Настоящее изобретение относится к новым лекарственным средствам для общего и местного применения и к композиции на их основе, которые используются при окислительном стрессе и/или в случаях эндотелиальной дисфункции.
Под окислительным стрессом понимают генерирование свободных радикалов или радикальных соединений, которые вызывают повреждение как клетки, так и окружающих тканей (Phathophysiology: The biological basis for disease in adults and children, McCance & Huether, 1998, стр. 48-54).
Под эндотелиальной дисфункцией понимают то, что относится к сосудистому эндотелию. Повреждение сосудистого эндотелия известно как одно из тех важных событий, которые могут вызвать серию патологических процессов, поражающих различные органы и аппараты тела, как описано ниже (Phathophysiology: The biological basis for disease in adults and children, McCance & Huether, 1998, стр. 1025).
Как известно, окислительный стресс и/или эндотелиальные дисфункции связаны с различными патологиями, как указано ниже. Окислительный стресс может также быть вызван токсичностью широкого ряда лекарственных средств, которая значительно влияет на их действие.
Вышеуказанные патологические события носят хронический, подрывающий здоровье характер и часто обычны для пожилых людей. Как уже говорилось, в вышеуказанных патологических условиях применяемые лекарства оказывают поразительно ухудшенное действие.
Примеры патологических ситуаций, вызванных окислительным стрессом и/или эндотелиальными дисфункциями или присутствующих у пожилых людей, являются следующими:
- Для сердечно-сосудистой системы: миокардиальная и сосудистая ишемия, гипертензия, приступ, артериосклероз и т.д.
- Для соединительной ткани: ревматоидный артрит и связанные с ним воспалительные заболевания и т.д.
- Для легочной системы: астма и связанные с ней воспалительные заболевания и т.д.
- Для желудочно-кишечной системы: язвенная и неязвенная диспепсии, кишечные воспалительные заболевания и т.д.
- Для центральной нервной системы: болезнь Альцгеймера и т.д.
- Для мочеполовой системы: импотенция, недержание.
- Для кожной системы: экзема, нейродерматит, угри.
- Инфекционные заболевания в общем (ссыл.: Schwarz-KB, Brady "Oxidative stress during viral infection: A review" Free radical Biol. Med., 21/5, 641-649, 1996).
Кроме того, процесс старения можно рассматривать как настоящее патологическое состояние (ссыл.: Pathophysiology: the biological basis for disease in adults and children, стр. 71-77).
Известные лекарства при введении пациенту, страдающему патологиями, связанными с окислительным стрессом и/или эндотелиальными дисфункциями, показывают низкую активность и/или высокую токсичность.
Это имеет место для лекарств, например, таких как противовоспалительные, сердечно-сосудистые лекарственные средства, лекарственные средства для дыхательного аппарата, лекарственные средства для центральной нервной системы, лекарственные средства для костной системы, антибиотики, урогенитальные, эндокринные лекарственные средства и т.д.
Исследования в области лекарственных средств направлены на обнаружение новых молекул, имеющих улучшенный терапевтический индекс (соотношение эффективность/токсичность) или пониженное соотношение риск/польза, в том числе для упомянутых выше патологических условий, причем терапевтический индекс большого ряда лекарств является пониженным. В действительности в вышеупомянутых условиях окислительного стресса и/или эндотелиальных дисфункций многие лекарства проявляют пониженную активность и/или более высокую токсичность.
Например, противовоспалительные лекарственные средства, такие как нестероидные противовоспалительные лекарства (НСПВЛ - NSAIDs) и лекарственные средства против колита, такие как 5-аминосалициловая кислота и ее производные, показывают следующие недостатки. НСПВЛ проявляют токсичность, особенно когда организм ослаблен или находится под влиянием болезненных условий, связанных с окислительным стрессом. Вышеупомянутые условия являются, например, следующими: возраст, ранее существовавшая язва, ранее существовавшие кровотечения в желудке, ослабляющие хронические заболевания, такие как, в частности, те, которые поражают сердечно-сосудистый, почечный аппараты, гематокризис и т.д. ("Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving non-steroidal anti-inflammatory drugs. A randomized, double blind, placebo-controlled trial." F.E. Silverstein et al., Ahh. Intern. Med. 123/4, 241-9, 1995; Martindale 31a ed., 1996, p.73, Current Medical Diagnosis and Treatment 1998, стр. 431 и 794).
Введение противовоспалительных лекарственных средств пациентам с вышеупомянутыми патологическими состояниями может быть проведено только в дозах, которые ниже, чем дозы, используемые в терапии, для того, чтобы избежать явлений сильной токсичности. Таким образом, противовоспалительная активность становится низкой.
Бета-блокаторы, используемые для лечения ангины, гипертензии и сердечной аритмии, проявляют побочные действия в отношении дыхательного аппарата (одышка, бронхостеноз) и, следовательно, они могут вызывать проблемы у пациентов, имеющих патологии у вышеупомянутых органов (астма, бронхит). Следовательно, бета-блокаторы также ухудшают такие респираторные заболевания как астма. Следовательно, для пациентов с астмой дозы вышеупомянутых лекарств должны быть уменьшены для того, чтобы не подвергать еще большей опасности дыхательную деятельность. Таким образом, эффективность бета-блокаторов становится очень низкой.
Антитромботические средства, такие как, например, дипиридамол, аспирин и т.д., используемые для профилактики тромботических явлений, имеют такие же недостатки. Для пациентов с патологиями, связанными с окислительным стрессом и/или эндотелиальными дисфункциями, терапевтическое действие или толерантность этих лекарств, как в случае аспирина, очень снижены.
Бронхолитические средства, например сальбутамол и т.д., используются при лечении астмы и бронхита и лекарственные средства, активные в отношении холинергической системы, используются в таких патологиях как недержание мочи. Их введение может вызвать похожие побочные эффекты, влияющие на сердечно-сосудистый аппарат, вызывая проблемы у пациентов с заболеваниями сердца и гипертензией. Заболевания сердца и гипертензия являются патологиями, связанными, как указано выше, с окислительным стрессом и/или эндотелиальными дисфункциями. Также эти лекарственные средства имеют те же недостатки, как указанные выше.
Отхаркивающие и муколитические лекарства, которые используются в терапии воспалительных состояний дыхательных органов, показывают недостатки на пациентах в связи с вышеупомянутыми условиями. Их введение может вызвать изжогу и раздражение желудка, особенно у пожилых.
Ингибиторы резорбции костей, такие как фосфонаты (например, алендронат) являются лекарствами, проявляющими высокую желудочно-кишечную токсичность. Следовательно, эти лекарства также могут проявлять такие же недостатки, как и указанные выше.
Ингибиторы фосфодиэстеразы, такие как, например, сильденафил, запринаст, используемые при заболеваниях сердечно-сосудистой и дыхательной систем, характеризуются сходными проблемами в отношении толерантности и/или эффективности в вышеупомянутых патологических условиях окислительного стресса и/или эндотелиальных дисфункций.
Противоаллергические лекарства, например цетиризин, монтелюкаст и т.д., имеют схожие недостатки в вышеуказанных патологических состояниях, в особенности это касается их эффективности.
Антиангиотензивные лекарства, ингибиторы ангиотензинконвертирующего фермента (АСЕ-ингибиторы), например эналаприл, каптоприл и т.д., и ингибиторы ангиотензиновых рецепторов, например лозартан, используются в лечении сердечно-сосудистых заболеваний. Их недостаток заключается в наличии побочных эффектов в отношении дыхательного аппарата (например, кашель и т.д.) в вышеупомянутых патологических условиях.
Антидиабетические лекарства, и инсулинового и гипогликемического типа, такие как, например, сульфонилурез, толбутамид, глипирид, гликлазид, глибурид, никотинамид и т.д., являются неэффективными в профилактике диабетических осложнений. Их введение может давать побочные эффекты, такие как, например, повреждения желудка. Эти явления могут стать более интенсивными в вышеуказанных патологических состояниях.
Антибиотики, например ампициллин, кларитромицин и т.д., и антивирусные лекарства, ацикловир и т.д., имеют проблемы в отношении своей переносимости, например они вызывают желудочно-кишечное раздражение.
Противоопухолевые лекарства, например доксорубицин, гидрохлорид рубомицина, цисплатин и т.д., являются высокотоксичными по отношению к различным органам, среди которых - желудок и кишечник. Вышеупомянутая токсичность также усугубляется в вышеуказанных патологиях окислительного стресса и/или эндотелиальных дисфункций.
Лекарства от слабоумия, например никотин и колиномиметики, отличаются низкой переносимостью, особенно при вышеупомянутых патологиях.
Ощущалась необходимость иметь лекарственные средства, показывающие улучшенное терапевтическое действие, то есть обеспечение и более низкой токсичности и/или более высокой эффективности, в результате чего они могли бы вводиться пациентам в болезненных состояниях окислительного стресса и/или эндотелиальных дисфункций, не вызывая при этом побочных эффектов лекарств предшествующего уровня техники.
В настоящее время с удивлением и неожиданно обнаружено, что вышеуказанные проблемы, демонстрируемые при введении лекарств пациентам, пораженных окислительным стрессом и/или эндотелиальными дисфункциями, и пожилым в принципе решаются новым классом лекарственных средств, как описано ниже.
Объектом настоящего изобретения являются соединения или их соли, имеющие следующую общую формулу (I):
A-(B)bo-C-N(O)2 (I),
где bo=0 или 1;
А=R-T1-, где R означает лекарственный радикал, как определено ниже, и имеет формулу R-T1-Z или R-T1-OZ, где Z представляет собой Н или C1-C5 алкил,
выбраны из следующих групп:
противовоспалительные лекарственные средства: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенак натрия, дифлунизал, этодолак, флуфенамовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенамовая кислота, мефенамовая кислота, мелоксикам, месаламин, напроксен, нифлюмовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенамовая кислота, толметин, зомепирак;
аналгезирующие лекарственные средства: ацетаминопрофен, ацетилсалицилсалициловая кислота, беноксапрофен, трамадол;
бронхолитические лекарственные средства: альбутерол, карбутерол, кленбутерол, фенотерол, метапротеренол, пирбутерол, салметерол, тербуталин;
отхаркивающие лекарственные средства: амброксол, бромгексин, гвайакол;
антиастматические лекарственные средства: цетиризин, левокабастин, терфенадин;
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл;
бета-блокаторы: альпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол;
антитромботические и вазоактивные лекарственные средства:
аргатробан, бенфуродил гемисукцинат, клопидогрел, дальтепарин, дипиридамол, эноксапарин, илопрост, озагрел, трифлузал;
антидиабетические лекарственные средства: никотинамид;
противоопухолевые лекарственные средства: антрамицин, даунорубицин, доксорубицин, эпирубицин;
противоязвенные лекарственные средства: циметидин, омепразол, пантопразол;
антигиперлипидемические лекарственные средства: аторвастатин, флувастатин, ловастатин, натриевая соль привастатина, симвастатин;
антибиотики: амоксициллин, ампициллин, азтреонам, биапенем, карбенициллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, меклоциклин, моксалактам, панипенем, бакампициллин;
противовирусные лекарственные средства: ацикловир, фамцикловир, ганцикловир, пенцикловир, зидовудин;
ингибиторы резорбции костей: алендроновая кислота, этидроновая кислота, памидроновая кислота;
лекарства против слабоумия: такрин.
T1=(CO), О, S, N или NR1C, где R1C означает Н или C1-C5 алкил,
В=-ТB-Х2-ТBI-,
где
ТB и ТBI равны или различны и могут быть выбраны из (СО), О, S, N или NR1C, где R1C имеет значения, как определено выше;
Х2 означает бивалентную мостиковую группу, как соответствующий предшественник В, имеющий формулу
Z'-TB-X2-TBI-Z",
где Z', Z" независимы и означают Н или ОН, выбраны из следующих соединений:
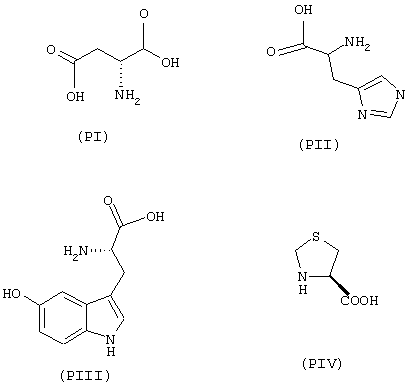
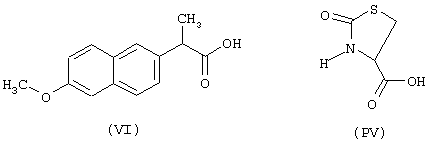
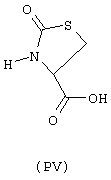
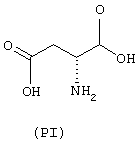
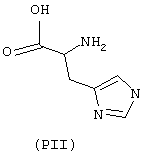
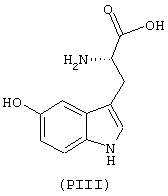
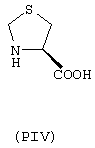
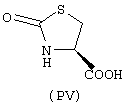
- аминокислоты: аспарагиновая кислота (PI), гистидин (PII), 5-гидрокситриптофан (PIII), 4-тиазолидинкарбоновая кислота (PIV), 2-оксо-4-тиазолидинкарбоновая кислота (PV)

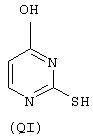
- моноспирты или тиолы: 2-тиоурацил (QI), 2-меркаптоэтанол (QII),
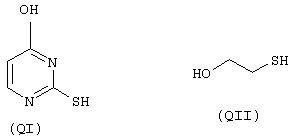
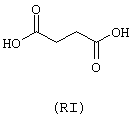
- янтарная кислота (RI)
С означает бивалентный -TC-Y- радикал,
где ТC=(СО), когда tx=О, ТC=X, когда txx=О, X является таким, как определено выше;
Y означает
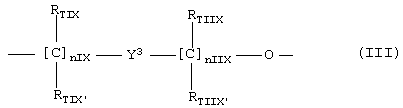
где nIX означает целое число между 0 и 3, предпочтительно 1;
nIIX означает целое число между 1 и 3, предпочтительно 1;
RTIX, RTIX', RTIIX, RTIIX' равны или отличны друг от друга, означают Н или линейный или разветвленный C1-C4 алкил, предпочтительно RTIX, RTIX', RTIIX, RTIIX' означают Н;
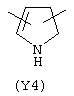
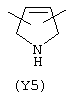
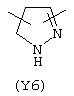
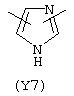
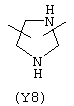
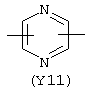
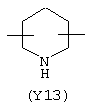
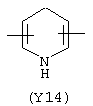
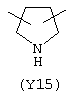
Y3 является насыщенным, ненасыщенным или ароматическим гетероциклическим кольцом, содержащим, по крайней мере, один атом азота, предпочтительно один или два атома азота, и вышеупомянутое кольцо содержит от 5 до 6 атомов, или Y означает Yo, выбранный из следующего:
- алкиленоксигруппа R’O, где R' является линейной или разветвленной, когда возможно C1-C20, предпочтительно имеющей от 1 до 6 атомов углерода, более предпочтительно 2-4, или циклоалкилен, имеющий от 5 до 7 атомов углерода, в циклоалкиленовом кольце один или более атомов углерода могут быть замещены гетероатомами, кольцо может иметь боковые цепи R'-типа, причем R’ является таким, как определено выше, или
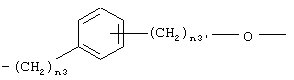
где n3 означает целое число от 0 до 3 и n3’ означает целое число от 1 до 3;
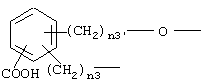
где n3 и n3' имеют вышеупомянутые значения;
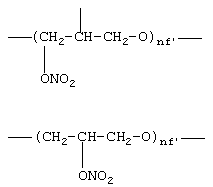
где nf’ означает целое число от 1 до 6;
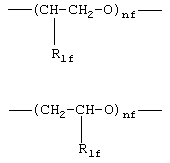
где R1f=Н, СН3 и nf означают целое число от 1 до 6,
при условии, что в соединениях формулы (I):
- когда bo=0 и С=-TС-Yo-, лекарства формулы A=R-T1-Z или A=R-T1-OZ, как определено выше, и относятся к группе АСЕ-ингибиторов;
- когда bo=R-T1-OZ не относятся к группе нестероидных противовоспалительных лекарственных средств;
- где Y=R'О, R' представляет собой C1-C6 алкил.
Неожиданно оказалось, что продукты изобретения формулы (I) имеют улучшенный терапевтический индекс в условиях окислительного стресса по сравнению с лекарствами-предшественниками.
Y3 в формуле (III) предпочтительно выбирают из следующих:
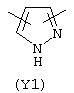
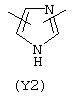
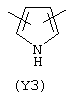
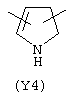
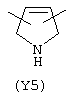
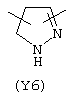
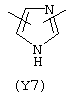
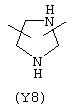
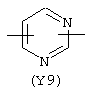
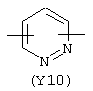
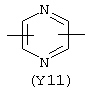
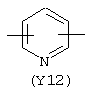
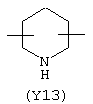
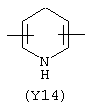
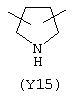
Наиболее предпочтительным из Y3 является Y12 (пиридил), замещенный в положениях 2 и 6. Эти связи также могут находиться в асимметричном положении, например Y (пиридил) может быть замещен также в положениях 2 и 3; Y1 (пиразол) может быть 3,5-дизамещенным.
Соединения согласно настоящему изобретению формулы (I) могут быть преобразованы в соответствующие соли. Например, одним из путей образования соли является следующий: когда в молекуле присутствует один атом азота, по существу являющийся основным, чтобы участвовать в реакции образования соли, посредством реакции в органическом растворителе, таком как, например, ацетонитрил, тетрагидрофуран, этот атом реагирует с эквимолекулярным количеством соответствующей органической или неорганической кислоты. Для образования соли предпочтительно в формуле изобретения присутствует Y или Y' формулы (III).
Примеры органических кислот являются следующими: щавелевая, винная, малеиновая, янтарная, лимонная кислоты.
Примеры неорганических кислот являются следующими: азотная, соляная, серная, фосфорная кислоты.
Производные согласно настоящему изобретению могут быть использованы в терапевтических показаниях лекарства-предшественника, позволяя получать преимущества, показанные ниже для некоторых групп этих лекарств:
- Противовоспалительные лекарственные средства НПЛС: соединения согласно настоящему изобретению являются переносимыми и эффективными, даже когда организм ослаблен и находится в условиях окислительного стресса. Вышеуказанные лекарства могут быть также использованы в тех патологиях, где воспаление играет значительную патогенную роль, в таких как, например, но не ограничиваясь этим, рак, астма, инфаркт миокарда.
- Адренергические блокаторы α- или β-блокаторного типа: спектр действия соединений формулы (I) приводит к более обширным результатам, чем исходные лекарственные средства; для прямого действия на гладкую мускулатуру применяют ингибирование нервных бета-адренергических сигналов, контролирующих сокращение кровяных сосудов. Побочные эффекты (одышка, бронхостеноз), влияющие на респираторную систему, являются сниженными.
- Антитромботические лекарственные средства: антитромбоцитная активность усиливается, и в случае производных аспирина переносимость желудком улучшается.
- Бронхолитические средства и лекарства, активные в отношении холинергической системы: побочные эффекты, влияющие на сердечно-сосудистый аппарат (тахикардия, гипертензия) снижаются.
- Отхаркивающие и муколитические лекарственные средства: улучшены результаты желудочно-кишечной толерантности.
- Дифосфонаты: токсичность в отношении желудочно-кишечного тракта очень снижена.
- Ингибиторы фосфодиэстеразы (ФДЭ) (бронхолитические средства): улучшена терапевтическая эффективность, причем при равной дозировке; следовательно, можно использовать соединения изобретения для введения более низких доз лекарства и уменьшения побочных эффектов.
- Антилейкотриеновые лекарства: улучшенная эффективность.
- АСЕ-ингибиторы: улучшенная терапевтическая эффективность и сниженные побочные эффекты (одышка, кашель) в отношении дыхательного аппарата.
Противодиабетические лекарства (инсулинового и гипогликемического типа), антибиотики, противовирусные, противоопухолевые, спазмолитические лекарственные средства, лекарства для терапии слабоумия: улучшенная эффективность и переносимость.
Примеры предшественников лекарственных средств, которые могут быть использованы, являются следующими.
Для противовоспалительных лекарственных средств могут быть приведены следующие примеры:
противовоспалительные лекарственные вещества: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенак натрия, дифлунизал, этодолак, флуфенамовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенамовая кислота, мефенамовая кислота, мелоксикам, месаламин, напроксен, нифлюмовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенамовая кислота, толметин, зомепирак;
аналгезирующие лекарственные средства: ацетаминофен, ацетаминосалол.
В качестве лекарств для респираторного и мочеполового аппаратов (бронхолитические лекарственные средства, отхаркивающие лекарственные средства, антиастматические лекарственные средства) могут быть упомянуты следующие:
бронхолитические лекарственные средства: альбутерол, карбутерол, кленбутерол, фенотерол, метапротеренол, пирбутерол, салметерол, тербуталин;
отхаркивающие лекарственные средства: амброксол, бромгексин, гвайакол;
антиастматические лекарственные средства: цетиризин, левокабастин, терфенадин.
В качестве сердечно-сосудистых лекарственных средств (АСЕ-ингибиторы, бета-блокаторы, антитромботические и сосудорасширяющие средства, антидиабетические и гипогликемические лекарства) могут быть упомянуты следующие:
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл;
бета-блокаторы: альпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол;
антитромботические и сосудорасширяющие лекарственные средства: аргатробан, бенфуродил гемисукцинат, клопидогрел, дальтепарин, дипиридамол, эноксапарин, илопрост, озагрел, трифлузал;
антидиабетические лекарственные средства: никотинамид.
Среди противоопухолевых лекарственных средств могут быть упомянуты следующие: антрамицин, даунорубицин, доксорубицин, эпирубицин.
Среди противоязвенных лекарственных средств могут быть упомянуты следующие: циметидин, омепразол, пантопразол.
Среди антигиперлипидемических лекарственных средств (статины) могут быть упомянуты следующие: аторвастатин, флувастатин, ловастатин, натриевая соль привастатина, симвастатин.
Среди антибиотиков/противовирусных препаратов могут быть упомянуты следующие:
антибиотики: амдиноциллин, амоксициллин, азтреонам, биапенем, карбенициллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, моксалактам, панипенем, бакампициллин;
противовирусные лекарственные средства: ацикловир, фамцикловир, ганцикловир, пенцикловир, видарабин, зидовудин.
Среди ингибиторов резорбции костей (дифосфонаты) могут быть упомянуты следующие:
алендроновая кислота, этидроновая кислота, памидроновая кислота.
Среди лекарственных средств против слабоумия могут быть упомянуты следующие: такрин.
Предпочтительными веществами являются следующие:
среди противовоспалительных препаратов: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенак натрия, дифлунизал, этодолак, флуфенамовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенамовая кислота, мефенамовая кислота, мелоксикам, месаламин, напроксен, нифлюмовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенамовая кислота, толметин, зомепирак;
среди аналгезирующих лекарственных средств: ацетаминофен.
Среди лекарств для респираторного и мочеполового аппаратов (бронхолитические лекарственные средства, отхаркивающие средства, лекарственные средства, антиастматические лекарственные средства):
бронхолитические лекарственные средства: альбутерол, карбутерол, кленбутерол, фенотерол, метапротеренол, пирбутерол, салметерол, тербуталин;
отхаркивающие лекарственные средства:
амброксол, бромгексин, гвайакол;
антиастматические лекарственные средства: цетиризин, левокабастин, терфенадин.
Среди сердечно-сосудистых лекарственных средств:
АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл;
бета-блокаторы: альпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол;
антитромботические и вазоактивные лекарственные средства: аргатробан, клопидогрел, дальтепарин, дипиридамол, эноксапарин, илопрост, озагрел, трифузал;
противодиабетические препараты: никотинамид.
Среди противоопухолевых лекарственных средств: антрамицин, даунорубицин, доксорубицин, эпирубицин.
Среди противоязвенных препаратов: циметидин, омепразол, пантопразол.
Среди антигиперлипидемических препаратов: ловастатин, натриевая соль привастатина, симвастатин.
Среди антибиотиков/противовирусных лекарственных веществ:
антибиотики: амоксициллин, азтреонам, биапенем, карбенециллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипинем, моксалактам, панипенем, бакампициллин;
противовирусные препараты: ацикловир, фамцикловир, гинцикловир, пенцикловир, видарабин, зидовудин.
Среди ингибиторов резорбции костей: алендроновая кислота, этидроновая кислота, памидроновая кислота.
Среди лекарств против слабоумия: такрин.
Вышеупомянутые вещества, предшественники А, получают согласно способам, известным в предшествующем уровне техники. Смотри, например, “The Merck Index”, 12a Ed.(1996), упомянутый здесь в качестве ссылки. Могут быть использованы соответствующие изомеры, включающие оптические изомеры, если они доступны.
Томоксипрол получают согласно способу, описанному в Европейском Патенте 12866.
Соединения формулы (I) получают способами синтеза, упомянутыми здесь ниже.
Выбор реакций для каждого способа зависит от реакционных групп, присутствующих в молекуле лекарства-предшественника, в соединении предшественника В или B1, который может быть, как упомянуто выше, бивалентным или моновалентным, и в соединении предшественника С.
Эти реакции проводят согласно способам, хорошо известным в предшествующем уровне техники, которые позволяют получить связи между лекарством-предшественником, лекарством-предшественником В или B1 и соединением-предшественником С, как определено выше.
Когда реакционная функция лекарства-предшественника (например, -СООН, -ОН) вступает в ковалентную связь, например, сложноэфирного, амидного, эфирного типа, вышеуказанная функция может быть восстановлена способами, хорошо известными в предшествующем уровне техники.
Некоторые схемы синтеза для получения соединений согласно изобретению представлены ниже:
А. Синтез соединений формулы (I).
1. Синтез соединения, полученного реакцией между лекарством-предшественником и соединением-предшественником В.
1а. Когда лекарство имеет общую формулу R-COOH и функциональная группа соединения-предшественника В, которая связывает саму себя с карбоксильной функцией лекарства, имеет формулу XZ, причем Х является таким, как определено выше, и Z=Н, реакции проводят в зависимости от природы второй реакционной группы, присутствующей в соединении-предшественнике В.
1a.1. Когда вторая реакционная группа, присутствующая в соединении-предшественнике В, является карбоксильной группой, общая схема синтеза предполагает начальное формирование галида R-COHHal кислоты (Hal=Сl, Вr) и последующую реакцию с НХ-группой соединения предшественника В:
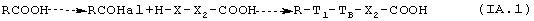
где Х2, T1, ТB такие, как определено выше.
Когда в двух реакционных соединениях присутствуют другие функциональные группы СООН и/или НХ, они должны быть защищены перед реакцией согласно методам, известным в данном уровне техники, например, как описано в книге Th. W. Greene: "Protective groups in organic synthesis". Harvard University Press, 1980.
Ацилгалид RCOHal получают согласно способам, известным в предшествующем уровне техники, например посредством тионил- или оксалилхлорида, РIII или Pv галидов в инертных растворителях в реакционных условиях, таких как, например, толуол, хлороформ, ДМФ и т.д.
В частности, если НХ-группа соединения-предшественника В означает NH2 или ОН, или SH, предшествующее лекарство формулы R-COOH сначала превращают в соответствующий ацилгалид RCOHal, как указано выше, и затем вводят в реакцию с НХ-группой предшествующего соединения В в присутствии органического основания, такого как триэтиламин, пиридин и т.д., используя инертный растворитель в реакционных условиях, такой как толуол, тетрагидрофуран и т.д., при температуре в интервале 0-25°С.
Альтернативно предыдущему синтезу лекарство-предшественник формулы R-COOH может быть обработано агентом, активирующим карбоксильную группу, выбранным из N,N'-карбонилдиимидазола (КДИ), N-гидроксибензотриазола и дициклогексилкарбодиимида, в растворителе, таком как, например, ДМФ, ТГФ, хлороформ и т.д., при температуре в интервале от -5 до -50°С, и полученному соединению позволяют реагировать in situ с реакционной функцией предшествующего соединения В для получения соединения формулы (IA.1).
1a. 2. Когда соединение-предшественник В содержит две функциональные группы XZ, одинаковые или отличные друг от друга, причем Х является таким, как определено выше, и Z=Н, лекарство-предшественник, имеющее формулу R-COOH, сначала обрабатывают агентом, активирующим карбоксильную группу, как описано выше в 1a.1, и затем соединением-предшественником В, после защиты одной или двух реакционных НХ-групп, например, реакцией с ацетилом или трет-бутилоксикарбонилом, предполагая инициирующую функцию в конце синтеза. Схема является следующей:

где X, T1, ТB, Х2 являются такими, как определено выше, и G является защитной группой НХ-функции.
2. Синтез нитроксипроизводного.
2а.1. Когда соединение, полученное в конце предыдущей стадии 1а, имеет формулу (IA.1), кислота может быть превращена в соответствующую натриевую соль и затем к ней можно применить способы, известные в данной области техники, для получения конечного соединения, например, согласно одной из следующих схем синтеза:
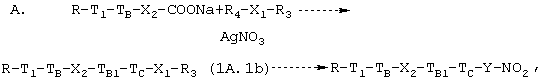
где T1, ТB, Х2, TBI, ТC являются такими, как определено выше, R4 выбран из Cl, Вr, Y такой, как определено выше, X1 означает Y-радикал, свободный от атома кислорода, R3 означает Сl, Вr, иод, ОН. Когда R3=ОН, соединение формулы (1A.1b) направляют на галогенирование, например, с РВr3, PCl5, SOCl2, PPh3 + I2, и затем в реакцию с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран. Если R3 означает Сl, Вr, иод, соединение формулы (1A.1b) реагирует прямо с AgNO3, как упомянуто выше.
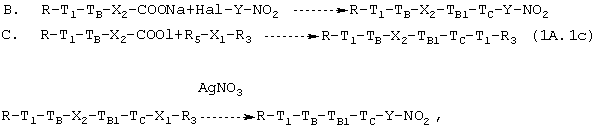
где R5=ОН или NHR1C, R1C, R3 и другие символы являются такими, как определено выше.
Показанные выше реакции хорошо известны в предшествующем уровне техники. Смотри, например, патентные заявки на имя данного Заявителя WO 94/12463, WO 95/09831 и WO 95/30641.
Когда X1 является линейным С4 алкилом, соответствующая кислота R-Т1-ТB-Х2-СООН реагирует с трифенилфосфином в присутствии галогенирующего агента, такого как СВr4 или N-бромсукцинимида, в тетрагидрофуране с получением соединения (1A.1c), где R3=Вr.
2а.2. Когда соединение, полученное в конце предыдущей стадии 1а, имеет формулу (IA.2), соответствующее нитроксипроизводное получают обработкой галогенкарбоновой кислоты формулы Hal-X1-COOH с таким X1, как определено выше, сначала с агентом, активирующим карбоксильную группу, как описано в 1А.1, и затем с соединением формулы (IA.2), получая галогенпроизводное, которое выделяют и затем растворяют в органическом растворителе (см. параграф 2а.1) и обрабатывают нитратом серебра. Общая схема реакции является следующей:
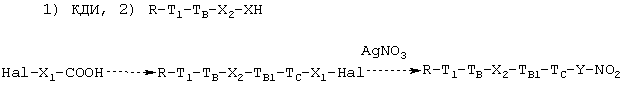
где T1, ТB, X2, TBI, TC, Y являются такими, как определено выше.
Альтернативно может быть использован галид Hal-X1-COCl, где Hal является предпочтительно бромом, который реагирует с соединением формулы (IA.2).
1b. Когда лекарство-предшественник имеет реакционную функцию НХ, где Х является таким, как определено выше, вместо карбоксильной группы две функциональные группы, присутствующие в соединении-предшественнике В, могут быть следующими.
1b.1. Карбоксильная группа, которая реагирует с НХ-функцией предшественника лекарства, и НХ-группа, причем последняя реакционная группа соединения предшественника В равна или отлична от функциональной группы лекарства-предшественника. Формула соединения предшественника В является Н-Х-Х2-СООН, тип, где Х и Х2 такие, как определено выше. Н-Х-функцию соединения предшественника В защищают согласно известному предшествующему уровню техники способу, и карбоксильная группа реагирует, как указано выше, согласно следующей схеме:
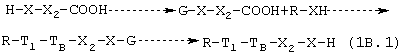
В конце реакции функция НХ соединения предшественника В восстанавливается.
1b.2. Когда соединение предшественника В содержит две карбоксильные группы, его обрабатывают эквимолярным количеством агента, активирующего карбоксильную группу, в условиях, предварительно описанных в 1a.1, и затем осуществляют реакцию с реакционной НХ-функцией молекулы лекарства-предшественника. Другие возможные реакционные функции НХ-типа, присутствующие в двух соединениях, должны быть защищены, как упомянуто ранее. В заключение получают соединение формулы R-T1-TB-X2-COOH (IB.2).
2b. Синтез нитроксипроизводного.
2b.1. Для получения конечного нитроксипроизводного, начиная с соединения формулы R-T1-ТB-Х2-Х-Н (1В.1), полученного в конце синтеза, описанного в 1b.1, соединение (1В.1) реагирует с галогенводородной кислотой формулы Hal-X1-COOH, которую обрабатывают, как предварительно описано в параграфе 1a.1, или с соответствующим хлоридом галогенводородной кислоты. Полученное соединение растворяют в органическом растворителе, например ацетонитриле или тетрагидрофуране, и осуществляют реакцию с нитратом серебра.
2b.2. Для получения конечного нитроксипроизводного, исходя из соединения формулы R-T1-TB-X2-COOH (1B.2), полученного в конце синтеза, описанного в 1b.2, кислоту превращают в соответствующую натриевую соль, она реагирует с соединением R4-X1-R3, предварительно определенным в реакции А схемы параграфа 2а.1, получая согласно такому же процессу, как там упомянуто, конечное нитроксипроизводное. Альтернативно, когда X1 означает линейный С4 алкил, кислота (1В.2) реагирует с трифенилфосфином в присутствии галогенирующего агента, такого как СВr4 или N-бромсукцинимид, в тетрагидрофуране, и полученное соединение, растворенное в органическом растворителе, например ацетонитриле, тетрагидрофуране, реагирует с нитратом серебра.
2b.3. Альтернативно процессу синтеза согласно 1b.1 и 2b.1 можно на первой стадии проводить реакцию НХ-функции соединения предшественника В НХ-Х2-СООН с ацилхлоридом галогенводородной кислоты формулы Hal-X1-CO-Cl, где Hal предпочтительно является Вr, и затем карбоксильной функции таким образом полученного соединения с лекарством-предшественником R-HX. На третьей и последней стадии Hal-группу замещают -ONO2 согласно процессу, описанному в 2b.1. Схема реакции является следующей:
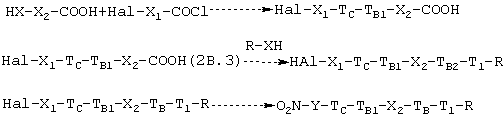
где ТC, ТB1, ТB, T1, Х2, X1, Y являются такими, как определено выше.
В предыдущей схеме нитрование может альтернативно проводиться с кислотой формулы (2В.3).
Следующие примеры служат для иллюстрации изобретения и не должны рассматриваться как ограничивающие его.
Пример 1
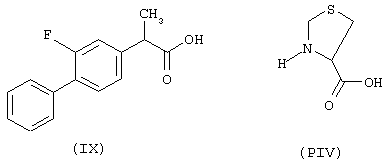
Синтез 4-нитроксибутилового эфира 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты (NO-флурбипрофен), соединение NCX 2002

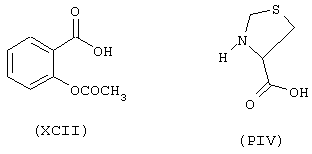
начиная с флурбипрофена (формула IX) и предшественника В в виде (L)-4-тиазолидинкарбоновой кислоты (формула PIV)
а) Синтез 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты
К раствору 2-фтор-α-метил-(1,1’-бифенил)-4-уксусной кислоты (10 г, 41 ммолей) в толуоле (100 мл) и N,N,-диметилформамиде (10 мл), охлажденному до 0°С, добавляют оксалилхлорид (3,52 мл, 82 ммоля). Через 2 часа при комнатной температуре раствор упаривают при пониженном давлении. Полученный остаток растворяют в ацетоне (50 мл) и раствор добавляют к раствору 4-тиазолидинкарбоновой кислоты (5,44 г, 41 ммоля) и триэтиламина (14,9 мл, 106 ммолей) в ацетоне (50 мл), охлажденному до 0°С. Через 2 часа раствор подкисляют 4 н. НСl, концентрируют под вакуумом, остаток обрабатывают этилацетатом и органическую фазу промывают сначала 2 н. НСl, затем водой. Органическую фазу обезвоживают сульфатом натрия и упаривают при пониженном давлении. Посредством кристаллизации с этилацетатом/н-гексаном получают 9,4 г ожидаемого продукта в форме белого твердого вещества, имеющего температуру плавления 142-147°С.
1Н-ЯМР (CDCl3): 7,74-7,62 (4Н, m), 7.35 (2Н, t), 7,18-7,13 (2Н, m), 5,06 (1H, m), 4.63 (1H, d), 4,42 (1H, d), 4,14 (1H, q), 3,13 (2H, m), 1,53 (2Н, d).
b) Синтез 4-бромбутилового эфира 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты
К раствору кислоты, полученному на предыдущей стадии а), (9,43 г, 26,24 ммоля) в тетрагидрофуране (150 мл) добавляют трифенилфосфин (13,76 г, 52,49 ммоля) и тетрабромид углерода (17,4 г, 52,49 ммоля). Реакционную смесь оставляют перемешиваться в течение 24 часов при комнатной температуре. Растворитель удаляют выпариванием при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле, разбавляя н-гексаном/этилацетатом 8/2. Получают 2,25 г сложного эфира в виде масла.
с) Синтез 4-нитроксибутилового эфира 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты.
К раствору сложного эфира, полученного в конце предыдущей стадии (2,6 г, 5,26 ммолей), в ацетонитриле (20 мл) добавляют нитрат серебра (1,07 г, 6,3 ммоля). Реакционную смесь нагревают с обратным холодильником в течение 4 часов в отсутствие света. Образовавшуюся соль удаляют фильтрованием и раствор упаривают при пониженном давлении. Полученный осадок очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом, 7/3. Получают 0,84 г 4-нитроксибутилового эфира 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты в виде масла.
1Н-ЯМР (CDCl3): 7,56-7,09 (8Н, m), 5,77 (1H, dd), 4,67 (2H, d), 4,51 (3Н, t), 4,24 (2Н, t), 4,15 (1H, q), 3,30-3,17 (2H, m), 1,74-1,70 (4Н, m), 1,52 (3Н, d).
Пример 2
Синтез 4-нитроксибутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)тиазолидона-4-карбоновой кислоты (NO-напроксен) (NCX 2001)
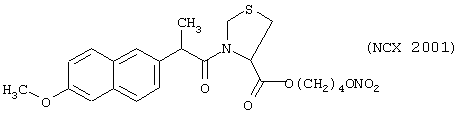
исходя из напроксена (формула VI) и предшественника В в виде (L)-4-тиазолидинкарбоновой кислоты (формула PIV)
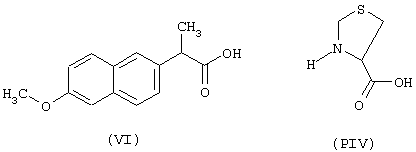
a) Синтез 3-(6-метокси-α-метил-2-нафтилацетил)тиазолидин-4-карбоновой кислоты
К раствору 6-метокси-α-метил-2-нафтилуксусной кислоты (4,02 г, 17,5 ммолей) в толуоле (30 мл) и N,N-диметилформамиде (0,3 мл), охлажденному до 0°С, добавляют оксалилхлорид (2,92 мл, 34,06 ммолей). Через 2 часа при комнатной температуре раствор выпаривают при пониженном давлении. Полученный остаток растворяют в ацетоне (50 мл) и этот раствор добавляют к раствору 4-тиазолидинкарбоновой кислоты (2,33 г, 17,5 ммолей) и триэтиламина (6,34 мл, 45,5 ммолей) в ацетоне (50 мл), охлажденному до 0°С. Через 2 часа раствор подкисляют 4 н. НСl, концентрируют под вакуумом, остаток обрабатывают этилацетатом и органическую фазу промывают сначала 2 н. НСl, затем водой. Органическую фазу обезвоживают сульфатом натрия и упаривают при пониженном давлении. Получают 4,43 г ожидаемого продукта в виде белого твердого вещества, имеющего температуру плавления 164-168°С.
1Н-ЯМР (CDCl3): 7,75-7,66 (3Н, m), 7,34 (1H, d), 7,14-7,11 (2Н, m), 5,14 (1H, m), 4,80-4,61 (2H, m), 4,07 (1H, q), 3,91 (3Н, s), 3,30-3,23 (2Н, m), 1.53 (3Н, d).
b) Синтез 4-бромбутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)тиазолидин-4-карбоновой кислоты
К раствору кислоты, полученной на предыдущей стадии а) (4 г, 11,6 ммолей), в тетрагидрофуране (50 мл) добавляют трифенилфосфин (6,07 г, 23,1 ммолей) и тетрабромид углерода (7,66 г, 23,2 ммолей). Реакционную смесь оставляют при перемешивании на 24 часа при комнатной температуре. Растворитель удаляют выпариванием при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом, 7/3. Получают 2,25 г сложного эфира в форме масла.
c) Синтез 4-нитроксибутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)тиазолидин-4-карбоновой кислоты
К раствору сложного эфира, полученного в конце предыдущей стадии (2 г, 4,16 ммолей) в ацетонитриле (20 мл) добавляют нитрат серебра (0,85 г, 5 ммолей). Реакционную смесь нагревают с обратным холодильником в течение 5 часов в отсутствие света. Образовавшуюся соль удаляют фильтрованием и раствор упаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом, 7/3. Получают 0,99 г 4-нитроксибутилового эфира 3-(6-метокси-α-метил-2-нафталинацетил)тиазолидин-4-карбоновой кислоты в виде масла.
1Н-ЯМР (СDС13): 7,66 (3Н, m), 7,38 (1Н, m), 7,15 (2Н, m), 5,06 (1Н, dd), 4,66 (2Н, d), 4,51 (2Н, t), 4,25 (2Н, t), 3,98 (1H, q), 3,92 (3Н, s), 3,13 (2Н, d), 1,84 (4Н, m), 1,53 (3Н, d).
Пример 3
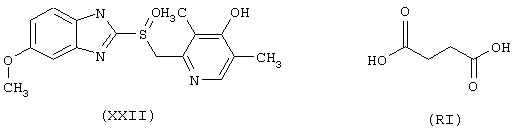
Синтез 4-нитроксибутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты (NCX 2150)
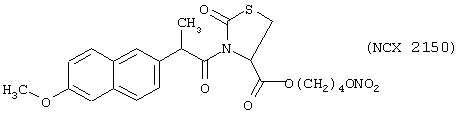
исходя из напроксена (формула VI) и предшественника В в виде (L)-2-оксо-4-тиазолидинкарбоновой кислоты (формула PV)
а) Синтез 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты
К раствору 6-метокси-α-метил-2-нафтилуксусной кислоты (7,0 г, 30,4 ммолей) в толуоле (100 мл) и N,N-диметилформамиде (10 мл), охлажденному до 0°С, добавляют оксалилхлорид (5,23 мл, 61 ммолей). Через 2 часа при комнатной температуре раствор упаривают при пониженном давлении. К раствору полученного осадка, растворенного в тетрагидрофуране (50 мл), добавляют смесь, состоящую из 2-оксотиазолидин-4-карбоновой кислоты (4,07 г, 27,6 ммолей), 4-диметиламинопиридина (0,84 г, 6,9 ммолей), триэтиламина (7,69 мл, 55,2 ммолей) в тетрагидрофуране (50 мл), охлажденную до -10°С. Смесь оставляют при комнатной температуре на 24 часа. Реакционную смесь промывают 5% НСl, затем водой. Органическую фазу обезвоживают сульфатом натрия и затем упаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле, элюируя метиленхлоридом/метанолом 95/5. Получают 6,79 г предполагаемого продукта в форме аморфного твердого вещества.
b) Синтез 4-бромбутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты
К раствору 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты (6,79 г, 18,9 ммолей) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (9,91 г, 37,8 ммолей) и тетрабромид углерода (12,53 г, 37,8 ммолей). Реакционную смесь оставляют перемешиваться в течение 16 часов при комнатной температуре, затем растворитель удаляют выпариванием при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 7/3. Получают 1,83 г сложного эфира в форме масла.
c) Синтез 4-нитробутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты
К раствору сложного эфира, полученного в конце предыдущей стадии (1,7 г, 3,44 ммоля) в ацетонитриле (20 мл) добавляют нитрат серебра (0,82 г, 4,81 ммолей). Реакционную смесь нагревают в течение 6 часов с обратным холодильником в отсутствие света. Образовавшуюся соль удаляют фильтрованием и раствор упаривают под давлением. Полученный остаток очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 7/3. Получают 0,77 г 4-нитроксибутилового эфира 3-(6-метокси-α-метил-2-нафтилацетил)-(R)-2-оксотиазолидин-4-карбоновой кислоты в виде масла.
1Н-ЯМР (CDCl3): 7,74-7,67 (3Н, m), 7,47 (1H, m), 7,14-7,10 (2H, m), 5,28 (1H, dd), 4,12-3,91 (5H, m), 3,90 (3Н, s), 3,63 (1H, dd), 3,33 (1H, dd), 1,55 (3Н, d), 1,30-1,23 (4H, m).
Пример 4
Синтез 4-нитроксибутилового эфира [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина
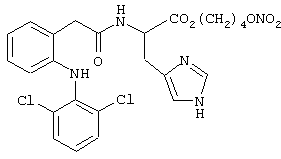
где лекарством-предшественником соединения изобретения является диклофенак формулы (XXIX) и соединением предшественника В является (L)-гистидин формулы (РII)
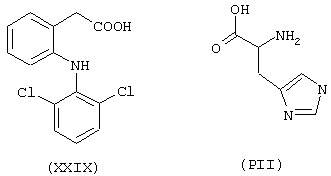
a) Синтез [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина
К раствору диклофенака (3 г, 10,13 ммолей) в тетрагидрофуране (50 мл), охлажденному до 0°С, добавляют при перемешивании 1,1’-карбонилдиимидазол (1,69 г, 10,13 ммолей). Через 10 минут раствор обрабатывают (L)-гистидином (1,57 г, 10,13 ммолей) и оставляют перемешиваться при комнатной температуре на 4 часа. Реакционную смесь концентрируют под вакуумом, обрабатывают метиленхлоридом и затем промывают последовательно 1% НСl и затем водой. Органическую фазу обезвоживают сульфатом натрия и упаривают под вакуумом. Полученный остаток очищают хроматографией на колонке с силикагелем, элюируя этилацетатом. Получают [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидин.
b) Синтез 4-бромбутилового эфира [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина
К раствору [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина (5 г, 11,54 ммолей) в тетрагидрофуране (100 мл) добавляют при перемешивании трифенилфосфин (9,08 г, 34,62 ммолей) и тетрабромид углерода (11,48 г, 34,62 ммоля). Реакционную смесь оставляют при комнатной температуре на 24 часа, затем растворитель удаляют выпариванием при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 1/1. Получают 4-бромбутиловый эфир (S)-[2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина.
с) Синтез 4-нитроксибутилового эфира [2-[(2,6-дихлорфенил)амино]бензолацетилокси]-(L)-гистидина
К раствору 4-бромбутилового эфира [2-[(2,6-дихлорфенил)амино]-бензолацетилокси]-(L)-гистидина (3 г, 5,28 ммолей) в ацетонитриле (30 мл) добавляют нитрат серебра (1,79 г, 10,56 ммолей). Реакционную смесь нагревают с обратным холодильником в течение 6 часов, пряча от света, образовавшуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный осадок очищают хроматографией на колонке с силикагелем, элиюруя н-гексаном/этилацетатом 1/1. Получают 4-нитроксибутиловый эфир [2-[(2,6-дихлорфенил)амино]бензолацетилокси)-(L)-гистидина. Выход 35%.
Пример 5
Синтез 5-[[4-оксо-(4-нитроксибутилокси)бутаноил]амино]-1,2,3,4-тетрагидроакридина
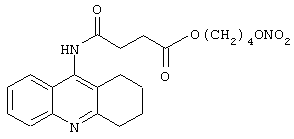
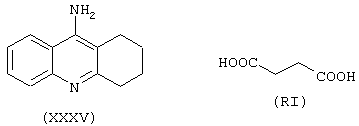
где лекарство-предшественник соединения согласно изобретению является такрином формулы (XXXV) и соединение предшественника связывающей группы В является янтарной кислотой формулы (RI)
a) Синтез 4-хлорбутилмоноэфира янтарной кислоты
К раствору янтарного ангидрида (2 г, 19,98 ммолей) в хлороформе (30 мл), охлажденному до 0°С, добавляют при перемешивании N,N’-дициклогексилкарбодиимид (4,2 г, 20,35 ммолей) и 4-диметиламинопиридин (100 мг, 0,8 ммолей). Через 30 минут добавляют 4-хлорбутанол (2,1 г, 19,35 ммолей). Реакционную смесь оставляют при комнатной температуре на 7 часов при перемешивании, затем ее подкисляют 5% НС1 и экстрагируют этилацетатом. Органическую фазу промывают солевым раствором, обезвоживают сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают хроматографией на колонке с силикагелем, элюируя метиленхлоридом/метанолом 8/2. Получают 4-хлорбутилмоноэфир янтарной кислоты.
b) Синтез 5-[[4-оксо-(4-хлорбутилокси)бутаноила]амино]-1,2,3,4-тетрагидроакридина
К раствору 4-хлорбутилмоноэфира янтарной кислоты (2,9 г, 10,02 ммоля) в N,N-диметилформамиде (30 мл), охлажденному до 0°С, добавляют при перемешивании N,N’-дициклогексилкарбодиимид (2,2 г, 10,66 ммолей) и 4-диметиламинопиридин (100 мг, 0,8 ммолей). Через 5 минут добавляют такрин (2 г, 10,08 ммоля). Реакционную смесь оставляют при комнатной температуре на 24 часа, затем подкисляют 5% НС1 и экстрагируют этилацетатом. Органическую фазу промывают солевым раствором, обезвоживают сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают хроматографией на силикагеле, элюируя метиленхлоридом/метанолом 8/2. Получают 5-[[4-оксо-(4-хлорбутилокси)бутаноил]амино]-1,2,3,4-тетрагидроакридин.
c) Cинтез 5-[[4-оксо-(4-нитроксибутилокси)бутаноила]амино]-1,2,3,4-тетрагидроакридина
К раствору 5-[[4-оксо-(4-хлорбутилокси)бутаноила]амино]-1,2,3,4-тетрагидроакридина (3 г, 7,71 ммоля) в ацетонитриле (50 мл) добавляют при перемешивании нитрат серебра (1,79 г, 10,56 ммоля). Реакционную смесь нагревают с обратным холодильником в течение 36 часов в отсутствие света, образовавшуюся соль удаляют фильтрованием и раствор упаривают при пониженном давлении. Полученный остаток очищают хроматографией на колонке с силикагелем, элюируя этилацетатом. Получают 5-[[4-оксо-(4-нитроксибутилокси)бутаноил]амино]-1,2,3,4-тетрагидроакридин. Выход 27%.
Пример 6
Синтез [4-амино-[4-оксо-(4-нитроксибутилокси)бутаноила]-1-гидроксибутилен]бифосфоновой кислоты
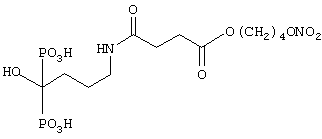
где лекарством-предшественником соединения изобретения является алендроновая кислота формулы (XXXVI) и соединением-предшественником связывающей группы В является янтарная кислота формулы (RI)

Данное соединение синтезируют, следуя процедуре синтеза, приведенной в примере 5. Выход 19%.
Пример 7
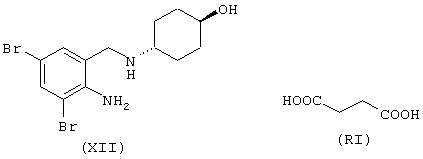
Синтез [4-оксо-(4-нитроксибутилокси)бутаноил-4-(2-амино-3,5-дибромфенил)метиламино]циклогексанолового эфира

где лекарством-предшественником соединения изобретения является амброксол формулы (XII) и соединением-предшественником мостиковой группы В является янтарная кислота формулы (RI)
a) Синтез 4-[(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]-транс-циклогексанола
К смеси 4-[(2-амино-3,5-дибромфенил)метиламино]циклогексанола (5 г, 13,22 ммолей) в диоксане (35 мл) и воде (50 мл)добавляют при перемешивании триэтиламин (3,31 мл, 23,7 ммолей) и ди-трет-бутилдикарбонат (3,46 г, 15,86 ммолей). Через 24 часа раствор концентрируют под вакуумом, обрабатывают 1% НС1 до нейтрального рН раствора и экстрагируют этилацетатом. Органическую фазу обезвоживают сульфатом натрия и упаривают под вакуумом. Получают 4-[(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанол, который используют без дальнейшей очистки.
b) Синтез [4-оксо-(4-хлорбутилокси)бутаноила]-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанолового эфира
К раствору 4-хлорбутилового моноэфира янтарной кислоты (4 г, 19,18 ммоль) в тетрагидрофуране (40 мл) добавляют при перемешивании 1,1’-карбонилдиимидазол (3,4 г, 20,96 ммолей). Через 10 минут раствор обрабатывают 4-[(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанолом (9,8 г, 20,5 ммоля) при комнатной температуре 4 часа. Реакционную смесь концентрируют под вакуумом, обрабатывают метиленхлоридом, промывают 1% НС1 и затем водой. Органическую фазу обезвоживают сульфатом натрия и упаривают под вакуумом. Полученный остаток очищают на колонке с силикагелем, элюируя н-гексаном/этилацетатом 1/1. Получают [4-оксо-(4-хлорбутилокси)бутаноила]-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексаноловый эфир.
c) Синтез [4-оксо-(4-нитроксибутилокси)бутаноил-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанолового эфира
К раствору [4-оксо-(4-хлорбутилокси)бутаноил-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанолового эфира (4 г, 5,98 ммолей) в ацетонитриле (70 мл) добавляют при перемешивании нитрат серебра (1,5 г, 8,83 ммоля). Реакционную смесь нагревают с обратным холодильником в течение 24 часов в отсутствие света, сформировавшуюся соль удаляют фильтрацией и раствор упаривают при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 7/3. Получают [4-оксо-(4-нитроксибутилокси)бутаноил-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексаноловый эфир.
d) Синтез [4-[4-оксо-(4-нитроксибутилокси)бутаноил](2-амино-3,5-дибромфенил)метиламино]циклогексанолового эфира
К раствору [4-оксо-(4-нитроксибутилокси)бутаноил-4-(2-трет-бутоксикарбониламино-3,5-дибромфенил)метиламино]циклогексанолового эфира (3,2 г, 4,6 ммолей) в этилацетате (50 мл), охлажденному до 0°С, добавляют при перемешивании этилацетат/5 н. НС1 (6,5 мл). Раствор оставляют на 4 часа при 0°С, осадок отфильтровывают. Полученный сырой продукт обрабатывают этилацетатом и 5% бикарбонатом натрия, затем водой. Органическую фазу обезвоживают сульфатом натрия и упаривают при пониженном давлении. Получают [4-[4-оксо-(4-нитроксибутилокси)бутаноил](2-амино-3,5-дибромфенил)метиламино]циклогексаноловый эфир. Выход 17%.
ФАРМАКОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
Пример
Острая токсичность
Острую токсичность оценивают введением группе из 10 крыс, весящих 20 г, одноразовой дозы каждого из проверяемых соединений через канюлю, перорально в водной суспензии 2% (мас./об.) карбоксиметилцеллюлозы.
За животными наблюдают в течение 14 дней. Ни у одного животного из группы не проявились симптомы токсичности даже после введения дозы 100 мг/кг.
Пример F1
Тест 1 - экспериментальная модель in vivo с N-этилмалеимидом (NEM): изучение желудочной толерантности некоторых лекарств, отобранных в качестве предшественников соединений настоящего изобретения.
Животные (крысы весом примерно 200 г) распределены по следующим группам (по 10 крыс на группу):
A) Контрольная группа:
10 группа: обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза 5 мл/кг, когда лекарство вводится перорально, физиологический раствор - когда парентеральным путем),
20 группа: обработка: носитель+NEM,
B) Группы, в которые вводится каждое лекарство:
Группа I: обработка: носитель+лекарство,
Группа II: обработка: носитель+лекарство+NEM.
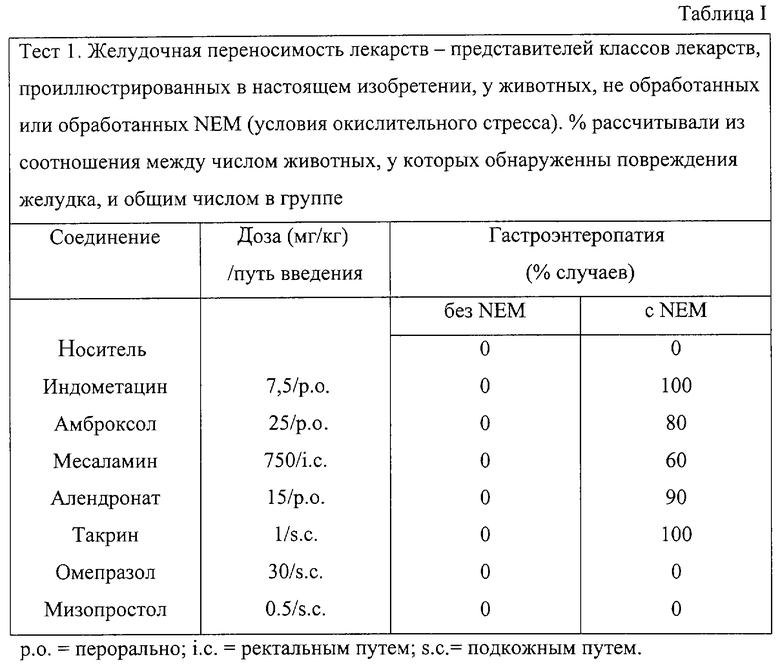
Лекарственные средства, анализируемые в этом эксперименте, являются следующими (Таблица I): индометацин, амброксол, месаламин, алендронат натрия, такрин, омепразол, мизопростол.
Индометацин, амброксол и алендронат вводят через рот, месаламин - ректальным путем и такрин, омепразол, мизопростол - подкожным путем.
Максимальная переносимая доза, определенная путем введения каждого вещества вышеуказанными путями в животных, не обработанных NEM, представлена в Таблице I. При более высоких дозах, чем те, которые представлены в Таблице I, у животных появляются энтеропатия, диарея, депрессия, тремор и седативный эффект.
В этой экспериментальной модели животных сначала обрабатывают NEM посредством подкожной инъекции при дозе 25 мг/кг в физиологическом растворе. Лекарство вводят через час в суспензии носителя. Животных умерщвляют через 24 часа и оценку повреждений желудочно-кишечной слизистой проводят путем подсчета числа крыс внутри каждой группы с повреждениями желудка при визуальном исследовании. Общее число вышеупомянутых крыс затем разделяют на общее число крыс в группе и умножают на 100. Полученные таким образом проценты представлены в Таблице I. Таблица I демонстрирует, что в группах крыс, обработанных вышеупомянутыми лекарствами без NEM, не обнаруживаются повреждения желудка.
Все крысы группы II (обработанные NEM) демонстрируют повреждения желудка после введения следующих лекарств: индометацин, амброксол, месаламин, алендронат натрия, такрин. Следовательно, вышеупомянутые лекарства могут быть использованы в синтезе продуктов изобретения.
Омепразол и мизопростол не могут использоваться взамен на основе результатов, представленных в тесте 1, для получения продуктов изобретения.
Пример F2
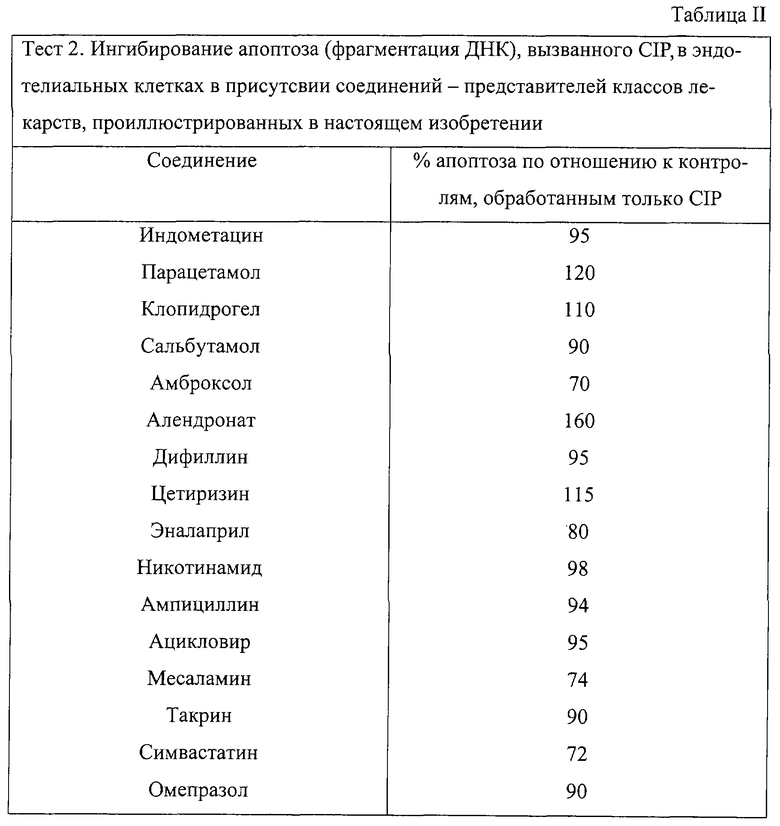
Тест 2 (in vitro) - ингибирование апоптоза (фрагментация ДНК), вызванного в эндотелиальных клетках посредством CIP в присутствии некоторых лекарств, отобранных в качестве предшественников соединений настоящего изобретения.
Были протестированы следующие лекарства-предшественники (Таблица II): индометацин, парацетамол, клопидогрел, сальбутамол, амброксол, алендронат натрия, дифиллин, сетиризин, эналаприл, никотинамид, ампициллин, ацикловир, месаламин, такрин, симвастатин, омепразол.
Эндотелиальныке клетки человека пупочной вены получают согласно стандартному способу. Свежие пупочные вены погружают в 0,1 мас.% коллагеназный раствор и инкубируют при 37°С в течение 5 минут.
Затем вены заливают средой М 199 (GIBCO, Grand Island, NY), pH 7,4, с 0,1% (мас./об.) коллагеназы, в которую добавлены 10 % бычьей фетальной сыворотки (10 мкг/мл), гепарина натрия (50 мкг/мл), тимидина (2,4 мкг/мл), глутамина (230 мкг/мл), пенициллина (100 ед/мл), стрептомицина (100 мкг/мл) и стрептомицина В (0,125 мкг/мл). Клетки собирают из перфузата центрифугированием на 800 об/мин и растят в культуральных колбах Т-75, предварительно обработанных фибронектином человека. Клетки затем растят в той же среде, добавив бычий фактор роста гипоталамуса (100 нг/мл). Когда клетки первичной культуры клеток (клетки, извлеченные непосредственно из ex vivo пупочной вены) образуют монослой слившихся клеток (примерно 8000000 клеток/колбу), рост останавливают и слои промывают и трипсинизируют. Клеточные суспензии переносят в ячейки культурального планшета, имеющего 24 ячейки, причем в половину из вышеупомянутых ячеек добавлена та же культуральная среда, содержащая лекарство с концентрацией 10-4 М, и растят в термостате при температуре 37°С при постоянной влажности (90%), СO2=5%. Когда лекарство нерастворимо в данной культуральной жидкости, его сначала растворяют в небольшом количестве диметилсульфоксида. Максимальное количество диметилсульфоксида, которое может быть добавлено в культуральную среду, составляет 0,5%. Только клетки, происходящие из этих первых подкультур, могут быть использованы для тестов с гидропероксидом кумена (CIP). Клетки идентифицируют как эндотелиальные посредством морфологического исследования и специфической иммунологической реакции против фактора VIII; эти культуры никогда не демонстрировали загрязнения миоцитами и фибробластами.
Перед началом теста удаляют среду клеточной культуры и клеточные слои тщательно промывают стандартным физиологическим раствором, забуференным фосфатом (0,1 М, рН 7,0), при температуре 37°С. Содержание каждой ячейки затем инкубируют в течение одного часа с суспензией CIP в культуральной среде с концентрацией 5 мМ. Оценку клеточного повреждения (апоптоз) проводят определением процента изменения фрагментации ДНК в культурах, содержащих лекарство+CIP, по отношению к контролям, обработанным только CIP. Вышеуказанный % изменения фрагментации ДНК определяют оценкой изменения флуоресценции посредством микроскопа ВХ60 Olympus microscope (Olympus Co., Roma), установленного на длину волны 405-450 нм, у образцов теста по отношению к оптической плотности контролей. Флуоресценцию каждого образца определяли для 5 репликатов. Статистическую оценку проводили с t-критерием Стьюдента (р<0,01).
Результаты представлены в Таблице II и демонстрируют, что индометацин, парацетамол, клопидогрел, сальбутамол, алендронат натрия, дифиллин, цетиризин, эналаприл, никотинамид, ампициллин, ацикловир, такрин, омепразол не ингибируют в занчительной степени апоптоз; следовательно, эти лекарства могут быть использованы для получения продуктов изобретения.
Напротив, амброксол, месаламин и симвастатин ингибируют апоптоз. Следовательно, на основе результатов теста 2 эти соединения не могут быть использованы для получения продуктов изобретения.
Пример F3
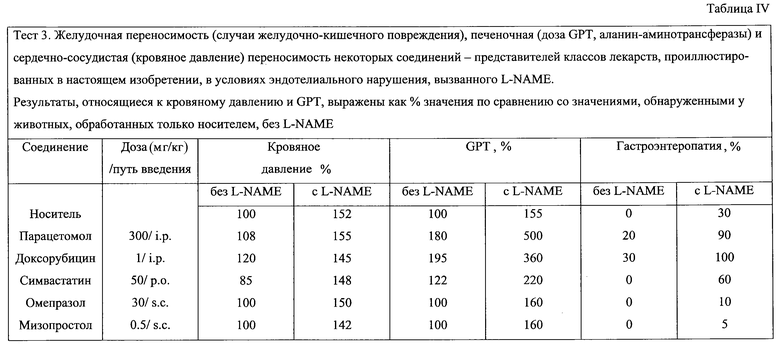
Тест 3 - экспериментальная модель in vivo с NW-нитро-L-аргинин-метиловым эфиром (L-NAME): желудочная толерантность (случай желудочно-кишечного повреждения), печеночная (доза GPT, аланин-аминотрансферазы) и сердечно-сосудистая (кровяное давление) толерантность некоторых лекарств, отобранных в качестве предшественников соединений изобретения.
Экспериментальная модель выбрана согласно J.Clin. Investigation 90, 278-281, 1992.
Эндотелиальную дисфункцию оценивают определением повреждения, вызванного введением L-NAME в желудочно-кишечную слизистую, повреждения печени (увеличение GPT) и повреждения сосудистого эндотелия или сердечно-сосудистого повреждения в виде кровяной гипертензии.
Животных (крысы, средний вес 200 г) подразделяют на группы, как здесь описано ниже. Группу, получающую L-NAME, обрабатывают в течение 4 недель вышеупомянутым соединением, растворенным с концентрацией 400 мг/л в питьевой воде. Создают следующие группы (по 10 животных в группе).
A) Контрольные группы:
10 группа: обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза 5 мл/кг, когда лекарство вводится через рот, физиологический раствор - когда парентеральным путем),
20 группа: обработка: носитель+L-NAME,
B) Группы, обработанные лекарством:
30 группа: обработка: носитель+лекарство,
40 группа: обработка: носитель+лекарство+L-NAME.
Лекарства, использованные в этом тесте, представляют собой парацетомол, доксорубицин, симвастатин, омепразол и мизопростол. Каждое лекарство вводится раз в день в течение 4 недель.
Максимальную переносимую дозу лекарства, вводимого животным, определяют оценкой в эксперименте на необработанных животных с повышением отдельной дозы появления у животных таких симптомов, как энтеропатия, диарея, депрессия, тремор, седативный эффект.
По окончании 4 недель прекращают подачу воды и через 24 часа животных умерщвляют.
За час до умерщвления определяют кровяное давление и увеличение кровяного давления используют как показатель повреждения, произошедшего в сосудистом эндотелии.
Повреждение слизистой желудка оценивают, как упомянуто ранее в тесте 1 (пример F1). Повреждение печени определяют оценкой после умерщвления аланин-аминотрансферазы (увеличение GPT).
Лекарство отвечает тесту 3 и, следовательно, может быть использовано для получения соединений изобретения, когда в группе крыс, обработанных L-NAME+лекарство+носитель, обнаруживается более сильное повреждение печени (более высокие значения GPT) и/или более сильное повреждение желудка, и/или более сильное сердечно-сосудистое повреждение (повышенное кровяное давление) по сравнению с группой, обработанной только носителем или группой, обработанной только носителем+лекарством, или группой, обработанной носителем+L-NAME.
Результаты теста представлены в Таблице IV. Процент повреждений желудка определяют, как в Тесте 1. Значения % GPT и % кровяного давления соотносят с соответствующими значениями, обнаруженными у животных 1-й группы контрольных групп.Среднее значение кровяного давления в этой группе составляет 105±8 мм рт.ст.
Полученные результаты демонстрируют, что парацетомол, доксорубицин и симвастатин вызывают повреждения печени и гастроэнтеропатию (значения GPT и повреждения желудка являются % более высокими по сравнению с соответствующими группами, обработанными лекарством, в отсутствие L-NAME и с контролями, обработанными L-NAME).
Следовательно, эти лекарства могут быть использованы для получения продуктов изобретения.
Омепразол и мизопразол не могут использоваться взамен на основании данного теста для получения продуктов изобретения.
Пример F4
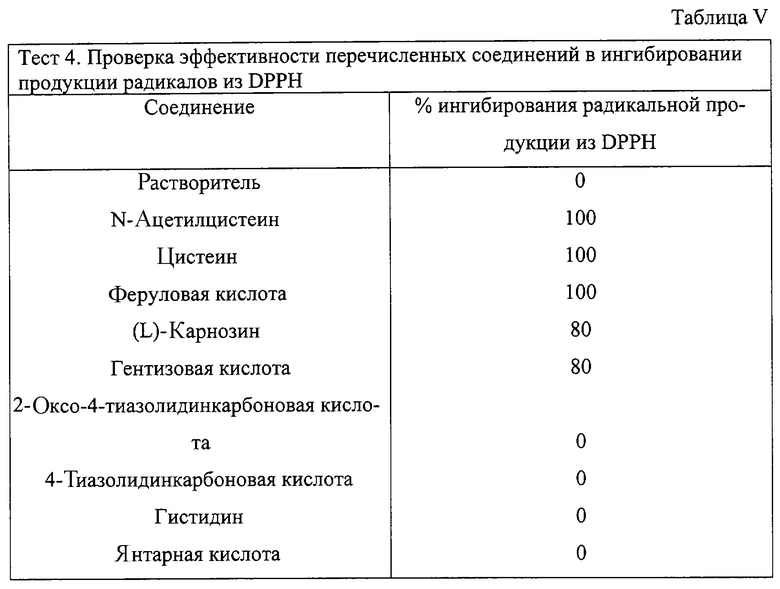
Тест 4 - ингибирование продукции радикалов из DDPH некоторыми веществами, используемыми в качестве предшественников В и В1 (см. формулы I и II настоящего изобретения).
Этот способ основан на колориметрическом тесте, в котором DPPH (2,2-дифенил-1-пикрил-гидразил) используется как соединение, образующее радикалы (M.S.Nenseter et al., Atheroscler. Tromb. 15, 1338-1344, 1995).
В начале готовят растворы тестируемых веществ в метаноле с конечной концентрацией 100 мкМ. 0,1 мл каждого из этих растворов добавляют к аликвотам 1 мл метанольного раствора 0,1 М DPPH и затем конечный объем доводят до 1,5 мл. После хранения растворов при комнатной температуре в отсутствие света в течение 30 минут измеряют абсорбцию при длине волны 517 нм. Определено, что абсорбция увеличивается по отношению к абсорбции раствора, содержащего такую же концентрацию DPPH.
Эффективность тестируемого соединения по ингибированию продукции радикалов или антирадикальной активности выражают следующей формулой:
(1-As/Ac)·100,
где As и Ас означают соответственно значения абсорбции раствора, содержащего тестируемое соединение+DPPH, и раствора, содержащего только DPPH.
Соединение, которое используют согласно настоящему изобретению, не отвечает тесту 4, если оно ингибирует продукцию радикалов, как определено выше, на 50% или выше.
В Таблице V представлены результаты, полученные для следующих веществ: N-ацетилцистеин, цистеин, феруловая кислота, (L)-карнозин, гентизовая кислота, 4-тиазолидинкарбоновая кислота и 2-оксо-4-тиазолидинкарбоновая кислота.
Таблица V демонстрирует, что
- N-ацетилцистеин, цистеин, феруловая кислота, (L)-карнозин, гентизовая кислота отвечают тесту 4, так как они ингибируют продукцию радикалов посредством DPPH в степени выше чем 50%, следовательно, они не могут быть использованы как предшественники В или B1 соединений изобретения;
- 4-тиазолидинкарбоновая кислота и 2-оксо-4-тиазолидинкарбоновая кислота не соответствуют тесту 4, так как они не ингибируют радикальную продукцию из DPPH в степени, равной или большей чем 50% и, следовательно, они могут быть использованы как предшественники соединений В или B1 согласно настоящему изобретению, предполагая, что они соответствуют следующему тесту 5.
Пример F5
Тест 5 - ингибирование продукции радикалов из FеII из соединений, использованных как предшественники В, B1 или С=-Tс-Y-H.
0,1 мл аликвоты 10-4 М метанольных растворов 4-тиазолидинкарбоновой кислоты и 2-оксо-4-тиазолидинкарбоновой кислоты добавляют в тестируемые пробирки, содержащие водный раствор, образованный смешением 0,2 мл 2 мМ дезоксирибозы, 0,4 мл фосфатного буфера, рН 7,4, 100 мМ и 0,1 мл 1 мМ FeII(NH4)2(S04)2 в 2 мМ НСl. Тестируемые пробирки затем выдерживают при температуре 37°С в течение одного часа. Затем в каждую тестируемую пробирку добавляют по очереди 0,5 мл 2,8% раствора трихлоруксусной кислоты в воде и 0,5 мл водного раствора 0,1 М тиобарбитуровой кислоты. Контроль сравнения создают, замещая указанные 0,1 мл аликвоты метанольных растворов тестируемых соединений 0,1 мл метанола. Тестируемые пробирки закрывают и нагревают в масляной бане при 100°С в течение 15 минут. Розовое окрашивание дает интенсивность, которая пропорциональна количеству дезоксирибозы, подвергшейся радикальной окислительной деградации. Растворы охлаждают до комнатной температуры и их абсорбцию на 532 нм измеряют против контроля.
Ингибирование, вызванное предшественником В или B1 или С=-Tc-Y-H (где свободная валентность насыщена так, как определено выше) в отношении радикальной продукции из FeII, определяют как процент посредством следующей формулы:
(1- As/Ac)·100,
где Аs и Ас являются соответственно значениями абсорбции раствора, содержащего тестируемое соединение+соль железа, и раствора, содержащего только соль железа. Результаты представлены в Таблице III, из которых следует, что обе кислоты соответствуют тесту 5, так как они ингибируют радикальную продукцию из FеII более чем на 50%. Следовательно, и 4-тиазолидинкарбоновая кислота и 2-оксо-4-тиазолидинкарбоновая кислота могут быть использованы как предшественники В, B1 или С=-Tc-Y-H для получения соединений настоящего изобретения.
Пример F6
Тест на желудочную переносимость соединений согласно настоящему изобретению по сравнению с соответствующими лекарствами-предшественниками в условиях эндотелиальной дисфункции, вызванной L-NAME (NW-нитро-L-аргинин-метиловый эфир).
Пример F3 повторяют и оценивают желудочную переносимость и для следующих лекарств-предшественников и для соответствующих производных согласно настоящему изобретению:
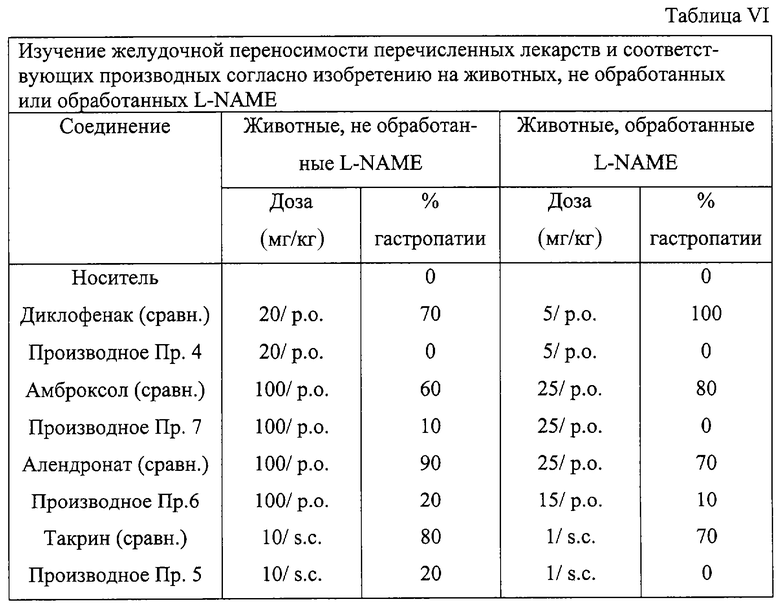
- Диклофенак и соответствующее производное согласно примеру 4.
- Амброксол и соответствующее производное согласно примеру 7.
- Алендронат и соответствующее производное согласно примеру 6.
- Такрин и соответствующее производное согласно примеру 5.
Результаты представлены в Таблице VI и демонстрируют, что при введении в одинаковых дозах соединений изобретения и соответствующего лекарства-предшественника результаты случаев гастропатии значительно уменьшены или исчезают в группах, обработанных соединениями изобретения.
Пример 8
Синтез 4-нитроксибутилового эфира 3-[2-(ацетилокси)бензоил]тиазолидин-4-карбоновой кислоты (формула XCI)
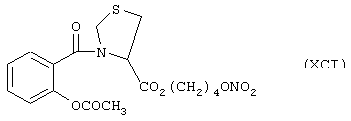
исходя из ацетилсалициловой кислоты (ХСII) и тиазолидин-4-карбоновой кислоты (формула PIV)
Соединение (ХСI) синтезируют согласно схеме, представленной в Примере 3. Выход 26%.
Элементный анализ
Расссчитано, %: С 49,51; Н 4,89; N 6,79; S 7,77.
Обнаружено, %: С 49,57; Н 4,94; N 6,70; S 7,73.
Пример 9
Синтез 2-(трет-бутиламино)-1-[4-гидрокси-3-[4-оксо-(4-нитроксибутилокси)бутирилокси]метилфенил]этанола формулы (ХСIII)
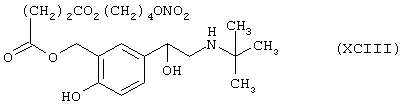
исходя из сальбутамола (XXV) и янтарной кислоты (формула RI)

Соединение (ХСIII) получают согласно процедуре Примера 7. Выход 14%.
Элементный анализ
Расссчитано, %: С 55,26; Н 7,06; N 6,14.
Обнаружено, %: С 55,20; Н 7,10; N 6,17.
Пример 10
Синтез 4-нитроксибутилового эфира 3-[[2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]ацетил]тиазолидин-4-карбоновой кислоты формулы (XCV)
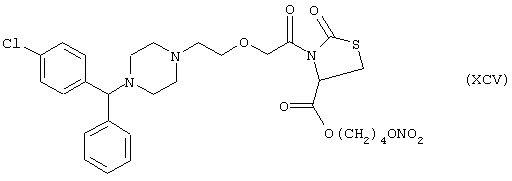
исходя из цетиризина (XIV) и 2-оксо-4-тиазолидинкарбоновой кислоты (формула PV)
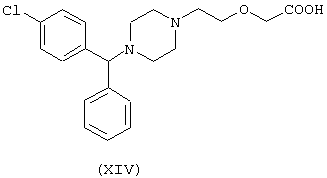
Соединение (ХСIII) получают согласно процедуре Примера 3. Выход 18%.
Элементный анализ
Расссчитано, %: С 55,44; Н 5,63; N 7,66; С 15,65.
Обнаружено, %: С 55,48; Н 5,60; N 7,61; С 16,71.
Пример 11
Синтез 4-нитроксибутилового эфира N[(S)1-[N[1-(этоксикарбонил)-3-фенилпропил]-L-аланил]-L-пролинил]гистидина формулы (XCVIII)
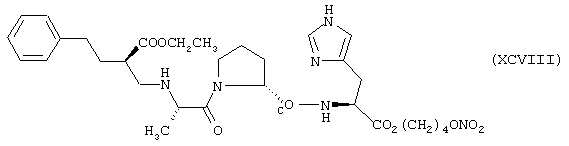
исходя из эналаприла формулы (XV) и гистидина формулы (РII)
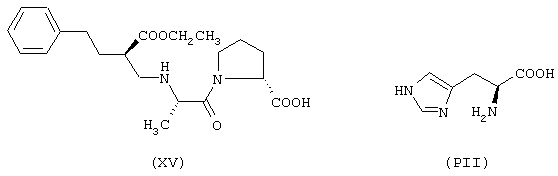
Соединение (XCVIII) получают согласно процедуре Примера 7. Выход 14%.
Элементный анализ
Расссчитано, %: С 57,75; Н 6,88; N 13,04.
Обнаружено, %: С 57,85; Н 6,95; N 13,01.
Пример 12
Синтез 1-[(1-метилэтил)амино]-3-(1-нафтилокси)-2-[4-оксо-(4-нитроксибутилокси)бутаноил]оксипропана формулы (ХСХ)
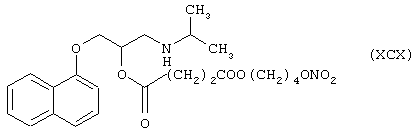
исходя из пропранолола формулы (XXIV) и янтарной кислоты формулы (RI)
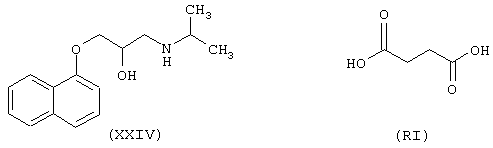
Соединение (ХСХ) получают согласно процедуре Примера 7. Выход 30%.
Элементный анализ
Расссчитано, %: С 60,49; Н 6,77; N 5,88.
Обнаружено, %: С 60,40; Н 6,75; N 5,91.
Пример 13
Синтез 4-нитроксибутилового эфира 3-[α-(2-хлорфенил)-6,7-дигидротиенол[3,2-с]пиридин-5(4Н)ацетил]тиазолидин-4-карбоновой кислоты формулы (ХСХII)
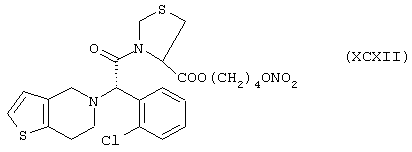
исходя из клопидогреля формулы (XI) и тиазолидин-4-карбоновой кислоты формулы (PIV)
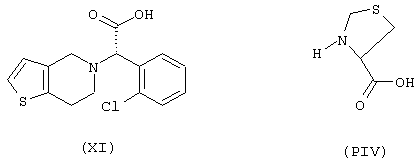
Соединение (ХСХII) получают согласно процедуре Примера 1. Выход 15%.
Элементный анализ
Расссчитано, %: С 51,15; Н 4,85; N 7,78; S 11,87; С 16,56.
Обнаружено, %: С 55,48; Н 5,60; N 7,61; S 11,85; C 16,59.
Пример 14
Синтез 4-нитроксибутилового эфира αN-[1-[5-(2,5-дигидро-5-оксо-3-фуранил)-3-метил-2-бензофуранил]этилокси-4-оксобутаноил]гистидина формулы (XCXIV)
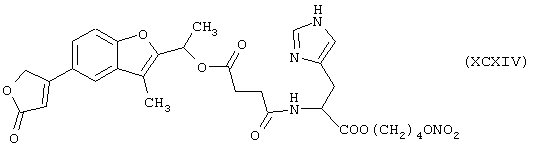
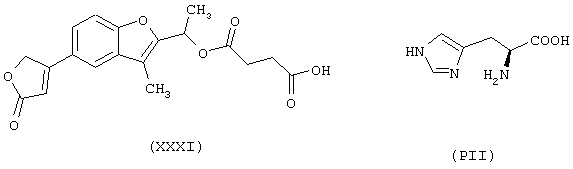
исходя из бенфуродилгемисукцината формулы (XXXI) и гистидина формулы (PII)
Соединение (XCXIV) получают, следуя процедуре, описанной в Примере 4. Выход 35%.
Элементный анализ
Расссчитано, %: С 56,86; Н 5,26; N 9,15.
Обнаружено, %: С 56,92; Н 5,29; N 9,10.
Пример 15
Синтез 4-нитроксибутилового эфира 3-никотиноилтиазолидинкарбоновой кислоты формулы (XCXVI)
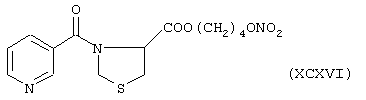
исходя из никотинамида формулы (ХХIII) и тиазолидин-4-карбоновой кислоты формулы (PIV)
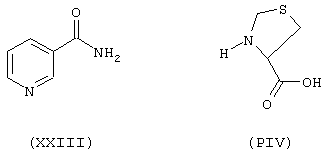
Соединение (ХХIII) синтезируют согласно процедуре, описанной в Примере 1, используя никотиновую кислоту. Выход 35%.
Элементный анализ
Расссчитано, %: С 47,32; Н 4,82; N 11,82; S 9,01.
Обнаружено, %: С 47,30; Н 4,79; N 1l,84; S 9,06.
Пример 16
Синтез 5-метокси-2-[[[4-оксо-4-(нитрокси)бутирилокси]-3,5-диметил-2-пиридинил]метил]сульфинил]-1Н-бензимидазола формулы (XCXVIII)
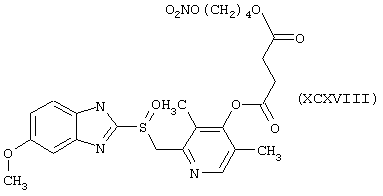
исходя из 4-гидроксиомепразола формулы (XXII) и янтарной кислоты формулы (RI)
Соединение (XCXVIII) получают, следуя процедуре, описанной в Примере 7. Выход 15%.
Элементный анализ
Расссчитано, %: С 52,64; Н 4,97; N 10,23; S 5,86.
Обнаружено, %: С 52,68; Н 5,01; N 10,15; S 5,81.
Пример 17
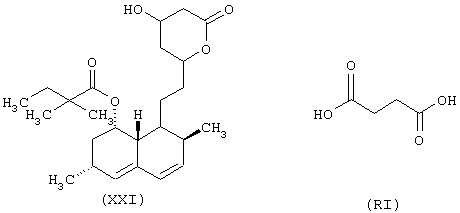
Синтез 1,2,3,7,8,8-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-[4-оксо-(4-нитроксибутилокси)бутирилокси]-6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира [1S-[1α, 3α, 7β, 8β, (2S*, 4S*)]]-2,2-диметилбутаноевой кислоты формулы (XCXIX)

исходя из симвастатина формулы (XXI) и янтарной кислоты формулы (RI)
Соединение (XCXIX) синтезируют, следуя процедуре Примера 7. Выход 12%.
Элементный анализ
Расссчитано, %: С 62,35; Н 7,77; N 2,20.
Обнаружено, %: С 62,50; Н 7,81; N 2,17.
Пример 18
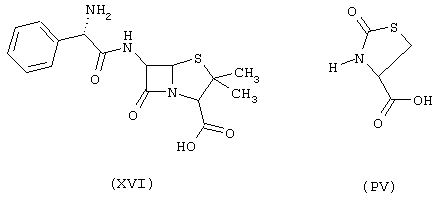
Синтез 4-нитроксибутилового эфира 3-[4-D-α-аминобензилпеницилламиноил]тиазолидинкарбоновой кислоты формулы (ХСХХ)

исходя из ампициллина формулы (XVI) и 2-оксо-4-тиазолидинкарбоновой кислоты формулы (PV)
Соединение (ХСХХ) получают, следуя процедуре Примера 3. Выход 19%.
Элементный анализ
Расссчитано, %: С 48,39; Н 4,91; N 11,76; S 10,77.
Обнаружено, %: С 48,43; Н 4,99; N 11,71; S 10,74.
Пример 19
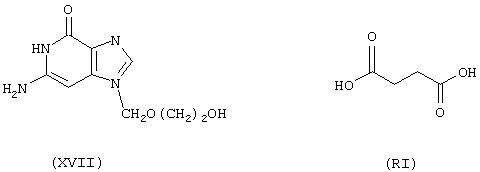
Синтез 9-[[2-[4-оксо-(4-нитроксибутилокси)бутирилокси]этокси]метил]гуанина формулы (XCXXI)
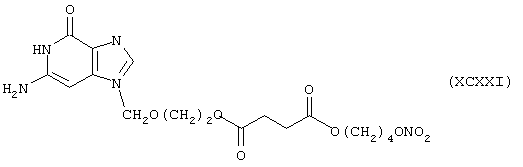
исходя из ацикловира формулы (XVII) и янтарной кислоты формулы (RI)
Соединение (XCXXI) синтезируют, следуя процедуре Примера 7. Выход 23%.
Элементный анализ
Расссчитано, %: С 46,26; Н 5,25; N 15,85.
Обнаружено, %: С 46,30; Н 5,28; N 15,84.
Пример 20
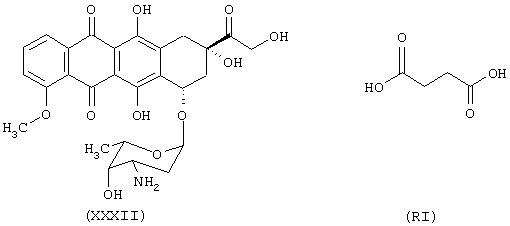
Синтез (8S-цис)-10-[(3-амино,2,3,6-тридезокси-α-L-ликсо-эксопиранозила)окси]-7,8,9,10-тетрагидро, 6,8,11-тригидрокси-8-[[4-оксо-(4-нитроксибутилокси)бутирилокси]метилоксо]-1-метокси-5,12-нафтацендиона формулы (ХСХХII)
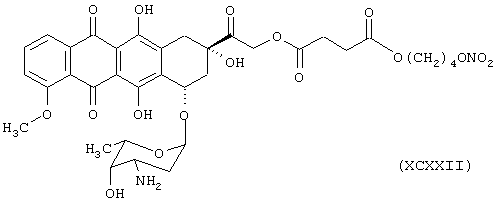
исходя из доксорубицина формулы (XXXII) и янтарной кислоты формулы (RI)
Соединение (ХСХХII) синтезируют согласно процедуре Примера 7. Выход 10%.
Элементный анализ
Расссчитано, %: С 55,26; Н 5,30; N 3,68.
Обнаружено, %: С 55,34; Н 5,32; N 3,65.
Пример F7
Повторяли Пример F1 с четырьмя группами крыс (каждая группа из 10 животных), причем все из них получают NEM, и проводили пероральное введение следующим образом:
а) контрольная группа: носитель, образованный водной суспензией 1% (мас./об.) карбоксиметилцеллюлозы,
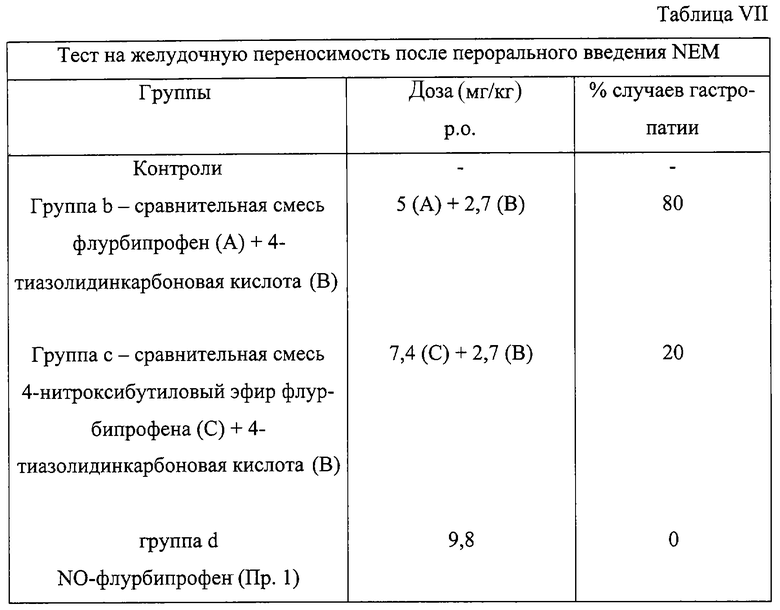
b) одной группе (группа b - сравнительная) вводили в то же время 5 мг/кг (0,02 ммолей/кг) флурбипрофена + 2,7 мг/кг (0,02 ммолей/кг) 4-тиазолидинкарбоновой кислоты в том же вышеуказанном носителе,
с) одной группе (группа с - сравнительная) вводили в то же время 7,4 мг/кг (0,02 ммолей/кг) 4-нитроксибутилового эфира флурбипрофена, синтезированного согласно способу, описанному в WO 94/12463, + 2,7 мг/кг (0,02 ммолей/кг) 4-тиазолидинкарбоновой кислоты в том же вышеуказанном носителе,
d) одной группе (группа d) вводили 9,8 мг/кг (0,02 ммолей/кг) 4-нитроксибутилового эфира 3-[2-фтор-α-метил-(1,1’-бифенил)-4-ацетил]тиазолидин-4-карбоновой кислоты, синтезированного как в Примере 1 (указанного как NO-флурбипрофен в Таблице VII), в том же вышеуказанном носителе.
Результаты представлены в Таблице VII и демонстрируют, что смеси, введенные соответственно в группы b и с (сравнительные), в отличие от соединения изобретения, введенного в группу d, почти неэффективны (группа b) или намного более эффективны (группа с) в уменьшении повреждений желудка.

Изобретение относится к органической химии и может найти применение в медицине. Описываются соединения или их соли, имеющие следующую общую формулу (I):
А-(В)bo-С-N(O)2 (I),
где bo=0 или 1; A=R-T1-, где R представляет собой радикал лекарственного вещества, такой, как определено в формуле изобретения, T1=(CO), О, S, N или NR1C, где R1C представляет собой Н или C1-C5 алкил; В=-ТB-Х2-ТBI, где ТB и tBI одинаковы или различны и выбраны из (СО), О, S, N или NR1C, где R1C такой, как определено выше, Х2 представляет собой бивалентную мостиковую группу, такую как соответствующий предшественник В, такой, как определено в формуле изобретения; С представляет собой бивалентный радикал -TC-Y-, где ТC=(СО), О, S, N или NR1C, где R1C такой, как определено выше, Y имеет значения такие, как представлено в формуле изобретения. Также описывается фармацевтическая композиция на основе соединений формулы (I) для использования в случаях окислительного стресса и/или эндотелиальных дисфункций. Технический результат - получены новые соединения и их соли, обладающие полезными биологическими свойствами. 2 н. и 5 з.п. ф-лы, 7 табл.
A-(B)bo-C-N(O)2, (I)
где bo=0 или 1;
A=R-T1-,
где R означает лекарственный радикал, как определено ниже, и имеет формулу R-T1-Z или R-T1-OZ, где Z представляет собой Н или C1-C5алкил, выбраны из следующих групп: противовоспалительные лекарственные средства: ацетилсалициловая кислота, 5-аминоацетилсалициловая кислота, карпрофен, диклофенак натрия, дифлунизал, этодолак, флуфенамовая кислота, флуниксин, флурбипрофен, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лорноксикам, локсопрофен, меклофенамовая кислота, мефенамовая кислота, мелоксикам, месаламин, напроксен, нифлюмовая кислота, олсалазин, пироксикам, салсалат, сулиндак, супрофен, теноксикам, тиапрофеновая кислота, толфенамовая кислота, толметин, зомепирак; аналгезирующие лекарственные средства: ацетаминофен, ацетилсалицилсалициловая кислота, беноксапрофен, трамадол; бронхолитические лекарственные средства: альбутерол, карбутерол, кленбутерол, фенотерол, метапротеренол, пирбутерол, салметерол, тербуталин; отхаркивающие лекарственные средства: амброксол, бромгексин, гвайакол; антиастматические лекарственные средства: цетиризин, левокабастин, терфенадин; АСЕ-ингибиторы: каптоприл, эналаприл, лизиноприл, рамиприл; Бета-блокаторы: альпренолол, атенолол, бупранолол, лабеталол, метипранолол, метопролол, пиндолол, пропранолол, тимолол; антитромботические и вазоактивные лекарственные средства: аргатробан, бенфуродил гемисукцинат, клопидогрел, дальтепарин, дипиридамол, эноксапарин, илопрост, озагрел, трифлузал; антидиабетические лекарственные средства: никотинамид; противоопухолевые лекарственные средства: антрамицин, даунорубицин, доксорубицин, эпирубицин; противоязвенные лекарственные средства: циметидин, омепразол, пантопразол; антигиперлипидемические лекарственные средства: аторвастатин, флувастатин, ловастатин, натриевая соль привастатина, симвастатин; антибиотики: амоксициллин, ампициллин, азтреонам, биапенем, карбенициллин, цефаклор, цефадроксил, цефамандол, цефатризин, цефокситин, диклоксациллин, имипенем, моксалактам, панипенем, бакампициллин; противовирусные лекарственные средства: ацикловир, фамцикловир, ганцикловир, пенцикловир, видарабин, зидовудин; ингибиторы резорбции костей: алендроновая кислота, этидроновая кислота, памидроновая кислота; лекарства против слабоумия: такрин;
T1=(CO), О, S, N или NR1C, где R1C означает Н или С1-С5 алкил,
В=-ТB-Х2-ТBI-,
где ТB и tBI равны или различны и могут быть выбраны из (СО), О, S, N или NR1C, где R1C имеет значения, как определено выше;
Х2 означает бивалентную мостиковую группу, как соответствующий предшественник В, имеющий формулу
Z'-TB-X2-TBI-Z'',
где Z', Z'' независимы и означают Н или ОН, выбраны из следующих соединений:
аминокислоты: аспарагиновая кислота (PI), гистидин (PII), 5-гидрокситриптофан (PIII), 4-тиазолидинкарбоновая кислота (PIV), 2-оксо-4-тиазолидинкарбоновая кислота (PV)
моноспирты или тиолы: 2-тиоурацил (QI), 2-меркаптоэтанол (QII),
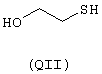
янтарная кислота (RI)

С означает бивалентный -Tc-Y- радикал, где
Тc=(СО), О, S, N или NR1C, R1C является таким, как определено выше;
Y означает:

где nIХ означает целое число между 0 и 3,
nIIX означает целое число между 1 и 3,
RTIX, RTIX', RTIIX, rTIIX' равны или отличны друг от друга, означают Н или линейный или разветвленный C1-C4 алкил;
Y3 является насыщенным, ненасыщенным или ароматическим гетероциклическим кольцом, содержащим, по крайней мере, один атом азота и вышеупомянутое кольцо содержит от 5 до 6 атомов;
или Y означает Yo, выбранный из следующего:
алкиленоксигруппа R'O, где R' является линейной или разветвленной, когда возможно C1-C20, алкил или циклоалкилен, имеющий от 5 до 7 атомов углерода, в циклоалкиленовом кольце один или более атомов углерода могут быть замещены гетероатомами, кольцо может иметь боковые цепи R' типа, причем R' является таким, как определено выше; или
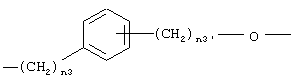
где n3 означает целое число от 0 до 3, n3' означает целое число от 1 до 3;
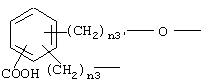
где n3 и n3' имеют вышеупомянутые значения;
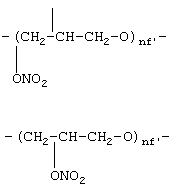
где nf’ означает целое число от 1 до 6;
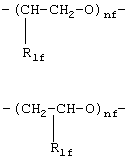
где R1f=H, СН3 и nf означают целое число от 1 до 6;
при условии, что в соединениях формулы (I):
когда bo=0 и С=-Tc-Yo-, лекарства формулы A=R-T1-Z или A=R-T1-OZ, как определено выше, не относятся к группе АСЕ-ингибиторов;
когда bo=0 и С=-Tc-y-, лекарства формулы A=R-T1-Z или A=R-T1-OZ не относятся к группе нестероидных противовоспалительных лекарственных средств.

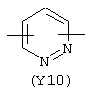

| Бензинораздаточная колонка | 1936 |
|
SU52910A1 |
| УСТРОЙСТВО для ОТДЕЛЕНИЯ СУСЛА от МЕЗГИ | 0 |
|
SU255164A1 |
| Способ консервации масляной системы турбоустановок | 1954 |
|
SU103320A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ПРОИЗВОДНЫЕ 2-(2,6-ДИГАЛОФЕНИЛАМИНО)ФЕНИЛУКСУСНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109009C1 |

