Изобретение относится к соединениям приведенной ниже формулы I, которые ингибируют фермент глицинамид рибонуклеотид формил трансферазу (GARFT). Данное изобретение также относится к фармацевтическим композициям, содержащим соединения формулы I, их применению для ингибирования GARFT и их применению для ингибирования роста и пролиферации клеток высших организмов или микроорганизмов, таких как бактерии, дрожжи и грибы. Данное изобретение также относится к получению таких соединений и промежуточным соединениям, используемым при их получении.
GARFT является фолат-зависимым ферментом в метаболическом пути биосинтеза пурина de novo. Этот путь является решающим для деления клеток и пролиферации. Известно, что блокирование этого пути имеет антипролиферативный эффект, в частности противоопухолевый. Таким образом, было синтезировано много аналогов фолата и изучена их способность ингибировать GARFT. В качестве прототипа известен [1] специфический образующий сильную связь ингибитор GARFT, 5,10-дидеазатетрагидрофолиевая кислота (DDATHF), проявляющий противоопухолевую активность.
Большой класс антипролиферативных агентов включает в себя соединения-антиметаболиты. Конкретный подкласс антиметаболитов, известных в качестве антифолатов или антифолев, представляют собой антагонисты витамина фолиевой кислоты. В типичном случае антифолаты сильно напоминают по структуре фолиевую кислоту и включают в себя характерную P-бензоил глутаматную часть фолиевой кислоты. Глутаматная часть фолиевой кислоты несет на себе двойной отрицательный заряд при физиологических pH, и поэтому это соединение и его аналоги имеют активную энергозависимую транспортную систему для прохождения через клеточную мембрану и проявляют метаболическое действие. Многие исследователи подтвердили, что фолиевая кислота в обеих ее формах, восстановленной и окисленной, а также ее аналоги активно транспортируются в клетки с помощью по меньшей мере двух различных транспортных механизмов. Эти транспортные белки называют транспортным белком восстановленного фолата, и он предпочтителен для восстановленных фолатов, хотя также транспортирует много производных фолиевой кислоты. Метотрексат (MTX) транспортируется через транспортную систему восстановленного фолата. Другой транспортный белок фолата называют мембранным фолат-связывающим белком или mFBP, и он предпочтителен для фолиевой кислоты [2].
Противораковые глутамат-содержащие антифолаты, используемые на сегодняшний день в клинике, включая MTX, поступают в клетку через транспортную систему восстановленного фолата с одним существенным исключением, 5,10-дидеазатетрагидрофолиевая кислота (DDATHF) является противоопухолевым ингибитором GARFT находящимся на стадии клинических исследований. Было показано, что DDATHF транспортируется в клетку как через транспортную систему восстановленного фолата, так и с помощью mFBP [3].
Было предположено, что нежелательная токсичность, в частности для фолат-истощенных млекопитающих, связана с тем, что DDATHF, известный ингибитор GARFT, имеет высокое сродство к mFBP, неконтролируемому во время дефицита фолата. Далее было предположено, что фолиевая кислота и другие молекулы, блокирующие mFBP, не позволяя транспортировать другие ингибиторы GARFT могут ослаблять токсичность таких ингибиторов [4-6].
Таким образом, задачей данного изобретения является получение соединений, являющихся сильными ингибиторами GARFT и обладающих пониженной токсичностью. Данная задача решается тем, что предложены антипролиферативные агенты данной ниже формулы 1, которые являются сильными ингибиторами GARFT, но не образуют сильной связи с mFBP. Такие соединения предпочтительно имеют константы связывания с mFBP по меньшей мере в 1000 раз меньшие чем для DDATHF, сохраняя при этом благоприятные свойства ингибирования GARFT и транспорт восстановленного фолата для противоопухолевой активности.
Как указано выше, соединения по изобретению обладают антипролиферативной активностью, то есть свойством, которое может само выразиться в форме противоопухолевой активности. Соединение по изобретению может быть активно само по себе или выступать в роли предшественника, in vivo превращающегося в активное соединение. Предпочтительные соединения по изобретению являются особенно активными при ингибировании фермента GARFT. Особенно предпочтительные соединения являются активными при ингибировании роста клеточной линии L1210, линии лейкемических клеток мыши, которые могут быть выращены в культуре ткани. Соединения по изобретению также могут быть активными при ингибировании роста бактерий, таких как грам-негативная бактерия Escherichia coli, которая может быть выращена в культуре.
Соединения по изобретению, а также их фармацевтически приемлемые соли, могут быть заключены в традиционные дозовые формы, такие как капсулы, таблетки и инъецируемые препараты. Могут быть использованы также твердые или жидкие фармацевтически приемлемые носители, разбавители или эксципиенты.
Твердые носители включают в себя крахмал, лактозу, дигидрат сульфата кальция, гипс, сахарозу, тальк, желатин, агар, пектин, гуммиарабик, стеарат магния и стеариновую кислоту. Жидкие носители включают в себя сироп, арахисовое масло, оливковое масло, солевой раствор и воду.
Носитель или разбавитель может включать в себя любой материал для пролонгированного высвобождения, такой как глицерил моностеарат или глицерил дистеарат, одиночные или с воском. При использовании жидкого носителя препарат может находиться в форме сиропа, элексира, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекции (например, раствора) либо неводной или водной жидкой суспензии.
Фармацевтические препараты готовят следуя традиционной технологии фармацевтической химии, включающей такие этапы, как смешивание, гранулирование и компрессование (при необходимости) в таблеточные формы, или смешивание, заполнение и растворение ингредиентов подходящим способом для получения желаемых продуктов для перорального, парэнтерального, местного, интравагинального, интраназального, внутрибронхиального, внутриглазного, внутриушного или ректального применения.
Композиции по изобретению также могут включать в себя одно или несколько фармацевтически активных соединений. Например, в композицию может быть включен один из следующих противоопухолевых агентов: митотические ингибиторы (например, винбластин); алкилирующие агенты; ингибиторы дигидрофолат редуктазы или TS ингибиторы; антиметаболиты (например, 5-фторурацил, цитозинрабинозид); интеркалирующие антибиотики (например, адриамицин, блеомицин); ферменты (например, аспарагиназа), ингибиторы топоизомеразы (например, этопозид); модификаторы биологического ответа (например, интерферон). Соединения по изобретению также могут быть использованы в комбинации с одним или несколькими антипролиферативными агентами или ингибиторами GARFT, в частности известными [7-8] . Композиции по изобретению также могут содержать один или несколько антибактериальных, антигрибных, противопаразитных, антивирусных, антипсориатических или антикокцидиальных агентов. Примерами антибактериальных агентов являются сульфонамиды, такие как сульфаметоксазол, сульфадиазин, сульфаметер и сульфадоксин; ингибиторы дигидрофолиевой редуктазы, такие как триметоприм, бромодиаприм и триметрексат; пенициллины; цефалоспорины; и хинолон карбоновые кислоты и их конденсированные с изотиазолом аналоги.
Другой аспект изобретения относится к терапевтическим способам ингибирования роста или пролиферации клеток высших организмов или микроорганизмов, при котором на хозяина (реципиента) воздействуют эффективным количеством соединения по изобретению. Соединения по изобретению, в частности, полезны при лечении млекопитающих хозяев (реципиентов), таких как человек, и при лечении птиц. Особенно предпочтительный терапевтический способ включает в себя воздействие на реципиента эффективным для ингибирования GARFT количеством соединения по изобретению.
Многие описанные здесь антипролиферативные соединения и их фармацевтически приемлемые соли могут быть использованы в терапевтическом способе по изобретению. Эти соединения могут быть применены в форме фармацевтически приемлемой композиции, содержащей разбавитель или носитель, как описано выше.
Доза композиции содержит по меньшей мере эффективное количество активного соединения и предпочтительно состоит из одной или нескольких фармацевтических дозовых единиц. "Эффективное количество" обозначает количество, достаточное для ингибирования метаболических путей фолата, и приводит к следующему из этого благоприятному эффекту при применении, например, одной или нескольких фармацевтических дозовых единиц.
Примерная суточная доза для позвоночных содержит до 1 г активного ингредиента на 1 кг веса тела реципиента, предпочтительно 0,5 г, более предпочтительно 100 мг и наиболее предпочтительно около 50 мг и ниже. Выбранной дозой можно воздействовать на теплокровного животного или млекопитающего, например человека, нуждающегося в лечении, осуществляемом через ингибирование метаболических путей фолата, с помощью любого подходящего способа применения такой дозы, включая местное, например в виде мази или крема; пероральное; ректальное, например в виде суппозитория; парэнтеральное путем инъекции; или непрерывное путем интравагинальной, интраназальной, внутрибронхиальной, внутриушной или внутриглазной инфузии.
Соединения по изобретению дают какой-либо один или несколько эффектов из числа следующих: антипролиферативный эффект, антибактериальный эффект, противопаразитный эффект, антивирусный эффект, антипсориазный эффект, антипротозойный эффект, антикокцидиальный эффект, противовоспалительный эффект, иммуносупрессивный эффект и антигрибной эффект. Эти соединения особенно полезны при получении противоопухолевого эффекта у позвоночных, имеющих опухоли.
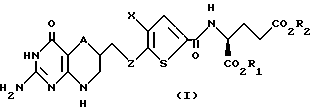
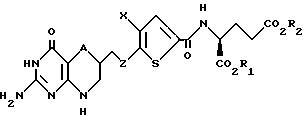
Данное изобретение относится, конкретно, к соединениям формулы I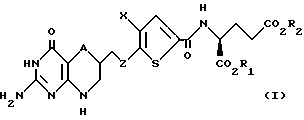
где A - CH2 или S;
Z - C1-C3 алкилен;
Х - C1-C6 алкил; и
R1 и R2 - водород.
Данное изобретение относится также к фармацевтически приемлемым солям соединения формулы I.
Хотя соединения формулы I и показаны в 4-оксоформе и именно о ней идет речь в данном описании, оксогруппа все же существует в таутомерном равновесии с соответствующей 4- гидроксигруппой. Поэтому должно быть понятно, что соединения формулы I включают в себя как изображенные на рисунках 4-оксо, так и таутомерные 4-гидроксиформы. Таким образом, изобретение также относится к фармацевтически приемлемым солям 4-гидрокси таутомеров соединения, изображаемого формулой I.
Соединения формулы I находятся в форме диастереоизомерных смесей. Должно быть понятно, что соединения, имеющие хиральные центры, находятся в форме смесей диастереоизомеров, если не указано иное.
В предпочтительном случае A является серой или CH2. Наиболее предпочтителен Z, являющийся CH2. Также предпочтителен незамещенный X. Наиболее предпочтителен X, являющийся метилом или этилом.
Предпочтительно, когда R1 и R2 каждый независимо является водородом.
В особо предпочтительном случае А является серой или CH2, Z является CH2 и X является метилом.
Предпочтительные примеры соединений формулы 1 включают в себя:
N-(5-[2-(2-амино-4(3Н)-оксо-5,6,7 8-тетрагидропиридо[2, 3-d] -пиримидин-6-ил)этил] -4-метилтиено-2-ил)-L-глутаминовая кислота; диэтиловый эфир N-(5-[2-(2-амино-4-оксо-4,6,7, 8-тетрагидро-3Н-пиримидо[5,4-6] [1,4] -тиазин-6-ил)этил] -4-метилтиено-2-ил)-L-глутаминовой кислоты; и
N-(5-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидо[5,4-6] [1,4]-тиазин-6-ил)этил]-4-метилтиено-2-ил)-L-глутаминовая кислота.
Соединения формулы I применимы в качестве ингибиторов GARFT. Соединения формулы I, в которой R1 и R2 каждый обозначает водород, являются особенно активными противоопухолевыми и антипролиферативными агентами.
Фармацевтически приемлемые соли по изобретению включают в себя, например, соли щелочного металла, щелочноземельного металла, других нетоксичных металлов, аммония и замещенного аммония глутаминовой кислоты соединений по изобретению. Примерами солей являются натриевая, калиевая, литиевая, кальциевая, магниевая соль, соль пиридина и замещенного пиридина данных соединений в форме свободной кислоты.
Соединения формулы I могут быть получены, как описано ниже.
Для получения соединений формулы I, где Z является CH2, в качестве исходного вещества подходит соединение формулы II: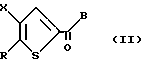
где R - галоген, предпочтительно бром;
X определен выше;
B - C1-C6алкокси.
Соединение формулы II подвергают взаимодействию с соединением формулы III: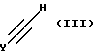
где Y обозначает CH2OH или защищенный пиридопиримидин формулы IV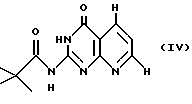
Далее синтез может идти одним или двумя путями в зависимости от того, является ли Y защищенным пиридопиримидином или CH2OH.
Если Y является защищенным пиридопиримилином формулы IV, сочетание соединений формулы II и III предпочтительно проводят в присутствии катализатора из переходного металла, предпочтительно палладия или никеля, в присутствии основания, предпочтительно ненуклеофильного вспомогательного основания, в растворителе, в котором по меньшей мере один из реагентов по меньшей мере частично растворим. Предпочтительными растворителями для сочетания соединений формул II и III являются диэтиламин, ацетонитрил, диметилформамид, диметилацетамид и триэтиламин. Основную среду для сочетания предпочтительно обеспечивают с помощью ненуклеофильного вспомогательного основания, являющегося основанием, способным нейтрализовать галогеноводородную кислоту, образующуюся при сочетании. Это основание предпочтительно является ди- или три-алкиламином, таким как диэтиламин, триэтиламин или диизопропилэтиламин. При возможности вместо отдельного растворителя и основания можно использовать основной растворитель.
Когда Y является пиридопиримидином, сочетание соединений формул II и III дает соединение формулы V: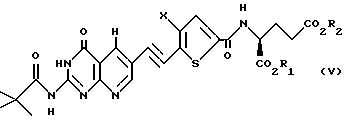
где X, R1 и R2 такие, как определено выше. Соединение формулы V подвергают взаимодействию с газообразным водородом, предпочтительно при 45-1000 пси, в присутствии подходящего катализатора из переходного металла, предпочтительно платины, палладия или родия на угле или другой подходящей подложке, в подходящем растворителе, предпочтительно уксусной кислоте или трифторуксусной кислоте, с получением соединения формулы VI: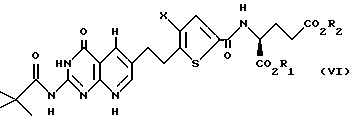
где X, R1 и R2 такие, как определено выше.
И наконец, соединение формулы VI гидролизуют с образованием свободной глутаминовой кислоты (R1 и R2 оба являются водородом) формулы I.
Если Y является CH2OH, взаимодействие соединений формул II и III дает соединение формулы VII: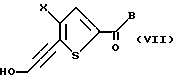
где Х и В такие, как определено выше.
Соединение формулы VII подвергают взаимодействию с газообразным водородом в присутствии подходящего металлического катализатора, предпочтительно палладия или платины, с получением соединения формулы VIII: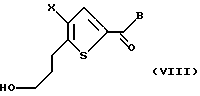
где X и В такие, как определено выше. Соединение формулы VIII подвергают взаимодействию с окисляющим агентом, предпочтительно перрутенатом тетрапропиламмония, с получением соединения формулы IX: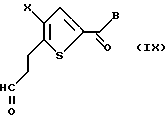
где Х и В такие, как определено выше.
Соединение формулы IX подвергают взаимодействию с донором метилена, предпочтительно метилен трифенилфосфораном, в подходящем растворителе, предпочтительно тетрагидрофуране, с получением соединения формулы X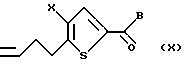
где Х и В такие, как определено выше.
Соединение формулы Х подвергают взаимодействию с дигидроксилирующим агентом, предпочтительно тетроксидом осмия, в присутствии подходящего окисляющего агента, предпочтительно N-метилморфолин-N-оксида, с получением соединения формулы XI: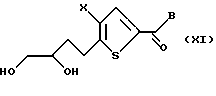
где Х и В такие, как определено выше.
Соединение формулы XI превращают в соединение формулы I, используя один из четырех описанных ниже способов.
В первом способе превращения соединение формулы XI подвергают взаимодействию с сульфонилирующим агентом, предпочтительно р-толуолсульфонил хлоридом или метансульфонил хлоридом, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, с получением промежуточного моно-сульфонилированного соединения. Это промежуточное соединение затем подвергают взаимодействию с сильным основанием, предпочтительно гидридом натрия, с получением соединения формулы XII:
где Х и В такие, как определено выше.
Эпоксид формулы XII подвергают взаимодействию с азотсодержащим нуклеофилом, предпочтительно азидом натрия, в присутствии мягкого катализатора из кислоты Льюиса, предпочтительно перхлората лития или перхлората магния, с получением промежуточного азида спирта. Восстановление азида спирта, предпочтительно газообразным водородом в присутствии металлического катализатора, с последующей защитой подходящей азотозащитной группой, предпочтительно трет-бутоксикарбонилом, бензоксикарбонилом или бензилом, дает соединение формулы XIII: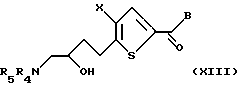
где Х и В такие, как определено выше, a R4 и R5 независимо друг от друга являются водородом или подходящей азотозащитной группой. Предпочтительными защитными группами являются трет-бутоксикарбонил, бензил-оксикарбонил и бензил.
Соединение формулы XIII подвергают взаимодействию с ацилирующим или сульфонилирующим агентом, предпочтительно метансульфонил хлоридом или р-толуолсульфонил хлоридом, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, в подходящем растворителе, в котором по меньшей мере один из реагентов является по меньшей мере частично растворимым, с получением активированной гидрокси группы. Активированную гидрокси группу замещают подходящим нуклеофилом, предпочтительно тиокислотной солью, более предпочтительно тиоацетатом калия, с получением соединения формулы XIV: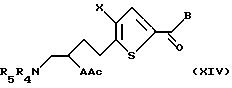
где A, X, В, R4 и R5 такие, как определено выше, а
Ac обозначает ацильную группу.
Предпочтительно Ас является ацетилом.
В альтернативном случае соединение формулы XIII может быть превращено в соединение формулы XIV в один химический прием с использованием трифенилфосфина, диэтила или диметил азадикарбоксилата и кислотного нуклеофила, предпочтительно тиоуксусной кислоты, в подходящем растворителе.
Соединение формулы XIV обрабатывают нуклеофильным основанием, предпочтительно карбонатом калия, карбонатом натрия, гидроксидом натрия или гидроксидом калия, в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, в присутствии алкилирующего агента, предпочтительно диметил или диэтил хлормалоната, с получением соединения формулы XV: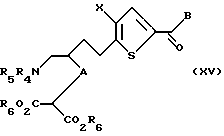
где A, X, B, R4 и R5 такие, как определено выше, а
R6 каждый независимо является водородом или группой, которая образует с присоединенной группой CO2 легко гидролизуемую эфирную группу. Предпочтительно R6 является C1-C6 алкилом, гидроксиалкилом, алкиларилом или аралкилом. Более предпочтительно R6 является C1-C2 алкилом.
Соединение формулы XV обрабатывают в условиях, подходящих для удаления либо R4 либо R5, либо обеих защитных групп с получением соединения формулы XVI: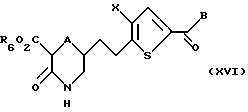
где A, X, В и R6 такие, как определено выше.
Если в качестве защитной группы использован трет-бутоксикарбонил, подходящими условиями является обработка трифторуксусной кислотой с последующей нейтрализацией.
Соединение формулы XVI подвергают взаимодействию с алкилирующим агентом, предпочтительно тетрафторборатом триметил или триэтил оксония, в подходящем растворителе, предпочтительно дихлорметане, с образованием промежуточного лактимного эфира. Промежуточный лактимный эфир подвергают взаимодействию с гуанидином в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с образованием соединения формулы XVII: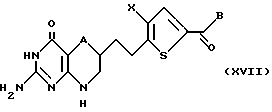
где A, Х и В такие, как определено выше.
В альтернативном случае соединение формулы XVI может быть превращено в соединение формулы XVII путем взаимодействия соединения формулы XVI с тиолирующим агентом, предпочтительно P2S5 или 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2, 4-дисульфидом, с образованием тиолактамного промежуточного соединения. Затем это промежуточное соединение алкилируют с помощью алкилирующего агента, предпочтительно метил иодида или тетрафторбората триметил или триэтил оксония, а затем гуанидином в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с получением соединения формулы XVII.
Если В является спиртовой функцией, то есть если группа, присоединенная с В, образует эфирную группу, то соединение формулы XVII гидролизуют в основных условиях с получением соединения формулы XVIII: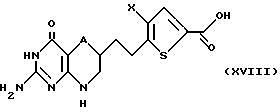
где A и X такие, как определено выше.
Соединение формулы XVIII соединяют [пептидной связью] с помощью известных из уровня техники средств с гидрохлоридом диэфира глутаминовой кислоты с получением диэфира формулы XIX: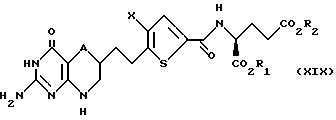
где A, X, R1 и R2 такие, как определено выше, за исключением того, что ни R1, ни R2 не являются водородом.
И наконец, если желательна форма свободной глутаминовой кислоты, соединение формулы XIX гидролизуют до образования соединения формулы I.
Во втором способе превращения соединение формулы XIV получают как описано выше. Это соединение обрабатывают кислотой, предпочтительно трифторуксусной, соляной или р-толуолсульфоновой, для удаления всех защитных групп (R4, R5 и Ac) с получением соединения формулы XX: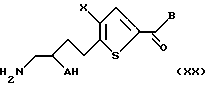
где A, X и B такие, как описано выше.
Соединение формулы XX в слабо-щелочном буфере, предпочтительно используя pH 7 фосфатный буфер, в подходящем растворителе, предпочтительно этаноле или метаноле, подвергают взаимодействию с соединением формулы XXI: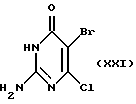
с получением соединения формулы XVII. Окончание второго способа, от соединения формулы XVII до соединения формулы I, осуществляют аналогично описанному выше.
В третьем способе превращения соединение формулы XI подвергают взаимодействию с подходящей гидроксил-защитной группой, предпочтительно триаалкилсилильной группой, более предпочтительно трет-бутилдиметилсилил хлоридом, в присутствии мягкого ненуклеофильного основания, предпочтительно триэтиламина, с получением соединения формулы XXII: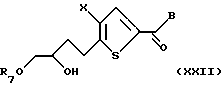
где Х и В такие, как определено выше, a R7 является подходящей гидроксил-защитной группой, предпочтительно триалкилсилильной группой.
Соединение формулы XXII затем подвергают взаимодействию с ацилирующим или сульфонилирующим агентом, предпочтительно метансульфонил хлоридом или р-толуолсульфонил хлоридом, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, в подходящем растворителе, в котором по меньшей мере один из реагентов по меньшей мере частично растворим, с получением активированной гидроксигруппы.
Активированную гидроксигруппу замещают подходящим нуклеофилом, предпочтительно тиокислотной солью, более предпочтительно тиоа-цетатом калия, с получением соединения формулы XXIII: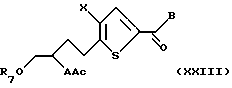
где А, X, В, R7 и Ас такие, как определено выше. В альтернативном случае соединение формулы XII может быть превращено в соединение формулы XIII в один химический прием с использованием трифенилфосфина, диэтила или диметил азадикар-боксилата и кислотного нуклеофила, предпочтительно тиоуксусной кислоты, в подходящем растворителе. Соединение формулы XXIII подвергают взаимодействию с нуклеофильным основанием или мягкой кислотой для селективного удаления ацильной группы или группировки А. Полученное промежуточное соединение подвергают взаимодействию с соединением формулы XXIV: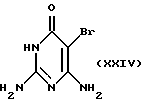
в присутствии ненуклеофильного основания, предпочтительно триэтиламина, диизопропилэтиламина или карбоната калия, с получением соединения формулы XXV: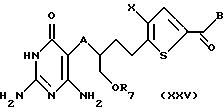
где A, X, В и R7 такие, как определено выше.
Защитную группу R7 на соединении формулы XXV удаляют путем обработки соответствующим реагентом с получением соединения формулы XXVI: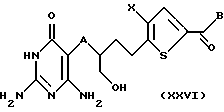
где A, Х и В такие, как определено выше.
Если R7 является триалкилсилилом, данным реагентом предпочтительно является соль фтора, более предпочтительно фторид калия, фторид тетрабутиламмония или фторид цезия.
Соединение формулы XXVI циклизуют с получением соединения формулы XVII путем активирования гидроксигруппы активирующим агентом, предпочтительно метансульфонил хлоридом, с последующей обработкой основанием. В альтернативном случае азот пиримидинона сначала защищают подходящей защитной группой, предпочтительно трет-бутоксикарбонилом, с последующей циклизацией и удалением защитной группы в кислотных условиях. Окончание этого способа, от соединения формулы XVII до соединения формулы I, осуществляют аналогично описанному выше.
В четвертом и предпочтительном способе превращения спиртовое соединение формулы XXVI получают как описано выше. Этот спирт подвергают взаимодействию с подходящим окисляющим агентом с получением альдегидной функции, которая циклизуется до соединения формулы XXVII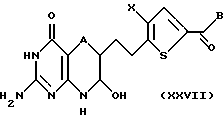
где A, Х и В такие, как определено выше.
Соединение формулы XXVII подвергают взаимодействию с восстанавливающим агентом, предпочтительно цианборогидридом натрия, в присутствии кислоты Льюиса, предпочтительно этерата трифторида бора, с получением соединения формулы XVII, определенной выше. Окончание этого способа, от соединения формулы XVII до соединения формулы I, осуществляют аналогично описанному выше.
Соединение формулы I, где Z является иным, чем CH2, может быть получено аналогично случаю, когда Z является CH2. В частности, соединение формулы I, где Z является иным, чем CH2, может быть получено с использованием олефина формулы XXXIV: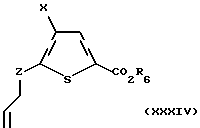
где Х и R6 такие, как определено выше, и Z такое, как определено для формулы I, но не CH2.
Если Z является C1-C3алкиленом, иным чем CH2, то соединение формулы XXXIV получают олефинированием альдегида формулы XXXVI: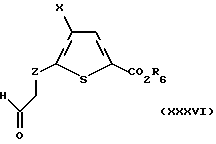
где Х и R6 такие, как определено выше, и Z является C1-C3 алкиленом, иным чем CH2. Альдегид формулы XXXVI может быть получен аналогично известному способу [9]. Олефинирование альдегида может быть осуществлено с использованием донора метилена, предпочтительно метилен-трифенилфосфорана.
Соединение формулы XXXIV подвергают взаимодействию с дигидроксилирующим агентом, предпочтительно тетроксидом осмия, в присутствии подходящего окисляющего агента, предпочтительно N-метилморфолин-N-оксида, с получением соединения формулы XXXVII: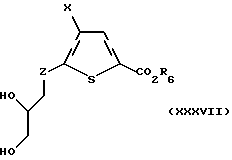
где X и R6 такие, как определено выше, и Z такое, как определено для формулы I, но не CH2.
Соединение формулы XXXVII подвергают взаимодействию с сульфонилирующим агентом, предпочтительно р-толуолсульфонилхлоридом или метансульфонил хлоридом, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, с получением промежуточного моносульфонидированного соединения. Это промежуточное соединение подвергают взаимодействию с сильным основанием, предпочтительно гидридом натрия, с получением соединения формулы XXXVIII: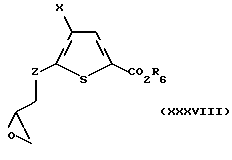
где Х и R6 такие, как определено выше, и Z такое, как определено для формулы I, но не CH2.
Эпоксид формулы XXXVIII подвергают взаимодействию с азотсодержащим нуклеофилом, предпочтительно азидом натрия, в присутствии катализатора из мягкой кислоты Льюиса, предпочтительно перхлората лития или магния, с получением промежуточного азида спирта. Это промежуточное соединение восстанавливают, предпочтительно газообразным водородом в присутствии металлического катализатора, с последующей защитой подходящей азотозащитной группой, предпочтительно трет-бутоксикарбонилом, бензоксикарбонилом или бензилом, с получением соединения формулы XVII':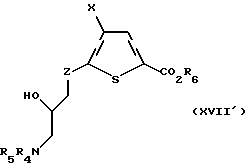
где X, R6 и R4 и R5 такие, как определено выше, a Z такое, как определено для формулы I, но не CH2.
Соединение формулы XVII' подвергают затем взаимодействию с ацилирующим или сульфонилирующим агентом, предпочтительно метансульфонил хлоридом или р-толуолсульфонил хлоридом, в присутствии ненуклеофильного основания, предпочтительно триэтиламина или диизопропилэтиламина, в подходящем растворителе, в котором по меньшей мере один из реагентов является по меньшей мере частично растворимым, с получением активированной гидроксигруппы. Активированную гидроксигруппу замещают подходящим нуклеофилом, предпочтительно тиокислотной солью, более предпочтительно тиоацетатом калия, с получением соединения формулы XVIII'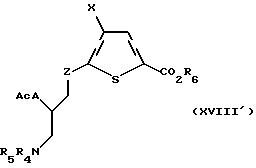
где A, X, R6, R4 и R5 и Ac такие, как определено выше, a Z такое, как определено для формулы I, но не CH2.
В альтернативном случае соединение формулы XVII' превращают в соединение формулы XVIII' в один химический прием с использованием трифенилфосфина, диэтила или диметил азадикарбоксилата и кислотного нуклеофила, предпочтительно тиоуксусной кислоты, в подходящем растворителе.
Соединение формулы XVIII' обрабатывают нуклеофильным основанием, предпочтительно карбонатом калия, карбонатом натрия, гидроксидом натрия или гидроксидом калия, в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, в присутствии алкилирующего агента, предпочтительно диметил или диэтил хлормалоната, с получением соединения формулы XIX':,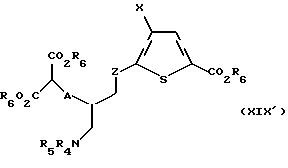
где A, X, R6, R4 и R5 такие, как определено выше, а Z такое, как определено для формулы I, но не CH2.
Соединение формулы XIX' обрабатывают в условиях, подходящих для удаления либо R4, либо R5, либо обеих защитных групп с получением соединения формулы XX':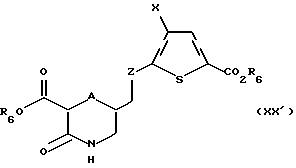
где A, Х и R6 такие, как определено выше, a Z такое, как определено для формулы I, но не CH2.
Если в качестве защитной группы использован трет-бутоксикарбонил, подходящими условиями для удаления этой группы является обработка трифторуксусной кислотой с последующей нейтрализацией с получением соединения формулы XX'.
Соединение формулы XX' подвергают взаимодействию с алкилирующим агентом, предпочтительно тетрафторборатом триметил или триэтил оксония, в подходящем растворителе, предпочтительно дихлорметане, с образованием промежуточного лактимного эфира. Этот промежуточный лактимный эфир подвергают взаимодействию с гуанидином в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с образованием соединения формулы XXI':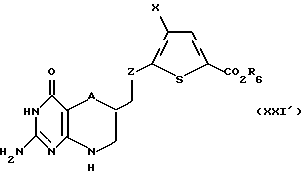
где A, X и R6 такие, как определено выше, a Z такое, как определено для формулы I, но не CH2.
В альтернативном случае соединение формулы XX' превращают в соединение формулы XXI' путем взаимодействия соединения формулы X' с тиолирующим агентом, предпочтительно P2S5 или 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидом, с образованием тиолактамного промежуточного соединения. Затем это промежуточное соединение алкилируют с помощью алкилирующего агента, предпочтительно метил иодида или тетрафторбората триметил или триэтил оксония, а затем гуанидином в спиртовом растворителе, предпочтительно метаноле, этаноле или изопропаноле, с получением соединения формулы XXI'.
Соединение формулы XXI' гидролизуют в основных условиях с получением соединения формулы XXII':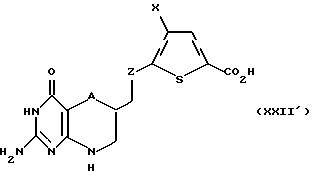
где A и Х такие, как определено выше, а Z такое, как определено для формулы I, но не CH2. Если R6 является водородом в соединении формулы XXI', тогда реакция гидролизации не является необходимой, и соединение формулы XXI' сочетают пептидной связью, как описано ниже.
Соединение формулы XXII' (или соединение формулы XXI', где R6 является водородом), находящееся в форме свободной карбоновой кислоты, может быть соединено [пептидной связью] (с помощью средств, известных из уровня техники) с гидрохлоридом диэфира глутаминовой кислоты с образованием диэфира формулы XXIII':
где A, Х и такие, как определено выше для формулы XXII', a R1 и R2 каждый независимо является группировкой, которая образует с присоединенным CO2 легко гидролизуемую эфирную группу, такую как C1-C6 алкил, гидроксиалкил, алкиларил или арилалкил.
И наконец, если желательна форма свободной кислоты, соединение формулы XXIII' гидролизуют с получением соединения формулы I, где R1 и R2 каждый является водородом.
Детальные примеры получения соединения формулы I приведены ниже.
Пример 1
N-(5- [2-(2-амино-4 (3Н)-оксо-5, 6,7,8-тетрагидропиридо[2, 3-d]пиримидин-6-ил)этил]-4-метилтиено-2-ил)-L-глутаминовая кислота (соединение 1)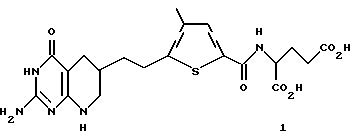
Синтез
Соединение 1 синтезируют следующим образом.
а) 5-бром-4-метилтиофен-2-карбоновая кислота: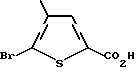
Это соединение получают известным [10] способом.
б) 6-этинил-2-(пивалоиламино)-4(3Н)-оксопиридо[2,3-d]пиримидин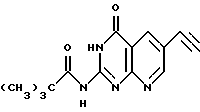
Это соединение получают известным [11] способом.
в) диэтил N-(5-бром-4-метилтиено-2-ил)-L-глутамат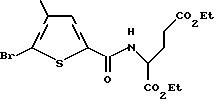
К перемешиваемому раствору 5-бром-4-метил-тиофен-2-карбоновой кислоты (3,32 г, 15 ммоль), 1-гидроксибензотриазола (2,24 г, 16,6 ммоль), гидрохлорида диэтилового эфира L-глутаминовой кислоты (3,98 г, 16,6 ммоль) и диизопропилэтиламина (2,9 мл, 2,15 г, 16,6 ммоль) в диметилформамиде (DMF) (40 мл) добавляют гидрохлорида 1- (3-диметиламинопропил)-3-этилкарбодиимида (3,18 г, 16,6 ммоль). Полученный раствор перемешивают в атмосфере аргона при комнатной температуре в течение 18 ч, выливают в солевой раствор (300 мл), разбавляют водой (100 мл) и экстрагируют эфиром (3 х 120 мл). Объединенные органические экстракты промывают водой (150 мл), высушивают над MgSO4 и концентрируют в вакууме с получением коричневой смолы, которую очищают флэш-хроматографией. Элюция смесью гексан : EtOAc (2:1) дает продукт в
виде оранжевого масла (5,05 г, 83% выход). Анализы показывают, что этот продукт является диэтил N-(5-бром-4- метилтиено-2-ил)-L-глутаматом.
ЯМР (CDCl3) δ: 7.22 (1H, s), 6.86 (1H, d, J = 7.5 Hz), 4.69 (1H, ddd, J = 4.8, 7.5, 9.4 Hz), 4.23 (2Н, q, J = 7.1 Hz), 4.12.(2Н, q, J = 7.1 Hz), 2.55 - 2.39 (2Н, m), 2.35 - 2.22 (1H, m), 2.19 (3H, s), 2.17 - 2.04 (1H, m), 1.29 (3Н, t, J = 7.1 Hz), 1.23 (3Н, t, J = 7.1 Hz).
Анал.: (C15H20NO5SBr) C, H, N, S, Br.
г) диэтил N-(5-[(2-[пивалоиламино]-4(3Н)-оксопиридо[2,3-d] пиримидин-6-ил)этинил]-4-метилтиено-2-ил) глутамат: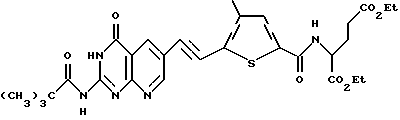
К перемешиваемому раствору диэтил N-(5-бром-4-метилтиено-2- ил) глутамата (4,21 г, 10,4 ммоль) в ацетонитриле (55 мл) в атмосфере аргона добавляют хлорид бис (трифенилфосфин) палладия (702 мг, 1,0 ммоль), иодид меди (200 мг, 1,1 ммоль), триэтиламин (1,5 мл, 1,09 г, 10,8 ммоль) и 6-этинил-2-(пивалоиламино)-4(3Н)- оксопиридо[2,3-d]пиримидин (5,68 г, 21 ммоль). Полученную суспензию нагревают с обратным холодильником в течение 6 ч. После охлаждения до комнатной температуры неочищенную реакционную смесь фильтруют и осадок промывают ацетонитрилом (50 мл) и этилацетатом (EtOAc) (2 х 50 мл). Объединенные фильтраты концентрируют в вакууме с получением коричневой смолы, которую очищают флэш- хроматографией. Элюция CH2Cl2:CH3OH (49:1) дает твердый оранжевый продукт (4,16 г, 67% выход). Анализы показывают, что этот продукт является диэтил N-(5-[(2-[пивалоиламино]-4(3Н)-оксопиридо[2,3-d]пиримидин-6-ил) этинил]-4-метилтиено- -2-ил) глутаматом.
ЯМР (CDCl3) δ:
8.95 (1H, d, J = 2.2 Hz), 8.59 (1H, d, J = 2.2 Hz), 7.33 (1H, s), 7.03 (1H, d, J = 7.4 Hz), 4.73 (1H, ddd, J = 4.8, 7.4, 9.5 Hz), 4.24 (2Н, q, J = 7.1 Hz), 4.13 (2Н, q, J = 7.1 Hz), 2.55 - 2.41 (2Н, m), 2.38 (3H, s), 2.35 - 2.24 (1H, m), 2.19 - 2.05 (1H, m), 1.34 (9Н, s), 1.30 (3H, t, J = 7.1 Hz), 1.24 (3H, t, J = 7.1 Hz).
Анал. (C29Н33N5O7S.0.75Н2O) C,H,N,S.
д) диэтил N-(5-[(2-[пивалоиламино]-4(3Н)-оксопиридо[2,3-d] пиримидин-6-ил)этил]-4-метилтиено-2-ил) глутамат: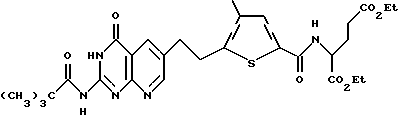
Суспензию диэтил N-(5-[(2-[пивалоиламино] -4(3H)-оксопиридо [2,3-d]пиримидин-6-ил)этинил] -4-метилтиено-2-ил) глутамата (959 мг, 1,6 ммоль) и 10% палладия на угле (1,5 г, 150% масс экв.) в трифторуксусной кислоте (30 мл) встряхивают при 50 пси H2 в течение 22 ч. Неочищенную реакционную смесь разбавляют CH2Cl2, фильтруют через подушечку Celite (диатомовая земля) и концентрируют в вакууме. Полученный остаток растворяют в CH2Cl2 (120 мл), промывают насыщенным NaHCO3 (2 х 100 мл), высушивают над Na2SO4 и концентрируют в вакууме с получением коричневой смолы, которую очищают флэш-хроматографией. Элюция смесью CH2Cl2:CH3OH (49:1) дает желтый твердый продукт (772 мг, 80% выход). Анализ показывает, что этот продукт является диэтил N-(5-[(2-[пивалоиламино] -4(3H)-оксопиридо[2,3-d]пиримидин-6-ил)этил]-4-метилтиено-2-ил) глутаматом.
(CDCl3) δ: 8.60 (1H, d, J > 2.2 Hz), 8.49 (1H, broad), 8.32 (1H, d, J = 2.2 Hz), 7.22 (1H, s), 6.78 (1H, d, J = 7.5 Hz), 4.72 (1H, ddd, J = 4.8, 7.5, 9.5 Hz), 4.23 (2Н, q, J = 7.1 Hz), 4.11 (2Н, q, J = 7.1 Hz), 3.12 - 3.00 (4H, m), 2.52 - 2.41 (2Н, m), 2.37 - 2.22 (1H, m), 2.16 - 2.04 (1H, m), 2.02 (3H, s), 1.33 (9Н, s), 1.29 (3H, t, J = 7.1 Hz), 1.23 (3H, t, J = 7.1 Hz).
Анал. (C29H37N5O7S.0.5H2O) C,H,N,S.
e) диэтил N-(5-[(2-[пивалоиламино]-4(3Н)-оксо-5,6,7,8- тетрагидропиридо[2,3-d]пиримидин-6-ил)этил]-4-метилтиено-2 -ил) глутамат: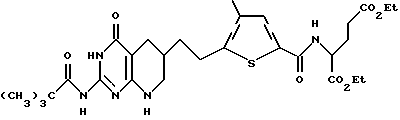
Суспензию диэтил N-(5-[(2-[пивалоиламино]-4(3H)-оксопиридо [2,3-d] пиримидин- 6-ил) этил]-4-метилтиено-2-ил) глутамата (2,98 г, 5 ммоль), 10% Pt на угле (1,5 г, 50% масс экв.) в трифторуксусной кислоте (170 мл) встряхивают при 800 пси H2 в течение 40 ч. Неочищенную реакционную смесь разбавляют CH2Cl2, фильтруют через подушечку Celite и концентрируют в вакууме. Полученный остаток растворяют в CH2Cl2 (150 мл), промывают насыщенным NаНСОз (2 х 150 мл), высушивают над Na2SO4 и концентрируют в вакууме с получением коричневой смолы, которую очищают флэш-хроматографией. Элюция смесью CH2Cl2: CH3OH (24: 1) сначала дает непрореагировавший субстрат, а затем желтый твердый продукт (293 мг, 10% выход). Анализ показывает, что этот продукт является диэтил N-(5-[(2-[пивалоиламино]-4(3Н)-оксо-5,6,7,8-тетрагидропиридо [2,3-d] пиримидин-6-ил) этил]-4-метилтиено-2-ил) глутаматом.
ЯМР (CDCl3) δ:
7.24 (1H, s), 6.75 (1H, d, J = 7.6 Hz), 5.57 (1H, broad), 4.72 (1H, ddd, J = 4.8. 7.6, 12.6 Hz), 4.22 (2Н, q, J = 7.1 Hz), 4.11 (2Н, q, J = 7.1 Hz), 3.43 - 3.36 (1H, m), 3.06 - 2.98 (1H, m), 2.89 - 2.68 (ЗН, m), 2.52 - 2.40 (3H, m),2.37 - 2.23 (1Н, m), 2.15: (3H, s), 2.14 - 2.03 (1H, m), 1.94 - 1.83 (IH, m), 1.73 - 1.63 (2Н, m), 1.32 (9H,s), 1.29 (3H, t, J= 7.1 Hz), 1.23 (3Н, t, J = 7.1 Hz).
Анал. (C29H41N5O7S.0.5H2O C,H,N,S.
ж) N-(5-[2-(2-амино-4(3Н)-оксо-5,6,7,8-тетрагидропиридо [2,3-d]пиримидин-6-ил)этил]-4-метилтиено-2-ил) глутаминовая кислота (соединение 1):
Раствор диэтил N-(5-[(2-[пивалоиламино] -4(3Н)-оксо-5,6,7,8 -тетрагидропиридо[2,3-d] пиримидин-6-ил)этил] -4-метилтиено-2-ил) глутамата (293 мг, 0,5 ммоль) в 1 N NaOH (25 мл) перемешивают при комнатной температуре в течение 90 ч, затем нейтрализуют 6 N НCl. Образовавшийся осадок собирают путем фильтрации и промывают водой (4 х 10 мл) с получением желтого твердого продукта (63 мг, 28% выход). Анализ показывает, что этот продукт является диэтил N-(5-[2- (2-амино-4(3Н)-оксо-5,6,7,8-тетрагидропиридо[2,3 -d]пиримидин-6-ил)этил]-4-метилтиено-2-ил) глутаминовой кислотой.
ЯМР (DMSO-d6) δ: 12.44 (2Н, broad), 9.89 (1H, broad), 8.42 (1H, d, J= 7.8 Hz), 7.57 (1H, s), 6.39 (1H, br s), 6.12 (2Н, br s), 4.30 (1H, ddd, J = 4.8, 7.8, 9.6 Hz), 3.26 - 3.18 (2Н, m), 2.83 - 2.74 (3Н, m), 2.31 (2Н, t, J = 7.4 Hz), 2.12 (3Н, s), 2.09 - 2.01 (1H, m), 1.94 - 1.80 (2Н, m), 1.68 - 1.47 (3H,m).
Анал. (C20H25N5O6S.1.1H2O). C,H,N,S.
Биологическая и биохимическая оценка
Определение констант ингибирования для GAR трансформилазы:
Известный метод анализа GAR-трансформилазы (GARFT) [12] был модифицирован и использован, как описано ниже. Реакционные смеси содержали каталитические домены человеческой GARFT, 0-250 нМ тестируемого соединения, 20 мкМ глицинамид рибонуклеотида (GAR), 10 или 20 мкМ N10 -формил-5,8-дидеазафолата (FDDF), 50 мкМ HEPES-KOH (pH 7.5) и 50 мкМ KC1. Реакцию инициировали добавлением фермента до конечной концентрации 11 нМ с последующим отслеживанием возрастания поглощения на 294 нм при 20oC (e294=18.9 мМ-1см-1).
Константу ингибирования GARFT (K1 определяли по зависимости установившейся скорости каталитической реакции от концентрации ингибитора и субстрата. По зависимости кажущейся Ki (Ki,app) от концентрации FDDF было определено, что наблюдаемый тип ингибирования является конкурентным по отношению к FDDF и описывается равенством Ki,app=Кi+(Ki/Km) [FDDF]. Константа Михаэлиса для FDDF, Km, была определена независимо по зависимости каталитической скорости от концентрации FDDF. Данные обоих определений, Km и Ki были подогнаны нелинейными методами к уравнению Михаэлиса или к уравнению Михаэлиса для конкурентного ингибирования соответственно. Были проанализированы данные, полученные в результате ингибирования сильной связи, и Ki определена путем подгонки этих данных нелинейными методами к уравнению сильной связи Моррисона [13].
Определение констант диссоциации для фолат-связывающего белка человека
Константу диссоциации (Kd) для фолат-связывающего белка (FBP) человека определяли в анализе на конкурентное связывание, используя соединенные с мембранами FBP, полученные из культивируемых KB клеток.
Приготовление мембранной фракции KB клеток:
Налипшие KB клетки соскребали с колб, промывали в ледяном PBS и центрифугировали при 5000 х g в течение 5 мин при 4oC. Осажденные клетки (2•108 клеток) ресуспендировали в 10 мл суспензионного буфера (KH2PO4-KOH pH 7,4: 10 мкМ EDTA: 10 мкм 2-меркаптоэтанола), быстро разрушали ультразвуком для завершения клеточного лизиса и центрифугировали при 12000 х g в течение 10 мин при 4oC. Осадок освобождали от эндогенно связанного фолата путем ресуспендирования в 20 мл кислотного буфера (50 мкМ KH2PO4-KOH pH 3,5: 10 мкМ EDTA: 10 мкМ 2-меркаптоэтанола) и центрифугировали, как и раньше. Затем осадок ресуспендировали в 5 мл суспензионного буфера при pH 7,4 без EDTA. Содержание белка определяли количественно с помощью метода Брадфорда, используя BSA в качестве стандарта. Типичный выход в этом способе составлял 4-5 мг общего мембранного белка на 2•108 клеток. Эту окончательную суспензию использовали как источник соединенного с мембранами человеческого FBP.
Анализ на конкурентное связывание FBP:
Ингибитор конкурировал с 3Н-фолиевой кислотой за связывание с FBP. Реакционные смеси содержали (в 1 мл 50 мМ KH2PO4-KOH pH 7,4: 10 мМ 2-меркаптоэтанола) 50-100 мг мембранного белка клеток, содержащего 3-6 пикомоль (3-6 нМ) FBP, 17,25 пмоль 3Н-фолиевой кислоты (17,25 нМ, 0,5 мкКи), различные концентрации конкурентов. Реакции проводили при 25oC. Из-за очень медленного высвобождения связанной 3Н-фолиевой кислоты конкурент подвергали предварительному связыванию в течение 30 мин в отсутствие 3Н-фолиевой кислоты. Затем добавляли 3Н-фолиевую кислоту и смесь оставляли приходить в равновесие на 2,5 ч. Реакционные смеси целиком пропускали через нитроцеллюлозные фильтры под вакуумом для отделения клеточных мембран со связанной 3Н-фолиевой кислотой. Отделенные мембраны затем промывали 4 раза за 1 мл реакционного буфера. Количество связанной 3Н-фолиевой кислоты измеряли путем подсчета сцинтилляций нитроцеллюлозной мембраны. Полученные данные нелинейно подгоняли, как описано выше. FBP Кd3Н-фолиевой кислоты, используемая для подсчета Кd конкурента, была получена путем прямого титрования FBP 3Н-фолатом с последующим нелинейным подгоном данных к уравнению Кd сильной связи.
Клеточные линии:
Используемые клеточные линии и их происхождение приведены в таблице 1. Условия выращивания и требования к среде для каждой клеточной линии приведены в таблице 2. Все культуры поддерживали при 37oC , 5% CO2 в воздухе, в увлажняющем инкубаторе.
Ингибирование роста in vitro:
Сток-растворы ингибиторов приготовили в 10 мМ бикарбонате натрия в воде и хранили в аликвотах по 1 мл при -20oC для экспериментов с культурами клеток. Ингибирование роста клеток измеряли с помощью модификации известного [14] метода.
Клетки каждой клеточной линии, находящиеся в середине логарифмической фазы роста, разводили до концентрации 18500 клеток/мл свежей RPMI ростовой средой (Mediatech, Washington, DC) с добавкой диализированной фетальной сыворотки теленка (Hyclone Laboratories Inc., Logan, UT), и затем их аликвоты помещали в колонки от 2-й до 12-й 96-луночных планшетов для микротитрования. Колонку 1 заполняли тем же объемом, 135 мл свежей среды без клеток, для использования в качестве бланка. Планшеты помещали в инкубатор на 37oC с 5% CO2 в воздухе. Через 1-4 ч планшеты вынимали из инкубатора, добавляли тестируемое соединение в 10 х финальной концентрации 15 мл/лунку в бинарных разведениях в колонки от 12-й до 4-й. Для обратного эксперимента гипоксантин (1,75 мМ) или AICA (1,75 мМ) включали во все растворы лекарства (конечная концентрация 175 мМ). Лунки, содержащие каждую концентрацию тестируемого соединения готовили на каждом планшете в четырех экземплярах. Пятнадцать миллилитров среды без тестируемого соединения добавляли в лунки колонки 1 планшетов. Затем клетки возвращали в инкубатор и оставляли там в покое на полный период инкубации. На 3-й день для L1210 и L1210/C1920 клеток или 5-й день для CCRF-CEM клеток во все лунки всех планшетов добавляли по 50 мл 0,8 мл/мг MTT бромида (4,5- диметилтиазол-2-ил)-2,5-дифенил тетразолия (каталог Sigma No М2128), растворенного в среде для культивирования ткани, после чего клетки возвращали в инкубатор. Через 4 ч все планшеты вынимали из инкубатора и центрифугировали при 1200 об/мин в течение 7 мин. Среды откачивали и во все лунки всех планшетов добавляли по 150 мл DMSO. Затем планшеты перемешивали при низкой скорости на вихревом миксере в течение 1 ч в темноте при комнатной температуре. Долю метаболизированного MTT измеряли спектрофотометрически при 540 нм на Molecular Devices Vmax кинетическом ридере для микропланшетов. Концентрация лекарства, необходимая для снижения клеточного роста на 50%, что измеряется по метаболизму MTT, была определена путем интерполяции между OD (минус бланк) непосредственно выше и ниже 50% контрольной OD (минус бланк).
Как показывают приведенные выше сравнительные данные, соединение 1 имеет Kd по отношению к фолат-связывающему белку примерно в 1400 раз меньшую, чем 6R-DDATHF.
Пример 2
N-(5-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо[5,4-6] [1,4]тиазин-6-ил)этил]-4-метилтиено-2-ил)-L-глутаминовая кислота (соединение 2):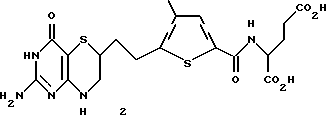
Соединение 2 готовят следующим образом.
а) метиловый эфир 5-бром-4-метилтиофен-2-карбоновой кислоты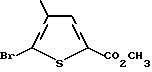
К раствору 5-бром-4-метилтиофен-2-карбоновой кислоты (20,32 г, 92 ммоль) в CH3OH (450 мл) добавляют концентрированную H2SO4 (4 мл). Полученный раствор нагревают с обратным холодильником в течение 18 ч. Растворитель удаляют путем концентрации в вакууме и полученный остаток разделяют в смеси насыщенного NaHCO3 (350 мл) и эфира (350 мл). Слои отделяют и водную фазу экстрагируют эфиром (3 х 150 мл). Объединенные органические экстракты высушивают над MgSO4 и концентрируют в вакууме с получением красного масла, которое очищают флэш-хроматографией. Элюция смесью гексан:этилацетат (9:1) дает продукт в виде желтого масла, отвердевающего при стоянии (18,34 г, 85% выход). Анализ показывает, что этот продукт является метиловым эфиром 5-бром-4- метилтиофен-2-карбоновой кислоты.
ЯМР (CDCl3) δ: 7,47 (1H, s), 3,86 (3H, s), 2,20 (3H, s).
Анал. (C7H7O2SBr) C,H,S,Br.
б) метиловый эфир 5-(3-гидроксипропинил)-4-метилтиофен-2- -карбоновой кислоты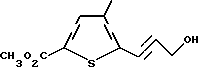
К перемешиваемому раствору метилового эфира 5-бром-4 -метилтиофен-2-карбоновой кислоты (5,18 г, 22 ммоль) в диэтиламине (60 мл) в атмосфере аргона добавляют хлорид бис(трифенилфосфин) палладия (77 мг, 0,11 ммоль), иодид меди (42 мг, 0,22 ммоль) и пропаргиловый спирт (1,5 мл, 1,44 г, 26 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч. Растворитель удаляют путем концентрации в вакууме и полученный остаток разбавляют водой (200 мл) и затем экстрагируют этилацетатом (3 х 100 мл). Объединенные органические экстракты промывают 0,5 N HCl (100 мл), высушивают над MgSO4 и концентрируют в вакууме с получением коричневого масла, которое очищают флэш- хроматографией. Элюция смесью гексан:EtOAc (2:1) дает продукт в виде оранжевого масла, отвердевающего при стоянии (4,07 г, 88% выход). Анализ показал, что этот продукт является метиловым эфиром 5-(3-гидроксипропинил)-4-метилтиофен-2-карбоновой кислоты.
ЯМР (CDCl3) δ: 7.52 (1H, s), 4,55 (2H, s), 3,87 (3H, s), 2,29 (3H, s)
Анал. (C10H10O3S) C, Н, S.
в) метиловый эфир 5-(3-гидроксипропил)-4-метилтиофен-2- карбоновой кислоты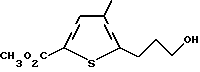
Суспензию метилового эфира 5-(3-гидроксипропинил)-4- метилтиофен-2-карбоновой кислоты (3,86 г, 18 ммоль) и 5% палладия на угле (0,72 г, 19% масс экв.) в EtOAc (110 мл) встряхивают при 50 пси H2 в течение 20 ч. Неочищенную реакционную смесь фильтруют через подушечку Celite и фильтрат концентрируют в вакууме с получением продукта в виде желтого масла (3,84 г, 98% выход). Анализ показывает, что этот продукт является метиловым эфиром 5-(3-гидроксипропил)-4-метилтиофен-2-карбоновой кислоты.
ЯМР (CDCl5) δ: 7,51 (1H, s), 3,84 (3H, s), 3,71 (2H, t, J=6,2 Гц), 2,86 (2H, t, J=7,6 Гц), 2,16 (3H, s), 1,92 (2H, tt, J=6,2 и 7,6 Гц)
Анал. (C10H14O3S) C, H, S.
г) метиловый эфир 4-метил-5-(3-оксипропил)-тиофен-2- карбоновой кислоты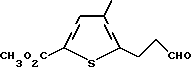
К перемешиваемой суспензии метилового эфира 5-(3- гидроксипропил)-4-метилтиофен-2-карбоновой кислоты (3,74 г, 17 ммоль), N-метилморфолин-N-оксида (3,00 г, 26 ммоль) и измельченного 4  молекулярного сита (4,5 г) в CH2Cl2 (50 мл) добавляют перрутенат тетрапропил аммония (300 мг, 0,85 ммоль). Полученную суспензию перемешивают при комнатной температуре 40 мин. Растворитель удаляют путем концентрации в вакууме и полученный остаток очищают флэш-хроматографией. Элюция смесью гексан: ЕtOAc (4:1) дает продукт в виде желтого масла (1,82 г, 49% выход). Анализ показывает, что этот продукт является метиловым эфиром 4-метил-5-(3-оксипропил)-тиофен-2-карбоновой кислоты.
молекулярного сита (4,5 г) в CH2Cl2 (50 мл) добавляют перрутенат тетрапропил аммония (300 мг, 0,85 ммоль). Полученную суспензию перемешивают при комнатной температуре 40 мин. Растворитель удаляют путем концентрации в вакууме и полученный остаток очищают флэш-хроматографией. Элюция смесью гексан: ЕtOAc (4:1) дает продукт в виде желтого масла (1,82 г, 49% выход). Анализ показывает, что этот продукт является метиловым эфиром 4-метил-5-(3-оксипропил)-тиофен-2-карбоновой кислоты.
ЯМР (CDCl3) δ: 9,83 (1H, t, J=0,8 Гц), 7,50 (1H, s), 3,84 (3H, s), 3,07 (2H, t, J=7,4 Гц), 2,83 (2H, dt, J=0,8 и 7,4 Гц), 2,17 (3H, s).
Анал. (C10H12O3S) C, H, S.
д) метиловый эфир 5-(3-бутенил)-4- метилтиофен-2-карбоновой кислоты
К перемешиваемой суспензии бромида метилтрифенилфосфора (3,14 г, 8,8 ммоль) в THF (30 мл) в атмосфере аргона при 0oC добавляют 2,5 М n-бутиллитий в гексане (3,4 мл, 8,5 ммоль). Полученный шлам перемешивают в течение 10 мин при 0oC, в течение 75 мин при комнатной температуре, и затем охлаждают до -65oC перед тем как добавить по каплям раствор метилового эфира 4-метил-5- (3-оксипропил)-тиофен-2-карбоновой кислоты (1,71 г, 8,1 ммоль) в THF (30 мл). Охлаждающую баню удаляют и реакционную смесь перемешивают в течение 90 мин, пока она постепенно не нагреется до комнатной температуры. Неочищенную реакционную смесь концентрируют в вакууме до объема 20 мл, разбавляют эфиром (200 мл), фильтруют через подушечку целита [броунмеллерита]. Фильтрат концентрируют в вакууме с получением оранжевого масла, которое очищают флэш- хроматографией. Элюция смесью гекcaн:EtOAc (95:5) дает продукт в виде желтого масла (772 мг, 46%). Анализ показывает, что этот продукт является метиловым эфиром 5-(3-бутенил)-4-метилтиофен- 2-карбоновой кислоты.
ЯМР (CDCl3) δ: 7.50 (1H, s), 5.84 (1H, ddt, J=10.2, 17.0, 6.6 Гц), 5.07 (1H, dd, J=1.6, 17.0 Гц) 5.02 (1H, dd, J=1.6, 10.2 Гц), 3.84 (3H, s).
Анал. (C11H14O2S) C, H, S.
е) метиловый эфир 5-(3,4-дигидробутил)-4-метилтиофен-2-карбоновой кислоты
К перемешиваемому раствору N-метилморфолин-N-оксида (735 мг, 6,3 ммоль) и тетроксида осмия (5 мг, 0,02 ммоль) в ацетоне (30 мл) добавили раствор метилового эфира 5-(3-бутенил)-4-метилтиофен-2- карбоновой кислоты (701 мг, 3,3 ммоль) в ацетоне (20 мл). Полученный раствор перемешивали в атмосфере аргона при комнатной температуре в течение 48 ч, затем фильтровали через подушечку Celite. Фильтрат подкисляли путем добавления 0,5 M H2SO4 (10 мл), и ацетон удаляли путем концентрации в вакууме. Водный остаток разбавляли водой (20 мл) и экстрагировали EtOAc (3 х 25 мл). Объединенные органические экстракты промывали водой (3 х 25 мл), высушивали над Na2SO4 и концентрировали в вакууме с получением коричневой смолы, которую очищали флэш-хроматографией. Элюция смесью CH2Cl2 : EtOAc (2:3) дала продукт в виде беловатого твердого вещества (577 мг, 71% выход). Анализ показал, что этот продукт представляет собой метиловый эфир 5-(3,4-дигидробутил)-4-метилтиофен-2-карбоновой кислоты.
ЯМР (CDCl3) δ: 7,50 (1H, s), 3,84 (3H, s), 3,79-3,72 (1H, m), 3,86 (1H, dd, J=3,2, 10,9 Гц), 3,48 (1H, dd, J=7,4, 10,9 Гц), 3,00-2,80 (2H, m).
Анал. (C11H16O4S) C, Н, S.
Приведенные выше примеры иллюстрируют различные аспекты изобретения. Понятно, что возможны соответствующие модификации, доступные специалисту в данной области.
Указанные здесь химические группы могут быть замещены, как принято в химии. В некоторых случаях эта возможность оговорена ссылкой, например замещенная или незамещенная C1-C3 алкильная группа.
Если указано несколько R6 групп (в любой формуле), то каждую из них выбирают независимо от других из предложенного ряда.
Литература
1. F. M. Muggia, "Folate antimetabolites inhibitor to de novo purine synthesis, " New Drugs, concepts and Results in Cancer Chemotherapy, Kluwer Academic Publishers, Boston (1992), 65-87.
2. A. C. Antony, "The Biological Chemistry of Folate Receptors," Blood, The Journal of the American Society of Hematology, vol. 79 (1992), 2807-2820.
3. G. Pizzorno et al. , "5,10-Dideazatetrahydrofolic Acid (DDATHF) Transport in CCRF-CEM and MA104 Cell Lines," The Journal of Biological Chemistry, vol. 268 (1993), 1017-1023.
4. T.Alati et al., "Evaluation of the Mechanism(s) of Inhibition of the Toxicity, But Not the Antitumor Activity of Lometrexol (DDATHF) by Folic Acid, " Proceedings of the American Association for Cancer Research, vol. 33 (1992), Abstract 2432, 407.
5. L. L. Habeck et al. , "A Novel Class of Monoglutamated Antifolates Exibits Tight-binding Inhibition of Human Glycinamide Ribonucleotide Formyltransferase and Potent Activity against Solid Tumors," Cancer Research, vol. 54 (1994), 1021-1026.
6. Патент США 5217974 (Grindey).
7. WO 94/13295, опубликовано 23.06.94.
8. WO 92/05153, опубликовано 02.04.92.
9. Chuan Shih et al. Journal of Medicinal Chemistry, vol. 35 (1992), 1109-1116.
10. N.Nemec, Collection Czechoslov. Chem. Соmmun., vol. 39 (1974), 3527.
11. E.C.Taylor & G.S.K.Wong, J.Org.Chem., vol.54 (1989), 3618.
12. Young et al. Biochemistry 23 (1984), 3979-3986.
13. Morrison, Biochem Biophys Acta 185 (1969), 269-286.
14. Mosmann, J.Immunol. Methods 65 (1983), 55-63.


Описываются новые соединения общей формулы I, где А - СН2 или S; Z - С1-С3алкилен; Х - С1-С6алкил и R1 и R2 - водород. Соединения формулы I, находящиеся в равновесии с их 4-гидрокси-таутомерами и находящиеся в форме диастереоизомерных смесей, и их фармацевтически приемлемые соли являются сильными ингибиторами GARFT. Эти соединения и их соли являются также полезными антипролиферативными агентами. Описывается также способ ингибирования и пролиферации и производные эфира тиофенкарбоновой кислоты. 3 с. и 11 з.п.ф-лы, 3 табл.
где A - CH2 или S;
Z - C1 - C3алкилен;
X - C1 - C6алкил;
R1 и R2 - водород,
или его фармацевтически приемлемые соли.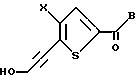
X - C1 - C6алкил;
B - C1 - C6алкокси.
| EP 0343801 A, 29.11.1989 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Ручной опыливатель | 1956 |
|
SU109381A1 |
| Ключевая схема с непосредственной стабилизацией | 1973 |
|
SU450353A1 |
| Устройство для рекуперации платиновых металлов | 1978 |
|
SU1170957A3 |
| EP 0530537 A1, 10.03.1994 | |||
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| Способ получения арилтрифторэтиламинов или их солей, или их оптических изомеров, или смесей их оптических изомеров | 1977 |
|
SU725561A3 |
| Способ получения производного пиридо[2,3- @ ]пиримидина, или его SS-, RS-изомеров, или смеси диастереомеров, или фармацевтически приемлемых солей с щелочными металлами | 1986 |
|
SU1676449A3 |
| CLARKE K | |||
| ET AL | |||
| Condensed isothiazoles | |||
| Part.B | |||
| J.Chem-Soc., Perkin Trans, 1 | |||
| - LETCHWORTH GB, 1980, p.1029-1037 | |||
| NEMEC M | |||
| ET AL | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Collection of Czechoslovak Chemical Communications | |||
| - PRAGE CS, 1974, v.39, p.3527-3561. | |||

