Образование гликозидных связей в растворе и твердой фазе. Настоящее изобретение относится к основном к способам, которые позволяют быстро конструировать олигосахариды и другие гликоконьюгаты. Более подробно, настоящее изобретение относится к способам для получения множества гликозидных связей в растворе в одну стадию. Настоящее изобретение обладает преимуществом в том, что было обнаружено, что относительная реакционная способность гликозидных остатков, содержащих аномерные сульфоксиды и нуклеофильные функциональные группы, может контролироваться. Другой аспект настоящего изобретения заключается в том, что реакционную способность аномерных сульфоксидов сахара используют в твердофазном способе для образования гликозидных связей. Раскрытые способы могут быть применены для получения конкретных олигосахаридов и других гликоконьюгатов, а также для получения гликозидных библиотек, содержащих смеси различных олигосахаридов, включая гликоконъюгаты, которые могут быть проверены на биологическую активность.
Олигосахаридные цепи гликопротеинов и гликолипидов играют важную роль в широком разнообразии биохимических процессов. Найдено, что как на поверхности клеток, так и циркулирующих в биологических жидкостях, эти гликозидные остатки выступают в качестве распознающих сигналов, которые служат связью ключевых событий в нормальном клеточном развитии и ее функции. Они включаются в оплодотворение, эмбриогенез, нейрональное развитие, гармональные активности, воспаление, клеточную пролиферацию и организацию различных типов клеток в конкретных тканях. Они также включаются во внутриклеточное классифицирование и секрецию гликопротеинов, а также в выведение плазмы гликопротеинов из циркуляции.
Дополнительно к их положительной роли в поддержании здоровья, олигосахариды также включаются в начало болезни. Например, олигосахариды на поверхности клетки выступают в качестве рецепторов вирусов и токсинов, а также более доброкачественных лигандов. Модифицированные карбогидраты поверхности клетки были вовлечены в онкогенезы и метастазы. Олигосахаридные структуры, которые связаны с промежуточным воспалением и помогают предотвратить инфекцию, могут, если образовались с избыточными уровнями, стимулировать развитие хронических воспалительных болезней. (Некоторые ссылки на роль олигосахаридов воспроизведенных зукариотами на состояние организма и болезни включают: Hakomori TIBS, 1984, 45; Feizi et., al. TIBS 1985, 24; Rademacher et.al. Annu. Rev. Biochem. 1988, 57, 785: Feizi TIBS, 1991, 84; Dennis and Laferte Cancer Res. 1985, 45, 6034; Fishman J.Membr.Biol.1982, 69,85; Markwell et. al. PNAS USA, 1981, 78, 5406; Wiley and Skehel J.Annu.Rev. Biochem., 1987, 56, 365; Kleinman et.al. PNAS USA, 1979, 76, 3367; Walz et.al. Scitnce 1990, 250).
Хотя бактерии не воспроизводят тот же тип олигосахаридов или других гликонуклеотидов как эукариоты, прикариоты, тем не менее воспроизводят широкое разнообразие гликозилированных молекул. Многие такие молекулы были выделены и было найдено, что они обладают противоопухолевой и антибиотиковой активностью. Бактериально воспроизведенные гликозилированные молекулы, обладающие потенциальной терапевтической пригодностью, включают хромацин, калихемицин, есперамицин и цикламицины. Во всех этих случаях карбогидратные цепи демонстрировали, что являются важными в биологической активности. Однако точные функции карбогидратных остатков до конца не понятны и являются непонятными связи структура-активность.
Из-за их разной функции в состоянии здоровья и болезни олигосахариды находятся в фокусе исследования. Широко принято, что развитие технологии 1) обнаружения и 2) блокирования или, иначе говоря, регулирования некоторых аномальных функций олигосахаридов будет приводить к значительному улучшению здоровья и благополучия.
Однако должно быть возможным эксплуатировать некоторые нормальные функции олигосахаридов (например, различные процессы узнавания) для других целей, включая высвобождение лекарства в данные типы клетки. Кроме того, возможна разработка новых противоопухолевых агентов из синтетических гликозилированных молекул, напоминающих гликолизированные бактериальные противоопухолевые агенты.
Продолжаются попытки разработки продуктов, имеющих отношение к олигосахаридам, включая диагностику котят, для обнаружения карбогидратов, связанных с различными заболеваниями вакцины для блокирования инфекции, вызванной вирусами, которые распознают карбогидраты поверхности клетки, носители для высвобождения лекарства, которые узнают карбогидратные рецепторы и моноклональные антитела, которые узнают аномальные карбогидраты, для использования в качестве лекарств. Своевременное развитие этих и других биомедицинских продуктов на основе карбогидрата, в свою очередь, зависит от доступности технологии получения олигосахаридов и других гликоконьюгатов быстро, эффективно и в практических количествах для основных и развиваемых исследований.
В частности, существует необходимость в способах, которые позволяют быстро получать гликозидные библиотеки, включающие смеси разливных олигосахаридов или других глюкоконъюгатов, которые могут быть затем проверены на конкретную биологическую активность. Было показано, например, что проверка смесей пептидов является эффективным путем идентификации активных соединений и объяснения связей структура-активность. Существует огромное количество путей генерирования химически разнообразных смесей пептидов и определения активных соединений. См. , например, Furka et.al., Int. J.Peptide Protein Res. 1992, 37, 487; Lam et. al. Nature 1991, 354, 82; Houghten Nature 1991, 254, 84; Zuckerman et.al., Proc. Natl.Acad. Sci. USA 1992, 89, 4505; Petithory Proc. Natl. Acad Sci. USA, 1984, 81, 3998; Houghten Proc.Natl. Acad. Sci. USA, 1985, 82, 5131; Fodor Science 1991, 251, 767. Мы не знаем эффективных способов генерирования различных смесей олигосахаридов и других гликоконьюгатов для тестирующих целей.
Антрациклины
Цикламицин 0 (формулы 1, приведенной ниже), антрациклиновый антибиотик выделенный из Streptomyces capoamus обладает высокой ингибирующей активностью in vitro против экспериментальных опухолей. Это лекарство содержит алгикон эпсилон-пирромицинона и трисахарид. См., Bieber et. al J.Antibiot. 1987, 40, 1335. Трисахарид содержит две повторяющиеся единицы 2-деокси-L-фукозы (A,B) и одну единицу кетосахара (C), L-цинерулозы. Все сахара связаны друг с другом через 1-4 аксиальную связь.
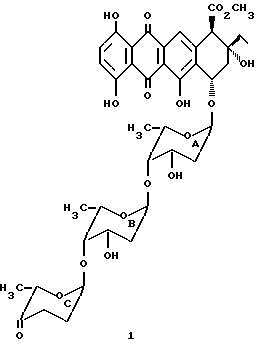
Хотя цикламин был открыт почти тридцать лет назад, мало известно о его функции, потому что недостаточные количества являлись доступными из натуральных источников. Следовательно, наилучшим путем получения цикламина в больших количествах и только пути получения его аналогов является химический синтез. Агликон цикламицина, епсилон-пирромицинон может быть получен дегликозилированием других легко доступных антибиотиков, таких как марцеломицин, музетамицин и цинерубин. В литературе существуют эффективные подходы для сочетания трисахарида с агликоном. См., например, Kolar et. al. Carbohydr. Res. 1990, 208,111. Однако способы конструирования трисахарида страдают от ограничений, связанных с общей легкостью и эффективностью.
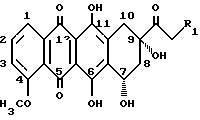
Антрациклиновые антибиотики выступают в качестве промежуточных соединений в метаболизме некоторых Streptomyces образцов. Они являются потенциальными химиотерапевтическими лекарствами, которые широко использовали при лечении различных твердых опухолей и лейкозов. См., Arcamone, F. Doxorubicin Anticancer Antibiotic; Academic Press: New York, 1981. Агликон всех антрациклинов состоит из трициклической хиноидной системы с функционализированным циклогексановым остатком. Различные примеры замещения, часто встречающиеся среди агликонов, приведены ниже.
Дауномиценон (R1 = H)
Адриамиценон (R1 = OH)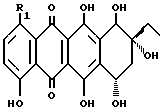
бета-Родомиценон (R1 = H)
1- OH-бета-Родомиценон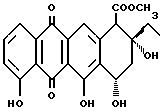
Аклавинон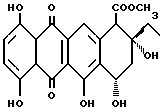
Эпсилон-пирромицинон
Общей чертой всех антрациклиновых антибиотиков является олигосахаридный остаток, присоединенный к C-7 гидроксильной группе агликона. Остаток сахара в этом положении может быть моно-, ди- или трисахаридом. Наиболее часто встречающиеся сахара включают даунозамин, родозамин, 2-деокси-L-фукозу и L-цинерулозу.
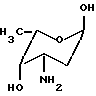
альфа-L-даунозамин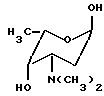
альфа-L-родозамин
альфа-L-деоксифукоза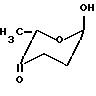
альфа-L-цинерулоза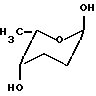
альфа- L-родиноза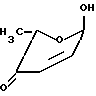
альфа-L-ацулоза
На основании некоторых исследований, проведенных на антрациклиновых антибиотиках дауномицина, адриамицина и аклациномицина, стало вполне очевидно, что олигосахаридные компоненты этих натуральных ДНК связок играют важную роль в ДНК связывании и узнавании. См., Bieber et.al. в печати. Однако мало известно о действительной функции сахаров, в частности потому, что возникают трудности при селективной модификации этих лекарств. Первый химический синтез цикламицина 0 был выполнен S.J.Danishefsky с соавторами. См. . Suzuki et.al., J.Am. Chem.Soc. 1990. 112, 8895.
Синтез 2-деоксиолигосахаридов
Комплексы гликоконьюгатов, подобных антрациклинам, и ауреолеиновых кислот представляют значительный научный и фармацевтический интерес и широко применялись в раковой химиотерапии. Общей структурной чертой в этих соединениях является присутствие 2-деоксиологисахаридов. Действительно, некоторые типы альфа- и бета-2-деокси-гликозидов часто находят в биологически активных молекулах натурального происхождения. Дополнительно к антибиотикам на основе ауреолеиновых кислот и антрациклиновым антибиотикам могут быть найдены кардиотонические гликозиды, авермектины, эритромицины и енидиновые антибиотики. Эффективное конструирование этих 2-деоксигликозидов, в частности 2-деокси-бета-гликозидов, являлось застарелой проблемой в химии карбогидратов. Регулирование бета-стереоселективности в 2-деоксисахарах является трудным, потому что не может быть стерео-направляющей анхимерной помощи от C-2 положения.
Вообще конкретный терапевтический эффект этих лекарств хотя и вызывают агликоном, в то же время сахара являются ответственнными за регулирование фармакокинетики. Надеются, что модификацией карбогидратного остатка возможно увеличить эффективность, а также уменьшить цитотоксичность этих лекарств.
Разработка аналогов сахара требует хороших синтетических способов для конструирования 2-деоксиолигосахаридов. К сожалению, способы гликозилирования, пригодные для синтеза 2-деоксиолигосахаридов являются в общем неудовлетворительными. Так как у 2-деоксигликозильных доноров отсутствует заместитель в C-2 положении, они являются нестабильными. Они быстро разлагаются в большинстве реакций гликозилирования, приводя тем самым к плохим выходам гликозидов. Фактически один из наилучших существующих способов конструирования 2-деоксиолигосахаридов - гликальный способ обходит эту проблему путем непосредственного использования неистинных 2-деоксигликозильных доноров. Этот способ, который является одним из наиболее широко используемых способов гликозилирования для конструирования 2-деоксигликозидов, включает двухстадийный процесс. В первой стадии, 1,2-ангидро сахар(гликаль) обрабатывают соответствующим электрофилом, E+, с образованием 1,2-ониевого промежуточного соединения. Руклеофильная атака с противоположной стороны дает гликозид с 1,2-транс конфигурированными связями. Во второй стадии заместитель у C-2 удаляют с образованием желаемого 2-деоксигликозида.
Способ получения олигосахаридов в растворе
В настоящее время существует два общих пути для получения олигосахаридов. Первый заключается в выделении из натуральных источников. Этот подход ограничивается олигосахаридами натурального происхождения, которые производят в больших количествах. Второй путь заключается в ферментативных или химических путях получения. Разнообразие олигосахаридов, доступных через ферментативный путь синтеза, ограничивается из-за того, что использованные ферменты могут принимать только определенные субстраты. Химические пути синтеза являются более гибкими, чем ферментативные пути синтеза, и обладают потенциалом для получения огромного разнообразия различных олигосахаридов. Проблема с химическими путями синтеза заключается в том, что они являются чрезвычайно дорогими что касается времени и усилий. Эта проблема является следствием того, что пути синтеза, по которым проводят химический синтез, олигосахаридов устарели.
Олигосахариды получают из моносахаридов, связанных гликозидными связями. При типичном химическом синтезе олигосахаридов полностью защищенный гликозильный донор активируют и позволяют взаимодействовать с гликозильным акцептором (обычно другим моносахаридом, содержащим незащищенную гидроксильную группу) в растворе. Реакция гликозилирования сама по себе протекает от нескольких минут до суток, в зависимости от использованного способа. Продукт сочетания затем очищают, химически модифицируют с превращением его в гликозильный донор. Химическая модификация может включать несколько стадий, каждая отдельная стадия требует последующей очистки. ("Одностадийным" определяют процесс химического превращения или ряда превращений, проведенных в "одном" реакционном сосуде без необходимости выделения промежуточного продукта или стадий очистки). Каждая очистка требует расхода времени и может приводить к значительным потерям вещества. Новый гликозильный донор, дисахарид, затем подвергают реакции сочетания с другим гликозильным акцептором. Затем выделяют и химически модифицируют как прежде. Не является необычным, что синтез трисахарида из компонентов моносахаридов требует десяти и более стадий. В одном из недавних примеров полностью защищенная трисахаридная боковая цепь противоракового антибиотика, названного цикламицином 0 была получена в 14 стадий с 9% выходом в расчете на компонент моносахаридов. См., Suzuki et.al. в печати. Таким образом, время и дороговизна, включенные в синтез олигосахаридов, были серьезным препятствием для разработки карбогидратных лекарств и других биохимических продуктов. Одним из путей повышения скорости и эффективности синтеза олигосахарида является разработка способов, которые позволяют конструировать множество гликозидных связей в одну стадию. До настоящего открытия заявители не знакомы с одностадийным способом, который включает региоселективное образование множества олигосахаридных связей и который обеспечивает быстрый, эффективный и с высоким выходом процесс для производства олигосахаридов.
Твердофазный синтез олигосахаридов
Кроме уменьшения числа стадий, включенных в синтез олигосахаридов, можно также увеличить скорость и эффективность процесса синтеза путем исключения необходимости выделения и очистки. Теоретически исключение необходимости выделения и очистки может быть достигнуто путем разработки твердофазного способа для синтеза олигосахаридов.
Благодаря важности потенциального преимущества твердофазного синтеза, ранее существовали попытки синтеза олигосахаридов в твердой фазе. Твердофазные способы синтеза исключали необходимость выделения и очистки, потому что избыток реагирующих веществ и продуктов разложения мог быть просто отмыт из продукта, связанного со смолой. Это преимущество преобразовывалось в огромное сохранение времени, усилий и выхода. (Преимущества твердофазных способов над способами в растворе для синтеза пептидов и нуклеиновых кислот были широко продемонстрированы. Эти преимущества конечно будут распространяться и на твердофазный синтез олигосахаридов. По твердофазному синтезу пептидов, см. , например, Barany G. and Merrifield, R.B. 1980, in The Peptides, Gross, E. and Meienhofer, J. Eds., Academic Press, New York, vol.2, pp.1-284).
Еще в 1971 году Frechet and Schuerch описали требования для твердофазного синтеза олигосахарида. См., Frecht and Schuerch J.Am.Chem.Soc.1971, 93, 492. Во-первых, смола должна быть совместима с условиями реакции. Во-вторых, твердая подложка должна содержать соответствующую функциональность для обеспечения связи с гликозидным центром (или в другом месте), связи, которая является инертной к условиям реакции, но может легко разрываться с удалением олигосахарида при окончании синтеза. В-третьих, схемы соответствующих защищающих групп должны работать таким образом, чтобы с данных гидроксильных групп могла быть селективно снята защита для следующей реакции сочетания. Другие гидроксильные группы должны быть защищены "перманентными" блокирующими группами, которые удаляют по окончании синтеза. В-четвертых, реакции гликолизирования должны быть эффективными, мягкими и протекать до конца во избежания неприятных последствий. В-пятых, стереохимия аномерных центров должна быть сохранена в процессе циклов сочетания и должна быть предсказуемой на основании результатов, полученных в растворе для любой данной пары донор/акцептор. В-шестых, разрыв перманентных блокирующих групп и связи с полимером должен сохранять олигосахарид неповрежденным.
К сожалению, хотя и было вообще принято, что твердофазный синтез олигосахарида является желаемой целью, и хотя Frechet and Schuerch (а также и другие) был способен описать стратегию твердофазного синтеза олигосахарида, ни один до настоящего открытия не был способен осуществить такую стратегию. В предыдущих попытках для синтеза олигосахаридов на нерастворимых смолах, выходы реакции сочетания были низкими и стереохимический контроль был неадекватен, в частности, для конструирования бета-гликозидных связей (т.е. 1,2-транс-гликозидных связей, в которых гликозидная связь в аномерном положении сахара является транс связью, несущей заместитель в C-2 положении сахара). Эти проблемы были приписаны тому факту, что кинетика реакции в твердой фазе медленнее, чем в она имеет место в растворе. См., Eby and Achuerch, Garbohydr. Res. , 1975, 39, 151. Последствие такой неблагоприятной кинетики состоит в том, что большинство реакций гликолизирования, которые приемлемо могут протекать в растворе, просто не протекают в твердой фазе, как в смысле стереохимического контроля, так и выхода. Таким образом, напрмиер, Frechet and Schuerch нашли, что две реакции гликолизирования, обе из которых включают замену аномерного галоида в присутствии катализатора, дают преимущественно бета-аномер (т. е. 1,2-транс продукт) в растворе, но дают смесь продуктов в твердой фазе. Frechet and Schuerch пришли к заключению, что для образования бета-гликозидных связей в твердой фазе будет необходимо использование участия соседней группы.
Вновь, однако, было найдено, что соседние участвующие группы (СУГр) часто дезактивируют гликозильные доноры до такой степени, что существующие способы гликозилирования не могут быть приняты для твердой фазы. См., Eby and Schuerch, в печати. В некоторых примерах смола также подвергалась разложению вследствие жестких условий, требуемых для гликозилирования. Кроме того, для многих СУГр эфирного типа, существует большая проблема, связанная с передачей ацила от гликозильных доноров к свободным гликозильным акцепторам на смоле. Эта побочная реакция приводит к тому, что закрывает смолу и предотвращает дальнейшую реакцию.
Frechet описывал проблемы, встречающиеся при попытке осуществления стратегии твердофазного синтеза олигосахаридов. См., Frechet, Polymer-supported Reactios in Organic Synthesis, p. 407, P.Hodge and D.C.Sherrington, Eds. , John Wiley and Sons, 1980. Он пришел к заключению, что твердофазный синтез олигосахаридов является пока еще неконкурентноспособным с синтезом в растворе "в основном, из-за отсутствия подходящих реакций гликозилирования",
Были предприняты некоторые усилия по преодолению неблагоприятной кинетики реакции, связанной с твердофазными реакциями путем использования растворимых смол. В наилучшем примере по данным Douglas et.al. использовал растворимую полиэтиленгликолевую смолу с янтарной кислотой в качестве линкера и достиг 85-95% выходов реакции сочетания, используя способ гликозилирования, известный в течение 80 лет (реакция Koenigs-Knorr) с превосходным регулированием агломерной стереохимии. См., Douglas et.al., J.Am. Chem. Soc. 1991, 113, 5095. Растворимые смолы могут обладать преимуществом для некоторых реакций гликозилирования, потому что они дают более "растворимо-подобное" окружение. Однако постадийный синтез на растворимых полимерах требует, чтобы промежуточное соединение было высажено после каждой стадии и подвергнуто кристаллизации до того как другой остаток сахара может быть подвергнут реакции сочетания. Однако некоторое добавление того же гликозилирующего агента обычно требуется для того, чтобы сдвинуть реакцию к завершению. В приведенном выше случае, например, Douglas et.al. повторял одну и ту же реакцию сочетания пять раз для достижения высокого выхода. Каждое повторение требует стадии осаждения с удалением реагирующих веществ путем промывания. Продукт может быть потерян в каждой стадии осаждения. Кроме того, повторение переосаждений делает процесс очень неэкономичным по времени. Таким образом, подход к синтезу олигосахарида с растворимой смолой не в состоянии обеспечить все потенциальные преимущества, связанные с твердофазным синтезом, использующим нерастворимые смолы.
Новый способ гликозилирования, включающий аномерные сульфоксиды сахара сообщался Kahne с соавторами. См., Kahne et.al. Chem. Soc. 1989, 111, 6881. Аномерные сульфоксиды сахара были активированы эквимолярными количествами ангидрида трифторметансульфоновой кислоты (трифлик ангидрида) в присутствии затрудненного основания. Гликозильный донор, активированный трифликовым ангидиридом, вполне обеспечивает реакционную способность в растворе и может быть использован для гликозилирования чрезвычайно нереакционноспособных субстратов в мягких условиях. Однако это сообщение ограничивалось реакциями в растворе и не было подтверждено, что твердофазные реакции могут быть проведены с любой приемлемой степенью пригодности.
Таким образом, уровень развития техники подчеркивает преобладающую и неосуществленную необходимость в способе гликозилирования, который обеспечивает быстрый, эффективный и с высоким выходом синтез олигосахаридов. Однако эффективный синтез олигосахаридов в твердой фазе, который обеспечивал все ранее упомянутые преимущества твердофазных способов и не был продемонстрирован.
Настоящее изобретение обеспечивает способы конструирования множеств гликозидных связей в растворе, используя аномерные сульфоксиды сахара в качестве гликозильных доноров, и конструирования последовательных гликозидных связей в твердой фазе, с регулированием стерехимической конфигурации аномерной связи. Таким образом, в зависимости от выбранных условий и исходных веществ могут быть получены альфа- и бета-аномеры в твердой фазе, используя аномерные сульфоксиды сахара в качестве гликозильных доноров.
Способы настоящего изобретения могут быть применены для получения определенных олигосахаридов или гликоконъюгатов или для получения смеси различных олигосахаридов или гликоконъюгатов для создания гликозидных библиотек, которые могут быть впоследствии испытаны для обнаружения соединений, обладающих желаемой биологической активностью.
Настоящее изобретение также относится к открытию того, что активация аномерных сульфоксидов каталитическими количествами активирующего агента обеспечивает очень хорошие выходы продукта конденсации в очень мягких условиях. Предпочтительно активирующий агент представляет сильную органическую кислоту, такую как трифторметансульфоновая кислота или "трифликовая" кислота (TfOH), п-толуолсульфоновая кислота (TsOH) или метансульфоновая кислота (MsOH), более предпочтительно TfOH. В частности, было найдено, что для конструирования 2-деоксигликозидов процедура каталитического гликозилирования, описанная здесь, рассматривает выбор способа.
Предпочтительный вариант этого аспекта изобретения, включающий синтез 2-деоксигликозидов через гликозилирование, катализированное трифликовой кислотой, описывают более детально ниже.
Другие цели настоящего изобретения будут очевидны специалисту при рассмотрении настоящего описания.
Фиг. 1 иллюстрирует способ синтезируемого в одну стадию защищенного трисахарида цикламицина 0 из компонентов моносахаридов.
Фиг. 2 иллюстрирует способ получения гомополимеров 2-деоксифукозы в одну стадию.
Фиг. 3 иллюстрирует способ синтезируемой смеси гликоконъюгатов, обладающих биологической активностью, включающий потенциальную связывающую активность ДНК. Гликонконъюгаты, полученные таким образом, могут быть впоследствии испытаны (например, на ДНК связующую активность) для оценки предпочтительной длины и предпочтительных остатков сахара олигосахаридной части гликоконъюгата на основании испытанной активности.
Фиг. 4 иллюстрирует способы образования и удаления двух примерных типов связей с твердой подложки (например, плистирольной смоле).
Фиг. 5 иллюстрирует аппараты использования для проведения твердофазного синтеза олигосахарида.
Фиг. 6 иллюстрирует общую схему синтеза бета-связанного дисахарида в твердой фазе.
Фиг. 7 иллюстрирует общую схему синтеза альфа-связанного дисахарида в твердой фазе.
Фиг. 8 иллюстрирует общую схему синтеза трисахарида в твердой фазе.
Фиг. 9 представляет 1H-ЯМР спектр моносахарида 1 фиг. 1.
Фиг. 10 представляет 1H-ЯМР спектр моносахарида 2 фиг. 1.
Фиг. 11 представляет 1H-ЯМР спектр моносахарида 3 фиг. 1.
Фиг. 12 представляет 1H-ЯМР спектр трисахарида 5 фиг. 1.
Фиг. 13 представляет расширенную область 1H-ЯМР спектра трисахарида 5, в котором отмечают аномерные протоны трисахарида.
Фиг. 14 представляет 1H-ЯМР спектр дисахарида 4 фиг. 1.
Фиг. 15 представляет схему синтеза цикламицина 0.
Фиг. 16 представляет схему синтеза трисахарида.
Фиг. 17 представляет схему синтеза выбранных дисахаридов.
За этим следует детальное описание предпочтительных вариантов настоящего изобретения.
Определения
Активирующий агент: Химический агент, который добавляют к гликозильному сульфоксиду, взаимодействует с аномерной сульфоксидной группой, превращая таким образом аномерный углерод в способный к нуклеофильной атаке. В случае бифункциональных сахаров или гликозидных остатков активирующий агент способен также к снятию защиты блокированной нуклеофильной группы в тех же условиях, использованных для активации аномерной сульфоксидной группы.
Акцептор кислоты: Такой химический агент как любое основание, которое принимает протоны, уменьшая тем самым побочные реакции, которые ускоряются в кислых условиях.
Акцептор сульфеновой кислоты: Химический агент, такой как метилпропионат, который конкретно принимает сульфеновую кислоту, обычно приводя к образованию нереакционноспособного монофенилсульфоксида. В отсутствие акцептора сульфеновой кислоты, сульфеновая кислота взаимодействует сама с собой с образованием дифенилдисульфидного моносульфоксида и воды. Вода мешает реакции гликозилирования.
Бифункциональный: Характеристика сахара или гликозидного остатка, который способен активировать как гликозильный донор, так и гликозильный акцептор в условиях одностадийного процесса настоящего изобретения.
Биологическая активность: Любая активность, проявляемая соединением или молекулой, которые обладают потенциальными физиологическими, фармакологичекими, диагностическими или терапевтическими применениям.
Карбогидратный рецептор: Любая молекула, которая связывает любой карбогидрат. Обычно молекула представляет такую молекулу как протеин или ДНК.
Гликоконъюгат: Любое соединение или молекула, которые ковалентно связываются с гликозидным остатком.
Гликозиды: Любой сахар, содержащий по крайней мере один остаток пентозы или гексозы, в котором аномерный углерод несет неводородный заместитель. Обычно, неводородный заместитель представляет гетероатом, такой как азот, кислород, фосфор, кремний или серу.
Гликозильный акцептор: Любое соединение, которое содержит по крайней мере одну нуклеофильную группу, которая в условиях одностадийного способа настоящего изобретения оказывается способной образовать ковалентную связь с аномерным углеродом гликозильного донора. Как отмечают здесь, гликозильный акцептор представляет любой сахар или гликоконъюгат, который содержит незащищенную гидроксильную, амино или меркапто группы или такие группы, которые могут быть удалены in situ, т.е. в условиях одностадийного способа настоящего изобретения.
Гликозильный донор: Сахар или гликозидный остаток, который несет сульфоксидную группу у аномерного углерода, группу, которая при активировании превращает аномерный углерод в способный к атаке нуклеофильной группой гликозильного акцептора с образованием гликозидной связи.
Гликозидные библиотеки: Смеси олигосахаридов различающихся последовательностей, которые могут быть подвергнуты процедуре испытаний для идентификации соединений или молекул, которые обнаруживают биологическую активность. Такие библиотеки могут также включать различные гликоконъюгаты.
Монофункциональный гликозильный акцептор: Гликозильный акцептор как он определен выше, с дополнительным условием, что способность выступать в качестве гликозильного донора в одно и то же время (т.е. в условиях одностадийного процесса настоящего изобретения) полностью исключается.
Монофункциональный гликозильный донор: Гликозильный донор, как он определен выше, с дополнительным условием, что способность выступать в качестве гликозильного акцептора в то же самое время (т.е. в условиях одностадийного процесса настоящего изобретения) полностью исключается.
Монофункциональная гликозильная единица: Сахар, который представляет либо гликозильный акцептор, либо гликозильный донор, но не обладает способностью выступать в обоих качествах при активации в условиях одностадийного процесса настоящего изобретения.
Олигосахариды: Гликозильный остаток, содержащий три или более моносахаридные единицы, соединенные гликозидными связями.
Потенциальный гликозильный акцептор: Любое соединение, содержащее по крайней мере одну нуклеофильную группу, которая потенциально способна к образованию ковалентной связи с аномерным углеродом гликозильного донора.
Одностадийная реакция: Одностадийную реакцию определяют как химическое превращение или набор превращений, проведенных в "одном" реакционном сосуде без необходимости выделения промежуточных соединений или стадий очистки (т. е. одностадийная или однореактивная реакция).
Временная защищающая группа: Блокирующая или защищающая группа, которая может быть удалена in situ, предпочтительно, но не обязательно, при тех же условиях, использованных для активации аномерной сульфоксидной группы.
Общие способы.
Следующие общие способы были разделены на две основных категории: первая касается реакций в растворе, включающих образование множества гликозидных связей, и вторая относится к синтезам олигосахаридов, в которых растущий олигомер связывают с твердой подложкой.
Образование множества гликозидных связей в растворе.
Один или более гликозильных доноров, содержащих алкильные или арильные сульфоксиды в аномерном положении и один или более гликозильных акцепторов, содержащих одну или более свободных гидроксильных групп и/или других нуклеофильных групп (например, амины) и/или гидроксильные группы, защищенные простым силильным эфиром, соединяют в реакционном сосуде. Полученная смесь может включать как монофункциональные гликозильные доноры и гликозильные акцепторы, так и бифункциональные гликозильные звенья, т.е. сахариды, которые могут одновременно выступать и как гликозильные доноры и как гликозильные акцепторы. Однако для того, чтобы получить больше чем одну гликозильную связь (т.е. для получения трисахарида или более длинного продукта), по крайней мере одно из реагирующих веществ должно быть бифункциональным гликозильным звеном.
Гликозильные акцепторы и доноры могут быть блокированы соответствующей защищающей группой, включающей, но не ограничивающейся ею, простой эфир, сложный эфир, ацетамидо, или тиоэфир защищающие группы, в одном или более положениях. Однако понятно, что сложноэфирная (или ацетамидо или тиоэфирная) защищающая группа в C-2 положении гликозильного донора будет влиять на стереохимический выход гликозилирования, давая 1,2-транс гликозидную связь.
Смесь гликозильных доноров и акцепторов растворяют в безводных условиях в ненуклиофильном растворителе, включающем, но не ограничивающимся им, толуол, эфир, тетрагидрофуран (ТГФ), метиленхлорид, хлороформ, пропионитрил или их смеси. Было найдено, что выбор растворителя влияет на стереохимический выход реакции гликозилирования, в которой не включается соседняя участвующая группа. В общем для данной пары донор/акцептор используют неполярный растворитель, такой как толуол, приводя к образованию с высоким выходом альфа изомера, в то время как использование более полярного растворителя, такого как пропионитрил, приводит к образованию с более высоким выходом бета аномера.
Реакцию инициируют добавлением эффективного количества активирующего агента. В данном варианте настоящего изобретения к реакционной смеси добавляют 0,5 эквивалента трифликового ангидрида, плюс 1,5 эквивалента основания (в качестве акцептора кислоты).
(Эквиваленты представляются относительно гликозильного сульфоксида). Каталитическое количество трифликовой кислоты (например, меньше 0,05 эквивалента) также может быть использовано предпочтительно вместе с избытком акцептора сульфоновой кислоты (например, около 20 эквивалентов метилпропиолата). Было найдено, что каталитическая трифликовая кислота является предпочтительной, если реакционная смесь содержит 2-деоксигликозильные доноры или если один из гликозильных акцепторов в реакции представляет силильный эфир. С другой стороны, трифликовый ангидрид является предпочтительным, если важным является максимальная реакционная способность гликозильных доноров. Однако необходимо отметить, что умеренно основные условия, которые получают с использованием трифликового ангидрида являются неэффективными для снятия защиты некоторых силильных эфиров (например, трет-бутилсилильных эфиров).
Однако хотя использование трифликового ангидрида плюс 2,6-дитретбутил-4-метилпиридин будет приводить к снятию защиты in situ триметилсилильных эфиров, использование трифликового ангидрида плюс более сильное основание (такое как основание Хаггиса) не будет приводить к снятию защиты. Таким образом, оба активирующих агента могут быть использованы в реакциях, включающих бифункциональное гликозильное звено, содержащее гидроксильную группу, защищенную силильным эфиром, хотя трифликовый ангидрид работает только при определенном наборе условий (выбор основания, выбор силильной защищающей группы). Иначе говоря оба способа активирования являются обычно взаимозаменяемыми.
Метилпропиолат или другой акцептор сульфеновой кислоты и/или активированные молекулярные сита могут быть добавлены к реакции либо до либо после добавления активирующего агента. Акцепторы сульфеновой кислоты значительно улучшают выход реакции гликозилирования, если в качестве активирующего агента используют каталитическую трифликовую кислоту.
Реакцию обычно проводят при низкой температуре (предпочтительно в области около -78oC до такой низкой, как -100oC), но реакция может протекать и при более высоких температурах, в некоторых случаях таких высоких, как при комнатной температуре.
Реакцию гасят добавлением водного бикарбоната и экстрагируют. Затем реакционную смесь подвергают процедуре очистки и/или снятию защиты с продукта если необходимо. Процедура может быть использована для конструирования определенных олигосахаридов или смесей различных олигосахаридов или других гликоконъюгатов для испытаний на биологическую активность.
В конкретных вариантах настоящего изобретения было обнаружено, что реакционная способность различных гликозильных доноров может быть изменена регулированием химической структуры и электронной природы аномерного сульфоксида. Такое регулирование, в частности, обусловлено тем, что стадией, лимитирующей скорость реакции гликозилирования, является активирование сульфоксида действием активирующего агента. Впоследствии было показано, что на реакционную способность гликозильных сульфоксидов можно влиять путем регулирования нуклеофильности сульфоксидного кислорода.
В общем более нуклеофильный сульфоксидный кислород больше ускоряет реакцию гликозилирования. Таким образом, электроно-донорные заместители в R' группе, активированные сульфоксидом, увеличивают нуклеофильность сульфоксидного кислорода и увеличивают скорость реакции. Напротив, электроно-акцепторные группы снижают нуклеофильность сульфоксидного кислорода и замедляют реакцию. Например, пербензилированный глюкозил п-метоксифенил сульфоксид взаимодействует быстрее, чем соответствующий незамещенный фенилсульфоксид, в то время как пербензилированный гликозил п-нитрофенилсульфоксид взаимодействует медленнее, чем соответствующий незамещенный фенилсульфоксид.
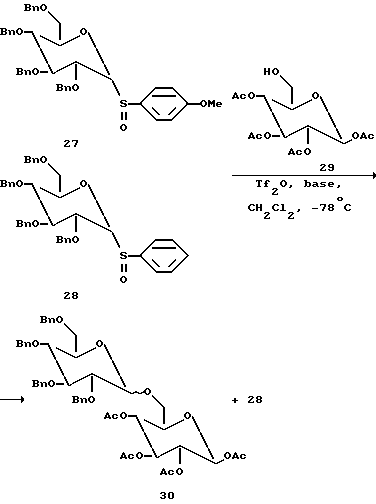
Способность нуклеофильности различных сульфоксидов оказывать влияние на реакцию и, следовательно, на управление реакционной способностью различных гликозильных доноров было использовано в данных вариантах настоящего изобретения. Например, эта способность позволяет проводить последовательные реакции гликозилирования растворе, как показано на фиг. 1. В других вариантах настоящего изобретения, множество гликозидных связей получают в растворе, используя силилированные гликозильные акцепторы. Силильные эфиры являются превосходными гликозильными акцепторами, если используют в качестве активируемого агента каталитическую трифликовую кислоты и триметилсилильные эфиры выступают как гликозильные акцепторы, если в качестве активирующего агента используют трифликовый ангидрид и 2,6-дитрет-бутил-4-метилпиридин используют как основание. Однако они должны быть незащищенными для проведения реакции сочетания.
(Следовательно, если илильные эфиры используют в качестве гликозильных акцепторов, то для реакции гликозилирования требуют слегка кислых условий). Поэтому силильные эфиры должны быть незащищенными для того, чтобы проводить сочетание, они реагируют более медленно, чем незащищенные спирты. Таким образом, было продемонстрировано, что можно изменять реакционную способность двух, иначе подобных, гликозильных акцепторов путем селективного использования силильных защищающих групп.
В выбранных вариантах настоящего изобретения на распределение длины олигосахаридов или гликозидных остатков полученных гликоконъюгатов можно влиять путем изменения отношения монофункциональных гликозильных акцепторов и монофункциональных гликозильных доноров к бифункциональным гликозильным звеньям в реакционной смеси. Например, было показано, что повышение отношений монофункциональных гликозильных акцепторов к бифункциональным гликозильным звеньям в реакционной смеси приводит к более короткой длине полимеров. Общая концентрация реагирующих веществ также влияет на длину распределения (см. Section 6.6 and 6.8 фиг. 2 и 3).
В конкретных вариантах настоящего изобретения может быть желательным включение только двух или трех различных типов сахаров в реакционную смесь и управление реакционной способностью гликозильных доноров и акцепторов таким образом, чтобы получить конкретные олигосахариды. Примеры этой процедуры даны в разделах 6.1. и 6.6 ниже.
Еще в других вариантах настоящего изобретения может быть желательным включение нескольких различных типов сахаров в реакционную смесь для того, чтобы генерировать химически разнообразные смеси олигосахаридов или продуктов гликоконъюгатов для создания библиотек, которые могут быть испытаны на биологическую активность. В качестве примера такого способа иллюстрируют в разделе 6.6 и фиг. 3.
На химическое разнообразие можно влиять путем изменения количества различных сахаров, включенных в смесь. Химическое разнообразие будет также функцией порядка, в котором взаимодействуют различные гликозильные пары донор/акцептор. Порядок, в котором взаимодействуют различные пары, будет зависеть, в свою очередь, от относительной реакционной способности различных пар донор/акцептор. Относительная реакционная способность различных пар донор/акцептор может управляться различными путями, как уже описано выше (например, путем управления структурой использованных сульфоксидных групп) и путем защиты некоторых гликозильных акцепторов силильными эфирами для снижения скорости, при которой они реагируют.
Другие факторы, которые влияют на относительную реакционную способность гликозильных доноров и акцепторов, такие как присутствие электроотрицательных защищающих групп на кольцах сахара, или может быть использовано также наличие стерических затруднений. См., например, Binkley Modern Carbohydrate Chemistry, Mercel Dekker, Inc., New York 1988; а также Paulsen Angew. Chem. Int. Ed. Engl. 1982, 22, 156. Следовательно, может быть принято во внимание потенциально много факторов в осуществлении раскрытого способа образования множества гликозидных связей с получением химически разнообразных смесей. Каталитическая активация аномерных сульфоксидов.
В другом варианте этого открытия была описана активация трифликовым ангидридом аномерных сульфоксидов. Трифликовый ангидрид взаимодействует с сульфоксидом с образованием трифлоксисульфониевой соли, которая является чрезвычайно реакционноспособной.
В присутствии основания, половины эквивалента трифликового ангидрида было достаточно для активирования полного эквивалента сульфоксида. Фенилтрифторметансульфенат (PhSOTf), генерированный в процессе протекания очевидно активирован остающимися 0.5 эквивалентами сульфоксида сахара (см. ниже).
Действительно были использованы другие эфиры сульфоната для активации тиогликозидов. Например, Ogawa et. al. фенилселенийтрифлат для активирования фенильных и алкильных тиогликозидов. См. Ito and Ogawa Tetrahedron Lett. 1987, 28, 2723.
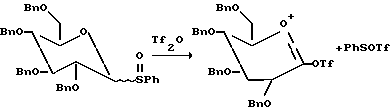
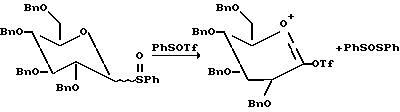
Однако в отсутствие основания меньше чем 0.05 эквивалентов трифликового ангидрида активирует полный эквивалент сульфоксида. Так как трифлоксифенилсульфонат (PhSOTf), соединенный с трифликовым ангидридом не дает количества больше чем 0.1 эквивалент, то активирующими сульфоксид в каталитическом цикле были некоторые другие соединения, генерированные в реакции. Было целесообразным считать, что предполагаемым катализатором была трифликовая кислота (TfOH). TfOH была использована другими авторами для активирования других гликозильных доноров. См., например, более недавнее сообщение Lonn Glycocjngugate J. 1987, 4, 117;l Mootoo et. al. J. Am. Chem. Soc. 1989, 111, 8540; Evans et. al, J. Am. Chem. Soc., 1990, 112, 7001; and Veeneman et. al., Tetraedron Lett. 1990, 31, 1331. Кроме того, было обнаружено, что только каталитические количества обычно требуются потому что кислота генерируется в реакции.
Для определения того, может ли TfOH активировать аномерные сульфоксиды были проведены следующие эксперименты с использованием пербензилированного сульфоксида глюкозы 1 в качестве гликозильного донора и С-6 первичный спирт 2a в качестве гликозильного акцептора (см. схему ниже).
Сульфоксид 1 (1.5 эквивалента) обрабатывали трифликовой кислотой (0.05 эквивалента) при -78oC в метиленхлориде. Эту стадию проводили с последующим добавлением нуклеофила (1.0 эквивалента) к реакционной смеси. Весь сульфоксид расходуется с образованием продукта, указывая на то, что трифликовая кислота в каталитических количествах активирует аномерные сульфоксиды.
Механизм
Хотя не хотелось бы ограничиваться теорией, предлагается следующая механическая интерпретация в помощь интересующегося читателя. Стехиометрический выход гликозилирования с использованием способа с каталитической трифликовой кислотой был идентичен тому, что было получено согласно способу, со стехиометрическим количеством трифликового ангидрида.
Предполагают, что обе реакции протекают через одни и те реакционноспособные промежуточные соединения, т.е. оксониевый ион или тесную ионную пару.
Однако в способе с TfOH выход желаемого дисахарида 3 был низким. В качестве побочных продуктов образовывалось значительное количество лактола (4) и 1,1-димера (5) (см. ниже). Природа этих побочных продуктов указывает по мнению заявителя на то, что в реакции присутствует вода. В частности, если оксониевый ион захватывается водой, будет образовываться лактол. Если аномерный лактол затем захватывает другой оксониевый ион, будет образовываться 1,1-димер гликозильного донора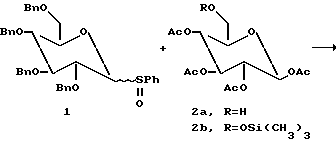
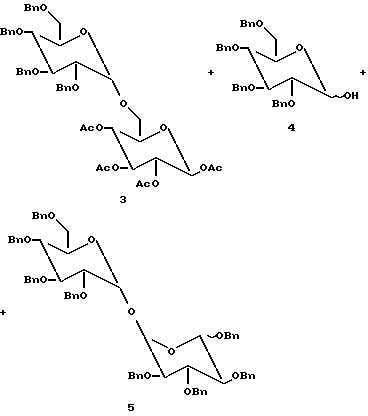
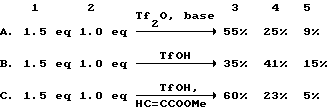
R = H with Tf2O;
R = Si(CH3)3 with TfOH
Для предотвращения образования воды, гликозилирование проводят в тщательно безводных условиях, используя активированные молекулярные сита. Несмотря на эти меры предосторожности, однако, наблюдали образование побочных продуктов, представляющих 40% массового баланса. Это последнее наблюдение подтверждают, что вода так или иначе образуется в процессе реакции, возможно при диизпропионировании фенилсульфоновой кислоты.
Как проиллюстрировано на схеме, приведенной в конце описания, первая стадия каталитического цикла заключается в пропионировании сульфоксида с образованием сульфониевой соли. Сульфониевая соль затем выделяет фенилсульфениевую кислоту (PhSOH) с образованием оксониевого иона и устойчивой ионной пары. Нуклеофильные ловушки оксониевого иона с последующим генерированием ThOH. В каждом каталитическом цикле одна молекула сульфоксида образует продукт и генерирует одну молекулу сульфеновой кислоты в качестве побочного продукта.
Сульфеновые кислоты являются классическими серуорганическими соединениями, которые не переносят выделения из-за их неустойчивости. Они обладают высокой реакционной способностью и как электрофилы и как нуклеофилы. Сульфеновые кислоты легко подвергаются диспропорционированию с тиосульфинатными эфирами и водой. Постулированный механизм диспропорционирования, включающий их двойственный электрофильный/нуклеофильный характер иллюстрируют ниже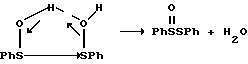
Добавление акцепторов сульфеновых кислот.
Сульфеновые кислоты легко присоединяются к электронодифицитным алкенам и алкинам с образованием винилсульфоксидов. Таким образом, становится возможным захватить эти соединения акцептором сульфеновой кислоты до их самоконденсации. Примеры алкенов и алкинов, часто используемых для улавливания сульфеновых кислот включает метилпропионат, метилпропиолат, стирол и диметилдикарбоксилат. Вышеуказанные соединения были испытаны в качестве потенциальных акцепторов и было найдено, что метилпропиолат являлся наиболее эффективным.
В типичной реакции 1 и 2 было позволено взаимодействие с TfOH в присутствии метилпропиолата (20 эквивалентов). Выход реакции улучшился с 35% (в отсутствие етилпропиолата) до 45% (в присутствии метилпропиолата). Хотя выход реакции улучшился, еще значительные количества 4 и 5 образовывались. Следовательно, были рассмотрены дальнейшие пути предотвращения диспропирционирования сульфеновой кислоты.
Использование силиловых эфиров в качестве нуклеофилов.
Ясно, что использование силиловых эфиров защищенных спиртов в качестве нуклеофилов может далее уменьшить образование воды. Силиловые эфиры могут взаимодействовать в мягких условиях реакции с образованием желаемого продукта конденсации дисахарида, TMSOTf и фенилсульфеновой кислоты. Затем ожидается, что сульфеновая кислота будет взаимодействовать с TMSOTf с образованием силилфенилсульфоната (PhSOSi(CH3)3), регенерируя тем самым трифликовую кислоту (см. ниже). Ясно, что так как силилированные сульфенаты являются более устойчивыми, чем сульфеновые кислоты, и можно ожидать, что не будут диспропорционировать так легко как силилированные сульфенаты, что может помочь снизить процесс образования воды. См., Nakamura J. Am. Chem. Soc. 1983, 105, 7172.
PhSOH + TMSOTf ---> PhSOSi(CH3)3 + TfOH
Таким образом, пербензилированный сульфоксид глюкозы 1 (1.5 эквивалента) был обработан трифликовой кислотой (0.05 эквивалента) в метиленхлориде при -78oC. В реакцию был добавлен силиловый эфир нуклеофила (2b). После взаимодействия желаемый трисахарид 3 был выделен в виде основного продукта с выходом 60%. (См. первую схему в разделе 5.4.1). Таким образом, используя силиловый эфир нуклеофила, выход реакции драматически улучшается, т.е. от 356% до 60%.
Применение каталитического способа для синтеза 2-деоксиолигосахаридов.
В последующих исследованиях был изучен способ каталитической трифликовой кислоты для активирования сульфоксидов. Таблица I показывает сравнение способов с каталитической трифликовой кислотой и стехиометрическим количеством трифликового ангидрида в реакции гликозилирования, используя ряд 2-деоксигликозильных сульфоксидов в качестве гликозильных доноров.
Установлено, что 2-деоксигликозильные сульфоксиды являются неустойчивыми и имеют тенденцию к образованию продукта сочетания с низким выходом (см. таблицу I). Способ со стехиометрическим количеством трифликового ангидрида для активирования сульфоксидов не всегда дает хорошие результаты с 2-деоксигликозильными донорами.
Однако способ с каталитической трифликовой кислотой работает очень хорошо, преимущественно потому, что мягкие условия реакции снижают разложение 2-деоксисульфоксидов. Фактически использование трифликовой кислоты улучшает выход гликозилирования по крайней мере на 50% для всех изученных случаев.
aR= H с трифликовым ангидридом R=OSi(CH3)3 с трифликовой кислотой. Как видно из результатов, приведенных в таблице I, выходы, полученные в каталитическом способе гликоилирования, являются сравнимыми с наилучшими выходами, сообщавшимися в литературе для различных гликальных способов. Кроме того, каталитическая TfOH может быть использована для активирования сульфоксидов даже в присутствии кислых чувствительных функциональных групп (таблица I, запись 2).
Аспекты способов каталитической TfOH и стехиометрического Tf2O для активирования сульфоксидов.
Два способа гликозилирования дополняют друг друга. Каталитический способ гликозилирования является преимущественным, если сульфокси неустойчив.
Условия реакции являются мягкими, поэтому разложение гликозильных доноров является минимальным. Кроме того, побочные реакции такие как трифляция (образование трифликовой кислоты) или сульфенирование нуклеофила, которые могут приводить к снижению выходов гликозилирования, не протекают в способе с каталитической TfOH. Однако способ с каталитической трифликовой кислотой для активирования сульфоксидов является значительно более медленным и требует слегка повышенных температур (-78oC до -30oC) по сравнению со способом с трифликовым ангидридом. Кроме того, каталитический способ является неэффективным, если на гликозильном доноре присутствуют электронооттягивающие защищающие группы. Наилучший способ гликозилирования может быть пригоден, если используют участие соседней группы с получением стереоселективности.
Важно отметить, что ни трифликовая кислота ни трифликовый ангидрид не активируют аномерные фенилсульфоксиды в использованных условиях реакции (таблица I, запись 5). Так как аномерные сульфиды могут быть легко превращены в сульфоксиды в чрезвычайно мягких условиях (mCPBA, CH2Cl2, -78oC до 0oC), оба способа дают сами по себе легко повторяемую стратегию для синтеза олигосахарида.
Таким образом, было показано, что аномерные сульфоксиды могут быть легко активированы для реакции гликозилирования каталитическим количеством сильной органической протонной кислоты, такой как трифликовая кислота. Реакция гликозилирования протекает в очень мягких условиях и дает следующие преимущества: i) разложение сульфоксидов является минимальным в условиях реакции; и ii) исключение проблем трифляции и сульфенирования нуклеофила. Эти преимущества являются значительными, особенно в контексте твердофазного синтеза олигосахарида, где сульфенирование или трифляция нуклеофила могут приводить к капсулированию растущей олигосахаридной цепи в смоле и, следовательно, прекращению синтеза.
Этот каталитический способ дополняет способ с трифликовым ангидридом и является особенно полезным для конструирования 2-деоксиолигосахаридов. Фактически способ с каталитической трифликовой кислотой был применен в эффективном конструировании 2-деокситрисахарида цикламицина 0, как описано в другой части настоящего изобретения. Наконец, мы продемонстрировали, что ни трифликовый ангидрид, ни трифликовая кислота не активируют аномерные фенилсульфоксиды в использованных условиях реакции; поэтому оба способа легко дают сами по себе дополнительную стратегию синтеза олигосахарида. Применение одностадийного способа гликозилирования для синтеза антрацилинового антибиотика.
Мы нашли, что порядок реакционной способности различных аномерных фенилсульфоксидов может быть контролирован путем изменения заместителей в пара положении фенильного кольца.
В результате мы добились успеха в синтезе трисахарида цикламицина 0, который был синтезирован стереоселективно с 25% выходом из моносахаридных компонентов в одну стадию. Синтетический подход представлен на фиг. 15. Спокойный ход синтеза включает применение способа гликозилирования с каталитической трифликовой кислотой для конструирования стереоселективно всех 2-деоксигликозидных связей. Кроме того, трисахарид несет фенилсульфоксид в аномерном центре кольца А. Аномерный фенилсульфоксид является устойчивым ("обезоруженным") в условиях, которые активируют аномерные сульфоксиды для гликозилирования. Они могут быть легко окислены в мягких условиях. Таким образом, сульфоксидный способ гликозилирования дает сам по себе дополнительную стратегию для синтеза олигосахарида. Сульфид в кольце A трисахарида цикламицина был окислен до соответствующего сульфоксида mCPBA и затем подвергнут сочетанию до агликона.
Было обнаружено, что стадией, ограничающей скорость реакции гликозилирования с образованием промежуточного сульфоксида, является трифляция сульфоксида.
Реакционная способность фенилсульфоксида может быть поэтому регулироваться путем изменения заместителя в пара положении фенильного кольца. Наблюдаемая реакционная способность располагается в следующей последовательности:
n-OMe выше n-H выше n-NO2
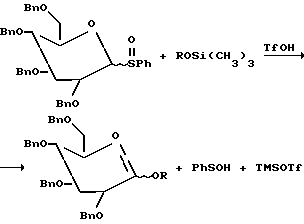
Следовательно, если пербензилированный пара-метоксифенилсульфоксид глюкозы 27 (2.0 эквив.) и пербензилированный фенилсульфоксид глюкозы 28 (2.0 эквив. ) предварительно смешивают вместе с CH2Cl2 и обрабатывают трифликовым ангидридом (1.0 эквив.), основанием (2.0 эквив.) и нуклеофилом 28 (2.0 эквив. ) при -78oC, то наблюдают по данным тонкослойной хроматографии (ТСХ), что пара-метоксифенилсульфоксид был селективно активирован.
Продукты, выделенные после хроматографии, включали дисахарид (80%) и непрореагировавший фенилсульфоксид (выход менее 60%). Однако если ту же реакцию проводят в присутствии избытка рифликового ангидрида, то оба сульфоксида 27 и 28 были активированы преимущественно последовательным путем с образованием продукта гликозилирования.
Однако дополнительные конкурирующие эксперименты показали, что относительная реакционная способность гликозильных акцепторов (нуклеофилов) также может управляться. Таким образом, пербензоилированные 2-деоксифенилсульфоксид фукозы 32 (2.0 эквив.), нуклеофил 31a (1.0 эквив.), силильный эфир 31b (1.0 эквив.) и основание (2,6-дитрет-бутил-4-метилпиридин) 2.0 экивив. - были предварительно смешаны в CH2Cl2 и охлаждены до -78oC. Эту реакционную смесь затем обрабатывали трифликовым ангидридом (1.0 эквив.). Реакционная смесь была подвергнута последующей ТСХ, которая указывает, что сульфоксид 32 и нуклеофил 31a израсходованы (с образованием дисахарида с 60% выходом) в то время как силильный эфир 31b остался непрорегагировавшим.
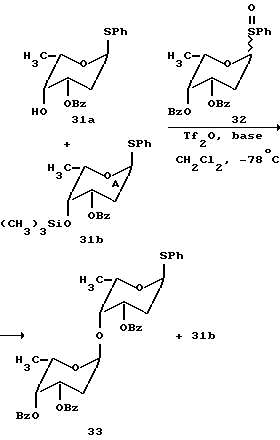
В другом эксперименте был использован избыток сульфоксида 32 (5.0 эквив. ); в этом случае сначала расходовался нуклеофил 31a с последующим расходованием силильного эфира 31b. Таким образом, силильные эфиры взаимодействуют более медленно, чем незащищенные спирты в качестве гликозильных акцепторов, преимущественно потому, что с них первых должна быть снята защита.
На основании продемонстрированной способности регулировать реакционную способность гликозильных доноров и гликозильных акцепторов, был проведен синтез трисахарида цикламицина согласно схеме на фиг. 15. Полагают, что n-метоксифенилсульфоксид B будет активироваться первым с последующим его взаимодействием с C-4 спиртом A с образованием дисахарида AB. Затем будет активирован фенилсульфоксид C и будет сочетаться с дисахаридом AB с образованием желаемого трисахарида ABC.
Одностадийный синтез трисахарида.
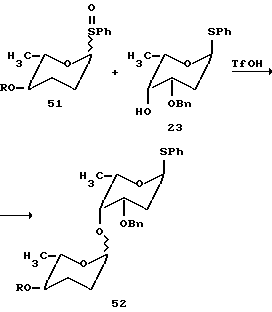
Сульфоксиды 21 и 23 и нуклеофил 23 были предварительно смешаны и растворены в 1: 1 смеси эфира-метиленхлорида при -78oC. К этому раствору были добавлены метилпропиолат (20 эквив. ) с последующим добавлением каталитического количества трифликовой кислоты (0,05 эквив.). Реакцию перемешивали при -70oC в течение получаса и затем гасили, выливая в насыщенный раствор бикарбоната натрия.
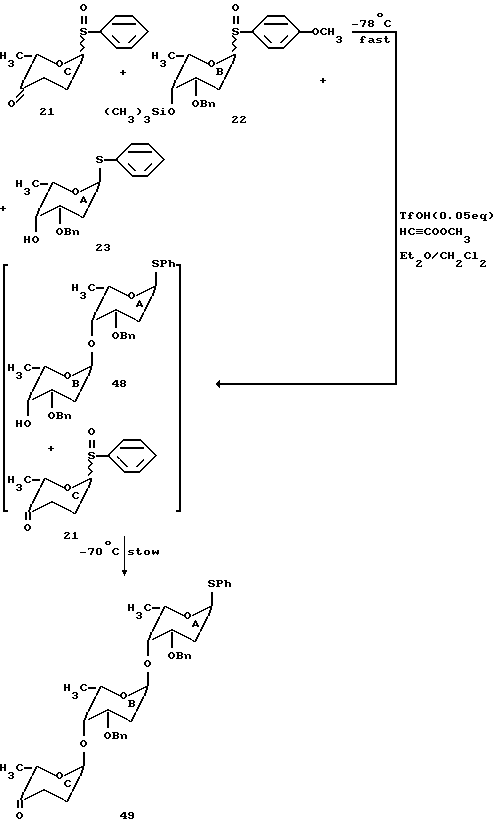
Желаемый трисахарид 49 был основной частью продукта и выделен с 25% выходом после флеш-хроматографии. Другие трисахариды не были выделены из реакционной смеси. Значительное количество другого продукта сочетания дисахарида 48, предшественника трисахарида, было выделено. Как полагают реакция последовательно с активированием в первую очередь n-метоксифенилсульфоксида B и затем взаимодействия со свободной C-4 гидроксильной группой нуклеофила A с образованием AB дисахарида (48). Впоследствии активируется фенилсульфоксид C и взаимодействует с дисахаридом AB с образованием желаемого трисахарида ABC (49). Учитывая предложенный механизм, если ту же реакцию проводят при -100oC (в бане гексан-жидкий азот), выделенные продукты являются силильным эфиром 47 дисахарида AB (60% выход) вместе с неактивированным сульфоксидом C. Выполняя опыты при низкой температуре, таким образом, подтверждают ступенчатую природу реакции.
Выход одностадийной реакции гликозилирования ограничивается не каким-либо нежелаемым перекрестным сочетанием, а нестабильностью гликозильных доноров, в частности кетосульфоксида C, который легко разлагается при комнатной температуре даже в отсутствие активирующего агента действительно менее 5% дисахарида из перекрестного сочетания фенилсульфоксида 21 и свободного спирта 22 было обнаружено. Присутствие кетофункциональной группы в пиразоновом кольце может вносить вклад в нестабильность этого сульфоксида.
В попытке увеличить общий выход гликозилирования проводили использование соответствующей защищенной формы кетосульфоксида.
Улучшение выхода сочетания BC.
Так как дисахарид АПВ 48 и нуклеофил 23 являются структурно очень похожими, нуклеофил 23 был выбран в виде модельного соединения для гликозильного акцептора в реакции гликозилирования с C. Предшественник кетосульфоксида, C-4 экваториальный спирт был выбран в качестве гликозильного донора. Влияние различных защищающих групп в C-4 центре было изучено.
Гликозильный донор Гликозильный акцептор
Использование соответственно защищенного C-4 спирта гликозильного донора драматически улучшает выход гликозилирования (40 - 60%). Для всех изученных случаев. Однако наблюдается потеря стереохимического контроля в аномерном центре, как проиллюстрировано ниже в таблице II.
Было изучено влияние присутствия соответствующей защищенной C-4 аксиальной гидроксильной группы в кольце C. В этом случае была получена желаемая альфа стереоселективность; однако требуются две дополнительные стадии последующего гликозилирования. Эти стадии включают снятие защиты с C-4 спирта с последующим окислением аксиального спирта до кетона. Эти дополнительные стадии приводят к уменьшению общего выхода. Таким образом, хотя 25% выход для одностадийного синтеза трисахарида оказывается скромным, отсутствие другой функциональной группы делает синтез эффективным.
Сочетание трисахарида с агликоном эпсилон-пирромицинона. Трисахарид содержал аномерный фенилсульфид в кольце A. Этот сульфид был окислен до сульфоксида с использованием mCPBA. Агликон эпсилон-0-пирромицинона 15 (1.0 эквив. ) и трисахарид сульфоксида 50 (3.0 эквив.) растворяли в 1:1 смеси эфир-метиленхлорид и охлаждали до -78oC. К реакционной смеси добавляли метилпропиолат (20 эквив.) с последующим добавлением каталитического количества (0.05 эквив. ) трифликовой кислоты. ТСХ, проведенная сразу после добавления трифликовой кислоты указывала на присутствие нового пятна несколько выше агликона. После окончания и очистки с помощью хроматографии это соединение, идентифицированное с помощью ЯМР спектроскопии было агликоном, подвергшимся сочетанию с трисахаридом (54). J H-H константа сочетания 3.0 Hz для аномерного протона согласуется с альфа стереохимией гликозидной связи.
Снятие защиты с цикламицина.
Для окончания синтеза цикламицина 0 требовалось удаления бензилового эфира защищающей группы в кольце A B. Бензиловые эфиры удаляли гидрогенолизом, используя Pd(OH) 2 на угле в качестве катализатора. К сожалению в этих условиях реакции дополнительно к бензиловым эфирам отщепляется также агликон. Ретроспективно это не было неожиданным, так как C-7 гидроксильная группа агликона, к которой присоединяют сахар, напоминает бензиловый эфир, таким образом, для получения неповрежденного цикламицина 0 не могут быть использованы условия гидрогенолиза. Для того, чтобы обойти эту проблему, защищающие группы на кольцах A и B сахара с необходимостью должны быть изменены. Параметоксибензиловые эфиры могут быть легко отщеплены в мягких условиях окисления, используя дихлор-5,6-дициано-1,4-бензохинон (DDQ). См., например, Iktmoto and Schreber J. Am. Chem. Soc., 1990, 112, 9657; Horita et al. , Tetrahedro, 1986, 42, 3021; Oikawa et al., Tet, Lett., 1984, 25, 5393; and Carbohydrates, Ed. Collins, P.M. Chapman and Hall: New York, 1987.
Таким образом, 1:1 смесь марцеломицина (который несет трисахарид в C-7 положении агликона) и кольца A (с защищающей группой п-метоксибензилового эфира в C-3) обрабатывают избытком DDQ. В этих условиях реакции гидролизовался только п-метоксибензиловый эфир в кольце A, в то время как марцеломицин оставался незатронутым. На основании этого результата защищающие группы на цикламициновом трисахариде могут быть предпочтительно изменены от бензила до п-метоксибензиловых эфиров.
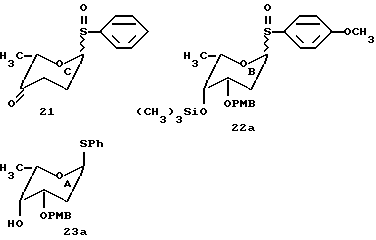
PMB=пара-метоксибензил.
Моносахариды 21, 22a и 23a могут быть использованы для синтеза желаемого цикламицинового трисахарида в одну стадию (20% выход), следуя обычной процедуре. Сульфид трисахарида окисляли до сульфоксида mCPBA и затем подвергали реакции сочетания с агликоном, используя каталитический способ гликозилирования с трифликовой кислотой.
Снятие защиты с продукта сочетания 54a требовало удаления пара-метоксибензиловых эфиров. Продукт сочетания 54a (1 мг) обрабатывали DDQ в метиленхлориде и перемешивали при комнатной температуре в течение 10 часов. Реакция протекала гладко с количественным образованием цикламицина 0.
Быстрый синтез олигосахаридов через контролированную полимеризацию: ограниченные библиотеки гомополимеров 2-деоксифукозы.
Сульфоксид B 2-деоксифукозы, использованный для одностадийного синтеза трисахарида цикламицина, представлял бифункциональный сахар. Он содержит живущую группу в аномерном центре (п-метоксифенилсульфоксид) и может служить в качестве гликозильного донора. Кроме того, он содержал силильный эфир в C-4 центре и мог также служить в качестве гликозильного акцептора. Наиболее обычные 2-деоксисахара, найденные в биоактивных природных продуктах, представляют 2,6-деоксисахара. Они часто встречаются как димеры или тримеры, присоединенные к агликону. При условии нашего успеха в синтезе осложненного трисахарида в одну стадию, мы удивились, если бы было возможно расширить эту идею для быстрого собирания олигосахаридов через контролированную полимеризацию. Бифункционально 2,6-деокси B кольцо представляет удобный случай для изучения возможности синтеза гомополимеров в одной реакции.
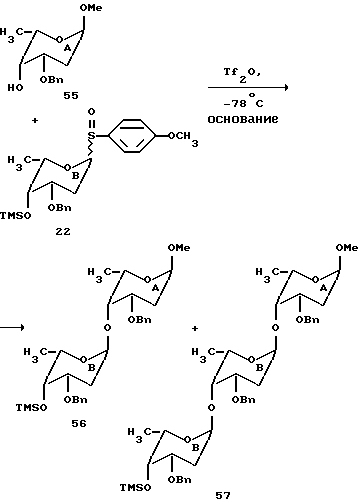
Сульфоксид 22 (2.0 эквив.) и нуклеофил 55 предварительно смешивают в 1:1 смеси эфир-метиленхлорид и охлаждают до -78oC. Реакционную смесь обрабатывают основанием (2.0 эквив.) и трифликовым ангидридом (1.0 эквив.) (см. выше). ТСХ, проведенная сразу после начала реакции, указывает на присутствие двух новых пятен, относящихся к продукту. После хроматографирования выделяют два продукта и идентифицируют с помощью ЯМР спектроскопии. Эти продукты были AB дисахаридом 56 (45% выход) и ABB трисахаридом 57 (20% выход). Константы взаимодействия J для аномерных протонов H-H согласовывались с альфа стереохимией для всех гликозидных связей. Этот результат указывал на то, что гомополимеры 2-деоксифукозы могут стереоселективно образовываться в одну стадию.
Для определения более высокого порядка полимеров 2-деоксифукозы, которые могут быть получены, увеличивают число эквивалентов сульфоксида B (22), используемого в гликозилировании.
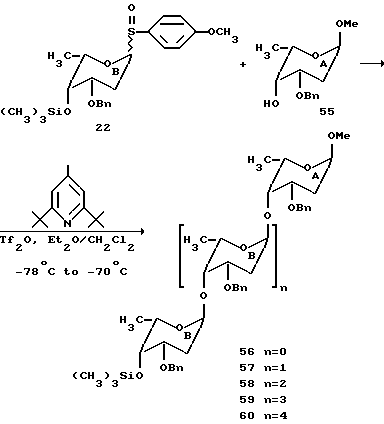
Если используют 5.0 эквивалентов B и 1.0 эквивалент A, то получают статистическую смесь ди, три, тетра, пента и гексасахаридов в одну реакцию, как описано выше. Таким образом, используя сульфоксидный способ возможно быстро синтезировать смесь гомополимеров 2-деоксифукозы через контролированную полимеризацию.
Сульфоксидный способ гликозилирования является гибким и мощным. Он может быть использован для быстрого синтеза осложненного трисахарида, подобного цикламицину, из моносахаридных компонентов в одну реакцию. Эта стратегия может быть распространена на синтез гексасахарида из дисахаридных компонентов в одну стадию. Кроме того, путем использования бифункциональных сахаров можно синтезировать смесь гомополимеров (различающихся длиной цепи) в одну стадию с помощью сульфоксидного способа. До сих пор мы изучали только 2-деоксифукозу в качестве субстрата для контролированных реакций полимеризации. Тем не менее эта стратегия может быть принципиально распространена также для включения других бифункциональных субстратов.
Путем использования комбинации одностадийного способа и стратегии контролированной полимеризации можно очень быстро синтезировать библиотеки олигосахаридов с различными присоединенными агликонами, как показано выше. Эти олигосахаридные библиотеки могут быть испытаны на ДНК связывание, например, путем использования ДНК афинной хроматографии. Такое исследование будет оказывать помощь в объяснении черт олигосахаридов, которые даются ДНК связыванием.
Сульфоксидный способ гликозилирования является быстрым, гибким и эффективным для конструирования олигосахаридов, используя обычные подходы. Реакционная способность гликозильных доноров может быть регулирована изменением заместителя в пара положении фенильного кольца. Электроно-донорные заместители увеличивают скорость реакции относительно случая незамещенных соединений, тогда как электроноакцепторные группы снижают скорость реакции. Реакционная способность гликозильных акцепторов также может регулироваться. Силильные эфиры взаимодействуют более медленно в реакциях гликозилирования, чем свободные спирты. Это позволяет осуществлять контролированное образование двух или более гликозидных связей в одной реакции. Эта стратегия была применена для синтеза цикламицина 0 стереоселективного трисахарида из моносахаридных компонентов в одну стадию. Кроме того, бифункциональные сахара могут быть использованы для синтеза библиотек олигосахаридов. Таким образом, сульфоксидный способ является гибким способом гликозилирования, который позволяет быстро синтезировать олигосахариды через контролированную олигомеризацию.
Образование гликозидных связей в твердой фазе.
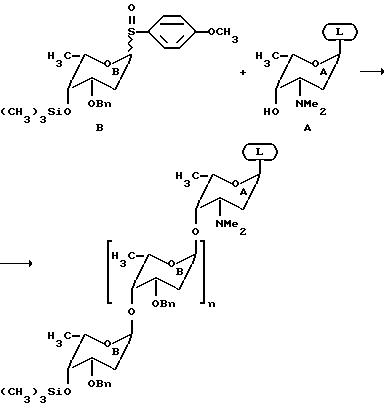
Потенциальный гликозильный акцептор присоединяют к нерастворимой подложке (далее называемой смола) через связь, которая может быть легко отщеплена по окончании синтеза, используя условия, которые не разрушают гликозидные связи. Смола может быть любым нерастворимым полимером, который набухает в органических растворителях и содержит боковые группы для присоединения гликозильного акцептора. Предпочтительные смолы включают, но не ограничиваются ими, полистирольные смолы, такие как смола Меррифилда, и полистирольные смолы, модифицированные ПЭГ, такие как TentaGell смолы.
Тип связи зависит от типа функциональных боковых групп, доступных в полимерной фазе, и от гликозильного акцептора. Так как смолы на основе полистирола могут быть легко функционализированы хлорметильными заместителями, связь обычно представляет бензильный эфир, полученный нуклеофильной заменой бензилхлорида на смоле свободной гидроксильной группой на гликозильном акцепторе. Или же может быть использован бензильный эфир, который получают нуклеофильной заменой бензилхлорида на смоле солью кислоты на гликозильном акцепторе (фиг. 4). Оба типа связей могут быть легко гидролизованы в аномерном углероде гликозильного акцептора обработкой смолы соединением Hg (II). Или же, эфирная связь может быть гидролизована метанолизом, как это делают для эфирных связей смолы в пептидном синтезе. Способ с Hg (II) является предпочтительным для обработки аликвот смолы с управлением хода реакции. Способ с Hg (II) также является предпочтительным, если в качестве конечного продукта желательным является лактол олигосахарида. Способ метанолиза является предпочтительным, если в качестве конечного продукта желательным является сульфид олигосахарида (фиг. 4).
Потенциальный гликозильный акцептор может быть любой молекулой, содержащей один или более потенциально реакционноспособных нуклеофила, включающих потенциально реакционноспособные гидроксильные группы, амины, и/или тиолы, при условии, что они также содержат пригодную для присоединения к смоле боковую группу. Потенциальный реакционноспособный нуклеофил представляет свободный нуклеофил или нуклеофил с временной защищающей группой, которая может быть легко удалена, если только гликозильный акцептор присоединяют к смоле. Потенциальный гликозильный акцептор также может содержать постоянно защищенные нуклеофилы, которые являются нуклеофилами с которых не может быть снята защита при условиях, которые используют для удаления временных защищающих групп. Потенциальный гликозильный акцептор может быть сахаром или какой-либо другой молекулой, несущей нуклеофил, включающей, но не ограничающейся или, стероиды, аминокислоты или пептиды, полярные липиды, полициклические ароматические соединения и им подобные. Схемы защищающих групп для сахаров, которые позволяют селективно защищать и снимать защиту в любых положениях хорошо известны.
См. , например, Bikley выше. Вслед за присоединением к смоле, с потенциально реакционноспособного нуклеофила селективно снимают защиту, если необходимо, и смолу, содержащую производные, лиофилизуют в течение ночи и хранят в эксикаторе до использования. Затем смолу предпочтительно помещают в специально сконструированный реакционный сосуд с простым стеклом. Любые отверстия герметизируют, например, каучуковыми перегородками (см., например, фиг. 5). Может быть очень много вариантов обычных аппаратов. Однако могут быть перечислены следующие важные черты: a) впускное отверстие для добавления растворителя и растворенных реагентов в реакционную камеру, которая пригодна для поддержания безводных условий; (В показанном аппарате каучуковая перегородка вокруг воронкообразного отверстия позволяет добавлять растворитель и растворенные реагенты с помощью канюли или сифоновой иглы, в то же время предотвращая контакт реакционной камеры с внешней атмосферой. В предпочтительном варианте реакционного сосуда это впускное отверстие также оборудуют Т-образным коннектором или аналогичным держателем, который позволяет делать впускное отверстие как вентиль для освобождения инертного газа, такого как азот или аргон, для предотвращения создания избыточного давления в пределах аппарата); б) реакционная камера для содержания смолы и раствора реагента, которую оборудуют пористым устройством или фильтром такой крупнозернистости или пористости, чтобы несвязанные компоненты, такие как непрореагировавшие растворенные вещества, но не смола, могли быть вымыты из реакционной камеры; с) отверстие, которое расположено на боковой стороне пористого устройства, которое находится напротив впускного бокового устройства, для введения инертного газа; газа прошедшего через пористое устройство, таким образом перемешивая реакционную смесь и оседая в пределах реакционной смеси, поддерживая таким образом безводную атмосферу внутри реакционной камеры. (Как видно из фиг. 5, аргон или азот проходит через смолу снизу противотоком потоку растворителя через пористое устройство и одновременно перемешивая смолу. В предпочтительном варианте это отверстие оборудуют Т-образным коннектором или аналогичным держателем, позволяя отверстию быть соединенным с аспиратором для удаления растворителя в вакууме).
Необходимо отметить также, что конфигурация аппарата является такой, что аппарат вплоть до уровня большей части реакционной камеры может быть погружен в охлаждающую баню. Следовательно, ниже отверстия аппарат может быть У-образной формы, как показано на фиг. 5, так что газовое отверстие может быть расположено выше охлаждающей среды.
Затем инертный газ, такой как аргон или азот, предпочтительно аргон, проходит через смолу в течение около 1 часа. Затем смолу суспендируют в 3-5 мл безводного растворителя, включающего, но не ограничивающегося ими, толуол, эфир, ТГФ, метиленхлорид, хлороформ, пропионитрил или их смеси. Из обсуждения в предыдущем разделе понятно, что выбор растворителя будет оказывать влияние на стереохимический выход реакций гликозилирования, в которых не включается участие соседней группы. Поток аргона регулируют для осторожного перемешивания смолы и предотвращения растворителя от перетекания через пористое устройство.
Затем гликозильный сульфоксид растворяют в безводных условиях в 2-4 мл безводного растворителя и переносят с помощью канюли в реакционный сосуд, содержащий смолу. Гликозильный сульфоксид может также содержать защищающую группу, присутствующую в другом месте в молекуле. Если сахаридная цепь является более расширенной, гликозильный сульфоксид должен также содержать по крайней мере одну временную защищающую группу. Обычно гликозильный сульфоксид добавляют в 2-4-кратном избытке по отношению к количеству гликозильного акцептора на смоле.
В зависимости от способа активации, использованного для инициирования реакции гликозилирования, ненуклеофильное основание, такое как 2,6-ди-третбутил-4-метилпиридин или основание Хаггиса (диизопропилэтиламин), может быть растворено с гликозильным сульфоксидом или добавлено в сосуд, содержащий смолу. Если используют, то основание присутствует предпочтительно в небольшом избытке по отношению к количеству добавленного гликозильного сульфоксида.
Затем реакционный сосуд, содержащий смолу, погружают в холодную баню при -78oC. Для активирования гликозильных доноров в реакции добавляют либо 0.05 эквив. (т.е. каталитическое) трифликовой кислоты или 0.5 эквив. трифликового ангидрида, разбавленных в большом объеме безводного растворителя, к реакционной смеси в безводных условиях. Молярные эквиваленты определяют относительно количества использованного гликозильного сульфоксида. Кроме того, разбавление в большом объеме обозначает, что объем чистого активирующего агента разбавляют по крайней мере 100-кратным добавлением соответствующего объема растворителя (например, 1 мкл чистого активирующего агента добавляют к по крайней мере 99 мкл растворителя до добавления донора).
Добавление активирующего агента может быть проведено, например, с помощью канюли. Другие активирующие агенты, пригодные в настоящем способе включают, но не ограничиваются ими, алкил- или арилсилилтрифлат (например, триметилсилилтрифлат), алкил- или арилсульфенилтрифлат и алкил- или арилселенилтрилат. Если в реакции генерируются протоны (когда используют 0.5 эквив. трифликового ангидрида для активации сульфоксида), то в смеси смолы должен присутствовать акцептор кислоты. Кроме того, если не используют активирующий агент в каталитических количествах (например, менее 0.1 эквив. относительно гликозильного сульфоксида), активирующий агент должен быть разбавлен в 100-кратном или более количестве до добавления. Было обнаружено, что в случае трифликового ангидрида такое разбавление является критическим, таким образом можно избежать трифляции гликозильных акцепторов на смоле.
Затем смолу осторожно перемешивают потоком аргона. Обычно реакции позволяют протекать в течение приблизительно 30 минут, после чего смолу неоднократно промывают для удаления продуктов и непрореагировавшего гликозильного донора. Если желательно, реакция может управляться путем удаления аликвот смолы, промывания смолы для удаления реагентов и затем гидролиза связи со смолой. Или же, если гликозильный акцептор является сахаром, который присоединяют к смоле через сульфидное производное, связанное с аномерным углеродом, связь аномерного углерода может быть гидролизована соединением Hg (II). Гидролиз Hg (II) является предпочтительным для регулирования степени реакции гликозилирования.
Продукты и протекание реакции могут быть анализированы с помощью тонкослойной хроматографии, используя стандарты для сравнения. Например, после гидролиза Hg (II) аликвот из реакционной смеси растворимые продукты анализируют с помощью ТСХ. Отсутствие моносахаридного остатка, который был связан со смолой, берется как указание того, что реакция прошла полностью.
Для получения продуктов смолу обычно неоднократно промывают метиленхлоридом с последующей промывкой метанолом (предпочтительно около 10 циклов). Реакция сочетания может быть повторена, если необходимо, для проведения реакции до конца. Или же, если сахаридная цепь является более расширенной, временные защищающие группы затем удаляют, смолу неоднократно промывают для удаления реагентов и добавляют другой гликозильный сульфоксидный остаток как прежде.
После окончания синтеза и промывания продуктов реакции для удаления реагентов, из смолы удаляют дисахариды, олигосахариды или гликоконъюгаты. Продукт может быть затем очищен и/или с него снята защита, если желательно. Или же дисахариды, олигосахариды или гликоконъюгаты могут быть использованы, хотя еще присоединены к смоле в процедурах для испытаний для оценки биологической активности.
Стратегически смеси олигосахаридов могут быть также получены с помощью твердофазного синтеза и испытаны на биологическую активность. Для получения смесей к смоле добавляют более чем один из различных типов гликозильных сульфоксидов при одном или более циклов синтеза. Было бы желательно менять сахара только в одном положении при синтезе для анализа структурных требований в этом положении. В этом случае связь структура-активность может быть легко оценена, в случаях, когда известны оба и конкретный карбогидрат и его рецептор. Или же возможно изменение сахаров в нескольких положениях в синтезе, дающее сложные смеси, которые могут быть испытаны на связывание с различными рецепторами. В любом случае, если обнаруживают активность, активное соединение(ния) может быть идентифицировано, используя способы, аналогичные тем, которые были использованы в пептидной области для идентификации активных пептидов от смесей, полученных при твердофазном синтезе. См., например, Furka et al., Int. J. Peptide Protein Res. 1992, 37, 487; Lam et al., Nature, 1991, 354, 83; Houghten, R.A. Nature 1991, 354, 84; Zuckermann et al., Proc. Natl, Acafd. Sci. USA 1992, 89, 4505; Pritihory Proc. Natl. Acad. Sci. USA 1991, 88, 11510; Geyse et al Proc. Natl. Acad. Aci. USA 1984, 81, 3998; Houghten Proc. Natl. Acad. Sci. USA 1985, 82, 5131; Fodor et al., Science 1991, 251, 767.
Примеры.
Следующие конкретные примеры оказывают помощь читателю в различных аспектах применения настоящего изобретения. Так как эти конкретные примеры являются больше иллюстративными, ничего в следующих описаниях не должно быть истолковано как ограничивающее изобретение ни в какой степени. Такие ограничения являются конечно определенными исключительно сопутствующей формулой изобретения.
Синтез трисахарида цикламицина 0 в одну стадию.
Фиг. 1 иллюстрирует один вариант способа образования множества гликозидных связей в растворе, в котором данный трисахарид, трисахарид цикламицина 0 синтезируют стереоспецифически в защищенной форме в одну стадию из моносахаридных компонентов. Моносахариды 1, 2 и 3 соединяют в отношении 3:2:1, как показано (417 мг, 7812 ммоля; 541 мг, 1.2 ммоля; 165 мг, 0.604 ммоля соответственно). Затем удаляют воду из смеси разгонкой азеотропной смеси из безводного толуола. (Эту стадию осушки проводят путем растворения сахарной смеси в толуоле (около 30 мл) и удаления толуола на роторном испарителе в вакууме. Безводную сахарную смесь затем используют непосредственно или хранят в атмосфере инертного газа до тех пор пока она не потребуется).
Безводную сахарную смесь затем растворяют в 20 мл безводного метиленхлорида в 50 мл склянке, высушенной в пламени. Затем добавляют 20 мл свежеперегнанного диэтилового эфира, содержащего 20 эквивалентов метилпропиолата. (Пропиолатный эфир используют для улавливания сульфоновой кислоты, которая образуется в реакции). Затем раствор охлаждают до -78oC. Затем по каплям добавляют каталитическое количество трифликовой кислоты (5.3 мкл, 0.05 эквив. ) и реакционной смеси позволяют нагреваться от -78oC до -7-oC в течение 45 минут и затем гасят насыщенным раствором NaHCO3. Двухфазную смесь затем экстрагируют CH2Cl2 (3 х 15 мл). Органические экстракты объединяют, сушат над безводным сульфатом натрия и концентрируют. Основной продукт, трисахарид 5 выделяют с 25% выходом, по отношению к моносахариду 3, после экстракции этилацетатом и флеш-хроматографирования на силикагеле (20% этилацетата в петролейном эфире). 1H-ЯМР спектр трисахарида 5 показан на фиг. 12 и 13. Достигнутая селективность является функцией пары донор-акцептор и условий гликозилирования (растворитель, температура). Мы нашли, что каталитическая трифликовая кислота не аномеризует гликозидные связи с заметной скоростью ниже -30oC. Других трисахаридов не образуется. Действительно только другой продукт сочетания в значительном количестве, обнаруженный после реакции, представляет дисахарид 4 (схема 1 фиг. 1, 15% выход; H-ЯМР, фиг. 14), предшественник трисахарида 5. Менее 5% дисахарида из реакции перекрестного сочетания фенилсульфоксида 1 и свободного спирта 3 обнаруживают даже в том случае, когда 1 присутствует в избытке; дисахарида из реакции перекрестного сочетания 1 и 2 не обнаруживается.
Таким образом, выход трисахарида 5 в реакции не ограничивается любой нежелательной реакцией перекрестного сочетания. Однако нестабильность гликозильного донора, особенно кетосульфоксида 1, который легко разлагается при комнатной температуре даже в отсутствие активирующего агента, может влиять на выход.
Продукты реакции указывают на то, что гликозилирование протекает последовательным путем с п-метоксифенилсульфоксидом 2, активирующим быстрее, чем фенилсульфоксид 1 и C-4 спиртом 3, взаимодействующим быстрее чем C-4 силильный эфир 2. В соответствии с этой последовательностью, если реакцию гасят при -100oC, то может быть выделен только силильный эфир дисахарида 4 (60%).
Таким образом, продукт одностадийной реакции, описанной выше, иллюстрирует принцип установленный настоящим изобретением; т.е. то, что реакционная способность обоих соединений гликозильных доноров и гликозильных акцепторов может быть эффективно регулирована таким образом, что гликозилирование протекает последовательным путем с образованием желаемого олигосахарида в одну стадию.
Наконец, необходимо отметить, что трисахарид (5), полученный в одностадийной реакции, содержит аномерный фенильный сульфоксид в кольце A. Аномерные фенильные сульфоксиды являются устойчивыми ("обезоруженными") в условиях, которые активируют аномерные фенильные сульфоксиды для гликозилирования, но они могут быть легко окислены в мягких условиях. См., Mootoo et al., J. Am. Chem. Soc. 1988, 110, 5583; Veeneman and van Boom Tet.Lett. 1990, 31, 275; and Mehta and Pinto Tet.Lett. 1991, 32, 4435. Таким образом, реакция сульфоксидного гликозилирования также приводит сама по себе к повторению стратегии синтеза олигосахарида. См., Friesen and Danishefsky J.Am.Chem.Soc., 1989, 11, 6656; Halcomb and Danichefsky J.Am.Chem.Soc., 1989, 11, 6661; Vootoo et. al., в печати; Veeneman and van Boom в печати; and Mehta and Pinto в печати; Nicolau et. al., J.Am.Chem.Soc., 1984, 106, 4189; Mootoo et. al. J. Am.Chem.Soc., 1989, 11, 8540; Barrett et. al., J.Am.Chem.Soc., 1989, 111, 1392.
Трисахарид 5 цикламицина окисляют до соответствующего сульфоксида с 80% выходом (1.2 эквив. mCPBA, CH2Cl2, -78oC до -50oC, 2 часа) и легко подвергают реакции сочетания с хромофором цикламицина.
Моносахарид 1 получают из L-рамнозы с общим 60% выходом (1H-ЯМР; фиг. 9). См. , Maethin et. al. , Carbohydr. Res. 1983, 115, 21. Моносахариды 2 (1H-ЯМР; фиг. 10) и 3 (1H-ЯМР, фиг. 11) получают из L-фукозы с общими выходами 47% и 52% соответственно. См., Giese et. al., Angew. Chem. Int. Ed. Engl. , 1987, 26, 233. Согласно другому способу настоящего изобретения получение индивидуальных исходных веществ, их организованная конденсация с образованием интересуемого трисахарида и их последующее сочетание с агликоном описывают более детально ниже.
Фенил-3-0-бензоил-2,6-дидеокси-1-тио-альфа-L-галактопиранозид (31a)
Соединение 37 (1.41 г, 5.9 ммоля) растворяют в дихлорметане и охлаждают до -78oC в аргоне. Добавляют гидрид натрия (566 мг, 23.6 ммоля) к этому раствору. Наблюдается оживленное вскипание. Через 10 минут добавляют бензоилхлорид (2.05 мл, 17.7 ммолей) к раствору. Реакционную смесь перемешивают при -78oC в течение получаса, постепенно нагревая до 60oC, и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученный раствор экстрагируют метиленхлоридом (3 x 15 мл); органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (20% этилацетат-петролейный эфир) дает продукт в виде белого твердого вещества (1.6 г, 70%). R = 0.3 (20% этилацетат-петролейный эфир).
1H-ЯМР (CDCl3, 270 Мц) дельта, 8,3 (м, 2H), 7.8-7.2 (м, 8H), 5,85 (д, J = 5.94 Гц, 1H, H-1), 5.55 (м, 1H, H-3), 4.7 (кв., J = 6.60 Гц, H-5), 4.1 (шир. 1H, H-4), 2.78 (д., J = 5.93, 12.86 Гц, 1H, H-2 акс.), 2.33 (дд, J = 4.95, 13.19 Гц, H-2 экв.), 2,12 (шир. 1H, OH), 1.41 (д., J = 6.60 Гц, 3H, CH3).
Здесь и далее: m-мультиплет; d-дублет; t-триплет; dd-двойной дублет; bs-широкий; q-квартет; s-синглет; dt-двойной триплет; Hz-Гц, MHz-МГц.
Фенил-3,4-0-бензоил-2,6-дидеокси-1-тио-альфа-L-галактопиранозид (32a)
Это соединение получают из фенил-3,4-0-диацетил-2,6-дидеокси-1- тио-альфа-L-галактопиранозида 36 в следующей двухстадийной последовательности:
i) NaOMe / CH3OH, амберлит IR (120) плюс ионообменная смола; и
ii) бензоил хлорид, пиридин. Rf (ТСХ)=0.3 (30% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 8.1-7.7 (m, 6H), 7.5-7.2 (m, 9H), 5.71 (m, 1H, H-3), 5.61 (bs, 1H, H-4), 5,48 (d, J=5.28 Hz, 1H), 4.33 (q, J = 6.27 Hz, 1H, H-5), 2.45 (dt, J=3,63, 12.54 Hz, 1H, H-2ax), 2.17 (dd, J = 4.95, 12,54 Hz, 1H, H-2eq), 1.25 (d, J=6.27 Hz, 3H, CH3). 13C NMR (CDCl3, 67.5 MHz) δ 169.49, 164.88, 134.11, 130.77, 130.00, 129.28, 129.13, 128.56, 128.44, 128.28, 128.12, 127.83, 83.33, 69.93, 67.81, 65.74, 53.22, 30.62, 16.17.
Фенил-3,4-0-бензоил-2,6-дидеокси-1-сульфинил-альфа-L- галактопиранозид (32)
Сульфид 32a (900 мг, 1.99 ммоля) растворяют в дихлорметане и охлаждают до -78oC (баня ацетон-сухой лед) в атмосфере аргона.
Добавляют mCPBA (483 мг, 2.80 ммоля) и реакцию перемешивают при -78oC в течение 1 часа. Реакционную смесь постепенно нагревают до 0oC в течение двух часов и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную двухфазную смесь экстрагируют метиленхлоридом (3 x 25 мл). Органические слои объединяют, промывают рассолом и сушат над безводным сульфатом натрия. Флеш-хроматография (40% этилацетат-петролейный эфир) дает сульфоксид 32 в виде кристаллического твердого вещества (690 мг, 80% выход). Rf (ТСХ)= 0.4 (40% этилацетат-петролейный эфир) 1H NMR (CDCl3, 270 MHz) δ 8,1-7.65 (m, 6H), 7.5-7.2 (m, 9H), 5.9 (m, 1H, H-3), 5.68 (d, J=2.64 Hz, 1H, H-4), 4.71 (d, J= 5.61 Hz, 1H, H-1), 4.63 (q, J=6,27 Hz, 1H, H-5), 2.80 (dd, J=5.28, 14.19 Hz, 1H, H-2eq), 2.55 (dt, J=5.94, 12.53 Hz, 1H, H-2ax), 1.27 (d, J= 6.27 Hz, 3H, CH3).
3,4-0-бензоил-2,6-дидеокси-галактопиранозил-альфа-(1-4)- фенил-3-0-бензоил-2,6-дидеокси-1-тио-альфа-L-галактопиранозид (33).
Сульфоксид 32 (177 мг, 0.4 ммоля), нуклеофил 31a (80 мг, 0.2 ммоля), силиловый эфир 31б (93 мг, 0.2 ммоля) и основание (82 мг, 0.4 ммоля) предварительно смешивают и подвергают азеотропной разгонке с толуолом три раза. В 25 мл склянку, высушенную в пламени под аргоном, добавляют свежеперегнанный хлористый метилен. Продукты азеотропной разгонки растворяют в 10 мл хлористого метилена и добавляют в склянку, которую затем охлаждают до -78oC. Через 10 минут добавляют трифликовый ангидрид (33.6 мкл, 0.2 ммоля). За протеканием реакции следят с помощью ТСХ (30% этитлацетатпетролейный эфир). Данные ТСХ указывают на то, что нуклеофил 341a реагирует полностью, в то время как силиловый эфир 31б остается непрореагировавшим. Реакционную смесь постепенно нагревают до -60oC в течение 2 часов и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют метиленхлоридом (3 x 15 мл), сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (30% этилацетат-петролейный эфир) дает дисахарид 33 в виде белого кристаллического твердого вещества (85 мг, 60% выход). Rf=0.4, (30% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 8,1-7.9 (m, 6H), 7.6-7.1 (m, 14H), 5.80 (d, J=5,28 Hz, 1H, H-1), 5.74 (m, 1H, H-3), 5.47 (m, 1H, H-3'), 5.43 (d, J=1.98 Hz, H-4), 5,21 (d, J=1.98 Hz, H-4'), 4.54 (q, J=6.27 Hz, H-5), 4.39 (q, J=6.60 Hz, 1H, H-5'), 4.20 (d, J=2.31 Hz, 1H, H-an), 2.96 (dt, J=5.61, 12.87 Hz, 1H, H-2'), 2.20 (m, 3H), 1.28 (d, J=6.60 Hz, 3H, CH3), 0.47 (d, J=6.27 Hz, 3H, CH3). 13C NMR (CDCl3, 67.5 MHz) δ 165.95, 165.90, 165.69, 134.94, 133.42, 133.11, 132.92, 131.06, 129.91, 129.88, 129.85, 129.76, 129.69, 129.57, 128.91, 128.58, 128.42, 128.23, 127.01 98.93, 83.79, 75.33, 70.34, 70.10, 67.60, 67.53, 65.71, 30.98, 30.21, 17.41, 16.08.
Синтез кольца A
Синтез кольца A начинается с L-фукозы и сопровождается шестистадийной реакцией с 60% выходом согласно схеме, приведенной ниже.
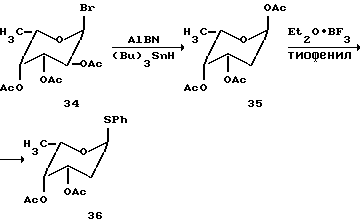
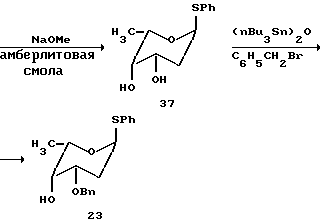
1,3,4,0-три-0-ацетил-2,6-дидеокси-альфа-L-галактопиранозид (35)
К раствору тетра-0-ацетил-фукозы (5,0 г, 15,05 ммоля) в 10 мл ледяной уксусной кислоты добавляют 15 мл гидробромной кислоты (30% по каплям и полученный раствор перемешивают при комнатной температуре. Реакцию заканчивают через два часа. Реакцию проводят в безводной атмосфере путем выливания реакционной смеси в склянку, содержащую 25 г безводного карбоната натрия (Nа2CO3), суспендированного в 200 мл четыреххлористого углерода. Полученную смесь перемешивают при комнатной температуре в течение 45 минут и фильтруют. Процедуру повторяют с фильтратом. Полученный раствор затем концентрируют в вакууме, получая неочищенный бромид 34 (5,2 г, 80%). Его используют без дальнейшей очистки в следующей стадии.
В двухлитровую трехгорловую склянку, оборудованную обратным холодильником и капельной воронкой, добавляют 1 литр свежеперегнанного бензола. Бромид фукозы 34 (5,2 г, 12,17 ммоля) растворяют в бензоле (15 мл) и добавляют в склянку. Полученную смесь кипятят с обратным холодильником. К этому раствору добавляют АИБН (200 мг, 1,21 ммоля). Через 30 минут добавляют по каплям гидрид трибутилолова (4,91 мл, 18,25 ммоля) в бензоле (100 мл) реакционной смеси и затем охлаждают до комнатной температуры и концентрируют в вакууме. Флеш-хроматография (25% этилацетат-петролейный эфир) дает продукт 35 (2,5 г, 81% в виде кристаллического белого твердого вещества. Rf (ТСХ) = 0,3 (25% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MНz) δ 6.24 (d, J = 2.64 Hz, 1H, H-1), 5.22 (m, 1H, H-3), 5.17 (t, J = 0.66 Hz, 1H, H-4), 4.11 (q, J = 5.94 Hz, 1H, H-5) 2.13 (s, 3H), 2.06 (s, 3H), 1.97 (s, 3H), 1.10 (d, J = 6.6 Hz, 3H, CH3);
13C NMR (CDCl3 67.9 MHz) δ 170.13, 169.65, 168.82, 91.51, 68.87, 66.93, 65.86, 28.37, 20.60, 20.42, 16.14, 16.01
Фенил-3,4,-0-диацетил-2,6-дидеокси- 1-тио-альфа-L-галактопираноза (36)
Соединение 35 (2,5 г, 9,11 ммоля) и тиофенол (1,15 мл, 10,94 ммоля) растворяют в дихлорметане (100 мл) и охлаждают под аргоном до -78oC. К раствору по каплям через сифон добавляют эфираттрехфтористый бор (5,6 мл, 45,5 ммоля). Реакционную смесь выдерживают при комнатной температуре в течение 1 часа и затем нагревают до 0oC и гасят, выливая в насыщенный водный раствор бикарбоната натрия. Полученную двухфазную смесь экстрагируют метиленхлоридом (3 х 50 мл); органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют.
Флеш-хроматография неочищенного продукта (15% этилацетат-петролейный эфир) дает сульфид 36 в виде белого твердого вещества (2,1 г, 71% выход).
Rf (ТСХ) = 0,3 (15% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MНz) δ 7.5-7.2 (m, 5H), 5.73 (d, J = 5.61 Hz, 1H, H-1), 5.29 (m, 1H, H-3), 5.23 (bs, 1H, H-3), 4.56 (q, J = 6.6 Hz, 1H, H-5), 2.49 (dt, J = 5.94, 12.84 Hz, 1H, H-2ax), 2.38 (s, 3H, OAc), 2.06 (m, 1H, H-2ед), 1.99 (s, 3H, OAc), 1.13 (d, J = 6.6 Hz, 3H, CH3).
13C NMR for α anоmer of sulfide (CDCl3, 67.9 MНz) δ 170.51, 169.87, 159.54, 134.33, 124.54, 114.54, 84.58, 69.69, 67.18, 65.51, 55.22, 20.79, 20.60, 16.34.
Фенил-2,6-дидеокси-1-тио-альфа-L-галактопиранозид (37)
Соединение 36 (2,1 г, 6,48 ммоля) растворяют в метаноле (50 мл) и к раствору добавляют метоксид натрия (420 мг, 7,78 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. ТСХ (40% этилацетат-петролейный эфир), проведенная по окончании этого времени свидетельствует о том, что реакция окончена. Раствор нейтрализуют амберлитом IR (120) плюс ионообменной смолой (1,0 г). Раствор фильтруют через шоттовский фильтр, промывают этилацетатом и концентрируют до получения диола 37. Это соединение используют без дальнейшей очистки в следующей стадии.
Фенил-3,0-бензил-2,6-дидеокси-1-тио- альфа-L-галактопиранозид (23)
Раствор 37 (923 мг, 3,84 ммоля) и оксида дибутилоолова (956 мг, 3,84 ммоля) в бензоле (60 мл) кипятят с обратным холодильником в склянке, снабженной аппаратом Дина Штарка. Через 15 часов реакционную смесь охлаждают до комнатной температуры и добавляют тетрабутиламмонийбромид (1,24 г, 3,84 ммоля) с последующим добавлением бензилбромида (5 мл, 8,4 ммоля). Полученную смесь далее кипятят с обратным холодильником в течение двух часов, затем охлаждают до комнатной температуры и концентрируют в вакууме. Флеш-хроматография сырого продукта (15% этилацетат-петролейный эфир) дает сульфид 23 в виде белого твердого вещества (1,10 г, 90% выход). Rf (ТСХ) = 0,5 (30% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MHz) α anоmer δ 7.40 - 7.41 (m, 10H), 5.87 (d, J = 5.61 Hz, 1H, H-1), 4.49 (s, 2H), 4.18 (q, J = 6.6 Hz, 1H, H-5), 3.74 (m, 1H, H-3), 3.70 (d, J = 3.63 Hz, 1H, H-4), 2.20 (dt, J = 5.61, 12.5 Hz, 1H, H-2), 2.14 (bs, 1H, OH), 1.95 (m, 1H, H-2'), 1.17 )d, J = 6.6 Hz, 3H, CH3), 13C NMR (CDCl3, 67.9 MHz) δ 137.72, 135.20, 130.76, 128.84, 128.52, 127.92, 127.67, 126.83, 83.93, 73.514, 70.13, 68.46, 66.83, 30.67, 16.61. HRMS m/e 330.1290 (M+) calcd for C19H22O3S 330.1290.
Синтез кольца B
Синтез кольца B сопровождается следующими реакциями: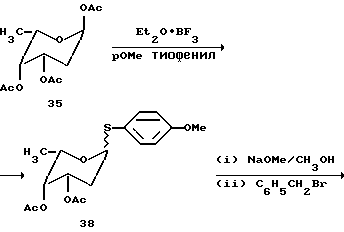
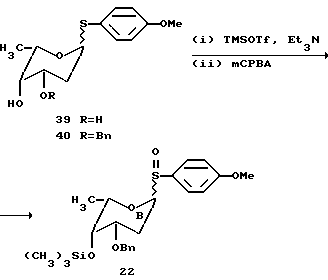
4-метоксифенил-3,4,0-диацетил-2,6-дидеокси-1-тио-L-галактопиранозид (38)
К раствору 35 (1,95 г, 7,14 ммоля) в разогнанном дихлорметане (100 мл) добавляют 4-метокситиофено (1 мл, 8,56 ммоля) и полученную смесь охлаждают до -78oC. К этой смеси по каплям добавляют эфират трехфтористого бора (4,4 мл, 35,70 ммоля), реакционную смесь перемешивают при низкой температуре в течение получаса и затем постепенно нагревают до -30oC и гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют хлористым метиленом (3 х 30 мл). Органические экстракты объединяют, сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Неочищенный продукт чистят с помощью флеш-хроматографии (20% этилацетат-петролейный эфир) с получением сульфида 38 (2,12 г, 85%), Rf (ТСХ) = 0,4 (20% этилацетат-петролейный эфир). 1NMR (CDCl3, 270 MHz) δ 7.40 (d, 2H). 6.8 (d, 2H), 5.51 (d, J = 5.61 Hz, 1H, H-1), 5.26 (m, 1H, H-3), 5.19 (t, J=2.97 Hz, 1H, H-4), 4.54 (q, J = 6.6 Hz, 1H, H-5), 3.77 (s, OCH3), 2.32 (dt, J = 5.61, 12.80 Hz, 1H, H-2), 2.12 (s, 3H), 2.02 (dt, J = 4.62, 12.80 Hz, 1H, H-2'), 1.98 (s, 3H), 1.07 (d, J = 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 67.9 MHz) δ 170.51, 169.87, 159.54, 134.33, 124.54, 114.54, 84.58, 69.69, 67.18, 65.51, 55.22, 30.24, 20.79, 20.60, 16.34; HRMS m/e 354.110 (M+), calcd for C17H22O6S 354.1137.
4-Метокси-2,6-дидеокси-1-тио-L-галактопиранозид (39)
К раствору 38 (1,0 г, 2,82 ммоля) в метаноле (100 мл) добавляют метоксид натрия (183 мг, 3,368 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение двух часов и затем нейтрализуют добавлением Амберлитовой смолы (1,0 г). Реакционную смесь фильтруют и концентрируют в вакууме с получением диола 39 (760 мг, 100%). Этот продукт используют без дальнейшей очистки в следующей стадии. Rf (ТСХ) = 0,1 (50% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MHz) α anоmer δ 7.35 (d, 2H), 6.80 (d, 2H), 5.41 (d, J = 5.28 Hz, 1H, H-1), 4.41 (q, J = 6.6 Hz, 1H, H-5), 3.95 (m, 1H, H-3), 3.76 (s, OCH3), 3.65 (d, J = 2.64 Hz, 1H, H-4), 2.01 (m 2H, H-2, 2'), 1.23 (d,. J = 6.6 Hz, 3H, CH3).
4-Метоксифенил-3-0-бензил-2,6- дидеокси- 1-тио-L-галактопиранозид (40)
Раствор 39 (2,13 г, 7,90 ммоля) и оксида дибутиолова (1,96 г, 7,90 ммоля) в бензоле (200 мл) кипятят с обратным холодильником в склянке, снабженной аппаратом Дина-Штарка. Через 15 часов реакционную смесь охлаждают до комнатной температуры и к ней добавляют бромид тетрабутиламмония (2,54 г, 7,90 ммоля) с последующим добавлением бензилбромида (2,82 мл, 23,7 ммоля). Полученную смесь далее кипятят с обратным холодильником в течение двух часов, затем охлаждают до комнатной температуры и концентрируют в вакууме. Флеш-хроматография неочищенного продукта (15% этилацетат-петролейный эфир) дает сульфид 40 в виде масла (2,5 г, 88%). Rf (ТСХ) = 0,25 (20% этилацетат-петролейный эфир). 1H ЯМР (CDCl3, 270 MHz), δ 7.5-7.25 (m, 7H), 6.81 (d, J= 8.91 Hz, 2H), 5.49 (d, J=5.61 Hz, 1H, H-1), 4.6 (s, 2H), 4.32 (q, J=6.59 Hz, 1H, H-5), 3.85 (m, 1H, H-3), 3.82 (d, J=3.3 Hz, 1H, H-4), 3.77 (s, OCH3), 2.25 (dt, J=5.94, 12.8 HZ, 1H, H-2), 2.2 (bs, OH), 1.72 (m, 1H, H-2'), 1.27 (d, J= 6.59 Hz, 3H, CH3). 13C NMR (CDCl3, 67.9 MHz) δ 159.25, 137.66, 134.03, 128.33, 127.70, 127.52, 124.98, 114.37, 84.81, 73.40, 69.87, 68.35, 66.55, 55.06, 30.24, 16.47. HRMS m/e 360/1385 (M+), calcd for C23H24O4S 360.1396.
4-Метоксифенил-3-0-бензил-2,6- дидеокси-1-тио-4-0-(триметилсилил) -L-галактопиранозид (41)
Раствор 40 (1.4 г, 3.88 ммоля) и триэтиламина (1.62 мл, 11,64 ммоля) в дихлорметане (100 мл) охлаждают до -78oC в атмосфере аргона. К этому раствору добавляют TMSOTf (825 мкл, 4.27 ммоля) по каплям. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют метиленхлоридом (3х30 мл). Органические экстракты объединяют, сушат над безводным сульфатом натрия и концентрируют. Флеш-хроматография (10% этилацетат-петролейный эфир) дает продукт 41 (1.3 г, 83%) в виде масла. Rf (ТСХ)=0,85 (15% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 7.32 (d, J = 8.58 Hz, 2H), 7.28 (m, 5H), 6.79 (d, J=8.91 Hz, 2H), 5.52 (d, J=5.28 Hz, 1H, H-1), 4.55 (d, J=0.99 Hz, 2H), 4.21 (q, J=6.60 Hz, 1H, H-5), 3.79 (bs, 1H, H-4), 3.76 (s, OCH3), 3.66 (m, 1H, H- 3), 2.32 (dt, J=5.61, 12.54 Hz, 1H, H-2), 1.97 (m, 1H, H-2), 1.14 (d, J=6.59 Hz, 3H, CH3 ), 0.11 (s, 9H, TMS). 13C NMR (CDCl3, 67.9 MHz) δ 138.19, 133.67, 128.15, 127.46, 127.39, 125.55 , 114.32, 85.16, 74.26, 71.05, 70.16, 67.76, 55.03, 30.11, 16.99, 0.5. HRMS m/e 432.1808 (M+), calcd for C23H32O4SSi 432.1790
4-Метоксифенил-3-0-бензил-2,6-дидеокси-1-сульфенил-4-9-(триметилсилил)-L- галактопиранозид (22)
К раствору сульфида 41 (402 мг, 0.93 ммоля) в дихлорметане (60 мл) добавляют избыток твердого бикарбоната натрия (1.0 г) и полученную смесь охлаждают до -78oC. К этой суспензии добавляют mCPBA (193 мг, 1.11 ммоля) и полученную смесь перемешивают при низкой температуре в течение 30 минут. Температуру реакционной смеси постепенно повышают до 0oC в течение одного часа и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют метиленхлоридом (3 х 30 мл), органические соли объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (30% этилацетат-петролейный эфир) дает сульфоксид 22 (400 мг, 96%) в виде белого твердого вещества. Rf (TCX)=0.4 (30% этилацетат-петролейный эфир). 1H NMR (270 MHz, CDCl3) δ 7.51 (d, J=8.58 Hz, 2H), 7.4-7.2 (m, 5H), 6.97 (d, J=8.58 Hz, 2H), 4.62 (d, JAB=11.87 Hz, 1H), 4.55 (d, JAB=11.88 Hz, 1H), 4.47 (d, J=5.27 Hz, 1H, H-1), 4.02 (q, J=6.6 Hz, 1H, H-5), 3.91 (m, 1H. H-3), 3.84 (bs, 1H, H-4), 3.83 (s, OCH3), 2.63 (dd, J=4.62, 13.86 Hz, 1H. H-2), 2.02 (dt, J=5.61, 13.85 Hz, 1H, H-2'), 1.12 (d, J=6.26 Hz, 3H, CH3), 0.09 (s, 9H, TMS).
Синтез кольца C
Кольцо C получают согласно следующей схеме реакции: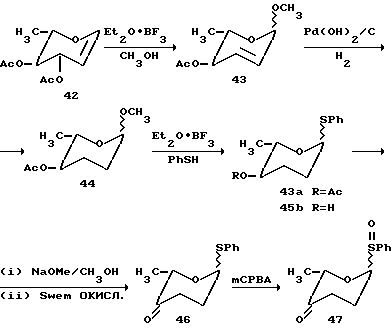
Метил-4-0-ацетил-2,3,6-тридеокси-L-эритро-гексапиранозид (44)
К раствору метил-4-0-ацетил-2,3,6-тридеокси-L-эритро-гекс-2-енопиранозида 43 (2.0 г, 0.01 ммоля) (см. Martin et al., Carbohyd. Res. 1983, 115, 21) в бензоле добавляют катализатор - гидроокись палладия на угле (200 мг) и полученную суспензию встряхивают в шейкере Парра в 2 атм. водорода (2.5 кг/см) Через 2 часа реакционную смесь фильтруют через целит и концентрируют в вакууме с получением 44 (2.0 г, 100% выход). Этот продукт используют в следующей стадии без дальнейшей очистки. Rf (TCX)=0.5 (20% этилацетат-петролейный эфир). 1H ЯМР (CDCl3, 270 MHz) δ 4.63 (s, 1H, H-4), 4.52 (bt, 1H, H-1), 4.32 (q, J = 6.27 Hz, 1H, H-5), 3.22 (s, OCH3), 2.02 (s, 3H, OAc), 1.9 (m, 1H, H-3'), 1.85-1.65 (m, 3H, H-2, 2', 3), 1.02 (d, J=6.27 Hz, 3H, CH3). 13C NMR (CDCl3, 67.9 MHz) δ 169.14, 96.78, 72.96, 65.85, 53.70, 28.68, 23.69, 20.29, 17.34. HRMS m/e 187.0971 (M+) calcd for C9H15O4 187.0970.
Фенил-4-0-ацетил-1-тио-2,3,5-тридеокси-бета-L-арабиногексапиранозил (45a)
К раствору 44 (2.3 г, 12.23 ммоля) и тиофенола (1.5 мл, 14.67 ммоля) в дихлорметане (100 мл), охлажденному до -78oC, добавляют эфират трехфтористого бора (4,5 мл, 36.69 ммоля) по каплям. Реакционную смесь перемешивают при низкой температуре в течение 30 минут, постепенно нагревая до -60oC, и гасят, выливая в насыщенный раствор бикарбоната натрия. Затем полученную смесь экстрагируют метиленхлоридом (3х25 мл), органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (15% этилацетат-петролейный эфир) дает сульфид 45а (3.0 г, 92%) в виде белого твердого вещества. Rf (TCX)=0.4 (15% этилацетат-петролейный эфир). 1H NMR (270 MHz, CDCL3) δ 7.55-7.2 (m, 5H), 5.52 (d, J=4.95 Hz, 1H, H-4), 4.59 (dt, J=4.62, 10.23 Hz, 1H, H-1), 4.30 (m, 1H, H-5), 2.2-2.1 (m, 2H, H-3, 3'), 2.09 (s, 3H, OAc), 1.9-1.75 (m, 2H, H-2, 2'), 1.17 (d, J=5.95 Hz, 3H, CH3). HRMS m/e 266.0970 (M+) calcd for C14H18O3S 266.0977.
Фенил-4-0-гидрокси-1-тио-2,3,5-тридеокси-бета-L-арабиногексапиранозид (45б)
К раствору 44а (37% г, 11.27 ммоля) в метаноле (100 мл) добавляют метоксид натрия (365 мг, 6.76 ммоля). Реакционную смесь перемешивают при комнатной температуре в течение двух часов и затем нейтрализуют добавлением Амберлитовой смолы (2.0 г) и перемешиванием в течение 15 минут. Реакционную смесь фильтруют и концентрируют в вакууме с получением спирта 45б (2752 г, 100%). Это соединение используют без дальнейшей очистки в следующей стадии. Rf (TCX)= 0.2 (25% этилацетат-петролейный эфир). NMR (270 MHz, CDCl3) δ 7.5-7.2 (m, 5H), 5.49 (d, J=4.62 Hz, 1H, H-1), 4.1 (m, 1H, H-4), 3.3 (m, 1H, H-5), 2.2-1.6 (m, 4H, H-2, 2', 3, 3'), 1.2 (d, J=5.49 Hz, 1H, CH3).
Окисление кетосульфида (46)
Раствор оксалилхлорида (5.5 мл, 11.16 ммоля) в дихлорметане (150 мл) обрабатывают ДМСО (1.5 мл, 22 ммоля) при -78oC. Через 10 минут к реакционной смеси добавляют раствор спирта 45б (2.27 г, 10.15 ммоля) в дихлорметане (15 мл) и триэтиламине (7 мл, 50.75 ммоля). Реакционную смесь перемешивают при низкой температуре в течение 30 минут, затем нагревают до 0oC и гасят, выливая в раствор бикарбоната натрия. Полученную смесь экстрагируют метиленхлоридом (3х30 мл); органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (15% этилацетат-петролейный эфир) дает кетон 46 (2.20 г, 97%) в виде светло-желтого масла. Rf (TCX) = 0.35 (15% этилацетат-петролейный эфир). 1H NMR (CDCl3, 500 MHz) δ 7.65-7.20 (m, 5H), 5.52 (t, J=6.59, 6.96 Hz, 1H, H-1), 4.49 (q, J=6.6 Hz, 1H, H-5), 2.65-2.40 (m, 3H, H-3, 3', 2), 1.93 (m, 1H, H-2'), 1.22 (d, J= 6.59Hz, 3H, CH3). 13C NMR (CDCl3, 67.9 MHz) δ 209.58, 134.40, 131.49, 128.91, 127.35, 82.78, 71.54, 34.98, 28.85, 14.63. HRMS m/e 222.0718 (M+) calcd for C12H14O2S 222.0715.
Окисление кетосульфида до сульфоксида (21).
К раствору сульфида 46 (418 мг, 1.88 ммоля) в дихлорметане (40 мл) добавляют избыток твердого бикарбоната натрия (1.0 г) и полученную смесь охлаждают до -78oC. К этому раствору добавляют mCPBA (455 мг, 2.63 ммоля) и реакционную смесь перемешивают при низкой температуре в течение 30 минут. Температуру реакционной смеси медленно повышают до 0oC и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют метиленхлоридом (3х30 мл); органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют в вакууме. Флеш-хроматография (40% этилацетат-петролейный эфир) дает сульфоксид 47 (380 мг, 85%) в виде светло-желтого масла. Rf (ТСХ)= 0.25 (40% этилацетат-петролейный эфир). 1H NMR (CDCl3, 500 MHz, ) δ 7.65-7.42 (m, 5H), 4.64 (t, J=5.93 Hz, 1H, 1H, H-1), 4.59 (q, 1H, H-5), 2.80-2.40 (m, 3H, H-3, 3', 2), 1.80 (m, 1H, H-2'), 1.29 (d. J=6.93 Hz, 3H, CH3).
3-0-бензил-2,6-дидеокси-4-0-(триметилсилил)-альфа-L- галактопиранозил-(1-4)-фенил-3-0-бензил-2,6-дидеокси-1-тио- альфа-L-галактопиранозид.
Сульфоксид 22 (350 мг, 0.78 ммоля) и нуклеофил 23 (129 мг, 0.39 ммоля) предварительно смешивают и подвергают азеотропной разгонке трижды и разогнанным толуолом. Свежеперегнанный диэтиловый эфир (9 мл) добавляют в склянку, предварительно высушенную в пламени в атмосфере аргона, и охлаждают до -78oC. Предварительно смешанные реагирующие вещества растворяют в 6 мл разогнанного дихлорметана и добавляют в склянку. Затем к смеси добавляют основание Хаггиса (136 мкл, 0.78 ммоля). После перемешивания в течение 5 минут к реакционной смеси добавляют трифликовый ангидрид (65.5 мкл, 0.19 ммоля). За протеканием реакции следят с помощью ТСХ (10% этилацетат-петролейный эфир). Реакционную смесь нагревают до -70oC и гасят, выливая в насыщенный раствор бикарбоната натрия. Полученный раствор экстрагируют метиленхлоридом (3х15 мл); органические слои объединяют и сушат над безводным сульфатом натрия. Раствор концентрируют в вакууме и чистят с помощью флеш-хроматографии (5% этилацетат-петролейный эфир) с получением дисахаридного продукта в виде масла (95 мг, 40% выход). Rf (ТСХ)=0.8 (10% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 7.45-7.10 (m, 10H), 5.68 (d, J=5.28 Hz, 1H), 5.07 (bs, 1H), 4.67 (d, JAB=12.54 Hz, 1H), 4.55 (d, JAB=12.54 Hz, 1H), 4.53 (s, 2H), 4.23 (q, J=6.60 Hz, 1H), 4.17 (q, J=6.60 Hz, 1H), 3.85 (d, J= 2.64 Hz, 1H), 3.73 (m, 2H), 3.68 (d, J-1.32 Hz, 1H), 2.33 (dt, J=6.93, 12.54 Hz, 1H), 2.02 (m, 1H), 2.19 (d, J=6.60 Hz, 3H), 0.89 (d, J=6.27 Hz, 3H), 0.06 (s, 9H). 13C NMR (CDCl3, 67.9 MHz) δ 138.63, 138.29, 135.32, 130.93, 128.90, 128.85, 128.59, 128.49, 128.20, 127.61, 127.55, 127.32, 126.89, 84.39, 74.26, 73.79, 73.76, 71.14, 70.25, 68.10, 67.43, 31.72, 29.54, 17.37, 17.23, 16.65, 0.63.
3-0-Бензил-2,6-дидеокси-альфа-L-галактопиранозил-(1-4)-фенил-3-0- бензил-2,6-дидеокси-1-тио-альфа-L-галактопиранозид (48)
Продукт дисахарида из предыдущей части (70 мг, 0.11 ммоля) растворяют в свежеперегнанном ТГФ. К раствору добавляют тетрабутиламмонийфторид (550 мкл, 5 ммолей). Реакцию заканчивают в течение одного часа. Продукт получают выливая реакционную смесь в раствор бикарбоната натрия и экстрагируя (3х15 мл) ТГФ. Органические слои объединяют и концентрируют в вакууме. Продукт 48 используют в следующей стадии без дальнейшей очистки. Rf (TCХ)=0.4 (этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 7.5-7.2 (m, 10H), 5.69 (d, J= 5.27 Hz, 1H), 5.03 (d, J=2.97 Hz, 1H), 4.66 (d, JAB=12.54 Hz, 1H), 4.60 (d, JAB=11.55 Hz, 1H), 4.56 (d, JAB=11.22 Hz, 1H), 4.50 (d, JAB= 11.22 Hz, 1H, 4.27 (q, J=6.93 Hz, 1H), 4.22 (q, J=6.60 Hz, 1H), 3.90 (m, 1H), 3.86 (bd, J=2.31 Hz, 1H), 3.74 (m, 1H), 3.72 (bs, 1H), 2.12 (bs, OH), 1.91 (m, 2H), 1.91 (d, J=6.60 Hz, 3H), 1.01 (d, J=6.60 Hz, 3H). 13C NMR (CDCl3, 67.9 MHz) δ 138.10, 138.01, 135.22, 130.86, 128.83, 128.46, 128.40, 127.79, 127.67, 127.63, 127.39, 126.88, 99.15, 84.30, 74.84, 73.64, 73.21, 70.34, 70.07, 68.30, 67.97, 65.86, 31.65, 29.83, 17.34, 16.62.
ABC трисахарид (49)
Сульфоксид 21 (60 мг, 0.22 ммоля) и нуклеофил 48 (61 мг, 0.11 ммоля) предварительно смешивают и трижды подвергают азеотропной разгонке с разогнанным толуолом. Свежеразогнанный дихлорметан (1 мл) и диэтиловый эфир (5 мл) добавляют в склянку, высушенную в пламени, и охлаждают в атмосфере аргона до -78oC. Предварительно смешанный сульфоксид и нуклеофил растворяют в дихлорметане (3 мл) и добавляют к охлажденной склянке. К этой смеси добавляют основание Хаггиса (40 мкл, 0.22 ммоля). Через 5 минут добавляют трифликовый ангидрид (18.5 мкл, 0.11 ммоля) в склянку с реакционной средой. Реакционную смесь перемешивают в течение двух часов при -78oC до -70oC. Затем реакцию гасят, выливая в насыщенный раствор бикарбоната натрия. Реакционную смесь экстрагируют хлористым метиленом (х15 мл), органические слои объединяют и сушат над безводным сульфатом натрия. Раствор концентрируют в вакууме и чистят с помощью флеш-хроматографии (20% этилацетат эфир) с получением трисахарида 49 (18 мг, 25% выход).
Одностадийный синтез трисахарида цикламицина 0.
Сульфоксиды 21 (417 мг, 1,812 ммоля, 3.0 эквив.), 22 (541 мг, 1.2 ммоля, 2.0 эквив.) и нуклеофил 23 (165 мг, 0.604 ммоля, 1.0 эквив.) предварительно смешивают и тщательно сушат путем тройной азеотропной разгонки с разогнанным толуолом. Затем исходные вещества растворяют в свежеперегнанном хлористом метилене (20 мл) и добавляют в 50 мл склянку, высушенную в пламени, в атмосфере аргона. К этой реакционной смеси добавляют 20 мл свежеперегнанного диэтилового эфира с последующим добавлением метиленпропиолата (9.06 ммоля, 15 эквив. ). Склянку охлаждают до -78oC, используя баню ацетон сухой лед. Через 5 минут к реакционной смеси по каплям добавляют трифликовую кислоту (5.3 мкл, 0.06 ммоля, 0.05 эквив.). За ходом реакции следят с помощью ТСХ (20% этилацетат-петролейный эфир). Реакционную смесь медленно нагревают до -70oC в течение получаса и затем гасят, выливая в насыщенный водный раствор бикарбоната натрия (30 мл). Полученную двухфазную смесь экстрагируют хлористым метиленом (3х15 мл). Объединенные органические экстракты сушат над безводным сульфатом натрия и концентрируют. Флеш-хроматография (20% этилацетат-петролейный эфир) дает трисахарид 49 (99 мг, 25%) в виде бесцветного масла. Rf (ТСХ)= 0.2 (20% этилацетат-петролейный эфир). 1H NMR (CDCl3, 500 MHz) δ 7.45-7.20 (m, 15H), 5.67 (d, J=4.85 Hz, 1H), 5.07 (d, J=2.64 Hz, 1H), 4.98 (t, J= 3.36 Hz, 1H0, 4.66 (d, J=1.32 Hz, 1H), 4.19 (q, J=6.6 Hz, 1H), 3.88 (m, 1H), 3.84 (bs, 1H), 3.73 (m, 1H), 2.52 (m, 1H), 2.28 (m, 1H), 2.23-2.02 (m, 2H), 1.19 (d, J=6.60 Hz, 3H), 0.90 (d, J=5.27 Hz, 3H), 0.88 (d, J=6.60 Hz, 3H). 13C MNR (CDCl3, 67.9 MHz) δ 211.13, 139.13, 138.94, 135.74, 131.74, 131.63, 129.20, 128.69, 128.60, 127.87, 127.81, 127.72, 127.69, 127.31, 99.57, 99.22, 84.80, 75.79, 74.69, 74.19, 73.40, 71.85, 70.45, 68.44, 67.59, 34.25, 31.88, 31.09, 29.89, 17.48, 17.35, 14.93.
Трисахарид цикламицина (49a)
Сульфоксиды 21 (190 мг, 0.8 ммоля, 5 эквив.), 22 (230 мг, 0.48 ммоля, 2.0 эквив.) и нуклеофил 23a (85 мг, 0.24 ммоля, 1.ю0 эквив.) предварительно смешивают и тщательно сушат путем тройной азеотропной разгонки с разогнанным толуолом. Затем исходные вещества растворяют в свежеперегнанном метиленхлориде (5 мл) и добавляют в 25 мл склянку, высушенную в пламени, в атмосфере аргона. К этой реакционной смеси добавляют 5 мл свежеперегнанного диэтилового эфира с последующим добавлением метилпропиолата (4.8 ммоля, 20 эквив.). Склянку охлаждают до -78oC, используя баню ацетон/сухой лед. Через 5 минут к реакционной смеси по каплями добавляют трифликолвую кислоту (4.8 ммоля, 20 эквив. ) За ходом реакции следят с помощью ТСХ (20% этилацетат-петролейный эфир). Реакционную смесь медленно нагревают до -70oC в течение получаса и затем гасят, выливая ее в насыщенный раствор бикарбоната натрия (30 мл). Полученную двухфазную смесь экстрагируют хлористым метиленом (3х15 мл). Объединенные органические экстракты сушат над безводным сульфатом натрия и концентрируют. Флеш-хроматография (30% этилацетат-петролейный эфир) дает трисахарид 49a (35 мг, 20%) в виде бесцветного масла. Rf (ТСХ)=0.3 (30% этилацетат-петролейный эфир).
Окисление сульфида до сульфоксида.
Сульфид трисахарида 49a (20 мг, 0.027 ммоля) растворяют в 15 мл свежеперегнанного хлористого метилена, помещенного в 25 мл склянку. К этому раствору добавляют твердый бикарбонат натрия (500 мг) с последующим добавлением mCPBA (7.8 мг, 0.045 ммоля). За ходом реакции следят с помощью ТСХ (40% этилацет-петролейный эфир). Реакционную смесь медленно нагревают до -60oC и гасят выливая в насыщенный раствор бикарбоната натрия. Полученную двухфазную смесь экстрагируют хлористым метиленом (3 x 10 мл); органические слои объединяют и сушат над безводным сульфатом натрия. Сульфоксид трисахарида 50a (сульфоксид трисахарида, защищенный РМВ) получают в виде масла (19 мг). Продукт используют для гликозилирования без дальнейшей очистки. Деструкция марцеломицина с получением агликона эпсилонпирромицинона. Марцеломицин 53, антрациклиновый антибиотик, выделенный из комплекса богемовой кислоты, содержит такой же агликроновый эпсилон-пирромицинон как цикламицин (см. ниже). (Марцеломицин великодушно подарен Bristol-Myers Squibb Company). Агликон может быть получен путем удаления трисахарида марцеломицина с помощью кислотного гидролиза.
Лекарство (75 мг) кипятят с обратным холодильником в метанольном растворе HCl (25 мл, 0.1N) в течение двух часов. По окончании этого времени марцеломицин (75 мг, 0.175 ммоля) растворяют в метанольном растворе HCl (25 мл, 0,1 N) и кипятят с обратным холодильником при 50oC в течение двух часов. По окончании этого периода реакционную смесь концентрируют в вакууме и чистят с помощью препаративной тонкослойной хроматографии (15% метанол-хлороформ). Агликон эпсилон-пирромицинона выделяют в виде ярко-красного твердого вещества (21 мг, 54% выход).
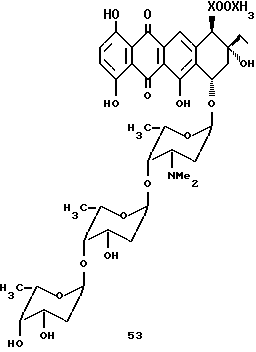
Реакция сочетания трисахарида с агликоном.
РВМ-защищенный трисахарид сульфоксида 50a (19 мг, 25 мкмоля) эпсилон-пирромицинон (6 мг, 14.15 мкмоля) и стильбен (2.5 мг, 14 мкмолей) предварительно смешивают и трижды подвергают азеотроопной разгонке с разогнанным толуолом. Свежеперегнанный эфир (2 мл) добавляют в 15 мл склянку, высушенную в пламени, и охлаждают в атмосфере аргона до -78oC. Продукты азеотропной разгонки растворяют в перегнанном дихлорметане (3 мл) и добавляют в склянку. Через 10 минут в склянку добавляют трифликовый ангидрид (0,118 мкл, 0.7 мкмоля) и за ходом реакции следят с помощью ТСХ (15% этилацетат-петролейный эфир). Реакционную смесь постепенно нагревают до -50oC и гасят, выливая в насыщенный раствор бикарбоната натрия. Полученную смесь экстрагируют дихлорметаном (3 x 5 мл), органические слои объединяют и сушат над безводным сульфатом натрия. Раствор концентрируют в вакууме и чистят с помощью флеш- хроматографии (15% эфир-метиленхлорид с последующей 10% метанол-метиленхлорид). Продукт выделяют в виде ярко-красного твердого вещества (0.75 мг, 16% выход).
Одностадийный синтез гомополимеров различной длины
Фиг. 1 иллюстрирует другой аспект настоящего изобретения, который позволяет синтезировать "гомополимеры" различной длины. Следовательно, альфа-связанные гомополимеры 2-деоксифукозы с различным распределением длин получают путем смешения в отдельных склянках различных соотношений бифункционального сульфоксида 4-метокси-фенил-3-0-бензил-4-0-триметилсилил-2-деокси- 1-сульфинил-альфа-L-фукопиранозида, B, с монофункциональным гликозильным акцептром метил-3-0-бензил-2-деокси-альфа-L- фукопиранозида, A, и основанием 2,6-дитретбутил-4-метилпиридином (2 эквивалента относительно сульфоксида). Таблица III показывает соотношение реагентов, которое используют для каждого из экспериментов 6.6.1-6.6.5. Сначала смеси тщательно сушат путем перегонки из толуола (предпочтительно три раза, как приведено выше).
Смеси затем каждую растворяют в 2.5-5 мл безводного метиленхлорида и добавляют в отдельные 25 мл склянки, высушенные в пламени в атмосфере аргона. К каждой реакционной смеси добавляют равный объем свежеперегнанного диэтилового эфира. Затем склянки охлаждают до -78oC, используя баню ацетон/сухой лед. Через 5 минут к реакционным смесям по каплям добавляют раствор трифликового ангидрида в метиленхлориде (1.0 эквив. относительно B). За ходом реакции следят с помощью тонкослойной хроматографии, используя 15% этилацетат/петролейный эфир в качестве элюента.
После нагревания до -70oC в течение периода около получаса реакционную смесь гасят насыщенным раствором бикарбоната натрия (приблизительно 30 мл каждому). Каждую из полученных двухфазных смесей экстрагируют метиленхлоридом (3 x 15 мл). Органические экстракты объединяют, сушат над безводным сульфатом натрия и концентрируют. Флеш-хроматографию(1:5 этилацетат-петролейный эфир) используют для выделения гликозилированных продуктов из каждой реакции. Распределение длин полученных "гомополимеров" находят с изменением отношения A к B, а также полной концентрацией реагирующих веществ в реакционной смеси, как показано в таблице III.
Более конкретно результаты реакции, приведенной выше, могут быть получены предварительным смешением сульфоксида (320 мг, 0.714 ммоля, 3.0 эквив.), нуклеофила (60 мг, 0.238 ммоля, 1.0 эквив.) и основания (147 мг, 0.714 ммоля, 3 эквив.). Полученную смесь затем сушат путем трехкратной азеотропной разгонки из толуола. Затем реагирующие вещества растворяют в 2.5 мл разогнанного хлористого метилена и добавляют в 25 мл склянку, высушенную в пламени, в атмосфере аргона. Затем склянку охлаждают до -78oC и к реакции добавляют 2.5 мл диэтилового эфира. (Концентрация сульфоксида составляет приблизительно 0.144 ммоля/л). Через 10 минут добавляют трифликовый ангидрид (1.5 эквив. 0.357 ммоля, 60 мкл). Реакционную смесь поддерживают при -78oC в течение получаса, затем позволяют нагреться до -70oC и перемешивают при этой температуре дополнительно полчаса. Реакцию гасят выливая реакционную смесь в насыщенный раствор бикарбоната натрия, с последующим экстрагированием 3 раза хлористым метиленом, сушат над безводным сульфатом натрия и концентрируют. Очистку достигают с помощью флеш-хроматографии: 15% этилацетат-петролейный эфир; 20% этилацетат-петролейный эфир. Структуры различных олигосахаридов доказывают с помощью данных протонного магнитного резонанса (270 МГц, CDCl3), в которых неконцевые B' отмечают X1, X2 или X3 и т.д.
Данные ЯМР спектров:
AB дисахарид: 1H NMR δ 5.08 bs, 1H, H1B; 4.81, d, 1H, H1A; 4.7-4.5, 4H benzyl methylenes; 4.2 q, 1H, H5B; 3.9-3.7, m, 5H, H3A, H4A, H4B, H3B, H5A; 3.3, s, 3H, methoxy; 1.8-2.1, 4H, H2A, H2B; 1,21, d, 3H, H6A (methyl); 0.9, d, 3H, H6B (methyl) ppm.
AXB трисахарид: 1H NMR δ 5.06, s, 1H, H1B; 5.01, s, 1H, H1X; 4.82, d, 1H, H1A; 4.75-4.45, m, 6H benzyl merhylene; 4.21, q, 2H, H5X, H5B; 3.91-3.64, m. 7G, H3A, H3X, H3B, H4A, H4B; 3.29, s, 3H, Al methoxy; 2.08-1.80, d, 3H, H6A; 1,23, d, 3H, H6A (methyl) δ 0.90, 3d, 6H, H6X, H6B (methyls) ppm.
AX1X2B тетрасахарид: 1H NMR δ 5.27, d, 1H,H1B; 4.99, d, 2H, H1X1, H12 ; 4.81, d, 1H, H1A; 4.72-4.45, m, BH benzyl methylenes; 4.2, m, 3H, H5X1, H52, H5B; 3.90-3.62, m, 9H, H5A, H4A, H4X1, H4X2, H4B, H3A, H3X1, H3X2, H3B; 3.29, s, 3H; 2.05-1.80, 8H, 2-deоxy, CH2A, CH2X1, CH2X2, CH2B; 1.22 d, 3H, H6A (methyl); 1.9-1.8, 9H, H6X1,Z H6X2, H6B (methyls) ppm.
AX1X2X3B пентасахарид: 1H NMR δ 5.06, s, 1H, H1B; 4.95, s, 3H, H1X1, H1X2, HX; 4,79, 1H, s, H1A; 4.7 - 4.4, m, 10H, benzyl methylenes; 4.15, m, 4H, HSX1, H5X2, H5X3, H5B; 3.3, s, 3H, Al methoxy; 1.2, 3H, d, H6A (methyl); 0.85, 12H, m, H6X1, H6X2, H6X3, H6B(methyls) ppm.
AX1X2X3X4B гексасахарид: 1H NMR δ 5.05, s, 1H, H1B, 4.98, s, 4H, HX1, H1X2, H1X3 H1X4; 4.7-4.4, 12H, benzyl methylenes; 4.2-4.1, 5H, H5X1, H5X2, H5X3, H5X4, H5B; 2.1-1.8, m, 12H, H2X1, H2X2, H2X3, H2X4, H2B; 1.2, d, 3H, H6A (methyl); 0.9 - 0.8, m, 15H, H6X1, H6X2, H6X3, H6X4, H6B (methyls) ppm.
Контролированные реакции полимеризации.
Согласно другому способу настоящего изобретения контролированную реакцию полимеризации проводят следующим образом: сульфоксид 22 (260 мг, 0.58 ммоля), нуклеофил 53 (73 мг, 0.29 ммоля) и основание 2,6-дитрет-бутил-4-метилпиридин (118 мг, 0.58 ммоля) предварительно смешивают и трижды подвергают азеотропной разгонке с перегнанным толуолом. К 25 мл склянку, высушенную в пламени, добавляют свежеперегнанный диэтиловый эфир (5 мл) и охлаждают в атмосфере аргона до -78oC. Продукты азеотропной разгонки растворяют разогнанном дихлорметане (5 мл) и добавляют в склянку. Через 10 минут в склянку добавляют трифликовый ангидрид (48,5 мкл, 0729 ммоля). Реакционную смесь перемешивают при -70oC в течение 45 минут и гасят выливая в насыщенный раствор бикарбоната натрия. Полученную двухфазную смесь экстрагируют хлористым метиленом (3 х 15 мл); органические слои объединяют и сушат над безводным сульфатом натрия. Раствор концентрируют в вакууме и чистят с помощью флеш-хроматографии (15% этилацетат-петролейный эфир). AB дисахарид 54 (70 мг, 45%) и ABB трисахарид 55 (40 мг, 20%) выделяют в виде масел. АВ дисахарид: Rf (TCX) = 0.4 (15% этилацетат-петролейный эфир). 1H ЯМР (CDCl3, 270 MHz,) δ 7.4-7.2 (m. 10H), 5.06 (bs, 1H, Han), 4.81 (d, J = 2.97) Hz, 1H, H-1'), 4.68 (d, JAB = 12.53 Hz, 1H), 4.54 (s, 2H), 4.50 (d, JAB = 10.89 Hz, 1H), 4.16 (q, J = 6.26 Hz, 1H, H-5), 3.9 (m, 1H, H-3), 3.8 (s, 1H, H-4), 3.75 (q, J = 5.94 Hz, 1H, H-5'), 3.70 (m, 1H, H-3'), 3.67 (d, J = 1.65 Hz, 1H, H-4), 3.27 (s, 3H, OCH3), 2.1 (m, 3H, H-2's), 1.8 (dd, J = 4.62, 12.21 Hz, 1H, H-2AX), 1.21 (d, J = 6.6 Hz, 3H, CH3), 0.9 (d, J = 6.27 Hz, 3H, CH3), 0.06 (s, 9H). 13C NMR (CDCl3, 67.9 MHz) δ 138.61, 128.24, 128.14, 127.57, 137.32, 127.27, 127.19, 99.27, 98.89, 74.17, 73.78, 72.91, 71.13, 70.22, 70.04, 67.28, 66.80, 54.67, 30.77, 29.51, 17.51, 17.21, 0.58.
Однореакторный синтез гликоконъюгатов с потенциально ДНК связывающей активностью.
Стратегия для формирования множества гликозидных связей в растворе может быть использована для синтеза в тех же реакциях некоторых гликоконъюгатов с потенциальной ДНК связующей активностью. В зависимости от ситуации гликоконъюгаты могут быть разделены и испытаны индивидуально на ДНК связующую активность или они могут быть испытаны в виде смесей. Например, смесь гликоконъюгатов, каждый включающий портенциальный интеркаллятор ДНК и олигосахаридную боковую цепь получают как в примере 6.6, но используя 4:1 отношение бифункционального донора к гликозильному акцептору. Конкретно 2-деокси-фукозилсульфоксидное производное B (294 мг, 0.48 ммоля) и 2,6-дитрет-бутил-4-метилпиридин (779 мг, 3.80 ммоля) объединяют, сушат путем тройной азеотропной разгонки из толуола и затем растворяют в 10 мл 1:1 смеси эфир-метиленхлорид (свежеперегнанные растворители). Раствор переносят под аргоном в склянку, высушенную в пламени. Склянку охлаждают до -78oC, используя баню ацетон/сухой лед. Через 5 минут к реакционной смеси по каплям добавляют 0.96 ммоля трифликового ангидрида. Реакционную смесь медленно нагревают до -70oC в течение получаса и затем гасят, выливая в насыщенный раствор бикарбоната натрия. Смесь экстрагируют метиленхлоридом (3 х 15 мл). Объединенные органические экстракты сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Продукт растворяют в 10 мл важного метиленхлорида и обрабатывают избытком дихлордицианхинона (DDQ) при комнатной температуре в течение 1 часа для удаления защищающих групп п-метоксибензилфенилового эфира. Затем удаляют растворитель в вакууме и компоненты разделяют с помощью флеш- хроматографии на силикагеле. Их относительно сродство к ДНК оценивают для определения предпочтительной длины боковой цепи олисахарида. Для идентификации олигосахаридов, которые связаны с конкретными рецепторами, может быть использована афинная хроматография. Например, смесь соединений пропускают через коллонку, содержащую твердую подложку, к которой присоединен рецептор интересующего лиганда или лиганд, если подвижная фаза содержит смесь потенциальных рецепторов. Соединения, которые связаны с рецептором, удерживаются на колонке дольше, чем соединения, не связанные с ним. Соединения могут расфоракционированы согласно их сродству к рецептору.
Таким образом, рецепторы, которые связывают карбогидраты, могут быть присоединены к твердой подложке. Карбогидратные рецепторы могут содержать ДНК (двухтяжную или однотяжную), РНК, протеин, олигосахариды или другие молекулы. Способы для присоединения нуклеиновых кислот, протеинов и олигосахаридов к твердым подложкам для использования в афинной хроматографии описаны. См. : a) Etemplate Chromatography of Nuclear Acids and roteins, Schott, H. Marcell Dekker, Inc. New York, 1984; b) Giycoconjugates: Composition, Structure and Fuction, Allth, H. J.; Kisailus, E.C.Eds.Marcel Dekker: NY 1992, и ссылки в нем. (Замечание: могут быть использованы также времена удерживания для оценки количественной способности для отдельного из соединений, прошедших афинную колонку).
В другом примере гликозильный акцептор А (фиг.3) предварительно смешивают с 2.3-п-метокси-бензил-4-триметилсилил рамнозилсульфоксидом C (фиг.3) и позволяют взаимодействовать в условиях (например, температура, растворитель, концентрация, отношение донор/акцептор), идентичных тем, которые описаны выше. После реакции и удаления п-метоксибензил защищающих групп DDQ, как указано выше, смесь гликоконъюгатов разделяют, используя флеш-хроматографию на силикагеле, и определяют относительно сродство различных соединений к ДНК. Гликоконъюгаты, полученные описанными выше способами сравнивают относительно их способности к связыванию с ДНК. В этом случае влияние различных сахаров на ДНК связывающую способность может быть сравнено с определенными предпочтительными сахарами.
Гликозильный акцептор A на фиг. 3 конструируют гликозилированием соответственно защищенного производного юглона (полученного процедурой Inhoffen et. al. Croatica Chem.Acta. 1957, 29, 329; Trost et.al. J.Am.Chem.Soc.1977, 99, 8116; and Strok and Hagedorn J.Am.Chem.Soc., 1978, 100, 3609) с соединением B (фиг. 3), используя трифликовый ангидрид-основание Ханига метиленхлорид/эфир 1:1 при низкой температуре. После стандартной обработки (включающей экстракцию, как описано в других примерах 6, приведенных здесь) и удаления растворителя смесь продуктов растворяют в метиленхлориде и обрабатывают избытком тетрабутиламмоний фторида при 0oC. Затем растворитель удаляют в вакууме и выделяют продукт с помощью флеш- хроматографии.
Общий процесс, описанный выше, может быть также применен для синтеза смесей гликоконъюгатов, содержащих несколько различных сахаров. В этом случае два или более гликозильных бифункциональных донора используют в реакции. После снятия защиты полученная смесь гликоконъюгатов может быть испытана на ДНК связывающую активность путем пропускания его через ДНК афинную колонку. Соединения могут быть расфракционированы согласно их временам удерживания на афинной колонке. Соединения с большими временами удерживания могут быть выделены и идентифицированы, используя стандартные способы для оценки структуры.
Дополнительные варианты, иллюстрирующие каталитический способ гликозилирования.
Следующие примеры относятся к способам гликозилирования, занимающим промежуточное положение, по каталитическим количествам сульфоновой кислоты, включая реакции, вовлекающие силилированные гликозильные акцепторы. 1,2,3,4-тетра-0-ацетил-6-0-(триметилсилил)-D- гликопираноза (2b) Следующая процедура является типичной для всех процессов силилирования нуклеофила. Спирт 2a (600 мг, 1.85 ммоля, 1.0 эквив.) и триэтиламин (775 мкл, 3.43 ммоля, 3.0 экивив.) растворяют в разогнанном метиленхлориде и охлаждают до -78oC в аргоне. Триметилсилилтрифлат (394 мкл. 2.03 ммоля, 1.1 экивив.) добавляют к реакционной смеси. За ходом реакции следят с помощью ТСХ. Через 30 минут реакцию гасят выливая в насыщенный раствор бикарбоната натрия. Полученную двухфазную смесь экстрагируют хлористым метиленом (3 х 25 мл). Органические слои объединяют и сушат над безводным сульфатом натрия. Очистка с помощью флеш- хроматографии дает силильный эфир 2b (700 мг, 90%) в виде белого твердого вещества. Rf(TCX) = 0.6 (40% этилацетат-петролейный эфир). (270 MHz, CDCl3) δ 5.69 (d, J = 8.25 Hz, 1H, H-1), 5.25 (t, J = 9.24, Hz, 1H, H-3), 5.15 (t, J = 9.24 Hz, 1H, H-4), 5.12 (t, J = 8.24, 9.25 Hz, 1H, H-2), 3.8-3.7 (m, 3H, H-5, H-6.6'), 2.1-2 (m, 12H, OAc), 0.5 (s, 9H, TMS).
Альфа-альфа-1,1-димер пербензилированной глюкозы (5)
1,1-димер пербензилированной глюкозы идентифицируют следующим образом:
Rf (ТСХ) = 0,8 (25% этилацетат-петролейный эфир).
1H NMR (270 MHz, CDCl3) δ 7.4-7.1 (m, 40H), 5.19 (d, J = 3.30 Hz, 1H), 5.16 (d, JAB = 11.55 Hz, 1H), 5.02 (d, JAB= 10.89 Hz, 1H), 4.97 (d, JAB = 12.21 Hz, 1H), 4.90 (d, JAB = 10.89 Hz, 1H), 4.89 (d, JAB = 10.89 Hz, 1H), 4.83 (d, JAB = 10.89 Hz, 1H), 4.82 (d, J = 3.30 Hz, 1Han), 4.81 (d, JAB= 11.54 Hz, 1H), 4.75 (d, JAB = 12.21 Hz, 1H), 4.69 (d, JAB = 12.21 Hz, 1H), 4.59 (bs, 2H), 4.55 (d, JAB = 11.88 Hz, 1H), 4.52 (d, JAB = 10.88 Hz, 1H), 4.49 (bs, 2H), 4.49 (bs, 2H), 4.34 (d, JAB=12.21 Hz, 1H), 4.21 (m, 1H), 4.15 (t, J = 9.24, 9.47 Hz, 1H), 3.82 (t, J = 9.24, 9.9, Hz, 1H), 3.7-3.45 (m, 7H).
Синтез 3,4,6-три-0-бензил-2-деокси-D-гликопиранозил-(1,6)-1,2,3,4-тетра-0-ацетил-бета-D-глюкопираноза (7).
Следующая процедура является типичной для всех реакций гликозилирования, проводимых в катализированных кислотой условиях, используя трифликовую кислоту: сульфоксид 6 (140 мг, 0,258 ммоля, 1,5 эквив.) и нуклеофил 2 (69 мг, 0,172 ммоля, 1,0 эквив.) предварительно смешивают и тщательно сушат с помощью тройной "азеотропии" с перегнанным толуолом. Затем исходные вещества растворяют в 8 мл свежеприготовленного метиленхлорида и добавляют в аргоне в 25 мл склянку, высушенную в пламени. Склянку охлаждают до - 78oC, используя баню с ацетон/сухой лед. К этому раствору добавляют метилпропиолат (230 мкл, 2,58 ммоля, 15 эквив.) в качестве акцептора сульфеновой кислоты.
Затем раствор перемешивают при -78oC в течение 2 минут, добавляют трифликовую кислоту (1,1 мкл, 0,0129 ммоля, 0,05 эквив.). За ходом реакции следят с помощью ТСХ (25% этилацетат-петролейный эфир). Реакционную смесь медленно нагревают до -30oC в течение периода около 2 часа и затем гасят выливая в насыщенный раствор бикарбоната натрия (25 мл). Полученную смесь экстрагируют метиленхлоридом (3 х 15 мл). Органические экстракты объединяют, сушат безводным сульфатом натрия и концентрируют. Флэш хроматография (5% этилацетат-петролейный эфир) дает дисахарид 7 (116 мг, 88%) в виде белого твердого вещества. Rf (ТСХ) = 0,3 (25% этилацетат-петролинейный эфир).
1H NMR (270 MHz, CDCl3) δ 7.4-7.2 (m, 15H), 5.75 (d, J = 8.25 Hz, 1H, H-a), 5.32 (t, J = 9.24 Hz, 1H, H-c), 5.21 (t, J = 9.57 Hz, 1H, H-d), 5.20 (t, J = 8.25, 9.24 Hz, 1H, H-b), 4.99 (d, J = 2.64 Hz, 1H, H-1), 4.95 (d, JAB = 11.21 Hz, 1H), 4.71 (bs, 2H), 4.66 (d, JAB = 11.87 Hz, 1H), 4.59 (d, JAB = 11.22 Hz, 1H), 4.55 (d, JAB = 11.88 Hz, 1H0, 4.10 (m, 1H, H-e), 3.85-3.50 (m, 7H), 2.36 (dd, J = 4.94, 12.86 Hz, 1H), 2.15 (s, 3H), 2.10 (s, 3H), 2.05 (s, 3H), 1.95 (s, 3H), 1.70 (m, 1H).
Метил-6-деокси-3,4-изопропилиден-2-0-(триметилсилил)-бета-D- галактопиранозид (8)
Соединение 8 синтезируют из D-фукозы путем следующей трехстадийной последовательности реакций: I) Метано, H+; II) ацетон, орто-фосфорная кислота (кат); III) TMSOTf, триэтиламин, метиленхлорид, -78oC. Rf (ТСХ) = 0,5 (15% этилацетат-петролейный эфир).
1H NMR (CDCl3 270 MHz) δ 4.03 (d, J = 8.24 Hz, 1H, H-1), 3.98 (dd, J = 5.28, 7.26 Hz, 1H), 3.98 (dd, J = 5.40, 1.98 Hz, 1H), 3.84 (dq, J = 1.98, 6.6 Hz, 1H, H-5), 3.49 (s, 3H, OCH3), 3.47 (dd, J = 7.59, 5.49 Hz, 1H), 1.48 (s, 3H, CH3), 1.38 (d, J = 6.6 Hz, 3H, CH3) 1.31 (s, 3H, CH3), 0.5 (s, 9H, TMS). 13C NMR (CDCl3, 67.9 MHz) δ 109.38, 103.70, 80.52, 76.48, 74.45, 68.58, 56.64, 28.02, 26.34, 16.49, 0.31.
3,4,6-три-0-бензил-2-D-глюкопиранозил-альфа-(1-2)-метил-6-деокси- 3,4-изопропилиден-бета-D-галактопиранозид (9) Rf (ТСХ) = 0,5 (25% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MHz) α anomer δ 7.4 -7.15 (m, 15H), 5.30 (d, J = 2.97 Hz, 1H), 4.86 (d, JAB = 10.89 Hz, 1H), 4.64 (d, J = 1.65 Hz, 2H), 4.62 (d, JAB = 12.20 Hz, 1H), 4.53 (d, JAB = 10.89 Hz, 1H), 4.45 (d, JAB = 12.20 Hz, 1H), 4.06 (d, J = 8.25 Hz, 1H), 3.70 (t, J = 9.56 Hz, 1H), 3.46 (s, OCH3), 2.02 (ddd, J = 0.99, 4.95, 12.87 Hz, 1H), 1.65 (dt, J = 3.62, 12.87 Hz, 1H), 1.40 (s, CH3), 1.36 (d, J = 6.6 Hz, 3H), 1.24 (s, 3H). 13C NMR (CDCl3, 67.9 MHz) δ 138.91, 138.83, 138.32, 128.28, 128.23, 127.92, 127.79, 127.45, 127.42, 127.38, 109.35, 103.95, 97.45, 78.39, 78.20, 77.70, 76.31, 76.12, 74.95, 73.45, 71.73, 70.53, 68.73, 68.48, 56.58, 35.47, 28.08, 26.34, 16.49.
Фенил-4-0-ацетил-2,3,6- тридеокси-1-сульфонил- альфа-L-эритро- гексопиранозид (10)
Соединение 10 синтезируют из L-рамнала в три стадии: а) водород (1 тм.), гидроокись палладия на угле, бензол; б) эфират трехфтористого бора, тиофенол, метиленхлорид, -78oC, до -60oC; c/mCPBA, метиленхлорид, -78oC до -60oC. Rf (ТСХ) = 0,4 (25% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MНz) β anomer (sulfide) δ 7.5-7.2 (m, 5H), 5.54 (d, J = 4.95 Hz, H-4), 4.81 (dd, J = 1.98, 12.1 Hz, 1H, H-1), 4.6 (m, 2H, H-3, 3'), 4.3 (m, 1H, H-5), 2.2 (m, 2H, H2,2'), 2.1 (s, 3H, OAc), 1.19 (d, J = 5.94 Hz, 3H, CH3). 13C NMR (CDCl3, 67.9 MHz) β anomer (sulfide) δ 169.80, 130.98, 130.73, 128.58, 128.49, 83.79, 67.15, 29.34, 25.34, 20.82, 18.00, 17.44.
4-0-ацетил-2,3,6-тридеокси-L-эритро-гексапиранозил-альфа-(1-6)- 1,2,3,4-тетра-0-ацетил-бета-D-глюкопираноза (11)
Rf 0,3 (25% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MHz) α anomer δ 5.63 (d, J = 7.92 Hz, 1H), 5.20 (t, J = 9.24 Hz, 1H), 5.19 (t, J = 9.24, 9.57 Hz, 1H), 5.09 (d, J = 7.91 Hz, 1H), 5.03 (t, J = 9.87 Hz, 1H), 4.61 (dd, J = 1.98, 8.58 Hz, 1H), 4.33 (ddd, J = 4.61, 10.23 Hz, 1H), 3.94 (dd, J = 3.96, 11.22 Hz, 1H), 3.71 (m, 1H), 3.56 (dd, J = 2.96, 11.22 Hz, 1H), 2.05 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.96 (s, 3H), 1.95 (s, 3H), 1.11 (d, J = 6.59 Hz, 3H), 13C NMR (CDCl3, 67.9 MHz) δ 170.19, 170.12, 169.18, 169.15, 168.95, 100.85, 91.79, 73.03, 72.96, 70.34, 68.60, 66.01, 29.54, 26.88, 21.07, 20.76, 20.64, 20.54, 20.50, 18.01, 17.72.
Фенил-3,4-бис-0-(4-метоксибензоил)-1-сульфинил-D-дигитоксоза (13)
Соединение 13 синтезируют из дигитоксозы путем следующей трехстадийной последовательности реакций: I) pOMeC6H4COCl, пиридин; II) тиофенол, эфираттрехфтористого бора, метиленхлорид; III) mCPBA. Rf (TCX)=0.2 (40% этилацетат-петролейный эфир).
1H NMR (CDCl3, 270 MHz) α anomer (sulfoxide) δ 8.09 (d, J=8.91 Hz, 2H), 7.87 (d, J= 9.24 Hz, 2H), 7.70 (m, 2H), 7.55 (m, 3H), 6.94 (d, J=8.90 Hz. 2H), 6.85 (d, J= 8.91 Hz, 2H), 5.84 (q, J=3.31 Hz, 1H), 5.10 (dd, J=2.63, 2.97, 1H), 5.04 (m, 1H), 4.49 (dd, J=1.64, 5.13 Hz, 1H), 3.83 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 1.28 (d, J=5.95 Hz, 3H).
Фенил-3,4,0-(4-метоксибензоил)-бета-D-дигитоксозил-(0)-N- гидроксиэтил-карбамат (14)
Rf (TCX)= 0.45 (сс. аномер, 40% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) α anomer δ 8.03 (d, J=8.91 Hz, 2H), 7.79 (d, J=9.24 Hz, 2H), 6.88 (d, J=9.24 Hz, 2H), 6.77 (d, J=9.01 Hz, 2H), 5.60 (q, 1H), 5.12 (d, J=4.62 Hz, 1H), 4.95 (dd, J=2.97, 10.23 Hz. 1H), 4.78 (m, 1H), 4.18 (q, 2H), 4.17 (m, 1H), 3.84 (s, OCH3), 3.79 (s, OCH3), 2.37 (dd, J=3.3, 15.51 Hz, 1H), 2.19 (m, 1H), 1.26 (t, J=6.93 Hz, 3H), 1.25 (d, J=6.27 Hz, 3H), 13C NMR (CDCl3, 67.9 MHz) δ 165.48, 165.17, 163.53, 163.45, 157.42, 131.97, 131.75, 122.65, 121.92, 113.62, 113.59, 100.69, 100.61, 72.21, 65.90, 63.65, 62.08, 55.40, 32.04, 17.53, 14.44
и Rf= (TCX)=0.5 (бета аномера 40% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) β anomer δ 7.92 (d, J=8.91 Hz, 2H), 7.78 (d, J=8.91 Hz, 2H), 6.89 (d, J=8.91 Hz, 2H), 6.77 (d, J=9.24 Hz, 2H), 5.74 (q, 1H), 5.23 (dd, J= 2.31, 9.23 Hz, 1H), 4.93 (dd, J=2.97, 9.23 Hz, 1H), 4.23 (q, 1H), 4.16 (q, 2H), 4.17 (m, 1H), 3.85 (s, OCH3), 3.79 (s, JCH3), 2.36 (m, 1H), 2.04 (m, 1H), 1.29 (d, J=6.27 Hz, 3H), 1.23 (t, J=6.93 Hz, 3H). 13C NMR (CDCl3, 67.9 MHz) δ 165.10, 164.91, 163.61, 163.55, 131.85, 122.18, 121.83, 113.74, 113.59, 101.77, 72.31, 69.21, 67.41, 62.19, 60.34, 55.44, 55.38, 33.29, 18.01, 14.38.
Фенил-2,3,-0-бензоил-6-деокси-1-тио-бета-L-галактопиранозид (15)
Соединение 15 синтезируют из L-фукозы путем следующей четырехстадийной последовательности реакций: I) этилацетат, пиридин; II) тиофенол, эфират трехфтористого бора; III) метоксид натрия, метанол, амберлитовая H+смола; IV) бензоилхолорид, DMAP. Rf (TCX)=0.6; (40% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 8.0-7.9 (m, 4H), 7.55-7.2 (m, 1H), 5.64 (t, J=9.89 Hz, 1H), 5.27 (dd, J=2.97, 9.9 Hz, 1H), 4.86 (d, J=9.9 Hz, 1H), 4.09 (d, J= 2.97 Hz, 1H), 3.87 (q, J=6.27 Hz, 1H), 1.40 (d, J=6.27 Hz, 3H).
Фенил-3,4-0-ацетил-2,6-дидеокси-1-сульфинил-L-галактопиранозид (16)
Соединение 16 получают из 1,3,4-три-0-ацетил-2-деокси-альфа-L-фукозы путем следующей двухстадийной последовательности реакций: I) тиофенол, эфират трехфтористого бора и II) mCPBA. Rf (TCХ для сульфида)=0.3 (15% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) a sulfide δ 7.5 - 7.2 (m, 5H), 5.73 (d, J=5.61 Hz, 1H, H-1), 5.29 (m, 1H, H-3), 5.23 (bs, 1H, H-3), 4.56 (q, J= 6.6 Hz, 1H, H-5), 2.49 (dt, J=5.94, 12.87 Hz, 1H, H-2ax), 2.38 (s, 3H, OAc), 2.06 (m, 1H. H-2eq), 1.99 (s, 3H, OAc), 1.13 (d, J=6.6 Hz. 3H, CH3). 13C NMR a sulfide (CDCl3, 67.9 MHz) δ 170.51, 169.87, 159.54, 134.33, 124.54, 114.54, 84.58, 69.69, 67.18, 65.51, 55.22, 20.79, 20.60, 16.3.
и Rf (TCX для сульфоксида)=0.25 (30% этилацетат-петролейный эфир) (и данные ЯМР спектров) с (для альфа аномера сульфоксида) 1HNMR α anomer sulfoxide (CDCl3, 270 MHz) δ 7.6-7.3 (m, 5H), 5.43 (m, 1H), 5.26 (bd, J=2.64 Hz, 1H), 4.52 (d, J=5.61 Hz, 1H), 4.32 (q, J=6.6 Hz, 1H), 2.47 (dd, J=5.28, 14.18 Hz, 1H), 2.14 (dt, J=5.61, 14.19 Hz, 1H), 2.10 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.12 (d, J=6.26 Hz, 3H).
3,4-0-ацетил-2,6-дидеокси-L-галактопиранозил-альфа-(1-4)фенил-3,4-0-бензоил- 6-деокси-1-тио-бета-L-галактопиранозид (17)
Rf(TCX)=0.4 (30% этилацетат-петролейный эфир). 1H NMR (CDCl3, 270 MHz) δ 7.9 (m, 4H), 7.55-7.25 (m, 1H), 5.55 (t, J=10.23 Hz, 1H), 5.20 (dd, J=2.96, 10.22 Hz, 1H), 5.12 (m, 1H), 5.02 (bd, J=1.98 Hz, 1H), 4.94 (bs, 1H), 4.84 (d, J= 9.9 Hz, 1H), 4.15 (d, J=9.97 Hz, 1H), 3.96 (q, J=6.27 Hz, 1H), 3.86 (q, J=6.27 Hz, 1H), 2.04 (s, 3H), 2.0 (s, 3H), 1.94 (m, 2H), 1.35 (d, J=6.27 Hz, 3H), 0.33 (d, J= 6.27 Hz, 3H). 13C NMR (CDCl3, 67.9 MHz) δ 170.44, 169.62, 166.05, 164.95, 133.51, 133.36, 133.01, 131.49, 129.78, 129.62, 129.12, 128.65, 128.44, 128.27, 128.18, 99.49, 85.42, 76.86, 75.27, 74.68, 69.56, 67.47, 66.50, 65.27, 29.70, 20.85, 20.56, 17.32, 15.77.
Синтез бета-связанного дисахарида A в твердой фазе.
К 10 мл раствора ДМФА, содержащего 0,338 (0.350 ммоля) 2,3,4-трибензил-6-тритилгалоктоза-1-п-гидрокси-фенилтиогликозид цезий ацетат (X, фиг. 6) добавляют 0.356 г (0.385 ммоля C1 эквив, 1.1 эквив.) смолы Меррифилда (BACHEM Bioscience). Эту смесь перемешивают за счет действия шейкера в течение 24 часов в атмосфере аргона при 75oC. В этом время полимер выливают в воронку Гоша с крупнозернистым фильтром и многократно промывают метанолом и метиленхлоридом. Затем воронку сушат в течение 4 часов в лиофилизующем сосуде при 20 миллиторах. Регистрируют изменение массы 0.244 г, которое расчитывают как 85% химический выход по отношению к соли цезия.
Полимер в воронке Гоша затем обрабатывают путем фильтрования в вакууме при умеренной скорости потока, 40 мл 10% трифторуксусной кислоты (ТФУК) в метиленхлориде. (ТФУК используют для удаления тритильных защищающих групп). Затем полимер многократно промывают метанолом и метиленхлоридом. Колонку вместе с связанным со смолой нуклеофилом сушат затем в течение 4 часов в лиофилизующем сосуде при 20 миллиторах до образования густой массы "massing". Впоследствии измеряют изменение массы 0.065 г, которое расчитывают как 83% химический выход по отношению к цезиевой соли. Затем расчитывают концентрацию нуклеофила, связанного со смолой (смола-X, фиг. 6), которая является равной 0.544 ммоля (200 мг), т.е. 0.11 ммоля модифицированной смолы лиофилизуют в течение ночи в реакционном сосуде и затем в течение 1 часа продувают аргоном. Затем смолу суспендируют в 5 мл метиленхлорида. Растворяют 4 эквивалента (0.44 ммоля) 2-пивалоил-3,4-бензил-6-п-метоксибензил-галактозилсульфоксида (Y, фиг. 6) и 6 эквивалентов (0.66 ммоля) 2,6-дитрет-бутил-4-метилпиридина в 5 мл метиленхлорида и добавляют с помощью канюли в реакционный сосуд. Смесь осторожно перемешивают потоком аргона в течение 30 минут при комнатной температуре и затем реакционный сосуд погружают в холодную баню и позволяют охлаждаться до -60oC. 2 эквивалента (0.22 ммоля) трифликового ангидрида разбавляют стократным количеством (объем/объем) метиленхлорида и медленно добавляют (в течение 15 минут) в реакционный сосуд. Полученную реакционную смесь осторожно перемешивают в течение 1 часа.
После окончания реакции, как показывает способ гидролиза Hg (II) и ТСХ анализом, растворитель и несвязанные реагенты удаляют из реакционного сосуда и смесь смолы многократно промывают метанолом и последующей промывкой метиленхлоридом. Впоследствии смесь смолы суспендируют в 15 мл метиленхлорида и затем обрабатывают избытком Hg(OCOCF3) в течение 8 часов для расщепления гликозидной связи со смолой. (Замечание: только 5 минут требуется для удаления достаточной части продукта из смолы с контролем за ходом реакции с помощью ТСХ анализа). Растворителю позволяют выделяться из смолы.
Затем используют дополнительный растворитель для промывания смолы. Затем фильтраты объединяют, трижды экстрагируют водой и концентрируют выпариванием. Желаемый бета-связанный дисахарид выделяют с помощью флеш- хроматографии на силикагеле. Не выделяют альфа-связанный дисахарид из реакции.
Тиосахар X на фиг. 6 получают из легкодоступной 1,6-дигидроглюкозы обработкой бензилбромидом с последующим кислотным гидролизом (серная кислота-тетрагидрофуран-вода), тритилированием (тритилхлорид-пиридин) более реакционноспособного первичного спирта и обработкой полученного лактола дисульфидом XX и три-н-бутилфосфином (т.е. стандартная процедура для получения тиофенилгликозидов из лактолов). Дисульфид XX получают взаимодействием дисульфида легко доступного 4-гидрокситиофенола с альфа-бромфенилацетатом.
Дисульфид XX
Сульфоксид Y на фиг.6 получают из легко доступной пента-ацетилированной галактозы, используя следующую последовательность реагирующих веществ: 1) BF3 эферет-тиофенол; 2) гидроксид; 3) ацетон-H+; 4) п-метоксибензилбромидгидрид натрия; 5) пивалоилхлорид; 6/nCPBA. Каждая стадия хорошо известна в литературе и реакции проводят в стандартных условиях (см. список "Standart References", в разделе 6.15, который вводится здесь ссылкой). Дисахарид, полученный из реакции X и Y подвергают метанолизу (для удаления его из смолы) и характеризуют с помощью 1H-ЯМР спектроскопии. Соответствующие данные включают: (CDCl3) 5.58 м.д. (д, J=5.3 Гц, H1 тиосахара), 5.47 м.д. (дд, J=7.9, 10.12 Гц, H2 C2 пивалоилированного сахара, 4.45 м.д. (д, частично перекрытый, H1 C2 пивалоилированного сахара).
Синтез альфа-связанного дисахарида в твердой фазе.
Натриевую соль гликозильного акцептора (X, фиг.7) присоединяют к смоле Мерифилда с помощью стандартного способа (ДМФА, 80oC, 24 часа). Затем проводят последующее сочетание и промывание (как описано в примере 6.10), смолу лиофилизуют и взвешивают. Нагрузку расчитывают при 0.52 ммоля/г из массы достигнутого увеличения. 200 мг (т.е. 0.1 ммоля) модифицированной смолы лиофилизуют в течение ночи в реакционном сосуде (фиг.7) и затем продувают в течение 1 часа арогоном. Затем смолу суспендируют в 5 мл метиленхлорида - эквивалента (0.4 ммоля) пербензилированного фукозилсульфоксида Y и 6 эквивалентов 2,6-дитрет-бутил-4-метилпиридина растворяют в 5 мл метиленхлорида и добавляют с помощью сифона в реакционный сосуд. Смесь осторожно перемешивают потоком аргона в течение 30 минут при комнатной температуре и затем реакционный сосуд погружают в холодную баню, позволяя охладиться до -60oC. 2 эквивалента (0.22 ммоля) трифликового ангидрида разбавляют метиленхлоридом в 100 раз и медленно добавляют сифоном (в течение 15 минут) в реакционный сосуд. Реакционную смесь осторожно перемешивают в течение 1 часа. Растворитель и непрореагировавшие реагенты затем убирают из реакционного сосуда и смесь смолы промывают многократно метанолом с последующей промывкой метиленхлоридом. После этого смесь смолы суспендируют в 15 мл метиленхлорида и обрабатывают избытком Hg(OCOCF3) в течение 8 часов с отщеплением гликозидной связи со смолой (требуется около 5 минут для удаления достаточного количества продукта из смолы с регистрированием хода реакции с помощью ТСХ анализа). Растворителю позволяют вытекать из смолы как ранее. Затем смолу промывают дополнительным количеством растворителя и фильтраты объединяют, экстрагируют трижды водой и концентрируют выпариванием. Флеш-хроматография на силикагеле дает только желаемый альфа-связанный дисахарид.
Тиосахарид X фиг.7 получают из легкодоступного соответствующего глюкозамина обработкой следующими реагентами: 1) фталевый ангидрид; 2) уксусный ангидрид; 3) тетрахлоротин-4-гидрокситиофенол; 4) гидроксид; 5) бензальдегид-H+; 6) гидрид натрия в условиях, которые являются стандартными в этой области (см. раздел 6.15).
Сульфоксид Y на фиг.7 получают из перацетилированной фукозы обработкой исходного вещества последовательно эфираттрехфтористого бора-тиофенол, с последующей обработкой гидроксидом и с последующей обработкой бензилбромидом и затем mCPBA. Все эти стадии являются стандартными и хорошо известны в литературе.
Дисахарид, полученный из реакции X и Y затем обрабатывают Hg(ОCОCF3) (для удаления его из смолы), характеризуют с помощью 1H-ЯМР спектров. Соответствующие данные: CDCl3 5.6 м.д. (очевидный триплет, J=7.6 Гц, H1 C2 фталимидосахара), 3.35 м. д. (д, J=7.6 Гц, лактол OH, т.е. фталимидо сахар после гидролитического удаления из смолы вес Hg (II), 4.9 м.д. (д, J=2.8 Гц, H1 производного фукозы).
Твердофазный синтез сахарида Льюисовского X.
Натриевую соль гликозильного акцептора (X, фиг.8) присоединяют к смоле Мерфилда, используя стандартный способ (ДМФА, 80oC, 24 часа). После использование обычной процедуры связывания, описанной детально в примере 6.10, безводную смолу суспендируют в 5 мл метиленхлорида. 4 эквив. 2,3,4,6-пивалоилированного галактозилсульфоксида Y и 6 эквив. основания (как выше) растворяют в 5 мл метиленхлорида.
Затем раствор реагентов добавляют к смоле и в реакционную смесь охлаждают до -60oC. Затем добавляют 3 эквивалента трифликового ангидрида, разбавленного в 100 раз (объем/объем) метиленхлоридом.
Через 30 минут смолу "дренируют" и многократно промывают метиленхлоридом и метанолом. Затем смолу суспендируют в 5 мл метиленхлорида и охлаждают до 0oC. Затем добавляют 5 мл раствора 1:2 трифторуксусная кислота/метиленхлорид и смолу осторожно перемешивают в течение 5 часов. Затем смолу "дренируют" и многократно промывают метиленхлоридом и метанолом. Вслед за последней промывкой безводным метиленхлоридом, смолу суспендируют в 5 мл безводного метиленхлорида. 4 эквивалента 2,3,4-триэтилсилилилфукозилсульфоксида Z и 6 эквивалентов затрудненного основания (т.е. такого, как использовано выше) растворяют в 5 мл метиленхлорида и добавляют к смоле. Реакционную смесь охлаждают до 60oC. 2 эквивалента трифликового ангидрида разбавляют в 100 раз метиленхлоридом и медленно добавляют с помощью сифона в реакцию. После осторожного перемешивания в течение 30 минут, смолу "дренируют" и промывают. Трисахарид затем удаляют и выделяют из смолы, используя Hg(OCOCF3), как описано выше в примере 6.10. Тиосахар X на фиг. 8 получают из легкого доступного глюкозамина обработкой 1) фталевым ангидридом; 2) уксусным ангидридом; 3) тетрахлоротин-4-гидрокситиофенолом; 4) бензилбромидом; 5) гидроксидом; 6) бензальдегидом-H+; 7) хлорметилметиловым эфиром (MOM хлорид); 8) вода H+; 9) пивалоилхлоридом; 10) гидрированием, гидроокись палладия: 11) гидридом натрия. Вновь все стадии являются стандартными, включая условия для снятия защиты бензильной защищающей группы на 4-гидроксифенилгликозиде, условия которой являются типичными для дебензилирования. В стадии дебензилирования не наблюдается расщепления связи сахара с серой. Сульфоксид Y на фиг. 8 получают обработкой перпивалоилированной галактозы эфиратом трехфтористого бора-тиофенолом с последующей обработкой mCPBA. Сульфоксид Z на фиг. 8 получают обработкой перацетилированной фукозы эфиратом трехфтористого бора-тиофенолом с последующей последовательной обработкой гидроксидом, триэтилсилилхлоридом и mCPBA.
Аналогично легко синтезируют другие сахара с Льюисовской группой крови, включая, но не ограничиваясь ими, группы Льюиса A и Льюиса B. Дополнительные эксперименты, проведенные в твердой фазе. Относительно фиг.16, в частности, цезий фторид (157.2 мл, 1.03 ммоля, 1.1 эквив.) и смолу Мерифилда (1 мол. эквив. C1 на грамм) (1.0975 г, 1.10 ммоля, 1.1 эквив.) добавляют к раствору свежеперегнанного 1 (803.5 мг, 0.904 ммоля, 1.0 эквив.) в ДМФА (20 мл). Полученную счуспензию механически встряхивают в атмосфере аргона при 60oC в течение 24 часов. Модифицированную сахаром смолу затем выделяют с помощью вакуумного фильтрования и промывают ДМФА (3х10 мл), метанолом (3х10 мл) и метиленхлоридом (5х10 мл) и затем сушат в вакууме в течение ночи с получением смолы 2 (1.4738 г), расчитанной на достигнутую массу увеличения, или 0.34 ммоля/г). ИК (КВr пластина) 1734 см-1 (v, C=O).
Смолу 2 затем промывают 10% трифторуксусной кислотой в метиленхлориде (20 мл) для удаления защищающей группы. Впоследствии смолу, с которой снята защита, 3 промывают метиленхлоридом (1010 мл) и сушат в вакууме в течение ночи. ИК (KBr пластина) 1734 см-1 (v, C=O), 3430 см-1 (шир., v O-H). Для получения смолы 5 смолу 3 (95.4 мг) добавляют к раствору сульфоксида 4 (122.1 мг, 0.156 ммоля, 1.0 эквив.) и 2,6-дитретбутил-4-метилпиридина (98.2 мг, 0.468 ммоля, 3.0 эквив.) в метиленхлориде (7.0 мл) в суспензию охлаждают до -78oC. Через 10 минут добавляют трифликовый ангидрид (13.1 мкл, 0.078 м моля, 0.5 эквив.) в метиленхлориде (5.0 мл) по каплям в течение 20 минут. Через 10 минут после окончания добавления ангидрида реакционную смесь нагревают до -60oC в течение 20 минут. Реакцию гасят добавлением насыщенного раствора бикарбоната натрия. Смолу собирают путем фильтрования отсасыванием и промывают метанолом (2 х 10 мл), водой (1х10 мл) и метиленхлоридом (10 х 10 мл). Смолу продукта 5 затем сушат в вакууме в течение ночи, после чего повторяют гликолизирование.
Смолу продукта 6 получают следующим образом: смолу 5 (112.7 мг) промывают 10% трифторуксусной кислотой в метиленхлориде (20 мл). И затем промывают метиленхлоридом (10 х 10 мл) и сушат в вакууме в течение ночи.
Затем смолу 7 получают из смолы 6 следующим образом: смолу 6 (110.7 мг) добавляют к раствору сульфоксида 4 (123.5 мг, 0.158 ммоля, 1.0 эквив.) и 2,6-дитретбутил-4-метилпиридина (99.3 мг, 0.474 ммоля, 3.; 0 эквив.) в метиленхлориде (37.0 мл) и суспензию охлаждают до -78oC. Через 10 минут добавляют трифликовый ангидрид (13.3 мкл, 0,079 ммоля, 0.5 эквив.) в метиленхлориде (5.0 мл) по каплям в течение 20 минут. Через 10 минут после окончания добавления реакционную смесь нагревают до -60oC в течение 20 минут. Реакцию гасят добавлением насыщенного раствора бикарбоната натрия. Смолу выделяют фильтрованием и промывают метанолом (1х10 мл) и затем метиленхлоридом (10 х 10 мл). Смолу продукта 7 сушат в вакууме в течение ночи, после чего повторяют гликозилирование.
Наконец, соединение 8 получают из смолы 7 следующим образом: смолу 7 (113.6 мг) промывают 1-10% трифторуксусной кислотой в метиленхлориде (20 мл). Затем ее промывают метиленхлоридом (10 х 10 мл) и суспендируют в пиридине (10 мл). К суспензии в атмосфере аргона добавляют уксусный ангидрид (183.7 мкл, 1.932 ммоля, 1.0 эквив.) и 4-диметиламинопиридин (4.7 мг, 0.039 ммоля, 0.02 эквив. ). Смесь перемешивают в течение ночи и затем гасят добавлением метанола (200 мкл). Смолу выделяют фильтрованием и промывают метанолом (2 х 10 мл) и метиленхлоридом (10 х 10 мл). Свободное соединение 8 выделяют из смолы обработкой бис(трифторацетатом) ртути (II) во влажном метиленхлориде, как описано ниже, например, для синтеза дисахарида 13. (Соединение 8) 1H-ЯМР (500 МГц, CDCl3). δ 4.42 (d, J = 8.06 Hz, H''1), 4.49 (d, J = 8.06 Hz, H'1), 6.27 (d, J = 3.67 Hz, H'1). 13C NMR (69 MHz, CDCl3) δ 91.6, 101.0, 101.3
(аномерный углерод).
Твердофазный синтез дисахаридов 13 и 16.
Согласно схеме фиг. 17 Pht-защищенный N-ацетилглюкозаминовый остаток 9 присоединяют к твердой подложке следующим образом: смолу Мерифилда (1.4075 г), сшитую на 1%, хлорметилированный стирол/дивинилбензольный сополимер, приблизительно 1 ммоль C1/г, Aldrich) добавляют к раствору свежеприготовленного 9 (1.42 г, 2.693 ммоля) в ДМФА (20 мл). Суспензию встряхивают механически при 60oC под аргоном в течение 24 часов. Смолу, модифицированную сахаром, выделяют фильтрованием, промывают ДМФА (5 х 10 мл), метанолом (3 х 10 мл) и метиленхлоридом (10 х 10 мл) и сушат в вакууме в течение ночи с получением 1.7466 г смолы, присоединившей 10. Расчитывают уровень нагрузки, который составляет 0.414 ммоль/г (на массу достигнутого увеличения). ИК (KBr) 1670, 716 см-1 (v, C = O), 3470 см-1 (v, C = O).
Смолу 10 подвергают реакции сочетания с сульфоксидом 11, фукозного звена, с получением 12 (альфа-гликозидная связь) следующим образом: соединение 10 (218.4 мг) добавляют к раствору сульфоксида 11 (204.1 мг, 0.3766 ммоля. 1.0 эквив.) и 2,6-дитретбутил-пиридина (236.7 мг, 1.1297 ммоля, 3.0 эквив. ) в метиленхлориде (7 мл). Суспензию охлаждают до -60oC и через 10 минут добавляют по каплям раствор трифликого ангидрида (31.7 мкл, 0.1883 ммоля, 0.5 эквив.) в метиленхлориде (5 мл) в течение 20 минут. Через 10 минут после окончания добавления реакционную смесь медленно нагревают до -30oC и через 30 минут гасят добавлением насыщенного раствора бикарбоната натрия. Смолу выделяют фильтрованием, промывают метанолом (2 х 10 мл), водой (2 х 10 мл), метанолом (10 мл) и метиленхлоридом (0 х 10 мл) и сушат в вакууме в течение ночи с получением 12.
Конечный продукт 13 получают следующим образом: Hg(OCOCF3)2 (120.5 мг, 0.2824 ммоля) и воды (несколько капель) добавляют к суспензии 12 (22.0 мг) в метиленхлориде (20 мл). Через 5 часов смесь фильтруют через хлопковую ткань. Фильтрат промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток чистят с помощью флеш- хроматографии (35% этилацетат-петролейный эфир) с получением 33.9 мг 13 (выход 59%) в виде бесцветного твердого вещества. Это твердое вещество может быть далее очищено с помощью RP-гель-проникающей тонкослойной хроматографии (C-18 колонка, 60 до 98% MeCN3 в воде в течение 30 минут), RT 15.7 мин. 1H-ЯМР (500 МГц, CDCl3) дельта 4.81 (д, J = 3.66 Гц, HI звена фукозы), 5.59 (д, J = 8.42 Гц звена глюкозамина). 13C-ЯМР (69 МГц, CDCl3) дельта 81.9, 93.7, 101.2 (атомы аномерного углерода).
Аналогично смола 10 может быть подвергнута реакции сочетания с галактозным звеном 14 с образованием смолы 15 (бета-гликозидная связь) следующим образом: соединение 10 (184.0 мг, 0.414 мэкв/г) добавляют к раствору сульфоксида 14 (208.0 мг, 0.325 ммоля) и 2,6-дитрет-бутилипиридина (204.3 мг, 0.975 ммоля) в метиленхлориде (7 мл). Суспензию охлаждают до -60oC и через 10 минут добавляют по каплям раствор трифликового ангидрида (31.7 мкл, 0.1883 ммоля, 0.5 эквив. ) в метиленхлориде (5 мл) в течение 20 минут. Через 10 минут после окончания добавления реакционную смесь медленно нагревают до -30oC и через 30 минут гасят добавлением насыщенного раствора бикарбоната натрия. Смолу выделяют фильтрованием, промывают метанолом (2 х 10 мл), водой (≈ 2 х 10 мл), метанолом (10 мл) и метиленхлоридом (10 х 10 мл) и сушат в вакууме в течение ночи получением 15. Для получения наилучших результатов, процедуру гликозилирования повторяют дважды.
Продукт удаляют из смолы, используя Hg (II) Hg(OCOCF3) (325.0 мг, 0.2824 ммоля) и воду (несколько капель) добавляют к суспензии 15 (222.0 мг) в метиленхлориде (20 мл). Через 5 часов реакции смесь фильтруют через хлопковую ткань. Фильтрат промывают насыщенным раствором бикарбоната, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток чистят с помощью флеш-хроматографии (35% этилацетат-петролейный эфир) с получением 19.4 мг 16 (28% выход). Этот продукт может быть очищен далее с помощью RP-гель-проникающей хроматографии (C-18 колонка, 60 - 95% MeCN в воде, в течение 30 минут), 1RT = 26.1 мин. H-ЯМР (500 МГц, CDCl3) дельта 4.69 (д, J = 7.69 Гц, H1 галактозного остатка), 5.32 (дд, J = 7.32, 8.8 Гц, H1 глюкозаминового остатка). 13C-ЯМР (69 МГц, CDCl3) дельта 93.5, 98,86 101.7 (аномерный углерод).
Стандартные ссылки.
Большинство превращений упомянутых выше (защита: бензилирование, бензилиденирование, ацетонирование, этерификация и карбо- или силилэтилирование; снятие защиты: дебензилирование, кислотный гидролиз бензилиденов или ацетонатов, основной гидролиз эфиров, удаление силильных групп с фторида или в кислых условиях) описывают в Binkley, R.W. Modern Carbohydrate Chemistry, MartcelDekker, Inc. New York, 198. Способы превращения лактолов или аномерных эфиров или аномерных эфиров с тиофенильными группами (с получением тиофенильных гликозидов) хорошо известны. См., например, Ferrier et al., Carbohydr. Res. 1973, 27, 55; Mukaiyama et al., Chem. Lett., 1979, 487; Van Cleave Carbohydr. Res. 1979, 70, 161; Hanessian et al., Carbohydr. Res. 1980, 80, C17.; Garegg et. al., Carbohydr. Res.& 1983, 116, 162; and Nicolau et al., J. Am. Chem. Soc., 1983, 105, 2430.
Из принципиально установленного здесь должно быть очевидно специалисту в этой области, что способность управлять реакционной способностью гликозильных доноров и гликозильных акцепторов для контроля порядка, в котором будет протекать гликозилирование, можно использовать синтез многих других олигосахаридов или гликоконъюгатов быстро, эффективно и с высоким выходом либо при гомогенных (в растворе) либо при гетерогенных (в твердой фазе) условиях.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФУКОЗИЛИРОВАННОГО УГЛЕВОДА, СПОСОБ ПОЛУЧЕНИЯ ФУКОЗИЛИРОВАННОЙ СИАЛИЛИРОВАННОЙ УГЛЕВОДНОЙ МОЛЕКУЛЫ, РЕАКЦИОННАЯ СИСТЕМА IN VITRO | 1992 |
|
RU2125092C1 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2608315C2 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГРИППА | 1997 |
|
RU2169145C2 |
| ДИАРИЛГИДАНТОИНЫ | 2021 |
|
RU2828689C2 |
| ДИАРИЛГИДАНТОИНЫ | 2011 |
|
RU2638833C2 |
| ТЕТРАГИДРОИНДЕНОИНДОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИОКСИДАНТНОЙ АКТИВНОСТЬЮ, ПРОТИВООКИСЛИТЕЛЬНАЯ КОМПОЗИЦИЯ И СПОСОБ СТАБИЛИЗАЦИИ СОЕДИНЕНИЙ, ВОСПРИИМЧИВЫХ К ОКИСЛЕНИЮ | 1990 |
|
RU2104270C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОЗИДОВ АКРИЛАТНЫХ ПРОИЗВОДНЫХ С ИСПОЛЬЗОВАНИЕМ ПОЛИСАХАРИДОВ И ГЛИКОЗИДАЗ ИЛИ ГЛИКОЗИЛТРАНСФЕРАЗ | 2011 |
|
RU2570556C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
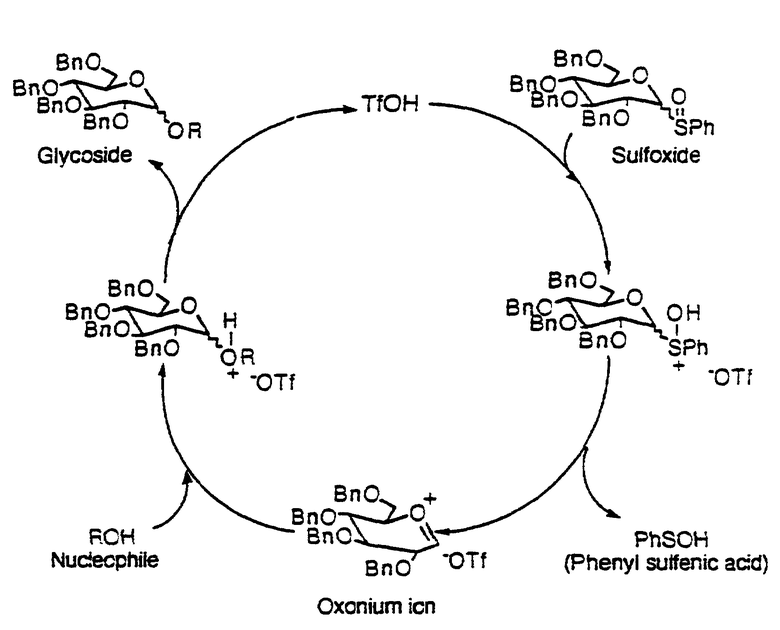
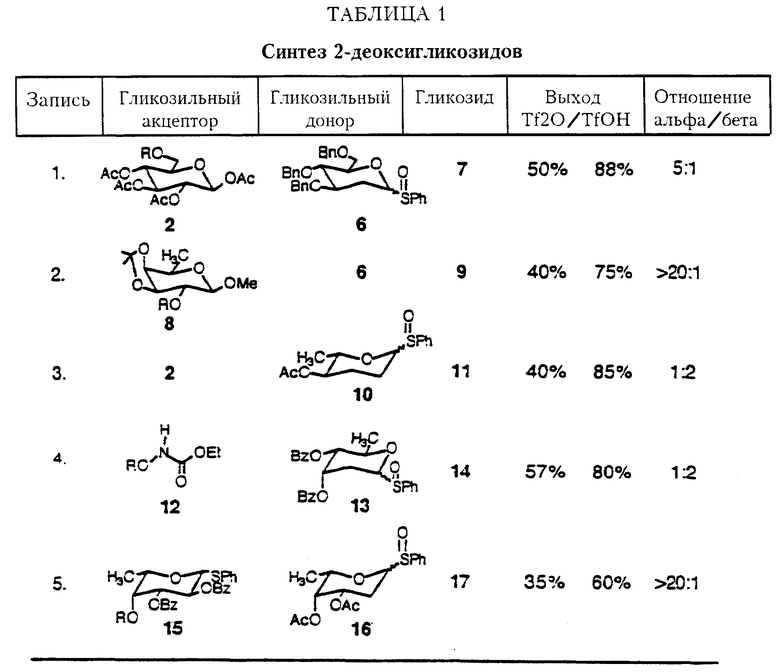
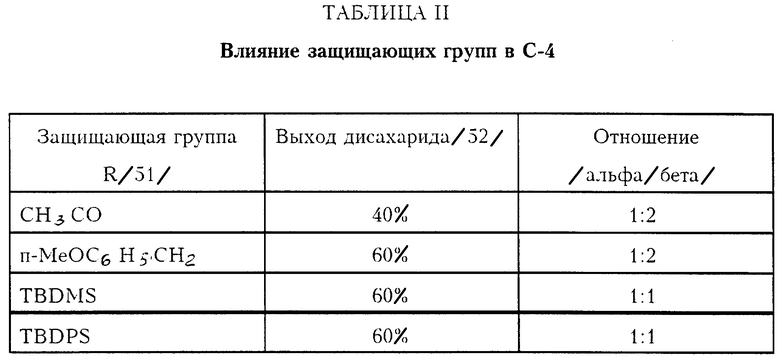
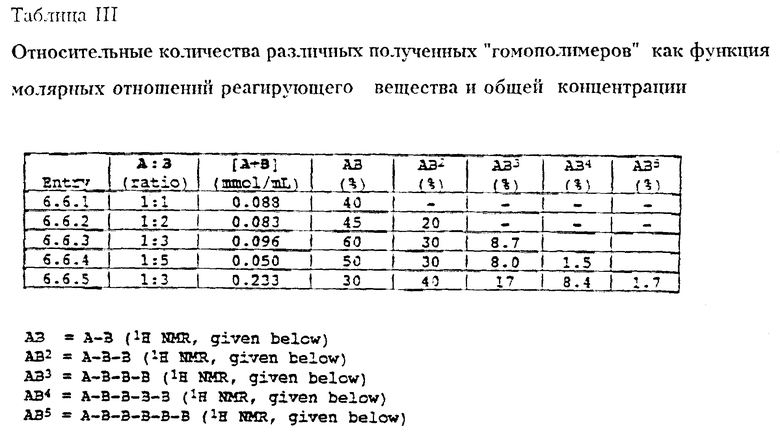
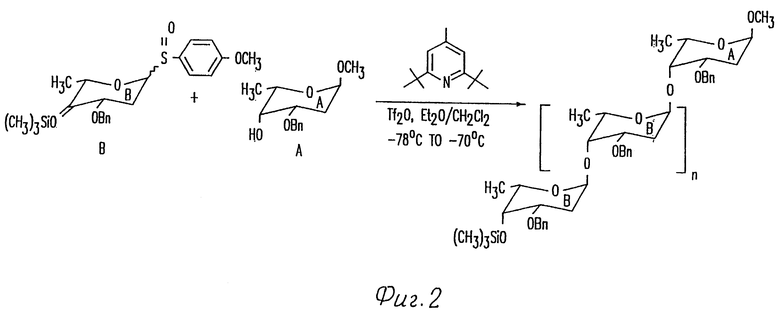
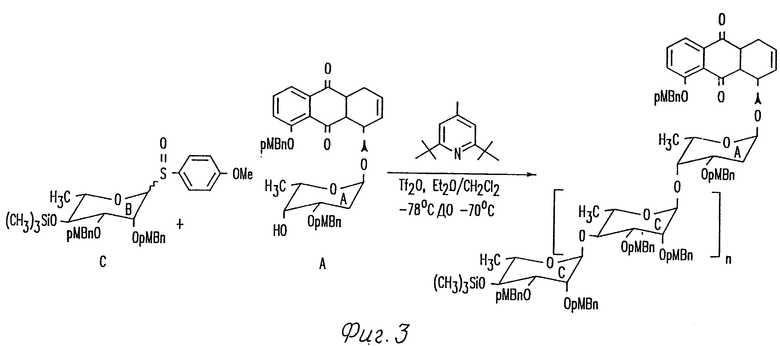
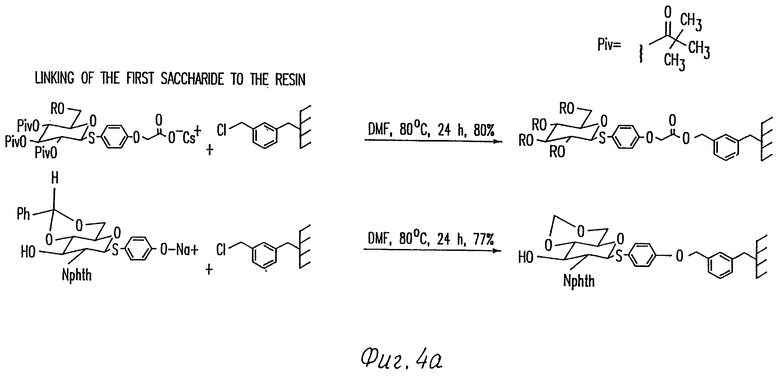
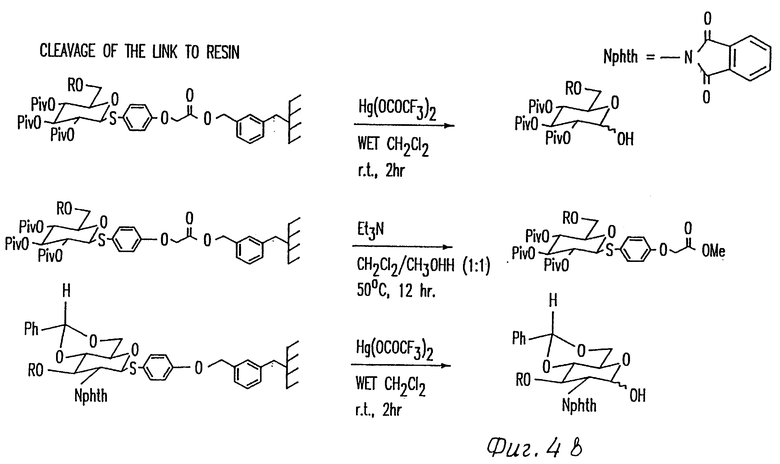
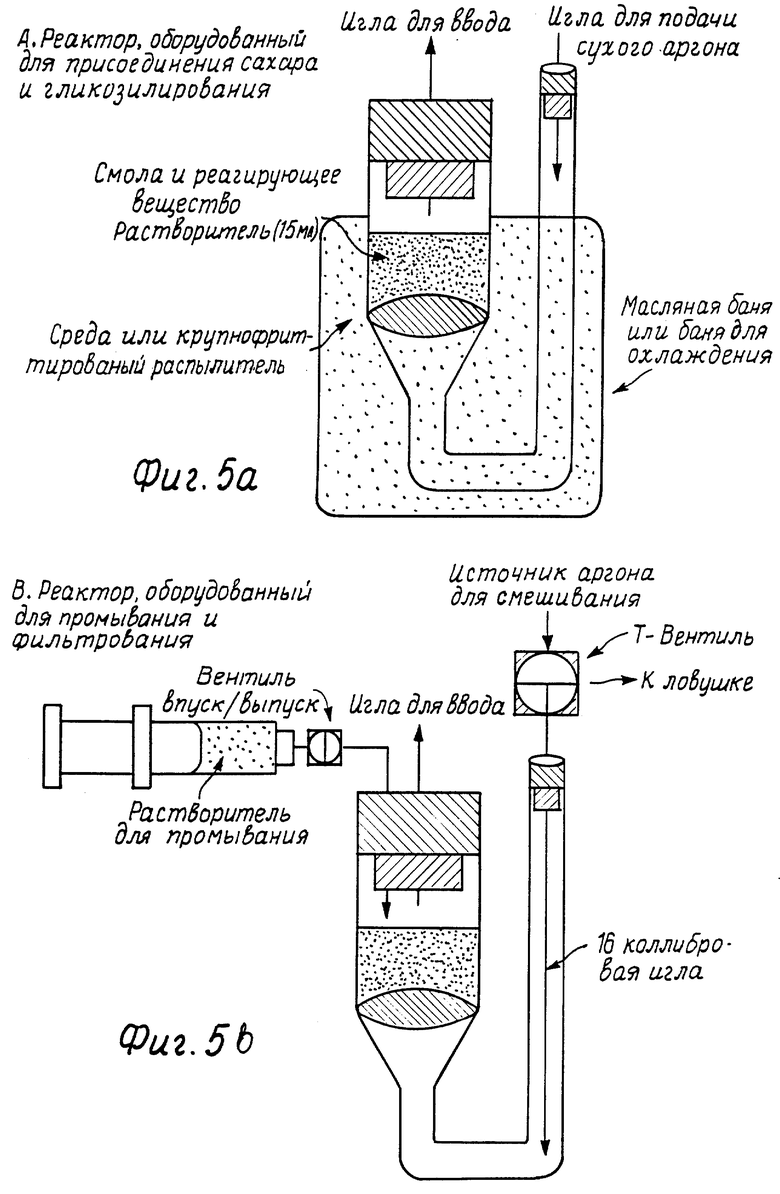
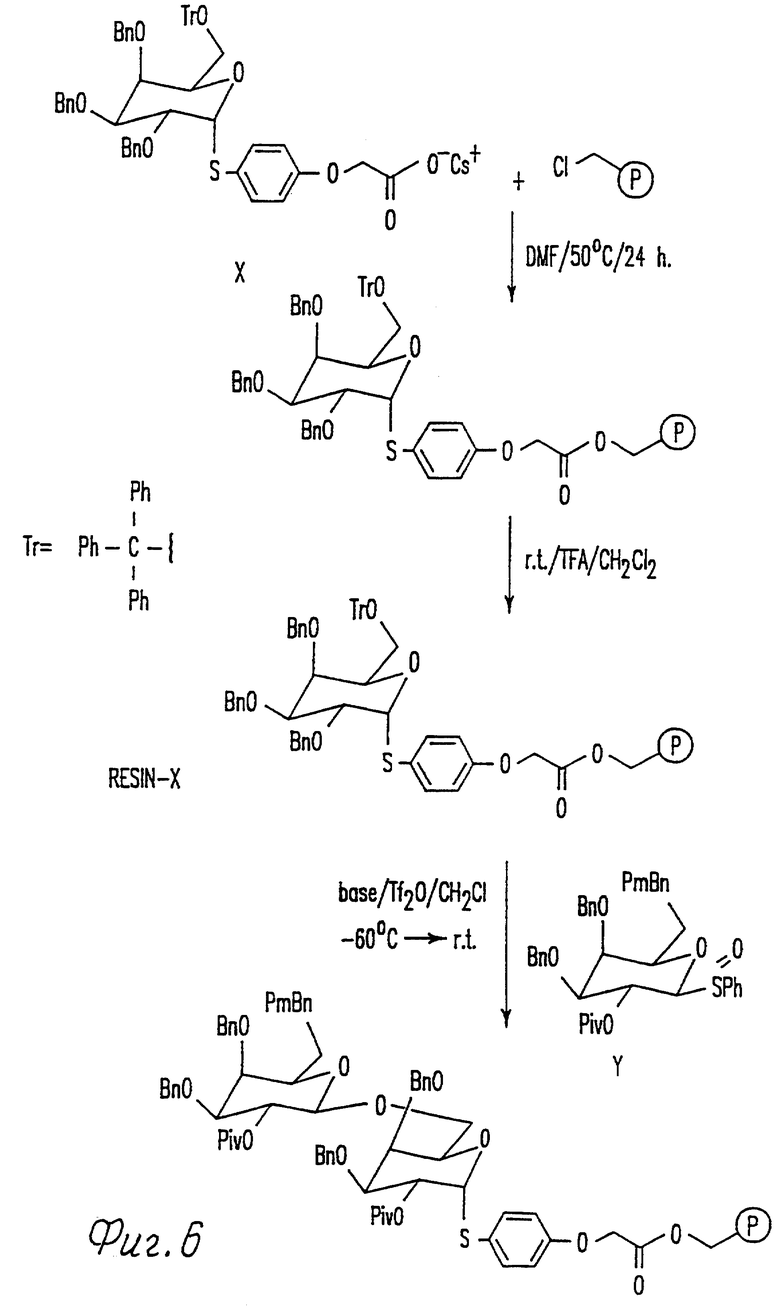

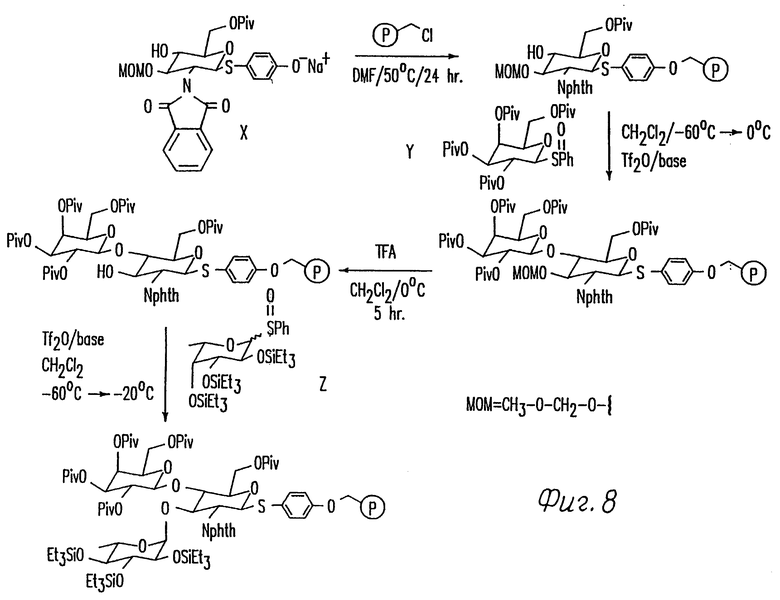
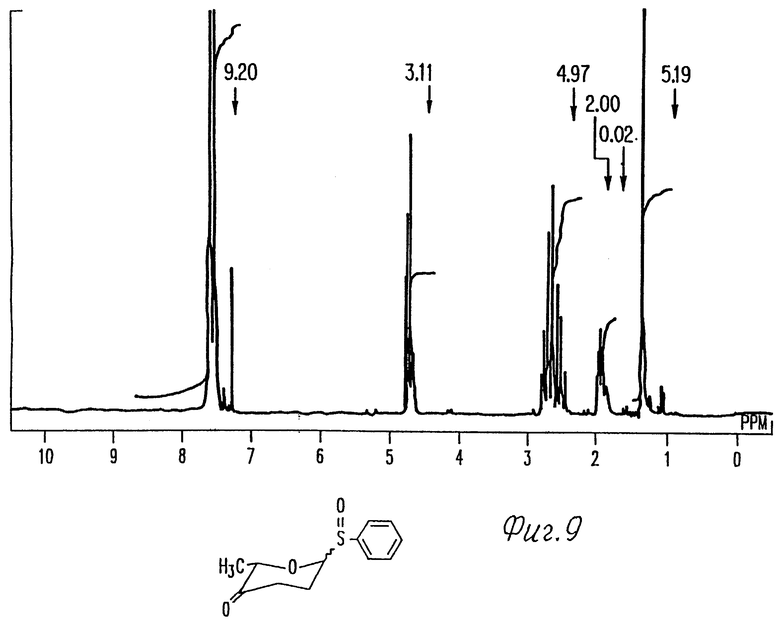
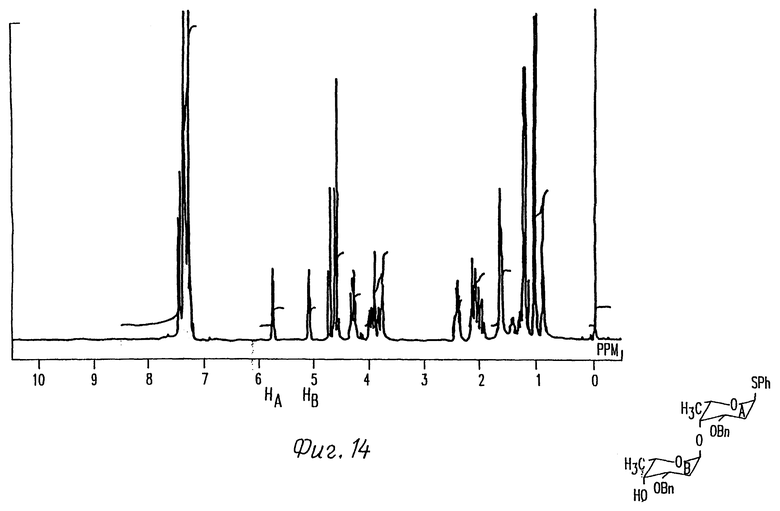
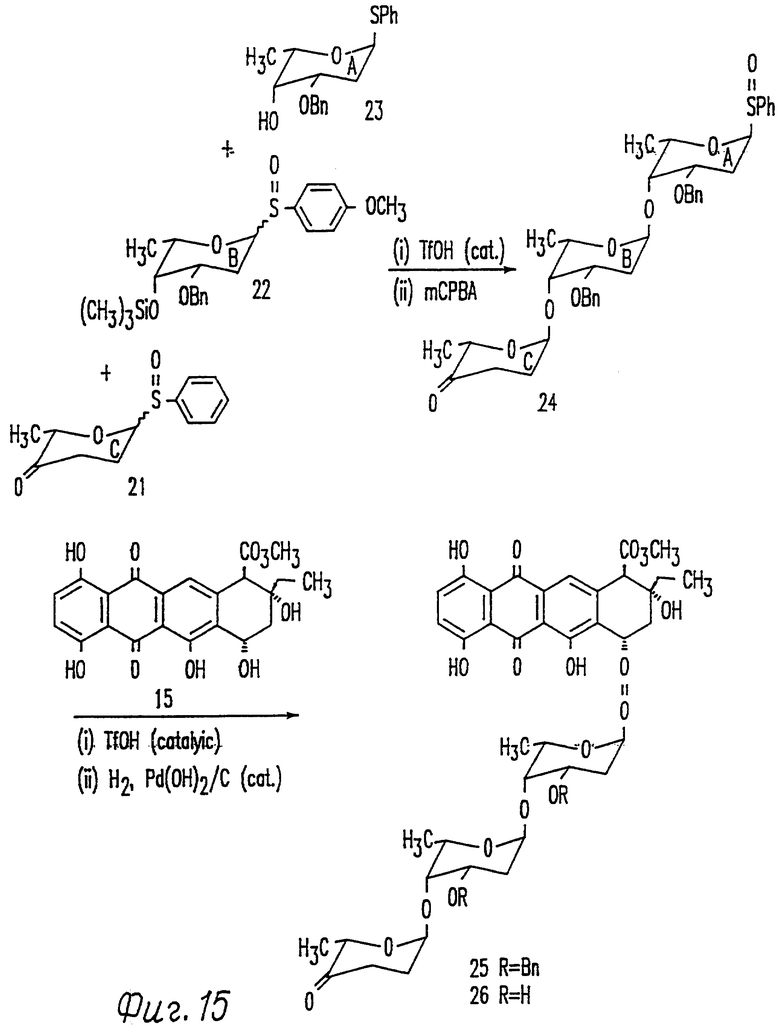
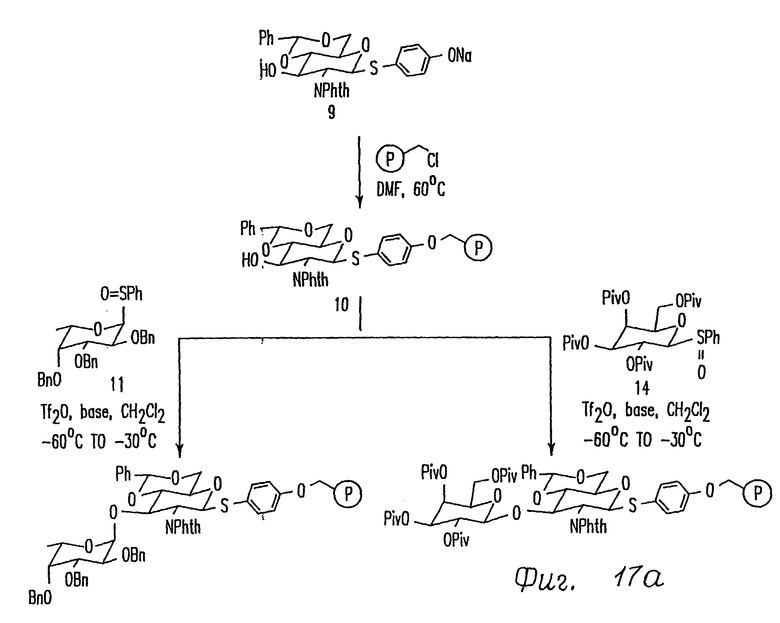
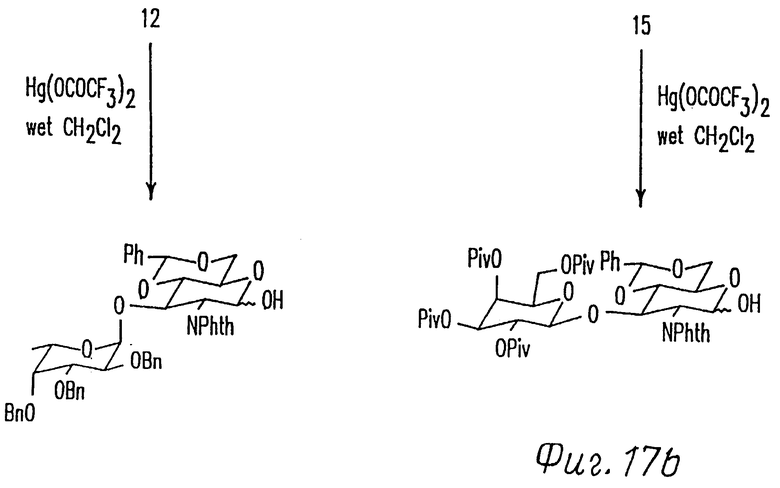
Описывается способ формирования множества региоселективных гликозидных связей, отличающийся тем, что проводят в одном реакторе без промежуточной очистки и выделения обработку первого бифункционального гликозида (ПГ), содержащего аномерный сульфоксидный заменитель, эффективным количеством активирующего агента (АА) в органическом растворителе с образованием ПГ, проявляющего гликозильные акцепторные и гликозильные донорные характеристики. Технический результат - упрощение пресса с сохранением высокого выхода целевого продукта. 7 с. и 43 з.п.ф-лы, 17 ил.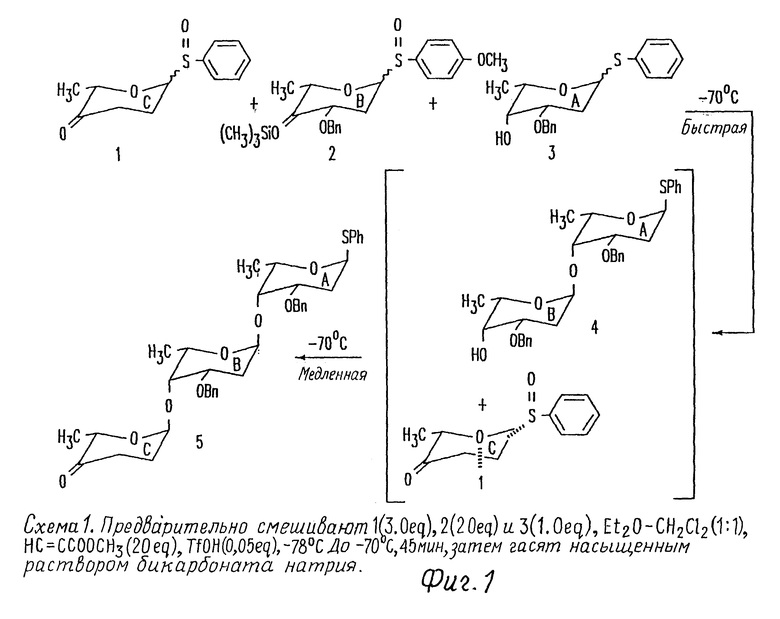
где X1, X2 или X3 независимо друг от друга могут быть водородом, азотом, кислородом или атомом серы;
R1 и R4 могут быть ароматической или алифатической группой, включающей 1 - 25 атомов углерода;
R1, R2 или R3 независимо друг от друга могут быть водородом, ароматической или алифатической группой, содержащей 1 - 25 атомов углерода, за исключением того, что если X1, X2 или X3 представляют водород, то соответствующие R1, R2 или R3 не присутствуют;
указанные ароматические или алифатические группы R1, R1, R2, R3 и R4 необязательно включают атомы азота, кислорода, фосфора, кремния или серы;
обладающий потенциальными гликозильными акцепторными и гликозильными донорными характеристиками: б) активирующий агент, способный вызывать проявление как гликозильных акцепторных, так и гликозильных донорных характеристик указанного гликозида.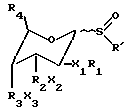
где X1, X2 или X3 независимо могут быть водородом, азотом, кислородом или атомом серы;
R' и R4 могут быть ароматической или алифатической группой, содержащей 1 - 25 атомов углерода;
R1, R2 или R3 каждый независимо может быть водородом, ароматической или алифатической группой, содержащей 1 - 25 атомов углерода, за исключением того, что если X1, X2 или X3 представляет водород, то соответствующие R1, R2 или R3 не присутствуют;
указанные ароматические или алифатические группы R', R1, R2, R3 или R4 необязательно включают атомы азота, кислорода, фосфора, кремния или серы; и если R3 не является водородом, то связь R3 - X3 является чувствительной к разрыву связи в тех же условиях, которые используют для активирования аномерной сульфоксидной группы, таким образом предусматривая проявление как гликозильных акцепторных, так и гликозильных донорных характеристик указанного гликозида.
Приоритет по пунктам:
23.02.93 - по пп.1 - 45;
18.02.94 - по пп.46 - 50.
