная группа;
p 0 или 1;
R5 группа, выбранная из С1 С6-алкила, С5 - С8-циклоалкила, необязательно замещенного С1 С3-алкильной группой, фенила или С1 С4-алкила, замещенного С1
С3-алкоксигруппой;
R2 метокси.
где R2 -метокси;
Rа водород или гидроксилзащитная группа,
или его соли, или его химически активного производного с этерифицирующим агентом, служащим для введения группы R1, значение которой определено в п.I, при необходимости замещения в полученном продукте гидроксилзащитной группы Rа водородом и/или выделение желаемого изомера соединения общей формулы I из смеси одного или более других изомеров этого соединения.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПОРШНЕВОЙ ПРЕСС НЕПРЕРЫВНОГО ДЕЙСТВИЯ | 0 |
|
SU381532A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

