Предметом настоящего изобретения являются нитроэфиры с противовоспалительной и/или антитромбоцитарной активностью, их фармацевтическое использование и способ их получения.
Предшествующий уровень знаний
Некоторые производные пропионовой кислоты, такие как, например 2-(3-бензоилфенил)пропионовая кислота, обычно известная как кетопрофен, длительное время использовали как фармацевтические препараты с противовоспалительной активностью и рекламировали на различных международных рынках много лет. Способ получения 2-(3-бензоилфенил)пропионовой кислоты был описан в Южно-африканском патенте N 68 00.524, соответствующем патенту США 3.641.127; в патенте Франции N M6444, а также в С.А. 75.5528 m (1971); G.A. PINNA et al., Farmaco Ed. Sci. 35,684 (1980); причем фармакокинетика человека описана в T. ISHIZAKI et. al., Eur. J. Clin. Pharmacol. 18,407 (1980). Использование производных пропионовой кислоты, таких как, например, кетопрофен, известно так же хорошо, как использование других продуктов, которые применяются как противовоспалительные агенты, включает, как известно, сильно неблагоприятные реакции, например, в гастроинтестинальном аппарате, также как возможные повреждения в печени и в почках.
Есть много экспериментальных доказательств [S. MONCADA, R.M.J. PALMER, E. A. HIGGS, Pharmacological Reviews, 43(2), 109 (1991); T.H. LUSHER, C.M. BOULANGER, Y. DOHI, Z. YANG, Hypertension, 19, 117 (1992)], на основании которых целостность сосудистого эндотелия, как предполагают, является основным барьером против атаки патологических процессов в некоторых органах и аппаратах от неблагоприятных реакций, которые типичны для противовоспалительных агентов.
Такой защитный барьер и, кроме того, целостность сосудистого эндотелия физиологически обеспечивается присутствием окиси азота и простациклина.
Лечение нестероидными препаратами, имеющими противовоспалительную активность, например, как 2-(3-бензилофенил)пропионовая кислота или кетопрофен, вызывают ингибирование циклооксигеназы - фермента, который синтезирует предшественник простациклина.
В заключение, если так ингибировать образование простациклина, то его запас в тканях заметно истощается, и, кроме того, целостность сосудистого эндотелия подвергается риску.
Как было сказано выше, из-за эндотелиального повреждения, обусловленного уменьшением простациклина, распространение диффузного патологического процесса поражает гастроинтестинальный аппарат, печень и почки.
Объекты изобретения
Объектом настоящего изобретения является группа продуктов, которые обеспечивают высокий уровень фармакологической активности противовоспалительных агентов, но при этом способны подавлять неблагоприятные реакции, вызываемые при лечении вышеупомянутыми агентами. Другой объект настоящего изобретения - это реализация процесса приготовления группы продуктов, имеющих противовоспалительную активность, но в то же время свободных от неблагоприятных реакций, которые типичны для противовоспалительных агентов.
Описание изобретения.
Эти и другие объекты и их преимущества, которые будут показаны в последующем описании, получены с помощью нитроэфиров, имеющих следующую общую формулу: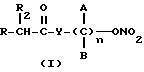
где A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, R выбран из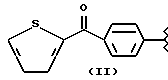
R2 выбран из водорода, метила, этила, алкила линейного или разветвленного с 3 - 12 атомами углерода, замещенного или незамещенного, Y выбран из кислорода, NH, NR1, где R1 - линейный или разветвленный алкил и n равно от 1 до 10.
Фактически установлено, что введение концевой нитроэфирной группы в общую формулу производных (I) приводит к увеличению фармакологической активности нестероидных противовоспалительных агентов, и в то же время уничтожает неблагополучную реакцию, появляющуюся при лечении этими агентами.
Кроме того, установлено, что производные (I) полезны также при таких болезненных состояниях, как, например, ревматические заболевания общего характера, иммунологические расстройства, а также для снятия болевых состояний слабой и средней тяжести любого характера.
Более того, производные (I), являющиеся предметом обсуждения этого изобретения, используются при лечении заболеваний сердечно-сосудистой системы, и в особенности при лечении миокарда и ишемии, а также артериальных тромбозов как антитромбоцитарные агенты.
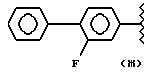
В соответствии с этим изобретением чрезвычайно полезным является нитроэфир общей формулы (I), где: водород выбран как A и B, метил выбран как R2 и R выбран как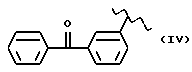
кислород выбран как Y и n равно 4 согласно следующей формуле: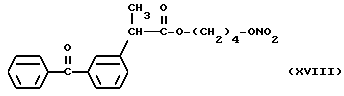
Также особенно полезным, согласно этому изобретению, является нитроэфир общей формулы I), где: водород выбран как A и B, R выбран как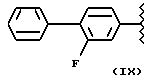
метил выбран как R, Y выбран как кислород и n равно четырем, согласно следующей формуле: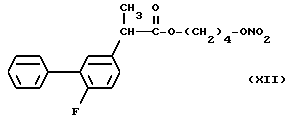
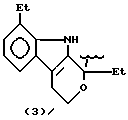
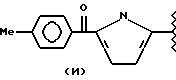
Более того, согласно настоящему изобретению, особенно полезны нитроэфиры общей формулы (I) где: водород выбран как A и B, R выбран как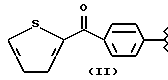
метил, этил и водород выбраны как R2, кислород выбран как Y и n равно четырем, согласно следующим формулам: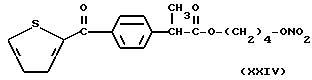
Для получения нитроэфиров общей формулы (I), объекта настоящего изобретения, особенно важен первый процесс, который, согласно изобретению, включает следующие стадии:
- Получение натриевой соли продуктов, имеющих следующую общую формулу: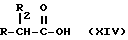
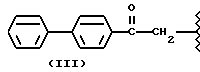
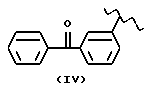
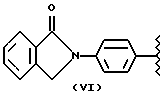
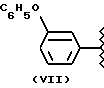
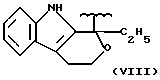
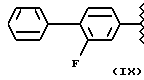
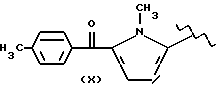
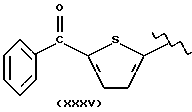
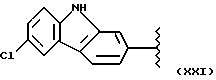
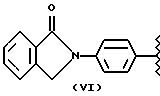
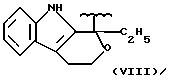
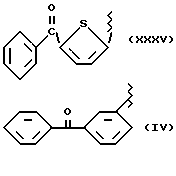
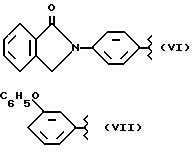
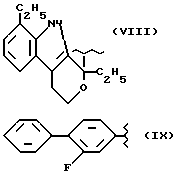
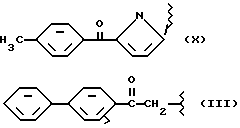
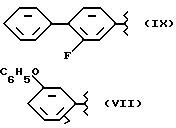
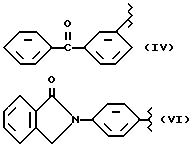
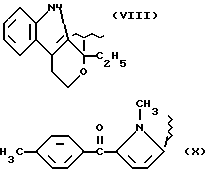
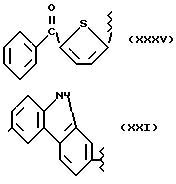
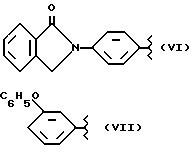
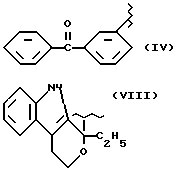
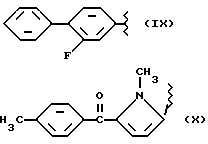
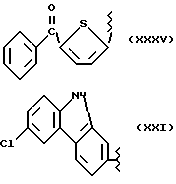
где R2 выбран из водорода, метила, этила, алкила линейного или разветвленного с 3 - 12 атомами углерода, замещенного или незамещенного, R выбран из: (II), (III), (IV), (VI), (VII), (VIII), (IX), (X), (XXI), (XXXV) или получение производных (XIV), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды и т.п.;
- Реакцию натриевой соли вышеуказанных производных (XIV) или вышеуказанных производных (XIV), замещенных по карбоксильной группе, с соединениями, имеющими следующую общую формулу: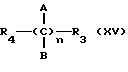
где R4 выбран из хлора, брома, NHR6, где R6 выбран из водорода, линейного или разветвленного алкила, A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, R3 выбран из хлора, брома, йода, и n равно от 1 до 10, с получением таким путем соответствующих эфиров или соответствующих амидов;
- Реакцию вышеуказанных эфиров или вышеуказанных амидов с нитрующим агентом, таким как AgNO3 или подобным для получения нитроэфиров (I).
- Второй процесс, являющийся также очень важным, согласно настоящему изобретению включает следующие стадии:
- Получение натриевой соли производных, имеющих следующую общую формулу: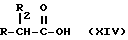
где R выбран из: (II), (III), (IV), (VI), (VII), (VIII), (IX), (X), (XXI), (XXXV), R2 выбран из водорода, метила, этила, линейного или разветвленного алкила с 3 - 12 атомами углерода, замещенного или незамещенного, или получение производных (XIV), замещенных по карбоксильной группе, таких как хлорангидриды, ангидриды или т.п.;
Реакцию натриевой соли вышеуказанных производных (XIV) или вышеуказанных производных (XIV), замещенных по карбоксильной группе, с соединением общей формулы: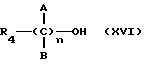
где R4 выбран из хлора, брома, NHR6, где R6 обозначает водород или линейный или разветвленный алкил, A и B выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила, и n равно от 1 до 10, с получением таким путем соответствующих эфиров или амидов;
- Реакцию вышеуказанных эфиров или вышеуказанных амидов с галоидирующим соединением PBr3 и/или подобным, с получением вышеуказанных эфиров или вышеуказанных амидов с концевой галоидной группой;
- Реакцию вышеуказанных эфиров или вышеуказанных амидов, имеющих концевую галоидную группу, с нитрующим агентом AgNO3 или подобным для получения нитроэфиров (I).
Растворители, используемые в процессах, являющихся предметом обсуждения настоящего изобретения, предпочтительно выбирают из хлороформа, хлористого метилена, ацетонитрила, диметилформамида, тетрагидрофурана, 1,4-диоксана и подобных.
Способы получения производных (I), которые являются предметом настоящего изобретения, состоят из нескольких стадий и позволяют получать продукты вышеупомянутых процессов за короткое время и с хорошими выходами даже в промышленном плане.
Согласно способам, рассматриваемым в этом изобретении, получают нитроэфир, имеющий следующую формулу:
который является особенно полезным, и который получают как описано в следующем примере, иллюстрирующем, но не ограничивающим данное изобретение.
Пример 1.
а) 2 г 2-фтор-альфа-метил-4-дифенилуксусной кислоты добавляют к раствору, содержащему 10 мл метанола и 0,23 г Na. Реакционную смесь перемешивают 5 минут, затем растворитель упаривают при пониженном давлении, получая натриевую соль 2-фтор-альфа-метил-4-дифенилуксусной кислоты.
б) Натриевую соль 2-фтор-альфа-метил-4-дифенилуксусной кислоты, полученной этим путем, суспендируют в 20 мл диметилформамида и 3 мл 1,4-дибромбутана, который добавляют прикапыванием к этой суспензии. Реакционную смесь перемешивают 22 часа при комнатной температуре, затем образующийся NaBr отфильтровывают и растворитель упаривают при пониженном давлении. Полученный остаток обрабатывают хлористым метиленом и, после отделения фильтрованием нерастворимого осадка, хлористый метилен выпаривают при пониженном давлении, получая 3 г сухого остатка, который очищают хроматографически на силикагеле, используя в качестве элюента смесь гексан/хлористый метилен 1/1 (по объему). Головную фракцию собирают, растворитель упаривают при пониженном давлении и получают 1,86 г 2-фтор-альфа-метил-4-дифенилацетата 4-бромбутила (XXII).
ИК (см-1): C = 0,1470
1H-ЯМР (300 МГц) (CDCl3): 1,51 м.д. (d, 3H); 1,56 м.д. (m, 4H); 3,35 м. д. (t, 2H); 3,61 м.д. (q, 1H); 4,1 м.д. (t, 2H), 7,05 м.д. (m, 1H); 7,17 м. д. (s, 1H); 7,3 - 7,55 м.д. (m, ароматический)
с) 1,2 г AgNO3, растворенного в 8,3 мл ацетонитрила, добавляют к 1,86 г (XXII), полученного как описано выше в б), растворенного в 7,5 мл ацетонитрила. Реакционную смесь перемешивают в течение 48 часов при комнатной температуре и затем отфильтровывают. Полученный раствор упаривают при пониженном давлении, получают остаток, который обрабатывают хлористым метиленом. Смесь, полученную таким путем, фильтруют снова и органическую фазу очищают хроматографически на силикагеле под давлением, используя в качестве элюента смесь диэтиловый эфир/гексан 3/7 (по объему). Фракции, содержащие продукты, собирают, упаривают растворитель при пониженном давлении и получают 1,2 г нитроэфира 2-фтор-альфа-метил-4-дифенилацетата 4-оксибутила.
ИК (см-1): C = 0,1737; ONO2, 1623, 1274.
1H-ЯМР (300 МГц) (CDCl3): 1,53 м.д. (d, 3H); 1,72 м.д. (m, 4H); 3,74 м. д. (q, 1H); 4,13 м.д. (t, 2H); 4,4 м.д. (t, 2H); 7,13 м.д. (t, 2H, ароматический); 7,32 - 7,42 м.д. (m, 4H, ароматический); 7,53 м.д. (m. 2H, ароматический).
Масс-спектрометрия: (M+1) 361; (M+1 - NO2) 316; 243; 199.
Согласно способу, который является предметом настоящего изобретения, также получают нитроэфир, имеющий следующую формулу: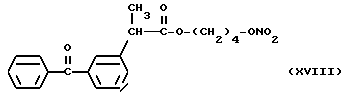
который является особенно полезным, его получение описано в примере, приведенном здесь ниже, и который дан для иллюстрации данного изобретения, но не ограничивает его объема.
Пример 2.
а) 10 г 2-(3-бензоилфенил)пропионовой кислоты добавляют к раствору, содержащему 1,19 г Na в 80 мл метанола. Реакционную смесь перемешивают 15 минут, затем растворитель упаривают при пониженном давлении, полученный остаток представляет собой натриевую соль 2-(3-бензоилфенил)пропионовой кислоты.
б) 100 мл диметилформамида и 28,1 г 1,4-дибромбутана добавляют к остатку, полученному таким путем. Реакционную смесь выдерживают 24 часа при комнатной температуре, а затем упаривают растворитель при пониженном давлении. Добавляют 40 мл воды и 60 мл хлористого этилена к остатку, полученному таким путем, органическую фазу экстрагируют, высушивают над сульфатом натрия и растворитель упаривают при пониженном давлении до получения сухого остатка. Остаток очищают хроматографически на силикагеле, используя в качестве элюента смесь диэтиловый эфир/гексан в соотношении 1/1 по объему). Головную фракцию собирают, растворитель упаривают при пониженном давлении и получают 8,8 г 2-(3-бензоилфенил)пропионат 4-бромбутила (XXIII).
1H-ЯМР (200 МГц) (CDCl3): 1,53 м.д. (d, 3H); 1,84 м.д. (m, 4H); 3,32 м. д. (t, 2H), 3,78 м.д. (q, 1H), 4,09 м.д. (t, 2H); 7,27 м.д. (m. 1H, ароматический); 7,38 - 7,99 м.д. (m, 8H, ароматический).
Масс-спектрометрия: 388 (M+); 309 (M+ - Br); 209.
с) 5,5 г AgNO3, растворенные в 38 мл ацетонитрила, добавляют к 8,8 г (XXIII), полученному как описано выше в б) и растворенному в 35 мл ацетонитрила. Реакционную смесь перемешивают 24 часа при комнатной температуре и, добавив 1,76 г AgNO3, перемешивают еще 24 часа при комнатной температуре и затем отфильтровывают. Из полученного раствора упаривают растворитель при пониженном давлении, получают остаток, который обрабатывают хлористым метиленом.
Смесь, полученную таким путем, снова фильтруют и органическую фазу очищают хроматографически на силикагеле под давлением, используя в качестве элюента смесь эфир/гексан в соотношении 3/7 (по объему)
Фракцию, содержащую продукт, собирают, растворитель упаривают при пониженном давлении и получают 3,4 г нитроэфира 2-(3-бензоилфенил)пропионата 4-оксибутила (XVIII).
ИК (см-1): C = 0,1737; ONO2, 1632, 1288; OCO, 1660.
1H-ЯМР (80 МГц) (CDCl3): 1,48 м.д. (d, 3H); 1,64 м.д. (m, 4H); 3,78 м.д. (q, 1H), 4,08 м.д. (m, 2H); 4,3 м.д. (m, 2H); 7,3 - 7,81 м.д. (m, ароматический).
Масс-спектрометрия: 371 (M+); 309 (M+ - ONO2); 255.
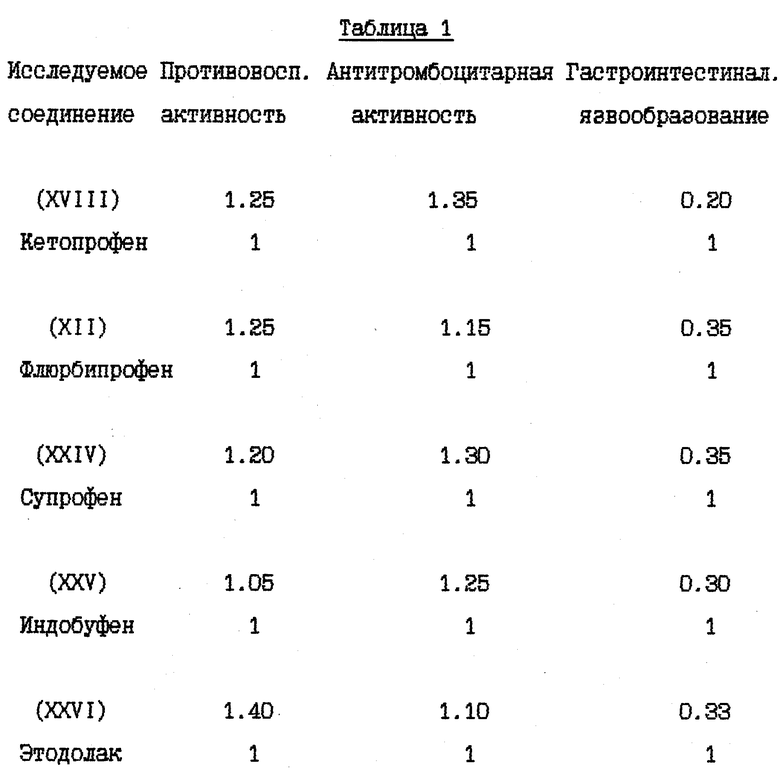
Противовоспалительная и антитромбоцитарная активность, а также гастроинтестинальная ульцерогенность, были изучены в результате биологических испытаний, имеющих следующие формулы: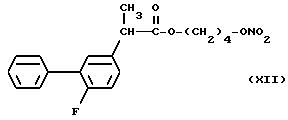
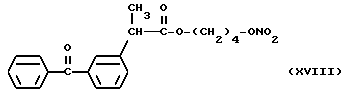
Противовоспалительная активность указанных выше нитроэфиров была изучена на крысах вида Вистар при использовании метода воздействия на индуцированный каррагином отек, как это описано C.A. WINTER, E. RISLEY, G.W. NUSS, Proc. Soc. Exp. Biol. Med. 111, 544 (1962), в то время как антитромбоцитарная активность вышеуказанных производных была изучена на человеческих тромбоцитах, стимулированных арахидоновой кислотой, согласно методу, описанному V. BERTELE и соавт., Science 220, 517 (1983).
Гастроинтестинальное язвообразование определялось оральным введением на крысах.
Противовоспалительная и антитромбоцитарная активность, а также гастроинтестинальная язвообразовательная активность вышеописанных соединений, даны в таблице 1, и показывают для каждого приведенного нитроэфира относительно высокую степень по сравнению с соответствующими кислотами, не относящимися к соединениям общей формулы (I), согласно настоящему изобретению. Каждая оценка представляет собой среднее значение, полученное при обработке 10 животных.
В частности, производные (XVIII) и (XII), представленные для дополнительного изучения фармакодинамических свойств, дали результаты, которые показаны в следующих примерах.
- Воздействие на индуцированный каррагином отек у крыс. Оба соединения (XVIII) и (XII) показали лучшие результаты по сравнению с соответствующими упоминаемыми лекарствами Кетопрофеном и Флюрбипрофеном, причем эффективные дозы в диапазоне от 1 до 10 мг/кг.
- Адъювант артрита крыс. Животные, обработанные в течение 19 дней подряд (с 3 по 21 день после инъекции адъюванта) в дозе 3 мг/кг или (XVIII) или (XII) и соответствующими им упоминаемыми соединениями, показали значительное снижение симптомов артрита по сравнению с контролем.
- Пароксизмы у мышей, индуцированные фенилхиноном. При дозах в диапазоне от 3 до 10 мг/кг, соединения (XVIII) и (XII) вполне эффективны и их эффективность почти сравнима с соответствующими упомянутыми соединениями.
- Тромбоцитарная агрегация in vivo. Если оба соединения XVIII) и Флюрбипрофен вводили дозой 20 мг/кг в крысу, то ингибировали коллагенвызванную тромбоцитарную агрегацию, причем первый (66% ингибирования в сравнении с контролем) был значительно более эффективен, чем последний (40%).
Биохимия.
- Синтез простагландина в воспалительном экссудате. Подкожная имплантация каррагиновой губки вызывает инфильтрацию воспаленных клеток, как сообщалось в Nature 284, 271 (1980). Оба соединения (XVIII) и (XII) при введении дозы 20 мг/кг ингибируют образование простагландина E2 в экссудате более чем на 75% в сравнении с контролем и показывают сравнительную эффективность с соответствующими вышеупомянутыми соединениями Кетопрофеном и Флюрбипрофеном.
- Синтез простагландинов. Оба соединения (XVIII) и (XII) были изучены для синтеза простагландинов при таких же дозах (5 - 20 мг/кг), как и для изучения желудочных поражений. Они значительно ингибировали синтез простагландинов E2 подобно соответствующим вышеупомянутым соединениям Кетопрофен и Флюрбипрофен, причем степень ингибирования больше 90% при максимальной дозе.
- Без выделения. Доказательство того, что соединения (XVIII) и (XII) выделяли оксид азота после их введения было получено при измерении нитрат/нитритных уровней плазмы, как сообщалось в J. Clin. Invest., 85, 264 (1990). Через 1 час после введения или (XVIII) или (XII) соединений нитрат/нитритные уровни плазмы значительно увеличивались более чем на 50%. Кетопрофен или Флюрбипрофен не оказывали значительного влияния на нитрат/нитритные уровни.
Кроме того, дополнительные биологические исследования были выполнены на производных (XII) и (XVIII); вышеупомянутые исследования показали следующие результаты.
Гастроинтестинальная толерантность.
- Повреждение слизистой оболочки крыс. (XVIII) и XII) были изучены при сравнении с соответствующими вышеупомянутыми соединениями Кетопрофеном и Флюрбипрофеном при дозах от 3 до 30 мг/кг, причем оба соединения (XII) и (XVIII) значительно лучше переносились чем вышеупомянутые соединения. Кетопрофен или Флюрбипрофен вызывали начало желудочных повреждений уже при дозе 3 мг/кг, тяжесть этих повреждений являлась зависимой от дозы, тогда как (XVIII) и (XII) хорошо переносились даже при дозе 30 мг/кг.
Гистологическая экспертиза подтвердила эти данные. Похожие различия в способности этих соединений вызывать желудочные и небольшие кишечные повреждения наблюдались также при повторном введении соединений.
Адгезия лейкоцитов/ диаметр сосудов желудка. Ранним этапом в патогенезе NSAID-индуцировнаного поражения слизистой оболочки является адгезия лейкоцитов с эндотелием посткапиллярных венул, как сообщалось в Gastroenterology 103, 146 (1992); Trends Pharmacol. Sci. 13, 129 (1992); Am. J. Physiol. 262, G903 (1992). При использовании интравитальной микроскопии адгезия лейкоцитов в мезентерических посткапиллярных венулах может быть первоначально количественно оценено в течение 1 часа после применения NSAID. В отличие от Кетопрофена или Флюрбипрофена (XVIII) или (XII) не вызывает значительной адгезии лейкоцитов, достоверно увеличивая диаметр сосудов. Изменений в кровяном давлении не обнаружено.
Общая фармакология.
Вторичная фармакологическая экспертиза соединений (XVIII) или (XII) была выполнена при сравнении с Кетопрофеном или Флюрбипрофеном. Не было обнаружено побочных реакций адгезии, поражающих центральную нервную систему, кардиоваскулярную, респираторную и гастроинтестинальную систему.
Токсикология
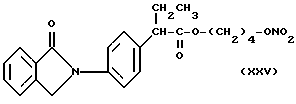
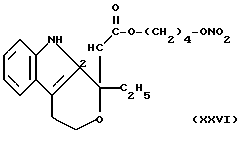
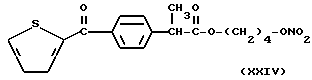
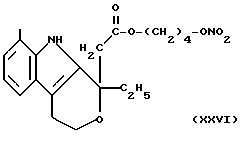
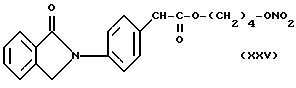
- Острая токсикология у грызунов. Острая токсичность вышеупомянутых производных (XVIII), (XXIV), (XXV), (XII) и (XXVI) была затем оценена при применении отдельных доз каждого соединения (XVIII), (XXIV), (XXV), (XII) и (XXVI), используя для каждого производного группы из 10 мышей породы Свисс. Доказательство смерти и начала симптомов интоксикации было описано для периода 14 дней.
Даже после применения дозы 100 мг/кг каждого соединения (XVIII), (XXIV), (XXV), (XII) и (XXVI), не установлено явных симптомов их токсичности при исследовании на животных.
В частности, предварительные исследования соединений (XVIII) или (XII) были представлены на мышах при двух методах введения. Доказательств токсичности не обнаружено у животных, которых лечили путем орального или интраперитонеального введения каждого соединения в дозе 300 мг/кг.
- Максимальная переносимая доза у грызунов. Предварительные исследования показывают, что соединения (XVIII) и (XII) хорошо переносились теми видами животных, которые, как известно, особенно чувствительны к этому классу соединений. У животных применялись постепенно увеличивающиеся дозы до 30 мг/кг каждого соединения, и при этом не было обнаружено явных симптомов, в то время как использование соединений Кетопрофена и Флюрбипрофена в дозе 10 мг/кг вызывало смерть животных.
Ниже приводятся физико-химические характеристики как сигналы 13CNMR специфических атомов углерода (идентифицированных маленькими буквами) в молекуле следующих соединений: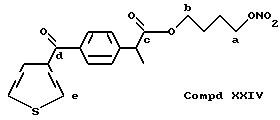
13CNMR typical signals, C-a 73,84, C-b 64,86, C-c 173,22, C-d 195,22, C-e 125,05.
для соединений XXV (см. выше) описание: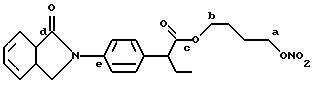
для соединений XXVI (см. выше) описание: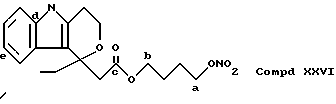
13CNMR typical signals, C-a 74,02, C-b 64,78, C-c 173,95, C-d 135,51, C-e 122,12.
Соединение, полученное благодаря радикалу (X) группы CH2COO(CH2)NO2 в предусмотренной позиции: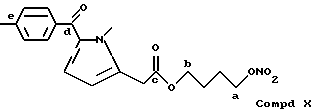
13CNMR typical signals, C-a 73,84, C-b 65,00, C-c 172,80, C-d 194,73, C-e 141,42.
Соединение, полученное благодаря радикалу (XXI) группы CH2(CH3)COO(CH2)4NO2 в предусмотренной позиции: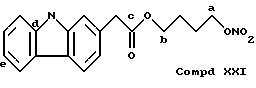
13CNMR typical signals, C-a 73,96, C-b 64,92, C-c 173,35, C-d 139,68, C-e 118,04.
В позиции e фенильного кольца слева отсутствует заместитель атома хлора для выявления атома углерода, выдающего сигнал 13C.
Соединение, полученное благодаря радикалу (XXXV) той же самой алифатической группы радикала (XXI) в предусмотренной позиции: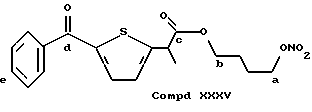
13CNMR typical signals, C-a 74,02, C-b 64,96, C-c 172,93, C-d 194,87, C-e 132,92.
Соединение, полученное благодаря радикалу (VII) той же самой алифатической группы радикала (XXI) в предусмотренной позиции: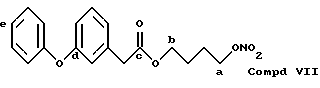
13CNMR typical signals, C-a 73,83, C-b 64,79, C-c 173,18, C-d 156,18, C-e 121,52.
Соединение, полученное благодаря радикалу (III) той же самой алифатической группы радикала (X) в предусмотренной позиции: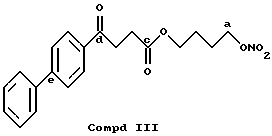
13CNMR typical signals, C-a 73,91, C-b 64,88, C-c 174,78, C-d 195,89, C-e 145,31.
Спектр 13CNMR был воспроизведен при комнатной температуре (295К) по спектрометру Varian Gemini 200 на частоте 50,3 MГц, оборудованному двойной экспериментальной головкой 5 мм. Дейтированный хлороформ был использован в качестве растворителя. Химические загрузки изученных соединений относились к хлороформу (77,0 ppm), Спектр 13CNMR был воспроизведен с использованием раствора 0,1 М с диапазоном спектра 14000 Гц, с импульсом 8μs (45oC) с продолжительностью процесса 0,6 с с точечным показателем 16К и с задержкой релаксации 0,5 с.
Пример 3.
Синтез N-(2-нитроксиэтил)-2-фтор-α-метил[1,1-бифенил]-4-ацетамид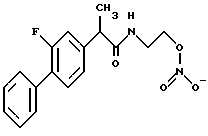
N-(2-хлорэтил)-2-фтор-aльфа-метил-[1,1-бифенил]-4-ацетамид.
К раствору 1,1'-карбонилдиимидазола (3,32 г, 20,4 ммоль) в хлороформе (50 мл) добавляют при комнатной температуре фторбипрофен (5 г, 20,4 ммоль). Полученную смесь обрабатывают частями 2-хлорэтиламин гидрохлоридом (2,36 г, 20,4 ммоль) и затем триэтиламином (2,84 мл, 20,4 ммоль). Смесь размешивают при комнатной температуре в течение 1 часа, разбавляют хлороформом и промывают водой. Органический слой просушивают сульфатом натрия и выпаривают в условиях вакуума и осадок очищают в хроматографической колонке (элюент хлороформ-метанол 10 - 0,2). Промежуточный продукт выделяют в чистом виде (5 г) как белое твердое вещество и используют для последующей реакции.
N-(2-нитроксиэтил)-2-фтор--α--метил-[1,1-бифенил]-4-ацетамид.
Смесь N-(2-хлорэтил)-2-фтор--α--метил-[1,1-бифенил]-4-ацетамида (3,4 г, 11,12 ммоль) и нитрата серебра (5,7 г, 33,35 ммоль) в ацетоннитриле (90 мл) орошают в течение 2 часов. После охлаждения твердое вещество фильтруют и растворитель выпаривают. Полученный осадок очищают в хроматографической колонке (элюент гексан-этилацетат 7-3). Полученное вещество выделяют в чистом виде как светлое желтое масло (1,8 г).
1H-NMR (CDCl3, ppm) 7,76 - 7,27 (8H, m), 7,10 (1H, t), 4,52 (2H, m), 3,60 (3H, m), 1,85 (3H, d).
Пример 4.
Синтез 2-фтор-α-метил-[1,1-бифенил] -4-уксусной кислоты 1-нитроокси-2-метил-2-пропил эфира.
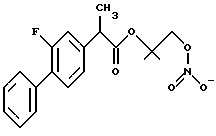
2-фтор-α-метил-[1,1-бифенил]-4-хлорида уксусной кислоты.
Раствор фторбипрофена (5 г, 20,4 ммоль) в толуоле (20 мл) обрабатывают при комнатной температуре диметилформамидом (2 мл). После охлаждения добавляют каплями хлорид оксалила (5,2 г, 40,9 ммоль) и полученный раствор размешивают при комнатной температуре в течение 4 часов. Растворитель выпаривают и сырой хлорид кислоты (5,37 г) используют для последующей реакции.
2-фтор-α-метил-[1,1-бифенил] -4-уксусная кислота-1-хлор-2-метил-2-пропил эфир.
Раствор 2-фтор-α-метил-[1,1-бифенил]-4-хлорид уксусной кислоты (5,37 г, 20,4 ммоль) в хлороформе (25 мл) обрабатывают триэтиламином (5,2 г, 51 ммоль). Полученную смесь обрабатывают при температуре 0oC, добавляя каплями 1-хлор-2-метил-2-пропанол (3,33 г). Смесь размешивают при комнатной температуре в течение 3 дней и промывают водой. Органический слой просушивают сульфатом натрия и выпаривают в условиях вакуума. Осадок очищают в хроматографической колонке (элюент гексан-этилацетат 9-1). Промежуточный продукт выделяют в чистом виде (3,7 г) и используют для последующей реакции.
2-фтор-α-метил[1,1-бифенил] -4-уксусная кислота 1-нитроокси-2-метил-2-пропил эфир.
Смесь 2-фтор-α-метил-[1,1-бифенил] -4-уксусная кислота 1-хлор-2-метил-2-пропил эфир (3,36 г, 10 ммоль) и нитрат серебра (2,39 г, 14 ммоль) в ацетоннитриде (30 мл) орошают 2 дня. После охлаждения твердое вещество фильтруют и растворитель выпаривают. Осадок очищают в хроматографической колонке (элюент гексан-этилацетат 9-1). Полученное вещество выделяют в чистом виде как светлое желтое масло (1,1 г).
1H-NMR (CDCl3, ppm) 7,52 (2H, m), 7,40 - 7,35 (4H, m), 7,11 (2H, t), 4,30 (2H, s), 3,74 (1H, q), 1,50 (3H, d), 1,26 (6H, s).
Нитроэфиры общей формулы I, где А и В - водород, замещенный или незамещенный алкил, R - остаток формулы (а-к), R2-водород, метил, этил, С3-12-алкил, Y - кислород, NH, NR1, где R1-алкил, n = 1-10, обладают противовоспалительной и/или антитромбоцитарной активностью. Установлено, что введение концевой нитроэфирной группы в общую формулу производных 1 приводит к увеличению фармакологической активности нестероидных противовоспалительных агентов и в то же время уничтожает неблагополучную реакцию, появляющуюся при лечении этими агентами. 2 с. и 9 з.п. ф-лы, 1 табл.
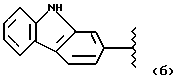
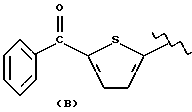
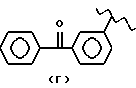

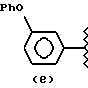
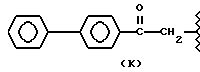
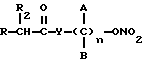
где А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила;
R выбран из: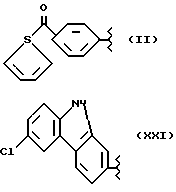
R 2 выбран из водорода, этила, алкила линейного или разветвленного с 3-12 атомами углеродами, замещенного или незамещенного;
Y выбран из кислорода, NH, NR1, где R1 означает линейный или разветвленный алкил;
n = 1 - 10.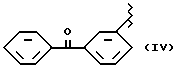
R2 означает метил; А и В означают водород; Y означает кислород и n = 4.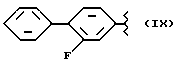
R2 означает метил; Y означает кислород; А и В означают водород и n = 4.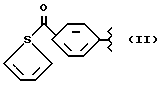
R2 означает метил; А и В означают водород; Y означает кислород и n = 4.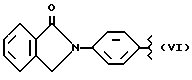
R2 означает этил; А и В означают водород; Y означает кислород и n = 4.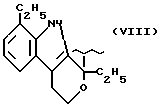
R2 означает водород; А и В означают водород; Y означает кислород и n = 4.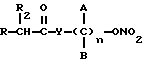
где А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила;
R выбран из: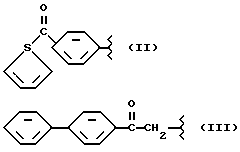
R2 выбран из водорода, метила, этила, алкила линейного или разветвленного с 3-12 атомами углерода, замещенного или незамещенного;
Y выбран из кислорода, NH, NR1, где R1 означает линейный или разветвленный алкил;
n = 1 - 10,
отличающийся тем, что включает следующие стадии: получение натриевой соли производных общей формулы XIV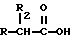
где R выбран из следующих структур: (II), (III), (IV), (VI), (VII), (VIII), (IX), (X), (XXI), (XXXV),
или получение производных XIV, замещенных по карбоксильной группе, таких, как хлорангидрид, ангидрид или т.п.; реакцию натриевой соли указанных производных (XIV) или указанных производных (XIV), замещенных по карбоксильной группе, с соединением общей формулы (XV)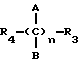
где R4 выбран из хлора, брома, NHR6, где R6 означает водород, линейный или разветвленный алкил;
А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила;
R3 выбран из хлора, брома или йода;
n = 1 - 10,
с получением соответствующих амидов;
реакцию указанных простых эфиров или указанных амидов с нитрующим агентом, таким, как AgNO3 или подобным, с получением нитроэфиров общей формулы (I).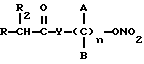
где А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила;
R2 выбран из водорода, метила, этила, алкила линейного или разветвленного с 3-12 атомами углерода, замещенного или незамещенного;
R выбран из: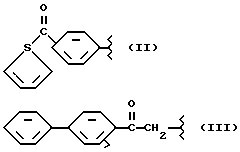
Y выбран из кислорода, NH, NR1, где R1 означает линейный или разветвленный алкил;
n = 1 - 10,
отличающийся тем, что включает следующие стадии: получение натриевой соли производных общей формулы XIV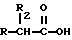
где R выбран из следующих структур: (II), (III), (IV), (VI), (VII), (VIII), (IX), (X), (XXI), (XXXV),
R2 выбран из водорода, метила, этила, алкила линейного или разветвленного с 3-12 атомами углерода, замещенного или незамещенного или получение производных (XIV), замещенных по карбоксильной группе, таких, как хлорангидрид, ангидрид и подобных;
реакцию натриевой соли вышеуказанных производных (XIV), замещенных по карбоксильной группе, с соединением общей формулы (XVI)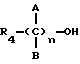
где R4 выбран из хлора, брома, NHR6, где R6 означает водород, линейный или разветвленный алкил;
А и В выбраны из водорода, линейного или разветвленного, замещенного или незамещенного алкила;
n = 1 - 10,
с получением соответствующих простых эфиров или соответствующих амидов;
реакцию указанных простых эфиров или амидов с галоидирующим соединением, например PBr3 или подобным, с получением указанных простых эфиров или амидов, характеризующихся наличием концевой галоидной группы;
реакцию указанных простых эфиров или амидов, характеризующихся наличием концевой галоидной группы, с нитрующим агентом, например AgNO3 или подобным, с получением нитроэфиров общей формулы (I).
| Способ получения -(аминофенил)-алифатических карбоновых кислот или их производных,или их солей | 1970 |
|
SU471714A4 |
| US 3641127 A, 1968. | |||

