Настоящее изобретение относится к новым термолизин-подобным нейтральным металло-протеазам и их применению, особенно для получения метилового эфира бензилоксикарбонил -α- -L-аспартил-L-фенилаланина.
Термолизин представляет собой ценный энзим, выпускаемый промышленностью и находящий применение в большом числе различных областей, например, в детергентных композициях, в процессах обработки пищи и в косметических рецептурах. Он также используется в синтезе метилового эфира бензилоксикарбонил -α- -L-аспартил-L-фенилаланина (далее в тексте на это вещество кратко ссылаются, как на Z-APM), который представляет собой предшественник аспартама, искусственного подслащивающего агента.
Описание уровня техники
Термолизин впервые был обнаружен в культуральном бульоне Bacillus thermoproteolyticus (Endo, s (1962) J. Fermentation Tech., 40, 346 - 353) и на этом объекте был проведен ряд исследований. Так, например, была установлена его аминокислотная последовательность (Titani. K. с сотр., (1972) Nature New Biol. 238. 35 - 37) и трехмерная структура энзима (Holmes M.A. и Mattews, B. W. , (1982) J. Mol.Biol. 160, 623-639). Между тем протеазный ген был клонирован из Bacillus thermoproteolyticus (EP-A-0418625) и аминокислотная последовательность зрелого энзима, выведенная из нуклеотидной последовательности указанного гена оказалась отличной от оригинальной первичной структуры, установленной Titani, по двум положениям. Так например, было сообщено, что 37-ой (считая от аминоконца) аминокислотный остаток зрелого энзима не является аспарагиновой кислотой, а представляет собой аспарагин, а 119-ый остаток является не глютаминовой кислотой а глютамином. Такая аминокислотная последовательность идентична последовательности закодированной nprM, одним из протеазных генов клонированных из Bacillus thermoproteolyticsu (Kubo, M с сотр., (1988) Journal of General Microb 134, 1883 - 1892).
В связи с этим, в настоящем описании на протеазу закодированную таким nprM геном или геном из Bacillus thermoproteolyticus ссылаются, как на "термолизин-подобную нейтральную металло-протеазу дикого типа".
Об изменении удельной активности и устойчивости термолизин-подобной нейтральной металло-протеазы сообщалось (Kubo M. c сотр. (1992) Applied and Environmental Microbiology, 58, 3779-3783). В этой статье были описаны мутанты, которые отличаются одним или более аминокислотными остатками в первичной структуре, особенно в положениях 93, 110, 114, 115, 136, 137, 143, 151, 157, 193, 211, 217 и 221. Однако, в этой ссылке активность измеряли только методом казеинового переваривания. Однако, ни один из таких мутантов не демонстрировал сколь-нибудь значительно улучшенной активности в отношении синтеза или переваривания Z-APM. В настоящее время (как это показано в примерах заявки на Европейский патент авторов настоящего изобретения, заявка N 93200773.5) также было установлено, что активность казеинового переваривания не коррелирует с активностью в синтезе Z-APM: оказалось, что даже если удельная активность в отношении переваривания казеина увеличивается, удельная активность в отношении синтеза Z-APM увеличивается не всегда.
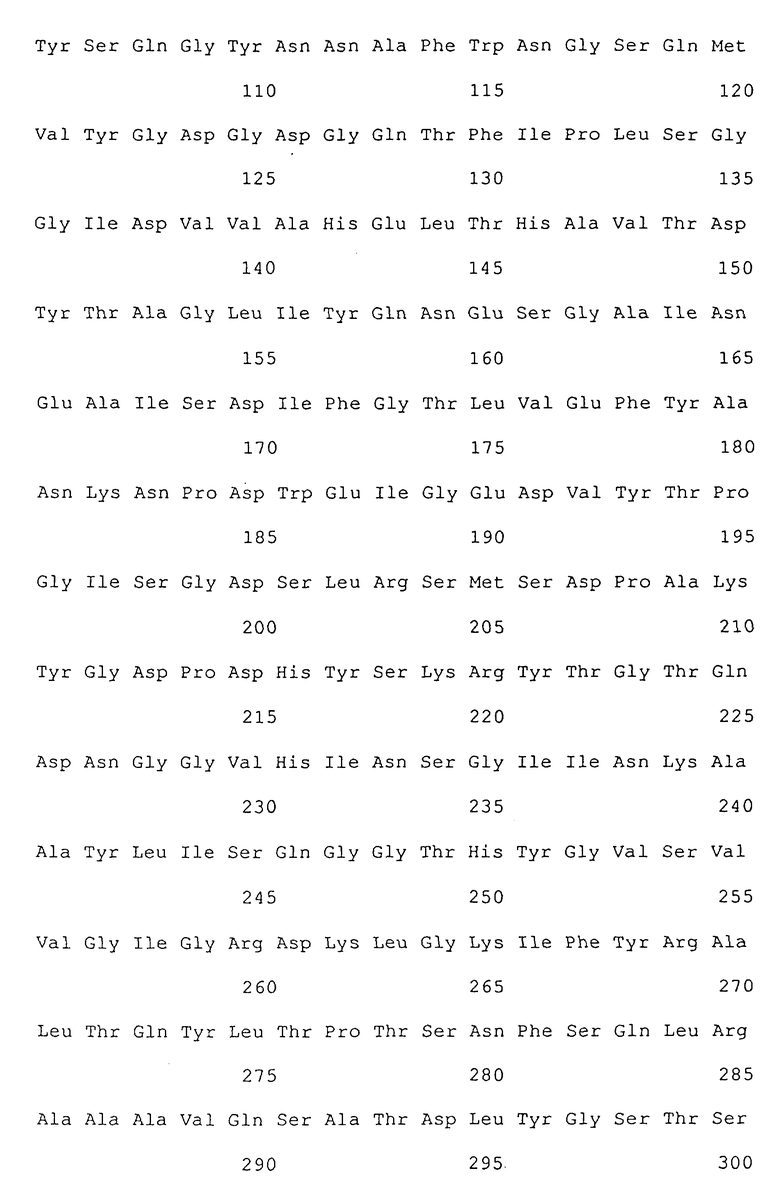
Кроме этого, авторы изобретения предварительно обнаружили, что ценные новые протеазы могут быть производными из термолизин-подобной нейтральной металло-протеазы, имеющей (дикий тип) аминокислотную последовательность SEQ 1D No:1, показанную ниже, в результате замены одного или более аминокислотных остатков в некоторых положениях другими аминокислотными остатками отличными от исходных.
(SEQ 1D No:1)
Заявители уже подали заявку на Европейский патент (Заявка N 93200773.5) на такие новые модифицированные протеазы, полученные из указанного дикого типа в результате замены, по крайней мере одного из следующих аминокислотных остатков на отличающиеся от них аминокислоты: 144-ый (лейцин), 150-ый (аспаргиновая кислота), 187-ой (глютаминовая кислота) и 227-ой (аспарагин) аминокислотные остатки.
Удельная активность модифицированных энзимов, упомянутых в указанных более ранних заявках на патент (которые не опубликованы к дате подачи настоящей заявки), имеющих единственную аминокислотную замену в одном из положений 144, 150, 187 и 227, не более чем в два раза превышает активность энзима дикого типа в отношении синтеза или переваривания Z-APM.
На основании этих наблюдений и поскольку имеются различные проблемы в энзиматическом синтезе Z-APM, например такие, как относительно низкая активность энзима, его инактивация во время реакции конденсации и гидролиза Z-APM и исходного материала - метилового эфира L- или D, L-фенилаланина (PM), вследствие длительного времени реакции и/или неблагоприятных значений pH, существует необходимость в разработке усовершенствованных энзимов, обладающих повышенной активностью в синтезе Z-APM. Разумеется, что при упоминании PM в настоящей заявке могут подразумеваться и его соли.
Краткое изложение сущности изобретения
В настоящее время было неожиданно установлено, что усиление активности модифицированного энзима, имеющего триптофановый остаток в положении 150 в последовательности SEQ 1Д N: 1 во много раз превосходит такой эффект по сравнению с ранее описанными модифицированными протеазами.
На основании этих наблюдений было реализовано настоящее изобретение, на базе которого обеспечиваются модифицированные протеазы, имеющие аминокислотную последовательность, указанную выше (SEQ 1Д N:1) но с заменой, по крайней мере, 150-го аминокислотного остатка аспаргиновой кислоты (дикий тип) на триптофан. Таким образом предусматривается существенное усиление активности Z-APM синтеза под действием термолизин-подобной нейтральной металло-протеазы, полученной из Bacillus stearothermophilus.
В связи с этим модифицированные протеазы согласно изобретению являются очень ценными веществами для крупномасштабного производства Z-APM.
Краткое описание чертежей
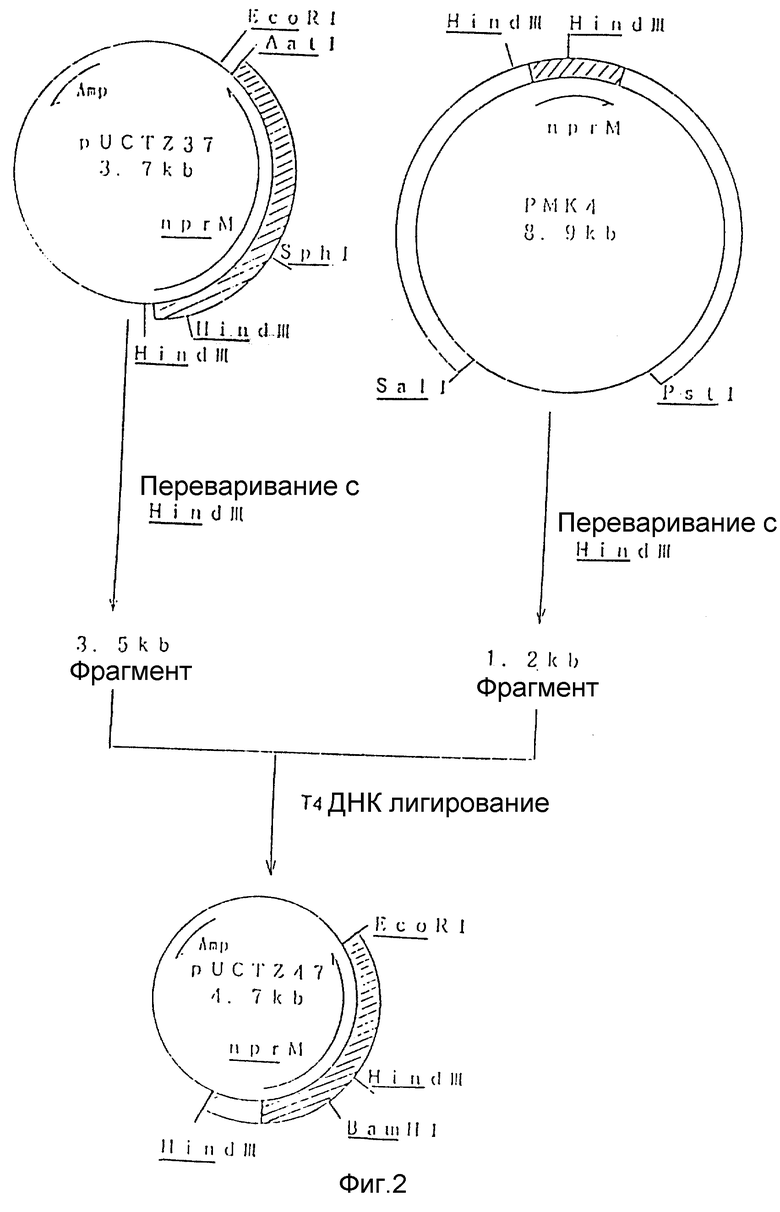
На рис. 1 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUCTZ37 из известной плазмиды pMK4.
На рис. 2 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUCTZ47 из плазмиды pMK 4 и плазмиды pUCTZ37.
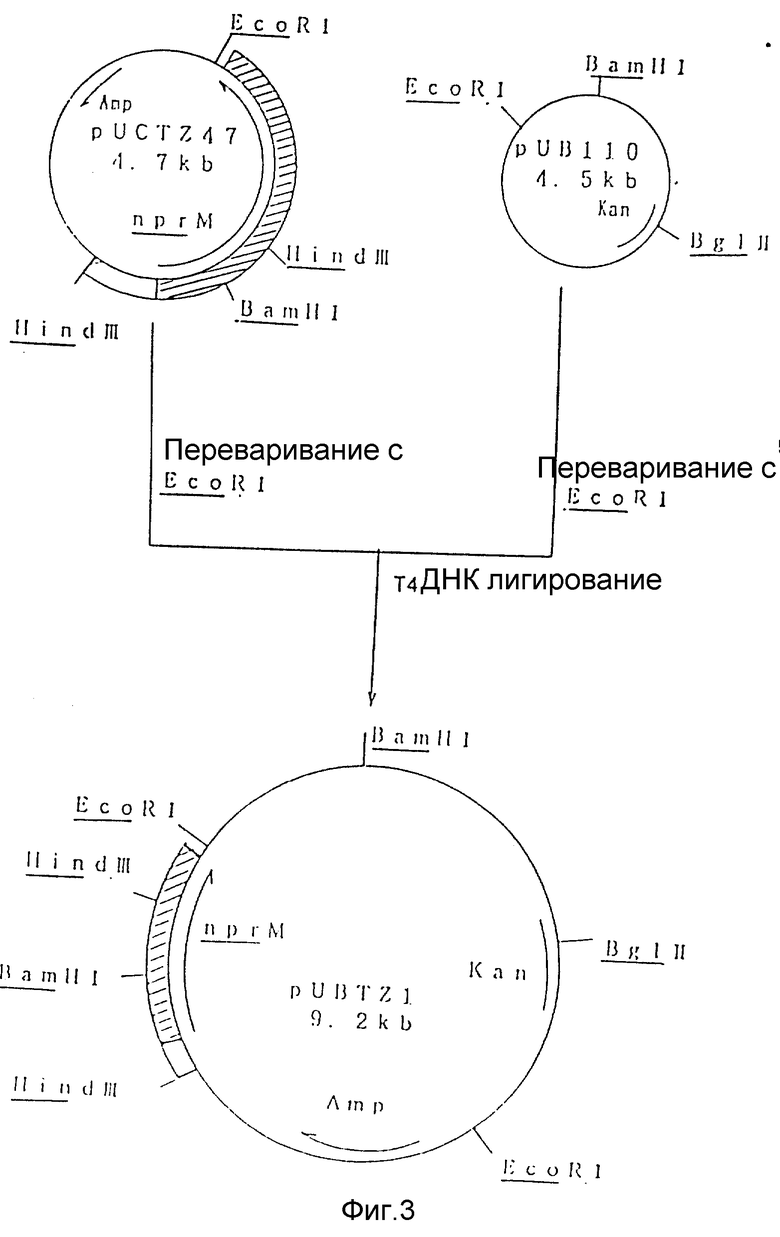
На рис. 3 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUBTZ1 из известной плазмиды pUCTZ47 и плазмиды pUB110.
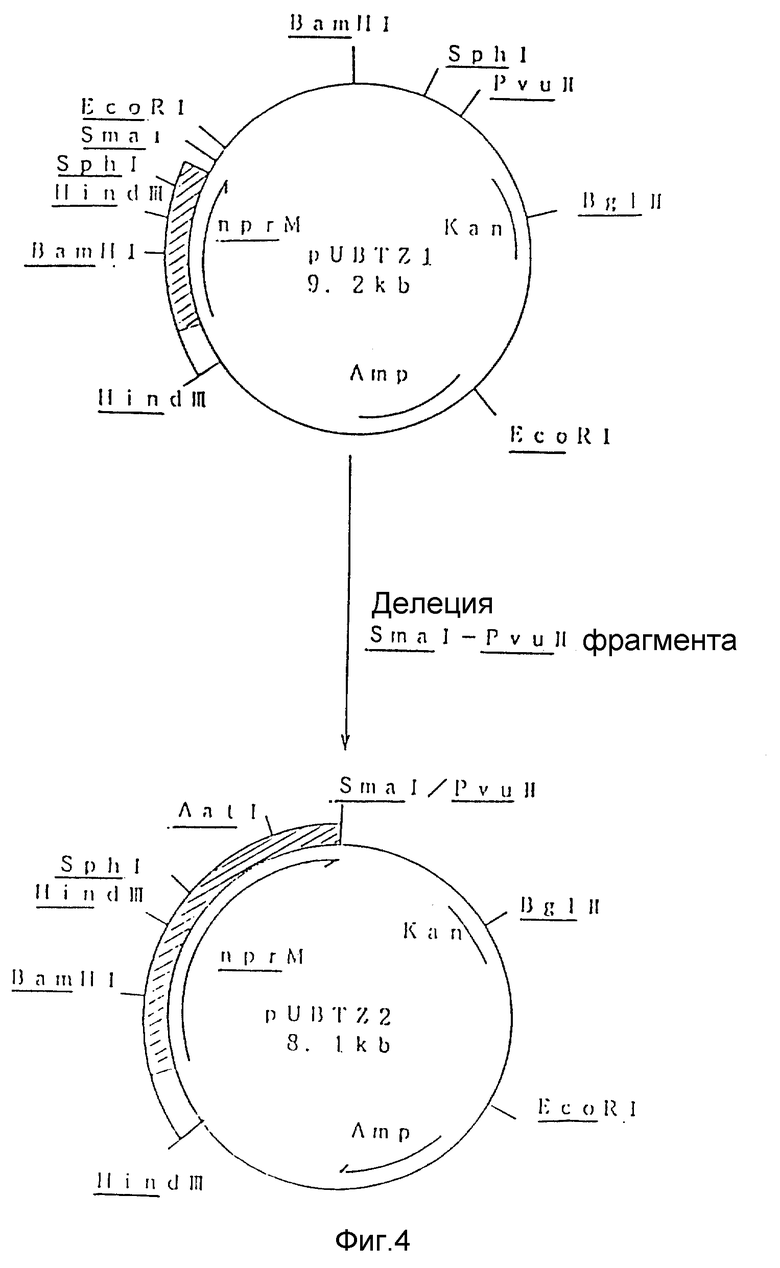
На рис. 4 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUBTZ2 из плазмиды pUBTZ1.
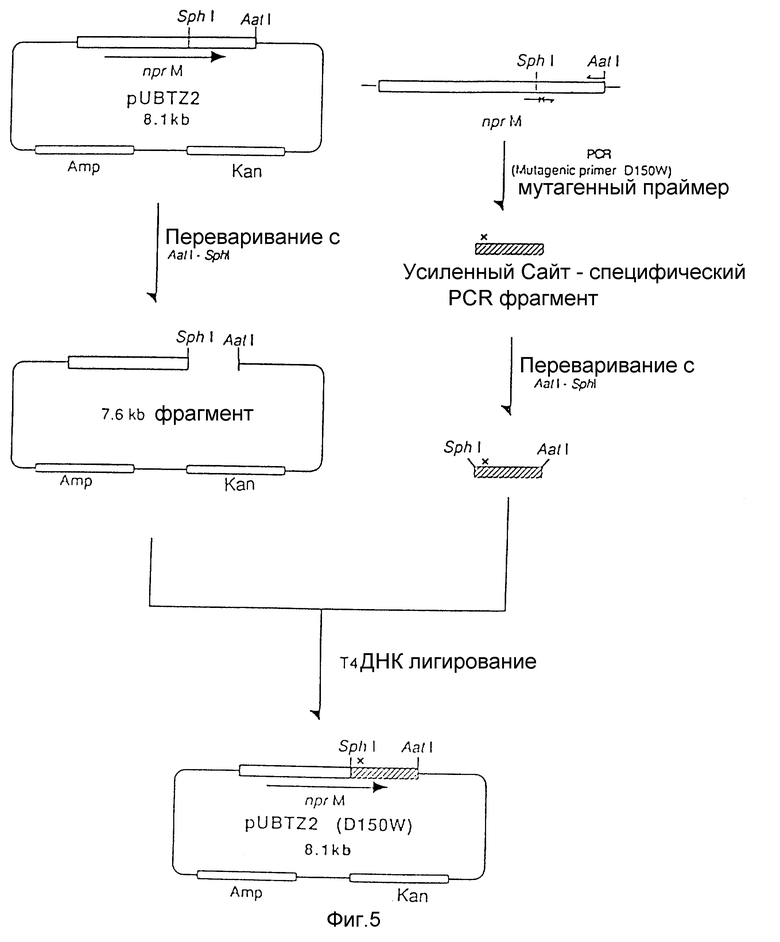
На рис. 5 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUBTZ2 (D150W) из плазмиды pUBTZ2 и фрагмента мутантной ДНК, полученного полимеразной цепной реакцией.
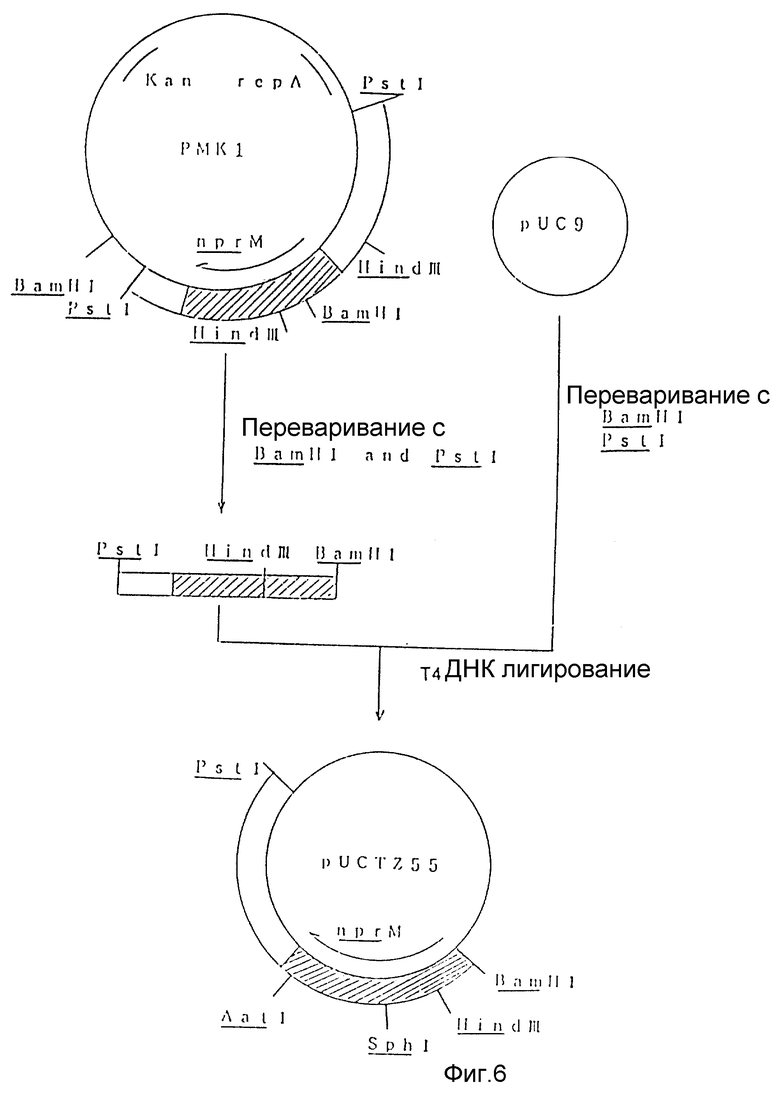
На рис. 6 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUCTZ55 из известной плазмиды pMK1.
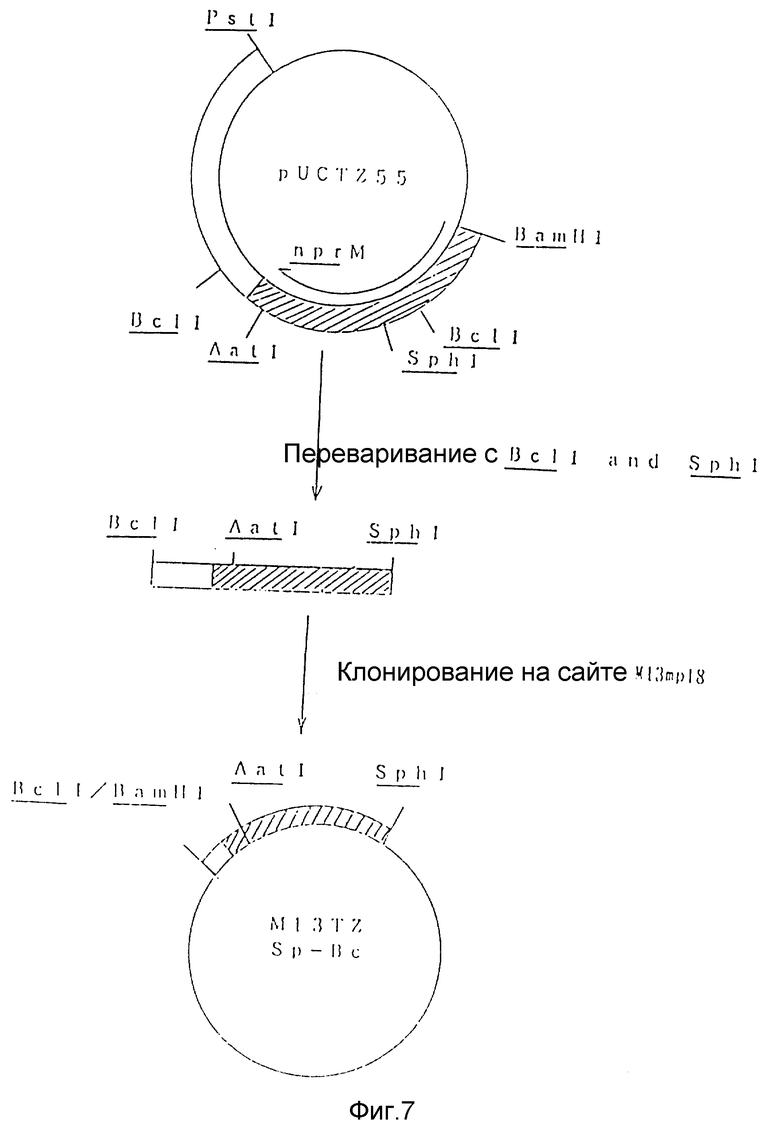
На рис. 7 показана схема, используемая для конструирования рекомбинантного M13 фага под названием M13TZSp-Bc из плазмиды pUCTZ55.
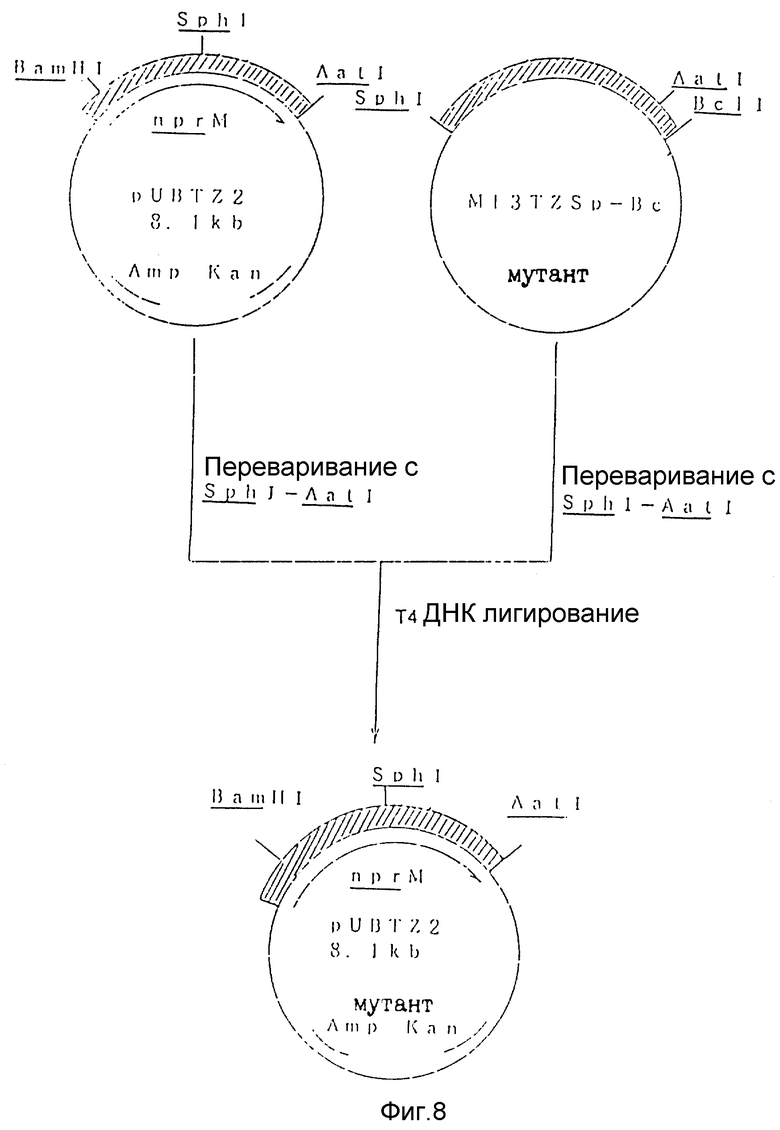
На рис. 8 показана схема, используемая для конструирования рекомбинантной плазмиды pUBTZ2 (N227H мутант) из плазмиды pUBTZ2 и M13TZSp-Bc (N227H мутант).
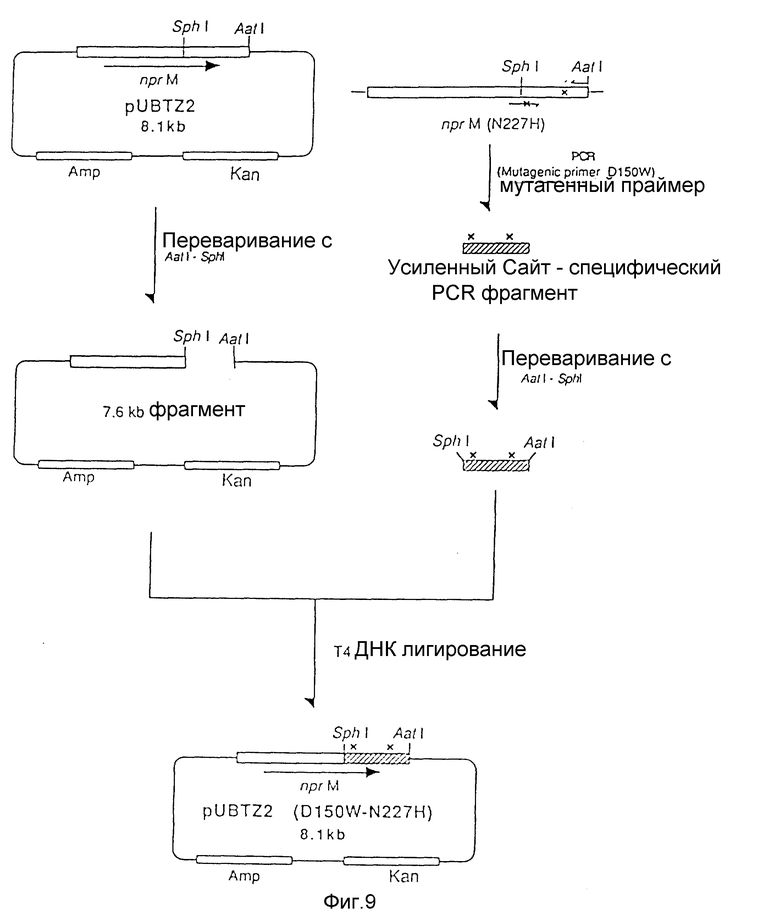
На рис. 9 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUBTZ2 (D150W - N227H) из плазмиды pUBTZ2 и фрагмента мутантной ДНК, полученного полимеразной цепной реакцией.
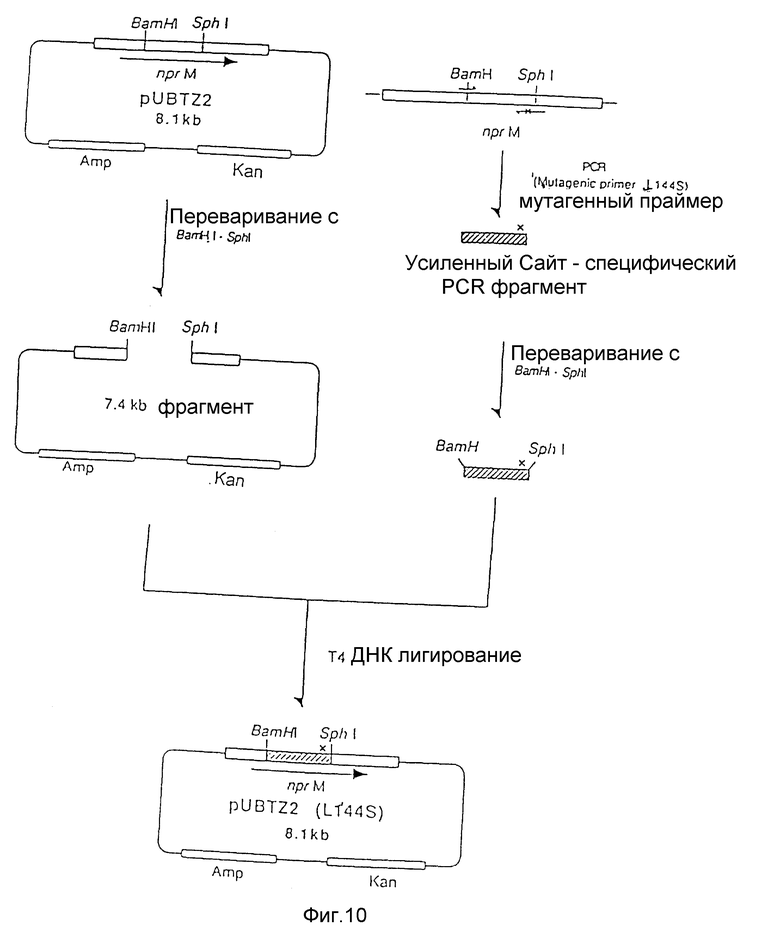
На рис. 10 показана схема, используемая для конструирования рекомбинантной плазмиды под названием pUBTZ2 (L144S) из плазмиды pUBTZ2 и фрагмента мутантной ДНК, полученного полимеразной цепной реакцией.
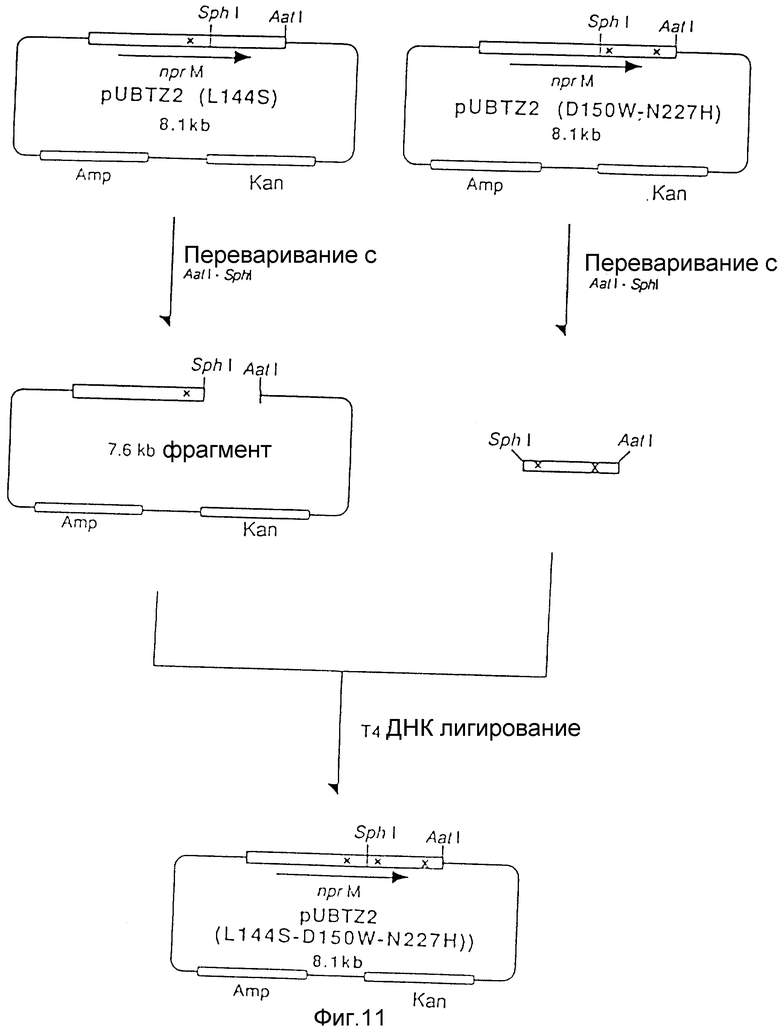
На рис. 11 показана схема, используемая для конструирования рекомбинатной плазмиды под названием UBTZ2 - (144S - D150W - N227H) из плазмиды pUBTZ2 (L144) и плазмиды pUBTZ2 (D150W N 227).
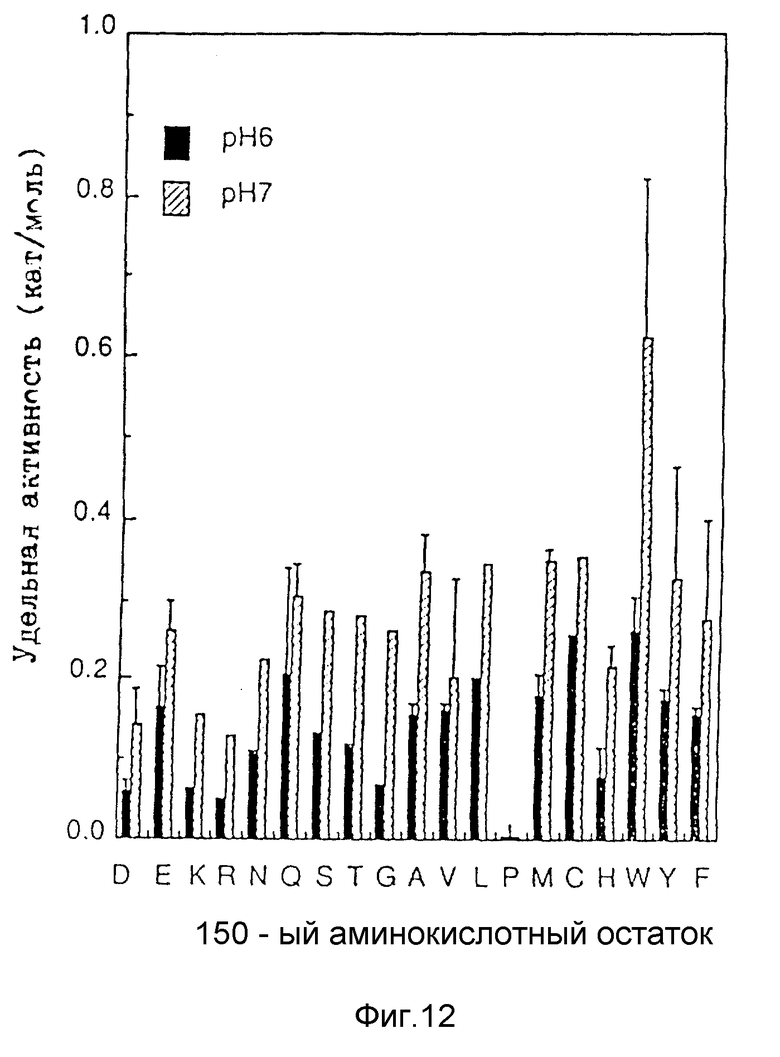
На рис. 12 показаны Z-APM синтетические активности модифицированных энзимов. Для аминокислот приняты однобуквенные сокращения. "D" в 150-ом аминокислотном остатке обозначает термолизин-подобную нейтральную металло-протеазу дикого типа. Энзиматическая активность, обеспечивающая синтез 1 моля Z-APM в секунду обозначена, как 1 катал (kat).
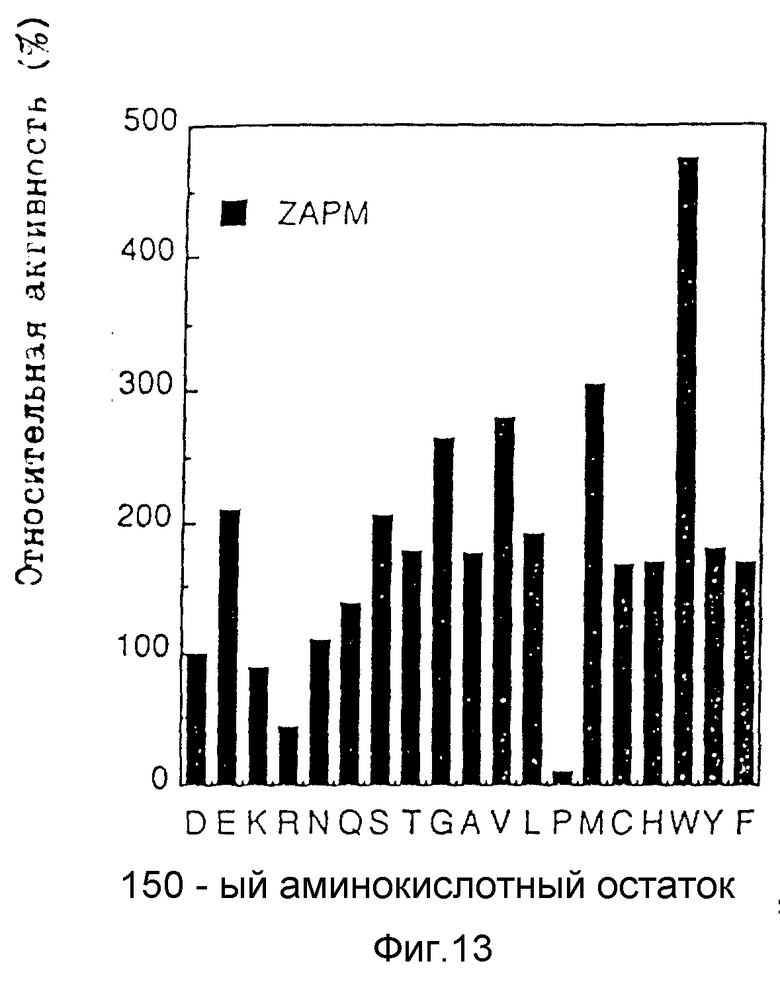
На рис. 13 представлены гидролитические активности модифицированных энзимов в отношении Z-APM.20.
Подробное описание изобретения
В упомянутой выше более ранней заявке на патент раскрываются модифицированные протеазы, в 150-ом положении которых аспаргиновая кислота заменена на аспарагин, гистидин и лизин. Активности таких модифицированных протеаз в сентезе или гидролизе Z-APM были в лучшем случае в 2 раза выше, чем активность термолизин-подобной нейтральной металло-протеазы дикого типа.
Новые модифицированные протеазы согласно настоящему изобретению представляют собой производные, имеющие триптофановой остаток вместо аспаргинокислотного остатка в 150-ом положении последовательности термолизин-подобной нейтральной металло-протеазы, SEQ 1Д N: 1 (Далее они обозначены, Как D150W). Главным образом такие новые протеазы обладают существенно усиленной активностью в синтезе и/или переваривании Z-APM. Пригодность протеаз, полученных согласно изобретению, может быть определена тестами на окончательную применимость. Обычно такое испытание проводится путем анализа активности в синтезе и/или переваривании Z-AMP и сравнения полученных активностей с активностью термолизин-подобной нейтральной металло-протеазы дикого типа, испытанной в аналогичных условиях. Такой метод описан в следующих ниже примерах.
Другие положения модифицированных протеаз (D150W) могут быть замены другими аминокислотными остатками. Так например, были синтезированы двух-сайтный мутант, в 150-ом положении которого была произведена замена аспаргиновой кислоты на триптофан, а в 227-ом положении - замена аспарагина на гистидин (D150W - N227H) и трех-сайтный мутант, в 114-ом положении которого лейцин заменяли на серин, в 150-ом положении аспаргиновую кислоту заменяли на триптофан, а в 227-ом положении аспарагин заменяли на гистидин (L144S - D150W - N227H) и было показано, что такие мутанты очень активны и стабильны в синтезе Z-AMP.
Модифицированные энзимы могут быть получены методами, известными perse специалистам в данной области.
Известны различные способы, которые могут использоваться для введения мутаций в клонированные ДНК. Так например, фрагменты мутантного nprM гена могут быть получены с использованием метода мутагенеза фаза M13 (Vandeyar с сотр. (1988) Gene, 65, 129).
Плазмидные и фаговые ДНК, используемые в качестве матриц в таком способе, могут быть производными известной плазмиды pMK1 (Kubo M., Imanaka T., (1989), J. Bacteriol, 171, 4080 - 4082). Некоторые рестриктазы могут использоваться для переваривания и клонирования фрагментов nprM гена в другую плазмиду или в фаговые векторы. Мутагенные праймеры должны быть комплементарны к одно-нитевой матричной ДНК, содержащей nprM ген, за исключением кодона (ов) для замененных аминокислотных остатков. Различные нуклеотидные последовательности также могут использоваться для таких целей. При использовании таких мутагеных праймеров, имеющих различные кодоны для замененных аминокислотных остатков, могут быть достигнуты любые желательные замены аминокислот.
С другой стороны, nprM ген может быть подвергнут мутации с помощью PCR техники (полимеразная цепная реакция) с использованием химически синтезированных праймеров (Higuchi R. , Krummel B., Saiki R. (1988), Nucleic Acids Res. 16, 7351 - 7367). В том случае, когда сайт рестрикционного энзима существует вблизи от сайта мутации, метод PCR оказывается особенно полезным. Поскольку, например, сайт расщепления для рестрикционного энзима Sphl находится вблизи кодона аспаргиновой кислоты в 150-ом положении термолизин-подобной нейтральной металло-протеазы дикого типа, мутагенные праймеры, содержащие такой Sphil сайт могут использоваться для продуцирования мутантов в 150-ом положении. Таким образом, мутагенный праймер используется в качестве смыслового праймера. В качестве обратно-направляющего праймера (антисмыслового) может использоваться олигонуклеотид, который комплиментарен nprM гену вправо, например, от сайта расщепления AatI nprM гена.
Два способа могут использоваться для осуществления мутагенеза на более, чем одном сайте. Один способ заключается в осуществлении одновременного мутагенеза на всех сайтах-мишенях, тогда как другой способ включает введение мутаций последовательно. Оба способа обеспечивают получение плазмид с мутациями на более, чем одном сайте.
Основной способ получения рекомбинантной термолизин-подобной нейтральной металло-протеазы описан в литературе (Kubo M., Imanaka T. (1989) J.Bacteriol, 171, 4080 - 4082) и он включает: вставку ДНК, кодирующий модифицированную термолизин-подобную нейтральную металло-протеазу, в вектор экспрессии, использование такого вектора для трансформации клетки-хозяина, культивирование трансформанта до аккумуляции модифицированной металло-протеазы в культуре и последующее отделение модифицированного энзима от культуры. Однако, плазмида pMK1, используемая в этой ссылке, имеет размер более 20 kb и поэтому довольно трудно трансформировать Escherichia coli с помощью указанной плазмиды. Кроме этого было обнаружено, что в случае Bacillus subtilis плазмида pMK1 в значительной мере выбирает на последней стадии культивирования.
В связи с этим, с целью снятия указанных проблем авторы изобретения сконструировали бифункциональные векторы, с помощью которых оба хозяина, Escherichia coli и Bacillus subtilis, способны подвергаться трансформации и которые способны экспрессировать nprM ген в этих хозяевах. Как показано на рис. 1 - 4 таким образом были сконструированы два бифункциональных вектора, содержащих nptM ген (pUBTZ1 и pUBTZ2). При их использовании для трансформации таких штаммов Escherichia coli, как HB101 и JM103, nprM ген экспрессируется в таких штаммах. Кроме того, трансформация таких штаммов разновидности Bacillus subtilis, как DB104, DB117 и MT-2, такими плазмидами приводит к успешной экспрессии nprM гена. Кроме этого, на последней стадии культивирования не наблюдалось их выхода из процесса.
Аналогичные результаты и преимущества использования таких бифункциональных векторов достигались при использовании модифицированных термолизин-подобных нейтральных металло-протеазных генов, вместо гена дикого типа.
Модифицированная термолизин-подобное нейтральная металло-протеаза может вырабатываться в рекомбинантных бактериях и секретироваться в культуральную среду. Такие протеазы могут быть извлечены осаждением с помощью сульфата аммония и очищены до гомогенности традиционными способами, например, хроматографией гидрофобного взаимодействия и/или гельфильтрацией.
Модифицированные протеазы могут использоваться для синтеза Z-AMP, являющегося предшественником аспартама, более эффективно, чем термолизин-подобная нейтральная металло-протеаза дикого типа. На этот факт указывает сравнение активностей таких модифицированных протеаз и энзимов дикого типа в переваривании Z-APM и синтезе Z-APM. Такие активности оказались значительно выше, чем у энзима дикого типа и у модифицированных протеаз, описанных в упомянутой выше заявке на Европейский патент (заявка N 93200773.5). Измеренные значения таких активностей будут приведены в следующих ниже примерах.
Как отмечалось выше, новые модифицированные протеазы согласно изобретению представляют собой термолизин-подобные нейтральные металло-протеазы с последовательностью SEQ 1D No:1, в которой 150-ый аспаргиновокислотный остаток заменен на триптофан (D150W).
Следует отметить, что активность казеинового переваривания не связана с активностью в синтезе или переваривании Z-APM. При сравнении активностей мутантных энзимов для казеина и Z-APM совершенно очевидно, что даже если активность в переваривании казеина понижается, то активность в синтезе и/или переваривании Z-APM может значительно увеличиваться. Следующие ниже примеры представлены лишь в целях иллюстрации настоящего изобретения и никоим образом не ограничивают его сферу.
Пример 1.
/Синтез модифицированной протеазы, в последовательности которой 150-ый кислотный остаток заменен с аспаргиновой кислоты на триптофан (D150W)/
a) Конструирование экспрессирующей плазмиды pUBTZ2, содержащей nprM ген дикого типа.
Из плазмиды pMK4 (Yamada с сотр., (1991) Gene, 99, 109 - 114), фрагмент ДНК размером примерно 1,0 kb, содержащий часть nprM гена, который был получен расширением BcLI клонировали в BamHI-сайт плазмиды pUC9 с целью конструирования плазмиды pUCTZ37 (Рис. 1).
Плазмида pUCTZ37 была неполной и не содержала 5'-концевого участка nprM гена. Плазмиду pUCTZ37 расщепляли рестрикционной эндонуклеазой HindIII и HindIII фрагмент pMK4 размером 1,2 kb клонировали в более крупный фрагмент pUCTZ37 для конструирования плазмиды pUCTZ47 (Рис. 2). Рекомбинантная плазмида pUCTZ47 содержит последовательность nprM полной длины и его транскрипционную промоторную последовательность.
Для конструирования бифункциональных векторов для Escherichia coli и Bacillus subtilis, как pUCTZ47, так и pUB110 (Keggins K.M. с сотр., Proc, Natl. Ac. Sci USA (1978), 75, 1423 - 1427) расщепляли EcoRI и лигировали T4 ДНК лигазой с целью конструирования плазмиды pUBTZ1, как это показано на Рис. 3.
Наконец, фрагмент ДНК между рестрикционными сайтами Smal и Pvull подвергали делеции из плазмиды pUBTZ1, как это показано на рис. 4, с целью конструирования плазмиды pUBTZ2.
Плазмида pUBTZ2 имеет единственные BamHI, SphI и AatI рестрикционные сайты в nprM гене.
b) Мутагенез сайта 150 Trp
Олигонуклеотиды, используемые для мутагенеза, синтезировали с использованием ДНК синтезатора модели 380B, выпускаемого Апплайд Биосистемс Ко. Лтд. Нуклеотидная последовательность праймера мутагенеза была следующей
(SEQ 1D No:2)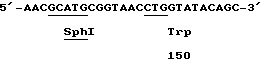
Кроме этого синтезировали обратно-направляющий праймер с нуклеотидной последовательностью, показанной ниже.
(SEQ 1D No:3)
5'-GAGATACCACTTTATTTCACCCCT-3'
1 нг плазмиды pUBTZ2 растворяли в 100 мкл реакционной смеси для PСR (67 мМ Трис-гидрохлорида (pH 8.8), 16,6 мМ сульфата аммония, 6.7 мМ MgCl2, 10 мМ 2-меркаптоэтанола, 0.05 мМ dATP, 0.05 мМ dTTP, 0.05 мМ dGTP, 0,05 мМ dCTP, 1 мкМ праймера мутагенеза, 1 мкМ обратно-направляющего праймера) и добавляли 1 единицу Tth ДНК-полимеразы. Раствор покрывали одной каплей минерального масла. Денатурацию в течение 1 минуты при 93oC, отжиг в течение 1 минуты при 45oC и удлинение в течение 45 секунд при 72oC повторяли 30 раз. После завершения реакции водный слой удаляли, экстрагировали фенолом и обрабатывали этанолом для выделения амплифицированной ДНК.
20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7.5, 10 мМ MgCl2, 0.1 M NaCl, 1 мМ ДТТ), содержащей половинное количество амплифицированной ДНК переваривали 5 единицами каждого из рестрикционных энзимов SphI и AatI при 37oC в течение 2 часов и инкубировали в течение 5 минут при 70oC. Мутированный SphI - AatI фрагмент лигировали с SphI - AatI фрагментом pUBTZ2 (7.6 kb) с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM103, используя традиционный способ с получением трасформанта JM103/pUBTZ2 (D150W). Замещенные аминокислоты подтверждали путем определения нуклеотидной последовательности такой плазмиды.
c) Приготовление очищенного мутантного энзима из рекомбинантной Bacillus subtilis
Описанную выше плазмиду ДНК экстрагировали быстрым щелочным-SDS методом (Maniatis T., Fritsch E.F., Sambrook Jr., (1989), Молекулярное клонирование: лабораторное руководство (2-ое изд.) Колд Спринг Харбор Лаборатори Пресс, Колд Спринг Харбор, Н-Й. США. 1.25 - 1.28). Трансформацию Bacillus subtilis MT-2 штамма осуществляли способом компетентных клеток (Harby K. (1985) в: Glover D. , ред. Клонирование ДНК, т. 11 (1-е изд.), IRL) Пресс Лимитед. Оксфорд. Англия, 1 - 17).
Одну колонию полученного таким образом трансформанта Bacillus subtilis MT-2/pUBTZ2 (D150W) переносили в 5 мл LB среды, содержащей канамицин (5 мкг/мл) и инкубировали в течение ночи при 37oC. Культуру переносили в 500 мл 2 L среды (2% триптона Бакто, 1% дрожжевого экстракта, 0.5% NaCl), содержащей канамицин (5 мкг/мл) и инкубировали в течение 20 часов при 37oC. Культуральный бульон центрифугировали в течение 30 минут со скоростью 8000 об/мин для удаления бактерий, к супернатанту добавляли сульфат аммония до достижения 60% насыщения и смесь перемешивали в течение ночи при 4oC.
Осадок удаляли центрифугированием и растворяли в 10 мл буфера A (20 мМ Трис-гидрохлорида при pH 9.0 10 мМ CaCl2). Раствор наносили на 20 мл Butyl-Toyopearl, после чего элюировали буфером A при скорости потока 1,5 мл/мин. Активные фракции объединяли и обессоливали с помощью 60% насыщенного сульфата аммония. Осадок собирали центрифугированием в течение 30 минут со скоростью 15000 об/мин и растворяли в 5 мл буфера B (10 мМ Трис-гидрохлорида при pH 7,0, 0,1 M NaCl, 10 мМ CaCl2). Энзимный раствор дополнительно пропускали через гель-фильтрационную колонку (TSK Гель G2000( 21,5 x 300 мм)), с последующим элюированием буфером B с объемной скоростью 1 мл/мин. Активные фракции объединяли с получением очищенного энзима.
На рис. 5 показана схема, используемая для конструирования рекомбинантной плазмиды pUBTZ2 (D150W).
Пример 2
/Синтез модифицированной протеазы с двойной заменой на 150-ом аминокислотном остатке аспаргиновой кислоты на триптофан и на 227-ом аминокислотном остатке аспарагина на гистидин (D150W - N227H)/
D150W - N227H двух-сайтный мутант термолизин-подобной нейтральной металло-протеазы конструировали следующим образом:
а) мутагенез сайта 227 His
1 мкг плазмиды рМК1, содержащей термолизин-подобный нейтральный металло-протеазный ген nprM, производный из Bacillus stearothermophilus MK 232 (Kubo M. , и Imanaka T (1989) J. Becteriol 171, 4080 - 4082) переваривали 5 единицами каждого из рестрикционных энзимов Pst I и BamHI в 20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 M NaCl, 1 мM ДДТ) при 37oC в течение 2 часов. Образец подвергали гельэлектрофорезу на 1% агарозе и фрагмент ДНК размером примерно 3,5 kb выделяли и очищали с помощью набора для очистки ДНК Био-101 Ген Клин.
Отдельно, 1 мкг плазмиды pUC9 переваривали 5 единицами каждого из рестрикционных энзимов PstI и BamHI в 20 мкл такой же реакционной среды, что указана выше в течение 2 часов при 37oC.
PstI - BamHI фрагмент nprM гена лигировали с PstI - BamHI фрагментом pUC9 с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM109 традиционным способом с получением рекомбинантной плазмиды (pUCTZ55), содержащей PstI - BamHI фрагмент nprM гена (рис. 6).
1 мкг рекомбинантной плазмиды pUCTZ55 переваривали 5 единицами каждого из SphI и BClI в 20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 M NaCl, 1 мМ ДДТ) при 37oC в течение 2 часов. Образец подвергали гельэлектрофорезу на 1% агарозе и фрагмент ДНК размером примерно 550 bp выделяли и очищали с использованием набора для очистки ДНК Био-101 Ген Клин.
Отдельно, 1 мкг фагового вектора M13mp18 переваривали 5 единицами каждого из рестрикционных энзимов SphI и BamHI в 20 мкл такой же реакционной среды, что указана выше, в течение 2 часов при 37oC.
SphI - BclI фрагмент nprM гена лигировали с SphI - BamHI фрагментом M13mp18 с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Esherichia coli JM109 традиционным способом с получением рекомбинантного фага (M13TZSp-Bc), содержащего SphI - BclI фрагмента nprM гена (рис. 7).
Однонитевую ДНК получали из M13TZSp-Bc традиционным способом и подвергали мутагенезу. Олигонуклеотиды, используемые для мутагенеза, получали с использованием ДНК-синтезатора фирмы Апплайд Биосистемс модель 380В.
Мутагенный олигонуклеотид, используемый для замены 227-го остатка (аспарагин на гистидин), показан ниже.
(SEQ 1Д No:4)
Мутагенез осуществляли с использованием USBT7-GEN in vitro мутагенного набора, после чего устанавливали последовательность ДНК для подтверждения мутации.
Двунитевую ДНК мутированной M13TZSp - Bc получали традиционным способом и 1 мкг двунитевой ДНК переваривали 5 единицами каждого из рестрикционных энзимов SphI и AatI в 20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 М NaCl, 1 мМ ДДТ) в течение 2 часов при 37oC и подвергали электрофорезу на 1% агарозном геле. Фрагмент ДНК размером около 550 bp выделяли из M13TZSp - Bc переваров и фрагмент ДНК очищали с использованием набора для очистки ДНК Био-101 Ген Клин.
Плазмиду pUBTZ2 переваривали рестрикционными энзимами SphI и AatI и выделяли фрагмент размером 7,6 kb. Мутированный SphI и AatI фрагмент nprM гена (около 550 bp) лигировали с помощью полученного таким образом SphI и AatI фрагмента pUBTZ2 с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM103 традиционным способом с получением рекомбинантной плазмиды pUBTZ2 (N227H) (рис. 8).
b) Мутагенез сайта 150 Trp и получение мутантного энзима (D150W - N 227H)
Плазмиду pUBTZ2 (N227H) использовали в качестве матрицы для полимеразной цепной реакции. Использовали мутагенезный праймер последовательности SEQ 1Д No:2 и обратно-направляющий праймер последовательности SEQ 1Д No:3.
1 нг плазмиды pUBTZ2 (N227H) растворяли в 100 мкл реакционной смеси PCR (67 мМ Трис-гидрохлорида при pH 8,8, 16,6 мМ сульфата аммония, 6,7 мМ MgCl2, 10 мМ 2-меркаптоэтанола, 0,05 мМ dATP, 0,05 мМ dTTP, 0,05 мМ dGTP, 0,05 мМ dCTP, 1 мкМ мутагенезного праймера, 1 мкм обратно-направляющего праймера) и добавляли 1 единицу Thh ДНК полимеразы. Раствор покрывали одной каплей минерального масла. Денатурацию в течение 1 минуты при 93oC, отжиг в течение 1 минуты при 45oC и удлинение в течение 45 секунд при 72oC повторяли 30 раз. После завершения реакции водный слой удаляли, экстрагировали фенолом и обрабатывали этанолом для извлечения амплифицированной ДНК (D150W - N227H).
20 мкл реакционной среды (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ ДДТ), содержащей половинное количество амплифицированной ДНК переваривали 5 единицами каждого из рестрикционных энзимов SphI и AatI при 37oC в течение 2 часов и инкубировали в течение 5 минут при 70oC. Мутированный SphI - AatI фрагмент лигировали с 7,6 kb SphI - AatI фрагментом pUBTZ2 с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM103 традиционным способом с получением трансформанта JM103/pUBTZ2 (D150W - N227H). Замещенную аминокислоту подтверждали определением нуклеотидной последовательности такой плазмиды.
Плазмидную ДНК использовали для трансформации Bacillus subtilis MT-2 и модифицированную протеазу (D150W - N227H) получали тем же способом, что описан в примере 1.
На рис. 9 показана схема, используемая для конструирования рекомбинантной плазмиды pUBTZ2 (D150W - N227H).
Пример 3
/Синтез модифицированной протеазы с тройным замещением: лейцина на серин на 144-ом аминокислотном остатке, аспаргиновой кислоты на триптофан на 150-ом аминокислотном остатке и аспарагина на гистидин на 227-ом аминокислотном остатке (L144S - D 150W - N227H)/
Трех-сайтный мутант термолизин-подобный нейтральный металло-протеазы конструировали следующим образом.
a) Мутагенез сайта 144 Ser.
Мутагенный олигонуклеотид, используемый для замены 144-го остатка (лейцина на серин), показан ниже.
(SEQ 1Д No: 5)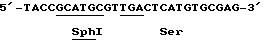
Кроме этого синтезировали смысловой праймер с нуклеотидной последовательностью, показанной ниже.
(SEQ 1Д No: 6)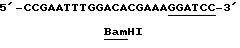
1 нг плазмиды pUBTZ2 растворяли в 100 мкл реакционной смеси для PCR (67 мМ Трис-гидрохлорида при pH 8,8, 16,6 мМ сульфата аммония, 6,7 мМ MgCl2, 10 мМ 2-меркаптоэтанола, 0,05 мМ dATP, 0,05 мМ dTTP, 0,05 мМ dGTP, 0,05 мМ dCTP, 1 мкМ мутагенезного праймера, 1 мкм смыслового праймера), и добавляли 1 единицу Tth ДНК полимеразы. Раствор покрывали одной каплей минерального масла. Денатурацию в течение 1 минуты при 93oC, отжиг в течение 1 минуты при 45oC и удлинение в течение 45 секунд при 72oC повторяли 30 раз. После завершения реакции водный слой удаляли, экстрагировали фенолом и обрабатывали этанолом для извлечения амплифицированной ДНК.
20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 М NaCl, 1 мМ ДДТ), содержащей половинное количество амплифицированной ДНК, переваривали 5 единицами каждого из рестрикционных энзимов BamI и SphI при 37oC в течение 2 часов, после чего инкубировали в течение 5 минут при 70oC. Мутированный BamI - SphI фрагмент лигировали с BamI - SphI фрагментом pUBTZ2 (7,4 kb) с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM103 традиционным методом с получением трансформанта JM103/pUBTZ2 (L144S). Замещенную аминокислоту подтверждали определением нуклеотидной последовательности такой плазмиды.
На рис. 10 приведена схема, используемая для конструирования рекомбинантной плазмиды .
b) Конструирование плазмиды pUBTZ2 (L144S - D150W - N227H)
20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 М NaCl, 1 мМ ДДТ), содержащей 1 мкг pUBTZ2 (D150W - N227H), полученной в примере 2, переваривали 5 единицами каждого из рестрикционных энзимов SphI и AatI в течение 2 часов при 37oC и инкубировали в течение 5 минут при 70oC. Мутантный SphI - AatI фрагмент лигировали с 7,6 kb SphI - AatI фрагментом pUBTZ2 (L144S) с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli JM103 традиционным способом с получением трансформанта JM103/pUBTZ2 (L144S - D150W - N227H). Замещенную аминокислоту подтверждали определением нуклеотидной последовательности такой плазмиды.
Плазмидную ДНК использовали для трансформации Bacillus subtilis MT-2 и модифицированную протеазу (L144S - D150W - N227H) получали тем же способом, что описан в примере 1.
На рис. 11 приведена схема, используемая для конструирования рекомбинантной плазмиды pUBTZ2 (L144S - D150W - N227H).
Пример 4.
/Определение активности модифицированных протеаз/
(1) Активность в синтезе Z-APM
Z-APM синтетическую активность определяли методом жидкостной хроматографии высокого давления (HPLC) после реакции конденсации бензилоксикарбонил-L-аспаргиновой кислоты (Z-Asp) с гидрохлоридом метилового эфира L-фенилаланина (L-PM). Мутантную протеазу инкубировали с 0,1 M Z-Asp и 0,1 M L-PM в 0,1 M Трис-малеатном буфере (pH 6 и 7) при 35oC в течение 30 минут. Реакцию обрывали добавлением равного объема 0,125 М ЕДТА. Количество синтезированной Z-APM определяли методом HPLC на колонке с космосил C-18 (Nakalai tesgue). HPLC осуществляли с помощью 60 мМ Триэтиламин-фосфатного буфера (pH 3,0), содержащего 40% ацетонитрила в качестве элюента, при скорости потока 1,0 мл/мин. и элюированный Z-APM определяли по поглощению при длине волны 224 нм. Активность синтеза 1 моля Z-APM в секунду определяли, как 1 катал (kat).
В целях сравнения авторы изобретения также синтезировали и исследовали все другие D150 мутанты с использованием случайного мутагенезного праймера.
Нуклеотидная последовательность случайного мутагенезного праймера была следующей: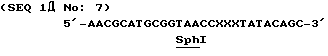
Кодон 150-ой аминокислоты где каждый Х, независимо друг от друга, представляет собой G, A, T или C.
Такой праймер имеет изменчивость на команде 150-го аминокислотного остатка и может вводить все 20 аминокислотных остатков в 150-е положение. Авторы изобретения ввели различные мутации в 150-е положение с использованием мутагенезного праймера и изучили их все, за исключением триптофана.
1 нг плазмиды pUBTZ2 растворили в 100 мкл реакционной смеси PCR (67 мМ трис-гидрохлорида при pH 8,8, 16,6 мМ сульфата аммония, 6,7 мМ MgCl2, 10 мМ 2-меркаптоэтанола. 0,05 мМ dATP 0,05 мМ dTTP, 0,05 мМ dGTP, 0,05 dCTP, 1 мкМ мутагенезного праймера, 1 мКм обратно-направляющего праймера) и добавляли 1 единицу Tth ДНК полимеразы. Раствор покрывали одной каплей минерального масла. Денатурацию в течение 1 минуты при 93oC, отжиг в течение 1 минуты при 45oC и удлинение в течение 45 секунд при 72oC повторяли 30 раз. После завершения реакции водный слой удаляли, экстрагировали фенолом и обрабатывали этанолом для извлечения амплифицированной ДНК.
20 мкл реакционной смеси (50 мМ Трис-гидрохлорида при pH 7,5, 10 мМ MgCl2, 0,1 M NaCl, 1 мМ ДТТ), содержащей половинное количество амплифицированной ДНК, переваривали 5 единицами каждого из SphI и AatI при 37oC в течение 2 часов и инкубировали в течение 5 минут при 70oC. Мутантный SphI - AatI фрагмент лигировали с 7,6 kb SphI - AatI фрагментом pUBTZ2 с использованием набора для лигирования ДНК Takara Shuzo. Лигирующую смесь использовали для трансформации Escherichia coli Jm103 традиционным способом с получением трансформанта JM103/pUBTZ2. Замещенную аминокислоту подтверждали определением нуклеотидной последовательности плазмиды.
Плазмидную ДНК, за исключением D150W мутанта, выделяли быстрым щелочным - SDS методом. Трансформацию штамма Bacillus subtilis МТ-2 проводили способом компетентных клеток.
Одну колонию каждого различного Bacillus subtilic МТ-2/pUBTZ2 (мутантного) трансформанта инокулировали в 5 мл LB среды, содержащей канамицин (5 мкг/мл), и инкубировали при 37oC в течение ночи. Культуру переносили в 500 мл 2L среды (2% Бакто триптона, 1% дрожжевого экстракта, 0,5% NaCl), содержащей канамицин (5 мкг/мл), и инкубировали при 37oC в течение 20 часов. Культуральный бульон центрифугировали со скоростью 8000 об/мин в течение 30 минут для удаления бактерий, к супернатанту добавляли сульфат аммония до достижения 60% насыщения и смесь перемешивали в течение ночи при 4oC.
Осадок извлекали центрифугированием и растворяли в 10 мл буфера A (20 мМ Трис-гидрохлорида при pH 9,0,10 мМ CaCl2). Энзимный раствор наносили на 20 мл Butil-Toypearl с последующим элюированием буфером A при скорости потока 1,5 мл/мин. Активные фракции объединяли и подвергали обработке 60% насыщенным сульфатом аммония. Осадок собирали центрифугированием со скоростью 15000 об/мин в течение 30 минут и растворяли в 5 мл буфера B (20 мМ Трис-гидрохлорида при pH 7,5, 10 мМ CaCl2). Энзимный раствор дополнительно пропускали через гель-фильтрационную колонку (TSK Гель G2000SW (21,5х300 мМ)), с последующим элюированием буфером B при скорости потока 1 мл/мин. Активные фракции объединяли с получения каждого очищенного энзима.
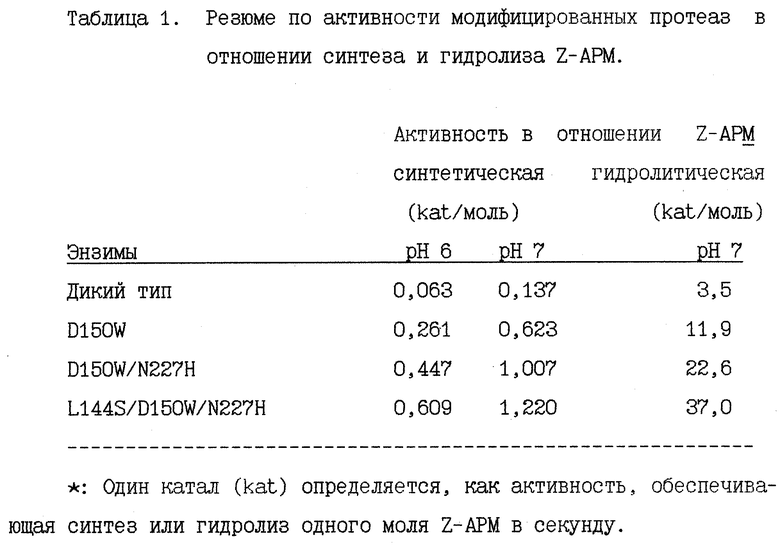
Синтетические активные модифицированных протеаз в которых 150-ый аспаргиново-кислотный остаток заменен на другие аминокислотные остатки (D150W мутанты) показаны на рис. 12. Мутант, у которого 150-ый аспаргиновокислотный остаток заменен на триптофан (D150W) проявляет явно более высокую удельную активность примерно в 4 раза более высокую, чем активность термолизина дикого типа (D), при этом большинство других мутантов проявляет более высокие активности, чем термолизин дикого типа (D), однако, эти активности значительно ниже, чем активность D150W. Совершенство очевидно, что триптофановый мутант обладает наивысшей активностью.
Активность, определенная для двух дополнительных мультисайтных мутантов, а именно, 2-сайтного мутанта D150W - N227H (т.е. при замене аспаргиновой кислоты на триптофан в положении 150 и аспарагина на гистидин в положении 227), а также 3-сайтного мутанта L144S - D150W - N227H (т.е. при замене лейцина на серин в положении 144, аспаргиновой кислоты на триптофан в положении 150 и аспарагина на гистидин в положении 227) еще выше, как это следует из данных, представленных в таблице 1. (2) Z-APM гидролитическая активность.
Гидролиз Z-APM в Z-Asp и PM в присутствии модифицированных протеаз измеряли, наблюдая уменьшение поглощения при длине волны 224 нм в соответствии с методом Inoue (Inoue K., (1992), J. Biochem 112, 335-340). Три мл 1 мМ Z-APM, растворенного в 0,1 М Трис-гидрохлоридном буфере (pH 7,0), инкубировали в присутствии модиицированных протеаз при 35oC и регистрировали уменьшение оптического спектрального поглощения при длине волны 224 нм. Количество гидролизованного Z-APM определяли по разнице в молярном коэффициенте поглощения Δε 224, которая согласно расчету составила - 493 (М-1. см-1).
Активности D150 мутантов показаны на рис. 13. D150W мутант обладает весьма высокой активностью, которая примерно в 4 раза выше активности термолизина дикого типа. Большинство других мутантов обладают активностями в гидролизе Z-APM, которые лишь в 1-3 раза выше активности термолизина дикого типа (D). Триптофановый мутант обладает явно более высокой активностью.
Активности D150W - N227H и L144S - D150W - Т227H также указаны в таблице 1. Их активности в отношении гидролиза Z-PMA в 6-7 раз и в 9-10 раз выше чем активность дикого типа.







Модифицированная протеаза термолизин-подобной нейтральной металлопротеазы имеет аминокислотную последовательность SEQ IDN:1, в которой 150-й остаток аспарагиновой кислоты замещен на триптофан. Возможно также проведение замещения 227-го остатка аспарагина на гистидин и/или 144-го остатка лейцина на серин. Модифицированная протеаза способна катализировать процесс синтеза и расщепления метилового эфира бензилоксикарбонил--α--L-аспартил-L-фенилаланина. 3 с. и 3 з.п.ф-лы, 13 ил., 1 табл.
| T.Jmanaka et al | |||
| "A new way of enhancing the thermostability of proteases" Nature, 1986, v | |||
| Телефонный аппарат, отзывающийся только на входящие токи | 1921 |
|
SU324A1 |
| 695 - 697 | |||
| Способ получения изделий типа стаканов с фасонной боковой поверхностью | 1975 |
|
SU616033A1 |
| Штамм бактерий RноDососсUS RноDоснRоUS - продуцент нитрилгидратазы | 1990 |
|
SU1731814A1 |
