Изобретение относится к новым замещенным 1-фенилпиразол-3-карбоксамидам, обладающим высоким сродством к рецепторам нейротензина человека, к способу их получения и к содержащим их в качестве действующих начал фармацевтическим композициям.
Первые потенциальные синтетические лекарственные средства непептидного типа, способные связываться с рецепторами нейротензина, описаны в европейском патенте 0477049. Речь идет об амидах пиразол-3-карбоновой кислоты, в которых аминокислоты по-разному замещены, которые вытесняют иодированный нейротензин из его рецептора, в дозах ниже микромоля, согласно опыту с мембранами головного мозга морской свинки. К этому классу веществ относится и соединение 2-[(1-(7-хлор-4-хинолил)-5-(2,6-диметоксифенил) пиразол-3-ил)карбониламино]адамантан-2-карбоновая кислота (SR 48692), обладающее высокой и селективной антагонистической активностью по отношению к нейротензину (D. Gully и др., Proc. Natl. Acad. Sci. USA, 90, 65-69 (1993)).
Характерной чертой этого класса соединений, описанной в европейском патенте 0477049, является наличие в положении 1 пиразольного цикла заместителя, в частности фенильной, нафтильной, 4-хинолильной группы, которые замещены или не замещены. Более конкретно, соединение SR 48692 в положении 1 пиразола содержит 7-хлор-4-хинолильную группу. Описанные в указанном патенте соединения, содержащие 1-нафтильную или 4-хлор-1-нафтильную группу в положении 1 пиразольного цикла, обладают чрезвычайно высоким сродством к рецептору нейротензина в случае морской свинки, так как их ИК50 составляет величину порядка 1-10 наномоль, тогда как их сродство к человеческому рецептору меньше, поскольку их ИК50 составляет величину 10-100 нмоль.
В настоящее время найдено, что, замещая фенильную группу в 1-фенилпиразол-3-карбоксамидах особыми группами, можно увеличить сродство к рецепторам нейротензина и, особенно, увеличить сродство к человеческим рецепторам нейротензина.
Кроме того, соединения по изобретению ин витро показывают более широкий спектр активности, чем соединения, описанные в европейском патенте 0477049, в качестве антагонистов рецепторов нейротензина.
Таким образом, изобретение относится, согласно одному из его объектов, к новым замещенным 1-фенилпиразол-3-карбоксамидам формулы (Iа):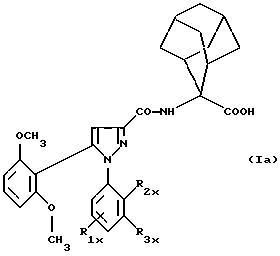
в которой R1x находится в положении 4 или 5 и обозначает группу -T-CONRaRb, в которой Т обозначает прямую связь или (C1-C7)-алкилен, NRaRb обозначает группу, выбранную из:
-NR9(CH2)sCR7R8(CH2)tNR5R6;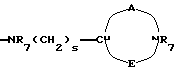
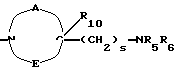
где R5 и R6 обозначают, независимо друг от друга, водород, (С1-С6)-алкил, (С3-С8)-алкенил или R5 и R6 вместе с атомом азота, с которым они связаны, обозначают гетероцикл, выбранный из пирролидина, пиперидина, морфолина, пиперазина, замещенного в положении 4 заместителем R9;
R7 обозначает водород, (C1-C4)-алкил или бензил;
R8 обозначает водород, (C1-C4)-алкил или R7 и R8 вместе с атомом углерода, с которым они связаны, образуют (С3-С5)-циклоалкан;
R9 обозначает водород, (C1-C4)-алкил, бензил или группу -X-NR'5R'6, где R'5 и R'6 обозначают, независимо друг от друга, (C1-C6)-алкил;
R10 обозначает водород, (C1-C4)-алкил;
s=0-3;
t=0-3 при условии, что (s+t) в одной и той же группе больше или равно 1;
двухвалентные радикалы А и Е вместе с атомом углерода и атомом азота, с которыми они связаны, образуют насыщенный гетероцикл, имеющий от 4 до 7 звеньев, который, кроме того, может быть замещен одним или несколькими (C1-C4)-алкилами;
R2x и R3x обозначают, независимо друг от друга, водород, (C1-С6)-алкил, (С3-С8)-циклоалкил, (С3-С8)-циклоалкилметил при условии, что R2x и R3x не обозначают одновременно водород, или R2x и R3x вместе образуют тетраметиленовую группу;
и их фармацевтически приемлемым солям.
Соли соединений согласно изобретению могут быть внутренними солями или солями со щелочными металлами, предпочтительно солями натрия или калия, со щелочноземельными металлами, предпочтительно солями кальция и солями с органическими основаниями, такими как диэтиламин, триметиламин, N-метил-D-глюкамин, лизин, аргинин, гистидин, холин или диэтаноламин, или солями с органическими оптически чистыми основаниями, такими как α-метилбензиламин.
Соли соединений формулы (I) согласно изобретению также представляют собой соли с неорганическими или органическими кислотами, которые позволяют осуществлять соответствующее разделение или кристаллизацию соединений формулы (I), с такими как пикриновая кислота, щавелевая кислота или оптически активная кислота, например миндальная кислота или камфорсульфокислота, и предпочтительно с такими, которые образуют фармацевтически приемлемые соли, такие как гидрохлорид, ацетат, гидросульфат, дигидрофосфат, метансульфонат, малеат, фумарат, 2-нафталинсульфонат, изетионат, бензолсульфонат, п-толуолсульфонат, тартрат, цитрат, этандисульфонат.
Предпочтительно изобретение относится к соединениям формулы (Iа), в которой:
Т обозначает прямую связь;
NRaRb обозначает: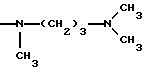
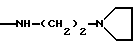
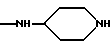
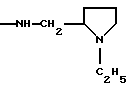
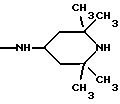
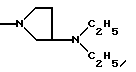
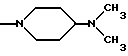
Одним из предпочтительных соединений по изобретению являются 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N- (3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота, ее внутренняя соль и ее фармацевтически приемлемые соли.
Изобретение относится, согласно другому из его объектов, к способу получения замещенных 1-фенилпиразол-3-карбоксамидов формулы (I) и их солей, заключающемуся в том, что:
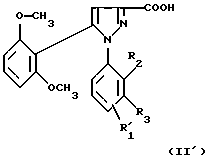
1) функциональное производное 1-фенилпиразол-3-карбоновой кислоты формулы (II) или (II'):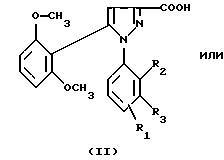
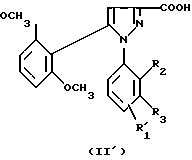
в которой R1, R2, R3 имеют значения, соответствующие значениям R1x, R2x и R3x, указанным для соединений формулы (Iа), а R'1 означает предшественник R1, выбираемый среди карбоксила, (C1-C4)-алкоксикарбонила, бензилоксикарбонила, обрабатывают аминокислотой, возможно защищенной с помощью обычных в пептидном синтезе защитных групп, формулы (III):
H-NН-AA(OH), (III)
в которой -NH-AA(OH) обозначает группу формулы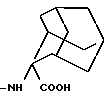
2) в случае необходимости, таким образом полученное функциональное производное кислоты формулы (I')
подвергают затем соответствующей обработке для превращения заместителя R'1, являющегося предшественником R1, в заместитель R1;
3) возможно, в полученном на стадии 1) или на стадии 2) соединении удаляют защитную группу с целью получения соответствующей свободной кислоты формулы (I)
4) и, при необходимости, получают соль таким образом полученного соединения формулы (I).
В качестве функционального производного 1-фенилпиразол-3-карбоновой кислоты формулы (II) или (II') можно использовать хлорангидрид, смешанный ангидрид с хлорформиатом изобутила или этила, (C1-C4)-алкиловый сложный эфир.
Аминокислоты формулы (III) могут быть использованы либо такими, какие есть, либо после предварительной зашиты карбоксильной группы с помощью обычных в пептидном синтезе групп, как описано, например, в руководстве "Защитные группы в органической химии", изд. J.F.W. McOmie, Plenum Press, 1973, с. 183 или в руководстве "Защитные группы в органическом синтезе", II-е издание, J. F.W. Greene и P.G.M. Wuts, John Willey and Sons, 1991, с. 224.
Так, на стадии 1) способа можно хлорангидрид 1-фенилпиразол-3-карбоновой кислоты, полученный путем реакции тионилхлорида с кислотой формулы (II) или (II'), ввести во взаимодействие с аминокислотой формулы (III) в растворителе, таком как ацетонитрил, тетрагидрофуран, диметилформамид или дихлорметан, в атмосфере инертного газа при комнатной температуре в течение времени от нескольких часов до нескольких дней в присутствии основания, такого как пиридин, гидроксид натрия или триэтиламин.
Один вариант осуществления стадии 1) состоит в получении хлорангидрида или смешанного ангидрида 1-фенилпиразол-3-карбоновой кислоты путем реакции изобутил- или этилхлорформиата с кислотой формулы (II) или (II') в присутствии основания, такого как триэтиламин, и во введении его во взаимодействие с N, O-бистриметилсилильным производным аминокислоты формулы (III), полученным путем реакции бис(триметилсилил)ацетамида или 1,3-бис(триметилсилил)мочевины, или бис(трифторметилсилил)ацетамида с аминокислотой формулы (III), в растворителях, таких как ацетонитрил, дихлорметан, в инертной атмосфере в течение времени от 1 часа до нескольких дней при температуре от комнатной до температуры кипения с обратным холодильником растворителя.
Другой вариант осуществления стадии 1) состоит во введении во взаимодействие смешанного ангидрида 1-фенилпиразол-3-карбоновой кислоты формулы (II) или (II') с аминокислотой формулы (III) в растворителе, таком как дихлорметан, в инертной атмосфере при комнатной температуре в течение времени от 1 дня до нескольких дней в присутствии основания, такого как триэтиламин.
Когда соединение формулы (I) содержит основную функцию и получается в форме свободного основания, солеобразование осуществляют путем обработки с помощью выбранной кислоты в органическом или водном растворителе. Путем обработки свободного основания, растворенного, например, в спирте, таком как изопропанол, с помощью раствора выбранной кислоты, в том же растворителе, получают соответствующую соль, которую выделяют обычными способами. Так, например, получают гидрохлорид, гидробромид, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, оксалат, малеат, фумарат, 2-нафталинсульфонат.
Когда соединение формулы (I) содержит основную функцию и его выделяют в форме одной из его солей, например в виде гидрохлорида или оксалата, свободное основание можно получать путем нейтрализации вышеуказанной соли с помощью неорганического или органического основания, такого как гидроксид натрия или триэтиламин, или с помощью карбоната или гидрокарбоната щелочного металла, такого как карбонат или гидрокарбонат натрия или калия.
Когда продукт формулы (I) получают в кислой форме, его можно превращать в соль металла, в частности щелочного металла, такую как соль натрия, или щелочноземельного металла, такую как соль кальция, согласно обычным способам.
Соединения формулы (Iа) можно подвергать дегидратации в присутствием ангидрида, например уксусного ангидрида, с целью получения производного оксазолона формулы (Iс):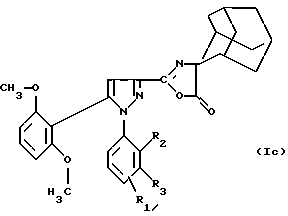
в которой R1 находится в положении 4 или 5 и обозначает группу -T-CONRaRb, в которой Т обозначает прямую связь или (C1-C7)-алкилен и NRaRb обозначает группу -NR9(CH2)sCR7R8(CH2)tNR5R6;
R2 обозначает (С1-С6)-алкил,
R3 обозначает водород,
R5 и R6 обозначают (С1-С6)-алкил;
R7 и R8 обозначают водород;
s равно числу от 0 до 3;
t равно числу от 0 до 3 при условии, что сумма (s+t) больше или равна 1.
Исходя из соединения формулы (Iс), снова получают соединение формулы (Iа) путем гидролиза в кислой или щелочной среде, например, в присутствии соли щелочного металла, такой как трет-бутилат калия.
Промежуточное получение соединения (Iс) может быть пригодным для проведения очистки соединения формулы (la).
1-Фенилпиразол-3-карбоновая кислота формулы (II) или (II'):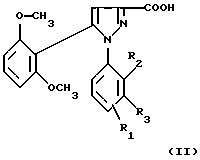
в которой R1, R2, R3 имеют значения, соответствующие значениям R1x, R2x, R3x, указанным для соединений формулы (Iа), а R'1 означает предшественник радикала R1, выбираемый среди карбоксила, (C1-C4)-алкоксикарбонила, бензилоксикарбонила, а также ее функциональные производные, выбранные из смешанного ангидрида с хлорформиатом изобутила или этила, хлорангидрида и сложного (C1-C4)-алкилового эфира, являются новыми соединениями и составляют отдельный предмет изобретения.
Кислоты формулы (II) и формулы (II'), хлорангидриды кислот формул (II) и (II'), (C1-C4)-алкиловые эфиры кислот формул (II) и (II'), которые могут быть также предшественниками вышеуказанных кислот, (в частности, метиловый, этиловый и трет-бутиловый эфиры) и смешанные ангидриды кислот формулы (II) и формулы (II') с изобутил- или этилхлорформиатом представляют собой наиболее предпочтительные промежуточные продукты.
Способ получения соединений формулы (II) или формулы (II') через сложные эфиры формулы (IIа) или (II'а) представлен схемой, приведенной в конце описания.
На первой стадии а) сильное основание, такое как алкоголят металла, вводят во взаимодействие с кетоном формулы 1, в которой R4 имеет вышеуказанное значение, затем (стадия b) проводят реакцию с эквимолярным количеством диэтилоксалата в алканоле, как, например, в метаноле или этаноле, согласно L. Claisen, Ber. , 42, 59 (1909). После осаждения в простом эфире, таком как диэтиловый эфир или диизопропиловый эфир, еноляты формулы 2 отфильтровывают. Можно также получать енолят лития согласно W.V. Murray и др., J. Heterocyclic Chem., 26, 1389 (1989).
Таким образом полученный анолят металла формулы 2 и производное фенилгидразина формулы 3 или его соль затем кипятят с обратным холодильником с уксусной кислотой (стадия с) для получения сложных эфиров формул (IIа) или (II'а).
Путем омыления сложных эфиров формулы (IIа) или (II'а) под действием щелочного агента, такого как, например, гидроксид калия, гидроксид натрия или гидроксид лития, затем подкисления получают кислоты формулы (II) или (II') (стадия d).
Среди соединений формулы 3 некоторые являются новыми и составляют объект изобретения.
Так, соединения формулы (3'):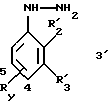
в которой R'2 и R'3, каждый, независимо друг от друга означают водород, (С1-С6)-алкил, (С3-С8)-циклоалкил или R'2 и R'3 вместе образуют тетраметиленовую группу;
Ry находится в положении 4 или в положении 5 и означает группу, выбираемую среди следующих групп: карбоксил, (C1-C4)-алкоксикарбонил, бензилоксикарбонил; при условии, что R'2 и R'3 одновременно не означают водород и что R'2 не является этилом, когда R'3 означает водород, a Ry означает метоксикарбонильную группу в положении 4,
а также их соли являются новыми соединениями и составляют следующий объект изобретения.
Производные фенилгидразина формулы 3 могут быть получены согласно методу Губен-Вейл, 1967, Х-2, 169. Например, можно осуществлять диазотирование соответствующего фениламина в присутствии нитрита натрия, затем восстановление соли диазония, например, путем воздействия хлорида двухвалентного олова. Когда фенил содержит электроноакцепторный заместитель, такой как циано- или нитрогруппа, также можно заместить фторфенильное производное гидразингидратом для получения соответствующего гидразинофенильного производного. Замещенные фениламины известны или их получают известными способами. Например, аминосульфокислоты получают согласно методу Губен-Вейл, "Методы органической химии", изд. 1955 г., том IX, с.450.
Производные фенилгидразина, замещенные группой R1=YCO2R7, получают из соответствующих производных анилина или нитрофенила.
Аминокислоты формулы (III) представляют собой промышленные продукты или они могут быть очень легко получены классическими способами. В частности, не имеющиеся в продаже аминокислоты формулы (III) получают согласно синтезу Strecker, Ann., 75, 27 (1850), или согласно синтезу Н.Т. Bucherer и др., J. Pract. Chem., 141, 5 (1934), с последующим гидролизом с целью получения аминокислот; например, 2-аминоадамантан-2-карбоновую кислоту и 9-аминобицикло(3.3.1)нонан-9-карбоновую кислоту получают согласно Н.Т. Naqasava и др., J. Med. Chem., 16 (7), 823 (1973).
Соединения формулы (Iа) и их соли обладают очень высоким сродством к человеческим рецепторам нейротензина в тестах, описанных D. Gully и др., Proc. Natl. Acad. Sci. USA, 90, 65-69 (1993).
Соединения формулы (Iа) и их соли исследовали ин виво. Работая по методике, описанной М. Ponceler и др., Naunyn Schmiedberq's Arch. Pharmacol., 60, 349-357 (1994), наблюдают, что соединение согласно изобретению, введенное перорально, противодействует контралатеральным вращениям, вызываемым интрастриатальной односторонней инъекцией нейротензина в случае мыши.
Кроме того, поступая согласно методике, описанной D. Nisato и др. в Life Sciences, 54, 7, 95-100 (1994), констатируют, что соединение согласно изобретению, введенное внутривенно, ингибирует повышение артериального давления, индуцируемое внутривенной инъекцией нейротензина в случае морской свинки.
Известные соединения, описанные в европейском патенте 0477049, в этих же тестах показывают активность ниже активности соединений согласно изобретению.
Соединения согласно изобретению являются слаботоксичными; в частности, их острая токсичность совместима с их использованием в качестве лекарственного средства. Для лечебного использования млекопитающим вводят эффективное количество соединения формулы (I) или одной из его фармацевтически приемлемых солей в целях лечения нейротензинозависимых патологий. Так, соединения согласно изобретению могут быть использованы для лечения нейропсихиатрических нарушений, в частности таких, которые связаны с дисфункцией допаминэргических систем, например таких, как психозы, в частности шизофрения, и заболевания двигательной системы, например болезнь Паркинсона (D.R. Handrich и др. , Brain Research, 231, 216-221 (1982) и C.B. Nemeroff, Biological Psychiatry, 15(2), 283-302 (1980)). Их можно использовать для диагностики и/или лечения канцерогенных заболеваний, например, как неоперабельные человеческие менингиомы (P. Mailleux, Peptides, 11, 1245-1253 (1990)), раковые заболевания простаты (I. Sehqal и др., Proc. Natl. Acad. Sci., 91, 4673-4677 (1994)), рак легких в малых клетках (T. Sethi и др., Cancer Res., 51, 3621-3623 (1991)). Их можно использовать для лечения нарушений перистальтики желудочно-кишечного тракта, а также нарушений желудочно-кишечного тракта секреторного язвенного и/или опухолевого происхождения (Обзор A. Shulkes в сборнике "Пептиды: биохимия и физиология", изд. J. Waish и G.J. Dockray, 1994). Соединения формулы (I) согласно изобретению также могут быть пригодны для лечения таких заболеваний, как синдром раздражимой ободочной кишки, диарея, колиты, язвы, опухоли желудочно-кишечного тракта, диспепсия, панкреатит, эзофагит. Они также могут представлять интерес в качестве модуляторов приема пиши (Веck В., Metabolism., 44, 972-975 (1995)). Соединения согласно изобретению можно применять в качестве диуретиков, также как в случае сердечно-сосудистых нарушений и в случае патологий, ассоциированных с высвобождением гистамина, таких как воспалительные процессы (D.E. Cochrane и др. , Faseb J., 8(7), 1195 (1994)). Эти соединения также могут представлять интерес в случае лечения некоторых нарушений, вызываемых стрессом, таких как мигрени, зуд нервного происхождения и интерстициальный цистит (Theoharides Т.C. и др., Endocrynol., 136, 5745-5750 (1995)). Соединения согласно изобретению могут также представлять интерес для использования в области анальгезии, воздействуя на эффекты морфина (М.О. Urban, J. Pharm. Exp. Ther., 265(2), 580-586 (1993)).
Таким образом, объектом изобретения являются фармацевтические композиции, содержащие в качестве действующих начал эффективное количество соединения формулы (I) или их возможные фармацевтически приемлемые соли.
В фармацевтических композициях согласно изобретению для перорального, подъязычного, подкожного, внутримышечного, внутривенного, чрескожного или ректального введения действующие начала можно вводить животным или людям в виде единичных лекарственных форм в смеси с классическими фармацевтическими носителями. Соответствующие лекарственные формы включают формы для перорального введения, такие как таблетки, желатиновые капсулы, порошки, гранулы и оральные растворы или суспензии; формы для введения путем ингаляции; формы для подъязычного введения или через рот; формы для подкожного, чрескожного, внутримышечного или внутривенного введения и формы для ректального введения.
Для достижения желательного эффекта доза действующего начала может изменяться от 0,5 до 1000 мг в день, предпочтительно от 2 до 500 мг.
Каждая разовая доза может содержать 0,5-250 мг действующего начала, предпочтительно 1-125 мг, в сочетании с фармацевтическим носителем. Эту разовую дозу можно вводить 1-4 раза в день.
Когда готовят твердую композицию в форме таблеток, то действующее начало смешивают с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или тому подобные наполнители. Таблетки можно покрывать сахарозой или другими соответствующими веществами или их можно обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заданное количество действующего начала.
Лекарственную форму в виде желатиновых капсул получают путем смешения действующего начала с разбавителем и внося полученную смесь в мягкие или жесткие желатиновые капсулы.
Лекарственная форма в виде сиропа или эликсира может содержать действующее начало вместе с подслащивающим агентом, предпочтительно некалорийным; метилпарабеном и пропилпарабеном в качестве антисептика, а также с агентом, придающим соответствующий вкус и цвет.
Диспергируемые в воде порошки или гранулы могут содержать действующее начало в смеси с диспергаторами или смачивателями, или суспендирующими агентами, такими как поливинилпирролидон и т.п., также как с подслащивающими агентами или улучшающими вкус агентами.
Для ректального введения используют суппозитории, которые получают с помощью связующих, плавящихся при ректальной температуре, например, как масло какао или полиэтиленгликоли.
Для парентерального введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат диспергаторы и/или смачиватели, которые фармакологически приемлемы, например пропиленгликоль или бутиленгликоль.
Действующее начало также может входить в состав лекарственной формы в виде микрокапсул, возможно с одним или несколькими носителями или добавками.
Для улучшения растворимости продуктов согласно изобретению соединения формулы (I) и их фармацевтически приемлемые соли также могут находиться в форме комплексов с циклодекстринами.
В описании и в примерах используют следующие сокращения:
МеОН: метанол
EtOH: этанол
эфир: диэтиловый эфир
изо-эфир: диизопропиловый эфир
солянокислый эфир: насыщенный раствор хлороводорода в эфире
солянокислый этанол: насыщенный раствор хлороводорода в этаноле
AcOEt: этилацетат
MeCN: ацетонитрил
ДХМ: дихлорметан
ДМФА: диметилформамид
ДМСО: диметилсульфоксид
ТГФ: тетрагидрофуран
НСl: хлороводород
Н2SO4: серная кислота
АсОН: уксусная кислота
ТФУК: трифторуксусная кислота
NaOH: гидроксид натрия
КОН: гидроксид калия
LiOH: гидроксид лития
NH4OH: гидроксид аммония
Na2SO4: сульфат натрия
NaHCO3: гидрокарбонат натрия
NaHSO3: гидросульфит натрия
Na2CO3: карбонат натрия
К2СО3: карбонат калия
P2O5: фосфорный ангидрид
NBS: N-бромсукцинамид
РОСl3: оксихлорид фосфора
NaNO2: нитрит натрия
SOCl2: тионилхлорид
SnCl2: хлорид двухвалентного олова
CuCN: цианид одновалентной меди
Ме, МеО: метил, метоксигруппа
Et: этил
iPr: изопропил
iBu: изобутил
n-Bu: н-бутил
t-Bu: трет.-бутил
Bz: бензил
т.пл.: температура плавления
ТК: комнатная температура
диоксид кремния Н: силикагель 60 Н, выпускаемый в продажу фирмой МЕРК (Дармштадт)
ЯМР: ядерный магнитный резонанс
За исключением противоположного, спектры ЯМР регистрируют при 200 МГц в дейтерированном диметилсульфоксиде. Химические сдвиги δ выражают в миллионных долях (м. д.) по отношению к тетраметилсилану в качестве внутреннего стандарта. В спектрах вводятся обозначения: с - синглет; уш.с - уширенный синглет; расщ.с - расщепленный синглет; д - дублет; д.д - двойной дублет; т - триплет; кд - квадруплет; кт - квинтет; сп - септет, м - массив; мт - мультиплет.
ПРЕПАРАТИВНЫЙ ПРИМЕР 1.1
Натриевая соль метил-4-(2,6-диметоксифенил)-4-оксидо-2-оксобут-3-еноата (соединение А)
Раствор 100 г 2,6-диметоксиацетофенона и 7,5 мл диэтилоксалата в 520 мл безводного метанола медленно добавляют к раствору метилата натрия, полученному из 12,7 г натрия и 285 мл безводного метанола. Кипятят с обратным холодильником в течение 7 часов и оставляют стоять в течение ночи при комнатной температуре. Реакционную смесь выливают в 2 л диизопропилового эфира и перемешивают в течение 15 минут. Получают целевой продукт путем отфильтровывания, промывки диизопропиловым эфиром и высушивания в вакууме; масса 120 г; т.пл.=178oС.
Калиевая соль этил-4-(2,6-диметоксифенил)-4-оксидо-2-оксобут-3-еноата (соединение А1)
К нагретому до температуры 50oС и перемешиваемому раствору 18 г 2,6-диметоксиацетофенона в 54 мл этанола в течение 6 минут добавляют раствор 13,4 г 95%-ного трет.-бутилата калия в 72 мл этанола. Нагревают до температуры кипения с обратным холодильником, добавляют 16,3 мл диэтилоксалата в течение 9 минут и продолжают кипятить с обратным холодильником в течение 1 часа. После этого отгоняют 40 мл этанола и оставляют охлаждаться при перемешивании в течение 2,5 часов. Осадок отфильтровывают, промывают с помощью 40 мл этанола и высушивают в вакууме при температуре 60oС в течение 17 часов, получая 31 г целевого продукта.
ЯМР: 1,2 (т, 3Н); 3,6 (с, 6Н); 4 (мт, 2Н); 5,5 (с, 1Н); 6,55 (д, 2Н); 2 7,1 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 1.2
Натриевая соль этил-4-[2-(циклопропилметилокси)-6-метоксифенил] -4-оксидо-2-оксобут-3-еноата
А) 2-(Циклопропилметилокси)-6-метоксиацетофенон
К раствору 26 г 2-гидрокси-6-метоксиацетофенона в 400 мл пропан-2-ола при комнатной температуре добавляют 32,7 мл 50%-ного раствора гидроксида цезия в воде и перемешивают в течение 15 минут при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают пропан-2-олом, концентрируют в вакууме, затем добавляют толуол и концентрируют в вакууме. Остаток растворяют в 200 мл ДМФА, добавляют 25,3 г циклопропилметилбромида и нагревают при 80oС в течение 2,5 часов. Концентрируют в вакууме, остаток обрабатывают водой, экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 32,7 г целевого продукта.
Б) Натриевая соль этил-4- [2-(циклопропилметилокси)-6-метоксифенил]-4-оксидо-2-оксобут-3-еноата
Раствор 32,6 г полученного в предыдущей стадии соединения и 20,1 мл диэтилоксалата в 100 мл этанола медленно добавляют к раствору этилата натрия, полученному из 3,4 г натрия и 60 мл этанола. Нагревают при температуре 60oС в течение ночи, оставляют охлаждаться до комнатной температуры и концентрируют в вакууме. Остаток обрабатывают пентаном, выпавший осадок отфильтровывают под вакуумом, промывают его пентаном и высушивают в вакууме. Получают 41,2 г целевого продукта.
Методики получения гидразинов формулы 3
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.1
Гидрохлорид 3-изопропил-4-гидразинобензойной кислоты
A) 2-Изопропилацетанилид
Это соединение описывается в Bull. Soc. Chim. France, 144 (1949).
Охлаждают льдом смесь, содержащую 300 мл толуола и 31 мл 2-изопропиланилина, и медленно добавляют 22 мл уксусного ангидрида. После перемешивания в течение 40 минут при комнатной температуре реакционную среду выпаривают, затем обрабатывают остаток петролейным эфиром. Выпавший осадок отфильтровывают под вакуумом. Получают 35,9 г целевого продукта (после кристаллизации из петролейного эфира). Т.пл.=81oС.
Б) 4-Бром-2-изопропилацетанилид
Это соединение описывается в J. Med. Chem., 17 (2), 221 (1974).
К смеси, содержащей 34,8 г полученного в предыдущей стадии соединения в 250 мл уксусной кислоты, медленно добавляют несколько капель раствора 10,1 мл брома в 180 мл уксусной кислоты, затем нагревают до 50oС; после охлаждения снова добавляют несколько капель раствора и нагревают до 50oС и все это продолжают до окончания всего добавления. Реакционную смесь постепенно нагревают до температуры кипения с обратным холодильником, затем оставляют на ночь для снижения температуры до комнатной. Выпавший осадок отфильтровывают, затем добавляют к разбавленному раствору NaHSO3. Снова отфильтровывают, промывают водой, потом сушат над Р2О5. Получают 27,4 г целевого продукта. Т. пл.=134oС.
Соединение, полученное на стадии Б, также может быть получено согласно нижеописываемой методике.
Б') 4-Бром-2-изопропилацетанилид
Готовят смесь, содержащую 117,6 г 2-изопропилацетанилида в 330 мл ДМФА, и в течение 25 минут добавляют раствор 117,6 г N-бромсукцинимида в 330 мл диметилформамида. Перемешивают при комнатной температуре в течение 5 часов, затем выливают в 1,5 л воды, охлаждая реакционную среду льдом. Выпавший осадок отфильтровывают, промывают водой, затем высушивают при температуре 50oС в вакууме. Фильтрат экстрагируют 2 раза дихлорметаном, промывают водой, затем сушат над сульфатом натрия для получения второй фракции целевого продукта. Объединяя различные очищенные фракции получают 158 г целевого продукта. Т.пл.=134oС.
В) 4-Циано-2-изопропилацетанилид
Смесь, содержащую 26,48 г полученного в предыдущей стадии продукта, 60 мл диметилформамида, 1 мл воды и 10,25 г цианида одновалентной меди, кипятят с обратным холодильником в течение 10 часов. После охлаждения ее выливают в раствор 50 г цианида натрия в 150 мл воды при температуре 40oС. Выпавший осадок отфильтровывают и промывают несколько раз водой. Получают 16,7 г целевого продукта. Т.пл.=134oС.
Г) Гидрохлорид 4-циано-2-изопропиланилина
Смешивают 16,13 г полученного в предыдущей стадии соединения, 65 мл 100%-ного этанола и 40 мл 1 н. соляной кислоты и перемешивают при кипячении с обратным холодильником в течение 19 часов. После выдерживания в течение ночи при комнатной температуре добавляют 10%-ный раствор NaOH до достижения рН 10. Реакционную среду экстрагируют 2 раза дихлорметаном, органическую фазу сушат над сульфатом натрия и концентрируют в вакууме. Остаток растворяют в эфире и добавляют солянокислый эфир. Выпавший осадок отфильтровывают и промывают его эфиром. Получают 15,32 г целевого продукта, который кристаллизуют из смеси диэтилового эфира с хлороводородом. Т.пл.=188oС.
Д) Гидрохлорид 4-амино-3-изопропилбензойной кислоты
Это соединение описывается в J. Med. Chem., 17(2), 221 (1974).
Смесь, содержащую 1 г полученного в предыдущей стадии соединения, 2,86 г размельченного диоксида калия, 6 мл воды и 0,5 мл диметоксиэтана, кипятят с обратным холодильником в течение 12 часов. После охлаждения добавляют концентрированную соляную кислоту до достижения рН 1, затем экстрагируют 2 раза дихлорметаном; органическую фазу сушат над сульфатом натрия и концентрируют. Получают 0,96 г целевого продукта. Т.пл.=128oС.
Е) Гидрохлорид 3-изопропил-4-гидразинбензойной кислоты
Смесь, содержащую 0,96 г полученного в предыдущей стадии продукта, 22 мл концентрированной соляной кислоты и 20 мл уксусной кислоты, охлаждают до -5oС, добавляют к ней раствор 0,36 г нитрита натрия в 4 мл воды, затем перемешивают при температуре 0oС в течение 1 часа 15 минут. Охлаждают до -10oС и добавляют 3,73 г дигидрата хлорида двухвалентного олова в 4 мл концентрированной соляной кислоты. Оставляют стоять для повышения температуры до 18oС, затем выпавший осадок отфильтровывают и промывают его с помощью 1 мл разбавленной соляной кислоты. Получают 0,96 г целевого продукта (после высушивания над Р2О5).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.1-БИС
Гидрохлорид 3-изопропил-4-гидразинбензойной кислоты также может быть получен согласно нижеописанной методике.
А) 4-Бром-2-изопропилацетанилид
В течение 10 минут и при поддержании температуры ниже 60oС 200 мл уксусного ангидрида добавляют к 300 мл 2-изопропиланилина. После перемешивания в течение 45 минут при комнатной температуре добавляют раствор одного эквивалента N-бромсукцинимида в 720 мл диметилформамида. После перемешивания в течение 2 часов смесь выливают в 5,7 л смеси воды с этилацетатом (в объемном соотношении 2: 1), декантируют, отделяют органическую фазу, сушат ее над сульфатом натрия и выпаривают в вакууме. Остаток сгущают в диизопропиловом эфире и отфильтровывают, получая 367 г целевого продукта.
Б) 4-Циано-2-изопропилацетанилид
10,25 г полученного на стадии А продукта и 1,2 эквивалента цианида одновалентной меди в 20 мл диметилформамида кипятят с обратным холодильником в течение 6 часов. Затем охлаждают до 20oС, выливают в смесь 200 мл этилацетата с 200 мл 20%-ного раствора аммиака. Органическую фазу промывают с помощью 50 мл 20%-ного раствора аммиака, затем 2 раза с помощью насыщенного раствора NaCl. После высушивания над сульфатом натрия выпаривают в вакууме, остаток обрабатывают диизопропиловым эфиром, отфильтровывают и высушивают при температуре 40oС в вакууме, получая 6,15 г целевого продукта.
Соединение, получаемое на стадии Б, также может быть получено согласно нижеприводимой методике.
Б') В перемешиваемую смесь из 22,16 г полученного на стадии А продукта и 0,6 эквивалента цианида цинка в 67 мл безводного диметилформамида пропускают путем барботирования аргон. Нагревают до 80oС и добавляют 2 г тетракис(трифенилфосфин)палладия-(0) в отсутствии света. После перемешивания в течение трех часов при температуре 80oС охлаждают до комнатной температуры и добавляют 120 мл 4%-ного раствора аммиака и 200 мл этилацетата, объединенные органические фазы промывают с помощью 4%-ного раствора аммиака, затем сушат над сульфатом натрия, выпаривают в вакууме, остаток обрабатывают диизопропиловым эфиром, отфильтровывают и высушивают в вакууме, получая 15 г целевого нитрила.
В) Гидрохлорид 4-амино-3-изопропилбензойной кислоты
Смесь 100 г продукта, полученного на стадии Б, с 500 мл концентрированной соляной кислоты и 500 мл уксусной кислоты кипятят с обратным холодильником в течение 10 часов. Концентрируют в вакууме, осадок отфильтровывают и высушивают в вакууме, получая 103,8 г целевого продукта.
Г) Гидрохлорид 3-изопропил-4-гидразинбензойной кислоты
К охлажденной до -5oС смеси из 59 г полученного на стадии В продукта вместе с 1050 мл уксусной кислоты и 1420 мл концентрированной соляной кислоты медленно добавляют раствор 27,7 г нитрита натрия в 250 мл воды. После перемешивания в течение 1 часа 20 минут при температуре 0oС смесь охлаждают до температуры -10oС и добавляют раствор 236 г дигидрата хлорида двухвалентного олова в 250 мл концентрированной соляной кислоты. Повышают температуру до комнатной, осадок отфильтровывают, промывают его концентрированной соляной кислотой и высушивают в вакууме, получая 56,36 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.2
Гидрохлорид 3-изопропил-4-гидразинбензолсульфокислоты
A) 4-Амино-3-изопропилбензолсульфокислота
5,7 мл Серной кислоты добавляют к 10 мл воды и нагревают до температуры 80oС, затем добавляют 13,5 г 2-изопропиланилина. Воду выпаривают путем нагревания в вакууме, затем постепенно, в течение полутора часов, температуру повышают до 260oС. После перемешивания в течение трех часов в вакууме при температуре 260oС смесь оставляют стоять для возврата температуры к комнатной и давления к атмосферному давлению, после чего в течение 30 минут нагревают в присутствии 15 мл NaOH и 100 мл воды для растворения реакционной среды. Отфильтровывают нерастворимую часть, охлаждают до 5oС, затем подкисляют с помощью концентрированной серной кислоты до рН 1. Выпавший осадок отфильтровывают, промывают с помощью 5 мл холодной воды и высушивают, получая 20 г целевого продукта.
ЯМР-спектр: 1,1 (д, 6Н); 2,95 (мт, 1Н); 6,85 (д, 1Н); 7,45 (д.д, 1Н); 7,6 (д, 1Н).
Б) Гидрохлорид 3-изопропил-4- гидразинбензолсульфокислоты
К раствору 10 г полученного в предыдущей стадии продукта в 10 мл 30%-ного раствора NаОН и 20 мл воды добавляют 10 мл льда и 3,2 г нитрита натрия. Этот раствор медленно выливают в раствор из 30 мл концентрированной соляной кислоты в 20 мл воды при температуре от -5oС до -15oС. Перемешивают в течение 1 часа при этой температуре, затем добавляют 26 г дигидрата хлорида двухвалентного олова в 40 мл концентрированной соляной кислоты при температуре от 0oС до -5oС. После перемешивания в течение 2,5 часов при комнатной температуре отфильтровывают, затем высушивают полученный продукт в вакууме в присутствии Р2O5. Таким образом получают 9,7 г целевого продукта.
ЯМР-спектр: 1,2 (д, 6Н); 3,15 (мт, 1Н); 6,8 (д, 1Н), 7,45 (д, 1Н); 7,55 (с, 1Н), 7,9 (уш.с, 1Н); 10 (уш.с, 3Н).
ПРЕПАРАТИВНЫИ ПРИМЕР 2.3
Гидрохлорид 4-гидразино-5,6,7,8-тетрагидронафталин-1-карбоновой кислоты
А) 4-Амино-5,6,7,8-тетрагидронафталин-1-карбоновая кислота
4-Нитро-5,6,7,8-тетрагидронафталин-1-карбоновая кислота описывается в Chem. Pharm. Bull., 32, 3968 (1984).
Осуществляют гидрирование 1,48 г этого нитропроизводного в метаноле в присутствии никеля Ренея®. После перемешивания в течение 4 часов катализатор отфильтровывают, фильтрат выпаривают досуха, остаток обрабатывают эфиром и отфильтровывают, получая 1 г целевого продукта. Т.пл.=180oС (разложение).
Б) Гидрохлорид 4-гидразино-5,6,7,8-тетрагидронафталин-1-карбоновой кислоты
К раствору 0,73 г 4-амино-5,6,7,8-тетрагидронафталин-1-карбоновой кислоты в 10 мл концентрированной соляной кислоты, охлажденному до -5oС, добавляют раствор 0,26 г нитрита натрия в 1 мл воды. После перемешивания в течение полутора часов при температуре -5oС при -5oС добавляют раствор 3,4 г дигидрата хлорида двухвалентного олова в 34 мл концентрированной соляной кислоты. Перемешивают в течение 1 часа при комнатной температуре, промывают концентрированной соляной кислотой, сушат в токе осушенного азота, получая 0,67 г целевого гидразина.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.4
Гидрохлорид 4-гидразино-5,6,7,8-тетрагидронафталин-1-сульфокислоты
А) 4-Амино-5,6,7,8-тетрагидронафталин-1-сульфокислота
К раствору 10 г 5,6,7,8-тетрагидронафталинамина в 100 мл 1,2-дихлорбензола добавляют нагретую суспензию 20 г сульфаминовой кислоты в 40 мл N-метилпирролидона. Нагревают в течение 7 часов при перемешивании при температуре 150oС, отфильтровывают, промывают дихлорбензолом, затем толуолом. Осадок снова суспендируют в 70 мл воды, нейтрализуют до рН 7 с помощью 5,5 мл 30%-ного раствора гидроксида натрия. Отфильтровывают нерастворимую часть, водную фазу экстрагируют эфиром. рН-Значение водной фазы доводят до 5 путем добавления соляной кислоты при 5oС, осадок отфильтровывают, промывают водой и высушивают, получая 7,5 г целевого продукта.
Б) Гидрохлорид 4-гидразино-5,6,7,8-тетрагидронафталин-1-сульфокислоты
К раствору 3 г полученной на стадии А кислоты в 10 мл воды и 2 мл 30%-ного раствора гидроксида натрия добавляют 1 г нитрита натрия. В течение 1 часа этот раствор выливают в 10 мл концентрированной соляной кислоты, охлажденной до 5oС. После перемешивания в течение трех часов при температуре 5oС медленно добавляют раствор 7,5 г дигидрата хлорида двухвалентного олова в 15 мл концентрированной соляной кислоты, поддерживая температуру 5oС. Перемешивают в течение полутора часов при комнатной температуре, отфильтровывают и высушивают в вакууме, получая 2,86 г целевого продукта.
ЯМР-спектр (D2O-NaOD): 1,6 (мт, 4Н); 2,25 (мт, 2Н); 2,9 (мт, 2Н); 6,75 (д, 1Н); 7,6 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.5
Гидрохлорид 4-гидразино-3-метилбензамида
A) 4-Амино-3-метилбензамид
Этот продукт получают путем каталитического гидрирования 3-метил-4-нитробензамида. Т.пл.=124oС.
Б) Гидрохлорид 4-гидразино-3-метилбензамида
0,5 г Полученного на стадии А соединения растворяют в 10 мл 1 н. соляной кислоты и 5 мл концентрированной соляной кислоты. Охлаждают до 0oС и добавляют раствор 230 мг нитрита натрия в 3 мл воды. После выдерживания в течение 15 минут при температуре -10oС добавляют раствор 1,5 г дигидрата хлорида двухвалентного олова в 5 мл концентрированной соляной кислоты. Спустя 1 час осадок отфильтровывают и высушивают в вакууме над Р2O5, получая 390 мг целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.6
Гидрохлорид 2,3-диметил-4-гидразинбензойной кислоты
К раствору 4,5 г 4-амино-2,3-диметилбензойной кислоты в 135 мл концентрированной соляной кислоты, охлажденному до температуры -5oС, медленно добавляют раствор 1,87 г нитрита в 7 мл воды. После перемешивания в течение двух часов при температуре -5oС при температуре -10oС добавляют раствор 25 г дигидрата хлорида двухвалентного олова в 250 мл концентрированной соляной кислоты, перемешивают в течение 30 минут при температуре -5oС, затем в течение двух часов при комнатной температуре. Осадок отфильтровывают, промывают с помощью 5 мл концентрированной соляной кислоты, сушат в токе осушенного азота, затем в вакууме, получая 5,5 г целевого продукта.
ЯМР-спектр: 2,1 (с, 3Н); 2,4 (с, 3Н); 6,8 (д, 2Н); 7,6 (д, 2Н); 8,2 (с, 1Н); 10 (уш.с, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.7
Гидрохлорид 4-гидразино-3-метоксибензойной кислоты
К раствору 5 г 4-амино-3-метоксибензойной кислоты в 50 мл концентрированной соляной кислоты, охлажденному до 0oС, медленно добавляют раствор 2,17 г нитрита натрия в 40 мл воды. После перемешивания в течение 1 часа 15 минут при температуре 0oС охлаждают до -10oС, в течение 30 минут добавляют раствор 23,6 г дигидрата хлорида двухвалентного олова в 20 мл концентрированной соляной кислоты и 20 мл воды. После перемешивания в течение полутора часов при температуре -10oС осадок отфильтровывают, промывают с помощью 50 мл пентана, получая, после высушивания, 6 г целевого продукта.
ЯМР-спектр: 3,8 (с, 3Н); 7 (д, 1Н); 7,4 (с, 1Н); 7,5 (д.д, 1Н); 8 (уш.с, 1Н); 10,6 (уш.с, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.8
Гидрохлорид 2-хлор-4-гидразинбензонитрила
При температуре -5oС смешивают 5 г 4-амино-2-хлорбензонитрила с 40 мл концентрированной соляной кислоты в 30 мл тетрагидрофурана, добавляют 2,26 г нитрита натрия в 30 мл воды и перемешивают в течение двух часов, после чего добавляют 30 г дигидрата хлорида двухвалентного олова в 30 мл концентрированной соляной кислоты и перемешивают в течение 30 минут при температуре -5oС. После возврата к комнатной температуре, отфильтровывают нерастворившуюся часть, добавляют NaCl и снова перемешивают. Целевой продукт кристаллизуется с NaCl, его поглощают этанолом, тогда как NaCI отфильтровывают. После выпаривания растворителей получают 4,25 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.9
Гидрохлорид 3-циклопропил-4-гидразинбензойной кислоты
A) 4-Ацетамидо-3-циклопропилбензонитрил
К раствору 4,3 г 4-бром-2-циклопропилацетанилида (получен согласно J. Am. Chem. Soc., 90, 3404 (1968)) в 100 мл диметилформамида добавляют 1,67 г CuCN и кипятят в течение 24 часов с обратным холодильником. Выливают в 30 мл воды, осадок отфильтровывают, промывают водой, затем осадок перемешивают в течение 30 минут в смеси из 59 мл воды и 25 мл этилендиамина. После экстракции с помощью 100 мл этилацетата, высушивания над сульфатом натрия и выпаривания в вакууме получают 2,39 г целевого продукта.
ЯМР-спектр: 0,6 (м, 2Н); 0,9 (м, 2Н); 1,9 (м, 1Н); 2,1 (с, 3Н); 7,3 (д, 1Н); 7,5 (д.д, 1Н); 7,8 (д, 1Н); 9,5 (уш.с, 1Н).
Б) Гидрохлорид 4-амино-3-циклопропилбензонитрил
Смесь 2,39 г полученного на стадии А продукта в виде раствора в 45 мл этанола с 36 мл воды и 5 мл концентрированной соляной кислоты перемешивают при кипячении с обратным холодильником в течение 12 часов. Этанол выпаривают в вакууме, осадок отфильтровывают, промывают его с помощью 1 мл воды и высушивают в вакууме, получая 1,5 г целевого продукта.
ЯМР-спекрт: 0,5 (м, 2Н); 0,9 (м, 2Н); 1,6 (м, 1Н); 6,7 (д, 1Н); 7,1-7,3 (мт, 2Н); 8 (уш.с, 2Н).
В) Гидрохлорид 4-амино-3-циклопропилбензойной кислоты
1,3 г Полученного на стадии Б продукта в 21 мл 50%-ного раствора КОН перемешивают при кипячении с обратным холодильником в течение 29 часов. После подкисления до рН 1 с помощью концентрированной соляной кислоты осадок отфильтровывают и высушивают в вакууме, получая 0,96 г целевого продукта.
ЯМР-спектр: 0,5 (м, 2Н); 0,9 (м, 2Н); 1,7 (м, 1Н); 5,9 (уш.с, 2Н); 6,6 (д, 1Н); 7,4 (с, 1Н); 7,5 (мт, 1Н); 12 (уш.с, 1Н).
Г) Гидрохлорид 3-циклопропил-4-гидразинбензойной кислоты
К раствору 0,95 г полученного на стадии В продукта в 22 мл концентрированной соляной кислоты и 21 мл уксусной кислоты, охлажденному до температуры -5oС, медленно добавляют раствор 0,38 г нитрита натрия в 4,5 мл воды и перемешивают в течение 1 часа 15 минут при температуре 0oС. Охлаждают до -10oС и медленно добавляют раствор 3,76 г дигидрата хлорида двухвалентного олова в 8 мл концентрированной соляной кислоты. После перемешивания в течение 4 часов при комнатной температуре осадок отфильтровывают, промывают с помощью 2 мл концентрированной соляной кислоты и высушивают в вакууме, получая 1 г целевого продукта.
ЯМР-спектр: 0,6 (м, 2Н); 1 (м, 2Н); 1,9 (м, 1Н); 7,1 (д, 1Н); 7,6 (с, 1Н); 7,8 (д, 1Н); 8,4 (с, 1Н); 10,7 (уш.с, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.10
Гидрохлорид 5-гидразино-2-хлорбензойной кислоты
К суспензии 5 г 5-амино-2-хлорбензойной кислоты в 50 мл концентрированной соляной кислоты, охлажденной до температуры -2oС, в течение 30 минут добавляют раствор 2,11 г нитрита натрия в 40 мл воды. Перемешивают в течение двух часов при температуре -3oС, охлаждают раствор до -10oС и в течение 30 минут добавляют раствор 23 г дигидрата хлорида двухвалентного олова в 20 мл концентрированной соляной кислоты и 20 мл воды. Перемешивают в течение полутора часов при 0oС, осадок отфильтровывают и высушивают, получая 4 г целевого продукта.
ЯМР-cпектр: 7,6 (уш.с, 2Н); 7,7 (уш.с, 1Н); 8,4 (уш.с, 1Н); 11,0 (уш.с, 3Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.11
Гидрохлорид 3-гидразино-4-метилбензойной кислоты
К раствору 5 г 3-амино-4-метилбензойной кислоты в 120 мл концентрированной соляной кислоты и 40 мл уксусной кислоты, охлажденному до температуры -5oС, в течение 30 минут добавляют раствор 2,74 г нитрита натрия в 28 мл воды и перемешивают в течение 1 часа 20 минут при 0oС. После охлаждения до -10oС медленно добавляют раствор 27,6 г дигидрата хлорида двухвалентного олова в 28 мл концентрированной соляной кислоты. После перемешивания в течение 1 часа при комнатной температуре, отфильтровывания и промывки осадка с помощью 5 мл 1 н. соляной кислоты, высушивания над фосфорным ангидридом в вакууме получают 6,15 г целевого продукта.
Следуя вышеуказанным методикам получения, из соответствующим образом замещенных производных анилина получают гидразины, описанные в таблице 1.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.18
Оксалат N-(4-гидразино-3-изопропилфенил)-4-метилбензолсульфонамида
A) N-(2-Бром-5-изопропил-4-нитрофенил)-4-метилбензолсульфонамид
Раствор 23,47 г N-(4-нитрофенил)-4-метилбензолсульфонамида в 230 мл тетрагидрофурана охлаждают до -30oС, добавляют 100 мл 2 М раствора изопропилмагнийхлорида в эфире и перемешивают в течение 30 минут при температуре -30oС. Затем добавляют 10,3 мл брома при температуре -30oС, перемешивают в течение 15 минут при этой температуре, после чего оставляют стоять до повышения температуры до 20oС. Затем добавляют 55 мл триэтиламина и перемешивают в течение 1 часа при комнатной температуре. Добавляют воду, подкисляют до рН 3-4 путем добавления 10%-ной соляной кислоты, декантируют органическую фазу, водную фазу экстрагируют эфиром и объединенные органические фазы сушат над сульфатом натрия. Органическую фазу перемешивают в присутствии животного угля, отфильтровывают и концентрируют в вакууме. Остаток обрабатывают этанолом и выкристаллизовавшийся продукт отфильтровывают под вакуумом. Получают 11,6 г целевого продукта. Т.пл.=132oС.
ЯМР-спектр: 1,0 (д, 6Н); 2,32 (с, 3Н); 3,12 (с, 1Н); 7,14 (с, 1Н); 7,36 (д, 2Н); 7,65 (д, 2Н); 8,08 (с, 1Н); 10,25 (уш.с, 1Н).
Б) N-(4-Амино-3-изопропилфенил)-4-метилбензолсульфонамид
В течение 7 часов при комнатной температуре и давления 1 бар гидрируют смесь 11,5 г полученного на предыдущей стадии соединения и 1 г 5%-ного палладия-на-угле в 200 мл метанола и 30 мл диметилформамида. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток обрабатывают водой, нейтрализуют до рН 7 путем добавления 10%-ного раствора NaOH, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 8 г целевого продукта.
ЯМР-спектр: 1,01 (д, 6Н); 2,4 (с, 3Н); 2,89 (мт, 1Н); 4,91 (уш.с, 2Н); 6,42-6,68 (м, 3Н); 7,35 (д, 2Н); 7,56 (д, 2Н); 9,38 (с, 1Н).
В) Оксалат N-(4-гидразино-3-изопропилфенил)-4- метилбензолсульфонамида
Перемешивают при температуре 0oС смесь 6,68 г полученного на предыдущей стадии соединения и 70 мл концентрированной соляной кислоты, добавляют раствор 1,48 г нитрита натрия в 5 мл воды и перемешивают в течение 1 часа при 0oС. Затем добавляют раствор 11,35 г дитионита натрия в 60 мл воды и продолжают перемешивание в течение 1 часа при температуре 0oС. После этого добавляют 120 г ацетата натрия в виде порошка, 300 мл воды и перемешивают в течение 30 минут при 0oС. Реакционную смесь экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия, отфильтровывают, к фильтрату добавляют раствор 1,98 г щавелевой кислоты в минимальном количестве этанола и все концентрируют в вакууме. Остаток обрабатывают диизопропиловым эфиром, перемешивают в течение 12 часов и выпавший осадок отфильтровывают под вакуумом. Получают 4,84 г целевого продукта, который используют таким, какой есть.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.19
Гидрофторид 1-гидразино-2-метил-4-нитробензола
Смесь 4,6 г 1-фтор-2-метил-4-нитробензола и 3 мл гидразингидрата в 45 мл пропан-2-ола кипятят с обратным холодильником в течение двух часов. Добавляют 3 мл гидразингидрата, продолжают кипятить с обратным холодильником в течение двух часов и перемешивают в течение ночи при комнатной температуре. Выпавший осадок отфильтровывают под вакуумом. Получают 3,53 г целевого продукта. Т.пл.=182oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.20
Гидробромид N-(4-гидразино-3-изобутилфенил)-4-метилбензолсульфонамида
А) N-(2-Бром-5-изобутил-4-нитрофенил)-4- метилбензолсульфонамид
Это соединение получают согласно методу, описанному на стадии А Препаративного примера 2.18, исходя из 10 г N-(4-нитрофенил)-4-метилбензолсульфонамида в 100 мл тетрагидрофурана и 42,6 мл 2 М раствора изобутилмагнийхлорида в эфире, затем 4,4 мл брома и 23,4 мл триэтиламина. Получают 5,9 г целевого продукта. Т.пл.=170oС.
ЯМР-спектр: 0,8 (д, 6Н); 1,7 (мт, 1Н); 2,35 (с, 3Н); 2,6 (д, 2Н); 7,2 (с, 1Н); 7,3 (д, 2Н); 7,4 (д, 2Н); 8,2 (с, 1Н); 10,25 (с, 1Н).
Б) N-(4-Амино-3-изобутилфенил)-4- метилбензолсульфонамид
Это соединение получают согласно методике, описанной на стадии Б Препаративного примера 2.18, исходя из 4,7 г полученного на предыдущей стадии соединения. Получают 2,8 г целевого продукта.
ЯМР-спектр: 0,8 (д, 6Н); 1,75 (мт, 1Н); 2,2 (д, 2Н); 2,4 (с, 3Н); 4,8 (с, 2Н); 6,45 (д, 1Н); 6,5 (д, 1Н); 6,65 (д.д, 1Н); 7,35 (д, 2Н); 7,55 (д, 2Н); 9,4 (с, 1Н).
В) Гидробромид N-(4-гидразино-3-изобутилфенил)-4-метилбензолсульфонамида
Это соединение получают согласно методике, описанной на стадии В Препаративного примера 2.18, исходя из 2,5 г полученного на предыдущей стадии соединения, 35 мл концентрированной соляной кислоты, 50 мл уксусной кислоты и 0,53 г нитрита натрия, затем 4,78 г дитионита натрия в 50 мл воды, 140 г ацетата натрия и 70 мл воды. После перемешивания в течение 30 минут при 0oС при температуре 0oС добавляют бромоводородную кислоту, выкристаллизовавшийся продукт отфильтровывают под вакуумом и высушивают его. Получают 1,9 г целевого продукта.
ЯМР-cпектр: 0,65 (д, 6Н); 1,6 (мт, 1Н); 2,05 (д, 2Н); 2,2 (с, 3Н); 6,05 (уш.с, 1Н); 6,4 (уш.с, 1Н); 6,6-6,8 (м, 2Н); 7,2 (д, 2Н); 7,4 (д, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.21
Оксалат N-(4-гидразино-3-циклопентилфенил)-4-метилбензолсульфонамида
А) N-(2-Бром-5-циклопентил-4-нитрофенил)-4-метилбензолсульфонамид
Это соединение получают согласно методике, описанной на стадии А Препаративного примера 2.18, исходя из 15 г N-(4-нитрофенил)-4-метилбензолсульфонамида в 100 мл тетрагидрофурана и 64 мл 2 М раствора циклопентилмагнийхлорида в эфире, затем 6,8 мл брома и 35 мл триэтиламина. Получают 6,1 г целевого продукта. Т.пл.=122oС.
ЯМР-спектр (ДМСО+ТФУК): 1,25 (мт, 2Н); 1,5-1,7 (м, 4Н); 1,95 (мт, 2Н); 2,36 (с, 3Н); 3,2 (кт, 1Н); 7,12 (с, 2Н); 7,4 (д, 2Н); 7,7 (д, 2Н); 8,08 (с, 1Н).
Б) N-(4-Амино-3-циклопентилфенил)-4-метилбензолсульфонамид
Это соединение получают согласно методике, описанной на стадии Б Препаративного примера 2.18, исходя из 6 г полученного на предыдущей стадии соединения. Получают 4,25 г целевого продукта. Т.пл.=128oС.
ЯМР-спектр (ДМСО+ТФУК): 1,25 (мт, 2Н); 1,5-1,75 (м, 4Н); 1,95 (мт, 2Н), 2,3 (с, 3Н); 3,0 (кт, 1Н); 7,0 (д.д, 1Н); 7,12 (д, 1Н); 7,2 (д, 1Н); 7,34 (д, 2Н); 7,65 (д, 2Н).
В) Оксалат N-(4-гидразино-3-циклопентилфенил)-4-метилбензолсульфонамид
Это соединение получают согласно методике, описанной на стадии В Препаративного примера 2.18, исходя из 3,35 г полученного на предыдущей стадии соединения, 20 мл серной кислоты, 50 мл уксусной кислоты, 10 мл воды и 0,69 г нитрита натрия, затем 5,5 г дитионита натрия в 50 мл воды и 200 г ацетата натрия и 300 мл воды. Получают 2,42 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.22
Гидрохлорид 4-гидразино-2-изопропилбензойной кислоты
А) 4-Иод-3-изопропилацетанилид
Это соединение получают согласно способу, описанному в Bull. Soc. Chim. Jap., 62, 1349 (1989).
К раствору 5 г 3-изопропилацетанилида в 150 мл уксусной кислоты при комнатной температуре добавляют 5,7 г хлорида цинка и 10,8 г бензилтриметиламмонийдихлориодата и перемешивают в течение двух дней. Концентрируют в вакууме, остаток обрабатывают с помощью 100 мл 5%-ного раствора гидросульфита натрия, доводят значение рН до 5-6 путем добавления 10%-ного раствора карбоната натрия, экстрагируют 4 раза по 200 мл хлороформом и органическую фазу сушат над сульфатом натрия. После отфильтровывания фильтрат хроматографируют на 100 г оксида алюминия, элюируя хлороформом. Получают 5,2 г целевого продукта.
ЯМР-спектр: 1,1 (д, 6Н); 2,0 (с, 3Н); 3,06 (м, 1Н); 7,28 (д.д, 1Н); 7,5 (д, 1Н); 7,72 (д, 1Н); 10,0 (уш.с, 1Н).
Б) 4-Иод-3-изопропиланилин
Смесь 5,1 г полученного на предыдущей стадии соединения в 40 мл 96%-ного этанола и 25 мл концентрированного раствора гидроксида натрия кипятят с обратным холодильником в течение 6 часов. Концентрируют в вакууме, экстрагируют эфиром, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 5 г целевого продукта в форме масла.
ЯМР-спектр: 1,16 (д, 6Н); 2,94 (м, 1Н); 5,2 (уш.с, 2Н); 6,2 (д.д, 1Н); 6,6 (д, 1Н); 7,4 (д, 1Н).
В) 4-Амино-2-изопропилбензойная кислота
К раствору 5 г полученного на предыдущей стадии соединения в 60 мл диметилформамида добавляют 40 мл воды и 11 г карбоната калия, затем раствор дегазируют в течение 10 минут путем барботирования азота. После этого добавляют 0,5 г ацетата палладия-(II), затем дегазируют в течение 10 минут путем барботирования азота. Реакционную смесь помещают в атмосферу монооксида углерода под давлением 1 бар на 10 часов и при перемешивании. Раствор фильтруют, фильтрат промывают 4 раза по 20 мл водой и концентрируют в вакууме. Остаток обрабатывают с помощью 50 мл воды и 10 мл насыщенного раствора хлорида натрия, водную фазу промывают эфиром, подкисляют до рН 3,5-4 путем добавления концентрированной соляной кислоты, экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают растворитель в вакууме. Получают 2 г сырого продукта, который обрабатывают в 20 мл насыщенного раствора газообразного хлороводорода в метаноле и в течение ночи кипятят с обратным холодильником. Концентрируют в вакууме, остаток обрабатывают 20 мл воды, подщелачивают до рН 8 путем добавления концентрированного раствора NaOH и экстрагируют с помощью 30 мл дихлорметана. К органической фазе добавляют 0,8 мл уксусного ангидрида, бикарбонат натрия и сульфат натрия и перемешивают. После отфильтровывания фильтрат концентрируют в вакууме и остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с эфиром в объемном соотношении 50: 50. Получают 0,8 г метилового эфира 4-ацетамидо-2-изопропилбензойной кислоты в виде масла. Смесь полученного продукта и 3 г КОН в 10 мл воды и 2 мл 1,2-диметоксиэтана кипятят с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакционную смесь промывают эфиром, подкисляют водную фазу до рН 3-4 путем добавления концентрированной соляной кислоты, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,6 г целевого продукта.
ЯМР-спектр: 1,17 (д, 6Н); 4,0 (сп, 1Н); 5,7 (уш.с, 2Н); 6,36 (д.д, 1Н); 6,80 (д, 1Н); 7,6 (д, 1Н); 11,80 (уш.с, 1Н).
Г) Гидрохлорид 4-гидразино-2-изопропилбензойной кислоты
Смесь 0,5 г полученного на предыдущей стадии соединения в 7 мл концентрированной соляной кислоты охлаждают до 0oС, добавляют раствор 0,23 г нитрита натрия в 4 мл воды и перемешивают в течение полутора часов при температуре 0oС. Охлаждают до -10oС и добавляют раствор 2,6 г дигидрата хлорида двухвалентного олова в 5 мл концентрированной соляной кислоты и 3 мл воды и перемешивают в течение двух часов при температуре 0oС. Выпавший осадок отсасывают, промывают концентрированной соляной кислотой и высушивают его при 50oС в вакууме. Получают 0,36 г целевого продукта.
ЯМР-спектр: 1,15 (д, 6Н); 3,98 (м, 1Н); 6,7 (д.д, 1Н); 6,96 (д, 1Н); 7,8 (д, 1Н); 8,2 (уш.с, 1Н); 13 (уш.с, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.23
Гидрохлорид 1-гидразино-4-нитро-5,6,7,8- тетригидронафталина
А) 1-Ацетамидо-5,6,7,8-тетрагидронафталин
Перемешивают в течение 1 часа при комнатной температуре смесь 24,39 г 1-амино-5,6,7,8-тетрагидронафталина и 18,6 г уксусного ангидрида в 230 мл дихлорметана. Концентрируют в вакууме, остаток обрабатывают эфиром и выпавший осадок отфильтровывают под вакуумом. Получают 26,8 г целевого продукта. Т.пл.=158oС.
Б) 1-Амино-4-нитро-5,6,7,8-тетрагидронафталин
Смесь 13 г полученного на предыдущей стадии соединения в 72 мл концентрированной серной кислоты охлаждают до 0oС, добавляют смесь 4,55 мл азотной кислоты (уд.вес=1,4) и 22 мл концентрированной серной кислоты и перемешивают в течение 45 минут при температуре 0oС. Реакционную смесь выливают на лед и выпавший осадок отфильтровывают под вакуумом. Осадок обрабатывают с помощью 145 мл этанола, 30 мл концентрированной соляной кислоты и 30 мл воды, затем кипятят с обратным холодильником в течение полутора часов. Реакционную смесь выпаривают на объем 70 мл, к оставшемуся раствору добавляют 220 мл воды, доводят рН-значение до 7 путем добавления концентрированного раствора гидроксида аммония, выпавший осадок отфильтровывают под вакуумом и высушивают его. Осадок обрабатывают с помощью 210 мл нитробензола, охлажденного до 0oС, и в течение 50 минут через полученный раствор барботируют ток газообразного хлороводорода. Выпавший осадок отфильтровывают под вакуумом и промывают эфиром. Осадок обрабатывают метанолом, нейтрализуют путем добавления концентрированного раствора гидроксида аммония, добавляют воду и осадок отфильтровывают под вакуумом. Получают 5,15 г целевого продукта (после высушивания). Т.пл.=114oС.
В) Гидрохлорид 1-гидразино-4-нитро-5,6,7,8-тетрагидронафталина
Смесь 3,8 г полученного на предыдущей стадии соединения в 70 мл концентрированной соляной кислоты охлаждают до 3oС, добавляют раствор 1,34 г нитрита натрия в 2 мл воды и перемешивают в течение двух часов при температуре 3oС. Затем добавляют раствор 18,2 г дигидрата хлорида двухвалентного олова в 90 мл концентрированной соляной кислоты и перемешивают в течение 30 минут при температуре 3oС, после чего оставляют температуру повышаться до комнатной. Выпавший осадок отфильтровывают под вакуумом и высушивают. Получают 6,3 г целевого продукта в смеси с солями олова.
ЯМР-спектр (ДМСО+ТФУК): 1,7 (мт, 4Н); 2,55 (мт, 2Н); 2,85 (мт, 2Н); 6,88 (д, 1Н), 7,85 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 2.24
3-Диэтиламнно-N-(4-изопропил-3- гидразинфенил)пропионамид
A) Оксалат 3-диэтиламино-N-(4-изопропил-3- нитрофенил)пропионамида
4 г 4-Изопропил-3-нитроанилина (получен согласно J. Orq. Chem., 19, 1067 (1954)) обрабатывают путем кипячения с обратным холодильником в течение 1 часа вместе с 8,14 мл бис(триметилсилил)ацетамида в 20 мл ацетонитрила, затем добавляют хлорангидрид кислоты, полученный из 4 г гидрохлорида 3-N,N-диэтиламинопропановой кислоты в дихлорметане, затем 8,9 мл триэтиламина. После перемешивания в течение 1 часа при комнатной температуре выпаривают досуха, остаток экстрагируют дихлорметаном, промывают экстракт водой, затем 5%-ным раствором гидроксида натрия. После высушивания над сульфатом натрия выпаривают в вакууме, полученное масло растворяют в минимальном количестве этанола, добавляют 1,2 г щавелевой кислоты. После перемешивания в течение двух часов отфильтровывают, получая 4,5 г целевого оксалата. Т.пл.=165oС.
Б) Оксалат 3-диэтиламино-N-(4-изопропил-3- аминофенил)пропионамида
Раствор 4,5 г полученного на предыдущей стадии нитропроизводного в 100 мл метанола и 10 мл диметилформамида гидрируют в течение 5 часов в присутствии никеля Ренея®. После отфильтровывания катализатора фильтрат концентрируют в вакууме, осаждают путем добавления диизопропилового эфира и перемешивают в течение 1 часа при комнатной температуре. После отфильтровывания получают 3 г целевого оксалата анилина. Т.пл.=127oС.
В) 3-Диэтиламино-N-(4-изопропил-3- гидразинфенил)пропионамид
К смеси 2,7 г полученного на предыдущей стадии амина и 30 мл концентрированной соляной кислоты, охлажденной до 0oС, добавляют 0,67 г нитрита натрия, растворенного в минимальном количестве воды, и перемешивают в течение 1 часа при температуре 0oС. Затем добавляют 5 г дитионита натрия, растворенного в минимальном количестве воды, и перемешивают еще 30 минут при 0oС. Добавляют 50 г ацетата натрия в виде порошка и перемешивают 30 минут при 0oС. Добавляют 50 г воды и экстрагируют некоторые примеси с помощью этилацетата. Водную фазу насыщают хлоридом натрия, повышают рН-значение до 9 с помощью 20%-ного раствора гидроксида аммония, экстрагируют дихлорметаном, сушат экстракт над сульфатом натрия и выпаривают в вакууме. Остаток выкристаллизовывается из пентана в течение ночи, его отфильтровывают, получая 3,26 г целевого гидразина. Т.пл.=77-80oС.
Методики получения сложных эфиров формул (IIа) и (II'а)
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.1
Метиловый эфир 1-(4-карбокси-2-изопропилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-CO2H; R2 - 2-изопропил; R3 - H; R4 - CH3)
Смесь, содержащую 0,96 г соединения, полученного в Препаративном примере 2.1, и 1,2 г соединения А, полученного в Препаративном примере 1.1, в 15 мл уксусной кислоты кипятят с обратным холодильником в течение 5 часов. После выдерживания в течение трех дней при комнатной температуре реакционную смесь выливают в смесь воды со льдом. Выпавший осадок отфильтровывают, промывают водой, затем сушат над фосфорным ангидридом. Получают 1,34 г целевого продукта. Т.пл.=228-230oС.
ЯМР-спектр: 0,85 (д, 6Н); 2,55 (сп, 1Н); 3,5 (с, 6Н); 3,75 (с, 3Н); 6,5 (д, 2Н); 6,75 (с, 1Н); 7,05-7,3 (м, 2Н); 7,7 (д, 1Н); 13,05 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.1-БИС
Этиловый эфир 1-(4-карбокси-2-изопропилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
Путем взаимодействия соединения, полученного согласно Препаративному примеру 2.1 или 2.1-бис, с соединением А1, согласно методике Препаративного примера 3.1, после перекристаллизации из этилацетата получают целевой этиловый эфир. Т.пл.=231oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.2
Метиловый эфир 1-[4-[N-метил-N-(3-N',N'- диметиламинопропил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(СН3)(СН2)3N(СН3)2; R2 - 2-изопропил; R3 - H; R4 - CH3)
A) Метиловый эфир 1-(4-хлорформил-2-изопропилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
Готовят смесь, содержащую 26 г соединения, полученного в Препаративном примере 3.1, и 170 мл тионилхлорида, перемешивают ее в течение 1 дня при комнатной температуре, затем выпаривают в вакууме, остаток обрабатывают дихлорметаном, выпаривают. Операцию повторяют 3 раза.
Б) Метиловый эфир 1-[4-[N-метил-N-(3-N',N'- диметиламинопропил)карбамоил] -2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
К 50 мл дихлорметана добавляют 9,3 мл триэтиламина и 9,9 мл N,N,N'-триметил-1,3-пропандиамина. В атмосфере азота добавляют полученный на предыдущей стадии эфир в 280 мл дихлорметана и перемешивают в течение трех с половиной часов при комнатной температуре. После промывки водой (2 раза) высушивают над сульфатом магния и выпаривают в вакууме. Остаток обрабатывают эфиром. После отфильтровывания нерастворившейся части и выпаривания остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении от 95:5:0,5 до 88:12:0,8. Получают 24,5 г целевого продукта.
ЯМР-спектр: 0,95 (д, 6Н); 1,7 (м, 2Н); 1,9-2,4 (м, 8Н); 2,95 (расщепленный синглет, 3Н); 3,5 (м, 2Н); 3,7 (с, 6Н); 3,9 (с, 9Н); 6,7 (д, 2Н); 6,9 (с, 1Н); 7,1-7,5 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.2-БИС
Этиловый эфир 1-[4-[N-метил-N-(3-N',N'- диметиламинопропил)карбамоил]-2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
A) Этиловый эфир 1-(4-хлорформил-2-изопропилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
К 50 мл тионилхлорида, охлажденным до 5oС, добавляют 26 г продукта, полученного в Препаративном примере 3.1 или 3.1-бис, и перемешивают в течение 5 часов при комнатной температуре при барботировании осушенного азота. После выпаривания в вакууме остаток обрабатывают дихлорметаном и выпаривают в вакууме; операцию повторяют 2 раза.
Также можно получить хлорангидрид кислоты согласно нижеописанной методике А'.
А') К раствору 5 г полученного в Препаративном примере 3.1 или 3.1-бис продукта в 50 мл дихлорметана добавляют 2,5 мл тионилхлорида и кипятят с обратным холодильником в течение трех часов, выпаривают в вакууме, остаток обрабатывают дихлорметаном и выпаривают в вакууме; операцию повторяют 2 раза.
Б) Этиловый эфир 1-[4-[N-метил-N-(3-N',N'- диметиламинопропил)карбамоил] -2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
К раствору 9 мл триэтиламина и 9,5 мл N,N,N'-триметил-1,3-пропандиамина в 50 мл дихлорметана в атмосфере осушенного азота добавляют раствор хлорангидрида кислоты, полученного на стадии А, в 220 мл дихлорметана и перемешивают в течение ночи. Реакционную смесь промывают 2 раза водой, водные фазы экстрагируют 2 раза дихлорметаном. Объединенные органические фазы сушат над сульфатом магния и выпаривают в вакууме. Остаток перемешивают с 300 мл эфира, отфильтровывают нерастворившуюся часть, фильтрат обесцвечивают с помощью животного угля и выпаривают, получая 28,8 г целевого продукта в виде масла.
ЯМР-спектр (ДМСО+ТФУК): 1 (д, 6Н); 1,3 (т, 3Н); 1,8-2,1 (м, 2Н); 2,65 (кт, 1Н); 2,7-3,05 (м, 9Н); 3,15 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 4,35 (кд, 2Н); 6,6 (д, 2Н); 6,85 (с, 1Н); 7,2-7,4 (м, 4Н).
Продукт стадии Б может быть получен также согласно нижеописанной методике.
Б') К раствору 1,32 г N,N,N'-триметил-1,3-пропандиамина в 37,5 мл 3 н. раствора гидроксида натрия добавляют раствор хлорангидрида кислоты, полученного на стадии А', в 37,5 мл дихлорметана. После перемешивания в течение 1 часа добавляют 25 мл хлороформа и 25 мл воды, декантируют, органическую фазу отделяют, сушат над сульфатом натрия и выпаривают в вакууме, получая 6,1 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.3
Метиловый эфир 1-[4-[N-(2-цианоэтил)карбамоил] -2- изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CONHCH2CH2CN; R2 - 2-изопропил; R3 - H; R4 - CH3)
0,935 г 3-Аминопропионитрил-полуфумарата смешивают с 3,7 мл 1,3 н. раствора гидроксида натрия, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и выпаривают растворитель в вакууме. Остаток обрабатывают с помощью 6 мл дихлорметана, добавляют 0,96 мл триэтиламина, затем медленно добавляют раствор 2,45 г соединения, полученного на стадии А Препаративного примера 3.2, в 30 мл дихлорметана. Перемешивают в течение ночи при комнатной температуре и в атмосфере азота. Отфильтровывают нерастворившуюся часть, фильтрат промывают водой, насыщенным раствором гидрокарбоната натрия, водой, сушат над сульфатом натрия и выпаривают растворитель в вакууме. Получают 2,24 г целевого продукта. Т.пл.=114-116oС (разложение).
ЯМР-спектр: 1 (д, 6Н); 2,7 (мт, 1Н); 2,8 (т, 2Н); 3,5 (к, 2Н); 3,65 (с, 6Н); 3,9 (с, 3Н); 6,7 (д, 2Н); 6,9 (с, 1Н); 7,3-7,4 (м, 2Н); 7,75 (д, 1Н); 7,9 (с, 1Н); 9 (т, 1Н).
Вводя во взаимодействие соответствующий первичный амин с соединением, полученным на стадии А Препаративного примера 3.2, и следуя методике, описанной в Препаративном примере 3.2, стадия Б, получают сложные эфиры, описанные в таблице 2.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.12
Метиловый эфир 5-(2,6-диметоксифенил)-1-(2-изопропил-4-сульфофенил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-SO3H; R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение 5 часов кипятят с обратным холодильником смесь, содержащую 0,82 г 3-изопропил-4- гидразинбензолсульфокислоты, полученной в Препаративном примере 2.2, и 1,26 г соединения А, полученного в Препаративном примере 1.1, в 15 мл уксусной кислоты. После выпаривания остаток растворяют в дихлорметане, полученный раствор промывают с помощью 1 н. соляной кислоты и обесцвечивают активным углем, затем сушат, выпаривают, остаток после выпаривания перемешивают при кипячении с обратным холодильником с диизопропиловым эфиром и отфильтровывают при нагревании. Получают 1,28 г целевого продукта.
ЯМР-спектр: 1 (д, 6Н); 2,6 (мт, 1Н); 3,6 (с, 6Н); 3,8 (с, 3Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7,15 (д, 1Н); 7,3 (т, 1Н); 7,4 (д, 1Н); 7,5 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.13
Метиловый эфир 5-(2,6-диметоксифенил)-1-[2-изопропил-4-(N-метил-N-(3-N', N'-диметиламинопропил)аминосульфонил) фенил] пиразол-3-карбоновой кислоты (формула (IIа); R1 - -4-SO2N(CH3)(СН2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - CH3)
А) Метиловый эфир 5-(2,6-диметоксифенил)-1-(4- хлорсульфонил-2-изопропилфенил)пиразол-3-карбоновой кислоты
1,08 г Полученного в Препаративном примере 3.12 продукта и 4 мл оксихлорида фосфора перемешивают при комнатной температуре в течение 24 часов, затем продолжают перемешивание при температуре 70oС в течение дополнительных 24 часов. Реакционную среду выпаривают с толуолом (2 раза) и получают 1,6 г целевого продукта.
Б) Метиловый эфир 5-(2,6-диметоксифенил)-1-[2- изопропил-4-(N-метил-N-(3-N', N'-диметиламинопропил)аминосульфонил)фенил]пиразол-3-карбоновой кислоты
К суспензии 1,6 г полученного на предыдущей стадии продукта в 10 мл толуола и 5 мл дихлорметана добавляют 1,8 мл N,N,N'-триметил-1,3-пропандиамина, затем 2 мл триэтиламина. Перемешивают в течение трех часов при комнатной температуре, затем в течение полутора часов при температуре 50oС. После отфильтровывания и концентрирования досуха остаток экстрагируют эфиром, затем этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают в вакууме. Получают 1,13 г целевого продукта.
ЯМР-спектр: 0,85 (д, 6Н); 1,45 (мт, 2Н); 2 (с, 6Н); 2,6 (с+мт, 4Н); 2,85 (т, 2Н); 3,5 (с, 6Н); 3,8 (с, 3Н); 6,5 (д, 2Н); 6,8 (с, 1Н); 7,2 (т, 1Н); 7,4 (д, 1Н); 7,5-7,6 (м, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.14
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-карбокси- 5,6,7,8-тетрагидронафт-1-ил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-CO2H; R2, R3 - -(CH2)4-; R4 - CH3)
Перемешивают в течение двух часов при кипячении с обратным холодильником смесь из 0,67 г гидразина, полученного в Препаративном примере 2.3, и 0,83 г соединения А в 6 мл уксусной кислоты. Экстрагируют дихлорметаном, промывают водой органическую фазу, сушат ее над сульфатом магния и выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:2, получая 0,7 г целевого продукта.
ЯМР-спектр: 1,4-2 (м, 4Н); 2,3-3,1 (м, 4Н); 3,4-4 (м, 9Н); 6,6 (д, 2Н); 6,8-7,6 (м, 4Н); 12,95 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.15
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-[N-(3-N', N'-диметиламинопропил)карбамоил] -5,6,7,8-тетрагидронафт-1- ил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CONH(СН2)3N(СН3)2; R2, R3 - -(CH2)4-; R4 - CH3)
В течение полутора часов при температуре 40oС нагревают раствор 0,7 г продукта, полученного в Препаративном примере 3.14, в 6 мл тионилхлорида и 30 мл дихлорметана. Выпаривают в вакууме, затем полученный хлорангидрид кислоты снова растворяют в 5 мл дихлорметана, охлаждают до 5oС, добавляют 0,225 мл N,N-диметилпропилендиамина и 0,225 мл триэтиламина. После перемешивания в течение двух часов при комнатной температуре смесь выпаривают в вакууме, остаток снова растворяют в дихлорметане, промывают полученный раствор водой, сушат над сульфатом магния и выпаривают в вакууме, получая 0,79 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.16
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-сульфо-5,6,7,8-тетрагидронафт-1-ил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-SO3H; R2, R3 - -(CH2)4 -; R4 - CH3)
В течение 4,5 часов кипятят с обратным холодильником смесь 0,5 г гидразина, полученного в Препаративном примере 2.4, и 0,62 г соединения А в 4 мл уксусной кислоты. Выпаривают в вакууме, остаток снова растворяют в дихлорметане, полученный раствор промывают 2 раза с помощью 1 н. соляной кислоты, сушат над сульфатом магния и выпаривают, получая 1 г целевого продукта.
ЯМР-спектр: 1,7 (уш.с, 4Н); 2,5 (м, 2Н); 3,2 (м, 2Н); 3,7 (с, 6Н); 3,95 (с, 3Н); 6,7 (д, 2Н); 6,8-6,9 (м, 2Н); 7,35 (т, 1Н); 7,55 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.17
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-N-метил-N-(2-N',N'-диметиламиноэтил)аминосульфонил] -5,6,7,8-тетрагидронафт-1-ил] пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-SO2N(СН3)(CH2)2N(СН3)2; R2, R3 - (CH2)4; R4 - CH3)
Смесь, содержащую 1 г кислоты, полученной в Препаративном примере 3.16, и 3 мл РОСl3, перемешивают в течение 5 часов при комнатной температуре, затем в течение 3,5 часов при температуре 70oС. Выпаривают в вакууме, затем добавляют толуол и выпаривают в вакууме (2 раза). Полученный таким образом в 10 мл дихлорметана раствор сульфонилхлорида добавляют к раствору 1,5 мл N,N, N'-триметилэтилендиамина и 1,5 триэтиламина в 10 мл дихлорметана при температуре 5oС. Перемешивают в течение четырех дней при температуре 10oС, отфильтровывают и выпаривают досуха. После растворения снова в дихлорметане, промывки водой, экстракции водной фазы дихлорметаном органическую фазу сушат над сульфатом магния и выпаривают в вакууме, получая 1,07 г целевого сульфонамида. Т.пл.=90oС.
ЯМР-спектр: 1,6-1,8 (м, 4Н); 2,1 (с, 6Н); 2,35 (т, 2Н); 2,4-2,6 (м, 2Н); 2,9 (с, 3Н); 3,1 (мт, 2Н); 3,2 (т, 2Н); 3,6 (с, 6Н); 3,9 (с, 3Н); 6,7 (д, 2Н); 6,9 (с, 1Н); 7,1 (д, 1Н); 7,4 (т, 1Н); 7,7 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.18
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-карбамоил-2-метилфенил)пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CONH2; R2 - 2-СН3; R3 - H; R4 - CH3)
Суспензию 0,39 г гидразина, полученного в Препаративном примере 2.5, и 450 мг соединения А в 5 мл уксусной кислоты кипятят с обратным холодильником в течение 8 часов. К реакционной среде добавляют 100 мл воды, выпавший осадок отфильтровывают, затем вносят в 10 мл диизопропилового эфира и кипятят с обратным холодильником в течение 30 минут, после чего отфильтровывают. Получают 300 мг целевого продукта. Т.пл.=219oС. Путем отфильтровывания водной фазы спустя 24 часа получают вторую порцию в количестве 70 мг целевого продукта в форме иголок.
ЯМР-спектр: 2,05 (с, 3Н); 3,5 (с, 6Н); 3,8 (с, 3Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7 (д, 1Н); 7,2 (т, 1Н); 7,4 (уш.с, 1Н); 7,6 (д, 1Н); 7,7 (с, 1Н); 7,9 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.19
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-карбокси-2,3-диметилфенил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-CO2H; R2 - 2-CH3; R3 - 3-CH3; R4 - CH3)
При перемешивании в течение трех часов кипятят с обратным холодильником смесь 4,5 г продукта, полученного в Препаративном примере 2.6, и 12 г соединения А в 50 мл уксусной кислоты. Осаждают путем выливания смеси на 300 мл воды со льдом, отфильтровывают выпавший осадок, промывают его с помощью 50 мл воды. После перемешивания в 50 мл эфира, отфильтровывания, высушивания в вакууме над Р2О5 получают 5 г целевого продукта. Т.пл.=240oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.20
Метиловый эфир 5-(2,6-диметоксифенил)-1-[2,3-диметил-4-[N-(2-N',N'-диметиламиноэтил)карбамоил]фенил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CONH(СН2)2N(СН3)3; R2 - 2-CH3; R3 - 3-CH3; R4 - CH3)
После нагревания в течение полутора часов при температуре 40oС раствора 2 г продукта, полученного в Препаративном примере 3.19, в 10 мл тионилхлорида и 40 мл дихлорметана этот раствор выпаривают в вакууме, полученный хлорангидрид кислоты снова растворяют в 10 мл дихлорметана и полученный раствор выливают в раствор 0,54 мл N,N-диметиламиноэтилендиамина в 10 мл дихлорметана. Добавляют 0,68 мл триэтиламина и перемешивают в течение двух часов при комнатной температуре. После выпаривания в вакууме, экстракции с помощью 100 мп дихлорметана, промывки водой, высушивания над сульфатом магния и выпаривания в вакууме получают 1,8 г целевого продукта.
ЯМР-спектр: 1,9 (с, 3Н); 2,2 (с+с, 9Н); 2,4 (т, 2Н); 3,4 (м, 2Н); 3,6 (с, 6Н); 3,8 (с, 3Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 6,9-7,1 (д.д, 2Н); 7,3 (т, 1Н); 8,2 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.21
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-карбокси-2-метоксифенил)пиразол-3-карбоновой кислоты (формула (II'а); R'1 - 4-CO2H; R2 - 2-OCH3; R3 - H; R4 - СН3)
Перемешивают в течение 6 часов при кипячении с обратным холодильником смесь 4,8 г продукта, полученного в Препаративном примере 2.7, и 5,6 г соединения А в 60 мл уксусной кислоты. После осаждения в 300 мл воды со льдом, перемешивания в течение 30 минут, отфильтровывания, промывки водой, затем пентаном и высушивания осадка получают 6 г целевого продукта. Т.пл.=210oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.22
Метиловый эфир 5-(2,6-диметоксифенил)-1-[-4-[N-метил-N-(2-N',N'-диэтиламиноэтил)карбамоил] -2-метоксифенил] пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(СН3)(СН2)2N(C2H5)2; R2 - 2-ОСН3; R3 - H; R4 - CH3)
В течение 3,5 часов кипятят с обратным холодильником раствор 2,5 г полученного в Препаративном примере 3.21 продукта в 30 мл дихлорметана и 5 мл тионилхлорида. После выпаривания в вакууме с последующими двумя азеотропными отгонками с 20 мл дихлорметана образовавшийся хлорангидрид кислоты снова растворяют в 40 мл дихлорметана, добавляют к раствору, содержащему 1,1 мл N, N-диэтил-N'-метилэтилендиамина и 1 мл триэтиламина в 40 мл дихлорметана, и перемешивают в течение 15 часов при комнатной температуре. После выпаривания в вакууме остаток хроматографируют на диоксиде кремния, элюируя градиентом растворителей от смеси дихлорметана с метанолом в объемном соотношении 90:10 до смеси дихлорметана с метанолом и водой в объемном соотношении 80:20:0,7, получая 1,73 г целевого продукта.
ЯМР-спектр: 07-1,1 (мт, 6Н); 2,2-3,7 (мт, 20Н); 3,85 (с, 3Н); 6,55 (д, 2Н); 6,8 (с, 1Н); 6,95 (мт, 2Н); 7,3 (мт, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.23
Этиловый эфир 1-(4-карбокси-2-хлорфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (II'а); R'1 - 4-CO2H; R2 - 2-Cl; R3 - H; R4 - CH3)
3-Хлор-4-гидразинбензойная кислота описана в патенте США 3959309. Эту кислоту в количестве 5,6 г и 8,75 г соединения А1 в 100 мл уксусной кислоты кипятят с обратным холодильником в течение 6 часов. Реакционную смесь выливают на 500 мл воды со льдом, отфильтровывают выпавший осадок, затем промывают этот последний водой, пентаном и диизопропиловым эфиром. После этого его высушивают в вакууме, получая 2 г целевого соединения.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.24
Этиловый эфир 5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N',N'-диэтиламиноэтил)карбамоил]-2-хлорфенил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(CH3)(CH2)2N(C2H5)2; R2 - 2-Cl; R3 - H; R4 - СН3)
В течение четырех часов кипятят с обратным холодильником раствор 2,63 г полученного в Препаративном примере 3.23 продукта в 30 мл дихлорметана и 4,5 мл тионилхлорида. После выпаривания в вакууме с последующими двумя азеотропными отгонками с 20 мл дихлорметана полученный хлорангидрид кислоты снова растворяют в 40 мл дихлорметана, затем выливают полученный раствор в раствор 1,1 мл N,N-диэтил-N'-метилэтилендиамина и 1 мл триэтиламина в 40 мл толуола и перемешивают в течение 15 часов при комнатной температуре. После выпаривания в вакууме, экстракции остатка с помощью 100 мл дихлорметана, двух промывок водой, высушивания над сульфатом натрия и выпаривания в вакууме остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 90:10, затем смесью дихлорметана с метанолом и водой в объемном соотношении 90:10:0,5, получая 2,6 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 2,8 (с, 3Н); 3-3,7 (м, 17Н); 6,6 (д, 2Н), 6,8 (с, 1Н); 7,05 (м, 2Н); 7,3 (м, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.25
Этиловый эфир 5-(2,6-диметоксифенил)-1-[4-(N-(2-морфолиноэтил)карбамоил)-2-хлорфенил] пиразол-3-карбоновой кислоты (формула (IIa); 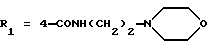 ; R2 - 2-Cl; R3 - H; R4 - CH3)
; R2 - 2-Cl; R3 - H; R4 - CH3)
Перемешивают в течение четырех часов при кипячении с обратным холодильником раствор 2,63 г продукта, полученного в Препаративном примере 3.23, в 4,5 мл тионилхлорида и 30 мл дихлорметана. После выпаривания в вакууме с последующими двумя азеотропными выпариваниями с 30 мл толуола образовавшийся хлорангидрид кислоты снова растворяют в 40 мл дихлорметана, добавляют полученный раствор к раствору 0,9 мл 4-(2-аминоэтил)морфолина и 1 мл триэтиламина в 4 мл толуола. После перемешивания в течение 15 часов при комнатной температуре, выпаривания в вакууме, растворения снова остатка в 200 мл дихлорметана, промывки с помощью 100 мл насыщенного раствора хлорида натрия, высушивания над сульфатом натрия и выпаривания в вакууме получают 3 г целевого продукта.
ЯМР-спектр: 1,3 (т, 3Н); 2,3-2,6 (мт, 6Н); 3,3 (мт, 2Н); 3,5 (мт, 10Н); 4,3 (кд, 2Н); 6,55 (д, 2Н); 6,8 (с, 1Н); 7,1-7,4 (мт, 3Н); 6,75 (д.д, 1Н); 6,9 (д.д, 1Н); 8,6 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.26
Метиловый эфир 5-(2,6-диметоксифенил)-1-(3-хлор-4-цианофенил)пиразол-3-карбоновой кислоты (формула (II'а); R'1 - 4-CN; R2 - 3-Cl; R3 - H; R4 - CH3)
На водяной бане в течение двух часов нагревают смесь, содержащую 4,2 г соединения, полученного в Препаративном примере 2.8, и 6 г соединения А в 10 мл уксусной кислоты. Реакционную смесь выливают в воду со льдом, образовавшийся осадок отфильтровывают, затем высушивают его, получая 3,78 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.27
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-карбокси-2-циклопропилфенил] пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-CO2H; R2 - 2-циклопропил; R3 - H; R4 - CH3)
В течение 7 часов при кипячении с обратным холодильником перемешивают смесь 1,28 г соединения А и 1 г продукта, полученного в Препаративном примере 2.9, в 12 мл уксусной кислоты. Осаждают в 120 мл воды со льдом, осадок отфильтровывают, промывают водой и высушивают в вакууме над фосфорным ангидридом, получая 1,27 г целевого продукта.
ЯМР-спектр: 0,5 (м, 2Н); 0,8 (м, 2Н); 1,3 (т, 3Н); 1,5 (м, 1Н); 3,6 (с, 6Н); 4,3 (кд, 2Н); 6,6 (д, 2Н); 6,9 (с, 1Н); 7,3 (м, 3Н); 7,7 (д, 1Н); 13 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.28
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N',N'-диэтиламиноэтил)карбамоил] -2-циклопропилфенил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(СН3)(СН2)2N(C2H5)2; R2 - 2-циклопропил; R3 - H; R4 - CH3)
В течение 5 часов нагревают при 100oС раствор 1,27 г продукта, полученного в Препаративном примере 3.27, в 28 мл толуола и 2 мл тионилхлорида. После выпаривания в вакууме с последующими двумя азеотропными отгонками с 30 мл толуола полученный хлорангидрид кислоты снова растворяют в 19 мл дихлорметана, медленно выливают в раствор 0,53 мл N,N-диэтил-N'-метилэтилендиамина и 0,61 мл триэтиламина в 1,9 мл толуола. После перемешивания в течение 12 часов при комнатной температуре, выпаривания в вакууме, экстракции остатка с помощью 100 мл дихлорметана, промывки экстракта водой, высушивания его над сульфатом натрия и выпаривания в вакууме получают 1,29 г целевого продукта.
ЯМР-спектр: 0,4-1 (м, 10Н); 1,2 (т, 3Н); 1,4 (мт, 1Н); 2,1-3 (м, 9Н); 3,5 (м, 8Н); 4,3 (кд, 2Н); 6,5 (д, 2Н); 6,6 (с, 1Н); 6,8 (с, 1Н); 6,9-7,2 (мт, 3Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.29
Метиловый эфир 5-(2,6-диметоксифенил)-1-[3-карбокси-4-хлорфенил] пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 3-CO2H; R2 - 4-Cl; R3 - Н; R4 - CH3)
При перемешивании в течение 5 часов кипятят с обратным холодильником смесь 4 г продукта, полученного в Препаративном примере 2.10, и 4,6 г соединения А в 60 мл уксусной кислоты. Выливают на 100 мл воды со льдом, осадок отфильтровывают, промывают с помощью 5 мл воды, затем с помощью 20 мл эфира, высушивают его в вакууме, получая 3 г целевого продукта. Т.пл.=206oС.
ЯМР-спектр: 3,55 (с, 6Н); 3,85 (с, 3Н); 6,7 (д, 2Н); 6,9 (с, 1Н); 7,35 (д.д, 1Н); 7,4 (т, 1Н); 7,55 (д, 1Н); 7,7 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.30
Метиловый эфир 5-(2,6-диметоксифенил)-1-[3-[N-метил-N-(2-N',N'-диметиламиноэтил)карбамоил] -4-хлорфенил] пиразол-3-карбоновой кислоты (формула (IIа); R1 - 3-CON(CH3)(CH2)2N(CH3)2; R2 - 4-Cl; R3 - Н; R4 - CH3)
При перемешивании в течение 3,5 часов при температуре 60oС нагревают раствор 1,5 г полученного в Препаративном примере 3.29 продукта в 20 мл дихлорметана и 2,6 мл тионилхлорида. После выпаривания в вакууме образовавшийся хлорангидрид кислоты снова растворяют в 10 мл дихлорметана и полученный раствор выливают в раствор 0,5 мл N,N,N'- триметилэтилендиамина и 0,6 мл триэтиламина в 2,5 мл толуола, затем перемешивают все в течение 15 часов при комнатной температуре. После выпаривания в вакууме, растворения остатка в 100 мл дихлорметана, промывки с помощью 100 мл воды, высушивания над сульфатом натрия и выпаривания в вакууме получают 0,8 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.31
Метиловый эфир 5-(2,6-диметоксифенил)-1-[5-карбокси-2-метилфенил]пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 5-CO2H; R2 - 2-CH3; R3 - H; R4 - CH3)
При перемешивании в течение полутора часов кипятят с обратным холодильником смесь 6,15 г продукта, полученного в Препаративном примере 2.11, и 7,76 г соединения А в 100 мл уксусной кислоты. После концентрирования в вакууме до объема 5 мл, осаждения в 20 мл воды со льдом, отфильтровывания осадка, промывки его с помощью 5 мл воды, высушивания над фосфорным ангидридом в вакууме при 80oC получают 9,55 г целевого продукта. Т.пл.=189oС.
ЯМР-спектр: 1,20 (т, 3Н); 3,55 (с, 6Н); 4,26 (кд, 2Н); 6,58 (д, 2Н); 6,80 (с, 1Н); 7,20 (т, 1Н); 7,60 (д, 1Н); 7,70 (д, 1Н); 7,80 (д.д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.32
Метиловый эфир 5-(2,6-диметоксифенил)-1-[5-[N-метил-N-(3-N',N'-диметиламинопропил)карбамоил] -2-метилфенил] пиразол-3-карбоновой кислоты (формула (IIа); R1 - 5-CON(CH3)(CH2)3N(CH3)2; R2 - CH3; R3 - H; R4 - CH3)
При комнатной температуре в течение 5 часов перемешивают раствор 2 г полученного в Препаративном примере 3.31 продукта в 10 мл тионилхлорида. После выпаривания в вакууме с последующими тремя азеотропными отгонками с 30 мл дихлорметана полученный хлорангидрид кислоты снова растворяют в 25 мл дихлорметана, выливают в раствор 0,81 мл N,N,N'-триметил-1,3-пропандиамина и 0,77 мл триэтиламина в 5 мл дихлорметана и перемешивают в течение двух часов при комнатной температуре. После двукратной промывки с помощью 20 мл воды, двукратной экстракции водных фаз с помощью 50 мл дихлорметана дихлорметановые фазы промывают два раза по 20 мл 5%-ным раствором гидрокарбоната натрия, затем с помощью 20 мл воды, после чего сушат над сульфатом магния и выпаривают в вакууме, получая 2,3 г целевого продукта (масло).
ЯМР-спектр: 1,9 (м, 2Н); 2,1 (с, 3Н); 2,6 (с, 3Н); 2,8 (с, 6Н); 2,9-3,2 (м, 2Н); 3,45 (м, 2Н); 3,6 (с, 6Н); 3,8 (с, 3Н); 6,6 (д, 2Н); 6,85 (с, 1Н); 7,05 (уш.с, 1Н); 7,2-7,4 (м, 3Н).
Следуя вышеописанным методикам, получают сложные эфиры формулы (IIа), описанные в таблице 3, либо из сложного эфира формулы (II'а), замещенного радикалом R'1, либо путем воздействия соответствующего гидразина на соединение А или соединение А1.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.57
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-N-метил-N-[3-N'-метил-N'-(трет. -бутоксикарбонил)амино]пропил]карбамоил]-2-изопропилфенил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(СН3)(СН2)3N(СН3)СОО-трет.-бутил; R2 - 2-изопропил; R3 - H; R4 - CH3)
А) Трет.-бутиловый эфир N-метил-N-(3-метиламинопропил)карбаминовой кислоты
Раствор 4,19 г N,N'-диметил-1,3-пропандиамина в 80 мл тетрагидрофурана охлаждают до 0oС, добавляют раствор 2,68 г ди-трет.-бутилдикарбоната в 25 мл тетрагидрофурана и перемешивают в течение 72 часов при комнатной температуре. Нерастворившуюся часть отфильтровывают и фильтрат концентрируют в вакууме. Остаток экстрагируют дихлорметаном, органическую фазу промывают три раза водой, сушат над сульфатом магния и выпаривают растворитель в вакууме. Получают 1,6 г целевого продукта в виде желтого масла.
Б) Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-[N-метил-N-[3-[N'-метил-N'-(трет.-бутоксикарбонил)амино]пропил] карбамоил]-2-изопропилфенил]пиразол-3-карбоновой кислоты
К раствору 1,31 г полученного на предыдущей стадии соединения и 0,9 мл триэтиламина в 4 мл дихлорметана при комнатной температуре и в атмосфере азота добавляют раствор 2,6 г соединения, полученного на стадии А Препаративного примера 3.2, и перемешивают в течение ночи при комнатной температуре. Реакционную смесь промывают два раза водой, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью толуола с этилацетатом в объемном соотношении от 65:35 до 60:40. Получают 2,85 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,0 (д, 6Н); 1,4 (д, 9Н); 1,6-1,9 (м, 2Н); 2,7 (мт, 1Н); 2,7-3,55 (д+уш.с+м, 10Н); 3,65 (с, 6Н); 3,9 (с, 3Н); 6,65 (д, 2Н); 6,9 (с, 1Н); 7,3-7,45 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.58
Этиловый эфир 5-(2,6-диметоксифенил)-1-[4-(4- метилфенилсульфониламино)-2-изопропилфенил]пиразол-3-карбоновой кислоты (формула (IIа); 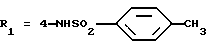 ; R2 - 2-изопропил; R3 - H; R4 - CH3)
; R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение 1 часа при температуре 70oС нагревают смесь 3,44 г соединения, полученного в Препаративном примере 2.18, и 2,6 г соединения А1 в 50 мл уксусной кислоты. Реакционную смесь выливают в воду, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:0,5. Получают 1,26 г целевого продукта.
ЯМР-спектр: 0,7 (д, 6Н); 1,22 (т, 3Н); 2,2-2,5 (м, 4Н); 3,45 (с, 6Н); 4,2 (т, 2Н); 6,46 (д. 2Н); 6,69 (с, 1Н); 6,75-6,9 (м, 2Н); 7,0 (д, 1Н); 7,15-7,3 (м, 3Н); 7,5 (д, 2Н); 10,2 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.59
Этиловый эфир 5-(2,6-диметоксифенил)-1-[4-[4- метилфенилсульфонил-N-(3-диэтиламинопропил)амино] -2-изопропилфенил] пиразол-3-карбоновой кислоты (формула (IIа); 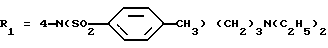 ; R2 - 2-изопропил; R3 - H; R4 - CH3)
; R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение двух часов при температуре 80oС нагревают смесь 0,65 г соединения, полученного в Препаративном примере 3.58, 0,338 г (3-хлорпропил)диэтиламина и 0,65 г карбоната калия в 5 мл диметилформамида. Реакционную смесь выливают в воду, экстрагируют этилацетатом, сушат этилацетатный слой над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:5. Получают 0,57 г целевого продукта.
ЯМР-спектр: 0,8 (уш. с, 6Н); 0,95 (т, 6Н); 1,4 (т, 3Н); 1,5 (кт, 2Н); 2,3-2,65 (м, 10Н); 3,5-3,8 (м, 8Н); 4,35 (кд, 2Н); 6,7 (д, 2Н); 6,75 (д, 1Н); 6,9 (с, 1Н); 7,05 (д.д, 1Н); 7,2-7,6 (м, 6Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.60
Этиловый эфир 5-[2-(циклопропилметилокси)-6-метоксифенил]-1-(4-карбокси-2-изопропилфенил)пиразол-3-карбоновой кислоты (формупа (II'a); R'1 - 4-CO2H; R2 - 2-изопропил; R3 - H; 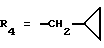 )
)
В течение 8 часов при температуре 80oС нагревают смесь 5,26 г соединения, полученного в Препаративном примере 1.2, и 3,9 г соединения, полученного в Препаративном примере 2.1, в 50 мл уксусной кислоты. Реакционную смесь выливают в смесь воды со льдом, выпавший осадок отфильтровывают под вакуумом и промывают его водой, затем пентаном. Осадок обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 5,2 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.61
Этиловый эфир 5-[2-(цикпопропилметилокси)-6-метоксифенил] -1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил]-2-изопропилфенил]пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CON(CH3)(CH2)3N(CH3)2; R2 - изопропил; R3 - H; 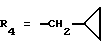 )
)
Это соединение получают согласно методикам, описанным в стадиях А и Б Препаративного примера 3.2, исходя из 5,2 г соединения, полученного в Препаративном примере 3.60, и 2.4 мл тионилхлорида, затем 1,39 г N,N,N'-триметил-1,3-пропандиамина и 1,5 мл триэтиламина в 10 мл толуола. Очистку осуществляют путем хроматографии на диоксиде кремния, элюируя дихлорметаном, затем смесью дихлорметана с метанолом в объемном соотношении 88:2. Получают 3,8 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.62
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-(4-метилфенилсульфониламино)-2-изобутилфенил]пиразол-3-карбоновой кислоты (формула (IIа); 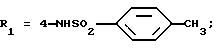 R2 - 2-изобутил; R3 - H; R4 - СН3)
R2 - 2-изобутил; R3 - H; R4 - СН3)
В течение 45 минут кипятят с обратным холодильником смесь 1,9 г соединения, полученного в Препаративном примере 2.20, и 1,6 г соединения А в 30 мл уксусной кислоты. После охлаждения до комнатной температуры реакционную смесь выливают в воду, выпавший осадок отфильтровывают под вакуумом и высушивают его. Получают 1 г целевого продукта после перекристаллизации из пропан-2-ола. Т.пл.=224oС.
ЯМР-спектр: 0,55 (д, 6Н); 1,5 (мт, 1Н); 1,9 (д, 2Н); 2,3 (с, 3Н); 3,45 (с, 6Н); 3,8 (с, 3Н); 6,5 (д, 2Н); 6,75 (с, 1Н); 6,8 (д, 1Н); 6,9 (д.д, 1Н), 7,05 (д, 1Н); 7,15-7,7 (м, 5Н); 10,25 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.63
Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-(4-метилфенилсульфониламино)-2-циклопентилфенил] пиразол-3-карбоновой кислоты (формула (IIа); 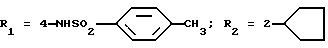 ; R3 - H; R4 - CH3)
; R3 - H; R4 - CH3)
В течение 1 часа при температуре 80oС нагревают смесь 2,42 г соединения, полученного в Препаративном примере 2.21, и 2,92 г соединения А в 50 мл уксусной кислоты. Реакционную смесь выливают в воду, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:1. Получают 0,95 г целевого продукта (после порошкования в эфире). Т.пл.=200-230oС.
ЯМР-спектр: 1,0-1,8 (м, 8Н); 2,37 (с, 3Н); 3,1-3,7 (м, 7Н); 3,82 (с, 3Н); 6,55 (д, 2Н); 6,79 (с, 1Н); 6,85-7,0 (м, 2Н); 7,05 (д, 1Н); 7,21-7,42 (м, 3Н), 7,58 (д, 2Н); 10,3 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.64
Этиловый эфир 5-(2,6-диметоксифенил)-1-(4-карбокси-3-изопропилфенил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-CO2H; R2 - 3-изопропил; R3 - H; R4 - CH3)
В течение 5 часов кипятят с обратным холодильником смесь 0,36 г соединения, полученного в Препаративном примере 2.22, и 0,48 г соединения А1 в 10 мл уксусной кислоты. Реакционную смесь выливают в 160 мл воды со льдом, выпавший осадок отфильтровывают под вакуумом, промывают его водой и высушивают. Получают 0,52 г целевого продукта. Т.пл.=180oС (разложение).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.65
Этиловый эфир 1-[4-[N-(2-диэтиламиноэтил)карбамоил]-3-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (IIа); R1 - 4-CONH(CH2)2N(C2H5)2; R2 - 3-изопропил; R3 - H; R4 - СН3)
Это соединение получают согласно методике, описанной в стадиях А и Б Препаративного примера 3.2, исходя из 0,5 г соединения, полученного в Препаративном примере 3.64, и 5 мл тионилхлорида в 20 мл хлороформа, затем 0,2 мл N, N-диэтилэтилендиамина и 0,8 мл триэтиламина в 20 мл хлороформа. Получают 0,37 г целевого продукта (после кристаллизации из этилацетата). Т.пл.=130oС (разложение).
ПРЕПАРАТИВНЫЙ ПРИМЕР 3.66
Метиловый эфир 5-(2,6-диметоксифенил)-1-(4-нитро- 5,6,7,8-тетрагидронафт-1-ил)пиразол-3-карбоновой кислоты (формула (II'a); R'1 - 4-NO2; R2, R3 - -(CH2)4-; R4 - CH3)
В течение двух часов кипятят с обратным холодильником смесь 6,3 г соединения, полученного в Препаративном примере 2.23, и 7,45 г соединения А в 150 мл уксусной кислоты. После охлаждения до комнатной температуры добавляют 100 мл воды и 30 мл метанола и выкристаллизовавшийся продукт отфильтровывают под вакуумом (отсасывают). Получают 4,6 г целевого продукта. Т.пл.=212oС.
ЯМР-спектр: 1,7 (мт, 4Н); 2,55 (мт, 2Н); 2,82 (мт, 2Н); 3,65 (с, 6Н); 3,85 (с, 3Н); 6,65 (д, 2Н); 6,9 (с, 1Н); 7,02 (д, 1Н); 7,33 (т, 1Н); 7,67 (д, 1Н).
ПРЕПАРАТИВНЫИ ПРИМЕР 3.67
Метиловый эфир 5-(2,6-диметоксифенил)-1-(5-(3-диэтиламино)пропаноиламино)-2-изопропилфенил)пиразол-3-карбоновой кислоты (формула (IIа); R1 - 5-NHCO(СН2)2N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - CH3)
Это соединение получают из гидразина, получаемого в Препаративном примере 2.24.
ЯМР-спектр (ДМСО+ТФУК): 0,65-1,35 (м, 12Н); 2,5 (кт, 1Н); 2,8 (т, 2Н); 3,15 (кд, 4Н); 3,36 (т, 2Н); 3,57 (с, 6Н); 3,8 (с, 3Н); 6,5 (д, 2Н); 6,78 (с, 1Н); 7,1-7,4 (м, 3Н); 7,8 (д, 1Н).
Методики получения кислот формул (II) и (II')
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.1
1-[4-[N-метил-N-(3-N',N'-диметиламинопропил)карбамоил]-2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(СН3)(СН2)3N(СН3)2; R2 - 2-изопропил; R3 - Н; R4 - CH3)
23 г Соединения, полученного в Препаративном примере 3.2, в 230 мл диоксана смешивают с 6,2 г гидроксида калия в 6 мл воды. Полученную смесь кипятят с обратным холодильником в течение 3,5 часов. После охлаждения реакционную смесь выпаривают, снова растворяют в минимальном количестве воды, промывают три раза эфиром, затем подкисляют до рН 4 путем добавления концентрированной соляной кислоты; водную фазу выпаривают, затем снова остаток растворяют в минимальном количестве этанола и отфильтровывают КСl (2 раза). После выпаривания получают 23,93 г целевого продукта в виде светло-желтой пены. Т.пл.=128oС (разложение).
ЯМР-спектр: 0,95 (д, 6Н); 1,95 (мт, 2Н); 2,45-3,3 (м, 12Н); 3,35-3,8 (м, 8Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7-7,5 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.1-БИС
Калиевая соль 1-[4-[N-метил-N-(3-N',N'-диметиламинопропил)карбамоил]-2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
К раствору 26,6 г полученного в Препаративном примере 3.2 продукта в 133 мл этанола добавляют раствор 8,07 г КОН в 133 мл воды. Раствор перемешивают в течение восьми часов, затем выдерживают при перемешивании в течение 15 часов и выпаривают в вакууме, получая целевую калиевую соль.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.2
1-[4-[N-(2-Цианоэтил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONHCH2CH2CN; R2 - 2-изопропил; R3 - H; R4 - CH3)
К раствору 3,04 г соединения, полученного в Препаративном примере 3.3, в 30 мл 1,4-диоксана с несколькими каплями метанола добавляют раствор 0,9 г КОН в 3 мл воды и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают водой, водную фазу промывают эфиром, подкисляют до рН 2 путем добавления 10%-ной соляной кислоты, экстрагируют дихлорметаном, органическую
фазу сушат над сульфатом натрия и выпаривают в вакууме растворитель. Получают 2,93 г целевого продукта. Т.пл.=128oС (разложение).
ЯМР-спектр: 1 (д, 6Н); 2,65 (мт, 1Н); 2,8 (т, 2Н); 3,5 (т, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7,2-7,4 (м, 2Н); 7,7 (д.д, 1Н); 7,8 (д, 1Н).
Исходя из сложных эфиров формулы (IIа), описанных в таблице 2, и следуя методике, описанной в Препаративном примере 4.1 или 4.2, получают кислоты формулы (II), описанные в таблице 4.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.11
5-(2,6-Диметоксифенил)-1-[2-изопропил-4-(N-метил-N-(3- N',N'-диметиламинопропил)аминосульфонил)фенил]пиразол-3-карбоновая кислота (формула (II); R1 - 4-SO2N(СН3)(СН2)3N(СН3)2; R2 - 2-изопропил; R3 - H; R4 - СН3)
В течение 6 часов при комнатной температуре перемешивают смесь, содержащую 1,1 г сложного эфира, полученного в Препаративном примере 3.13, и 280 мг гидроксида калия в 10 мл воды. Реакционную среду концентрируют в вакууме до получения объема 5 мл, затем перемешивают в присутствии 100 мл эфира и 3 мл воды. Водную фазу нейтрализуют до рН 6 путем добавления 1 н. соляной кислоты, отфильтровывают, затем сушат над P2O5, получая 860 мг целевого продукта.
ЯМР-спектр: 0,85 (д, 6Н); 1,5 (мт, 2Н); 2,15 (с, 6Н); 2,3 (т, 2Н); 2,6 (с+мт, 4Н); 2,9 (т, 2Н); 3,5 (с, 6Н); 6,5 (д, 2Н); 6,7 (с, 1Н); 7,2 (т, 1Н); 7,4 (д, 1Н); 7,5 (м, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.12
5-(2,6-Диметоксифенил)-1-[4-[N-[3-(N',N'-диметиламино) пропил]карбамоил] -5,6,7,8-тетрагидронафт-1-ил] пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONH(CH2)3N(CH3)2; R2, R3 - -(CH2)4-; R4 - СH3)
В течение трех часов при температуре 40oС нагревают смесь 0,98 г соединения, полученного в Препаративном примере 3.15, и 0,16 г гидроксида лития в 5 м.л метанола и 1 мл воды, затем ее выпаривают в вакууме, нейтрализуют до рН 6 с помощью 1 н. соляной кислоты, после чего экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и выпаривают в вакууме, получая 0,47 г целевого продукта.
ЯМР-спектр: 1,5-2,1 (м, 6Н); 2,3-4 (м, 20Н); 6,5-7,6 (м, 6Н); 8,4 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.13
Калиевая соль 5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N', N'-диметиламиноэтил)аминосульфонил] -5,6,7,8-тетрагидронафт-1-ил]пиразол-3-карбоновой кислоты (формула (II); R1 - 4-SO2N(СН3)(CH2)2N(CH3)2; R2, R3 - -(CH2)4-; R4 - метил)
В течение восьми часов при комнатной температуре перемешивают раствор 0,87 г сложного эфира, полученного в Препаративном примере 3.17, в 5 мл диоксана с 320 мг КОН в 0,5 мл воды. Выпаривают в вакууме и остаток экстрагируют смесью из 10 мл воды, 5 мл этанола и 100 мл эфира. После декантации полученную смолу порошкуют три раза в эфире, продукт кристаллизуется. Его отфильтровывают, получая 0,9 г целевой соли.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.14
4-(2,6-Диметоксифенил)-1-(4-карбамоил-2- метилфенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONH2; R2 - 2-CH3; R3 - H; R4 - CH3)
При комнатной температуре в течение двух часов перемешивают раствор, содержащий 0,4 г соединения, полученного в Препаративном примере 3.18, в 5 мл диоксана и 220 мг КОН в 1 мл воды. Раствор подкисляют до рН 1 путем добавления концентрированной соляной кислоты, затем концентрируют в вакууме. Добавляют 5 мл воды, после чего образовавшуюся смолу перемешивают с 50 мл дихлорметана и отфильтровывают выпавший осадок. Получают 330 мг целевого продукта. Т.пл.=275-276oС.
ЯМР-спектр: 2,05 (с, 3Н); 3,55 (с, 6Н); 6,55 (д, 2Н); 6,7 (с, 1Н); 7 (д, 1Н); 7,2 (т, 1Н); 7,4 (уш.с, 1Н); 7,55 (д, 1Н); 7,7 (с, 1Н); 7,9 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.15
5-(2,6-Диметоксифенил)-1-[2,3-диметил-4-[N-(2-N', N'-диметиламиноэтил)карбамоил] фенил]пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONH(CH2)2N(CH3)2; R2 - 2-CH3; R3 - 3-CH3; R4 - CH3)
В течение двух часов при температуре 40oС перемешивают раствор 1,8 г продукта, полученного в Препаративном примере 3.20, и 0,32 г гидроксида лития в 10 мл метанола и 2 мл воды. Доводят значение рН до 6 с помощью 1 н. соляной кислоты и выпаривают в вакууме. После экстракции остатка с помощью 50 мл дихлорметана и выпаривания получают 1,3 г целевого продукта.
ЯМР-спектр: 1,9 (с, 3Н); 2,2 (с+с, 9Н); 2,5 (т, 2Н); 3,4 (кд, 2Н); 3,7 (с, 6Н); 6,6-6,7 (м, 3Н); 6,9-7,1 (м, 2Н); 7,3 (т, 1Н); 8,3 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.16
5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(2-N',N'-диэтиламиноэтил)карбамоил]-2-метоксифенил]пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(CH3)(CH2)3N(C2H5); R2 - 2-OCH3; R3 - H; R4 - CH3)
В течение 6 часов при перемешивании кипятят с обратным холодильником смесь 1,73 г продукта, полученного в Препаративном примере 3.22, 0,3 г гидроксида лития в 400 мл метанола и 6 мл воды, затем подкисляют до рН 2 с помощью концентрированной соляной кислоты. После выпаривания в вакууме, перемешивания в течение 30 минут при комнатной температуре остаточного масла с 400 мл хлороформа, декантации, отделения, высушивания органической фазы над сульфатом натрия и выпаривания получают 1,2 г целевого продукта.
ЯМР-спектр: 1,05-1,4 (мт, 6Н); 3 (уш.с, 3Н); 3,1-3,9 (мт, 13Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7-7,4 (мт, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.17
5-(2,6-Диметоксифенил)-1-[4- [N-метил-N-(2-N', N'- диэтиламиноэтил)карбамоил] -2-хлорфенил]пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(CH3)(CH2)2N(C2H5)2; R2 - 2-Cl; R3 - H; R4 - CH3)
В течение трех дней при комнатной температуре перемешивают смесь 2,6 г продукта, полученного в Препаративном примере 3.24, растворенного в 100 мл этанола, и 0,53 г КОН в 15 мл воды. После подкисления до рН 3 с помощью концентрированной соляной кислоты, выпаривания в вакууме, порошкования остатка в 10 мл воды, отфильтровывания и высушивания в вакууме над фосфорным ангидридом получают 1,55 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.18
5-(2,6-Диметоксифенил)-1-[4-(N-(2-морфолиноэтил)карбамоил)-2-хлорфенил] пиразол-3-карбоновая кислота (формула (II); 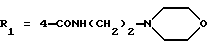 ; R2 - 2-Сl; R3 - Н; R4 - CH3)
; R2 - 2-Сl; R3 - Н; R4 - CH3)
В течение 1 часа при перемешивании при температуре 60oС нагревают раствор 1,5 г продукта, полученного в Препаративном примере 3.25, в 75 мл этанола вместе с раствором 0,38 г КОН в 10 мл воды. После подкисления до рН 4,5 с помощью концентрированной соляной кислоты и выпаривания в вакууме получают 4 г смеси целевого продукта и хлорида калия.
ЯМР-спектр (ДМСО+ТФУК): 2,9-4 (мт, Н); 6,5 (д, 2Н); 6,8 (с, 1Н); 7,1-7,4 (мт, 2Н); 7,8 (д.д, 1Н); 8 (д, 1Н); 9,1 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.19
5-(2,6-Диметоксифенил)-1-(3-хлор-4-цианофенил)пиразол-3-карбоновая кислота (формула (II'); R'1 - 4-CN; R2 - 3-Cl; R3 - H; R4 - CH3)
На водяной бане в течение трех часов нагревают смесь, содержащую 0,5 г полученного в Препаративном примере 3.26 продукта и 60 мг гидроксида лития в 5 мл водного метанола. После охлаждения снижают значение рН до 5 путем добавления 1 н. соляной кислоты. Выпавший осадок отфильтровывают и высушивают, получая 0,36 г целевого соединения.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.20
5-(2,6-Диметоксифенил)-1-(4-карбамоил-3-хлорфенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONH2; R2 - 3-Сl; R3 - Н; R4 - СН3)
В течение 24 часов при комнатной температуре перемешивают смесь, содержащую 0,87 г соединения, полученного в Препаративном примере 4.19, 390 мг карбоната калия и 0,4 мл 30%-ного пероксида водорода в 5 мл диметилсульфоксида. Подкисляют до рН 3 путем добавления 1 н. соляной кислоты, затем добавляют воду, выпавший осадок отфильтровывают и высушивают, получая 0,73 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.21
5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(2-N',N'-диэтиламиноэтил)карбамоил] -2-циклопропилфенил] пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(CH3)(СН2)2N(С2Н5)2; R2 - 2-циклопропил; R3 - H; R4 - CH3)
В течение 22 часов при комнатной температуре перемешивают раствор 1,29 г продукта, полученного в Препаративном примере 3.28, в 26 мл этанола с раствором 0,33 г КОН в 4 мл воды. После подкисления до рН 3 с помощью концентрированной соляной кислоты, выпаривания и азотропного разделения со 100 мл толуола, затем со 100 мл пентана полученный остаток порошкуют в пентане, отфильтровывают и высушивают, получая 1,4 г смеси целевого продукта с хлоридом калия.
ЯМР-спектр: 0,4-1,2 (мт, 14Н); 1,5 (м, 1Н); 2,1-3,8 (мт, 13Н); 6,5 (д, 2Н), 6,7 (уш.с, 2Н); 7 (с, 2Н); 7,2 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.22
5-(2,6-Диметоксифенил)-1-[3-[N-метил-N-(2-N', N'- диметиламиноэтил)карбамоил] -4-хлорфенил]пиразол-3-карбоновая кислота (формула (II); R1 - 3-CON(CH3)(CH2)2N(CH3)2; R2 - 4-Cl; R3 - H; R4 - CH3)
В течение 6 часов при комнатной температуре перемешивают раствор 0,8 г продукта, полученного в Препаративном примере 3.30, в 10 мл диоксана вместе с 0,22 г КОН в 2 мл воды. После выпаривания в вакууме остаток растворяют в 5 мл воды, нейтрализуют полученный раствор до рН 5 с помощью концентрированной соляной кислоты, затем насыщают хлоридом натрия. Экстрагируют 2 раза по 100 мл дихлорметаном, органическую фазу сушат над сульфатом натрия и выпаривают в вакууме, получая 0,43 г целевого продукта.
ЯМР-спектр: 1,9-3,15 (м, 13Н); 3,6 (с, 6Н); 6,75 (д, 2Н); 6,85 (расщ.с, 1Н); 7,1-7,35 (м, 2Н); 7,45 (т, 1Н); 7,57 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.23
5-(2,6-Диметоксифенил)-1-[5-[N-метил-N-(3-N', N'- диметиламинопропил)карбамоил]-2-метилфенил]пиразол-3-карбоновая кислота (формула (II); R1 - 5-CON(CH3)(СН2)3N(СН3)2; R2 - 2-CH3; R3 - H; R4 - CH3)
В течение 15 часов при комнатной температуре перемешивают раствор 2,28 г продукта, полученного в Препаративном примере 3.32, в 10 мл диоксана вместе с раствором 0,65 г КОН в 1,5 мл воды, выпаривают в вакууме, затем остаток растворяют в 20 мл воды и экстрагируют 3 раза по 50 мл эфиром. После подкисления водной фазы до рН 4 путем добавления 1 н. соляной кислоты, азеотропной перегонки с этанолом, остаток порошкуют с помощью 20 мл этанола, отфильтровывают хлорид калия, затем фильтрат выпаривают в вакууме. Повторяют операцию удаления хлорида калия и фильтрат выпаривают в вакууме, получая 2 г целевого продукта.
ЯМР-спектр: 1,9 (м, 2Н); 2,2 (с, 3Н); 2,4-3 (м, 11Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,65 (д, 2Н); 6,85 (с, 1Н); 7,1 (уш.с, 1Н); 7,3-7,5 (м, 3Н).
Следуя вышеописанным методикам, получают кислоты формулы (II), описанные в таблице 5.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.40
1-[4-[N-Этил-N-(2-N', N'-диэтиламиноэтил)карбамоил]-2- изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(C2H5)(CH2)2N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - СН3)
Раствор 0,48 г соединения, полученного в Препаративном примере 4.32, и 0,28 мл этилиодида в 3 мл тетрагидрофурана охлаждают до температуры 5oС, затем порциями добавляют 0,063 г гидрида натрия в виде 60%-ной дисперсии в масле и перемешивают 24 часа при комнатной температуре. Добавляют 0,48 мл смеси тетрагидрофурана с водой в объемном соотношении 50:50 и реакционную смесь концентрируют в вакууме. Остаток обрабатывают водой, водную фазу промывают 2 раза пентаном, водную фазу подкисляют до рН 1 путем добавления 1 н. соляной кислоты, экстрагируют этилацетатом, затем дихлорметаном, органические фазы сушат над сульфатом натрия и выпаривают в вакууме растворители. Получают 0,14 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 0,8-1,3 (м, 15Н); 2,6 (м, 1Н); 3,0-3,8 (м, 16Н); 6,5 (д, 2Н); 6,75 (с, 1Н); 7,2 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.41
Гидрохлорид 1-[4-[[3-(диэтиламино)пирролидин-1-ил] карбонил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (II); 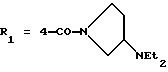 ; R2 - 2-изопропил; R3 - Н; R4 - CH3)
; R2 - 2-изопропил; R3 - Н; R4 - CH3)
К раствору 0,44 г соединения, полученного в Препаративном примере 3.51, в 4 мл диоксана при комнатной температуре добавляют раствор 0,11 г КОН в 0,5 мл воды и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают водой, промывают водную фазу два раза эфиром, подкисляют водную фазу до рН 2 путем добавления 1,2 н. соляной кислоты, добавляют этанол и концентрируют в вакууме. Остаток обрабатывают этанолом, отфильтровывают хлорид калия и фильтрат концентрируют в вакууме. Получают 0,39 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,0 (д, 6Н); 1,15-1,35 (м, 6Н); 2,1-2,45 (м, 2Н); 2,65 (мт, 1Н); 2,9-4,1 (3 м+с, 15Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7,2-7,5 (мт, 3Н); 7,55 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.42
1-[4-[N-(Пропан-2-ил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CONHCH2СН=CH2; R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение 7 часов при комнатной температуре перемешивают смесь 1,72 г соединения, полученного в Препаративном примере 3.54, и 0,78 г гидрата гидроксида лития в 10 мл метанола и 1 мл воды. Концентрируют в вакууме, остаток обрабатывают водой, водную фазу промывают два раза эфиром, подкисляют до рН 2-3 путем добавления 1,2 н. соляной кислоты, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 1,64 г целевого продукта.
ЯМР-спектр: 1,0 (д, 6Н); 2,7 (кт, 1Н); 3,7 (с, 6Н); 3,95 (т, 2Н); 5,1-5,3 (м, 2Н); 5,8-6,1 (м, 1Н); 6,65 (д, 2Н); 6,85 (с, 1Н); 7,25-7,4 (м, 2Н); 7,75 (д, 1Н); 7,9 (с, 1Н); 8,8 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.43
1-[4-[N-метил-N-[3-[N'-метил-N'-(трет. -бутоксикарбонил)амино] пропил]-карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-CON(CH3)(СН2)3N(СН3)СОО-трет.-бутил; R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение 3,5 часов при комнатной температуре перемешивают смесь 2,85 г соединения, полученного в Препаративном примере 3.57, и 0,98 г гидрата гидроксида лития в 20 мл метанола и 1 мл воды. Концентрируют в вакууме, остаток обрабатывают водой, подкисляют до рН 2 путем добавления буферного раствора с рН 2, выпавший осадок отфильтровывают под вакуумом и промывают водой. Получают 2,47 г целевого продукта (после высушивания над фосфорным ангидридом). Т.пл.=112-114oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.44
1-[4-(4-Метилфенилсульфониламино)-2-изопропилфенил] -5-(2,6-диметоксифенил]пиразол-3-карбоновая кислота (формула IIа);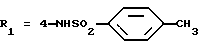 ;
;
R2 - 2-изопропил; R3 - H; R4 - CH3)
В течение трех часов при температуре 60oС нагревают смесь 1,05 г соединения, полученного в Препаративном примере 3.58, и 0,33 г гидрата гидроксида лития в 5 мл метанола и 0,5 мл воды. Реакционную смесь выливают в воду, подкисляют до рН 2-3 путем добавления 10%-ного раствора соляной кислоты, выпавший осадок отфильтровывают под вакуумом и высушивают. Получают 0,92 г целевого продукта.
ЯМР-спектр: 0,7 (д, 6Н); 2,3-2,6 (м, 4Н); 3,55 (с, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 6,85-7,01 (м, 2Н); 7,11 (д, 1Н); 7,25-7,42 (м, 3Н); 7,6 (д, 2Н); 10,3 (с, 1Н); 12,75 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.45
1-(4-Амино-2-изопропилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II'); R'1 - 4-NH2; R2 - 2-изопропил; R3 - H; R4 - метил)
В течение 10 минут кипятят с обратным холодильником смесь 0,9 г соединения, полученного в Препаративном примере 3.58, 11 мл уксусной кислоты и 25 мл 70%-ной хлорной кислоты. Реакционную смесь выливают в смесь воды со льдом, отфильтровывают нерастворимую часть, рН-значение фильтрата доводят до 5 путем добавления 10%-ного раствора гидроксида натрия, отфильтровывают, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и выпаривают растворитель в вакууме. Остаток обрабатывают эфиром и выпавший осадок отфильтровывают под вакуумом. Получают 0,54 г целевого продукта. Т. пл.=190oС (разложение).
ЯМР-спектр: 0,92 (д, 6Н); 2,42 (мт, 1Н); 3,65 (с, 6Н); 5,42 (уш.с, 2Н); 6,3 (д.д, 1Н); 6,4 (д, 1Н); 6,6 (д, 2Н); 6,67 (с, 1Н); 6,85 (д, 1Н); 7,3 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.46
1-[4-[3-(Диэтиламино)пропаноиламино] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-NHCO(CH2)2N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - метил)
В течение 1 часа кипятят с обратным холодильником смесь 0,22 г хлорангидрида 3-диэтиламинопропановой кислоты и 2 мл тионилхлорида в 2 мл дихлорметана, затем концентрируют в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 1 часа при температуре 70oС нагревают смесь 0,47 г соединения, полученного в Препаративном примере 4.45, и 0,95 мл бис(триметилсилил)ацетамида в 5 мл ацетонитрила. После охлаждения до комнатной температуры добавляют вышеполученный хлорангидрид кислоты в виде раствора в дихлорметане, затем 0,17 мл триэтиламина и все перемешивают в течение 1 часа при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают водой, рН-значение водного раствора доводят до 5 путем добавления 10%-ного раствора гидроксида натрия, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток обрабатывают эфиром и выпавший осадок отфильтровывают под вакуумом. Получают 0,27 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 0,91 (д, 6Н); 1,2 (мт, 6Н); 2,55 (мт, 1H); 2,8 (т, 2Н); 3,1-3,22 (м, 4Н); 3,35 (т, 2Н); 3,6 (с, 6Н); 6,58 (д, 2Н); 6,75 (с, 1Н); 7,1-7,3 (м, 2Н); 7,4-7,55 (м, 2Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.47
1-[4-[(3-Диэтиламинопропил)амино] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-NH(CH2)3N(С2Н5)2; R2 - 2-изопропил; R3 - H; R4 - метил)
В течение 10 минут кипятят с обратным холодильником смесь 0,55 г соединения, полученного в Препаративном примере 3.59, 6,5 мл уксусной кислоты и 14 мл 70%-ной хлорной кислоты. Реакционную смесь выливают в воду, доводят рН до 5 путем добавления 10%-ного раствора гидроксида натрия, экстрагируют этилацетатом, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,5 г целевого продукта.
ЯМР-спектр: 1,0 (мт, 6Н); 1,25 (т, 6Н); 1,9 (мт, 2Н); 2,55 (с, 1Н); 3,0-3,3 (м, 8Н); 3,7 (с, 6Н); 5,9 (с, 1Н); 6,3-6,55 (м, 2Н); 6,65 (д, 2Н); 6,75 (с, 1Н); 7,0 (д, 1Н); 7,3 (т, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.48
1-[4-[N-Ацетил-N-(3-диэтиламинопропил)амино] -2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-N(COCH3)(CH2)3N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - метил)
В течение 1 часа при температуре 60oС нагревают смесь 0,38 г соединения, полученного в Препаративном примере 4.47, и 0,36 мл бис(триметилсилил)ацетамида в 10 мл толуола. Добавляют 0,052 мл ацетилхлорида, затем 0,1 мл триэтиламина и перемешивают 2 часа при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают насыщенным раствором хлорида натрия, экстрагируют этилацетатом, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,39 г целевого продукта.
ЯМР-спектр: 0,95 (д, 6Н); 1,25 (т, 6Н); 1,6-2,0 (м, 5Н); 2,65 (сп, 1Н); 3,0-3,3 (м, 6Н); 3,65 (с, 6Н); 3,75 (т, 2Н); 6,65 (д, 2Н); 6,85 (с, 1Н); 7,2-7,6 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.49
Калиевая соль 1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил]-2-изопропилфенил] -5-[2-(цихлопропилметилокси)-6-метоксифенил] пиразол-3-карбоновой кислоты (формула (II); R1 - 4-CON(СН3)(CH2)3N(СН3)2; R2 - 2-изопропил; R3 - H; 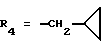 )
)
В течение 20 часов при комнатной температуре перемешивают смесь 3,8 г соединения, полученного в Препаративном примере 3.61, и 0,92 г КОН в 76 мл этанола и 12 мл воды. Концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 3,9 г целевого продукта.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.50
1-(2-Метил-4-нитрофенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула ( II'); R'1 - 4-NO2; R2 - 2-метил; R3 - H; R4 - метил)
В течение ночи перемешивают смесь 3,5 г соединения, полученного в Препаративном примере 3.56, и 0,44 г гидрата гидроксида лития в 20 мл метанола и 4 мл воды. Концентрируют в вакууме, остаток обрабатывают водой, подкисляют до рН 3 путем добавления 10%-ной соляной кислоты, выпавший осадок отфильтровывают под вакуумом и высушивают. Получают 3,2 г целевого продукта.
ЯМР-спектр: 2,9 (с, 3Н); 3,58 (с, 6Н); 6,6 (д, 2Н); 6,88 (с, 1Н); 7,2-7,38 (м, 2Н); 7,95 (д.д, 1Н); 8,22 (д, 1Н),
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.51
1-(4-Амино-2-изобутилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II'); R'1 - 4-NH2; R2 - 2-изобутил; R3 - H; R4 - метил)
Это соединение получают согласно методике, описанной в Препаративном примере 4.45, исходя из 0,5 г соединения, полученного в Препаративном примере 3.62, 6 мл уксусной кислоты и 14 мл 70%-ной хлорной кислоты. Получают 0,3 г целевого продукта.
ЯМР-спектр: 0,65 (д, 6Н); 1,55 (мт, 1Н); 1,9 (д, 2Н); 3,5 (с, 6Н); 5,05 (с, 2Н); 6,0-7,3 (м, 7Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.52
1-[4-[3-(Диэтиламино)пропаноиламино] -2-изобутилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-NHCO(CH2)2N(C2H5)2; R2 - 2-изобутил; R3 - H; R4 - метил)
Это соединение получают согласно методике, описанной в Препаративном примере 4.46, исходя из 0,133 г гидрохлорида 3-диэтиламинопропановой кислоты и 1 мл тионилхлорида в 1 мл дихлорметана и 0,29 г соединения, полученного в Препаративном примере 4.51, и 4 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. Получают 0,15 г целевого продукта.
ЯМР-спектр: 0,7 (д, 6Н); 1,1 (т, 6Н); 1,65 (мт, 1Н); 2,0 (д, 2Н); 2,75 (т, 2Н); 3,1 (кд, 4Н); 3,3 (т, 2Н); 3,6 (с, 6Н); 6,5 (д, 2Н); 6,7 (с, 1Н); 7,1 (д, 1Н); 7,2 (т, 1Н); 7,3-7,5 (м, 2Н); 10,3 (с, 1Н); 15 (с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.53
1-(4-Амино-2-циклопентилфенил)-5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II'); R'1 - 4-NH2; 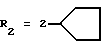 ; R3 - H; R4 - метил)
; R3 - H; R4 - метил)
Это соединение получают согласно методике, описанной в Препаративном примере 4.45, исходя из 0,9 г соединения, полученного в Препаративном примере 3.63, 11 мл уксусной кислоты и 27 мл 70%-ной хлорной кислоты. Получают 0,52 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,18-1,9 (м, 8Н); 2,6 (мт, 1Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.54
1-[4-[3-(Диэтиламино)пропаноиламино] -2-циклопентилфенил] -5-(2,6-диметоксифенил)пиразол-3-карбоновая кислота (формула (II); R1 - 4-NHCO(CH2)2N(C2H5); 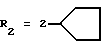 ; R3 - H; R4 - метил)
; R3 - H; R4 - метил)
Это соединение получают согласно методике, описанной в Препаративном примере 4.46, исходя из 0,22 г гидрохлорида 3-диэтиламинопропановой кислоты, 2 мл тионилхлорида в 5 мл дихлорметана, 0,5 г соединения, полученного в Препаративном примере 4.53, и 0,73 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. Получают 0,32 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,1-1,8 (м, 14Н); 2,5 (мт, 1Н); 2,72 (т, 2Н); 3,02 (м, 4Н); 3,22 (мт, 2Н); 3,58 (с, 6Н); 6,5 (д, 2Н); 6,65 (с, 1Н); 7,03 (д, 1Н); 7,2 (т, 1Н); 7,35 (д.д, 1Н); 7,5 (д, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.55
Калиевая соль 1-[4-[N-(2-Диэтиламиноэтил)карбамоил]-3-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты (формула (II); R1 - 4-CONH(CH2)2N(C2H5)2; R2 - 3-изопропил; R3 - H; R4 - метил)
В течение двух дней при комнатной температуре перемешивают смесь 0,34 г соединения, полученного в Препаративном примере 3.65, и 0,073 г КОН в 6 мл диоксана и 2 мл воды. Концентрируют в вакууме, остаток обрабатывают толуолом и концентрируют в вакууме. Получают 0,37 г целевого продукта. Т.пл. выше 260oС.
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.56
5-(2,6-Диметоксифенил)-1-(4-нитро-5,6,7,8-тетрагидронафт-1-ил)пиразол-3-карбоновая кислота (формула (II'); R'1 - 4-NO2; R2, R3 - -(CH2)4-; R4 - метил)
В течение двух часов при температуре 70oС нагревают смесь 4,2 г соединения, полученного в Препаративном примере 3.66, вместе с 0,8 г гидрата гидроксида лития в 95 мл этанола и 5 мл воды. После охлаждения до комнатной температуры добавляют воду, подкисляют до рН 3 путем добавления 10%-ной соляной кислоты, выпавший осадок отфильтровывают под вакуумом и высушивают. Получают 4,16 г целевого продукта. Т.пл.=130oС.
ЯМР-спектр: 1,7 (мт, 4Н); 2,55 (мт, 2Н); 2,82 (мт, 2Н); 3,65 (с, 6Н); 6,65 (д, 2Н); 6,85 (с, 1Н); 7,05 (д, 1Н);7,35 (т, 1Н); 7,7 (д, 1Н); 12,95 (уш.с, 1Н).
ПРЕПАРАТИВНЫЙ ПРИМЕР 4.57
5-(2,6-Диметоксифенил)-1-[5-(3-диэтиламинопропаноиламино)-2-изопропилфенил]пиразол-3-карбоновая кислота (формула (II); R1 - 5-NHCO(CH2)2N(С2Н5)2; R2 - 2-изопропил; R3 - H; R4 - метил)
Это соединение получают из сложного метилового эфира, полученного в Препаративном примере 3.67. После перекристаллизации из метанола температура плавления составляет 195-198oС.
ЯМР-спектр (ДМСО+ТФУК): 0,65-1,35 (м, 12Н); 2,5 (кт, 1Н); 2,82 (т, 2Н); 3,15 (мт, 4Н); 3,36 (т, 2Н); 3,6 (с, 6Н); 6,55 (д, 2Н); 6,76 (с, 1Н); 7,15-7,4 (м, 3Н); 7,8 (д, 1Н).
ПРИМЕР 1
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(СН3)(СН2)3N(СН3)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
А) Гидрохлорид хлорангидрида l-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
В атмосфере азота и при комнатной температуре в течение 5 часов перемешивают 1,07 г кислоты, полученной в Препаративном примере 4.1, в 2 мл тионилхлорида. Выпаривают, затем остаток обрабатывают 3 раза дихлорметаном, получая целевой продукт, который используют таким, какой есть, в следующей стадии.
Хлорангидрид кислоты можно также получать по нижеприводимой методике.
А') Гидрохлорид хлорангидрида 1-[4-[N-метил-N-(3- диметиламинопропил)карбамоил] -2-изопропилфенил]-5-(2,6-диметоксифенил)пиразол-3-карбоновой кислоты
Калиевую соль, полученную в Препаративном примере 4.1-бис, растворяют в 130 мл этанола, добавляют 50 мл солянокислого этанола, неорганическое вещество отфильтровывают и фильтрат выпаривают в вакууме. Остаток снова растворяют в 100 мл дихлорметана, медленно добавляют 11 мл тионилхлорида и кипятят с обратным холодильником в течение 4 часов. Выпаривают в вакууме, остаток снова растворяют в 30 мл дихлорметана и опять выпаривают в вакууме, повторяя эту операцию 3 раза.
Б) Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
В атмосфере азота в течение 40 минут кипятят с обратным холодильником смесь, содержащую 0,37 г 2-аминоадамантан-2-карбоновой кислоты (соединение Б), 5 мл ацетонитрила и 0,82 мл бис(триметилсилил)ацетамида. После возврата к комнатной температуре добавляют 0,3 мл триэтиламина и полученный на предыдущей стадии продукт, растворенный в 15 мл ацетонитрила. Перемешивают при комнатной температуре в течение 1 недели, растворители удаляют путем концентрирования реакционной смеси. Путем добавления эфира вызывают кристаллизацию. Кристаллы перемешивают в смеси из 1,5 мл толуола и 1,5 мл ацетонитрила. Отфильтровывают нерастворимую часть, промывают и растворители выпаривают. Остаток перемешивают в водном метаноле, затем снова выпаривают и обрабатывают этанолом. Экстрагируют дихлорметаном, органическую фазу промывают насыщенным раствором хлорида натрия, затем хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 92:8: 0,7. Получают 0,18 г целевого продукта после порошкования в эфире. Т.пл.= 185oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 0,95 (д, 6Н); 1,6-2,2 (м, 14Н); 2,4-3 (м, 12Н), 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,5 (м, 4Н).
Альтернативно стадию Б можно осуществлять следующим образом.
В атмосфере азота в течение 40 минут кипятят с обратным холодильником смесь 8,79 г соединения Б, 22 мл бис(триметилсилил)ацетамида в 120 мл безводного ацетонитрила и охлаждают до комнатной температуры. Затем добавляют раствор хлорангидрида кислоты, полученного согласно стадии А из 23,83 г кислоты, полученной в Препаративном примере 4.1, и 140 мл тионилхлорида в 300 мл безводного ацетонитрила. После перемешивания в течение 15 часов при комнатной температуре и выпаривания в вакууме остаток снова растворяют в 180 мл метанола, медленно добавляют 180 мл воды, перемешивают в течение часа, отфильтровывают нерастворимое вещество и фильтрат после добавления этанола выпаривают в вакууме. После перемешивания остатка в 200 мл 1 н. соляной кислоты отфильтровывают, осадок промывают 1 н. соляной кислотой и высушивают в вакууме над фосфорным ангидридом, получая 29,8 г продукта примера 1. Т.пл.= 211oС (разложение) (после перекристаллизации из пропан-2-ола).
ПРИМЕР 1'
Внутренняя соль 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
Из гидрохлорида соединения, полученного в примере 1, высвобождают внутреннюю соль следующим образом.
0,97 г Продукта примера 1 растворяют в 10 мл воды, повышают значение рН до 7 путем добавления 1,3 н. раствора гидроксида натрия. Отфильтровывают, осадок промывают водой и высушивают в вакууме над Р2O5, получая 0,86 г внутренней соли, которую перекристаллизуют из 3 мл ацетонитрила, получая 0,5 г целевой внутренней соли.
ЯМР-спектр (ДМСО+ТФУК): 1 (мт, 6Н); 1,4-2,3 (м, 14Н); 2,3-3,4 (м, 14Н); 3,5 (м, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,5 (м, 4Н).
После перекристаллизации из пропан-2-ола т.пл.=238oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,1-7,5 (м, 4Н).
Можно также получать продукт примера 1' без выделения продукта примера 1 согласно следующей методике.
В атмосфере азота в течение двух часов кипятят с обратным холодильником смесь 9,7 г соединения Б и 27 мл бис(триметилсилил)ацетамида в 100 мл безводного дихлорметана. После охлаждения таким образом полученный раствор выливают в раствор продукта из стадии А' примера 1 в 100 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Выпаривают в вакууме, остаток обрабатывают путем перемешивания в течение трех часов со 100 мл метанола и 100 мл воды и повышают рН до 7-7,5 путем добавления насыщенного раствора гидрокарбоната натрия. После перемешивания в течение 1 часа отфильтровывают, получая 22,1 г целевого продукта (чистота согласно ВЭЖХ 98,5%).
Внутреннюю соль также можно превратить в ее гидрохлорид (продукт примера 1) согласно следующей методике.
6,85 г Внутренней соли в смеси из 3,5 мл концентрированной соляной кислоты и 40 мл воды нагревают при перемешивании. После растворения охлаждают при перемешивании, отфильтровывают и высушивают в вакууме, получая 6,5 г гидрохлорида.
Из внутренней соли можно получить ее гидрохлорид следующим образом.
При нагревании 0,3 г внутренней соли растворяют в 3 мл метанола и 2 мл дихлорметана, охлаждают до комнатной температуры, добавляют 0,5 мл 1,2 н. соляной кислоты, концентрируют в вакууме до объема 0,5 мл, охлаждают до -20oС и отфильтровывают, получая 0,2 г продукта примера 1.
ПРИМЕР 2
2-[1-[4-[N-(2-Цианоэтил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONHCH2CH2CN; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 5 часов при комнатной температуре перемешивают смесь 2,6 г соединения, полученного в Препаративном примере 4.2, и 20 мл тионилхлорида. Концентрируют в вакууме, остаток обрабатывают дихлорметаном и растворитель выпаривают в вакууме. Полученный таким образом хлорангидрид кислоты используют таким, какой есть. С другой стороны, в атмосфере азота в течение 30 минут кипятят с обратным холодильником смесь 1,09 г 2-аминоадамантан-2-карбоновой кислоты, 4,2 мл бис(триметилсилил)ацетамида в 20 мл ацетонитрила. После охлаждения медленно добавляют раствор хлорангидрида кислоты, полученного выше, в 40 мл ацетонитрила и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают метанолом, добавляют несколько капель воды и перемешивают 2 часа при комнатной температуре. Осадок отфильтровывают под вакуумом, промывают метанолом и высушивают. Осадок хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:3, затем смесью дихлорметана с метанолом и уксусной кислотой в объемном соотношении 100:3:0,5. После кристаллизации из эфира получают 1,96 г целевого продукта. Т.пл.=269oС.
ЯМР-спектр: 1 (д, 6Н); 1,4-2,1 (м, 12Н); 2,4-2,8 (м, 5Н); 3,4 (мт, 2Н); 3,5 (с, 6Н); 6,5 (д, 2Н); 6,6 (с, 1Н); 7,1-7,4 (м, 2Н); 7,5 (д, 1Н); 7,8 (с, 1Н); 8,9 (т, 1Н).
ПРИМЕР 3
2-[1-[4-[N-(3-Аминопропил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH(CH2)3NH2; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение ночи при комнатной температуре и при атмосферном давлении гидрируют смесь 0,3 г полученного в примере 2 соединения в присутствии 0,03 г никеля Ренея® в 10 мл метанола, затем катализатор отфильтровывают, промывают метанолом и фильтрат частично концентрируют в вакууме. Образовавшиеся кристаллы отфильтровывают и промывают этанолом, получая первую партию целевого продукта. Фильтрат частично концентрируют в вакууме и перемешивают при комнатной температуре. Образовавшиеся кристаллы отфильтровывают под вакуумом и промывают этанолом, получая вторую партию целевого продукта. Путем объединения обеих партий получают 0,045 г целевого продукта. Т.пл.=280oС (разложение).
ЯМР-спектр: 1,1 (д, 6Н); 1,5-2,2 (м, 14Н); 2,4-3 (м, 5Н); 3,3 (мт, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,2-7,4 (м, 2Н); 7,6 (д, 1Н); 7,7 (с, 1Н).
ПРИМЕР 4
2-[1-[4-[N-(2-Амидиноэтил)карбамоил] -2-изопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH(CH2)2C(-NH)NH2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Стадия А: На бане со льдом охлаждают раствор 0,37 г соединения, полученного в примере 2, в 10 мл этанола и 10 мл безводного эфира, затем в течение 50 минут через раствор барботируют газообразный хлороводород. Выдерживают в течение трех дней при температуре +5oС, затем концентрируют в вакууме, получая гидрохлорид промежуточного имидата (R1 - 4-CONH(CH2)2C(=NH)OC2H5).
Стадия Б: Остаток обрабатывают с помощью 20 мл безводного этанола, полученный раствор охлаждают на бане со льдом и пропускают через него в течение 35 минут аммиак. Перемешивают в течение 30 минут при комнатной температуре, концентрируют в вакууме, остаток обрабатывают водой и оставляют кристаллизоваться. После отфильтровывания под вакуумом, затем высушивания кристаллов их перекристаллизуют из горячего этанола. Получают 0,3 г целевого продукта. Т.пл.=257oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,5-2,2 (м, 14Н); 2,4-2,8 (м, 3Н); 3,4-3,7 (м+с, 8Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,2-7,4 (м, 2Н); 7,6 (д.д, 1Н); 7,8 (уш.с, 1Н).
ПРИМЕР 5
Дигидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(2-дигидроимидазол-2-илэтил) карбамоил] -2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); 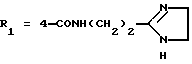 ; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 30 минут перемешивают смесь, содержащую 0,49 г промежуточного имидата, полученного в примере 4, стадия А, и 5 мл 1,2-диаминоэтана. Реакционную среду выпаривают. За счет добавления воды выпадает осадок, который отфильтровывают, затем промывают водой. Этот продукт суспендируют в этаноле и добавляют солянокислый эфир. После выпаривания растворителя продукт порошкуют в эфире, отфильтровывают, промывают эфиром, затем высушивают при 60oС над фосфорным ангидридом. Получают 0,3 г целевого продукта. Т.пл.=220oС (разложение).
ЯМР-спектр: 1,05 (д, 6Н); 1,5-2,2 (м, 12Н); 2,5 (уш.с, 2Н); 2,6-2,8 (м, 3Н); 3,55 (мт, 2Н); 3,65 (с, 6Н); 3,8 (с, 1Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,2-7,4 (д.д, 1Н); 7,8 (д, 1Н).
ПРИМЕР 6
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(3-N', N'- диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CONH(CH2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Получают смесь, содержащую 1,45 г соединения, полученного в Препаративном примере 4.3, 30 мл дихлорметана, 0,38 мл изобутилхлорформиата, 0,82 мл триэтиламина и 17 мл ацетонитрила, которую перемешивают при комнатной температуре в течение 5,5 часов. С другой стороны, смешивают 0,57 г 2-аминоадамантан-2-карбоновой кислоты с 10 мл ацетонитрила и 2,2 мл бис(триметилсилил)ацетамида и кипятят с обратным холодильником в атмосфере азота в течение 30 минут. После охлаждения добавляют вышеполученный смешанный ангидрид и перемешивают при комнатной температуре в течение 1 дня. Нерастворимое вещество отфильтровывают и удаляют; растворители выпаривают, затем добавляют воду, перемешивают в течение 30 минут и отфильтровывают выпавший осадок. Вторую фракцию получают из фильтрата после добавления этанола, экстракции дихлорметаном (2 раза), высушивания экстракта над сульфатом
магния и выпаривания растворителей. Обе объединенные фракции хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении сначала 90:10:0,8, затем 88:12:1. Получают 40 мг целевого продукта после порошкования в диизопропиловом эфире и отфильтровывания. Т.пл.=220oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,75 (с+мт, 7Н); 3,1 (м, 2Н); 3,3 (м, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,2-7,4 (м, 2Н); 7,6 (д.д, 1Н); 7,8 (д, 2Н).
Исходя из кислот формулы (II), описанных в таблице 4, и следуя методике, описанной в примере 1, получают соединения формулы (I) согласно изобретению, описанные в таблице 6.
ПРИМЕР 14
Гидрохлорид 2-[5-(2,6-диметоксифенил)-4-[4-[N-метил-N-(3-N', N'-диметиламинопропил)аминосульфонил] -2- изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 4-SO2N(CH3)(CH2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 5,5 часов при комнатной температуре перемешивают 850 мг полученной в Препаративном примере 4.11 кислоты и 3 мл тионилхлорида. Добавляют 10 мл толуола, затем реакционную среду выпаривают в вакууме (операцию повторяют 2 раза). Таким образом получают 1 г хлорангидрида кислоты, полученной в Препаративном примере 4.11. В течение 4,5 часов перемешивают смесь, содержащую 0,41 г 2-амино-адамантан-2-карбоновой кислоты и 3,5 мл бис(триметилсилил)ацетамида в 5 мл ацетонитрила. К этой реакционной среде добавляют раствор вышеполученного хлорангидрида кислоты в 5 мл ацетонитрила и 1 мл триэтиламина и перемешивают в течение четырех дней при комнатной температуре. Добавляют 3 мл воды, 5 мл метанола и перемешивают 4 часа при комнатной температуре, затем отфильтровывают и выпаривают в вакууме. Остаток порошкуют в 6 мл 1 н. соляной кислоты, затем добавляют этанол и выпаривают в вакууме. Остаток перемешивают в 200 мл дихлорметана и 5 мл воды, декантируют, отделяют органическую фазу, сушат ее над сульфатом натрия и выпаривают в вакууме, получая 1,26 г неочищенного продукта. Его перекристаллизуют из 5 мл CH3CN, охлаждая до -20oС, и отфильтровывают 0,6 г целевого продукта. Т. пл.=211oС.
ЯМР-спектр: 1 (д, 6Н); 1,4-2,1 (м, 14Н); 2,4-2,5 (мт, 2Н); 2,5-2,65 (мт, 1Н); 2,6 (с, 3Н); 2,65 (с, 6Н); 2,9 (мт, 4Н); 3,5 (с, 6Н); 6,5 (д, 2Н); 6,7 (с, 1Н); 7,15-7,4 (м, 3Н); 7,45-7,6 (м, 2Н).
ПРИМЕР 15
2-[5-(2,6-Диметоксифенил)-1-[4- [N-[3-(N',N'- диметиламино)пропил]карбамоил] -5,6,7,8-тетрагидронафт-1-ил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH(CH2)3N(CH3)3; R2, R3 - -(CH2)4-; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение полутора часов при температуре 40oС нагревают раствор 0,47 г соединения, полученного в Препаративном примере 4.12, в 5 мл тионилхлорида и 30 мл дихлорметана. Выпаривают в вакууме, получая хлорангидрид кислоты, который снова растворяют в 5 мл ацетонитрила и добавляют к раствору, полученному путем кипячения с обратным холодильником в течение двух часов смеси 0,28 г соединения Б, 0,69 мл бис(триметилсилил)ацетамида и 3 мл ацетонитрила. Добавляют 0,26 мл триэтиламина и перемешивают в течение двух часов при комнатной температуре. Выпаривают в вакууме, остаток порошкуют в 2 мл насыщенного раствора хлорида натрия, отфильтровывают и высушивают в вакууме, получая 0,77 г продукта. Его хроматографируют на силикагеле 60 Н, элюируя с помощью смеси дихлорметана с метанолом и водой в объемном соотношении 80:20: 2,5. Выпаривают, порошкуют остаток в эфире и отфильтровывают, получая 0,11 г целевого продукта. Т.пл.=200oС (смола).
ЯМР-спектр: 1,4-2,05 (м, 18Н); 2,1 (с, 6Н); 2,2 (т, 3Н); 2,4-2,8 (м, 6Н); 3,2 (кд, 2Н); 3,6 (с, 6Н); 6,6 (2Н, д); 6,65 (с, 1Н); 6,8-7 (д.д, 2Н); 7,2-7,3 (м, 2Н); 8,2 (т, 1Н).
ПРИМЕР 16
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N', N'-диметиламиноэтил)аминосульфонил] -5,6,7,8-тетрагидронафт-1-ил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-SO2N(CH3)(CH2)2N(CH2)2; R2, R3 - -(СН2)4-; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 15 часов при комнатной температуре перемешивают 0,5 г соли, полученной в Препаративном примере 4.13, в 10 мл тионилхлорида, затем выпаривают с толуолом, получая хлорангидрид кислоты, который смешивают с раствором силилированного соединения Б, полученного путем перемешивания в течение 6 часов при комнатной температуре смеси из 0,279 г соединения Б, 2 мл бис(триметилсилил)ацетамида и 8 мл ацетонитрила. После перемешивания в течение 20 дней при комнатной температуре реакционную среду выпаривают в вакууме, затем остаток перемешивают в течение 1 часа при комнатной температуре с 5 мл воды и 5 мл метанола; отфильтровывают и фильтрат выпаривают в вакууме, после чего остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и уксусной кислотой. Остаток порошкуют в эфире и отфильтровывают, получая 0,31 г целевого продукта. Т.пл. выше 260oС.
ЯМР-спектр (ДМСО+ТФУК): 1,6-2,3 (м, 16Н); 2,6 (м, 2Н); 2,9 (уш.с, 9Н); 3,1 (мт, 2Н); 3,4 (мт, 2Н); 3,6 (мт, 2Н); 3,7 (с, 6Н); 6,7 (д, 2Н); 6,85 (с, 1Н); 7,15 (д, 1Н); 7,4 (1Н, т); 7,5 (уш.с, 1Н); 7,6 (д, 1Н).
ПРИМЕР 17
2-[5-(2,6-Диметоксифенил)-1-(2-метил-4-карбамоилфенил)пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH2; R2 - 2-метил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение часа кипятят с обратным холодильником суспензию 220 мг соединения Б с 0,4 мг бис(триметилсилил)ацетамида в 10 мл ацетонитрила. Кроме того, готовят раствор, содержащий 330 мг соединения, полученного в Препаративном примере 4.14, и 0,15 мл триэтиламина в 10 мл ацетонитрила, охлаждают до -5oС, добавляют 0,13 мл изобутилхлорформиата, затем перемешивают в течение 1 часа при комнатной температуре и полученный смешанный ангидрид добавляют к раствору силилированного соединения Б, полученному выше. Выдерживают при комнатной температуре в течение восьми дней, затем отфильтровывают нерастворимое вещество, фильтрат выпаривают досуха, после чего остаток растворяют в 5 мл дихлорметана. Полученную органическую фазу промывают 1,2 н. соляной кислотой, сушат над сульфатом натрия и выпаривают; добавляют к остатку 5 мл дихлорметана, выпавший осадок отфильтровывают и растворяют его в 1 мл метанола. Образовавшиеся кристаллы отфильтровывают, получая 100 мг целевого продукта. Т.пл.=290oС (разложение).
ЯМР-спектр: 1,4-2,3 (м, 15Н); 2,55 (м, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,05 (д, 1Н); 7,3 (т, 1Н); 7,4 (с, 2Н); 7,6 (д.д, 1Н); 7,75 (с, 1Н); 7,9 (с, 1Н).
ПРИМЕР 18
2-[5-(2,6-Диметоксифенил)-1-[2,3-диметил-4-[N-(2-N',N'- диметиламиноэтил)карбамоил] фенил] пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH(CH2)2N(CH3)2; R2 - 2-метил; R3 - 3-метил; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение полутора часов кипятят с обратным холодильником раствор 1,3 г продукта, полученного в Препаративном примере 4.15, в 10 мл тионилхлорида и 50 мл дихлорметана. Выпаривают в вакууме, получая хлорангидрид кислоты, который снова растворяют в 10 мл ацетонитрила, и этот раствор добавляют к раствору, полученному после кипячения с обратным холодильником в течение двух часов смеси из 0,82 г соединения Б, 2 мл бис(триметилсилил)ацетамида в 10 мл ацетонитрила. Добавляют 0,77 мл триэтиламина и перемешивают в течение 15 часов при комнатной температуре. После выпаривания в вакууме, порошкования в 10 мл насыщенного раствора хлорида натрия, отфильтровывания и высупивания в вакууме продукт хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:10:1. После выпаривания растворителей и порошкования остатка в эфире отфильтровывают, получая 0,7 г целевого продукта. Т.пл.=210oС.
ЯМР-спектр: 1,6-2,2 (м, 12Н); 2 (с, 3Н), 2,25 (с, 3Н); 2,35 (с, 6Н); 2,5-2,7 (м, 2Н + 2Н); 3,4 (кд, 2Н); 3.7 (с, 6Н); 6,65 (д, 2Н); 6,8 (с, 1Н); 7,0-7,2 (м, 2Н); 7,3-7,45 (м, 2Н); 8,4 (т, 1Н).
ПРИМЕР 19
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N', N'-диэтиламиноэтил)карбамоил] -2-мeтoкcифeнил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(СН2)2N(С2Н5)2; R2 - 2-OCH3; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают 1,2 г продукта, полученного в Препаративном примере 4.16, в 12 мл тионилхлорида и 12 мл дихлорметана. После выпаривания в вакууме с двумя последующими азеотропными перегонками с 30 мл толуола полученный хлорангидрид кислоты снова растворяют в 10 мл ацетонитрила и полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение 1 часа 15 минут смеси из 0,43 г соединения Б, 1,1 мл бис(триметилсилил)ацетамида и 20 мл ацетонитрила. Кипятят с обратным холодильником при перемешивании в течение 4 часов, выпаривают в вакууме, остаток перемешивают в 4 мл метанола и 0,5 мл воды; выпаривают в вакууме, затем хроматографируют на силикагеле 60 Н, элюируя с помощью смесей дихлорметана с метанолом и 20%-ным гидроксидом аммония в объемных соотношениях 95:5:0,5; затем 90:10:0,5 и 85:15:0,5, получая 0,3 г целевого продукта. Т.пл.=170oС (разложение).
ЯМР-спектр: 0,8 (мт, 3Н); 1 (мт, 3Н); 1,5-3,6 (мт, 34Н); 6,5 (д, 2Н); 6,65 (с, 1Н); 6,95 (мт, 2Н); 7,3 (мт, 3Н).
ПРИМЕР 20
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(2-N', N'-диэтиламиноэтил)карбамоил] -2-хлорфенил] пиразол-3- илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(CH2)2N(C2H5)2; R2 - 2-Cl; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают 1,3 г продукта, полученного в Препаративном примере 4.17, в 12 мл дихлорметана и 12 мл тионилхлорида. После выпаривания в вакууме с последующими двумя азеотропными перегонками с 30 мл толуола полученный хлорангидрид кислоты снова растворяют в 10 мл ацетонитрила и полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение 1 часа 15 минут смеси из 0,46 г соедирения Б, 1,2 мл бис(триметилсилил)ацетамида в 20 мл ацетонитрила. Кипятят с обратным холодильником при перемешивании в течение 4 часов, выпаривают в вакууме, остаток порошкуют в 4 мл метанола и 2 мл воды, перемешивают в течение 30 минут при комнатной температуре и выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и 20%-ным гидроксидом аммония в объемном соотношении 80:20:0,5, получая 0,3 г целевого продукта. Т.пл.=160oС (разложение).
ПРИМЕР 21
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(2- морфолиноэтил)карбамоил] -2-хлорфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); 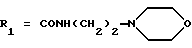 ; R2 - 2-Cl; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-Cl; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 18 часов при комнатной температуре перемешивают смесь 1,4 г продукта, полученного в Препаративном примере 4.18, 14 мл тионилхлорида и 80 мл дихлорметана. После выпаривания в вакууме с последующими двумя азеотропными перегонками с 30 мл толуола и растворения снова полученного хлорангидрида кислоты в 20 мл ацетонитрила полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение 4 часов суспензии 0,53 г соединения Б в 1,33 мл бис(триметилсилил)ацетамида и 30 мл ацетонитрила. После кипячения в течение 4 часов с обратным холодильником и при перемешивании выпаривают в вакууме, остаток перемешивают с 10 мл метанола и 1 мл воды и отфильтровывают. После перемешивания в течение 1 часа полученного осадка с 5 мл воды и 5 каплями концентрированной соляной кислоты, отфильтровывания, промывки с помощью 1 мл воды, 5 мл пентана и 5 мл диизопропилового эфира получают 0,7 г целевого продукта. Т.пл.=200oС.
ЯМР-спектр: 1,5-2,2 (м, 12Н); 2,6 (уш.с, 2Н); 2,85 (уш.с, 4Н); 3,3-3,8 (мт, 20Н); 6,6 (д, 2Н); 6,75 (1Н, с); 7,3 (т, 1Н); 7,45 (мт, 2Н); 7,8 (д.д, 1Н); 7,95 (д, 1Н); 8,85 (уш.с, 1Н).
ПРИМЕР 22
2- [5-(2,6-Диметоксифенил)-1-(3-хлор-4-цианофенил)пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CN; R2 - 3-Cl; R3 - H; R4 - CH3; AA(OH) - 2-карбоксиадамант-2-ил)
В течение трех дней при комнатной температуре перемешивают смесь, содержащую 0,36 г соединения, полученного в Препаративном примере 4.19, 0,145 мл изобутилхлорформиата и 0,145 мл триэтиламина в 5 мл дихлорметана. Кроме того, в течение 1 часа кипятят с обратным холодильником смесь, содержащую 0,23 г соединения Б и 0,34 мл бис(трифторметилсилил)ацетамида в 2 мл ацетонитрила. Смешивают таким образом полученные два раствора и перемешивают при комнатной температуре в течение 48 часов. После отфильтровывания и промывки метанолом растворители выпаривают, затем остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом и уксусной кислотой в объемном соотношении 100:1:0,5, получая 120 мг целевого продукта. Т. пл.=292oС.
ПРИМЕР 23
2-[5-(2,6-Диметоксифенил)-1-(4-карбамоил-3-хлорфенил)пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH2; R2 - 3-Сl; R3 - H; R4 - CH3; AA(OH) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают смесь, содержащую 0,73 г соединения, полученного в Препаративном примере 4.20, 0,263 мл изобутилхлорформиата и 0,26 мл триэтиламина в 5 мл дихлорметана. Кроме того, в течение 1 часа кипятят с обратным холодильником смесь, содержащую 0,34 г соединения Б и 0,65 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. Смешивают два таким образом полученных раствора и перемешивают при комнатной температуре в течение 4 дней. После отфильтровывания, промывки 1 н. соляной кислотой, затем этанолом и высушивания над сульфатом магния остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и уксусной кислотой в объемном соотношении 100:2:1. Т.пл.=293oС.
ПРИМЕР 24
Гидрохлорид 2-[1-[4-[N-метил-N-(2-N',N'- диэтиламиноэтил)карбамоил]-2-циклопропилфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(СН2)2N(C2H5)2; R2 - 2-циклопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают 0,5 г продукта, полученного в Препаративном примере 4.21, в 5 мл дихлорметана и 1 мл тионилхлорида. После выпаривания в вакууме с двумя последующими азеотропными перегонками с 30 мл толуола образовавшийся хлорангидрид кислоты снова растворяют в 5 мл ацетонитрила, полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение 2,5 часов суспензии из 0,19 г соединения Б в 0,5 мл бис(триметилсилил)ацетамида и 8,5 мл ацетонитрила, и перемешивают в течение 12 часов при комнатной температуре. Выпаривают в вакууме, остаток в течение 45 минут перемешивают с 3,5 мл метанола и 1 мл воды, затем прикапывают 2,5 мл воды и отфильтровывают. После выпаривания фильтрата в вакууме и высушивания остатка в течение 24 часов при температуре 60oС в вакууме получают 0,14 г целевого продукта. Т.пл.=135oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 0,6 (м, 2Н); 0,9 (м, 2Н); 1,2 (м, 6Н); 1,5-2,2 (м, 12Н); 2,6 (м, 2Н); 2,9 (уш.с, 3Н); 3,2 (м, 6Н); 3,6 (с, 6Н); 3,7 (м, 2Н); 6,6 (д, 2Н); 6,75 (c, 1H); 6,85 (c, 1H); 7,1-7,4 (м, 3Н).
ПРИМЕР 25
Гидрохлорид 2-[1-[3-N-метил-N-(2-N',N'- диметиламиноэтил)карбамоил]-4-хлорфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 3-CON(CH3)(CH2)2N(CH3)2; R2 - 4-Cl; R3 - H; R4 - CH3; AA(OH) - 2-карбоксиадамант-2-ил)
В течение 15 часов при комнатной температуре перемешивают раствор 0,43 г продукта, полученного в Препаративном примере 4.22, в 10 мл дихлорметана и 6 мл тионилхлорида. После выпаривания в вакууме с последующими двумя азеотропными перегонками с 30 мл толуола полученный хлорангидрид кислоты снова растворяют в 3 мл ацетонитрила, полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение трех часов суспензии 0,17 г соединения Б в 0,5 мл бис(триметилсилил)ацетамида и 10 мл ацетонитрила. Перемешивают в течение трех часов при кипячении с обратным холодильником, затем в течение 15 часов при комнатной температуре. Выпаривают в вакууме, остаток перемешивают в течение часа с 12 мл метанола и 6 мл воды. Метанол выпаривают в вакууме, остаток экстрагируют 2 раза по 50 мл дихлорметаном, органическую фазу сушат над сульфатом натрия и выпаривают в вакууме, получая 0,16 г целевого продукта. Т.пл.=206oС (разложение).
ЯМР-спектр: 1,5-2,3 (м, 12Н); 2,6-3,8 (м, 21Н); 6,7 (д, 2Н); 6,8 (с, 1Н); 7,1-7,7 (м, 4Н).
ПРИМЕР 26
2-[1-[5-[N-метил-N-(3-N', N'-диметиламинопропил)карбамоил]-2-метилфенил] -5-(2,6-диметоксифенил)пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 5-CON(CH3)(СН2)3N(CH3)2; R2 - 2-CH3; R3 - Н; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 5,5 часов при комнатной температуре перемешивают раствор 1,7 г полученного в Препаративном примере 4.23 продукта в 15 мл тионилхлорида. После выпаривания в вакууме с последующими тремя азеотропными перегонками с 30 мл дихлорметана образовавшийся хлорангидрид кислоты снова растворяют в 30 мл ацетонитрила, полученный раствор добавляют к раствору, полученному путем кипячения с обратным холодильником в течение 1 часа суспензии 0,69 г соединения Б в 1,75 мл бис(триметилсилил)ацетамида и 6 мл ацетонитрила. После перемешивания в течение 15 часов при комнатной температуре и выпаривания в вакууме полученный остаток растворяют в 13 мл метанола, медленно добавляют 12 мл воды и перемешивают в течение 30 минут при комнатной температуре. После выпаривания в вакууме, порошкования остатка в 5 мл 1 н. соляной кислоты, декантации, трех экстракций остаточной смолы с помощью 100 мл дихлорметана, высушивания экстрактов над сульфатом магния выпаривают в вакууме, затем растворяют остаток в 15 мл воды. Подщелачивают с помощью 30%-ного раствора гидроксида натрия до рН 8, затем кристаллизуют под действием ультразвука. После отфильтровывания кристаллы перекристаллизуют из толуола и высушивают в вакууме при температуре 60oС, получая 1,32 г целевого продукта. Т.пл.=165oС.
ЯМР-спектр (ДМСО+ТФУК): 1,6-2,2 (м, 14Н); 2,2 (с, 3Н); 2,5-3,2 (м, 13Н); 3,5 (мт, 2Н); 3,7 (с, 6Н); 6,65 (д, 2Н); 6,8 (с, 1Н); 7,3-7,6 (м, 3Н).
Исходя из кислот формулы (II), описанных в таблице 5 или в Препаративных примерах, и следуя вышеприведенным методикам, получают соединения согласно изобретению, представленные в таблице 7.
ПРИМЕР 37
Гидросульфат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
К раствору 0,08 г серной кислоты в 5 мл метанола при комнатной температуре добавляют 0,5 г соединения, полученного в примере 1', этот раствор выливают в 150 мл охлажденного до температуры 5oС эфира и выпавший осадок центрифугируют. Получают 0,54 г целевого продукта. После перекристаллизации из воды т.пл.=212oС (разложение). После перекристаллизации из изопропан-2-ола т.пл.=263oС.
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,1-7,5 (м, 4Н).
Продукт примера 37 также можно получать по нижеприводимой методике.
К суспензии 3,4 г внутренней соли, полученной в примере 1', в 34 мл воды при перемешивании медленно добавляют 22 мл концентрированной серной кислоты и нагревают при температуре 40oС до изменения внешнего вида суспензии. В течение 4 часов охлаждают до комнатной температуры при перемешивании, осадок отфильтровывают и высушивают, получая 3,8 г целевого гидросульфата.
ПРИМЕР 38
Бензолсульфонат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2- изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
Смесь 0,5 г соединения, полученного в примере 1', и 0,16 г бензолсульфокислоты в 5 мл метанола выливают в 75 мл эфира, охлажденного до температуры 5oС, и выпавший осадок отфильтровывают под вакуумом. Получают 0,06 г целевого продукта. Т.пл.=170oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1 (д, 6Н); 1,4-2,2 (м, 14Н); 2,45 (уш.с, 2Н); 2,5-3,2 (м, 12Н); 3,4 (мт, 2Н); 3,55 (с, 6Н); 6,5 (д, 2Н); 6,65 (с, 1Н); 7-7,4 (м, 7Н); 7,5 (мт, 2Н).
ПРИМЕР 39
Цитрат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
К раствору 0,3 г соединения, полученного в примере 1', в 5 мл этанола и 3 мл дихлорметана при комнатной температуре добавляют 0,084 г лимонной кислоты и перемешивают в течение двух часов при комнатной температуре. Концентрируют в вакууме и остаток кристаллизуют из пропан-2-ола. Получают 0,26 г целевого продукта. Т.пл.=168oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,3 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3,3 (м, 16Н); 3,3-3,8 (с+м, 8Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,1-7,5 (м, 4Н).
ПРИМЕР 40
Малеат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
При нагревании 0,1 г соединения, полученного в примере 1', и 0,017 г малеиновой кислоты растворяют в 2,3 мл пропан-2-ола и концентрируют в вакууме. Остаток растворяют в 0,3 мл этанола, этот раствор выливают в 30 мл эфира и образовавшийся осадок отфильтровывают под вакуумом. Получают 0,04 г целевого продукта. Т.пл.=260oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,55-2,2 (м, 14Н); 2,5 (уш.с, 2Н): 2,6-3 (м, 10Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,3 (с, 2Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,15-7,45 (м, 4Н).
ПРИМЕР 41
Соль (S)-(+)-аргинина 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
0,1 г Соединения, полученного в примере 1', и 0,03 г (S)-(+)-аргинина растворяют при нагревании в 4 мл метанола, этот раствор концентрируют до объема 1 мл и выливают в 10 мл эфира, охлажденного до 5oС. Получают 0,055 г целевого продукта (после отфильтровывания под вакуумом и высушивания над фосфорным ангидридом). Т.пл.=176oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 20Н); 2,55 (уш.с, 2Н); 2,6-3,55 (м, 16Н); 3,65 (с, 6Н); 3,95 (т, 1Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,15-7,4 (м, 4Н).
ПРИМЕР 42
Этандисульфонат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
A) 1,2-Этандисульфокислота
Раствор 3 г динатриевой соли 1,2-этандисульфокислоты в 10 мл воды медленно выливают на 200 мл смолы Dowex® 50WX8, затем элюируют с помощью 200 мл деминерализованной воды. Элюат разбавляют путем добавления этанола и концентрируют в вакууме. Получают 3,35 г целевого продукта в виде масла, которое кристаллизуется при комнатной температуре.
Б) Этандисульфонат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
0,05 г Соединения, полученного в примере 1', и 0,04 г соединения, полученного в стадии А, растворяют при нагревании в 2 мл пропан-2-ола и концентрируют в вакууме. Остаток растворяют в 0,3 мл воды с 8-мью каплями диоксана и оставляют кристаллизоваться при комнатной температуре. Выкристаллизовавшийся продукт отфильтровывают под вакуумом, промывают водой и высушивают в вакууме при 90oС. Получают 0,042 г целевого продукта. Т.пл.=266oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м+с, 14Н); 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,15-7,45 (м, 4Н).
ПРИМЕР 43
Натриевая соль 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
0,206 г Соединения, полученного в примере 1', и 0,026 г метилата натрия растворяют в 1 мл метанола с несколькими каплями дихлорметана, затем этот раствор выливают в 50 мл эфира, охлажденного до 5oС. Образовавшийся студнеобразный осадок отфильтровывают под вакуумом и высушивают в вакууме над фосфорным ангидридом. Получают 0,15 г целевого продукта. Т.пл.=191oС.
Это соединение также можно получать согласно нижеописанной методике.
К раствору 0,1 г соединения, полученного в примере 1', в 5 мл метанола и 4 мл дихлорметана добавляют 0,7 мл раствора 0,104 г гидроксида натрия в 10 мл метанола и затем все концентрируют в вакууме. Остаток растворяют в 1 мл пропан-2-ола, этот раствор выливают в 75 мл эфира, охлажденного до температуры 5oС, и выпавший осадок отфильтровывают под вакуумом. Получают 0,05 г целевого продукта.
ЯМР-спектр: 1 (д, 6Н); 1,4-2,3 (м, 20Н); 2,55 (уш.с, 2Н); 2,6 (мт, 1Н); 2,85 (д, 3Н); 3,1 и 3,4 (2 мт, 4Н); 3,6 (с, 6Н); 6,55 (с, 1Н); 6,6 (с, 2Н); 6,95 (с, 1Н); 2 7-7,35 (м, 4Н).
Снятый в присутствии ДМСО+ТФУК ЯМР-спектр слегка отличается: 1,05 (д, 6Н); 1,5-2,3 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,5 (мт, 6Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 44
Фумарат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
0,1 г Соединения, полученного в примере 1', и 0,017 г фумаровой кислоты растворяют в 1,5 мл этанола с 1,5 мл дихлорметана и 4 мл метанола и перемешивают в течение 10 минут при комнатной температуре. Частично концентрируют в вакууме и оставляют кристаллизоваться. Получают 0,025 г целевого продукта (после отфильтровывания под вакуумом и промывки этанолом). Т.пл.=243oС.
ЯМР-спектр (ДМСО+ТФУК): 1 (д, 6Н); 1,5-2,3 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,4 (мт, 2Н); 3,6 (с, 6Н); 6,5-6,7 (д+с, 3Н); 6,75 (с, 1Н); 7,1-7,4 (м, 5Н).
ПРИМЕР 45
Соль N-метил-(D)-глюкамина 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил]-2-изопропилфенил]пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
Раствор 0,07 г соединения, полученного в примере 1', в 5 мл этанола с 1 мл дихлорметана кипятят с обратным холодильником, добавляют 0,02 г N-метил-(D)-глюкамина и перемешивают в течение полутора часов при комнатной температуре. Частично концентрируют в вакууме, выливают в 15 мл эфира и выпавший осадок отфильтровывают под вакуумом. Получают 0,032 г целевого продукта. Т.пл.=90oС (смола).
ЯМР-спектр (ДМСО+ТФУК): 1 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5-2,6 (мт, 5Н); 2,6-3,2 (м, 14Н); 3,2-3,7 (м, 13Н); 3,85 (мт, 1Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 46
Соль диэтаноламина 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил ] -2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
0,1 г Полученного в примере 1' соединения растворяют в 1,5 мл этанола с 1,5 мл дихлорметана, добавляют 0,015 г диэтаноламина, перемешивают в течение 30 минут при комнатной температуре и оставляют стоять в течение ночи при температуре 5oС. Выкристаллизовавшийся продукт отфильтровывают под вакуумом. Получают 0,03 г целевого продукта. Т.пл.=200oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3,25 (м, 16Н); 3,3-3,8 (м+с, 12Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 47
(L)(+)-Тартрат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
Кипятят с обратным холодильником смесь из 0,1 г соединения, полученного в примере 1', и 0,022 г (L)(+)-винной кислоты в 1,5 мл этанола с 1,5 мл дихлорметана, затем добавляют 8 мл этанола и продолжают кипятить с обратным холодильником в течение 5 минут. После охлаждения до комнатной температуры реакционную среду частично концентрируют в вакууме, выливают в 10 мл эфира и выпавший осадок отфильтровывают под вакуумом. Получают 0,07 г целевого продукта после высушивания над фосфорным ангидридом. Т.пл.=154oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,4 (м, 14Н); 2,55 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 4,35 (с, 2Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,15-7,5 (м, 4Н).
ПРИМЕР 48
Соль холина 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение 15 минут при температуре 35oС перемешивают раствор 1 мл дихлорметана, содержащий 0,05 соединения, полученного в примере 1', и 0,025 мл 45%-ного раствора гидроксида холина в метаноле, затем концентрируют в вакууме. Остаток порошкуют в 5 мл эфира и образовавшийся осадок отфильтровывают под вакуумом. Получают 0,03 г целевого продукта. Т.пл.=150oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,4-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,4-3 (м, 10Н); 3,1 (уш.с, 11Н); 3,4 (мт, 2Н); 3,5 (мт, 2Н); 3,6 (с, 6Н); 3,8 (мт, 2Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 49
Изетионат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
Кипятят с обратным холодильником смесь 0,1 г соединения, полученного в примере 1', в 3 мл пропан-2-ола, добавляют 0,022 г 83%-ной изетионовой кислоты (получают путем элюирования изетионата натрия на смоле Dowex®-50WX8 в Н+-форме) и оставляют кристаллизоваться в течение ночи. Выкристаллизовавшийся продукт отфильтровывают под вакуумом. Получают 0,055 г целевого продукта. Т.пл.=230oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,55 (м, 14Н); 2,55 (уш.с, 2Н); 2,6-3,05 (м, 12Н); 3,1 (м, 2Н); 3,5 (мт, 2Н); 3,6-3,7 (с+мт, 8Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,15-7,45 (м, 4Н).
ПРИМЕР 50
Калиевая соль 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение ночи выдерживают при комнатной температуре раствор 0,15 г соединения, полученного в примере 1', и 0,03 г трет.-бутилата калия в 6,5 мл пропан-2-ола, затем концентрируют в вакууме. Остаток растворяют в 0,5 мл метанола и этот раствор выливают в 25 мл диизопропилового эфира, охлажденного до температуры -20oС. Выпавший осадок отфильтровывают под вакуумом и сушат над фосфорным ангидридом при температуре 80oС. Получают 0,09 г целевого продукта. Т.пл.=222oС.
Это соединение также можно получать, следуя нижеописанной методике.
К раствору 0,1 г соединения, полученного в примере 1', в 4 мл дихлорметана добавляют 1 мл раствора из 0,129 г КОН в 10 мл метанола, затем концентрируют в вакууме. Остаток растворяют в 0,5 мл пропан-2-ола, этот раствор выливают в 75 мл эфира, охлажденного до 5oС, и выпавший осадок отфильтровывают под вакуумом. Получают 0,015 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,3 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (т, 2Н); 3,5 (т, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,5 (м, 4Н).
ПРИМЕР 51
Дигидрофосфат 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение 1 часа при комнатной температуре перемешивают смесь 0,1 г соединения, полученного в примере 1', и 0,017 г 85%-ной ортофосфорной кислоты в 2 мл дихлорметана с 3 мл этанола. Концентрируют частично в вакууме, выливают в 10 мл эфира, охлажденного до 5oС, и выпавший осадок отфильтровывают под вакуумом. Получают 0,04 г целевого продукта после высушивания при 60oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3,3 (м, 12Н); 3,5 (мт, 2Н); 3,6 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 52
2-Нафталинсульфонат 2-[5-(2,6-диметоксифенил)-1-[4-[N- мeтил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты
Раствор 0,5 г соединения, полученного в примере 1', и 0,16 г 2-нафталинсульфокислоты в 5 мл метанола выливают в 25 мл охлажденного до 5oС эфира, выпавший осадок отфильтровывают под вакуумом и фильтрат концентрируют. Осадок растворяют в 2 мл метанола, этот раствор выливают в 50 мл охлажденного до 5oС эфира, выпавший осадок отфильтровывают под вакуумом и получают 0,2 г целевого продукта. Первый фильтрат выливают в 50 мл охлажденного до 5oС эфира, отфильтровывают под вакуумом выпавший осадок и получают 0,27 г второй порции целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,5 (уш.с, 2Н); 2,6-3 (м, 10Н); 3,1 (мт, 2Н); 3,5 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,6 (м, 7Н); 7,7 (д, 1Н); 7,8-8,1 (м, 2Н); 8,2 (с, 1Н).
ПРИМЕР 53
2- [5-(2,6-Диметоксифенил)-1-[4-[N-(2-диизопропиламиноэтил)карбамоил]-2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONH(CH2)2N(изопропил)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 4 часов при комнатной температуре перемешивают смесь 0,53 г соединения, полученного в Препаративном примере 4.30, и 2 мл тионилхлорида. Концентрируют в вакууме, остаток обрабатывают дихлорметаном и выпаривают в вакууме, остаток обрабатывают дихлорметаном и растворитель выпаривают в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 35 минут в атмосфере азота кипятят с обратным холодильником смесь 0,19 г соединения Б и 0,49 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. После охлаждения до комнатной температуры добавляют раствор вышеполученного хлорангидрида кислоты в 8 мл ацетонитрила и перемешивают в течение ночи при комнатной температуре. Выпаривают в вакууме, остаток перемешивают с 8,8 мл метанола, добавляют 8,8 мл воды и выпаривают в вакууме. Остаток обрабатывают 1,2 н. соляной кислотой, выпавший осадок отфильтровывают под вакуумом. Осадок обрабатывают с помощью 10 мл воды, подщелачивают до рН 8 путем добавления 1,3 н. раствора гидроксида натрия, осадок отфильтровывают под вакуумом и промывают водой. Получают 0,475 г целевого продукта (после кристаллизации при нагревании из 40 мл ацетонитрила). Т.пл.=196-198oС.
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,3 (д, 12Н); 1,6-2,2 (м, 12Н); 2,55 (м, 2Н); 2,7 (м, 1Н); 3,2 (м, 2Н); 3,5-3,8 (м+с, 10Н); 6,6 (д, 2Н); 6,75 (с, 1Н); 7,2-7,4 (мт, 2Н); 7,65 (д, 1Н); 7,85 (с, 1Н).
ПРИМЕР 54
2-[5-(2,6-Диметоксифенил)-1-[4-[N, N-бис(2-диэтиламиноэтил) карбамоил]-2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CON(CH2CH2N(C2H5)2)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают смесь 0,4 г соединения, полученного в Препаративном примере 4.34, и 2,5 мл тионилхлорида. Концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 45 минут кипятят с обратным холодильником смесь 0,18 г соединения Б и 0,5 мл бис(триметилсилил)ацетамида в 4 мл ацетонитрила. После охлаждения до комнатной температуры добавляют раствор вышеполученного хлорангидрида кислоты в 3 мл ацетонитрила и перемешивают в течение 72 часов при комнатной температуре. Добавляют 3 мл метанола и реакционную смесь концентрируют в вакууме. Остаток растворяют в 3 мл 1,2 н. соляной кислоты, промывают три раза этилацетатом, водную фазу нейтрализуют до рН 6 путем добавления концентрированного раствора гидроксида натрия и декантируют образовавшийся смолистый продукт. Этот смолистый продукт хроматографируют на силикагеле 60Н, элюируя смесью дихлорметана с метанолом и гидроксидом аммония в объемном соотношении 75:25:1,2. Получают 0,4 г целевого продукта (после порошкования в эфире). Т.пл.=169oС.
ПРИМЕР 55
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(пиперид-4-ил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); 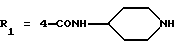 ; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 5 дней при комнатной температуре, затем в
течение 4 дней при температуре 50oС и при атмосферном давлении гидрируют смесь 0,3 г соединения, полученного в примере 12, 0,05 г 10%-ного палладия-на-угле и 0,033 мл концентрированной соляной кислоты в 10 мл метанола с 4 мл диметилформамида. Катализатор отфильтровывают на целите® и фильтрат выпаривают в вакууме. Остаток обрабатывают эфиром и выкристаллизовавшийся продукт отфильтровывают под вакуумом. После высушивания над фосфорным ангидридом при температуре 70oС в вакууме получают 0,121 г целевого продукта. Т. пл.=252oС.
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,5-2,2 (м, 16Н); 2,4-3,5 (3 мт+уш. с, 7Н); 3,6 (с, 6Н); 3,9-4,15 (м, 1Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (мт, 2Н); 7,6 (д, 1Н); 7,8 (с, 1Н).
ПРИМЕР 56
2-[5-(2,6-Диметоксифенил)-1-[4-[N-(1-этилпирролидин-2-ил)метил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); 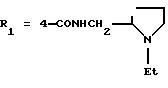 ; R2 - 2-изопропил; R3 - Н; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - Н; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 0,48 г соединения, полученного в Препаративном примере 4.35, и 2 мл тионилхлорида, затем 0,18 г соединения Б и 0,46 г бис(триметилсилил)ацетамида в 2 мл ацетонитрила. Получают 0,2 г целевого продукта (после кристаллизации из пропан-2-ола). Т.пл.=212oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 0,9 (уш.с, 6Н); 1,05 (м, 3Н); 1,3-2,1 (м, 14Н); 2,3 (уш. с, 2Н); 2,5 (м, 1Н); 2,9 (м, 2Н); 3,2-3,6 (м+с, 11Н); 6,4 (д, 2Н); 6,5 (уш.с, 1Н); 7-7,3 (м, 2Н); 7,45 (д, 1Н); 7,65 (уш.с, 1Н).
ПРИМЕР 57
2-[5-(2,6-Диметоксифенил)-1-[4-[N-(2,2,6,6-тетраметилпиперид-4-ил)карбамоил] -2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); 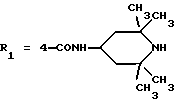 ; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 0,43 г соединения, полученного в Препаративном примере 4.36, и 2 мл тионилхлорида, затем 0,15 г соединения Б и 0,39 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. После перемешивания реакционной смеси в течение ночи при комнатной температуре выпавший осадок отфильтровывают под вакуумом и промывают его ацетонитрилом. Осадок обрабатывают с помощью 4 мл метанола, затем постепенно добавляют 4 мл воды и после этого концентрируют в вакууме. Остаток обрабатывают 1,2 н. соляной кислотой и после порошкования осадок отфильтровывают под вакуумом. Осадок обрабатывают с помощью 3 мл воды, подщелачивают до рН 9 путем добавления 1,3 н. раствора гидроксида натрия, выпавший осадок отфильтровывают под вакуумом и промывают его водой. Получают 0,24 г целевого продукта (после высушивания над фосфорным ангидридом). Т.пл.=270-272oС.
ЯМР-спектр (ДМСО+ТФУК): 1,5 (д, 6Н); 1,2 (с, 6Н); 1,4 (с. 6Н); 1,5-2,2 (2 м, 16Н); 2,65 (мт, 1Н); 3,6 (с, 6Н); 4,2-4,4 (м, 1Н); 6,55 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 2Н); 7,6 (д, 1Н); 7,8 (с, 1Н).
ПРИМЕР 58
2-[5-(2,6-Диметоксифенил)-1-[4-[[3-(диэтиламино)пирролидин-1-ил] карбонил]-2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); 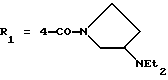 ; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 0,39 г соединения, полученного в Препаративном примере 4.41, и 2 мл тионилхлорида, затем 0,13 г соединения Б и 0,34 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. После перемешивания реакционной смеси в течение ночи при комнатной температуре отфильтровывают нерастворимое вещество и фильтрат концентрируют в вакууме. Остаток обрабатывают с помощью 5 мл метанола, добавляют 5 мл воды и концентрируют в вакууме. Остаток обрабатывают 1,2 н. соляной кислотой и образовавшиеся кристаллы отсасывают. Кристаллы растворяют в воде, раствор подщелачивают до рН 9 путем добавления 1,3 н. раствора гидроксида натрия и выпавший осадок отфильтровывают под вакуумом. Получают после перекристаллизации из ацетонитрила, 0,07 г целевого продукта. Т.пл.=175oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,0 (д, 6Н); 1,1-1,3 (м, 6Н); 1,5-2,8 (4 м, 17Н); 2,8-4,2 (3 м+с, 15Н); 6,6 (д, 2Н); 6,7 (с, 1H); 7,2-7,4 (м, 3Н); 7,5 (уш.с, 1Н).
ПРИМЕР 59
2-[5-(2,6-Диметоксифенил)-1-[4-[[-4- (диметиламино)пиперид-1-ил] карбонил] -2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); 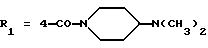 ; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 0,45 г соединения, полученного в Препаративном примере 4.37, и 2 мл тионилхлорида, затем 0,17 г соединения Б и 0,43 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила. После кристаллизации при нагревании из ацетона, затем метанола получают 0,26 г целевого продукта. Т. пл.=200oС (разложение).
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,4-2,3 (2 м, 16Н); 2,5 (уш.с, 2Н); 2,7 (с+мт, 7Н); 2,8-3,8 (2 м+с, 10Н); 4,4-4,8 (м, 1Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 4Н).
ПРИМЕР 60
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(2- цианоэтил)карбамоил]-2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота 2 (формула (I); R1 - 4-CON(CH3)(CH2)2CN; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 3,48 г соединения, полученного в Препаративном примере 4.38, и 20 мл тионилхлорида, затем 1,43 г соединения Б и 3,6 мл бис(триметилсилил)ацетамида в 25 мл ацетонитрила. После перемешивания реакционной смеси в течение ночи при комнатной температуре ее концентрируют в вакууме, остаток обрабатывают с помощью 64 мл метанола, добавляют 64 мл воды и все концентрируют в вакууме. Остаток обрабатывают 1,2 н. соляной кислотой, выпавший осадок отфильтровывают под вакуумом и промывают его 1,2 н. соляной кислотой. Осадок обрабатывают с помощью 5 мл метанола, кипятят с обратным холодильником, охлаждают до комнатной температуры и осадок отфильтровывают под вакуумом. Получают 3,78 г целевого продукта (после высушивания над Р2O5). Т.пл.=249oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 12Н); 2,42-3,0 (м, 8Н); 3,3-3,75 (м, 8Н); 6,58 (д, 2Н); 6,73 (с, 1Н); 7,1-7,42 (м, 4Н).
ПРИМЕР 61
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(3- аминопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CON(CH3)(CH2)3NH2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 4 часов при комнатной температуре и при атмосферном давлении гидрируют смесь 1 г соединения, полученного в примере 60, 10 мл 20%-ного раствора гидроксида аммония и 0,1 г никеля Ренея® в 20 мл этанола. Катализатор отфильтровывают на целите®, промывают этанолом, затем метанолом и фильтрат частично концентрируют. Выкристаллизовавшийся продукт отфильтровывают под вакуумом, а фильтрат концентрируют в вакууме. Выкристаллизовавшийся продукт и остаток после концентрирования обрабатывают 1,2 н. соляной кислотой, осадок отфильтровывают под вакуумом и промывают его 1,2 н. соляной кислотой. Осадок растворяют в воде, водную фазу нейтрализуют до рН 7 путем добавления 1,3 н. раствора гидроксида натрия, выпавший осадок отфильтровывают под вакуумом, промывают пропан-2-олом, кипятят с обратным холодильником, охлаждают до комнатной температуры и осадок отфильтровывают под вакуумом. Получают 0,54 г целевого продукта (после высушивания). Т.пл.=239-241oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,2 (м, 14Н); 2,4-3,8 (м, 8Н); 3,5 (мт, 2Н); 3,64 (с, 6Н); 6,6 (д, 2Н); 6,72 (с, 1Н); 7,1-7,45 (м, 4Н).
ПРИМЕР 62
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(2-карбамоилэтил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-CON(CH3)(CH2)2CONH2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 3,5 часов при комнатной температуре перемешивают смесь 0,2 г соединения, полученного в примере 60, 0,12 мл 30%-ного раствора пероксида водорода в воде и 0,18 мл 6 н. раствора гидроксида натрия в 10 мл 95%-ного этанола. Затем добавляют 0,06 мл 30%-ного раствора пероксида водорода и 0,06 мл 6 н. раствора гидроксида натрия и продолжают перемешивание в течение полутора часов при комнатной температуре. Нерастворимое вещество отфильтровывают, к фильтрату добавляют воду, водную фазу промывают два раза дихлорметаном, подкисляют до рН 3 путем добавления 1,2 н. соляной кислоты, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью толуола с метанолом в объемном соотношении 90:10. Получают 0,018 г целевого продукта (после порошкования в эфире). Т.пл.=164-166oС.
ПРИМЕР 63
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(2-карбоксиэтил)-карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 3-CON(CH3)(CH2)2СO2H; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Смесь 0,4 г соединения, полученного в примере 60, и 0,042 мл 4-метоксибензилового спирта в 3 мл дихлорметана охлаждают до 0oС, в течение 30 минут через нее пропускают ток газообразного хлороводорода, реакционную смесь разбавляют с помощью 17 мл дихлорметана и перемешивают в течение двух часов при температуре 0oС. Концентрируют в вакууме, полученный промежуточный имидат обрабатывают с помощью 9 мл ацетона, добавляют 2 мл 1,2 н. соляной кислоты и перемешивают 5 дней при комнатной температуре. Добавляют 6 мл диметилформамида и 1 мл 1,2 н. соляной кислоты, в течение трех дней кипятят с обратным холодильником и перемешивают 72 часа при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают дихлорметаном, органическую фазу экстрагируют насыщенным раствором гидрокарбоната натрия, водную фазу промывают дихлорметаном, подкисляют до рН 1 путем добавления концентрированной соляной кислоты, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. После порошкования в эфире, затем высушивания при 60oС над фосфорным ангидридом получают 0,03 г целевого продукта. Т.пл.=166-168oС.
ЯМР-спектр (ДМСО+ТФУК): 1 (д, 6Н); 1,5-1,9 (м, 12Н); 1,9-2,8 (м, 5Н); 2,8-3,0 (мт, 3Н); 3,2-3,6 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,0-7,5 (м, 4Н).
ПРИМЕР 64
2-[5-(2,6-Диметоксифенил)-1-[4-[N-(пропен-2-ил)карбамоил] -2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CONHCH2CH=CH2; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 53, исходя из 1,49 г соединения, полученного в Препаративном примере 4.42, и 25 мл тионилхлорида, затем 0,65 г соединения Б и 1,6 мл бис(триметилсилил)ацетамида в 10 мл ацетонитрила. После перемешивания в течение ночи при комнатной температуре отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток обрабатывают с помощью 10 мл метанола, добавляют 10 мл воды, твердый продукт отфильтровывают под вакуумом и промывают его метанолом. Твердый продукт обрабатывают ацетонитрилом, кипятят с обратным холодильником, оставляют охлаждаться до комнатной температуры, осадок отфильтровывают под вакуумом и сушат над фосфорным ангидридом. Получают 1,6 г целевого продукта. Т.пл.=304oС.
ЯМР-спектр: 1,1 (д, 6Н); 1,5-2,2 (м, 12Н); 2,5 (уш.с, 2Н); 2,65 (кт, 1Н); 3,65 (с, 6Н); 3,9 (т, 2Н); 5,0-5,2 (м, 2Н); 5,8-6,0 (м, 1Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,4 (м, 3Н); 7,6 (д, 1Н); 7,85 (с, 1Н); 8,7 (т, 1Н).
ПРИМЕР 65
Иодид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3- триметиламмонийпропил)карбамоил] -2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); 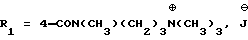 ; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 24 часов при комнатной температуре перемешивают смесь 0,1 г соединения, полученного в примере 1', и 0,04 г метилиодида в 6 мл дихлорметана. Концентрируют в вакууме, остаток порошкуют в эфире и выпавший осадок отфильтровывают под вакуумом. Получают 0,12 г целевого продукта. Т.пл.=22oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,5-2,3 (м, 14Н); 2,5 (уш.с, 2Н); 2,7 (кт, 1Н); 2,75-3,7 (м, 22Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,5 (м, 4Н).
ПРИМЕР 66
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-[3-[N'-метил-N'-(трет. -бутоксикарбонил)амино] пропил] карбамоил] -2- изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-CON(CH3)(СН2)3N(CH3)COO-трет. -бутил; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Раствор 1,17 г соединения, полученного в Препаративном примере 4.43, и 0,3 мл триэтиламина в 3 мл диметилформамида охлаждают до -10oС, в атмосфере азота добавляют 0,21 мл этилхлорформиата и перемешивают в течение 15 минут при температуре -10oС. С другой стороны, в течение 45 минут при температуре 80oС нагревают смесь 0,77 г соединения Б и 2 мл бис(триметилсилил)ацетамида в 3 мл диметилформамида. После охлаждения до комнатной температуры этот раствор добавляют к полученному выше раствору смешанного ангидрида и перемешивают в течение трех дней при комнатной температуре. Отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток обрабатывают с помощью 32 мл метанола, добавляют постепенно 32 мл воды и концентрируют в вакууме. Остаток обрабатывают водой, выкристаллизовавшийся продукт отфильтровывают под вакуумом, промывают его водой и высушивают. Кристаллы обрабатывают дихлорметаном, отфильтровывают нерастворимую часть и фильтрат хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом в объемном соотношении от 100:0,5 до 100:2,5. После порошкования в пентане получают 1 г целевого продукта. Т.пл.=118-120oС.
ПРИМЕР 67
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-метиламинопропил)карбамоил]-2-изопропилфенил]пиразол-3- илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(CH2)3NH(CH3); R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 20 минут при комнатной температуре перемешивают смесь 0,6 г соединения, полученного в примере 66, и 4,2 мл концентрированной соляной кислоты в 2,7 мл метанола с 1,8 мл воды. Добавляют этанол и реакционную смесь концентрируют в вакууме. Остаток обрабатывают этанолом и растворитель выпаривают в вакууме. Остаток обрабатывают эфиром, выпавший осадок отфильтровывают под вакуумом и промывают его эфиром. После высушивания в вакууме при температуре 60oС получают 0,51 г целевого продукта. Т.пл.=240oС.
ЯМР-спектр (ДМСО+ТФУК): 1,1 (д, 6Н); 1,5-2,4 (м, 14Н); 2,6 (д, 3Н); 2,7 (мт, 1Н); 2,8-3,6 (м+с, 7Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,7 (с, 1Н); 7,1-7,45 (м, 4Н).
ПРИМЕР 68
2-[5-(2,6-Диметоксифенил)-1-[4-(4-метилфенилсульфониламино)-2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); 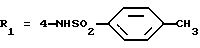 ; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 1 часа при температуре 40oС нагревают смесь 0,92 г соединения, полученного в Препаративном примере 4.44, и 7 мл тионилхлорида в 7 мл дихлорметана. Концентрируют в вакууме и таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 1 часа кипятят с обратным холодильником смесь 0,54 г соединения Б и 1,35 мл бис(триметилсилил)ацетамида в 5 мл ацетонитрила. После охлаждения до комнатной температуры этот раствор добавляют к вышеполученному хлорангидриду кислоты, добавляют 0,25 мл триэтиламина и перемешивают в течение двух часов при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают с помощью 10%-ной соляной кислоты, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:3:0,5. Получают 0,9 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 0,85 (д, 6Н); 1,52-2,25 (м, 12Н); 2,34 (с, 3Н); 2,45-2,06 (м, 3Н); 3,55 (с, 6Н); 6,55 (д, 2Н), 6,65 (с, 1Н); 6,84 (д.д, 1Н); 6,95-7,05 (м, 2Н); 7,23-7,36 (м, 3Н); 7,58 (д, 2Н).
ПРИМЕР 69
2-[5-(2,6-Диметоксифенил)-1-[4-[3- (диэтиламино)пропаноиламино ] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-NHCO(CH2)2N(С2Н5)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 68, исходя из 0,27 г соединения, полученного в Препаративном примере 4.46, в 5 мл тионилхлорида и 5 мл дихлорметана, с одной стороны, и 0,155 г соединения Б и 0,39 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила с 0,14 мл триэтиламина, с другой стороны. Получают 0,13 г целевого продукта. Т.пл.=180oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,25 (т, 6Н); 1,55-2,22 (м, 12Н); 2,5-2,72 (м, 3Н); 2,85 (т, 2Н); 3,2 (кд, 4Н); 3,4 (мт, 2Н); 3,68 (с, 6Н); 6,6 (д, 2Н); 6,72 (с, 1Н); 7,15 (д, 1Н); 7,25-7,5 (м, 2Н); 7,58 (д, 1Н).
ПРИМЕР 70
2-[5-(2,6-Диметоксифенил)-1-[4-[N-ацетил-N-(3- диэтиламинопропил)амино] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-N(COOH)3(СН2)3N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 68, исходя из 0,38 г соединения, полученного в Препаративном примере 4.48, и 3 мл тионилхлорида в 3 мл дихлорметана, затем 0,164 г соединения Б и 0,36 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила с 0,075 мл триэтиламина. Получают 0,24 г целевого продукта. Т.пл.=220oС.
ЯМР-cпектр (ДМСО+ТФУК): 1,0 (д, 6Н); 1,5 (т, 6Н); 1,45-2,2 (м, 17Н); 2,5-2,72 (м, 3Н); 2,9-3,15 (м, 6Н); 3,6 (с, 6Н); 3,7 (т, 2Н); 6,59 (д, 2Н); 6,74 (с, 1Н); 7,15-7,42 (м, 5Н).
ПРИМЕР 71
2-[5-(2,6-Диметоксифенил)-1-[4-[(3-диэтиламинопропил)амино] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-NH(CH2)3N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 16 часов кипятят с обратным холодильником смесь 0,2 г соединения, полученного в примере 70, и 1 мл концентрированной соляной кислоты в 5 мл воды с 5 мл этанола. Добавляют воду, доводят рН до 5 путем добавления 10%-ного раствора гидроксида натрия и осадок отфильтровывают под вакуумом. Получают 0,145 г целевого продукта. Т.пл.=180oС.
ЯМР-спектр (ДМСО+ТФУК): 1,05 (д, 6Н); 1,19 (т, 6Н); 1,4-2,2 (м, 14Н); 2,4-2,63 (м, 3Н); 2,98-3,3 (м, 8Н); 3,62 (с, 6Н); 6,5-6,85 (м, 5Н); 7,05 (д, 1Н); 7,3 (т, 1Н).
ПРИМЕР 72
Гидрохлорид 2-[5-[2-(циклопропилметилокси)-6-метоксифенил] -1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(CH2)3N(CH3)2; R2 - 2-изопропил; R3 - Н;  ; АА(ОН) - 2-карбоксиадамант-2-ил)
; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение восьми часов при температуре 60oС нагревают смесь 3,87 г соединения, полученного в Препаративном примере 4.49, и 2,4 мл тионилхлорида в 50 мл дихлорметана. Концентрируют в вакууме, остаток обрабатывают толуолом и выпаривают растворитель в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 3 часов при температуре 80oС нагревают смесь 1,27 г соединения Б и 2,65 г бис(триметилсилил)ацетамида в 80 мл ацетонитрила. Затем добавляют раствор вышеполученного хлорангидрида кислоты в 80 мл ацетонитрила и нагревают при 60oС в течение трех часов. Отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток обрабатывают с помощью 16 мл метанола, добавляют 16 мл воды и концентрируют в вакууме. Остаток обрабатывают водой, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:5:0,5. Получают 2,1 г целевого продукта.
ПРИМЕР 73
Гидрохлорид 2-[5-(2-гидрокси-6-метоксифенил)-1-[4-[N-метил-(3-диметиламинопропил)карбамоил] -2- изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CON(CH3)(CH2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - H; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение 5 часов при температуре 60oС нагревают смесь 1 г соединения, полученного в примере 72, и 20 мл метанола с 20 мл соляной кислоты. Концентрируют в вакууме, остаток обрабатывают толуолом и концентрируют в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 90:10, затем смесью дихлорметана с метанолом и гидроксидом аммония в объемном соотношении 80:20:2. Получают 0,6 г целевого продукта. Т.пл. выше 250oС.
ПРИМЕР 74
2-[5-(2,6-Диметоксифенил)-1-[4-[3-(диэтиламинопропанол)амино] -2-метилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-NHCON(CH2)2N(C2H5)2; R2 - 2-метил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
А) 2-[5-(2,6-Диметоксифенил)-1-(2-метил-4- нитрофенил)пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в примере 68, исходя из 3,2 г полученного в Препаративном примере 4.50 соединения и 20 мл тионилхлорида в 40 мл дихлорметана, затем 2,4 г соединения Б и 6 мл бис(триметилсилил)ацетамида в 15 мл ацетонитрила и затем 1,1 мл триэтиламина. После перемешивания в течение ночи при комнатной температуре реакционную среду концентрируют в вакууме. Остаток обрабатывают смесью ацетона с водой, выпавший осадок отфильтровывают под вакуумом и высушивают. Осадок хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:0,3:0,2. Получают 4,3 г целевого продукта. Т.пл.=150oС.
ЯМР-спектр: 1,6-2,2 (м, 12Н); 2,25 (с, 3Н); 2,62 (мт, 2H); 3,63 (с, 6Н); 6,68 (д, 2H); 6,88 (с, 1Н); 7,03-7,43 (м, 2H); 7,58 (с, 1Н); 8,05 (д.д, 1Н), 8,28 (д, 1Н); 12,4 (уш.с, 1Н).
Б) 2-[5-(2,6-Диметоксифенил)-1-(4-амино-2- метилфенил)пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота
В течение 4 часов при комнатной температуре и при атмосферном давлении гидрируют смесь 4,2 г соединения, полученного на предыдущей стадии, в присутствии 0,5 г никеля Ренея® в 40 мл метанола и 2 мл диметилформамида. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток обрабатывают эфиром и отфильтровывают под вакуумом выпавший осадок. Получают 3,37 г целевого продукта. Т.пл.=205oС.
ЯМР-спектр: 1,42-2,1 (м, 15Н); 2,52 (мт, 2H); 3,57 (с, 6Н); 5,1 (уш.с, 2H); 6,1 (д.д, 1Н); 6,22 (д, 1Н); 6,42-6,68 (м, 4Н); 7,17-7,25 (м, 2H).
В) 2-[5-(2,6-Диметоксифенил)-1-[4-[3-(диэтиламинопропаноил)амино] -2-метилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота
В течение 45 минут при температуре 35oС нагревают смесь 0,3 мл гидрохлорида 3-диэтиламинопропановой кислоты и 3 мл тионилхпорида в 6 мл дихлорметана, затем концентрируют в вакууме. Таким образом полученный хлорангидрид кислоты добавляют к раствору 0,87 г соединения, полученного на предыдущей стадии, и 0,157 мл триэтиламина в 5 мл дихлорметана. Концентрируют в вакууме и остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:5:0,5. Получают 0,5 г целевого продукта. Т.пл.=190oС.
ЯМР-спектр: 1,28 (т, 6Н); 1,6-2,2 (м, 15Н); 2,5-3,2 (м, 10Н); 3,6 (с, 6Н); 6,63 (д, 2Н); 6,75 (с, 1Н), 7,05 (д, 1Н); 7,28-7,48 (м, 3Н); 7,55 (д, 1Н); 10,18 (с, 1Н).
ПРИМЕР 75
2-[5-(2,6-Диметоксифенил)-1-[4-[[-3(пиперид-1-ил)пропаноил]амино]-2-метилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I);  ; R2 - 2-метил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R2 - 2-метил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 74, стадия В, исходя из 0,1 г 3-(пиперид-1-ил)пропановой кислоты и 1 мл тионилхлорида в 2 мл дихлорметана, затем 0,337 г соединения, полученного на стадии Б примера 74, и 0,17 мл триэтиламина в 5 мл дихлорметана. Получают 0,2 г целевого продукта. Т.пл.=240oС.
ЯМР-спектр: 1,22-2,1 (м, 21Н); 2,22-2,38 (м, 10Н); 3,58 (c, 6H); 6,5 (д, 2Н); 6,6 (с, 1Н); 6,9 (д, 1Н); 7,18-7,3 (м, 3Н); 7,38 (д, 1Н); 10,1 (с, 1Н).
ПРИМЕР 76
2-[5-(2,6-Диметоксифенил)-1-[4-[3-(диэтиламино)пропаноиламино] -2-изобутилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 4-NHCO(CH2)2N(C2H5)2; R2 - 2-изобутил; R3 - H; R4 - метил, АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 68, исходя из, с одной стороны, 0,15 г соединения, полученного в Препаративном примере 4.52, и 2 мл тионилхпорида в 2 мл дихлорметана и, с другой стороны, 0,084 г соединения Б и 0,21 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила и 0,79 мл триэтиламина. Получают 0,014 г целевого продукта. Т.пл.= 180-200oС.
ЯМР-спектр: 0,75 (д, 6Н); 1,15 (т, 6Н); 1,4-2,25 (м, 15Н); 2,5 (с, 2Н); 2,8 (т, 2Н); 3,1 (кд, 4Н); 3,3 (т, 2Н); 3,55 (с, 6Н); 6,5 (д, 2Н); 6,6 (с, 1Н); 6,8-7,6 (м, 5Н); 10,3 (с, 1Н).
ПРИМЕР 77
2-[5-(2,6-Диметоксифенил)-1-[4-[3-(диэтиламино)пропаноиламино] -2-циклопентилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота (формула (I); R1 - 4-NHCO(CH2)2-N(C2H5)2; 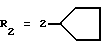 ; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в примере 68, исходя из, с одной стороны, 0,32 г соединения, полученного в Препаративном примере 4.54, и 2 мл тионилхлорида в 5 мл дихлорметана и, с другой стороны, 0,17 г соединения Б, 0,42 мл бис(триметилсилил)ацетамида в 2 мл ацетонитрила и 0,154 мл триэтиламина. Получают 0,035 г целевого продукта. Т.пл.=175-185oС.
ЯМР-спектр (ДМСО+ТФУК): 1,1-2,55 (м, 26Н); 2,5-2,75 (м, 5Н); 3,15 (мт, 4Н); 3,35 (мт, 2Н); 3,62 (с, 6Н); 6,55 (д, 2Н); 6,65 (с, 1Н); 7,03 (д, 1Н); 7,25 (т, 1Н); 7,35 (д.д, 1Н); 7,55 (д, 1Н).
ПРИМЕР 78
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(2-диэтиламиноэтил)карбамоил] -3-изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-CONH(CH2)2N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
В течение ночи при комнатной температуре перемешивают смесь 0,36 г соединения, полученного в Препаративном примере 4.55, и 5 мл тионилхлорида в 15 мл хлороформа. Концентрируют в вакууме, остаток обрабатывают толуолом и концентрируют в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 30 минут кипятят с обратным холодильником смесь 0,123 г соединения Б и 0,315 мл бис(триметилсилил)ацетамида в 10 мл ацетонитрила. Этот раствор добавляют к раствору вышеполученного хлорангидрида кислоты в 15 мл ацетонитрила и в течение трех часов кипятят с обратным холодильником. Концентрируют в вакууме, остаток обрабатывают с помощью 15 мл метанола и 5 мл воды и перемешивают 2 часа при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют хлороформом, экстракт промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из хлороформа получают 0,35 г целевого продукта. Т.пл.=210oС (разложение) (продукт кристаллизуется с 1 молем хлороформа).
ПРИМЕР 79
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-(2-аминоацетиламино)-5,6,7,8-тетрагидронафт-1-ил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты (формула (I); R1 - 4-NHCOCH2NH3; R2, R3 - -(CH2)4-; R4 - метил; АА(ОН) - 2-карбоксиадамант-2-ил)
А) 2-[5-(2,6-Диметоксифенил)-1-[4-нитро-5,6,7,8- тетрагидронафт-1-ил]-пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в примере 15, исходя из 4 г соединения, полученного в Препаративном примере 4.56, и 20 мл тионилхлорида в 20 мл дихлорметана, затем 2,74 г соединения Б и 6,86 мл бис(триметилсилил)ацетамида в 20 мл ацетонитрила и 0,8 мл триэтиламина. После концентрирования в вакууме остаток обрабатывают этанолом и выпавший осадок отфильтровывают под вакуумом, его обрабатывают метанолом, отфильтровывают под вакуумом и промывают эфиром. Получают 5,3 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,5-2,25 (м, 16Н); 2,42-2,65 (м, 4Н); 2,8 (мт, 2Н); 3,6 (с, 6Н), 6,61 (д, 2Н); 6,75 (с, 1Н); 7,06 (д, 1Н); 7,3 (т, 1H); 7,4 (c, 1Н), 7,65 (д, 1Н).
Б) 2-[5-(2,6-Диметоксифенил)-1-(4-амино-5,6,7,8- тетрагидронафт-1-ил)пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота
При комнатной температуре и при атмосферном давлении гидрируют смесь 3 г полученного на предыдущей стадии соединения и 0,5 г никеля Ренея® в 200 мл диметилформамида. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Остаток обрабатывают водой и выпавший осадок отфильтровывают под вакуумом. Получают 2,16 г целевого продукта (после высушивания).
ЯМР-спектр (ДМСО+ТФУК): 1,42-2,2 (м, 16Н); 2,3-2,8 (м, 6H); 3,6 (с, 6Н); 6,55 (д, 2Н); 6,7 (с, 1Н); 6,98 (д, 1Н); 7,12 (д, 1Н); 7,25 (т, 1Н).
В) 2-[5-(2,6-Диметоксифенил)-1-[4-[2-(трет. -бутоксикарбониламино)ацетиламино] -5,6,7,8-тетрагидронафт-1-ил]пиразол-3-илкарбониламино] адамантан-2-карбоновая кислота
В течение 1 часа при температуре 60oС нагревают смесь 0,3 г полученного на предыдущей стадии соединения и 0,258 мл бис(триметилсилил)ацетамида в 2 мл толуола. После охлаждения до комнатной температуры добавляют 0,64 мл Вос-глицин-N-карбоксангидрида, 0,006 мл N-метилморфолина и перемешивают в течение ночи при комнатной температуре. Добавляют буферный раствор с рН 4, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении от 100:1 до 100:5. Получают 0,14 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,32 (с, 9Н); 1,45-2,12 (м, 16Н); 2,35-2,6 (м, 6Н); 3,55 (с, 6Н); 3,65 (с, 2Н); 6,5 (д, 2Н); 6,62 (с, 1Н); 6,83 (д, 1Н); 7,13-7,3 (м, 3Н).
Г) Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-(2- аминоацетиламино)-5,6,7,8-тетрагидронафт-1-ил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение 30 минут при комнатной температуре перемешивают смесь 0,14 г полученного на предыдущей стадии соединения и 5 мл концентрированной соляной кислоты в 5 мл метанола. Добавляют воду, отфильтровывают под вакуумом выпавший осадок и высушивают его. Получают 0,06 г целевого продукта. Т.пл.= 220oС.
ЯМР-спектр: 1,59-2,5 (м, 16Н); 2,42-2,75 (м, 6Н); 3,7 (с, 6Н); 3,88 (мт, 2Н); 6,68 (д, 2Н); 6,75 (с, 1Н); 6,95 (д, 1Н); 7,2-7,48 (м, 3Н); 8,2 (мт, 1Н); 9,85 (с, 1Н); 12,4 (уш.с, 1Н).
ПРИМЕР 80
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[(3-диэтиламинопропаноил)амино] -5,6,7,8-тетрагидронафт-1- ил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 4-NHCO(CH2)2N(C2H5); R2, R3 - -(CH2)4-; R4 - CH3; AA(OH) - 2-карбоксиадамант-2-ил)
В течение 1 часа при температуре 40oС нагревают смесь 0,16 г гидрохлорида 3-диэтиламинопропановой кислоты и 2 мл дихлорметана. Концентрируют в вакууме, остаток обрабатывают дихлорметаном и при комнатной температуре полученный раствор добавляют к раствору 0,5 г соединения, полученного на стадии Б примера 79, и 0,124 мл триэтиламина в 3 мл дихлорметана. После перемешивания в течение ночи при комнатной температуре добавляют воду, экстрагируют дихлорметаном, органические фазы сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле 60 Н, элюируя смесью дихлорметана с метанолом в объемном соотношении 100:3. Получают 0,11 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,21 (т, 6Н); 1,41-2,2 (м, 16Н); 2,35-2,7 (м, 6Н); 2,84 (т, 2Н); 3,01-3,12 (м, 4Н); 3,35 (т, 2Н); 3,6 (с, 6Н); 6,55 (д, 2Н); 6,7 (с, 1Н); 6,9 (д, 1Н); 7,12-7,3 (м, 2Н).
ПРИМЕР 81
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-(2-аминоэтилсульфониламино)-5,6,7,8-тетрагидронафт-1- ил] пиразол-3-илкарбониламино] адамантан-2-карбоновой кислоты (формула (I); R1 - 4-NHSO2(СН2)2NH2; R2, R3 - -(СH2)4-; R4 - мeтил; АА(ОН) - 2-карбоксиадамант-2-ил)
А) Калиевая соль 2-фталимидоэтансульфокислоты
Это соединение и соединение стадии Б получают согласно J. Am. Chem. Soc. , 69, 1393-1401 (1947).
В течение 10 минут нагревают до температуры кипения с обратным холодильником смесь, содержащую 30 г таурина, 25 г ацетата калия и 90 мл уксусной кислоты, затем добавляют 37,8 г фталевого ангидрида. В течение 2,5 часов кипятят с обратным холодильником, затем осадок отфильтровывают, промывают уксусной кислотой, потом пропан-2-олом; затем промывают эфиром и высушивают в вакууме, получая 59,14 г целевого продукта.
Б) Хлорангидрид 2-фталимидоэтансульфокислоты
В течение 1 часа кипятят с обратным холодильником 60 г полученного на стадии А соединения в 300 мл толуола в присутствии 30,7 г пентахлорида фосфора. Снова добавляют 30,7 г пентахлорида фосфора и кипятят с обратным холодильником в течение 90 минут. Добавляют 280 г льда в реакционную среду, перемешивают, отфильтровывают нерастворимую часть, затем промывают холодной водой. Остаток высушивают над фосфорным ангидридом, после чего перекристаллизуют из дихлорэтана, получая 32 г целевого продукта. Т.пл.=160oС.
В) 2-[5-(2,6-Диметоксифенил-1-[4-(2- фталимидоэтансульфонил)-5,6,7,8-тетрагидронафт-1-ил]пиразол-3-ил-карбониламино]адамантан-2-карбоновая кислота
В течение 1 часа при температуре 70oС перемешивают смесь, содержащую 0,5 г соединения, полученного в примере 79, стадия Б, 0,43 мл бис (триметилсилил)ацетамида и 5 мл ацетонитрила. Выдерживают для возврата температуры к комнатной, затем добавляют 0,63 г полученного на стадии Б соединения и 0,30 мл триэтиламина. После перемешивания в течение двух часов при комнатной температуре подкисляют с помощью 10%-ной соляной кислоты. Осадок отфильтровывают, затем высушивают над фосфорным ангидридом, получая 0,9 г целевого продукта в неочищенной форме. Его перекристаллизуют из этанола (100%-ного) и обеспечивают животным углем в дихлорметане. Полученный продукт хроматографируют на силикагеле 60 Н, эпюируя смесью дихлорметана с метанолом и водой в объемном соотношении 100:2:0,2, получая 0,28 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,45-2,15 (м, 16Н); 2,4-2,6 (м, 4Н); 2,75 (мт, 2Н); 3,48 (с, 6Н); 3,95-4,15 (м, 4Н); 6,46 (д, 1Н); 6,75 (с, 1Н); 6,9 (д, 1Н); 7,1-7,3 (м, 2Н); 7,7-7,85 (м, 4Н).
Г) Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-(2- аминоэтилсульфониламино)-5,6,7,8-тетрагидронафт-1- ил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение двух часов кипятят с обратным холодильником смесь, содержащую 0,24 г полученного на предыдущей стадии соединения, 2 мл 95%-ного этанола и 23 мкл гидразингидрата. Реакционную среду разбавляют метанолом, затем кристаллы отфильтровывают, после чего их нагревают до температуры кипения в воде и раствор фильтруют горячим. Полученные кристаллы сушат над фосфорным ангидридом. Эти кристаллы снова растворяют в метаноле, добавляют солянокислый эфир, выпаривают досуха, после чего обрабатывают эфиром и пентаном. Отфильтровывают, получая 60 мг целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 1,48-2,18 (м, 16Н); 2,18-2,62 (м, 4Н); 2,7 (мт, 2Н); 3,19 (мт, 2Н); 3,41 (мт, 2Н); 3,62 (с, 6Н); 6,58 (д, 1Н); 6,7 (с, 1Н); 6,82 (д, 1Н); 7,08 (д, 1Н); 7,28 (т, 1Н); 7,39 (с, 1Н).
ПРИМЕР 82
(R)-2-Циклогексан-2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] уксусная кислота (формула (I); R1 - 4-CON(CH3)(CH2)3N (СН3)2; R2 - 2-изопропил: R3 - H; R4 - метил; AA(OH) - (R)-(α-карбокси)циклогексилметил)
Смешивают 1,2 г гидроксида натрия в 20,2 мл воды и 1,62 г (D)-циклогексилглицинтрифторацетата. Прикапывают раствор 1,58 г хлорангидрида кислоты, полученного в примере 1, стадия А, в 40 мл безводного тетрагидрофурана и перемешивают в течение 48 часов при комнатной температуре. Среду концентрируют, добавляют лед и доводят рН до значения 7 путем добавления концентрированной соляной кислоты. Отфильтровывают, промывают водой, затем пентаном и сушат в вакууме. Продукт размельчают, затем перемешивают в смеси воды с дихлорметаном. Отфильтровывают, затем водную фазу экстрагируют дихлорметаном и сушат полученный экстракт над сульфатом натрия. Концентрируют, перемешивают остаток в пентане и снова отфильтровывают. Получают 380 мг целевого продукта. Т.пл.=160oС.
ПРИМЕР 83
Гидрохлорид (S)-2-циклогексил-2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино]уксусной кислоты (формула (I); R1 - 4-CON(CH3)(CH2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - CH3; AA(OH) - (S)-(α-карбокси)циклогексилметил)
В течение трех часов при температуре 80oС нагревают смесь, содержащую 0,57 г (S)-циклогексилглицина и 1,49 г бис(триметилсилил)ацетамида в 39 мл ацетонитрила и прикапывают раствор 1,93 г полученного в примере 1, стадия А, хло-рангидрида кислоты в 39 мл ацетонитрила. После выдерживания в течение трех часов при температуре 60oС температуру смеси оставляют возвращаться к комнатной, после чего отфильтровывают реакционную среду и фильтрат концентрируют. К остатку добавляют 8 мл метанола, 3 мл воды и перемешивают в течение 30 минут. Добавляют 5 мл воды и концентрируют. Полученное масло поглощают в дихлорметане, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют. Остаток обрабатывают диизопропиловым эфиром и отфильтровывают, получая 1,12 г целевого соединения. Т.пл.=160oС.
ПРИМЕР 84
2-[5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2-изопропилфенил] пиразол-3-илкарбониламино] бицикло[3.3.1] нонан-9-карбоновая кислота (формула (I); R1 - 4-CON(CH3)(СН2)3N(CH3)2; R2 - 2-изопропил; R3 - H; R4 - метил; АА(ОН) - 9-карбоксибицикло[3.3.1]нонан-9-ил)
585 мг 9-Амино[3.3.1]бициклононан-9-карбоновой кислоты смешивают с 1,5 мл бис(триметилсилил)ацетамида в 39 мл ацетонитрила и нагревают в течение трех часов при температуре 80oС. Прикапывают раствор 1 эквивалента хлорангидрида кислоты, полученного в примере 1, стадия А, в 39 мл ацетонитрила и нагревают при 60oС в течение трех часов. После выдерживания в течение 12 часов при комнатной температуре отфильтровывают нерастворимую часть, затем концентрируют фильтрат и остаток перемешивают с 8 мл метанола и 8 мл воды. Снова концентрируют, потом экстрагируют дихлорметаном, получая затем 900 мг целевого продукта. Т.пл.=160oС.
ПРИМЕР 85
2-[5-(2,6-Диметоксифенил)-1-[5-[(3-диэтиламинопропаноил)амино] -2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновая кислота (формула (I); R1 - 5-NHCO(CH2)2N(C2H5)2; R2 - 2-изопропил; R3 - H; R4 - CH3; АА(ОН) - 2-карбоксиадамант-2-ил)
Хлорангидрид кислоты получают из 0,95 г соединения, полученного в Препаративном примере 4.57, в 5 мл тионилхлорида и 15 мл дихлорметана путем кипячения с обратным холодильником в течение 1 часа, затем выпаривания. В течение 1 часа кипятят с обратным холодильником смесь 0,55 г соединения Б, 1,37 мл бис(триметилсилил)ацетамида в 5 мл ацетонитрила и хлорангидрида кислоты в виде раствора в 5 мл дихлорметана и 0,5 мл триэтиламина. После перемешивания в течение двух часов при комнатной температуре выпаривают досуха, затем остаток перемешивают с 10 мл воды, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом магния, выпаривают досуха и остаток кристаллизуют из ацетона, получая 0,55 г целевого продукта.
ЯМР-спектр (ДМСО+ТФУК): 0,8-1,35 (м, 12Н); 1,5-2,4 (м, 12H); 2,4-2,6 (м, 3Н); 2,8 (т, 2Н); 3,15 (кд, 4Н); 3,3 (т, 2Н); 3,5 (с, 6Н); 6,55 (д, 2Н); 6,65 (с, 1Н); 7,15-7,4 (м, 3Н); 7,75 (д, 1Н).
ПРИМЕР 86
Внутренняя соль 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил]-2-изопропилфенил]пиразол-3-илкарбониламино]-адамантан-2-карбоновой кислоты
Соединение также может быть получено из соединения примера 61 согласно следующей методике.
В течение 30 минут при температуре 100oС нагревают смесь, содержащую 0,2 г соединения примера 61, 0,33 мл муравьиной кислоты и 0,11 мл формальдегида. После выдерживания в течение двух часов при комнатной температуре добавляют 1 мл 2 н. соляной кислоты, затем добавляют дихлорметан и метанол для растворения образовавшейся смолы. Растворители выпаривают, остаток обрабатывают водой, затем нейтрализуют 1,3 н. раствором гидроксида натрия вплоть до достижения рН 7, охлаждая среду льдом. Осадок отфильтровывают, промывают водой, затем сушат над фосфорным ангидридом, получая 0,13 г целевого продукта.
ПРИМЕР 87
Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-(3- диметиламинопропил)карбамоил] -2-изопропилфенип] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
Ниже описывается другой способ получения соединения примера 1.
A) 2-(Бензилоксикарбониламино)адамантан-2-карбоновая кислота
В течение полутора часов кипятят с обратным холодильником 1,015 г 2-аминоадамантан-2-карбоновой кислоты и 6 мл бис(триметилсилил)ацетамида в 10 мл дихлорметана. Добавляют 0,75 мл бензилоксикарбонилхлорида и в течение 15 минут нагревают при 50oС. Реакционную среду охлаждают до -70oС, затем разлагают путем добавления льда и экстрагируют этилацетатом. Промывают экстракт водой (2 раза), насыщенным раствором хлорида натрия, сушат органическую фазу над сульфатом магния и выпаривают в вакууме. Продукт кристаллизуют из гексана. Получают 1,164 г.
ЯМР-спектр (ДМСО+ТФУК): 1,5 (д, 2Н); 1,8 (м, 6Н); 2 (т, 4Н); 2,4-2,5 (м, 2Н); 5 (с, 2Н); 7,3 (уш.с, 5Н).
Б) трет. -Бутиловый эфир 2- (бензилоксикарбонил)аминоадамантан-2-карбоновой кислоты
1,164 г Полученного на предыдущей стадии соединения растворяют в 15 мл дихлорметана, добавляют 100 г гидратиро-ванной п-толуолсульфокислоты, затем смесь охлаждают до температуры -78oС и добавляют раствор изобутилена в 15 мл дихлорметана. Оставляют стоять для возврата температуры к комнатной и перемешивают в течение 24 часов. Добавляют 50 мкл концентрированной серной кислоты для растворения твердого вещества, спустя 5 часов среду охлаждают, затем добавляют насыщенный раствор гидрокарбоната натрия; сушат над сульфатом магния и выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью гексана с этилацетатом в объемном соотношении 80:20, и получают 612 мг целевого соединения.
ЯМР-спектр (дейтерохлороформ): 1,4 (с, Н); 1,5-1,9 (м, 8Н); 2 (т, 4Н); 2,5 (с, 2Н); 4,9 (с, 1Н); 5,1 (с, 2Н); 7,2-7,4 (м, 5Н).
В) Гидрохлорид трет.-бутилового эфира 2-аминоадамантан-2-карбоновой кислоты
600 мг Полученного на предыдущей стадии продукта растворяют в 40 мл этанола, добавляют 150 мкл концентрированной соляной кислоты, затем 80 г палладия-на-угле, потом среду гидрируют. Спустя 1 час катализатор отфильтровывают и выпаривают из фильтрата растворитель, получая 503 мг целевого продукта.
ЯМР-спектр (дейтерированный метанол): 1,6 (с, 9Н); 1,8-2 (м, 8Н); 2-2,2 (м, 4Н); 2,4 (с, 2Н).
Г) Метиловый эфир 5-(2,6-диметоксифенил)-1-[4-[N-метил-[3-[N'-метил-N'-(бензилоксикарбонил)амино] пропил] карбамоил]-2-изопропилфенил]пиразол-3-карбоновой кислоты
0,33 г Соединения, полученного в Препаративном примере 3.57, растворяют в 20 мл солянокислого метанола. После перемешивания в течение 72 часов растворители выпаривают. Полученный гидрохлорид растворяют в 5 мл дихлорметана, затем добавляют 0,5 мл триэтиламина и 150 мкл бензилоксикарбонилхлорида. Спустя 1 час реакционную среду концентрируют в вакууме, затем остаток хроматографируют на диоксиде кремния, элюируя смесью толуола с ацетоном в объемном соотношении от 80:20 до 70:30. Получают 252 мг целевого продукта.
Д) 5-(2,6-Диметоксифенил)-1-[4-(N-метил-N-[3-[N'-метил-N'-(бензилоксикарбонил)амино]пропил]карбамоил]-2-изопропилфенил]пиразол-3-карбоновая кислота
252 мг Полученного на предыдущей стадии соединения растворяют в 2,5 мл диоксана и 90 мкл водного раствора гидроксида калия (1 г/мл). После перемешивания в течение 24 часов среду подкисляют с помощью 1 мл концентрированной соляной кислоты. Экстрагируют этилацетатом, органическую фазу сушат над сульфатом магния, получая затем 236 мг целевого соединения.
Е) трет.-Бутиловый эфир[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-[3-[N'-метил-N'- (бензилоксикарбонил)амино] пропил]карбамоил]-2-изопропилфенил]пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
Полученную на предыдущей стадии кислоту растворяют в 2 мл ацетонитрила, добавляют 0,5 мл тетрахлорида углерода и 158 мг трифенилфосфина и перемешивают в течение двух часов. К полученному хлорангидриду кислоты добавляют 110 мг полученного на стадии В соединения и 100 мкл триэтиламина. Осаждается триэтиламингидрохлорид и перемешивают еще 15 минут. Добавляют воду, затем экстрагируют дихлорметаном; органическую фазу сушат над сульфатом магния и концентрируют в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью толуола с ацетоном в объемном соотношении 80:20, получая затем 323 мг целевого продукта.
Ж) Гидрохлорид трет. -бутилового эфира [5-(2,6- диметоксифенил)-1-[4-[N-метил-N-[3-[N'- метиламино]пропил]карбамоил]-2-изопропилфенил]-пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
В течение 24 часов в атмосфере водорода перемешивают смесь, содержащую 323 мг полученного на предыдущей стадии соединения, 2 мг палладия-на-угле и 40 мкл концентрированной соляной кислоты в 15 мл этанола. Катализатор отфильтровывают и фильтрат выпаривают в вакууме. Среду обрабатывают эфиром и перемешивают. Выпавший осадок белого цвета отфильтровывают, получая 190 мг целевого продукта.
ЯМР-спектр (дейтерированный метанол): 1,1 (д, 6Н); 1,5 (с, 9Н); 1,7-1,9 (м, 8Н); 2,2-2,3 (м, 6Н); 2,6 (с, 2Н); 2,7-2,9 (к+с, 4Н); 3 (с, 3Н); 3,1 (т, 2Н); 3,7 (с+мт, 8Н); 6,6 (д, 2Н); 6,8 (с, 1Н); 7,2-7,6 (м, 5Н).
З) Гидрохлорид 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбамоил] -2- изопропилфенил] пиразол-3-илкарбониламино]адамантан-2-карбоновой кислоты
190 мг Полученного на предыдущей стадии соединения суспендируют в 50 мкл ацетонитрила, добавляют 0,5 мл раствора метилиодида в толуоле (89 мкл метилиодида в 100 мл толуола) и 7,6 мг карбоната серебра. Отфильтровывают нерастворимую часть, затем растворитель выпаривают в вакууме. Среду обрабатывают с помощью 2 мл муравьиной кислоты и 0,2 мл концентрированной соляной кислоты и перемешивают в течение ночи. После выпаривания в вакууме и порошкования остатка в эфире получают 90 мг целевого продукта.
ПРИМЕР 88
Оксазолон соединения примера 1':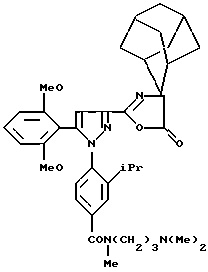
В течение 4,5 часов перемешивают раствор 0,23 г соединения примера 1' в 2 мл дихлорметана и 0,5 мл уксусного ангидрида. Выпаривают в вакууме, остаток порошкуют в пентане, отфильтровывают и высушивают, получая 230 мг целевого оксазолона. Т.пл.=129oС (разложение).
ИК-спектр (KBr): 1800 см-1.
Масс-спектр: М 667,9.
ЯМР-спектр: 1 (д, 6Н); 1,5-1,9 (м, 8Н); 2 (уш.с, 8Н); 2,1-2,5 (м, 6Н); 2,65 (кт, 1Н); 2,9 и 3 (2с, 3Н); 3,1 (мт, 2Н); 3,4 (мт, 2Н); 3,65 (с, 6Н); 6,6 (д, 2Н); 6,9 (с, 1Н); 7,1-7,4 (м, 4Н).
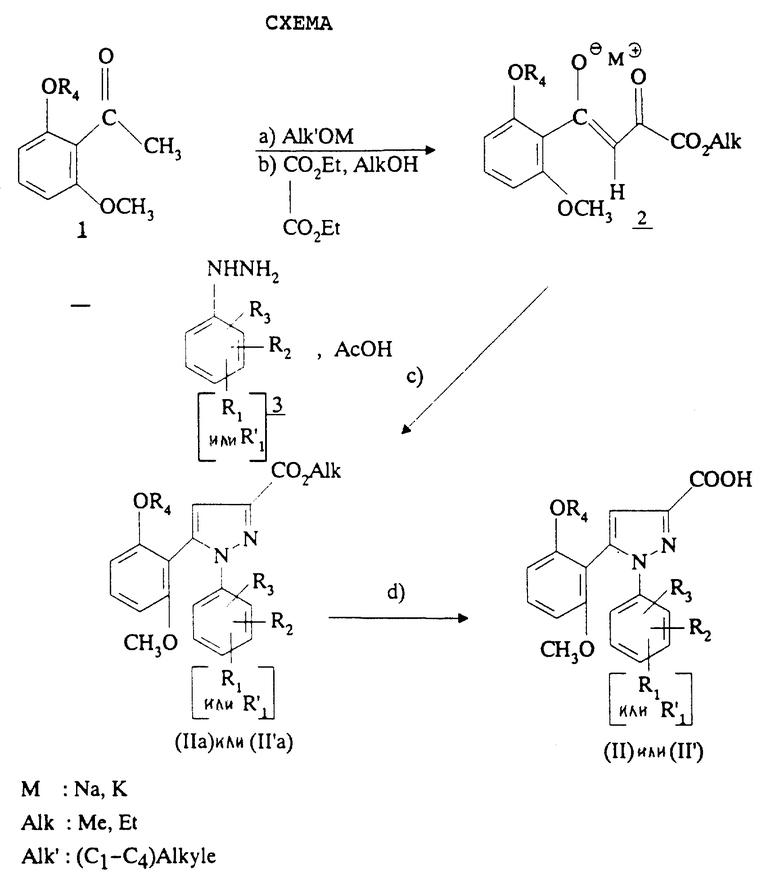
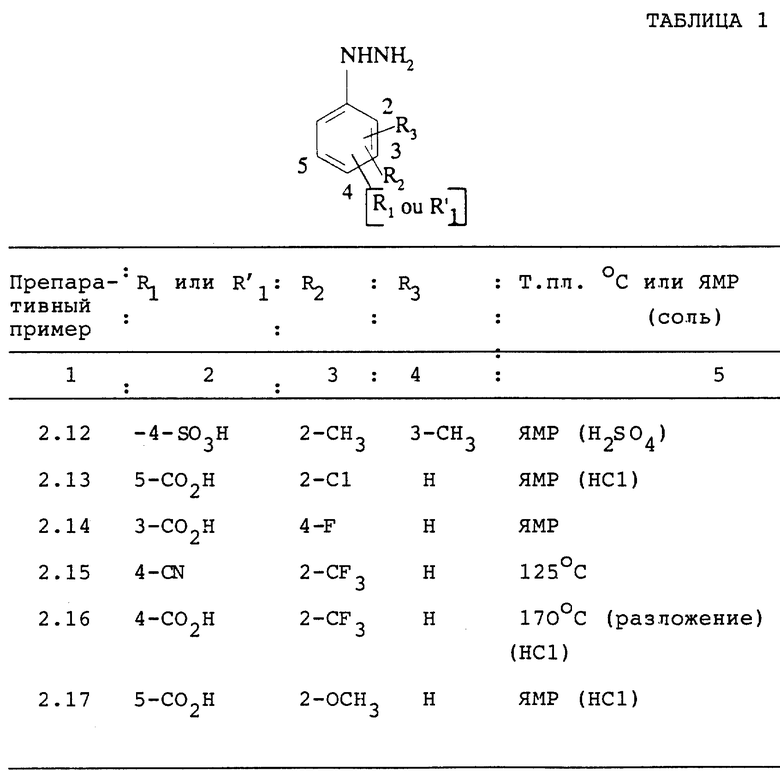

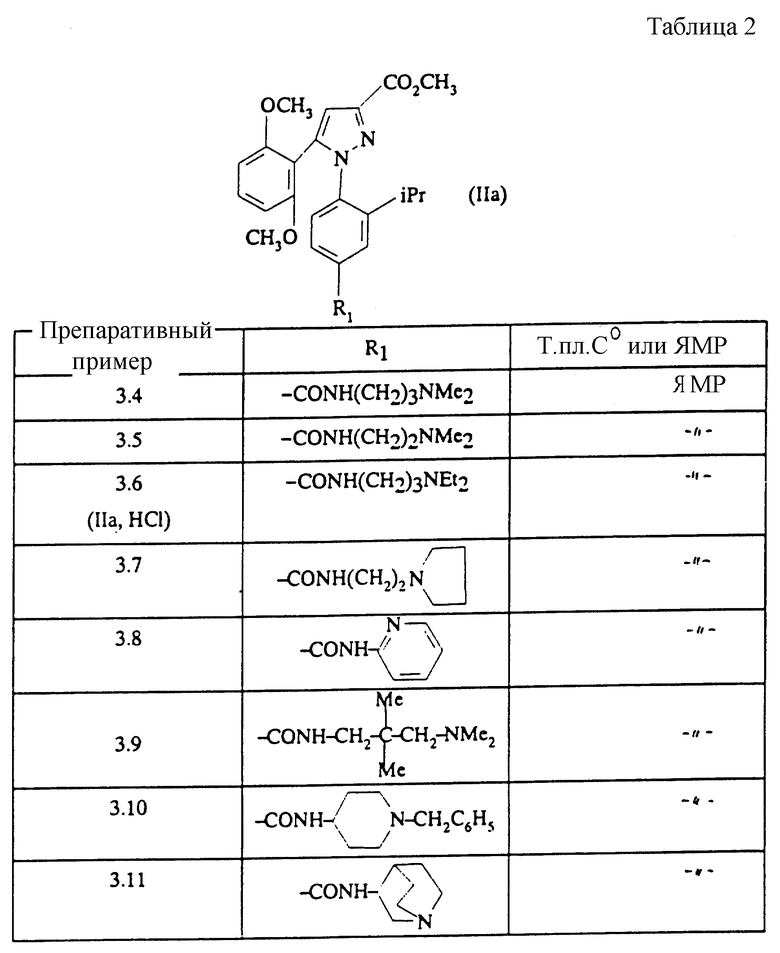


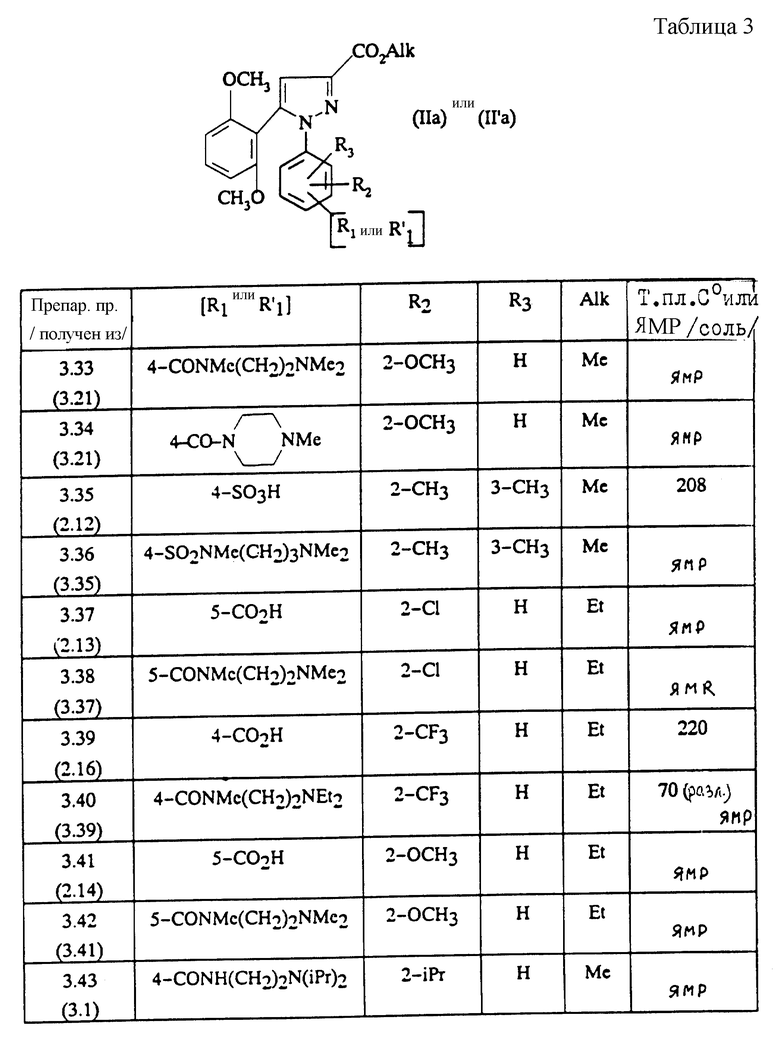
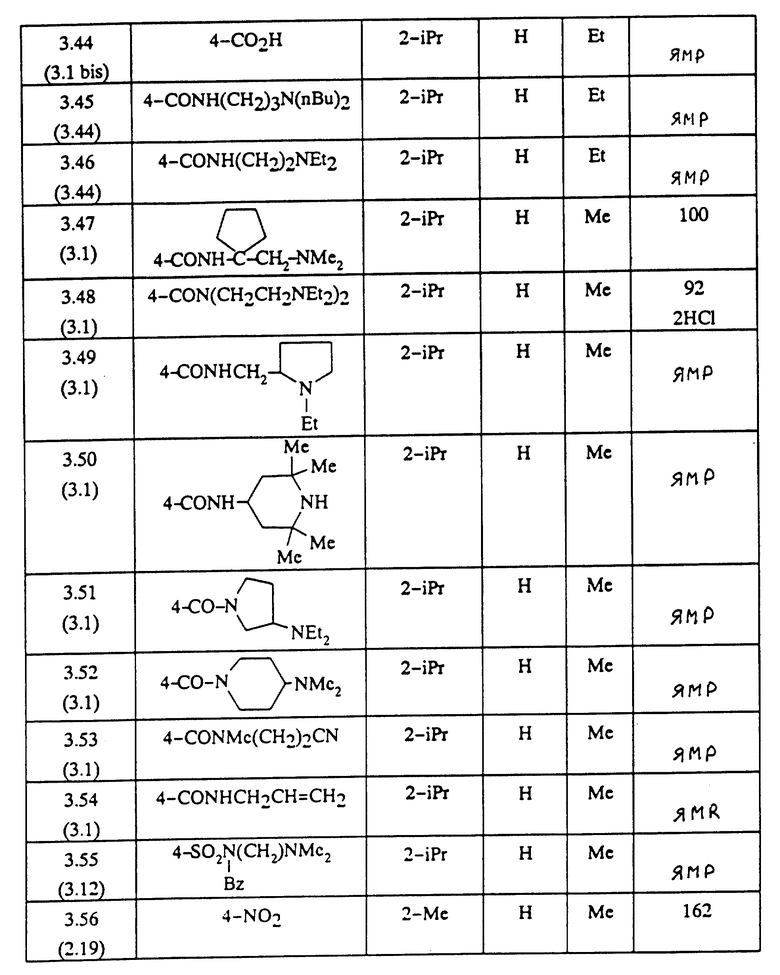



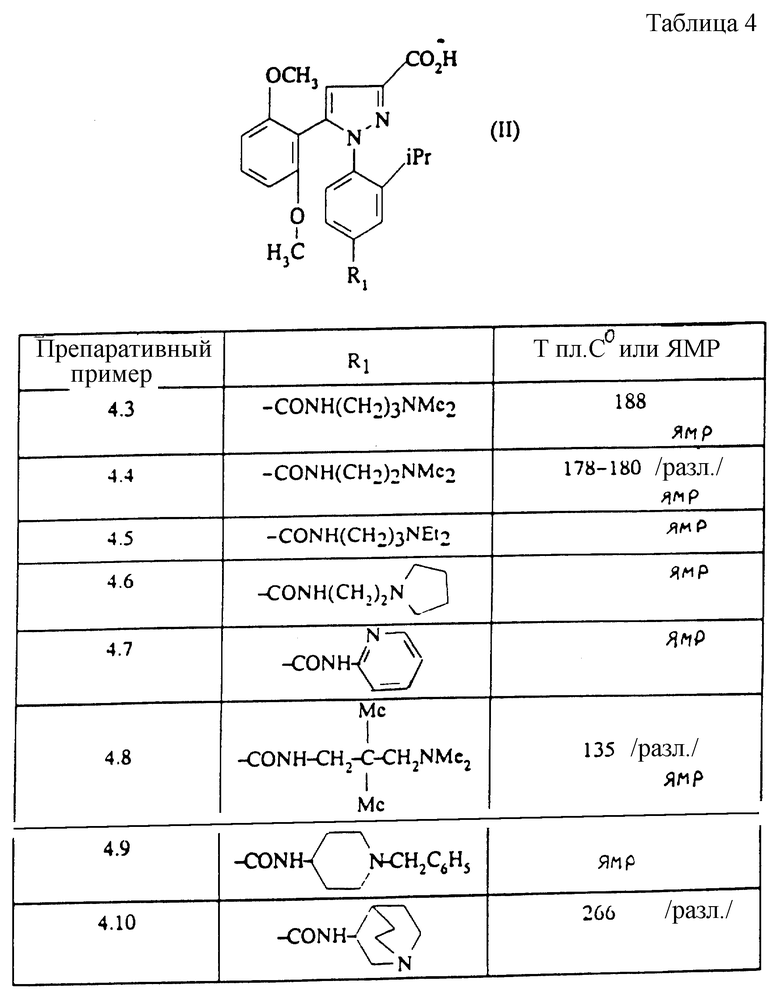


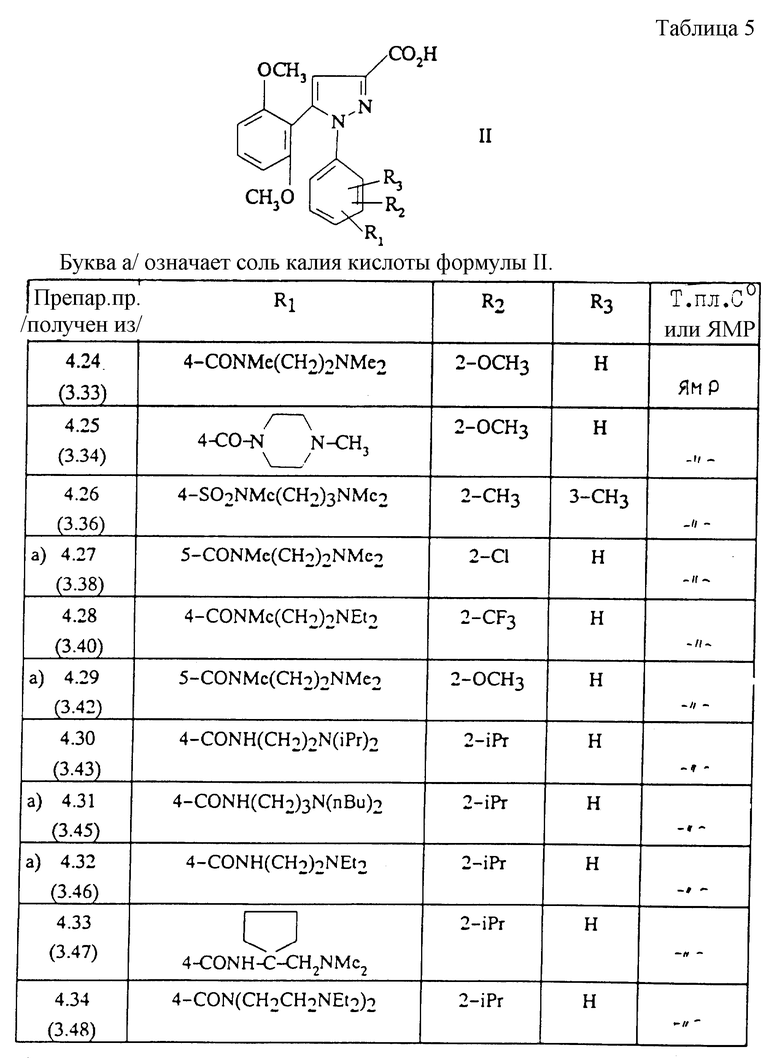
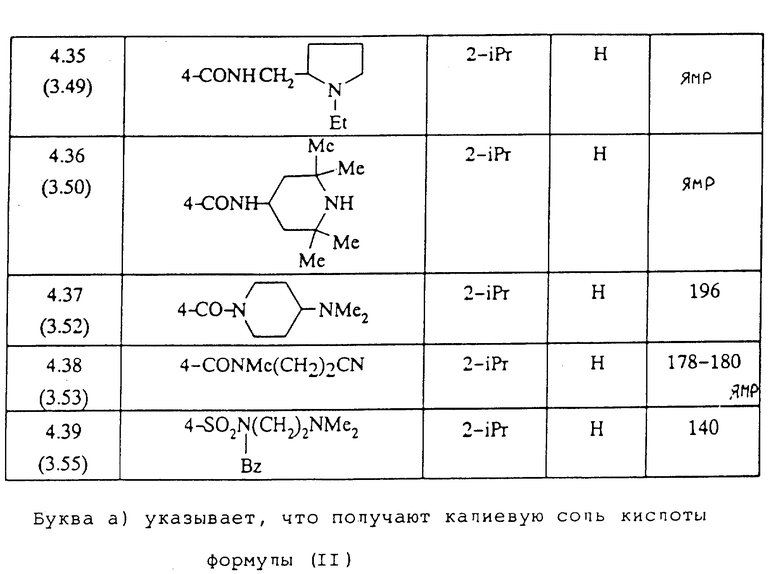


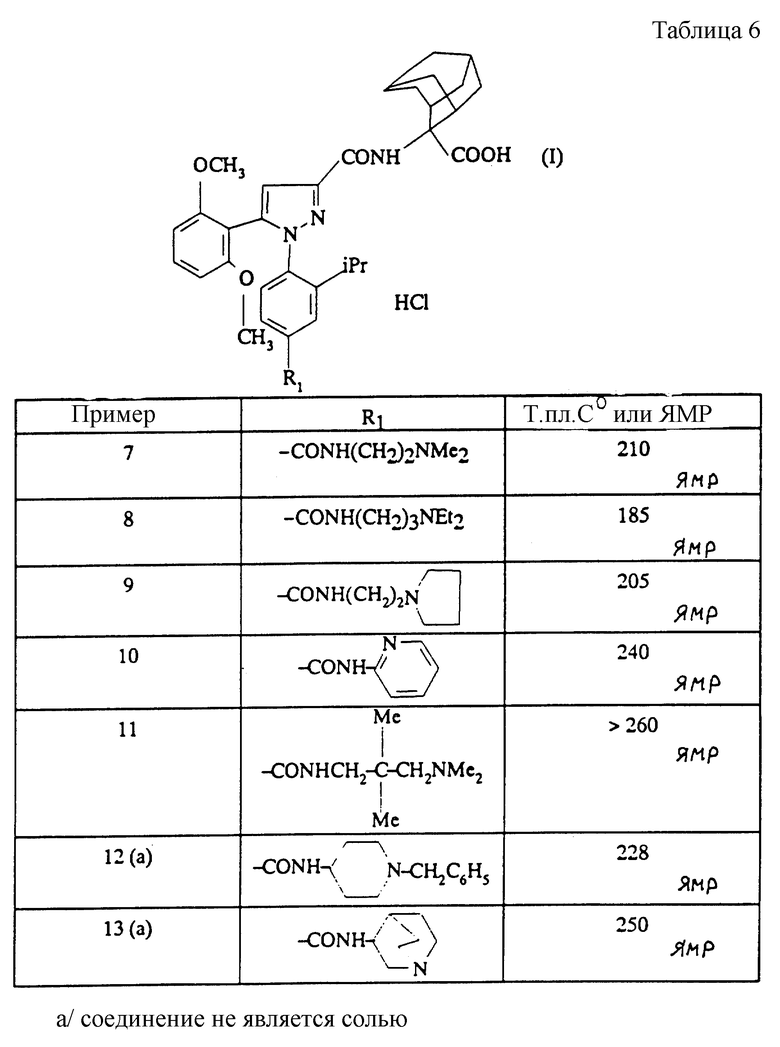


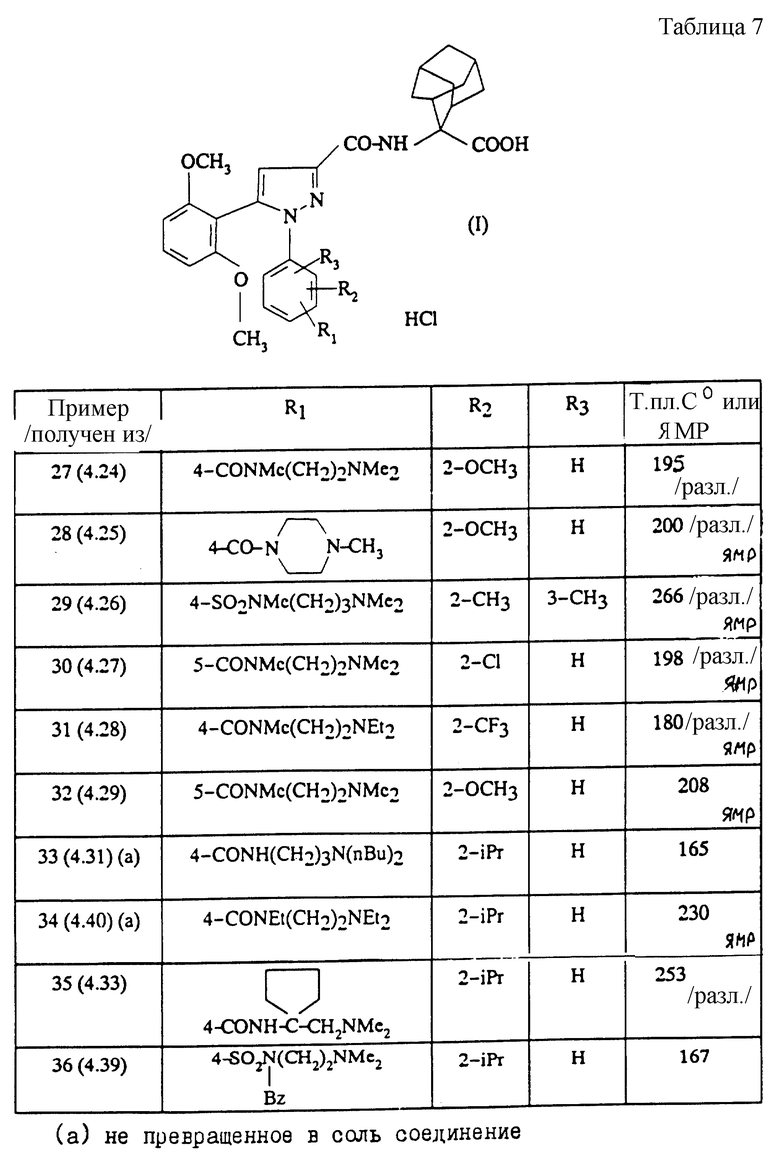

Изобретение относится к замещенным 1-фенилпиразол-3-карбоксамидам формулы (Iа), в которой R1x находится в положении 4 или 5 и обозначает группу -T-CONRaRb, в которой Т обозначает прямую связь или (C1-C7)-алкилен; NRaRb обозначает группу, выбранную из (а), (б), (в); R5 и R6 обозначают, независимо друг от друга, водород, (C1-C6)-алкил, (С3-С8)-алкенил или R5 и R6 вместе с атомом азота, с которым они связаны, обозначают гетероцикл, выбранный из пирролидина, пиперидина, морфолина, пиперазина, замещенного в положении 4 заместителем R9; R7 обозначает водород, (С1-С4)-алкил или бензил; R8 обозначает водород, (С1-С4)-алкил или R7 и R8 вместе с атомом углерода, с которым они связаны, образуют (С3-С5)-циклоалкан; R9 обозначает водород, (C1-C4)-алкил, бензил или группу -X-NR'5R'6, в которой R'5 и R'6 обозначают, независимо друг от друга, (C1-C6)-алкил; R10 обозначает водород, (С1-С4)-алкил; s= 0-3; t=0-3 при условии, что (s+t) в одной и той же группе больше или равно 1; двухвалентные радикалы А и Е вместе с атомом углерода и атомом азота, с которыми они связаны, образуют насыщенный гетероцикл, имеющий от 4 до 7 звеньев, который, кроме того, может быть замещен одним или несколькими (С1-С4)-алкилами; R2x и R3x обозначают, независимо друг от друга, водород, (С1-С6)-алкил, (С3-C8)-циклоалкил, (С3-С8)-циклоалкилметил при условии, что R2x и R3x не обозначают одновременно водород или R2x и R3x вместе образуют тетраметиленовую группу; и их фармацевтически приемлемым солям. Кроме того, предложены промежуточные соединения 1-фенилпиразол-3-карбоновой кислоты формул (II) и (II'), в которых R1, R2, R3 имеют значения, соответствующие значениям R1x, R2x и R3x, указанными для формулы (Iа), способ получения соединений формулы (Iа), исходя из указанных промежуточных соединений, и промежуточные производные фенилгидразина для получения соединений формул (II) и (II'). Соединения формулы (Iа) обладают сродством к человеческим рецепторам нейротензина и являются основой фармацевтической композиции. 6 с. и 2 з.п.ф-лы, 7 табл.

-NR9(CH2)sCR7R8(CH2)tNR5R6 (a)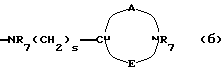
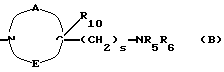
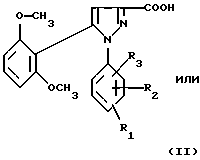
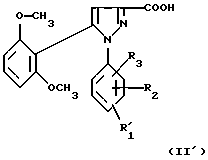
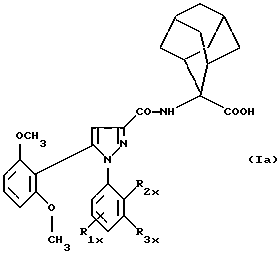
в которой R1x находится в положении 4 или 5 и обозначает группу -T-CONRaRb, в которой Т обозначает прямую связь или (C1-C7)-алкилен;
NRaRb обозначает группу, выбранную из
-NR9(CH2)sCR7R8(CH2)tNR5R6

где R5 и R6 обозначают, независимо друг от друга, водород, (С1-С6)-алкил, (С3-С8)-алкенил; или R5 и R6 вместе с атомом азота, с которым они связаны, обозначают гетероцикл выбранный из пирролидина, пиперидина, морфолина, пиперазина, замещенного в положении 4 заместителем R9;
R7 обозначает водород, (C1-C4)-алкил или бензил;
R8 обозначает водород, (C1-C4)-алкил, или R7 и R8 вместе с атомом углерода, с которым они связаны, образуют (С3-С5)-циклоалкан;
R9 обозначает водород, (C1-C4)-алкил, бензил или группу -X-NR'5R'6;
R'5 и R'6 обозначают, независимо друг от друга, (C1-С6)-алкил;
R10 обозначает водород, (C1-C4)-алкил;
s= 0-3;
t= 0-3, при условии, что (s+t) в одной и той же группе больше или равно 1;
двухвалентные радикалы А и Е вместе с атомом углерода и атомом азота, с которыми они связаны, образуют насыщенный гетероцикл, имеющий от 4 до 7 звеньев, который, кроме того, может быть замещен одним или несколькими (C1-C4)-алкилами;
R2x и R3х обозначают, независимо друг от друга, водород, (C1-С6)-алкил, (С3-С8)-циклоалкил, (С3-С8)-циклоалкилметил, при условии, что R2x и R3х не обозначают одновременно водород, или R2x и R3х вместе образуют тетраметиленовую группу;
и их фармацевтически приемлемые соли.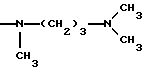
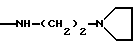
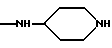
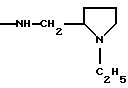
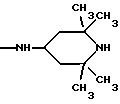
3. Соединение по п. 1, отличающееся тем, что оно представляет собой 2-[5-(2,6-диметоксифенил)-1-[4-[N-метил-N-(3-диметиламинопропил)карбомоил] -2-изопропилфенил] пиразол-3-илкарбониламино] адамантан-2-карбоновую кислоту, ее внутреннюю соль, и ее фармацевтически приемлемые соли.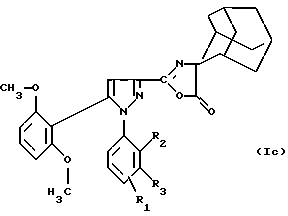
в которой R1 находится в положении 4 или 5 и обозначает группу -T-CONRaRb, в которой Т обозначает прямую связь или (C1-С7)алкилен;
NRaRb обозначает группу -NR9(CH2)sCR7R8(CH)tNR5R6;
R2 обозначает (C1-C6)алкил;
R3 обозначает водород;
R5 и R6 обозначают (C1-C6)алкил;
R7 и R8 обозначают водород;
s = 0-3;
t = 0-3, при условии, что сумма (s+t) больше или равна 1.
1) функциональное производное 1-фенилпиразол-3-карбоновой кислоты формулы (II) или (II')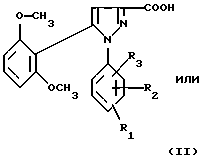

которой R1, R2, R3 имеют значения, соответствующие значениям R1x, R2x и R3х, указанным для соединения формулы (Iа) в п. 1,
R'1 означает предшественник R1, выбираемый среди карбоксила, (C1-C4)-алкоксикарбонила, бензилоксикарбонила,
обрабатывают аминокислотой, возможно защищенной с помощью обычных в пептидном синтезе защитных групп, формулы (III)
H-HN-AA(OH) (III)
в которой -NH-AA(OH) обозначает группу формулы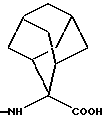
2) в случае необходимости, таким образом полученное функциональное производное кислоты формулы (I')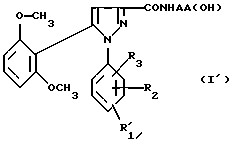
подвергают затем соответствующей обработке для превращения заместителя R'1, являющегося предшественником R1, в заместитель R1;
3) возможно, в полученном на стадии 1) или на стадии 2) соединении удаляют защитную группу с целью получения соответствующей свободной кислоты формулы (I);
4) и, при необходимости, получают соль таким образом полученного соединения формулы (I).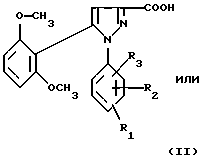
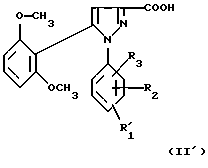
в которой R1, R2, R3 имеют значения, соответствующие значениям R1x, R2x и R3х, указанным для соединений формулы (1а) в п. 1;
R'1 означает предшественник радикала R1, выбираемый среди карбоксила, (C1-C4)-алкоксикарбонила, бензилоксикарбонила,
а также ее функциональные производные, выбранные из хлорангидрида и сложного (C1-C4)-алкилового эфира.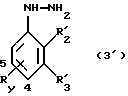
в которой R'2 и R'3 независимо друг от друга, означают водород, (C1-С6)-алкил, (С3-С8)-циклоалкил; или R'2 и R'3 вместе образуют тетраметиленовую группу;
Ry находится в положении 4 или в положении 5 и означает группу, выбираемую среди следующих групп: карбоксил, (C1-C4)-алкоксикарбонил, бензилоксикарбонил, при условии, что R'2 и R'3 одновременно не обозначают водород и что R'2 отличается от этила, когда R'3 обозначает водород, а Ry обозначает метоксикарбонильную группу в положении 4;
и его соли.
| Способ получения катионного флокулянта | 1973 |
|
SU447049A1 |
| Способ уплотнения волокнистого материала | 1976 |
|
SU576357A1 |
| RU 2066317 С, 10.09.1996. | |||

