Изобретение относится к способу получения цис-(+)-гидрокси-5-[2-(диметиламино)этил]-2,З-дигидро-2-(4- метоксифенил)-бензотиазепин-4(5H)-она, имеющего структуру (I)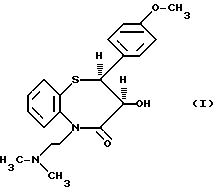
цис-(+)-Гидрокси-5-[2-(диметиламино)этил] -2,3- дигидро-2-(4-метоксифенил)-бензотиазепин-4(5H)-он является подходящим промежуточным соединением при получении дилтиазема (II).
Дилтиазем в настоящее время используется при лечении сердечно- сосудистых заболеваний, в частности при лечении стенокардии.
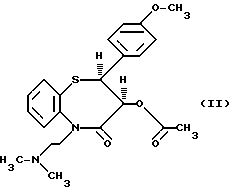
Получение дилтиазема описывается в патенте США 3582257, где в качестве исходного материала используется цис-(+)-3-гидрокси-2- (4-метоксифенил)-2,3-дигидро-1,5-бензо-тиазепин-4(5H)-он (III)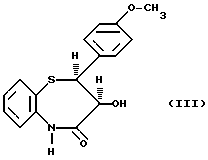
В процессе, описанном в упомянутом патенте, данное соединение взаимодействует с 2-хлорэтилдиметиламином (IV)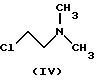
образуя промежуточное соединение (I).
В качестве возможных растворителей в данной реакции N- алкилирования описываются диметилсульфоксид, толуол, ксилол и диоксан. Основанием, используемым в реакции, описанной в патенте США 3582257, является металлический натрий, амид натрия или гидрид натрия. Согласно патенту, особенно хорошо подходят для данной реакции, в частности диметилсульфоксид в сочетании с гидридом натрия. В связи с данной реакцией существует очевидный риск взрыва (Chem. Eng. News, 44 (15), 48 (1966), и, кроме того, диметилсульфоксид трудно регенерировать, так как он полностью растворяется в воде и имеет высокую температуру кипения.
Европейский патент ЕР 81234 описывает усовершенствованный способ N-алкилирования, с помощью которого удается избежать недостатки способа вышеупомянутого патента. В данном процессе соединение (III) взаимодействует с соединением (IV) с образованием соединения (I). Реакцию осуществляют в присутствии гидроксида калия в ацетоне или в присутствии карбоната калия в растворителе, который выбирают среди ацетона, низшего алкилацетата, смеси воды и ацетона или смеси низшего алкилацетата и воды. Однако при применении в промышленном масштабе данный процесс имеет серьезные недостатки. Во всех процессах, описанных в примерах, продукт после множества стадий обработки кристаллизуют из этанола в виде его гидрохлорида, чтобы можно было получить достаточно чистое промежуточное соединение. Когда в качестве растворителя применяют ацетон, последующая обработка осложняется, так как ацетон полностью смешивается с водой, и обычное удаление солей не может быть осуществлено с применением воды. Сначала необходимо заменить растворитель, например, толуолом, который затем после промывки от солей заменяют этанолом, чтобы можно было выкристаллизовать продукт и посредством этого освободить его от примесей. Во всех примерах, описанных в упомянутом патенте, в реакции алкилирования используют 8-10-кратное количество (в мл) растворителя на грамм исходного вещества, подвергаемого алкилированию. В описательном разделе патента количество растворителя ограничивается 5-15- кратным относительно исходного материала.
Лабораторные эксперименты заявителей показывают, что те примеры по патенту, в которых растворитель представляет собой ацетон или этилацетат и основание представляет собой карбонат калия, не являются воспроизводимыми. Время реакции в примерах патента варьирует от 3 часов в примере 2 до 30 часов в примере 7. Если принять во внимание время, требуемое для реакции, количество растворителя, используемое в реакции, и громоздкую и сложную последующую обработку, такие способы требуют больших производственных мощностей, большого количества рабочего времени и больших расходов энергии. Кроме того, этилацетат гидролизуется в щелочных условиях, и, следовательно, его регенерация для повторного применения является проблематичной.
В патенте США 3582257 алкилирование промежуточного соединения (III) происходит при применении соединения (IV) в форме основания, так же, как и в заявке на патент ЕР 594101, в которой реакцию N-алкилирования осуществляют в толуоле с карбонатом натрия в качестве основания.
Данная реакция также описывается в международной заявке на патент 92/10485, в которой растворителем является толуол вместе со вспомогательным растворителем - диметилформамидом или N-метилпирролидин-2-оном и водой, а основанием является карбонат калия. Однако данная реакция требует применения катализатора переноса фаз.
Заявители обнаружили, к удивлению, что в процессе согласно настоящему изобретению реакция N-алкилирования протекает быстро и с отличным выходом, когда растворителем является смесь толуола и N-метилпирролидин-2-она, и основанием является тонко измельченный карбонат натрия. Толуол требуется в количестве, только в четыре раза превышающем количество исходного вещества. Воду, образовавшуюся при реакции, удаляют из смеси отделением. Время реакции составляет 1/2 - 7 час, в зависимости от температуры, при которой проводится реакция. N-Метилпирролидин-2-он требуется для растворения 2-хлордиметиламина и для обеспечения быстрого протекания реакции применяется в количестве 0,5 г на один грамм исходного вещества. Обработка после реакции значительно проще, так как соли можно вымыть из реакционной смеси, просто добавляя к реакционной смеси воду и разделяя слои. Такая процедура возможна, так как толуол отделяется от воды в отличие от ацетона. Преимуществом является также то, что полученное в качестве продукта промежуточное соединение (I) является очень чистым, и отдельной кристаллизации не требуется; после отделения водного слоя можно продолжить процесс непосредственно с промежуточным соединением (I) путем отгонки толуола для повторного использования и подвержения остатка после перегонки взаимодействию с ангидридом уксусной кислоты с образованием дилтиазема без растворителя.
Способ согласно настоящему изобретению характеризуется тем, что соединение формулы (III)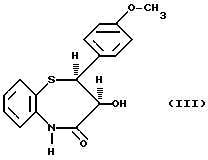
подвергают взаимодействию с соединением формулы (IV)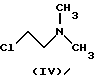
в смеси толуола и N-метилпирролидин-2-она в присутствии тонко измельченного карбоната натрия, в результате чего получают нужный промежуточный пункт (I)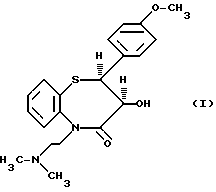
из которого можно получить дилтиазем (II) непосредственно при воздействии промежуточного соединения (I), после удаления растворителя, с ангидридом уксусной кислоты.
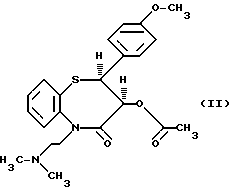
При получении соединения (I) по способу настоящего изобретения соединение (III) подвергают взаимодействию с соединением (IV) в смеси N-метилпирролидин-2-она с толуолом, которая содержит толуол в количестве 50-95%, предпочтительно-88%, и N- метилпирролидин-2-он в количестве 5-50%, предпочтительно - 12%, в присутствии тонко измельченного карбоната натрия, имеющего размер частиц 2-30 мкм, предпочтительно - 8 мкм, при температуре 80-120oC, предпочтительно - 115oC, причем время реакции составляет 1/2 - 7 часов, наиболее предпочтительно - 1 час. Растворитель реакции N-алкилирования может быть удален перегонкой, и остаток от перегонки вводят во взаимодействие с ангидридом уксусной кислоты - без растворителя, с образованием дилтиазема (II), из которого затем можно получить соответствующую хлористоводородную соль известным способом.
Способ, соответствующий настоящему изобретению, не требует отдельной установки для освобождения гидрохлорида 2- хлорэтилдиметиламина, в противоположность заявке на патент ЕР 594101, в которой данное канцерогенное и высокотоксичное вещество высвобождают в форме свободного основания из его хлористоводородной соли в отдельном реакторе перед проведением реакции N-алкилирования. В последнем случае упомянутое соединение после регулирования величины pH экстрагируют толуолом из воды, в которой неизбежно будет оставаться часть этого токсичного и хорошо растворимого в воде хлорированного амина, что вызывает значительные проблемы обработки отходов. В процессе настоящего изобретения лишь необходимое количество амина в виде его гидрохлорида загружают непосредственно в реакционный сосуд. В процессе согласно заявке на патент ЕР 594101 данного канцерогенного вещества требуется почти на 70% больше - на один килограмм исходного вещества-, чем в процессе настоящего изобретения, и аналогичным образом карбоната натрия на один килограмм исходного вещества также требуется приблизительно на 60% больше. Кроме того, процесс настоящего изобретения требует на 45% меньше толуола. Таким образом, с точки зрения экономики, требований производительности и защиты окружающей среды, способ согласно настоящему изобретению превосходит вышеупомянутый способ.
В процессе согласно заявке на патент WO 92/10485, толуола требуется на 125% больше, чем в процессе настоящего изобретения, и, кроме того, токсичного гидрохлорида 2-хлор-этилдиметиламина требуется в количестве на 15% больше. Этот способ предполагает применение дорогостоящего катализатора переноса фаз.
Следующие ниже примеры представлены для иллюстрации настоящего изобретения.
Пример 1. Смесь, содержащую 96 г цис-(+)-(4-метоксифенил)-3- гидрокси-2,3-дигидро-1,5-бензотиазепин-4(5H)-она, 52,8 г гидрохлорида 2-хлорэтилдиметиламина, 90 г тонко измельченного карбоната натрия, 384 мл толуола и 48 мл N-метилпирролидин-2-она нагревают с отделением воды до +115oC, и смесь перемешивают при этой температуре в течение 1 часа, после чего смесь охлаждают до 40oC, затем добавляют к ней 240 мл воды, и слои разделяют. Водный слой отбрасывают. Продукт находится в толуольном слое, который еще раз промывают водой. Процесс продолжают, отгоняя толуол для повторного использования и добавляя к остатку после перегонки ангидрид уксусной кислоты. После обработки ангидридом уксусной кислоты продукт экстрагируют толуолом, из которого его выкристаллизовывают путем добавления раствора этанола и HCl. Продукт (1) реакции алкилирования не выделяют, но определяют точный выход. После отгонки толуола остается 121,2 г промежуточного соединения. Из данного количества отвешивают образец точно 2 г, и этот образец очищают с помощью MPLC-хроматографии (жидкостная хроматография среднего давления). Выход чистого продукта составляет 1,825 г (93,2%). Чистота промежуточного продукта проверялась с помощью ЯМР- спектров.
Пример 2. Смесь, содержащую 32 г цис-(+)-(4-метоксифенил)- 3-гидрокси-2,3-дигидро-1,5-бснзотиазепин-4(5H)-она, 17,6 г гидрохлорида 2-хлорэтилдиметиламина, 30 г тонко измельченного карбоната натрия, 128 мл толуола и 16 мл N-метилпирролидин-2-она, нагревают под вакуумом -0,42 бар до +93oC, при которой смесь кипит с отделением воды. Затем вакуум отключают, смесь охлаждают до +40oC и перемешивают при этой температуре в течение 3 часов, добавляют 84 мл воды и разделяют слои. Водный слой отбрасывают. Продукт находится в толуольном слое, который еще раз промывают водой. Процесс продолжают, как в примере 1. Выход в реакции определяют так же, как в примере 1, взвешивая остаток после упаривания, который составляет 40,73 г, и очищают точно 2 г с помощью MPLC. Выход чистого промежуточного продукта после хроматографической очистки составляет 1,827 г (94,1%). Чистоту промежуточного соединения проверяют с помощью ЯМР-спектров.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3-ГИДРОКСИ-5-[2-(ДИМЕТИЛАМИНО)-ЭТИЛ]-2,3-ДИГИДРО-4-(МЕТОКСИФЕНИЛ)-1,5-БЕНЗ ОТИАЗЕПИН-4(5Н)-ОНА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОТИАЗЕПИНА, СПОСОБ ПОЛУЧЕНИЯ ЦИС-(+)-3-АЦЕТОКСИ-5-[2-(ДИМЕТИЛАМИНО)-ЭТИЛ]-2,3-ДИГИДРО-2-(4-МЕТОКСИФЕНИЛ )-1,5-БЕНЗОТИАЗЕПИН-4(5Н)-ОНА | 1995 |
|
RU2141957C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3,4-ДИГИДРОКСИ-5-НИТРОБЕНЗАЛЬДЕГИДА | 1993 |
|
RU2098405C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3,4-ДИГИДРОКСИ-5-НИТРОБЕНЗАЛЬДЕГИДА | 1995 |
|
RU2130449C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ИМИДАЗОЛА | 1992 |
|
RU2045519C1 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ С ЩЕЛОЧНЫМИ ИЛИ ЩЕЛОЧНО-ЗЕМЕЛЬНЫМИ МЕТАЛЛАМИ | 1992 |
|
RU2068844C1 |
| Способ получения производных 1,5-бензотиазепина или их фармацевтически приемлемых кислотно-аддитивных солей | 1988 |
|
SU1709908A3 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ИМИДАЗОЛА ИЛИ ЕГО НЕТОКСИЧНОЙ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 1990 |
|
RU2021263C1 |
| Способ получения производных 8-хлор-1,5-бензотиазепина или их фармацевтически приемлемых аддитивных солей кислот | 1985 |
|
SU1362401A3 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
Описывается способ получения дилтиазема по реакции цис-(+)-гидрокси-5-[2-(диметиламино)-этил] -2,3-дигидро-2-(4-метоксифенил)-бензотиазепин-4(5Н)-она, получаемого при взаимодействии цис-(+)-3-гидрокси-2-(4-метоксифенил)-2,3-дигидро-1,5-бензотиазепин-4(5Н)-она с 2-хлорэтилдиметиламином в смеси толуола и N-метилпирролидин-2-она в присутствии тонко измельченного карбоната натрия с уксусным ангидридом. Технический результат - упрощение процесса и увеличение выхода целевого продукта. 4 з.п.ф-лы.
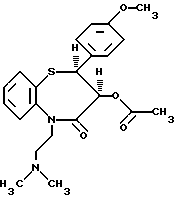
включающий реакцию соединения формулы III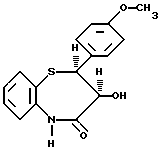
с соединением формулы IV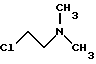
в среде растворителя в присутствии основания с последующей реакцией получаемого цис-(+)-гидрокси-5-[2-(диметиламино)-этил] -2,3-дигидро-2-(4-метоксифенил)-бензотиазепин-4(5Н)-она формулы I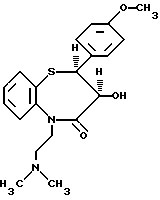
с ангидридом уксусной кислоты, отличающийся тем, что реакцию соединения III с соединением IV проводят с использованием в качестве растворителя смеси толуола и N-метилпирролидин-2-она и в качестве основания - тонко измельченного карбоната натрия в безводных условиях, после промывки реакционной смеси водой и отделения водного слоя растворители удаляют с помощью перегонки и остаток от перегонки вводят в реакцию с уксусным ангидридом.
| Способ получения производных бензотиазепина | 1982 |
|
SU1176839A3 |
| Устройство для стабилизации распределения нагрузок между двумя параллельно работающими электрическими машинами постоянного тока | 1949 |
|
SU81234A1 |
| ПОРТАЛЬНАЯ БУРОВАЯ КАРЕТКА | 0 |
|
SU353032A1 |
| УСТРОЙСТВО ПРЕОБРАЗОВАНИЯ СИГНАЛА ДЛЯ ПЕРЕДАЧИ ИНФОРМАЦИИ | 1971 |
|
SU424214A1 |
| РЕГУЛЯТОР ТЕМПЕРАТУРЫ | 0 |
|
SU338892A1 |
| УСТРОЙСТВО для ЭЛЕКТРОХИМИЧЕСКОЙ ОБРАБОТКИ ВРАЩАЮЩИМСЯ ЭЛЕКТРОДОМ-ИНСТРУМЕНТОМ _ | 0 |
|
SU320361A1 |
| Огнетушитель | 0 |
|
SU91A1 |
