Изобретение относится к соединениям приведенной ниже формулы 1, которые ингибируют фермент глицинамид рибониклеотид формил трансферазу (GARFT). Данное изобретение также относится к промежуточным соединениям для получения этих соединений, к фармацевтическим композициям, содержащим эти соединения, их применению для ингибирования GARFT и их применению для ингибирования роста и пролиферации клеток высших организмов или микроорганизмов, таких как бактерии, дрожжи и грибы. Эти соединения обладают противоопухолевой, противовоспалительной, антипсориазной и/или иммуносупрессивной активностью. Данное изобретение также относится к получению таких соединений.
Большой класс антипролиферативных агентов включает в себя соединения-антиметаболиты. Конкретный подкласс антиметаболитов, известных в качестве антифолатов или антифолев, представляют собой антагонисты витамина фолиевой кислоты. В типичном случае антифолаты сильно напоминают по структуре фолиевую кислоту и включают в себя характерную P-бензоил глутаматную часть фолиевой кислоты. Глутаматная часть фолиевой кислоты несет на себе двойной отрицательный заряд при физиологических pH. Поэтому это соединение и его аналоги имеют активную энергозависимую транспортную систему для прохождения через клеточную мембрану и проявляют метаболическое действие.
GARFT является фолатзависимым ферментом в метаболическом пути биосинтеза пурина de novo. Этот путь является решающим для деления клеток и пролиферации. Известно, что блокирование этого пути имеет антипролиферативный эффект, в частности противоопухолевый. Таким образом, было синтезировано много аналогов фолата и изучена их способность ингибировать GARFT. Сообщается [F. M. Muggia, "Folate antimetabolites inhibitor to de novo purine synthesis, " New Drugs, concepts and Results in Cancer Chemotherapy, Kluwer Academic Publishers, Boston (1992) , 65-87] , что являющийся прототипом специфический образующий сильную связь ингибитор GARFT, 5,10-дидеазатетрагидрофолиевая кислота, проявляет противоопухолевую активность.
Описаны [PCT/US 94/00418, подана 18.01.94] конденсированные гетероциклические производные глутаминовой кислоты, применяемые в качестве антипролиферативных агентов или ингибиторов GARFT. Настоящее изобретение продолжает эту работу путем дальнейшей разработки полезных антипролиферативных агентов и ингибиторов GARFT.
Резюме
Настоящее изобретение относится к соединениям определенной ниже формулы 1. Эти соединения являются эффективными для ингибирования глицинамид рибонуклеотид формил трансферазы (GARFT) и ингибирования роста и пролиферации клеток высших организмов или микроорганизмов, таких как бактерии, дрожжи и грибы. Данное изобретение далее относится к фармацевтическим композициям, содержащим эти соединения, и применению этих соединений в качестве ингибиторов фермента GARFT.
Как указано выше, соединения по изобретению обладают антипролиферативной активностью, то есть свойством, которое может само выразиться в форме противоопухолевой активности. Соединение по изобретению может быть активно само по себе или выступать в роли предшественника, in vivo превращающегося в активное соединение. Предпочтительные соединения по изобретению являются особенно активными при ингибировании фермента GARFT. Особенно предпочтительные соединения являются активными при ингибировании роста клеточной линии L1210, линии лейкемических клеток мыши, которые могут быть выращены в культуре ткани. Соединения по изобретению также могут быть активными при ингибировании роста бактерий, таких как грамнегативная бактерия Escherichia coli, которая может быть выращена в культуре.
Соединения по изобретению, а также их фармацевтически приемлемые соли могут быть заключены в традиционные дозовые формы, такие как капсулы, таблетки и инъецируемые препараты. Могут быть использованы также твердые или жидкие фармацевтически приемлемые носители, разбавители или эксципиенты.
Твердые носители включают в себя крахмал, лактозу, дигидрат сульфата кальция, гипс, сахарозу, тальк, желатин, агар, пектин, гуммиарабик, стеарат магния и стеариновую кислоту. Жидкие носители включают в себя сироп, арахисовое масло, оливковое масло, солевой раствор и воду.
Носитель или разбавитель может включать в себя любой материал для пролонгированного высвобождения, такой как глицерил моностеарат или глицерил дистеарат, одиночные или с воском. При использовании жидкого носителя препарат может находиться в форме сиропа, элексира, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекции (например раствора) либо неводной или водной жидкой суспензии.
Фармацевтические препараты готовят, следуя традиционной технологии фармацевтической химии, включающей такие этапы как смешивание, гранулирование и прессование (при необходимости) в таблеточные формы, или смешивание, заполнение и растворение ингредиентов подходящим способом для получения желаемых продуктов для перорального, парентерального, местного, интравагинального, интраназального, внутрибронхиального, внутриглазного, внутриушного или ректального применения.
Композиции по изобретению также могут включат в себя одно или несколько фармацевтически активных соединений. Например, в композицию может быть включен один из следующих противоопухолевых агентов: митотические ингибиторы (например винбластин); алкилирующие агенты; ингибиторы дигидрофолат редуктазы или TS ингибиторы; антиметаболиты (например 5-фторурацил, цитозинрабинозид); интеркалирующие антибиотики (например адриамицин, блеомицин); ферменты (например аспарагиназа), ингибиторы топоизомеразы (например этопозид); модификаторы биологического ответа (например интерферон). Композиции по изобретению также могут содержать другой ингибитор GARFT или антипролиферативный агент, в частности известные из WO 94/13295 (опубликовано 23.06.94) или WO 92/05153 (опубликовано 02.04.92). Композиции по изобретению также могут содержать один или несколько антибактериальных, антигрибных, противопаразитных, антивирусных, антипсориатических или антикокцидиальных агентов. Примерами антибактериальных агентов являются сульфонамиды, такие как сульфаметоксазол, сульфадиазин, сульфаметер и сульфадоксин; ингибиторы дигидрофолиевой редуктазы, такие как триметоприм, бромодиаприм и триметрексат; пенициллины; цефалоспорины; и хинолон карбоновые кислоты и их конденсированные с изотиазолом аналоги.
Другой аспект изобретения относится к терапевтическим способам ингибирования роста или пролиферации клеток высших организмов или микроорганизмов, при котором на хозяина (реципиента) воздействуют эффективным количеством соединения по изобретению. Соединения по изобретению, в частности, полезны при лечении млекопитающих хозяев (реципиентов), таких как человек, и при лечении птиц. Особенно предпочтительный терапевтический способ включает в себя воздействие на реципиент эффективным для ингибирования GARFT количеством соединения по изобретению.
Многие описанные здесь антипролиферативные соединения и их фармацевтически приемлемые соли могут быть использованы в терапевтическом способе по изобретению. Эти соединения могут быть применены в форме фармацевтически приемлемой композиции, содержащей разбавитель или носитель, как описано выше.
Доза композиции содержит по меньшей мере эффективное количество активного соединения и предпочтительно состоит из одной или нескольких фармацевтических дозовых единиц. "Эффективное количество" обозначает количество, достаточное для ингибирования метаболических путей фолата, и приводит к следующему из этого благоприятному эффекту при применении, например, одной или нескольких фармацевтических дозовых единиц.
Примерная суточная доза для позвоночных содержит до 1 г активного соединения на 1 кг веса тела реципиента, предпочтительно 0,5 г, более предпочтительно 100 мг и наиболее предпочтительно около 50 мг и ниже. Выбранной дозой можно воздействовать на теплокровного животного или млекопитающего, например человека, нуждающегося в лечении, осуществляемом через ингибирование метаболических путей фолата, с помощью любого подходящего способа применения такой дозы, включая местное, например в виде мази или крема; пероральное; ректальное, например в виде суппозитория; парентеральное путем инъекции; или непрерывное путем интравагинальной, интраназальной, внутрибронхиальной, внутриушной или внутриглазной инфузии.
Соединения по изобретению дают какой-либо один или несколько эффектов из числа следующих: антипролиферативный эффект, антибактериальный эффект, противопаразитный эффект, антивирусный эффект, антипсориазный эффект, антипротозойный эффект, антикокцидиальный эффект, противовоспалительный эффект, иммуносупрессивный эффект и антигрибной эффект. Эти соединения особенно полезны при получении противоопухолевого эффекта у позвоночных, имеющих опухоли.
Детальное описание изобретения
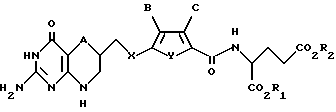
Данное изобретение относится, конкретно, к антипролиферативным производным глутаминовой кислоты формулы I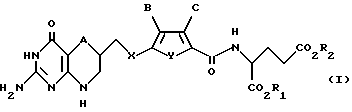
где A обозначает серу;
X обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород;
R1 и R2 каждый независимо обозначает водород или низший алкил.
Предпочтительно соединение, в котором R1 и R2 каждый независимо выбран из водорода и этила.
Более предпочтительно соединение, в котором R1 и R2 каждый является водородом.
Наиболее предпочтительными соединениями являются
диэтиловый эфир (4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н- пиримидо[5,4-b] [1,4]-тиазин-6-ил)-этил]-2,5-тиеноиламино-L-глутаминовой кислоты); и
4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидо[5,4-b][1,4]- тиазин-6-ил)-этил]-2,5-тиеноиламино-L-глутаминовой кислоты.
Данное изобретение, в частности, относится к производному глутаминовой кислоты формулы: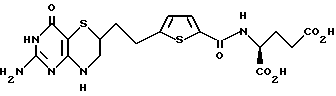
Это соединение обладает свойствами ингибитора роста и пролиферации клеток.
Предпочтительны соединения, имеющее следующую структуру:
и
Данное изобретение относится также к фармацевтической композиции, обладающей свойством ингибировать рост и пролифирацию клеток, включающая активное начало и фармацевтически приемлемый носитель, разбавитель или эксципиент, содержащей в качестве активного начала эффективное количество указанного выше соединения.
Также предложен способ ингибирования роста или пролиферации клеток микроорганизмов или высших организмов, при котором на реципиента, млекопитающего или птицу, воздействуют эффективным количеством указанного выше соединения.
Согласно данному изобретению предложены также следующие промежуточные соединения для получения указанного выше соединения.
Соединение формулы VI: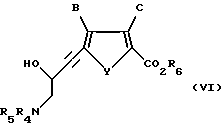
где Y обозначает серу;
B обозначает водород;
C обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу; и
R4 и R5 обозначает водород или легко удаляемую азотозащитную группу.
Предпочтительно, когда R6 является низшим алкилом; и R4 и R5 независимо представляют собой водород или трет-бутоксикарбонил.
Наиболее предпочтителен 4-N-(трет-бeтоксикарбонил)-3-гидрокси- 1-(2-карбометокси-5-тиофен)-бутин.
Соединение формулы VII''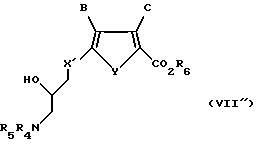
где X' обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу; и
R4 и R5 обозначает водород или легко удаляемую азотозащитную гpуппу.
Предпочтительно, когда R6 является низшим алкилом; и R4 и R5 независимо представляют собой водород или трет-бутоксикарбонил.
Наиболее предпочтителен метил-5-[(4-N-(трет-бутоксикарбонил)- амино-3-гидрокси)-бутил]-тиенил-карбоксилат.
Соединение формулы VIII''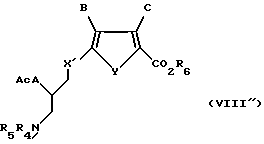
где X' обозначает CH2;
A обозначает серу;
Y обозначает серу;
B обозначает водород;
C обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу;
R4 и R5 обозначают водород или легко удаляемую азотозащитную группу; и
Ac обозначает низший ацил.
Предпочтительно, когда R6 является низшим алкилом; и R4 и R5 независимо представляют собой водород или трет-бутоксикарбонил.
Наиболее предпочтителен метил-5-[(4-N-(трет-бутоксикарбонил)- амино-3-ацетилтио)-бутил]-2-тиофен-карбоксилат.
Соединение формулы IX'':
где A обозначает серу;
X' обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую гриппу; и
R4 и R5 обозначают водород или легко удаляемую азото-защитную гриппу.
Предпочтительно, когда R6 является низшим алкилом; и R4 и R5 независимо представляют собой водород или трет-бутоксикарбонил.
Наиболее предпочтителен метил-5-[(4-N-(трет-бутоксикарбонил)- амино-З-(диметилмалонил)тио)-бутил]-2-тиофен-карбоксилат.
Соединение формулы X'':
где A обозначает серу;
X' обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород; и
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу.
Предпочтительно, когда R6 является низшим алкилом.
Наиболее предпочтителен 6-[(5-карбометокситиен-2-ил)-этил]-2- карбометокси-3-оксо-3,4,5,6-тетрагидро-[1,4]-тиазин.
Соединение формулы XI''
где A обозначает серу;
X' обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород; и
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу.
Предпочтительно, когда R6 является низшим алкилом.
Наиболее предпочтителен метил-[2-(2-амино-4-оксо-4,6,7,8- тетрагидро-3Н-пиримидо [5,4-b][1,4]тиазин-6-ил)-этил]-2,5-тиеноат.
Соединение формулы XII''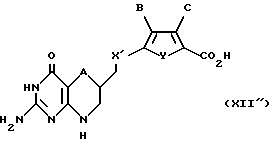
где A обозначает серу;
X' обозначает CH2;
Y обозначает серу;
B обозначает водород;
C обозначает водород.
Предпочтительна [2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н- пиримидо[5,4-b][1,4]тиазин-6-ил)-этил]-2,5-тиеновая кислота.
Хотя соединения формулы 1 и показаны в 4-оксоформе и именно о ней идет речь в данном описании, оксогруппа все же существует в таутомерном равновесии с соответствующей 4-гидроксигруппой. Поэтому должно быть понятно, что "соединения формулы I" включают в себя как изображенные на рисунках 4-оксо, так и таутомерные 4-гидроксиформы. Таким образом, изобретение также относится к фармацевтически приемлемым солям 4-гидрокситаутомеров соединения, изображаемого формулой I.
Соединения формулы I применимы в качестве ингибиторов GARFT. Соединения формулы I, в которой R1 и R2 каждый обозначает водород, являются особенно активными противоопухолевыми и антипролиферативными агентами. Соединения формулы I, в которой R1 и R2 каждый является остатком, который образует легко гидролизуемую эфирную группу с присоединенным CO2 предпочтительно этильную группу, являются полезными промежуточными соединениями для образования форм данного соединения со свободной глутаминовой кислотой и могут быть также гидролизованы in vivo и служить, таким образом, в качестве пролекарств.
Соединения по изобретению могут быть получены, как описано ниже. Для получения соединений формулы I, где X является CH2, в качестве исходного вещества подходит соединение формулы II: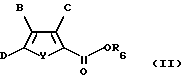
где B, C и Y такие, как определено в формуле I;
D обозначает Cl, Br или I;
R6 обозначает водород или группу, которая образует с присоединенным CO2 легко гидролизуемую эфирную группу. В предпочтительном случае D является бромом или иодом. R6 в предпочтительном случае обозначает водород, С1-С6 алкил, гидроксиалкил, алкиларил или арилалкил, наиболее предпочтительно водород или С1-С2 алкил.
Соединение формулы II подвергают взаимодействию с соединением формулы III:
где R3 является замещенной или незамещенной C1-C6 алкильной группой или тризамещенной силильной группой. Предпочтительно, когда R3 является CH2OH или триметилсилилом.
Взаимодействие соединений формулы II и III проводят в присутствии подходящего катализатора из переходного металла, предпочтительно палладия или никеля, ненуклеофильного вспомогательного основания, предпочтительно замещенного амина, в растворителе, в котором по меньшей мере один из реактантов по меньшей мере частично растворим при условиях, подходящих для получения соединения формулы IV: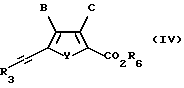
где B, C и Y такие, как определено в формуле I;
R6 такое, как определено в формуле II;
R3 такое, как определено в формуле III.
Когда R3 является тризамещенной силильной группой, силильную группу предпочтительно удаляют с помощью нуклеофильного основания, такого как метанольный или этанольный карбонат калия, или соли фтора, такой как фторид калия, фторид цезия или фторид тетрабутиламмония, в растворителе, в котором по меньшей мере один из реактантов по меньшей мере частично растворим (например метаноле, диметилформамиде, этаноле, диметилацетамиде, диметилсульфоксиде или изопропаноле) с получением соединения формулы V: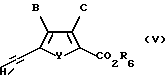
где B, C и Y такие, как определено выше для формулы I, и
R6 такое, как определено в формуле II.
Соединение формулы V подвергают взаимодействию с электрофилом, предпочтительно N-защищенным глициналем, более предпочтительно N-трет-бутоксикарбонилглициналем или бис-N-трет-бутоксикарбонилглициналем, в основных условиях с использованием ненуклеофильного основания, предпочтительно бис-триметилсилиламида лития, бис-триметилсилиламида калия, бис-триметилсилиламида натрия или диизопропиламида лития, при пониженной температуре, предпочтительно от -90oC до 25oC, в подходящем растворителе, в котором один из реактантов по меньшей мере частично растворим, предпочтительно тетрагидрофуране, диэтиловом эфире или диоксане, с получением соединения формулы VI: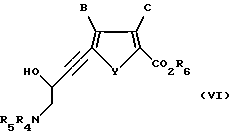
где B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II;
R4 и R5 каждый независимо выбран из водорода или легко удаляемой азотозащитной группы.
В предпочтительном случае R4 и R5 представляют собой водород, трет-бутоксикарбонил, бензилоксикарбонил или бензил.
Затем соединение формулы VI восстанавливают, предпочтительно газообразным водородом в присутствии подходящего металлического катализатора, с получением соединения формулы VII: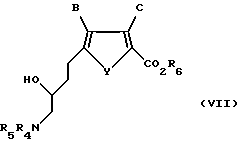
где B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II;
R4 и R5 такие, как определено выше для формулы VI.
Соединение формулы VII подвергают взаимодействию с ацилирующим или сульфонилирующим агентом, предпочтительно метансульфонилхлорида или р-толуолсульфонил хлорида, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, в подходящем растворителе, в котором по меньшей мере один из реактантов по меньшей мере частично растворим, с получением активированной гидроксигруппы. Эту активированную гидроксигруппу замещают подходящим нуклеофилом, предпочтительно тиоуксусной солью, более предпочтительно тиоацетатом калия, с получением соединения формулы VIII: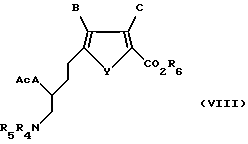
где A, B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II;
R4 и R5 такие, как определено выше для формулы VI;
Ac обозначает ацильную группу, предпочтительно ацетил.
В альтернативном случае соединение формулы VII превращают в соединение формулы VIII в одну химическую стадию, используя трифенилфосфин, диэтиловый или диметиловый эфир азадикарбоновой кислоты, и кислотный нуклеофил, предпочтительно тиоуксусную кислоту, в подходящем растворителе.
Соединение формулы VIII обрабатывают нуклеофильным основанием, предпочтительно карбонатом калия, карбонатом натрия, гидроксидом натрия или гидроксидом калия, в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, в присутствии алкилирующего агента, предпочтительно диметилового или диэтилового эфира хлормалоновой кислоты, с получением соединения формулы IX: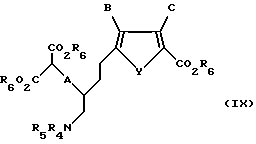
где A, B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II;
R4 и R5 такие, как определено выше для формулы VI.
Соединение формулы IX обрабатывают в условиях, подходящих для удаления либо одной, либо обеих защитных групп R4 и R5, с получением соединения формулы X: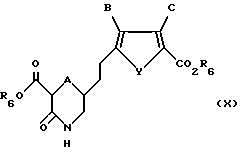
где A, B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II.
Если защитной группой является трет-бутокси-карбонил (t-BOC), то условиями для удаления этой группы является обработка трифторуксусной кислотой с последующей нейтрализацией с получением соединения формулы X.
Соединение формулы X подвергают взаимодействию с алкилирующим агентом, предпочтительно триметил или триэтил оксоний тетрафторборатом, в подходящем растворителе, предпочтительно дихлорметане, с образованием промежуточного лактимного эфира. Промежуточный лактимный эфир подвергают взаимодействию с гуанидином в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с получением соединения формулы XI: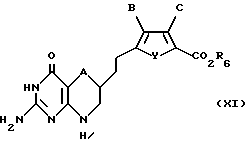
где A, B, C и Y такие, как определено выше для формулы I;
R6 такое, как определено выше для формулы II.
В альтернативном случае соединение формулы X превращают в соединение формулы XI путем реакции соединения формулы X с тиолирующим агентом, предпочтительно P2S5 или 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидом, с образованием тиолактамного промежуточного соединения. Затем это соединение можно алкилировать с помощью алкилирующего агента, предпочтительно метилиодида или триметил или триэтил оксоний тетрафторбората, а затем гуанидина в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с получением соединения формулы XI. Соединение формулы XI гидролизуют в основных условиях с получением соединения формулы XII:
где A, B, C и Y такие, как определено выше для формулы I.
Если R6 в формуле XI является водородом, то реакция гидролиза не является необходимой, и соединение формулы XI сочетают пептидной связью как описано ниже.
Соединение формулы XVII (или соединение формулы XI, где R6 является водородом), находящееся в форме свободной карбоновой кислоты, может быть соединено пептидной связью (с помощью известных из уровня техники средств) с гидрохлоридом диэфира глутаминовой кислоты с получением диэфира формулы XIII: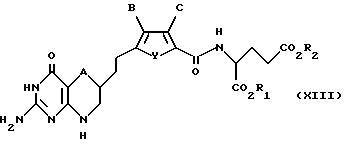
где A, B, C и Y такие, как определено выше для формулы I;
R1 и R2 каждый независимо является группировкой, которая образует с присоединенным CO2 легко гидролизуемую эфирную группу, такую как C1-C6 алкил, гидроксиалкил, алкиларил или арилалкил.
И наконец, при желании, соединение формулы XIII гидролизуют до образования формы свободной глутаминовой кислоты соединения формулы I (R1 и R2 каждый является водородом).
Соединения по изобретению содержат один или несколько хиральных центров. Данное изобретение охватывает рацемические смеси и смеси диастереоизомеров, а также оптически активные соединения, такие, как соединения, по существу свободные от других оптических изомеров, эти оптически активные соединения могут быть получены известными из уровня техники средствами.
Детальные примеры получения предпочтительных соединений формулы I приведены ниже.
Пример 1
4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидо[5,4-b] [1,4] тиазин- 6-ил)этил]-2,5-тиеноиламино-L-глутаминовая кислота (соединение 1)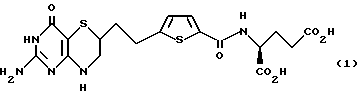
Получение метил-5-бром-2-тиофенкарбоксилата 2: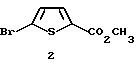
Соединение 2 (CAS регистр. номер [62224-19-5]) получают известным [S. Gronowitz, Ark. Kemi 8, 1955, 87 и S.O.Lawesson, Ark.Kemi 11, 1957, 337] способом.
Получение 5-этинил-2-карбометокси тиофена 3:
К перемешиваемому раствору 5 г (22,7 ммоль) бромида 3, 160 мг (0,23 ммоль) трис-трифенилфосфина хлорида палладия (II) и 65 мг (0,34 ммоль) иодида меди в 75 мл диэтиламина добавляют 4,8 мл (34 ммоль) триметилсилил ацетилена. Полученный раствор перемешивают при 25oC. Через 18 ч растворитель удаляют при пониженном давлении и необработанный остаток растворяют в 300 мл диэтилового эфира (Et2O) и экстрагируют 100 мл 1N HCl и 100 мл насыщенного раствора бикарбоната. Органический слой высушивают (Na2SO4) и растворитель удаляют при пониженном давлении. Необработанный остаток подвергают флэш-хроматографии на силикагеле со смесью 5% Et2O/гексан с получением 4,4 г (81% выход) силилзащищенного продукта в виде желтого твердого вещества. Этот материал непосредственно используют на следующей стадии.
К раствору 4,4 г (18,5 ммоль) упомянутого выше силилзащищенного ацетилена в 50 мл диметилформамида (DMF) добавляют 3,3 г (35,2 ммоль) моногидрата фторида калия. Через 15 мин выдерживания при 25oC смесь выливают в 500 мл Et2O и экстрагируют 4 раза 100 мл воды (4 х 100 мл H2O). Органический раствор высушивают (Na2SO4) и удаляют растворитель при пониженном давлении. Необработанный остаток подвергают флэш-хроматографии на силикагеле со смесью 10% Et2O/гексан с получением 2,65 г (86% выход) желаемого продукта 3 в виде твердого оранжевого вещества.
ЯМР (CDCl3) δ: 3,45 (s, 1H), 3,88 (s, 3H), 7,22 (d, 1H, J = 3,5 Гц), 7,65 (d, 1H, J = 3,5 Гц).
Анал. Вычисл. для C8H6SO2: C, 57,81; H, 3,64; S, 19,29.
Найдено: C, 57,91; H, 3,71; S, 19,18.
Может быть использован альтернативный способ удаления силильной группы. К перемешиваемой суспензии 3,7 г (0,02 моль) безводного K2CO3 в 700 мл метанола добавляют 59,7 г приготовленного ранее силил-ацетилена в виде твердого вещества. Суспензию перемешивают в течение 2 ч при 35oC, а затем выпаривают метанол. Остаток добавляют к 50 мл воды и смесь экстрагируют 3 х 150 мл Et2O, объединенные органические фракции высушивают (Na2SO4) и выпаривают. Необработанный остаток подвергают хроматографии на флэш силикагеле, используя смесь 7% Et2O/гексан с получением желаемого продукта 3 с 98% выходом (38,72 г).
Получение N-(трет-бутоксикарбонил)-глициналя 4: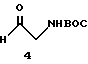
К перемешиваемому раствору 2 г (12,4 ммоль) N-(трет-бутоксикарбонил)- 2-гидрокси-этиламина (полученного от Sigma Chemicals) в 80 мл CH2Cl2 при -78oC добавляют 1,3 мл (18,6 ммоль) диметил сульфоксида (DMSO) и 1,2 мл (13,7 ммоль) оксалил хлорида. Через 5 мин добавляют 5,4 мл (38,4 ммоль) триэтиламина и позволяют раствору нагреться до 25oC. Смесь выливают в 200 мл Et2O и экстрагируют 100 мл воды, 100 мл 0,5 N HCl и 100 мл насыщенного раствора бикарбоната. Органические фракции высушивают (Na2SO4) и удаляют растворитель при пониженном давлении. Необработанный альдегид растворяют в 100 мл бензола и снова удаляют растворитель при пониженном давлении. Этот необработанный материал, который оказался нестабильным, немедленно используют на следующей стадии.
Получение 4-N-(трет-бутоксикарбонил)-3-гидрокси-1-(2-карбометокси- 5-тиофен)-бутина 5:
К раствору 1,7 г (10,2 ммоль) ацетилена 3 в 10 мл сухого тетрагидрофурана (THF) при -78oC добавляют 14,3 мл (14,0 ммоль) 1М раствора бис-триметилсилила лития в THF. Через 10 мин необработанный ранее приготовленный альдегид 4 по каплям добавляют в 5 мл сухого THF. Раствор перемешивают при -78oC в течение 10 мин и позволяют нагреться до 0oC за 20 мин. Добавляют к нему 5 мл насыщенного водного раствора хлорида аммония. Смесь выливают в 200 мл Et2O и промывают 100 мл H2O и затем 50 мл насыщенного раствора NaCl. Органический слой высушивают (Na2SO4) и удаляют растворитель при пониженном давлении. Необработанный остаток подвергают флэш-хроматографии на силикагеле со смесью 10-40% EtOAc/гексан с получением 0,96 г (29% выход) желаемого спирта 5 в виде слегка желтого масла.
ЯМР (CDCl3) δ: 1,46 (s, 9H), 3,38 и 3,58 (AB, 2H, J = 3, 7 Гц), 3,88 (s, 3H), 4,71 (dt, 1H, J = 2,9, 5,2 Гц), 5,05 (brs, 1H), 7,15 (d, 1H, J = 3,9 Гц), 7,64 (d, 1H, J = 3,9 Гц).
ИК (чистый): 2978,3, 1729,5, 1685, 1523, 1452, 1368, 1286, 1167, 1098, 959, 822, 750,4 см-1.
Масс спектрометрия высокого разрешения:
Вычисл. для C15H19NO5S: M+Cs+, 458,0038. Найдено: 458,0051.
Получение метил-5-[(4-N-(трет-бутоксикарбонил)-амино-З-гидрокси)- бутил] -тиенил-2-карбоксилата 6: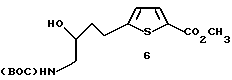
К перемешиваемому раствору 940 мг (2,89 ммоль) полученного ацетилена 5 в 50 мл этилацетата (EtOAc) добавили 320 мг 5% Pd/C. Смесь поместили в 40 пси газообразного водорода и перемешивали при 25oC в течение 17 ч. Смесь фильтровали через Celite (диатомированная земля) и удаляли растворитель при пониженном давлении. Необработанный остаток подвергали флэш-хроматографии на силикагеле со смесью 20% EtOAc/CH2Cl2 с получением 800 мг (84% выход) желаемого спирта в виде желтого масла.
ЯМР (CDCl3) δ: 1,44 (s, 9H), 1,81 (m, 2H), 2,28 (brs, 1H), 2,99 (m, 2H), 3,10 и 3,29 (AB, 2H, J = 3, 6,9 Гц), 3,74 (m, 1H), 3,86 (s, 3H), 4,89 (brs, 1H), 6,82 (d, 1H, J = 3,7 Гц), 7,63 (d, 1H, J = 3,7 Гц).
Анал. Вычисл. для C15H23NO5S: C, 54,69: H, 7,04; N, 4,25; S 9,73.
Найдено: C, 54,79; H, 7,02; N, 4,29; S, 9,63.
Возможен следующий альтернативный способ получения спирта 6 с использованием бис-трет-бутоксикарбонил аллиламина 14. Получение бис-трет-бутоксикарбонил аллиламина 14.
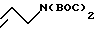
К раствору 57,10 г (1,0 моль) аллиламина и 1,2 г (0,01 моль) диметиламинопиридина (DMAP) в 500 мл ацетонитрила добавляют раствор 220 г (1 моль) (t-BOC)2O в 100 мл ацетонитрила и полученную смесь перемешивают в течение 6 ч. Реакционную смесь разбавляют толуолом (100 мл) и выпаривают растворители при пониженном давлении при 60oC. Полученное масло повторно растворяют в ацетонитриле (400 мл) и добавляют еще 1,2 г (0,01 моль) DMAP, к этой смеси медленно добавляют раствор 220 г (1 моль) (t-BOC)2O в 100 мл ацетонитрила. Реакционную смесь перемешивают в течение 12 ч при 60oC, растворитель выпаривают при пониженном давлении при 60oC и добавляют NaHCO3 (100 мл). Эту смесь экстрагируют 3 х 150 мл CH2Cl2, объединенные органические слои промывают рассолом, высушивают (Na2SO4) и выпаривают. Необработанный остаток очищают хроматографически на флэш-силикагеле, элюируя градиентом 5-20% EtOAc/гексан с получением 156,9 г (63% выход) желаемого продукта 14 в виде прозрачного кристаллического твердого вещества (т.пл. 43-44oC).
1ЯМР (CDCl3) δ: (ppm): 1,5 (s, 18H), 4,18 (dd, 2H, J = 15 Гц, J = 1 Гц, 5,14 (ddd, 2H, J = 15 Гц, J = 10 Гц, J = 1 Гц), 5,85 (ddt, 2Н, J = 10 Гц, J = 5 Гц, J = 1 Гц).
ИК (КВг): 2978, 2935, 2860, 1724, 1689, 1342, 1130 см-1.
Анал. Вычисл. для C13H23NO4: C, 60,68; H, 9,01; N, 5,44;
Найдено: C, 60,78; H, 9,04; N, 5,50.
Получение 1-(бис-трет-бутоксикарбониламино)-2-этаналя 15: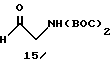
Раствор 0,60 г (2,34 ммоль) бис-трет-бутоксикарбонил аллиламина 14 в 20 мл CH2Cl2 озонировали (40 вольт, 500 ампер, 1,0 л/мин O2 @ 3 пси) при -78oC пока сохраняется голубой цвет, добавляют диметилсульфид и смесь перемешивают в течение 14 ч при 25oC. Смесь добавляют к солевому раствору и экстрагируют CH2Cl2 (3 х 50 мл), высушивают органические фракции (Na2SO4) и выпаривают. Хроматография с использованием флэш-силикагеля дает 603 мг (99% выход) желаемого продукта 15 в виде прозрачного кристаллического твердого вещества (т.пл. 37-39oC).
1ЯМР (CDCl3) δ: (ppm): 1,50 (s, 18H), 4,38 (s, 2H), 9,55 (s, 1H).
ИК (KBr): 2984, 2935, 2724, 1792, 1734, 1699, 1362, 1153 см-1.
Анал. Вычисл. для C12H21NO5: C, 55,58; H, 8,16; N, 5,40.
Найдено: C, 55,20; H, 8,19; N, 5,19.
Получение метил-5-[(4-N-(трет-бутоксикарбонил)-амино-3-гидрокси)- бутил] -тиенил-2-карбоксилата 6:
К раствору 9,64 г (59 ммоль) 5-этинил-2-карбометокси тиофена 3 в THF (250 мл) при -78oC добавили 65,6 мл (60 ммоль, 0,9М) гексаметил дисилазида лития (LiHMDS) и перемешивали смесь в течение 2 ч при -78oC. Раствор 15,6 г (60 ммоль) 1-(бис-трет-бутоксикарбониламино)-2-этаналя 15 в THF (40 мл) добавляли через канюлю в реакционную смесь и смесь перемешивали в течение 8 ч при -78oC. Раствор 3,4 мл (60 ммоль) уксусной кислоты в метаноле (10 мл) добавляли для остановки реакции, смесь перемешивали в течение 10 мин, нагревая до 0oC, и добавляли воду (60 мл). Эту смесь экстрагировали EtOAc (3 х 100 мл) и органические экстракты промывали NaHCO3 и высушивали (Na2SO4). Растворитель удаляли при пониженном давлении до около 50 мл. К полученному остатку добавляли EtOAc (125 мл) и этот раствор добавляли в сосуд Парра, содержащий 7,38 г (30 вес.% ацетиленового исходного материала) 5% Pd на угле и гидрировали при 55 пси H2 в течение 24 ч. Необработанный продукт гидрогенизации фильтровали через Celite и осадок с фильтра промывали метанолом (100 мл) и EtOAc (100 мл). Растворитель выпаривали при пониженном давлении, необработанный остаток фильтровали через диоксид кремния, элюируя 50% EtOAc в гексане, и выпаривали растворитель. Остаток сольватировали в 20 мл метанола (MeOH), азеотропированного с бензолом (50 мл), и повторно растворяли в сухом MeOH (20 мл). Эту смесь добавляли к свежеприготовленному раствору 60 мл (2М) метоксида натрия в метаноле. Реакционную смесь перемешивали в течение 45 мин, добавляли HCl (0,1М, 5 мл) и экстрагировали эту смесь EtOAc (3 х 75 мл). Объединяли органические фракции и промывали буфером pH 7 (1М), высушивали (Na2SO4) и выпаривали. Необработанный остаток подвергали хроматографии на флэш-силикагеле, элюируя 40% EtOAc/гексан с получением 9,32 г (48% выход) желаемого продукта 6.
Получение метил-5-[(4-N-(трет-бутоксикарбонил)-амино-З-ацетилтио)- бутил]-2-тиофенкарбоксилата 7: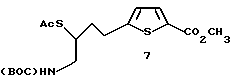
К перемешиваемому раствору 9,26 г (28,1 ммоль) спирта 6 в 100 мл THF при 0oC добавляли 5,9 мл (42 ммоль) триэтиламина (TEA) и 2,4 мл (31 ммоль) метансульфонил хлорида. Через 20 мин добавляли 100 мл DMF и 12,8 г (110 ммоль) тиоацетата калия, полученному раствору позволяли нагреться до 25oC. Через 3 дня смесь выливали в 500 мл воды и экстрагировали 800 мл Et2O. Органический слой промывали 200 мл воды, 200 мл 1N HCI, 200 мл насыщенного раствора бикарбоната и 100 мл насыщенного раствора NaCl. Органический слой высушивали (MgSO4) и растворитель удаляли при пониженном давлении. Необработанный остаток подвергали флэш-хроматографии на силикагеле со смесью 25% EtOAc/гексан с получением 10,3 г (95% выход) желаемого тиоацетата 7 в виде желтого масла.
ЯМР (CDCl3) δ: 1,44 (s, 9H), 1,91 и 2,04 (ABm, 2H), 2,37 (s, 3H), 2,96 (m, 2H), 3,36 (m, 2H), 3,61 (m, 1H), 3,86 (s, 3H), 4,74 (brs, 1H), 6,79 (d, 1H, J = 3,6 Гц), 7,62 (d, 1H, J = 3,6 Гц).
ИК (чистый): 3366, 2976,4, 1713, 1520, 1462,1, 1366, 1290,5, 1267,3, 1169, 1098, 752, 631 см-1.
Масс спектрометрия высокого разрешения: Вычисл. для C17H25NO5S2: M+Cs+, 520,0229. Найдено: 520,0240.
Получение метил-5-[(4-N-(трет-бутоксикарбонил)-амино-3- (диметилмалонил)тио)-бутил]-2-тиофенкарбоксилата 8: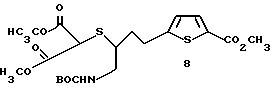
К перемешиваемому раствору 10,2 г (26 ммоль) тиоацетата 7 в 200 мл сухого метанола при 0oC добавляют 7,2 г (52 ммоль) K2CO3 и 3,7 мл (29 ммоль) диметил хлормалоната. Через 3 ч смесь вылили в 500 мл воды и экстрагировали Et2O (3 х 500 мл). Объединенный органические слои промывали 500 мл воды и затем 200 мл насыщенного раствора NaCl и высушивали (MgSO4). Растворитель удаляли при пониженном давлении и необработанный остаток подергали флэш-хроматографии на силикагеле со смесью 30% EtOAc/гексан с получением 11,46 г (93% выход) желаемого тиоэфира 8 в виде слегка желтого масла.
ЯМР (CDCl3) δ: 1,44 (s, 9H), 1,85 и 2,00 (ABm, 2H), 3,03 (s, 3H), 3,30 (m, 2H), 3,80 (s, 6H), 3,86 (s, 3H), 5,10 (brs, 1H), 6,82 (d, 1H, J = 3,8 Гц), 7,63 (d, 1H, J = 3,8 Гц).
ИК (чистый): 3442, 2974, 2955, 1734, 1713, 1512, 1460, 1435, 1366, 1291, 1267, 1167, 1098, 1022, 752 см-1.
Масс спектрометрия высокого разрешения:
Вычисл. для C20H29NO8S2: M+Cs+, 608,0389. Найдено: 608,0370.
Получение 6-[(5-карбометокситиен-2-ил)-этил] -2-карбометокси-3- оксо-3,4,5,6-тетрагидро-[1,4]-тиазина 9:
К перемешиваемому раствору 470 мг (0,99 ммоль) тиоэфира 8 в 12 мл CH2Cl2 при 0oC добавили 4 мл трифторуксусной кислоты (TFA). Через 1,5 ч смесь вылили в 50 мл насыщенного раствора бикарбоната и экстрагировали CH2Cl2 (4 х 100 мл). Растворитель удалили при пониженном давлении и необработанный остаток растворили в 10 мл метанола. После перемешивания при 25oC в течение 1,5 ч растворитель удалили при пониженном давлении и необработанный остаток подвергли флэш-хроматографии на силикагеле со смесью 40% EtOAc/CH2Cl2 с получением 298 мг (88% выход) желаемого лактама 9 в виде бесцветного масла.
ЯМР (CDCl3) δ: 1,94 (m, 2H), 3,01 (m, 2H), 3,4-3,7 (m, 4H), 3,80 и 3,82 (s, 3H), 3,87 (s, 3H), 6,25 (brs, 1H), 6,83 (d, 1H, J = 3,7 Гц), 7,64 (d, 1H, J = 3,7 Гц).
ИК (чистый): 2951, 1732, 1669, 1462, 1294, 1267, 1194, 1157, 1098, 1005, 752 см-1.
Анал. Вычисл. для C14H17NO5S2: С, 48,96; H, 4,99; N, 4,08, S, 18,62.
Найдено: C, 49,06; H, 4,93; N, 4,09, S, 18,60.
Получение метил-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н- пиримидо[5,4-b][1,4]тиазин-6-ил)-этил]-2,5-тиеноат 10: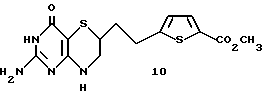
К перемешиваемому раствору 1,9 г (5,5 ммоль) лактама 9 в 150 мл сухого CH2Cl2 одной порцией добавили 1,06 г (7,2 ммоль) тетрафторбората триметилоксония. Раствор перемешивали в течение 6 ч при 25oC. Полученную смесь вылили в 50 мл 10% водного K2CO3 и экстрагировали CH2Cl2 (3 х 200 мл). Объединенные органические слои высушивали (MgSO4) и удаляли растворитель при пониженном давлении. Полученный материал непосредственно использовали на следующей стадии.
В отдельную колбу поместили 1,58 г (16,6 ммоль) сухого гуанидин гидрохлорида. Добавили туда 600 мл сухого метанола. Сухой аргон пропускали через этот раствор в течение 10 мин, после чего добавили 926 мг (17,1 ммоль) сухого метоксида натрия. К полученной смеси через канюлю добавили необработанный лактимный эфир, полученный ранее, в 20 мл MeOH. Раствор кипятили с обратным холодильником в атмосфере аргона в течение 20 ч. Подвели pH до 4 с помощью 1N HCl и удалили растворитель при пониженном давлении. Необработанный остаток растворили в 500 мл CHCl3 и промыли 2 х 100 мл воды. Органический слой высушили (MgSO4) и удалили растворитель при пониженном давлении. Необработанный остаток подвергли флэш-хроматографии на силикагеле с 5-10% MeOH/CH2Cl2 с получением 413 мг беловатого твердого вещества. Этот материал затем растворили в 25 мл горячего MeOH и медленно охладили до 4oC за 18 ч. Твердое вещество собрали путем фильтрования с получением 180 мг (9% выход) желаемого пиримидинона 10 в виде слегка желтого твердого вещества.
ЯМР (d6-DMSO) δ: 1,72 (m, 1H), 1,88 (m, 1H), 2,8-3,22 (m, 4Н), 3,50 (dt, 1H, J = 2, 8 Гц), 5,99 (brs, 2H), 6,64 (brs, 1H), 6,98 (d, 1H, J = 3,8 Гц), 7,62 (d, 1H, J = 3,8 Гц), 10,02 (brs, 1H).
Масс спектрометрия высокого разрешения:
Вычисл. для C14H16N4O3S2: M+Na+, 375,0562. Найдено: 375,0550.
Может быть также использован следующий альтернативный способ получения пиримидинона 10. К перемешиваемому раствору 500 мг (1,46 ммоль) лактама 9 в 20 мл сухого THF добавили 648 мг (1,60 ммоль) реагента Лавессона (2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан- 2,4-дисульфид). После того, как эту смесь перемешивали при 25oC в течение 20 ч, ее выливают в 50 мл насыщенного раствора бикарбоната и экстрагируют 2 х 200 мл EtOAc. Объединенные органические слои промывают 50 мл насыщенного раствора NaCl и высушивают (MgSO4) и удаляют растворитель при пониженном давлении. Необработанный тиолактам подвергают флэш-хроматографии на силикагеле с 8% EtOAc/CH2Cl2 с получением 525 мг соответствующего тиолактама 11:
Этот материал используют немедленно на следующей стадии.
К 525 мг тиолактама 11 в смеси 10 мл сухого THF и 10 мл сухого метанола добавляют 1,6 мл 1N NaOH. К этой смеси добавляют 0,1 мл (1,6 ммоль) метил иодида для получения метилированного тиолактама, который используют как указано ниже, без очистки. В отдельную колбу помещают 1,39 г (14,6 ммоль) сухого гуанидин гидрохлорида. К нему добавляют 300 мл сухого метанола. Сухой аргон пропускают через этот раствор в течение 10 мин, после чего добавляют 796 мг (14,7 ммоль) сухого метоксида натрия.
К полученной смеси через канюлю добавляют необработанный метилированный тиолактам, полученный ранее. Этот раствор кипятят с обратным холодильником в атмосфере аргона в течение 48 ч. Подводят pH до 4 с помощью 1N HCl и при пониженном давлении удаляют растворитель. Необработанный остаток растворяют в 500 мл CHCl3 и промывают 2 х 100 мл воды. Органический слой высушивают (MgSO4) и удаляют растворитель при пониженном давлении. Необработанный остаток подвергают флэш-хроматографии на силикагеле с 5-10% MeOH/CH2Cl2. Этот материал затем растворяют в 20 мл горячего MeOH и медленно охлаждают до 4oC за 18 ч. Твердое вещество собирают путем фильтрации с получением 131 мг (9% выход) желаемого пиримидинона 10.
Получение [2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н- пиримидо[5,4-b][1,4] тиазин-6-ил)-этил]-2,5-тиеновой кислоты 12: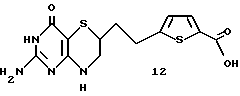
К 180 мг (0,51 ммоль) пиримидинона 10 добавляют 5 мл 1N NaOH в воде. Полученный раствор перемешивают при 25oC в течение 20 ч. После охлаждения до 0oC подводят pH до 2 с помощью 1N HCl. Твердое коричневое вещество отфильтровывают и промывают водой и высушивают под вакуумом с получением 111 мг (64% выход) желаемой кислоты 12 в виде слегка коричневого твердого вещества.
ЯМР (d6-DMSO) δ: 1,72 (m, 1H), 1,92 (m, 1H), 2,78-3,68 (m), 6,07 (brs, 2Н), 6,68 (brs, 1H), 6,92 (d, 1H, J = 3,7 Гц), 7,52 (d, 1H, J = 3,7 Гц), 10,02 (brs, 1H), 12,89 (brs, 1H).
Масс спектрометрия высокого разрешения:
Вычисл. для C13H14N4O3S2: M+, 338,0507. Найдено: 338,0517.
Получение диэтилового эфира 4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро- 3Н-пиримидо[5,4-b] [1,4] тиазин-6-ил)-этил] -2,5-тиеноиламино-L-глутаминовой кислоты 13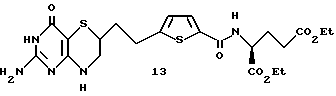
К перемешиваемому раствору 110 мг (0,33 ммоль) полученной ранее кислоты 12 в 5 мл сухого DMF добавляют 48 мг (0,36 ммоль) гидрата 1-гидрокси-бенэотриазола, 62 мкл (0,36 ммоль) диизопропилэтиламина, 86 мг (0,36 ммоль) гидрохлорида диэтилового эфира глутаминовой кислоты и 106 мг (0,36 ммоль) метиодида 1-(3-диметиламинопропил)-3- этилкарбодиимида. Полученный раствор перемешивают в атмосфере аргона при 25oC в течение 20 ч и затем выливают в 50 мл воды. Воду экстрагируют 2 х 150 мл EtOAc. Объединенные органические слои экстрагируют обратно 3 х 50 мл воды и затем 10 мл насыщенного раствора NaCl и высушивают (MgSO4). Растворитель удаляют при пониженном давлении и необработанный остаток подвергают флэш-хроматографии на силикагеле с 0-10% MeOH/CH2Cl2 с получением 120 мг (71% выход) желаемого амида 13 в виде слегка желтого аморфного твердого вещества.
ЯМР (d6-ацетон) δ: 1,20 (m, 6H), 2,2 (m, 1H), 2,46 (t, 1H, J = 7,6 Гц), 2,78 (s, 1H), 2,82 (s, 1H), 2,92-3,1 (m, 2H), 3,41 (m, 1H), 3,72 (dt, 1H, J = 3,0, 12,7 Гц), 4,02-4,2 (m, 4H), 4,58 (m, 1H), 5,90 (brs, 1H), 6,05 (brs, 1H), 6,90 (d, 1H, J = 3,8 Гц), 7,58 (d, 1H, J = 3,8 Гц), 7,78 (d, 1H, J = 8 Гц).
Масс спектрометрия высокого разрешения:
Вычисл. для C22H29N5O6S2: M+H+, 524,1638. Найдено: 524,1650.
Получение 4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н- пиримидо[5,4-b] [1,4]тиазин-6-ил)-этил]-2,5-тиеноиламино-L-глутаминовой кислоты 1:
К 115 мг (0,22 ммоль) диэтилового эфира 13 добавили 3 мл 1N раствора NaOH в воде. Полученную смесь перемешивали при 25oC в течение 14 ч. После охлаждения до 0oC подводили pH до 3,5 с помощью водной HCl. Твердое вещество отфильтровывали, промывали водой и высушивали под вакуумом с получением 82 мг (80% выход) желаемой кислоты в виде беловатого твердого вещества.
ЯМР (d6-DMSO) δ: 1,62-2,02 (m, 4H), 2,27 (m, 2H), 2,78-3,00 (m, 3H), 4,27 (dd, 1H, J = 6, 6,8 Гц), 6,00 (brs, 2H), 6,63 (brs, 1H), 6,88 (d, 1H, J = 3,7 Гц), 7,63 (d, 1H, J = 3,7 Гц), 8,37 (d, 1H, J = 7,4 Гц), 10,05 (brs, 1H), 12,85 (brs, 1H).
ИК (KBr): 3371, 1700, 1643, 1543, 1345 см-1.
Масс спектрометрия высокого разрешения:
Вычисл. для C18H21N5O6S2: M+H+, 468,1012. Найдено: 468,1025.
Анал. Вычисл. для C18H21N5O6S2• H2O: C, 43,71; H, 4,89; N, 14,16, S, 12,97. Найдено: С, 43,50; H, 4,67; N, 14,07, S, 12,72.
Пример 2
Получение оптически чистых производных тиофена
(Varney et al., "Protein Structure-Based Design, Synthesis, and Biological Evaluation of 5-Thia-2,6-diamino-4(3H)-oxipyrimidines: Potent Inhibitors of Glycinamide Ribonucleotide Transformylase with Potent Cell Growth Inhibition" J. Med. Chem., 1997, 40, No.16, 2506-7,2515)
Синтез начинается с катализируемого палладием сочетания оптически чистой формы ацетилена (34)
с этиловым эфиром 2-бромтиофен-5-карбоновой кислоты и 3-метилпроизводным с последующим восстановлением и кислотным гидролизом для получения диольного промежуточного соединения (35).
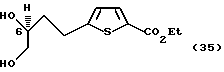
Для C-6(R) соединений атом азота в 8-положении вводили путем монотозилирования первичного спирта с последующим замещением азидом натрия. Для C-6(S) соединений гидроксигруппу инвертировали путем защиты первичного спирта в виде силильного эфира, мезилирования вторичного спирта, удаления защитной группы и образования инвертированного эпоксида с гидридом натрия. Этот эпоксид селективно открывали в наименее заторможенном первичном положении азидом натрия. Азидоспирты восстанавливали в присутствии BOC (трет-бутоксикарбонил) ангидрида с получением спиртов (36).

Атом серы в 5 положении вводили путем замещения соответствующего мезилата соединением KSAc. Ацетилзащитные группы удаляли в основных условиях в присутствии диметилхлормалоната, и как только BOC-защитня группа была удалена, свободный амин спонтанно циклизовался в лактам (37).
Лактам алкилировали тетрафторборатом триметилоксония и полученный лактимный эфир обрабатывали 3 эквивалентами гуанидина в виде свободного основания в дефлегмирующем этаноле с получением 2-амино-4(3Н)-оксопиримидо[5,4-b] [1,4] -тиазинов. Гидролиз этиловых эфиров, пептидное сочетание и гидролиз завершали синтез глутаминовых кислот (16-17).

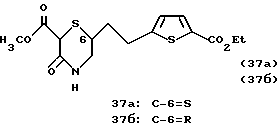
2(S)-[[[5-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидо[5,4-b] [1,4] -тиазин-6-(R)-ил)этил]тиофен-2-ил]карбонил]амино]пентандиоевая кислота (16).
Получена в виде беловатого твердого вещества.
Т.пл. 191-194oC, пенится;
[α]589 +61,9 (c=0,65, 1N NaOH);
ИК (KBr) 3389, 3235, 3086, 2924, 1701, 1624, 1545, 1340 см-1;
1H ЯМР (DMSO-d6) δ 1,70-2,04 (m, 4H), 2,29 (t, 2H, J = 7,3 Гц), 2,90 (m, 2H), 3,13-3,53 (m, 3H, частично перекрыт H2O), 4,29 (m, 1H), 6,30 (s, 2H), 6,77 (s, 1H), 6,89 (d, 1H, J = 3,7 Гц), 7,66 (d, 1H, J = 3,7 Гц), 8,50 (d, 1H, J = 8,1 Гц), 10,30 (brs, 1H).
Анал. (C18H21N5O6S2•1,8 H2O) C, H, N, S.
2(S)-[[[5-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидо[5,4-b] [1,4] -тиазин-6-(S)-ил)этил]тиофен-2-ил]карбонил]амино]пентандиоевая кислота (17).
Получена в виде беловатого твердого вещества.
Т.пл. 220oC, разложение;
[α]589 -57,1 (c=0,61, 1N NaOH);
ИК (KBr) 3353, 3094, 2926, 1711, 1641, 1605, 1559, 1454, 1400, 1333, 1279 см-1;
1H ЯМР (DMSO-d6) δ 1,70-2,05 (m, 4H), 2,29 (t, 2H, J = 7,4 Гц), 2,87 (m, 2H), 3,15-3,48 (m, 3H, частично перекрыт H2O), 4,29 (m, 1H), 6,03 (s, 2H), 6,66 (s, 1H), 6,89 (d, 1H, J = 3,7 Гц), 7,65 (d, 1H, J = 3,7 Гц), 8,50 (d, 1H, J = 7,7 Гц), 10,05 (s, 1H), 12,50 (brs, 2H).
Анал. (C18H21N5O6S2•1,4H2O) C, H, N, S.
Биологическая и биохимическая оценка
Определение констант ингибирования для GAR трансформилазы:
Известный метод анализа GAR-трансформилазы (GARFT) [Young et al., Biochemistry 23 (1984), 3979-3986 был модифицирован и использован как описано ниже. Реакционные смеси содержали каталитические домены человеческой GARFT, 0-250 нМ соединения 1, 20 мкМ глицинамид рибонуклеотида (GAR), 10 или 20 мкМ N10-формил-5,8-дидеазафолата (FDDF), 50 мкМ HEPES-KOH (pH 7.5) и 50 мкМ KCl. Реакцию инициировали добавлением фермента до конечной концентрации 11 нМ с последующим отслеживанием возрастания поглощения на 294 нм при 20oC (e294= 18,9 мМ-1см-1).
Константу ингибирования GARFT (Ki) определяли по зависимости установившейся скорости каталитической реакции от концентрации ингибитора и субстрата. По зависимости кажущейся Ki (Ki,app) от концентрации FDDF было определено, что наблюдаемый тип ингибирования является конкурентным по отношению к FDDF и описывается равенством Ki,app=Ki+(Ki/Km) [FDDF]. Константа Михаэлиса для FDDF, Km, была определена независимо по зависимости каталитической скорости от концентрации FDDF. Данные обоих определений, Km и Ki, были подогнаны нелинейными методами к уравнению Михаэлиса или к уравнению Михаэлиса для конкурентного ингибирования соответственно. Были проанализированы данные, полученные в результате ингибирования сильной связи, и Ki определена путем подгонки этих данных нелинейными методами к уравнению сильной связи Моррисона [Morrison, Biochem Biophys Acta 185 (1969), 269-286].
Клеточные линии:
Используемые клеточные линии и их происхождение приведены в таблице 1. Условия выращивания и требования к среде для каждой клеточной линии приведены в таблице 2 (см. табл. 1-5 в конце описания). Все культуры поддерживали при 37oC, 5% CO2 в воздухе, в увлажняющем инкубаторе.
Ингибирование роста in vitro:
Сток-растворы ингибиторов приготовили в 10 мМ бикарбонате натрия в воде и хранили в аликвотах по 1 мл при -20oC для экспериментов с культурами клеток. Ингибирование роста клеток измеряли с помощью модификации известного [Mosmann, J.Immunol.Methods 65 (1983), 55-63] метода.
Клетки каждой клеточной линии, находящиеся в середине логарифмической фазы роста, разводили до концентрации 18500 клеток/мл свежей RPMI ростовой средой (Mediatech, Washington, DC) с добавкой диализированной фетальной сыворотки теленка (Hyclone Laboratories Inc., Logan, UT), и затем их аликвоты помещали в колонки от 2-й до 12-й 96-луночных планшетов для микротитрования. Колонку 1 заполняли тем же объемом, 135 мл, свежей среды без клеток, для использования в качестве бланка. Планшеты помещали в инкубатор на 37oC с 5% CO2 в воздухе. Через 1-4 ч планшеты вынимали из инкубатора, добавляли соединение 1 в 10 x финальной концентрации 15 мл/лунку в бинарных разведениях в колонки от 12-й до 4-й. Для обратного эксперимента гипоксантин (1,75 мМ) или AICA (1,75 мМ) включали во все растворы лекарства (конечная концентрация 175 мМ). Лунки, содержащие каждую концентрацию соединения 1, готовили на каждом планшете в четырех экземплярах. Пятнадцать миллилитров среды без тестируемого соединения добавляли в лунки колонки 1 планшетов. Затем клетки возвращали в инкубатор и оставляли там в покое на полный период инкубации. На 3-й день для L1210 и L1210/CI920 клеток или 5-й день для CCRF-CEM клеток во все лунки всех планшетов добавляли по 50 мл 0,8 мл/мг МТТ бромида (4,5-диметилтиазол-2-ил)-2,5-дифенил тетразолия (каталог Sigma N М2128), растворенного в среде для культивирования ткани, после чего клетки возвращали в инкубатор. Через 4 ч все планшеты вынимали яз инкубатора и центрифугировали при 1200 об/мин в течение 7 мин. Среды откачивали и во все лунки всех планшетов добавляли по 150 мл DMSO. Затем планшеты перемешивали при низкой скорости на вихревом миксере в течение 1 ч в темноте при комнатной температуре. Долю метаболизированного MTT измеряли спектрофотометрически при 540 нм на Molecular Devices Vmax кинетическом ридере для микропланшетов. Концентрация лекарства, необходимая для снижения клеточного роста на 50%, что измеряется по метаболизму MTT, была определена путем интерполяции между OD (минус бланк) непосредственно выше и ниже 50% контрольной OD (минус бланк).
Противоопухолевая активность in vivo:
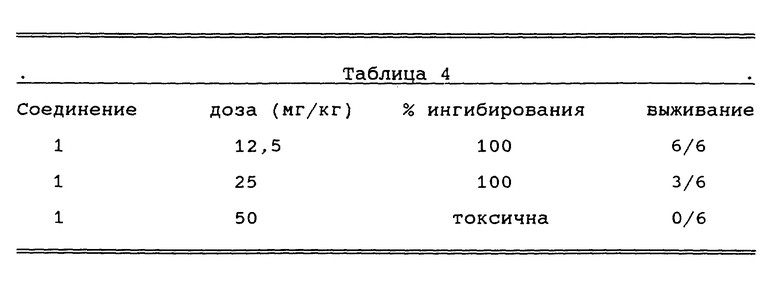
В нулевой день 2 мм2 троакаровые фрагменты 6C3HED лимфоцитомы имплантировали подкожно в подмышечную область в группы из шести C3H/Не самцов мышей. Начиная с дня 1, тестируемое соединение вводили внутрибрюшинно в 40% Encapsin, даваемый один раз в день в течение 9 дней. Контрольных животных подвергали идентичной обработке без тестируемого соединения. Приведенный ниже в Таблице 4 процент ингибирования был рассчитан путем сравнения массы контрольных опухолей (получавшие только разбавитель) на 11-й день с опухолями животных, получавших тестируемое соединение.
В нулевой день 2 мм2 троакаровые фрагменты C3H/BA аденокарциномы молочной железы имплантировали подкожно в подмышечную область в группы из шести C3H/He самцов мышей. Начиная с дня 1, тестируемое соединение вводили внутрибрюшинно в 40% Encapsin, даваемый один раз в день в течение 9 дней. Контрольных животных подвергали идентичной обработке без тестируемого соединения. Процент ингибирования был рассчитан путем сравнения массы контрольных опухолей (получавшие только разбавитель) на 14-й день с опухолями животных, получавших тестируемое соединение. Результаты суммированы в таблице 5.



Описываются новые производные глутаминовой кислоты формулы I, где A обозначает серу; X обозначает CH2; Y обозначает серу; B обозначает водород; C обозначает водород; R1 и R2 каждый независимо обозначает водород или низший алкил, которые ингибируют фермент глицинамид рибонуклеотид формил трансферазу (GARFT). Описываются промежуточные соединения для получения соединений формулы I, фармацевтические композиции, содержащие соединения формулы I, их применение для ингибирования GARFT и их применение для ингибирования роста и пролиферации клеток высших организмов или микроорганизмов, таких как бактерии, дрожжи и грибы. Вышеуказанные соединения обладают противоопухолевой, противовоспалительной, антипсориазной и/или иммуносупрессивной активностью. Изобретение также относится к получению соединений формулы I. 11 с. и 21 з.п. ф-лы, 5 табл.
где А обозначает серу;
Х обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R1 и R2 каждый независимо обозначает водород или низший алкил.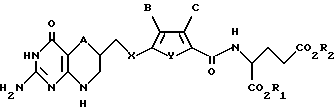
где А обозначает серу;
Х обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R1 и R2 каждый независимо обозначает водород или низший алкил.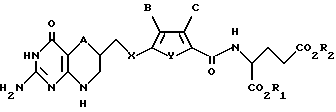
где А обозначает серу;
Х обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R1 и R2 каждый независимо обозначает водород или низший алкил.
где Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу;
R4 и R5 независимо представляют собой водород или легко удаляемую азотозащитную группу.
где X' обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу;
R4 и R5 независимо представляют собой водород или легко удаляемую азотозащитную группу.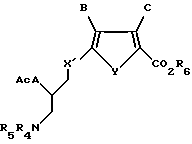
где X' обозначает CH2;
А обозначает серу;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу;
R4 и R5 независимо представляют собой водород или легко удаляемую азотозащитную группу;
Ас обозначает низший ацил.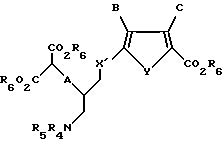
где А обозначает серу;
X' обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу;
R4 и R5 независимо представляют собой водород или легко удаляемую азотозащитную группу.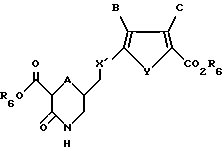
где А обозначает серу;
X' обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу.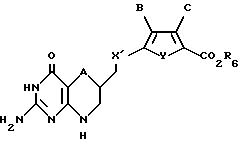
где А обозначает серу;
X' обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород;
R6 обозначает группу, которая образует вместе с CO2 легко гидролизуемую группу.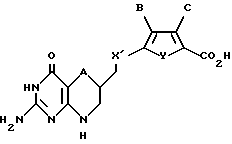
где А обозначает серу;
X' обозначает CH2;
Y обозначает серу;
В обозначает водород;
С обозначает водород.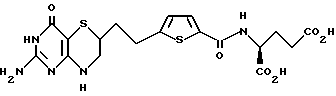
30. Соединение по п.29, обладающее свойствами ингибитора роста и пролиферации клеток.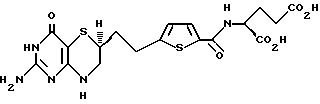
32. Соединение по п.30, имеющее следующую структуру:
| Способ получения арилтрифторэтиламинов или их солей, или их оптических изомеров, или смесей их оптических изомеров | 1977 |
|
SU725561A3 |
| Способ получения производного пиридо[2,3- @ ]пиримидина, или его SS-, RS-изомеров, или смеси диастереомеров, или фармацевтически приемлемых солей с щелочными металлами | 1986 |
|
SU1676449A3 |
| EP 530537 A1, 10.03.97 | |||
| Устройство для рекуперации платиновых металлов | 1978 |
|
SU1170957A3 |
| EP 431953 A2, 12.06.91 | |||
| EP 530579 A1, 10.03.93 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| US 4123550 A, 31.10.78. | |||

