Предметом настоящего изобретения являются производные 2-аминопиразин-5-карбоксамида, способы их получения и использование в терапии.
Известны [Машковский М.В., Лекарственные средства, Харьков, "Торсинг", 1998, с. 259-261] производные пиперазина, празозин и диоксазозин, обладающие избирательной антагонистической активностью по отношению к α1- адренергическим рецепторам. Их применяют при гипертонической болезни, застойной сердечной недостаточности, а также при аденоме простаты. Однако при их применении возможно развитие толерантности к препарату и имеют место побочные явления.
Настоящее изобретение решает задачу создания новых соединений, обладающих антагонистической активностью по отношению к α1- адренергическим рецепторам на уровне нижнего мочевого аппарата.
Производные 2-аминопиразин-5-карбоксамида по изобретению соответствуют общей формуле (I)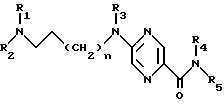
где n = 0 или 1;
R1 обозначает метильную группу и в этом случае R2 обозначает фенокси(С1-С4)алкильную группу, в которой феноксигруппа замещена метокси- и этoкcигpуппами или R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 4-(феноксиметил) пиперид-1-ил группу, в которой феноксигруппа замещена одной или двумя (С1-С4) алкильными группами, или 4-фенилпиперазин-1-ил группу, в которой фенильная группа замещена одним или двумя заместителями, такими как галоген, метокси, этокси и (С1-С4) алкильная группа;
R3 обозначает атом водорода или метильную группу;
R4 - атом водорода;
R5 - атом водорода или группу общей формулы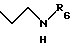
где R6 обозначает атом водорода, трет-бутилоксикарбонильную группу, 4-карбамоилпиримидин-2-ил группу или 5-карбамоилпиразин-2-ил группу.
Соединения согласно изобретению могут существовать в форме оснований или кислотно-аддитивных солей.
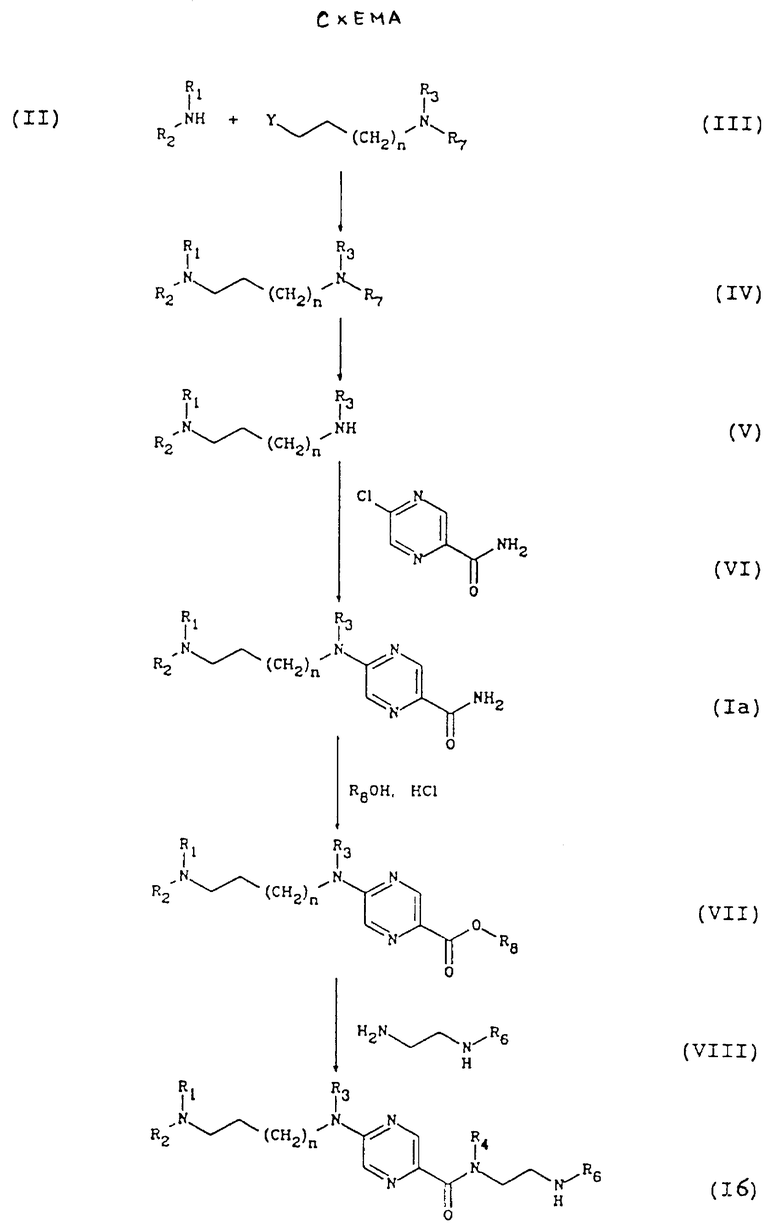
По изобретению, соединения общей формулы (I) могут быть получены по способу, представленному на схеме, представленной в конце описания
Амин общей формулы (II), где R1 и R2 описаны ранее, обычно в виде соли, взаимодействует с галогенизированным реагентом общей формулы (III), где Y обозначает атом галогена, n - как определялось выше, и либо R3 - как определено выше, a R7 обозначает защитную группу амина, например, трифенилметил, либо R3 и R7 совместно образуют, в совокупности с атомом азота, к которому они присоединены, фталимидную группу, как описано в [1].
Реакция протекает в апротонном растворителе, например, диметилформамиде, в присутствии неорганического основания, например, карбоната калия, при 40 - 80oC.
В результате получают продукт общей формулы (IV), от концевого алкиламина которого отщепляют защитную группу, затем в случае, когда R является трифенилметильной группой, проводят обработку газообразной соляной кислотой в алифатическом спирте, например, метаноле, при 0 - 60oC, в случае, когда R3 и R7 образуют вместе фталимидную группу, проводят обработку, аналогичную описанной в вышеуказанной литературе, например, гидразином.
В результате получают амин общей формулы (V), который взаимодействует с 2-хлорпиразин-5-карбоксамидом с формулой (VI) в апротонном растворителе, например, N,N-диметилформамиде, в присутствии основания, например, карбоната калия, при температуре от 20 до 40oC, с образованием производного 2-аминопиразин-6-карбоксамида с общей формулой (Iа), которое соответствует общей формуле (I), где R4 и R5 каждый обозначают атом водорода.
Для получения соединений общей формулы (I), где R5 обозначает группу общей формулы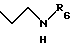
амид общей формулы (Iа), в которой n, R1, R2 и R3 определены выше, переводят в эфир с общей формулой (VII), где R8 обозначает C1-C4,-алкильную группу, путем реакции с С1-С4-алифатическим спиртом, например, метанолом, в присутствии кислоты, например, газообразной соляной кислоты, при 0 - 60oC, затем полученный таким образом эфир взаимодействует с диамином общей формулы (VIII), где R6 обозначает защитную группу амина, например, трет-бутилоксикарбонильную группу, в алифатическом спирте, например, метаноле или n-бутаноле, при температуре от 0 до 100oC, с образованием соединения общей формулы (Iб), в которой R обозначает трет-бутилоксикарбонильную группу.
Для получения соединений с общей формулой (I6), в которой R6 обозначает 4-карбамоилпиримидин-2-ил- либо 5-карбамоилпиразин-2-ил- группу, у полученного ранее соединения проводят отщепление радикала по известному способу, например, с трифторуксусной кислотой в дихлорметане, что приводит к образованию соединения общей формулы (Iб), где R6 обозначает водород, последнее взаимодействует с 2-хлорпиримидин-4-карбоксамидом либо 2-хлорпиразин-5-карбоксамидом, в апротонном растворителе, например, N,N-диметилформамиде, в присутствии основания, например, карбоната калия, при 20 - 40oC.
Амины общей формулы (II) могут быть получены по способам, аналогичным описанным в [2] для случая феноксиалкиламинов, [3] и [4] для случая феноксиметилпиперидинов.
Галогенизированный реагент общей формулы (III) является либо коммерчески доступным продуктом, в случае, когда R3 и R7 совместно образуют фталимидную группу, либо, в случае, когда R3 является H либо CH3, может быть получен способом, аналогичным описанному в [5].
2-Хлорпиразин-5-карбоксамид с формулой (VI) может быть получен способом, аналогичным описанному в [6,7,8,9].
2-Хлорпиримидин-4-карбоксамид может быть получен по способу, аналогичному описанному в [5].
Диамины с одной защитной группировкой общей формулы (VII) могут быть получены способами, аналогичными описанным в [10].
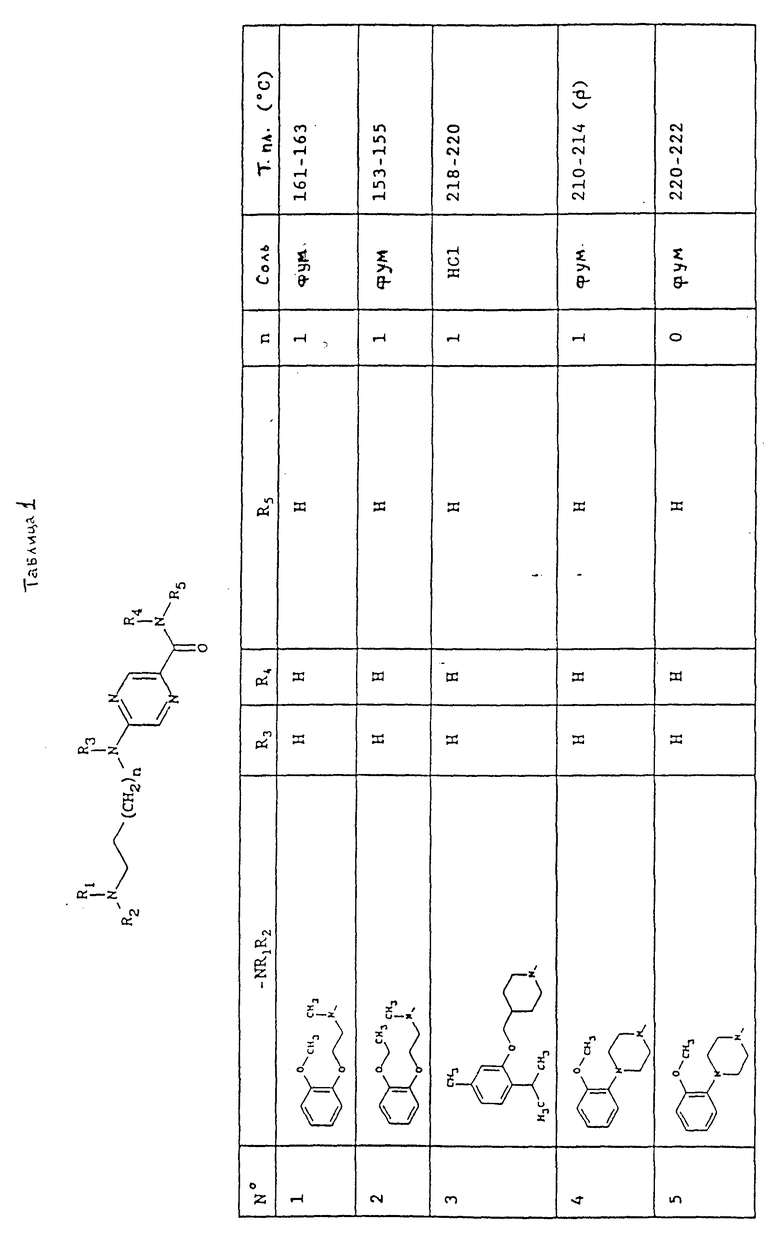
Следующие примеры подробно иллюстрируют получение отдельных соединений по изобретению. Элементные микроанализы и ЯМР и ИКР спектры подтверждают структуры полученных соединений.
Номера, указанные в скобках в заголовках, соответствуют номерам в первой колонке приводимой ниже таблицы.
Пример 1 (соединение N1).
2-[[3-[[2-(Метоксифенокси)этил] метиламино] пропил] -амино] пираэин-5-карбоксамид (Е)-бут-2-ендиоат (1:1).
1.1 N-[2-(2-Метоксифенокси)этил] -N-метил-N'-(трифенилметил-1, З-пропандиамин.
8,05 г (0,0370 моль) N-метил-2-(2-метоксифенокси)этиламин гидрохлорида, 15,5 г (0,0407 моль) N-трифенилметил-3-бромпропиламина (0,0925 моль) карбоната калия и 75 мл N,N-диметилформамида вводятся, под аргоном, в трехгорлую круглодонную колбу на 500 мл. Смесь перемешивается в течение 15,5 часов при 90oC. Реакционную смесь обрабатывают смесью воды со льдом и экстрагируют этилацетатом. Органическую фазу промывают водой, высушивают над сульфатом натрия и концентрируют под пониженным давлением.
В результате получают 18,2 г оранжевого масла, которое очищают путем хроматографии на силикагеле, в качестве элюента используют 98/2 смесь дихлорметан/метанол. Получают 13,7 г масла, используемого на следующей стадии.
1.2. N-[2-(2-Метоксифенокси)этил]-N-метил-1,3-пропандиамин.
12.9 г (0.0268 моль) N-[2-(2-метоксифенокси)этил]-N-метил-N'-(трифенилметил) -1,3-пропандиамина и 250 мл метанола смешивают в круглодонной колбе на 1 л. Пропускают струю газообразной соляной кислоты в течение 15 минут при охлаждении смесью воды и льда. Затем смесь нагревают до комнатной температуры, доводят до температуры дефлегмации и выдерживают в течение 7,5 часов. Смесь концентрируют до сухости, остаток переносят в этанол и снова концентрируют. Остаток переносят в воду, смесь подщелачивают, верхний слой масла переносят в разбавленную соляную кислоту и экстрагируют диэтиловым эфиром. Кислую водную фазу затем обрабатывают гидроксидом натрия до щелочного pH и проводят экстракцию дихлорметаном. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. В результате получают 5,6 г желтого масла, используемого на следующей стадии.
1.3. 2-[[3-[[2-(2-Meтоксифенокси)этил] -метиламино]-пропил]амино]-пиразин- 5-карбоксамид (Е)-бут-2-ендиоат.
5,0 г (0,021 моль) N-[2-(2-метоксифенокси)-этил]-N-метил-1,3-пропандиамина, 3,3 г (0,021 моль) 2-хлорпиразин-5-карбоксамида, 100 мл ацетонитрила и несколько кристаллов иодида натрия вводят под аргоном в круглодонную колбу на 250 мл. Добавляют 2,9 г ( 0,021 моль ) карбоната калия и снова выдерживают смесь при температуре дефлегмации в течение 30 часов.
Смесь охлаждают до комнатной температуры, осадок собирают посредством фильтрации и очищают хроматографически на колонке с силикагелем, в качестве элюата используют смесь дихлорметан/метанол от 100/0 до 90/10.
Полученную твердую фазу перекристаллизовывают из ацетонитрила и получают 2,82 г (0,00785 моль) основания.
Фумарат получают из 2,82 г основания, растворенного в 50 мл метанола путем добавления 0,91 г (0,00785 моль) фумаровой кислоты в раствор. Раствор концентрируют при пониженном давлении и проводят перекристаллизацию из этанола. Получают 3,32 г белого соединения.
Т. пл. 161-163oC.
Пример 2 (соединение N3).
2-[3-[4-[[5-метил-2-(1-метилэтил)фенокси] метил] -пиперид-1-ил] пропил]- амино]пиразин-5-карбоксамид гидрохлорид (1:1).
2.1. 2-[3-[4-[[5-Метил-2-(1-метилэтил)фенокси] -метил] пиперид -1-ил] пропил] -1H-изоиндол-1,3(2H)-дион.
11,35 г (0,04 моль) 4-[[5-метил-2-(1-метилэтил)фенокси]метил]пиперидин гидрохлорида, 10,72 r (0,04 моль) 2-(3-бромпропил)-1H-изоиндол-1,3(2H)-диона и 13,8 г (0,1 моль) карбоната калия взаимодействуют в 113 мл N,N-диметилформамида. Смесь перемешивают в течение 3 часов при 100oC. Ее вливают в охлажденную льдом воду. Раствор экстрагируют этилацетатом и промывают водой. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт используют на следующей стадии.
2.2 4-[[5-Метил-2-(1-метилэтил)фенокси] метил] -пиперидин-1-пропиламин. 17,35 г (0,04 моль) 2-[3-[4-[[5-метил-2-(1-метилэтил)фенокси]метил]пиперид-1-ил]пропил]- 1H-изоиндол-1,3(2H)-диона взаимодействуют в 340 мл этанола с 3,9 мл (0,08 моль) гидрата гидразина. Смесь прогревают при температуре дефлегмации в течение 3 часов. Смесь фильтруют, твердую фазу промывают небольшим количеством этанола, фильтрат концентрируют и переводят в диэтиловый эфир. Нерастворимую фазу снова отделяют путем фильтрации и фильтрат снова концентрируют. Нерастворимые фазы переносят в круглодонную колбу и добавляют к ней 25 мл концентрированной соляной кислоты и 75 мл воды. Смесь подвергают дефлегмации в течение 2 часов при перемешивании. Ее охлаждают, нерастворимую фазу отделяют путем фильтрации, промывают водой, проводят подщелачивание концентрированным водным аммиаком и осуществляют трехкратную экстракцию диэтиловым эфиром. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученное соединение используют на следующей стадии.
2.3. 2-[[3-[4-[[5-метил-2-(1-метилэтил)фенокси] -метил] пиперид-1-ил] пропил]амино]пиразин-5-карбоксамид гидрохлорид (1:1).
7,45 г (0,0245 моль) 4-[[5-метил-2-(1-метилэтил)фенокси]метил]пиперидин-1-пропиламина, 3,86 г (0,0246 моль) карбоната калия взаимодействуют в 100 мл ацетонитрила. Смесь прогревают в течение 28 часов при температуре дефлегмации и выпаривают растворитель при пониженном давлении.
Полученную твердую фазу очищают путем хроматографии на колонке с силикагелем, в качестве элюента используют смесь от 100/0 до 80/20 дихлорметан/метанол. Полученную твердую фазу перекристаллизовывают из этилацетата и получают 1,07 г (0,0025 моль) основания.
Гидрохлорид получают из 1,07 г основания, растворенного в 20 мл 2-пропанола, путем добавления 25 мл 0,1 N соляной кислоты в 2-пропаноле, затем растворитель выпаривают при пониженном давлении. Осадок перекристаллизовывают из 2-пропанола и получают 0,7 г белого вещества.
Т.пл. 218-220oC.
Пример 3 (соединение N5).
2-[[2-[4-(2-Метоксифенил)пиперазин-1-ил] этил] амино]-пиразин-5-карбоксамид (Е)-бут-2-ендиоат (1:1).
3.1. 2-[4-(2-Метоксифенил)пиперазин-1-ил]-N-(трифенилметил) этанамин.
10 г (0,273 моль) 2-бром-N-(трифенилметил)этанамина, 200 мл ацетонитрила, 5,15 г (0,0273 моль) 1-(2-метоксифенил)пиперазина, 5,6 г безводного карбоната калия, несколько кристаллов иодида натрия и 1 мл диметилформамида смешивают в круглодонной колбе емкостью 500 мл, снабженную дефлегматором, и помещают в атмосферу азота. Смесь выдерживают при температуре дефлегмации в течение 15 часов, растворители выпаривают, добавляют воду и дихлорметан, сушат над сульфатом натрия и выпаривают растворитель при пониженном давлении. В результате получают вязкое масло, очищают его посредством хроматографии на колонке с силикагелем, в качестве элюента используют смесь этилацетата и дихлорметана. Получают 9,24 г продукта, используемого на следующей стадии.
3.2. 2-[4-(-Метоксифенил)пиперазин-1-ил)-N-(трифенилметил)этанамин тригидрохлорид.
9,24 г 2-[4-(2-метоксифенил)пиперазин-1-ил)-N-(трифенилметил)этанамина растворяют в 400 мл метанола и, после гомогенизации, через раствор в течение 10 минут пропускают струю газообразной соляной кислоты. Осадок собирают, промывают метанолом и сушат под вакуумом. В результате получают 5,33 г белого вещества.
3.3. 2-[[2-[4-(2-метоксифенил)пиперазин-1-ил] этил] -амино] пираэин-5-карбоксамид (Е)-бут-2-ендиоат.
5,7 г (0,0242 моль) 2-[4-(2-метоксифенил)-пиперазин-1-ил]этанамина, 3,82 г (0,0242 моль) 2-хлорпиразин-5-карбоксамида, 200 мл ацетонитрила и 3,75 г (0,0242 моль) карбоната натрия смешивают в круглодонной колбе емкостью 500 мл, снабженную дефлегматором, и помещают в атмосферу азота. Смесь прогревают при температуре дефлегмации в течение 22 часов, охлаждают и выпаривают растворитель при пониженном давлении. Остаток очищают посредством хроматографии на колонке с силикагелем, в качестве элюента используют смесь от 100/0 до 85/15 дихлорметан/метанол, полученную твердую фазу перекристаллизовывают из этилацетата. В результате получают 0,97 г белого твердого вещества.
Т.пл.: 220-222oC.
Пример 4 (соединение N12).
2-[[3-[4-(2-Циклопропилфенил)пиперазин-1-ил] пропил] -метиламино] пиразин-5-карбоксамид (Е)-бут-2-ендиоат (1:1).
4.1. 3-[4-(2-Циклопропилфенил)пиперазин-1-ил)-N-метилпропанамин тригидрохлорид.
9,0 г (0,0444 моль) 1-(2-циклопропилфенил)пиперазина, 200 мл диметилформамида, 17,5 г (0,0444 моль) 3-бром-N-(трифенилметил)пропанамина и 9 г карбоната калия смешивают в круглодонной колбе емкостью 500 мл, снабженной дефлегматором, помещают в атмосферу азота, смесь трижды по 6 часов прогревают при температуре 96oC. Растворитель выпаривают при пониженном давлении, остаток извлекают водой и дихлорметаном, органическую фазу отделяют, промывают водой, сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении. В результате получают 4,17 г 3-[4-(2-циклопропилфенил)пиперазин-1-ил] -N-метил-N-(трифенилметил) пропанамина в виде масла, растворяют его в 200 мл метанола, пропускают через него в течение 10 минут поток газообразной соляной кислоты, смесь концентрируют, выдерживают в течение 2 дней и отделяют осадок путем фильтрации. В результате получают 2,94 г соединения.
4.2. 2-[[3-[4-(2-Циклопропиламино] пиразин-5-карбоксамид (Е)-бут-2-ендиоат (1:1).
3,77 г (0,0138 моль) 3-[4-(2-циклопропилфенил)пиперазин-1-ил]-N-метилпропанамина, 2,17 г (0,0138 моль) 2-хлорпиразин-5-карбоксамида, 1,9 г (0,0138 моль) карбоната калия и 100 мл ацетонитрила вводят в круглодонную колбу емкостью 500 мл, снабженную дефлегматором, и помещают в атмосферу азота. Смесь прогревают при температуре дефлегмации в течение 18 часов, охлаждают, растворитель выпаривают при пониженном давлении, а остаток очищают путем хроматографии на колонке с силикагелем, в качестве элюента используют смесь от 100/0 до 90/10 дихлорметан/метанол. Полученное вещество перекристаллизовывают из этилацетата и получают 2,37 г (0,006 моль) основания.
Фумарат получают из 2,37 г основания, растворенного в 50 мл метанола, и 0,7 г (0,06 моль) фумаровой кислоты, растворенной в 50 мл метанола. Смесь концентрируют при пониженном давлении и кристаллизуют продукт. В результате получают 1,7 г белого вещества.
Т.пл.: 184-186oC.
Пример 5 (соединение N10).
2-[[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил] пропил] амино] пиразин-5-карбоксамид (Е)-бут-2-ендиоат (1:1).
5.1. 2-[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил]-пропил]-1H-изоиндол- 1,3(2H)-дион.
17,16 г (0,05236 моль) 1-(5-хлор-2-метоксифенил)пиперазин (Е)-бут-2-ендиоата (1: 1), 14,04 г (0,05236 моль) 2-(3-бромпропил)-1H-изоиндол-1,3(2H)-диона и 7,24 г (0,05236 моль) карбоната калия в виде суспензии в 150 мл диметилформамида вносят в круглодонную колбу емкостью 500 мл и прогревают смесь в течение 4 часов при 90oC. Реакционную смесь вливают в 300 мл воды и проводят экстракцию этилацетатом (2•150 мл). Органическую фазу промывают водой (3•150 мл), затем высушивают над сульфатом натрия, фильтруют и выпаривают растворители под пониженным давлением. Сырой остаток перекристаллизовывают из диэтилового эфира и в результате получают 14,7 г твердого вещества.
Т.пл. 130-131oC.
5.2. 3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил]-пропанамин.
19,7 г (0,05105 моль) 2-[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил]пропил] -1H-изоиндол- 1,3(2H)-диона, растворенного в 300 мл этанола, вносят в круглодонную колбу емкостью 1 л, затем туда добавляют 5,11 г (0,1021 моль) гидрата гидразина и смесь прогревают при температуре дефлегмации в течение 4 часов. Растворитель выпаривают при пониженном давлении, затем к сырому остатку добавляют 100 мл воды и 17 мл концентрированной соляной кислоты и вновь проводят прогревание в течение 3 часов при температуре дефлегмации растворителя.
Нерастворимую фракцию отделяют путем фильтрации, фильтрат подщелачивают 30%-ным раствором гидроксида натрия, а затем экстрагируют дихлорметаном. Органическую фазу промывают водой, сушат над сульфатом натрия, фильтруют, а растворители затем выпаривают под пониженным давлением и в результате получают 13,76 г масла, используемого на следующей стадии.
5.3. 2-[[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил] пропил]амино]пиразин- 5-карбоксамид (Е)-бут-2-ендиоат (1:1).
13,67 г ( 0,04817 моль) 3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил] пропанамина, 8,65 г (0,062 моль) карбоната калия и 7,59 г (0,04817 моль) 2-хлорпиразин-5-карбоксамида в виде суспензии в 200 мл диметилформамида вносят в круглодонную колбу емкостью 500 мл, смесь выдерживают при комнатной температуре в течение 48 часов при непрерывном перемешивании.
Растворитель выпаривают при пониженном давлении, остаток очищают путем перекристаллизации из этилацетата и в результате получают 12,6 г основании.
Фумарат получают из 1,58 г (0,039 моль) основания, растворенного в 50 мл этанола, и 0,47 г (0,039 моль) фумаровой кислоты, растворенной в 50 мл этанола. Смесь концентрируют и продукт перекристаллизовывают из смеси метанол/метанол. В результате получают 1,08 г (0,00207 моль) белого порошка.
Т.пл.: 219-223oC (разложение).
Пример 6 ( Соединение N13).
1 1,1-Диметилэтил 2-[[[2-[[3-[4-(5-Хлор-2-метоксифенил) пиперазин-1-ил] пропил]амино]пиразин-5-карбоксилат.
6.1 Метил 2-[[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил]пропил]амино] пиразин -5-карбоксилат.
9,7 г (0,024 моль) 2-[[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил] пропил]амино]пиразин-5-карбоксамида, растворенного в 400 мл метанола, вносят в круглодонную колбу емкостью 1 л, в течение нескольких минут пропускают струю газообразной соляной кислоты, а затем проводят прогревание в течение 5 часов при температуре дефлегмации метанола.
Растворитель выпаривают при пониженном давлении, к остатку добавляют 200 мл дихлорметана и охлаждают полученную смесь до 0oC. Затем смесь подщелачивают насыщенным водным раствором гидрокарбоната натрия, проводят ее разделение путем отстаивания, органическую фазу высушивают над сульфатом натрия, фильтруют, а растворитель выпаривают при пониженном давлении.
После хроматографии на колонке с силикагелем (элюент: от 100/0 до 90/10 смесь дихлорметан/метанол) с последующей перекристаллизацией из циклогексана получают 8,47 г (0,020 моль) соединения.
Т.пл.: 120-122oC.
6.2. 1,1-Диметилэтил 2-[[[2-[[3-[4-(5-Хлор-2-метоксифенил)пиперазин-1-ил]пропил] амино]-пиразин-5-ил] карбонил]амино]этилкарбамат.
4 г (0,0095 моль) метил 2-[[3-[4-(5-Хлор-2-метоксифенил) пиперазин-1-ил] пропил] амино] -пиразин-5-карбоксилата и 3,05 г (0,02 моль) 1,1-диметилэтил 2-аминоэтилкарбамата растворяют в 10 мл 2-пропанола в круглодонной колбе емкостью 500 мл и смесь прогревают при температуре дефлегмации в течение 2 дней.
Растворитель выпаривают при пониженном давлении, в результате очистки с помощью хроматографии на колонке с силикагелем (элюент: от л 100/0 до 90/10 дихлорметан/метанол) получают желтое масло, которое кристаллизуют путем растирания в порошок в диэтиловом эфире. В результате получают 1,5 г (0,00274 моль) соединения.
Т.пл.: 159-161oC.
Пример 7 (соединение N14).
N-(2-аминоэтил)-2-[[3-[4-(5-Хлор-2-метоксифенил)-пиперазин-1-ил] пропил] амино]пиразин-5-карбоксамид.
2 г (0,00365 моль) 1,1-диметилэтил 2-[[2-[[3-[4-(5-хлор-2- метоксифенил)пиперазин-1-ил] пропил] амино]пиразин-5-ил]карбонил] амино]этилкарбамата растворяют в 10 мл воды в круглодонной колбе емкостью 0,25 л, а затем по каплям добавляют 10 мл концентрированной соляной кислоты. Смесь охлаждают до 0oC смесью лед/соль/вода и порциями добавляют 30% раствор гидроксида натрия, до тех пор, пока рН не приобретет щелочное значение. Проводят экстракцию дихлорметаном, органическую фазу сушат над сульфатом натрия, фильтруют, растворители выпаривают при пониженном давлении и в результате получают 1,32 г (0,00295 моль) аморфного вещества.
Т.пл.: 45-55oC.
Пример 8 (соединение N15 ).
N-[2-[[4-(Аминокарбонил)пиримидин-2-ил] амино] этил]-2-[[3- [4-(5-хлор-2-метоксифенил)пиперазин-1-ил]-пропил]амино] пиразин-5-карбоксамид.
1,32 г (0,00295 моль) N-(2-аминоэтил)-2-[[3-[4-(5-хлор-2- метоксифенил)пиперазин-1-ил] -пропил]амино]пиразин-5-карбоксамида, 0,5 г (0,00317 моль) 2-хлорпиримидин-4-карбоксамида и 0,6 г (0,00434 моль) карбоната калия растворяют в 50 мл диметилформамида в круглодонной колбе емкостью 0,25 л, смесь прогревают при 40oC в течение 40 часов.
Растворитель выпаривают при пониженном давлении, а сырой остаток очищают с помощью хроматографии на колонке с силикагелем, в качестве элюента используют смесь от 98/2 до 80/20 дихлорметан/метанол. После перекристаллизации из ацетона получают 0,99 г (0,00174 моль) соединения.
Т.пл.: 197-199oC.
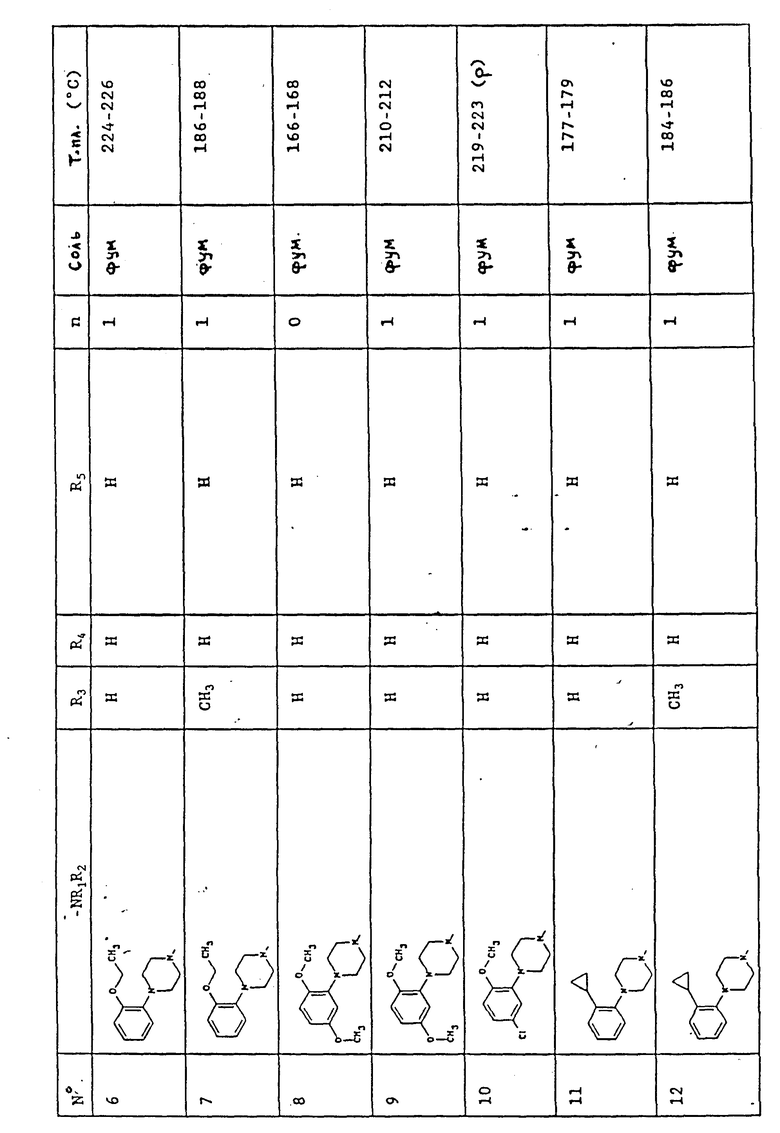
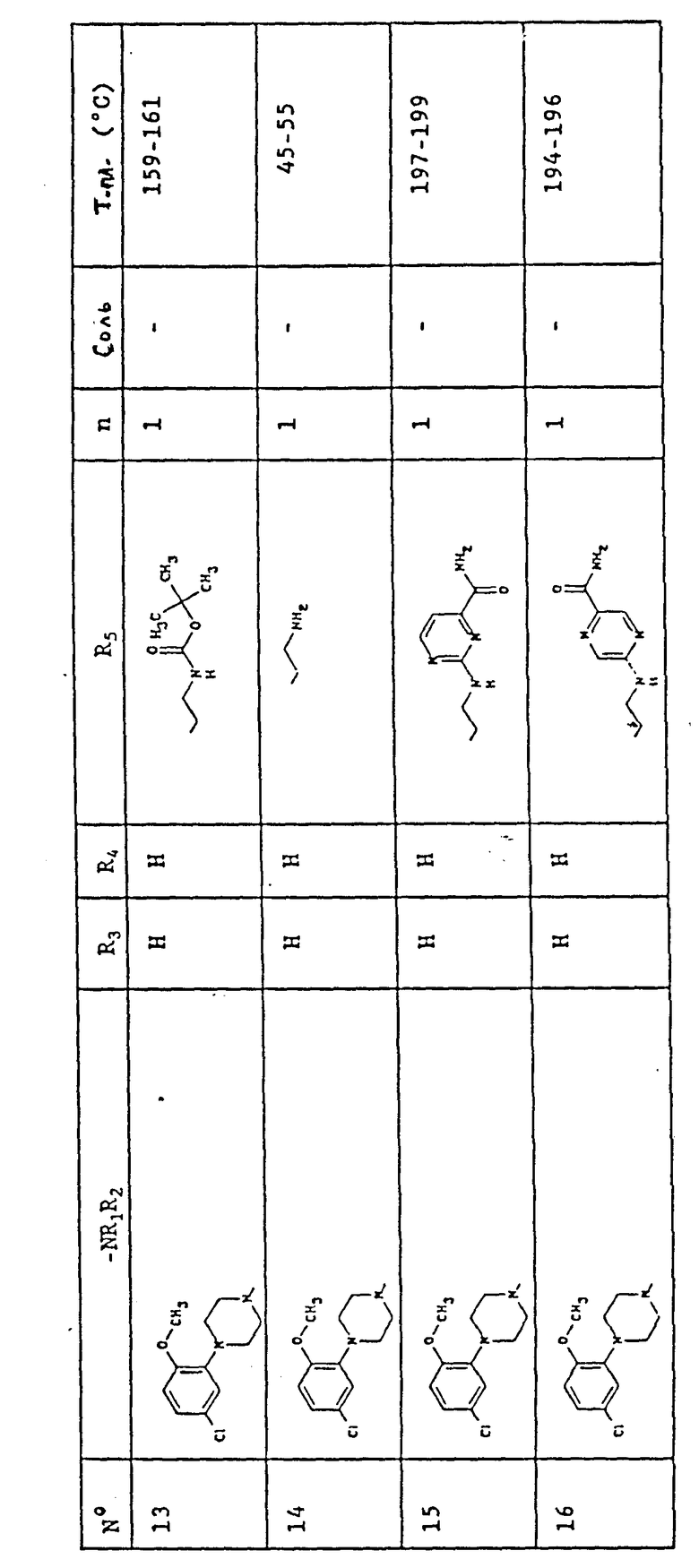
В табл. 1 представлены химические структуры и физические свойства некоторых соединений по изобретению.
Пояснения к табл. 1:
В колонке "Соль", "фум" обозначает (Е)-бут-2-ендиоат (1:1) (фумарат), "HCl" обозначает гидрохлорид (1:1) и "-" обозначает, соединение в основной форме. В колонке "Т.пл. (oC)" "р" обозначает температуру плавления с разложением.
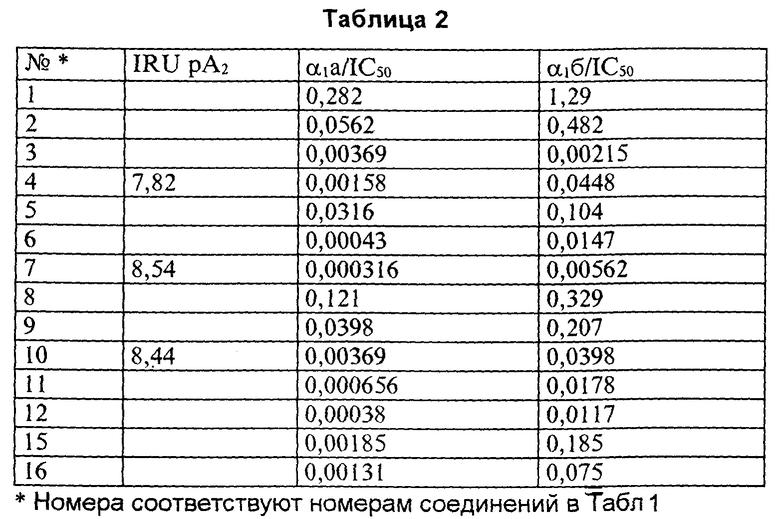
Соединения по изобретению явились предметом исследований относительно их антагонистической активности по отношению к α1 адренергическим рецепторам на уровне нижнего мочевого аппарата.
Их активность in vitro изучали на изолированной кроличьей уретре (IRU).
Уретральные кольца взрослых самцов кроликов обрабатывали по способу [11] , а затем, после сенсибилизации к норадреналину, снимали кривую зависимости реакции на фенилефрин от его концентрации в отсутствии и в присутствии исследуемого соединения.
Величину α1 адренергического антагонизма каждого соединения определяли путем вычисления рА2, антилогарифма молярной концентрации антагониста, при наличии которой концентрация агониста должна быть удвоена, чтобы вызвать тот же эффект, что и в его отсутствие.
Значения рА2 соединений представлены в табл. 2 в графе IRU рА2.
Сродство к подгруппам α1 а и α1 б α1- рецепторов исследовали на слюнных железах крысы α1 а) и на печени крысы α1 б).
Исследования проводили согласно C.Faure et al. European Journal of Pharmacology, Molecular Pharmacological section, 268 (1994), 141-149.
1. Получение тканевой мембраны.
Мембраны из слюнной железы крысы и печени крысы были получены гомогенизацией и центрифугированием дважды при 37000 g в течение 10 мин в 50 мас.% Tris-HCI буфере, pH 7,4.
2. Связывание радиолиганда.
Исследования связывания лиганда проводили в 50 мМ Tris-HCI буфере, pH 7,4. Препараты клеточных мембран (4 г prot) инкубировали с [3H]-празозином при 25oC в течение 30 мин при общем объеме 2 мл. Тканевые мембраны инкубировали с [3H]-празозином в течение 1 ч при 25oC при общем объеме 2 мл. Фентоламин (10 мкМ) использовали для определения неспецифического связывания. Инкубирования заканчивали скоростной фильтрацией, используя коллектор клеток Бранделла, через стекловолокнистые фильтры Whatman GF/B. Фильтры промывали 2х25 мл буфера, сушили и измеряли радиактивность посредством жидкостной сцинтилляционной спектрометрии. Данные насыщения и конкурентного ингибирования анализировали с использованием программ нелинейного регрессионного анализа. Результаты выражали как 1С50) (концентрация соединения, при которой происходит ингибирование связывания радиолиганда на 50%). Результаты приведены в табл. 2.
In vivo активность соединений по изобретению исследовали по их воздействию на уретральную гипертонию, вызываемую стимуляцией симпатических волокон подчревного нерва у анестезированных котов.
Взрослых котов анестезировали пентабарбиталом натрия, обрабатывали по методу [12] , вызывая уретральную гипертонию путем стимуляции симпатических волокон подчревного нерва. До и после внутривенного введения исследуемых соединений в кумулятивных дозах от 1 до 1000 мкг/кг регистрировали сокращения уретры в ответ на электрическую стимуляцию подчревного нерва.
Величину α1- адренергического антагонизма каждого соединения оценивали путем вычисления ID50 дозы, которая ингибирует уретральную гипертонию на 50%.
Значения ID50 соединений по изобретению находятся в интервале от 0,001 до 1 мг/кг.
Результаты тестов показывают, что соединения по изобретению проявляют in vitro антагонистическую активность по отношению к адренергическим рецепторам гладких мускулов нижнего мочевого аппарата (уретры), стимулированных α1 адренергическим агонистом (фенилефрином). In vivo они ингибируют уретральную гипертонию, вызываемую стимуляцией симпатического нерва.
Соединения по изобретению можно использовать для симптоматического применения при заболеваниях и жалобах, которые включают в себя гиперактивность α1 адренергической системы на уровне нижнего мочевого аппарата, и особенно для лечения уринарных расстройств, связанных с доброкачественной гипертрофией простаты, таких как дизурия и поллакиурия.
Наконец, их можно использовать во всех формах, приемлемых для энтерального и парентерального введения, в сочетании с фармацевтическими эксципиентами, например в форме таблеток, покрытых сахаром пилюль, капсул, включая желатиновые капсулы, растворов или суспензий для питья или инъекций, или суппозиториев, причем в таком количестве, чтобы обеспечить суточную дозу от 0,1 до 500 мг активного вещества. Ниже приведены примеры фармацевтических композиций, подходящих для соединений по настоящему изобретению. Для их получения используют традиционные способы.
Получение гранулята для капсул или таблеток
Ингредиенты, %:
Активное вещество - 0,1 - 20
Лактоза - 40 - 80
Гидроксипропилметилцеллюлоза - 5 - 10
Микрокристаллическая целлюлоза - 15 - 25
Карбоксиметилкрахмал, натриевая соль - 2 - 4
Коллоидный диоксид кремния - 0,1 - 0,3
Стеарат магния - 2 - 3 - 100%
Раствор для инъекций
Ингредиенты, мг:
Активное вещество - 4 - 6 мг
Глюкоза - 250
Апирогенная вода для инъекций в количестве, достаточном, чтобы довести объем до, мл - 5
Флакон, мл - 5,
Изобретение решает задачу создания новых соединений, обладающих антагонистической активностью по отношению к α1- адренергическим рецепторам на уровне нижнего мочевого аппарата. Производные 2-аминопиразин-5-карбоксамида общей формулы I, где n = 0 или 1, R1 обозначает метильную группу, и в этом случае R2 обозначает фенокси(С1-С4)-алкильную группу, в которой феноксигруппа замещена метокси- и этоксигруппами, или R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 4-(феноксиметил)пиперид-1-илгруппу, в которой феноксигруппа замещена одной или двумя (С1-С4)-алкильными группами, или 4-фенилпиперазин-1-илгруппу, в которой фенильная группа замещена одним или двумя заместителями, такими как галоген, метокси, этокси и (С1-С4)-алкильная группа; R3 обозначает атом водорода или метильную группу; R4 - атом водорода, R5 - атом водорода или группа общей формулы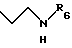
где R6 обозначает атом водорода, трет-бутилоксикарбонильную группу, 4-карбомоилпиримидин-2-илгруппу или 5-карбамоилпиразин-2-илгруппу, в форме основания или кислотно-аддитивной соли. Описан способ получения соединений формулы I, а также медицинский препарат и фармацевтическая композиция, содержащие соединения формулы I, обладающие вышеуказанной активностью. 5 с.п. ф-лы, 1 ил., 2 табл.
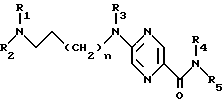
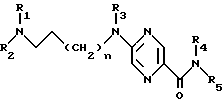
где n = 0 или 1;
R1 обозначает метильную группу и в этом случае R2 обозначает фенокси (С1-С4) алкильную группу, в которой феноксигруппа замещена метокси- и этоксигруппами, или R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 4-(феноксиметил)пиперид-1-илгруппу, в которой феноксигруппа замещена одной или двумя (С1-С4) алкильными группами, или 4-фенилпиперазин-1-илгруппу, в которой фенильная группа замещена одним или двумя заместителями, такими, как галоген, метокси, этокси и (С1-С4) алкильная группа;
R3 обозначает атом водорода или метильную группу;
R4 - атом водорода,
R5 - атом водорода или группу общей формулы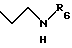
где R6 обозначает атом водорода, трет-бутилоксикарбонильную группу, 4-карбамоилпиримидин-2-илгруппу или 5-карбамоилпиразин-2-илгруппу,
в форме основания или кислотно-аддитивной соли.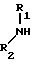
где R1 и R2 определены выше,
обычно в виде соли, подвергают взаимодействию с галогенизированным реагентом общей формулы III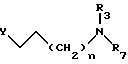
где Y - атом галогена;
n определено в п.1;
R3 либо как определено в п.1, а R7 обозначает трифенилметильную группу, либо R3 и R7 вместе образуют совместно с атомом азота, к которому они присоединены, фталимидную группу,
в апротонном растворителе в присутствии неорганического основания при 40 - 80oC с образованием диамина общей формулы IV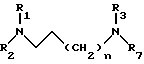
концевой алкиламин которого лишен защитной группы, затем в случае, когда R7 является трифенилметильной группой, проводят обработку полученного диамина газообразной соляной кислотой в алифатическом спирте при 0 - 60oC, а когда R3 и R7 совместно образуют фталимидную группу, проводят обработку гидразином, получая амин общей формулы V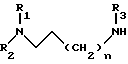
который подвергают взаимодействию с 2-хлорпиразин-5-карбоксамидом в апротонном растворителе в присутствии основания при 20 - 40oC с образованием производного 2-аминопиразин-5-карбоксамида общей формулы Iа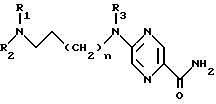
которая соответствует общей формуле I,
где R4 и R5 - атом водорода,
затем при необходимости, чтобы получить соединение общей формулы I, где R5 обозначает группу общей формулы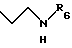
амид общей формулы Iа переводят в эфир общей формулы VII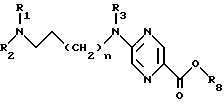
где R8 обозначает С1-С4 алкильную группу,
путем проведения реакции с С1-С4 -алифатическим спиртом в присутствии кислоты при 0 - 60oC, после чего полученный таким образом эфир подвергают взаимодействию с диамином общей формулы VIII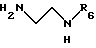
где R6 обозначает трет-бутилоксикарбонильную группу,
в алифатическом спирте при 0 - 100oC с получением соединения общей формулы Iб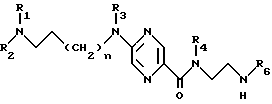
где R6 обозначает трет-бутилоксикарбонильную группу,
затем при необходимости, чтобы получить соединения общей формулы Iб, где R6 обозначает 4-карбамоилпиримидин-2-ил или 5-карбамоилпиразин-2-илгруппу, от полученного соединения отщепляют защитную группу с помощью трифторуксусной кислоты в дихлорметане, что приводит к получению соединения общей формулы Iб, где R6 - водород, которое подвергают взаимодействию с 2-хлорпиримидин-4-карбоксамидом или 2-хлорпиразин-5-карбоксамидом в апротонном растворителе в присутствии основания при 20 - 40oC. адренергическим рецепторам, отличающаяся тем, что она содержит 0,1 - 500,0 мг соединения по п.1 в сочетании с инертным наполнителем.
адренергическим рецепторам, отличающаяся тем, что она содержит 0,1 - 500,0 мг соединения по п.1 в сочетании с инертным наполнителем.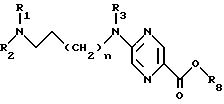
где n, R1, R2 и R3 определены в п.1;
R8 обозначает С1-С4 -алкильную группу,
как важный промежуточный продукт способа по п.2.
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч.1, с.271-274 | |||
| SU, 1627084 A3, 1991 | |||
| EP, 0435749 A, 1992. |
