Дитерпены со структурой таксана, в частности таксола, как известно, обладают противоопухолевым действием в отношении ряда опухолей человека. Однако применение этих лекарств, особенно таксола, имеет некоторые недостатки вследствие нежелательных побочных эффектов. По этой причине и вследствие того, что эти противоопухолевые препараты быстро индуцируют устойчивость к их действию, разработка новых веществ, использование которых снижает проблемы, наблюдаемые при клиническом применении, представляет интерес.
В заявке WO 93/02067 (Nippon Steel) от 4 февраля 1993 года описывается, например, 10-альфа-ацетилтаксол, экстрагированный из культуры ткани белка вида Taxus и обладающий значительной цитотоксической активностью.
Данное изобретение относится к новым производным со структурами таксана, полученным полусинтетическим способом и обладающим сильной противоопухолевой активностью. Производные данного изобретения имеют структуру формулы (1):
Их можно подразделить на две группы:
a) производные таксана, содержащие двойную олефиновую связь в положении 11, 12 и гидроксильную или ацетилоксигруппу в положении 10a (таксаны формулы 1a)
(1a) (R3 = H; R4 = OH или ацилоксигруппа)
b) производные таксана, содержащие одинарную связь между атомами углерода в положении 11 и 12, причем метил в положении 12 альфа-ориентирован, и гидроксильную или ацилоксигруппу в положении 10-бета (таксан формулы 1b).
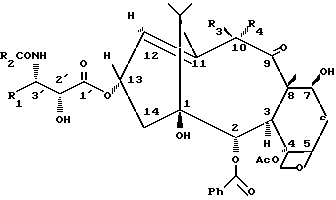
(1b) (R3 = OH или ацилоксигруппа; R4 = H)
В таксанах общей формулы (1) R1 и R2, которые могут быть одинаковыми или разными, представляют собой C1-C8 алкил, C2-C8 алкенил, арил (предпочтительно фенил) или гетероарильную группу. R2 может также представлять собой трет-бутокси группу.
В соединениях формулы (1a) R3 - водород, a R4 - гидроксильная или C2-C8 ацилоксигруппа.
В соединениях формулы (1b) R3 - гидроксильная или С2-C8 ацилоксигруппа и R4 - водород.
Таксаны формулы (1) получают этерификацией в положении 13 новых синтонов формулы (2), используя активированные подходящим образом цепочки изосерина в качестве ацилирующих агентов, в соответствии со способом, который описан в литературе для полусинтеза таксола и его аналогов (см., например, EP-A-400971, 1992, Fr. dem.86, 10400; E. Didier et al., Tetrahedron Letters 35, 2349, 1994; E. Didier et al., ibid. 35, 3063, 1994).
В формуле (2)
где когда двойная олефиновая связь находится в положении 11, 12, R3 представляет собой атом водорода, R4 и R5 - гидроксильные, C2-C8-ацилокси-, алкилсилилокси или 2,2,2-трихлорэтоксикарбонилокси-группы;
когда двойная олефиновая связь отсутствует в положении 11, 12, метил в положении 12 является альфа-ориентированным, R4 - атом водорода, R3 и R5 - гидроксильные группы, C2-C8-ацилокси-, алкилсилилокси или 2,2,2-трихлорэтоксикарбонилокси-группы.
В частности, синтоны формулы (2a) используются для синтеза новых таксанов формулы (1a). С другой стороны, синтоны формулы (2b) используются для синтеза новых таксанов формулы (1b).
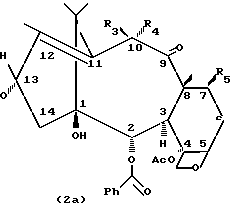
В синтонах (2a) двойная олефиновая связь находится в положении 11, 12, и C2-C8 ацилоксигруппа или необязательно защищенная гидроксильная группа находятся в положении 10. Следовательно, в синтонах (2a) R3 - атом водорода, R4 и R5 представляют собой гидроксильные, ацилокси, алкилсилилокси (например, триэтилсилилокси, O-TES) или 2,2,2-тpиxлopэтоксикарбонилокси (O-CO-O-CH2CCl3, O-TROC) группы.

В синтонах (2b) атомы углерода в положениях 11 и 12 связываются одинарной связью, метил в положении 12 является альфа-ориентированным, и ацилоксигруппа или необязательно защищенная гидроксильная группа находятся в 10d-положении. Следовательно, в синтонах (2b), R4 - атом водорода, R3 и R5 - представляют собой гидроксильные группы, ацилокси-, алкилсилилокси- (такие как триэтилсилилокси, O-TES) или 2,2,2-трихлорэтоксикарбонилокси (O-CO-O-CH2CCl3, O-TROC) группы.
После этерификации в положение 13 синтонов (2) изосериновой цепью защитные группы удаляют обычными способами, описанными в литературе, в результате получают новые таксаны формулы (1).
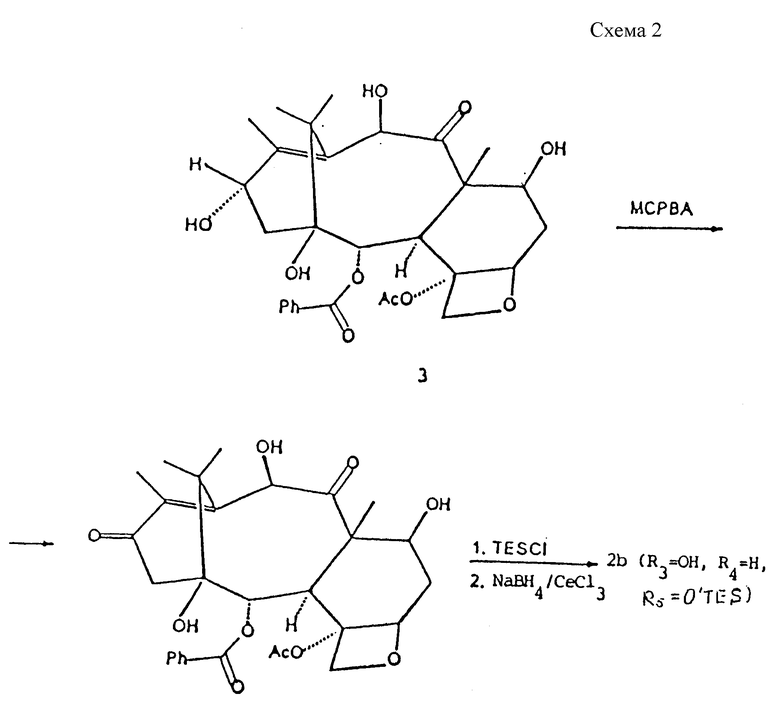
10-Деацетилбаксатин III (3), который может быть выделен из листьев Taxus Baccata (G. Chauviere et coll., C.R. Acad. Sc. Ser. III, 293; 591 [1981]), используется как единственное исходное вещество для получения синтонов (2a) и (2b).
Синтоны формулы (2a), которые не описаны в литературе, получают (Схема 1) из соединения (3) окислением в положение 10 ацетатом меди (II) с получением дикетона (4) и последующим восстановлением боргидридом натрия в присутствии солей церия (III).
Образующийся продукт (2a, R3 = H, R4 = R5 = OH), который представляет собой эпимер в положении 10 соединения (3), содержит подходящие защитные группы в положениях 7 и 10 и используется для синтеза таксанов (1a).
Новый секотаксан (5) получают в качестве побочного продукта реакции, представленной на Схеме 1.

Секотаксан (5) может быть использован для синтеза других таксанов с потенциальной противоопухолевой активностью.
Данное изобретение относится также к новым производным со структурой секотаксана, полученным полусинтетическим способом и обладающим сильной противоопухолевой активностью. Указанные производные имеют строение формулы (5a)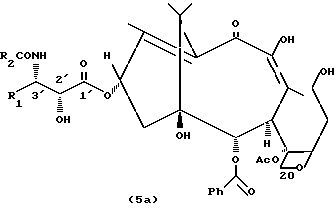
где R1 и R2, которые могут быть одинаковыми или разными, представляют собой C1-C20 алкил, C2-C8 алкенил, арил (предпочтительно фенил) или гетероарил. R2 может также представлять собой трет-бутоксигруппу.
Таксаны формулы (5a) получают этерификацией соединения формулы (5) в положение 13, используя в качестве ацилирующих агентов подходящим образом активированные изоцериновые цепи, в соответствии с описанным в литературе получением полусинтетического таксола и его аналогов (см., например, EP-A-400971; Е. Didier et al., Tetrahedron Letters 35, 2349, 1994; Е. Didier et al. , ibid. 35, 3063, 1994). Гидроксильные группы соединения (5) могут необязательно защищаться подходящими защитными группами в соответствии с общепринятыми способами.
После этерификации соединения (5) в положение 13 изосериновой цепочкой защитные группы удаляют в соответствии с общеизвестными способами, описанными в литературе, посредством чего получают секотаксаны формулы (5a).
Синтоны формулы (2b), которые не описаны в литературе, также получают из 10-деацетилбаксатина III (3) (Схема 2). Было установлено, что окислением соединения (3) м-хлорпербензойной кислотой (MCPBA) получают соответствующее 13-кетопроизводное (6). После введения в качестве защитной группы для гидроксильной группы в положении 7 триэтилсилилхлорида (TESCl), восстановлением боргидридом натрия в присутствии солей церия (III) получают синтон (2b) (R3 = OH, R4 = H, R5 = O-TES), который может быть полезным в синтезе таксанов формулы (1b). Альфа-ориентация метильной группы в положении 12 в синтонах (2b) устанавливается тщательным изучением спектра ядерного магнитного резонанса.
Продукты данного изобретения были подвергнуты скринингу на их цитотоксическое действие в отношении клеточных линий различных опухолей, при сравнении их активности с активностью таксола. В табл. 1 и 2 представлены IC50 таксола и соединений данного изобретения: 13-[(2R,3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил] - 10-эпи-10-деацетилбаккатина III (1a, R1 = Ph, R2 = трет-бутокси, R3 = H, R4 = OH), 13-[(2R,3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил] -10-деацетил-11,12-дигидробаккатина III (1b, R1 = Ph, R2 - трет-бутокси, R3 = OH, R4 = H), 13-[(2R,3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламино-пропаноил] -C-секо-10-деацетилбаккатина III (5a, R1 = Ph, R2 = трет-бутокси) и 13-[(2R,3S)-3-изобутил-2-гидрокси-3-капроиламино-пропаноил] -C-секо-10- деацетилбаккатина (5a, R1 = изобутил, R2 = пентил).
Соединения с различными заместителями в изосериновой цепи ведут себя аналогично. Соединения проявляют неожиданное преимущество по сравнению с таксолом в отношении линий клеток, устойчивых к другим противоопухолевым веществам, таким как адриамицин и цис-платинум. Различия между таксолом и этими веществами еще более очевидны в опытах in vivo, таких как имплантация атимической (лишенной тимуса) голой мыши опухоли человека. Было установлено, что соединения данного изобретения, в которых R2 - алкильная или алкенильная группа, неожиданно не проявляют кардиотоксической активности в отличие от таксола и его известных производных, следовательно, они могут успешно применяться пациентами с патологией сердца, которые не могут использовать для лечения таксол и его известные производные.
Соединения данного изобретения подходят для включения в подходящие фармацевтические рецептуры для парентерального и перорального введения. Для внутривенного введения используются, главным образом, смеси полиэтоксилированного касторового масла и этанола или липосомные препараты, полученные с натуральным фосфатидилхолином, или смеси натуральных фосфолипидов в присутствии холестерина.
Приведенные ниже примеры дополнительно иллюстрируют данное изобретение.
Пример 1. Получение 10-дегидро-10-деацетилбаккатина III (4).
10 г 10-деацетилбаккатина III (3) суспендируют в 350 мл метанола, куда добавляют 65 г Cu(OAc)2. Суспензию непрерывно перемешивают при комнатной температуре в течение 120 часов. Соли отфильтровывают и раствор хроматографируют на 100 г силикагеля, элюируя смесью гексан/этилацетат (6:4). После перекристаллизации из лигроина получают 9.5 г (4), M+ при m/z 542.
Пример 2. Получение 10-деацетил-10-эпибаккатина III (2a, R3 = H, R4 = R5 = OH) и C-секо-10-деацетилбаккатина III (5).
К раствору 300 г (4) в 5 мл метанола добавляют один эквивалент CeCl3 • 3H2O. Перемешивают при комнатной температуре в течение 5 минут, затем добавляют 80 мл NaBH4. Раствор обрабатывают раствором NH4Cl экстрагируют этилацетатом и хроматографируют на силикагеле, элюируя смесью гексан/этилацетат (3: 7). Получают 98 мг соединения (2a) (M+ при m/z 544) и 120 мг (5) (M+ при m/z 546).
10-Деацетил-10-эпибаккатин III имеет следующий ЯМР-спектр (CDCl3): H2, д 5.68 J 6.8; H3, д 4.26 J 6.8; H5, д 5.03 J 7.1; H7/13, m 4.76; H10, уш. с 5.20; 10 OH, уш. с 3.44; H16, с 1.14; H17, с 1.68; H18, с 2.22; H19, с 1.13; H20a, д 4.33; H20b, д 4.18; Ac, с 2.31; Bnz, уш. 8.12 J 8, уш. т 7.60 J 8, уш. т 17.49 J 8.
Пример 3. Получение 10-деацетил-13-дегидробаккатина III (6).
3 г мета-хлорпербензойной кислоты и 1 г ацетата натрия добавляют в суспензию 1 г 10-деацетилбаккатина III (3) в 100 мл CH2Cl2. Суспензию непрерывно перемешивают в течение 120 часов при комнатной температуре и затем разбавляют 5%-ным водным раствором Na2CO3. Органическую фазу промывают 5%-ным Na2CO3 и упаривают досуха. Остаток очищают на силикагеле, элюируя смесью гексан/этилацетат (3:7). Получают 789 г соединения (6), M+ при m/z 542.
Пример 4. Получение 10-деацетил-11,12-дигидро-7- триэтилсилилбаккатина III (2b, R3 = OH, R4 = H, R5 = O-TES).
1.6 г соединения (6) растворяют в метиленхлориде и смешивают с 370 мг 4-диметиламинопиридина и 2.5 мл триэтилсилилхлорида. После выдерживания смеси в течение 2 часов при комнатной температуре ее разбавляют метиленхлоридом и промывают водой. Органическую фазу упаривают досуха. Получают 1.72 г остатка, который смешивают со 150 мл 95%-ного этанола и обрабатывают 9 г NaBH4. Спустя 3 часа смесь разбавляют раствором NH4Cl и продукт экстрагируют этилацетатом. После хроматографии на силикагеле (элюент: гексан/этилацетат (7:3)), получают 800 мг (2b) (R3 = OH, R4 = H, R5 = O-TES).
C35H52O10Si, мол. вес. = 660
CI-MC (NH3): 678 (M+NH4)+ (80).
IK (Kbr табл): 3500, 1745, 1710, 1270, 1245, 1110, 980, 705 см-1
1H-ЯМР (400 МГц, CDCl3, комн. темп.): 8,15 (Bz AA'), 7,60 (Bz, C), 7,47 (Bz, BB'), 5,61 (д, J = 8,2 Гц, H-2), 5,10 (д, J = 8,3 Гц, H-5), 4,97 (дд, J = 8,6, 4,9 Гц, H-10), 4,34 (дд, J = 11,9, 8,5 Гц, H-7), 4,29 (д, J = 8,7 Гц, H-20a), 4,27 (д, J = 8,7 Гц, H-20b), 4,03 (уш.с, H-13), 3,33 (д, J = 8,2, Гц, H-3), 2,48 (м, H-12), 2,24 (с, OAc), 1,71 (с, H-19), 1,23 (д, J = 7,0 Гц, H-18), 1,11 (с, H-17), 1,02 (с, H-16), 0,93 (т, J = 7,9 Гц, ТЭС), 0,54 (кв, J = 7,9 Гц, ТЭС).
13C ЯМР (50 МГц, CDCl3, комн. темп.): 215,1 (с), 169,9 (с), 167,7 (с), 133,4 (д), 130,1 (д), 129,8 (с), 128,5 (д), 83,8 (д), 80,1 (с), 79,7 (c), 77,0 (т), 73,4 (д), 71,6 (д), 70,9 (д), 69,4 (д), 60,5 (д), 56,4 (с), 40,6 (д), 40,3 (с), 36,4 (т), 35,4 (т), 32,5 (кв), 32,4 (д), 25,6 (кв), 22,4 (кв), 15,6 (кв), 9,1 (кв), 5,3 (т).
Пример 5. Получение 11,12-дигидро-7-TES-баккатина III (2b, R3 = OH, R4 = H, R5 = O-TES) и 11,12-дигидробаккатина III (2b, R3 = OAc, R4 = H, R5 = OH)
500 мг 10-деацетил-11,12-дигидро-7-триэтилсилилбаккатина III (2b, R3 = OH, R4 = H, R5 = O-TES) взаимодействуют в безводном пиридине с 3 эквивалентами ацетилхлорида при температуре 0oC в течение 6 часов. Реакционную смесь разбавляют водой и экстрагируют метиленхлоридом. После выпаривания растворителя остаток перекристаллизовывают из смеси ацетон/гексан. Получают 510 мг 11,12-дигидро-7-TES-баккатина III, M+ при m/z 702 III. Продукт растворяют в метаноле и обрабатывают разбавленной HCl до полного десилилирования. Реакционную смесь разбавляют водой, экстрагируют этилацетатом и перекристаллизовывают из водного метанола. Получают 400 мг 11,12-дигидробаккатина 11, M+ при m/z 588.
Пример 6. Получение 13-[(2R,3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил] - 11,12-дигидробаккатина III (1b, R1 = Ph, R2 = трет-BuO, R3 = OAc, R4 = H).
500 мг 11,12-дигидробаккатина III (2b, R3 = OAc, R4 = H, R5 = O-TES) растворяют в 20 мл толуола с 0.45 г (4S,5R)-N-трет-бутоксикарбонил-2,2-диметилфенил-5-оксазолидинкарбоновой кислоты, дициклогексилкарбодиимида (1.03 экв. ) и N,N-диметиламинопиридином (0.2 экв.) при температуре 80oC в течение 2 часов. Реакционную смесь промывают водой до тех пор, пока избыток реагентов не будет удален, затем упаривают досуха. Остаток обрабатывают метанолом, содержащим 1% муравьиной кислоты, в течение 4 часов при комнатной температуре. Метанольный раствор разбавляют водой, нейтрализуют и экстрагируют этилацетатом, органическую фазу упаривают досуха и остаток обрабатывают раствором, содержащим 1.5 экв. ди-трет-бутилкарбоната и бикарбоната натрия в 15 мл тетрагидрофурана. Реакционную смесь разбавляют водой, экстрагируют этилацетатом и гетеро-уксусную фазу упаривают досуха. Остаток смешивают с подкисленным соляной кислотой метанолом для полного десилилирования. Раствор затем разбавляют водой и экстрагируют этилацетатом. Остаток, полученный в результате упаривания гетеро-уксусной фазы, хроматографируют на силикагеле, элюируя смесью ацетон/гексан (1: 1) для удаления примесей. Получают 580 г продукта, M+ при m/z 851.
Пример 7. Получение 13-[(2R,3S)-3-бензоиламино-3-фенил-2-гидроксипропаноил]-11,12- дигидробаккатина III (1b, R1 = R2 = Ph, R3 = OAc, R4 = H).
500 мг 11,12-дигидро-7-TES-баккатина (2b, R3 = OAc, R4 = H, R5 = O-TES) растворяют в 20 мл толуола и вместе с 1.5 г (4S,5R)-N-бензоил-2,2-диметил-4-фенил-5-оксазолидинкарбоновой кислоты, дициклогексилкарбодиимидом (1.03 экв.) и N,N-диметиламинопиридином (0.2 экв.), смесь выдерживают при температуре 80oC в течение 2 часов. Реакционную смесь промывают водой до тех пор, пока избыток реагентов не будет удален, затем упаривают досуха. Остаток обрабатывают метанолом, содержащим 1% муравьиной кислоты, в течение 4 часов при комнатной температуре. Метанольный раствор разбавляют водой, нейтрализуют и экстрагируют этилацетатом. Органическую фазу упаривают досуха и остаток соединяют с метанолом, подкисленным соляной кислотой для полного десилилирования. Раствор затем разбавляют водой и экстрагируют этилацетатом. Остаток, полученный после упаривания гетеро-уксусной фазы, хроматографируют на силикагеле, элюируя смесью ацетон/гексан (1:1) для удаления примесей. Получают 530 мг продукта, M+ при m/z 855.
Пример 8. Получение 13-[(2R,3S)-3-фенил-2-гидрокси-3-трет- бутоксикарбониламино-пропаноил] -10-эпи-10-деацетилбаккатина III (1a, R1 = Ph, R2 = трет-BuO, R3 = H, R4 = OH).
500 мг 10-деацетил-10-эпибаккатина III (2a, R3 = H, R4 = R5 = OH) растворяют в 15 мл безводного пиридина и обрабатывают в течение 5 минут при температуре 80oC тремя эквивалентами трихлорэтоксикарбонилхлорида (TROC-Cl) и затем охлаждают до комнатной температуры. Для разложения избытка TROC-Cl добавляют 1 мл метанола. Раствор разбавляют ледяной водой и экстрагируют хлороформом, органическую фазу промывают разбавленным раствором соляной кислоты. Органическую фазу упаривают досуха и остаток обрабатывают при комнатной температуре в течение 24 часов раствором толуола, содержащим три эквивалента (4S, 5R)-N-трет-бутоксикарбонил-2,2-диметил-4-фенил-5- оксазолидинкарбоновой кислоты, 3 эквивалента дициклогексилкарбодиимида и 0.2 эквивалента N,N-диметиламинопиридина. Реакционную смесь промывают водой и органическую фазу упаривают досуха под вакуумом. Остаток поглощают с метанолом и обрабатывают одним эквивалентом п-толуолсульфокислоты в течение 48 часов, после чего разбавляют водой и экстрагируют этилацетатом. Органическую фазу упаривают в вакууме, остаток переносят в 200 мл смеси уксусная кислота/этилацетат (1: 1) и обрабатывают в течение 3 часов при температуре 30oC 11-ю эквивалентами порошкообразного цинка. Твердый осадок отфильтровывают, раствор разбавляют водой, экстрагируют этилацетатом и хроматографируют на силикагеле смесью этилацетат/гексан (1:4). Получают 512 мг продукта (1a), M+ при m/z 807.
Пример 9. Получение 7,9-дитриэтилсилил-C-секо-10- деацетилбаккатина III.
В раствор соединения (5) (200 мг, 0.37 ммоля) в безводном диметилформамиде (ДМФА) (5 мл) добавляют имидазол (75 мг, 1.11 ммоля, 3 мол.экв.) и триэтилсилилхлорид (TES) (186 мл, 167.3 мг, 1.11 ммоля, 3 экв.мол), реакционную смесь перемешивают при комнатной температуре в течение 10 минут. Анализ реакционной смеси проводят методом тонкослойной хроматографии (гексан:этилацетат = 3:7, Rf исходной массы 0.10, Rf продукта 0.80). Реакцию гасят добавлением воды и CeliteR (броутмиллерита), осадок фильтруют и промывают водой для удаления ДМФА, затем CHCl3 для удаления продукта. После очистки колоночной хроматографией (гексан/этилацетат 9:1 для элюирования силанола, затем гексан/этилацетат 4:6 для элюирования продукта) получают 146 мг указанного в заглавии продукта (51%).
C41H64O10Si2, мол. вес = 772
CI-MC (NH3): 790 (M+NH4), (50).
ИК (KBr табл.): 3453, 1709, 1689, 1240, 1109, 710 см-1.
1H-ЯМР (200 МГц, CDCl3, комн.темп.): 8,02 (Bz AA'), 7,59 (Bz, C), 7,45 (Bz, BB'), 5,58 (д, J = 9 Гц), H-2), 5,10 (уш.с., H-5), 4,92 (уш.м., H-13), 4,15 (уш. м. , H-13), 3,60 (2H, уш.с.), 1,98, 1,95 (с, H-18 и H-19), 1,16, 1,15 (с, H-16 и H-17), 1,00-0,60 (м, 2 • ТЭС).
13C-ЯМР (CDCl3, комн.темп.): 192,0, 169,2, 167,1, 143,1, 141,8, 133,6, 129,5, 128,8, 128,7, 85,8, 80,2, 75,6, 74,6, 67,6 58,4, 44,1, 42,6, 41,2. Сигналы от групп метила очень широкие и представляют собой мультиплетные пики.
Пример 10. Получение 13-[(2R, 3S)-3-фенил-2-гидрокси-3-трет- бутоксикарбониламинопропаноил-C-секо-10-деацетилбаккатина III (5a, R1 = Ph, R2 = трет-BuO).
К раствору продукта, полученного в примере 9 (126 мг, 0,16 ммоля), в безводном толуоле (5 мл) добавляют 67,5 мг дициклогексилкарбодиимида (0.327 ммоля, 2 мол. экв.), 105 мг (4S,5R) -N-Boc-(2,4-диметоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты (0.327 ммоля, 2 мол. экв.) и 5 мг 4-диметиламинопиридина. Смесь нагревают до 60oC и выдерживают при этой температуре в течение 24 часов, разбавляют насыщенным водным раствором NaHCO3 и этилацетатом. Остаток очищают методом колоночной хроматографии, элюируя смесью гексан/этилацетат (8:2), получают 175 мг сложного эфира (95%). Остаток поглощают 50 мл метанол/HCI (0,01%) и реакционную смесь оставляют на 1 час при комнатной температуре. Раствор подщелачивают до pH 5 и упаривают досуха под вакуумом. Остаток хроматографируют на колонке с силикагелем, элюируя смесью метиленхлорид/метанол (98:2). После перекристаллизации из этилацетата получают 85 мг указанного в заглавии соединения.
C43H53NO14, Мол. вес = 807
Cl-MC (NH3): 825 (М + NH4)+ (20).
1H-ЯМР (400 МГц, CDCl3, 60oC): 8,05 (Bz AA'), 7,60 (Bz, C), 7,48 (Bz, BB'), 7,41 (AA', o-Ph), 7,37 (BB', m-Ph), 7,29 (с, p-Ph), 6,45 (с, 9-OH), 6,23 (ддкв, J = 10,0, 7,0, 1,5 Гц, H-13), 5,64 (уш.д. JJ = 9,0 Гц, H-2), 5,49 (д, J = 9,5 Гц, NH), 5,32 (дд, J = 10,0, 3,0 Гц, H-3'), 5,26 (уш.д. J = 11,5 Гц, H-5), 5,16 (д, J = 8,0 Гц, H-20a), 4,67 (д, 3,0 Гц, H-2'), 4,35 (уш. д. J = 9,0 Гц, H-З), 4,29 (д, J = 8,0 Гц, H-20 уш.), 3,86 (ддд, J = 11,0, 6,0, 6,0 Гц, H-7a), 3,70 (ддд, J = 11,0, 6,5, 6,5 Гц, H-7b), 2,80 (уш. дд, J = 16,0, 7,0 Гц, H-14a), 2,52 (м, H-6a), 2,44 (уш.д. J = 16,0, 10,0 Гц, H-14b), 2,18 (с, 1-OH), 2,10 (м, H-6b), 1,93 (с, OAc), 1,86 (уш.с. H-19), 1,85 (д, J = 1,5 Гц, H-18), 1,31 (с, BOC), 1,28 (с, H-17), 1,12 (с, H-16).
Пример 11. Получение 13-[(2R,3S)-3-изобутил-2-гидрокси-3- капроиламинопропаноил-C-секо-10-деацетилбаккатина III (5a, R1 = Ph, R2 = пентил).
К раствору продукта, полученного в примере 9 (126 мг, 0.16 ммоля), в безводном толуоле (5 мл) добавляют 67.5 мг дициклогексилкарбодиимида (0.327 ммоля, 2 моль. экв.), 140 мг (4S,5R)-N-капроил-2-(2,4-диметоксифенил)-4-изобутил-5- оксазолидинкарбоновой кислоты (0.327 ммоля, 2 мол. экв.) и 5 мг 4-диметиламинопиридина. Смесь нагревают до 60oC и выдерживают при этой температуре в течение 24 часов, затем разбавляют насыщенным водным раствором NaHCO3 и этилацетатом. Остаток очищают методом колоночной хроматографии (элюент: гексан/этилацетат (8: 2)), в результате получают 175 мг 13-эфира (95%). Остаток поглощают в 50 мл смеси метанол/HCl (0.01%) и реакционную смесь оставляют при комнатной температуре на 1 час. Раствор подщелачивают до pH 5 и упаривают досуха в вакууме. Остаток хроматографируют на колонке с силикагелем, элюируя смесью метиленхлорид-метанол (98:2). После перекристаллизации из этилацетата получают 88 мг указанного в заглавии соединения.
Примеры фармкомпозиций
Пример А: Раствор для парэнтерального введения
Соединение по пр. 11 - 15 мг
Кремофор - 175 -"-
Абсолют. спирт - 0,4 -"-
Пример Б: Таблетки
Соединение по пр. 8 - 50 мг
Поперечно-сшитая натрий-карбоксиметилцеллюлоза - 12 -"-
Лактоза (сушка распылением) - 134 -"-
Микрокристаллическая целлюлоза - 50 мг
Двуокись кремния коллоидная - 1 -"-
Стеарат магния - 3 -"-.



Полусинтетический таксан формулы 1, где R1, R2 - C1-8-алкил или фенил, R2 может также представлять собой трет-бутоксигруппу; когда двойная связь присутствует в 11, 12 положениях, R3 - водород, R4 - гидроксил или С2-8-ацилоксигруппа; когда двойная связь отсутствует в 11, 12 положениях, метил в положении 12 имеет альфа-ориентацию, R3-гидроксильная группа или С2-8-ацилоксигруппа; R4 - атом водорода, и фармацевтическая композиция на его основе обладает противоопухолевой активностью. 7 с. и 5 з.п.ф-лы, 2 табл.

где R1 и R2, которые могут быть одинаковыми или разными, представляют собой C1-C8 алкил или фенил, R2 может также представлять собой трет-бутоксигруппу;
когда двойная связь присутствует в положениях 11, 12, R3 представляет собой атом водорода, R4 представляет собой гидроксильную группу или C2-C8 ацилоксигруппу;
когда двойная связь отсутствует в положениях 11, 12, метил в положении 12 имеет альфа-ориентацию, R3 представляет собой гидроксильную группу или C2-C8 ацилоксигруппу и R4 представляет собой атом водорода,
при условии, что когда R1 и R2 являются фенилами, а положения 11 и 12 связаны двойной связью, R4 не является ацетилом.
где R1 - фенил, R2 - трет-бутокси, R3 - H, R4 - OH.
где R1 - фенил, R2 - трет-бутокси, R3 - ацетоксигруппа и R4 - атом водорода.
где когда двойная олефиновая связь присутствует в положении 11, 12, R3 представляет собой атом водорода, R4 и R5 представляют собой гидроксильные группы, C2-C8 ацилокси-, триэтилсилилокси- или 2,2,2-трихлорэтоксикарбонилоксигруппы;
а когда двойная олефиновая связь отсутствует в положении 11, 12, метил в положении 12 является альфа-ориентированным, R4 представляет собой атом водорода, R3 и R5 - гидроксильные группы, C2-C8-ацилокси-, триэтилсилилокси- или 2,2,2-трихлорэтоксикарбонилоксигруппы,
подвергают этерификации в соответствии с известными способами подходящим образом активированным и/или защищенным производным изосерина, посредством чего в положение 13 вводят ацильную группу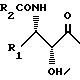
где R1 и R2 принимают значения, определенные в п.1,
с последующим удалением защитных групп известными способами.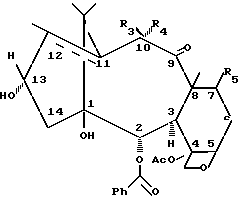
где когда двойная связь присутствует в положении 11, 12,
R3 представляет собой атом водорода,
R4 и R5 представляют собой гидроксильные группы, C2-C8 ацилокси-, триэтилсилилокси- или 2,2,2-трихлорэтоксикарбонилоксигруппы,
а когда двойная олефиновая связь отсутствует в положении 11, 12, метил в положении 12 является альфа-ориентированным, R4 представляет собой атом водорода, R3 и R5 представляют собой гидроксильные группы, C2-C8 ацилокси-, триэтилсилилокси- или 2,2,2-трихлорэтоксикарбонилоксигруппы,
в качестве промежуточного продукта.
в качестве промежуточного продукта.
где R1 и R2, которые могут быть одинаковыми или разными, представляют собой C1-C20 алкил или фенил;
R2 может представлять собой также трет-бутоксигруппу.
где R1 - фенил,
R2 - трет-бутоксигруппа.
подвергают этерификации общепринятыми способами, производными, подходящим образом активированными и/или защищенными в изосериновой цепи, посредством чего в положении 13 вводится ацильная группа
где R1 и R2 принимают значения, определенные в п.7,
с последующим удалением защитных групп обычными способами.
Приоритет по пунктам:
26.07.94 - по пп.1-12;
19.06.95 - по пп.1-12 разновидности радикалов.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| 0 |
|
SU400971A1 | |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Экономайзер | 0 |
|
SU94A1 |

