Изобретение касается нового ряда химических соединений, полезных как ингибиторы протеазы ВИЧ, и использования таких соединений в качестве антивирусных агентов.
Синдром приобретенного иммунодефицита (СПИД) - относительно недавно распознанная болезнь или состояние. СПИД вызывает как постепенное ослабление человеческой иммунной системы, так и прогрессивное ухудшение центральной и периферийной нервных систем. С момента первоначального распознавания в начале 1980-х годов СПИД быстро распространился и теперь достиг эпидемических размеров среди относительно ограниченной части населения. Интенсивное исследование привело к открытию агента, ответственного за это заболевание, - человеческого Т-лимфотропного ретровируса III (HTLV-III), теперь более часто упоминаемого как вирус иммунодефицита человека или ВИЧ.
ВИЧ - член класса вирусов, известных как ретровирусы. Ретровирусный геном составлен из РНК, которая преобразуется в ДНК обратной транскрипцией. Эта ретровирусная ДНК затем стабильно интегрируется в хромосому клетки хозяина и, используя репликативные процессы клеток хозяина, производит новые ретровирусные частицы и распространяет инфекцию к другим клеткам. ВИЧ, по-видимому, обладает специфической аффинностью к человеческим T4-лимфоцитам, которые играют жизненно важную роль в иммунной системе человека. Инфекция ВИЧ этих белых кровяных клеток исчерпывает их популяцию. В конечном счете иммунная система становится бездействующей и неэффективной в борьбе с различными сопутствующими заболеваниями, среди которых такие, как pneumocystic carini пневмония, саркома Капоши и рак лимфатической системы.
Хотя точный механизм формирования и работы вируса ВИЧ не установлен, идентификация вируса привела к некоторому прогрессу в управлении болезнью. Например, было обнаружено, что лекарственный препарат азидотимидина (АЗТ) эффективен для ингибирования обратной транскрипции ретровирусного генома вируса ВИЧ, что позволяет контролировать течение болезни, но не излечивать пациентов, страдающих СПИД. Поиск лекарственных средств, которые могут излечивать или по меньшей мере обеспечивать улучшенную меру контроля над смертоносным вирусом ВИЧ продолжается.
Ретровирусная репликация обычно подразумевает посттрансляционный процессинг полипротеинов. Этот процессинг осуществляется закодированным в вирусе ферментом - протеазой ВИЧ. В результате процессинга образуются зрелые полипептиды, впоследствии помогающие формированию и функционированию инфекционного вируса. Подавление этого молекулярного процессинга ограничивает нормальное продуцирование ВИЧ. Следовательно, ингибиторы протеазы ВИЧ могут действовать как антиВИЧ-агенты.
Протеаза ВИЧ является одним из транслируемых продуктов pol гена ВИЧ. Эта ретровирусная протеаза специфично расщепляет другие структурные полипептиды в отдельных сайтах, активируя эти вновь синтезированные структурные белки и ферменты и, таким образом, делая вирион способным к репликации. Следовательно, ингибирование протеазы ВИЧ сильнодействующими соединениями может как предотвращать провирусную интеграцию в инфицированные T-лимфоциты в ранней фазе жизненного цикла ВИЧ-1, так и ингибировать вирусный протеолитический процессинг в поздней его стадии. Дополнительными преимуществами ингибиторов протеазы могут быть большая доступность, большая продолжительность действия на вирус и меньшая токсичность по сравнению с доступными в настоящее время лекарственными препаратами, благодаря их специфичности к ретровирусной протеазе.
В соответствии с настоящим изобретением предлагается новый класс химических соединений, способных ингибировать и/или блокировать действие протеазы ВИЧ, помогающей пролиферации ВИЧ, фармацевтические составы, содержащие эти соединения, и использование этих соединений в качестве ингибиторов протеазы ВИЧ.
Настоящее изобретение касается соединений, описываемых приведенной ниже формулой (1), и их фармацевтически приемлемых солей, ингибирующих протеазу, кодируемую вирусом иммунодефицита человека (ВИЧ) типа 1 (ВИЧ-1) или типа 2 (ВИЧ-2). Эти соединения полезны при лечении инфекции, вызываемой ВИЧ, и синдрома приобретенного иммунодефицита (СПИД). Соединения, их фармацевтически приемлемые соли и фармацевтические составы, предусмотренные изобретением, могут использоваться самостоятельно или в сочетании с другими антивирусными средствами, иммуномодуляторами, антибиотиками или вакцинами. Соединения по настоящему изобретению, могут также использоваться как пролекарства. Обсуждаются методы лечения СПИД, методы лечения ВИЧ-инфекции и методы ингибирования протеазы ВИЧ.
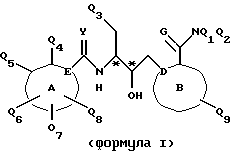
Соединения по настоящему изобретению описываются формулой I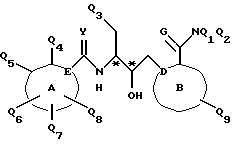
где Q3 представляет собой арил или -S-арил, возможно замещенные галогеном, где арил - это карбоциклический ароматический 5-14- членный моноциклический или полициклический остаток;
A представляет собой карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток; пиридинил, пиридинил-N-оксид; хинолинил; изохинолинил; индолил: индолинил; тиазолил-1,1-диоксид; тиенил; или тиенил-1,1-диоксид;
B представляет собой карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток; насыщенный 5-14-членный моноциклический или полициклический остаток, включающий гетероатом азота; пиридилметилпиперазинил; октагидротиено[3,2-c]пиридинил; или октагидротиено[3,2-c] пиридинил-1,1-диоксид;
Q1 и Q2 независимо представляют собой атом водорода или алкил;
Q4-Q8 независимо представляют собой атом водорода, гидроксил, галоген, нитро-, амино-, алкилсульфониламино-, алкиламино-, алкил, возможно замещенный галогеном, алкоксил, группу -O-J (где J обозначает отгидролизовываемую группу) или группу L6C(O)L4 (где L6 обозначает простую связь или -O, a L4 обозначает алкил или алкоксил);
Y и G представляют собой атомы кислорода;
D представляет собой атом углерода или азота, причем D соединен простой связью с каждым из смежных атомов кольца;
E представляет собой атом углерода;
Q9 представляет собой атом водорода;
или его фармацевтически приемлемая соль.
Особо предпочтительны те соединения формулы (I), в которых Q3 выбран из замещенных галогеном и незамещенных фенила, -S-фенила, нафтила и -S-нафтила; или Q3 выбран из замещенных галогеном и незамещенных фенила и -S-фенила; или Q3 выбран из замещенного галогеном и незамещенного фенила; или Q3 выбран из замещенного галогеном и незамещенного -S-фенила.
Предпочтительны также соединения, в которых один из Q1 и Q2 является трет-бутилом; Q3 обозначает -S-фенил или фенил; и Q5 обозначает гидроксильную группу или группу -O-J, или фармацевтически приемлемая соль этого соединения; либо один из Q1 и Q2 является трет-бутилом, а другой - атомом водорода; Q3 обозначает -S-фенил или фенил; Q4 обозначает метил; Q5 обозначает гидроксильную гриппу или группу -O-J; Q6, Q7 и Q8 обозначают атомы водорода; D обозначает атом азота; A обозначает фенил; и B обозначает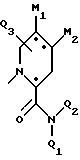
где M1 и M2 представляют собой атомы водорода, или M1 и M2 могут образовывать часть кольца, имеющего до 10 членов; в частности M1 и M2 независимо содержат от нуля до восьми неводородных атомов.
Также предпочтительны соединения, в которых один из Q1 и Q2 является трет-бутилом, а другой - атомом водорода; Q4 обозначает метил; Q5 обозначает гидроксильную группу или -O-J; Q6, Q7 и Q8 обозначают атомы водорода; D обозначает атом азота; A обозначает фенил; и B обозначает декагидроизохинолинил или октагидротиено[3,2-c] пиридинил или фармацевтически приемлемая соль этого соединения.
В этом случае Q3 может являться -S-арильной группой.
Предпочтительно, когда один из Q1 и Q2 является трет-бутилом, а другой - атомом водорода; Q3 обозначает -S-арил; D обозначает атом азота; B обозначает декагидроизохинолинил или октагидротиено[3,2-c]пиридинил.
При этом более предпочтительны соединения, в которых A представляет собой карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток; Q4 обозначает гидроксильную группу или алкоксил или алкил; и Q5 обозначает гидроксильную группу, группу -O-J или алкоксил; или конкретнее: A обозначает фенил; Q4 обозначает алкил; и Q5 обозначает гидроксильную группу или группу -OPO(OH)2; при этом Q3 может являться -S-фенилом.
Предпочтительными являются соединения, в которых по меньшей мере один из Q1 и Q2 является алкилом; Q4-Q8 независимо выбраны из атома водорода, гидроксила, галогена, группы -O-J (где J обозначает отгидролизовываемую гриппу), алкоксила, амино-, алкила, возможно замещенного галогеном, и группы L6C(O)L4 (где L6 обозначает простою связь или -O, а L4 обозначает алкил); D обозначает атом азота; A представляет собой карбоциклический ароматический 5-7-членный моноциклический или полициклический остаток; пиридинил, пиридинил-N-оксид; хинолинил; изохинолинил; индолил; индолинил; тиазолил-1,1-диоксид; тиенил; или тиенил-1,1-диоксид. B представляет собой насыщенный 8-12-членный моноциклический или полициклический остаток, включающий гетероатом азота; пиридилметилпиперазинил; октагидротиено[3,2-c]пиридинил; или октагидротиено[3,2-c]пиридинил-1,1-диоксид.
При этом более предпочтительно, чтобы Q3 был выбран из замещенных галогеном и незамещенных фенила, -S-фенила, нафтила и -S-нафтила; или замещенных галогеном и незамещенных фенила и -S-фенила; или замещенного галогеном и незамещенного фенила; или же из замещенного галогеном и незамещенного -S-фенила.
При этом предпочтительно, чтобы один из Q1 и Q2 являлся алкилом, а другой - атомом водорода; Q4 обозначал алкил; Q5 обозначал гидроксильную группу или группу -O-J, где J - отгидролизовываемая группа, или алкоксил или аминогруппа; E обозначал атом углерода; A представляет собой карбоциклический ароматический 5-6-членный моноциклический или полициклический остаток; пиридинил, пиридинил-N-оксид; хинолинил; изохинолинил; индолил; индолинил; тиазолил-1,1-диоксид; тиенил; или тиенил-1,1-диоксил; B представляет собой насыщенный 8-10-членный моноциклический или полициклический остаток, включающий гетероатом азота; пиридилметилпиперазинил; октагидротиено[3,2-c]пиридинил; или октагидротиено[3,2-c]пиридинил-1,1-диоксид.
Более предпочтительно, чтобы один из Q1 и Q2 - третичный алкил, а другой - атом водорода; Q4 - метил; Q5 - гидроксильная группа, аминогруппа или группа -O-J, где J - отгидролизовываемая группа; A - фенил; и B представляет собой насыщенный 9-10-членный бициклический остаток, включающий гетероатом азота; октагидротиено[3,2-c]пиридин; или октагидротиено[3,2-c]пиридин-1,1-диоксид.
При этом еще более предпочтительно, когда B - это декагидроизохинолинил или октагидротиено[3,2-c]пиридинил.
Предпочтительны также соединения, в которых один из Q1 и Q2 является алкилом, а другой - атомом водорода; Q4-Q8 независимо выбраны из атома водорода, гидроксила, галогена, группы -O-J (где J обозначает отгидролизовываемую группу), алкоксила, амино-, алкила, возможно замещенного галогеном, и группы L6C(O)L4 (где L6 обозначает простою связь или -O, а L4 обозначает алкил или алкоксил); A представляет собой карбоциклический ароматический 5-7-членный моноциклический остаток; пиридинил, пиридинил-N-оксид; тиазолил-1,1-диоксид; тиенил; или тиенил-1,1-диоксид; B представляет собой насыщенный 8-10-членный полициклический остаток, включающий гетероатом азота; октагидротиено[3,2-c]пиридинил; или октагидротиено[3,2-c]пиридинил-1,1-диоксид.
И конкретнее, B представляет собой насыщенный 9-10-членный бициклический остаток, включающий гетероатом азота; пиридилметилпиперазин; октагидротиено[3,2-c]пиридин; или октагидротиено[3,2-c]пиридин-1,1-диоксид.
Также возможно, чтобы B обозначал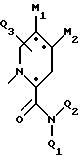
где M1 и M2 представляют собой атомы водорода, или M1 и M2 могут образовывать часть кольца, имеющего до 10 членов.
Все значения температур, заявленные здесь, даны в градусах Цельсия (oC). Все единицы измерения, используемые здесь, даются в единицах веса, кроме жидкостей, которые измеряются в единицах объема.
Используемый здесь термин "алкил" относится к группам с линейной или разветвленной цепью, предпочтительно содержащей от одного до восьми, более предпочтительно - от одного до шести и наиболее предпочтительно - от одного до четырех атомов углерода. Термин "C1-C6-алкил" обозначает линейную или разветвленную алкильную цепь, содержащую от одного до шести углеродных атомов. Типичные C1-C6-алкильные группы включают метил, этил, н-пропил, изопропил, бутил, изобутил, в-бутил, т-бутил, пентил, нео-пентил, гексил, изогексил и т.п. Термин "C1-C4-алкил" подпадает под термин "C1-C6-алкил".
Термин "циклоалкил" обозначает насыщенное или частично насыщенное моно- или поликарбоциклическое кольцо, предпочтительно содержащее 5-14 атомов углерода. Типичные циклоалкилы включают моноциклические кольца, состоящие из 3-7, предпочтительно 3-6, атомов углерода, как например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п. Типичным циклоалкилом является C5-C7-циклоалкил, имеющий структуру насыщенного углеводородного кольца, содержащего от пяти до семи атомов углерода.
Термин "алкоксил" обозначает -O-алкил. Примером алкоксила является C1-C6-алкоксил, который представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до шести углеродных атомов, соединенных с атомом кислорода. Типичные C1-C6-алкоксильные группы включают метоксил, этоксил, пропоксил, изопропоксил, бутоксил, в-бутоксил, т-бутоксил, пентоксил, гексоксил и т.п. C1-C4-алкоксил подпадает под определение C1-C6-алкоксила.
Используемый здесь термин "арил" относится к карбоциклическому или гетероциклическому ароматическому 5-14-членному моноциклическому или полициклическому кольцу. Типичные арилы включают фенил, нафтил, антрил, фенантрил, тиенил, пирролил, имидазолил, пиразолил, фурил, изотиазолил, фуразанил, изоксазолил, тиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, триазинил, бензо[b] тиенил, нафто[2,3-b]тиантренил, изобензофуранил, хроменил, ксантенил, феноксантенил, индолизинил, изоиндолил, индолил, индазолил, пуринил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хинолинил, бензотиазолил, бензимидазолил, тетрагидрохинолинил, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, фенотиазинил и феноксазинил.
Термин "арилоксил" обозначает -O-арил.
Термин "отгидролизовываемая группа" обозначает группу, которая, будучи присоединенной к атому кислорода, образует эфир, способный гидролизоваться in vivo с образованием гидроксильной группы. Типичные отгидролизовываемые группы, которые могут быть замещены, включают ацильные, сульфонатные и фосфатные группы. Например, такие отгидролизовываемые группы включают блокированные или свободные аминокислотный, хемисукцинатный и никотинатный остатки.
Термин "атом галогена" обозначает атомы хлора, фтора, брома или иода. Термин "гало" обозначает хлор-, фтор-, бром- или иод-.
Термин "карбоцикл" относится к ароматическому или насыщенному либо частично насыщенному 5-14-членному моноциклическому или полициклическому кольцу, например 5-7-членному моноциклическому или 7-10-членному бициклическому кольцу, в которых все атомы кольца являются атомами углерода.
Термин "гетероцикл" относится к ароматическому или насыщенному либо частично насыщенному 5-14-членному моноциклическому или полициклическому кольцу, например 5-7-членному моноциклическому или 7-10-членному бициклическому кольцу, содержащему от одного до трех гетероатомов, выбранных из атомов азота, кислорода и серы, в котором каждый из гетероатомов азота и серы может быть окислен, а любой гетероатом азота может быть в виде катиона. Гетероциклическое кольцо может быть присоединено к любому подходящему гетероатому или атому углерода. Примерами таких гетероциклов являются декагидроизохинолинил, октагидротиено[3,2-c]пиридинил, пиперидинил, пиперазинил, азепинил, пирролил, пирролидинил, пиразолил, пиразолидинил, имидазолил, изобензофуранил, фуразанил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, тиантренил, триазинил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, хроменил, ксантенил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоазолил, фурил, тетрагидрофурил, тетрагидропиранил, тиенил, бензотиенил, бензо[b] тиенил, нафто[2,3-b]тиенил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, оксадиазолил, триазолил, тетрагидрохинолинил, тетрагидроизохинолинил, феноксатиенил, индолизинил, изоиндолил, индазолил, пуринил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хинолинил, тетрагидрохинолинил, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, изотиазолил, фенотиазинил и феноксазинил.
Термин "тиоэфир" включает S-арил, например фенилтио- и нафтилтиогруппу; S-гетероцикл, при этом гетероцикл насыщен или частично насыщен; S-(C5-C7)-циклоалкил; и S-алкил, например C1-C6-алкилтиогруппу. В тиоэфире -арил, -гетероцикл, -циклоалкил и -алкил могут быть замещены. Примером тиоэфира является "C1-C6-алкилтио", который представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до шести атомов углерода, присоединенных к атому серы. Типичные C1-C6-алкилтиогруппы включают метилтио-, этилтио-, пропилтио-, изопропилтио-, бутилтио-, в-бутилтио-, т-бутилтио-, пентилтио-, гексилтиогруппы и т.п.
Термин "меркапто" обозначает -SH.
Термин "амино" обозначает -NL1L2, где L1 и L2 предпочтительно независимо выбраны из атома кислорода, карбоцикла, гетероцикла, алкила, сульфонила и атома водорода; или NC(O)L3, где L3 - предпочтительно алкил, алкоксил, атом водорода или - NL1L2. Арильные, алкильные и алкоксигруппы могут быть замещены. Примером аминогрупп является C1-C4-алкиламиногруппа, которая представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода, присоединенных к аминогруппе. Типичные C1-C4-алкиламиногруппы включают метиламино-, этиламино-, пропиламино-, изопропиламино-, бутиламино-, в-бутиламиногруппы и т. п. Другим примером аминогруппы является ди(C1-C4)алкиламиногруппа, представляющая собой две линейные или разветвленные алкильные цепи, каждая из которых содержит от одного до четырех атомов углерода, соединенных с общей аминогруппой. Типичные ди(C1-C4)алкиламиногруппы включают диметиламино-, этилметиламино-, метилпропиламино-, этилизопропиламино-, бутилметиламино-, в-бутилметиламиногруппы и т. п. Примером аминогруппы является C1-C4-алкилсульфониламиногруппа, имеющая линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода, соединенных с сульфониламинной частью. Типичные C1-C4-алкилсульфониламиногруппы включают метилсульфониламино-, этилсульфониламино-, пропилсульфониламино-, изопропилсульфониламино-, бутилсульфониламино-, в-бутилсульфониламино-, т-бутилсульфониламиногруппы и т.п.
Термин "ацил" обозначает L6C(O)L4, где L6 - одинарная связь -O или -N и, кроме того, где L4 - предпочтительно алкил, аминогруппа, гидроксильная группа, алкоксильная группа или атом водорода. Алкильные и алкоксильные группы могут быть замещены. Типичным ацилом является C1-C4-алкоксикарбонил, представляющий собой линейную или разветвленную алкоксильную цепь, содержащую от одного до четырех углеродных атомов, соединенных с карбонильной частью. Типичные C1-C4-алкоксикарбонильные группы включают метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил и т. п. Другим типичным ацилом является карбоксигруппа, отличающаяся тем, что L6 представляет собой одинарную связь, а L4 - алкоксил, атом водорода или гидроксильную группу. Следующим типичным ацилом является N-(C1-C4)алкилкарбамоил (L6 - одинарная связь, а L4 - аминогруппа), представляющий собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода, соединенных с атомом азота карбамоильной части. Типичные N-(C1-C4)алкилкарбамоильные группы включают N-метилкарбамоил, N-этилкарбамоил, N-пропилкарбамоил, N-изопропилкарбамоил, N-бутилкарбамоил, N-т-бутилкарбамоил и т.п. Еще одним типичным ацилом является N,N-ди(C1-C4)алкилкарбамоил, который имеет две линейные или разветвленные алкильные цепи, каждая из которых содержит от одного до четырех атомов углерода, соединенных с атомом азота карбамоильной части. Типичные N,N-ди(C1-C4)алкилкарбамоильные группы включают N, N-диметилкарбамоил, N,N-этилметилкарбамоил, N,N-метилпропилкарбамоил, N,N-этилизопропилкарбамоил, N, N-бутилметилкарбамоил, N,N-втор-бутилэтилкарбамоил и т.п.
Термин "сульфинил" обозначает -SO-L5, где L5 - предпочтительно алкил, аминогруппа, арил, циклоалкил или гетероцикл. Алкил, арил, циклоалкил и гетероцикл могут все быть замещены.
Термин "сульфонил" обозначает -SO2-L5, где L5 - предпочтительно алкил, арил, циклоалкил, гетероцикл или аминогруппа. Алкил, арил, циклоалкил и гетероцикл могут все быть замещены. Примером сульфонила является C1-C4-алкилсульфонил, представляющий собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода, соединенных сульфонильной частью. Типичные C1-C4-алкилсульфонильные группы включают метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, в-бутилсульфонил, т-бутилсульфонил и т.п.
Как отмечено выше, многие из групп могут быть замещены. Для всех представленных здесь формул все химические группы могут быть замещены или незамещены в зависимости от того, позволяют ли валентности этих групп такие замещения, даже если определения химических групп явно не устанавливают замещенность или незамещенность групп. Например, если группа просто определена как алкил, это может быть замещенный или незамещенный алкил. Примеры заместителей для алкила и арила включают меркапто-, тиоэфирную, нитро-(NO2), аминогруппу, арилоксил, атом галогена, гидроксильную группу, алкоксил и ацил, равно как арил, циклоалкил и насыщенные и частично насыщенные гетероциклы. Примеры заместителей для гетероцикла и циклоалкила включают тот же список вышеупомянутых заместителей для алкила и арила, равно как сами арил и алкил.
Типичные замещенные арилы включают фенильное или нафтильное кольцо, замещенное одним или большим числом заместителей, предпочтительно от одного до трех, независимо выбранных из гало-, гидроксильной группы, морфолино(C1-C4)алкоксикарбонила, пиридил(C1-C4)алкоксикарбонила, гало(C1-C4)алкила, C1-C4-алкила, C1-C4-алкоксигруппы, карбоксигруппы, C1-C4-алкоксикарбонила, карбамоила, N-(C1-C4)алкилкарбамоила, амино-, C1-C4-алкиламино-, ди(C1-C4)алкиламиногруппы или группы формулы -(CH2)a-R7, где a = 1, 2, 3 или 4; а R7 является гидроксильной группой, C1-C4-алкокси-, карбоксигруппой, C1-C4-алкоксикарбонилом, аминогруппой, карбамоилом, C1-C4-алкиламино- или ди-(C1-C4)алкиламиногруппой.
Другим замещенным алкилом является гало(C1-C4)алкил, который представляет собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода с 1-3 атомами галогена, присоединенными к ней. Типичные гало(C1-C4)алкильные группы включают хлорметил, 2-бромэтил, 1-хлоризопропил, 3-фторпропил, 2,3-дибромбутил, 3-хлоризобутил, иод-т-бутил, трифторметил и т.п.
Другим замещенным алкилом является гидрокси-(C1-C4)алкил, представляющий собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода с присоединенными к ним гидроксильными группами. Типичные гидрокси(C1-C4)алкильные группы включают гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 2-гидроксиизопропил, 4-гидроксибутил и т.п.
Еще одним замещенным алкилом является C1-C4-алкилтио(C1-C4)алкил, представляющий собой линейную или разветвленную C1-C4-алкильную группу с присоединенной к ней C1-C4-алкилтиогруппой. Типичные C1-C4-алкилтио(C1-C4)алкильные группы включают метилтиометил, этилтиометил, пропилтиопропил, в-бутилтиометил и т.п.
Еще одним типичным замещенным алкилом является гетероцикл(C1-C4)алкил, представляющий собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода с присоединенным к ним гетероциклом. Типичные гетероцикл(C1-C4)алкилы включают пирролилметил, хинолинилметил, 1-индолилэтил, 2-фурилэтил, 3-тиен-2-илпропил, 1-имидазолилизопропил, 4-тиазолилбутил и т.п.
Еще одним замещенным алкилом является арил(C1-C4)алкил, представляющий собой линейную или разветвленную алкильную цепь, содержащую от одного до четырех атомов углерода с присоединенной к ним арильной группой. Типичные арил(C1-C4)алкильные группы включают фенилметил, 2-фенилэтил, 3-нафтилпропил, 1-нафтилизопропил, 4-фенилбутил и т.п.
Гетероцикл может, например, быть замещен 1, 2 или 3 заместителями, независимо выбранными из галогруппы, гало(C1-C4)алкила, C1-C4-алкила, C1-C4-алкоксигруппы, карбоксигруппы, C1-C4-алкоксикарбонила, карбамоила, N-(C1-C4)алкилкарбамоила, амино-, C1-C4-алкиламино-, ди(C1-C4)алкиламиногруппы или группы, имеющей структуру -(CH2)a-R7, где a = 1, 2, 3 или 4 и R7 является гидроксильной, C1-C4-алкоксильной, карбоксигруппой, C1-C4-алкоксикарбонилом, аминогруппой, карбамоилом, C1-C4-алкиламино- или ди(C1-C4)алкиламиногруппой.
Примеры замещенных гетероциклов включают 3-N-т-бутилкарбоксамиддекагидроизохинолинил, 6-N-т-бутилкарбоксамидоктагидротиено[3,2-c] -пиридинил, 3-метилимидазолил, 3-метоксипиридил, 4-хлорхинолинил, 4-аминотиазолил, 8-метилхинолинил, 6-хлорхиноксалинил, 3-этилпиридил, 6-метоксибензимидазолил, 4-гидроксифурил, 4-метилизохинолинил, 6,8-дибромхинолинил, 4,8-диметилнафтил, 2-метил-1,2,3,4-тетрагидроизохинолинил, N-метилхинолин-2-ил, 2-т-бутоксикарбонил-1,2,3,4-изохинолин-7-ил и т.п.
Типичные гетероциклические кольцевые системы, изображенные в виде А или Б, включают: (1) 5-членные моноциклические кольцевые группы типа тиенила, пирролила, имидазолила, пиразолила, фурила, изотиазолила, фуразанила, изоксазолила, тиазолила и т.п.; (2) 6-членные моноциклические группы типа пиридила, пиразинила, пиримидинила, пиридазинила, триазинила и т.п.; и (3) полициклические гетероциклические кольцевые группы типа декагидроизохинолинила, октагидро-тиено[3,2-c] пиридинила, бензо[b] тиенила, нафто[2,3-b]тиантренила, изобензофуранила, хроменила, ксантенила, и полностью или частично насыщенные их аналоги. Циклоалкил может быть дополнительно замещен 1, 2 или 3 заместителями, независимо выбранными из галогруппы, гало(C1-C4)алкила, C1-C4-алкила, C1-C4-алкокси-, карбоксигруппы, C1-C4-алкоксикарбонила, карбамоила, N-(C1-C4)алкилкарбамоила, амино- , C1-C4-алкиламино-, ди(C1-C4)алкиламиногруппы или группы, имеющей структуру -(CH2)a-R7, где a = 1, 2, 3 или 4 и R7 является гидроксильной группой, C1-C4-алкокси-, карбоксигруппой, C1-C4-алкоксикарбонилом, аминогруппой, карбамоилом, C1-C4-алкиламино- или ди(C1-C4)алкиламиногруппой. Типичные замещенные циклоалкильные группы включают 3-метилциклопентил, 4-этоксициклогексил, 5-карбоксициклогептил, 6-хлорциклогексил и т.п.
Типичные замещенные отгидролизовываемые группы включают N-бензилглицил, N-Cbz-L-валил и N-метилникотинат.
Соединения по настоящему изобретению имеют по крайней мере два асимметричных центра, обозначенные звездочкой в формуле I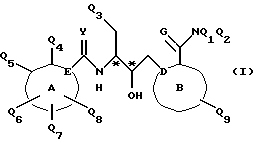
Вследствие этого соединения по настоящему изобретению могут существовать в любой из возможных стереоизомерных форм и могут быть использованы в виде смесей стереоизомеров, оптически активных или рацемических, или могут использоваться в виде существенно чистых стереоизомеров, то есть стереоизомеров по меньшей мере 95%-ой чистоты. Все асимметричные формы, индивидуальные стереоизомеры и их комбинации являются предметом настоящего изобретения.
Индивидуальные стереоизомеры могут быть получены из их соответствующих предшественников методами, описанными выше, путем разделения рацемических смесей или выделения диастереомеров. Разделение может быть осуществлено в присутствии разделяющего агента, с помощью хроматографии, посредством перекристаллизации или некоторой комбинацией этих известных специалистам методов. Дальнейшие детали относительно методов разделения можно найти в Jacques et. al. Enantiomers, Racemates and Resolutions. John Wiley & Sons, 1981 [1]. Предпочтительная чистота соединений по настоящему изобретению превышает 50%. Более предпочтительной является по меньшей мере 75%-ная чистота. Еще более предпочтительны соединения с чистотой более 90%. Еще более предпочтительна по меньшей мере 95%-ная чистота, более предпочтительна по меньшей мере 97%-ная чистота и наиболее предпочтительна по меньшей мере 99%-ная чистота.
Как упомянуто выше, изобретение включает фармацевтически приемлемые соли соединений, описываемых формулой I. Соединение по настоящему изобретению может содержать существенно кислотные, существенно основные или оба этих вида функциональных групп и соответственно реагировать с любыми неорганическими или органическими основаниями и неорганическими и органическими кислотами, образуя фармацевтически приемлемую соль.
Используемый здесь термин "фармацевтически приемлемая соль", относится к солям соединений вышеупомянутой формулы, которые являются существенно нетоксичными для живых организмов. Типичные фармацевтически приемлемые соли включают соли, получаемые в реакции соединений по настоящему изобретению с минеральной или органической кислотой или неорганическим основанием. Реактанты в общем случае объединяются во взаимном растворителе типа диэтилового эфира или бензола для солей кислот либо воды или спиртов для солей оснований. Соли обычно выпадают в осадок из раствора в течение периода времени продолжительностью от приблизительно одного часа до десяти дней и могут быть выделены фильтрованием или другими обычными методами. Такие соли известны как соли кислот и оснований.
Кислоты, которые можно использовать для образования соответствующих солей - это неорганические кислоты типа соляной, бромисто-водородной, иодисто-водородной, серной, фосфорной кислоты и т.п., и органические кислоты типа п-толуолсульфоновой, метансульфоновой, щавелевой, п-бромфенилсульфоновой, угольной, янтарной, лимонной, бензойной, уксусной кислоты и т.п.
Примеры фармацевтически приемлемых солей: сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, однозамещенный фосфат, двузамещенный фосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капроат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, цебакат, фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, сульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, g-гидроксибутират, гликолат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и т.п.
Предпочтительными фармацевтически приемлемыми солями кислот являются соли, образованные такими минеральными кислотами, как соляная и бромисто-водородная кислоты и соли, образованные такими органическими кислотами, как малеиновая и метансульфоновая кислоты.
Соли оснований включают таковые, образованные неорганическими и органическими основаниями, такими как аммоний или гидроксиды, карбонаты, бикарбонаты и т.п. щелочных или щелочно-земельных металлов. Подобные основания, полезные в получении солей соединений по настоящему изобретению, таким образом, включают гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид кальция, карбонат кальция и т.п. Особо предпочтительными являются формы солей, содержащих калий и натрий.
Следует признать, что индивидуальный противоион, образующий часть любой соли по настоящему изобретению, не является критичным, пока соль в целом фармакологически приемлема и пока противоион не вносит нежелаемых свойств в соль в целом.
Соединения формулы I могут представлять собой пролекарства. Например соединения, в которых по меньшей мере один из Q4-Q8 является -O-J, как определено выше, можно использовать в качестве пролекарств с такими улучшенными фармацевтическими свойствами, как например фармакокинетические свойства, а именно улучшенной биодоступностью или растворимостью. Приготовление пролекарств может осуществляться в результате взаимодействия соединения формулы I в котором по меньшей мере один из Q4-Q8 является -O-H, с например, активированной аминоацильной, фосфорильной или гемисукцинильной производной.
Настоящее изобретение, кроме того, охватывает прописи фармацевтических препаратов, включающих эффективное количество соединения формулы I или его фармацевтически приемлемой соли в комбинации с фармацевтически приемлемым носителем типа разбавителя или эксипиента.
Настоящее изобретение далее охватывает способ лечения СПИД, включающий введение носителю вируса или пациенту, такому как примат, эффективного количества соединения по настоящему изобретению.
Настоящее изобретение, кроме того, охватывает способ ингибирования репликации ВИЧ, включающий введение в ВИЧ-инфицированную клетку, в клетку, восприимчивую к ВИЧ-инфекции, или носителю вируса или пациенту, такому как примат, эффективного количества соединения по настоящему изобретению.
Настоящее изобретение охватывает новые соединения, подпадающие под формулу I, описанную выше, которые являются полезными для лечения ВИЧ-инфекции и/или СПИД.
Предпочтительными реализациями формулы I являются: [3S-(3R*,4aR*,8aR*, 2'S*,3'S*)] -2-[2'- гидрокси-3'-фенилтиометил- 4'-аза-5'-оксо-5'-(2''- метил-3''-гидроксифенил) пентил] декагидроизохинолин -3-N-т-битилкарбоксамид, и его фармацевтически приемлемые соли, особенно соль метансульфоновой кислоты, и его пролекарственные аналоги, в которых 3''-гидроксильная группа преобразована в -O-J, как определено выше, особенно гидрохлорид однозамещенного фосфата; и [6S-(6R*, 3aS*, 7aR*,2'S*,3'S*)]- 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] октагидротиено[3,2-c]пиридин-6-N-т-бутилкарбоксамид и его фармацевтически приемлемые соли, особенно соль метансульфоновой кислоты, и его пролекарственные аналоги, в которых 3''-гидроксильная группа преобразована в -O-J, как определено выше.
Предпочтительными соединениями являются: 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид: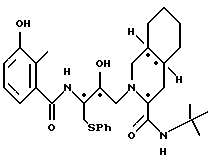
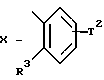
где T2 - атом водорода или метил;
Z1 - группа, имеющая структуру: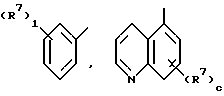
где R7 - атом водорода, C1-C4-алкил, гало-, нитро-, амино-, гидроксильная группа;
a = 1, 2 или 3;
c = 1;
или их фармацевтически приемлемую соль. Из этих соединений более предпочтительными являются те соединения, где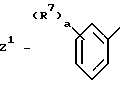
R7 - атом водорода, метил, этил, гидроксильная группа, аминогруппа, атом хлора; R1 - представляет собой -S-фенил или -S-нафт-2-ил и R3 представляет собой -C(O)NR4R4, или их фармацевтически приемлемая соль.
Особо предпочтительными из этих соединений являются те соединения, где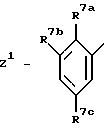
где R7a - атом водорода, метил, этил, атомы хлора, брома или фтора; R7b - атом водорода, гидроксильная группа, атом хлора или аминогруппа; R7c - атом водорода, гидроксильная группа или аминогруппа; R3 представляет собой -C(O)NH(т-бутил), или их фармацевтически приемлемая соль.
Предпочтительными соединениями являются: 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид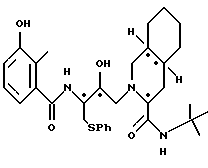
2-[2'-гидрокси-3'-фенилтиометил-4'-aзa-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамида метансульфоновой кислоты соль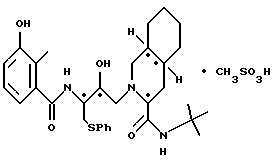
3''-однозамещенкый фосфат 2-[2'-гидрокси-3'-фенилтиометил-4'-аза- 5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил] декагидроизохинолин-3- N-т-бутилкарбоксамида гидрохлорида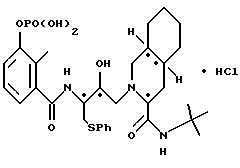
2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил]-октагидро-тиено[3,2-c]пиридин-6-N-т-бутилкарбоксамид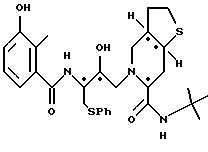
и 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] -октагидро-тиено[3,2-c]пиридин-6-N-т-бутилкарбоксамида метансульфоновой кислоты соль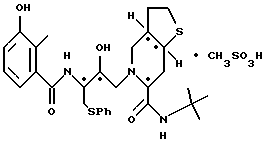
Каждая из вышеупомянутых пяти формул имеет пять асимметричных центров и таким образом определяет соединение, выбранное из 32 индивидуальных стереоизомеров и любой смеси из двух или более стереоизомеров.
Предпочтительные стереоизомеры этих соединений: [3S-(3R*,4aR*,8aR*,2'S*, 3'S*)] - 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид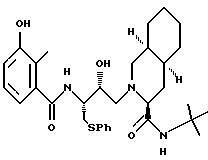
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'-гидрокси-3'-фенилтиометил- 4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил] декагидроизохинолин- 3-N-т-бутилкарбоксамида метансульфоновой кислоты соль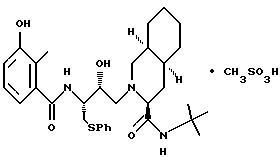
3''-однозамещенный фосфат [3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)]-2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'- оксо-5'-(2''-метил-3''-гидроксифенил)пентил]декагидроизохинолин-3-N- т-бутилкарбоксамида гидрохлорида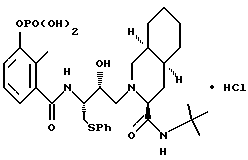
[6S-(6R*, 3aS*, 7aR*, 2'S*,3'S*)]- 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамид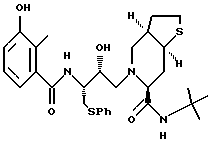
и [6S-(6R*, 3aS*,7aR*,2'S*,3'S*)]- 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] октагидро-тиено[3,2-c] пиридин-6-N-т-бутилкарбоксамида метансульфоновой кислоты соль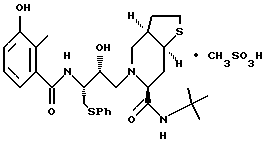
Другие соединения настоящего изобретения включают: [3S-(3R*,4aR*,8aR*, 2'S*, 3'R*)] - 2-[2'-гидрокси-3'-фенилметил-4'- аза-5'-оксо-5'-(2''-пропил-3''- гидроксифенил)пентил] декагидроизохинолин-3- N-т-бутилкарбоксамид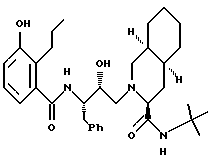
[2S-(2R*, 2'S*, 3'S*)]-1-[2'-гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-(3''-гидрокси-2''-метилфенил)пентил] - 4-пирид-3''-илметилпиперазин-2-N-т-бутилкарбоксамид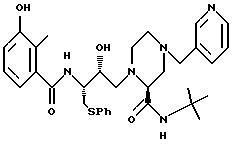
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)]- 2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(1'', 2'', 3'',4''- тетрагидрохинолин-5''-ил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид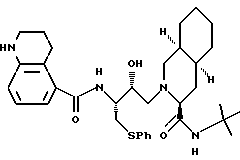
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид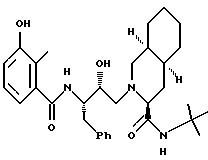
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-этил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид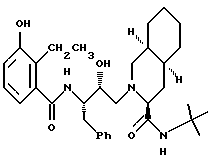
[2'R-(2'R*, 3'S*)] -N-т-бутил-2-[2'-гидрокси-3'- нафт-2-илтиометил-4'-аза-5'-оксо-5'-(1'',2'',3'',4''-тетрагидрохинолин- 5''-ил)пентил]бензамид;
[2'R-(2'R*, 3'S*)] -N-т-бутил-2-[2'-гидрокси-3'-нафт-2- илтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил]бензамид;
[2'R-(2'R*, 3'S*)] -N-т-бутил-2-[2'-гидрокси-3'-нафт-2- илтиометил-4'-аза-5'-оксо-5'-(2''-метил-3'',5''-диаминофенил)пентил]бензамид;
[2'R-(2'R*, 3'S*)] -N-т-бутил-2-[2'-гидрокси-3'-нафт- 2-илтиометил-4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил]-1- нафтиламид; и
[2'R-(2'R*, 3'S*)] -N-т-бутил-2-[2'-гидрокси-3'- нафт-2-илтиометил-4'-аза-5'-оксо-5'-(2''-хлор-3''-аминофенил)пентил]-1- нафтиламид, или фармацевтически приемлемую соль любого из вышеупомянутых наиболее предпочтительных соединений.
Соединения формулы I могут быть получены согласно следующей реакции I.
Реакция I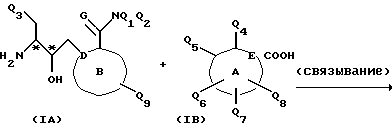
где обозначения для формулы I определены выше.
Реакция I представляет собой стандартную реакцию связывания, обычно применяемую в синтезе амидов или пептидов, которая осуществляется посредством взаимодействия подходяще замещенного амина формулы IА с подходяще замещенной карбоновой кислотой формулы IБ в апротонном растворителе или смеси растворителей. Обычно реакция протекает в присутствии или отсутствие активирующего агента, предпочтительно в присутствии активирующего агента, и в присутствии связывающего реагента. Типичными апротонными растворителями для этой реакции являются тетрагидрофуран и диметилформамид, или смесь таких растворителей. Обычно реакцию проводят при температуре приблизительно от -30oC до приблизительно 25oC. Аминный реактант в основном используется в эквимолярных соотношениях относительно карбоново-кислотного реактанта в присутствии эквимолярного количества либо небольшого избытка связывающего реагента. Типичные связывающие реагенты включают карбодиимиды, такие как дициклогексилкарбодиимид (DCC) и N,N'-диэтилкарбодиимид; имидазолы, такие как карбонилдиимидазол; а также такие реагенты, как бис(2-оксо-3-оксазолидинил)фосфинхлорид (BOP-Cl) или N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ). Предпочтительным связывающим реагентом для этой реакции является DCC. Для проведения этой реакции предпочтительно использовать активирующий агент; предпочтительным активирующим агентом является гидроксибензотриазол моногидрат (HOBT•H2O).
По окончании реакции соединение может быть при желании выделено с помощью известных специалистам методов, например закристаллизовано и затем собрано фильтрованием, или растворитель, использованный в реакции, может быть удален экстракцией, упариванием или декантацией. Если необходимо, соединение может быть далее очищено общими методами, такими как кристаллизация или хроматография на таких твердых носителях, как силикагель или оксид алюминия.
Исходные соединения формулы IА могут быть получены в соответствии со способами, показанным на реакционной схеме А (см. в конце описания).
Реакционная схема А выполняется последовательным проведением реакций 1-7. По окончании каждой из реакций промежуточное соединение может быть при желании выделено с помощью известных специалистам методов, например соединение может быть закристаллизовано и затем собрано фильтрованием или растворитель, использованный в реакции, может быть удален экстракцией, упариванием или декантацией. Если необходимо, перед осуществлением следующего этапа реакционной схемы промежуточное соединение может быть далее очищено общими методами, такими как кристаллизация или хроматография на таких твердых носителях, как силикагель или оксид алюминия.
Реакция A.1 представляет собой преобразование защищенной по аминогруппе карбоновой кислоты, имеющей структуру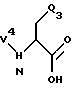
к соответствующему смешанному ангидриду в условиях, известных специалистам. Например, защищенная по аминогруппе карбоновая кислота может реагировать с C1-C6-алкилхлорформиатом, таким как изобутилхлорформиат, предпочтительно в присутствии акцептора кислоты. Предпочтительными акцепторами кислоты являются триалкиламины, предпочтительно триэтиламин. Реакция в типичном случае выполняется в апротонном растворителе типа этилацетата. Выбор растворителя не критичен, пока используемый растворитель инертен к протекающей реакции и реактанты достаточно солюбилизированы, чтобы произвести желаемую реакцию. Получающийся смешанный ангидрид используется в реакции А.2 преимущественно без дополнительных стадий выделения или очистки.
Реакция А.2 выполняется в два этапа. Сначала раствор гидроксида натрия, покрытый слоем эфира, предпочтительно диэтилового эфира, реагирует с большим избытком N-метил-N-нитро-N-нитрозогуанидина с образованием диазометанового реактанта. Гидроксид натрия предпочтительно используется в виде водного раствора с концентрацией приблизительно от четырех до шести моль/л. По окончании реакции органический слой высушивают над высушивающим агентом типа гидроксида калия. Этот раствор затем реагирует со смешанным ангидридом, полученным в описанной выше реакции А.1 с образованием соответствующего α-диазокарбонильного соединения. Диазометановый реактант предпочтительно используется в этой реакции без выделения или очистки. Реакция обычно проводится при температуре от приблизительно -50oC до приблизительно -10oC, предпочтительно при температуре около -20oC.
В реакции А.3 α-диазокарбонильное соединение, полученное по реакции А.2, взаимодействует с кислотой формулы H-ZZ, где ZZ является галогруппой, обычно в апротонном растворителе, например диэтиловом эфире, с образованием α-галокарбонильного соединения. Предпочтительным кислотным реактантом является соляная кислота, выступающая в роли соответствующего α-хлоркарбонильного соединения. Реакция обычно проводится при температуре от приблизительно -30oC до приблизительно 0oC. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Кислотный реактант обычно добавляется в форме безводного газа малыми порциями до существенно полного завершения реакции. Ход реакции можно контролировать тонкослойной хроматографией.
В реакции А.4 карбонильная часть соединения, полученного в реакции А.3, восстанавливается в известных стандартных условиях с образованием соответствующего α-хлоргидроксильного соединения. Например соединение, полученное в реакции А. 3, смешивают с восстанавливающим агентом в смеси растворителей. Типичные восстанавливающие агенты включают боргидрид натрия, боргидрид лития, боргидрид цинка, диизобутилалюмогидрид и бис(2-метоксиэтокси)алюмогидрид натрия. Предпочтительным восстанавливающим агентом является боргидрид натрия. Типичные смеси растворителей включают протонные и апротонные смеси типа тетрагидрофуран/вода. Выбор растворителя не критичен, пока используемый растворитель инертен к протекающей реакции и реактанты достаточно солюбилизированы, чтобы произвести желаемую реакцию. Реакция обычно проводится при температуре приблизительно -10oC, предпочтительно при температуре около 0oC.
В реакции А.5 α-хлоргидроксильное соединение, полученное в реакции А.4, обрабатывают сильным основанием для образования соответствующего эпоксида в стандартных, хорошо известных специалистам в этой области условиях. Например α-хлоргидроксильное соединение может взаимодействовать со смесью гидроксид калия/этанол в спиртовом растворителе, таком как этанол. Обычно реакцию проводят в интервале температур от приблизительно 0oC до температуры дефлегмации растворителя. Предпочтительно реакцию осуществляют при комнатной температуре.
В реакции А.6 эпоксид, полученный в реакции А.5, взаимодействует с гетероциклическим реактантом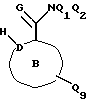
обычно в спиртовом растворителе при температуре в пределах от приблизительно 20oC до 100oC. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Типичные растворители для этой реакции включают спирты, предпочтительно изопропанол или этанол. Предпочтительно реакцию осуществляют при температуре около 80oC.
Реакция А.7 - стандартная реакция снятия защиты с аминогруппы с использованием методов, известных специалистам в данной области, с целью получения соответствующего амина, который применяется в вышеуказанной реакции I. Этот амин можно вводить в реакцию без очистки, но предпочтительно, чтобы сначала он был очищен.
Соединения формулы IА, где Q3 представляет собой -S-арил, обычно получают в результате сначала взаимодействия защищенного по аминогруппе серина с трифенилфосфином и диэтилазодикарбоксилатом (DEAD) в апротонном растворителе при температуре приблизительно от -80oC до 0oC с образованием соответствующего β-лактона. Обычно реакцию проводят в эфире, таком как тетрагидрофуран, при температуре приблизительно от -80oC до -50oC. Затем раскрывают лактоновое кольцо, обеспечивая получение соединения структуры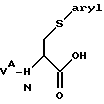
обычно путем взаимодействия лактона с подходяще замещенным тиоанионом, имеющим структуру -S-арила. Тиоанионное соединение получают преимущественно при взаимодействии соответствующего тиола с сильным основанием, таким как гидрид натрия или гидрид калия. Обычно эту реакцию проводят в апротонном растворителе при температуре от около 0oC до приблизительно 40oC и в инертной атмосфере, например в азоте. Типичными растворителями для этой реакции являются эфиры, предпочтительно тетрагидрофуран.
В альтернативном случае соединения формулы IА, где Q3 представляет собой -S-арил, могут быть получены с использованием способов, детально описанных в Photaki. JACS, 85, 1123 (1963) [2], Sasaki N.A. et. al. Tetrahedron Letters, 21, 6069, (1987) [3]. Например, соединения могут быть получены в результате реакции дважды защищенного серина (защищенного по карбоксильной и аминогруппам) с толуолсульфонилхлоридом в присутствии диметиламинопиридина (DMAP) и акцептора кислоты типа пиридина в апротонном растворителе типа метиленхлорида с образованием соответствующего толуолсульфоната, который может затем реагировать с подходяще замещенным тиоанионом, имеющим структуру -S-арила. Тиоанион предпочтительно образуется, как описано выше, в результате реакции соответствующего тиола с сильным основанием. Карбоксилзащитная группа может быть удалена из полученного дважды защищенного арилтиоаланина в условиях, известных специалистам.
Гетероциклические реактанты формулы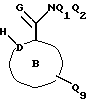
примененные в реакции А.6, могут быть получены с использованием способов и методов, известных специалистам. Например, их обычно получают из соответствующих защищенных по аминогруппе аминокислот путем кислотной активации с последующей обработкой алкиламином. Эта реакция обычно осуществляется в присутствии акцептора кислоты типа N-метилморфолина. После удаления аминозащитных групп стандартными химическими способами получают желаемые гетероциклические реактанты. В частности, [3S-(3R*,4aR*,8aR*)]- декагидроизохинолин-3-N-т-бутилкарбоксамид получали, используя 2S-1,2,3,4-тетрагидро-3-изохинолинкарбоновую кислоту, следующим способом:
1) защита аминогруппы (t-Boc);
2) кислотная активация/взаимодействие с т-бутиламином;
3) каталитическое гидрирование;
4) снятие защиты с аминогруппы.
Пиперазиновые реактанты могут быть получены путем превращения подходяще замещенного пиразинового соединения в соответствующее пиперазиновое соединение способами, известными специалистам, преимущественно с использованием каталитического гидрирования. Например, гидрирование может быть выполнено путем смешения пиразинового реактанта с катализатором в атмосфере водорода в апротонном растворителе при температуре от приблизительно 0oC до приблизительно 60oC. Подходящими катализаторами являются палладий на угле, металлическая платина, окись платины и т.п. Предпочтительным катализатором является окись платины. Типичные растворители для этой реакции включают тетрагидрофуран, диметилформамид или смесь тетрагидрофурана и диметилформамида.
Атом азота в полученном пиперазиновом реактанте может быть проалкилирован известными специалистам способами. Например может быть проведена реакция между пиперазиновым реактантом и гало(C1-C4)алкилом или галометилпиридином, такими как йодистый метил или хлорметилпиридин. Предпочтительными галозаместителями являются хлор, бром и иод. Реакцию проводят при температурах от приблизительно 0oC до 60oC во взаимно инертном растворителе и в присутствии акцептора кислоты. Предпочтительным акцептором кислоты является карбонат калия. Типичные растворители включают смесь протонных и апротонных растворителей, таких как ацетонитрил и вода. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции.
В альтернативном случае алкилированный пиперазиновый реактант может быть получен с использованием восстановительного аминирования. Например может быть проведена реакция между полученным выше пиперазиновым реактантом и альдегидом (а именно, 3-пиридинкарбоновым альдегидом, этаналем, пропаналем) или кетоном в присутствии восстанавливающего агента и кислоты. Обычно реакция проводится в спиртовом растворителе, таком как метанол, этанол или изопропанол. Типичные восстанавливающие агенты включают боргидрид натрия, цианборгидрид лития, цианборгидрид натрия и т.п. Предпочтительным восстанавливающим агентом является цианборгидрид натрия. Типичные кислоты включают любую кислоту, такую как соляная, серная, метансульфоновая и уксусная кислоты. Предпочтительной кислотой является уксусная кислота.
Промежуточный реактант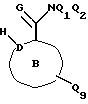
также может быть получен в виде соединения формулы 2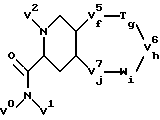
где V0 и V1 - независимо атомы водорода, C1-C6-алкил или гидрокси(C1-C6)алкил;
V2 - атом водорода, аминозащитная группа или группа формулы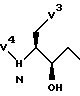
V3 представляет собой -(CH2)t-V3';
t = 0, 1, 2, 3 или 4;
V3' является арилом, -O-арилом или -S-арилом;
V4 - атом водорода или аминозащитная группа; f, h и j каждый независимо 0, 1 или 2; g и i каждый независимо 0 или 1;
V5 представляет собой -CH2-, -CHV5'- или -CV5'V5'-;
V6 представляет собой -VH2-, -CHV6'- или -CV6'V6'-;
V7 представляет собой -CH2-, -CHV7'- или -CV7'V7'-;
каждый из V5', V6' и V7' независимо выбран из галогруппы, гидроксильной группы, C1-C6-алкила, гало(C1-C6)алкила, гидрокси(C1-C6)алкила, C1-C6-алкокси-, C1-C6-алкилтио-, амино- или цианогруппы;
T и W - независимо -S-, -S(O)-, -S(O)2-, -O-, -NH- или -(V9)-; и
V9 представляет собой C1-C6-алкил, арил(C1-C6)алкил, арил или ацил;
с условиями, что:
g и i не могут оба быть 0;
сумма f, g, h, i и j должна быть 2, 3, 4 или 5;
если V5 представляет собой -CV5'V5'-, то V6 должен быть -CH2- или - CHV6'-; а V7 должен быть -CH2- или -CHV7'-;
если V6 представляет собой -CV6'V6'-, то V5 должен быть -CH2- или - CHV5'-; а V7 должен быть -CH2- или -CHV7'-;
если V7 представляет собой -CV7'V7'-, то V5 должен быть -CH2- или - CHV5'-; а V6 должен быть -CH2- или -CHV6'-;
или его фармацевтически приемлемая соль.
Соединения формулы 3 могут быть получены в соответствии с реакционной схемой II (см. в конце описания).
Реакционная схема II выполняется последовательным проведением реакций 1-3 (или 1-5). По окончании каждой из реакций промежуточное соединение может быть при желании выделено с помощью известных специалистам методов, например закристаллизовано и затем собрано фильтрованием или растворитель, использованный в реакции, может быть удален экстракцией, упариванием или декантацией. Если необходимо, перед осуществлением следующего этапа реакционной схемы, промежуточное соединение может быть далее очищено общими методами типа кристаллизации или хроматографии на таких твердых носителях, как силикагель или оксид алюминия.
Реакция II. 1 обычно осуществляется путем активации карбоново-кислотной части с использованием, например DCC или смешанного ангидрида, такого как изобутил, с последующей реакцией с первичным или вторичным амином, имеющим формулу NV0V1, где V0 и V1 аналогичны вышеупомянутым для формулы (2). Обычно реакцию проводят в неполярном апротонном растворителе или смеси растворителей в присутствии или отсутствие акцептора кислоты при температуре от приблизительно -20oC до приблизительно 25oC с получением соответствующего амида. Подходящие растворители для этой реакции включают эфиры и хлорированные углеводороды, предпочтительно диэтиловый эфир, хлороформ или метиленхлорид. Предпочтительно эту реакцию проводят в присутствии акцептора кислоты типа третичного амина, предпочтительно триэтиламина. Амид, получаемый в этой реакции, может быть выделен или использован в реакции далее, как показано в реакции II.2.
Реакцию II. 2 обычно проводят с соединением, полученным в реакции II.1, используя методы, детально описанные в Heteroatom Manipulation. In: Comprehensive Organic Synthesis. Vol. 6 (Barry M. Trost, ed.), pp. 736-746, (1991) [4] . В общем, соответствующе замещенное моноциклическое кольцо реагирует с альдегидом, типа формальдегида или трихлорацетальдегида в присутствии кислоты. Кислота может использоваться как растворитель. Типичные кислоты включают соляную, бромисто-водородную, серную, уксусную, трифторуксусную и т.п. К реакционной смеси может дополнительно быть добавлен сорастворитель. Выбор сорастворителя не критичен, пока применяемый сорастворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Типичные растворители для этой реакции включают галогенированные растворители, такие как метиленхлорид, трихлорэтан, четыреххлористый углерод и т.п. В альтернативном случае альдегид может быть получен in situ с использованием, например, диметоксиметана и подходящей кислоты.
В реакции II. 3 соединение, выделенное после проведения реакции II.2, восстанавливают, как показано выше, с получением насыщенного гетероциклического соединения. Предпочтительным методом восстановления является каталитическое гидрирование. Типичные катализаторы включают катализаторы на основе палладия, катализаторы на основе родия (например родий на алюминии) и катализаторы на основе рения. Предпочтительными катализаторами являются катализаторы в виде палладия на угле. Подходящие растворители для этой реакции включают C1-C4-спирты, тетрагидрофуран, уксусную кислоту в спирте, этилацетат и т.п. Предпочтительным растворителем является этанол. Реакцию обычно проводят в атмосфере водорода при давлении от приблизительно 1000 до приблизительно 4000 пси, при температуре от примерно 25oC до примерно 150oC. Предпочтительно реакцию проводят в атмосфере водорода при давлении в интервале от 2000 до 3000 psi и температуре в интервале от 50oC до 100oC. Катализатор в общем случае используют в количествах, лежащих в пределах от примерно эвимолярных до примерно двенадцатикратного избытка (по весу) над реактантом, предпочтительно от шести- до десятикратного избытка (по весу) катализатора над субстратом.
Реакции II.4 и II.5 можно использовать для получения соединений формулы (3), которые соответствуют соединениям формулы (2), где V2 представляет собой группу формулы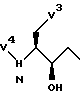
и V3 и V4 определены так же, как и ранее для формулы (2), включая определения для V3' и t.
Реакция II. 4 представляет собой стандартную реакцию снятия защит с аминогрупп с использованием способов и методов, известных специалистам, приводящую к получению соответствующего амина, который затем используется в реакции II. 5. Предпочтительными являются химические способы снятия защит. Например, защитные группы с соединения, выделенного после проведения реакции II. 3, могут быть сняты с использованием триметилсилилиодида (TMSI) в апротонном растворителе или смеси растворителей при температуре от приблизительно 10oC до 60oC, предпочтительно при температуре от приблизительно 20oC до 40oC. Типичные растворители включают метиленхлорид, ацетонитрил, трихлорэтан и т.п.
В реакции II.5 эпоксид, полученный ранее в реакции А.5, в котором группа Q3 заменена на V3, реагирует с соединением, полученным в реакции II.4, в спиртовом растворителе при температуре от приблизительно 20oC до 100oC. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Типичные растворители для этой реакции включают спирты, предпочтительно изопропанол и этанол. Реакция предпочтительно выполняется при температуре приблизительно 80oC.
С соединения, полученного в реакции II.5, могут, кроме того, быть сняты защитные группы, что приводит к получению соединения формулы (3), в котором VA является атомом водорода.
Эпоксид, используемый в реакции II.5, может быть синтезирован с использованием вышеупомянутой реакционной схемы А, в которой Q3 заменен на V3.
Карбоново-кислотный реактант формулы (IБ)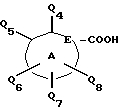
используемый в реакционной схеме I, не всегда коммерчески доступный, может быть получен с использованием известных процедур. В частности, этот реактант может быть получен дальнейшим замещением и/или окислением коммерчески доступного карбоциклического или гетероциклического соединения. Например, карбоциклические или гетероциклические соединения формулы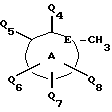
могут быть окислены с применением известных специалистам способов. Более конкретно соединение формулы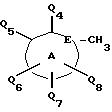
может взаимодействовать с окисляющим агентом типа диоксида селена или перманганата калия при температурах от приблизительно 0oC до 200oC во взаимно инертном растворителе, таком как вода или дифениловый эфир.
Второй метод получения соединений формулы (IБ) включает защиту подходяще замещенной карбоксилированной карбоциклической или гетероциклической группы с помощью карбоксизащитной группы с последующим замещением карбоциклической или гетероциклической группы известными специалистам способами. Карбоксизащитная группа может затем быть удалена, известными способами с получением желаемого карбоново-кислотного реактанта формулы (IБ).
Термин "карбоксизащитная группа", введенный в спецификации терминов, относится к заместителям карбоксильной группы, в общем случае используемым для ее блокирования или защиты при проведении реакций с другими функциональными группами соединения. Примеры таких карбоксизащитных групп включают метил, п-нитробензил, п-метилбензил, п-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2', 4,4'-тетраметоксибензгидрил, т-бутил, т-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4',4''-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, т-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, b-(ди(н-бутил)метилсилил)этил, п-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)проп-1-ен-3-ил и подобные им остатки. Предпочтительный метод защиты карбоксильных групп, обеспечивающий желаемое их замещение, включает превращение карбоксильной части в амидную с последующим обратным гидролизом амида. Дополнительные примеры таких групп имеются в Haslam E. Protective Groups in Organic Chemistry. J.G.W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapter 5 [5], Greene T.W. Protective Groups in Organic Synthesis. John Wiley & Sons, New York, N.Y., 1981, Chapter 5 [6].
Предпочтительная процедура защиты карбоксильной части включает кислотную активацию карбоксильной части с последующим образованием амида. Например карбоксильная часть может быть превращена в ацилгалогенид, ацилангидрид, ацилимидазол и т.п. предпочтительно в присутствии акцептора кислоты с образованием активизированной карбоксильной группы. Обычно используемый коммерчески доступный кислый хлорид устраняет необходимость в дальнейшей кислотной активации. Предпочтительными акцепторами кислоты являются триалкиламины, предпочтительно триэтиламин. Реакцию обычно проводят в апротонном растворителе типа диэтилового
эфира, метиленхлорида или подобным им растворителям. Предпочтительный растворитель - метиленхлорид. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Активированная карбоксильная часть затем реагирует с амином, R11-NH2, например с анилином, в апротонном растворителе, образуя амидный реактант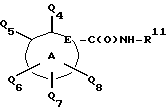
который может затем быть далее замещен в соответствии с известными методами.
Амидный реактант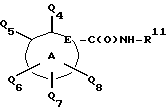
может быть далее замещен путем орто-депротонирования группы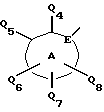
с получением соответствующего аниона с последующей реакцией с разнообразными реагентами типа алкилгалогенидов или галоге- нирующих агентов типа брома. Амидный реактант в общем случае депротонируют дважды, используя два эквивалента сильного основания типа н-бутиллития или в-бутиллития относительно амидного реактанта, при необходимости в присутствии металл-координирующего агента типа тетраметилэтилендиамина (TMEDA). Реакцию обычно проводят в апротонном растворителе, предпочтительно эфире типа диэтилового эфира, тетрагидрофурана или подобного им растворителя при температуре от приблизительно -78oC до приблизительно 25oC.
Полученное соединение может затем быть гидролизовано с использованием известных специалистам методов для получения желаемого замещенного карбоново-кислотного реактанта формулы (IБ). Например подходящий гидролиз включает действие на амидный реактант сильной минеральной кислоты, органической кислоты или смеси минеральной и органической кислот при температуре от приблизительно 100oC до приблизительно 160oC. Типичные кислоты, которые могут использоваться в этой реакции, включают бромисто-водородную кислоту, уксусную кислоту, соляную кислоту и т.п. Проведение реакции в запаянной пробирке можно дополнительно использовать для увеличения скорости реакции.
Третий метод получения замещенного карбоново-кислого реактанта формулы (IБ) включает диазотирование анилина с последующей реакцией (гашением) получающейся в результате реакции соли диазония. Более конкретно, аминогруппу анилинового реактанта превращают в соль диазония посредством реакции с азотистой кислотой. Азотистая кислота может быть получена in situ обработкой нитрита натрия водным раствором сильной кислоты типа соляной или серной. Эта реакция обычно проводится при температуре равной или ниже 5oC. Соль диазония затем гасится в реакции с подходящим реагентом с получением желаемой замещенной ароматической системы. Типичные гасящие реагенты включают воду, цианид, галогенид, водную серную кислоту и т.п. Обычно для облегчения протекания реакции реакционная смесь нуждается в нагревании.
Имеется большое число разнообразных реакций, известных специалистам, которые могут быть использованы для осуществления желаемых замещений в карбоциклических и гетероциклических соединениях. Например имеется большое число разнообразных реакций электрофильного и нуклеофильного замещения в ароматических соединениях March J. Advanced Organic Chemistry, 3rd. ed., Wiley 1985, Chapter 11, 13 [7].
Кроме того, соединения формулы (IБ) могут быть получены карбоксилированием подходяще замещенного карбоциклического или гетероциклического соединения. Карбоксилирование может быть выполнено с использованием ряда различных реагентов. Например карбоциклический или гетероциклический реагенты могут реагировать с фосгеном, оксалилхлоридом, гидрохлоридом мочевины или N, N-диэтилкарбамоилхлоридом в присутствии катализаторов Фриделя - Крафтса. Разновидность этого метода включает взаимодействие карбоциклического или гетероциклического реагента с алкилтиохлорформиатом (RSCOCl) или карбамоилхлоридом (H2NCOCl) с целью получения соответственно амида и тиолового эфира. Амид и тиоловый эфир могут затем быть гидролизованы для получения желаемой карбоксильной группы March J. Advanced Organic Chemistry, 3rd. ed., Wiley 1985, p. 491 [8].
Примеры катализаторов Фриделя-Крафтса включают кислоты Льюиса типа бромида алюминия (AlBr3), хлорида алюминия (AlCl3), хлорида трехвалентного железа (FeCl3), трихлорида бора (BCl3), трифторида бора (BF3) и т.п. См. также Olah. Friedel - Crafts and Related Reactions, Interscience, New York, 1963-1965 [9], Olah. Friedel-Crafts Chemistry, Wiley, New York, 1973 [10].
Дополнительно карбоново-кислотные реактанты хинолина могут быть получены в результате взаимодействия подходяще замещенного анилина с глицерином с использованием реакции Скраупа, изложенной в Bradford L. et. al. J. Chem. Soc. , p. 437 (1947) [11]. Например 3-аминобензойная кислота может реагировать с глицерином в присутствии окисляющего агента типа м-нитробензолсульфоновой кислоты или м-нитробензолсульфоната натрия в 60-75% водном растворе серной кислоты с получением желаемого карбоксизамещенного хинолина. Реакцию обычно проводят при температуре от приблизительно 35oC до температуры дефлегмации в течение одного - шести часов, предпочтительно при температуре от приблизительно 50oC до температуры дефлегмации от двух до четырех часов.
Получающиеся в результате реакции реактанты могут затем быть восстановлены или гидрированы с использованием известных процедур. См. например, March J. Advanced Organic Chemistry, 3rd ed., Wiley, 1985, стр. 700 [12]. Предпочтительная процедура включает каталитическое гидрирование, например путем смешения хинолинкарбоново-кислотного реактанта с газообразным водородом в присутствии катализатора. Предпочтительный катализатор - палладий на угле. Типичные растворители, подходящие для использования в этой реакции, включают любой органический растворитель, такой как этилацетат. Выбор растворителя не критичен, пока применяемый растворитель инертен к протекающей реакции. Реакция в общем случае заканчивается в течение приблизительно от 1 до 24 часов, в том случае когда проводится при температуре в диапазоне от приблизительно 25oC до приблизительно 100oC.
В соответствии с другим способом соединения формулы IА, в которых Q3 заменен на R1, могут быть получены согласно реакционной схеме Б (см. в конце описания).
Реакционная схема Б осуществляется проведением реакций 1-6 в последовательном порядке. По окончании каждой из реакций промежуточное соединение может быть при желании выделено с помощью известных специалистам методов, например соединение может быть закристаллизовано и затем собрано фильтрованием или растворитель, использованный в реакции, может быть удален экстракцией, упариванием или декантацией. Если необходимо, перед осуществлением следующего этапа реакционной схемы, промежуточное соединение может быть далее очищено общими методами типа кристаллизации или хроматографии на таких твердых носителях, как силикагель или оксид алюминия.
Реакция Б. 1 обычно осуществляется путем активации, т.е. превращения подходяще замещенной группы формулы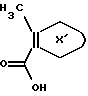
в соответствующий ацилхлорид или ацилбромид в реакции с тионилхлоридом, тионилбромидом, трихлоридом фосфора, трибромидом фосфора, пентабромидом фосфора или пентахлоридом фосфора согласно известным способам и в условиях, известных специалистам. Подходящие соединения
являются коммерчески доступными или получаются в соответствии с известными стандартными методами.
В реакции Б. 2 ацилхлорид или ацилбромид, полученный в реакции Б.1, обычно реагирует с аммиаком или первичным либо вторичным амином формулы H-NR4R4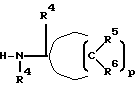
или
где R4, R5, R6 и p определены как и выше для формулы IБ, в неполярном апротонном растворителе или смеси растворителей в присутствии или отсутствие акцептора кислоты с образованием соответствующего амида. Реакция обычно выполняется при температуре от приблизительно -20oC до приблизительно 25oC. Типичные растворители для этой реакции включают эфиры и хлорированнные углеводороды, предпочтительно диэтиловый эфир, хлороформ или метиленхлорид. Реакцию предпочтительно проводят в присутствии акцептора кислоты типа третичного амина, предпочтительно триэтиламина.
В реакции Б.3 амид, полученный в реакции Б.2, взаимодействует с сильным основанием в присутствии солюбилизирующего агента с образованием соответствующего аниона, который затем реагирует в реакции Б.4 с Weinreb амидом, образуя кетон. Реакция Б.3 обычно выполняется в апротонном растворителе при температуре от приблизительно -78oC до приблизительно 0oC. Типичные основания, используемые в реакции Б.3, включают амидлитиевые и алкиллитиевые основания, предпочтительно C1-C4-алкиллитиевые основания и ди(C1-C4)алкиламидлитиевые основания.
Типичными солюбилизирующими агентами для реакции 3 являются тетраметил(C1-C4)алкилендиамины, предпочтительно тетраметилэтилендиамин. Реакцию Б. 4 обычно проводят в апротонном растворителе при температуре от приблизительно -80oC до приблизительно -40oC. Типичные растворители для реакций Б.3 и Б.4 включают эфиры, предпочтительно тетрагидрофуран. В реакции Б.4 анион в общем случае используется в количестве, лежащем в пределах от приблизительно эквимолярного до приблизительно трехмолярного избытка аниона, предпочтительным является приблизительно двумолярный избыток аниона по отношению к амидному реактанту Weinreb.
В реакции Б.5 кетон, полученный в реакции Б.3, восстанавливают до соответствующего спирта, используя подходящий восстанавливающий агент. Реакцию проводят в протонном растворителе при температуре от приблизительно -25oC до приблизительно 25oC. Типичные восстанавливающие агенты для этой реакции включают боргидрид натрия, боргидрид лития, диизобутилалюмогидрид и бис(2-метоксиэтокси)алюмогидрид натрия. Предпочтительный восстанавливающий агент - боргидрид натрия. Типичные протонные растворители для этой реакции включают спирты, предпочтительно этанол.
Реакция Б.6 - стандартная реакция снятия защит с аминогрупп, использующая способы и методы, известные специалистам, в результате которой получается соответствующий амин, используемый в описанной выше реакции I. Этот амин можно использовать в реакции без очистки, но предпочтительно, чтобы он был предварительно очищен.
Weinreb амид, применяемый в качестве реактанта в реакции Б.4, обычно получается в результате реакции защищенной по аминогруппе аминокислоты с N-метокси-N-метиламином присутствии активирующего агента, акцептора кислоты и связывающего агента. Реакцию обычно проводят в апротонном растворителе или смеси растворителей при температуре от приблизительно -25oC до 25 oC. Предпочтительным активирующим агентом для этой реакции является HOBT•H2O. Предпочтительные акцепторы кислоты - третичные алкиламины, предпочтительно триэтиламин или N-метилморфолин. Предпочтительный связывающий реагент - гидрохлорид этилдиметиламинопропилкарбодиимида. Предпочтительно, чтобы Weinreb амид, получаемый в этой реакции, был выделен перед его использованием в реакции Б.4.
Соединения формулы IА, в которых R1 заменяет Q3 и R1 представляет собой -S-арил, получают по схеме Б на первом этапе в реакции защищенного по аминогруппе серина с трифенилфосфином и диэтилазодикарбоксилатом (DEAD) в апротонном растворителе при температуре от приблизительно -80oC до 0oC, с образованием соответствующего β-лактона. Реакцию обычно проводят в эфире типа тетрагидрофурана при температуре от приблизительно -80oC до 50oC. На втором этапе лактоновое кольцо раскрывают, получая соединение, имеющее структуру
взаимодействием лактона с подходяще замещенным тиоанионом, имеющим структуру -S-арила. Тиоанион предпочтительно получают в реакции соответствующего тиола с сильным основанием типа гидрида натрия или гидрида калия. Эту реакцию обычно проводят в апротонном растворителе при температуре от приблизительно 0oC до приблизительно 40oC в атмосфере инертного газа, такого как азот. Типичные растворители для этой реакции включают эфиры, предпочтительно тетрагидрофуран. Желаемый амидный реактант затем образуется в результате реакции полученного карбоново-кислотного реактанта с N-метокси-N-метиламином в присутствии активирующего агента, акцептора кислоты и связывающего агента так же, как описано выше.
В альтернативном случае, соединения формулы (IА), в которых R1 заменяет Q3 и в которых R1 представляет собой -S-арил, могут быть получены по схеме Б с использованием способов, детально описанных в [2, 3]. Например эти соединения могут быть получены в результате реакции дважды защищенного серина (защищенного по карбоксильной и аминогруппам) с толуолсульфонилхлоридом в присутствии диметиламинопиридина (DMAP) и акцептора кислоты типа пиридина в апротонном растворителе, таком как метиленхлорид, с образованием соответствующего толуолсульфоната, который может затем реагировать с подходяще замещенным тиоанионом, имеющим структуру -S-арила. Тиоанион, как описано выше, преимущественно получают в реакции соответствующего тиола с сильным основанием. Карбоксизащитная группа может затем быть удалена из полученного дважды защищенного арилтиоаланина в условиях, известных специалистам.
В некоторых случаях промежуточное соединение для получения соединений по настоящему изобретению получают следующим образом. Промежуточное соединение имеет формулу 4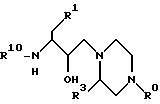
где R1 - арил или -S-арил;
R10 - атом водорода или аминозащитная группа;
R0 представляет собой C1-C4-алкил или -CH2-пиридил;
R3 - группа, имеющая структуру:
1) -C(O)-NR4R4,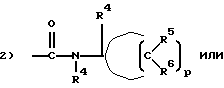
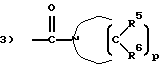
p = 4 или 5;
R4 в каждом случае - независимо атом водорода, C1-C6-алкил или гидрокси(C1-C4)алкил; и
R5 и R6 независимо выбраны из атома водорода, гидроксильной группы, C1-C6-алкила, C1-C6-алкоксигруппы или гидрокси(C1-C4)алкила;
или его фармацевтически приемлемая соль. Промежуточное соединение, имеющее формулу 4, обычно получают в процессе, включающем:
(а) восстановление соединения формулы
с получением пиперазинового соединения;
(б) алкилирование пиперазинового соединения с получением соединения формулы
(в) взаимодействие пиперазинового соединения, полученного на этапе (б), с эпоксидом формулы
где Rb - аминозащитная группа;
в спиртовом растворителе при температуре от приблизительно 20oC до 100oC с образованием соединения формулы 4, в котором R10 представляет собой аминозащитную группу; и (г) возможное удаление аминозащитной группы, с образованием соединения формулы 4, в котором R10 - атом водорода.
Приведенные ниже подготовки и примеры иллюстрируют аспекты изобретения. Данные примеры приводятся в иллюстративных целях и их не следует рассматривать как ограничивающие сферу изобретения.
Сокращения для терминов, обозначающих точку плавления, спектры ядерного магнитного резонанса, электронно-ударные масс-спектры, масс-спектры с ионизацией при полевой десорбции, масс-спектры с ионизацией быстрыми атомами, инфракрасные спектры, ультрафиолетовые спектры, элементный анализ, высокоэффективную жидкостную хроматографию и тонкослойную хроматографию, соответственно: т. п. , ЯМР, EIMS, масс-спектр (FD), масс-спектр (FAB), ИК, УФ, анализ, ВЖХ и ТСХ. Кроме того, абсорбционные максимумы, приведенные для ИК-спектров - это не все наблюдаемые максимумы, а только те, которые представляют интерес.
В приводимых ЯМР-спектрах использованы следующие сокращения: синглет (s), дублет (d), дублет дублетов (dd), триплет (t), квартет (q), мультиплет (m), дублет мультиплетов (dm), широкий синглет (br.s), широкий дублет (br. d), широкий триплет (br.t) и широкий мультиплет (br.m). J обозначает константу спин-спинового взаимодействия, выраженную в герцах (Гц). Если особо не отмечено, данные ЯМР относятся к свободному основанию рассматриваемого соединения.
Спектры ЯМР были получены на 270 МГц приборе Bruker Corp. или на 300 МГц приборе General Electric QE-300. Химические сдвиги выражены в величинах δ (ppm ниже от тетраметилсилана). Масс-спектры (FD) получены на Varian-MAT 731 Спектрометре с использованием углеводородных дендритных эмиттеров. EIMS спектры снимали на приборе марки CEC 21-110 Consolidated Electrodynamics Corporation. Масс-спектры (FAB) были получены на VG ZAB-3 Спектрометре. ИК спектры снимали на приборе модели 281 фирмы Perkin-Elmer. УФ спектры были получены на приборе модели 118 фирмы Сагу. ТСХ проводили на пластинах силикагеля фирмы E. Merck. Точки плавления нескорректированы.
Подготовка 1
А. [3S-(3R*, 4aR*,8aR*,2'S*,3'R*)]- 2-[3'-N-(Бензилоксикарбонил)амино-2'-гидрокси-4'-фенил] бутилдекагидроизохинолин-3-N-т-бутилкарбоксамид
Раствор [1'S-(1'R*, 1R*)] -1-[1'-N-(бензилоксикарбонил) амино-2-(фенил)этил] оксирана и [3S-(3R*,4aR*,8aR*)]- декагидроизохинолин-3-N-т-бутилкарбоксамида в абсолютном этаноле нагревали при 80oC в течение ночи. Реакционную смесь упаривали досуха при пониженном давлении, получая остаток. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 10-50% градиент этилацетата в метиленхлориде), получая 6,47 г беловатой пены.
Выход: 75%.
1H ЯМР (CDCl3): δ 1,29 (s, 9H), 1,25-2,05 (m, 2H), 2,20-2,35 (m, 2H), 2,55-2,70 (m, 11H), 2,85-3,10 (m, 3H), 3,24 (br.s, 1H), 3,82 (br.s, 1H), 3,98 (br. s, 1H), 4,99 (br. s, 2H), 5,16-5,18 (m, 1H), 5,80 (br.s, 1H), 7,05-7,38 (m, 10H).
ИК (CHCl3): 3600-3100 (br.), 3031, 2929, 1714, 1673, 1512, 1455, 1368, 1232, 1199, 1047 см-1.
Масс-спектр (FD): m/e 536 (M+).
Б. [3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[3'-Амино-2'-гидрокси-4'-фенил] бутилдекагидроизохинолин-3-N-т- бутилкарбоксамид
Быстро перемешиваемую суспензию 6,37 г (11,91 ммоль) поименованного соединения подготовки 1А и 1,2 г 10% палладия на угле в 200 мл абсолютного этанола помещали в атмосферу водорода. По истечении приблизительно 48 часов реакционную смесь фильтровали через целит и упаривали досуха при пониженном давлении, получая 5,09 г желаемого поименованного соединения. Это соединение использовали без дальнейшей очистки.
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 1,40-1,95 (m, 10H), 2,25-2,48 (m, 2H), 2,59-2,75 (m, 3H), 2,80-3,40 (m, 7H), 3,75-3,90 (m, 1H), 6,19 (br.s, 1H), 7,18-7,35 (m, 5H).
ИК (CHCl3): 3600-3100 (br.), 2929, 2865, 1671, 1515, 1455, 1367, 1245, 1047 см-1.
Масс-спектр (FD): m/e 402 (M+, 100).
Подготовка 2
А. 2R-N(Бензилоксикарбонил)амино-3-нафт-2-илтиопропановая кислота
К раствору 1,28 г (8,00 ммоль) нафталин-2-тиола в 30 мл тетрагидрофурана медленно добавляли 1,77 г (8,16 ммоль) 60% гидрида натрия в атмосфере азота. После перемешивания в течение приблизительно 15 минут к смеси медленно добавляли раствор N(бензилоксикарбонил)серин β-лактона в 20 мл тетрагидрофурана. Реакцию проводили в течение приблизительно одного часа при комнатной температуре, после чего реакционную смесь упаривали при пониженном давлении до получения остатка. Этот остаток растворяли в этилацетате и промывали последовательно 0,5н. бисульфатом натрия и насыщенным солевым раствором. Образовавшиеся слои разделяли и органический слой высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью флэш-хроматографии, получая 2,08 г бледно-желтого твердого вещества.
Выход: 68%.
1H ЯМР (CDCl3): δ 3,42-3,61 (br.m, 2H), 5,53-5,76 (br.s, 1H), 4,85-5,08 (br.m, 2H), 5,54-5,76 (br.s, 1H), 7,06-7,97 (m, 12H).
[α]D -55,72o (c=1,0, MeOH).
ИК (KBr): 3348, 3048, 1746, 1715, 1674, 1560, 1550, 1269, 1200, 1060 см-1.
Масс-спектр (FD): m/e 381 (M+), 381 (100).
Анализ для C20H19NO4S:
Расчет: C 66,12; H 5,02; N 3,67.
Обнаружено: C 66,22; H 5,04; N 3,86.
Б. 3R-1-Диазо-2-оксо-3-N-(бензилоксикарбонил)амино-4-(нафт-2- илтио)бутан
К холодному раствору (-30oC) 15,38 г (40,3 ммоль) поименованного соединения подготовки 2А в 230 мл этилацетата медленно, используя шприц, добавляли 5,62 мл (40,3 ммоль) триэтиламина в атмосфере азота. К полученному раствору из шприца добавляли 7,84 мл (60,5 ммоль) изобутилхлорформиата. В отдельной колбе 10 г N(метил)-N(нитро)-N(нитрозо)гуанидина осторожно добавляли к двуслойной смеси, состоящей из 170 мл диэтилового эфира и 170 мл 5н. раствора гидроксида натрия, в результате чего бурно выделялся газ. По окончании реакции органический слой декантировали от водного в гидроксид калия и высушивали. Эту реакцию диазометанового образования и присоединения повторяли, используя идентичные количества диэтилового эфира и гидроксида натрия и 30 г N(метил)-N(нитро)-N(нитрозо)гуанидина. Полученный диазометановый реактант добавляли к раствору смешанного ангидрида, приготовленному ранее, и реакцию осуществляли на холоду (-30oC) в течение приблизительно 20 минут. По завершении реакции, по данным ТСХ, для устранения непрореагировавшего диазометана через раствор пробулькивали азот, используя оплавленную на огне пастеровскую пипетку, затем раствор упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 10% этилацетат в метиленхлориде), получая 13,62 г желтого масла.
Выход: 83%.
1H ЯМР (CDCl3): δ 3,32-3,46 (m, 2H), 4,40-4,67 (m, 1H), 5,00-5,09 (m, 2H), 5,44 (s, 1H), 5,76 (d, J = 7,8 Гц, 1H), 7,25-7,86 (m, 12H).
В. 3R-1-Хлор-2-оксо-3-N-(бензилоксикарбонил)амино-4-(нафт-2- илтио)бутан
Безводный хлористый водород (газ) пропускали в течение приблизительно двух секунд через холодный раствор (-20oC) 13,62 г (33,59 ммоль) поименованного соединения подготовки 2Б в 230 мл диэтилового эфира, в результате чего выделялся газ. Эту процедуру повторяли, избегая образования избытка хлористо-водородной кислоты. По завершении реакции, по данным ТСХ, раствор упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 10% этилацетат в метиленхлориде), получая 12,05 г твердого вещества бледной рыжевато-коричневой окраски.
Выход: 87%.
1H ЯМР (CDCl3): δ 3,41 (dd, J = 12, 6 Гц, 1H), 3,53 (dd, J = 12, 6 Гц, 1H), 4,18 (AB q, J = 41,9 Гц, J = 15,9 Гц, 2H), 4,77 (dd, J = 9, 3 Гц, 1H), 5,04 (AB q, J = 12 Гц, J = 10,4 Гц, 2H), 5,59 (d, J = 7 Гц, 1H), 7,24-7,85 (m, 12H).
[α]D -80,00o (c = 1,0, MeOH).
ИК (CHCl3): 3426, 3031, 3012, 1717, 1502, 1340, 1230, 1228, 1045 см-1.
Масс-спектр (FD): m/e 413 (M+), 413 (100).
Анализ для C22H20NO3SCl:
Расчет: C, 63,84; H, 4,87; N, 3,38;
Обнаружено: C, 64,12; H, 4,95; N, 3,54.
Г. [3R-(3R*, 4S*)]-1-Хлор-2-гидрокси-3-N- (бензилоксикарбонил)амино-4-(нафт-2-илтио)бутан
К холодному раствору (0oC) 530 мг (1,28 ммоль) поименованного соединения подготовки 2В в 10 мл тетрагидрофурана и 1 мл воды добавляли 73 мг (1,92 ммоль) боргидрида натрия. По завершении реакции, по данным ТСХ, pH раствора подводили к 3, используя 10 мл водного насыщенного раствора хлорида аммония и 500 мкл 5н. раствора соляной кислоты. Полученный раствор экстрагировали дважды метиленхлоридом, объединенные органические слои промывали водой, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (элюирующий раствор - метиленхлорид), получая 212 мг твердого вещества рыжевато-коричневой окраски.
Выход: 40%.
1H ЯМР (CDCl3): δ 3,40 (s, 2H), 3,61-3,71 (m, 2H), 3,97-3,99 (m, 2H), 4,99 (s, 2H), 5,16 (br.s, 1H), 7,21-7,83 (complex, 12H).
Масс-спектр (FD): m/e 415 (M+), 415 (100).
[α]D -47,67o (c = 0,86, MeOH).
ИК (CHCl3): 3630, 3412, 3011, 1720, 1502, 1236, 1044 см-1.
Анализ для C22H22NO3ClS:
Расчет: C 63,53; H 5,33; N 3,37.
Обнаружено: C 63,72; H 5,60; N 3,64.
Д. [1'R-(1'R*, 1S*)]-1-[(1'-N-(Бензилоксикарбонил)амино- 2'-(нафт-2-илтио)этил]оксиран
Раствор 31 мг (0,55 ммоль) гидроксида калия в 1 мл этанола добавляли к раствору 190 мг (0,46 ммоль) поименованного соединения подготовки 2Г в 6 мл раствора этанол/этилацетат в отношении 1:2. По завершении реакции, по данным ТСХ, реакционную смесь выливали в смесь воды с метиленхлоридом. Образовавшиеся слои разделяли, органический слой промывали водой, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (элюирующий раствор - 10% этилацетат в метиленхлориде), получая 172 мг твердого вещества светлой рыжевато-коричневой окраски.
Выход: 99%.
1H ЯМР (CDCl3): δ 2,76 (br.s, 2H), 3,01 (br.s, 1H), 3,31 (d, J = 5 Гц, 2H), 3,77 (br. s, 1H), 5,05 (s, 2H), 5,22 (d, J = 6 Гц, 1H), 7,25-7,85 (complex, 12H).
[α]D -125,42 (c = 0,59, MeOH).
Масс-спектр (FD): m/e 379 (M+), 379 (100).
ИК (CHCl3): 3640, 3022, 2976, 1720, 1502, 1235, 1045 см-1.
Анализ для C22H21NO3S:
Расчет: C 69,63; H 5,58; N 3,69.
Обнаружено: C 69,41; H 5,53; N 3,64.
Е. [2S-(2R*,2'R*,3'S*)]-1-[2'-Гидрокси- 3'-(N-бензилоксикарбонил)амино-4'-(нафт-2-илтио)бутил]пиперидин-2- N-(т-бутил)карбоксамид
Раствор 0,51 г (1,34 ммоль) поименованного соединения подготовки 2Д и 0,26 г (1,41 ммоль) поименованного соединения подготовки 4В в 25 мл изопропанола нагревали при 55oC в течение приблизительно сорока восьми часов. Полученную реакционную смесь охлаждали и упаривали при пониженном давлении, получая неочищенный материал. Этот материал очищали с помощью радиальной хроматографии (4 мм пластина; элюирующий раствор - 10% ацетон в метиленхлориде), получая 104 мг белой пены.
Выход: 14%.
1H ЯМР (CDCl3): δ 1,29 (s, 9H), 1,44-1,82 (m, 6H), 2,19 (m, 1H), 2,40 (m, 1H), 2,68 (m, 2H), 3,09 (m, 1H), 3,46 (m, 2H), 4,00 (m, 2H), 5,01 (s, 2H), 5,73 (d, 1H), 6,01 (br.s, 1H), 7,23-7,34 (m, 5H), 7,45 (m, 3H), 7,72-7,83 (m, 4H).
Масс-спектр (FD): m/e 563 (M+, 100).
Ж. [2S-(2R*,2'S*,3'S*)]-1-[2'-Гидрокси-3'- амино-4'-(нафт-2-илтио)бутил] пиперидин-2-N-(т-бутил)карбоксамид
Раствор, содержащий 1,05 г (0,18 ммоль) поименованного соединения подготовки 2 в 10 мл 30% бромисто-водородной кислоты в уксусной кислоте, реагировал в течение приблизительно одного часа. Реакционную смесь упаривали, азеотропно перегоняли три раза с толуолом, перерастворяли в метаноле, содержащем по 4,5 мл диэтиламина и гидроксида аммония, и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (1 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде, содержащем 1% уксусную кислоту), получая 64 мг белой пены.
Выход: 80%.
1H ЯМР (CDCl3): δ 1,29 (s, 9H), 1,52-1,73 (m, 6H), 1,84 (m, 1H), 2,31-2,43 (m, 2H), 2,75-3,04 (m, 5H), 3,17 (m, 1H), 3,41 (m, 1H), 3,71 (m, 1H), 6,22 (br.s, 1H), 7,47 (m, 3H), 7,73-7,82 (m, 4H).
Масс-спектр (FD): m/e 430 (M+, 100).
Подготовка 3
А. 2S-N-(Бензилоксикарбонил)-2-пирролидинкарбоксилата пентафторфениловый эфир
К холодному раствору (0oC) 30 г (0,12 моль) 2S-N-(бензилоксикарбонил)-2-пирролидинкарбоновой кислоты и 25,8 г (0,14 моль) пентафторфенола в 450 мл тетрагидрофурана добавляли в виде одной порции 27,7 г (0,14 моль) 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC) с последующим добавлением 150 мл метиленхлорида. Полученную реакционную смесь нагревали до комнатной температуры и реакцию проводили в течение приблизительно четырех часов. По завершении реакции, по данным ТСХ, реакционную смесь упаривали при пониженном давлении до получения остатка. Этот остаток растворяли в 500 мл этилацетата и промывали последовательно водой, карбонатом калия, 1н. соляной кислотой и солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении досуха, получая твердое вещество. Это твердое вещество повторно растворяли в гексане и промывали карбонатом калия, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 45,95 г желаемого поименованного соединения.
Выход: 92%.
1H ЯМР (CDCl3): δ 1,95-2,15 (m, 2H), 2,20-2,35 (m, 1H), 2,35-2,50 (m, 1H), 3,50-3,75 (m, 2H), 4,65-4,75 (m, 1H), 5,02-5,30 (m, 2H), 7,20-7,45 (m, 5H).
Б. 2S-N-(Бензилоксикарбонил)пирролидин-2-N(т-бутил)карбоксамид
К холодному раствору (0oC) 45,90 г (0,111 ммоль) поименованного соединения подготовки 3А в 100 мл безводного метиленхлорида медленно добавляли 100 мл (0,952 ммоль) т-бутиламина. Реакционную смесь нагревали до комнатной температуры, реакцию проводили в течение приблизительно одного часа, после чего реакционную смесь разбавляли 1000 мл метиленхлорида и затем промывали последовательно 1н. карбонатом калия, 1н. соляной кислотой, 1н. карбонатом калия и солевым раствором, высушивали над сульфатом натрия и далее фильтровали через пробку, используя 50% этилацетат в гексане, получая 37,74 г желаемого соединения, которое использовали без дальнейшей очистки.
1H ЯМР (CDCl3): δ 0,95-1,50 (m, 9H), 1,70-2,40 (m, 4H), 3,30-3,60 (m, 2H), 4,10-4,30 (m, 1H), 4,95-5,35 (m, 2H), 5,65 (br.s, 0,5H), 6,55 (br.s, 1H), 7,20-7,50 (m, 5,5H).
В. 2S-Пирролидин-2-N-(т-бутил)карбоксамид
С соединения, поименованного в подготовке 3Б (2,71 г, 8,9 ммоль) снимали защиту так же, как детально описано в подготовке 1Б, используя 500 мг 10% палладия на угле и газообразный водород (1 атмосфера) в 200 мл этанола.
Выход: 1,53 г (100%).
1H ЯМР (CDCl3): δ 1,35 (s, 9H), 1,60-1,75 (m, 2H), 1,76-1,90 (m, 1H), 2,00-2,15 (m, 1H), 2,58 (br.s, 1H), 2,80-3,05 (m, 2H), 3,55-3,65 (m, 1H), 7,45 (br.s, 1H).
Г. [2S-(2R*, 2'S*, 3'R*)]-1-[3'-N(Бензилоксикарбонил)- амино-2'-гидрокси-4'-фенилбутил]пирролидин-2-N-(т-бутил)карбоксамид
Раствор, содержащий 122 мг (0,72 ммоль) поименованного соединения подготовки 3В и 200 мг (0,68 ммоль) [1S-(1R*,1'R*)]- 1-[(1'-N-(бензилоксикарбонил)амино-2'-фенил)этил] оксирана в 10 мл метанола, перемешивали в течение ночи. По завершении реакции, по данным ТСХ, реакционную смесь упаривали при пониженном давлении. Желаемое соединение очищали с помощью колоночной хроматографии (элюирующий раствор - 2-4% градиент метанола в метиленхлориде), получая 232,2 мг прозрачного аморфного твердого вещества.
Выход: 55%.
[α]D -56,97o (c = 0,27, MeOH).
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 1,55-1,95 (m, 4H), 2,05-2,25 (m, 1H), 2,40-2,55 (m, 1H), 2,65-2,75 (m, 2H), 2,80-3,00 (m, 3H), 3,15-3,30 (m, 1H), 3,65-3,75 (m, 1H), 3,85-3,95 (m, 1H), 4,86 (br.d, J = 1,1 Гц, 1H), 5,03 (s, 2H), 6,95 (m, 1H), 7,15-7,40 (m, 10H).
ИК (CHCl3): 3700-3100 (br.), 3434, 3031, 2976, 1720, 1664, 1604, 1512, 1455, 1394, 1367, 1343, 1233, 1156, 1107, 1063, 1028, 911 см-1.
Масс-спектр (FD): m/e 468 (M+, 100).
Д. [2S-(2R*, 2'S*,3'R*)]-1-[3'-Амино-2'- гидрокси-4'-фенил-бутил]пирролидин-2-N-(т-бутил)карбоксамид
С соединения, поименованного в подготовке 3Г (222 мг, 0,47 ммоль), снимали защиту так же, как детально описано в подготовке 1Б, используя 67 мг 10% палладия на угле и газообразный водород (1 атмосфера) в 15 мл этанола. Желаемое соединение очищали с помощью колоночной хроматографии (элюирующий раствор - 10% изопропанол в метиленхлориде, содержащем 0,75% гидроксид аммония), получая 80 мг беловатого твердого вещества.
Выход: 51%.
[α]D -55,26o (c = 0,23, MeOH).
1H ЯМР (CDCl3): δ 0,80-3,70 (m, 25H), 6,90-7,40 (m, 6H).
ИК (CHCl3): 3692, 3600-3200 (br.), 2975, 1657, 1603, 1522, 1497, 1479, 1455, 1393, 1366, 1232, 1198, 1137, 1049, 882 см-1.
Масс-спектр (FD): m/e 334 (M+, 100).
Подготовка 4
А. 2S-N-(т-Бутоксикарбонил)пиперидин-2-карбоновая кислота
Раствор 1,64 г карбоната натрия в 15 мл воды добавляли к холодному раствору (0oC) 2,0 г (15,5 моль) 2S-пиперидинкарбоновой кислоты в 50 мл диоксана. По истечении приблизительно десяти минут к смеси добавляли 3,7 г (17,0 моль) ди-т-бутилдикарбоната. Полученная реакционная смесь взаимодействовала в течение приблизительно шести часов, после чего ее концентрировали до одной четвертой части от первоначального объема и затем подкисляли до pH 2, используя 1 М однозамещенный сульфат натрия и этилацетат. Образовавшиеся слои разделяли и органические слои промывали насыщенным солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали досуха при пониженном давлении, получая 2,67 г белого кристаллического твердого вещества.
Выход: 75%.
[α]D -55,26o (c = 0,23, MeOH).
1H ЯМР (CDCl3): δ 1,20-1,80 (m, 15H), 2,15-2,30 (m, 1H), 2,85-3,10 (m, 1H), 3,90-4,10 (m, 2H), 4,70-5,00 (m, 1H).
ИК (CHCl3): 3700-1800 (br.), 3025, 3018, 3011, 2980, 2947, 2865, 1716, 1685, 1449, 1394, 1368, 1280, 1252, 1162, 1147, 1129 см-1.
Масс-спектр (FD): m/e 229 (M+, 100).
Анализ для C27H37N3O4:
Расчет: C 57,63; H 8,35; N 6,11.
Обнаружено: C 57,90; H 8,35; N 6,19.
Б. 2S-N-(т-Бутоксикарбонил)пиперидин-2-карбоксилат, пентафторфениловый эфир
К холодному раствору (0oC) 2,53 г (11,03 моль) поименованного соединения подготовки 4А и 2,34 г (12,7 моль) пентафторбензойной кислоты в 50 мл тетрагидрофурана добавляли 2,42 г (12,7 моль) EDC. Полученную реакционную смесь нагревали до комнатной температуры и реакцию проводили в течение приблизительно двух часов. Смесь затем упаривали при пониженном давлении, получая твердое вещество. Это твердое вещество повторно растворяли в метиленхлориде и раствор промывали последовательно карбонатом калия и солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали досуха при пониженном давлении, получая 3,85 г прозрачного масла, которое отвердевало при стоянии.
Выход: 88%.
1H ЯМР (CDCl3): δ 1,20-1,90 (m, 15H), 2,30-2,40 (m, 1H), 2,90-3,15 (m, 1H), 3,90-4,15 (m, 1H), 5,05-5,35 (m, 1H).
В. 2S-N-(т-Бутоксикарбонил)пиперидин-2-N-т-бутилкарбоксамид
К холодному раствору (0oC) 3,8 г (9,6 ммоль) поименованного соединения подготовки 4Б в 200 мл метиленхлорида медленно добавляли 2,53 мл (24,0 ммоль) т-бутиламина. Реакционная смесь взаимодействовала в течение приблизительно четырех часов, после чего ее упаривали при пониженном давлении до получения остатка. Этот остаток повторно растворяли в метиленхлориде и затем промывали последовательно 1 М карбонатом калия и солевым раствором, высушивали над сульфатом натрия, фильтровали и очищали с помощью колоночной хроматографии (элюирующий раствор - 10-20% градиент этилацетата в гексане), получая 2,52 г белого твердого вещества.
Выход: 92%.
[α]D -41,47o (c = 0,506, MeOH).
1H ЯМР (CDCl3): δ 1,10-1,70 (m, 15H), 2,20-2,35 (m, 1H), 2,65-2,82 (m, 1H), 3,90-4,10 (m, 1H), 4,62 (br.s, 1H).
ИК (CHCl3): 3600-3300 (br.), 2978, 2945, 2869, 1677, 1512, 1455, 1413, 1394, 1367, 1317, 1280, 1255, 1162, 1144, 1127, 1078, 1042, 868 см-1.
Масс-спектр (FD): m/e 284 (M+, 100).
Анализ для Cl5H28N2O3:
Расчет: C 63,35; H 9,92; N 9,85.
Обнаружено: C 63,10; H 9,66; N 9,92.
Г. 2S-Пиперидин-2-N-T-бутилкарбоксамид
Раствор, содержащий 1,0 г (3,5 моль) поименованного соединения подготовки 4В и 3,5 мл трифторуксусной кислоты в 25 мл метиленхлорида, перемешивали при комнатной температуре в течение приблизительно двух часов. Реакционную смесь концентрировали и один раз азеотропно перегоняли с толуолом. Полученную реакционную смесь затем распределяли между метиленхлоридом и бикарбонатом натрия. Образовавшиеся слои разделяли и органический слой высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 641 мг поименованного соединения.
Выход: 99%.
[α]D -22,45o (с = 0,95, MeOH).
1H ЯМР (CDCl3): δ 1,20-1,50 (m, 12H), 1,51-1,62 (m, 1H), 1,64 (s, 1H), 1,75-1,88 (m, 1H), 1,90-2,00 (m, 1H), 2,60-2,72 (m, 1H), 2,98-3,10 (m, 2H), 6,63 (br.s, 1H).
ИК (CHCl3): 3363, 3002, 2969, 2940, 2860, 1738, 1660, 1522, 1480, 1455, 1398, 1367, 1324, 1295, 1230, 1129, 1110, 852 см-1.
Масс-спектр (FD): m/e 184 (M+, 100).
Д. [2S-(2R*, 2'S*,3'R*)]-N-[3'-(N- Бензилоксикарбонил)-амино-2'-гидрокси-4'-фенил]бутилпиперидин-2-N-т- бутилкарбоксамид
Раствор, содержащий 195 мг (1,06 ммоль) поименованного соединения подготовки 4Г и 300 мг (1,01 ммоль) [1S-(1R*,1'R*)]-1-[(1'-N- (бензилоксикарбонил)амино-2'-фенил)этил]оксирана в 10 мл изопропанола, перемешивали при 55oC в течение приблизительно сорока восьми часов. По завершении реакции, по данным ТСХ, реакционную смесь концентрировали при пониженном давлении. Желаемое соединение очищали, используя колоночную хроматографию (элюирующий раствор - 1-5% градиент изопропанола в метиленхлориде).
Выход: 395 мг (81%).
[α]D -55,64o (c = 0,22, MeOH).
1H ЯМР (CDCl3): δ 1,32 (s, 9H), 1,45-1,90 (m, 6H), 2,25-2,50 (m, 2H), 2,70-3,20 (m, 5H), 3,30-3,40 (m, 1H), 3,75-4,05 (m, 2H), 4,95-5,10 (m, 3H), 6,15 (br.s, 1H), 7,18-7,40 (m, 10H).
ИК (CHCl3): 3700-3100 (br.), 3623, 3021, 2976, 1668, 1603, 1511, 1456, 1313, 1047, 878 см-1.
Масс-спектр (FD): m/e 482 (M+, 100).
Е. [2S-(2R*, 2'S*,3'R*)]-N-[3'-амино-2'- гидрокси-4'-фенил]бутилпиперидин-2-N-т-бутилкарбоксамид
С соединения, поименованного в подготовке 4Д (371 мг, 0,77 ммоль), снимали защиту так же, как детально описано в подготовке 1Б, используя 110 мг 10% палладия на угле и газообразный водород в 20 мл этанола и получая 260 мг белой пены.
Выход: 97%.
[α]D -64,92o (c = 0,39, MeOH).
1H ЯМР (CDCl3): δ 1,35 (s, 9H), 1,45-1,90 (m, 6H), 2,25-2,35 (m, 1H), 2,50-2,90 (m, 5H), 3,00-3,40 (m, 3H), 3,85-3,98 (m, 1H), 6,29 (s, 1H), 7,15-7,38 (m, 5H).
ИК (CHCl3): 3693, 3650-3100 (br.), 2943, 2862, 1671, 1603, 1517, 1497, 1455, 1394, 1367, 1233, 1185, 1049, 887 см-1.
Масс-спектр (FD): m/e 348 (M+, 100).
Подготовка 5
А. Пиразин-2-N-(т-бутил)карбоксамид
К суспензии 50 г (0,403 моль) пиразин-2-карбоновой кислоты в 600 мл тетрагидрофурана и 100 мл диметилформамида добавляли 65,9 г (0,407 моль) карбонилдиимидазола. Полученная реакционная смесь взаимодействовала при 50oC до тех пор, пока не прекращалось выделение газа. После охлаждения реакционной смеси к ней медленно добавляли 73,5 г (1,00 моль) бутиламина. Реакцию проводили в течение приблизительно тридцати минут, после чего реакционную смесь упаривали при пониженном давлении, перерастворяли в 500 мл метиленхлорида и затем промывали последовательно водой, соляной кислотой (pH 2), насыщенным бикарбонатом натрия, водой, 1 М гидроксидом калия и солевым раствором, высушивали над сульфатом натрия и концентрировали, получая 68,5 г белого твердого вещества.
Выход: 95%.
1H ЯМР (CDCl3): δ 1,51 (s, 9H), 7,73 (br.s, 1H), 8,49 (m, 1H), 8,72 (m, 1H), 9,38 (s, 1H).
Б. (+/-)-Пиперазин-2-N-(т-бутил)карбоксамид
Смесь 68,5 г (0,382 моль) поименованного соединения подготовки 5А и 70 г (0,308 моль) окиси платины в 186 мл этанола нагревали в течение ночи при 40oC в атмосфере водорода (60 psi). Полученный неочищенный материал фильтровали и фильтрат концентрировали, получая 65 г белого твердого вещества.
Выход: 95%.
Масс-спектр (FD): m/e 185 (M+, 100).
В. (+/-)-4-(Пирид-3'-илметил)пиперазин-2-N-(т-бутил)карбоксамид
К раствору 5,0 г (0,027 моль) поименованного соединения подготовки 5Б в 160 мл смеси воды и ацетонитрила в отношении 1:1 добавляли 18,65 г (0,135 моль) карбоната калия. Полученную смесь энергично перемешивали во время добавления 4,43 г (0,027 моль) гидрохлорида 3-хлорметилпиридина и реакцию проводили в течение ночи. Реакционную смесь концентрировали при пониженном давлении, суспендировали в растворе 20% изопропанола в хлороформе, промывали последовательно водой и солевым раствором, высушивали над сульфатом натрия, фильтровали и затем концентрировали до получения-остатка. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 5% метанол в метиленхлориде, содержащем 1% гидроксид аммония), получая 1,34 г прозрачного желтого масла.
Выход: 18%.
1H ЯМР (CDCl3): δ 1,10 (s, 9H), 1,89-2,01 (m, 2H), 2,35 (m, 1H), 2,57-2,74 (m, 4H), 3,09 (m, 1H), 3,27 (s, 2H), 6,71 (br.s, 1H), 7,03 (m, 1H), 7,44 (m, 1H) 8,26 (m, 2H).
ИК (KBr): 3691, 3611, 3366, 2974, 1666, 1602, 1521, 1479, 1456, 1427, 1393, 1366, 1324, 1139, 1047, 839 см-1.
Масс-спектр (FD): m/e 276 (M+, 100).
Г. [2S-(2R*,2'S*,3'R*)]-1-[2'-Гидрокси-3'- (N-бензилоксикарбонил)амино-4'-фенилбутил] -4-(пирид-3''-илметил) пиперазин-2-N-(т-бутил)карбоксамид
Раствор, содержащий 0,377 г (1,27 ммоль) [1S-(1R*,1'R*)]- 1-[(1'-N-бензилоксикарбонил)амино-2'-фенил)этил] оксирана и 0,350 г (1,27 ммоль) поименованного соединения подготовки 5В в 12 мл изопропанола, взаимодействовал при 45oC в течение приблизительно сорока восьми часов. Реакционную смесь охлаждали и затем упаривали при пониженном давлении, получая неочищенный материал. Этот материал очищали с помощью радиальной хроматографии (6 мм пластина; элюирующий раствор - 5-10% градиент изопропанола в метиленхлориде), получая 120 мг изомера А и 68 мг изомера Б.
Выход: общий 26%.
Изомер А:
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 2,26-2,89 (m, 13H), 3,29 (m, 1H), 3,45 (s, 2H), 2,79-3,95 (m, 3H), 4,73 (br.s, 1H), 4,97 (br.s, 2H), 5,20 (m, 1H), 7,14-7,29 (m, 6H) 7,57 (m, 1H), 7,82 (br.s, 1H), 8,53 (m, 2H).
ИК (KBr): 3692, 3434, 2970, 2829, 1714, 1661, 1604, 1579, 1512, 1455, 1427, 1393, 1365, 1231, 1149, 1029, 909 см-1.
Масс-спектр (FD): m/e 573 (M+, 100).
Д. [2S-(2R*, 2'S*, 3'R*)] -1-[2'-Гидрокси-3'-амино- 4'-фенил]бутил-4-(пирид-3''-илметил)пиперазин-2-N-(т-бутил)карбоксамид
Раствор, содержащий 0,062 г (0,11 ммоль) поименованного соединения подготовки 5Г (изомер А), перемешивали в течение приблизительно девяноста минут в 1,5 мл раствора 30% бромисто-водородной кислоты в уксусной кислоте. Полученную смесь концентрировали, азеотропно перегоняли три раза с толуолом, повторно растворяли в метаноле, содержащем по 1 мл диэтиламина и гидроксида аммония, и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (2 мм пластина; элюирующий раствор - 15-25% градиент метанола в метиленхлориде, содержащем 1% гидроксид аммония), получая 13 мг белого твердого вещества.
Выход: 28%.
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 2,36-3,21 (m, 15H), 3,47 (d, 2H), 3,75 (m, 1H), 7,19-7,30 (m, 6H), 7,57 (m, 2H), 8,52 (m, 2H).
Масс-спектр (FD): m/e 440 (M+, 100).
Подготовка 6
А. [2S-(2R*, 2'S*, 3'S*)]-1-[3'-N- (Бензилоксикарбонил)амино-2'-гидрокси-4'-фенилтиобутил] -4-[пирид-3''- илметил]пиперазин-2-N-т-бутилкарбоксамид [изомер Б]
Раствор 596 мг (1,81 ммоль) [1S-(1R*,1'S*)]-1- [1'-N-(бензилоксикарбонил)амино-2'-(фенилтио)этил] оксирана и 500 мг (1,81 ммоль) поименованного соединения подготовки 5В в 15 мл изопропанола нагревали при 43oC в течение приблизительно сорока восьми часов. За реакцией следили, используя ТСХ (10% изопропанол в метиленхлориде, содержащем 1% гидроксид аммония; изомер А Rf) = 0,7; изомер Б Rf = 0,6). По завершении реакции реакционную смесь упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (6 мм пластина; элюирующий раствор - 5-15% градиент изопропанола в метиленхлориде, содержащем 1% гидроксид аммония), получая 200 мг изомера А в виде рыхлой пены рыжевато-коричневой окраски и 119 мг беловатой пены (изомер Б).
Изомер А:
Выход: 18%.
1H ЯМР (CDCl3): δ 1,31 (s, 9H), 2,25-2,62 (m, 7H), 2,78-2,95 (m, 2H), 2,98-3,08 (m, 1H), 3,10-3,25 (m, 2H), 3,40-3,55 (m, 2H), 3,72-3,85 (m, 1H), 3,90-4,00 (m, 1H), 5,05 (s, 2H), 7,01 (br.s, 1H), 7,10-7,40 (m, 11H), 7,62 (d, J = 7,8 Гц, 1H), 8,49 (s, 2H).
Масс-спектр (FD): m/e 606 (M+, 100).
Анализ для C33H43N5O4S:
Расчет: C 65,42; H 7,15; N 11,56.
Обнаружено: C 65,38; H 7,27; N 11,36.
Изомер Б:
Выход: 11%.
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 2,25-2,85 (m, 8H), 3,20-3,32 (m, 3H), 3,47 (s, 2H), 3,78-3,95 (m, 2H), 5,06 (s, 2H), 5,30-5,38 (m, 1H), 7,10-7,42 (m, 12H), 7,55-7,85 (m, 2H), 8,50-8,60 (m, 2H).
Масс-спектр (FD): m/e 606 (M), 497 (100).
HR Масс-спектр (FAB) для C33H44N5O4S:
Расчет: 606,3114.
Обнаружено: 606,3141.
Б. [2S-(2R*, 2'S*,3'S*)]-1-[2'-Гидрокси-3'- амино-4'-фенилтиобутил]-4-[пирид-3''-илметил]пиперазин-2-N-т-бутилкарбоксамид
Раствор 110 мг (0,18 ммоль) изомера Б из подготовки 6А в 5 мл 30% бромисто-водородной кислоты в уксусной кислоте перемешивали при комнатной температуре в течение приблизительно 1 часа. Реакционную смесь упаривали при пониженном давлении до получения остатка. Этот остаток перерастворяли в 4 мл гидроксида аммония. Полученный раствор экстрагировали четыре раза порциями по 10 мл раствора 10% изопропанола в хлороформе. Объединенные органические слои высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (2 мм пластина; элюирующий раствор - 10-30% градиент метанола в метиленхлориде, содержащем 1% гидроксид аммония), получая 65 мг рыхлой желтой пены.
Выход: 72%.
1H ЯМР (CDCl3): δ 1,25 (s, 9H), 2,25-2,78 (m, 7H), 3,00-3,32 (m, 4H), 3,47 (s, 2H), 3,60-3,75 (m, 1H), 4,18-4,35 (m, 1H), 6,90-7,65 (m, 9H), 8,40-8,60 (m, 2H).
Масс-спектр (FD): m/e 473 (M+, 100).
Подготовка 7
А. [3S-(3R*,4aR*,8aR*,2'S*,3'S*)]- 2-[3'-N-(Бензилоксикарбонил) амино-2'-гидрокси-4'-(нафт-2-илтио)]бутилдекагидроизохинолин-3- N-(т-бутил)карбоксамид
Готовили раствор, содержащий 165 мг (0,40 ммоль) поименованного промежуточного соединения подготовки 2Д и 94 мг (0,43 ммоль) 3-(1-N-(т-бутил)амино-1-оксометил)октагидро-(2H)-изохинолина в 5 мл этанола. Реакцию осуществляли при 80oC в течение приблизительно 19 часов. Раствор затем охлаждали до комнатной температуры и упаривали при пониженном давлении до получения остатка. Этот остаток очищали с помощью радиальной хроматографии (элюирующий раствор - 10% этилацетат в метиленхлориде), получая 103 мг беловатой пены.
Выход: 42%.
1H ЯМР (CDCl3): δ 1,10-1,73 (m, 20H), 2,13-2,31 (m, 2H), 2,44-2,53 (m, 1H), 2,56-2,68 (m, 1H), 2,86-2,97 (m, 1H), 3,52 (br.s, 2H), 4,02 (br.s, 2H), 4,98 (s, 2H), 5,65 (s, 1H), 5,94 (s, 1H), 7,25-7,83 (complex, 13H).
Масс-спектр (FD): m/e 629 (M+), 138 (100).
[α]D -92,45o (c = 1,06, MeOH).
ИК (CHCl3): 3429, 3010, 2929, 1713, 1670, 1514, 1455, 1047 см-1.
Анализ для C35H47N3O4S:
Расчет: C 69,98; H 7,67; N 6,80.
Обнаружено: C 69,86; H 7,78; N 6,58.
Б. [3S-(3R*,4aR*,8aR*,2'S*,3'S*)]- 2-[3'-амино-2'-гидрокси-4'-(нафт-2-илтио)]бутилдекагидроизохинолин-3-N- (т-бутил)карбоксамид
Готовили раствор, содержащий 50 мг (0,081 ммоль) поименованного промежуточного соединения подготовки 7А и 1 мл 38% водного раствора бромисто-водородной кислоты в уксусной кислоте. Реакцию осуществляли при комнатной температуре в течение приблизительно 1 часа, после чего реакционную смесь упаривали при пониженном давлении до получения остатка. Этот остаток суспендировали в толуоле и затем упаривали при пониженном давлении, получая 61 мг желаемого поименованного промежуточного соединения. Полученное соединение использовали без очистки в примере 9.
1H ЯМР (CDCl3): δ 1,14 (s, 1H), 1,17-2,07 (complex, 15H), 2,66-2,87 (m, 2H), 3,21-3,25 (m, 2H), 3,75 (d, J = 12 Гц, 1H), 3,85 (d, J = 6 Гц, 1H), 4,36-4,47 (m, 1H), 6,73 (s, 1H), 7,39-7,90 (complex, 7H).
Масс-спектр (FD): 483 (M+), 483 (100).
Подготовка 8
А. 2R-2-N(Бензилоксикарбонил)амино-3-фенилтиопропановая кислота
Желаемое поименованное промежуточное соединение получали в виде остатка в соответствии с методикой, детально описанной в подготовке 2А, используя 13,1 мл (127 ммоль) тиофенола, 4,6 г (117 ммоль) 60% раствора гидрида натрия и 25,6 г (116 ммоль) L-N(бензилоксикарбонил)серин-β-лактона в 450 мл тетрагидрофурана. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 0-2% градиент уксусной кислоты в смеси метиленхлорид/этилацетат в отношении 4:1), получая 27,9 г белого твердого вещества.
Выход: 72%.
1H ЯМР (CDCl3): δ 7,55-7,18 (m, 10H), 5,55 (d, J = 7 Гц, 1H), 5,08 (s, 2H), 4,73-4,60 (m, 1H), 3,55-3,30 (m, 2H).
ИК (KBr): 3304, 3035, 1687, 1532, 736 см-1.
Масс-спектр (FD): m/e 332, 288, 271, 181.
Анализ для C17H17NO4S:
Расчет: C 61,61; H 5,17; N 4,23.
Обнаружено: C 61,69; H 5,22; N 4,47.
Б. 3S-1-Диазо-2-оксо-3-N-(бензилоксикарбонил)амино-4-фенилтиобутан
Желаемое поименованное соединение получали в виде остатка в соответствии с методикой, детально описанной в подготовке 2Б, используя 12,1 г (37 ммоль) поименованного соединения подготовки 8А, 5,09 мл (37 ммоль) триэтиламина, 7,13 мл (55 ммоль) изобутилхлорформиата, 146 ммол раствора диазометана. Раствор диазометана готовили, используя 100 мл диэтилового эфира, 150 мл 5н. раствора гидроксида натрия и 21 г (146 ммоль) N(метил)-N(нитро)-N(нитрозо)гуанидина, как описано в подготовке 2Б. Полученный остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 0-5% градиент этилацетата в метиленхлориде), получая желтое масло.
Выход: 73%.
1H ЯМР (CDCl3): δ 7,50-7,19 (m, 10H), 5,62 (d, J = 7 Гц, 1H), 5,47 (br. s, 1H), 5,11 (s, 2H), 4,50-4,32 (m, 1H), 3,33 (d, J = 6 Гц, 1H).
ИК (KBr): 3012, 2115, 1720, 1501, 1367, 1228 см-1.
Масс-спектр (FD): m/e 356, 328, 242.
В. 3R-1-Хлор-2-оксо-3-N-(бензилоксикарбонил)амино-4-фенилтиобутан
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 2В, используя 22,3 г (63 ммоль) поименованного соединения подготовки 8Б и небольшие количества хлористого водорода (газа) в 400 мл диэтилового эфира, получая 21 г белого твердого вещества. Это твердое вещество использовали без дальнейшей очистки.
1H ЯМР (CDCl3): δ 7,50-7,15 (m, 10H), 5,56 (dd, J = 26,7 Гц, 1H), 5,11 (s, 2H), 4,78-4,67 (m, 1H), 4,20 (d, J = 15,9 Гц, 1H), 4,12 (d, J = 15,9 Гц, 1H), 3,48-3,23 (m, 2H).
ИК (KBr): 3349, 1732, 1684, 1515, 1266 см-1.
Масс-спектр (FD): m/e 363 (M+).
Анализ для C18H18NO3SCl:
Расчет: C 59,42; H 4,99; N 3,85.
Обнаружено: C 59,57; H 5,09; N 4,13.
Г. [2S-(2R*, 3S*)] -1-Хлор-2-гидрокси-3- N(бензилоксикарбонил)амино-4-фенилтиобутан
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 2Г, используя 21 г (58 ммоль) поименованного соединения подготовки 8В и 2,4 г (63 ммоль) боргидрида натрия в 300 мл тетрагидрофурана. Полученный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 0-2% градиент метанола в метиленхлориде) с последующей флэш-хроматографией (элюирующий раствор - 0-2% градиент этилацетата в хлороформе), и затем перекристаллизовывали из метиленхлорида при -78oC, получая 8,3 г поименованного соединения.
Выход: 39%.
1H ЯМР (CDCl3): δ 7,47-7,19 (m, 10H), 5,22-5,03 (m, 1H), 5,09 (s, 2H), 4,01-3,89 (m, 2H), 3,75-3,58 (m, 2H), 3,32 (d, J = 4 Гц, 2H).
ИК (KBr): 3321, 2951, 1688, 1542, 1246, 738 см-1.
Масс-спектр (FD): m/e 366 (M+), 119.
Анализ для C18H20NO3SCl:
Расчет: C 59,09; H 5,51; N 3,83.
Обнаружено: C 59,03; H 5,50; N 3,96.
Д. [1'R-(1'R*, 1S*)] -1-((1'-N-(Бензилоксикарбонил) амино-2'- фенилтио)этилоксиран
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 2Д, используя 8,3 г (23 ммоль) поименованного соединения подготовки 8Г, 1,4 г (25 ммоль) гидроксида калия в 400 мл этанола. Полученный остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 0-2% градиент этилацетата в метиленхлориде), получая 6,4 г белого твердого вещества.
Выход: 85%.
1H ЯМР (CDCl3): δ 7,45-7,15 (m, 10H), 5,12 (s, 1H), 5,08 (s, 2H), 3,77-3,62 (m, 1H), 3,21 (d, J = 6 Гц, 2H), 2,99 (m, 1H), 2,77 (m, 2H).
ИК (KBr): 3303, 3067, 1694, 1538, 1257, 741 см-1.
Масс-спектр (FD) m/e 329.
Анализ для C32H45N3O4S:
Расчет: C 65,63; H 5,81; N 4,25.
Обнаружено: C 65,48; H 5,82; N 4,29.
Е. [3S-(3R*, 4aR*,8aR*,2'S*,3'S*)]- 2-[3'-N-(бензилоксикарбонил)амино-2'-гидрокси-4'-(фенил)тио) бутилдекагидроизохинолин-3-N-т-бутилкарбоксамид
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 2Е, используя 6,3 г (19 ммоль) поименованного соединения подготовки 8Д, 5 г (21 ммоль) (3S-(3R*,4aR*,8aR*))-декагидроизохинолин- 3-N-т-бутилкарбоксамида в 300 мл этанола. Полученный остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 0-20% градиент этилацетата в метиленхлориде), получая 4,3 г белого твердого вещества.
Выход: 40%.
1H ЯМР (CDCl3): δ 7,41-7,11 (m, 10H), 5,90 (d, J = 5 Гц, 1H), 5,64 (s, 1H), 5,05 (d, J = 4 Гц, 2H), 4,08-3,90 (m, 2H), 3,40 (d, J = 6, 2H), 3,05 (s, 1H), 2,95-2,85 (m, 1H), 2,62-2,45 (m, 2H), 2,28-2,15 (m, 2H), 2,05-1,88 (m, 2H), 1,78-1,10 (m, 7H), 1,29 (s, 9H).
ИК (KBr): 3330, 2925, 2862, 1706, 1661, 1520, 1454, 1246, 738, 694 см-1.
Масс-спектр (FD): m/e 568 (M+), 467.
Анализ для C32H45N3O4S:
Расчет: C 67,69; H 7,99; N 7,40.
Обнаружено: C 67,64; H 8,20; N 7,45.
Ж. [3S-(3R*, 4aR*, 8aR*, 2'S*, 3'S*))- 2-[3'-амино-2'-гидрокси-4'-(фенил)тио]бутилдекагидроизохинолин-3-N- т-бутилкарбоксамид
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 2Ж, используя 1 г (1,8 ммоль) поименованного соединения подготовки 8 и 40 мл 30% бромисто-водородной кислоты в растворе уксусной кислоты, за исключением того, что неочищенный материал растворяли в 30 мл метанола. К полученному раствору добавляли 2 мл диэтиламина и 2 мл концентрированного гидроксида аммония и затем смесь упаривали при пониженном давлении до получения остатка. Этот остаток повторно растворяли в воде и этилацетате. Образовавшиеся слои разделяли и органический слой промывали последовательно водным раствором бикарбоната натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали досуха при пониженном давлении до получения остатка. Этот остаток очищали с помощью флэш-хроматографии (элюирующий раствор - 0-10% градиент метанола в хлороформе, содержащем 3 капли гидроксида аммония на 1000 мл хлороформа), получая 0,54 г белой пены.
Выход: 71%.
1H ЯМР (CDCl3): δ 7,41-7,16 (m, 5H), 6,07 (s, 1H), 3,78-3,70 (m, 1H), 3,45-3,38 (m, 1H), 3,03-2,84 (m, 3H), 2,38-2,20 (m, 3H), 2,0-1,05 (m, 12H), 1,33 (s, 9H).
ИК (KBr): 2924, 2862, 1660, 1517, 1454, 1439, 737, 691 см-1.
Масс-спектр (FD): m/e 434 (M+), 293.
Подготовка 9
А. 3-Метокси-N-фенилбензамид
Раствор 13,4 мл (147 ммоль) анилина в 30,7 мл триэтиламина медленно добавляли к раствору, содержащему 25,1 г (147 ммоль) 3-метоксибензилхлорида в метиленхлориде. Реакцию проводили в течение приблизительно тридцати минут, после чего реакционную смесь разбавляли 1н. бикарбонатом натрия. Образовавшиеся слои разделяли и органический слой промывали последовательно водой, 1 М гидроксидом натрия и затем солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 31,6 г беловатого твердого вещества.
Выход: 95%
Б. 3-Метокси-2-метил-N-фенилбензамид
К холодному раствору (-70oC) 4,54 г (20 ммоль) поименованного соединения подготовки 9А и 5,11 г (44 ммоль) TMEDA в 70 мл безводного тетрагидрофурана, добавляли 26,9 мл 1,56 М раствора н-бутиллития в гексане. Полученную реакционную смесь нагревали до - 15oC и перемешивали в течение приблизительно 45 минут, получая желтую суспензию. Эту суспензию повторно охлаждали до -70oC и к ней добавляли 2,89 г (20 ммоль) йодистого метила, что приводило к образованию белого осадка. Реакционную смесь перемешивали в течение ночи при комнатной температуре, реакцию останавливали насыщенным хлоридом аммония и реакционную смесь разбавляли диэтиловым эфиром. Образовавшиеся слои разделяли и органическую фазу последовательно промывали насыщенным хлоридом аммония, водой, насыщенным бикарбонатом натрия и солевым раствором. Органические экстракты затем высушивали над сульфатом натрия и концентрировали, получая белое твердое вещество, которое очищали перекристаллизацией из раствора этилацетат/гексан в отношении 2:1, получая 4,00 г игольчатых кристаллов.
Выход: 99%.
1H ЯМР (CDCl3): δ 2,36 (s, 3H), 3,88 (s, 3H), 3,89 (s, 1H), 6,90-7,70 (m, 8H).
ИК (CHCl3): 3424, 3013, 2963, 2943, 2840, 1678, 1597, 1585, 1519, 1463, 1438, 1383, 1321, 1264, 1240, 1178, 1083, 1069 см-1.
Масс-спектр (FD): m/e 241 (M+, 100).
Анализ для C15H15NO2:
Расчет: C 74,67; H 6,27; N 5,80.
Обнаружено: C 74,65; H 6,29; N 5,82.
В. 3-Гидрокси-2-метилбензойная кислота
Смесь 1,21 г (5,00 ммоль) поименованного соединения подготовки 9Б, 35 мл 5н. соляной кислоты и 20 мл 30% раствора бромисто-водородной кислоты в уксусной кислоте кипятили с обратным холодильником в течение 24 часов. После охлаждения реакционную смесь разбавляли 100 мл этилацетата и 100 мл воды. Образовавшиеся слои разделяли, и органический слой промывали один раз водой и затем подщелачивали до pH 11, используя 0,5н. гидроксид натрия; образовавшиеся слои разделяли и водный слой подкисляли до pH 1, используя 5н. соляную кислоту. Желаемое соединение экстрагировали из этого водного слоя этилацетатом. Этилацетатные экстракты промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и затем концентрировали до получения остатка, составившего после двух упариваний из гексана 750 мг белого твердого вещества.
Выход: 98%.
1H ЯМР (ДМСО-d6): δ 2,26 (s, 3H), 6,98 (d, J = 8,03 Гц, 1H), 7,02 (t, J = 7,69 Гц, 1H), 7,15 (d, J = 7,37 Гц, 1H), 9,55 (br.s, 1H).
ИК (CHCl3): 3600-2100 (br.), 3602, 2983, 1696, 1588, 1462, 1406, 1338, 1279, 1174, 1154, 1075, 1038, 920, 892, 854, 816 см-1.
Масс-спектр (FD): m/e 152 (M+, 100).
Анализ для C8H8O3:
Расчет: C 63,15; H 5,30.
Обнаружено: C 63,18; H 5,21.
В альтернативном случае желаемое поименованное соединение получали, добавляя небольшими порциями 22,6 г (0,33 моль) нитрита натрия к охлажденному раствору (-10oC) 45 г (0,30 моль) 3-амино-2-метилбензойной кислоты и 106 г (58 мл; 1,08 моль) концентрированной серной кислоты в 400 мл воды при поддержании температуры ниже 7oC. Полученную реакционную смесь перемешивали в течение приблизительно 30 минут при -10oC, выливали в раствор, содержащий 240 мл концентрированной серной кислоты в 1,2 л воды, и затем медленно нагревали до 80oC (бурное газовыделение происходит между температурами 40-60oC). Когда выделение газа заканчивалось, реакционную смесь охлаждали до комнатной температуры и поименованное соединение экстрагировали пятикратно этилацетатом (600 мл). Полученные органические фазы объединяли с 500 мл водного раствора насыщенного карбоната натрия. Образовавшиеся слои разделяли и водный слой подкисляли до pH 2 концентрированной соляной кислотой. Названное соединение затем экстрагировали, используя этилацетат (500 мл), и объединенные органические фазы промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении, получая неочищенный материал. Этот материал очищали с помощью двух перекристаллизаций из смеси этилацетат/хлороформ, получая 23,2 г рыхлого оранжевого порошка.
Выход: 52%.
Подготовка 10
А. 2-Этил-3-метокси-N-фенилбензамид
Поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 9Б, используя 13,5 мл (21 ммоль) 1,56 М н-бутиллития, 2,27 г (10,0 ммоль) поименованного соединения подготовки 9А, 2,56 г (22,0 ммоль) TMEDA и 1,56 г (10,0 ммоль) йодистого этила в 50 мл безводного тетрагидрофурана. Полученный неочищенный материал очищали перекристаллизацией из раствора этилацетат/гексан в отношении 3:1, получая 1,57 г игольчатых кристаллов.
Выход: 62%.
1H ЯМР (CDCl3): δ 1,22 (t, J = 7,4 Гц, 3H), 2,81 (q, J = 7,4 Гц, 2H), 3,88 (s, 3H), 6,96 (d, J = 8,2 Гц, 1H), 7,05 (d, J = 7,6 Гц, 1H), 7,10-7,45 (m, 4H), 7,50 (s, 1H), 7,62 (d, J = 7,95 Гц, 1H).
Масс-спектр (FD): m/e 255 (M+, 100).
Анализ для C16H17NO2:
Расчет: C 75,27; H 6,71; N 5,49.
Обнаружено: C 75,39; H 6,72; N 5,43.
Б. 2-Этил-3-гидроксибензойная кислота
Раствор, содержащий 180 мг (0,71 ммоль) поименованного соединения подготовки 10А, 3 мл 5н. соляной кислоты и 3 мл раствора 30% бромисто-водородной кислоты в уксусной кислоте нагревали при 155oC в течение 20 часов в запаянной пробирке. После охлаждения реакционную смесь разбавляли этилацетатом и водой. Образовавшиеся слои разделяли, и органический слой экстрагировали один раз водой и затем подщелачивали до pH 11, используя 0,5н. гидроксид натрия. Образовавшиеся слои разделяли и водный слой подкисляли до pH 1, используя 5н. соляную кислоту. Желаемое соединение экстрагировали из этого водного слоя этилацетатом. Этилацетатные экстракты промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и затем концентрировали, получая 103 мг твердого вещества бледно-красной окраски.
Выход: 88%.
1H ЯМР (ацетон-d6): δ 1,16 (t, J = 7,4 Гц, 3H), 2,98 (q, J = 7,4 Гц, 2H), 7,00-7,15 (m, 2H), 7,32-7,36 (m, 1H), 8,48 (br.s, 1H).
Масс-спектр (FD): m/e 166 (M+, 100).
Подготовка 11
А. 2-Фтор-3-метокси-N-фенилбензамид
Желаемое поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 9Б, добавляя раствор 3,15 г (10,0 ммоль) N-фторбензолсульфонимида в 5 мл тетрагидрофурана к раствору, содержащему 13,5 мл (21,0 ммоль) 1,56 М н-бутиллития, 2,27 г (10,0 ммоль) поименованного соединения подготовки 9А и 2,56 г (22,0 ммоль) TMEDA в 50 мл безводного тетрагидрофурана. Полученный неочищенный материал дважды перекристаллизовывали из раствора этилацетат/гексан в отношении 2:1 и затем очищали далее с помощью радиальной хроматографии (6 мм, 0,5% этилацетат в метиленхлориде), получая 540 мг беловатого твердого вещества.
Выход: 22%.
1H ЯМР (CDCl3): δ 3,94 (s, 3H), 7,05-7,80 (m, 8H), 8,35-8,50 (m, 1H).
Масс-спектр (FD): m/e 245 (M+, 100).
Б. 2-Фтор-3-гидроксибензойная кислота
Поименованное соединение получали в соответствии с методикой, детально описанной в подготовке 9В, используя раствор 255 мг (1,02 ммоль) поименованного соединения подготовки 11А, 3 мл 5н. соляной кислоты и 5 мл 30% раствора бромисто-водородной кислоты в уксусной кислоте и получая 134 мг белого твердого вещества.
Выход: 86%.
1H ЯМР (ацетон-d6): δ 7,05-7,50 (m, 5H).
Масс-спектр (FD): m/e 156 (M+, 100).
Подготовка 12
А. 4-N-(Фенил)карбамоилпиридин
Раствор 22,8 мл (250 ммоль) анилина в 104,5 мл (750 ммоль) триэтиламина медленно добавляли к раствору 44,5 г (250 ммоль) 4-хлорформилпиридина гидрохлорида в 500 мл хлороформа. Полученную реакционную смесь перемешивали в течение ночи и затем кипятили с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь разбавляли 600 мл воды, в результате чего происходило образование осадка. После добавления к смеси 200 мл изопропанола образовавшиеся слои разделяли и органический слой промывали последовательно 0,1н. гидроксидом натрия, водой и затем солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении при 70oC, получая белое твердое вещество с коричневым оттенком. Это твердое вещество промывали 200 мл этилацетата, получая 38,9 г желаемого поименованного соединения.
Выход: 78%.
Б. 4-N-(Фенил)карбамоилпиридина N-оксид
К горячему раствору (85-90oC) 19,8 г (100 ммоль) поименованного соединения подготовки 12А в 60 мл ледяной уксусной кислоты, помещенному за защитным экраном, медленно добавляли 51 мл перекиси водорода. Полученная реакционная смесь взаимодействовала в течение приблизительно четырех часов при 90oC, после чего ее охлаждали до комнатной температуры, разбавляли приблизительно 60 мл смеси изопропанола и хлороформа и затем подщелачивали до pH 12. Полученные слои разделяли и объединенные органические экстракты высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая твердое вещество бледно-желтой окраски. Это твердое вещество растирали с 250 мл метиленхлорида и упаривали досуха, получая 15,95 г беловатого твердого вещества.
Выход: 75%.
В. 2-Хлор-4-N-(фенил)карбамоилпиридин
К раствору, содержащему 20,2 г (97,0 ммоль) пентахлорида фосфора в 27 мл (289 ммоль) оксихлорида фосфора, добавляли 14,4 г (67,2 ммоль) поименованного соединения подготовки 12Б. Полученную реакционную смесь медленно нагревали до 130oC и реакцию осуществляли в течение приблизительно 40 минут. Реакционную смесь охлаждали до комнатной температуры и затем упаривали при пониженном давлении до получения остатка. Этот остаток повторно растворяли в 80 мл воды и разбавляли 80 мл водного карбоната калия, что приводило к образованию желтого осадка. Осадок отделяли фильтрованием, растворяли в 250 мл горячего этанола и фильтровали в горячем состоянии, получая темно-желтый раствор. Этот раствор упаривали при пониженном давлении приблизительно до объема 160 мл, после чего опять фильтровали в горячем состоянии перед добавлением приблизительно 50-60 мл воды. Полученный раствор охлаждали и желаемое соединение выделяли перекристаллизацией, получая 8,0 г игольчатых кристаллов бледно-желтой и белой окраски.
Выход: 51%.
Г. 2-Метокси-4-N-(фенил)карбамоилпиридин
К суспензии, содержащей 4,09 г (18,0 ммоль) поименованного соединения подготовки 12В в 30 мл метанола, добавляли 2,92 г (42,0 ммоль) метоксида натрия. Полученную реакционную смесь кипятили с обратным холодильником в течение приблизительно восемнадцати часов, охлаждали и упаривали при пониженном давлении, получая твердое вещество. Это твердое вещество промывали водой и растирали с холодным бензолом, получая 1,8 г твердого вещества. Анализ этого твердого вещества показал, что реакция не прошла полностью, поэтому к полученному веществу в метаноле дополнительно добавляли 10,01 г (144 ммоль) метоксида натрия. Полученную реакционную смесь кипятили в метаноле с обратным холодильником в течение пятнадцати часов и обрабатывали тем же способом, что и ранее, получая 300 мг твердого вещества. Это твердое вещество очищали с помощью радиальной хроматографии (2 мм пластина; элюирующий раствор - 40% этилацетат в гексане) с последующей перекристаллизацией из горячего гексана, получая 140 мг желаемого соединения.
Выход: 3%.
Д. 2-Метокси-3-метил-4-N-(фенил)карбамоилпиридин
Поименованное соединение получали в соответствии с процедурой, детально описанной в подготовке 9Б, используя 260 мг (1,17 ммоль) поименованного соединения подготовки 12Г, 404 мл (2,68 ммоль) TMEDA, 1,78 мл (2,68 ммоль) н-бутиллития и 329 мл (5,61 ммоль) йодистого метила в 2 мл тетрагидрофурана. Неочищенный материал очищали с помощью радиальной хроматографии (2 мм пластина; элюирующий раствор - 40% этилацетат в гексане) с последующей перекристаллизацией из горячего гексана, получая 140 мг желаемого поименованного соединения.
Е. 3-Метил-2-пиридон-4-карбоновая кислота
Суспензию, содержащую 150 мг (0,598 ммоль) поименованного соединения подготовки 12Д в 4 мл 5н. соляной кислоты (водной), кипятили с обратным холодильником в течение приблизительно пяти часов. После охлаждения реакционную смесь упаривали при пониженном давлении, получая желтое масло. Это масло растворяли в 15 мл воды и pH полученного раствора подводили к 8, используя гидроксид калия, после чего его разбавляли 10 мл толуола. Образовавшиеся слои разделяли и водный слой подкисляли до pH 3,5, используя 5н. соляную кислоту, упаривали при пониженном давлении, получая твердое вещество желтого цвета. Полученное твердое вещество суспендировали в 2 мл горячего этанола и фильтровали через хлопковую набивку. Фильтрат упаривали досуха при пониженном давлении, получая 130 мг твердого вещества. Это твердое вещество промывали 5 мл горячей 10% уксусной кислоты в этилацетате, получая 17 мг твердого вещества, которое кристаллизовали из этанола, получая 6,8 мг желаемого поименованного соединения.
Выход: 6%.
Подготовка 13
2,6-Дихлор-3-гидроксибензойная кислота
Газообразный хлор (20 г, 282 ммоль) медленно пробулькивали через холодный раствор (-70oC) 20 г (145 ммоль) 3-гидроксибензойной кислоты в 100 мл метанола в атмосфере азота, что приводило к повышению температуры примерно до -5oC. Реакционную смесь повторно охлаждали и по истечении приблизительно тридцати минут газообразный хлор вытесняли азотом. Реакционную смесь нагревали до комнатной температуры и разбавляли 100 мл воды. Желаемое названное соединение выделяли перекристаллизацией, получая твердое вещество белого цвета. Это твердое вещество очищали перекристаллизацией из 90 мл воды с последующей перекристаллизацией из 250 мл бензола, содержащего 10 мл ацетона, получая 4,8 г желаемого названного соединения.
Выход: 16%.
Подготовка 14
2-Хлор-3-гидроксибензойная кислота
Газообразный хлор (10,3 г; 147 ммоль) медленно пробулькивали через холодный раствор 20 г (145 ммоль) 3-гидроксибензойной кислоты в 100 мл метанола в атмосфере азота, поддерживая температуру ниже -60oC. Приблизительно через тридцать минут газообразный хлор вытесняли азотом, реакционную смесь оставляли нагреваться до комнатной температуры и разбавляли 100 мл воды. Желаемое названное в заголовке соединение выделяли перекристаллизацией, получая белое твердое вещество. Это твердое вещество очищали перекристаллизацией из 50 мл воды с последующей перекристаллизацией из 130 мл бензола, содержащего 10 мл ацетона, получая желаемое названное в заголовке соединение.
Подготовка 15
А. 2-Метил-3-метоксибензоата метиловый эфир
Суспензию 306 мг (2,00 ммоль) поименованного соединения подготовки 9В, 1,06 мл (20,0 ммоль) метилиодида и 1,38 г (10,0 ммоль) карбоната калия в 8 мл ацетона кипятили с обратным холодильником в течение приблизительно 3 часов. Так как реакция не прошла до конца, к реакционной смеси дополнительно добавляли 2 мл (37,7 ммоль) метилиодида, 2 г (14,5 ммоль) карбоната калия и 10 мл ацетона. После кипячения смеси с обратным холодильником в течение приблизительно шестнадцати часов смесь фильтровали. Фильтрат затем концентрировали при пониженном давлении до получения остатка. Этот остаток растворяли в этилацетате, промывали водой и затем упаривали досуха при пониженном давлении, получая 188 мг материала, который содержал 88% желаемого продукта.
Б. 2-Метил-3-метокси бензойная кислота
Раствор 116 мг (4,86 ммоль) гидроксида лития в 1 мл воды добавляли к раствору 175 мг (0,97 ммоль) поименованного соединения подготовки 15А в 3 мл тетрагидрофурана. Образовавшуюся реакционную смесь интенсивно перемешивали. По завершении реакции, по данным ТСХ, реакционную смесь концентрировали при пониженном давлении до получения остатка. Этот остаток повторно растворяли в 10 мл гексана, 25 мл воды и 3 мл 1н. гидроксида натрия. Образовавшиеся слои разделяли, водный слой разбавляли этилацетатом и затем подкисляли до pH 1, используя 1 М соляную кислоту. Образовавшиеся слои разделяли и этилацетатный слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 73 мг желаемого поименованного в заголовке соединения.
Подготовка 16
А. 2-Бутил-3-метокси-N-фенилбензамид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 9Б, используя 11,95 мл 1,51 М н-бутиллития в гексане (18,04 ммоль), 1,95 г (8,95 ммоль) поименованного соединения подготовки 9А, 2,19 г (18,89 ммоль) TMEDA и 1,60 г (9,45 ммоль) бутилиодида в 30 мл безводного тетрагидрофурана. Полученный неочищенный материал очищали, используя радиальную хроматографию (4 мм пластина; элюирующий раствор - 15% этилацетат в гексане), получая 83 мг прозрачного, бесцветного масла.
Выход: 3,5%.
1H ЯМР (CDCl3): δ 0,89 (t, J=7,27 Гц, 3H), 1,36 (m, 2H), 1,56 (m, 2H), 2,78 (m, 2H), 3,84 (s, 3H), 6,92 (d, J=7,98 Гц, 1H), 7,00 (d, J=7,36 Гц, 1H), 7,11-7,22 (m, 2H), 7,35 (t, 2H), 7,59 (m, 2H).
ИК (CHCl3): 3691, 3619, 3424, 3024, 3010, 2963, 2874, 1677, 1602, 1580, 1517, 1459, 1437, 1315, 1265, 1177, 1055, 877 см-1.
Macc-спектр (FD): m/e 283 (M+, 100).
Б. 2-Бутил-3-гидроксибензойная кислота
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 10Б, используя 80 мг (0,28 ммоль) поименованного соединения подготовки 16А в 2 мл 5н. соляной кислоты и 2 мл 30% бромистоводородной кислоты в уксусной кислоте, получая 44 мг неочищенного материала, который использовали без дальнейшей очистки.
Выход: 60% (по данным 1H ЯМР).
1H NNR (CDCl3): δ 0,96 (t, J=8,09 Гц, H), 1,44 (m, 2H), 1,59 (m, 2H), 3,03 (m, 2H), 6,99 (d, J=8,03 Гц, 1H), 7,15 (t, J=7,77 Гц, 1H), 7,59 (d, J= 6,85 Гц, 1H).
Подготовка 17
А. 3-Метокси-2-пропил-N-фенилбензамид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 9Б, используя 2,5 г (11,0 ммоль) поименованного соединения подготовки 9А, 2,81 г (24,2 ммоль) TMEDA, 15,23 мл (23,13 ммоль) н-бутиллития и 1,33 г (11,0 ммоль) аллилбромида в 30 мл тетрагирофурана, получая 2,5 г неочищенного материала. Этот материал растворяли в 30 мл абсолютного зтанола в присутствии 0,5 г 10% палладия на угле, после чего смесь реагировала в атмосфере водорода в течение приблизительно двенадцати часов. Смесь затем фильтровали через целит и фильтрат концентрировали при пониженном давлении, получая оранжевое масло. Это масло очищали, используя радиальную хроматографию (6 мм пластина; элюирующий раствор - 10% этилацетат в гексане), получая 438 мг белой пены.
Выход: 15%.
1H ЯМР (CDCl3): δ 0,94 (t, J=7,35 Гц, 3H), 1,62 (m, 2H), 2,75 (m, 2H), 3,84 (s, 3H), 6,92 (d, J=8,06 Гц, 1H), 7,00 (d, J=7,39 Гц, 1H), 7,16 (m, 2H), 7,34 (t, 2H), 7,59 (d, 2H), 7,69 (br.s, 1H).
Б. 3-Гидрокси-2-пропилбензойная кислота
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 10Б, используя 438 мг (1,62 ммоль) поименованного соединения подготовки 17А в 7 мл 5н. соляной кислоты и 7 мл 30% бромисто-водородной кислоты в уксусной кислоте, получая твердое вещество рыжевато-коричневой окраски. Это твердое вещество очищали перекристаллизацией из горячего толуола, получая 84 мг твердого вещества рыжевато-коричневой окраски.
Выход: 29%.
1H ЯМР (CDCl3): δ 1,01 (t, J=7,33 Гц, 3H), 1,63 (m, 2H), 2,98 (m, 2H), 6,98 (d, J=7,97 Гц, 1H), 7,14 (t, J=7,86 Гц, 1H), 7,57 (d, J= 7,28 Гц, 1H).
ИК (KBr): 3383, 3047, 2962, 2872, 2641, 1698, 1458, 1412, 1341, 1296, 1278, 1223, 1174, 1086, 929, 815, 752 см-1.
Macc-спектр (FD): m/e 180 (M+, 100).
Подготовка 18
А. 2-Изопропил-3-метоксибензонитрил
К смеси 2,76 г (0,115 моль) магния в 75 мл диэтилового эфира медленно добавляли 24,31 г (0,143 моль) изопропилиодида. Образовавшейся реакционной смеси позволяли реагировать до тех пор, пока весь магний не был использован. Затем в течение девяноста минут к смеси добавляли раствор 15,0 г (0,92 моль) 2,3-диметоксибензонитрила в 75 мл диэтилового эфира. Образовавшаяся реакционная смесь реагировала в течение ночи при комнатной температуре, после чего ее кипятили с обратным холодильником в течение четырех часов. Полученную реакционную смесь затем охлаждали до 0oC, и верхний слой декантировали в насыщенный хлорид аммония во льду. Образовавшиеся слои отделяли и органический слой промывали последовательно разбавленным раствором гидроксида натрия, водой и разбавленным раствором соляной кислоты, высушивали над сульфатом натрия, фильтровали и затем концентрировали, получая оранжевое масло. Это масло перегоняли при пониженном давлении (5 дюймовая vigreux колонка; 0,2 мм Hg), получая 6,25 г оранжевого масла.
Выход: 39%.
1H ЯМР (CDCl3): δ 1,37 (d, J=6,47 Гц, 6H), 3,55 (m, 1H), 3,83 (s, 3H), 7,04 (d, J=7,79 Гц, 1H), 7,18 (m, 2H).
ИК (CDCl3): 3690, 3617, 3019, 2968, 2939, 2841, 2228, 1577, 1470, 1457, 1440, 1387, 1363, 1265, 1100, 1070, 1045, 878 см-1.
Масс-спектр (FD): m/e 175 (M+, 100).
Б. 3-Гидрокси-2-изопропилбекзойная кислота
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 10Б, используя 330 мг (1,88 ммоль) поименованного соединения подготовки 18А в 2 мл 5н. соляной кислоты и 30% бромисто-водородной кислоты в уксусной кислоте. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде, содержащем 1% уксусную кислоту), получая 125 мг твердого вещества розового цвета.
Выход: 37%.
1H ЯМР (CDCl3): δ 1,40 (d, J=6,92 Гц, 6H), 3,62 (m, 1H), 6,83 (d, J=7,86 Гц, 1H), 7,06 (t, J=7,89 Гц, 1H), 7,24 (d, J=7,55 Гц, 1H).
ИК (CHCl3): 3599, 3025, 2965, 2876, 1695, 1603, 1584, 1466, 1454, 1404, 1360, 1275, 1234, 1166, 1148, 1086, 1057, 926 см-1.
Macc-спектр (FD): m/e 180 (M+, 100).
Анализ для C10H12O3:
Расчет: C 66,65; H 6,71.
Обнаружено: C 66,53; H 6,84.
Подготовка 19
3-Метилизоникотиновая кислота
К горячему раствору (155oC) 10,7 г (0,1 моль) 3,4-лутидина в 100 мл дифенилового эфира добавляли порциями 18 г (0,16 моль) диоксида селена. По прошествии приблизительно 20 минут реакционную смесь нагревали до 185oC и реакцию проводили в течение примерно тридцати минут. После охлаждения реакционную смесь разбавляли водой и фильтровали. Фильтрат экстрагировали хлороформом и хлороформные экстракты затем концентрировали при пониженном давлении, получая 6,0 г твердого вещества бледно-коричневой окраски.
Выход: 44%.
1H ЯМР (CDCl3): δ 2,43 (s, 3H), 7,61 (d, J=4,98 Гц, 1H), 8,49 (d, J=4,99 Гц, 1H), 8,53 (s, 1H).
13C ЯМР (CDCl3): δ 17,91, 123,21, 132,81, 138,15, 148,12, 152,71, 167,89 ppm.
ИК (KBr): 3425, 2418, 1724, 1606, 1445, 1387, 1303, 1278, 1235, 1100, 1072, 850 см-1.
Macc-спекто (FD): m/e 138 (M+, 100).
Подготовка 20
5-Хинолинкарбоновая кислота
К раствору, содержащему 15 г (0,1 моль) м-аминобензойной кислоты, 27 г (0,13 моль) м-нитробензолсульфоната и 25 г (0,4 моль) глицерина, добавляли 125 г 70% серной кислоты. Полученную реакционную смесь кипятили с обратным холодильником приблизительно 2,5 часа, разбавляли 125 мл воды, подщелоченной до pH 9 с помощью гидроксида аммония, перемешивали в течение ночи с 5 г угля и затем фильтровали. Фильтрат кипятили с 5 г угля, фильтровали, охлаждали до 50oC, подкисляли до pH 5 с помощью ледяной уксусной кислоты (15 мл) и фильтровали, получая коричневое твердое вещество. Это твердое вещество кипятили в 300 мл воды, содержащей 10 мл уксусной кислоты, и фильтровали в горячем состоянии, получая неочищенный материал. Этот материал очищали, используя перекристаллизацию из кипящей уксусной кислоты, получая 6,1 г твердого вещества бледно-коричневой окраски.
Выход: 32%.
1H ЯМР (CDCl3): δ 7,62 (m, 1H), 7,81 (t, J=7,82 Гц, 1H), 8,20 (m, 2H), 8,93 (d, J=3,79 Гц, 1H), 9,24 (d, J=8,58 Гц, 1H).
ИК (KBr): 2772, 2431, 1906, 1708, 1610, 1589, 1507, 1363, 1323, 1269, 1235, 1211, 1141, 1076, 1034, 999, 866, 807 см-1.
Масс-спектр (FD): m/e 173 (M+, 100).
Подготовка 21
1,2,3,4-Тетрагидро-5-хинолинкарбоновая кислота
Раствор, содержащий 1,03 г (5,95 ммоль) соединения подготовки 20, 1,87 г (29,77 ммоль) формиата аммония в 100 мл этанола, продували азотом в течение 10 минут. К этому раствору добавляли 0,5 г палладиевой черни и полученную реакционную смесь нагрели до 65oC. После приблизительно трех часов реакционную смесь фильтровали; полученный фильтрат концентрировали при пониженном давлении до получения остатка. Этот остаток распределяли между водой (pH=4) и раствором 10% изопропанола в хлороформе. Образовавшиеся слои разделяли и органический слой промывали водой (pH=4), высушивали над сульфатом натрия, фильтровали и концентрировали, получая неочищенный материал. Этот материал очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор 5-10% градиент метанола в метиленхлориде, содержащем 1% уксусную кислоту), получая 87 мг твердого вещества рыжевато-коричневой окраски.
Выход: 8%.
1H ЯМР (CDCl3): δ 1,04 (m, 2H), 2,16 (t, 2H), 2,40 (m, 2H), 5,81 (d, J= 8,05 Гц, 1H), 6,09 (t, J=7,78 Гц, 1H), 6,23 (d, J=7,96 Гц, 1H).
ИК (KBr): 3296, 2965, 2929, 1691, 1597, 1474, 1461, 1443, 1350, 1305, 1279, 1236, 1184, 1159, 1106, 1073, 1022, 827 см-1.
Macc-спектр (FD): m/e 177 (M+, 100).
Анализ для C10H11NO2:
Расчет: C 67,78; H 6,26; N 7,90.
Обнаружено: C 67,96; H 6,10; N 7,88.
Подготовка 22
А. 3-Амино-2-метилбензоата метиловый эфир
Раствор 10 г (66,2 ммоль) 3-амино-2-метилбензойной кислоты и 20 г п-толуолсульфоновой кислоты моногидрата в 400 мл метанола кипятили с обратным холодильником в течение ночи и затем разбавляли смесью этилацетата и 1 М карбоната калия. Образовавшиеся слои охлаждали и разделяли. Органический слой промывали последовательно 1 М карбонатом калия и солевым раствором, высушивали над сульфатом натрия, фильтровали и концентрировали, получая 9,23 г оранжевого масла.
Выход: 85%.
1H ЯМР (CDCl3): δ 2,34 (s, 3H), 3,73 (br.s, 2H), 3,88 (s, 3H), 6,81 (d, J=7,96 Гц, 1H), 7,05 (t, J=7,78 Гц, 1H), 7,19-7,30 (m, 1H).
ИК (CDCl3): 3406, 3027, 3012, 2978, 2953, 1718, 1621, 1467, 1435, 1315, 1301, 1265, 1196, 1159, 1108, 1066, 1045, 810 см-1.
Масс-спектр (FD): m/e 165 (M+, 100).
Б. 3-N-(Метилсульфонил)амино-2-метилбензоата метиловый эфир
К холодному раствору (0oC) 1,07 г (6,48 ммоль) поименованного соединения подготовки 22А в 50 мл безводного метилен-хлорида добавляли 1,18 г (6,80 ммоль) метилсульфонового ангидрида.
Полученная реакционная смесь реагировала в течение ночи при комнатной температуре, после чего ее разбавляли 100 мл метиленхлорида, промывали дважды раствором бикарбоната натрия, высушивали над сульфатом натрия, фильтровали, концентрировали, перерастворяли в гексане и затем опять концентрировали до получения остатка. Этот остаток трижды растирали в гексане, после чего упаривали досуха при пониженном давлении, получая 1,46 г твердого вещества розового цвета. Это твердое вещество затем перекристаллизовывали, используя 20 мл смеси 30% гексан/50% этилацетат/20% метанол.
Выход: 57%.
1H ЯМР (ДМСО-d6): δ 2,25-2,45 (m, 4,5H) 2,97 (s, 1,5H), 3,80 (s, 3H), 7,23-7,63 (m, 3H), 9,24 (s, 1H).
ИК (KBr): 3900-2400 (br. ), 3298, 1713, 1466, 1320, 1290, 1265, 1248, 1210, 1183, 1156, 1047, 971, 964, 752, 563, 519 см-1.
Macc-спектр (FD): m/e 243 (M+, 100).
Анализ для C10H13NO4S:
Расчет: C 49,37; H 5,39; N 5,76.
Обнаружено: C 49,15; H 5,54; N 5,80.
В. 3-N-(Метилсульфонил)амино-2-метилбензойная кислота
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 15Б, используя 400 мг (1,64 ммоль) соединения, поименованного в заголовке подготовки 22Б и 118 мг (4,93 ммоль) гидроксида лития в 20 мл тетрагидрофурана и 8 мл воды, получая 206 мг белого твердого вещества.
Выход: 55%.
1H ЯМР (ДМСО-d6): δ 2,43 (s, 3H), 2,97 (s, 3H), 7,26 (t, J=7,87 Гц, 1H), 7,43 (d, J=7,79 Гц, 1H), 7,60 (d, J=7,17 Гц, 1H).
ИК (KBr): 3800-2200 (br. ), 3252, 1685, 1404, 1334, 1309, 1277, 1149, 982, 965, 914, 780, 763, 748, 632, 518, 498 см-1.
Macc-спектр (FD): m/e 243 (M+, 100).
Подготовка 23
А. 3-метокси-М-фенилбензамид
Раствор 13,4 мл (147 ммоль) анилина в 30,7 мл триэтиламина медленно добавляли к раствору, содержащему 25,1 г (147 ммоль) 3-метоксибензоилхлорида в метиленхлориде. Полученная реакционная смесь реагировала в течение приблизительно тридцати минут, после чего ее разбавляли 1н. бикарбонатом натрия. Образовавшиеся слои разделяли и органический слой промывали последовательно водой, 1 М гидроксидом натрия и затем солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 31,6 г беловатого твердого вещества.
Выход: 95%.
Б. 3-Метокси-2-метил-N-фенилбензамид
К холодному раствору (-70oC) 4,54 г (20 ммоль) соединения поименованного в заголовке подготовки 23А и 5,11 г (44 ммоль) TMEDA в 70 мл безводного тетрагидрофурана добавляли 26,9 мл раствора 1,56 М н-бутиллития в гексане. Полученную реакционную смесь нагревали до -15oC и перемешивали в течение приблизительно 45 минут, получая желтую суспензию. Суспензию затем повторно охлаждали до -70oC и к ней добавляли 2,89 г (20 ммоль) метилиодида, в результате чего формировался белый осадок. Реакционную смесь перемешивали в течение ночи при комнатной температуре, реакцию останавливали, добавляя насыщенный хлорид аммония, и содержимое реакционной смеси разбавляли диэтиловым эфиром. Образовавшиеся слои разделяли и органическую фазу промывали последовательно насыщенным хлоридом аммония, водой, насыщенным бикарбонатом натрия и солевым раствором. Органические экстракты затем высушивали над сульфатом натрия и концентрировали, получая белое твердое вещество, которое очищали перекристаллизацией из раствора гексана/этилацетат в отношении 2:1, получая 4,00 г игольчатых кристаллов.
Выход: 99%.
1H ЯМР (CDCl3): δ 2,36 (s, 3H), 3,88 (s, 3H), 3,89 (s, 1H), 6,90-7,70 (m, 8H).
ИК (CHCl3): 3424, 3013, 2963, 2943, 2840, 1678, 1597, 1585, 1519, 1463, 1438, 1383, 1321, 1264, 1240, 1178, 1083, 1069 см-1.
Масс-спектр (FD): m/e 241 (M+, 100).
Анализ для C15H15NO2:
Расчет: C 74,67; H 6,27; N 5,80.
Обнаружено: C 74,65; H 6,29; N 5,82.
В. 2-Метил-3-гидроксибензойная кислота
Смесь 1,21 г (5,00 ммоль) поименованного соединения подготовки 23Б, 35 мл 5н. соляной кислоты и 20 мл 30% раствора бромисто-водородной кислоты в уксусной кислоте кипятили с обратным холодильником в течение 24 часов. После охлаждения реакционную смесь разбавляли, добавляя 100 мл этилацетата и 100 мл воды. Образовавшиеся слои разделяли и органический слой промывали один раз водой и затем его pH подводили к 11, используя 0,5н. раствор гидроксида натрия. Образовавшиеся слои разделяли и pH водного слоя подводили к 1, используя 5н. соляную кислоту. Желаемое соединение затем экстрагировали из этого водного слоя с использованием этилацетата. Этилацетатные экстракты затем промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали до получения остатка, который после двух упариваний из гексана давал 750 мг белого твердого вещества.
Выход: 98%.
1H ЯМР (ДМСО-d6): δ 2,26 (s, 3H), 6,98 (d, J=8,03 Гц, 1H), 7,02 (t, J= 7,69 Гц, 1H), 7,15 (d, J=7,37 Гц, 1H), 9,55 (br.s, 1H).
ИК (CHCl3): 3600-2100 (br.), 3602, 2983, 1696, 1588, 1462, 1406, 1338, 1279, 1174, 1154, 1075, 1038, 920, 892, 854, 816 см-1.
Масс-спектр (FD): m/e 152 (M+, 100).
Анализ для C8H8O3:
Расчет: C 63,15; H 5,30.
Обнаружено: C 63,18; H 5,21.
Альтернативная подготовка для получения 2-метил-3-гидроксибензойной кислоты
К холодной суспензии (0oC) 0,54 г (3,3 ммоль) 2-метил-3- аминобензойной кислоты в 5 мл воды, содержащей 0,65 мл концентрированной серной кислоты, добавляли 0,25 г (3,6 ммоль) твердого нитрита натрия. По истечении приблизительно 15 минут реакционную смесь выливали в 20 мл теплой воды, содержащей 4 мл концентрированной серной кислоты. Образовавшуюся реакционную смесь медленно нагревали до 90oC, в результате чего выделялся газ. После окончания выделения газа раствор охлаждали до комнатной температуры и экстрагировали этилацетатом. Органические слои объединяли, промывали 0,5н. соляной кислотой, высушивали и концентрировали при пониженном давлении. Неочищенный остаток очищали посредством быстрой фильтрации через силикагель (элюирующий раствор - 5% метанол в метиленхлориде), получая 350 мг белого твердого вещества (т.п. 137-138oC).
Выход: 69%.
1H ЯМР (CDCl3): δ 8,18 (br.s, 1H), 7,42 (d, J=7,7 Гц, 1H), 7,13 (t, J= 7,9 Гц, 1H), 6,93 (d, J=7,9 Гц, 1H), 2,46 (s, 3H).
Анализ для C8H8O3:
Расчет: C 63,15; H 5,29.
Обнаружено: C 63,32; H 5,36.
Подготовка 24
А. N-(т-бутил)-2-метилбензамид
К холодному раствору (0oC) 139,2 г (0,9 моль) хлорида о-толуоила в 1200 мл метиленхлорида при 25oC в атмосфере азота медленно добавляли 180,0 г (1,8 моль) триэтиламина с последующим добавлением по каплям раствора, содержащего 73,14 г (1,0 моль) т-бутиламина в 200 мл метиленхлорида. Образовавшуюся реакционную смесь нагревали до комнатной температуры и оставляли реагировать в течение 2,5 часов. После этого реакционную смесь разбавляли с помощью 1800 мл воды. Образовавшиеся органические и водные слои разделяли и органический слой промывали последовательно 2н. гидроксидом натрия, 1,0н. соляной кислотой и солевым раствором, высушивали над сульфатом магния, фильтровали и затем упаривали досуха при пониженном давлении, получая 167,6 г желаемого поименованного соединения в виде беловатого твердого вещества (т.п. 77-78oC).
Выход: 97%.
1H ЯМР (CDCl3): δ 1,41 (s, 9H), 2,41 (s, 3H), 5,54 (br.s, 1H), 7,13-7,30 (m, 4H).
ИК (CHCl3): 3430, 3011, 2971, 2932, 1661, 1510, 1484, 1452, 1393, 1366, 1304, 1216, 876 см-1.
Масс-спектр (FD): m/e 191 (M+), 191 (100).
Анализ для C12H17NO:
Расчет: C 75,35; H 8,76; N 7,32.
Обнаружено: C 75,10; H 9,11; N 7,20.
Б. S-N-т-Бутил-2-(3-(N-бензилоксикарбонил)амино-2-оксо-4- фенилбутил)бензамид
К раствору 7,0 г (36,5 ммоль) поименованного соединения подготовки 24А в 200 мл безводного тетрагидрофурана из шприца добавляли 12,1 мл (80,3 ммоль) N, N,N',N'-тетраметилэтилендиамина (TMEDA). Образовавшийся раствор охлаждали до -78oC, после чего к нему по каплям из шприца добавляли 55,9 мл в-бутиллития, поддерживая температуру реакционной смеси ниже -60oC. Образовавшийся реакционный раствор затем перемешивали в течение приблизительно 1 часа при -78oC, прежде чем добавить, используя cannula, раствор, содержащий 5,00 г (14,6 ммоль) S-N-метокси-N- метил-2-(N-бензилоксикарбонил)амино-3-фенилпропанамид в 50 мл безводного тетрагидрофурана, поддерживая температуру реакционной смеси ниже -65oC. Образовавшуюся реакционную смесь нагревали до -20oC, реакцию останавливали, используя 20 мл насыщенного хлорида аммония и затем разводили 200 мл диэтилогого эфира. Органические и водные слои разделяли и органический слой промывали последовательно водой, 0,2н. однозамещенным сульфатом натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая бесцветное масло. Это масло очищали, используя флэш-хроматографию (элюирующий раствор - 25% этилацетат в метиленхлориде), получая 6,08 г бесцветной пены.
Выход: 88%.
[α]D -289,26o (с=0,12, MeOH).
1H ЯМР (CDCl3): δ 1,38 (s, 9H), 2,99 (dd, J=15; 6 Гц, 1H), 3,24 (dd, J= 15; 6 Гц, 1H), 3,89 (d, J=18 Гц, 1H), 4,16 (d, J=18 Гц, 1H), 4,72 (dd, J=15, 6 Гц, 1H), 5,00-5,09 (m, 2H), 5,56 (d, J=6 Гц, 1H), 5,93 (br.s, 1H), 7,03-7,40 (m, 14H).
ИК (CHCl3): 3431, 3027, 3012, 2973, 1713, 1658, 1511, 1454, 1383, 1366, 1307, 1231, 1046 см-1.
Macc-спектр (FD): m/e 472 (M+), 218 (100).
Анализ для C29H32N2O4:
Расчет: C 73,70; H 6,82; N 5,93.
Обнаружено: C 73,41; H 6,98; N 5,83.
В. [2R-(2R*, 3S*)] -N-т-Бутил-2-(3-(N-бензилоксикарбонил) амино-2-гидрокси-4-фенилбутил)бензамид
К раствору 6,96 г (14,7 ммоль) поименованного соединения подготовки 24Б в 200 мл абсолютного этанола в атмосфере азота добавляли 2,78 г (73,5 ммоль) боргидрида натрия. По завершении реакции, по данным тонкослойной хроматографии (ТСХ), реакционную смесь разбавляли 200 мл этилацетата и реакцию останавливали, добавляя по каплям 20 мл насыщенного хлорида аммония. Органический и водный слои затем разделяли и органический слой промывали последовательно 1н. соляной кислотой, насыщенным раствором бикарбоната натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха при пониженном давлении, получая 6,4 г бесцветного масла. Это масло очищали, используя флэш-хроматографию (элюирующий раствор - 2-10% градиент метиленхлорида в этилацетате), получая 5,12 г поименованного соединения.
Выход: 74%.
[α]D +10,38o (с=0,10, MeOH).
1H ЯМР (CDCl3): δ 1,40 (s, 9H), 2,79 (dd, J=12, 3 Гц, 1H), 2,90-2,98 (m, 2H), 3,04 (44, J=12, 3 Гц, 1H), 3,70-3,81 (m, 1H), 3,97 (m, 1H), 4,96-5,08 (m, 2H), 5,10 (d, J=9 Гц, 1H), 5,88 (d, J=6 Гц, 1H), 5,93 (s, 1H), 7,13-7,42 (m, 14H).
ИК (CHCl3): 3431, 3028, 3012, 2971, 1773, 1643, 1515, 1454, 1367, 1229, 1028 см-1.
Macc-спектр (FD): m/e 475 (M+), 475 (100).
Анализ для C29H34N2O4:
Расчет: C 73,39; H 7,22; N 5,99.
Обнаружено: C 73,12; H 7,48; N 5,62.
Г. [2R-(2R*,3S*) 1-N-т-Бутил-2-(3-амино-2-гидрокси-4-фенилбутил)бензамид
Готовили суспензию, содержащую 41,0 г (120 ммоль) поименованно соединения подготовки 24В и 500 кг 10% палладия на угле в 150 мл абсолютного этанола. Эту суспензию встряхивали в атмосфере водорода под давлением в 60 psi в аппарате Парра. Катализатор - 10 % палладий, на угле удаляли фильтрованием. Фильтрат упаривали досуха при пониженном давлении, получая 31,1 г светло-желтой пены. Это соединение использовали без дальнейшей очистки.
Выход: 96%.
[α]D +34,68o (с=1,0, MeOH).
1H ЯМР (CDCl3): δ 1,46 (s, 9H), 2,71 (dd, J=13,7; 9,5 Гц, 1H), 2,84 (dd, J= 13,3; 2,51 Гц, 1H), 2,95-3,06 (m, 2H), 3,23-3,29 (m, 1H), 3,84-3,90 (m, 1H), 6,23 (s, 1H), 7,19-7,37 (m, 12H).
ИК (CHCl3): 3440, 3382, 3007, 2970, 2934, 1643, 1516, 1454, 1367, 1213 см-1.
Macc-спектр (FD): m/e 341 (M+), 341 (100).
Подготовка 25
А. 2R-2-N(т-Бутоксикарбонил)амино-3-нафт-2илтиопропановая кислота
К раствору 2,14 г (13,4 ммоль) 2-нафталинтиола в 40 мл безводного тетрагидрофурана при комнатной температуре добавляли суспензию 0,54 г (13,5 ммоль) гидрида натрия в минеральном масле. По истечении приблизительно 15 минут к смеси добавляли по каплям раствор 2,5 г (13,4 ммоль) S-N-бутоксикарбонил)-серин-bлактона в 30 мл тетрагидрофурана. Смесь реагировала в течение приблизительно одного часа, после чего ее концентрировали при пониженном давлении, получая липкое твердое вещество. Его очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в этилацетате), получая 4,35 г белого твердого вещества.
Выход: 94%.
1H ЯМР (CDCl3): δ 10,25 (s, 1H), 7,89 (s, 1H), 1,78 (m, 3H), 7,46 (m, 3H), 5,39 (d, 1H), 4,61 (m, 1H), 3,49 (m, 2H), 1,37 (s, 9H).
Б. 2R-N(Meтoкcи)-N(метил)[2-N(т-бутoкcикapбoнил)aминo- 3-нафт-2-илтио] пропанамид
К холодному раствору (0oC), содержащему 4,3 г (12,4 кмол) поименованного промежуточного соединения подготовки 25А, 1,58 г (16,15 ммоль) N,O-диметилгидроксиламина гидрохлорида, 2,18 г (16,15 ммоль) 1-гидроксибензотриазола моногидрата (HOBT• H2O), 2,24 мл (16,15 ммоль) триэтиламина и 2,73 мл (24,86 ммоль) N-метилморфолина в 100 мл метилекхлорида, добавляли 2,62 г (13,67 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (EDC). Образовавшейся реакционной смеси позволяли реагировать при комнатной температуре в течение ночи. Реакционную смесь разбавляли 100 мл гексана, промывали последовательно 200 мл насыщенного раствора бикарбоната натрия и 200 мл солевого раствора. Образовавшиеся слои разделяли и органический слой высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении, получая прозрачное желтое масло.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,80 (m, 3H), 7,49 (m, 3H), 5,41 (d, 1H), 4,92 (m, 1H), 3,59 (S, 3H), 3,18-3,46 (m, 2H), 3,05 (s, 3H), 1,42 (s, 9H).
Масс-спектр (FD): m/e 391 (M+), 390 (100).
В. 3R-N(т-Бутил)-2-[2'-оксо-3'-N(т-бутоксикарбонил)амино -4'-нафт-2-илтио]бутилбензамид
К холодному раствору (-78oC), содержащему 8,60 г (45 ммоль) поименованного соединения подготовки 24А и 14,2 мл (95 ммоль) TMEDA в 100 мл безводного тетрагидрофурана, в атмосфере инертного газа медленно добавляли 111 мл (95 ммоль) 0,85 М раствора в-бутиллития в гексане, используя шприц. За внутренней температурой реакционного сосуда следили в течение добавления в-бутиллития, обеспечивая не превышение -57oC. После проведения реакции в течение приблизительно одного часа при -78oC к реакционной смеси по каплям добавляли раствор 7,90 г (20 ммоль) поименованного промежуточного соединения подготовки 2Б в 80 мл тетрагидрофурана. По окончании добавления реакционную смесь нагревали до -20oC, после чего реакцию останавливали добавлением насыщенного раствора хлорида аммония. Полученную смесь затем разбавляли 600 мл диэтилового эфира. Образовавшиеся слои разделяли и органический слой промывали последовательно 1 М раствором бисульфата натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая желтое масло. Это масло очищали, используя флэш-хроматографию (элюирующий раствор - 10-50% градиент этилацетата в гексане), получая 8,5 г желаемого поименованного промежуточного соединения.
Выход: 82%.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,79 (t, 3H), 7,48 (m, 3H), 7,40 (d, 1H), 7,29 (m, 2H), 7,05 (d, 1H), 5,94 (br. s, 1H), 5,65 (m, 1H), 4,65 (d, 1H), 4,24 (d, J=17 Гц, 1H), 3,86 (d, J=17 Гц, 1H), 3,66 (m, 1H), 3,40 (m, 1H), 1,42 (s, 9H), 1,39 (s, 9H).
Масс-спектр (FD): m/e 521 (M+), 521 (100).
Г. [(2R-(2R*3R*)] - N(т-Бутил)-2-[2'-гидрокси-3'-N-(т-бутoксикарбонил)амино-4' -нафт-2-илтио]бутилбензамид
К раствору 3,49 г (6,7 ммоль) поименованного промежуточного соединения подготовки 25В в 150 мл абсолютного этанола добавляли 0,51 г (13 ммоль) боргидрида натрия и образовавшейся реакционной смеси позволяли реагировать в течение ночи при комнатной температуре. Реакционную смесь затем охлаждали до 0oC, реакцию останавливали добавлением насыщенного раствора хлорида аммония и реакционную смесь разбавляли 550 мл метиленхлорида. Образовавшиеся слои разделяли и органический слой промывали последовательно 1н. соляной кислотой, 2н. гидроксидом натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении, получая бесцветную пену. Эту пену очищали, используя флэш-хроматографию (элюирующий раствор - 10-25% градиент гексана в этилацетате), получая 2,78 г желаемого поименованного промежуточного соединения.
Выход: 78%.
1H ЯМР (CDCl3): δ 7,84 (s, 1H), 7,73 (m, 3H), 7,41 (m, 3H), 7,29 (t, 2H), 7,16 (t, 2H), 6,53 (s, 1H), 5,32 (d, 1H), 3,86 (m, 2H), 3,33 (m, 2H), 2,83 (m, 2H), 1,40 (s, 9H).
Масс-спектр (FD): m/e 523 (M+), 522 (100).
Анализ для C30H38N2O4S:
Расчет: C 68,94; H 7,33; N 5,36.
Обнаружено: C 68,65; H 7,34; N 5,15.
Д. [(2R-(2R*,3R*)]-N(т-Бутил)-2-[2'-гидрокси- 3'-амино-4'-нафт-2-илтио] бутилбензамид
К холодному раствору (0oC) 2,89 г (5,53 ммоль) поименованного промежуточного соединения подготовки 25Г в 100 мл метиленхлорида добавляли 18 мл трифторуксусной кислоты. Образовавшейся реакционной смеси позволяли реагировать в течение приблизительно одного часа. Реакционную смесь затем упаривали; при пониженном давлении, получая пену. Ее суспензировали в толуоле и упаривали при пониженном давлении, получая пену, которую затем очищали, используя флэш-хроматографию (элюирующий раствор - 5% метанол в метиленхлориде), получая 1,71 г белой пены.
Выход: 74%.
1H ЯМР (CDCl3): δ 7,75-7,85 (m, 4H), 7,24-7,51 (m, 7H), 6,06 (s, 1H), 3,75 (m, 1H), 3,61 (m, 1H), 3,07 (m, 2H), 2,95 (m, 2H), 1,47 (s, 9H).
Масс-спектр (FD): m/e 423 (M+), 422 (100).
Подготовка 26
А. N-т-Бутил-2-метил-1-нафтиламид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 24А. Неочищенный материал очищали перекристаллизацией из гексана/этилацетата, получая 20,99 г бесцветных игольчатых кристаллов (т.п. 124-126oC).
Выход: 68%.
1H ЯМР (CDCl3): δ 1,54 (s, 9H), 2,50 (s, 3H), 5,50-5,65 (br.s, 1H), 7,23-7,54 (m, 3H), 7,74 (d, J=10 Гц, 1H), 7,78 (d, J=10 Гц, 1H), 7,87 (d, J= 10 Гц, 1H).
ИК (CHCl3): 3424, 3010, 2969, 1660, 1512, 1503, 1454, 1366, 1291, 1263, 1221 см-1.
Масс-спектр (FD): m/e 241 (М+), 241(100).
Анализ для C16H19NO:
Расчет: C 79,63; H 7,94; N 5,80.
Обнаружено: C 79,90; H 8,11; N 5,76.
Б. S-N-T-Бутил-2-(3-(N-бензилоксикарбонил)амино-4-фенил-2 -оксобутил)-1-нафтиламид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 24А. Остаток очищали, используя флэш-хроматографию (злюирующий раствор - 10-30% градиент этилацетата в гексане), получая 7,43 г бесцветной пены.
Выход: 86%.
[α]D -6,86o (с=0,10, MeOH).
1H ЯМР (CDCl3): δ 1,45 (s, 9H), 3,03 (dd, J=15, 8 Гц, 1H), 3,18 (dd, J= 15, 5 Гц, 1H), 3,91 (d, J=16 Гц, 1H), 4,04 (d, J= 16 Гц, 1H), 4,70-4,80 (m, 1H), 4,94-5,06 (m, 2H), 5,41 (d, J= 8 Гц, 1H), 6,12-6,20 (br.s, 1H), 7,10-7,38 (m, 11H), 7,42-7,58 (m, 2H), 7,76-7,85 (m, 2H), 7,93 (s, J=9 Гц, 1H).
ИК (CHCl3): 3420, 3029, 3012, 2970, 1713, 1658, 1505, 1455, 1367, 1232, 1045 см-1.
Масс-спектр (FD): m/e 522 (M+), 522 (100).
Анализ для C33H34N2O4:
Расчет: C 75,84; H 6,56; N 5,36.
Обнаружено: C 75,56; H 6,74; N 5,17.
В. [2R-(2R*, 3S*)] -N-т-Бутил-2-(3-(N-бензилоксикарбонил) амино-3-(фенилметил-2гидроксипропил)-1-нафтиламид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 24В. Материал очищали, используя флэш-хроматографию (элюирующий раствор - 2-10% градиент этилацетата в метиленхлориде), получая 5,50 г бесцветной пены.
Выход: 74%.
[α]D +11,55o (с=0,20, MeOH).
1H ЯМР (CDCl3): δ 1,54 (s, 9H), 2,85-3,15 (m, 4H), 3,85- 3,95 (m, 1H), 4,00-4,13 (m, 2H), 4,90-5,34 (m, 3H), 5,85-5,95 (m, 1H), 7,05-7,60 (m, 15H), 7,81 (d, J=9 Гц, 2H), 7,91 (d, J=9 Гц, 2H).
ИК (CHCl3): 3420, 3012, 2970, 1713, 1643, 1515, 1454, 1367, 1219, 1209, 1028 см-1.
Масс-спектр (FD): m/e 524 (M+), 524 (100).
Анализ для C33H36N2O4:
Расчет: C 75,55; H 6,92; N 5,34.
Обнаружено: C 75,41; H 7,16; N 5,14.
Г. [2R-(2R*,3S*)] -N-т-Бутил-2-(3-амино-2-гидроксипропил)-1-нафтиламид
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 24Г. Неочищенный фильтрат концентрировали, получая 1,30 г бесцветной пены, которую использовали без какой-либо дальнейшей очистки.
Выход: 92%.
Подготовка 27
А. 2-Иод-4-гидроксиметилтолуол
К раствору 5,0 г (19,1 ммоль) 2-иод-3-метилбензойной кислоты в 50 мл безводного тетрагидрофурана медленно добавляли 22 мл 1 М раствора борана в тетрагидрофуране. Реакция протекала в течение приблизительно девяноста минут, после чего ее останавливали добавлением этанола, вызывающим выделение газообразного водорода. Смесь разбавляли этилацетатом. Образовавшиеся слои разделяли и органический слой промывали последовательно бикарбонатом натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и кристаллизовали из смеси гексан/этилацетат, получая 120 мг желаемого поименованного соединения.
В. 2-Метил-5-гидроксиметилбензойная кислота
Смесь 142 мг (5,92 ммоль) гидроксида лития и 249 мг (1,48 ммоль) поименованного соединения подготовки 27А в смеси тетрагидрофуран/вода в отношении 3: 1 реагировала в течение приблизительно двадцати четырех часов. По окончании реакции, по данным ТСХ, реакционную смесь упаривали при пониженном давлении и подкисляли 1н. соляной кислотой. Смесь разбавляли этилацетатом и образовавшиеся слои разделяли. Органический слой промывали солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали досуха, получая 70 мг желаемого поименованного в заголовке соединения.
Подготовка 28
2-Метил-3-метиламинобензойная кислота
К раствору 500 мг (2,5 ммоль) метилового эфира 2-метил-3- аминобензоата в 5 мл диметилформамида добавляли 387 мг (2,7 ммоль) йодистого метила и 700 мг (5,4 ммоль) диизопропилэтиламина. Реакционную смесь нагревали до 70oC в течение приблизительно двух часов и затем выливали в 10 мл 1н. гидроксида калия. По истечении приблизительно шестнадцати часов смесь подкисливали 2н. соляной кислотой до pH 6. Желаемое названное в заголовке соединение экстрагировали в этилацетат, высушивали и упаривали досуха при пониженном давлении, получая 343 мг белого твердого вещества (т.п. 165-167oC).
Выход: 84%.
1H ЯМР (CDCl3): δ 12,52 (br.s, 1H), 7,38 (d, J=7,8 Гц, 1H), 7,25 (t, J= 7,9 Гц, 1H), 6,93 (d, J=7,8 Гц, 1H), 2,92 (s, 3H), 2,21 (s, 3H).
Анализ для C9H11NO2:
Расчет: C 65,44; H 6,71; N 8,48.
Обнаружено: C 65,62; H 6,84; N 8,26.
Подготовка 29
А. 2-Метил-5-аминобензойная кислота
Желаемое названное в заголовке соединение получали, восстанавливая 2-метил-5-нитробензойную кислоту, используя смесь олова с соляной кислотой (т. п. 142-144oC).
Выход: 75%.
1H ЯМР (ДМСО-d6): δ 12,67 (br.s, 1H), 7,23 (s, 1H), 7,04 (d, J=8,2 Гц, 1H), 6,82 (d, J=7,9 Гц, 1H), 3,25 (s, 2H), 2,40 (s, 3H).
Анализ для C8H9NO2:
Расчет: C 63,57; H 6,00; N 9,27.
Обнаружено: C 63,81; H 6,24; N 9,06.
Б. 2-Метил-5-гидроксибензойная кислота
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в альтернативной подготовке 23В, используя соединение подготовки 29А.
Выход: 65% (т.п. 136-139oC).
1H ЯМР (DMSO): δ 12,77 (br.S, 1H), 9,46 (br. s, 1H), 7,26 (s, 1H), 7,12 (d, J=8,3 Гц, 1H), 6,85 (d, J=8,l Гц, 1H), 2,41 (s, 3H).
Анализ для C8H8O3:
Расчет: C 63,15; H 5,29.
Обнаружено: C 63,27; H 5,22.
Подготовка 30
А. 5-Цианизохинолин
К холодному раствору (0oC) 10,0 г (61,4 ммоль) 5-амино-изохинолина в 288 мл 1,5н. соляной кислоты добавляли 15 мл 5,2 М раствора нитрита натрия в воде. Приблизительно через 5 минут к реакционной смеси добавляли охлажденный насыщенный раствор бикарбоната натрия до тех пор, пока реакционный раствор показывал отрицательную реакцию в тесте с использованием иодида и крахмальной бумаги. Раствор выливали в холодную (0-5oC) двуфазную смесь, содержащую 300 мл толуола и 150 мл водного раствора, включающего 8,4 г (177 ммоль) цианида натрия и 7,6 г (85 ммоль) цианида меди. Полученную реакционную смесь нагревали до комнатной температуры и реакцию проводили в течение приблизительно 1 часа, затем реакционную смесь разбавляли смесью этилацетата и воды. Образовавшиеся слои разделяли и органическую фазу высушивали над сульфатом натрия, фильтровали и затем упаривали досуха при пониженном давлении, получая 5,9 г желтого твердого вещества.
Выход: 56%.
1H ЯМР (CDCl3): δ 9,38 (s, 1H), 8,76 (d, J=5,89 Гц, 1H), 8,25 (d, J=8,29 Гц, 1H), 8,13 (d, J=8,30 Гц, 1H), 8,03 (d, J= 8,59 Гц, 1H), 7,71 (t, J=7,78 Гц, 1H).
ИК (KBr): 3433, 3090, 3026, 2924, 2226, 1618, 1574, 1495, 1433, 1373, 1277, 1225, 1034, 829, 766, 714.
Б. 5-Карбоксиизохинолин
Раствор 6,5 г (42 ммоль) поименованного соединения подготовки 30А в 55 мл концентрированной соляной кислоты нагревали до 155oC в запечатанной пробирке в течение 5,5 часов, охлаждали до комнатной температуры и упаривали досуха, получая твердое вещество. Это твердое вещество повторно растворяли в 300 мл воды и pH раствора подводили к 6, используя разбавленный раствор гидроксида аммония, в результате чего выпадал твердый осадок коричневого цвета. Этот осадок отделяли фильтрованием, азеотропно перегоняли с бензолом и затем высушивали при пониженном давлении при 130oC в течение приблизительно 3 часов, получая 5,7 г мелкозернистого темного порошка рыжевато-коричневой окраски (т.п. 270-272oC).
Выход: 78%.
1H ЯМР (DMSO): δ 13,4 (dr. s, 1H), 8,69 (d, 1H, J=6,00 Гц), 8,58 (d, 1H, J=4,6 Гц), 8,40 (d, 1H, J=7,37 Гц), 8,36 (d, 1H, J=8,12 Гц), 7,74 (t, 1H, J= 7,76).
ИК (KBr): 3460, 3014, 2930, 2851, 2777, 2405, 1912, 1711, 1622, 1574, 1493, 1427, 1375, 1264, 1211, 1152, 1044.
В. 5-Карбоксиизохинолинпентафторфениловый эфир
К холодному раствору (0oC) 1,53 г (7,39 ммоль) 1,3- дициклогексилкарбодиимида (DCC) в 60 мл этилацетата добавляли 1,28 г (7,39 ммоль) поименованного соединения подготовки 30Б и 4,08 г (22,17 ммоль) пентафторфенола в 30 мл этилацетата. Реакционная смесь реагировала в течение приблизительно 6 часов при 0oC, после чего ее фильтровали через целит. Фильтрат промывали последовательно 1н. гидроксидом натрия, водой и солевым раствором и затем упаривали при пониженном давлении, получая белое твердое вещество. Это твердое вещество очищали, используя колоночную хроматографию (диоксид кремния; элюирующий раствор - 33% этилацетат в гексане), получая 1,80 г желаемого поименованного соединения (т.п. 142-144oC).
Выход 72%.
1H ЯMP (CDCl3): δ 9,38 (s, 1H), 8,74 (m, 3H), 8,34 (d, J=8,1 Гц, 1H), 7,78 (t, J=7,7 Гц, 1H).
ИК (KBr): 3422, 3021, 2089, 1752, 1622, 1522, 1215, 758.
Анализ для C16H6NO2F5• 0,3CH2Cl2:
Расчет: C 57,30; H 2,17; N 4,03.
Обнаружено: C 57,40; H 2,10; N 4,33.
Подготовка 31
5-Карбоксихинолинпентафторфениловый эфир
Желаемое поименованное в заголовке соединение получали в соответствии с методикой, детально описанной в подготовке 30В, используя 0,236 г (1,36 ммоль) 5-карбоксихинолина, 0,746 г (4,05 ммоль) пентафторфенола и 0,571 г (2,76 ммоль) DCC в 25 мл этилацетата, за исключением того, что реакцию проводили в течение 48 часов. Полученный неочищенный материал очищали, используя колоночную хроматографию, получая 0,40 г белого твердого вещества.
Выход 87%.
1H ЯМР (CDCl3): δ 9,33 (d, J=8,54 Гц 1H), 9,03 (dd, J=4,16, 1,28 Гц, 1H), 8,63 (d, J=7,25 Гц, 1H), 8,47 (d, J=8,53 Гц, 1H); 7,87 (t, J=7,96 Гц, 1H), 7,61 (dd, J=8,76, 4,18 Гц, 1H).
ИК (KBr): 3472, 2667, 2461, 1749, 1520, 1319, 1259, 1182, 1145, 1105, 1005, 947, 812.
Подготовка 32
1H-Индолин-4-карбоновая кислота
К холодному раствору (10oC), содержащему 100 мг (0,62 ммоль) индолин-4-карбоновой кислоты в 5 мл уксусной кислоты, добавляли 390 мг (6,2 ммоль) твердого цианборгидрида натрия. Полученная смесь реагировала в течение приблизительно 16 часов при комнатной температуре, после чего ее разбавляли водой. Желаемое соединение экстрагировали из этого раствора с помощью метиленхлорида и органические экстракты высушивали над сульфатом натрия и фильтровали. Неочищенный материал очищали, используя колоночную хроматографию (диоксид кремния; элюирующий раствор - 1% метанол в метиленхлориде), получая 12 мг названного соединения (т.п. 97-98oC).
Выход: 12%.
1H ЯМР (CDCl3): δ 7,48 (d, J=8,8 Гц, 1H), 7,34 (t, J=8,6 Гц, 1H), 6,88 (d, J=8,8 Гц, 1H), 3,59 (m, 4H).
Анализ для C9H9NO2:
Расчет: C 66,25; H 5,56; N 8,58.
Обнаружено: C 66,36; H 5,82; N 8,42.
Подготовка 33
А. 2,3-Диметокси-6-хлортолуол
К смеси 25 г (0,16 ммоль) 1-метил-2,3-диметоксибензола в 25 мл уксусной кислоты медленно добавляли 26,4 г (0,33 ммоль) 1- хлорметилметилового эфира. Полученная реакционная смесь реагировала в течение ночи при 30oC, после чего ее разбавляли холодной водой, что приводило к образованию осадка. Этот осадок очищали перекристаллизацией из горячего гексана и затем упаривали досуха при пониженном давлении, получая 20,3 г белого твердого вещества (т.п. 69-70oC).
Выход: 62%.
1H ЯМР (CDCl3): δ 7,01 (d, J=6,1 Гц, 1H), 6,75 (d), 4,62 (s, 2H), 3,85 (s, 3H), 3,76 (s, 3H), 2,37 (s, 3H).
Анализ для C10H13O2Cl:
Расчет: C 59,93; H 6,54.
Обнаружено: C 59,87; H 6,43.
Б. 2-Метил-3,4-диметоксибензойная кислота
К смеси 3,0 г (15 ммоль) поименованного соединения подготовки 33А в 150 мл воды добавляли 3,2 г (20 ммоль) твердого перманганата калия и 3,0 г (36 ммоль) карбоната натрия. Реакционную смесь нагревали до 80oC и реакцию проводили в течение приблизительно 24 часов. После охлаждения реакционную смесь фильтровали и разбавляли этилацетатом. Образовавшиеся слои разделяли и водный слой подкисляли, используя 2н. соляную кислоту, что приводило к образованию осадка. Этот осадок отделяли фильтрацией и промывали холодным гексаном, получая 1,7 г белого твердого вещества (т.п. 179-180oC).
Выход: 58%.
1H ЯМР (ДМСО-d6): δ 12,49 (br.s, 1H), 7,71 (br.s, 1H) 6,99 (br.s, 1H), 3,89 (s, 3H), 3,77 (s, 3H), 2,45 (s, 3H).
Анализ для C10H12O4:
Расчет: C 61,28; H 6,17.
Обнаружено: C 61,36; H 6,24.
В. 2-Метил-3,4-дигидроксибензойная кислота
К холодной смеси (0oC) 250 мл (1,3 ммоль) поименованного соединения подготовки 33Б в 5 мл метиленхлорида, добавляли 6,4 мл 6,4 ммол/1,0 М раствора трибромида бора в метиленхлориде. Полученная реакционная смесь реагировала в течение приблизительно 90 минут, после чего ее разбавляли 25 мл 2н. соляной кислоты. Желаемое соединение экстрагировали, используя этилацетат, и органические экстракты высушивали над сульфатом натрия, фильтровали и упаривали, получая 197 мг твердого вещества рыжевато-коричневой окраски (т.п. 200-201oC).
Выход: 92%.
1H ЯМР (ДМСО): δ 12,14 (br.s, 1H), 9,96 (br.s, 1H), 8,34 (dr.s, 1H), 7,27 (d, J=7,0 Гц, 1H), 6,67 (d, J=6,7 Гц, 1H), 2,37 (s, 3H).
Анализ для C8H8O4:
Расчет: C 57,14; H 4,80.
Обнаружено: C 57,34; H 4,76.
Пример 1
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2'' -фтор-3''гидроксифенил) пентил] декагидроизохинолин-3-N-т- бутилкарбоксамид
К холодному раствору (-10oC), содержащему 80 мг (0,20 ммоль) поименованного соединения подготовки 1В, 31 мг (0,20 ммоль) подготовки 11Б и 27 мг (0,20 ммоль) 1-гидроксибензотриазола моногидрата (HOBT•H2О) в 3 мл безводного тетрагидрофурана, добавляли 41 мг (0,20 ммоль) 1,3-дициклогексилкарбодиимида (DCC). Реакционную смесь перемешивали в течение 36 часов при комнатной температуре и затем упаривали при пониженном давлении. Полученный остаток повторно растворяли в этилацетате, фильтровали через целит, промывали последовательно насыщенным бикарбонатом натрия и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 2-5% градиент метанола в метиленхлориде), получая 79 мг белой пены.
Выход: 73%.
[α]D -90,80o (с=0,333, MeOH).
1H ЯМР (CDCl3): δ 1,24 (s, 9H), 1,16-2,05 (m, 14H), 2,20-2,40 (m, 2H), 2,55-2,70 (m, 2H), 2,90-3,04 (m, 2H), 3,10-3,25 (m, 1H), 4,03 (br.s, 1H), 4,51 (dr.s, 1H), 6,01 (s, 1H), 6,90-7,35 (m, 9H).
ИК (CHCl3): 3580, 3550-3100 (br.), 2929, 2865, 1662, 1596, 1521, 1472, 1455, 1394, 1368, 1293, 1157, 1047, 879, 839 см-1.
Масс-спектр (FD): m/e 540 (M+, 100).
HR Масс-спектр (FAB): m/e для C31H43N3O4F:
Расчет: 540,3238.
Обнаружено: 540,3228.
Пример 2
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]-2- [2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-хлорпиридил- 3''-ил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 31 мг (0,20 ммоль) 2-хлорникотиновой кислоты, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 0-5% градиент метанола в метиленхлориде), получая 58 мг беловатой пены.
Выход: 54%.
[α]D -70,64o (c=0,224, MeOH).
1H ЯМР (CDCl3): δ 1,16 (s, 9H), 1,17-2,10 (m, 12H), 2,25-2,37 (m, 2H), 2,52-2,70 (m, 2H), 2,97-3,06 (m, 2H), 3,44-3,53 (m, 2H), 4,05 (br.s, 1H), 4,60-4,70 (m, 1H), 5,64 (s, 1H), 7,18-7,38 (m, 7H), 7,60-7,63 (m, 1H), 8,38-8,40 (m, 1H).
ИК (CHCl3): 3618, 3428, 3650-3100 (br.), 2929, 1667, 1583, 1515, 1455, 1401, 1368, 1247, 1071, 1046, 877 см-1.
Macc-спектр (FD): m/e 541 (M+), 440 (100).
Анализ для C30H41N4O3Cl:
Расчет: C 66,59; H 7,64; N 10,35; Cl 6,55.
Обнаружено: C 66,60; H 7,58; N 10,17; Cl 6,84.
Пример 3
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-этил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 35 мг (0,21 ммоль) поименованного соединения подготовки 10Б, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3-5% градиент метанола в метиленхлориде), получая 71 мг беловатой пены.
Выход: 65%.
[α]D: -76,29o (c=0,291, MeOH).
1H ЯМР (CDCl3): δ 1,03 (t, J=7,42 Гц, 3H), 1,21 (s, 9H), 1,22-2,10 (m, 11H), 2,24-2,35 (m, 2H), 2,44-2,70 (m, 4H), 2,96 -3,05 (m, 2H), 3,26-3,40 (m, 1H), 3,96-4,23 (m, 2H), 4,53 (br. s, 1H), 5,80 (s, 1H), 6,30-6,56 (m, 3H), 6,77 (d, J=7,77 Гц, 1 H), 6,88 (t, J=7,75 Гц, 1H), 7,19-7,39 (m, 5H).
ИК (CHCl3): 3700-3100 (br.), 3429, 3327, 3011, 2971, 2930, 2867, 1662, 1604, 1585, 1514, 1455, 1394, 1368, 1278, 1155, 1087, 1046, 910 см-1.
NS (FD): m/e 550 (M+, 100).
HR Масс-спектр (FAB): m/e для C33H48N3O4:
Расчет: 550,3645.
Обнаружено: 550,3664.
Пример 4
[2S-(2R*, 2'S*, 3'R*)]-1-[2'-Гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил) пентил]пирролидин-2-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 55 мг (0,16 ммоль) поименованного соединения подготовки 3Д, 25 мг (0,16 ммоль) по именованного соединения подготовки 9Б, 33 мг (0,16 ммоль) DCC и 22 мг (0,16 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 4-8% градиент метанола в метиленхлориде), получая 52 мг белого твердого вещества.
Выход: 69%.
[α]D: -72,15o (c=0,211, MeOH).
1H ЯМР (CD3OD): δ 1,33 (s, 9H), 1,70-1,90 (m, 4H), 2,06-2,20 (m, 1H), 2,45-3,30 (m, 8H), 3,60-3,70 (m, 1H), 4,25-4,38 (m, 1H), 6,48 (d, J=8,8 Гц, 1H), 6,74 (d, J=7,7 Гц, 1H), 6,93 (t, J=7,7 Гц, 1H), 7,15-7,32 (m, 5H).
ИК (CHCl3): 3600-2700 (br.), 3450, 3255, 2968, 2928, 1653, 1632, 1588, 1513, 1454, 1364, 1291, 1233, 1064, 884, 836 см-1.
Масс-спектр (FD): m/e 468 (M+, 100).
Анализ для C27H37N3O4:
Расчет: C 69,35; H 7,98; N 8,99.
Обнаружено: C 69,54; H 8,10; N 9,19.
Пример 5
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(пирид-3''-ил-N- оксидил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 28 мг (0,20 ммоль) N-оксида никотиновой кислоты, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 5-10% градиент метанола в метиленхлориде), получая 81 мг белой пены.
Выход: 76%.
[α]D: -104,39o (с=0,213, MeOH).
1H ЯМР (ДМСО-d6): δ 1,19 (s, 9H), 1,19-2,10 (m, 14H), 2,50-2,60 (m, 1H), 2,65-2,79 (m, 1H), 2,95- 3,10 (m, 2H), 3,83 (br.s, 1H), 4,22-4,32 (m, 1H), 4,87 (d, J=5,5 Гц, 1H), 7,06-7,11 (m, 1H), 7,17-7,22 (m, 2H), 7,33-7,44 (m, 3H), 7,57 (d, J=8,0 Гц, 1H), 8,26 (d, J=6,4 Гц, 1H), 8,44-8,48 (m, 2H).
ИК (CHCl3): 3600-3100 (br.), 3428, 2930, 2864, 1669, 1603, 1515, 1479, 1455, 1432, 1394, 1368, 1300, 1279, 1245, 1135, 1083, 1046, 1017 см-1.
Macc-спектр (FD): m/e 522 (M+, 100).
Пример 6.
[2S-(2R*, 2'S*, 3'R*)]-1-[2'-Гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил] пиперидин-2-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 100 мг (0,29 ммоль) поименованного соединения подготовки 4Е, 44 мг (0,29 ммоль) поименованного соединения подготовки 9Б, 59 мг (0,29 ммоль) DCC и 39 мг (0,29 ммоль) HOBT•H2О в 5 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 1,5-7% градиент метанола в метиленхлориде), получая 57 мг беловатой пены.
Выход: 41%.
[α]D: -58,90o (c=0,163, MeOH).
1H ЯМР (CD3OD): δ 1,29 (s, 9H), 1,50-2,20 (m, 10H), 2,60-2,75 (m, 4H), 3,10-3,35 (m, 4H), 3,85-4,02 (m, 2H), 4,10-4,35 (m, 2H), 4,85 (s, 1H), 3,85-4,02 (m, 2H), 4,10-4,35 (m, 2H), 4,85 (s, 1H), 6,55 (d, J=7,29 Гц, 1H), 6,75 (d, J=7,83 Гц, 1H), 6,90-6,96 (m, 1H), 7,15-7,35 (m, 5H).
ИК (CDCl3): 3601, 3600-3100 (br.), 3428, 3340, 3008, 2941, 2861, 1661, 1601, 1587, 1514, 1455, 1394, 1367, 1284, 1156, 1086, 1047, 832 см-1.
Масс-спектр (FD): m/e 482 (M+, 100).
Анализ для C28H39N3O4:
Расчет: C 69,83; H 8,16; N 8,72.
Обнаружено: C 69,84; H 8,46; N 8,50.
Пример 7
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]-2- [2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'- (2''-метил-3''- фторфенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 31 мг (0,20 ммоль) 3-фтор-2-метилбензойной кислоты, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 1,5-3% градиент метанола в метиленхлориде), получая 40 мг белой пены.
Выход: 37%.
[α]D: -80,10o (с=0,132, MeOH).
1H ЯМР (CDCl3): δ 1,13 (s, 9H), 1,13-2,10 (m, 16H), 2,20- 2,35 (m, 2H), 2,50-2,70 (m, 2H), 2,95-3,05 (m, 2H), 3,53-3,58 (m, 1H), 3,98 (s, 1H), 4,03-4,10 (m, 1H), 5,68 (s, 1H), 6,83-7,07 (m, 3H), 7,10-7,40 (m, 5H).
ИК (CHCl3): 3650-3150 (br.), 3429, 3030, 3008, 2930, 2863, 1672, 1608, 1514, 1455, 1394, 1368, 1277, 1046, 910, 830 см-1.
Macc-спектр (FD): m/e 538 (M+, 100).
HR Масс-спектр (FAB): m/e для C32H45N3O3F:
Расчет: 538,3445.
Обнаружено: 538,3469.
Пример 8
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-хлор-3'', 5''- дигидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 100 мг (0,25 ммоль) поименованного соединения подготовки 1Б, 47 мг (0,25 ммоль) 2-хлор-3,5-дигидроксибензойной кислоты, 51 мг (0,25 ммоль) DCC и 34 мг (0,25 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2-10% градиент метанола в метиленхлориде), получая 47 мг белого твердого вещества.
Выход: 33%.
[α]D: -53,79o (c=0,097, MeOH).
1H ЯМР (CDCl3): δ 0,5-3,10 (m, 32H), 3,70-4,60 (m, 2H), 6,00-7,50 (m, 8H).
ИК (CHCl3): 3700-3100 (br.), 2930, 2865, 1658, 1604, 1521, 1455, 1368, 1246, 1156, 1047, 1014, 856 см-1.
Macc-спектр (FD): 572 (M+, 100).
Анализ для C31H42N3O5Cl:
Расчет: C 65,08; H 7,40; N 7,34.
Обнаружено: C 65,30; H 7,35; N 7,43.
Пример 9
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'', 5''-диаминофенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 100 мг (0,25 ммоль) поименованного соединения подготовки 1Б, 41 мг (0,25 ммоль) 3,5-диамино-2-метилбензойной кислоты, 51 мг (0,25 ммоль) DCC и 34 мг (0,25 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана и 0,5 мл диметилформамида. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 1- 10% градиент метанола в метиленхлориде), получая 30 мг рыхлой оранжевой пены.
Выход: 22%.
[α]D -89,27o (c=0,137, MeOH).
1H ЯМР (CDCl3): δ 1,21 (s, 9H), 1,30-2,02 (m, 16H), 2,19- 2,35 (m, 2H), 2,48-2,70 (m, 2H), 2,90-3,07 (m, 2H), 3,10-3,23 (m, 1H), 3,50 (br.s, 4H), 3,94 (br. s, 1H), 4,40- 4,50 (m, 1H), 5,70 (s, 1H), 5,89-5,95 (m, 2H), 6,30 (d, J=8,4 Гц, 1H), 7,15-7,33 (m, 5H).
ИК (CHCl3): 3600-3100 (br.), 3029, 3005, 2928, 2865, 1664, 1621, 1513, 1455, 1392, 1367, 1276, 1244, 1171, 1047, 841 см-1.
Масс-спектр (FD): m/e 550 (M+, 100).
HR Масс-спектр (FAB): m/e для C32H48N5O3:
Расчет: 550,3757.
Обнаружено: 550,3762.
Пример 10
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'', 5''-динитрофенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 100 мг (0,25 ммоль) поименованного соединения подготовки 1Б, 56 мг (0,25 ммоль) 3,5-динитро-2-метилбензойной кислоты, 51 мг (0,25 ммоль) DCC и 34 мг (0,25 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 0-3% градиент метанола в метиленхлориде), получая 61 мг беловатой пены.
Выход: 41%.
[α]D 105,96o (c=0,302, MeOH).
1H ЯМР (CDCl3): δ 1,02 (s, 9H), 1,02- 2,60 (m, 20H), 2,90 -3,06 (m, 2H), 3,21 (br. s, 1H), 3,60-3,75 (m, 1H), 4,05-4,20 (m, 1H), 4,65-4,80 (m, 1H), 5,47 (s, 1H), 7,20-7,50 (m, 5H), 8,00-8,20 (m, 2H), 8,56 (s, 1H).
ИК (CHCl3): 3621, 3500-3100 (br.), 3428, 3024, 2977, 2931, 1665, 1615, 1539, 1455, 1347, 1278, 1245, 1047, 878 см-1.
Macc-спектр (FD): me 610 (M+, 100).
HR Масс-спектр (FAB): m/e для C32H44N5O7:
Расчет: 610,3241.
Обнаружено: 610,3240.
Пример 11
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-хлор-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 116 мг (0,29 ммоль) поименованного соединения подготовки 1Б, 50 мг (0,29 ммоль) названного соединения подготовки 14, 60 мг (0,29 ммоль) DCC и 39 мг (0,29 ммоль) HOBT•H2О в 4 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 2,5-5% градиент метанола в метиленхлориде), получая 83 мг белого твердого вещества.
Выход: 51%.
[α]D -74,29o (c=0,140, MeOH).
1H ЯМР (CDCl3): δ 1,19 (s,9H), 1,19-2,80 (m, 16H), 2,90- 3,15 (m, 2H), 3,35 (br. s, 1H), 4,06 (br.s, 1H), 4,56 (br.s, 1H), 5,85 (dr.s, 1H), 6,60-6,70 (m, 1H), 6,90-7,35 (m, 8H).
ИК (CHCl3): 3621, 3600-3100 (br), 3429, 2977, 2929,1 1671, 1584, 1515, 1445, 1394, 1368, 1292, 1182, 1046, 878, 823 см-1.
Масс-спектр (FD): m/e 556 (M+, 100).
Анализ для C31H42N3O4Cl:
Расчет: C 66,95; H 7,61; N 7,56.
Обнаружено: C 66,76; H 7,72; N 7,69.
Пример 12
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'' -гидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 261 мг (0,65 ммоль) поименованного соединения подготовки 1Б, 100 мг (0,65 ммоль) поименованного соединения подготовки 9Б, 134 мг (0,65 ммоль) DCC и 88 мг (0,65 ммоль) HOBT•H2О в 6 мл безводного тетрагидрофурана и 0,2 мл безводного диметилформамида. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 1-5% градиент метанола в метиленхлориде), получая 304 мг белого твердого вещества.
Выход: 87%.
[α]D -75,00o (с=0,200, MeOH).
1H ЯМР (CDCl3): δ 1,18 (s, 9H), 1,19-2,05 (m, 18H), 2,20- 2,35 (m, 2H), 2,50-2,70 (m, 2H), 2,90-3,05 (m, 2H), 3,22-3,35 (m, 1H), 3,96-4,05 (m, 1H), 4,45-4,55 (m, 1H), 5,77 (s, 1H), 6,53 (d, J=7,4 Гц, 2H), 6,75 (d, J=7,8 Гц, 1H), 6,85-6,90 (m, 1H), 7,15-7,35 (m, 6H).
ИК (CHCl3): 3606, 3600-3100 (br), 3429, 3011, 2929, 2865, 1663, 1604, 1587, 1514, 1455, 1367, 1277, 1200, 1156, 1046, 910 см-1.
Масс-спектр (FD): m/e 537 (M+, 100).
HR Масс-спектр (FAB): m/e для C32H46N3O4:
Расчет: 536,3488.
Обнаружено: 536,3488.
Пример 13
[3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил- 4'-аза-5'-оксо-5'-(2''-метил-3''-метоксифенил)пентил] декагидроизохинолин- 3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 33 мг (0,20 ммоль) поименованного соединения подготовки 15Б, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2,5% метанол в метиленхлориде), получая 93 мг белой пены.
Выход; 84%.
1H ЯМР (CDCl3): δ 1,17 (s, 9H), 1,17-2,05 (m, 12H), 2,05 (s, 3H), 2,25- 2,38 (m, 2H), 2,50-2,75 (m, 2H), 2,95-3,10 (m, 2 H), 3,35-3,50 (m, 1H), 3,79 (s, 3H), 3,98-4,15 (m, 2H), 4,59-4,65 (m, 1H), 5,72 (s, 1H), 6,47 (br.d, J= 8,21 Гц, 1H), 6,63 (d, J=7,7 Гц, 1H), 6,82 (d, J=8,12 Гц, 1H), 7,08 (t, J= 7,0 Гц, 1H), 7,15-7,45 (m, 5H).
Анализ для C33H47N3O4:
Расчет: C 72,10; H 8,62; N 7,64.
Обнаружено: C 71,84; H 8,49: N 7,67.
Пример 14
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2'', 3'' -дихлорфенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 38 мг (0,20 ммоль) 2,3-дихлорбензойной кислоты, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2O в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (2 мл пластина; элюирующий раствор - 2,5-5% градиент метанола в метиленхлориде), получая 95 мг белой пены.
Выход: 84%.
1H ЯМР (CDCl3): δ 1,16 (s, 9H), 1,17-2,05 (m, 12H), 2,20- 2,38 (m, 2H), 2,50-2,75 (m, 2H), 2,95-3,10 (m, 2H), 3,40-3,55 (m, 1H), 3,69 (s, 1H), 4,00-4,10 (m, 1H), 4,58-4,72 (m, 1H), 5,77 (s, 1H), 6,98-7,47 (m, 9H).
Масс-спектр (FD): m/e 574 (М+), 473 (100).
Пример 15
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''- трифторметилфенил)пентил]декагидроизохинолин-3-N-т- бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 80 мг (0,20 ммоль) поименованного соединения подготовки 1Б, 38 мг (0,20 ммоль) 2-трифторметилбензойной кислоты, 41 мг (0,20 ммоль) DCC и 27 мг (0,20 ммоль) HOBT•H2О в 3 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2,5-5% градиент метанола в метиленхлориде), получая 72 мг белой пены.
Выход: 63%.
1H ЯМР (CDCl3): δ 1,10 (s, 9H), 1,16-2,05 (m, 14H), 2,15- 2,35 (m, 2H), 2,45-2,70 (m, 2H), 2,92-3,05 (m, 2H), 3,38- 3,55 (m, 1H), 3,70 (br.s, 1H), 3,98-4,10 (m, 1H), 4,58-4,70 (m, 1H), 5,90 (s, 1H), 7,00-7,65 (m, 10H).
Масс-спектр (FD): m/e 573 (M+, 100).
Анализ для C32H42N3O3F3:
Расчет: C 67,00; H 7,38; N 7,32.
Обнаружено: C 67,11; H 7,09; N 7,10.
Пример 16
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидpoкcи-3'-фeнилмeтил-4'-аза-5'-оксо-5'-(2''-оксо-3''- метилпирид-4''-ил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 14,7 мг (0,037 ммоль) поименованного соединения подготовки 1Б, 5,6 мг (0,037 ммоль) поименованного соединения подготовки 12Е, 7,6 мг (0,037 ммоль) DCC и 4,9 мг (0,037 ммоль) HOBT•H2О в 1,3 мл безводного диметилформамида. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 10% метанол в метиленхлориде), получая 6,5 мг белого твердого вещества.
Выход: 34%.
1H ЯМР (CDCl3): δ 1,00-3,40 (m, 32H), 4,00-4,70 (m, 3H), 5,90-6,10 (m, 1H), 6,90-7,40 (m, 8H).
Масс-спектр (FD): m/e 537 (M+, 100).
Пример 17
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'- (2'',6''-дихлор-3''- гидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 48 мг (0,12 ммоль) поименованного соединения подготовки 1Б, 25 мг (0,12 ммоль) поименованного соединения подготовки 13, 2,5 мг (0,12 ммоль) DCC и 16 мг (0,12 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор -2-5% градиент метанола в метиленхлориде), получая 14 мг желаемого названного соединения.
1H ЯМР (CDCl3): δ 0,9-2,15 (m, 23H), 2,22-2,85 (m, 4H), 2,95- 3,10 (m, 2H), 3,30-3,58 (m, 1H), 3,98-4,12 (m, 1H), 4,56- 4,75 (m, 1H), 5,60-5,82 (m, 1H), 6,60-6,79 (m, 1H), 6,90-7,40 (m, 6H).
ИК (CHCl3): 3010, 2937, 1644, 1606, 1605, 1497, 1474, 1454, 1433, 1417, 1341, 1313, 1274, 1252, 1161, 1093, 1074, 1027, 991 см-1.
Масс-спектр (FD): m/e 590 (M+, 100).
Пример 18
[3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)]- 2-[2'-Гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''- аминофенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 100 мг (0,25 ммоль) поименованного соединения подготовки 1Б, 38 мг (0,25 ммоль) 3-амино-2-метилбензойной кислоты, 34 мг (0,25 ммоль) HOBT•H2О, 52 мг (0,25 ммоль) DCC в 3 мл безводного тетрагидрофурана за исключением того, что реакцию проводили в присутствии 76 мг (0,75 ммоль) триэтиламина. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2-5% градиент метанола в метиленхлориде), получая 78 мг беловатой пены.
Выход: 58%.
1H ЯМР (CDCl3): δ 1,19 (s, 9H), 1,20-2,08 (m, 15H), 2,20- 2,35 (m, 2H), 2,50-2,70 (m, 2H), 2,92-3,05 (m, 2H), 3,28-3,38 (m, 1H), 3,61 (br.s, 1H), 3,93-4,20 (m, 2H), 4,45-4,58 (m, 1H), 5,80 (s, 1H), 6,44 (d, J=7,5 Гц, 2H), 6,63 (d, J=7,9 Гц, 1H), 6,90 (t, J=7,7 Гц, 1H), 7,17-7,36 (m, 6H).
Масс-спектр (FD): m/e 535 (M+, 100).
Пример 19
[2S-(2R*, 2'S*, 3'S*)]-1-[2'- Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(3''-гидрокси-2''- метилфенил)пентил]-4-пирид-3''-илметилпиперазин-2-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 50 мг (0,11 ммоль) поименованного соединения подготовки 6Б, 16 мг (0,11 ммоль) поименованного соединения подготовки 9В, 14 мг (0,11 ммоль) HOBT• H2О и 22 мг (0,11 ммоль) DCC в 2 мл безводного тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 5-10 % градиент метанола в метиленхлориде), получая 35 мг беловатой пены.
Выход: 55%.
1H ЯМР (CDCl2): δ 1,29 (s, 9H), 2,18 (s, 3H), 2,23-2,33 (m, 1H), 2,45-2,85 (m, 7H), 3,20-3,35 (m, 3H), 3,45 (s, 1H), 4,00-4,10 (m, 1H), 4,25-4,35 (m, 1H), 5,00-5,40 (br.s, 1H), 6,61 (d, J=7,6 Гц, 1H), 6,76-6,80 (m, 2H), 6,92 (t, J= 7,7 Гц, 1H), 7,12-7,43 (m, 7H), 7,57-7,62 (m, 1H), 7,78 (br.s, 1H), 8,48-8,58 (m, 2H).
Масс-спектр (FD): m/e 606 (M+, 100).
Пример 20
[3S-(3R*, 4aR*,8aR*,2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4-аза-5'-оксо-5'-(2''-изопропил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 55 мг (0,137 ммоль) поименованного соединения подготовки 1Б, 24,7 мг (0,137 ммоль) поименованного соединения подготовки 18Б, 28,25 мг (0,137 ммоль) DCC и 18,5 мг (0,137 ммоль) HOBT•H2О в 8 мл тетрагидрофурана. Неочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде), получая 46 мг белой пены.
Выход: 60%.
[α]D -84,61o (с=2,60, MeOH).
1H ЯМР (CDCl3): δ 1,19 (d, J=3,7 Гц, 3H) 1,21 (d, J=3,75 Гц, 3H), 1,23 (s, 9H), 1,27-1,51 (m, 7H), 1,61-2,00 (m, 6H), 2,26-2,35 (m, 2H), 2,56-2,65 (m, 2H), 2,91-3,03 (m, 3H), 3,19-3,27 (m, 1H), 3,96 (m, 1H), 4,51 (m, 1H), 5,82 (br. s, 1H), 5,93 (br.s, 1H), 6,23 (d, J=8,53 Гц, 1H), 6,46 (d, J=7,15 Гц, 1H), 6,66 (d, J=7,17 Гц, 1H), 6,86 (t, J=7,74 Гц, 1H), 7,21-7,31 (m, 5H).
ИК (CDC[3): 3427, 3322, 3028, 3008, 2930, 2868, 1660, 1603, 1582, 1513, 1455, 1393, 1366, 1304, 1278, 1245, 1088, 1059 см-1.
Масс-спектр (FD): m/e 564 (М+, 100).
Анализ для C34H49N3O4:
Расчет: C 72,43; H 8,76; N 7,45.
Обнаружено: C 72,13; H 8,85; N 7,30.
Пример 21
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''- бутил-3''-гидроксифенил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 91 мг (0,227 ммоль) поименованного соединения подготовки 1Б, 44 мг (0,227 ммоль) поименованного соединения подготовки 16Б, 46,7 мг (0,227 ммоль) DCC и 30,6 мг (0,227 ммоль) HOBT•H2О в 10 мл тетрагидрофурана. Неочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 4-7% градиент метанола в метиленхлориде), получая 72 мг белой пены.
Выход: 55%.
[α]D -77,36o (с=0,36, MeOH).
1H ЯМР (CDCl3): δ 0,84 (t, J=7,2 Гц, 3H), 1,20 (s, 9H), 1,29-2,00 (m, 18H), 2,27 (m, 2H), 2,48-2,69 (m, 4H), 2,99 (m, 2H), 3,29 (m, 1H), 3,99 (m, 1H), 4,49 (m, 1H), 5,85 (s, 1H), 6,45 (m, 2H), 6,75 (d, J=7,19 Гц, 1H), 6,86 (t, J=7,67 Гц, 1H), 7,21-7,31 (m, 5H).
ИК (KBr): 3303 (br.), 3087, 3029, 2927, 2862, 1647, 1583, 1520, 1455, 1366, 1281, 1209, 1108, 735, 698 см-1.
Масс-спектр (FD): m/e 578 (M+, 100).
HR Масс-спектр (FAB): m/e для C35H51N3O4:
Расчет: 578,3958.
Обнаружено: 578,3962.
Пример 22
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-пропил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 67 мг (0,167 ммоль) поименованного соединения подготовки 1Б, 30 мг (0,167 ммоль) поименованного соединения подготовки 17Б, 34 мг (0,167 ммоль) DCC и 23 мг (0,167 ммоль) HOBT•H2О в 4 мл тетрагидрофурана. Неочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде), получая 75 мг белой пены.
Выход: 80%.
[α]D -43,75 (c=0,160, MeOH).
1H ЯМР (CDCl3): δ 0,87 (t, 3H), 1,18 (s, 9H), 1,21-2,04 (m, 15H), 2,24-2,33 (m, 2H), 2,49- 2,58 (m, 3H), 2,66 (m, 1H), 2,98 (m, 2H), 3,37 (m, 1H), 3,99 (m, 1H), 4,52 (m, 1H), 5,07 (m, 1H), 5,70 (s, 1H), 6,43 (d, J=8,32 Гц, 1H), 6,56 (d, J=7,32 Гц, 1H), 6,76 (d, J=7,12 Гц, 1H), 6,95 (t, J=7,78 Гц, 1 H), 7,20-7,33 (m, 5H).
ИК (KBr): 3287 (br.), 3086, 2932, 2868, 1681, 1558, 1456, 1368, 1334, 1291, 1261, 1218, 1169, 1101, 1042, 776, 734, 552 см-1.
Macc-спектр (FD): m/e 564 (M+, 100).
HR Масс-спектр (FAB): m/e для C34H50N3O4:
Расчет: 564,3801.
Обнаружено: 564,3789.
Пример 23
[3S-(3R*, 4aR*,8aR*,2'S*,3'S*)]- 2-[2'-Гидрокси-3-фенилтиометил-4'-аза-5'-оксо-5'-(2''- метил-3''-оксифенил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 70 мг (0,16 ммоль) поименованного соединения подготовки 8Ж, 24,6 мг (0,16 ммоль) поименованного соединения подготовки 9В, 33 мг (0,16 ммоль) DCC и 22 мг (0,16 ммоль) HOBT•H2O в 4 мл тетрагидрофурана. Hеочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде), получая 54 мг белой пены.
Выход: 60%.
[α]D -119,23o (с=0,26, MeOH).
1H ЯМР (CDCl3): δ 1,09 (s, 9H), 1,12-1,79 (m, 12H), 1,93-2,02 (m, 2H), 2,17-2,30 (m, 2H), 2,31 (s, 3H), 2,43-2,61 (m, 2 H), 2,91 (m, 1H), 3,42 (m, 1H), 3,78 (m, 1H), 4,07 (m, 1H), 4,47 (m, 1H), 5,37 (m, 1H), 5,51 (br.s, 1H), 6,84 (m, 1H), 7,06 (m, 2H), 7,17-7,32 (m, 4H), 7,45 (m, 2H).
ИК (KBr): 3297, 2925, 2862, 1627, 1586, 1530, 1482, 1466, 1439, 1366, 1287, 1221, 1156, 1119, 1026, 801, 735, 689 см-1.
Macc-спектр (FD): m/e 568 (M+, 100).
HR Масс-спектр (FAB) для C32H46N3 O4S:
Расчет: 568,3209.
Обнаружено: 568,3182.
Пример 24
[3S-(3R*, 4aR*,8aR*,2'S*,3'S*)]- 2-(2'-Гидрокси-3'-(нафт-2-илтиометил)-4'-аза-5'-оксо-5'- (2'' -метил-3''-гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 70 мг (0,145 ммоль) поименованного соединения подготовки 7Б, 22 мг (0,145 ммоль) поименованного соединения подготовки 9В, 29 мг (0,145 ммоль) DCC и 19 мг (0,145 ммоль) HOBT•H2О в 4 мл тетрагидрофурана. Неочищенное вещество, получающееся в результате реакции, очищали, используя флэш-хроматографию (элюирующий раствор - 5- 15% градиент ацетона в метиленхлориде), получая 65 мг белого твердого вещества.
Выход: 73%.
[α]D -112,00o (с=0,25, MeOH).
1H ЯМР (CDCl3): δ 1,10 (s, 9H), 1,15-1,80 (m, 12H), 1,93- 2,06 (m, 1H), 2,17-2,28 (m, 2H), 2,29 (s, 3H), 2,42-2,61 (m, 2 H), 2,94 (d, 1H), 3,51 (m, 1H), 3,83-3,92 (m, 1H), 4,10 (m, 1H), 5,36 (br.s, 1H), 5,53 (br.s, 1H), 6,79 (m, 1H), 6,93 (m, 2H), 7,21 (d, J=8,83 Гц, 1H), 7,40-7,53 (m, 3H), 7,73 (m, 3H), 7,90 (s, 1H).
ИК (KBr): 3427, 3311 (br), 2929, 2864, 1703, 1661, 1587, 1514, 1456, 1393, 1366, 1276, 1200, 1177, 1146, 1119, 1070, 1042 см-1.
Macc-спектр (FD): m/e 618 (M+, 100).
Анализ для C34H49N3O4:
Расчет: C 69,98; H 7,67; N 6,80.
Обнаружено: C 69,92; H 7,72; N 6,75.
Пример 25
[2S-(2R*, 2'S*,3'S*)]-1-[2'-Гидрокси-3'- (нафт-2-илтиометил)-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] пиперидин-2-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 28 мг (0,065 ммоль) поименованного соединения подготовки 2Ж, 10 мг (0,065 ммоль) поименованного соединения подготовки 9В, 13,5 мг (0,065 ммоль) DCC и 9 мг (0,065 ммоль) HOBT•H2О в 2 мл тетрагидрофурана. Hеочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 2% метанол в метиленхлориде), получая 23 мг белой пены.
Выход: 63%.
[α]D -233,33o (с=0,09, MeOH).
1H ЯМР (CDCl3): δ 1,17 (s, 9H), 1,26 (m, 1H), 1,56-1,73 (m, 6H), 2,19-2,23 (m, 2H), 2,25 (s, 3H), 2,42 (m, 1H), 2,62-2,73 (m, 2H), 3,11-3,19 (m, 1H), 3,50-3,72 (m, 2H), 4,10 (m, 1H), 4,45 (m, 1H), 5,89 (s, 1H), 6,77-6,87 (m, 3H), 7,00 (d, J=8,65 Гц, 1H), 7,43- 7,51 (m, 3H), 7,72-7,80 (m, 3H), 7,88 (s, 1H).
ИК (KBr): 3329, 2934, 2857, 1646, 1586, 1522, 1457, 1364, 1284, 1223, 1133, 1072, 944, 835, 811, 744, 474 см-1.
Масс-спектр (FD): m/e 564 (M+, 100).
HR Масс-спектр (FAB) для C32H42N3O4S:
Расчет: 564,2896.
Обнаружено: 564,2916.
Пример 26
[2S-(2R*, 2'S*,3'R*)]-1- [2'-Гидрокси-3'-фенилметил-4'-аза- 5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил] -4-(пирид-3'''- илметил)пиперазин-2-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 65 мг (0,148 ммоль) поименованного соединения подготовки 5Д, 22,5 мг (0,148 ммоль) поименованного соединения подготовки 9В, 30,5 мг (0,148 ммоль) DCC и 20 кг (0,148 ммоль) HOBT•H2О в 5 мл тетрагидрофурана. Неочищенное вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3% метанол в метиленхлориде), получая 64 мг белой пены.
Выход: 75%.
1H ЯМР (CDCl3): δ 1,33 (s, 9H), 1,86 (s, 3H), 2,30 (m, 1H), 2,49-2,58 (m, 11H), 3,33 (m, 1H), 3,46 (m, 1H), 4,02 (m, 1H), 4,46 (m, 1H), 6,29 (d, J-9,16 Гц, 1H), 6,46 (d, J=7,23 Гц, 1H), 6,73 (d, J=7,79 Гц, 1H), 6,83 (t, J= 7,84 Гц, 1H), 7,17-7,31 (m, 7H), 7,60 (m, 1H), 7,95 (br.s, 1H), 8,50-8,55 (m, 2H).
Масс-спектр (FD): m/e 574 (M+, 100).
HR Масс-спектр (FAB): m/e для C33H44N5O4:
Расчет: 574,3393.
Обнаружено: 574,3373.
Пример 27
[3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)]- 2-[2'- Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''- этил-3''-гидроксифенил)пентил]дeкагидpoизoxинoлин-3- N-т-бутилкapбoкcaмидa мономезилатная соль
К холодному раствору (0oC) 35,1 мг (0,064 ммоль) названного соединения Примера 3 в 2 мкл безводного метиленхлорида добавляли по каплям 134 мл (0,067 ммоль) 0,5 М раствора метансульфоновой кислоты в метиленхлориде. Образующееся в результате реакции вещество упаривали досуха при пониженном давлении (0,2 - 0,1 Torr), получая 38 мг светло-желтой (неочищенной) пены.
Выход: 90%.
1H ЯМР (CD3OD): δ 0,91 (t, J=7,39, 3H), 1,29 (s, 9H), 1,30-3,20 (m, 21H), 4,00-4,40 (m, 2H), 6,47 (d, J=7,30 Гц, 1H), 6,73 (d, J=7,78 Гц, 1H), 6,91 (t, J=7,78 Гц, 1H), 7,15- 7,32 (m, 5H).
Пример 28
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамида мономезилатная соль
Названное соединение получали в соответствии с методикой, детально описанной в примере 27, используя 125 мг (0,23 ммоль) названного соединения Примера 13 в 5 мл безводного метиленхлорида и 240 мкл (0,24 ммоль) 1,0 М раствора метансульфоновой кислоты в метиленхлориде, получая 136 мг беловатой (неочищенной) пены.
Выход: 95%.
1H ЯМР (CD3OD): δ 1,12 (s, 9H), 1,10-2,20 (m, 16H), 2,60-2,75 (m, 4H), 3,10-3,50 (m, 6H), 3,60-3,70 (m, 1H), 3,90-4,30 (m, 3H), 6,53 (d, J=7,35 Гц, 1H), 6,55 (t, J=7,87 Гц, 1H), 6,89 (t, J=7,82 Гц, 1H).
Пример 29
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'- Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-фенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 15 мг (0,034 ммоль) поименованного соединения подготовки 8Ж, 4,7 мг (0,034 ммоль) о-толуиловой кислоты, 7,13 мг (0,034 ммоль) DCC и 4,7 мг (0,034 ммоль) HOBT•H2О в 2,5 мл тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 10% ацетон в метиленхлориде), получая 16 мг белой пены.
Выход; 84%.
[α]D -80,00o (c=0,15).
1H ЯМР (CDCl3): δ 1,04 (s, 9H), 1,08-1,80 (m, 11H), 1,93 (m, 3H), 2,22 (m, 4H), 2,44 (m, 1H), 2,49 (s, 3H), 2,58 (m, 1H), 2,94 (m, 1H), 3,47 (m, 1H), 3,84 (m, 1H), 4,03 (m, 1H), 4,50 (m, 1H), 5,45 (br.s, 1H), 7,12-7,32 (m, 7H), 7,45 (m, 2H), 7,51 (d, J=7,51 Гц, 1H).
ИК (KBr): 3327, 2928, 2852, 1627, 1574, 1535, 1481, 1364, 1311, 1275, 1225, 1088, 737 см-1.
HR Масс-спектр (FAB) для C32H46N3O3S:
Расчет: 552,3260.
Обнаружено: 552,3272.
Пример 30
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'-Гкдрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-[3''-метилпирид-4''-ил)] пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 15 мг (0,034 ммоль) поименованного соединения подготовки 8Ж, 6,69 мг (0,048 ммоль) названного соединения подготовки 19, 7,13 мг (0,034 ммоль) DCC и 4,7 мг (0,034 ммоль) HOBT•H2О в 1,5 мл тетрагидрофурана и 1 мл диметилформамида. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - градиент 3-5% метанола в метиленхлориде), получая 10 мг белой пены.
Выход: 52%.
[α]D -95,65o (c=0,115).
1H ЯМР (CDCl3): δ 1,00 (s, 9H), 1,20-1,77 (m, 12H), 1,99 (m, 1H), 2,17 (m, 2H), 2,44 (m, 5H), 2,92 (m, 1H), 3,41 (m, 1H), 3,84 (m, 1H), 4,13 (m, 1H), 4,56 (m, 1H), 5,39 (s, 1H), 7,20-7,46 (m, 6H), 7,75 (d, J=8,94 Гц, 1H), 8,46 (m, 2H).
ИК (KBr): 3307, 2925, 2860, 1653, 1542, 1481, 1439, 1391, 1365, 1281, 1224, 1058, 1041, 738, 691, 669 см-1.
HR Масс-спектр (FAB) для C31H45N4O3S:
Расчет: 553,3212.
Обнаружено: 553,3222.
Пример 31
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'- Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(хинолин- 5''-ил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 15 мг (0,034 ммоль) поименованного соединения подготовки 8Ж, 6,0 мг (0,034 ммоль) названного соединения подготовки 20, 7,13 мг (0,034 ммоль) DCC и 4,7 мг (0,034 ммоль) HOBT•H2O в 2 мл тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3-5% градиент метанола в метиленхлориде), получая 15 мг белой пены.
Выход: 74%.
[α]D -99,50o (с=0, 201).
1H ЯМР (CDCl3): δ 0,74 (s, 9H), 1,15-1,79 (m, 12H) 1,97 (m, 1H), 2,17 (m, 2H), 2,36 (m, 1H), 2,54 (m, 1H), 2,90 (m, 1H), 3,45 (m, 1H), 3,99 (m, 1H), 4,16 (m, 1H), 4,62 (m, 1H), 5,29 (s, 1H), 7,18-7,32 (m, 3H), 7,40-7,50 (m, 3H), 7,70 (m, 1 H), 7,89 (m, 2H), 8,17 (m, 1H), 8,91 (m, 2H).
ИК (KBr): 3299, 2923, 2862, 1644, 1546, 1481, 1439, 1390, 1327, 1279, 1222, 1207, 1037, 810, 735, 689 см-1.
HR Масс-спектр (FAB) для C34H45N4 O3S:
Расчет: 589,3212.
Обнаружено: 589,3237.
Пример 32
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)]- 2-[2'-Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'- (1'', 2'', 3'',4''-тетрагидрохинолин-5''-ил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 18 мг (0,04 ммоль) поименованного соединения подготовки 8Ж, 7,38 мг (0,04 ммоль) названного соединения подготовки 21, 8,56 мг (0,04 ммоль) DCC и 5,61 мг (0,04 ммоль) HOBT•H2О в 2 мл тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3-5% градиент метанола в метиленхлориде), получая 12 мг белой пены.
Выход: 50%.
[α]D -98,59o (c=0,142).
1H ЯMP (CDCl3): δ 1,13 (s, 9H), 1,14-2,04 (m, 15H), 2,19 (m, 2H), 2,45 (m, 1H), 2,57 (m, 1H), 2,75 (m, 1H), 2,90-3,09 (m, 2H), 3,26 (m, 2H), 3,44 (m, 2H), 3,75 (m, 1H), 4,01-4,14 (m, 2H), 4,42 (m, 1H), 5,56 (s, 1H), 6,49 (d, J= 7,96 Гц, 1H), 6,80 (d, J=7,40 Гц, 1H), 6,93 (t, J=7,72 Гц, 1H), 7,08 (d, J=8,39 Гц, 1H), 7,18 (m, 1H), 7,27 (m, 2H), 7,42 (d, 2H).
ИК (KBr): 3327, 2928, 2852, 1629, 1590, 1519, 1481, 1449, 1364, 1310, 1275, 1229, 1087, 738, 690 см-1.
Масс-спектр (FAB) для C34H49N4O3S:
Расчет: 593,3525.
Обнаружено: 593,3552.
Пример 33
[2S-(2R*, 2'S*, 3'S*)]-1-[2'-Гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-(1'', 2'', 3'',4''- тетрагидрохинолин-5''-ил)пентил]-4-(пирид-3'''- илметил)пиперазин-2-N-т-бутилкарбоксамид
К охлажденному раствору (-10oC), содержащему 45 мг (0,10 ммоль) поименованного соединения подготовки 6Б, 18 мг (0,10 ммоль) 1,2,3,4-тетрагидрохинолин-5-карбоновой кислоты, 30 мг (0,30 ммоль) триэтиламина и 14 мг (0,10 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана, добавляли 22 мг (0,11 ммоль) DCC. Получившуюся реакционную смесь перемешивали в течение приблизительно 24 часов при комнатной температуре и затем упаривали при пониженном давлении до получения остатка. Этот остаток повторно растворяли в этилацетате и фильтровали через целит. Фильтрат затем экстрагировали последовательно насыщенным бикарбонатом натрия (дважды) и солевым раствором, высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Неочищенный материал очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 2,5-5% градиент метанола в метиленхлориде), получая 33 мг беловатой пены.
Выход: 62%.
1H ЯМР (CDCl3): δ 1,29 (s, 9H), 1,79-1,97 (m, 2H), 2,26-3,00 (m, 11H), 3,20-3,50 (m, 9H), 3,95-4,05 (m, 1H), 4,23 - 4,35 (m, 1H), 6,43-6,62 (m, 2H), 6,89 (t, J=7,8 Гц, 1H), 7,12 - 7,35 (m, 6H), 7,41 (d, J=7,7 Гц, 2H), 7,57-7,70 (m, 2H), 8,50 - 8,58 (m, 2H).
Масс-спектр (FD): m/e 631 (M+, 100).
Пример 34
[2S-(2R*, 2'S*, 3'S*)]-1-[2'-Гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-(хинолин-5''-ил)пентил] -4- (пирид-3'''-ил-метил)пиперазин-2-N-т-бутилкарбоксамид
Названное соединение получали из примера 33.
Выход: 13 мг беловатой пены.
1H ЯМР (CDCl3): δ 1,18 (s, 9H), 2,27-2,90 (m, 9H), 3,17-3,60 (m, 5H), 4,07-4,19 (m, 1H), 4,40-4,55 (m, 1H), 4,75-4,95 (m, 1H), 6,90-7,68 (m, 11H), 8,16 (d, J=8.1 Гц, 1H), 8,48 -8,60 (m, 2H), 8,80 (d, J=8,4 Гц, 1H), 8,89-8,97 (m, 1H).
Масс-спектр (FD): m/e 527 (M+, 100).
Пример 35
[2S-(2R*, 2'S*, 3'S*)]-1-[2'-Гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-[3''-метилпирид-4''- ил)] пентил]-4-(пирид-3'''-илметил)пиперазин-2-N-т- бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 20,3 мг (0,148 ммоль) названного соединения подготовки 19, 70 мг (0,148 ммоль) поименованного соединения подготовки 19, 31 мг (0,148 ммоль) DCC и 20 мг (0,148 ммоль) HOBT•H2О в тетрагидрофуране, содержащем 62 мл триэтиламина. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2,5-15% градиент метанола в метиленхлориде), получая 48 мг белой пены.
Выход: 55%.
1H ЯМР (CDCl3): δ 1,23 (s, 9H), 2,30-2,90 (m, 12H), 3,16-3,50 (m, 5H), 4,02-4,10 (m, 1H), 4,30-4,42,41 (m, 1H), 4,85 (br.s, 1H), 6,90-7,60 (m, 10H), 8,38-8,57 (m, 3H).
Масс-спектр (FAB): m/e 591,4 (M+, 100).
Пример 36
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'- Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-[2''-метил-3'' -N-(метилсульфонил)аминофенил)] пентил]декагидроизохинолин-3- N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 70 мг (0,17 ммоль) поименованного соединения подготовки 1Б, 40 мг (0,17 ммоль) названного соединения подготовки 22, 35 мг (0,17 ммоль) DCC и 123 мг (0,17 ммоль) HOBT•H2О в 2 мл безводного тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 1-5% градиент метанола в метиленхлориде), получая 72 мг беловатого твердого вещества.
Выход: 69%.
1H ЯМР (CDCl3): δ 1,14 (s, 9H), 1,19-2,38 (m, 19H), 2,50- 2,70 (m, 2H), 2,92-3,06 (m, 4H), 3,43-3,55 (m, 1H), 4,01-4,10 (m, 1H), 4,58-4,70 (m, 1H), 5,66 (s, 1H), 6,37 (br.s, 1H), 6,82-6,93 (m, 2H), 7,10-7,39 (m, 6H), 7,48 (d, J=8,16 Гц, 1H).
ИК (KBr): 3691, 3600-3300 (br.), 2929, 2866, 1672, 1603, 1513, 1455, 1393, 1368, 1327, 1277, 1154, 1047, 972, 909, 877 см-1.
Масс-спектр (FD): m/e (M+, 100).
Пример 37
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)]- 2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-[(1'',2'',3'',4''- тетрагидрохинолин-5''-ил)]пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 18,5 мг (0,046 ммоль) поименованного соединения подготовки 1Б, 8,14 мг (0,046 ммоль) названного соединения подготовки 20, 9,48 мг (0,046 ммоль) DCC и 6,21 мг (0,046 ммоль) HОВТ•H2О в 2 мл безводного тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 2-5% градиент метанола в метиленхлориде), получая 11 мг пены.
Выход: 43%.
1H ЯМР (CDCl3): δ 1,20 (s, 9H), 1,25-2,02 (m, 15H), 2,28 (m, 2H), 2,46-2,70 (m, 4H), 2,99 (m, 2H), 3,21 (m, 1H), 3,35 (m, 1H), 3,98 (m, 1H), 4,49 (m, 1H), 5,75 (br.s, 1H), 6,38 (m, 3H), 6,83 (t, 1H), 7,21-7,33 (m, 5H).
Пример 38
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'- Гидрокси-3'-фенил-тиометил-4'-аза-5'-оксо-5'-[6'' -метил-(1'', 2'',3'',4''-тетрагидрохинолин-5''-ил)]пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 15 мг (0,035 ммоль) поименованного соединения подготовки 8Ж, 6,5 мг (0,035 ммоль) 6- метил-1,2,3,4-тетрагидро-5-хинолинкарбоновой кислоты, 7,15 мг (0,035 ммоль) DCC и 4,7 мг (0,035 ммоль) HOBT•H2О в 2 мл тетрагидрофурана и 1 мл диметилформамида. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 3-5% градиент метанола в метиленхлориде), получая 12,5 мг белого твердого вещества.
Выход: 60%.
HR Масс-спектр (FAB) для C35H47N4O3S:
Расчет: 603,3369.
Обнаружено: 603,3384.
Пример 39
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)]- 2-[2'-Гидрокси-3'-фенил-тиометил-4'-аза-5'-оксо-5'-[2'',6'' -диметил-3''-гидроксифенил]пентил)декагидроизохинолин-3- N-т-бутилкарбоксамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 1, используя 20 мг (0,046 ммоль) поименованного соединения подготовки 8Ж, 11,53 мг (0,0694 ммоль) 2,6-диметил-3-гидроксибензойной кислоты, 9,54 мг (0,046 ммоль) DCC и 6,25 мг (0,046 ммоль) HOBT•H2О в 3 мл тетрагидрофурана. Вещество, получающееся в результате реакции, очищали, используя радиальную хроматографию (1 мм пластина; элюирующий раствор - 4% метанол в метиленхлориде), получая 14 мг белого твердого вещества.
Выход: 52%.
HR Масс-спектр (FAB) для C33H48N3O4S:
Расчет: 582,3375.
Обнаружено: 582,3373.
Пример 40
[2R'-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''-гидроксифенил)пентил] бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 40, используя 100 мг (0,29 ммоль) поименованного соединения подготовки 24Г, 44 мг (0,29 ммоль) поименованного соединения подготовки 23В, 60 мг (0,29 ммоль) DCC и 39 мг (0,29 ммоль) 1-гидроксибензотриазола моногидрата (HOBT•H2О) в 4 мл безводного тетрагидрофурана. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 2-5% градиент метанола в метиленхлориде), получая 58 мг белого порошка.
Выход: 42%.
[α]D 2,34 (с=3,4, MeOH).
1H ЯМР (CD3OD): δ 1,47 (s, 9H), 1,88 (s, 3H), 2,70-2,80 (m, 1H), 2,95-3,10 (m, 3H), 3,25-3,30 (m, 1H), 3,85-3,95 (m, 1H), 4,35-4,45 (m, 1H), 4,84 (s, 1H), 6,55-6,58 (m, 1H), 6,74 (d, J=8,0 Гц, 1H), 6,94 (t, J=7,8 Гц, 1H), 7,15-7,45 (m, 11H).
ИК (CHCl3): 3580, 3550-3100 (br.), 2929, 2865, 1662, 1596, 1521, 1472, 1455, 1394, 1368, 1293, 1157, 1047, 879, 839 см-1.
Macc-спектр (FD): 475 (M+, 100).
HR Масс-спектр (FAB): m/e для C29H35N2O4:
Расчет: 475,2597.
Обнаружено: 475,2610.
Пример 41
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидpoкcи-3'- фeнилмeтил-4'-аза-5'-оксо-5'-(2''-метил-5''- гидроксиметилфенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 40, используя 95 мг (0,28 ммоль) поименованного соединения подготовки 24Г, 65 мг (0,28 ммоль) поименованного соединения подготовки 27Б, 58 мг (0,28 ммоль) DCC и 38 мг (0,28 ммоль) HOBT•H2O в 2 мл тетрагидрофурана, содержащего 0,2 мл диметилформамида. Неочищенный продукт очищали, используя радиальную хроматографию (2 мм пластина; элюирующий раствор - 4% метанол в метиленхлориде), получая 64,6 мг желаемого названного соединения.
Выход: 47%.
[α]D -0,003o (с=1,02, MeOH).
1H ЯМР (CDCl3): δ 1,44 (s, 9H), 1,98 (s, 3H), 2,70-2,85 (m, 1H), 3,00-3,12 (m, 2H), 3,25-3,35 (m, 1H), 3,85- 3,97 (m, 1H), 4,00-4,10 (m, 2H), 4,35-4,46 (m, 1H), 4,50 (s, 2H), 6,98- 7,43 (m, 11H), 8,06-8,18 (m, 1H).
Масс-спектр (FD): m/e (M++1, 490).
Анализ для C30H36N2O4:
Расчет: C 73,74; H 7,43; N 5,52.
Обнаружено: C 74,00; H 7,49; N 5,68.
Пример 42
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидpoкcи-3'- нaфт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-3'' -аминофенил)пентил]бензамид
К холодному раствору (0oC) 50 мг (0,12 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида добавляли 22 мг (0,14 ммоль) 2-метил-3-аминобензойной кислоты, 16 мг (0,12 ммоль) HOBT, 22 мг (0,12 ммоль) EDC И 0,081 мл (0,58 ммоль) триэтиламина. Полученную реакционную смесь перемешивали при 0oC в течение приблизительно одного часа и затем в течение шестнадцати часов при комнатной температуре. Реакцию останавливали добавлением воды и реакционную смесь экстрагировали этилацетатом. Образовавшиеся слои разделяли и органический слой высушивали, фильтровали и упаривали при пониженном давлении до получения неочищенного остатка. Этот остаток очищали, используя флэш-хроматографию (элюирующий раствор - 3% метанол в метиленхлориде), получая 52 мг белого твердого вещества (т.п. 105- 106oC).
Выход: 80%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,75 (m, 3H), 7,40 (m, 7H), 6,86 (t, J= 9,0 Гц, 1H), 6,12 (s, 1H), 5,93 (s, 1H), 4,51 (m, 1H), 4,02 (m, 1H), 3,68 (br.s, 2H), 3,51 (m, 3H), 3,12 (s, 2H), 3,04 (dd, J=13,4, 10,1 Гц, 1H), 2,92 (dd, J=13,4, 3,3 Гц, 1H), 2,23 (s, 3H), 1,50 (s, 9H).
ИК (KBr): 3304, 3068, 1633, 1516, 1321, 1221, 1076, 746 см-1.
Анализ для C33H37N3O3S:
Расчет: C 71,32; H 6,71; N 7,56.
Обнаружено: C 71,54; H 6,83; N 7,32.
Пример 43
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- N(метил)аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 42 мг (0,26 ммоль) названного соединения подготовки 28, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш- хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 102 мг белого твердого вещества (т.п. 111 -113oC).
Выход: 76%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,75 (m, 2H), 7,52-7,21 (m, 9H), 7,00 (t, J=7,9 Гц, 1H), 6,62 (t, J=7,4 Гц, 1H), 6,41 (d, J=9,1 Гц, 1H), 6,09 (d, J= 5,8 Гц, 1H), 5,91 (s, 1H), 4,48 (m, 1H), 4,01 (m, 1H), 3,69 (s, 1H), 3,50 (m, 2H), 3,01 (m, 2H), 2,85 (s, 3H), 2,15 (s, 3H), 1,45 (s, 9H).
Анализ для C34H49N3O3S:
Расчет: C 71,67; H 6,89; N 7,37.
Обнаружено: C 71,92; H 6,74; N 7,42.
Пример 44
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси- 3'-нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-хлор-3'' -аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 48 мг (0,28 ммоль) 2-хлор-3-аминобензойной кислоты, 32 мг (0,23 ммоль) HОВТ, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 97 мг белого твердого вещества (т.п. 107- 108oC).
Выход: 72%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,78 (m, 2H), 7,61-7,23 (m, 9H), 6,95 (t, J=7,8 Гц, 1H), 6,78 (m, 1H), 6,52 (d, J=7,9 Гц, 1H), 6,05 (d, J=6,0 Гц, 1H), 5,92 (s, 1H), 4,51 (m, 1H), 4,21 (s, 2H), 4,16 (m, 1H), 3,51 (m, 2H), 3,01 (m, 3H), 1,49 (s, 9H).
Анализ для C32H34ClN3O3S:
Расчет: C 66,71; H 5,95; N 7,29.
Обнаружено: C 66,85; H 6,06; N 7,42.
Пример 45
[2'R-(2'R*, 3'S*)]-N-Бутил-2-[2'-гидрокси- 3'-нафт-2-илтиометил-4'-аза-5'-оксо-5'-(2''-бром-3'' -аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 61 мг (0,28 ммоль) 2-бром-3-аминобензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 102 мг белого твердого вещества (т.п. 110 - 112oC).
Выход: 71%.
1H ЯМР (CDCl3): δ 7,88 (s, 1H), 7,78 (m, 2H), 7,60- 7,25 (m, 9H), 6,95 (t, J= 7,8 Гц, 1H), 6,78 (m, 1H), 6,52 (d, J=7,9 Гц, 1H), 6,1 (d, J=6,1 Гц, 1H), 5,90 (s, 1H), 4,52 (m, 1H), 4,21 (s, 2H), 4,15 (m, 1H), 3,50 (m, 2H), 3,00 (m, 3H), 1,49 (s, 9H).
Анализ для C32H34BrN3O3S:
Расчет: C 61,93; H 5,52; N 6,77.
Обнаружено: C 61,82; H 5,83; N 6,63.
Пример 46
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-3'' -гидроксифенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 75 мг (0,18 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 32 мг (0,21 ммоль) поименованного соединения подготовки 23В, 24 мг (0,18 ммоль) HOBT, 34 мг (0,18 ммоль) EDC и 0,12 мл (0,88 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 52 мг белого твердого вещества (т.п. 119- 120oC).
Выход: 53%.
ИК (KBr): 3297, 1636, 1518, 1284, 1221, 1073, 746 см-1.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,76 (m, 3H), 7,48 (m, 6H), 6,79 (m, 4H), 6,52 (d, J=9,2 Гц, 1H), 6,23 (s, 1H), 5,92 (s, 1H), 4,50 (m, 1H), 4,02 (m, 1H), 3,49 (m, 3H), 3,03 (dd, J =13,4, 10,2 Гц, 1H), 2,97 (dd, J=13,4, 3,4 Гц, 1H), 2,25 (s, 3H), 1,49 (s, 9H).
Анализ для C33H36N2O4S:
Расчет: C 71,19; H 6,52; N 5,03.
Обнаружено: C 70,95; H 6,59; N 4,87.
Пример 47
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил- 5''-аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 44 мг (0,28 ммоль) названного соединения подготовки 29, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 101 мг белого твердого вещества (т.п. 106 - 107oC).
Выход: 79%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,76 (m, 3H), 7,40-7,25 (m, 7H), 6,85 (t, J=9,0 Гц, 1H), 6,62 (d, J=7,7 Гц, 1H), 6,43 (d, J=9,0 Гц, 1H), 6,08 (d, J= 5,8 Гц, 1H), 5,89 (s, 1H), 4,51 (m, 1H), 4,02 (m, 1H), 3,70 (br.s, 2H), 3,50 (m, 3H), 3,04 (dd, J=13,3, 10,1 Гц, 1H), 2,92 (dd, J=13,3, 3,2 Гц, 1H), 2,21 (s, 3H), 1,50 (s, 9H).
Анализ для C33H37N3O3S:
Расчет: C 71,32; H 6,71; N 7,56.
Обнаружено: C 71,64; H 6,93; N 7,45.
Пример 48
[2'R-(2'R*,3'S*)]-N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил- тиометил-4'-аза-5'-оксо-5'-(2''-метил-3''- гидроксифенил)пентил]-1-нафтиламид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,21 ммоль) поименованного соединения подготовки 26Г в 2,0 мл диметилформамида, 35 мг (0,23 ммоль) поименованного соединения подготовки 23В, 29 мг (0,21 ммоль) HOBT, 40 мг (0,21 ммоль) EDC и 0,15 мл (1,10 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1,5% метанол в метиленхлориде), получая 106 мг белого твердого вещества (т.п. 115-117oC).
Выход: 82%.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,76 (m, 2H), 7,53-7,24 (m, 11H), 6,85 (t, J=7,6 Гц, 1H), 6,73 (m, 1H), 6,63 (d, J=5,7 Гц, 1H), 6,51 (d, J=9,2 Гц, 1H), 6,10 (s, 1H), 5,90 (s, 1H), 4,50 (m, 1H), 4,09 (m, 1H), 2,48 (m, 2H), 3,10 (dd, J=12,9, 9,7 Гц, 1H), 2,88 (dd, J=12,9, 3,2 Гц, 1H), 2,13 (s, 3H), 1,46 (s, 9H).
Анализ для C37H38N2O4S:
Расчет: C 73,24; H 6,31; N 4,62.
Обнаружено: C 73,46; H 6,70; N 4,35.
Пример 49
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-хлор-3''- аминофенил)пентил]-1-нафтиламид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,21 ммоль) поименованного соединения подготовки 26Г в 2,0 мл диметилформамида, 39 мг (0,23 ммоль) 2-хлор-3-аминобензойной кислоты, 29 мг (0,21 ммоль) HOBT, 40 мг (0,21 ммоль) EDC и 0,15 мл (1,10 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1,5% метанол в метиленхлориде), получая 97 мг белого твердого вещества (т.п. 110-112oC).
Выход: 74%.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,81 (m, 4H), 7,75-7,21 (m, 9H), 6,95 (t, J=7,8 ГЦ, 1H), 6,75 (m, 1H), 6,51 (d, J=8,2 Гц, 1H), 6,12 (d, J=5,9 Гц, 1H), 5,95 (s, 1H), 4,50 (m, 1H), 4,21 (s, 2H), 4,15 (m, 1H), 3,51 (m, 2H), 3,00 (m, 3H), 1,49 (s, 9H).
Анализ для C36H36ClN3O3S:
Расчет: C 69,05; H 5,79; N 6,71.
Обнаружено: C 69,21; H 5,85; N 6,54.
Пример 50
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(3''- аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 38 мг (0,28 ммоль) 3-аминобензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 90 мг белого твердого вещества (т.п. 101-102oC).
Выход: 72%.
1H ЯМР (CDCl3): δ 7,87 (s, 1H), 7,78 (m, 2H) 7,61-7,22 (m, 10H), 6,96 (c, J=7,7 Гц, 1H), 6,76 (m, 1H), 6,52 (d, J=7,8 Гц, 1H), 6,04 (d, J=6,1 Гц, 1H), 5,91 (s, 1H), 4,5 (m, 1H), 4,20 (s, 2H), 4,15 (m, 1H), 3,50 (m, 2H), 3,01 (m, 3H), 1,49 (s, 9H).
Анализ для C32H35N3O3S:
Расчет: C 70,95; H 6,51; N 7,76.
Обнаружено: C 71,21; H 6,72; N 7,72.
Пример 51
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(3''- гидроксифенил]пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 50 мг (0,12 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 20 мг (0,14 ммоль) 3-гидроксибензойной кислоты, 16 мг (0,12 ммоль) HOBT, 22 мг (0,12 ммоль) EDC и 0,081 мл (0,58 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 50% этилацетат в гексане), получая 36 мг белого твердого вещества (т.п. 125-128oC).
Выход: 57%.
1H ЯМР (CDCl3): δ 7,87 (s, 1H), 7,73 (m, 3H), 7,20-7,50 (m, 7H), 6,95-7,15 (m, 4H), 6,80 (m, 1H), 6,80 (m, 1H), 6,50 (s, 1H), 6,30 (m, 1H), 5,95 (s, 1H), 4,53 (m, 1H), 4,10 (m, 1H), 3,45 (m, 2H), 3,03 (dd, J=13,4, 10,5 Гц, 1H), 2,90 (dd, J=13,4, 3,5 Гц, 1H), 1,46 (s, 9H).
HR Масс-спектр для C32H34N2O4S:
Расчет: m/e 675,1294.
Обнаружено: m/e 675,1311.
Пример 52
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидpoкcи-3'- нaфт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''- метилфенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 50 мг (0,12 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 19 мг (0,14 ммоль) 2-метилбензойной кислоты, 16 мг (0,12 ммоль) HOBT, 22 мг (0,12 ммоль) EDC и 0,081 мл (0,58 ммоль) триэтиламина. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 40% этилацетат в гексане), получая 33 мг белого твердого вещества (т.п. 85-87oC).
Выход: 52%.
1H ЯМР (CDCl3): δ 7,89 (d, J=1,0 Гц, 1H), 7,76 (m, 3H), 7,15-7,52 (m, 11H), 7,02 (t, J= 7,4 Гц, 1H), 6,48 (d, J=9,0 Гц, 1H), 6,08 (d, J=6,1 Гц, 1H), 5,89 (s, 1H), 4,53 (m, 1H), 4,02 (m, 1H), 3,48 (d, J=6,8 Гц, 2H), 3,00 (dd, J=13,4, 10,2 Гц, 1H), 2,92 (dd, J=13,4, 3,6 Гц, 1H), 2,46 (s, 3H), 1,45 (s, 9H).
HR Масс-спектр для C33H36N2O3S:
Расчет: m/e 673,1501.
Обнаружено: m/e 673,1504.
Пример 53
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'- гидрокси-3'-нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2'' -метил-3'',5''-диаминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 50 мг (0,12 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида, 23 мг (0,14 ммоль) 2-метил-3,5-диаминобензойной кислоты, 16 мг (0,12 ммоль) HOBT, 22 мг (0,12 ммоль) EDC и 0,081 мл (0,58 ммоль) триэтиламина. Неочищенное масло очищали флэш-хроматографией (элюирующий раствор - 5% метанол в метиленхлориде), получая 28 мг беловатого порошка (т.п. 125-128oC).
Выход: 42%.
1H ЯMP (CDCl3): δ 7,90 (d, J=1,2 Гц, 1H), 7,77 (m, 3H), 7,20-7,53 (m, 10H), 6,35 (d, J= 9,3 Гц, 1H), 6,15 (br.m, 1H), 6,01 (d, J=2,1 Гц, 1H), 5,92 (s, 1H), 5,83 (d, J=2,1 Гц, 1H), 4,50 (m, 1H), 3,96 (m, 1H), 3,50 (m, 4H), 3,03 (dd, J=13,4, 10,2 Гц, 1H), 2,91 (dd, J=13,4, 3,5 Гц, 1H), 2,10 (s, 3H), 1,47 (s, 9H).
HR Масс-спектр для C33H38N4O3S:
Расчет: m/e 703,1719.
Обнаружено: m/e 703,1733.
Пример 54
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2'',2''- дихлорфенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 75 мг (0,18 ммоль) поименованного соединения подготовки 25Д в 1,0 мл диметилформамида, 40 мг (0,21 ммоль) 2,3-дихлорбензойной кислоты, 24 мг (0,18 ммоль) HOBT, 34 мг (0,18 ммоль) EDC и 0,12 мл (0,88 ммоль) триэтиламина. Неочищенное масло очищали, используя флэш-хроматографию (элюирующий раствор - 25-50% градиент этилацетата в гексане), получая 75 мг белого твердого вещества (т.п. 116-119oC).
Выход: 74%.
1H ЯМР (CDCl3): δ 7,90 (s, 1H), 7,75 (m, 3H), 7,20-7,52 (m, 9H), 7,13 (dd, J= 7,9, 1,2 Гц, 1H), 7,00 (t, J=7,8 Гц, 1H), 6,64 (d, J=9,9 Гц, 1H), 5,88 (br. s, 1H), 4,52 (m, 1H), 4,03 (m, 1H), 3,50 (d, J=6,0 Гц, 2H), 3,00 (m, 2H), 1,44 (s, 9H).
Анализ для C32H32Cl2N2O3S:
Расчет: C 64,53; H 5,42; N 4,70.
Обнаружено: С 64,54; H 5,50; N 4,73.
Пример 55
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-хлор-5'' -аминофенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 75 мг (0,18 ммоль) поименованного соединения подготовки 25Д в 1,0 мл диметилформамида, 36 мг (0,21 ммоль) названного соединения подготовки 29, 24 мг (0,18 ммоль) HOBT, 34 мг (0,18 ммоль) EDC и 0,12 мл (0,88 ммоль) триэтиламина. Неочищенное масло очищали, используя флэш-хроматографию (элюирующий раствор - 50% этилацетат в гексане), получая 90 мг белого твердого вещества (т.п. 109-110oC).
Выход: 90%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,75 (m, 3H), 7,21-7,52 (m, 10H), 7,04 (d, J= 8,3 Гц, 1H), 6,73 (m, 1H), 6,55 (m, 2H), 5,92 (br.s, 1H), 4,50 (m, 1H), 3,99 (m, 1H), 3,52 (d, J=5,6 Гц, 2H), 3,02 (m, 2H), 1,45 (s, 9H).
Анализ для C32H34ClN3O3S:
Расчет: C 66,71; H 5,95; N 7,29.
Обнаружено: C 66,94; H 6,34; N 6,92.
Пример 56
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидpoкcи-3'- нaфт-2-ил-тиометил-4'-аза-5'-оксо-5'- (2''-хлор-3'' -гидроксифенил)пектил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 42, используя 75 кг (0,18 ммоль) поименованного соединения подготовки 25Д в 1,0 мл диметилформамида, 36 мг (0,21 ммоль) названного соединения подготовки 14, 24 мг (0,18 ммоль) HOBT, 34 мг (0,18 ммоль) EDC и 0,12 мл (0,88 ммоль) триэтиламина. Неочищенное масло очищали, используя флэш-хроматографию (элюирующий раствор - 25-50% градиент этилацетата в гексане), получая 71 мг белого твердого вещества (т.п. 104-105oC).
Выход: 71%.
1H ЯМР (CDCl3): δ 7,90 (d, J=1,0 Гц, 1H), 7,7 (m, 3H), 7,19-7,52 (m, 8H), 7,00 (m, 2H), 6,87 (m, 1H), 6,64 (d, J=9,1 Гц, 1H), 5,89 (s, 1H), 4,52 (m, 1H), 4,04 (m, 1H), 3,50 (d, J= 6,1 Гц, 1H), 3,05 (dd, J=13,4, 10,2 Гц, 2H), 2,94 (dd, J=13,4, 3,6 Гц, 1H), 1,45 (s, 9H).
Анализ для C32H33ClN2O4S:
Расчет: C 66,59; H 5,76; N 4,85.
Обнаружено: C 66,64; H 5,90; N 4,93.
Пример 57
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(изохинолин-5''- ил)пентил]бензамид
К раствору 0,40 г (0,95 ммоль) поименованного соединения подготовки 25Д и 134 мкл (1,22 ммоль) N-метилморфолина в 15 мл тетрагидрофурана добавляли 0,45 г (1,33 ммоль) поименованного соединения подготовки 30В. Реакцию проводили в течение приблизительно 8 часов, после чего реакционную смесь разбавляли этилацетатом. Образовавшиеся слои разделяли и органический слой промывали последовательно водой и солевым раствором и затем концентрировали, получая неочищенный материал. Этот неочищенный материал очищали, используя флэш-хроматографию (диоксид кремния; элюирующий раствор - 4% метанол в метиленхлориде), получая 0,53 г белого твердого вещества (т.п. 109-112oC).
Выход: 97%.
1H ЯМР (CDCl3): δ 9,19 (s, 1H), 8,50 (d, J=4,6 Гц, 1H), 8,23 (d, J=5,9 Гц, 1H), 7,92 (m, 2H), 7,76 (m, 3H), 7,56 (m, 3 H), 7,43 (m, 3H), 7,32 (m, 2H), 7,24 (m, 1H), 6,88 (d, J=9,0 Гц, 1H), 6,05 (br.s, 1H), 5,93 (s, 1H), 4,64 (m, 1H), 4,12 (m, 1H), 3,51 (d, J=6,3 Гц, 2H), 3,01 (m, 2H), 1,40 (s, 9H).
ИК (неразбавленный тонкий слой): 3428, 3019, 2978, 1647, 1514, 1215, 758 см-1.
HR Масс-спектр для C35H36N3O3S (MH+):
Расчет: 578,2477.
Обнаружено: 578,2468.
Анализ для C35H35N3O3S•0,17CH2Cl2:
Расчет: C 71,33; H 6,02; N 7,10; S 5,41.
Обнаружено: C 71,35; H 6,00; N 7,09; S 5,44.
Пример 58
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3' -нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(1'',2'',3'',4'' -тетрагидроизохинолин-5''-ил)пентил]бензамид
К раствору 0,15 г (0,26 ммоль) названного соединения Примера 57 в 6 мл уксусной кислоты добавляли 0,08 г (1,27 ммоль) цианборгидрида натрия. Реакцию проводили в течение приблизительно 1 часа, после чего ее останавливали добавлением насыщенного раствора бикарбоната натрия. Желаемое соединение затем экстрагировали, используя этилацетат, и органические экстракты промывали последовательно водой и солевым раствором и затем упаривали при пониженном давлении до получения пены. Эту пену очищали, используя флэш-хроматографию (диоксид кремния; элюирующий раствор - 4% метанол в метиленхлориде), получая 0,10 г белого аморфного твердого вещества (т.п. 197-199oC).
Выход: 66%.
1H ЯМР (CDCl3): δ 7,85 (s, 1H), 7,75 (m, 3H), 7,50-7,20 (m, 7H), 7,06 (m, 1H), 6,95 (m, 2H), 6,59 (d, J=9,1 Гц, 1H); 6,02 (s, 1H), 4,48 (br.s, 1H), 4,00 (br.s, 1H), 3,98 (s, 2H), 3,45 (m, 2H), 3,01 (s, 1H), 2,98 (d, J= 6,0 Гц, 3H), 2,89 (m, 3H), 1,44 (s, 9H), OH не наблюдали.
ИК (неразбавленный тонкий слой): 3418, 3281, 3019, 1632, 1516, 1215, 756.
HR Масс-спектр для C35H40N3O3S:
Расчет: 582,2790.
Обнаружено: 582,2792.
Анализ для C35H35N3O3S•0,17CH2Cl2:
Расчет: C 70,85; H 6,65; N 7,05; S 5,38.
Обнаружено: C 70,85; H 6,74; N 7,16; S 5,42.
Пример 59
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'- гидрокси-3'-нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-[2-N (метил)-1'', 2'', 3'', 4''-тетрагидроизохинолин-5''- ил)пентил]бензамид
К горячему раствору (60oC) 0,11 г (0,19 ммоль) названного соединения примера 57 в 3 мл тетрагидрофурана добавляли 53 мг (1,40 ммоль) боргидрида натрия и 75 мкл муравьиной кислоты. После приблизительно 1 часа реакцию останавливали добавлением насыщенного раствора бикарбоната натрия. Желаемое соединение экстрагировали, используя этилацетат, и органические экстракты промывали последовательно водой и солевым раствором и затем концентрировали, получая пену. Эту пену очищали, используя флэш-хроматографию (диоксид кремния; элюирующий раствор - 5% метанол в метиленхлориде), получая 0,05 г белого аморфного твердого вещества (т.п. 110-113oC).
Выход: 44%.
1H ЯМР (CDCl3): δ 7,86 (s, 1H), 7,75 (m, 3H), 7,50-7,20 (m, 7H), 7,00 (m, 3H), 6,46 (d, J=9,0 Гц, 1H), 6,13 (d, J=5,0 Гц, 1H), 5,96 (s, 1H), 4,45 (m, 1H), 3,97 (m, 1H), 3,54 (S, 2H), 3,46 (m, 2H), 3,20-2,90 (m, 4H), 2,60 (t, J=5,9 Гц, 2H), 2,40 (s, 3H), 1,44 (s, 9H).
ИК (неразбавленный тонкий слой): 3432, 3019, 2976, 1645, 1516, 1215, 756 см-1.
HR Масс-спектр для C36H42N3O3S (MH+):
Расчет: 596,2947.
Обнаружено: 596,2939.
Анализ для C36H41N3O3S•0,32CH2Cl2:
Расчет: C 70,02; H 6,74; N 6,75; S 5,15.
Обнаружено: C 70,03; H 6,74; N 6,81; S 5,24.
Пример 60
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси- 3'-фенилметил-4'-аза-5'-оксо-5'-(1'',2'',3'',4'' -тетрагидроизохинолин-5''-ил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 58.
1H ЯМР (CDCl3): δ 7,42 (m, 10H), 7,00 (m, 3H), 6,28 (d, J =9,4 Гц, 1H), 5,95 (s, 1H), 4,60 (m, 1H), 3,95 (bs, 3H), 2,80-3,20 (m, 7H), 2,62 (m, 1H), 1,47 (s, 9H).
Анализ для C31H37N3O3• MeOH:
Расчет: C 72,29; H 7,77; N 7,90.
Обнаружено: C 72,61; H 7,58; N 7,61.
Пример 61
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(нафт-1''-ил)пентил]бензамид
К холодному раствору (0oC) 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д в 2,0 мл диметилформамида добавляли 45 мг (0,26 ммоль) нафталин-1-карбоновой кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина. Реакцию проводили в течение приблизительно 1 часа при 0oC и 16 часов при комнатной температуре, после чего реакционную смесь разбавляли 10 мл этилацетата. Полученную смесь промывали водой, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении до получения остатка. Этот остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 82 мг белого твердого вещества (т.п. 92-95oC).
Выход, 63%.
1H ЯМР (CDCl3): δ 8,35 (br.s, 1H), 7,95-7,68 (m, 7H), 7,62-7,30 (1m, 10H), 6,71 (d, J=8,9 Гц, 1H), 6,10 (d, J=6,2 Гц, 1 H), 5,89 (s, 1H), 4,61 (m, 1H), 4,26 (m, 1H), 3,51 (d, J= 8,9 Гц, 2H), 3,0 (m, 2H), 1,51 (s, 9H).
Анализ для C36H36N2O3S:
Расчет: C 74,97; H 6,29; N 4,86.
Обнаружено: C 75,13; H 6,45; N 4,49.
Пример 62
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(индол-4''-ил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) соединения подготовки 25Д, 42 мг (0,26 ммоль) названного соединения подготовки 32, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 43 мг белого твердого вещества (т.п. 109-110oC).
Выход: 35%.
1H ЯМР (CDCl3): δ 8,45 (br.s, 1H), 7,90 (s, 1H), 7,76 (m, 3H), 7,57-7,23 (m, 10H), 7,19-6,89 (m, 3H), 6,24 (d, J=6,2 Гц, 1H), 5,97 (s, 1H), 4,63 (m, 1H), 4,13 (m, 1H), 3,51 (m, 2H), 3,01 (m, 2H), 1,49 (s, 9H).
Анализ для C34H36N3O3S:
Расчет: C 72,18; H 6,24; N 7,43.
Обнаружено: C 72,31; H 6,37; N 7,22.
Пример 63
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нaфт-2-ил-тиометил-4'-аза-5'-оксо-5'-(хинолин-5''-ил) пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 57, используя 0,060 г (0,15 ммоль) поименованного соединения подготовки 25Д, 42 мкл (0,38 ммоль) N-метилморфолина и 0,074 г (0,38 ммоль) названного соединения подготовки 31 в 2 мл тетрагидрофурана, получая 0,045 г белого твердого вещества.
Выход: 54%.
1H ЯМР (CDCl3): δ 8,85 (m, 1H), 8,75 (m, 1H), 8,75 (d, J= 8,21 Гц, 1H), 8,07 (m, 2H), 7,95 (s, 1H), 7,76 (m, 3H), 7,64 (m, 2H), 7,54 (m, 2H), 7,44 (m, 2H), 7,38 (m, 3H), 7,25 (m, 1H), 4,88 (s, 2H), 4,45 (m, 1H), 4,05 (m, 1H), 3,69 (dd, J=14, 3,09 Гц, 1H) 3,23 (m, 1H), 3,05 (m, 2H), 1,32 (s, 9H).
ИК (KBr): 3485, 3429, 3279, 3061, 2964, 1638, 1543, 1454, 1364, 1319, 1219, 1072, 806, 746 см-1.
HR Масс-спектр для C35H36N3O3S (MH+):
Расчет: 578,2477.
Обнаружено: 578,2491.
Анализ для C35H35N3O3S•0,6H2O:
Расчет: C 71,42; H 6,20; N 7,14; S 5,45.
Обнаружено: C 71,44; H 6,16; N 7,19; S 5,41.
Пример 64
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(1'',2'',3'',4''- тетрагидрохинолин-5''-ил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 58, используя 0,023 г (0,36 ммоль) цианборгидрида натрия, 0,041 г (0,07 ммоль) названного соединения Примера 63 и 2 мл уксусной кислоты, получая 0,024 г белого аморфного твердого вещества.
Выход: 60%.
1H ЯМР (CDCl3): δ 7,88 (s, 1H), 7,75 (m, 3H), 7,42 (m, 6H), 6,79 (t, J= 7,73 Гц, 1H), 6,54 (d, J=7,28 Гц, 1H), 6,44 (d, J=8,15 Гц, 2H), 6,10 (br. 1H), 5,91 (br.s, 1H), 4,45 (m, 1H), 4,05 (m, 1H), 3,48 (m, 2H), 3,24 (t, J= 5,50 Гц, 2H), 2,89 (m, 4H) 1,85 (m, 2H), 1,46 (s, 9H).
ИК (KBr): 3450, 2972, 1638, 1618, 1591, 1512, 1454, 1309, 1119, 1134, 1086, 814, 698, 621 см-1.
HR Масс-спектр для C35H40N3O3S (MH+):
Расчет: 582,2790.
Обнаружено: 582,2792.
Пример 65
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-aзa-5'-oкco-5'-(индoлин-4''-ил)пентил] бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 42 мг (0,26 ммоль) названного соединения подготовки 32, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1,5% метанол в метиленхлориде), получая 12 мг белого твердого вещества (т.п. 83-84oC).
Выход: 9%.
1H ЯМР (CDCl3): δ 7,99 (s, 1H), 7,76 (m, 3H), 7,69-7,23 (m, 10H), 7,10 (d, J=8,8 Гц, 1H), 6,60 (d, J=8,9 Гц, 1H), 5,99 (d, J=6,2 Гц, 1H), 5,89 (s, 1H), 4,53 (m, 1H), 4,11 (m, 1 H), 3,44 (m, 6H), 3,01 (m, 2H), 1,49 (s, 9H).
Анализ для C34H37N3O3S:
Расчет: C 71,92; H 6,57; N 7,40.
Обнаружено: C 72,21; H 6,72; N 7,26.
Пример 66
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нaфт-2-ил-тиометил-4'-аза-5'-оксо-5'-(хинолин-4''- ил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 45 мг (0,26 ммоль) хинолин-4-карбоновой кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1,5% метанол в метиленхлориде), получая 42 мг белого твердого вещества (т.п. 88-92oC).
Выход: 32%.
1H ЯМР (CDCl3): δ 8,59 (s, 1H), 8,33 (d, J=7,9 Гц, 1H), 8,09 (d, J=8,4 Гц, 1H), 7,93 (s, 1H), 7,80-7,71 (m, 4H), 7,69- 7,25 (m, 8H), 7,15 (s, 1H), 6,88 (d, J=8,4 Гц, 1H), 5,99 (s, 1H), 5,85 (s, 1H), 4,63 (m, 1H), 4,21 (m, 1H), 3,51 (d, 6,2 Гц, 2H), 3,02 (m, 2H), 1,39 (s, 9H).
Анализ для C35H35N3O3S:
Расчет: C 72,76; H 611; N 7,27.
Обнаружено: C 72,91; H 6,33; N 7,36.
Пример 67
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-3'' -нитрофенил]пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 47 мг (0,26 ммоль) 2-метил-3-нитробензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 100 мг белого твердого вещества (т.п. 80-81oC).
Выход: 74%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,75 (m, 3H), 7,65-7,25 (m, 9H), 7,10 (d, J= 7,9 Гц, 1H), 6,63 (d, J=8,9 Гц, 1H), 5,97 (d, J=6,0 Гц, 1H), 5,87 (s, 1H), 4,53 (m, 1H), 4,11 (m, 1H), 3,44 (m, J=6,3 Гц, 2H), 3,03 (dd, J=13,3, 10,2 Гц, 1H), 2,28 (dd, J=13,5, 2,8 Гц, 1H), 2,53 (s, 3H), 1,47 (s, 9H).
Анализ для C33H35N3O5S:
Расчет: C 67,67; H 6,02; N 7,17.
Обнаружено: C 67,83; H 5,93; N 7,05.
Пример 68
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(3''-нитро-6'' -метилфенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 47 мг (0,26 ммоль) 2- метил-5-нитробензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 102 мг белого твердого вещества (т.п. 85-88oC).
Выход: 75%.
1H ЯМР (CDCl3): δ 8,17 (s, 1H), 8,07 (d, J=8,4 Гц, 1H), 7,78 (m, 2H), 7,59-7,22 (m, 10H), 6,71 (d, J=8,9 Гц, 1H), 6,03 (d, J=6,1 Гц, 1H), 5,9 (s, 1H), 4,52 (m, 1H), 4,13 (m, 1H), 3,45 (d, J=6,2 Гц, 2H), 3,03 (dd, J=13,3, 9,61 Гц, 1H), 2,9 (dd, J=13,3, 3,72 Гц, 1H), 2,55 (s, 3H), 1,43 (s, 9H).
Анализ для C33H35N3O5S:
Расчет: C 67,67; H 6,02; N 7,17.
Обнаружено: C 67,92; H 6,22; N 7,02.
Пример 69
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(1''-N(метил)индoл- 4''-ил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 46 мг (0,26 ммоль) 1-N- метил-4-карбоновокислого индолина, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 1% метанол в метиленхлориде), получая 42 мг белого твердого вещества (т.п. 86-89oC).
Выход: 31%.
1H ЯМР (CDCl3): δ 7,88 (s, 1H), 7,79-7,65 (m, 3H), 7,53-6,95 (m, 13H), 6,22 (d, J=6,3 Гц, 1H), 5,99 (s, 1H), 4,67 (m, 1H), 4,13 (m, 1H), 3,75 (s, 3H), 3,51 (m, 2H), 3,03 (m, 2H), 1,49 (s, 9H).
Анализ для C35H35N3O3S:
Расчет: C 72,51; H 6,43; N 7,25.
Обнаружено: C 72,83; H 6,51; N 7,15.
Пример 70
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-3'', 4''-дигидроксифенил)пентил]бензамид
Названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 44 мг (0,26 ммоль) поименованного соединения подготовки 33В, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2,5% метанол в метиленхлориде), получая 76 мг белого твердого вещества (т.п. 121-123oC).
Выход: 58%.
1H ЯМР (CDCl3): δ 7,89 (s, 1H), 7,75 (m, 2H), 7,55- 7,22 (m, 10H), 6,85 (t, J=7,9 Гц, 1H), 6,72 (m, 2H), 6,61 (d, J=5,7 Гц, 1H), 6,50 (d, J=9,4 Гц, 1H), 6,13 (s, 1H), 5,92 (s, 1H), 4,51 (m, 1H), 4,09 (m, 1H), 3,51 (m, 2H), 3,12 (dd, J=13,1, 10 Гц, 1H), 2,87, (dd, J=13,l, 3,1 Гц, 1H), 2,13 (s, 3H), 1,46 (s, 9H).
Анализ для C33H36N2O5S:
Расчет: C 69,21; H 6,34; N 4,89.
Обнаружено: C 69,43; H 6,72; N 4,72.
Пример 71
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(3''- гидроксифенил)пентил]бензамид
Желаемое названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 45 мг (0,26 ммоль) 2-хлор-4-аминобензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 2% метанол в метиленхлориде), получая 92 мг белого твердого вещества (т.п. 102-104oC).
Выход: 69%.
1H ЯМР (CDCl3): δ 7,88 (s, 1H), 7,77 (m, 2H), 7,61-7,23 (m, 9H), 6,95 (t, J=7,7 Гц, 1H), 6,75 (m, 1H), 6,51 (d, J=7,8 Гц, 1H), 6,06 (d, J=6,l Гц, 1H), 5,90 (s, 1H), 4,51 (m, 1H), 4,20 (s, 2H), 4,12 (m, 1H), 3,50 (m, 2H), 3,01 (m, 3H), 1,48 (s, 9H).
Анализ для C32H34ClN3O3S:
Расчет: C 66,71; H 5,95; N 7,29.
Обнаружено: C 66,92; H 5,97; N 7,16.
Пример 72
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(2''-метил-5'' -гидроксифенил)пентил]бензамид
Желаемое названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 47 мг (0,26 ммоль) поименованного соединения подготовки 29Б, 32 мг (0,23 ммоль) HOBT, 40 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 3% метанол в метиленхлориде), получая 86 мг белого твердого вещества (т.п. 104-106oC).
Выход: 67%.
1H ЯМР (CDCl3): δ 7,85 (s, 1H), 7,72 (m, 3H), 7,60-7,22 (m, 9H), 6,92 (t, J=7,5 Гц, 1H), 6,72 (m, 1H), 6,50 (d, J=7,6 Гц, 1H), 5,96 (s, 1H), 5,90 (s, 1H), 4,50 (m, 1H), 4,15 (m, 1H), 4,02 (m, 1H), 2,51 (m, 2H), 3,01 (m, 3H), 2,36 (s, 3H), 1,45 (s, 9H).
Анализ для C33H37N3O3S:
Расчет: C 71,32; H 6,71; N 7,56.
Обнаружено: C 71,56; H 6,76; N 7,52.
Пример 73
[2'R-(2'R*, 3'S*)] -N-т-Бутил-2-[2'-гидрокси-3'- нафт-2-ил-тиометил-4'-аза-5'-оксо-5'-(3''-гидрокси-4'' -аминофенил)пентил]бензамид
Желаемое названное соединение получали в соответствии с методикой, детально описанной в примере 61, используя 100 мг (0,23 ммоль) поименованного соединения подготовки 25Д, 40 мг (0,26 ммоль) 3-гидрокси-4-аминобензойной кислоты, 32 мг (0,23 ммоль) HOBT, 45 мг (0,23 ммоль) EDC и 0,16 мл (1,20 ммоль) триэтиламина в 2,0 мл диметилформамида. Неочищенный остаток очищали, используя флэш-хроматографию (элюирующий раствор - 3% метанол в метиленхлориде), получая 43 мг белого твердого вещества (т.п. 119-122oC).
Выход: 34%.
1H ЯМР (CDCl3): δ 7,91 (s, 1H), 7,75 (m, 2H) 7,60-7,20 (m, 10H), 6,96 (t, J= 7,9 Гц, 1H), 6,75 (m, 1H), 6,55 (d, J=7,8 Гц, 1H), 6,1 (s, 1H), 5,95 (s, 1H), 4,51 (m, 1H), 4,23 (s, 2H), 4,12 (m, 1H), 3,52 (m, 2H), 3,00 (m, 3H), 1,48 (s, 9H).
Анализ для C32H35N3O4S:
Расчет: C 68,92; H 6,33; N 7,53.
Обнаружено: C 69,12; H 6,57; N 7,32.
Реакционная схема III (см. в конце описания) демонстрирует структуры соединений в приведенных ниже примерах 74(А - M).
Пример 74
Пример A
N-(Бензилоксикарбонил-3-(2-тиенил)-D,L-аланин
В колбу емкостью 500 мл помещали 3,0 г 3-(2-тиенил)-D, L-аланина (оптически активный материал в L-форме выпускается фирмами Aldrich или SIGMA и может применяться для получения оптически активного продукта) в смеси, состоящей из 75 мл H2O и 60 мл диоксана, и к ним добавляли 5,6 г K2CO3 с последующим добавлением 2,85 мл карбобензилоксихлорида. Смесь быстро перемешивали в течение 1 часа. ТСХ (21/7/7/9, EtOAc/AcOH/ CH3CN/H2O) показала, что исходный материал реакции исчерпан. Наблюдали появление нового продукта с более высоким значением Rf. Диоксан упаривали и водный слой промывали Et2O (75 мл). Водный слой смешивали с CH2Cl2 (150 мл) и значение его pH подводили к 2,0 с помощью 5н. HCl. Желаемый N-(бензилоксикарбонил)-3-(2-тиенил)-D,L-аланин экстрагировали с помощью CH2Cl2. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали, получая 5,05 г желаемого N-(бензилоксикарбонил)-3-(2-тиенил)-D,L-аланина (выход 98 %).
1H ЯМР (300 МГц, CDCl3): δ 7,37 (m, 5H), 7,18 (d, J=4 Гц, 1H), 6,95 (m, 1H), 6,83 (m, 1H), 5,35 (d, J=8 Гц, 1H), 5,15 (s, 2H), 4,7 (m, 1H) и 3,4 (m, 2H).
Пример Б
N-(Бензилоксикарбонил)-3-(2-тиенил)-L-аланин-т-бутиламид
В колбу емкостью 500 мл помещали 8,06 г поименованного соединения примера А, N-(бензилоксикарбонил)-3-(2-тиенил)-L- аланина в 130 мл THF. Соединение охлаждали до 0oC. В колбу добавляли N-метилморфолин (4,23 мл) с последующим добавлением в течение двух минут изобутилхлорформиата (4,04 мл). Смесь перемешивали в течение 15-20 минут, после чего к ней добавляли 3,74 мл т-бутиламина. Охлаждение убирали и смесь перемешивали при комнатной температуре в течение двух часов. Смесь упаривали на роторном испарителе и остаток растворяли в этилацетате. Остаток промывали последовательно H2O, HCl и насыщенным раствором NaHCO3. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и концентрировали до состояния масла. Масло растворяли в 100 мл горячего гексана и охлаждали в рефрижераторе в течение ночи, получая твердое вещество. Гексан декантировали, после чего осадок высушивали, получая 9,25 г твердого N-(карбобензилокси)-3-(2-тиенил)-L- аланин-т-бутиламида (выход 97%).
1H ЯМР (300 МГц, CDCl3): δ 7,37 (s, 5H), 7,2 (d, J=4 Гц, 1H), 6,95 (dd, J= 4 Гц, 8 Гц, 1H), 6,87 (d, J=4 Гц, 1H), 5,52 (m, 2H), 5,12 (s, 2H), 4,27 (m, 1H), 3,27 (m, 2H) и 1,23 (s, 9H).
Пример В
N-т-Бутил-5-бензилоксикарбонил-(4,5,6,7)- тетрагидро-тиено [3,2-c] пиридин-6S-N- т-бутилкарбоксамид
В колбу емкостью 50 мл помещали 500 мг поименованного соединения примера Б, N-(бензилоксикарбонил)-3-(2-тиенил)-L- аланин-т-бутиламида в 12 мл 1,1,2-трихлорэтана. К содержимому колбы добавляли 2 мл TFA с последующим добавлением 2 мл диметоксиметана. Смесь кипятили с обратным холодильником, контролируя прохождение реакции с помощью ТСХ каждые пять минут. По истечении 15 минут ТСХ показала, что исходный материал реакции исчерпан. По получении в основном желаемого продукта, нагревание убирали и содержимое колбы выливали в 30 мл H2О, содержащей 3,5 г K2CO3 и 40 мл CH2Cl2. Продукт переносили в делительную воронку, органическую фазу отделяли, высушивали над Na2SO4, фильтровали и концентрировали до состояния масла. Продукт очищали флэш-хроматографией, используя 25 г (SiO2) и 3% EtOAc/CH2Cl2. В результате получали 357 мг N-т-бутил-5-бензилоксикарбонил (4,5,6,7)-тетрагидро-тиено [3,2-c]пиридин-6S-N-т-бутилкарбоксамида (выход 69%).
Соблюдение интервала времени в пятнадцать минут от начала дефлегмации до удаления источника нагревания и немедленное последующее смешивание очень важны для избежания побочных реакций.
1H ЯМР (300 МГц, d6 ДМСО): δ 7,35 (m, 7H), 6,83 (m, 1H), 5,15 (m, 2H), 4,98 (m, 1H), 4,35 (m, 2H), 3,10 (m, 2H) и 1,10 (s, 9H).
Масс-спектр: m/e 372 (М+).
Пример Г
[6S-(6R*,3aS*,7aR*)]-N-(Бензилоксикарбонил)- октагидро-тиено[3,2-c]пиpидин-6-N-т-бутилкapбoкcaмид
В сосуд для гидрирования под высоким давлением помещали поименованное соединение примера В, N-т-бутил-5-бензилоксикарбонил- (4,5,6,7)-тетрагидро-тиено[3,2-с] пиридин-6S-N-т- бутилкарбоксамид (10,5 г), и 105 г 5% палладия на угле в 1100 мл THF и 525 мл EtOH. Смесь помещали в атмосферу H2 (3000 psi) при 80oC на 24 часа. Реакционную смесь охлаждали, катализатор отфильтровывали и промывали 20% MeOH/CHCl3. Органические фазы объединяли и концентрировали до состояния неочищенного масла. Масло растворяли в CH2Cl2 и подвергали флэш-хромотографии на 250 г (SiO2), элюируя 2% MeOH/CH2Cl2. Желаемый цис-изомер (основной) в результате хроматографии получается в очищенном виде с небольшой примесью минорного изомера. Эту смесь перекристаллизовывали, растворяя в 1,5 мл MeOH, добавляя 20 мл Et2O, с последующим добавлением 120 мл гексана, после чего смесь помещали в рефрижератор на ночь. Полученные кристаллы отделяли фильтрованием, промывали холодным гексаном и высушивали под вакуумом, получая 2,54 г цис-изомера [6S-(6R*,3aS*,7aR*)]-N- (бензилоксикарбонил) октагидро-тиено[3,2,-с] пиридин-6-N-т- бутилкарбоксамида (выход 24%).
1H ЯМР (300 МГц, CDCl3): δ 7,37 (s, 5H), 6,0 и 5,5 (br.s, 1H), 5,18 (br. s, 2H), 4,22 (m, 2H), 3,40 (m, 1H), 2,87 (m, 3H), 2,48 (m, 1H), 2,15 (m, 2H), 1,70 (m, 1H) и 1,15 (br.s, 9H).
Масс-спектр: m/e 377 (М+ +1).
Пример Д
[6S-(6R*,3aS*,7aR*)] -Октагидротиено[3,2-c]пиридин-6-N- т-бутилкарбоксамид
В колбу емкостью 100 мл помещали 2,41 г поименованного соединения примера Г, [6S-(6R*, 3aS*,7aR*)] -Т-(бензилоксикарбонил)- октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамида в 12 мл смеси 1:1 CH3CN/CH2Cl2. К раствору добавляли первую порцию триметилсилилиодида (TMSI) (1,9 мл) и перемешивали в течение 10 минут. Затем добавляли вторую порцию TMSI (0,94 мл) и перемешивали в течение 10 минут. Добавляли третью порцию TMSI (0,48 мл) и перемешивали в течение 30 минут. ТСХ (5% EtOAc/CH2Cl2) показала, что исходный материал реакции исчерпан. Реакционную смесь разбавляли 30 мл диэтилового эфира, 40 мл H2O и 6 мл 1н. HCl. Слой эфира отделяли и промывали 15 мл 0,1н. HCl. Объединенные эфирные слои отбрасывали, а водные промывки объединяли. К ним добавляли насыщенный раствор NaHCO3, для подведения pH водного слоя до 8. Водный слой экстрагировали дважды 200 мл CH2Cl2, органические слои объединяли и высушивали над Na2SO4. Раствор фильтровали и концентрировали, получая 1,3 г (выход 84%) желаемого [6S-(6R*,3aS*,7aR*)]-октагидротиено[3,2-с] пиридин-6-N-т-бутилкарбоксамида.
1H ЯМР (300 МГц, CDC13): δ 6,43 (s, 1H), 3,22 (m, 2H), 2,95 (m, 4H), 2,17 (m, 3H), 2,0 (m, 1H), 1,55 (m, 2H) и 1,32 (s, 9H).
[α]D(EtOH) = -179,1o (при 25oC).
Пример Е
[6S-(6R*,3aS*,7aR*, 2'S*,3'S*)]-5-[2-Гидрокси-4- фенилтио-3-(бензоксикарбонил)-аминобутил] -октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамид
В колбу емкостью 100 мл помещали 1,45 г [1'R-(1'R*,1S*)]-1-[(1' -N-(бензилоксикарбонил)амино-2'-(фенилтио)этил] оксирана (полученного после подготовки 8Д, [1'R-(1'R*, 1S*)] -1-[(1'-N (бензилоксикарбонил)амино-2'-(фенилтио)этил]оксиран может также быть получен как показано в примере Н ниже)) и 1,07 г поименованного соединения примера 75, [6S-(6R*,3aS*, 7aR*)] октагидротиено[3,2-с] пиридин-6-N-т-бутилкарбоксамида в 30 мл EtOH и смесь нагревали до 65oC в течение 60 часов. Реакционную смесь концентрировали до состояния пены и очищали на хроматотроне (4000 микронная пластина), элюируя 1% MeOH/CH2Cl2. Нужные фракции концентрировали, получая 1,8 г желаемого [6S-(6R*, 3aS*,7aR*,2'S*,3'S*)]-5-[2-гидрокси-4- фенилтио-3-(бензоксикарбонил)- аминобутил] -октагидротиено[3,2-с] пиридин-6-N-т-бутил- карбоксамида. Несколько фракций в начале разделения, содержащих смесь веществ, объединяли, получая 326 мг смеси, которую снова очищали хроматографически в тех же самых условиях на 2000 микронной пластине. В результате получали дополнительные 228 мг желаемого [6S-(6R*,3aS*,7aR*,2'S*, 3'S*)]-5-[2-гидрокси-4- фенилтио-3-(бензоксикарбонил)-аминобутил]октагидротиено[3,2- с]пиридин-6-N-т-бутилкарбоксамида. Общий выход полученного [6S-(6R*,3aS*,7aR*,2'S*, 3'S*)] -5-[2-гидрокси-4-фенилтио-3-(бензоксикарбонил)аминобутил] -октагидротиено[3,2-c]пиpидин-6-N-т-бутилкapбoкcaмидa составил 80,5%.
1H ЯМР (300 МГц, CDCl3): δ 7,30 (m, 10H), 5,80 (m, 2H), 5,08 (АВ, 2H), 3,95 (m, 2H), 3,42 (m, 2H), 3,17 (m, 3H), 2,90 (m, 2H), 2,67 (m, 1H), 2,58 (m, 1H), 2,48 (m, 1H), 2,35 (m, 2H), 1,98 (m, 4H) и 1,30 (s, 9H).
Пример Ж
[6S-(6R*, 3aS*,7aR*,2'S*,3'S*)] -5-[2-Гидрокси-4- фенилтио-3-аминобутил] -октагидротиено[3,2-c]пиридин-6-N-т- бутилкарбоксамид
В колбу емкостью 100 мл помещали 1,8 г поименованного соединения примера Е, [6S-(6R*,3aS*,7aR*,2'S*,3'S*)] -5-[2-гидрокси-4-фенилтио-3-(бензоксикарбонил)-аминобутил] октагидротиено [3,2-с] пиридин-6-N-т-бутилкарбоксамида в 10 мл каждого из CH2Cl2 и CH3CN. К раствору добавляли первую порцию (TMSI) (1,14 мл) и перемешивали в течение 10 минут. Затем добавляли вторую порцию TMSI (0,72 мл) и перемешивали в течение 10 минут. Добавляли третью порцию TMSI (0,24 мл) и перемешивали в течение 15 минут. Реакционную смесь разбавляли 40 мл Et2O и выливали в 30 мл 0,1н. HCl и 60 мл Et2O. Этанольный слой отделяли и отбрасывали. Водный слой подщелачивали насыщенным раствором NaHCO3 и экстрагировали CH2Cl2 (2 X 100 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и концентрировали, получая 1,18 г [6S-(6R*, 3aS*,7aR*,2'S*,3'S*)] -5-[2-гидрокси-4-фенилтио-3-аминобутил]октагидротиено[3,2- с]пиридин-6-N-т-бутилкарбоксамида (86% выход) в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3): δ 7,38 (m, 2H); 7,28 (m, 2H); 7,20 (m, 1H); 6,23 (s, 2H); 3,65 (s, 1H); 3,28 (m, 3H); 2,90 (m, 4H); 2,70 (m, 2H); 2,58 (m, 1H); 2,43 (m, 1H); 2,34 (m, 1H); 2,05 (m, 4H); 1,80 (m, 3H) и 1,32 (s, 9H).
ИК (CHCl3): 3430; 3005; 2973; 1670; 1514; 1456; 1366 и 1090 см-1.
Масс-спектр: m/e 437 (M+).
Пример З
[6S-(6R*, 3aS*, 7aR*, 2'S*,3'S*)] -2-[2'-Гидpoкcи-3'-фeнилтиометил-4'-аза-5'-оксо-5'-(2''- метил-3''-гидрокси-фенил)пентил] -октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамид
В колбу емкостью 25 мл помещали 40 мг поименованного соединения примера Ж, [6S-(6R*,3aS*,7aR*,2'S*,3'S*)] -5-[2-гидрокси-4-фенилтио-3-аминобутил]-октагидротиено[3,2-с] пиридин- 6-N-т-бутилкарбоксамида, 14 мг 3-гидрокси-2-метилбензойной кислоты и 12,6 мг HOBT в 2 мл THF и реакционную смесь охлаждали до -10oC. К смеси добавляли DCC (18,7 мг), нагревали до комнатной температуры и перемешивали в течение 85 часов. Реакционную смесь разбавляли 2 мл Et2O и фильтровали через хлопковую набивку, фильтрат концентрировали и остаток элюировали на хроматотроне (2000 микронная пластина) с помощью 3% MeOH/CHCl3. Желаемые фракции концентрировали, получая 44 мг [6S-(6R*,3aS*, 7aR*, 2'S*, 3'S*)] -2-[2'-гидрокси-3'-фенилтиометил- 4'-аза-5'-оксо-5' -(2''-метил-3''-гидроксифенил) пентил] -октагидротиeнo [3,2-c]пиpидин-6-N-т- бутилкарбоксамида (выход 85%).
Пример И
[6S-(6R*, 3aS*, 7aR*, 2'S*,3'S*)] -2-[2'-Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-3''-гидроксифенил)пентил] -октагидротиено[3,2- с] пиридин-6-N-т-бутилкарбоксамида соль метансульфоновой кислоты
В колбу емкостью 50 мл помещали 330 мг поименованного соединения примера 3, [6S-(6R*,3aS*,7aR*,2'S*,3'S*)] -2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-3''-гидроксифенил)пентил] -октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамида в смеси CH2Cl2/CH2CN (4 мл/2 мл), и к нему, используя микролитровый шприц, добавляли 37,5 мл MeSO3H. Смесь становилась непрозрачной. Ее разбавляли 1 мл CH2Cl2, добавляли Et2O и гексан и концентрировали. Остаток с гексаном и концентрировали два раза, получая 385 мг желаемой соли метансульфоновой кислоты [6S-(6R*,3aS*,7aR*,2'S*,3'S*)] -2-(2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''- метил-3''гидроксифенил)пентил] -октагидротиено[3,2-с] пиридин-6- N-т-бутилкарбоксамида (выход 100%).
Пример К
[6S-(6R*,3aS*,7aR*,2'S*,3'R*)] -5-[2-Гидрокси- 4-фенил-3-(бензоксикарбонил)-аминобутил] -октагидротиено[3,2- c] пиридин-6-Т-т-бутилкарбоксамид
В колбу емкостью 50 мл помещали 145 мг [1'S-(1'R*,1R*)]-1-[(1'-N-(бензилоксикарбонил)амино-2'- (фенил)этил] оксирана (может быть получен так же, как и в реакционной схеме А (шаги от 1 до 5), приведенной ниже) и 118 мг поименованного соединения примера Д, [6S-(6R*, 3aS*,7aR*)]-октагидротиено[3,2-с] пиридин- 6-N-т-бутилкарбоксамида, в виде смеси энантиомеров в 3 мл EtOH. Смесь нагревали до 65oC и выдерживали при этой температуре в течение 20 часов. Далее реакционную смесь концентрировали и неочищенный остаток очищали с помощью хроматотрона на 2000 микронной пластине, элюируя 1% MeOH/CHCl3 и получая 98 мг [6S-(6R*,3aS*,7aR*,2'S*,3'R*)] -5-[2-гидрокси-4-фенил-3- (бензоксикарбонил)-аминобутил] октагидротиено[3,2-c]пиридин- 6-N-т-бутилкарбоксамида (выход 37%) и 109 мг диастереомера [6S-(6R*,3aS*,7aR*, 2'S*, 3'R*)] -5-[2-гидрокси-4-фенил-3- (бензоксикарбонил)-аминобутил]октагидротиено[3,2-c]пиридин-6-N -т-бутилкарбоксамида.
Использование существенно энантиомерно чистого (6S-(6R*,3aS*,7aR*)]-октагидротиено[3,2-с] пиридин- 6-N-т-бутилкарбоксамида вместо [6S-(6R*,3aS*, 7aR*)] -октагидротиено[3,2- с] пиридин-6-N-т-бутилкарбоксамида в виде смеси энантиомеров должно приводить к более высокому выходу [6S-(6R*,3aS*,7aR*, 2'S*,3'R*)] -5-[2-гидрокси-4-фенил-3-(бензоксикарбонил)-аминобутил]- октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамида (см., например, пример Е выше).
Пример Л
[6S-(6R*, 3aS*,7aR*,2'S*,3'R*)] -5-[2-Гидрокси-4- фенил-3-аминобутил]-октагидротиено[3,2-с]пиридин-6-N-т- бутилкарбоксамид
В колбу емкостью 25 мл помещали 85 мг поименованного соединения примера К, [6S-(6R*,3aS*,7aR*,2'S*,3'R*)] -5-[2-гидрокси-4-фенил-3-(бензоксикарбонил)-аминобутил] октагидротиено [3,2-с] пиридин-6-N-т-бутилкарбоксамида в смеси CH3CN/CH2Cl2. К раствору порциями в 56, 34 и 11 микролитров соответственно каждые десять минут добавляли TMSI и перемешивали в течение 1,5 часов. Смесь разбавляли Et2O (5 мл) и выливали в 15 мл 1н. HCl и Et2O (20 мл). Органическую фазу отделяли и отбрасывали. Водный слой обрабатывали 30 мл насыщенного раствора NaHCO3 и экстрагировали CH2Cl2 (2 X 50 мл). Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали до состояния масла, которое кристаллизовали, получая 64 мг [6S-(6R*,3aS*, 7aR*,2'S*,3'R*)] -5-[2-гидрокси-4-фенил-3-аминобутил]-октагидротиено [3,2-с] пиридин-6-N-т-бутилкарбоксамида (выход 100%).
1H ЯМР (300 МГц, CDCl3): δ 7,28 (m, 5H), 6,38 (s, 1H), 3,75 (m, 1H), 3,32 (m, 2H), 3,12 (m, 1H), 2,93 (m, 2H), 2,78 (m, 2H), 2,58 (m, 3H), 2,38 (m, 1H), 2,12 (m, 5H), 1,83 (m, 2H) и 1,35 (s, 9H).
Пример M
[6S-(6R*, 3aS*, 7aR*, 2'S*,3'R*)] -2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'- (2''- метил-3''-гидроксифенил)пентил] -октагидротиено[3,2- c] пиpидин-6-N-т-бутилкарбоксамид
В колбу емкостью 25 мл помещали 64 мг поименованного соединения примера Л, [6S-(6R*, 3aS*,7aR*,2'S*,3'R*)] -5-[2-гидрокси-4-фенил-3-аминобутил]-октагидротиено[3,2-с] пиридин-6- N-т-бутилкарбоксамида, 24 мг 3-гидрокси-2-метилбензойной кислоты (получена методом, изложенным в Подготовке 23В) и 22 мг HOBT•H2O в 2 мл THF и смесь охлаждали до -10oC. После этого к смеси добавляли DCC (32 мг), смесь нагревали до комнатной температуры и перемешивали в течение 60 часов. Реакционную смесь разбавляли 2 мл Et2O, фильтровали через хлопковую набивку, фильтрат концентрировали и остаток элюировали на хроматотроне (2000 микронная пластина) градиентом от 1,5% MeOH/CHCl3 до 4% MeOH/CHCl3. Нужные фракции концентрировали, получая 72 мг [6S-(6R*,3aS*, 7aR*, 2'S*, 3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5' -(2''-метил-3''-гидроксифенил)пентил] - октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамида (выход 85%).
Пример 75
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)] -2-[2'-Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-3''-гидроксифенил)пентил]-декагидроизохинолин-3-N-т- бутилкарбоксамида соль метансульфоновой кислоты
Это соединение получали так же, как в примере 23, за исключением того, что пункты подготовки 8А и 8Г были изменены, как изложено ниже в пункте (1) и, кроме этого, был добавлен изложенный ниже этап образования соли (2).
(1). В колбу емкостью 2 л добавляли Ph3P (109,6 г) в 500 мл CH2Cl2 и смесь охлаждали до -70oC. К смеси по каплям в течение 25 минут добавляли раствор диэтилазидодикарбоксилата (66 мл) в 60 мл THF. По истечении 25 минут к смеси по каплям в течение 45 минут добавляли раствор N-карбобензилокси-L-серина (100 г) в 400 мл THF и позволяли смеси нагреться в водяной бане до комнатной температуры в течение двух часов. После этого к смеси добавляли 150 мл THF. В другой колбе раствор тиофенола (46 г) в 1 л THF охлаждали в ледяной бане до 0oC и обрабатывали порциями измельченного NaH (10 г), получая вязкий раствор. По прошествии одного часа к раствору тиолата в течение 30 минут добавляли по каплям, используя капельную воронку, неочищенный раствор лактона. Через 12 часов отфильтровывали белый осадок, который промывали THF. Твердое вещества растворяли в 0,4н. NaHSO4 и EtOAc, разделяли фазы и органический слой промывали солевым раствором, высушивали и упаривали, получая 85 г 2R-2-N-(бензилоксикарбонил)амино-3-фенилтиопропановой кислоты в виде вязкого масла.
Первоначально полученное твердое вещество, по-видимому, является натриевой солью желаемого продукта. Таким образом, выход реакции и простота выделения могут быть улучшены путем выделения непосредственно натриевой соли.
Неочищенный хлоркетон 3R-1-хлор-2-оксо-3-N-(бензилоксикарбонил)амино-4- фенилтиобутан (16,87 г, 46,4 ммоль) добавляли к 1 л абсолютного EtOH и 200 мл THF и раствор охлаждали в CO2-ацетоновой бане (Tвн = -78oC), после чего к нему по каплям добавляли в течение 1 часа раствор NaBH4 (2,63 г, 69,5 ммоль) в 200 мл абсолютного EtOH (Твн < -75oC). Анализ с помощью ТСХ, проведенный по окончании добавления, показал, что реакция прошла полностью. Реакционную смесь разбавляли 300 мл эфира и реакцию останавливали медленным добавлением 0,4N NaHSO3 с перемешиванием, которое вызывало выделение газа. Полученную смесь упаривали при пониженном давлении для удаления большей части EtOH и в нее добавляли дополнительное количество воды. Смесь экстрагировали эфиром, объединенные органические слои промывали насыщенным водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали, получая 15,7 г беловатого твердого вещества. Этот материал растирали в порошок с кипящим гексаном (300 мл) и гексан тщательно декантировали в горячем состоянии. Данную процедуру повторяли 10 раз (каждый раз в 300 мл), получая 10,35 г беловатого твердого вещества (один чистый изомер по данным ТСХ). Фильтрат гексана концентрировали, получая 6 г белого твердого вещества, которое откладывали. Растертое в порошок твердое вещество нагревали в 50 мл CH2Cl2 и примерно 6 мл гексана и фильтровали в горячем виде. Прозрачному раствору позволяли охладиться до 25oC, после чего его помещали в морозильник. Полученное твердое вещество фильтровали и промывали с гексаном, получая 7,157 г белого твердого вещества. Фильтрат объединяли с гексановым фильтратом, полученным ранее, и неочищенным продуктом, полученным при проведении двух мелкомасштабных экспериментов (по 500 мг исходного кетона в каждом), и объединенный материал хроматографировали на SiO2 (система растворителей от 2:1 гексан-эфир до 1: 1 гексан-эфир, насыщенных CH2Cl2), получая дополнительно 2,62 г продукта. Общее количество полученного чистого изомера [2S-(2R*,3S*)] -1-хлор- 2-гидрокси-3-N-(бензилоксикарбонил)амино-4-фенилтиобутана составило 10,31 г (выход 50% исходя из кислоты).
[α]D -63,6o (с=1, MeOH).
(2). Образование соли
[3S-(3R*, 4aR*,8aR*,2'S*,3'S*)] -2-[2'-Гидрокси- 3'-фенилтиометил-4'-аза -5'-оксо-5'-(2''-метил -3''- гидроксифенил)пентил] декагидроизохинолин-3-N -т-бутилкарбоксамид (3,34 г) растворяли в 30 мл MeOH и 30 мл CH2Cl2 и к раствору по каплям добавляли раствор метансульфоновой кислоты (596 мг) в 10 мл CH2Cl2. По прошествии 10 минут реакционную смесь концентрировали до состояния пены. Неочищенную соль растворяли в 5 мл THF и медленно добавляли к смеси, состоящей из 175 мл этилового эфира и 25 мл гексана, с перемешиванием до получения мелкой суспензии. Суспензию охлаждали в морозильнике, фильтровали в холодном состоянии и промывали несколько раз этиловым эфиром с последующим высушиванием в вакуумной печи, получая 3,75 г (96%) соли метансульфоновой кислоты [3S-(3R*,4aR*,8aR*,2'S*,3'S*)] -2-[2'-гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'- (2''-метил-3''- гидроксифенил) пентил]-декагидроизохинолин -3-N-т-бутилкарбоксамида в виде белого порошка.
Пример 76
3-[Бисбензоксифосфинил)окси-2-метилбензойная кислота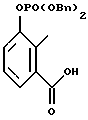
К охлажденному (0oC) перемешиваемому раствору, содержащему 706 мг (4,67 ммоль) 3-гидрокси-2-метилбензойной кислоты в 30 мл пиридина добавляли по каплям в течение 5 минут 10,3 мл (10,21 ммоль) 1,0 М раствора гексаметилдисилазида лития. После перемешивания в течение 5 минут к смеси одной порцией добавляли 3,0 г (5,57 ммоль) тетрабензилпирофосфата и реакционную смесь нагревали до комнатной температуры в течение 30 минут. Реакционную смесь концентрировали и остаток распределяли между 2,5н. HCl (200 мл) и смесью 50/50 этилацетат/гексан (200 мл). Слои разделяли и водный слой экстрагировали дважды раствором этилацетат/гексан в отношении 50/50. Органические слои объединяли, промывали солевым раствором и высушивали над сульфатом натрия. Очистка неочищенного продукта флэш-хроматографией (элюирование градиентом - 50-70% этилацетат/гексан/2% уксусная кислота) дала 910 мг светло-желтого масла, которое представляет собой 3- (бисбензоксифосфинил)окси-2-метилбензойную кислоту.
Выход: 47%.
1H ЯМР (CDCl3): δ 2,49 (s, 3H), 5,14 (d, J=8,60 Гц, 4H), 7,10- 7,40 (m, 11H), 7,48 (d, J=8,09 Гц, 1H), 7,81 (d, J=7,80 Гц, 1H).
ИК (CHCl3): 3700-2350 (br), 1700, 1457, 1382, 1273, 1240, 1179, 1082, 1034, 1023, 1001, 966, 881, 851 см-1.
Масс-спектр: (FD): m/e 413 (M+, 100).
Пример 77
[3S-(3R*, 4aR*,8aR*,2'S*,3'R*)] -2-[2'-Гидрокси-3' -фенилметил-4'- аза-5'-оксо-5'-(2''-метил-3'' -(бисбензоксифосфинил)оксифенил) пентил]декагидроизохинолин -3- N-т-бутилкарбоксамид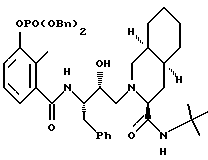
К охлажденному (-10oC) раствору, содержащему 95 мг (0,23 ммоль) поименованного соединения примера 76, 3-(бисбензоксифосфинил) окси-2-метилбензойной кислоты, 92 мг (0,23 ммоль) [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-(3'-амино-2'-гидрокси-4'- фенил] бутилдекагидроизохинолин-3-N-т-бутилкарбоксамида (см. например подготовку 1Б) и 31 мг (0,23 ммоль) HOBT в 5 мл безводного THF добавляли одной порцией 48 мг (0,23 ммоль) DCC. После перемешивания в течение 3 дней при комнатной температуре реакционную смесь разбавляли этилацетатом и фильтровали через хлопковую набивку. Полученный фильтрат дважды экстрагировали насыщенным карбонатом натрия, промывали солевым раствором и высушивали над сульфатом натрия. Очистка продукта радиальной хроматографией (2 мм пластина; элюирующий раствор - 2,5-5% градиент метанола в метиленхлориде) дала 1,00 мг белой пены, которая представляет собой [3S-(3R*,4aR*,8aR*,2'S*, 3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'' -(бисбензоксифосфинил)оксифенил)пентил]-декагидроизохинолин- 3-N-т-бутилкарбоксамид.
Выход: 52%.
1H ЯМР (CDCl3): δ 1,13 (s, 9H), 1,14-2,10 (m, 15H), 2,23- 2,36 (m, 2H), 2,50-2,70 (m, 2H), 2,92-3,05 (m, 2H), 3,39-3,50 (m, 1H), 3,80- 4,10 (m, 2H), 4,52-4,62 (m, 1H), 5,03-5,13 (m, 4 H), 5,65 (s, 1H), 6,62 (d, J=8,51 Гц, 1H), 6,83 (d, J=7,60 Гц, 1H), 7,02 (t, J=8,10 Гц, 1H).
ИК (CHCl3): 3690, 3600-3100 (br), 3009, 2929, 2866, 1672, 1603, 1513, 1456, 1368, 1277, 1239, 1182, 1037, 1023, 1001, 967, 880 см-1.
Масс-спектр (FD): m/e 796 (M+, 100).
Анализ для C46H58N3O7P1:
Расчет: C 69,41; H 7,34; N 5,28.
Обнаружено: C 69,57; H 7,33; N 5,20.
Пример 78
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)] -2-[2'-Гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'' -гидроксифенил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамида
3''-однозамещенный фосфат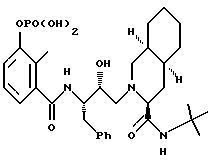
Смесь 86 мг (0,108 ммоль) поименованного соединения примера 77, [3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)] -2-(2'-гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3'' -(бисбензоксифосфинил)оксифенил)пентил] декагидроизохинолин-3 -N-т-бутилкарбоксамида и 23 мг 10% палладия на угле в 16 мл метанола перемешивали под одной атмосферой водорода в течение 1 часа. Реакционную смесь фильтровали через целит и концентрировали, получая 61 мг белого твердого вещества, которое представляет собой 3''-однозамещенный фосфат [3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5' -оксо-5'-(2''- метил-3''- гидроксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамида.
Выход: 96%.
1H ЯМР (Метанол-d6): δ 1,32 (s, 9H), 1,33-2,21 (m, 14H), 2,60, 2,75 (m, 1H), 3,18-3,49 (m, 5H), 3,56- 3,70 (m, 1H), 3,95 - 4,35 (m, 3H), 5,47 (s, 1H), 6,71 (d, J=7,26 Гц, 1H), 7,02 (t, J=8,24 Гц, 1H), 7,15-7,35 (m, 5H), 7,40 (d, J=8,18 Гц, 1H).
ИК (KBr): 3800-2400 (br), 1673, 1545, 1456, 1395, 1368, 1222, 1185, 1077, 942, 857, 792 см-1.
Масс-спектр (FAB): m/e 616,3 (M+, 100).
Пример 79
[3S-(3R*, 4aR*, 8aR*, 2'S*,3'S*)] -2-[2'-Гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-3''-(бисбензоксифосфинил)оксифенил)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид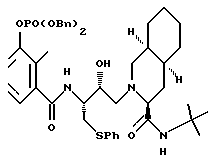
К охлажденному (0oC) перемешиваемому раствору, содержащему 478 мг (1,16 ммоль) поименованного соединения примера 76, 3- (бисбензоксифосфинил)окси-2-метилбензойной кислоты, 500 мг (1,16 ммоль) [3S-(3R*,4aR*,8aR*,2'S*,3'S*)] -2-[3'-амино-2'-гидрокси-4'-(фенил)тио] бутилдекагидроизохинолин-3 -N-т-бутилкарбоксамида (см. , например, подготовку 8Ж или подготовку 8Ж с модификациями подготовки 8А и 8Г как в примере 75), 352 мг (3,48 ммоль), триэтиламина и 166 мг (1,23 ммоль) HOBT в 8 мл безводного THF, добавляли одной порцией 254 мг (1,23 ммоль) DCC. После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрировали, к остатку добавляли этилацетат и фильтровали через хлопковую набивку. Полученный фильтрат дважды экстрагировали насыщенным карбонатом натрия, промывали солевым раствором и высушивали над сульфатом натрия. Очистка продукта радиальной хроматографией (6 мм пластина; элюирующий раствор - 30% градиент этилацетата в гексане) дала 644 мг белой пены, которая представляет собой [3S-(3R*,4aR*,8aR*,2'S*, 3'S*)] -2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2''- метил-3'' -(бисбензоксифосфинил)оксифенил)пентил]декагидроизохинолин- 3-N-т-бутилкарбоксамид.
Выход: 67%.
1H ЯМР (CDCl3): δ 1,04 (s, 9H), 1,15-2,61 (m, 19H), 2,89- 3,00 (m, 1H), 3,39-3,50 (m, 1H), 3,67 (s, 1H), 3,75-3,85 (m, 1 H), 4,03-4,15 (m, 1H), 4,43-4,58 (m, 1H), 5,00-5,20 (m, 4H), 5,47 (s, 1H), 7,10- 7,55 (m, 19H).
ИК (CHCl3): 3600-3150 (br), 3010, 2975, 2929, 2867, 1670, 1517, 1457, 1440, 1368, 1277, 1239, 1082, 1035, 1025, 1001, 968, 879 см-1.
Масс-спектр (FAB): 828,4 (M+, 100).
Анализ для C46H58N3O7S1P1:
Расчет: C 66,73; H 7,06; N 5,07; S 3,87.
Обнаружено: C 66,56; H 7,29; N 4,82; S 3,62.
Пример 80
[3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)] -2-[2'-Гидрокси-3'-фенилтиометил-4' -аза-5'-оксо-5'-2''- метил-3''-гидроксифенил)пентил] декагидроизохинолин-3-N-т- бутилкарбоксамида
3''-однозамешенный фосфат гидрохлорид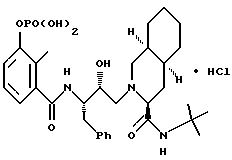
Смесь, содержащую 505 мг (0,61 ммоль) поименованного соединения примера 79, [3S-(3R*, 4aR*, 8aR*,2'S*,3'S*)] -2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(2'' -метил-3''-(бисбензоксифосфинил)оксифенил) пентил]декагидроизохинолин-3-N-т-бутилкарбоксамида и 500 мг 10% палладия на угле в 20 мл метанола перемешивали под одной атмосферой водорода в течение 24 часов. Реакционную смесь фильтровали через целит и концентрировали, получая 380 мг неочищенного продукта, который очищали с помощью ВЖХ (колонка Waters Nova pack C18 RCM (40х10 см); скорость потока 40 мл/мин; элюирующий раствор - 45% (1% HCl) вода, 15% ацетонитрил, 40% метанол), получая 230 мг белой пены, которая представляет собой 3''-однозамещенный фосфат [3S-(3R*,4aR*,8aR*, 2'S*, 3'S*)] -2-[2'-гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5' -(2''-метил-3''- гидроксифенил)пентил] декагидроизохинолин-3- N-т- бутилкарбоксамида.
Выход: 58%.
1H ЯМР (Метанол-d4): δ 1,10-2,30 (m, 25H), 2,39 (s, 3H), 2,95- 3,65 (m, 4H), 3,90-4,25 (m, 3H), 7,15-7,50 (m, 8H), 7,99 (s, 1H).
ИК (KBr): 3700-2100 (br), 1674, 1547, 1458, 1440, 1395, 1368, 1241, 1182, 1074, 1025, 966, 867 см-1.
Масс-спектр (FAB): m/e 648,3 (M++1, 100).
Анализ для C32H41N3O9 S1Cl1P1:
Расчет: C 53,37; H 7,14; N 5,83.
Обнаружено: C 53,44; H 6,76; N 5,84.
Пример 81
3-(Ацетил)гидрокси-2-метилбензойная кислота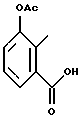
К гетерогенному раствору, содержащему 3,06 г (30 ммоль) уксусного ангидрида и 1,53 г (10 ммоль) 3-гидрокси-2-метилбензойной кислоты добавляли одну каплю концентрированной серной кислоты. Смесь нагревали при помощи heat gun в течение 2 мин и затем выливали в 14 мл холодной воды. Получившийся в результате осадок собирали с помощью вакуумного фильтрования, промывали дважды водой и высушивали в течение ночи в вакуумной печи. Перекристаллизация из 20% этилацетата/гексана (7 мл) дала 595 мг белого твердого вещества, представляющего собой 3-(ацетил) гидрокси-2-метилбензойную кислоту.
Выход: 31%.
ИК (CHCl3): 3700-2300 (br), 1765, 1698, 1460, 1404, 1372, 1299, 1273, 1172, 1081, 1041, 1012, 933, 913, 865, 823 см-1.
Масс-спектр: (FD): m/e 194 (M+, 100).
Пример 82
[3S-(3R*, 4aR*, 8aR*,2'S*,3'R*)] -2-[2'-Гидрокси-3'- фенилметил-4'-аза-5'-оксо-5'-(2''-метил-3''- (ацетил)гидроксифенил) пентил]декагидроизохинолин-3- N-т- бутилкарбоксамид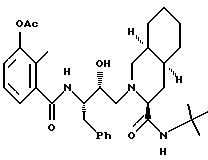
К охлажденному (-10oC) перемешиваемому раствору, содержащему 34 мг (0,174 ммоль) поименованного соединения примера 81, 3-(ацетил)гидрокси-2-метилбензойной кислоты, 70 мг (0,174 ммоль) [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[3'-амино-2'-гидрокси-4'-фенил] бутилдекагидроизохинолин-3-N-т- бутилкарбоксамида и 24 мг (0,174 ммоль) HOBT в 3 мл безводного THF добавляли одной порцией 36 мг (0,174 ммоль) DCC. После перемешивания в течение 2 дней при комнатной температуре, реакционную смесь разбавляли этилацетатом и фильтровали через хлопковую набивку. Полученный фильтрат экстрагировали один раз насыщенным карбонатом натрия, один раз солевым раствором и высушивали над сульфатом натрия. Очистка продукта с помощью радиальной хроматографии (1 мм пластина; элюирующий раствор - 0-5% градиент метанол/метиленхлорид) дала 65 мг белой пены, представляющей собой [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилиетил-4'-аза5'-оксо-5'-(2''-метил-3'' -(ацетил)гидроксифенил)пентил]декагидроизохинолин-3-N-т-бутилкарбоксамид.
Выход: 65%.
1H ЯМР (CDCl3): δ 1,15 (s, 9H) 1,16-2,37 (m, 21H) 2,50-2,70 (m, 2H), 2,93-3,05 (m, 2H), 3,39-3,50 (m, 1H), 3,99-4,10 (s, 1H), 4,53-4,64 (m, 1H), 3,99-4,10 (m, 1H), 4,53- 4,54 (m, 1H), 5,69 (s, 1H), 6,64 (d, J=8,45 Гц, 1H), 6,91 (d, J=7,47 Гц, 1H), 7,00 (d, J=7,57 Гц, 1H), 7,11 (t, J=7,75 Гц, 1H), 7,19-7,40 (m, 5H).
ИК (CHCl3): 3700-3100 (br), 3008, 2929, 2865, 1762, 1671, 1604, 1514, 1455, 1394, 1368, 1303, 1277, 1175, 1121, 1082, 1047, 910 см-1.
Масс-спектр: (FD): m/e 578 (M+, 100).
Пример 83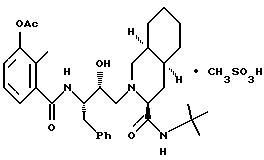
К холодному раствору (0oC), содержащему 35 мг (0,061 ммоль) поименованного соединения примера 82, [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2'' -метил-3''-(ацетил)гидроксифенил)пентил]-декагидроизохинолин- 3-N-т-бутилкарбоксамида в 2 мл безводного метиленхлорида, добавляли по каплям 128 микролитров (0,064 ммоль) 0,5 М раствора метансульфоновой кислоты в метиленхлориде. Образующуюся реакционную смесь упаривали досуха при пониженном давлении (0,2-0,1 Torr), получая 40,5 мг светло-желтой (неочищенной) пены, представляющей собой соль метансульфоновой кислоты [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5' -оксо-5'-(2''-метил-3''-(ацетил)гидроксифенил) пентил]декагидроизохинолин-3-N-т-бутилкарбоксамида.
Выход: 98%.
Пример 84
N-Вос-4-тио-L-пролин (может быть получен из фирмы Sigma) (1,5 г) растворяли в 3 мл метанола и охлаждали до 0oC в ледяной бане. В отдельной колбе растворяли 5,8 г "OXONE" в 5 мл H2O и полученный раствор добавляли по каплям к реакционной смеси. По истечении 30 минут реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи, после чего разбавляли CHCl3/H2O, разделяли фазы и экстрагировали с помощью CHCl3 (3 X 100 мл). Органические слои объединяли, высушивали над Na2SO4 и концентрировали в вакууме, получая соединение приведенной выше формулы (700 мг, выход 41%) в виде белого твердого вещества.
Пример 85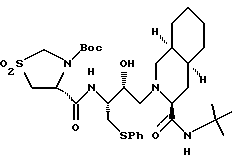
Соединение формулы, приведенной в примере 84, и [3S-(3R*,4aR*,8aR*,2'S*, 3'S*)] -2-[3'-амино-2'-гидрокси-4'- (фенил)тио]бутилдекагидроизохинолин-3-N-т-бутилкарбоксамид сшивали в соответствии с методикой, описанной выше в примере 79. Неочищенный материал очищали флэш-хроматографией (3% MeOH/CH2Cl2), получая 40 мг (выход 51%) соединения приведенной выше формулы.
Пример 86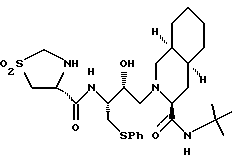
Соединение формулы, приведенной в примере 85 (20 мг), растворяли в 1 мл CH2Cl2 и обрабатывали 1 мл трифторуксусной кислоты. По истечении 30 минут при комнатной температуре продукт реакции упаривали в вакууме, получая соединение приведенной выше формулы, представляющее собой [3S-(3R*,4aR*,8aR*, 2'S*, 3'S*, 4''S)] - 2-[2'-гидрокси-3'-фенилтиометил-4' -аза-5'-оксо-5'-(тиазолино-4''- ил-1'', 1''-диоксид)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамид.
Pandex IC50 = 244 нг/мл.
Пример 87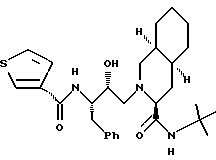
3-Карбоновокислый тиофен (может быть получен из фирмы Aldrich) и [3S-(3R*, 4aR*, 8aR*, 2'S*, 3'R*)] -2-[3'-амино- 2'-гидрокси-4'-фенил]бутилдекагидроизохинолин-3-N-т- бутилкарбоксамид сшивали в соответствии с методикой, описанной выше в примере 77. Неочищенный материал очищали флэш-хроматографией (2% MeOH/CH2Cl2), получая 70 мг (выход 63%) соединения, приведенной выше формулы, представляющего собой [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(тиено-3''-ил) пентил]декагидроизохинолин-3 -N-т-бутилкарбоксамид.
Pandex IC50 = 25% при 1,000 нг/мл.
Пример 88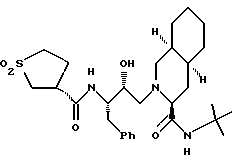
3-Карбоновокислый тетрагидротиофен-1,1-диоксид и [3S-(3R*, 4aR*,8aR*, 2'S*, 3'R*)] -2-[3'-амино-2'-гидрокси-4'- фенил]бутилдекагидроизохинолин-3-N-т-бутилкарбоксамид сшивали в соответствии с методикой, описанной выше в примере 77. Неочищенный материал очищали флэш-хроматографией (3% MeOH/CH2Cl2), получая 50 мг (42% выход) соединения, приведенной выше формулы [3S-(3R*, 4aR*, 8aR*, 2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4' -аза-5'-оксо-5'-(тетрагидротиено-3''-ил-1'', 1'' -диоксид)пентил] декагидроизохинолин-3-N-т-бутилкарбоксамида, в виде смеси диастереомеров.
Pandex IC50 = 28% при 20 нг/мл.
Пример 89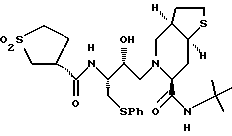
3-Карбоновокислый тетрагидротиофен-1,1-диоксид и [6S-(6R*, 3aS*,7aR*, 2'S*, 3'S*)] -5-[2-гидрокси-4-фенилтио-3- (бензоксикарбонил)аминобутил] октагидротиено[3,2-c] пиридин-6-N -т-бутилкарбоксамид сшивали в соответствии с методикой, описанной выше в примерах 74 Ж и З. Неочищенный материал очищали флэш-хроматографией (3-4% MeOH/CH2Cl2), получая 30 мг (выход 57%) [6S-(6R*, 3aS*, 7aR*,2'S*,3'S*)] -2-[2'-гидрокси-3'- фенилтиометил-4'-аза-5'-оксо-5'-(тетрагидротиено-3'' -ил-1'', 1''-диоксид)пентил]октагидротиено[3,2-c]пиридин- 6-N-т-бутилкарбоксамида в виде смеси диастереоизимеров.
CEM IC95 = 98 нм.
Pandex IC50 = 50 нг/мл (0,9).
Пример 90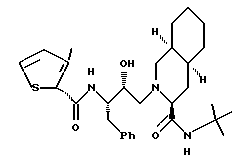
3-Метил-2-карбоновокислый тиофен и [3S-(3R*,4aR*,8aR*,2'S*,3'S*)] -2-[3'-амино-2'-гидрокси-4'-(фенил)тио] бутилдекагидpoизoxинoлин-3-N-т- бутилкapбoкcaмид сшивали в соответствии с методикой, описанной выше в примере 79, получая 39 мг (выход 76 %) соединения вышеуказанной формулы, представляющего собой [3S-(3R*, 4aR*,8aR*,2'S*,3'S*)] -2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо-5'-(3'' -метил-тиено-2''-ил)пентил] -декагидроизохинолин-3-N-т-бутилкарбоксамид.
Пример 91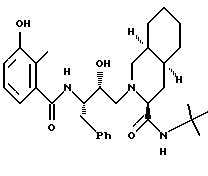
[6S-(6R*, 3aS*, 7aR*, 2'S*,3'R*)] -2-[2'-гидрокси-3'-фенилметил-4'-аза-5'-оксо-5'-(2'' -метил-3'-гидроксифенил)пентил] -октагидротиено[3,2-c] пиридин-6-N-т-бутилкарбоксамид (см., например, пример 74 М) (30,5 мг) растворяли в 2 мл MeOH. В другой колбе "OXONE" (51 мг) растворяли в 1 мл воды и приготовленный раствор добавляли к содержимому первой колбы. После 6 часов перемешивания добавляли вторую порцию "OXONE" (17 мг) и реакционную смесь перемешивали в течение 42 часов. Реакционную смесь разбавляли CH2Cl2 и промывали водой. Органический слой высушивали над Na2SO2, фильтровали и концентрировали. Неочищенный остаток очищали радиальной хроматографией (1000 микронная пластина; 3-9% MeOH/CH2Cl2), получая 5 мг [6S-(6R*,3aS*,7aR*,2'S*, 3'R*)] -2-[2'-гидрокси-3'-фенилтиометил-4'-аза-5'-оксо- 5'-(2'' -метил-3''- гидроксифенил)пентил] - октагидротиено[3,2-с]пиридин-1,1-диоксид-6- N-т-бутилкарбоксамида.
Пример 92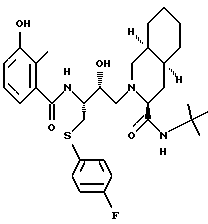
Соединение, приведенное выше, [3S-(3R*,4aR*,8aR*,2'S*,3'R*)] -2-[2'-гидрокси-3'- (4'''-фтор)-фенилтиометил-4'- аза-5'-оксо-5'-(2'' -метил-3''-гидроксифенил)пентил] декагидроизохинолин-3- N-т-бутилкарбоксамид, получали, используя способы, аналогичные приведенным в примере 23, за исключением того, что тиофенол был заменен на 4-фтортиофенол в Подготовке 8А.
Полученный продукт использовали аналогично использованию продукта Подготовки 8А, приведенному в примере 23.
Пример 93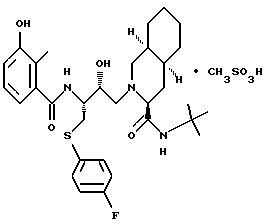
Приведенное выше соединение, соль метансульфоновой кислоты [3S-(3R*, 4aR*, 8aR*, 2'S*, 3'R*)] -2-[2'-гидрокси-3'-(4'''- фтор)-фенилтиометил-4'-аза-5'- оксо-5'-(2''-метил-3'' -гидроксифенил)пентил] декагидроизохинолин-3-N-т- бутикарбоксамида, получали методом, аналогичным приведенному выше в примере 75 (шаг 2).
Как отмечалось выше, соединения по настоящему изобретению полезны в качестве ингибиторов протеазы ВИЧ, фермента, связанного с образованием и сборкой вирусных компонентов. Существенной составляющей настоящего изобретения является метод лечения ВИЧ-инфекции, заключающийся во введении носителю вируса или пациенту, таким как приматы, эффективного количества соединения формулы (1) или его фармацевтически приемлемой соли. Другая существенная составляющая настоящего изобретения - способ лечения СПИДа, заключающийся во введении носителю вируса или пациенту эффективного количества соединения формулы (1) или его фармацевтически приемлемой соли. Следующая существенная составляющая настоящего изобретения - способ ингибирования протеазы ВИЧ, заключающийся во введении ВИЧ-инфицированным клеткам или носителю вируса либо пациенту, таким как приматы, инфицированному вирусом ВИЧ, эффективного количества соединения формулы (1) или его фармацевтически приемлемой соли.
Термин "эффективное количество" означает то количество соединения формулы (1) или его фармацевтически приемлемой соли, которое является эффективным для ингибирования опосредованного протеазой ВИЧ образования и сборки вирусных компонентов. Конкретная доза соединения, вводимого согласно настоящему изобретению для получения терапевтического или ингибиторного воздействий, будет, конечно, определяться специфическими обстоятельствами, сопутствующими заболеванию, включая, например, конкретное вводимое соединение, способ введения, состояние лечащегося больного и индивидуальные особенности лечащегося вирусоносителя или пациента. Примерная ежедневная доза (вводимая однократно или отдельными дозами) составляет приблизительно от 0,01 мг до 50 мг соединения по изобретению на кг веса тела. Предпочтительные ежедневные дозы обычно составляют примерно от 0,05 мг/кг до 20 мг/кг и более предпочтительные - примерно от 0,1 мг/кг до 10 мг/кг.
Соединения по изобретению могут вводиться множеством способов, включая оральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный способы и через носовое отверстие. Соединения по изобретению предпочтительно вводятся в состав лекарственного средства перед их употреблением. Следовательно, другой существенной составляющей настоящего изобретения является фармацевтический состав или пропись лекарственного средства, содержащего эффективное количество соединения формулы (1) или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, такой как разбавитель или эксипиент.
Содержание активного ингредиента предпочтительно составляет от 0,1% до 99,9% веса лекарственного средства. Под термином "фармацевтически приемлемый" подразумевается, что носитель, такой как разбавитель или эксипиент, является совместимым с другими ингредиентами лекарственного средства и не является вредным для вирусоносителя или пациента.
Фармацевтические лекарственные средства могут быть приготовлены из соединений, предусмотренных изобретением, при помощи известных методик, использующих легко доступные ингредиенты. В процессе приготовления составов по настоящему изобретению активный ингредиент обычно смешивают с носителем, или разбавляют носителем, или включают внутрь носителя, который может быть выполнен в форме капсулы, облатки, бумаги или другого подходящего контейнера. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует как наполнитель, эксипиент или среда для активного компонента. Таким образом составы могут быть изготовлены в форме таблеток, пилюль, порошков, лепешек, облаток, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как твердое вещество или жидкая сера), мазей (содержащих, например до 10% активного соединения по весу), мягких или твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов, стерильно упакованных порошков и т. п.
Следующие примеры описей приведены только для иллюстрации и не ограничивают область применения изобретения. Термин "активный ингредиент" означает соединение, представленное формулой (1), или фармацевтически приемлемую соль этого соединения.
Пропись 1
Приготовление твердых желатиновых капсул с использованием следующих ингредиентов, мг/капсула:
Активный ингредиент - 250
Крахмал высушенный - 200
Стеарат магния - 10
Итого, мг - 460
Пропись 2
Приготовление таблетки с использованием приведенных ниже ингредиентов, мг/таблетка:
Активный ингредиент - 250
Целлюлоза микрокристаллическая - 400
Белая сажа - 10
Стеариновая кислота - 5
Итого, мг - 665
Компоненты смешивают и прессуют до образования таблеток весом 665 мг каждая.
Пропись 3
Приготовление раствора аэрозоля с использованием следующих ингредиентов, вес.%:
Активный ингредиент - 0,25
Этанол - 25,75
Пропеллант 22
(хлордифторметан) - 74,00
Итого - 100,00
Активное соединение смешивают с этанолом, смесь добавляют к порции пропелланта 22, охлаждают до -30oC и переносят в заполняющее устройство. Затем требуемым количеством смеси заполняют контейнер из нержавеющей стали и разбавляют остатком пропелланта. Далее в контейнер устанавливают элементы клапана.
Пропись 4
Таблетки, содержащие 60 мг активного ингредиента каждая, готовят следующим образом:
Состав - мг/таблетка
Активный ингредиент - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон
(в виде 10% водного раствора) - 4
Натриевая соль карбоксиметилкрахмала - 4,5
Стеарат магния - 0,5
Тальк - 1
Итого - 150
Активный ингредиент, крахмал и целлюлозу пропускают через сито N 45 меш U. S. и тщательно перемешивают. Водный раствор, содержащий поливинилпирролидон, смешивают с образующимся порошком и смесь пропускают через сито N 14 меш U.S. Полученные таким образом гранулы сушат при 50oC и пропускают через сито N 18 меш U.S. Натриевую соль карбоксиметилкрахмала, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш U.S., добавляют к гранулам, которые после смешивания прессуют на машине для приготовления таблеток, получая таблетки весом 150 мг каждая.
Пропись 5
Капсулы, содержащие 80 мг активного ингредиента каждая, готовят следующим образом:
Состав - мг/капсула
Активный ингредиент - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Итого - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито N 45 меш U.S. и этой смесью в количестве 200 мг заполняют твердые желатиновые капсулы.
Пропись 6
Суппозитории, содержащие по 225 мг активного ингредиента, готовят следующим образом:
Состав - количество мг
Активный ингредиент - 225
Глицериды насыщенных жирных кислот - 2000
Итого - 2225
Активный ингредиент пропускают через сито N 60 меш U.S. и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных с использованием минимально необходимого количества теплоты. Затем смесь заливают в суппозиторийную форму номинальной вместимостью 2 г и оставляют для охлаждения.
Пропись 7
Суспензии, содержащие по 50 мг активного ингредиента на 5 мл дозы каждая, готовят следующим образом:
Активный ингредиент, мг - 50
Натриевая соль карбоксиметилцеллюлозы, мг - 50
Сироп, мл - 1,25
Раствор бензойной кислоты, мл - 0,10
Ароматизатор - q.v.
Краситель - q.v.
Очищенная вода до общего объема, мл - 5
Активный ингредиент пропускают через сито N 45 меш U.S. и смешивают с натриевой солью карбоксиметилцеллюлозы и сиропом до образования однородной пасты. Раствор бензойной кислоты, ароматизатор и краситель разбавляют частью воды и добавляют при перемешивании. Затем добавляют оставшуюся воду до получения требуемого объема.
Пропись 8.
Лекарственные препараты для внутривенного введения готовят следующим образом:
Активный ингредиент, мг - 100
Изотонический солевой раствор, мл - 1000
Раствор вышеупомянутых ингредиентов обычно вводят субъекту внутривенно со скоростью 1 мл в минуту.
Скрининг активности
Для исследования биологической активности соединений, ингибирующих протеазу ВИЧ, использовали ряд тестов. Например, использовали тесты для анализа скоростей протеолитического ингибирования и антивирусного действия на ВИЧ-инфицированные клеточные линии. Методики этих экспериментов описаны ниже. Результаты анализов просуммированы в таблице 1, представленной ниже, или приведены в указанных выше примерах.
I. Первичный скрининг препаратов антиВИЧ-соединений, выполненный в Южном Научно-исследовательском институте (SRI) (результаты, внесенные в таблицу 1, обозначены "SRI CEM (нг/мл)" или "SRI МТ2 (нг/мл)")
А. Принцип МТТ-анализа:
SRI обладает учрежденной программой для первичного антивирусного анализа соединений посредством микротитр-анализов, которые измеряют способность выбранного соединения ингибировать гибель ВИЧ-индуцированных клеток. Этот анализ заключается в превращении тетразолиевого красителя МТТ в окрашенный формазановый продукт под действием митохондриальных ферментов в метаболически активных клетках. Эта аналитическая система используется в SRI для проверки свыше 30000 соединений в год. Вкратце данный анализ заключается в инфицировании CEM или МТ2 клеток в круглодонных 96-луночных планшетах. Интересующее соединение добавляют непосредственно перед инфицированием. После 6 дней инкубации при 37oC планшеты окрашивают МТТ. Результаты анализа определяют количественно спектрофотометрически на аппарате для чтения планшетов (Molecular Devices Vmax). Данные анализируют методом линейной регрессии, используя собственную программу для вычисления антивирусной активности (IC25, IC50, IC95) и токсичности (TC25, TC50, TC95), а также других величин.
Первичные антивирусные анализы обычно выполняют на CEM или МТ2-клетках. В SRI было обнаружено, что все активные соединения проявляются на CEM клетках, в то время как в экспериментах, выполненных на МТ2-клеточной линии, небольшая часть активных соединений не проявляет своего действия.
Б. Стандартные скрининг-анализы на CEM и MT2-клетках
1. Разведение соединений и их нанесение на планшеты
Лекарственные средства растворяют в подходящем растворителе, таком как дистиллированная вода или в случае необходимости ДМСО. Латексные перчатки, лабораторную одежду и маски используют в продолжение всех стадий выполнения процесса для предотвращения контакта с потенциально вредными агентами. Лекарственный препарат готовят в необходимой концентрации и хранят при -20oC до использования в скрининг-лаборатории. Первое разведение каждого соединения делают в пробирке для разведения со средой таким образом, чтобы получилась концентрация, в два раза превышающая самую высокую тестируемую концентрацию. Затем используют стерильные пробирки для титрования, производя последовательные полулогарифмические разведения каждого соединения. После этого разведенное соединение добавляют в соответствующую лунку 96-луночного планшета для микротитрования. На одном планшете можно удобно проанализировать до 12 разведений в трех параллелях со всеми необходимыми контролями, включая контроли на клетку, вирус, токсичность, цвет препарата, контроль на среду и контроль на пластмассу планшета (фон). Если проводят анализ только шести разведений, то на одном планшете для титрования можно проанализировать два препарата. Препараты наносят на планшет в конечном объеме 100 мкл.
2. Клетки и вирус
Пока готовят разведения препарата, клетки промывают и подсчитывают. Жизнеспособность контролируют методом исключения красителя трипана синего и испытания не проводят, если жизнеспособность падает ниже 90%. Клетки поддерживают в экспоненциальной фазе роста и разделяют 1:2 в день, предшествующий проведению анализа для обеспечения экспоненциальной скорости роста.
Для первичного скрининга используют клеточные линии CEM и MT2. Если специально не указано, то используют среду RPMI 1640 с 10% раствором термоинактивированной фетальной сыворотки теленка (FBS), глутамином и антибиотиками.
Клетки выращивают при 37oC в атмосфере 5% CO2 в воздухе. Вирус, использованный в данной работе, представляет собой ВИЧ-1 изоляты IIIВ и/или RF, полученные с помощью острого инфекционного процесса.
Вкратце, инфицированные вирусом клетки ежедневно собирают центрифугированием, начиная с третьего дня после инфекции, до тех пор, пока вирус не уничтожит все клетки в культуре. Для идентификации пулов с самым большим количеством вируса используют измерение активности обратной транскриптазы и метод р24 ELISA.
Эти 24-часовые сборы клеток объединяют, фильтруют и замораживают при -90oC. Инфекционный пул вируса оттитровывают на всех доступных клеточных линиях до его использования в анализе с целью определения количества вируса, требуемого для антивирусного анализа.
Обычно пулы, полученные методом острой инфекции, требуют добавления одного микролитра инфекционного вируса на лунку, в результате чего при скрининге препаратов множественность инфекции составляет 0,01. Этим способом готовят и замораживают вирус в достаточном количестве для заполнения свыше одной тысячи планшетов для титрования, позволяющих тестировать до двух тысяч соединений из одной линии инфекционного вируса. Использование одной линии вируса в течение продолжительного периода тестирования оказывает очень благоприятное влияние на воспроизводимость результатов измерительных систем.
Вирусная инфекция CEM и МТ2-клеток при проведении антивирусного анализа осуществляется в процессе инфицирования в объеме. Необходимое число клеток, требуемое для осуществления теста, смешивают с инфекционным вирусом в конической центрифужной пробирке в небольшом конечном объеме, равном 1-2 мкл.
После 4-часового инкубирования инфицированные клетки доводят до нужной конечной концентрации 5•104 клеток на миллилитр с помощью свежей питательной среды для культуры тканей и 100 мкл добавляют в лунки, соответствующие эксперименту и контролю на вирус. Неинфицированные клетки в той же самой концентрации помещают на планшеты для проведения контролей на токсичность и контролей на клетки. Можно также выполнять анализы, используя метод инфекции в лунках. В этом случае препарат, клетки и вирус добавляют в лунку по отдельности. В каждом случае MOI подбирают так, чтобы получить полное уничтожение клеток в лунках контроля на вирус на шестой день.
3. Оценка CPE-ингибирования
После добавления клеток и лекарственных препаратов в планшет для микротитрования последний инкубируют в течение 6 дней при 37oC. Опыт показал, что инкубация в течение более длительных периодов времени (7-8 дней) или использование более высоких начальных количеств клеток (1•104) приводит к значительному уменьшению жизнеспособности в клеточном контроле и к сужению различий в оптической плотности между клеточным и вирусным контролями при окрашивании с помощью МТТ.
Метод оценки антивирусного анализа включает добавление 20 микролитров тетразолиевой соли МТТ в концентрации 5 мг/мл в каждую лунку планшета на 4-8 часов. После этого инкубационного периода клетки разрушают добавлением 50 микролитров 20% SDS в 0,01н. HCl.
Наличие метаболической активности у жизнеспособных клеток в культуре приводит к появлению окрашенного продукта реакции, который измеряют спектрофотометрически в аппарате для чтения планшетов (Molecular Devices Vmax) при 570 нм. Значение оптической плотности (O.D.) является функцией количества формазанового продукта, которое пропорционально числу жизнеспособных клеток.
Аппарат для чтения планшетов подключен к установленному в скрининг-лаборатории микрокомпьютеру, который оценивает и подсчитывает данные, полученные с планшетов. Отчет о полученных на планшетах результатах представляет краткое изложение всей имеющей отношение к делу информации, включая необработанные значения O.D., расчетные значения O.D. и процентное уменьшение в вирусном CPE, а также вычисления, включающие TC50, IC50, антивирусный индекс и индекс специфичности. Наконец, результаты включают график, который визуально отображает влияние соединения на неинфицированные клетки (токсичность) и эффект защиты или незащиты соединением инфицированных клеток.
II. Скрининг на целых клетках антиВИЧ-соединений в Eli Lilly (результаты, внесенные в таблицу 1, обозначены "Целая клетка IC50 нМ" или "Целая клетка IC90 нМ")
А. Цель и материалы
Цель: определить IC50 и CC50 для соединений:
Реактивы и материалы
Среда А
Среда А [1% ДМСО] (100 мкл ДМСО + 9,9 мл среды А)
SN 123 используют для инфицирования клеток (15 мл на 6 планшетов) (10 мл на 4 планшета)
CEM-клетки @ [1•104] клеток/мл (4 планшета = 40 мл) (6 планшетов = 60 мл)
ДМСО (необходимо 5 мл)
35В в [10 мМ] (необходимо 70 мкл каждого соединения)
A-D в [10 мМ] в 100% ДМСО
4 или 6 96-луночных планшета с u-образным дном
4 96-луночных планшета с плоским дном для разведений
8-10 коробок стерильных costar наконечников
Приблизительно 10 подносов для реактивов
Costar 12-канальная пипетка
Вспомогательная информация:
1000 клеток/лунка = 1•104 клеток/мл = 1000 мкл
200 мкл = общий объем в лунке
Конечная концентрация ДМСО = 0,25%
Конечное разведение Sn123 = 1:64
Последовательно разведенные соединения 35В, A-D, 1:3
Б. Методика
1. Подготовка клеток и нанесение их на планшеты, среда А и среда А (1% ДМСО)
а. Пронумеруйте 96-луночный планшет для культуры ткани на каждое тестируемое соединение, один планшет для контроля и один для контрольного соединения.
Планшет # - Описание
1 - Контроль отриц. и полож.
2 - 35В
3 - A
4 - B
5 - C
6 - D
б. Подсчитайте клетки на гемоцитометре и ресуспендируйте их в 40 мл или 80 мл Среды А в концентрации [1•104] клеток/мл.
Подсчет клеток на гемоцитометре:
Пометьте две nunc пробирки вместимостью 1,8 мл метками 1 и 2.
Поместите 0,5 мл хорошо перемешанных CEM-клеток (в фазе роста) в пробирку 1.
Поместите 50 мкл PBS и 40 мкл трипана синего в пробирку 2.
Хорошо перемешайте клетки в пробирке 1, затем отберите 10 мкл клеток и поместите их в пробирку 2.
Перемешайте хорошо содержимое пробирки 2, затем отберите 10 мкл окрашенных клеток и поместите их в гемоцитометр.
Подсчитайте число клеток в центральном квадрате гемоцитометра при помощи микроскопа при увеличении 10Х.
Концентрация исходной суспензии CEM, выраженная в клетках/мл определяется следующим образом:
Количество подсчитанных клеток • 1•105= Концентрация CEM в [клетках/мл].
в. Добавьте 200 мкл среды А в:
А1 планшетов 2-6.
Это - бланки.
А4-H4 планшета 1.
Это - бланки.
г. Добавьте 5 мкл среды А во все лунки рядов A-D в планшетах 2-6 за исключением лунки А1 (верхняя половина каждого планшета).
д. Добавьте 50 мкл среды А в лунки A1-D3 планшета 1 (верхняя половина планшета).
е. Добавьте 50 мкл среды А [1% ДМСО] во все лунки колонок 1-3 планшета 1.
ж. Добавьте 100 мкл [1•104] клеток/мл во все лунки колонок 1-3 планшета 1 и во все лунки (за исключением А1, которая является бланком) других планшетов. Это вносит 1000 клеток на лунку.
з. Поместите планшеты в инкубатор на время подготовки разведений лекарственного препарата.
2. Подготовка контрольного и тестируемого лекарственных препаратов
(а) Подготовка последовательных разведений (35В, A-D) 1:3 в планшете с 100% ДМСО.
(1) Поместите 60 мкл ДМСО во все лунки колонок 2-12, рядов A-E.
(2) Поместите 70 мкл 35B [10 мМ] в 100% ДМСО в лунку A1.
(3) Поместите 70 мкл A [10 мМ] в 100% ДМСО в лунку B1.
(4) Поместите 70 мкл B [10 мМ] в 100% ДМСО в лунку C1.
(5) Поместите 70 мкл C [10 мМ] в 100% ДМСО в лунку D1.
(6) Поместите 70 мкл D [10 мМ] в 100% ДМСО в лунку E1.
(7) Последовательно разведите (35B, A-D) 1:3 до колонки 12, перенося 30 мкл из колонки 1 в колонку 2, затем из колонки 2 в колонку 3 и т.д. до колонки 12. Меняйте наконечники перед каждым разведением.
(б) Подготовка планшета для разведения 1:10 в среде A:
(1) В рядах A-E другого планшета сделайте ряд для первого разведения 1: 10 таким образом, чтобы он соответствовал ряду, содержащему 100% ДМСО для каждого соединения.
35B в ряд A для первого разведения 1:10.
A в ряд B для первого разведения 1:10.
B в ряд C для первого разведения 1:10.
C в ряд D для первого разведения 1:10.
D в ряд E для первого разведения 1:10.
(2) Поместите 180 мкл среды A во все лунки рядов A-E, соответствующие рядам, содержащим 100% ДМСО. Для подготовки каждого ряда необходимо 2,5 мл среды.
(3) Отберите по 20 мкл из всех лунок каждого ряда в рядах, содержащих 100% ДМСО, и перенесите их в соответствующие ряды для разведения 1:10.
(в) Подготовка планшета для разведения 1:100 в Среде A:
(1) Подготовьте планшет для каждых 3 исследуемых соединений.
(2) Поместите 225 мкл среды A во все лунки рядов A, B, D, E, G и H, оставляя ряды C и F пустыми. Используйте 20 мл среды A на планшет.
(3) Перенесите 25 мкл каждого соединения из ряда для разведения 1:10 в соответствующие два ряда на планшете для разведения 1:100, меняя наконечники перед каждым перенесением (см. таблицу 4).
3. Добавление вируса Sn123 в планшеты
а. Разморозьте Sn123 в водяной бане при 37oC приблизительно за 10 минут,
б. Разведите Sn123 в соотношении 1:16, добавляя 1 мл Sn123 к 15 мл среды A.
в. Добавьте 50 мкл Sn123 [1:16] в лунки E1-H12 на планшетах 2-6 и в лунки E1-H3 на планшете 1.
4. Добавление лекарственных препаратов в планшеты
а. Добавьте 50 мкл контрольного и тестируемых лекарственных препаратов из рядов планшетов с разведением 1:100 в соответствующие ряды в конечном планшете (меняя наконечники перед каждым перенесением). Из одного ряда планшета 1:100 будет получаться 4 ряда на конечном планшете. Оставьте лунку A1 пустой.
б. Инкубируйте все планшеты 7 дней при 37oC в 5% CO2.
в. Выполните Xtt-протокол на 7 день следующим образом.
г. Подготовка Xt-t/PMS раствора:
(на 4 планшета = 20 мл)
(на 6 планшетов = 30 мл)
(1) Рецепт для 2 мМ PMS:
15,3 мг PMS + 0,5 мл PBS = PMS в конц. [100 мM]
100 мкл [100 мM] PMS +4,9 мл PBS = PMS] в концентрации [2 мМ]
(2) Прогрейте 500 мл H2O в микроволновой печи в течение 5 минут.
(3) Поместите 20 или 30 мл фенолового красного RPMI в центрифужную пробирку, вместимостью 50 мл.
(4) Поместите RPMI в мензурку с горячей водой.
(5) Добавьте 20 или 30 мг XTT к нагретому RPMI. Конечная концентрация XTT = [1 мг/мл].
(6) Подождите пока растворится XTT, затем добавьте 200 мкл [2 мМ] PMS на 10 мл раствора XTT.
д. Добавление XTT/PMS в планшет:
(1) Добавьте 50 мкл раствора XTT/PMS во все лунки всех планшетов.
(2) Закройте планшеты и инкубируйте их 4 часа при 37oC в 5% CO2.
(3) Удалите планшеты из инкубатора и замените крышки на пластмассовые защитные покрытия для планшетов.
(4) Перемешайте содержимое планшетов.
(5) Прочитайте планшеты при тестируемой длине волны 450 нМ и эталонной длине волны 650 нМ.
III. Флюоресцентный анализ на ингибирование протеазы ВИЧ-1 для проверки ингибирования протеазы ВИЧ (результаты, внесенные в таблицу 1, обозначены "Pandex (нг/мл)'')
Использованные здесь сокращения определены следующим образом:
BSA - бычий сывороточный альбумин
BOC - т-бутоксикарбонил
BrZ - 2-бромбензилоксикарбонил
2-ClZ - 2-хлорбензилоксикарбонил
DCC - дициклогексилкарбодиимид
DIEA - диизопропилэтиламин
DTT - дитиотреитол
EDTA - этилендиаминтетрауксусная кислота
FITC - флюоресцеин изотиокарбамила
HEPES - 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота
MES - 4-морфолинэтансульфоновая кислота
РАМ - фенилацетимидометил
TAPS - 3-[трис(гидроксиметил)метил]амино-1-сульфоновая кислота
TRIS - трис(гидроксиметил)аминометан
TOS - п-толуолсульфонил (тозил)
A. Подготовка протеазной и GAG-фракций
1. Культура E.coli К12 L507/pHP10D
Лиофилизованные культуры штамма E.coli К12 L507/pHP10D были получены из Северной Региональной Научно-исследовательской лаборатории, Peoria, Штат Иллинойс 61604 под каталожным номером NRRL В-18560 (штамм депонирован 14 ноября 1989 г.). Лиофилизованные бактериальные клетки декантировали в пробирки, содержащие 10 мл LB-среды (10 г бактотриптона, 5 г бактодрожжевого экстракта и 10 г водного хлористого натрия на литр), подводили pH до 7,5 и инкубировали при 32oC в течение ночи.
Небольшую часть ночной культуры высевали на чашки с LB-агаром (LB-среда с 15 г/л бактоагара), содержащие 12,5 микрограмм/мл тетрациклина с целью получения отдельных изолированных колоний штамма E.coli К12 L507/pHP10D. Полученную отдельную колонию инокулировали в 10 мл LB-среды, содержащей 12,5 микрограмм/мл тетрациклина, и инкубировали в течение ночи при 32oC с энергичным встряхиванием. 10 мл ночной культуры инокулировали в LB-среду, содержащую 12,5 микрограмм/мл тетрациклина, и инкубировали при 32oC с энергичным встряхиванием до достижения середины логарифмической фазы роста.
2. Культура E.coli К12 L507/pHGAG
Лиофилизованные культуры штамма E.coli К12 L507/pHGAG были получены из NRRL под каталожным номером NRRL В- 18561 (штамм депонирован 14 ноября 1989 г.). Очищенную колонию E.coli К12 L507/pHGAG выделяли и использовали как посевной материал для культуры, которую выращивали до середины логарифмической фазы роста в полном соответствии с приведенной выше в разделе А методикой для штамма E.coli К12 L507/pHP10D.
3. Подготовка протеазной фракции
Культуру штамма E.coli К12 L507/pHP10D выращивали до середины логарифмической фазы при 32oC в LB-среде, содержащей 12,5 микрограмм/мл тетрациклина. Температуру культивирования быстро поднимали до 40oC с целью индуцирования экспрессии генов и клетки оставляли расти при этой температуре в течение 2,5 часов, после чего культуру быстро охлаждали во льду. Клетки осаждали центрифугированием и клеточный осадок ресуспендировали в 20 мл 50 ммоль MES-буфера (pH 6,0), содержащего 1 ммоль EDTA, 1 ммоль DTT, 1 ммоль PMSF и 10% глицерин ("Буфер A"). Клетки лизировали ультразвуком, используя клеточный дезинтегратор (Модель 300, Fischer) и зонд с микронаконечником. После центрифугирования при 27000xg надосадочную жидкость разбавляли Буфером A до конечного объема 60 мл и наносили на колонку с QAE-сефарозой размером 2,0х19 см (1 мл/мин, 4oC), которую уравновешивали Буфером A. Колонку промывали изократно в течение 180 мин и затем элюировали градиентом 0-1,0 M водного раствора хлорида натрия в Буфере A в течение 120 мин. Ферментативную активность измеряли с помощью ВЖХ, используя синтетический пептид Ser-Gln-Asn-Tyr-Pro-Ile-Val, как описано в [13]; измеряли образование pl-пептида (Ser-Gln-Asn-Tyr).
Активные фракции объединяли, подводили к pH...1,2 М... в сульфате аммония и наносили колонку размером 2,0х18 см с гексилагарозой, уравновешенную Буфером A, содержащим 1,2 M сульфат аммония. Образец наносили при скорости протекания 1 мл/мин и 4oC, промывали уравновешивающим буфером в течение 240 мин (1 мл/мин) и затем элюировали, используя обратный линейный градиент сульфата аммония 1,2 - 0 М в Буфере A в течение 120 мин при той же самой скорости потока. Колонку затем промывали изократно Буфером A в течение 120 мин.
Активные фракции объединяли, концентрировали до 10 мл, используя ячейку с перемешиванием (Amicon) и мембрану YM-10, и затем наносили на MonoS-катионообменную колонку (1,0х10 см), уравновешенную Буфером A. Образец наносили при скорости потока 1 мл/мин и 25oC. После промывки изократно в течение 30 мин протеазу элюировали в течение 40 мин, используя линейный градиент 0 - 0,45 M водного раствора хлорида натрия в Буфере A. Колонку промывали изократно Буфером A, содержащим 0,45 М водный раствор хлорида натрия, в течение 30 минут.
Активные фракции объединяли и концентрировали до 200 микролитров, используя ячейку с перемешиванием (Amicon) и мембрану YM-10, после чего наносили на колонку с Superose 6 для гель-фильтрации, уравновешенную Буфером A, содержащим 0,1 М водный раствор хлорида натрия. Колонку промывали изократно этим буфером при скорости потока 0,5 мл/мин и ВИЧ-протеазу элюировали в виде единственного пика.
QAE-сефароза и гексил-агароза были приобретены у химической компании Sigma. Superose 6 и MonoS были приобретены у Pharmacia. Буферы и реактивы были получены из Sigma.
4. Подготовка GAG-фракции
Аналогичным способом культуру E.coli К12 507/pHGAG выращивали до середины логарифмической фазы при 32oC, затем температуру повышали до 40oC примерно на 4-5 часов. Культуру охлаждали во льду и центрифугировали, осадок ресуспендировали в 8 мл лизирующего буфера, содержащего 5 мг/мл лизоцима. Лизирующий буфер включал 50 мМ трис-HCl (pH 7,8), 5 мМ EDTA, 1 мМ DTT, 100 мМ NaCl, 1 микрограмм/мл Е64 и 2 микрограмма/мл апротинина. Культуру инкубировали примерно от 30 до 60 минут при 4oC, затем быстро лизировали ультразвуком при помощи клеточного дезинтегратора (Branson @) при 60% мощности тремя 20-секундными импульсами с охлаждением перед каждым импульсом. После этого культуру осаждали центрифугированием при 15000xg. Надосадочную жидкость, содержащую непроцессированный gag- белок, частично очищали гель-фильтрацией на колонке с сефадексом G-50 и хранили при -20oC в 50% глицерине и лизирующем буфере.
Б. Подготовка субстрата: Na-Биотин-Gly-Ser-Gln-Asn-Tyr- Pro-Ile-Val-Gly-Lys(Ne-FITC)-OH
(а = Альфа, e = Эпсилон)
1. Подготовка пептида, биотинилированного по аминоконцу
Смолу, содержащую защищенный пептид, Na-Boc-Gly-Ser-Gln-Asn-Tyr(BrZ) -Pro-Ile-Val-Gly-Lys (2-ClZ) -OCH2-PAM-смолу, синтезировали на синтезаторе пептидов (Advanced Chemtech, Модель 200) в количестве 1,5 ммоль, используя стандартный алгоритм двойного связывания. Аминоконцевую т-Boc-группу удаляли 50% трифторуксусной кислотой в метиленхлориде и образующуюся в результате смолу нейтрализовали 5% диизопропилэтиламином (DIEA) в метиленхлориде. Затем к пептидной смоле добавляли 1,1 г (4,5 ммоль) биотина в 20 мл диметилсульфоксида с последующим добавлением 4,5 ммоль дициклогексилкарбодиимида (DCC) в 9 мл метиленхлорида. Полученную реакционную смесь разбавляли до конечного объема 40 мл, используя 1,1 мл метиленхлорида, после чего реакцию проводили в течение приблизительно 5 часов. Реакционный раствор концентрировали, смолу промывали последовательно диметилсульфоксидом, диметилформамидом и метиленхлоридом и затем нейтрализовали 5% DIEA в метиленхлориде. Эту реакцию повторяли дважды, увеличивая время реакции до 12 часов. Анализ смолы с помощью нингидрина свидетельствовал о завершении реакции биотина с аминогруппой глицина. Полученную в итоге пептидную смолу интенсивно промывали диметилформамидом и метиленхлоридом и высушивали, получая 4,3 г смолы (выход 98%).
2. Снятие защиты
С пептида снимали защиту и отделяли его от смолы, обрабатывая 50 мл раствора фтористо-водородной кислоты/м-крезола при 0oC в течение 1 часа. После удаления фтористо-водородной кислоты вакуумной перегонкой м-крезол экстрагировали из реакционной смеси, используя 100 мл диэтилового эфира. Затем пептид солюбилизировали в 50% водной уксусной кислоте, замораживали и лиофильно высушивали, получая 2,14 г пептида.
3. Очистка
Неочищенный пептид, биотинилированный по концевой аминогруппе, растворяли в 200 мл 5% водного раствора ацетонитрила, содержащего 0,1% трифторуксусную кислоту, и затем фильтровали через 0,22 микронный фильтр. Полученный раствор наносили на 2,2х25 см обращенно-фазную колонку с октадецил-силикагелем (Vydac C-18), уравновешенную тем же самым буфером. Пептид элюировали, используя 855-минутный линейный градиент 7,5-25% ацетонитрила, при скорости потока 2 мл/минуту, производя сбор фракций. Эти фракции анализировали, используя аналитическую ВЖХ. ВЖХ выполняли на 4,6х250 мм колонке Vydac C-18 в аналогичных условиях по буферу. Фракции, содержащие желаемый материал, объединяли, замораживали и лиофильно высушивали, получая 1,206 г пептида (выход 62%).
Аминокислотный анализ полученного биотинилированного пептида показал следующие результаты, соответствующие теоретически рассчитанным: Asn 1,1; Ser 0,96; Gln 1,1; Pro 1,1; Gly 2,1; Val 0,80; Ile 0,78; Tyr 1,1; Lys 1,1. FAB-масс-спектрометрия дала значение массы молекулярного иона равное 1288, что соответствует теоретически рассчитанному.
4. Введение метки
Очищенный биотинилированный пептид затем метили флюоресцентным маркером по C-концевой группе для использования в Pandex-анализе. Сначала биотинилированный пептид (1,206 г, 0,936 ммоль) растворяли в 100 мл 0,1 M бората натрия, pH 9,5. Затем к нему добавляли десятью одинаковыми порциями в течение двух часов раствор, содержащий 3 г (7,7 ммоль) флюоресцеинизотиоцианата в 15 мл диметилсульфоксида. Реакцию проводили в течение одного часа после последнего добавления. pH раствора подводили к 3, используя 5н. соляную кислоту, что приводило к образованию осадка, который удаляли центрифугированием.
Затем pH раствора пептида подводили к значению 7,8, используя 5н. гидроксид натрия, после чего раствор разбавляли до 200 мл добавлением 0,1 М ацетата аммония, pH 7,5. Полученный раствор фильтровали через 0,22 микронный фильтр и наносили на 2,2х25 см колонку Vydac C-18, уравновешенную 5% ацетонитрилом в 0,1 M ацетате аммония (pH 7,5). Пептид элюировали с колонки, используя 855-минутный линейный градиент 5-25% ацетонитрила, при скорости потока 2 мл/минуту со сбором фракций. Для анализа фракций использовали аналитическую ВЖХ. Фракции, содержащие желаемый продукт, объединяли, замораживали и лиофильно высушивали, получая 190,2 мг пептида (12%).
Аминокислотный анализ очищенного пептида показал следующие результаты, соответствующие теоретически рассчитанным: Asn 1,1; Ser 1,0; Gln 1,1; Pro 1,1; Gly 2,1; Val 0,8; Ile 0,8; Tyr 1,1; Lys 1,0. FAB-масс-спектрометрия дала значение массы молекулярного иона равное 1678, что соответствует теоретически рассчитанному.
5. Флюоресцентный анализ на ингибирование протеазы ВИЧ-1
Следующие буферы и растворы использовали во флюоресцентном анализе на ингибирование ВИЧ-1-протеазы:
MES-ALB-буфер:
0,05 М 4-морфолинэтан
сульфоновая кислота, pH 5,5
0,02 M NaCl
0,002 М EDTA
0,001 М DTT
1,0 мг/мл BSA
TBSA-буфер:
0,02 M TRIS
0,15 M NaCl
1,0 мг/мл BSA
Раствор покрытых авидином шариков:
0,1% раствор Fluoricon авидиновых тест-частиц (авидин, конъюгированный с твердыми полистирольными шариками диаметром 0,6-0,8 мкм в TBSA-буфере)
Раствор фермента:
27 IU/мл очищенной HIV-1-протеазы в MES-ALB-буфере (1 IU соответствует количеству фермента, требуемому для гидролиза 1 мкмоль субстрата в минуту при 37oC)
К каждой лунке круглодонного 96-луночного планшета добавляют 20 мкл раствора фермента с последующим добавлением 10 мкл анализируемого соединения в 20% водном растворе диметилсульфоксида. Очищенную протеазу ВИЧ-1 получали, как описано выше. Полученный раствор инкубируют в течение одного часа при комнатной температуре, после чего в каждую лунку добавляют по 20 мкл раствора, содержащего субстрат, приготовленный ранее, в MES-ALB-буфере (1,5 мкл). Растворы затем инкубируют в течение 16 часов при комнатной температуре, после чего содержимое каждой лунки разбавляют 150 мкл MES-ALB-буфера.
К каждой лунке второго круглодонного 96-луночного Pandex планшета добавляют по 25 мкл раствора покрытых авидином шариков. Затем к каждой лунке добавляют по 25 мкл приготовленных ранее разбавленных инкубационных растворов. Растворы тщательно перемешивают и планшеты помещают в Pandex-машину, промывают, опорожняют и прочитывают. Детектирование образцов проводили, используя возбуждение при 485 нМ, прочитывая возникающую в результате эпифлюоресценцию при 535 нМ.
Результаты IC50, полученные во флюоресцентном анализе для соединений по настоящему изобретению, указаны в таблицах 1, 2 и 3. Все значения были отнесены к положительному контролю, которым является [1S-(1R*,4R*,5S*)]-N-(1-(2-амино-2- оксоэтил)-2-oкco-3-аза-4-фенилметил-5-гидpoкcи-6-(2-(1-т-бутиламино- 1-оксометил)фенил)гексил)-2-хинолинилкарбоксамид.
Иллюстративные данные по активности для соединений, охватываемых настоящим изобретением, представлены в таблицах 1, 2 и 3 и в предшествующих Примерах. Результаты, данные в круглых скобках, приведены для Примера 1 из опубликованной заявки на Европейский патент 0526009 A1 = 35B в том же самом анализе.
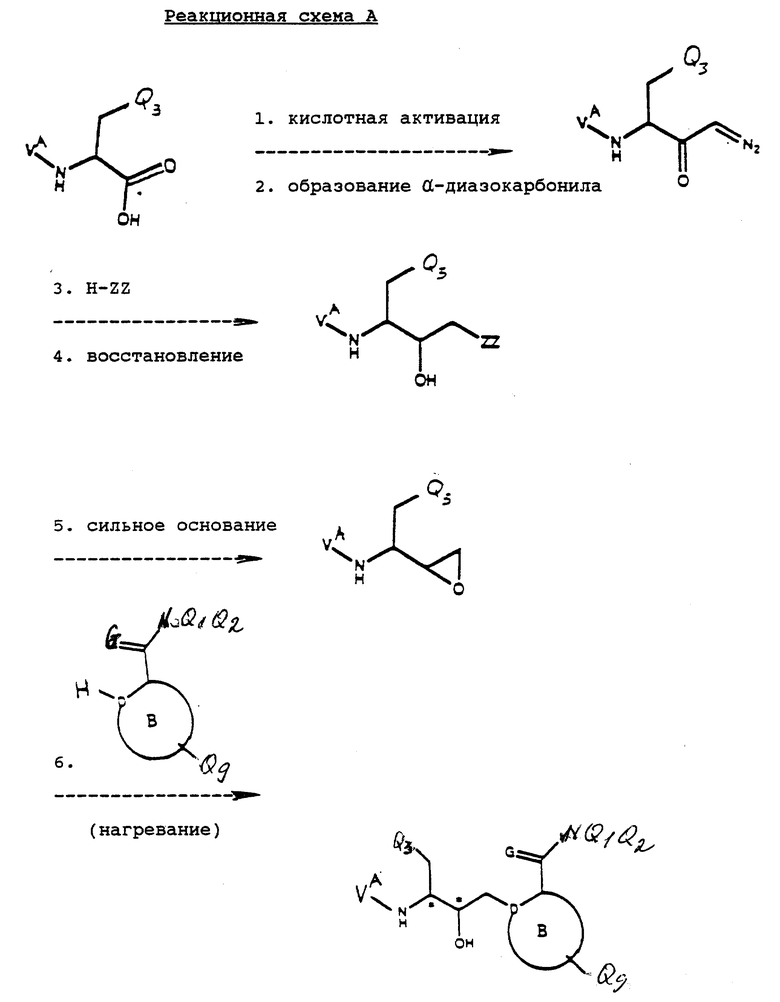
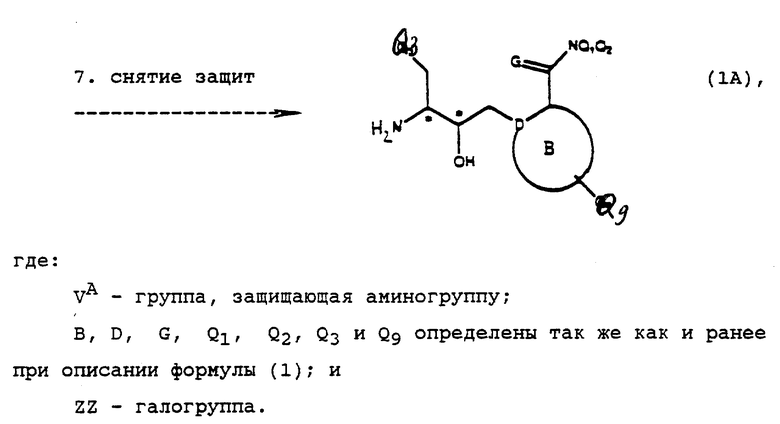
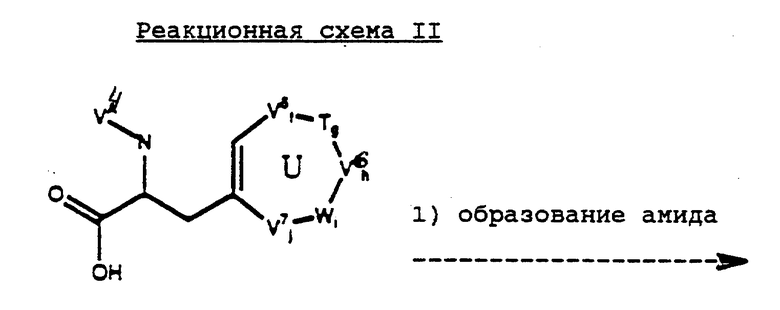

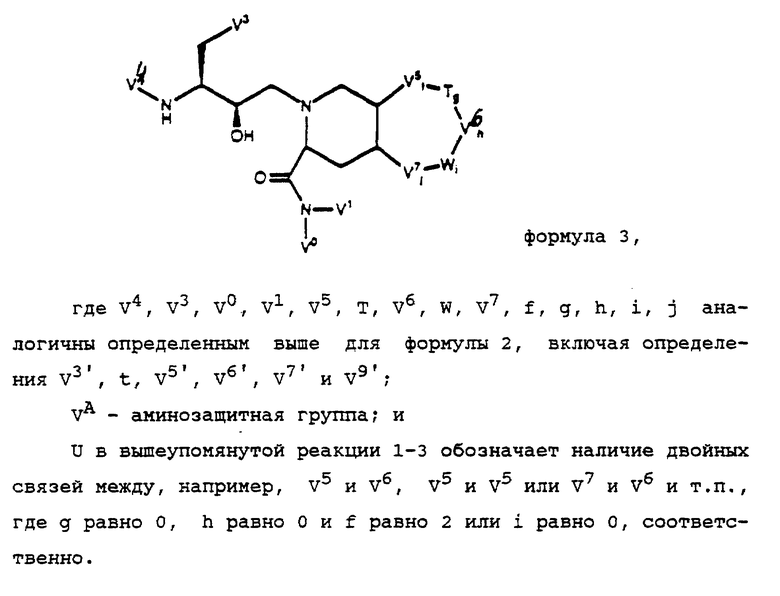
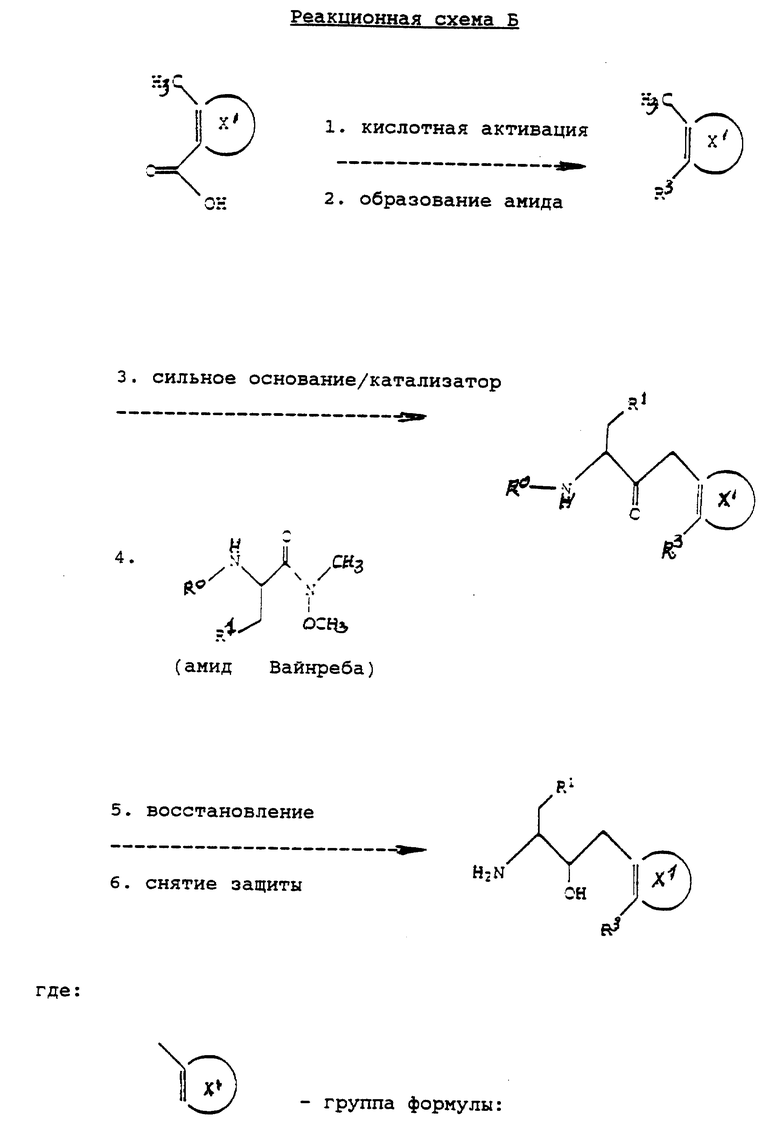
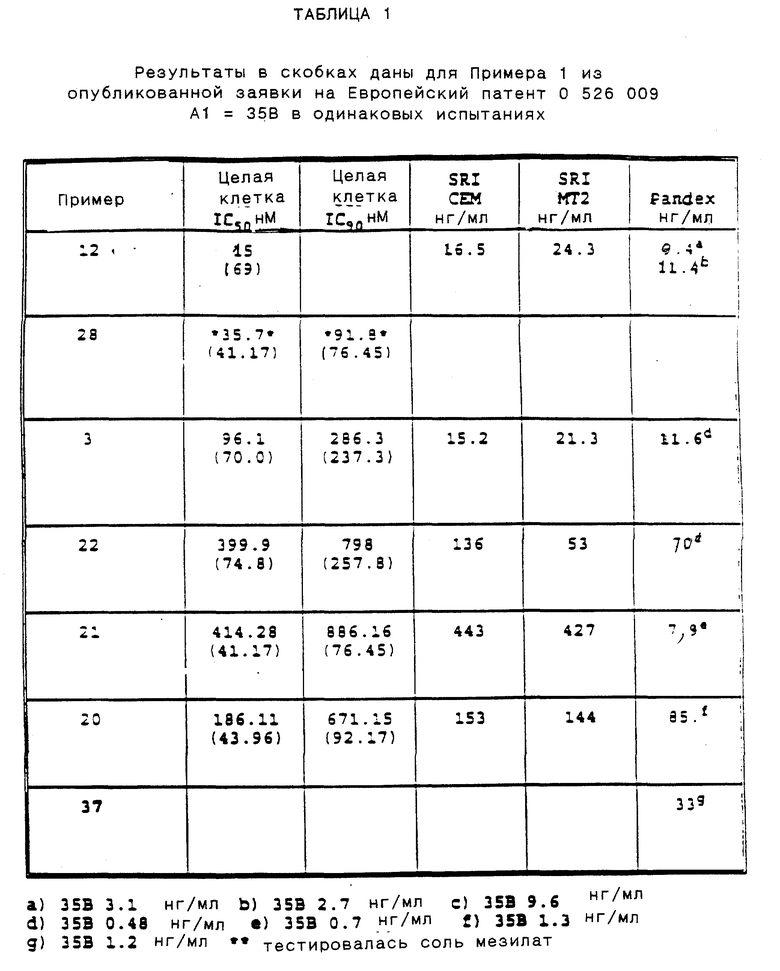
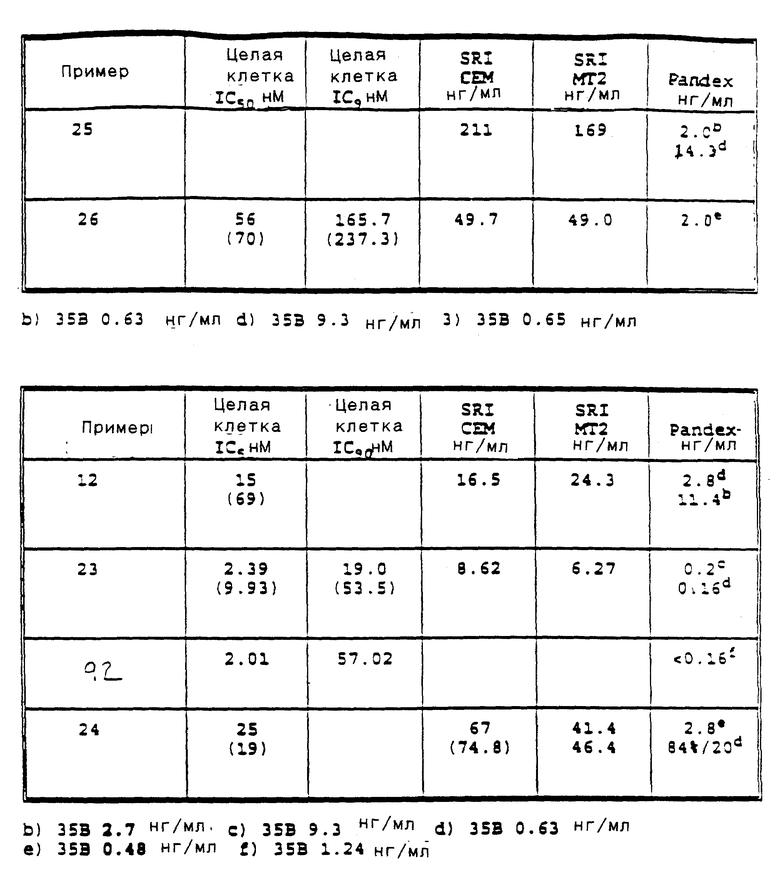
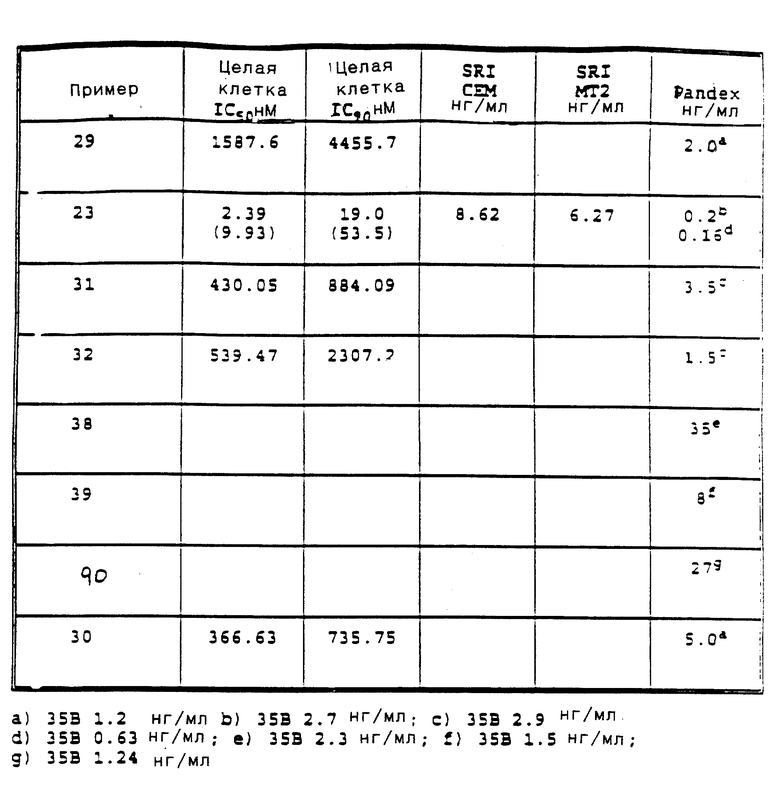
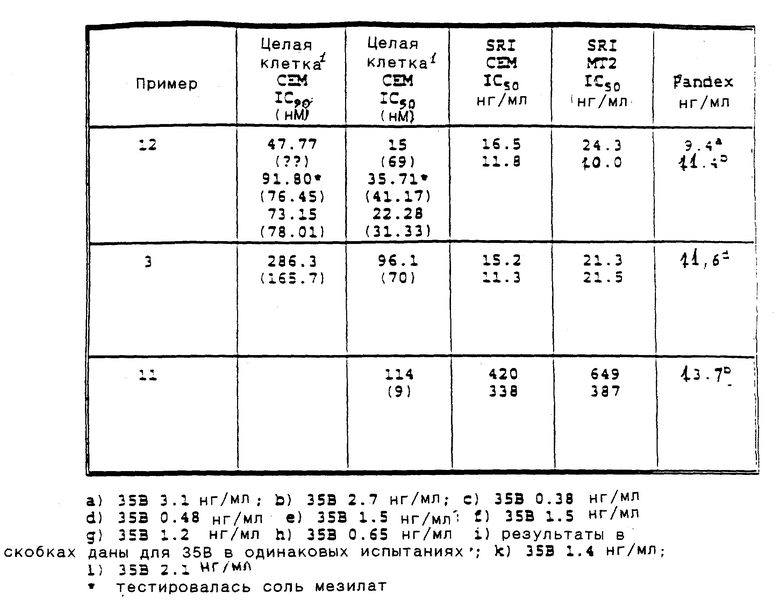
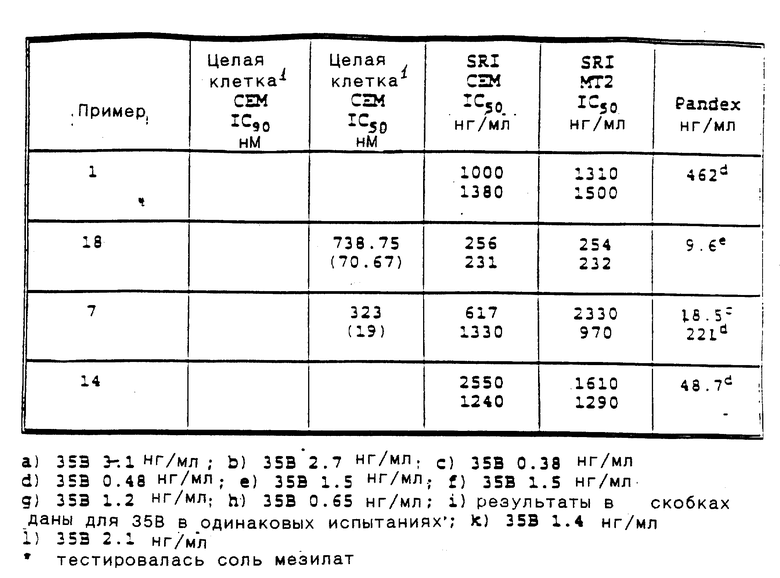
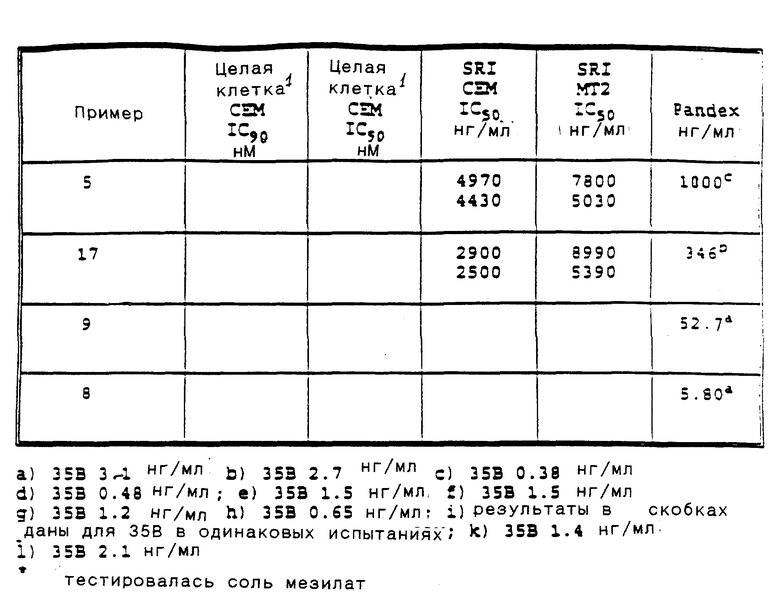
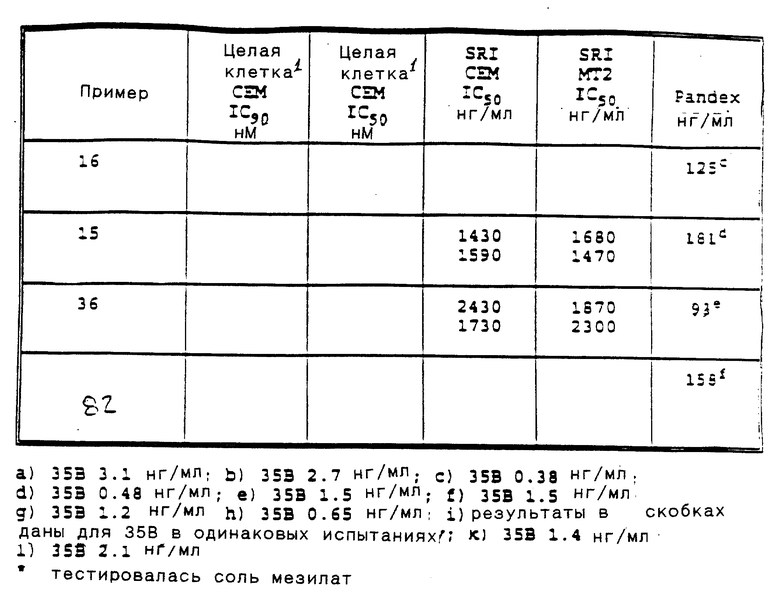
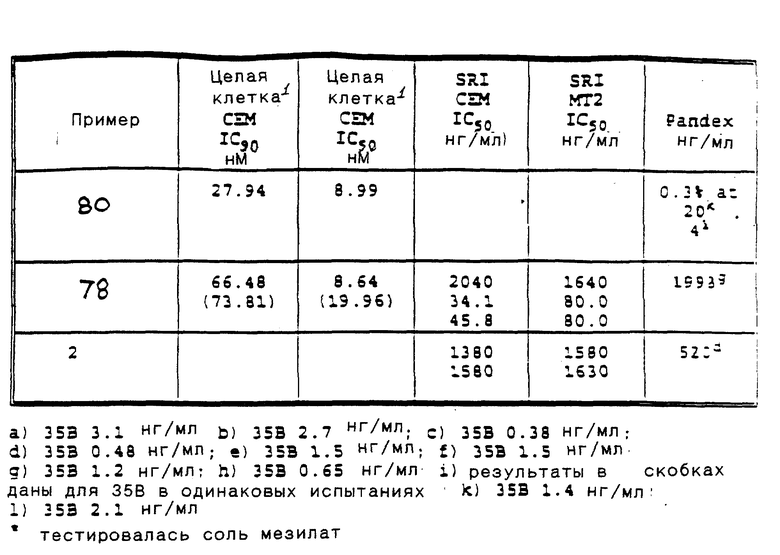
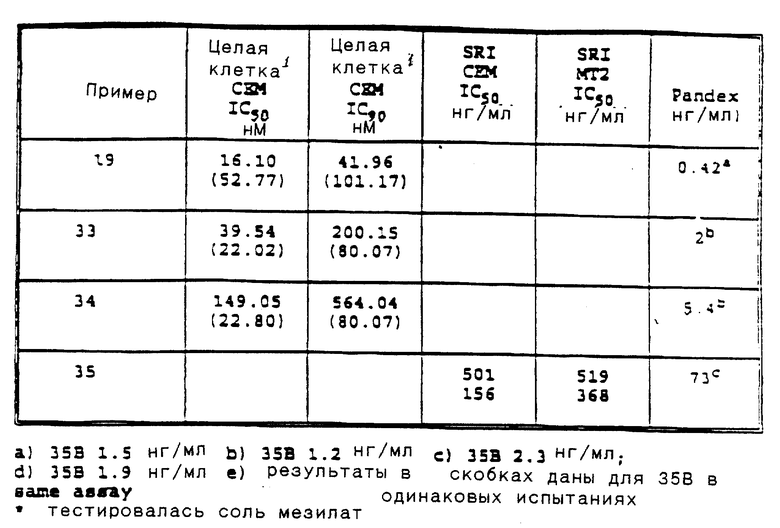
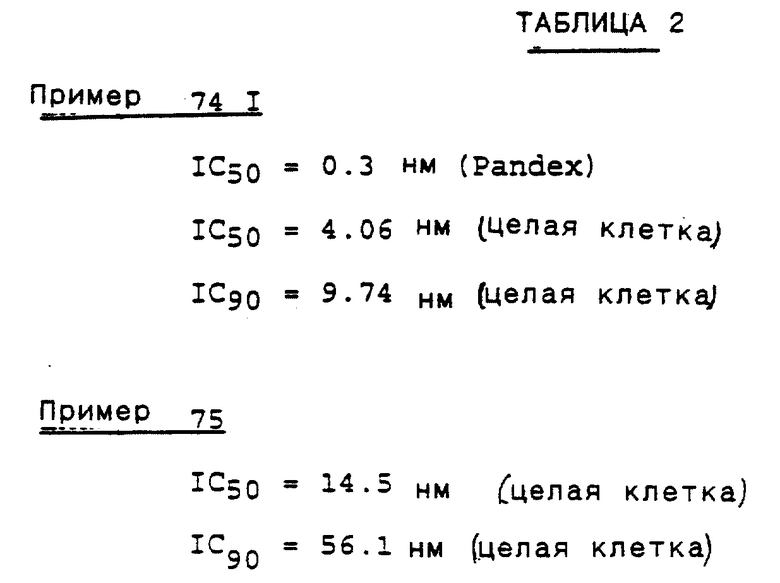

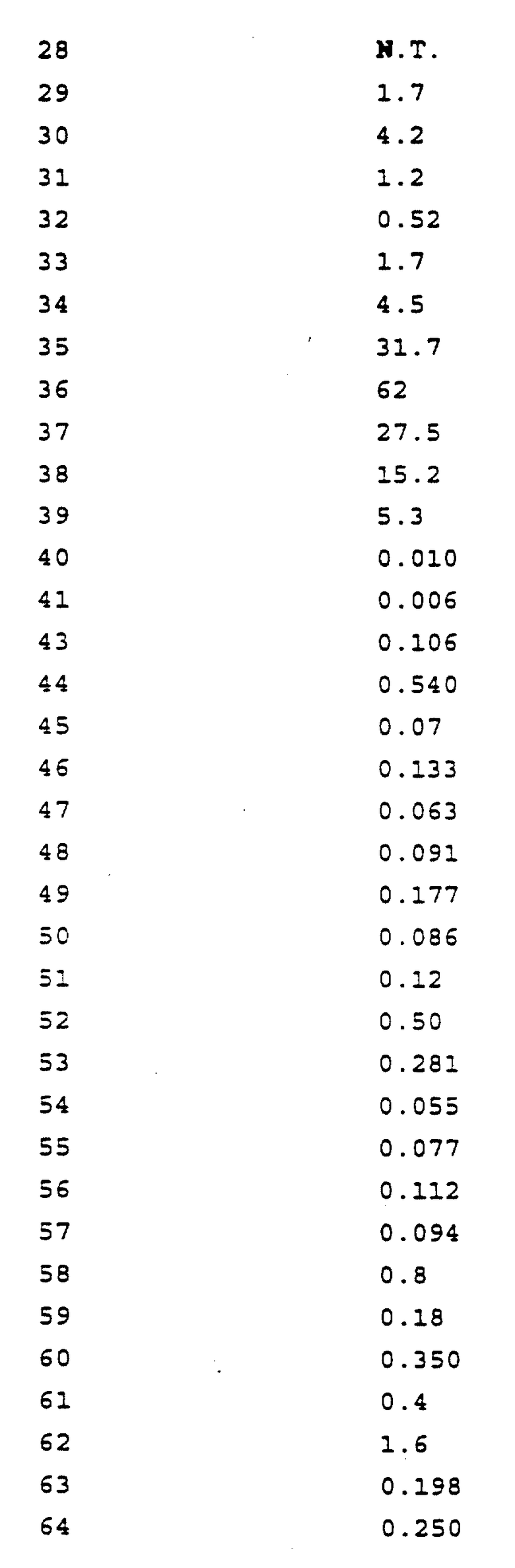

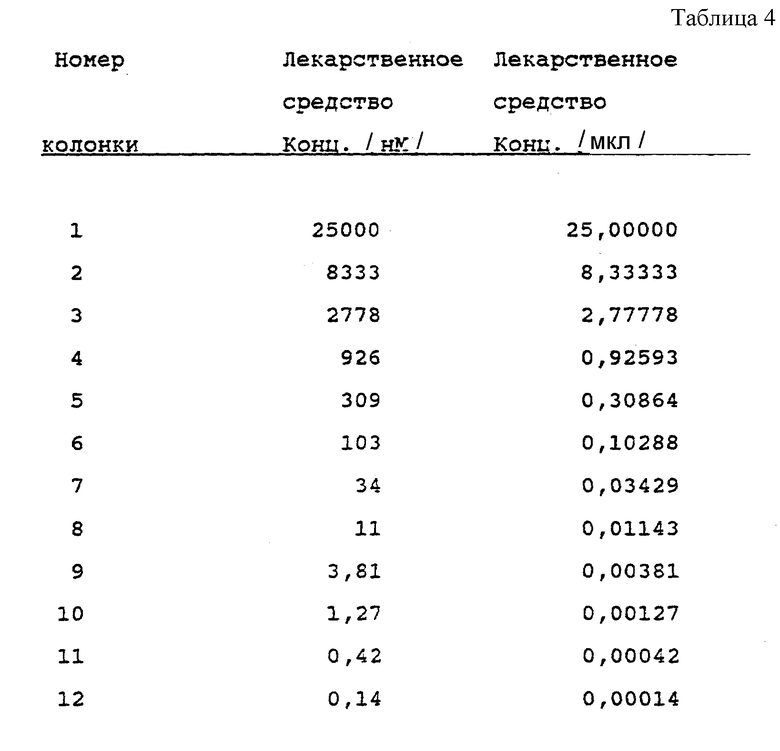
Соединение формулы I, приведенной в тексте описания, где Q3 - арил или S-арил, возможно замещенные галогеном, арил - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток; А - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, пиридин, пиридин-N-оксидил, хинолин, изохинолин, индол, индолин, тиазол-1,1-диоксид, тиофен или тиофен-1,1-диокси; В - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, включающий гетероатом азота, пиридилметилпиперазин, октагидротиено[3,2-с]пиридин или октагидротиено[3,2-с] пиридин-1,1-диоксид; Q1, Q2 независимо представляют атом водорода или алкил; Q4-8 независимо представляют атом водорода, гидроксил, галоген, нитро, амино, сульфониламино, алкиламино, алкил, возможно замещенный галогеном, алкоксил, группу -О-J (где J - отгидролизовываемая группа) или группу L6C(O)L4 (где L6 - одинарная связь или -О, a L4 - алкил или алкоксил); Y и G - атом кислорода, D - атом углерода или азота, причем атом D соединен одинарной связью с каждым из смежных атомов кольца; Е - атом углерода; Q9 - водород, или его фармацевтически приемлемая соль или фармацевтическая композиция на его основе ингибирует ВИЧ-протеазу. 10 с. и 42 з.п. ф-лы, 4 табл.
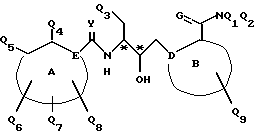
где Q3 - арил или -S-арил, возможно замещенные галогеном, где арил - это карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток;
А - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, пиридинил, пиридинил-N-оксид, хинолинил, изохинолинил, индолил, индолинил, тиазолил-1,1-диоксид, тиенил, или тиенил-1,1-диоксид;
В - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, насыщенный 5-14-членный моноциклический или полициклический остаток, включающий гетероатом азота, пиридилметилпиперазинил, октагидротиено[3,2-с]пиридинил или октагидротиено[3,2-с]пиридинил-1,1-диоксид;
Q1 и Q2 независимо - атом водорода или алкил;
Q4 - Q8 независимо - атом водорода, гидроксил, галоген, нитро-, амино-, алкилсульфониламино-, алкиламино-, алкил, возможно замещенный галогеном, алкоксил, группа -O-I (где I обозначает отгидролизовываемую группу) или группа L6C(O)L4 (где L6 обозначает простую связь или - О, а L4 - алкил или алкоксил);
Y и G - атомы кислорода;
D - атом углерода или азота, причем D соединен простой связью с каждым из смежных атомов кольца;
E - атом углерода;
Q9 - атом водорода;
или его фармацевтически приемлемая соль.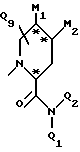
где М1 и М2 - атомы водорода или М1 и М2 могут образовывать часть кольца, имеющего до 10 членов,
или фармацевтически приемлемая соль этого соединения.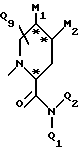
где М1 и М2 - атомы водорода или М1 и М2 могут образовывать часть кольца, имеющего до 10 членов.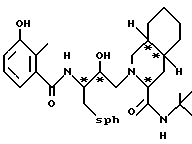
или его пролекарство, или его фармацевтически приемлемая соль.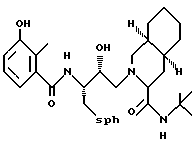
или его пролекарство, или его фармацевтически приемлемая соль.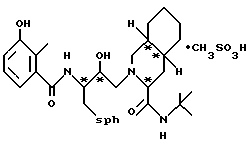
39. Соединение по п. 38, отличающееся тем, что оно представляет собой стереоизомер и имеет формулу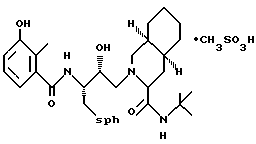
40. Соединение по п. 39, отличающееся тем, что оно представляет собой существенно чистый стереоизомер.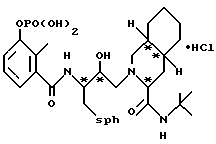
42. Соединение по п. 41, отличающееся тем, что оно представляет собой стереоизомер и имеет формулу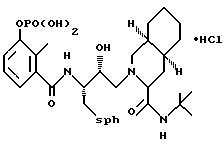
43. Существенно чистый стереоизомер по п.42.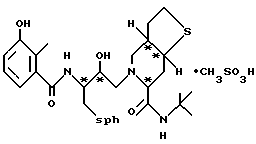
45. Соединение по п. 44, отличающееся тем, что оно представляет собой стереоизомер и имеет формулу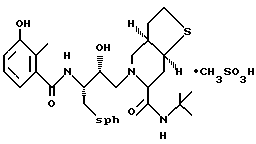
46. Соединение по п. 45, отличающееся тем, что оно представляет собой существенно чистый стереоизомер.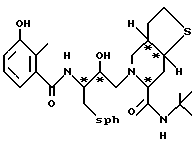
или его пролекарство, или его фармацевтически приемлемая соль.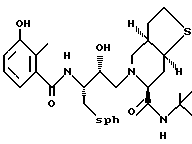
или его пролекарство, или его фармацевтически приемлемая соль.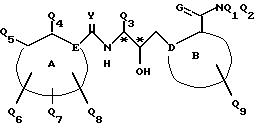
где Q3 - арил или -S-арил, возможно замещенные галогеном, где арил - это карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток;
А - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, пиридинил, пиридинил-N-оксид, хинолинил, изохинолинил, индолил, индолинил, тиазолил-1,1-диоксид, тиенил или тиенил-1,1-диоксид;
В - карбоциклический ароматический 5-14-членный моноциклический или полициклический остаток, насыщенный 5-14-членный моноциклический или полициклический остаток, включающий гетероатом азота, пиридилметилпиперазинил, октагидротиено[3,2-c] пиридинил, или октагидротиено[3,2-c]пиридинил-1,1-диоксид,
Q1 и Q2 независимо - атом водорода или алкил;
Q4 - Q8 независимо - атом водорода, гидроксил, галоген, нитро- амино-, алкилсульфониламино-, алкиламино-, алкил, возможно замещенный галогеном, алкоксил, группу -O-I (где I обозначает отгидролизовываемую группу) или группу L6C(O)L4 (где L6 обозначает простую связь или -О, а L4 - алкил или алкоксил);
Y и G - атомы кислорода;
D - атом углерода или азота, причем D соединен простой связью с каждым из смежных атомов кольца;
Е - атом углерода;
Q9 - атом водорода,
или его фармацевтически приемлемой соли.
Приоритет по пунктам:
02.02.94 при В - октагидротиено[3,2-c] пиридин и В - октагидротиено[3,2-c]пиридин-1,1-диоксид, п.1-33, 38-50, 51 и 52.
| Способ получения пептидов или их фармацевтически приемлемых солей | 1985 |
|
SU1676454A3 |
| SU 1287532 A, 1990 | |||
| СПОСОБ ПРЕССОВАНИЯ ЛИСТОВ ИЛИ ПЛЕНОК | 0 |
|
SU346847A1 |
| ЗАМОК ДЛЯ РАЗРЕЗНЫХ УПРУГИХ УПЛОТНИТЕЛЬНЫХ КОЛЕЦ | 0 |
|
SU361341A1 |
| Штамп для изготовления деталей из шпона | 1973 |
|
SU490667A1 |
| Огнетушитель | 0 |
|
SU91A1 |
