Данное изобретение относится к новым соединениям бензимидазола, фармацевтическим составам, содержащим эти соединения, способам их применения в лечебной практике и способу получения таких соединений бензимидазола. Новые соединения полезны для лечения заболеваний и нарушений центральной нервной системы, чувствительных к модуляции ГАМКA-рецепторного комплекса, таких, как, например, состояние тревоги, расстройства сна, нарушения памяти и эпилепсия или другие судорожные синдромы.
Предпосылки изобретения
Рецепторы γ -аминомасляной кислоты (ГАМК), ГАМКA-рецепторы, являются наиболее распространенными рецепторами ингибирования в мозге млекопитающих. Структурно ГАМКA-рецептор представляет собой макромолекулярный гетеропентамерный комплекс (сочетание α , β и γ/δ белковых субъединиц). Благодаря применению современных молекулярно-биологических подходов описано несколько субтипов подобных ГАМКA-рецепторов. Любой ГАМКA-рецепторный комплекс содержит канал для хлоридного иона, контролирующий поток хлоридных ионов через мембрану нервной клетки, и многочисленные сайты узнавания для небольших молекул модуляторов типа бенздиазепинов, барбитуратов, пикротоксина и некоторых стероидов. Когда ГАМК взаимодействует со своим рецептором, ионный канал открыт, приток хлоридных ионов усилен, мембрана гиперполяризована и клетка становится менее чувствительной к возбуждающему стимулу. Этот индуцированный ГАМК ионный поток может регулироваться разнообразными агентами, в том числе агентами, взаимодействующими с рецептором или сайтом узнавания бенздиазепина.
Агенты, связывающиеся или взаимодействующие с модуляторными сайтами ГАМКA-рецепторного комплекса, такими, как например, бенздиазепиновый рецептор, могут оказывать либо усиливающий эффект на действие ГАМК, то есть позитивный модуляторный эффект рецептора (агонисты, частичные агонисты), либо ослабляющий эффект на действие ГАМК, то есть осуществлять негативную модуляцию рецептора (обратные агонисты, частичные обратные агонисты), либо они могут конкурентно блокировать влияние как агонистов, так и обратных агонистов (антагонисты или лиганды без внутренней активности).
Агонисты, как правило, оказывают миорелаксантное, гипнотическое, успокаивающее, транквилизирующее и/или противосудорожное воздействие, в то время как обратные агонисты оказывают проконвульсивное, противоалкогольное и анксиогенное воздействие. Частичные агонисты характеризуются как соединения, оказывающие транквилизирующее воздействие, но не обладающие или обладающие незначительным миорелаксантным, гипнотическим и успокаивающим действием, тогда как применение частичных обратных агонистов считается полезным для усиления познавательной способности.
В течение последних трех десятилетий было синтезировано большое число соединений, принадлежащих к различным группам соединений, обладающих сродством к бенздиазепиновым рецепторам. Однако несмотря на то, что сайты рецептора бензодиазепина по-прежнему считаются очень привлекательными биологическими сайтами для воздействия на ЦНС с целью лечения различных нарушений и заболеваний, почти все синтезированные ранее соединения, действующие на эти рецепторные сайты, не выдержали клинических испытаний в силу оказываемых ими неприемлемых побочных эффектов.
В настоящем изобретении предлагаются новые соединения бензимидазола, взаимодействующие с бенздиазепиновым рецептором ГАМКA-рецепторного комплекса. Соединения по настоящему изобретению являются полезными модуляторами ГАМКA-рецепторного комплекса.
Задача изобретения
Задачей настоящего изобретения является создание новых соединений бензимидазола и их фармацевтически приемлемых солей, получаемых в результате присоединения кислоты, которые полезны при лечении нарушений, заболеваний или недомоганий центральной нервной системы, чувствительных к модуляции ГАМКA-рецепторного комплекса и, в особенности, к позитивной модуляции ГАМКA-рецепторного комплекса.
В задачу настоящего изобретения также входит создание фармацевтических составов, содержащих новые соединения бензимидазола, полезных для вышеуказанных применений. Еще одной задачей настоящего изобретения является создание нового способа лечения с помощью новых соединений бензимидазола.
Настоящим изобретением также решается задача создания способа получения новых фармацевтических составов.
Дополнительные задачи станут очевидны из следующего далее описания, а остальные будут понятны специалистам в данной области.
Резюме
Таким образом, согласно данному изобретению, inter alia, предлагается одно или в сочетании:
соединение формулы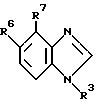
или его фармацевтически приемлемые соль либо оксид,
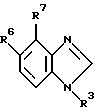
где R3 представляет собой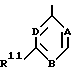
где каждый из A, B и D представляет собой CH или один либо два из A, B и D представляют собой N, а остальные являются CH;
R11 представляет собой фенил, бензимидазолил или моноцик лический гетероарил, которые могут быть все замещены один или несколько раз заместителями, выбранными иэ алкила, алкоксигруп- пы, галогена, CF3, амино-, нитро-, цианогруппы, ациламиногруп- пы, ацила, фенила и моноциклического гетероарила;
один из R6 и R7 представляет собой водород, а другой группу -CR'=NOR", где каждый из R' и R" независимо друг от друга представляет собой водород, алкил, алкенил, алкинил или фенил;
приведенное выше соединение, которое представляет собой O-метилоксим 1-(3-(З-фуранил)фенил)-5-формил-бензимидазола, O-этилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола, оксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола, O-метилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола, O-этилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола или O-метилоксим 5-ацетил-1-(3-(2-тиазолил)фенил)бензимидазола, либо его фармацевтически приемлемые соль или оксид;
фармацевтический состав, содержащий эффективное количество любого вышеприведенного соединения либо его фармацевтически приемлемых соли или оксида вместе по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем;
применение любого вышеприведенного соединения для изготовления лекарственного средства для лечения расстройства или заболевания живого организма животного, включая человека, причем расстройство или заболевание чувствительно к модуляции ГАМКA-рецепторного комплекса центральной нервной системы;
применение любого вышеприведенного соединения для изготовления лекарственного средства для лечения расстройства или заболевания живого организма животного, включая человека, причем расстройство или заболевание чувствительно к позитивной модуляции ГАМКA-рецепторного комплекса центральной нервной системы;
применение любого вышеприведенного соединения для изготовления лекарственного средства для лечения нарушения или заболевания, выбранного из состояния тревоги, расстройств сна, нарушений памяти, эпилепсии и любого другого судорожного синдрома;
способ лечения расстройства или заболевания живого организма животного, включая человека, причем расстройство или заболевание чувствительно к модуляции ГАМКA-рецепторного комплекса центральной нервной системы, при котором на указанный живой организм животного, включая человека, воздействуют, в случае необходимости, терапевтически эффективным количеством любого вышеприведенного соединения;
упомянутый выше способ, при котором проводят лечение расстройства или заболевания, чувствительного к позитивной модуляции ГАМКA-рецепторного комплекса;
упомянутый выше способ, при котором проводят лечение состояния тревоги, расстройств сна, нарушений памяти, эпилепсии или любого другого судорожного синдрома; и
упомянутый выше способ, при котором активный ингредиент вводят в форме его фармацевтического состава, в котором он присутствует вместе с фармацевтически приемлемым носителем или разбавителем.
Галоген представляет собой фтор, хлор, бром или иод.
Алкил означает прямую цепь или разветвленную цепь, состоящую из атомов углерода в количестве от одного до восьми, или циклический алкил, состоящий из атомов углерода в количестве от трех до семи, включая, но не ограничиваясь этим, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил, циклопропил, циклобутил, циклопентил, циклогексил; предпочтительными группами являются метил, этил, пропил, изопропил и трет-бутил.
Алкенил означает прямую цепь или разветвленную цепь, состоящую из атомов углерода в количестве от двух до шести, содержащую одну двойную связь, включая, но не ограничиваясь этим, этенил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил и 3-бутенил.
Алкинил означает прямую цепь или разветвленную цепь, состоящую из атомов углерода в количестве от двух до шести, содержащую одну тройную связь, включая, но не ограничиваясь этим, этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил и 3-бутинил.
Алкоксил означает O-алкил, в котором алкил такой, как определено выше.
Ацил означает -(C=O)-H или -(C=O)-алкил, в котором алкил такой, как определено выше.
Ациламиногруппа представляет собой ацил-NH-, где ацил такой, как определено выше.
Аминогруппа предсталяет собой -NH2 или -NH-алкил, или -N-(алкил)2, где алкил такой, как определено выше.
Моноциклический гетероарил представляет собой 5- или 6-членную гетероциклическую моноциклическую группу. Подобная моноциклическая гетероарильная группа включает, например окса-зол-2-ил, оксазол-4-ил, оксазол-5-ил, изоксазол-3-ил, изоксазол-4-ил, иэоксазол-5-ил, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил, иэотиазол-3-ил, изотиазол-4-ил, иэотиазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,5-оксадиазол-4-ил, 1,2,5-тиадиазол-3-ил, 1,2,5-тиадиазол-4-ил, 1-имидазолил, 2-имидазолил, 4-имидазолил, 1-пирролил, 2-пирролил, 3-пирролил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-пиримидил, 3-пиридазинил, 4-пиридазинил, 2-пиразинил, 1-пиразолил, 3-пиразолил и 4-пиразолил.
Примеры фармацевтически приемлемых солей, полученных присоединением кислоты, включают в себя соли, полученные присоединением неорганических и органических кислот, например гидрохлорид, гидробромид, фосфат, нитрат, перхлорат, сульфат, цитрат, лактат, тартрат, малеат, фумарат, манделат, бензоат, аскорбат, циннамат, бензолсульфонат, метансульфонат, стеарат, сукцинат, глутамат, гликоллят, толуол-п-сульфонат, формиат, малонат, нафталин-2-сульфонат, салицилат и ацетат.
Другие кислоты, как например щавелевая кислота, хотя и не находятся среди фармацевтически приемлемых, могут быть использованы в процессе приготовления солей, полезных в качестве промежуточных производных при получении соединений по изобретению и их фармацевтически приемлемых солей, образованных в результате присоединения кислоты. Такие соли образуются согласно способам, хорошо известным специалистам.
Кроме того, соединения по данному изобретению могут существовать в несольватированной, равно как и в сольватированной, формах с фармацевтически приемлемыми растворителями типа воды, этанола и им подобным. Для решения задач данного изобретения сольватированные и несольватированные формы рассматриваются в основном как эквивалентные.
Некоторые из соединений по настоящему изобретению существуют в (+) и (-)-формах, равно как и в рацемических формах.- Рацемические формы можно разделить на оптические изомеры известными методами, например разделением их диастереомерных солей, полученных посредством обработки оптически активной кислотой, и высвобождением оптически активного аминного соединения в результате обработки основанием. Другой способ разделения рацематов на оптические изомеры основан на хроматографии на оптически активной матрице. Таким образом, рацемические соединения по настоящему изобретению можно разделить на их оптические изомеры, например, фракционной кристаллизацией d- или l-солей (например тартратов, манделатов или камфорсульфонатов). Соединения по данному изобретению могут также быть разделены путем образования диастереомерных амидов в реакции соединений по настоящему изобретению с оптически активной активированной карбоновой кислотой типа кислоты, полученной из (+) или (-)-фенилаланина, (+) или (-)-фенилглицина, (+) или (-)-камфановой кислоты, или путем образования диастереомерных карбаматов в реакции соединений по настоящему изобретению с оптически активным хлорформиатом или подобным ему соединением.
Могут быть использованы дополнительные известные методы разделения оптических изомеров, очевидные для специалистов в данной области (J.Jaques, A. Collet, and S. Wilen "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New York (1981)).
Кроме того, в тех случаях, когда соединения по изобретению представляют собой оксимы, они могут существовать в двух формах, Z- и E-формах, в зависимости от расположения заместителей вокруг двойной связи -C=N-. Настоящее изобретение охватывает как Z-, так и E-форму соединений по изобретению, равно как и их смеси.
Соединения по изобретению могут быть получены разнообразными способами.
Таким образом, соединения по изобретению и их фармацевтически приемлемые производные могут быть получены любым известным из уровня техники способом получения соединений аналогичной структуры и так, как продемонстрировано в приведенных далее примерах.
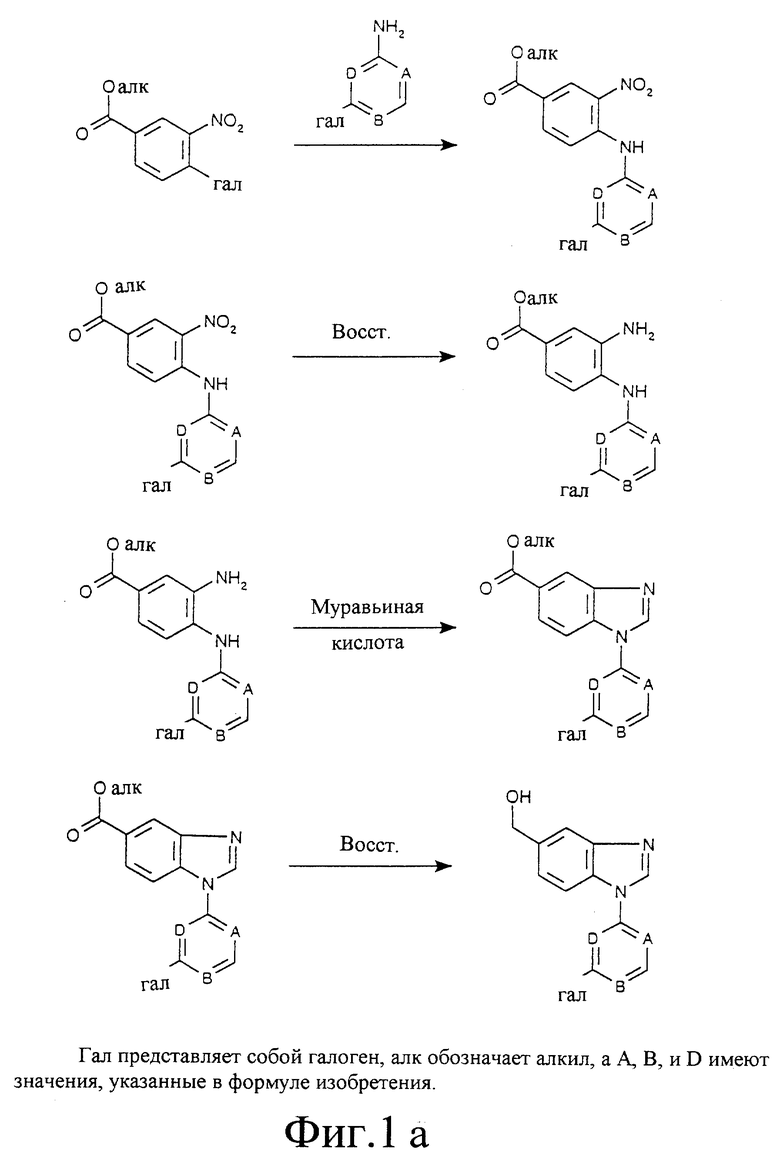
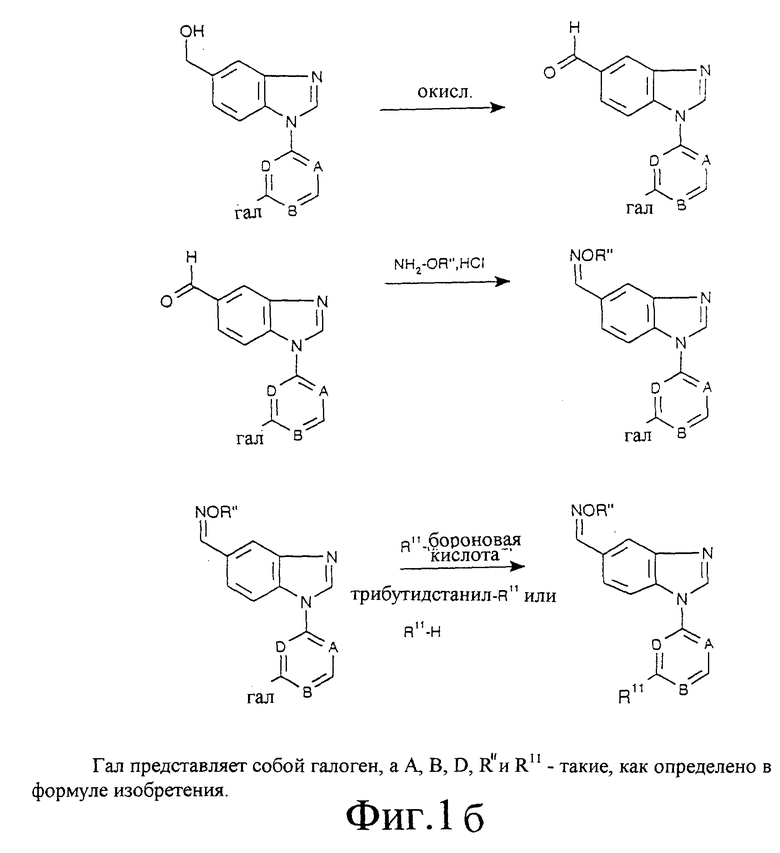
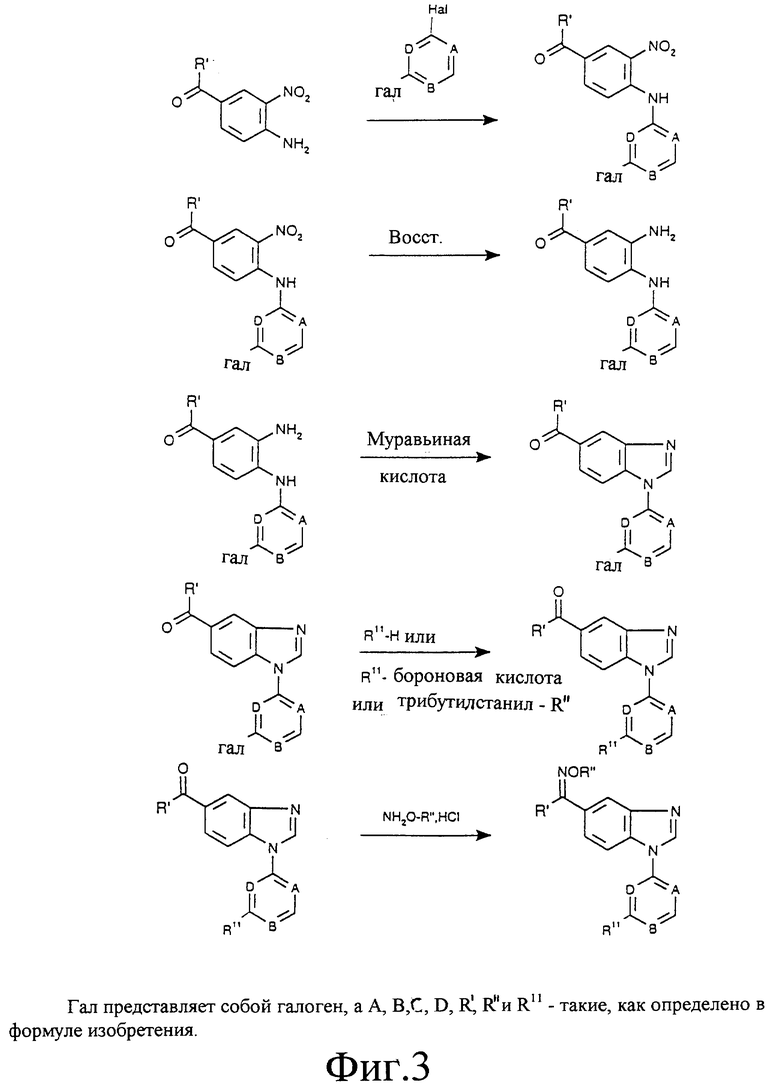
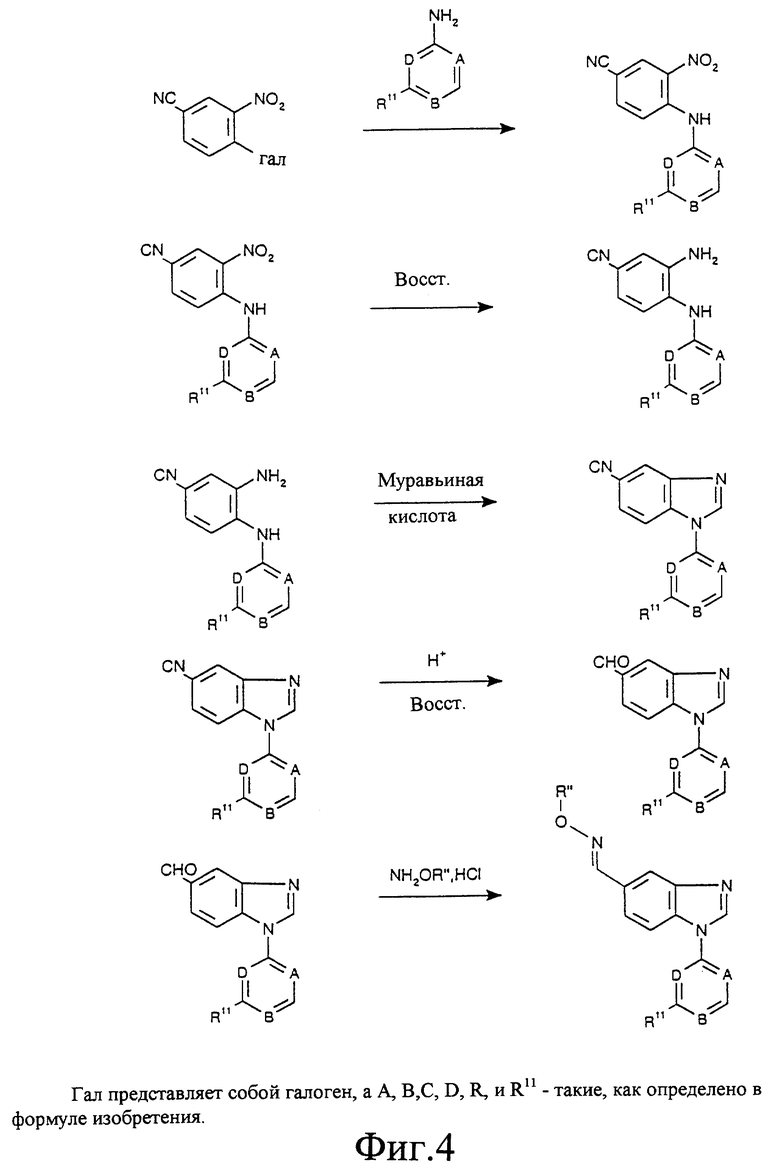
Фиг. 1а, 1б, 2, 3 и 4 отображают способы получения соединений по изобретению, в которых R6 представляет собой оксим, a R7 является водородом. Аналогичным образом могут быть синтезированы соединения по изобретению, в которых R7 представляет собой оксим, a R6 является водородом.
Исходные материалы для процессов, описанных в настоящей заявке на изобретение, известны или могут быть получены известными способами из имеющихся в продаже химических реактивов.
Продукты описанных здесь реакций выделяют обычными способами, как например экстракцией, кристаллизацией, перегонкой, хроматографией и им подобными.
Биология
4-Аминомасляная кислота (ГАМК) является главным ингибирующим нейромедиатором, и ее действие, как было установлено ранее, реализуется как через центральную, так и через периферическую нервную систему. В настоящее время известны два типа ГАМК-рецепторов, ГАМКA- и ГAMKB-рецепторы. Современными молекулярно-биологическими методами продемонстрировано, что ГАМКA-рецепторы могут быть подразделены на многочисленные субрецепторы в соответствии с селективными и/или частными фармакологическими эффектами, наблюдаемыми для определенных лигандов бенздиазепинового рецептора, в противоположность неселективным эффектам, наблюдаемым для классических лигандов бенздиазепинового рецептора, каким является например диазепам. Активация ГАМК-рецепторов приводит к изменениям мембранного потенциала (гиперполяризации). ГАМКA-рецепторы связаны с поступлением хлоридных ионов через располагающийся в них хлоридный канал, в то время как активация ГАМКB-рецепторов опосредованно влияет на изменение проницаемости каналов для ионов калия и кальция, а также изменяет выработку вторичных переносчиков. Сайты узнавания ГАМКA могут активироваться ГАМК, мусцимолом и, например изогувацином, но не ГAMKB-агонистами, таким, как баклофен. Модуляторный сайт узнавания ГАМКA в бензодиазепиновых рецепторных сайтах может быть селективно помечен радиоактивным изотопом 3H-флунитразепамом. Это дает возможность оценивать сродство различных возможных лигандов к сайтам бензодиазепинового рецептора, определяя способность тестируемых соединений вытеснять 3H-флунитразепам.
Способ
Препарат ткани: Препараты готовят при температуре 0-4oC кроме случаев, отмеченных особо. Кору головного мозга особей мужского пола крыс линии Wistar (150-200 г) гомогенизируют в течение 5-10 с в 20 мл Трис-HCl (30 мМ, pH 7,4), используя гомогенизатор Ultra-Turrax. Суспензию центрифугируют при 27000•g в течение 15 мин и осадок три раза промывают буферным раствором (центрифугируя каждый раз при 27000•g по 10 мин). Промытый осадок гомогенизируют в 20 мл буферного раствора и инкубируют на водяной бане (37oC) в течение 30 мин для удаления эндогенной ГАМК, после чего центрифугируют в течение 10 мин при 27000•g. По окончании осадок ресуспендируют в 30 мл буферного раствора, замораживают и хранят при -20oC.
Анализ: Мембранный препарат размораживают и центрифугируют при 2oC, 27000•g в течение 10 мин. Осадок дважды промывают 20 мл 50 мМ Трис-цитрата, pH 7,1, применяя гомогенизатор Ultra-Turrax и центрифугируя по 10 мин при 27000•g. По окончании осадок ресуспендируют в 50 мМ Трис-цитрате, pH 7,1 (500 мл буферного раствора на 1 г исходной ткани) и далее используют для анализа связывания. Аликвоты материала ткани объемом 0,5 мл добавляют к 25 мкл раствора тестируемого соединения и 25 мкл 3H-FNM (конечная концентрация 1 нМ), перемешивают и инкубируют при 2oC в течение 40 мин. Степень неспецифического связывания определяют, используя клоназепам (конечная концентрация 1 мкМ). По окончании инкубации образцы добавляют к 5 мл охлажденного во льду буферного раствора, выливают непосредственно на Whatman GF/C стекловолоконные фильтры, фильтруют с использованием разрежения и немедленно промывают 5 мл охлажденного во льду буферного раствора. Количество радиоактивного материала на фильтрах определяют обычными методами жидкостной сцинтилляции. Под специфическим связыванием понимают общее связывание за вычетом неспецифического связывания.
Измеряемая величина рассчитывается в виде ИК50 (концентрация (нМ) тестируемого соединения, ингибирующая специфическое связывание 3H-FNM на 50%).
С результатами тестирования отдельных соединений по настоящему изобретению можно ознакомиться по приведенным ниже данным:
Тестируемое соединение: ИК50 (нМ)
O-метилоксим 1-(3-(3-фуранил)фенил)-5-формил-бензимидазола - 1,5
O-этилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола - 1,4
O-метилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола - 0,6
O-этилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола - 1,2
O-метилоксим 5-ацетил-1-(3-(2-тиазолил)фенил)бензимидазола - 1,7
Фармацевтические составы
Поскольку для применения в терапии оказывается возможным введение соединения по изобретению в виде неочищенного химического вещества, то предпочтительно представить активный ингредиент как фармацевтический препарат.
Таким образом, согласно данному изобретению предложен фармацевтический препарат, содержащий соединение по изобретению или его фармацевтически приемлемую соль либо его производное вместе с одним или более чем одним фармацевтически приемлемым носителем и, возможно, другими ингредиентами для терапии и/или профилактики. Носитель (и) должен быть "приемлемым" в смысле совместимости с другими ингредиентами препарата и безвредности для реципиента.
Фармацевтические препараты включают в себя препараты, пригодные для перорального, ректального, назального, местного (включая трансбуккальное или подъязычное), вагинального или парентерального (включая внутримышечное, подкожное и внутривенное) введения, или форму, пригодную для введения путем ингаляции или инсуффляции.
Соединения по изобретению вместе с традиционными адьювантом, носителем или разбавителем могут, следовательно, быть представлены в форме фармацевтических составов и в их стандартных дозах и в такой форме могут применяться в виде твердых форм, например таблеток или заполненных капсул, либо жидкостей, например растворов, суспензий, эмульсий, эликсиров или заполненных указанными жидкостями капсул (все вышеуказанное - для перорального применения), в форме суппозиториев для ректального введения, либо в форме стерильных инъекционных растворов для парентерального (включая подкожное) применения. Подобные фармацевтические составы и их формы в виде стандартных доз могут содержать традиционные ингредиенты в обычных пропорциях с дополнительными активными соединениями или составляющими либо без них, и такие формы стандартных доз могут содержать любое подходящее эффективное количество активного ингредиента, соразмерное с назначенной суточной дозой, которую необходимо использовать. Препараты, содержащие один (1) миллиграмм активного ингредиента или, более широко, от 0,01 до ста (100) миллиграмм на таблетку, представляют поэтому наиболее приемлемые стандартные дозы.
Соединения по настоящему изобретению могут применяться в виде большого разнообразия дозированных форм для перорального и парентерального введения. Специалистам будет ясно, что следующие дозированные формы могут содержать в виде активного компонента как соединение по изобретению, так и его фармацевтически приемлемую соль.
При получении фармацевтических составов из соединений по настоящему изобретению фармацевтически приемлемые носители могут быть как в твердом, так и в жидком виде. Препараты в твердой форме представляют собой порошки, таблетки, драже, капсулы, крахмальные облатки, суппозитории и диспергированные гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые могут также действовать как разбавители, корригенты, солюбилизаторы, смазывающие вещества, суспендирующие средства, связующие, консерванты, разрыхлители таблеток или инкапсулирующий материал.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое присутствует в смеси с тонкоизмельченным активным компонентом.
В таблетках активный компонент в приемлемых пропорциях смешан с носителем, обладающим необходимой связывающей способностью, и спрессован до получения желаемой формы и размера.
Предпочтительно порошки и таблетки содержат от одного до приблизительно семидесяти процентов активного соединения. Приемлемыми носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, карбоксиметилцеллюлозы натриевая соль, воск с низкой температурой плавления, масло какао и им подобные. Термин "препарат" обозначает состав, содержащий активный компонент вместе с инкапсулирующим материалом, используемым в качестве носителя, обеспечивающим получение капсулы, в которой активный компонент, с носителями или без них, находится в окружении носителя, который таким образом оказывается ассоциированным с ним. Подобным же образом это относится к крахмальным облаткам и лепешкам. Таблетки, порошки, капсулы, драже, крахмальные облатки и лепешки можно применять как твердые формы, подходящие для перорального введения.
Для получения суппозиториев сначала расплавляют имеющий низкую температуру плавления воск типа смеси глицеридов жирной кислоты или масла какао и гомогенно диспергируют в нем активный компонент, например с помощью перемешивания. Расплавленную гомогенную смесь далее разливают в формы удобного размера, ей дают охладиться и тем самым отвердеть.
Препараты, подходящие для вагинального введения, могут быть представлены в виде вагинальных суппозиториев, тампонов, кремов, гелей, паст, пенок или аэрозолей, содержащих в дополнение к активному ингредиенту известные из уровня техники соответствующие носители.
Препараты в жидкой форме представляют собой растворы, суспензии и эмульсии, например водные растворы или растворы в водных растворах пропиленгликоля. К примеру, инъекционные жидкие препараты для парентерального введения могут быть приготовлены как растворы в водном растворе полиэтиленгликоля.
Соединения по настоящему изобретению могут, таким образом, быть приготовлены в виде препаратов для парентерального введения (например для инъекции, такой как инъекция ударной дозы вещества или непрерывная инфузия) и могут быть представлены в виде формы стандартной дозы в ампулах, заранее заполненных шприцах, сосудах для инфузии малого объема или сосудах с многократной дозой с добавкой консерванта. Данные составы могут принимать форму суспензий, растворов или эмульсий в масле либо водных растворителях и могут содержать агенты типа суспендирующих, стабилизирующих и/или диспергирующих. С другой стороны, активный ингредиент может быть в форме порошка, полученного в результате асептического выделения стерильного твердого содержимого или лиофилизации из раствора, предназначенного для растворения перед применением в подходящем растворителе, например стерильной апирогенной воде.
Водные растворы, пригодные для перорального применения, могут быть приготовлены путем растворения активного компонента в воде и добавления при желании подходящих красителей, корригентов, стабилизирующих и загущающих агентов.
Водные суспензии, пригодные для перорального применения, могут быть получены путем диспергирования тонкоизмельченного активного компонента в воде с вязким материалом, таким, как природные или синтетические смолы, полимеры, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, и другими хорошо известными суспендирующими агентами.
Изобретение также охватывает препараты в твердой форме, которые предназначены для перевода непосредственно перед употреблением в препараты жидкой формы для перорального введения. Подобные жидкие формы представляют собой растворы, суспензии и эмульсии. Данные препараты в дополнение к активному компоненту могут содержать красители, корригенты, стабилизаторы, буферы, искусственные или природные подсластители, диспергирующие, загущающие и солюбилизирующие агенты и им подобные вспомогательные вещества.
Для местного нанесения на эпидермис соединения по изобретению могут быть приготовлены в виде мазей, кремов или лосьонов, либо в виде диадерматического пластыря. Мази и кремы могут, например, быть приготовлены на водной или масляной основе с добавлением подходящих загущающих и/или гелеобразующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и обычно также содержат один или несколько эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей.
Препараты, пригодные для местного введения в ротовую полость, представляют собой лепешки, содержащие активный агент, заключенный в корригент, являющийся обычно сахарозой и аравийской камедью или трагакантом; пастилки, содержащие активный ингредиент, заключенный в инертную основу типа желатина и глицерина или сахарозы и аравийской камеди; и жидкости для полоскания рта, содержащие активный ингредиент в соответствующем жидком носителе.
Растворы или суспензии непосредственно наносят в носовую полость традиционными способами, например с помощью капельницы, пипетки или аэрозоля. Могут быть предложены препараты в форме однократной или множественной дозы. В последнем случае при использовании капельницы или пипетки это может быть достигнуто введением пациенту целесообразного, предварительно определенного объема раствора или суспензии. В случае аэрозоля это может быть достигнуто, например, при помощи дозированной распылительной подачи аэрозоля.
Введение в дыхательные пути может также быть достигнуто посредством применения препарата в аэрозольной форме, активный ингредиент в котором представлен в герметичной упаковке с приемлемым пропеллантом типа хлорфторуглерода (CFC), например дихлордифторметаном, трихлорфторметаном или дихлортетрафторэтаном, углекислым или другим подходящим газом. Аэрозоль может также содержать поверхностно-активное вещество, как например лецитин. Дозу лекарственного средства можно контролировать, установив дозирующий клапан.
С другой стороны, активные ингредиенты могут быть представлены в виде сухого порошка, например порошкообразной смеси данного соединения в подходящей порошковой основе типа лактозы, крахмала, производных крахмала, таких, как гидроксипропилметилцеллюлоза и поливинилпирролидин (PVP). Хорошо, если порошкообразный носитель будет образовывать в носовой полости гель. Состав в виде порошка может быть представлен в форме стандартной дозы, например в капсулах или картриджах, в частности, из желатина, либо в блистерных упаковках, порошок из которых может быть введен с помощью ингалятора.
Размер частиц соединения в препаратах, предназначенных для введения в дыхательные пути, обычно будет составлять, например порядка 5 микрон или менее. Подобный размер частиц может быть достигнут с помощью известных специалистам способов, например, использованием микронной мельницы.
При желании могут быть использованы препараты, адаптированные для длительного высвобождения активного ингредиента.
Фармацевтические препараты предпочтительно представлены в формах стандартной дозы. В такой форме препарат подразделен на стандартные дозы, содержащие соответствующие количества активного компонента. Форма стандартной дозы может представлять собой упакованный препарат, причем упаковка содержит разделенные количества препарата, как, например, упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, форма стандартной дозы сама может быть представлена в виде капсулы, таблетки, крахмальной облатки или лепешки, либо она может составлять соответствующее количество любой из них в упакованном виде.
Предпочтительными составами являются таблетки и капсулы для перорального введения и жидкости для внутривенного введения.
Способ применения
Соединения по настоящему изобретению чрезвычайно полезны при лечении расстройств и заболеваний живого организма животного ввиду их сродства к сайту связывания бенздиазепина ГАМКA-рецептора. Данное свойство делает соединения по настоящему изобретению чрезвычайно полезными при лечении конвульсий, состояния тревоги, расстройств сна, нарушений памяти, равно как и других заболеваний, чувствительных к модуляции ГАМКA-рецептора. Соединения по данному изобретению могут поэтому вводиться субъекту, включая человека, нуждающемуся в лечении, облегчении или ликвидации расстройства или заболевания, связанного с ГАМКA-рецепторами. В особенности это касается конвульсий, состояния тревоги, расстройств сна и нарушений памяти.
Приемлемая доза лежит в интервале 0,01-100 миллиграммов в день, 0,1-50 миллиграммов в день и, в особенности, 0,1-30 миллиграммов в день, как обычно в зависимости от способа введения, вводимой формы лекарственного средства, указания к применению, субъекта и веса субъекта, для лечения которого применяется лекарственное средство, а также мнения и опыта лечащего врача или ветеринара.
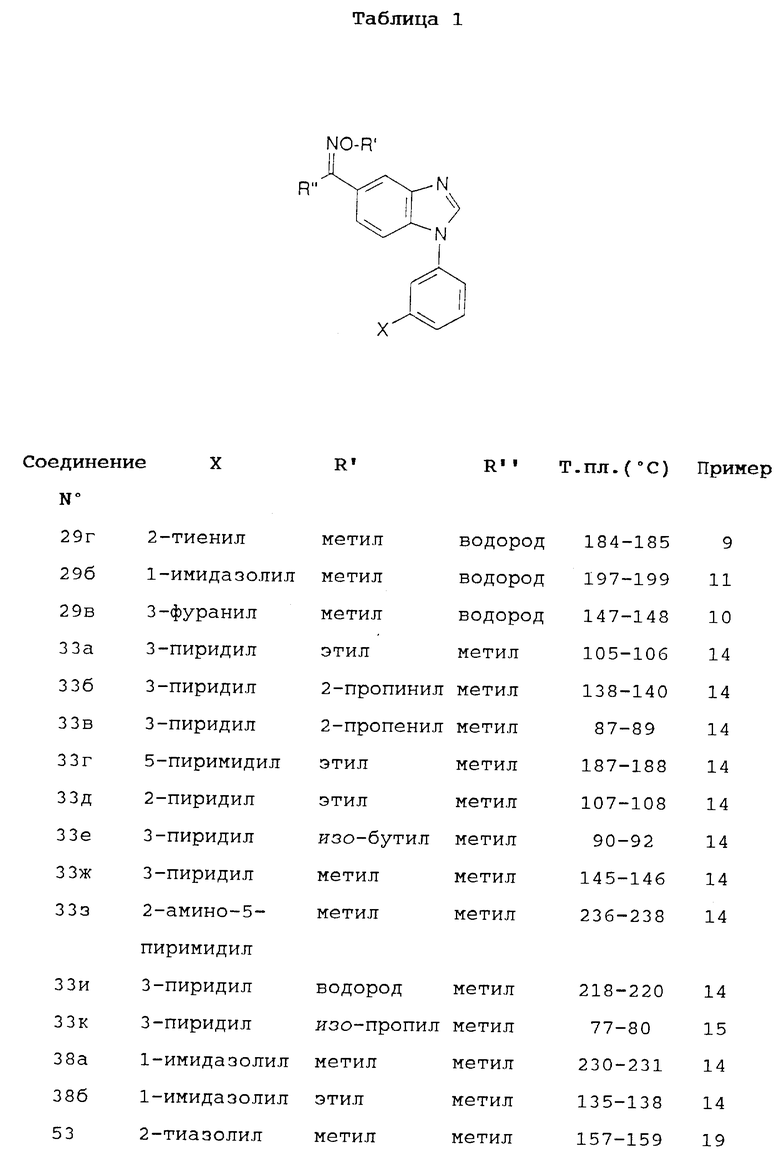
Следующие далее примеры приведены для дальнейшей иллюстрации изобретения, однако они не должны истолковываться как ограничивающие объем изобретения. Соединения по изобретению, полученные в описанных ниже примерах, приведены в таблице 1.
Пример 1
4-Фтор-3-нитроацетофенон (1ж): Концентрированную серную кислоту (200 мл) охлаждают до 5oC. К ней добавляют 4-фторацетофенон (20 мл; 0,16 моль), поддерживая температуру ниже 10oC. Смесь охлаждают до 0-5oC и к ней в течение 2 часов порциями добавляют нитрат калия (25 г; 0,25 моль), поддерживая температуру в указанном интервале. По окончании добавления смесь перемешивают на холоду дополнительные 2 часа. Указанную смесь выливают в лед (600 г) и отфильтровывают неочищенный продукт. Очистка колоночной хроматографией на силикагеле с использованием в качестве элюента смеси этилацетата и петролейного эфира (1:9) позволяет получить чистое соединение 1ж (18,2 г; 60 %). Т. пл. 48-49oC.
4-Амино-3-нитроацетофенон (1з): Суспензию 4-аминоацетофенона (25 г; 184 ммоль) в 250 мл уксусного ангидрида перемешивают без термостатирования в течение 30 мин. Смесь охлаждают до 0oC и к ней по каплям добавляют концентрированную азотную кислоту (18 мл), поддерживая температуру в интервале от -5 до +5oC. По окончании добавления смесь оставляют нагреваться до комнатной температуры и перемешивание без термостатирования продолжают в течение ночи. Смесь выливают в ледяную воду (700 мл) и продукт, N-(4-ацетил-2-нитрофенил)ацетамид, отфильтровывают, тщательно промывают водой и высушивают. Выход: 26,5 г (65%). Указанный продукт добавляют к горячей смеси воды (50 мл) и концентрированной серной кислоты (100 мл) и полученную смесь перемешивают в течение 15 мин. Соединение 1з осаждается из охлажденной смеси по мере разбавления ее водой. Продукт отфильтровывают, промывают водой и высушивают. Выход 91%. Т.пл. 121-123oC.
Пример 2
3-(3-Пиридил)анилин (2а): Смесь диэтил-3-пиридилборана (16,3 г; 0,11 моль), 3-броманилина (12,2 мл; 0,11 моль), карбоната калия (45,8 г; 0,33 моль) и тетракис(трифенилфосфин)-палладия (0) (80 мг) в смеси воды (40 мл) и диметоксиметана (80 мл) нагревают при 80oC с продувкой азотом в течение ночи. После охлаждения смесь разбавляют водой и этилацетатом и фильтруют через складчатый бумажный фильтр. Слои разделяют и водный слой один раз экстрагируют этилацетатом. Объединенные органические фазы высушивают над сульфатом натрия и концентрируют при пониженном давлении. Остаток растворяют в этаноле. Добавляют воду и смесь упаривают досуха. Остаток кристаллизуется в процессе растирания с ледяной водой. Кристаллы собирают, высушивают и промывают петролейным эфиром, что позволяет получить чистое соединение 2а (16,3 г; Т.пл. 75-76oC).
3-(5-Пиримидил)анилин (2б): Суспензию, содержащую 5-бромпиримидин (15 г; 94,3 ммоль), гемисульфат 3-аминофенилбороновой кислоты (19,3 г; 104 ммоль), бикарбонат натрия (39,6 г; 472 ммоль) и тетракис(трифенилфосфин)палладий(0) (1 г) в смеси воды (75 мл) и диметоксиэтана (150 мл), нагревают в течение ночи при 80oC с продувкой азотом. После охлаждения смесь выливают в ледяную воду. Осадок отфильтровывают, промывают водой и высушивают, получая соединение 2б (15 г; 93%). Т.пл. 164-165oC.
3-(1-Имидазолил)анилин (2г): Смесь 1-иод-3-нитробензола (90 г; 0,36 моль), имидазола (54 г; 0,79 моль), карбоната калия (54 г; 0,39 моля) и тонкоизмельченного порошка меди (1 г) нагревают до 200oC. Расплав перемешивают в атмосфере азота в течение 2 часов. Пары воды, выделяющиеся в ходе реакции, улавливают с помощью молекулярных сит, помещенных между реакционным сосудом и холодильником. По окончании реакции смесь охлаждают до 100oC и к ней добавляют воду. Смесь оставляют охлаждаться до комнатной температуры и неочищенный продукт отфильтровывают и высушивают. Перекристаллизация из толуола (200-250 мл) позволяет получить чистый 3-(1-имидазолил)нитробензол (54,2 г; 79 %). Т.пл. 101-102oC.
К 3-(1-имидазолил)нитробензолу (51,6 г; 0,27 моль) в уксусной кислоте (500 мл) добавляют палладиевый катализатор (5 г 5%-ного Pd на активированном угле) и смесь гидрируют под давлением (Pначальное: 4 бар) до тех пор, пока не прекратится поглощение водорода. Смесь фильтруют через броунмиллерит и фильтрат упаривают досуха, получая соединение 2 г в виде масла светло-коричневой окраски. Выход: 40,4 г (93%).
N-Aцeтил-3-(1-имидaэoлил)aнилин (2д): Соединение 2 г (5,88 г; 37 ммоль) перемешивают без термостатирования в уксусном ангидриде (30 мл) в течение 1 часа. Смесь выливают в ледяную воду и подщелачивают, добавляя водный гидроксид натрия (12 М). Продукт отфильтровывают, промывают водой и высушивают, получая соединение 2д (6,34 г; 85%). Т.пл. 181-183oC.
3-(2-Пиридил)анилин (2е): К раствору 2-(3-нитрофенил)пиридина (полученному согласно (J. Chem. Soc. 1958 р. 1759)) (12,7 г; 63,5 ммоль) в абсолютном этаноле добавляют палладиевый катализатор (1,3 г 5%-ного Pd на активированном угле) и смесь гидрируют под атмосферным давлением до тех пор, пока не прекратится поглощение водорода. Смесь фильтруют через броунмиллерит и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетата и петролейного эфира (9:1), получая соединение 2е (9,5 г; 88%) в виде масла светло-коричневой окраски.
3-(2-Аминопиримид-5-ил)анилин (2l): Смесь, содержащую 2-(ацетамино)-5-бромпиримидин (5,4 г; 25 ммоль), гемисульфат 3-аминофенилбороновой кислоты (5,58 г; 30 ммоль), карбонат калия (10,4 г; 75 ммоль), 1,3-пропандиол (9 мл; 0,13 моль) и тетракис(трифенилфосфин)палладий(0) (0,5 г) в смеси воды (25 мл) и диметоксиэтана (50 мл), перемешивают с продувкой азотом в течение ночи при 80oC. После охлаждения смесь выливают в ледяную воду. Продукт (деацетилированный в ходе реакции) отфильтровывают, промывают водой и высушивают, получая соединение 2l (4,19 г; 90%). Т.пл. 171-172oC.
Пример 3
изо-Пропил-4-(3-бромфенил)амино-3-нитробензоат (24): Смесь изо-пропил-4-хлор-3-нитробензоата (25,88 г; 0,11 моль), 3-броманилина (17,36 мл; 0,16 моль) и карбоната калия (14,63 г; 0,11 моль) в N-метил-2-пирролидоне (25 мл) нагревают при 150oC в течение 3 дней. После охлаждения смесь выливают в разбавленную соляную кислоту (300 мл; 1 М). Осадок отфильтровывают, промывают водой и высушивают (37,4 г). Данный неочищенный продукт промывают горячим 2-пропанолом, что позволяет получить чистое соединение 24 (26,25 г; 65%). Т. пл. 162-165oC.
Пример 4
изо-Пропил-3-амино-4-(3-бромфенил)аминобензоат (25): Соединение 24 из примера 3 (3,79 г; 10 ммоль) суспендируют в смеси этанола (30 мл) и дихлорметана (30 мл). Добавляют влажный никель Ренея (Raney) (0,5 г) и смесь гидрируют до тех пор, пока не прекратится поглощение водорода. Смесь фильтруют через броунмиллерит и растворитель удаляют при пониженном давлении, получая соединение 25 с количественным выходом. Т.пл. 82-85oC.
5-Ацетил-2-((3-бромфенил)амино)анилин в смеси с 5-ацетил-2-((3- иодфенил)амино)анилином (35) получали аналогичным образом из соединения 34 (пример 16). Продукт был выделен в виде масла.
Пример 5
5-Ацетил-1-(3-(3-пиридил)фенил)бензимидазол (32а): Раствор соединения 31а из примера 13 (7 г; 23,1 ммоль) в муравьиной кислоте (20 мл) перемешивают без термостатирования в течение ночи. Смесь выливают в воду (750 мл) и подщелачивают концентрированным водным аммиаком. Осадок отфильтровывают, промывают водой и высушивают. Неочищенный продукт растворяют в этаноле при температуре дефлегмации. К раствору добавляют воду до тех пор, пока не начнется образование осадка. Смесь оставляют охлаждаться. Продукт отфильтровывают и высушивают, получая 4,3 г (60%). Т.пл. 200-202oC.
1-(3-Бромфенил)-5-(и-пропилкарбокси)бензимидазол (26) получали аналогичным образом из соединения 25 (пример 4). Выход: 85%. Т.пл. 102-104oC.
5-Ацетил-1-(3-иодфенил)бензимидазол в смеси с 5-ацетил(3-бромфенил)бензимидазолом (36) получали аналогичным образом из соединения 35 (пример 4). Выход: приблизительно 91%. (2 стадии, начиная с соединения 34).
5-Ацетил-1-(3-(5-пиримидил)фенил)бензимидазол (32б) получали аналогичным образом из соединения 31б (пример 13). Выход: 71%. Т.пл. 253-254oC.
5-Aцeтил-1-(3-(2-пиpидил)фeнил)бeнзимидaзoд (32в) получали аналогичным образом из соединения 31в (пример 13). Выход: 91%. Т.пл. 158-159oC.
5-Ацетил-1-(3-(2-аминопиримид-5-ил)фенил)бензимидазол (32г) получали аналогичным образом из соединения 31г (пример 13). Выход: 84%. Т.пл. 275-278oC.
Пример 6
1-(3-Бромфенил)-5-(гидроксиметил)бензимидазол (27): Перемешиваемую суспензию соединения 26 из примера 5 (18 г; 50,18 ммоль) в 300 мл сухого диэтилового эфира выдерживают при комнатной температуре с продувкой азотом. К суспензии порциями добавляют LiAlH4 (1,9 г; 50 ммоль) и смесь перемешивают в течение ночи. Полученную эмульсию фильтруют через броунмиллерит и производят разделение фаз. Водную фазу один раз экстрагируют этилацетатом. Объединенные органические фазы промывают водой, высушивают над сульфатом натрия и концентрируют при пониженном давлении. Остаток хроматографируют на силикагеле, последовательно используя в качестве элюентов этилацетат и смесь этилацетата и метанола (9: 1). Выход соединения 27: 7,69 г (51%). Т.пл. 107-109oC.
Пример 7
1-(3-Бромфенил)-5-формилбензимидазол (28): Смесь соединения 27 из примера 6 (3,9 г; 12,9 ммоль) и бензолселеновой кислоты (3,04 г; 16,1 ммоль) в толуоле перемешивают при 70oC в течение ночи. Продукт выпадает в осадок в процессе охлаждения. Осадок отфильтровывают, промывают петролейным эфиром и высушивают. Последующие промывки водным карбонатом натрия и водой позволяют получить чистое соединение 28. Выход 2,99 г (77%). Т.пл. 179-181oC.
Пример 8
O-Метилоксим 1-(3-бромфенил)-5-формилбензимидазола (29а): К суспензии соединения 28 из примера 7 (2,95 г; 9,8 ммоль) в абсолютном этаноле (100 мл) добавляют гидрохлорид метоксиламина (1,23 г; 14,7 ммоль) и смесь нагревают до 70oC. К смеси в течение 20 мин порциями добавляют бикарбонат натрия (1,23 г; 14,7 ммоль). По окончании добавления смесь перемешивают при 70oC дополнительные 2 часа. После охлаждения растворитель удаляют при пониженном давлении. К остатку добавляют воду и продукт отфильтровывают, промывают водой и высушивают. Выход 2,77 г (86%). Т.пл. 119-120oC.
Пример 9
O-Метилоксим 1-(3-(2-тиенил)фенил)-5-формилбензимидазола (29г): Смесь соединения 29а из примера 8 (0,7 г; 2,1 ммоль), 2-(трибутилстаннил)тиофена (1,59 г; 4,3 ммоль) и дихлорида трис(трифенилфосфин)палладия (50 мг) в DMF (диметилформамиде)(5 мл) нагревают при 80oCв течение ночи. Охлажденную реакционную смесь разбавляют 4 объемами воды и экстрагируют этилацетатом. Объединенные органические экстракты высушивают над сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетата и петролейного эфира (1:1). Выход: 0,56 Г (80%). Т.пл. 184-185oC.
Пример 10
O-Метилоксим 1-(3-(3-фуранил)фенил)-5-формилбензимидазола (29в): Смесь O-метилоксима 1-(3-бромфенил)-5-формилбензимидазола (29а) из примера 8 (0,7 г; 2,1 ммоль), 3-фуранилбороновой кислоты (0,26 г; 2,34 ммоль), бикарбоната натрия (0,89 г; 10,6 ммоль) и тетракис(трифенилфосфин)палладия(0) (50 мг) в смеси воды (5 мл) и диметоксиэтана (10 мл) перемешивают в атмосфере азота в течение ночи при 80oC. После охлаждения к смеси добавляют воду и экстрагируют этилацетатом. Экстракт высушивают над сульфатом натрия и концентрируют при пониженном давлении. Остаток элюируют через силикагель с помощью смеси этилацетата и петролейного эфира (1:1). Фракции, содержащие чистый продукт, упаривают досуха. Растирание с петролейным эфиром позволяет получить кристаллический продукт белого цвета. Выход: 0,42 г (63%). Т.пл. 147-148oC.
Пример 11
O-Метилоксим 1-(3-(1-имидазолил)фенил)-5-формилбензимидазола (29б): Смесь O-метилоксима 1-(3-бромфенил)-5-формилбензимидазола (29а) из примера 8 (0,7 г; 2,13 ммоль), имидазола (0,33 г; 4,85 ммоль), карбоната натрия (0,29 г, - 2,13 ммоль) и каталитических количеств медной бронзы в 5 мл N-метил-2-пирролидона нагревают при 140oC с продувкой азотом в течение 24 часов. После охлаждения смесь выливают в воду. Добавляют небольшое количество метанола и смесь экстрагируют дихлорметаном. Экстракт высушивают над сульфатом натрия и концентрируют при пониженном давлении. Остаток элюируют через силикагель с помощью смеси дихлорметана и этанола (10:1), получая чистое соединение 29б. Выход: 0,23 г (34%). Т.пл. 197-199oC.
5-Ацетил-1-(3-(1-имидазолил)фенил)бензимидазол (37): получали аналогичным образом из соединения 36 (пример 5). Выход: приблизительно 26%. Т.пл. 205-206oC.
Пример 12
4-Ацетил-2-нитро-N-(3-(3-пиридил)фенил)анилин (30а): Смесь соединения (1ж) из примера 1 (5 г; 27,3 ммоль) и соединения (2а) из примера 2 (4,62 г; 27,2 ммоль) в сухом N-метил-2-пирролидоне (10 мл) перемешивают при температуре 40-50oC в течение ночи. Полученную твердую реакционную смесь суспендируют в ледяной воде и подщелачивают, добавляя водный карбонат натрия (1 М). Продукт отфильтровывают, промывают водой и высушивают, получая 7,68 г соединения 30а (85%). Т.пл. 112-113oC.
4-Ацетил-2-нитро-N-(3-(5-пиримидил)фенил)анилин (30б) получали аналогичным образом из соединения 1ж (пример 1) и соединения 2б (пример 2). Выход: 65%. Т.пл. 131-132oC.
4-Ацетил-2-нитро-N-(Э-(2-пиридил)фенил)анилин (30в) получали аналогичным образом из соединения 1ж (пример 1) и соединения 2е (пример 2). Выход: 87%. Т.пл. 195-196oC.
4-Ацетил-2-нитро-N-(3-(2-аминопиримид-5-ил)(фенил)анилин (30г) получали аналогичным образом из соединения 1ж (пример 1) и соединения 21 (пример 2). Выход: 80%. Т.пл. 233-236oC.
Пример 13
5-Aцeтил-2-(3-(3-пиpидил)фeнилaминo)aнилин (31а): Соединение 30а из примера 12 (2 г; 6 ммоль) суспендируют в смеси этанола (50 мл) и дихлорметана (10 мл) и гидрируют при атмосферном давлении, используя в качестве катализатора палладий (5%-ный на активированном угле). Фильтрование полученного раствора через броунмиллерит с последующим упариванием растворителя дает масло, растирание которого со смесью диэтилового эфира и петролейного эфира (1: 1) позволяет получить 1,46 г чистого соединения (31а) (80%). Т.пл. 175-176oC
5-Ацетил-2-(3-(5-пиримидил)фениламино)анилин (31б) получали аналогичным образом из соединения 30б (пример 12). Неочищенный продукт в виде масла использовали на следующей стадии (пример 5) без дополнительной очистки.
5-Ацетил-2-(3-(2-пиридил)фениламино)анилин (31в) получали аналогичным образом из соединения 30в (пример 12). Выход: 92%. Т.пл. 145-146oC.
5-Ацетил-2-(3-(2-аминопиримид-5-ил)фениламино) анилин (31г) получали аналогичным образом из соединения 30г (пример 12). Выход: 71%. Т.пл. 228-230oC.
Пример 14
O-Этилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33а): Соединение (32а) из примера 5 (5 г; 15,97 ммоль) суспендируют в абсолютном этаноле (50 мл) и нагревают до 70oC. Добавляют гидрохлорид O-этилгидроксиламина (2,4 г; 24,61 ммоль) и смесь кипятят с обратным холодильником в течение 1,5 часа. После охлаждения растворитель удаляют упариванием, а остаток перемешивают с водным гидроксидом натрия (50 мл; 1 М). Неочищенный продукт отфильтровывают. Этап очистки с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента смеси этилацетата и этанола (9:1) позволяет получить чистое соединение 33а. Выход: 3,7 г (65%). Т.пл. 105-106oC.
Следующие соединения получали аналогичным образом из соединения 32а (пример 5) и соответствующих гидрохлоридов O-алкил-, O-алкенил- и O-алкинилгидроксиламина:
O-Пропаргилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33б). Выход: 49%. Т.пл. 138-140oC.
O-Аллилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33в). Выход: 73%. Т.пл. 87-89oC.
O-изо-Бутилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33е). Выход: 38%. Т.пл. 90-92oC.
O-Метилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33ж). Выход: 59%. Т.пл. 145-146oC.
Оксим 5-ацетил-1-(3-(3-пиридил)фенил)бензимидазола (33и). Выход: 82%. Т. пл. 218-220oC.
Следующие соединения получали аналогичным образом из соответствующих гидрохлоридов O-алкилгидроксиламина и соединений 326, 32в и 32г (пример 5), соответственно:
O-Этилоксим 5-ацетил-1-(3-(5-пиримидил)фенил)бензимидазола (33г). Выход: 54%. Т.пл. 187-188oC.
O-Этилоксим 5-ацетил-1-(3-(2-пиридил)фенил)бензимидазола (33д). Выход: 53%. Т.пл. 107-108oC.
O-Метилоксим 5-ацетил-1-(3-(2-аминопиримид-5-ил)фенил)бензимидазола (33з). Выход: 62%. Т.пл. 236-238oC.
Следующие соединения получали аналогичным образом из соединения 37 (пример 11) и гидрохлорида O-метилгидроксиламина и гидрохлорида O-этилгидроксиламина, соответственно:
O-Метилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола (38а). Выход: 67 %. Т.пл. 230-231oC.
O-Этилоксим 5-ацетил-1-(3-(1-имидазолил)фенил)бензимидазола (38б). Выход: 67%. Т.пл. 135-138oC.
Пример 15
O-изо-Пропилоксим 5-ацетил-1-(3-(3-пиридил)фенил)бензинидазола (33к): К суспензии соединения (33и) (пример 14) (0,4 г; 1,22 ммоль) в сухом DMF (5 мл) добавляют гидрид натрия (50 мг 60%- ной дисперсии в минеральном масле). Смесь перемешивают при температуре 30-40oC в течение 30 мин. Добавляют 2-бромпропан (0,14 мл; 1,49 ммоль) и смесь перемешивают при 40oC в течение ночи. После охлаждения смесь разбавляют 4 объемами воды и экстрагируют дихлорметаном. Органический экстракт концентрируют и элюируют через силикагель с помощью этилацетата, получая 0,15 г соединения 33к (33%). Т.пл. 77-80oC.
Пример 16
4-Aцeтил-N-(3-бpoмфeнил)-2-нитpoaнилин в смеси с 4-ацетил-N-(3-иодфенил)-2-нитроанидином (34): Смесь 4-ацетил-2-нитроанилина (1з) из примера 1 (15,6 г; 86,7 ммоль), З-бром-1-иодбензола (13,3 мл; 104 ммоль), карбоната калия (12 г; 87 ммоль) и каталитических количеств иодида меди и меди-бронзы нагревают с перемешиванием при 180oC с продувкой азотом в течение 24 часов. Смесь оставляют охлаждаться до 70oC и смолистый реакционный осадок дважды экстрагируют горячим этилацетатом. Объединенные экстракты очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетата и петролейного эфира (3:7), получая 9 г смеси продуктов.
Пример 17
2-(Трибутилстаннил)тиазол (50): К раствору тиазола (0,71 мл; 10 ммоль) в сухом THF (20 мл) по каплям в атмосфере арго на при температуре -78oC добавляли 1,6 М BuLi в гексане (6,9 мл; 11 ммоль). Реакционную смесь перемешивали при -78oC в течение 0,5 часа, после чего к ней по каплям добавляли Bu3SnCl (3,1 мл; 11 ммоль). По окончании перемешивания в течение 1 ч при -78oC и 1 ч при комнатной температуре смесь концентрировали, растирали с водой (50 мл) и экстрагировали диэтиловым эфиром (100 мл х 3). Экстракт промывали солевым раствором, высушивали и концентрировали при пониженном давлении, получая соединение (50) в виде бесцветного масла. (3,7 г; колич.).
Пример 18
5-Ацетил-1-(3-(2-тиазолил)фенил)бензимидазол (51): К раствору соединения (50) из примера 17 (3,6 г; 9,7 ммоль) в сухом THF (20 мл) в атмосфере аргона добавляли 5-ацетил-1-(3-бромфенил)бензимидазол (52) (1,5 г; 4,8 ммоль) и (PPh3)2PdCl2 (340 мг; 0,48 ммоль). Реакционную смесь в загерметизированном сосуде емкостью 50 мл, перемешивали при температуре 80oC в течение 24 ч. После охлаждения смесь концентрировали, растирали с водой (100 мл) и экстрагировали CH2Cl2 (200 мл х 3). Экстракт промывали рассолом, высушивали и концентрировали при пониженном давлении. Остаток промывали эфиром, что позволяло получить соединение (51) в кристаллическом виде (1,5 г; 89%).
5-Ацетил-1-(3-бромфенил)бензимидазол (52) получали следующим образом:
4-Ацетил-2-нитроанилин: К смеси воды и концентрированной серной кислоты, 150 мл (1: 2), добавляли N-(4-ацетил-2-нитрофенил)ацетамид (26,5 г; 11,94 ммоль). По истечении 15 минут смесь выливали в воду. Продукт отфильтровывали, промывали водой и высушивали.
N-(3-6poмфенил)-4-ацетил-2-нитроанилин: Смесь 4-ацетил-2-нитроанилина (3,41 г; 18,94 ммоль), 1,3-дибромбензола (4,6 мл; 38,06 ммоль), карбоната калия (2,62 г; 19 ммоль) и каталитических количеств медной бронзы нагревают при 180oC с продувкой азотом и перемешиванием в течение 2 дней. После охлаждения твердый реакционный осадок экстрагируют смесью дихлорметана и метанола (9: 1). Экстракт концентрируют при пониженном давлении. Остаток экстрагируют этилацетатом. Данный экстракт концентрируют при пониженном давлении и остаток элюируют через силикагель с помощью смеси петролейного эфира и этилацетата (4: 1), что позволяет получить чистый продукт. Выход: 0,67 г (10,6%). Т.пл. 142-144oC.
5-Ацетил-1-(3-бромфенил)бензимидазол (52): N-(3-бромфенил)-4-ацетил-2-нитроанилин (9,0 г; 26,63 ммоль) суспендировали в 99%-ном этаноле (100 мл). Добавляли никель Ренея и смесь гидрировали при атмосферном давлении в течение 20 часов. Добавляли хлороформ, смесь фильтровали через броунмиллерит и упаривали в вакууме, получая 8,03 г масла. К полученному маслу добавляли 80 мл муравьиной кислоты и смесь нагревали при 80oC в течение 1,5 часов. Избыток муравьиной кислоты удаляли в вакууме. Остаток перемешивали в воде и подщелачивали водным гидроксидом натрия. Продукт отфильтровывали, промывали водой и высушивали.
Пример 19
O-Метилоксин 5-ацетил-1-(3-(2-тиазолил)фенил)бензимидазола (53). Соединение (51) из примера 18 (300 мг; 0,94 ммоль) добавляли к смеси этанола (5 мл), гидрохлорида метоксиламина (300 мг; 3,5 ммоль) и триэтиламина (0,17 мл; 1,2 ммоль) и смесь перемешивали при 60oC в течение 0,5 ч. После охлаждения смесь выливали в 5%-ный водный раствор NaHCO3 (100 мл), осадок отфильтровывали, промывали водой и высушивали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя в качестве элюента смесь CH2Cl2 и метанола (100:1), получая соединение (53) (270 мг; 86%). Т.пл. 157-159oC.
Пример 20
5-Циано-1-(3-(2-тиазолил)фенил)бензимиидазол (54) синтезировали так же, как описано в примере 18, используя 5-циано-1-(3-иодфенил)бензимидазол (2,0 г; 5,9 ммоль) вместо соединения (52), (PPh3)2PdCl2 (100 мг; 0,14 ммоль) и соединение (50) (3,6 г; 9,7 ммоль). В результате реакции получали 5-циано-1-(3-(2-тиазолил)фенил)бенэимидазол (1,5 г; 86%).
5-Циано-1-(3-иодфенил)бензимидазол получали, как описано ниже:
N-(3-Иодфенил)-4-циано-2-нитроанилин: К раствору 4-хлор-3-нитробензонитрила (1,82 г; 10 ммоль) в сухом DMF (25 мл) добавляют триэтиламин (1,54 мл; 11 ммоль) и 3-иоданилин (1,2 мл; 10 ммоль) и смесь нагревают до 80-100oC в течение ночи. После охлаждения смесь выливают в четырехкратный объем ледяной воды. Осадок отфильтровывают, промывают водой и высушивают. Данный неочищенный продукт промывают теплым этанолом, получая 2,1 г (58%) поименованного в заголовке соединения. Т.пл. 211- 212oC.
2-Аминo-(N-(3-иoдфeнил))-4-циaнoaнилин: К суспензии N-(3-иодфенил)-4-циано-2-нитроанилина (2,1 г; 5,75 ммоль) в метаноле (50 мл) добавляют хлорид аммония (0,92 г; 17,25 ммоль) и сульфид натрия нонагидрат (4,14 г; 17,25 ммоль) и смесь кипятят с обратным холодильником в течение 1,5 часов. После охлаждения смесь выливают в ледяную воду (200 мл), продукт отфильтровывают, промывают водой и высушивают, получая 1,8 г (93%) поименованного в заголовке соединения. Т.пл. 170-172oC.
5-циано-1-(3-иодфенил)бензимидазол: Суспензию 2-амино-(N-(3-иодфенил))-4-цианоанилина (1,8 г; 5,36 ммоль) в муравьиной кислоте (20 мл) нагревают до 80-100oC в течение 1,5 часов. Реакционную смесь в горячем состоянии фильтруют через хлопковую подложку в ледяную воду (100 мл). Осадок отфильтровывают, промывают водой и высушивают. Неочищенный продукт растворяют в дихлорметане и, добавляя петролейный эфир, добиваются его выпадения в осадок. Продукт отфильтровывают и высушивают. Выход поименованного в заголовке соединения: 1,38 г (75%). Т.пл. 177-179oC.
Пример 21
5-Формил-1-(3-(2-тиазолил)фенил)бензимидазол (55): К раствору соединения (54) (1,5 г; 4,9 ммоль) в смеси HCO2H (18 мл) и воды (6 мл) добавляли никель Ренея (2,0 г). Смесь перемешивали в атмосфере аргона при 110oC в течение 1 часа. После охлаждения смесь фильтровали. Фильтрат концентрировали, распределяли между 5%-ным водным раствором NaHCO3 и этилацетатом. Органическую фазу промывали рассолом, высушивали и концентрировали при пониженном давлении, получая соединение (55) (1,3 г; 87%).
Пример 22
Оксим 5-формил-1-(3-(2-тиазолил)фенил)бензимидазола: Соединение (55) (700 мг; 2,29 ммоль) добавляли к смеси этанола (25 мл) и NH2OH•Cl (600 мг; 6,9 ммоль). Смесь перемешивали при 90oC в течение 1 ч. После охлаждения смесь выливали в воду (100 мл), осадок отфильтровывали, промывали водой и высушивали при пониженном давлении, получая оксим 5-формил-1-(3-(2-тиазолил)фенил)бензимидазола (690 мг; 94%).
O-метилоксим 5-формил-1-(3-(2-тиазолил)фенил)бензимидазола (56) получали аналогичным образом, используя NH2OMe•HCl вместо NH2OH•HCl. Т.пл. 154-160oC.
Пример 23
4-(3-Нитрофенил)пиримидин (57): Смесь 4-фенилпиримидина (10 г; 64 ммоль) и конц. H2SO4 (33 мл) добавляли при температуре 0oC к смеси конц. H2SO4 (22 мл) и конц. HNO3 (16 мл). Полученную смесь перемешивали при 0oC в течение 2 ч, выливали в дробленый лед и экстрагировали CH2Cl2. Экстракт промывали 5%-ным водным раствором NaHCO3, высушивали на MgSO4 и концентрировали при пониженном давлении. Остаток растирали с изопропанолом, осадок отфильтровывали и высушивали при пониженном давлении, получая соединение (57) (6,4 г; 50%).
Пример 24
4-(3-Аминофенил)пиримидин (58): К суспензии соединения (57) (6,3 г; 31 ммоль) в смеси MeOH (60 мл) и THF (30 мл) добавляли 5%-ный палладий на активированном угле (300 мг) и смесь гидрировали при атмосферном давлении в течение 1 ч. Смесь фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, используя в качестве элюента смесь гексана и этилацетата (3:1), и получали соединение (58) (5,1 г,- 96%).
Пример 25
N-(3-(4-Пиримидил)фенил)-4-циано-2-нитроанилин (59): К смеси 4-хлор-3-нитробензнитрила (5,5 г; 30 ммоль) и соединения (58) (5,1 г; 30 ммоль) в THF (120 мл) добавляли гидрид натрия (2,3 г; 50%-ная суспензия в минеральном масле). После перемешивания при комнатной температуре в течение 2 дней смесь выливали в воду и экстрагировали CH2Cl2. Экстракт концентрировали при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение (59) в кристаллическом состоянии (9,2 г; 96%).
Пример 26
N-(3-(4-пиримидил)фенил)-4-циано-2-аминоанилин (60): синтезировали так же, как описано в примере 24, используя соединение (59) (9,2 г; 29 ммоль) вместо соединения (57) и 600 мг катализатора. В результате реакции получали соединение (60) (8,3 г; колич.).
Пример 27
5-Циано-1-(3-(4-пиримидил)фенил)бензимидазол (61): Смесь соединения (60) (3,0 г; 10 ммоль) и HCO2H (20 мл) перемешивали при температуре 110oC в течение 1 ч. Смесь концентрировали в вакууме. Остаток распределяли между 5%-ным водным NaHCO3 и CH2Cl2. Органическую фазу высушивали над MgSO4 и растворитель удаляли упариванием. Остаток растирали с этилацетатом, получая соединение (61) в кристаллическом состоянии (2,6 г; 85%).
Пример 28
5-Формил-1-(3-(4-пиримидил)фенил)бензимидазол (62): синтезировали так же, как описано в примере 21, используя соединение (61) (2,5 г; 8,4 ммоль) вместо соединения (54) и 1,5 г никеля Ренея. В результате реакции получали соединение (62) (1,9 г; 73%).
Пример 29
Оксим 5-формил-1-(3-(4-пиримидил)фенил)бензимидазола (63): синтезировали так же, как описано в примере 22, используя соединение (62) (150 мг; 0,50 ммоль) вместо соединения (55) и NH2OH•HCl (100 мг; 1,5 ммоль). В результате реакции получали соединение (63) (120 мг; 76%). Т.пл. 220-221oC.
O-Метилоксим 5-формил-1-(3-(4-пиримидил)фенил)бензимидазола (64): синтезировали так же, как описано в примере 22, используя соединение (62) (200 мг; 0,66 ммоль) вместо соединения (55) и NH2OMe•HCl (250 мг; 2,0 ммоль). В результате реакции получали соединение (64) (61 мг; 28%). Т.пл. 180-182oC.
Пример 30
N-(3-(2-пиpимидил)фенил)-4-циaнo-2-нитpoaнилин (65): синтезировали так же, как описано в примере 25, используя 2-(3-аминофенил) пиримидин (4,50 г; 26,3 ммоль) вместо соединения (58). В результате реакции получали соединение (65) (5,08 г; 61%).
Пример 31
N-(3-(2-пирмидил)фенил-4-циано-2-аминоанилин (66): синтезировали так же, как описано в примере 24, используя соединение (65) (1,2 г; 3,8 ммоль) вместо соединения (57). В результате реакции получали соединение (66) (1,0 r; 93%).
Пример 32
5-Циано-1-(3-(2-пиримидил)фенил)бензимидазол (67): синтезировали так же, как описано в примере 27, используя соединение (66) (1,0 г: 3,5 ммоль) вместо соединения (60). В результате реакции получали соединение (67) (840 мг; 80%).
Пример 33
5-формил-1-(3-(2-пиримидил)фенил)бензимидазол (68): Раствор DIBAL-H в толуоле (2,5 мл; 1,0 М) при температуре -78oC добавляли к смеси соединения (67) (367 мг; 1,2 ммоль) и CH2Cl2 (60 мл). Реакционную смесь перемешивали в течение 1 ч при -78oC и 1 ч при комнатной температуре. К реакционной смеси добавляли насыщенный раствор NH4Cl и перемешивание продолжали в течение 0,5 ч. Смесь выливали в воду и экстрагировали CH2Cl2. Органический экстракт высушивали над MgSO4, растворитель упаривали при пониженном давлении, получая соединение (68) (142 мг; 38%).
Пример 34
O-метилоксим 5-формил-1-(3-(2-пиримидил)фенил)бензимидазола (69): синтезировали так же, как описано в примере 22, используя соединение (68) (210 мг; 0,70 ммоль) вместо соединения (55) и NH2OMe•HCl (300 мг; 3,6 ммоль) вместо NH2OH•HCl. В результате реакции получали соединение (69) (192 мг; 83%). Т.пл. 158-159oC.
Соединения, полученные в приведенных выше примерах, перечислены в следующей ниже таблице 1.2


Производное бензимидазола формулы 1, где R3 - остаток формулы (а), R11 - пиридил, имидазол, пиримидинил, фуранил, тиенил или тиазолил, возможно замещенный аминогруппой; один из R6 или R7 - водород, а другой представляет -CR'= NOR'', в котором каждый из R' и R'' - водород, алкил, алкенил или алкинил, или его фармацевтически приемлемая соль либо оксид или фармацевтическая композиция на его основе обеспечивает модуляцию ГАМКA-рецепторного комплекса центральной нервной системы живого организма. 3 с. и 5 з.п. ф-лы, 1 табл. 1 ил. 
или его фармацевтически приемлемая соль либо оксид,
где R3 представляет собой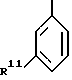
где R11 представляет собой пиридил, имидазолил, пиримидинил, фуранил, тиенил или тиазолил, возможно замещенный аминогруппой;
один из R6 и R7 представляет собой водород, а другой представляет собой -CR1= NOR11-, в котором каждый из R1 и R11 независимо представляет собой водород, C1-C6-алкил, C1-C6-алкенил или C1-C6-алкинил, причем, если R11 является водородом или C1-C6-алкилом, то R1 не является водородом.
Приоритет по пунктам и признакам:
21.04.95 по п.2: 0-этилоксим 5-ацетил-1-[3-(1-имидазолил)фенил]бензимидазол, по пп.1, 4, 5, 7 и 8;
27.06.95 при 0-этилоксим 5-ацетил-1-[3-(3-пиридил)фенил]бензимидазол;
19.04.96 при 0-метилоксим 1-[3-(3-фуранил)фенил]-5-формил-бензимидазол, оксим 5-ацетил-1-[3-(3-пиридил)фенил]бензимидазол, 0-этилоксим 5-ацетил-1-[3-(1-имидазолил)фенил] бензимидазол, 0-этилоксим 5-ацетил-1-[3-(2-тиазолил)фенил]бензимидазол, по пп.3 и 6.
| ОКСИАЛКИЛФУРАНОВЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, ИЛИ ИХ СТЕРЕОХИМИЧЕСКИЕ ИЗОМЕРНЫЕ ФОРМЫ, ОБЛАДАЮЩИЕ АНТИАЛЛЕРГИЧЕСКОЙ АКТИВНОСТЬЮ И АНТИАЛЛЕРГИЧЕСКАЯ КОМПОЗИЦИЯ | 1990 |
|
RU2030415C1 |
| Искусственная нога | 1927 |
|
SU12291A1 |
| ЭЛЕКТРОЛИЗЕР С ТРУБЧАТЫМИ ДИАФРАГМАМИ | 1935 |
|
SU48218A1 |
| ПРЕДОХРАНИТЕЛЬНЫЙ ОТ ПОРЕЗОВ ЩИТОК ДЛЯ ДВУХЛЕЗВЕННЫХ БРИТВ С КЛИНКАМИ ОБЫЧНОЙ ФОРМЫ | 1927 |
|
SU6831A1 |
| ПУЛЬТ ДЛЯ ВВОДА ЦИФРОВОЙ ИНФОРМАЦИИ | 0 |
|
SU393738A1 |
| EP 0616805 A1, 1994. | |||

