Настоящее изобретение касается новых изохинолинов, способа их получения, их применения в качестве фармацевтических средств, а также фармацевтических составов, содержащих указанные изохинолины.
Конкретно, одним из аспектов настоящего изобретения является соединение, соответствующее формуле (I)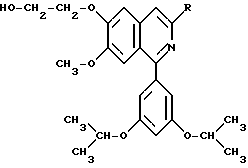
в которой R является этильной или н-пропильной группой, или его физиологически гидролизуемый и приемлемый эфир, а также соль, образующаяся в результате добавления кислоты к указанному соединению или эфиру.
В предпочтительном случае R в формуле (I) является этильной группой.
Под термином "физиологически гидролизуемый и приемлемый эфир" в настоящем описании подразумевают эфир, представляющий собой соответствующее формуле (I) соединение, этерифицированное по его гидроксильной группе и гидролизуемое при физиологических условиях с получением кислоты, физиологически приемлемой в необходимых для применения количествах. Таким образом, указанный термин следует понимать как обозначение обычных форм предшественника медикамента. Примерами такого рода эфиров являются, в частности, ацетаты и бензоаты соединений, соответствующих формуле (I).
Как уже было отмечено выше, соединения, соответствующие формуле (I), а также их эфиры существуют как в свободной форме, так и в форме соли, образующейся в результате добавления к ним кислоты. К числу солей, образующихся в результате добавления фармацевтически приемлемых кислот и предназначенных для фармацевтического использования в соответствии с настоящим изобретением, относятся, в частности, гидрохлориды, кислые фумараты, кислые малеаты и кислые оксалаты.
Предусмотренные настоящим изобретением соединения, эфиры и соли подпадают под область рассмотрения изобретения, описанного и определенного в патентах [UK 2213482, US 4980359], а также в соответствующих патентах и заявках различных стран. Соединения, эфиры и соли, предусмотренные настоящим изобретением, являются новыми, а по сравнению с соединениями, эфирами и солями, непосредственно описанными в указанных патентах, неожиданно проявляют более выгодные свойства, в особенности в плане их предполагаемого фармацевтического применения, в частности, описанного ниже.
Еще одним аспектом настоящего изобретения является способ получения соединения, соответствующего формуле (I), или его физиологически гидролизуемого и приемлемого эфира, а также соли, образующейся в результате добавления кислоты к указанному соединению или эфиру, при котором
а) для получения соединения, соответствующего формуле (I), проводят снятие защиты и/или дегидрогенирование соединения, описываемого формулой (II)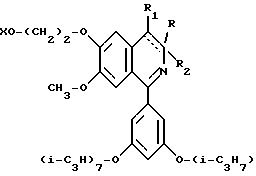
в которой X обозначает атом водорода,
а R1 и R2 представляют собой дополнительную связь, изображенную в виде пунктирной линии, или
X соответствует группе, защищающей гидроксильный радикал, а
каждая из групп R1 и R2 представляет собой атом водорода или вместе обозначают дополнительную связь, изображенную в виде пунктирной линии;
б) для получения физиологически гидролизуемого и приемлемого эфира соединения, соответствующего формуле (I), проводят этерификацию соответствующего формуле (I) соединения и выделяют продукт этапа а) или б) в свободной форме или в форме соли, образованной в результате добавления к нему кислоты.
В соответствии с этапом а) удаление группы, защищающей гидроксильный радикал, или дегидрогенирование может быть осуществлено с помощью хорошо известных способов. Удобный вариант этапа а) предусматривает как снятие защиты, так и дегидрирование, например, используя соединение, соответствующее формуле (II), в которой X обозначает бензильную защитную группу, а каждый из радикалов R1 и R2 представляет собой атом водорода, и обеспечивая отщепление бензильной группы и дегидрирование в ходе реакции, осуществляющейся в одном сосуде, например, при помощи обработки палладиево-угольным катализатором при повышенной температуре в инертной атмосфере, в инертном растворителе или разбавителе, например, как это описано ниже в Примере 1.
В соответствии с этапом б) этерификацию также можно проводить с помощью стандартных способов, например, путем приведения соединения, соответствующего формуле (I), во взаимодействие с галогенидом или ангидридом приемлемой кислоты в присутствии основания, в том числе амина или карбоната щелочного металла. Реакцию удобно проводить в инертном растворителе или разбавителе, например, при температуре от 0 до 120oC в условиях инертной атмосферы.
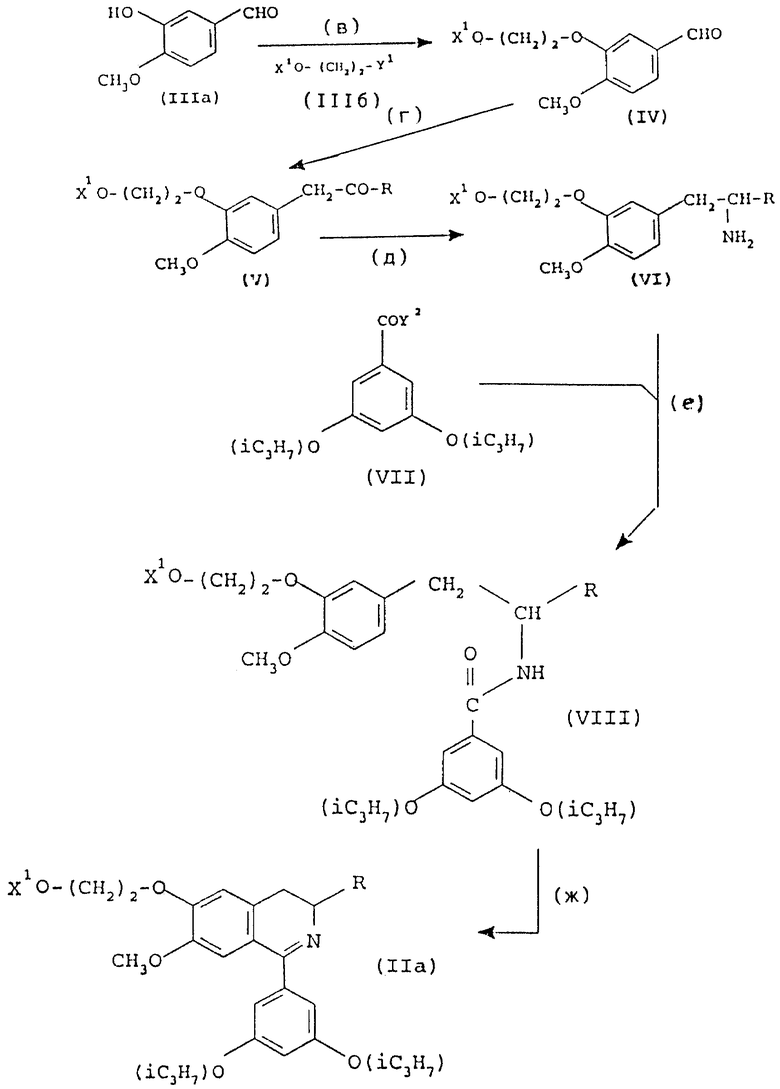
Исходные вещества для этапа а) описанного выше способа могут быть получены в соответствии со схемой реакции, приведенной в конце описания, в которой X1 представляет собой группу, защищающую гидроксильиый радикал, например, бензильную группу, а каждый из радикалов Y1 и Y2 обозначает отщепляемую группу. Удобными отщепляемыми радикалами Y1 могут служить атом иода или тозильная группа, причем тозильная группа является предпочтительной в случае более масштабного производства. Удобным радикалом Y2 служит атом хлора. Этапы в) - ж) могут быть осуществлены в соответствии со стандартными способами, например, описанными в прилагающихся Примерах. Снятие защиты с соединений, отвечающих формуле (IIа), приходит к образованию соединений, описываемых формулой (II), в которой X представляет собой атом водорода. Дегидрирование соединений, соответствующих формуле (IIа), приводит к образованию соединений, отвечающих формуле (II), в которой радикалы R1 и R2 вместе обозначают дополнительную связь.
Соединения, соответствующие формулам (IlIa), (IIIб) и (VII), являются коммерчески доступными, хорошо известными веществами, или их можно получить по аналогии с известными соединениями. Подобные вещества, соответствующие формуле (VII), можно получить на основе метилового эфира 2,5-дигидрокси бензойной кислоты посредством его алкилирования изопропил иодидом в присутствии основания, такого как K2CO3, с использованием метил-этилкетона в качестве растворителя, гидролиза полученного метилового эфира 3,5-ди-изопропокси бензойной кислоты, например, с помощью обработки NaOH в метаноле в качестве растворителя, и, наконец, превращения полученной 3,5-ди-изопропокси бензойной кислоты в, например, соответствующий галоидангидрид, в частности, в хлорангидрид кислоты с помощью взаимодействия с SOCl2.
Следующие Примеры иллюстрируют предусмотренный настоящим изобретением способ.
Пример 1.
Получение 1-(3,5- диизопропоксифенил)-3-этил- 6-(2-гидроксиэтокси)-7-метокси-изохинолина (Формула I: R = - C2H5)
1) Этап а) предусмотренного настоящим изобретением способа - снятие защиты и дегидрирование
13,5 г 6-бензилоксиэтокси-1-(3,5-диизопропоксифенил)-3- этил-7-метокси-3,4-дигидро-изохинолина (Формула IIа: X1 = бензильная группа, R = этильная группа), 1,3 г 10%-го палладиево-угольного катализатора и 500 мл декагидронафталина перемешивают в течение 5 часов при 200oC в атмосфере аргона. Полученную реакционную смесь охлаждают до комнатной температуры, фильтруют через Hyflo и промывают этилацетатом. Проводят отгонку декагидронафталина при 50oC в вакууме, а полученный продукт реакции очищают с помощью хроматографии на силикагеле (  0,04-0,06 мм) с получением искомого соединения:
0,04-0,06 мм) с получением искомого соединения:
т.пл. свободного основания = 116-118oC.
гидрохлорида 218-222oC
кислого фумарата = 82,5oC
кислого оксалата = 106-107oC
кислого малеата = 87-96oC
Исходные вещества для указанного способа можно получить следующим образом:
2) 3-(2-Бензилоксиэтокси)-4-метокси- бензальдегид (формула IV: X1 = бензильная группа)
20 г изованиллина (формула IlIa), 41,3 г 2-бензилоксиэтил иодида (формула IIIб) и 21,8 г карбоната калия в 200 мл этилметилкетона перемешивают в течение 12 часов в условиях дефлегмации. Полученную суспензию охлаждают до комнатной температуры, отфильтровывают образующийся осадок, промывают ацетоном и выпаривают. Остаток отбирают в этилацетат, трижды экстрагируют водой и один раз - насыщенным раствором соли. Органическую фазу высушивают, фильтруют и выпаривают с получением искомого соединения в виде масла.
3) 3-(2-Бензилоксиэтокси)-4- метоксибензилэтилкетон (формула V: X1 = бензильная группа, R = этильная группа)
24 г продукта, получаемого в результате этапа 2), 21,5 г этилового эфира бромбутировой кислоты и 30 мл т-бутил-метилового эфира добавляют по каплям в течение 50 мин при 5oC к заранее приготовленной суспензии метилата натрия в 25 мл т-бутил-метилового эфира. Реакционную смесь перемешивают в течение 45 мин при температуре приблизительно 5oC, после чего перемешивают в течение еще 12 ч при комнатной температуре. Добавляя ледяную уксусную кислоту, доводят pH до 5, а полученную суспензию разбавляют водой и трижды экстрагируют т-бутил-метиловым эфиром. Органическую фазу экстрагируют NaHCO3, затем насыщенным раствором соли, после чего выпаривают до объема, приблизительно равного 100 мл. По каплям добавляют 13,5 мл 50%-ного водного NaOH и перемешивают полученную смесь в течение 2 ч при 40oC, затем разбавляют 50 мл воды и перемешивают в течение еще 30 мин при комнатной температуре. Отделяют органическую фазу, pH водной фазы доводят до 1 с помощью 15 мл концентрированной HCl при температуре не выше 40oC. Полученную реакционную смесь перемешивают при 40oC в течение еще 1,5 ч, охлаждают до комнатной температуры и экстрагируют толуолом. Органическую фазу промывают NaHCO3 и насыщенным раствором соли, высушивают над Na2SO4, фильтруют и выпаривают с получением искомого соединения в виде масла.
4) 1-[3-(2-Бензилоксиэтокси)-4-метоксифенил] -2-аминобутан (формула VI: X1 = бензильная группа, R = этильная группа)
20,6 г продукта, получаемого в результате этапа в), 48,4 г ацетата аммония и 3,9 г цианоборогидрида натрия в 235 мл CH3OH в присутствии 12,1 г молекулярного сита 0,4 мм  перемешивают в течение ночи при комнатной температуре в инертной атмосфере. Полученную реакционную смесь фильтруют через Hyflo и промывают CH3OH. Концентрируют фильтрат, остаток отбирают в этиловый эфир и экстрагируют 15%-ным NaOH, водой и водным раствором соли. Органическую фазу высушивают над Na2SO4, фильтруют и выпаривают с получением искомого соединения в виде масла.
перемешивают в течение ночи при комнатной температуре в инертной атмосфере. Полученную реакционную смесь фильтруют через Hyflo и промывают CH3OH. Концентрируют фильтрат, остаток отбирают в этиловый эфир и экстрагируют 15%-ным NaOH, водой и водным раствором соли. Органическую фазу высушивают над Na2SO4, фильтруют и выпаривают с получением искомого соединения в виде масла.
5) N-(3, 5- Диизопропоксибензоил)-1-[3-(2-бензилоксиэтокси)-4-метоксифенил] - 2-аминобутан (Формула VIII: X1 = бензильная группа, R = этильная группа)
19,9 г 3,5-диизопропокси бензоил хлорида в 100 мл CH2Cl2 добавляют по каплям в течение 1,5 ч к 17,0 г продукта, получаемого в результате этапа 4), 15,6 г триэтиламина и 630 мг N,N-диметиламинопиридина в 150 мл CH2Cl2 и перемешивают реакционную смесь в течение 12 ч при температуре в пределах от 0oC до комнатной. Концентрируют полученную смесь, остаток отбирают в этилацетат и экстрагируют 1N соляной HCl, 10%-ным раствором NaHCO3 и насыщенным раствором соли. Органическую фазу высушивают над Na2SO4, фильтруют и концентрируют. Начинающий кристаллизоваться остаток разбавляют этиловым эфиром и фильтруют с получением искомого соединения. Его используют в следующей реакции без дополнительной очистки.
6) 6-Бензилоксиэтокси-1-(3, 5-диизопропоксифенил)-3-этил-7-метокси- 3,4-дигидроизохинолин
15,3 г продукта, получаемого в результате этапа 5), и 12,4 г POCl3 в 250 мл ацетонитрила перемешивают в течение 5 ч в условиях дефлегмации. Концентрируют реакционную смесь, остаток отбирают в этилацетат и экстрагируют раствором Na2SO4 и насыщенным раствором соли. Органическую фазу высушивают над Na2SO4, фильтруют и выпаривают, а остаток очищают с помощью хроматографии на силикагеле (  0,04-0,06 мм) с получением искомого соединения в виде масла.
0,04-0,06 мм) с получением искомого соединения в виде масла.
Пример 2
Получение 1-(3, 5-диизопропоксифенил)-6-(2- гидроксиэтокси)-7-метокси-3-н-пропил-изохинолина (Формула I: R = н-пропил)
Искомое соединение получают аналогично Примеру 1: свободное основание получают в виде пены:
т.пл. кислого оксалата = 154-156oC.
Соединения, соответствующие формуле (I), их физиологически гидролизуемые и приемлемые эфиры, а также фармацевтически приемлемые соли, образующиеся в результате добавления кислоты к указанным соединениям или эфирам (в дальнейшем совокупно называемые предусмотренными настоящим изобретением агентами) проявляют фармакологическую активность и рекомендуются к использованию в качестве фармацевтических средств, в том числе в терапевтических целях при лечении заболеваний и симптомов, перечисляемых ниже.
Конкретно, предусмотренные настоящим изобретением агенты обладают активностью, ингибирующей изофермент циклическую нуклеотидфосфодиэстеразу (ФДЭ), оказывающей избирательный эффект по отношению к IV типу изофермента, а также проявляющей существенно и неожиданно большую специфичность по отношению к изоферменту IV типа, нежели известные соединения, например, описанные в вышеупомянутых патентах [VK, 2213482, US, 4980359].
Предусмотренные настоящим изобретением агенты обладают противовоспалительными, снижающими гиперреактивность дыхательных путей, а также бронхорасширяющими свойствами. Кроме того, они проявляют иммуносупрессивную, подавляющую секрецию TNF α и прочие фармакологические активности, что можно продемонстрировать стандартными тестовыми методами, в частности, следующими.
(Все описываемые ниже эксперименты адекватно проведены с использованием кислой щавелевокислой соли тестируемого соединения, например, соединения из Примера 1. В случае соединения из Примера 1 можно приготовить стабильный 30 мМ сток-раствор с 10%-ным Tween в 80%-ном C2H5OH. Для использования в фармакологических экспериментах его следует разбавлять по меньшей мере в соотношении 1:10000.)
ИНГИБИРОВАНИЕ ИЗОФЕРМЕНТА ФДЭ
Тест A: Тест на ингибирование изофермента ФДЭ человека
Все препараты изоферментов получены из человеческих тканей. Препараты изоферментов III и IV типа получены с помощью перечисленных ниже способов, используя преобладание изофермента III типа в тромбоцитах, а изофермента IV типа - в нейтрофилах.
Клетки и ткани гомогенизируют на ледяной бане в 10 мМ трис-HCI (pH 7,4), содержащем сахарозу (250 мМ), ЭДТА (1 мМ), дитиотрейтол (1 мМ), лейпептин и пепстатин (по 1 мкг/мл каждого) и фенил-метил-сульфонил фторид (ФМСФ, 0,17 мг/мл, добавляют непосредственно перед гомогенизацией). Нейтрофилы (тип IV) и тромбоциты (типы II и III) получают из крови человека и гомогенизируют под действием ультразвука (Branson probe, 4х15 сек). Легкое человека (типы I и V) получают от больных, претерпевающих хирургическое вмешательство, и гомогенизируют с помощью гомогенизатора Polytron (две обработки по 30 сек).
Препараты изоферментов: препараты ФДЭ III и IV (субстрат -1 мкМ цАМФ) состоят из низкоскоростных супернатантов соответственно тромбоцитов и нейтрофилов. Изоферменты типа 1 (субстрат - 1 мкМ цАМФ, 0,5 мМ Ca2+, 125 нМ кальмодулин), II (100 мкМ цАМФ) и V (1 мкМ цГМФ) разделяют с помощью анион-обменной хроматографии (Q-Сефароза) с использованием градиента NaCI в буфере для гомогенизации, не содержащем сахарозы и ФМСФ (0-0,1 М NaCI в 2,5 объемах колонки, 0,1 - 0,45 М в 24 объемах колонки). ФДЭ 1: фракции, в которых гидролиз 1 мкМ цАМФ можно стимулировать смесью Ca2+ кальмодулин (0,5 мМ и 125 нМ, соответственно); элюция при 0,17- 0,18 М NaCI. ФДЭ II: фракции, в которых существенная активность, гидролизующая цАМФ, наблюдается при 100 мкМ, но не 1 мкМ субстрата: элюция при 0,31-0,32 М NaCl. ФДЭ V: фракции, избирательно гидролизующие 1 мкМ цГМФ, а не 1 мкМ цАМФ; элюция при 0,20-0,24 М NaCl.
Активность ФДЭ определяют в присутствии или в отсутствие тестируемого вещества в различных концентрациях с помощью метода ионно-обменной колонки, описанного Томпсоном с соавторами [Nucleotide Res., 10, 69-92, 1979], с использованием в качестве субстрата 1 мкМ [3H]-циклического АМФ.
В этом тестовом методе предусмотренные настоящим изобретением агенты преимущественно ингибируют тизоферменты III, IV и V типов, оказывая сравнительно слабое влияние на изоферменты I и II типов. В пределах группы изоферментов III, IV и V типов предусмотренные настоящим изобретением агенты обладают заметно повышенной избирательностью ингибирования IV типа изоферментов ФДЭ по сравнению с другими известными ингибиторами изоферментов ФДЭ и могут быть охарактеризованы как специфичные по отношению к изоферментам IV типа. Так, в одном из проведенных тестов показано, что соединение из Примера 1 в форме кислой щавелевокислой соли проявляет по меньшей мере в 180 раз большую ингибирующую активность по отношению к изоферменту IV типа, нежели к другим исследованным препаратам изофермента.
ПРОТИВОВОСПАЛИТЕЛЬНАЯ АКТИВНОСТЬ
Тест Б: Подавление активации эозинофилов формил-метионин- лейцин-фенилаланином (ФМЛФ)
Очищенные эозинофилы человека (104/лунку в 0,2 мл HBSS) стимулируют с помощью ФМЛФ (1 мкМ) в присутствии люцигенина (25 мкМ). Подавление всплеска окисления (измеряемого по изменениям в хемолюминисценции) определяют на основании кривых дозового ответа с помощью логистического уравнения.
Предусмотренные настоящим изобретением агенты проявляют активность в вышеупомянутом тестовом методе в концентрациях порядка 0,001- 0,5 мкМ.
г) Тест В: Подавление секреции ТNF-α
900 мкл клеток ТНР-1 (0,5х106 клеток вместе с 100 Ед. γ -интерферона/0,9 мл) раскапывают в 24-луночные планшеты для культивирования, после чего добавляют 100 мкл тестируемого вещества. Через 3 часа инкубирования при 37oC в атмосфере 5% CO2 и 95% воздуха добавляют 10 мкл LPS в концентрации 5 мкг/мл и продолжают инкубирование в течение еще 40 часов. Кроме того, проводят необходимые контроли. Затем отбирают полученную среду и проводят осаждение с помощью центрифугирования при 1000 g в течение 10 мин. К лункам добавляют по 1,0 мл 0,01%-ного дигитонина для лизирования клеток, разрыхленных путем растирания резиновым скребком, и оставляют при 4oC на 10 минут. Непосредственно вслед за этим измеряют активность лактат дегидрогеназы, после чего хранят образцы при -20oC до тех пор, пока не станет возможным провести другие определения. Исследуемыми пробами являются: IL-1 β (среда), TNF- α (среда) и ДНК (лизаты). Пробы на IL-1 β и TNF- α осуществляют с использованием коммерчески доступных наборов для ELISA.
Для исследования ДНК используют метод Капусцинского с соавторами [Anal. Biochem. 83, 252-257, 1977]. Образцы клеточных лизатов в 0,01%-ном дигитонине объемом 300 мкл смешивают с 750 мкл трис-HCl буфера pH 7,0 (содержащего 13,2 мМ Na2SO4), 300 мкл воды и 150 мкл 2 мкг/мл ДАФИ (4',6-диамидино-2-фенилиндол•2HCl). После этого с помощью флюориметра Perkin Elmer 3000 измеряют флюоресценцию при 372 нм (возбуждение) и 454 нм (эмиссия). Образцы сравнивают со стандартной кривой ДНК тимуса теленка (0,5-10 мкг/мл), исследуемой в это же время.
Тестирование лактат дегидрогеназы проводят следующим образом: образцы объемом 50 мкл (среда или клеточные лизаты в 0,01%-ном дигитонине) вносят в 96-луночные планшеты для микротитрования, после чего добавляют 200 мкл 0,3 мМ НАДН / 1мМ пирувата натрия в натрийфосфатном (62 мМ) буфере pH 7,5. Осторожно покачивают планшет с помощью механического встряхивателя для микротитровальных планшетов и помещают в спектрофотометр Twinreader (Flow Laboratories). В течение 11 минут через каждую минуту автоматически измеряют значения молярной экстинкции при 340 нм и с помощью компьютерной программы автоматически высчитывают активность фермента. Так как ферментативная активность утрачивается при замораживании и оттаивании, тестирование проводят на свежеполученных образцах.
Добавляют тестируемые соединения с γ -интерферона в различной концентрации и оставляют их с клетками в течение эксперимента.
В описанном выше тестовом методе предусмотренные настоящим изобретением соединения оказывают мощное подавление секреции TNF- α , находясь в концентрациях порядка 0,001 - 0,5 мкМ. Подавление секреции интерлейкина-1 β наблюдается при существенно более высоких концентрациях.
Тест Г: Подавление образования лейкотриена (SRS-A)
За 24 ч до эксперимента морских свинок пассивно иммунизируют путем внутривенного введения 1 мл гомологичной анти-овальбуминовой антисыворотки. Перед применением антигена тестовым животным предварительно внутривенно вводят 0,32 мг/кг пропанола (ингибирование эндогенных катехоламинов), 3,2 мг/кг мепирамина (антагонист рецептора H1) и 3,2 мг/кг индометацина (ингибирование цикло-оксигеназы). Воздействие аллергеном осуществляют путем внутривенного введения 32 мкг/кг овальбумина, наблюдаемый в результате этого ответ сужающей мышцы на устойчивость дыхательных путей используют в качестве функционального показателя активности лейкотриена. Отдельные тестируемые группы получают 10 мг/кг FLP 55712 (антагонист рецептора LTD4) внутривенно за 1 минуту до воздействия или различные концентрации тестируемого вещества путем внутривенного вливания, начинаемого за 16 минут до воздействия. Группы, получившие FLP 55712, проявляют отсутствие сужения бронхов, что подтверждает участие лейкотриена в качестве посредника в рассматриваемом ответе.
В вышеописанном тестовом методе предусмотренные настоящим изобретением агенты подавляют продукцию лейкотриена, что видно из ослабления ответа сужающей мышцы при внутривенном вливании доз в пределах от 5 до 100 мкг/кг/мин.
Тест Д: Летальный эффект, индуцированный у морских свинок бактериальным эндотоксином (LРS).
Морских свинок анестезируют посредством внутрибрюшинной инъекции фенобарбитона натрия (100 мг/кг) с добавкой пентобарбитона натрия (30 мг/кг) и парализуют с помощью внутримышечной инъекции галламина (10 мг/кг). Животным осуществляют искусственное дыхание через трахейную канюлю смесью воздуха и кислорода (объемное соотношение 40:60; 8 мл/кг, 1 Гц). За протеканием искусственного дыхания ведут наблюдение с помощью пневмотахографа, соединенного с передатчиком дифференциального давления. Соответствующие изменения давления внутри грудной клетки измеряют через грудную канюлю с использованием передатчика дифференциального давления таким образом, что разницу давления между трахеей и грудной клеткой можно измерять и выводить на дисплей. Данные о кровяном давлении и частоте сердечных сокращений снимают с сонной артерии с помощью передатчика давления, а также вводят канюлю в правую яремную вену для обеспечения внутривенного вливания тестируемого вещества как за минуты до, так и сопутствующего вливанию LPS, производимого с постоянной скоростью (3,0 мл/ч с получением конечной концентрации 10 мг/кг/ч) через вливающий насос. В левую яремную вену вставляют канюлю для введения (±) пропанолола (1 мг/кг), инъецируемого во внутривенный просвет. На основании измерений потока воздуха и легочного давления после каждого дыхательного цикла рассчитывают значения RL и Cdyn с использованием цифровой электронной системы слежения за дыханием, представляющей данные о кровяном давлении, давлении внутри грудной клетки и воздушном потоке, а также вычисляющей для настоящего времени значения RL и Cdyn, предназначенные для выведения на визуальный дисплей. Полученные в экспериментах данные последовательно сохраняют, а по окончании эксперимента вычерчивают экспериментальные кривые или обработанные данные.
Вливание [(±) пропанолола] подтверждает четкую чувствительность к LPS. У анестезированных животных, предварительно обработанных (±) пропанололом, вливание LPS в рассматриваемой модели вызывает прогрессивное затруднение дыхания. Смерть в результате эндотоксинового шока обычно наступает в течение приблизительно 1 ч после прекращения вливания LPS.
В вышеописанной тестовой модели предусмотренные настоящим изобретением агенты при внутривенном введении в дозах порядка 1 - 500 мг/кг/мин предохраняют животных от затруднения дыхания, вызванного эндотоксином (LPS) в течение эксперимента, а также от вызванного LPS летального эффекта,
Тест Е: Контактный дерматит, вызванный у мышей раздражителем в виде арахидомовой кислоты
Самкам мышей линии NMRI (вес тела приблизительно 30 г) производят локальную обработку наружной и внутренней частей правого уха смесью диметилсульфоксида, ацетона и этанола (в соотношении 2:4:4), содержащей различные концентрации тестируемого соединения. По прошествии 30 минут правое ухо локально обрабатывают с обеих сторон 1 мг арахидоновой кислоты в 10 мкл ацетона. Через 30 минут животных умервщляют, удаляют уши по линии хряща и проводят их взвешивание. Высчитывают разницу в весе между левым и правым ушами и определяют процент подавления раздражения по сравнению с контрольной группой, обработанной только арахидоновой кислотой.
Предусмотренные настоящим изобретением агенты подавляют развитие контактного дерматита в вышеописанной тестовой модели, находясь в концентрации порядка 3,0-300 мМ.
БРОНХОРАСШИРЯЮЩАЯ АКТИВНОСТЬ
Тест Ж: Расслабление бронха человека
Образцы человеческого легкого, удаленного в ходе хирургического лечения рака, получают в течение трех дней с момента удаления. Отделяют мелкие бронхи (внутренний диаметр приблизительно 2-5 мм), разрезают на сегменты и помещают в ампулы для хранения жидкого азота емкостью 2 мл, заполненные фетальной телячьей сывороткой (ФТС), содержащей 1,8 М диметилсульфоксид (ДМСО) и 0,1 М сахарозу в качестве криопротекторов. Указанные ампулы помещают в полистирольную коробку (размером 11х11х22 см) и медленно замораживают со средней скоростью охлаждения приблизительно 0,6oC/ мин в морозильнике, поддерживающем температуру -70oC. Через 3 - 15 ч ампулы переносят в жидкий азот (-196oC), где их хранят до момента использования. Перед использованием ткани выдерживают в течение 30-60 мин при температуре -70oC, после чего проводят их оттаивание, помещая ампулы на 2,5 мин в водяную баню с температурой 37oC. В дальнейшем бронхиальные сегменты ополаскивают, помещая их в чашку, содержащую раствор Кребса- Генслейта (состав в мМ: NaCl - 118; KCl - 4,7; MgSO4 - 1,2; CaCl2 -1,2; KH2PO4 - 1,2; NaHCO3 - 25, глюкоза - 11; ЭДТА -0,03) при 37oC, нарезают на кольца и суспендируют в ванночках для органов емкостью 10 мл с целью регистрации изометрического растяжения в условиях предварительной нагрузки приблизительно в 1 г. Кривые "доза-эффект" получают путем кумулятивного добавления, каждую концентрацию добавляют по достижении максимального эффекта, вызываемого предыдущей концентрацией. По окончании снятия кривой "доза-эффект" добавляют папаверин (300 мкМ) для индукции полного расслабления бронхиальных колец. Получаемый при этом результат рассматривают в качестве 100%-ного расслабления.
В вышеописанной тестовой модели предусмотренные настоящим изобретением агенты вызывают зависимое от концентрации расслабление препаратов бронхиальных колец человека, находясь в концентрации от 0,001 до 1,0 мкМ.
Тест 3: Подавление сужения бронхов, вызванного лейкотриеном
Морских свинок (самцов линии Dunkin-Hartley, весом 400-600 г) анестезируют путем внутрибрюшинного введения фенобарбитала (100 мг/кг) и пентобарбитала (30 мг/кг) и парализуют с помощью внутримышечной инъекции галламина (100 мг/кг). Через трахейную канюлю животным осуществляют искусственное дыхание смесью воздуха и кислорода (объемное соотношение 45:55, 8 мл/кг, 1 Гц). Данные о кровяном давлении и частоте сердечных сокращений снимают с сонной артерии. Наблюдение за искусственным дыханием проводят с помощью передатчика потока Флейша, подсоединенного к дыхательному кругу. При измерении потока соответствующие изменения давления в грудной клетке регистрируют непосредственно через внутригрудной троакар, что позволяет выявлять связанное с трахеей дифференциальное давление. На основании этой информации, исходя из данных о потоке и дифференциальном давлении, с помощью цифрового анализатора дыхания рассчитывают показатели устойчивости (R1) и лабильности (Cdyn) для каждого дыхательного цикла.
За 24 часа до тестирования подопытных животных пассивно иммунизируют путем внутривенного введения гомологичной анти- овальбуминовой антисыворотки (1 мл). Перед применением аллергена животным предварительно внутривенно вводят пропанолол (0,32 мг/кг) для ингибирования эндогенных катехоламинов, мепирамин (3,2 мг/кг) для блокирования рецепторов H1 гистамина, а также индометацин (3,2 мг/кг) для ингибирования циклооксигеназы. Воздействие аллергеном осуществляют путем введения овальбумина (ОА), а наблюдаемый в результате этого ответ сужающей бронх мышцы используют в качестве функционального показателя активности лейкотриена.
Проводят два эксперимента:
1) В первом эксперименте животным внутривенно вводят ОА (32 мг/ кг) и исследуют эффекты, производимые внутривенной инъекцией лейкотриена FLP 55712 (антагонист рецептора D4, 10 мг/кг за 1 минуту до введения ОА) и внутривенным вливанием тестируемого соединения (1, 10 и 100 мг/кг/мин с началом за 15 минут до введения ОА).
2) Во втором эксперименте животных обрабатывают ОА (1,0 или 1,8 мг/мл, ингалируемые в течение 60 дыханий) и измеряют эффект, производимый тестируемым соединением, вводимым в разных концентрациях в легкое путем покапельного вливания через трахею.
В первом эксперименте эффект сужения бронха под действием ОА снимается последующим применением FLP 55712, что хорошо согласуется с посреднической ролью лейкотриена в контроле рассматриваемого ответа. Предусмотренные настоящим изобретением агенты вызывают зависящее от дозы подавление сужающего бронх ответа.
Во втором эксперименте предусмотренные настоящим изобретением агенты проявляют ингибирующую активность в дозах порядка 0,001-10 мг/кг/мин, вводимых путем покапельного закапывания через трахею.
Тест И: Подавление вызванного бомбезином сужения бронхов
Животных (морских свинок) подготавливают так же, как это описано выше для Теста 3. Бомбезин вводят путем равномерного внутривенного вливания с интенсивностью 100 мг/кг/мин, вызывая тем самым устойчивый спазм бронхов. Тестируемое вещество вводят внутривенно с физиологическим раствором или внутрикожно с физиологическим раствором, содержащим этанол (1 объемный процент по отношению к весу). Эффект расширения бронхов измеряют через 1 и 3 мин после внутривенного введения или через 16 и 64 мин после внутрикожного введения и выражают в виде процента подавления исходного ответа с использованием показателя RL в качестве характеристики функционирования легкого.
В вышеописанной тестовой модели предусмотренные настоящим изобретением агенты проявляют заметную бронхорасширяющую активность при внутривенном введении в дозах порядка 0,001-0,1 мг/кг или при внутрикожном введении в дозах порядка 0,1-5,0 мг/кг.
Тест К: Подавление вызванного метахолином (MeX) сужения бронхов у макаки-резус
Самцов макаки-резус (вес тела 10,3-13,2 кг) исходно анастезируют с помощью внутримышечной инъекции кетамина (20 мг/кг), а в ходе эксперимента поддерживают анестезию путем внутривенного введения тиопентала (8 мг/кг/ч) через катетер, вживленный в левую подкожную вену. Животным позволяют произвольно дышать и укладывают их в лежачее положение на левый бок. Анестезируют гортань, надгортанник и глотку местным введением ксилокаина, что позволяет ввести и разместить оснащенную манжетой педиатрическую эндотрахейную трубку диаметром 4,5 мм.
MeX применяют в виде аэрозоля (физиологический раствор в качестве носителя: аэрозоль создают пульверизатором, работающим при воздушном потоке 6 л/мин; средний размер частиц 3,5 мкм) с 2-минутной экспозицией при вдохах и выдохах. Во всех тестах используют раствор, содержащий 0,6 мг/мл Мех или 2,5 мг/мл MeX в случае слабого ответа конкретных особей. Тестирования обусловленного MeX сужения бронхов производят через получасовые промежутки времени, за 15 минут до воздействия MeX вводят исследуемое соединение (в составе 1 мл суспензии на основе лактозного носителя [1 мг/мл], применяемой в условиях бронхоскопического контроля, на расстоянии 1 см выше киля).
Исследуемые соединения применяют кумулятивным способом. Бронхорасширяющую активность оценивают как процент подавления ответа, заключающегося в сужении бронхов в результате действия MeX, по отношению к резистентности.
Предусмотренные настоящим изобретением агенты активны в вышеописанной тестовой модели при использовании в дозах порядка 10-500 нг/кг.
ПОДАВЛЕНИЕ ГИПЕРРЕАКТИВНОСТИ ДЫХАТЕЛЬНЫХ ПУТЕЙ
Тест Л: Гиперреактивность, вызываемая у морских свинок иммунным комплексом
Морских свинок анестезируют и подготавливают к регистрации легочной функции, как в случае описанного выше Теста Е. Аллергическую реакцию вызывают с помощью трехкратного внутривенного введения предварительно образованных иммунных комплексов (полученных путем добавления 30 мкг γ -глобулина быка в составе 0,05 мл физиологического раствора к 0,05 мл антисыворотки, вырабатываемой у морской свинки в ответ на γ-глобулин быка) с интервалами 10 минут. Последующее внутривенное введение гистамина (1-3,7 мкг/кг с интервалами в 10 минут) позволяет определить чувствительность дыхательных путей до и после введения иммунного комплекса. Гиперреактивность дыхательных путей выражается в виде пoпарной разницы между максимальными значениями RL в ответ на гистамин до и после введения иммунного комплекса. Тестируемые соединения в различных концентрациях вводят через трахею вслед за индукцией гиперреактивности.
Предусмотренные настоящим изобретением агенты проявляют активность, снимающую или снижающую гиперреактивность дыхательных путей в вышеописанном тестовом методе, при их введении через трахею в дозах порядка 0,5-50,0 мкг/кг.
ИММУНОСУПРЕССОРНАЯ АКТИВНОСТЬ
Тест М: Реакция смешанных лимфоцитов мыши
Приблизительно 0,5х106 лимфоцитов, выделенных из селезенки самок мышей (8-10 недель) линии Balb/c, инкубируют в течение 5 дней в 0,2 мл среды для роста клеток, содержащей приблизительно 0,5х106 лимфоцитов, выделенных из селезенки самок мышей (8-10 недель) линии CBA. К среде добавляют различные концентрации тестируемого вещества. О наличии активности судят по способности подавлять связанный с пролиферацией синтез ДНК, выявляемый по включению радиоактивно меченного тимидина.
Предусмотренные настоящим изобретением агенты подавляют включение тимидина при их использовании в концентрациях порядка 0,1-50,0 нМ.
Кроме того, показано, что предусмотренные настоящим изобретением агенты подавляют in vitro пролиферативные ответы мононуклеарных клеток периферической крови человека, например, по отношению к туберкулину, и, в частности, проявляют синергический эффект подавления при совместном использовании с агентами, обладающими иммуносупрессивной активностью, например, с иммуносупрессивными циклоспоринами, такими как циклоспорин A, а также кортикостероидами.
В дополнение к вышесказанному общее фармакологическое тестирование, свидетельствует о том, что по сравнению с ранее известными соединениями предусмотренные настоящим изобретением агенты обладают ощутимыми и неожиданно более выгодными свойствами в плане предлагаемого ниже терапевтического использования, например, пониженным влиянием на поведенческую реакцию и/или, в частности, пониженным кардиоваскулярным эффектом в отношении гемодинамических параметров (влияния на частоту сердечных сокращений, индукции сужения сосудов и т.д.).
Так, в сериях экспериментов с использованием теста Допплера по определению аортного потока у кроликов показано, что соединение из Примера 1 в форме кислой щавелевокислой соли, образующейся в результате добавления кислоты, не вызывает побочных эффектов при использовании в дозах вплоть до приблизительно 0,3 мг/кг, и лишь слабое уменьшение частоты сердечных сокращений, происходящее вследствие сужения сосудов, наблюдается при использовании концентраций порядка 1 мг/кг.
Кроме того, показано, что указанное соединение в форме кислой щавелевокислой соли, образующейся в результате добавления кислоты, не оказывает никакого влияния или вызывает лишь минимальное изменение среднего артериального давления, частоты сердечных сокращений, а также концентрации глюкозы в плазме крови, в частности, при внутривенном или пероральном введении бодрствующим собакам в дозах, например, до 0,3 или 0,6 мг/кг, соответственно, что обеспечивает существенное и длительное подавление ФДЭ IV, добавляемой к плазме. Общее перенесение организмом всех доз - также хорошее.
Как уже было отмечено выше, в качестве ингибиторов IV типа изоферментов ФДЭ предусмотренные настоящим изобретением агенты также характеризуются четкой и более высокой специфичностью. Кроме того, им свойственно заметно более продолжительное время полужизни метаболической активности.
Учитывая их противовоспалительную активность, влияние на гиперреактивность дыхательных путей, а также их способность к ингибированию изофермента ФДЭ, особенно в качестве селективных ингибиторов IV типа, предусмотренные настоящим изобретением агенты предназначены для использования при лечении, в частности при профилактике затрудненного дыхания и воспалительных заболеваний дыхательных путей, например, путем постоянного и регулярного применения в течение продолжительных промежутков времени, призванного создать упреждающую защиту от повторного сужающего бронхи или иного симптоматического приступа, возникающего в результате затрудненного дыхания или воспалительного заболевания дыхательных путей, или же обеспечить контроль, ослабление или вылечивание основного состояния указанного заболевания.
Судя по их бронхорасширяющей активности, предусмотренные настоящим изобретением агенты могут быть использованы в качестве бронхорасширяющих агентов, например, для лечения хронического или острого сужения бронхов, в частности, для лечения симптомов затрудненного дыхания или воспалительных заболеваний дыхательных путей.
В настоящем описании и формуле изобретения понятие "лечение", используемое в отношении затрудненного дыхания и воспалительных заболеваний дыхательных путей, следует понимать таким образом, что оно охватывает как профилактическое, так и симптоматическое направления терапии.
В соответствии с вышесказанным, настоящее изобретение также предусматривает
А. Способ
а) лечения гиперреактивности дыхательных путей,
б) расширения бронхов или, в частности,
в) лечения затрудненного дыхания или воспалительных заболеваний дыхательных путей у пациента, нуждающегося в таком лечении, основанный на введении указанному пациенту эффективного количества предусмотренного настоящим изобретением агента.
К числу затрудняющих дыхание или воспалительных заболеваний дыхательных путей, на которые распространяется действие настоящего изобретения, относятся астма, пневмокониоз, хронически затрудненное дыхание, вызванное заболеваниями дыхательных путей или легких, синдром нарушения дыхания у взрослых, а также гиперреактивность дыхательных путей, развившаяся в результате терапевтического применения других медикаментов, например аспирина или β- агонистов.
Настоящее изобретение может быть использовано при лечении астмы вне зависимости от характера ее возникновения, включая врожденную и приобретенную астму. Оно может быть использовано при лечении аллергической (атонической или опосредованной иммуноглобулином E) астмы. Оно также может быть использовано при лечении неатопической астмы, включая, например, бронхитную, вызванную нагрузкой и оккупационную астму, астму, развившуюся в результате бактериальной инфекции, а также другие неаллергические астмы. Оно также может быть использовано при лечении синдрома детской астмы (детская, начальная стадия астмы).
Настоящее изобретение может быть использовано при лечении пневмокониозов вне зависимости от характера их возникновения, включая, например, алюминозы, антракозы, асбестозы, халикозы, птилозы, сидерозы, силикозы, табакозы и биссинозы.
Настоящее изобретение может быть использовано при лечении хронически затрудненного дыхания, вызванного заболеваниями дыхательных путей или легких, включая хронический бронхит, энфизему легких или связанную с ней одышку.
Настоящее изобретение также может быть использовано при лечении бронхита вне зависимости от характера его возникновения, включая, например, острый, арахидный, катарральный, хронический, крупозный или фтиноидный бронхит и т. д.
Принимая во внимание их активность в качестве селективных ингибиторов выделения TNF- α, предусмотренные настоящим изобретением агенты также могут быть предназначены для использования при понижающей регуляции или подавлении выделения TNF- α, например, при лечении заболеваний или симптомов, при которых выделение TNF- α происходит непосредственно или играет опосредующую роль, в том числе заболеваний и состояний, в этиологию которых вовлечено или принимает участие патологическое, например, нежелательное, случайное или нерегулируемое выделение TNF- α , в частности, при лечении кахексии или эндотоксинового шока, а также при лечении СПИД.
Предусмотренный настоящим изобретением способ может быть применен при лечении кахексии, связанной с патологическим выделением TNF- α или с высокими уровнями содержания TNF-α в кровяной сыворотке вне зависимости от характера их происхождения, включая кахексию, развивающуюся в результате, например, бактериальной, вирусной или паразитической инфекции, либо в результате нарушения или ухудшения гуморальной функции или (функции органов, в том числе почки. В частности, он может быть использован при лечении кахексии, обусловленной раком, малярией или глистами, либо развивающейся в результате нарушения гипофиза, щитовидной или зобной желез, а также при лечении уремической кахексии. Его можно, в частности, использовать при лечении кахексии, обусловленной СПИД, в том числе кахексии, возникающей в результате или связанной с инфекцией ВИЧ.
Предусмотренный настоящим изобретением способ может быть также использован при лечении эндотоксинового шока. В отношении этого аспекта следует подчеркнуть, что настоящее изобретение предусматривает способ лечения эндотоксинового шока, а также симптомов, появляющихся в результате или связанных с эндотоксиновым шоком, например, синдрома нарушения дыхания у взрослых.
Предусмотренный настоящим изобретением способ может быть также применен при лечении заболеваний, возникающих в результате ВИЧ-инфекции, например, СПИД, в том числе для ослабления или контроля развития указанных заболеваний.
Принимая во внимание их свойства в плане ингибирования изоферментов ФДЭ и/или подавления выделения TNF- α, а также их иммуносупрессивную активность, предусмотренные настоящим изобретением агенты можно рекомендовать также для использования в качестве иммуносупрессивных агентов, например, при лечении аутоиммунных заболеваний, в частности для лечения аутоиммунных заболеваний, сопровождающихся или возникающих в связи с воспалительными процессами, либо в качестве противовоспалительных агентов для лечения воспалительных заболеваний, в частности для лечения воспалительных заболеваний, сопровождающихся или возникающих в связи с аутоиммунными процессами.
К числу указанных заболеваний, по отношению к которым можно использовать настоящее изобретение, принадлежат аутоиммунные гематологические заболевания (например, гемолитическая анемия, апластическая анемия, анемия красных кровяных телец, а также идиопатическая тромбоцитопения), эритрематоз системной волчанки, полихондрит, склеродома, грануломатоз Вегенера, дерматомиозит, хронический активный гепатит, базедова болезнь, синдром Стивена-Джонсона, идиопатическое спру, аутоиммунное воспалительное заболевание кишечника (например, язвенный колит и болезнь Крона), эндокринная офтальмопатия, болезнь Грэйва, саркоидоз, альвеолит, хроническая гиперчувствительная пневмония, множественный склероз, первичный желчный цирроз, ювенильный диабет (diabetes mellitus, I тип), ювеит (передний и задний), сухой и весенний кератоконъюнктивиты, интерстициальный фиброз легких, псориатический артрит и гломерулонефрит (с наличием или отсутствием нефротического синдрома, в том числе включая идиопатический нефротический синдром или нефропатию минального перерождения), а также воспалительные и/или гиперпролиферативные заболевания кожи, такие как псореазный атопический дерматит, пузырьчатая сыпь и, в частности, контактный дерматит, например, аллергический контактный дерматит.
Предусмотренные настоящим изобретением агенты рекомендуются в частности для использования при лечении артрита и других ревматических и воспалительных заболеваний, особенно для лечения ревматоидного артрита.
В качестве иммуносупрессантов предусмотренные настоящим изобретением агенты также рекомендуются для использования в целях предотвращения отторжения пересаженных тканей, например, для поддержания аллогенных трансплантантов органов или т.п., в том числе имеющих отношение к трансплантантам почки, печени, легкого, сердца, сердца и легкого, кишечника, костного мозга, кожи или роговицы,
Принимая во внимание их противовоспалительную активность, особенно по отношению к активации эозинофилов, предусмотренные настоящим изобретением агенты рекомендуются также для использования при лечении связанных с эозинофилами заболеваний, в том числе эозинофилии, в особенности - связанных с эозинофилами заболеваний дыхательных путей (например, сопровождающихся патологическим проникновением эозинофилов в ткани легких), включая гиперэозинофилию, поскольку она оказывает влияние на дыхательные пути и/или легкие, а также, например, связанных с эозинофилами заболеваний дыхательных путей, развивающихся в результате или одновременно с синдромом Леффлера, эозинофильной пневмонией, заражением паразитами (особенно многоклеточными), включая тропическую эозинофилию, бронхо-легочным аспергиллезом, нодозным полиартритом (включая синдром Хурга-Штраусса), эозинофильной гранулемой и связанными с эозинофилами заболеваниями, поражающими дыхательные пути в результате реакции на лекарственный препарат.
Учитывая их свойства в плане ингибирования изоферментов ФДЭ, особенно их способность выступать в качестве селективных ингибиторов IV типа, предусмотренные настоящим изобретением агенты рекомендуются также для использования в качестве ингибиторов IV типа ФДЭ, например, для лечения заболеваний, сопровождающихся вымыванием кальция из тканей, в частности - дегенеративных заболеваний костей и окружения, сопровождающихся потерей кальция, особенно - остeопороза. В этом плане указанные агенты рекомендуются также для использования при лечении аллергических воспалительных заболеваний, таких как ринит, конъюнктивит, атопический дерматит, крапивница и желудочно-кишечные аллергии, связанные с кишечными заражениями: в качестве сосудорасширяющих агентов, например, при лечении ангины, повышенного давления, сердечной недостаточности и угрозы множественных инфарктов; а также для лечения иных симптомов, при которых показано подавление ФДЭ IV, например, депрессии, симптомов и заболеваний, связанных с ослаблением психической функции, включая болезнь Альцхеймера, паркинсонизм и паралич.
Принимая во внимание их способность к синергическому взаимодействию с иммуносупрессивными и/или противовоспалительными лекарственными препаратами, предусмотренные настоящим изобретением агенты рекомендуются также для использования в качестве терапевтических агентов, предназначенных для совместного применения с указанными препаратами, например, в качестве усилителей терапевтической активности указанных препаратов или в целях уменьшения необходимых доз или возможных побочных эффектов, вызываемых указанными препаратами. К числу лекарственных препаратов, с которыми удобно совместное применение предусмотренных настоящим изобретением агентов, принадлежат, в частности, циклопептидные, циклопептолидные или макролидные иммуносупрессивные или противовоспалительные лекарственные препараты, например, препараты, относящиеся к классу циклоспоринов, в том числе циклоспорины A или G, лекарственные препараты такролимус (известный также под названием FK 506), аскомицин и рапамицин, различные известные родственные им препараты и их производные, а также глюкокортикостероидные препараты. К числу заболеваний, к которым может быть применена указанная совместная терапия, принадлежат любые заболевания или симптомы, требующие терапии иммуносупрессивными или противовоспалительными препаратами, в том числе перечисленные выше. В частности, предусмотренные настоящим изобретением агенты удобны для использования при вышеописанной совместной терапии, осуществляемой в целях, например, иммуносупрессивного, противовоспалительного или противоастматического лечения, в том числе с получением эффекта экономии циклоспорина, например, циклоспорина A, макролида или стероида.
В соответствии с вышесказанным, настоящее изобретение также предусматривает
Б. Способ
а) понижающей регуляции или подавления выделения TNF-α,
б) ингибирования активности изофермента ФДЭ IV,
в) получения иммуносупрессии,
г) лечения воспалительных заболеваний или
д) лечения любых конкретных вышеперечисленных симптомов или заболеваний у пациента, нуждающегося в таком лечении, основанный на введении указанному пациенту эффективного количества предусмотренного настоящим изобретением агента.
Настоящее изобретение также предлагает
В. Агент, предусмотренный настоящим изобретением для применения в качестве фармацевтического средства, например, предназначенного для применения в любом способе или при лечении любого вышеперечисленного заболевания или симптома, в том числе вышепоименованных в пунктах А и Б.
Безусловно, дозы, используемые при реализации настоящего изобретения, могут варьировать, например, в зависимости от конкретного излечиваемого заболевания или симптома, конкретного применяемого агента, предусмотренного настоящим изобретением, способа применения и желаемого вида терапии. Однако, в целом, для получения удовлетворительных результатов, например, при лечении вышеперечисленных заболеваний рекомендуется пероральное применение в дозах порядка 0,01-2,0 мг/кг, а рекомендуемая дневная доза в случае перорального применения составляет соответственно приблизительно 0,75-150 мг, указанную дозу удобно применять однократно или в виде дробных доз 2-4 раза в день, или в форме, обеспечивающей отсроченное высвобождение агента. Таким образом, каждая форма дозирования, предназначенная для перорального применения, в приемлемом случае содержит приблизительно от 0,2 до 75 или 150, в том числе приблизительно от 0,2 или 2,0 до 50. 75 или 100 мг предусмотренного настоящим изобретением агента вместе с его фармацевтически приемлемым разбавителем или носителем.
Для использования при лечении хронических заболеваний дыхательных путей или затрудненного дыхания, например астмы, предусмотренные настоящим изобретением агенты предпочтительно применяют путем ингаляции. Используемые при этом дозы также могут варьировать, например, в зависимости от конкретного заболевания или симптома, конкретного применяемого агента, предусмотренного настоящим изобретением, конкретного способа применения (в том числе, производят ли ингаляцию сухим порошком или иначе) и желаемого эффекта. Однако, в целом, рекомендуемая для ингаляции дневная доза должна находиться в пределах от приблизительно 2,5 до приблизительно 130,0 мкг/кг/день, в том числе от приблизительно 13,0 до приблизительно 60,0 мкг/кг/день, а рекомендуемая дневная доза, предназначенная для применения путем ингаляции, например, в случае лечения астмы, должна находиться в пределах от приблизительно 0,2 до приблизительно 10,0 мг, в том числе от приблизительно 1 до приблизительно 5 мг, указанную дозу удобно применять однократно или же в 2 или 3 отдельных приема в течение дня. Таким образом, приемлемая доза, применяемая за один прием, должна находиться в пределах от приблизительно 200 мкг до 3,3 мг в случае 3-кратного приема в течение дня, который удобно осуществлять с использованием приспособления, предназначенного для ингаляции сухим порошком, посредством серий из 2-8 распылений в течение каждого приема.
Кроме того, предусмотренные настоящим изобретением агенты можно применять с использованием любого другого приемлемого способа, в том числе вливания, например, при лечении эндотоксинового шока: закапывания в нос, например, при лечении ринита: закапывания в глаза, например, при лечении аутоиммунных заболеваний глаза; наружного применения, т.е. непосредственного местного нанесения на кожу, например, в случае лечения кожных заболеваний или псореаза; или ректального введения, в том числе с помощью клизмы или суппозитория, например, для лечения воспалительного заболевания кишечника. Приемлемые дозы, предназначенные для введения с помощью указанных способов, в целом, должны быть приблизительно в 10-100 раз меньшими, нежели в случае перорального применения.
Фармацевтические составы, включающие в себя предусмотренные настоящим изобретением агенты, могут быть получены с использованием традиционных разбавителей или эксипиентов, а также приемов, хорошо известных в области галогеновых препаратов. Так, формы, предназначенные для перорального приема доз, могут включать в себя таблетки, капсулы и т.п. Средства, предназначенные для наружного применения, могут быть представлены в форме кремов, мазей, гелей или систем, обеспечивающих подкожное проникновение, в том числе пластырей, а в дополнение к инертным разбавителям и носителям могут содержать агенты, способствующие проникновению через кожу, также хорошо известные в этой области.
Используемые для ингаляции средства могут включать в себя аэрозоль или другие распыляемые составы, а также составы, предназначенные для ингалирования сухим порошком, содержащие или не содержащие разбавитель и применяемые с помощью любой известной системы, используемой для ингаляции сухим порошком. Для получения форм, предназначенных для ингаляции сухим порошком, соединения, соответствующие формуле (I), или их физиологически гидролизуемые или приемлемые эфиры удобно использовать в форме их фармацевтически приемлемой соли, образующейся в результате добавления к ним кислоты. В случае соединения из Примера 1 наиболее удобной солью является гидрохлорид (т. пл. 218-222oC). Указанную форму соли удобно измельчать, в том числе с использованием воздушной струи или керамической мельницы, позволяющих получать тонко измельченный порошок, например, со средним диаметром частиц, приблизительно равным 2-3 мкм. По меньшей мере 90% полученного вещества должно состоять из частиц со средним диаметром в приемлемом случае менее 7,8 мкм, в более предпочтительном случае - менее 4,8 мкм. Для того, чтобы быть уверенным в получении надлежащего и стойкого продукта, состоящего из частиц и пригодного для применения путем ингаляции в форме сухого порошка, может оказаться предпочтительным проводить измельчение активного ингредиента, например, гидрохлорида соединения из Примера 1, предварительно смешанного с соответствующей пригодной для ингаляции несущей средой, в том числе лактозой, в условиях пониженной температуры.
В соответствии с вышесказанным, настоящее изобретение также предлагает: фармацевтический состав, включающий в себя предусмотренный настоящим изобретением агент вместе с его фармацевтически приемлемым разбавителем или носителем, в частности, предназначенный для использования любым из вышеописанных способов.
Предусмотренные настоящим изобретением агенты, представляющие собой соли, образующиеся в результате добавления фармацевтически приемлемой кислоты, проявляют такую же активность и толерантность, как и вышеописанные соединения, соответствующие формуле (I), или их физиологически гидролизуемые и приемлемые эфиры.
Изохинолины формулы I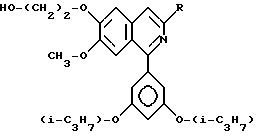
где R - этильная или н-пропильная группа, или их физиологически гидролизуемые и приемлемые сложные эфиры или кислотноаддитивные соли таких соединений или сложных эфиров, обладают противовоспалительными, снижающими гиперреактивность дыхательных путей, а также бронхорасширяющими свойствами. 3 с. и 3 з.п. ф-лы.
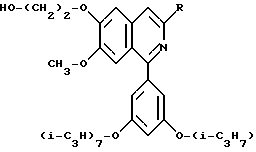
где R - этильная или н-пропильная группа, или их физиологически гидролизуемые и приемлемые сложные эфиры или кислотноаддитивные соли таких соединений или сложных эфиров.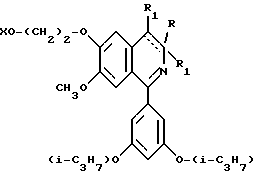
где X - атом водорода, R имеет указанные в п.1 значения, R1 и R2 обозначают дополнительную связь, изображенную в виде пунктирной линии, или X соответствует группе, защищающей гидроксильный радикал, а каждая из групп R1 и R2 представляет собой атом водорода или вместе обозначают дополнительную связь, изображенную в виде пунктирной линии, дегидрируют и/или снимают защиту или для получения физиологически гидролизуемого и приемлемого эфира соединения формулы I, проводят его этерификацию и выделяют целевой продукт в свободном виде или в виде соли, образованной в результате добавления к нему кислоты.
| 6,7-Диэтокси-1-(4-этокси-3-оксибензил)-3,4-дигидроизохинолин,обладающий действием на сердечно-сосудистую систему | 1982 |
|
SU1097621A1 |
| Пресс-прокладка для ламинирования древесных плит | 1985 |
|
SU1407685A1 |
| GB 1400425, 16.07.75. | |||
