Изобретение относится к применению селективно действующих соединений-агонистов человеческого рецептора CB2 для получения лекарственных иммуномодуляторов. Объектом изобретения являются также новые соединения-агонисты человеческого рецептора СВ2 и фармацевтическая композиция на их основе, а также способы их получения.
Известно, что Δ9-ТНС является основным активным компонентом, экстрагируемым из Cannabis sativa (Tuner, 1985; In Marijuana 1984; Ed Harvey, DY, IRL Press Oxford).
Многочисленные публикации описывают не только психотропные эффекты каннабиноидов, но и влияние последних на иммунную функцию (Hollister L.E., J. Psychoact. Drugs, 24 (1992), 159-164). Большинство исследований in vitro обнаружило иммуносупрессорные эффекты каннабиноидов: ингибирование пролиферативных реакций лимфоцитов Т и В, индуцированных митогенами (Luo Y.D. et al. , Int. J. Immunopharmacol. (1992) 14, 49-56; Schwartz, H. et al., J. Neuroimmunol. (1994) 55, 107-115), ингибирование активности цитотоксических клеток Т (Klein et al., J. Toxicol. Environ Health (1991) 32, 465-477), ингибирование микробиоцидной активности макрофагов и синтеза TNFα (Arata, S. et al. , Life Sci. (1991) 49, 473-479; Fisher-Stenger et al. J., Pharm. Exp. Ther. (1993) 267, 1558-1565), ингибирование цитолитической активности гранулолимфоцитов (Kusher et al., Cell Immun. (1994) 154, 99-108). В некоторых исследованиях наблюдали эффект амплификации: увеличение биологической активности интерлейкина-1 под воздействием выделенных из мышей макрофагов или дифференцированных клеточных макрофагоцитарных линий, обусловленное возросшим уровнем TNFα (Zhu et аl., J. Pharm. Exp. Ther. (1994) 270, 1334-1339; Shivers, S.C. et al., Life Sci. (1994) 54, 1281-1289).
Описанные эффекты каннабиноидов обусловлены их взаимодействием с высокоафинными специфическими рецепторами, присутствующими как на центральном уровне (Devane et al., Molecular Pharmacology (1988) 34, 605-613), так и на периферическом (Nye et al. , The Journal of Pharmacology and Experimental Therapeutics (1985) 234, 784-791; Kaminski et al., Molecular Pharmacology (1992) 42, 736-742; Munro et al., Nature (1993) 365, 61-65).
Центральные эффекты обусловлены каннабиноидными рецепторами первого типа (CB1), присутствующими в мозге. С другой стороны, Munro с сотр. (Nature (1993) 365, 61-65) клонировали второй тип каннабиноидных рецепторов, спаренных с протеином G, получивших наименование СВ2, присутствующих только на периферии, преимущественно на клетках иммунного происхождения. Присутствие каннабиноидных рецепторов СВ2 на лимфоцитных клетках может объяснить эффект иммуномодуляции, вызываемой агонистами вышеупомянутых каннабиноидных рецепторов.
Известные до настоящего времени агонисты рецепторов каннабиноидов представляют собой смешанные агонисты, т.е. действующие как на центральные рецепторы (CB1), так и на периферические (СВ2). Из указанного следует, что при лечении иммунной системы известными агонистами рецепторов каннабиноидов всегда имеет место заметный побочный эффект, а именно психотропный эффект.
Следующие патенты описывают соединения-агонисты неселективного действия: ЕР 0570920, W094-12466, описывающий анандамид; US 4371720, описывающий агонист СР 55940.
Кроме того, рецептор СВ2 известен с 1993 г., однако многочисленные патенты, относящиеся к каннабиноидным соединениям, не приводят каких-либо сведений об их селективности. Среди последних можно указать патенты US 5081122, US 5292736, US 5013837, ЕР 0444451, описывающие соединения индольной или инденовой структуры.
Другие производные индола, проявляющие активность каннабиноидного типа, описаны в статье J. W. Hufman с сотр. (Biorg. Med. Chem. Lett. (1994) 4, 563).
В настоящее время установлено, что специфические агонисты человеческого рецептора СВ2, имеющие повышенное сродство к указанному рецептору, являются сильными иммуномодуляторами, которые могут быть использованы без риска проявления указанного выше побочного эффекта.
В настоящем описании термином "повышенное сродство к человеческому рецептору СВ2" обозначают сродство, характеризуемое константой сродства, меньшей или равной 10 нМ, а термином "специфические" - обозначают соединения, константа сродства которых к рецептору СВ2 имеет величину, по меньшей мере в 30 раз ниже соответствующей константы для рецептора CB1, и для которых константа сродства к рецептору CB1 равна или выше 100 нМ. Кроме того, специфичность соединений согласно изобретению проявляется также по отношению к другим рецепторам: так, соединения согласно изобретению имеют константу ингибирования к человеческим рецепторам, отличным от рецепторов каннабиноидного типа, превышающую 1 мкМ.
Таким образом, настоящее изобретение относится к применению специфических агонистов человеческого рецептора СВ2 для получения лекарств иммуномодулирующего действия.
В качестве примера специфических агонистов по отношению к рецептору СВ2, которые подходят для целей данного изобретения, можно назвать соединения формул (I) и (I'), описанных ниже. Соединениями согласно изобретению являются соединения формулы (I) или (I') в виде чистых энантиомеров или в виде рацемических форм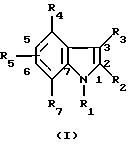
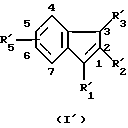
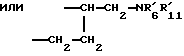
где R1 - радикал, выбранный из группы -CH2CHR10NR6R11; -(CH2)2NR'6R'11; -CHR9CH2NR'6R'11; -(CH2)nZ и -COR8;
R'1 означает группу -CH2CHR10NR6R11 или -(СН2)2NR'6R'11;
R2 и R'2 - водород, галоген или (C1-C4) алкил;
R3 - водород (C1-C4)алкил или группа, выбранная из группы -CH2CHR10NR6R11; -(CH2)2NR'6R11 и -COR8;
R'3 - группа =CR6R8;
R4 имеет одно из значений, указанных для радикала R5, или означает группу -COR8;
R5 - водород, (C1-C4)алкил, (C1-C4)алкокси, галоген, группа -СF3, -ОСF3, (C1-C4)алкилтио;
R'5 имеет одно из значений, указанных для радикала R5, и находится в положении 5 или 6 инденового цикла;
R6 - водород или (C1-C4)алкил;
R'6 - (C1-C4)алкил;
R7 имеет одно из значений, указанных для радикала R5 или R7 и R9 вместе образуют группу -Y-CH2-, соединенную через звено Y с индольным циклом в положении 7;
R8 - означает фенил, замещенный 1-4 раза заместителем, выбранным из галогена (C1-C4)алкила или (C1-C4)алкоксила; полицикл, выбранный из нафт-1-ила, нафт-2-ила, 1,2,3,4-тетрагидронафт-1-ила, 1,2,3,4-тетрагидронафт-5-ила, антрила, бензофурила, бензотиен-2-ила, бензотиен-3-ила, 2-,3-,4- или 8-хинолила, причем указанные полициклические радикалы не замещены или замещены 1-2 раза заместителем, выбранным из группы: (C1-C4)алкил, (C1-C4)алкокси, (C1-C4)алкилтио, галоген, циано, гидроксил, трифторметил и имидазол-1-ил;
R10 и R11 вместе обозначают группу, выбранную из группы -CH2-O-CH2-CR12R13- и -(CH2)p-CR12R13-, в которых атом углерода, замещенный радикалами R12 и R13, связан с атомом азота;
R'11 - алкил (C1-C4), или R'11 и R'6 образуют с атомом азота, с которым они связаны, группу, выбранную из морфолин-4-ила, тиоморфолин-4-ила, пиперидин-1-ила, пиролидин-1-ила;
R12 и R13 - означают каждый, независимо друг от друга, атом водорода или алкил (C1-C4);
n означает 2, 3, 4 или 5;
р означает 2 или 3;
Z - метил или галоген;
Y означает метиленовую группу или атом кислорода.
При условии, что в формуле (I) любой один из заместителей R1, R3 или R4 означает группу -COR8 и что:
если R1 = -COR8, то R3 представляет группу -CH2CHR10NR6R11 или -(СН2)2NR'6R'11, а R4 имеет одно из значений R5;
если R3 = -COR8, то R1 означает группу, выбранную из групп: -CH2CHR10NR6R11, -CHR9CH2NR'6R'11, -(CH2)2NR'6R'11 и -(CH2)nZ, R4 имеет одно из значений R5 и по меньшей мере один из радикалов R4, R5 и R7 является водородом;
если R4 = -COR8, то R1 означает группу, выбранную из групп: -CH2CHR10NR6R11, -CHR9CH2NR'6R'11, -(СН2)2NR'6R'11 и -(CH2)nZ, и R3 означает водород или алкил (C1-C4);
а также их фармацевтически приемлемые соли.
Из соединений общей формулы (I) и (I') предпочтительными являются соединения, в которых R2 или R'2 означают водород или метил.
Предпочтительные соединения формул (I) и (I') включают также соединения, в которых R8 означает радикал нафт-1-ил, незамещенный или замещенный в положении 4 атомом фтора, хлора, брома, группой метил-, циано-, метокси, группой имидазол-1-ил; радикал нафт-2-ил; группу бензофур-4-ил или бензофур-7-ил. Предпочтительными согласно изобретению являются также соединения формул (I) и (I'), в которых R5 или R'5 означают атом водорода.
Также предпочтительными являются соединения общих формул (I) и (I'), в которых группа NR'6R'11 означает морфолин-4-ил.
Особенно предпочтительными соединениями общей формулы (I) являются соединения, в которых:
R2 - водород или метил;
R8 - нафт-1-ил, незамещенный или замещенный в положении 4 атомом фтора, хлора, брома, группой метил-, циано-, метокси-, имидазол-1-ил, нафт-2-ил, бензофур-4-ил или бензофур-7-ил;
R5 - водород;
-NR'6R'11 означает морфолин-4-ил;
R1, R3, R4 и R7 имеют указанные выше значения.
Особенно предпочтительными соединениями общей формулы (I') являются те, в которых:
R'2 - водород или метил;
R8 - нафт-1-ил, незамещенный или замещенный в положении 4 атомом фтора, хлора, брома, группой метил-, циано-, метокси-, имидазол-1-ил, нафт-2-ил, бензофур-4-ил или бензофур-7-ил;
R'5 - водород;
-NR'6R'11 означает морфолин-1-ил;
R'1 и R'3 имеют указанное выше значение.
Из соединений формул (I) и (I') наиболее предпочтительными являются соединения, в которых R2 и R'2 означают метил.
Соединения общей формулы (I) представляют собой производные индола, замещенные в положении 1, 3 или 4 ацильной группой (-СОR8). В зависимости от положения ацильной группы соединения (I) можно разделить на три подгруппы соответственно с общими формулами (Ia), (Ib) и (Iс), приведенными ниже.
Соединения формулы (Iа) являются индолами, ацилированными в положении 1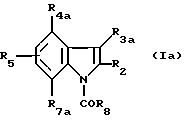
В данной формуле:
R3a означает -CH2CHR10NR6R11 или -(СН2)2NR'6R'11;
R4a - водород (C1-C4)алкил, (C1-C4)алкокси, атом галогена, группа -СF3, группа -ОСF3 или (C1-C4)алкилтио;
R7a - водород, (C1-C4)алкил, (C1-C4)алкокси, атом галогена, группа -СF3, группа -ОСF3 или (C1-C4)алкилтио;
R2, R5, R8, R6, R'6, R10, R11 и R'11 имеют значения, указанные выше для соединений формулы (I).
Из производных индола формулы (Iа) предпочтительными являются соединения, в которых:
R2 - водород или метил;
R3a- одна из нижеуказанных групп: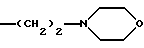
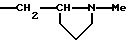
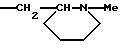
R4a, R5, R7a, каждый, означает водород.
Индолы, ацилированные в положении 3, образуют группу соединений формулы (Ib)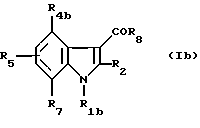
где R1b означает -CH2CHR10NR6R11, -(СН2)2NR'6R'11, -CHR9CH2NR'6R'1 и -(CH2)nZ;
R4b - водород, (C1-C4)алкил, (C1-C4)алкокси, атом галогена, группа -СF3, группа -ОСF3 или (C1-C4)алкилтио;
R2, R5, R6, R9, R10, R11, R'6, R'11, n, Z, R7 и R8 - имеют значения, указанные выше для соединений формулы (I).
Из производных индола формулы (Ib) предпочтительными являются соединения, в которых:
R1b - одна из нижеуказанных групп: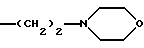
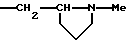
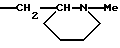
или R1b означает группу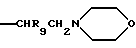
в которой R9 образует вместе с R7 группу -Y-CH2, в которой Y означает О или -СН2-, в этом случае R1b означает:
группу формулы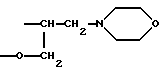
в которой кислород связан в положении 7 индольного цикла,
или группу формулы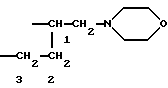
в которой углерод С3 связан в положении 7 индольного цикла;
R2 - водород или метил;
R8 - нафт-1-ил, незамещенный или замещенный в положении 4 атомом фтора, хлора, брома, группой метил-, циано-, метокси-, имидазол-1-ил; нафт-2-ил; бензофур-4-ил или бензофур-7-ил;
R4b, R5 и R7 имеют указанные выше значения.
Индолы, ацилированные в положении 4, образуют группу соединений формулы (Iс)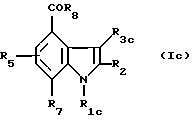
где R1c означает группу - CH2CHR10NR6R11, -(CH2)2NR'6R'11, CHR9CH2NR'6R'11 или -(CH2)nZ;
R3с - водород или (C1-C4)алкил;
R2, R5, R6, R'6, R7, R8, R9, R11, R'11, n, Z имеют значения, определенные выше для соединений формулы (I).
Из производных индола формулы (Ic) предпочтительными являются соединения, в которых:
R3с и R5 означают каждый водород;
R1c, R2, R7 и R8 - имеют указанные выше значения.
Из соединений формулы (I) особенно предпочтительными являются 1-(2-(4-морфолинил)этил)-2-метил-3-(1- нафтикарбонил)-7-метоксииндол; 1-(2-(4-морфолинил)этил)-2-метил-3-(4-хлор -1-нафтилкарбонил)-7-метоксииндол; 1-н-пентил-2-метил-3-(4-хлор-1 -нафтилкарбонил)-7-метоксииндол.
Соединения формул (I) и (I') согласно изобретению могут быть получены различными методами синтеза, включающими, в частности, стадии присоединения алкиламиногруппы, ацилирования, а также циклизации, хорошо известных специалисту.
Наиболее подходящие способы получения соединений согласно изобретению описаны, в частности, в патентах NL 7308094, US 5109135, US 4939138, US 5-081122, US 4840950, ЕР 0278265, US 5292736 и US 4581354.
Эти способы кратко изложены ниже.
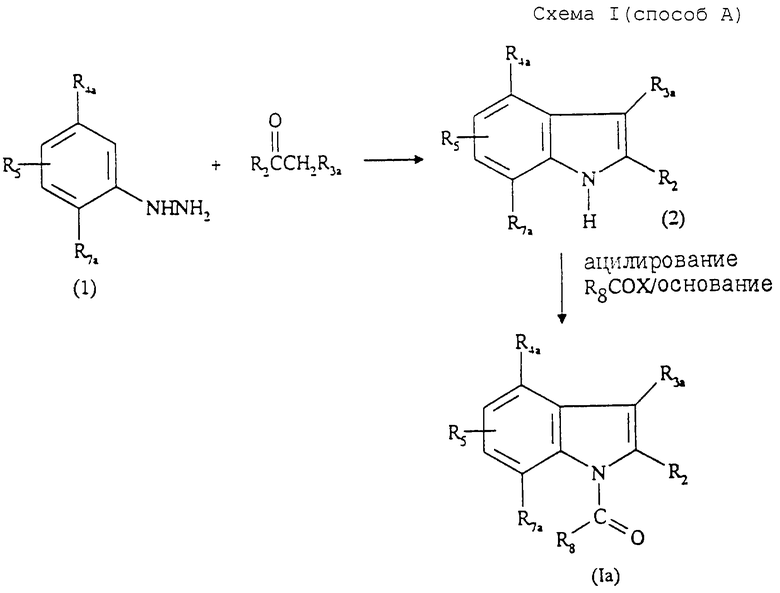
Так, соединения общей формулы (Iа) могут быть получены "способом А", представленным на cхеме I в конце описания.
Данный способ А, описанный в патенте NL 7308094, состоит в том, что:
1) осуществляют взаимодействие гидразина формулы (I) с кетоном формулы R2COCH2R3a, в котором R2 и R3а имеют значения, указанные выше, и получают соединение формулы (2);
2) ацилируют получение соединения формулы (2) с образованием соединения формулы (Iа);
3) при необходимости превращают полученное соединение формулы (Iа) в одну из фармацевтически приемлемых солей.
Стадия 1) способа А является реакцией Фишера, которую преимущественно проводят в инертном растворителе, в таком как метанол, этанол, изопропанол или уксусная кислота, в присутствии кислотного катализатора, такого как серная кислота, соляная, ледяная уксусная кислота, хлорид цинка, при температуре от 20oС до 150oС.
Стадия 2) способа А является реакцией ацилирования, протекающей преимущественно при использовании галогенангидрида кислоты формулы R8COX в присутствии основания, такого как гидроксид, гидрид, амид или алкоголят щелочного металла в инертном органическом растворителе. Подходящими растворителями для этой реакции являются, например, толуол, ксилол, ДМФ. Реакция может осуществляться при температуре от 0oС до температуры кипения используемого растворителя.
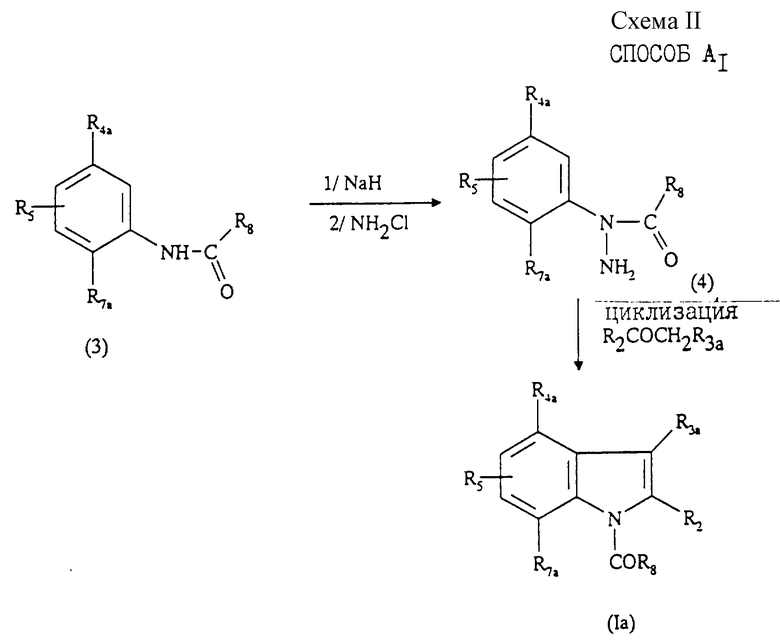
Соединения формулы (Iа) могут быть также получены "способом A1", представленным на схеме II в конце описания.
Данный способ A1 состоит в том, что:
1) осуществляют взаимодействие соединения формулы (3) последовательно с гидридом щелочного металла, таким как гидрид натрия, затем с хлоридом аммония с образованием замещенного гидразина формулы (4);
2) осуществляют взаимодействие полученного гидразина с кетоном формулы R2COCH2R3a с образованием путем циклизации соединения формулы (Iа);
3) при необходимости превращают полученное соединение формулы (Iа) в одну из фармацевтически приемлемых солей.
Стадию 1) способа A1 осуществляют преимущественно в инертном растворителе, таком как диэтиловый эфир или ТФГ при 25oС.
Стадия 2) способа A1 является циклизацией, протекающей в тех же условиях, которые описаны выше для осуществления стадии 1 способа А.
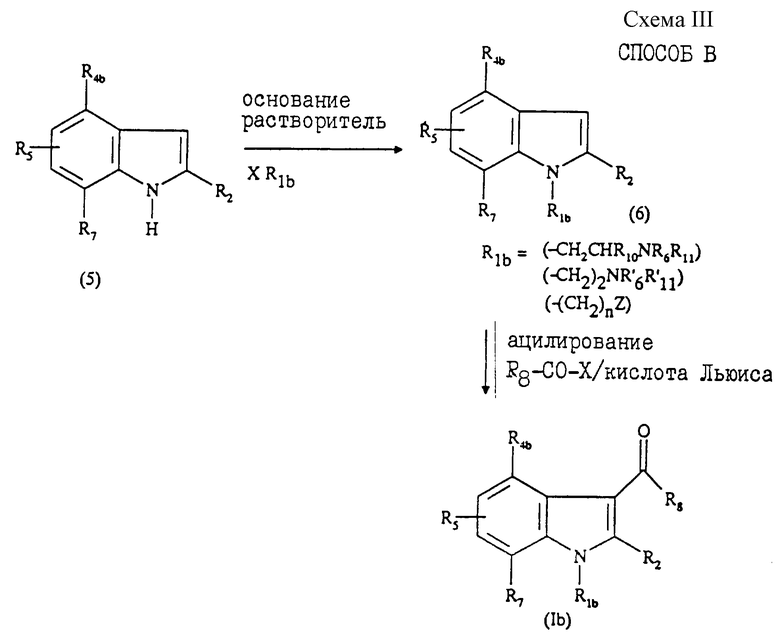
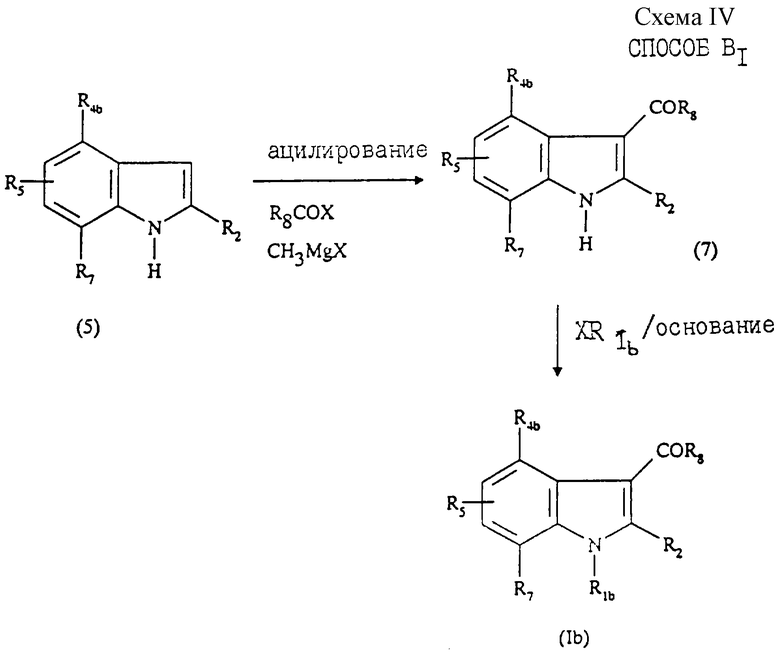
Соединения общей формулы (Ib), в которой R1b - радикал, выбранный из группы -CH2CHR10NR6R11, -(CH2)2NR'6R'11 или -(CH2)nZ, могут быть получены "способом В", представленным на схеме III в конце описания.
Данный способ, описанный, в частности, в патенте US 4581354, состоит в том, что:
1) осуществляют взаимодействие производного индола формулы (5), замещенного в положении 2 радикалом R2, охарактеризованном выше, с галогенидом формулы: XCH2CHR10NR6R11, X(CH2)2NR'6R'11 или Х(СН2)nZ, в которых R6, R10, R11, R'6, R'11, n и Z имеют значения, указанные выше, а Х - галоген, например, хлор, бром или йод, с образованием соединения формулы (6), в которой R1b - радикал -CH2CHR10NR6R11, -(CH2)2NR'6R'11 или -(СН2)nZ;
2) ацилируют полученное соединение формулы (6) галоидангидридом формулы R8-CO-X, в которой Х - галоген, например хлор или бром, a R8 - радикал, описанный выше, с образованием соединения общей формулы (Ib);
3) при необходимости превращают соединение формулы (Ib) в одну из фармацевтически приемлемых солей.
Стадия 1) способа В преимущественно протекает в присутствии основания в инертном органическом растворителе в описанных условиях реакции. В качестве основания можно использовать карбонат щелочного металла, например карбонат натрия или калия, гидрид, например гидрид натрия, или гидроксид щелочного металла, например гидроксид калия. Использование гидроксида калия является наиболее предпочтительным.
В качестве растворителей можно использовать, например, толуол, диметилформамид (ДМФА) или диметилсульфоксид (ДМСО), причем последний более предпочтителен. Реакцию проводят в интервале от 0oС до температуры кипения растворителя.
В частном случае, когда R1b означает группу -(CH2)nZ, реакцию осуществляют в присутствии трис-[2-(2-метоксиэтокси)этил]амина (TDA-1).
Стадия 2) способа В представляет собой ацилирование согласно реакции Фриделя - Крафтса, осуществляемой в присутствии кислоты Льюиса, такой как хлорид алюминия, в инертном растворителе, таком как 1,2-дихлорэтан или дисульфид углерода. Реакцию ацилирования можно также осуществлять в присутствии такой кислоты Льюиса, как хлорэтилмагнийхлорид в инертном растворителе, таком как дихлорметан, согласно способу, описанному в J. Med.Chem., 1995, 38, 3094.
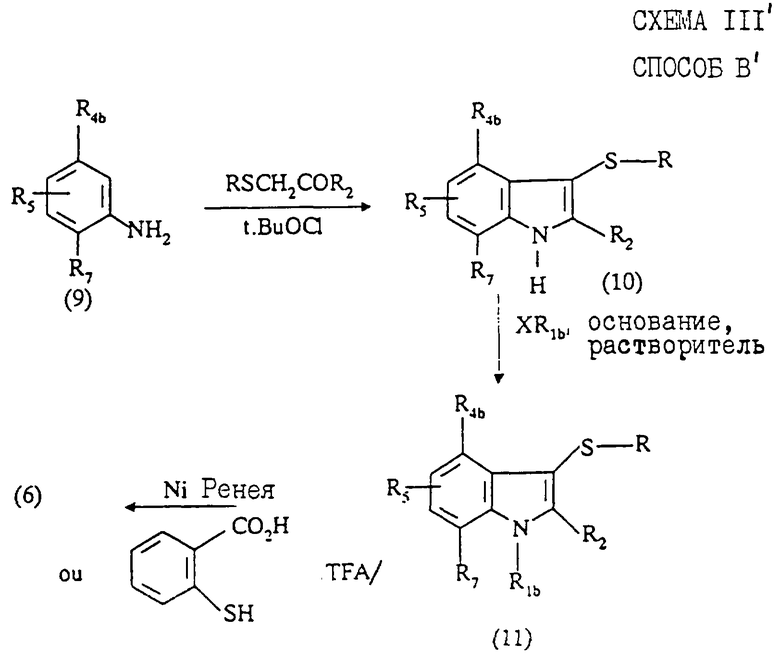
Согласно варианту стадии 1) способа В (способ В'), соединение формулы (6) можно также получить в три стадии, представленные на схеме III' в конце описания. Этот вариант заключается в том, что анилин формулы (9) подвергают взаимодействию с кетоном формулы RSCH2COR2, где R означает метил или фенил, в присутствии гипохлорита трет-бутила (t-BuOCl) согласно методике, описанной в J. Am.Chem.Soc., 1974, 96, 5495.
Полученное соединение формулы (10) затем алкилируют в условиях, аналогичных описанным выше для стадии 1) способа В, с получением соединения формулы (11). Удаляют затем сульфидную группу реакцией с никелем Ренея или взаимодействием с 2-меркаптобензойной кислотой в трифторуксусной кислоте согласно методике, описанной в Tetrahedron Lett., 1993, 34 (13), 2059-2062, и получают соединение формулы (6).
Согласно варианту, также описанному в патенте US 4581354 (способ B1, схема IV, приведенная в конце описания), осуществляют сначала ацилирование соединения взаимодействием с метилмагнийгалогенидом и галогенангидридом формулы R8COX в простом эфире с образованием соединения общей формулы (7), затем осуществляют присоединение заместителя R1b реакцией соединения формулы (7) с галогенидом XR1b в присутствии основания в условиях, аналогичных описанным выше для стадии 1) способа В.
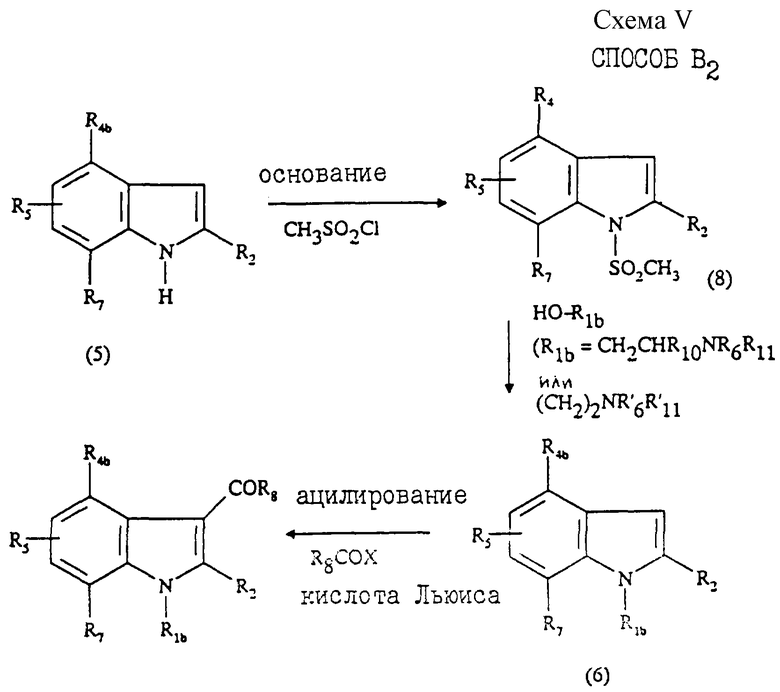
Согласно другому варианту, описанному в патенте ЕР 0444451 (способ В3, схема V, приведенная в конце описания), обрабатывают соединение общей формулы (5) основанием, таким как гидрид натрия или К2СО3, затем подвергают взаимодействию с мезилхлоридом с образованием соединения общей формулы (8). Затем осуществляют присоединение заместителя R1b взаимодействием соединения (8) с гидроксиалкиламином формулы R1bOH. Полученное соединение (6) ацилируют с образованием соединения общей формулы (Ib).
Соединения формулы (Ib), в которых R1b представляет собой группу -CHR9CH2NR'6R'11 , a R9 образует с R7 группу -СН2-O- таким образом, что радикал R1b означает группу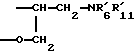
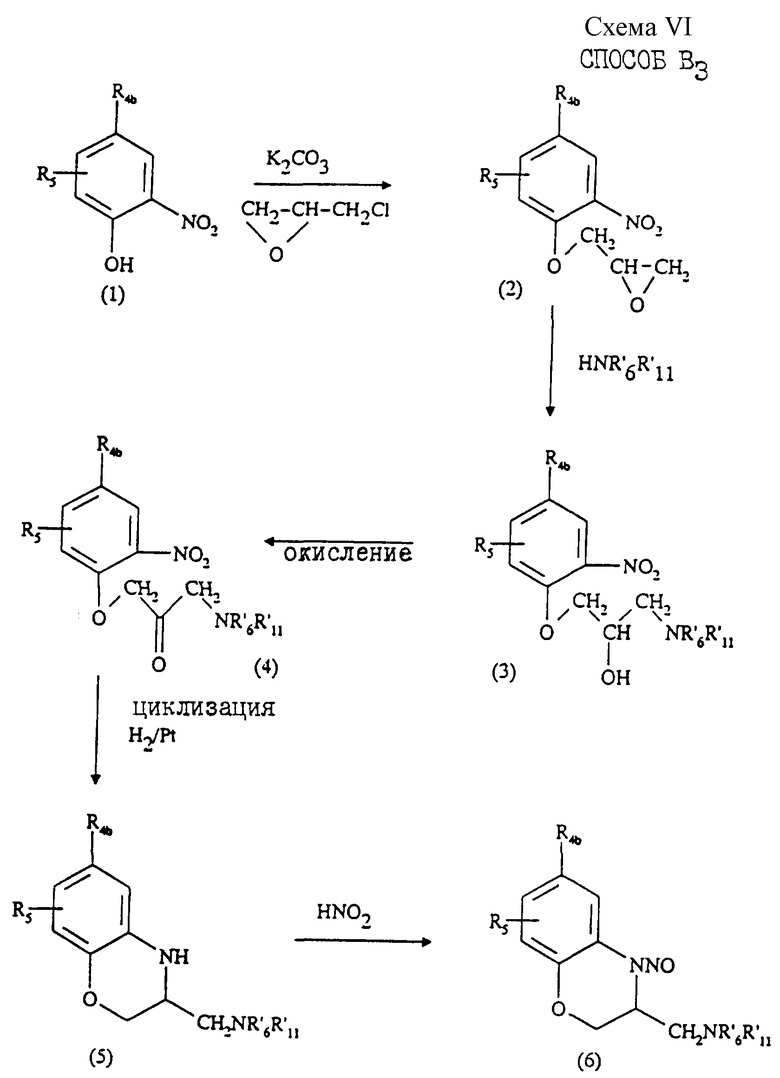
могут быть получены "способом В3" (см. схему VI, приведенную в конце описания).
Этот способ, который описан в патентах US 5109135 и US 4938138 заключается в том, что:
1) смесь соединения формулы (1) и карбоната калия нагревают с эпихлоргидрином с получением соединения формулы (2);
2) соединение (2) подвергают взаимодействию с соответствующим амином формулы HNR'6R'11 с образованием соединения формулы (3);
3) соединение (3) окисляют с получением соединения (4);
4) соединение формулы (4) восстанавливают, затем подвергают циклизации в присутствии платины в качестве катализатора с образованием соединения формулы (5);
5) полученное соединение (5) подвергают взаимодействию с нитритом щелочного металла в кислой водной среде при 0-10oС, в результате чего образуется соединение формулы (6);
6) соединение формулы (6) восстанавливают водородом в присутствии металлического катализатора или гидридом алюминия и щелочного металла, например LiAlH4, в инертном растворителе, например тетрагидрофуране (ТГФ), при температуре от 0o до температуры кипения используемого растворителя с образованием соединения формулы (7).
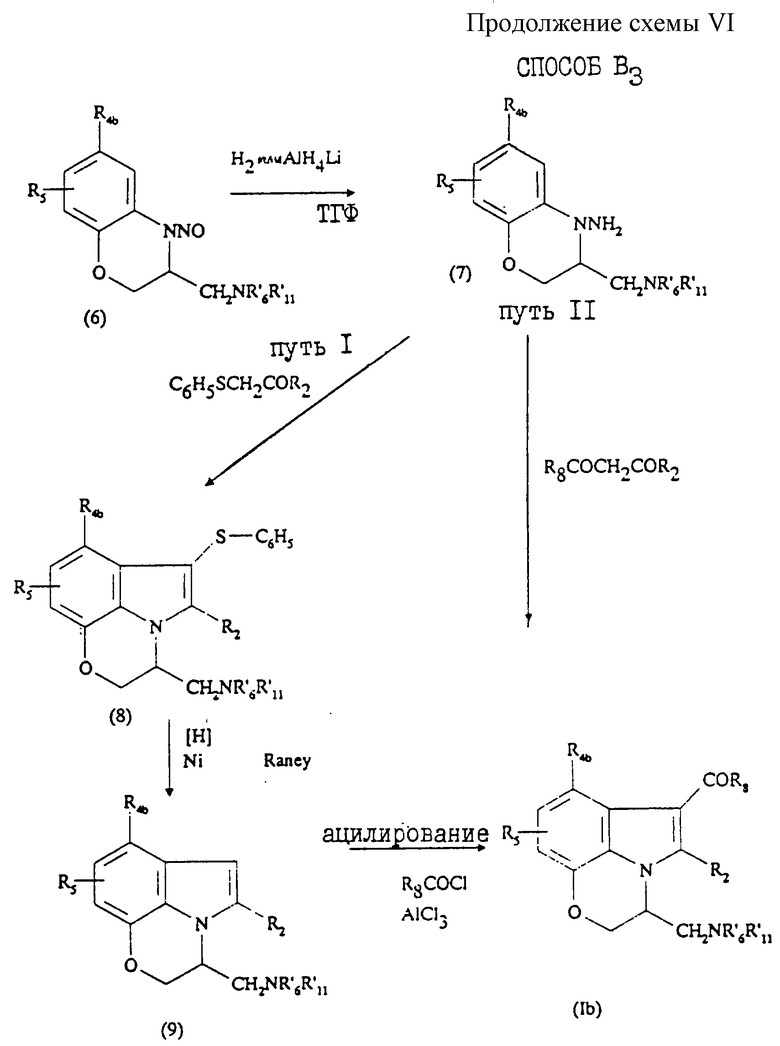
Исходя из соединения (7), можно получить соединения общей формулы (Ib) двумя путями, обозначенными как путь I и путь II (см. продолжение схемы VI в конце описания).
Путь I заключается в том, что:
7) получают соединение формулы (8) из соединения формулы (7), используя индольный синтез по Фишеру, т.е. взаимодействием последнего соединения с кетоном формулы С6Н5SСН2СОR2. Эта реакция осуществляется при 20-150oС в инертном органическом растворителе, например метаноле, в присутствии кислотного катализатора, например серной или ледяной уксусной кислоты (последняя предпочтительнее);
8) проводят отщепление тиофенильной группы от соединения (8), нагревая последнее в органическом растворителе в присутствии никеля Ренея при температуре кипения органического растворителя с обратным холодильником и получают соединение формулы (9);
9) получают конечный продукт (Ib) взаимодействием соединения (9) с галоидангидридом R8COCl в присутствии кислоты Льюиса, например АlСl3, в инертном органическом растворителе.
Путь II позволяет непосредственно получить соединение формулы (Ib) и заключается в том, что соединение (7) подвергают реакции с дикетоном формулы R8COCH2COR2 согласно реакции Фишера, описанной на стадии 7 пути I.
Для получения соединения (Ib) в оптически активной форме осуществляют разделение энантиомеров соединения (5), согласно способу, описанному в патенте US 4939138.
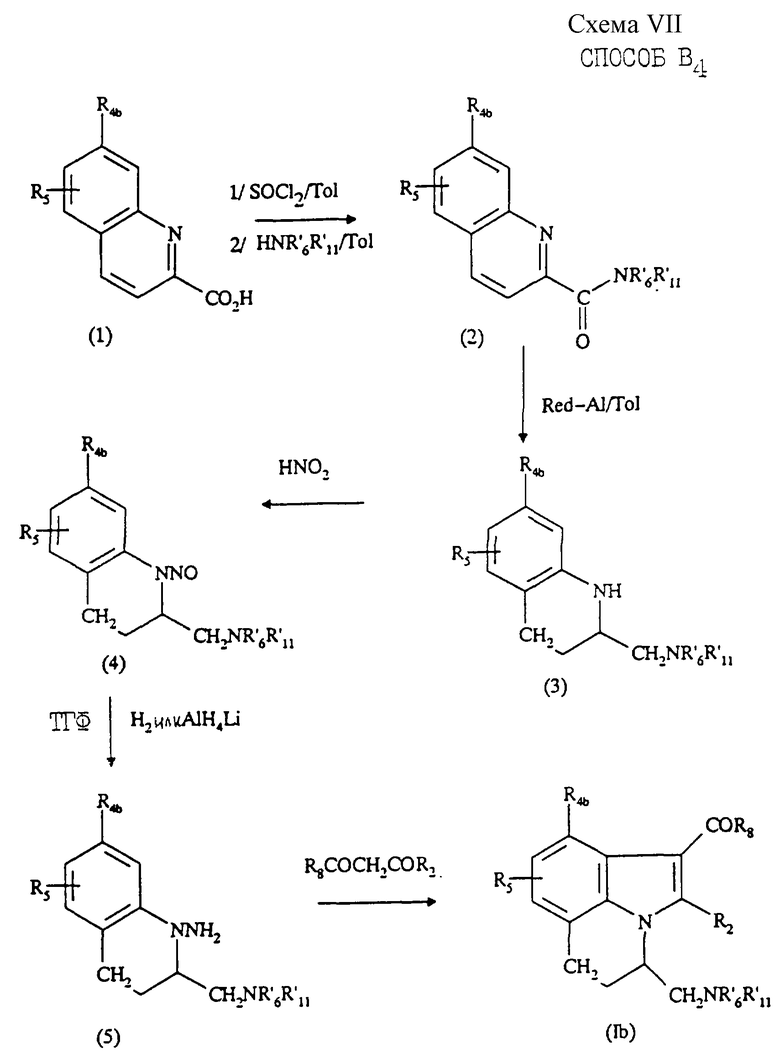
Соединения формулы (Ib), в которой R1b означает группу -CHR9CH2NR'6R'11, a R9 образует с R7 группу -CH2-CH2 - таким образом, что R1b означает группу 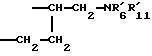 могут быть получены "способом B4", представленным на схеме VII в конце описания.
могут быть получены "способом B4", представленным на схеме VII в конце описания.
Этот способ заключается в том, что
1) осуществляют реакцию хинальдиновой кислоты (1) с хлористым тионилом в толуоле с последующим добавлением к реакционной смеси соединения формулы HNR'6R'11, что приводит к образованию соединения (2);
2) восстанавливают соединение (2) в толуоле в присутствии катализатора Red.Al с образованием соединения (3); затем
3) обрабатывают соединение (3) по схеме способа В3 (5-я и последующие стадии) с получением конечного соединения формулы (Ib).
Две первых стадии способа В4 описаны Stanton'ом с сотр. (J. Med.Chem. (1983), 26, 986-989).
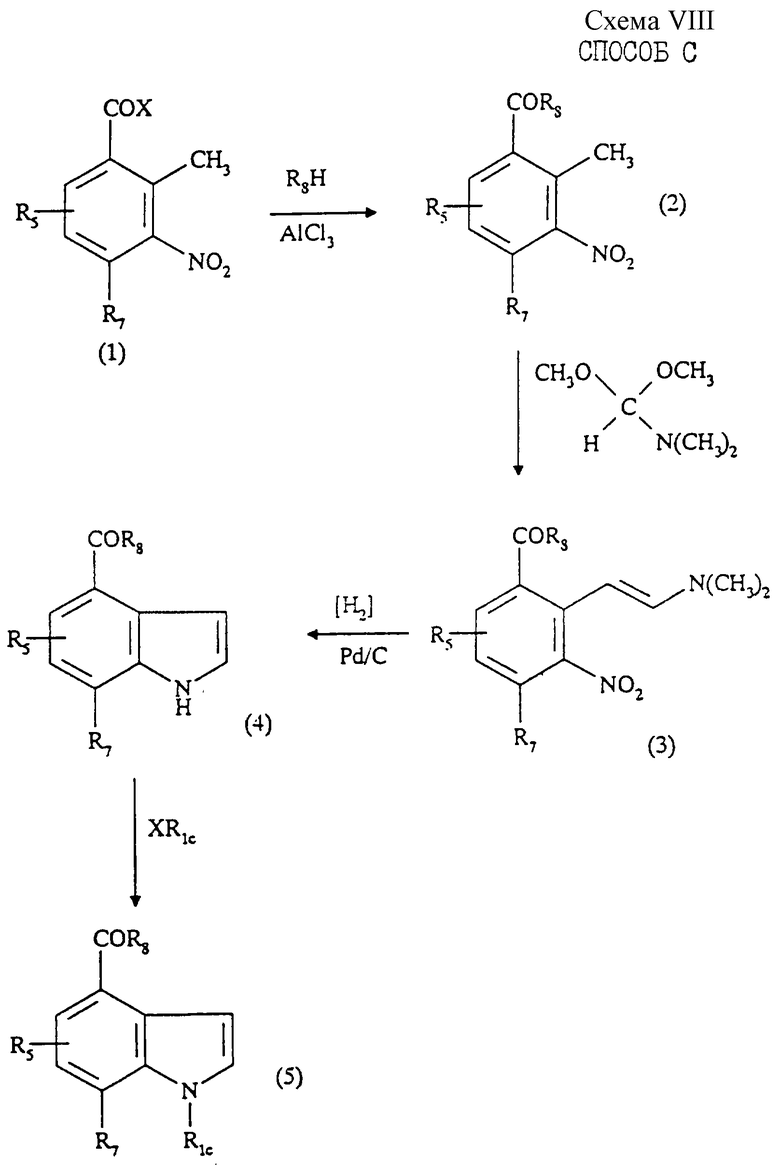
Соединения формулы (Ic), в которой R1c означает группу -CH2CHR10NR6R11, -(CH2)2NR'6R'11 или -(CH2)nZ и R2c, R3с - атомы водорода, могут быть получены "способом С" (схема VIII, приведенная в конце описания).
Данный способ, описанный, в частности, в патентах US 4840950 и ЕР 0278265, состоит в том, что:
1) осуществляют взаимодействие галогенида 2-метил-3-нитробензоила формулы (1) с соединением формулы R8H, в которой R8 охарактеризован выше с образованием соединения формулы (2);
2) полученное соединение (2) подвергают взаимодействию с диметилацеталем диметилформамида с образованием соединения (3);
3) циклизуют соединение (3) с образованием соединения (4);
4) осуществляют взаимодействие соединения (4) с галоидным соединением формулы XR1c, в котором R1c охарактеризован выше, с образованием соединения (5).
Стадия 1 способа С является реакцией Фриделя - Крафтса, проводимая в инертном органическом растворителе, таком как хлористый метилен, в присутствии АlСl3. При осуществлении стадии желательно довести все реагенты до комнатной температуры, затем нагреть смесь до температуры кипения растворителя.
Стадию 2 способа С предпочтительно проводить при нагревании с обратным холодильником раствора соединения (2) с 2-4-мольным избытком диметилацеталь диметилформамида в инертном органическом растворителе, например в диметилформамиде или диоксане.
Стадия 3 способа С является реакцией циклизации соединения (3), протекающая преимущественно в инертном органическом растворителе, таком как этилацетат или этанол, при комнатной температуре. Реакцию осуществляют при давлении водорода от 50 до 100 p.s.i.g. Применяемыми катализаторами для этого типа реакции обычно являются никель Ренея или палладий на угле.
Стадия 4 способа С является реакцией взаимодействия (4) с соответствующим соединением XR1c в присутствии сильного основания, такого как гидрид натрия. Реакцию преимущественно осуществляют в инертном органическом растворителе, таком как ДМФА, при температуре между комнатной и кипения используемого растворителя.
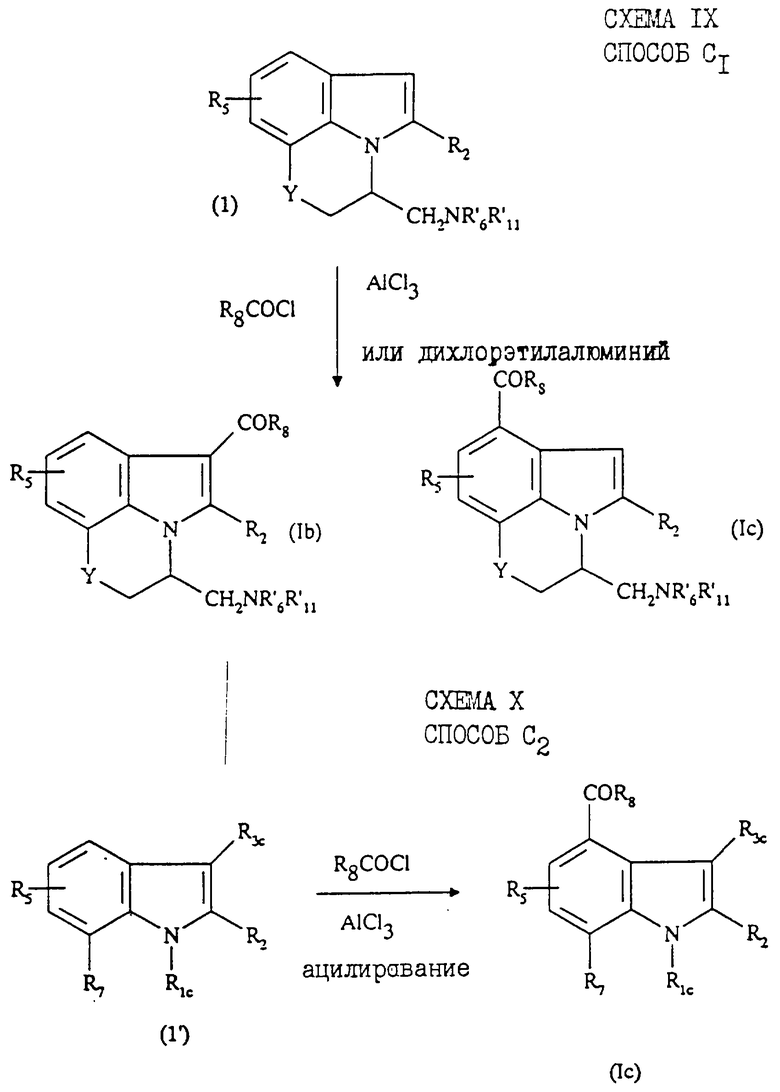
Соединения формул Ib и Ic, индолы, ацилированные соответственно в положении 3 и 4, в которых R1b и R1c являются группой -CHR9CH2NR'6R'11, a R7 и R9 образуют вместе группу -Y-СН2-, в которой Y означает О или -СН2-, таким образом, что радикалы R1b и R1c означают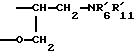
могут быть синтезированы по схеме IX ("способ C1"), приведенной в конце описания.
Указанный способ заключается в ацилировании соединения (1) галогенангидридом R8COCl в присутствии кислоты Льюиса, например дихлорэтилалюминия или избытка АlСl3. Получают индолы, ацилированные в положении 3 и 4, которые затем разделяют.
Соединения формулы (Iс), в которых R3с означает алкил C1-C4, могут быть получены по способу С2 (схема X в конце описания), согласно которому соединение формулы (1') ацилируют в рабочих условиях способа В при умеренных значениях параметров способа.
Способ C1 получения соединений, в которых Y является кислородом, описан в патенте US 4939138.
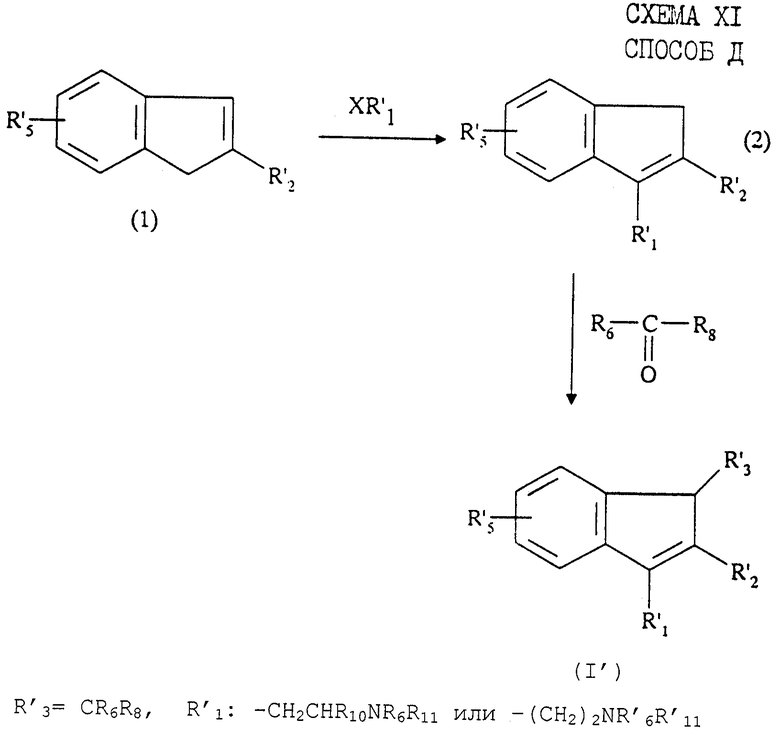
Соединения формулы (I') могут быть синтезированы "способом D", представленным на схеме XI в конце описания.
Этот способ, описанный, в частности, в патенте US 5292736, заключается в том, что:
1) инден формулы (1) обрабатывают сильным основанием, таким как н-бутиллитий, в инертной атмосфере и инертном растворителе при температуре от комнатной до температуры кипения растворителя, затем полученное соединение подвергают взаимодействию с соответствующим галогенидом формулы XR'1, взятых в эквимолярном соотношении, при температуре от 0o до температуры кипения смеси в инертной атмосфере с образованием соединения (2);
2) соединение (2) обрабатывают сильным основанием, таким как метилат натрия, затем это соединение подвергают взаимодействию с соответствующим кетоном или альдегидом общей формулы R6-(СО)-R8 с образованием соединения (I').
Указанную реакцию проводят преимущественно в инертном растворителе при температуре от комнатной до температуры кипения растворителя.
Энантиомеры соединений формул (I) и (I') могут быть получены классическими методами, хорошо известными специалисту.
Среди соединений формул (I) и (I'), описанных выше, соединения формул (Iа) и (Iс), а также соединения формул (Ib1), (Ib2), (Ib3) и (I'а), описанные ниже, являются новыми и составляют другой объект изобретения.
Этими соединениями являются:
А) Соединения формулы (Ib1)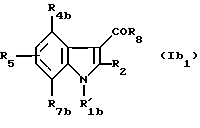
в которой R'1b означает группу -CH2CHR10NR6R11 или -(СН2)2NR'6R'11;
R4b означает водород, (C1-C4)алкил, (C1-C4)алкоксил, атом галогена, группу -СF3, -ОСF3 или (C1-C4)алкилтиогруппу;
R7b означает водород, (C1-C4)алкил, (C1-C4)алкокси, атом галогена, группу -СF3, -ОСF3 или (C1-C4)алкилтиогруппу;
R2, R5, R6, R'6, R8, R10, R11, R'11 - имеют значения, указанные выше для соединений формулы (I),
при условии, что:
1) когда -CH2CHR10NR6R11 является группой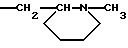
R4b, R5 и R7b - атомы водорода, а R2 - Н или метил, тогда R8 не является 1-нафтилом;
2) когда -СН2СНR10NR6R11 является группой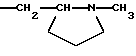
R4b, R5 и R7b - атомы водорода, а R2 - метил, тогда R8 не является 1-нафтилом;
3) когда -(СН2)2NR'6R'11 является группой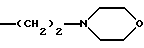
R7b - метоксил, a R2 - метил, R4b и R5 - атомы водорода, тогда R8 не является 1-нафтилом;
4) когда -(CH2) 2NR'6R'11 является группой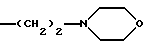
а R2, R4b, R5 и R7b - атомы водорода, тогда R8 не является 1-(4-бромнафтилом).
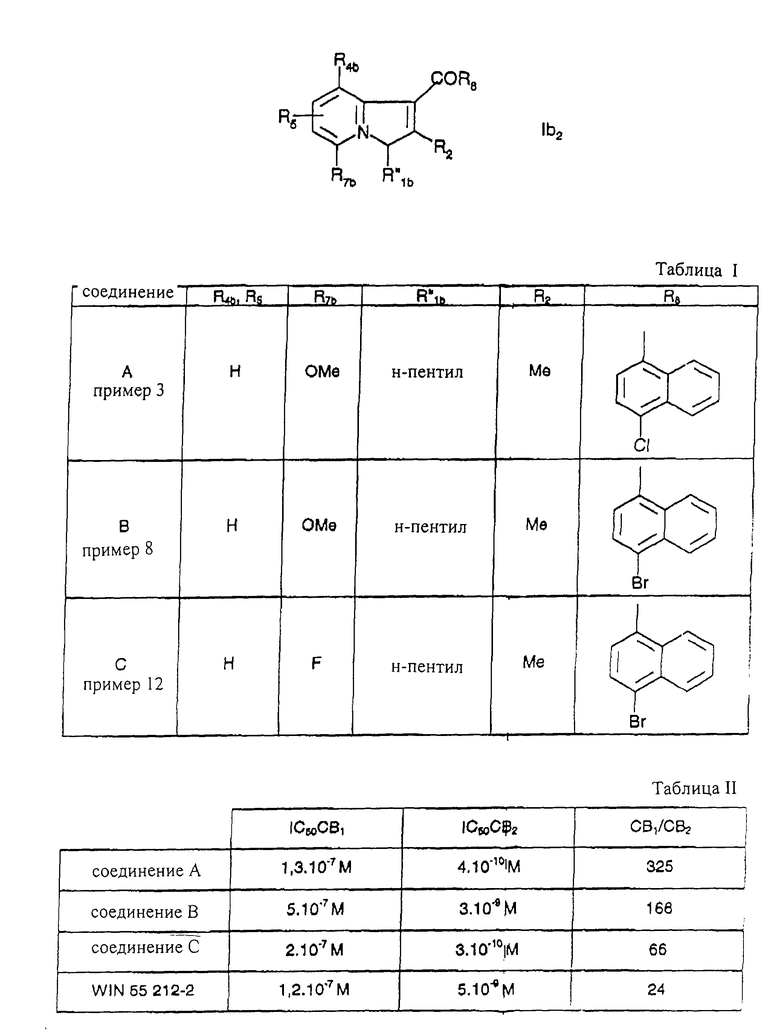
В) Соединения формулы (Ib2)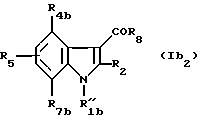
в которой R"1b означает группу -(CH2)nZ;
R4b означает водород, (C1-C4)алкил, (C1-C4)алкоксил, атом галогена, группу -СF3, -ОСF3 или (C1-C4)алкилтиогруппу;
R7b означает водород, (C1-C4)алкил, (C1-C4)алкоксил, атом галогена, группу -СF3, -ОSF3 или (C1-C4)алкилтиогруппу;
R2, R5, R6, R'6, R8, R10, R11 и R'11 - имеют значения, указанные выше для соединений формулы (I),
при условии, что, когда Z - бром, n равно 3 или 4, R4b, R5 и R7b - водороды, а R2 - метил, тогда R8 не является 1-нафтилом или 4-метоксифенилом.
С) Соединения формулы (Ib3)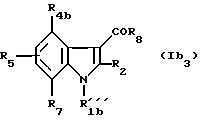
в которой R'''1b - группа -CHR9CH2NR'6R'11;
R4b - водород, (C1-C4)алкил, (C1-C4)алкокси, атом галогена, группу -СF3, -OSF3 или (C1-C4)алкилтиогруппу;
R2, R5, R6, R'6, R7, R8, R9, R10, R11 и R'11 - имеют значения, указанные выше для соединений формулы (I),
при условии, что, когда -NR'6R'11 представляет собой группу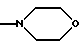
Y - кислород, а R2 - метил, R4b и R5 - атомы водорода, тогда R8 не является 1-нафтилом, 1-(4-бром)нафтилом и 1-(5,7-дибром)нафтилом.
D) Соединения формулы (I'а) представляют собой соединения формулы (I'), охарактеризованной выше, при условии, что:
1) когда R'1 означает группу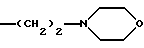
a R'2, R'5 и R6 - атомы водорода, тогда R8 не является 1-(4-метокси)нафтилом, 1-(4-гидрокси)нафтилом и 9-антрилом;
2) когда R'1 означает группу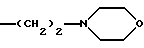
а R'2 - метил, R'5 и R6 - атомы водорода, тогда R8 не является 1-нафтилом, 1-(4-метокси)нафтилом.
Среди этих соединений особенно предпочтительными являются следующие:
1-(2-(4-морфолинил)этил)-2-метил-3-(4-фтор-1-нафтилкарбонил)-7-метоксииндол;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол;
1-н-пентил-2-метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол;
метансульфонат (-)-3-(4-морфолинилметил)-5-метил-7-(5,7 - дибромонафтилкарбонил)-2,3-дигидропиррол [1,2,3-de]-1,4-бензоксазин;
метансульфонат (+)-(2-метил-4-(4-морфолинилметил)-5,6дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)нафталин-1-ил-метанон;
метансульфонат 1-(1-нафтилкарбонил)-3-(2-(4-морфолинил)этил)индол;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол;
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол;
1-(2-(4-морфолинил)этил)-2-метил-3-(1-нафтилкарбонил)-7-(трифторметил)индол;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-(трифторметил)индол;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол;
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол;
1-(2-(4-морфолинил)этил)-2,7-диметил-3-(4-бром-1-нафтилкарбонил)индол.
Соединения, пригодные для изготовления лекарственных средств согласно изобретению, обычно вводят в единичных дозах. Единичные дозы предпочтительно входят в состав фармацевтических композиций, в которых активное начало смешано с фармацевтическим экципиентом.
Другим объектом изобретения является, таким образом, фармацевтическая композиция, содержащая в качестве активного начала соединение формулы (I) или (I'), имеющее высокое сродство к рецептору СВ2 человека, характеризуемой константой ингибирования Ki, меньшей или равной 10 нМ, определенной в исследованиях по связыванию лиганда; более конкретно, настоящее изобретение относится к фармацевтической композиции, содержащей в качестве активного начала соединение общей формулы (Ia), (Ib1), (Ib2), (Ib3), (Iс) или (I'а).
Соединения формул (I) или (I') или их фармацевтически приемлемые соли вводятся в эффективных количествах. Суточные дозы этих соединений составляют от 0,1 до 100 мг/кг массы тела млекопитающего, предпочтительно в дозах 0,2-50 мг/кг. Для человека доза может варьироваться от 0,5 до 1000 мг/сутки, предпочтительно 1-500 мг, в зависимости от возраста пациента или от цели приема: профилактического или в терапевтических целях.
Соединения согласно изобретению и их фармацевтические соли пригодны для лечения таких заболеваний, которые имеют аутоиммунный, инфекционный и аллергический характер. В частности, можно назвать следующие аутоиммунные заболевания: рассеянная красная волчанка, болезни соединительных тканей или коннективиты, синдром Сьогрена, анкилозирующий спондилоартрит, реактивный артрит, невыраженный спондилоартрит, болезнь Бехсетса, гемолитические аутоиммунные анемии. В качестве аллергических заболеваний можно назвать реактивную гиперчувствительность, а также астму. Также соединения согласно изобретению и их фармацевтически приемлемые соли могут применяться для лечения васкулитов, паразитарных инфекцих, амилоидоза, а также болезней, затрагивающих плазмоцитарную линию.
В фармацевтических композициях согласно изобретению активное начало в смеси с традиционными фармацевтическими носителями может вводиться в виде единичных доз в формах, годных для введения людям или животным орально, под язык, подкожно, внутримышечно, внутривенно, трансдермально, локально, ректально, путем ингаляции. Соответствующие фармацевтические формы выбирают в зависимости от болезней, подлежащих лечению: для орального применения этими формами могут быть таблетки, желатиновые капсулы, порошки, гранулы, растворы или суспензии; соответствующие формы для ингаляции, для приема под язык, для питья, для подкожного введения, для внутримышечного, внутривенного, для введения через нос, глаза, а также формы для ректального введения. Предпочтительными формами введения в рамках настоящего изобретения являются формы для орального введения, внутривенного и ингаляционного.
При изготовлении твердой композиции в виде таблеток активное начало смешивают с наполнителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик и т.п. Возможно покрытие таблеток сахарозой или другими подходящими веществами, или возможно обрабатывать таблетки с целью придания им эффекта пролонгированного или отсроченного действия или для непрерывного высвобождения активного начала в заданном количестве.
Препарат в виде желатиновых капсул получают путем смешивания активного компонента с разбавителем с последующим введением полученной смеси в размягченную или твердую желатиновую капсулу.
Препарат в виде сиропа или эликсира может содержать активное начало вместе с подсластителем, предпочтительно бескалорийным; с метилпарабеном или пропилпарабеном в качестве антисептика, а также с вкусовой добавкой и красителем.
Формы в виде порошков или диспергируемых в воде гранул могут содержать активное начало в смеси с агентами-диспергаторами или смачивателями или с суспенгаторами, например с поливинилпирролидоном, а также с подсластителями или корректорами вкуса.
Для ректального введения применяют свечи, изготовленные из связующего, плавящегося при ректальной температуре, например из масла какао или полиэтиленгликоля.
Для парентального, интраназального или внутриглазного введения применяют водные суспензии, изотонические солевые растворы; растворы, предназначенные для инъекций, содержащие фармакологически совместимые диспергаторы и/или смачиватели, например пропиленгликоль или бутиленгликоль.
Для ингаляционного введения используют аэрозоль, содержащий, например, триолеат сорбитола или олеиновую кислоту, а также трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой другой биологически совместимый газ-носитель.
Активное начало может находиться в составе микрокапсул, возможно с одним или несколькими носителями или добавками.
В каждой единичной форме активное начало формулы (I) или (I') находится в количествах, соответствующих предусмотренным суточным дозам. Как правило, каждая единичная форма подбирается в зависимости от дозы и пути введения, например таблетки, капсулы и им подобные формы, пакетики, ампулы, сиропы и им подобные, капли, и таким образом, чтобы такая единичная форма содержала от 0,5 до 1000 мг активного начала, предпочтительно 1-500 мг, более предпочтительно от 1 до 200 мг при ежедневном приеме 1-4 раза.
Вышеуказанные композиции могут также содержать другие активные вещества, терапевтически полезные, например кортикостероиды и β2-агонисты.
Благодаря своему очень высокому сродству к рецептору СВ2 человека и высокой селективности, соединения согласно изобретению могут применяться в виде радиомеченых соединений в качестве реактивов для лабораторных исследований. Например, они позволяют провести характеристику, идентификацию и локализацию рецептора СB2 человека в срезах тканей или рецептора CB2 животного методом ауторадиографии.
Соединения согласно изобретению позволяют также осуществить сортировку или скрининг молекул в зависимости от сродства к рецептору СВ2 человека. Для этого используют реакцию смещения меченного согласно изобретению лиганда из своего рецептора СВ2 человека.
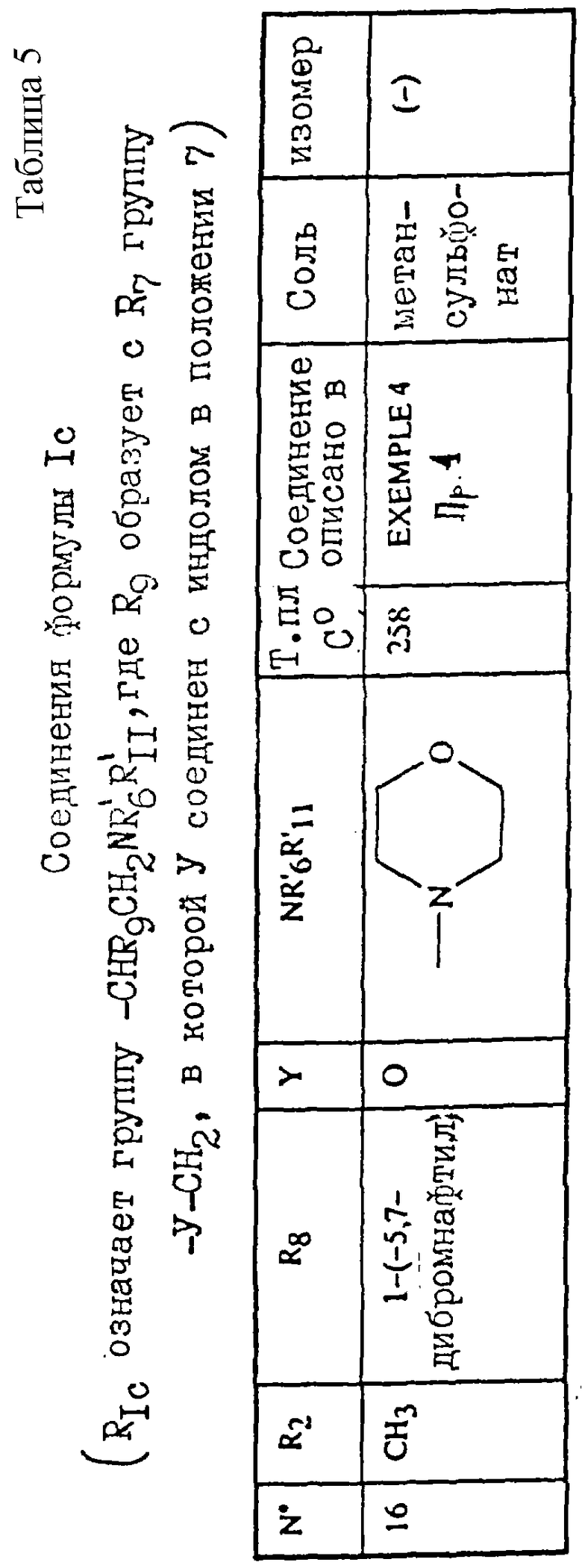
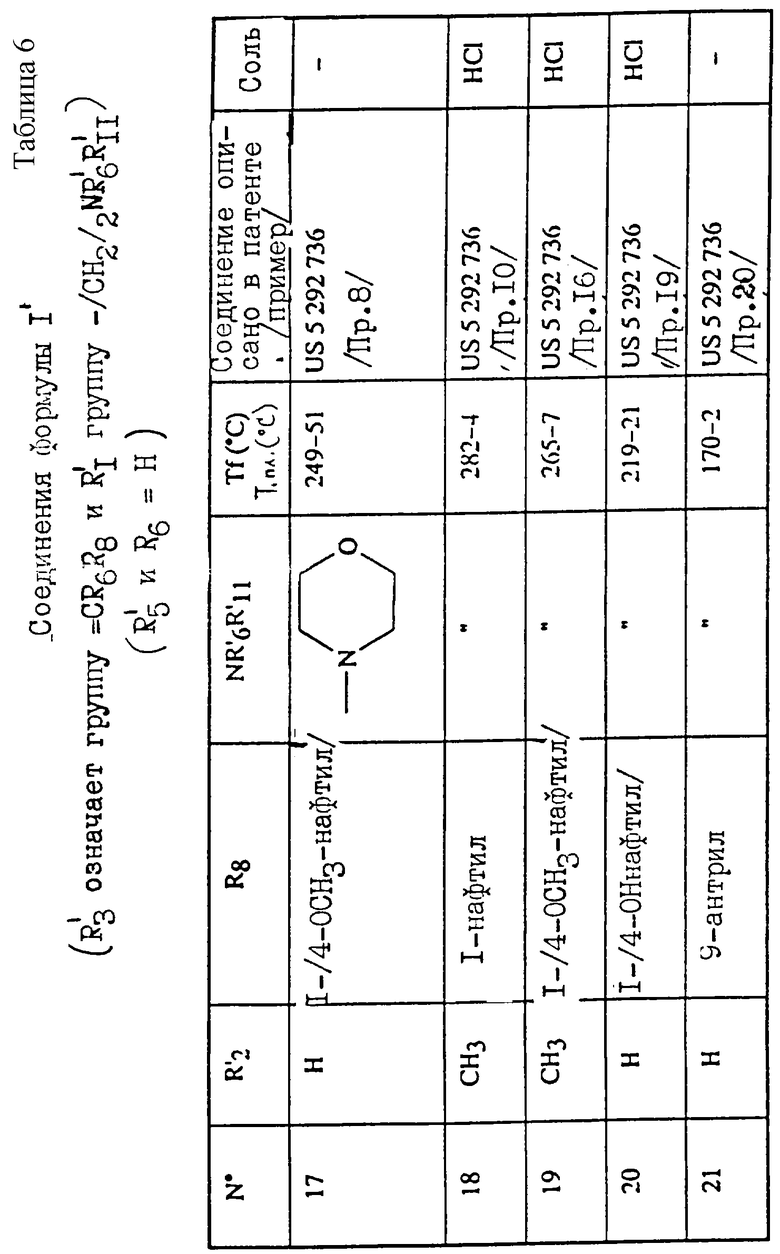
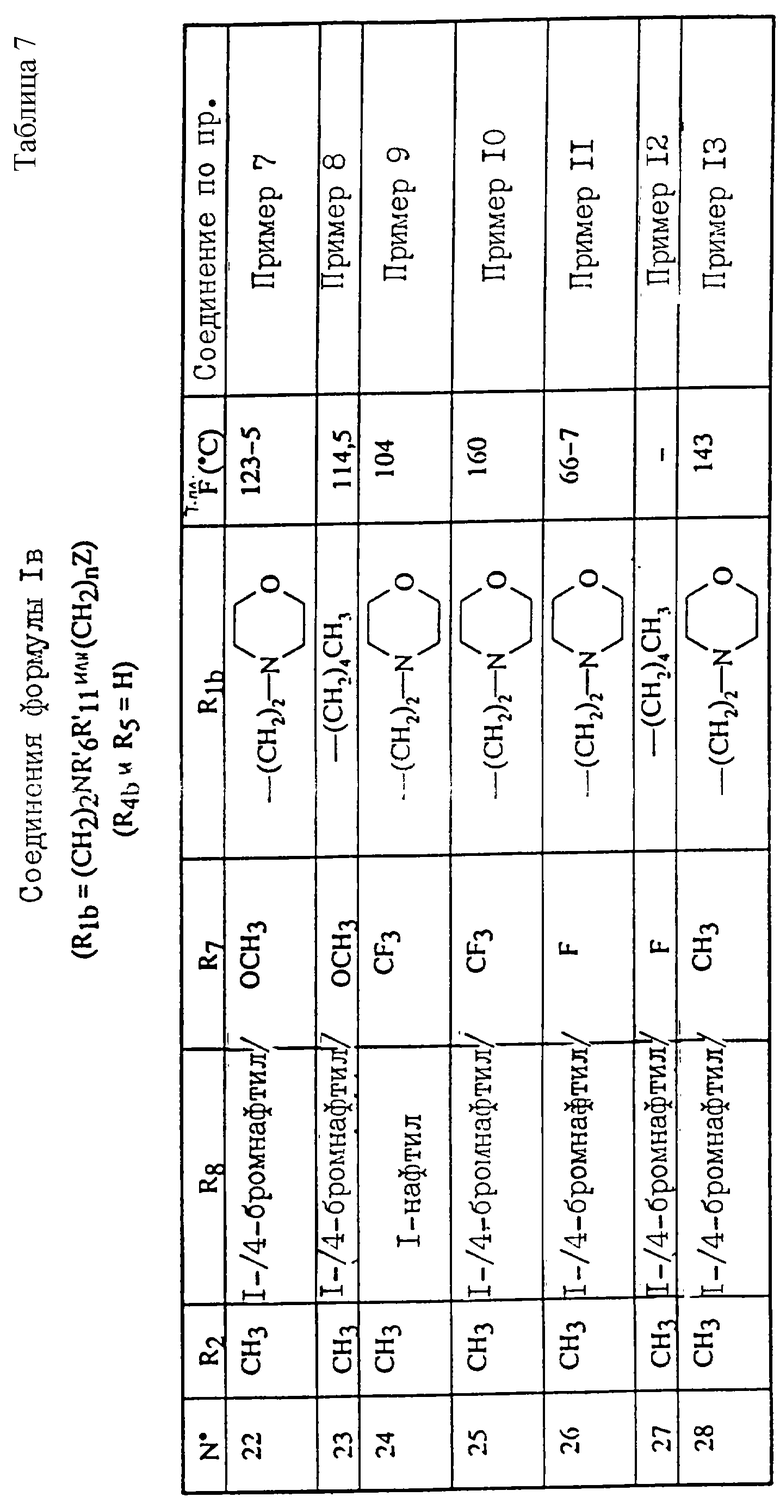
Примеры соединений согласно изобретению подробно описаны ниже в примерах 1-13, а также в таблицах 1-7, соединения по примерам 1-13 сведены также в указанные таблицы.
В нижеследующих примерах используются следующие сокращения:
Т.пл. - температура плавления
Pd/C - палладий на угле
Pt - платина
ДХМ - дихлорметан
ТФГ - тетрагидрофуран
ДМФА - диметилформамид
ДМСО - диметилсульфоксид
ЭА - этилацетат
Me - метил
МеОН - метиловый спирт
Эфир - диэтиловый эфир
IPr - изопропил
Вu - н-бутил
TFA - трифторуксусная кислота
SOCl2 - хлористый тионил
TDA-1: трис[(2-(2-метоксиэтокси)этил]амин
Red-Al -бис(2-метоксиэтокси)натрийалюминий гидрид
Изоэфир - диизопропиловый эфир
Эфир: диэтиловый эфир
s - синглет; t - триплет; m - мультиплет
Пример 1
1-(2-(4-морфолинил)этил)-2-метил-3-(4-фтор-1-нафтилкарбонил)-7-метоксииндол
А) 2-Метил-3-(4-фтор-1-нафтилкарбонил)-7-метоксииндол
Раствор 1,02 г 2-метил-7-метоксииндола в 5 мл эфира добавляют по каплям в 2,60 мл 3 М раствора метилмагнийбромида в эфире. Реакционную смесь разбавляют в 6 мл эфира и охлаждают до 0oС. Смесь перемешивают в течение 1 ч при комнатной температуре, затем охлаждают до 0oС. Прикапывают к полученной смеси суспензию хлорангидрида 4-фтор-1-нафтойной кислоты в растворе, состоящем из 6 мл эфира и 4 мл ТФГ.
Затем полученную смесь перемешивают 16 ч при комнатной температуре, затем 2 ч при кипячении с обратным холодильником.
Затем реакционную смесь гидролизуют 50 мл ледяной воды, к которой добавлено 50 мл насыщенного раствора хлорида аммония.
Растворители выпаривают в вакууме и водную фазу экстрагируют ДХМ, затем промывают водой и сушат над MgSO4.
Растворители выпаривают, полученный продукт очищают хроматографией на силикагеле, элюируя ДХМ. Получают 0,48 г целевого продукта (т.пл. 170oС).
В) 1-(2-(4-морфолинил)этил)-2-метил-3-(4-фтор-1-нафтилкарбонил)-7-метоксииндол
0,28 г гидрида натрия (60% суспензия в масле) добавляют в раствор 0,67 г полученного выше продукта в 7 мл ДМФА. Смесь перемешивают 10 мин при комнатной температуре, затем к смеси добавляют суспензию хлоргидрата 4-(2-хлорэтил)морфолина в 3 мл ДМФА. Полученную смесь нагревают 16 ч при 100oС, затем выливают в 100 мл насыщенного раствора NH4Cl при 0oС. Смесь экстрагируют ДХМ, промывают водой, сушат над MgSO4.
Растворители выпаривают, продукт очищают хроматографией на силикагеле, элюируя смесью ЭА-толуол (градиент от 1:1 до 6:4), и получают 0,43 г целевого продукта (т.пл. 175oС).
Пример 2
1-(2-(4-морфолинил)этил)-2-метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол
А) 2-Метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол
Операции осуществляют так, как описано выше (пример 1, стадия А), но в качестве хлорангидрида кислоты применяют хлорангидрид 4-хлор-1-нафтойной кислоты, и получают целевой индол (т.пл. 184oС).
В) 1-(2-(4-морфолинил)этил)-2-метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол
Работают так, как описано выше (пример 1, стадия В), используя в качестве исходного соединения продукт, полученный на предыдущей стадии А, и получают целевой продукт (т.пл. 149oС).
Пример 3
1-н-пентил-2-метил-3-(4-хлор-1-нафтилкарбонил)-7-метоксииндол
Добавляют 0,13 г NaH (60% суспензия в масле) в раствор 0,77 г индола (полученного на стадии А примера 2) в 10 мл ДМФА. Смесь перемешивают 10 мин, затем добавляют 0,43 мл 1-йодпентана и смесь нагревают при 100oС в течение 16 ч, после чего выливают в 100 мл насыщенного раствора NH4Cl при 0oС. Органическую фазу промывают водой, сушат над MgSO4. Продукт очищают хроматографией на силикагеле, элюируя толуолом. Продукт кристализуют из смеси ДХМ - iPr2O и получают 0,34 г целевого продукта (т.пл. 112oС).
Пример 4
Метансульфонат (-)-3-(4-морфолинилметил)-5-метил-7-(5,7 дибромнафтилкарбонил)-2,3-дигидропиррол [1,2,3-de]-1,4-бензоксазина
Добавляют по каплям 20 мл этилалюминийхлорида в раствор, содержащий 4,09 г (+)-3-(4-морфолинилметил)-5-метил-2,3-дигидропиррол-[1,2,3-de] 1,4-бензоксазина (полученного в условиях приготовления 5В патента US 4939138) и 5,19 г хлорангидрида 5,7-дибром-1-нафтойной кислоты в 100 мл ДХМ, охлажденный до 10oС. Смесь перемешивают 30 мин при 10oС, затем выливают в 100 мл ледяной воды, подщелоченной 35% раствором NaOH. Полученную смесь экстрагируют ДХМ, промывают водой, сушат над МgSO4 и растворители выпаривают. После очистки хроматографией на силикагеле (элюэнт эфир-гексан 70:30) получают 1,0 г менее полярного продукта, который растворяют в минимальном объеме ацетонитрила. Добавляют 2 г метансульфоновой кислоты, предварительно растворенной в 1 мл эфира. Полученные кристаллы фильтруют, затем перекристаллизовывают в смеси СНСl3/МеОН с получением 0,37 г целевого продукта ([α]D=82,6o (1%, ДМФА); т.пл. 258oС).
Пример 5
Метансульфонат (+)-(2-метил-4(4-морфолинилметил)-5,6-дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)нафталин-1-ил-метанона
А) 2-(4-морфолинилкарбонил)хинолин
Это соединение получают согласно методике, описанной в J. Med.Chem., 26, 986 (1983), с использованием хинальдиновой кислоты в качестве исходного соединения (т.пл. 105oС).
В) (+)2-(4-морфолинилметил)-1,2,3,4-тетрагидрохинолин
Добавляют по каплям 101 мл 3,4М раствора Red-Al в толуоле в раствор 16,56 г амида, полученного выше, растворенного в 350 мл толуола. Смесь кипятят с обратным холодильником 16 ч. После охлаждения в ледяной бане, к смеси добавляют 250 мл полунасыщенного раствора соли Рошеля (тартрат Na и К), затем перемешивают 30 мин.
Смесь экстрагируют эфиром, промывают водой, сушат над MgSO4 и выпаривают растворители.
После кристаллизации в этаноле получают 11,76 г твердого вещества желтого цвета, которое растворяют в дибензоилвинной кислоте, как описано в патенте US 5109135 (т.пл. 88oС; [α]D = +99o (1% ДМФА)).
С) 1-амино-2-(4-морфолинилметил)-1,2,3,4-тетрагидрохинолин
Это соединение получают по методике, описанной в патенте US 5109135 (приготовления 2 и 3), исходя из амина, полученного на предыдущей стадии. Этот продукт используют без очистки на последующей стадии.
D) Метансульфонат (+)-(2-метил-4-(4-морфолинилметил)-5,6- дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)нафталин-1-ил-метанона
Соединение получают реакцией вышеполученного гидразина с 4-(1-нафтил)-2,4-бутандионом по методике, описанной в патенте US 5109135 (пример 2) с последующим солеобразованием реакцией с метансульфоновой кислотой в эфире и кристаллизацией в этаноле ([α]D = +11,7o (1%, ДМФА), т.пл. 250oС).
Пример 6
Метансульфонат 1-(1-нафтилкарбонил)-3-(2-(4-морфолинил)этил)индола
Это соединение получают реакцией 3-[2-(4-морфолинил)этил]индола (DL. Nelson с сотр. , Adv. Biochem. Psychopharmacol. (1993) 37, 337) с хлорангидридом 1-нафтилкарбоновой кислоты в присутствии NaH в ДМФА по методике, описанной в патенте NL 7308094 (пример1), получают продукт с т.пл. 187oС.
Пример 7
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол
А) 2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол
14,7 мл 3 М раствора метилмагнийбромида в эфире, разбавленного 30 мл эфира, охлаждают до 0oС, добавляют к последнему по каплям раствор 5,6 г 2-метил-7-метоксииндола в 25 мл эфира, затем оставляют на 1 ч при перемешивании при комнатной температуре. В смесь добавляют раствор 12,3 г хлорангидрида 4-бром-1-нафтойной кислоты в 28 мл эфира и 19 мл ТФГ, греют с обратным холодильником 1 ч и оставляют на 16 ч при перемешивании при комнатной температуре. Затем реакционную смесь выливают в 350 мл ледяной воды, добавляют 40 г NH4Cl и концентрируют под вакуумом растворители. Водную фазу экстрагируют эфиром, органическую фазу промывают 50 мл насыщенного раствора NH4Cl, затем 100 мл насыщенного раствора NaCl сушат над MgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле (элюент ДХМ). Получают 2 г продукта после перекристаллизации в толуоле.
В) 1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол
Это соединение получают по методике, описанной на стадии В примера 1, исходя из 0,8 г соединения, полученного на предыдущей стадии, 0,28 г NaH (60%-ный раствор в масле), 7 мл ДМФА и суспензии 0,64 г хлоргидрата 4-(2-хлорэтил)морфолина в 5 мл ДМФА. Получают 0,86 г целевого продукта после перекристаллизации в диизопропиловом эфире (т.пл. 123-125oС).
Пример 8
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол
Смесь 0,46 г соединения, полученного на стадии А примера 7, с 0,49 г 1-йодпентана, 0,16 г TDA-1 и 0,16 г КОН в 7 мл толуола нагревают в течение 2 ч при 90oС. Реакционную смесь выливают в 20 мл воды, после декантации органическую фазу промывают 10% раствором HCl, водой, затем насыщенным раствором NaCl, сушат над МgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле (элюент смесь ЭА:толуол 50/50 об/об). Получают 0,5 г целевого продукта (т.пл. 114,5oС).
Пример 9
1-(2-(4-морфолинил)этил)-2-метил-3-(1-нафтилкарбонил)-7-(трифторметил)индол
А) 2-метил-3-(метилтио)-7-(трифторметил)индол
Охлаждают до -65oС в атмосфере азота раствор 15 г 2-(трифторметил)анилина в 300 мл ДХМ, к последнему по каплям добавляют раствор 11,5 мл третбутилгипохлорита в 30 мл ДХМ, смесь оставляют на 10 мин при перемешивании. Добавляют затем при -65oС раствор 9,66 г метилтиоацетона в 30 мл ДХМ и оставляют на 2 ч при перемешивании при - 65oС. Доводят температуру смеси до -40oС, добавляют раствор 12,9 мл триэтиламина в 30 мл ДХМ и оставляют при перемешивании до достижения комнатной температуры. Смесь гидролизуют добавлением 200 мл воды, после декантации органическую фазу промывают водой, затем насыщенным раствором NaCl, сушат над МgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюент - толуол. Получают 9,9 г целевого продукта.
В) 2-метил-7-(трифторметил)индол
К раствору 9,9 г соединения, полученного выше на стадии А, в 100 мл TFA добавляют при комнатной температуре 12,4 г 2-меркаптобензойной кислоты и оставляют на 1 ч при перемешивании при комнатной температуре. Реакционную смесь концентрируют под вакуумом, остаток экстрагируют ЭА, органическую фазу промывают 1 н. раствором NaOH, водой, насыщенным раствором NaCl, сушат над МgSO4 и выпаривают растворитель под вакуумом. Получают 7,36 г целевого продукта.
С) 1-(2-(4-морфолинил)этил)-2-метил-7-(трифторметил)индол
К раствору 2,8 г хлоргидрата 4-(2-хлорэтил)морфолина в 15 мл ДМСО добавляют при комнатной температуре 2,25 г тонкоизмельченного КОН и оставляют на 5 мин при перемешивании. Добавляют затем по каплям раствор 2 г соединения, полученного на предыдущей стадии, в 15 мл ДМСО, оставляют на 2 ч при перемешивании при комнатной температуре, затем нагревают при 100oС в течение 18 ч. Реакционную смесь выливают в 300 мл ледяной воды, экстрагируют ДХМ, органическую фазу промывают водой, насыщенным раствором NaCl, сушат над МgSO4 и выпаривают растворитель под вакуумом. Остаток хроматографируют на силикагеле, используя в качестве элюента смесь ЭА:гексан в соотношении от (30/70 об/об) до (40/60 об/об). Получают 2,42 г целевого продукта, (т.пл. 72oС).
D) 1-(2-(4-морфолинил)этил)- 2-метил-3-(1-нафтилкарбонил)-7 - (трифторметил)индол
Охлаждают до 0oС в атмосфере азота раствор 0,4 г соединения, полученного на предыдущей стадии, и 0,2 мл хлорангидрида 1-нафтойной кислоты в 10 мл ДХМ, к смеси по каплям добавляют 1,54 мл 1,8 М раствора дихлорэтилалюминия (EtAlCl2) в толуоле и оставляют на 24 ч при перемешивании при комнатной температуре. Реакционную смесь выливают в 100 мл ледяной воды, экстрагируют ДХМ, органическую фазу промывают 5%-ным раствором Nа2СО3, водой, затем насыщенным раствором NaCl, сушат над МgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюент - смесь ЭА:гексан (30/70 об/об). Получают 0,3 г целевого продукта после кристаллизации в диизопропиловом эфире (т.пл. 104oС).
Пример 10
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-(трифторметил)индол
Это соединение получают по методике, описанной на стадии D примера 9, исходя из 0,8 г соединения, полученного на стадии С примера 9, 0,92 г хлорангидрида 4-бром-1-нафтойной кислоты в 20 мл ДХМ и 3,1 мл 1,8 М раствора EtAlCl2 в толуоле. Получают 0,9 г целевого продукта (т.пл. 160oС).
Пример 11
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол
А) 2-метил-3-(метилтио)-7-фториндол
Это соединение получают по методике, описанной на стадии А примера 9, исходя из 20 г 2-фторанилина в 600 мл ДХМ, 23,4 г трет-бутилгипохлорита, 22,5 г метилтиоацетона и 30 мл триэтиламина. Получают 20,5 г целевого продукта.
В) 2-метил-7-фториндол
К раствору 10 г соединения, полученного на предыдущей стадии, в 100 мл TFA добавляют при комнатной температуре 16 г 2-меркаптобензойной кислоты и оставляют на 2 ч при перемешивании при комнатной температуре. Нерастворимые вещества отфильтровывают, фильтрат концентрируют под вакуумом. Остаток обрабатывают 100 мл смеси ЭА:вода (50/50 об/об), органическую фазу промывают три раза 10%-ным раствором NaOH, водой, затем 10%-ным раствором НСl, насыщенным раствором NaCl, сушат над MgSO4 и испаряют под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюент - толуол. Получают 3,8 г целевого продукта.
С) 1-(2-(4-морфолинил)этил)-2-метил-7-фториндол
Смесь 1,3 г соединения, полученного на стадии В, 2,76 г хлоргидрата 4-(2-хлорэтил)морфолина, 2,05 г тонкоизмельченного КОН и 0,13 г KI в 13 мл ДМСО, нагревают в течение 16 ч при 100oС. После охлаждения до комнатной температуры реакционную смесь выливают в 100 мл воды, экстрагируют толуолом, органическую фазу промывают водой, затем насыщенным раствором NaCl, сушат над МgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюент - смесь ЭА:толуол (50:50 об/об). Получают 1,3 г целевого продукта.
D) 1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол
Это соединение получают по методике, описанной на стадии D примера 9, исходя из 1,3 г соединения, полученного выше на стадии С, 1,8 г хлорангидрида 4-бром-1-нафтойной кислоты в 50 мл ДХМ и 6 мл 1,8 М раствора EtAlCl2 в толуоле. Остаток хроматографируют на силикагеле, элюент - смесь ЭА:толуол (75/25 об/об). Получают 1,0 г целевого продукта (т.пл. 66-67oС).
Пример 12
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол
А) 1-н-пентил-2-метил-7-фториндол
Смесь 1 г соединения, полученного на стадии В примера 11, 1,6 г 1-йодпентана, 0,21 г TDA-1 и 0,75 г тонкоизмельченного КОН в толуоле нагревают при 95oС в течение 5 ч. После охлаждения до комнатной температуры, добавляют 30 мл воды, декантируют, органическую фазу промывают 10%-ным раствором НСl, водой, насыщенным раствором NaCl, сушат над МgSO4 и выпаривают под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюент - смесь циклогексан:толуол (60/40 об/об). Получают 0,58 г целевого продукта.
В) 1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол
Это соединение получают по методике, описанной на стадии D примера 9, исходя из 0,58 г соединения, полученного на предыдущей стадии 0,95 г хлорангидрида 4-бром-1-нафтойной кислоты в 25 мл ДХМ и 3,17 мл 1,8 М раствора EtAlCl2 в толуоле. Остаток хроматографируют на силикагеле, элюент - ЭА:толуол (50/50 об/об). Получают 0,27 г целевого продукта.
ЯМР (200 МГц; м.д.): ДМСО (2,5 м.д.); DOH (3,3 м.д.); 0,85 (t, 3Н); 1,3 (m, 4H); 1,75 (m, 2H); 2,35 (s, 3H); 4,3 (t, 2H); 6,8-8,4 (m, 9Н).
Пример 13
1-(2-(4-морфолинил)этил)-2,7-диметил-3(4-бром-1 нафтилкарбонил)индол
А) 2,7-диметил-3-(фенилтио)индол
Охлаждают до -70oС в атмосфере азота раствор 21,4 г 2-метиланилина в 600 мл ДХМ, прикапывают 21,7 трет-бутилгипохлорита и оставляют на 5 мин при перемешивании. Затем добавляют при -70oС раствор 24,9 г фенилтиоацетона в 50 мл ДХМ и оставляют на 2 ч при перемешивании при -65oС. При той же температуре добавляют 25,3 г триэтиламина и оставляют при перемешивании до подъема температуры до комнатной. Проводят гидролиз добавлением 250 мл воды, декантируют, органическую фазу промывают сначала 10%-ным раствором НСl, затем насыщенным раствором NaCl, водой, сушат над МgSO4 и испаряют растворитель под вакуумом. Остаток хроматографируют на силикагеле, элюент - толуол. Получают 19,69 г целевого продукта.
В) 1-(2-(4-морфолинил)этил)-2,7-диметил-3-(фенилтио)индол
Смесь 2,4 г соединения, полученного на предыдущей стадии А, 3 г 4-(2-хлорэтил)морфолина, 2,23 тонкоизмельченного КОН и 0,2 г KI в 24 мл ДМСО нагревают 16 ч при 100oС. Реакционную смесь выливают в 50 мл воды, экстрагируют толуолом, органическую фазу промывают водой, затем насыщенным раствором NaCl, сушат над МgSO4 и испаряют растворитель под вакуумом. 0,7 г остатка хроматографируют на силикагеле, элюент - смесь ЭА:толуол (50/50 об/об). Получают 0,56 г целевого продукта (т.пл. 129,5oС).
С) 1-(2-(4-морфолинил)этил)-2,7-диметилиндол
Смесь 2,4 г соединения, полученного на предыдущей стадии В, и 2,2 г 2-меркаптобензойной кислоты в 24 мл ТФУ оставляют на 3 ч при перемешивании при комнатной температуре. Реакционную смесь концентрируют под вакуумом, остаток поглощают смесью ЭА: вода, органическую фазу промывают 10%-ным раствором NaOH, водой, затем насыщенным раствором NaCl, сушат над МgSO4 и испаряют под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюант - ЭА: толуол (50/50 об/об). Получают 1,3 г целевого продукта.
D) 1-(2-(4-морфолинил)этил)-2,7-диметил-3-(4-бром-1-нафтилкарбонил)индол
Охлаждают до 0oС в атмосфере азота раствор 1,2 г соединения, полученного на предыдущей стадии и 1,68 г хлорангидрида 4-бром-1-нафтойной кислоты в 50 мл ДХМ, добавляют 5,7 мл 1,8 М раствора EtAlCl2 в толуоле, смесь оставляют на 36 ч при перемешивании при комнатной температуре. Реакционную смесь выливают в 100 мл воды, экстрагируют ДХМ, органическую фазу промывают 10%-ным раствором NaOH, водой, насыщенным раствором NaCl, сушат над МgSO4 и испаряют под вакуумом растворитель. Остаток хроматографируют на силикагеле, элюант - смесь ЭА: толуол (70/30 об/об). Получают 0,36 г целевого продукта (т.пл. 143oС).
Биохимические тесты
Было установлено, что при наномолярных концентрациях соединения согласно изобретению, например такие как метансульфонат 3-(4-морфолинилметил)-5-метил-6-(1-нафтилкарбонил)-2,3-дигидропирроло[1,2,3-de] -1,4-бензоксазина и 1-(2-(4-морфолинил)этил)-2-метил-3-(1-нафтилкарбонил)-7-метоксииндол, способны заметно увеличить уровень синтеза ДНК человеческих клеток В, состимулируемых антителами anti-Ig (увеличение абсорбции тинидина примерно на 40%).
При применении селективного антагониста рецептора CB1 (SR 141716А) в широком диапазоне концентраций одновременно с соединением СР 55940 (или Δ9-ТНС, или WIN 55212-2) при концентрации 10-9 М никакого эффекта блокирования обнаружено не было.
Феномен увеличения роста клеток В можно было наблюдать, используя другой путь активации, заключающийся в стимуляции клеток В приведением в контакт антигена СД40 с моноклональными антителами, продуцированными клетками L CD W 32.
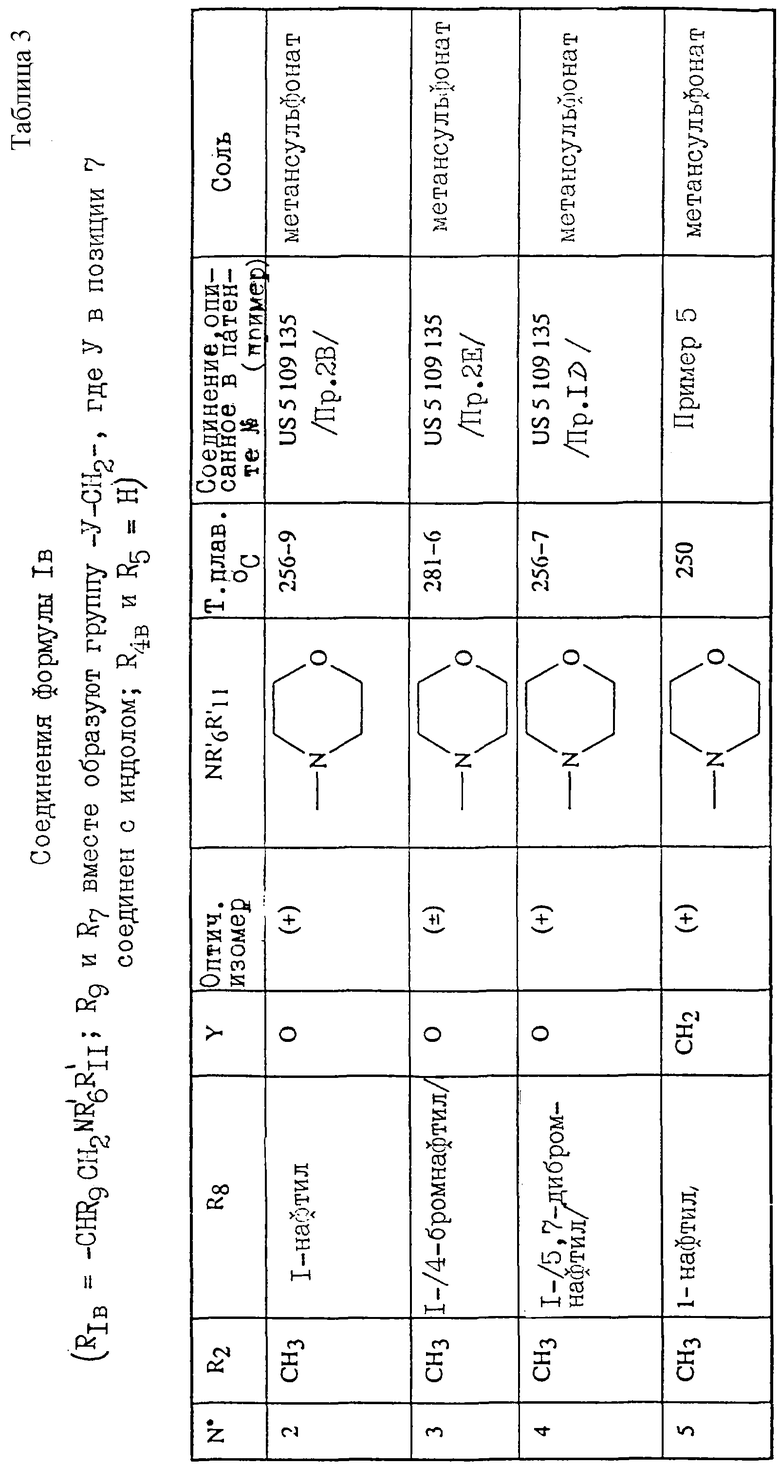
Соединения согласно изобретению общих формул (I) и (I') и их возможные соли показали сродство (в тесте in vitro) в 30-1000 раз более высокое в отношении человеческих переферийных рецепторов каннабиноидов (CB2), чем к человеческим рецепторам центральной нервной системы (CB1), экспрессированные в клетки яичника китайского хомяка (СНО). Исследования аффиной связи были выполнены в экспериментальных условиях, описанных Devane'ом с сотр. (Molecular Pharmacology (1988), 34, 605-613), на мембранах, выделенных из клеточных линий, в которые были экспримированы рецепторы CB1 и СВ2 (Munro с сотр., Nature (1993), 365-561-565).
Предпочтительными соединениями являются следующие соединения: 1-(2-(4-морфолинил)этил)-2-метил-3-(1-нафтилкарбонил)-7-метоксииндол; метансульфонат 1-(1-нафтилкарбонил)-3-(2-(4-морфолинил)этил)индола; 2-метил-1-[2-(1-метил-2-пиперидинил)метил]-3-(1-нафтилкарбонил)индол;
метансульфонат (+)-(2-метил-4-(4-морфолинилметил)-5,6-дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)нафталин-1-ил-метанона;
4-[2-[1-[1-нафтил)метилен]-1-метил-инден-3-ил]этил]морфолин;
4-[2-[1-[1-(4-метоксинафтил)метилен]-1-метил-инден-3-ил]этил]морфолин;
4-[2-[1-[(9-антрил)метилен]-lH-инден-3-ил]этил]морфолин;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол;
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол;
1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-(трифторметил)индол;
1-н-пентил-2-метил-3-(4-бром-1-нафтилкарбонил)-7-фториндол.
Особенно предпочтительным является соединение 1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндол, сродство которого к рецептору CB1 превышает 1000 нМ, а к рецептору СВ2 составляет 1,8 нМ.
С другой стороны, соединения согласно изобретению ведут себя в тесте in vitro как специфические агонисты человеческих рецепторов каннабиноидов СВ2 в противоположность к CB1, экспримированные в клетки СНО. Действительно, специфически связываясь с рецепторами СВ2, они уменьшают продуцирование АМРс, стимулируемое форсколином, ингибируя при этом аденилатциклазу. Соответствующие испытания были осуществлены в экспериментальных условиях, описанных Matsuda с сотр. (Nature, 1990, 346, 561-564).
В качестве примера для 1-(2-(4-морфолинил)этил)-2-метил-3-(4-бром-1-нафтилкарбонил)-7-метоксииндола были рассчитаны следующие значения концентраций, ингибирующие на 50% аденилатциклазу IC50:
в случае рецептора CB2 IC50 = 1 нМ;
в случае рецептора CB1 IC50 = 1 мкМ.
Соединения согласно изобретению имеют также сродство в тесте in vitro к рецепторам каннабиноидов, находящихся на уровне мышиной селезенки, при введении соединений внутривенно, интраперитонально или орально. Испытания были проведены по методике, описанной Rinaldi-Carmona с сотр. (Life Sciences, 1995, 56, 1941-47).
Изобретение относится к медицине. Предложено новое средство иммуномодулирующего действия. Средство содержит селективный агонист, имеющий константу сродства к рецептору СВ2 по меньшей мере в 30 раз ниже константы сродства к рецептору CB1. В качестве такого агониста могут служить известные или новые соединения общих формул (I) или (I') со значениями радикалов, приведенными в формуле. Изобретение расширяет арсенал средств заявленного назначения. Помимо иммуномодулирующего дейстия некоторые соединения дополнительно обладают анальгетическим действием. 3 с. и 10 з.п. ф-лы, 7 табл.
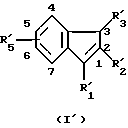
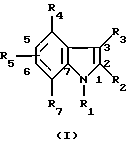
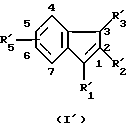
где R1 - радикал, выбранный из группы -CH2CHR10NR6R11; (CH2)2NR'6R'11; -CHR9CH2NR'6R'11; -(CH2)nZ и -COR8;
-R'1 означает группу -CH2CHR10NR6R11 или группу -(CH2)2NR'6R'11;
R2 и R'2 - водород, галоген или (С1-С4)алкил;
R3 - водород, (С1-C4) алкил или группа, выбранная из групп: -CH2CHR10-NR6R11; -(CH2)2NR'6R'11 и -COR8;
R'3 - группа = CR6R8;
R4 имеет одно из значений, указанных для радикала R5, или означает группу -COR8;
R5 - водород, (С1-C4) алкил, (С1-C4)алкокси, галоген, СF3, ОСF3, (С1-C4)алкилтио;
R'5 имеет одно из значений, указанных для радикала R5, и находится в положении 5 или 6 инденового цикла;
R6 - водород или (С1-C4) алкил;
R'6 - (С1-C4) алкил;
R7 - имеет одно из значений, указанных для радикала R5, или R7 и R9, вместе образуют группу Y-CH2-, связанную с индольным циклом в положении 7 через группу Y;
R8 - фенил, замещенный 1-4 заместителями, выбранными из галогена, группы (С1-C4) алкил или (С1-C4)алкоксил; полицикл, выбранный из нафт-1-ила, нафт-2-ила, 1,2,3,4-тетрагидронафт-1-ила, 1,2,3,4-тетрагидронафт-5-ила, антрила, бензофурила, бензотиен-2-ила, бензотиен-3-ила, 2-, 3-, 4- и 8-хинолила, причем указанные полициклические радикалы не замещены или замещены 1-2 заместителями, выбранными из группы: (С1-C4)алкил, (С1-C4) алкоксил, (С1-C4)алкилтио, галоген, циано, гидроксил, трифторметил, имидазол-1-ил;
R10 и R11 вместе составляют группу, выбранную из группы: -СН2-О-СН2-CR12R13 - и -(CH2)p-CR12R13, в которых атом углерода, замещенный радикалами R12 и R13, связан с атомом азота;
R'11 - алкил (С1-C4), либо R'11 и R'6 образуют с атомом азота, с которым они связаны, группу, выбранную из группы: морфолин-4-ил, тиоморфолин-4-ил, пиперидин-1-ил, пиролидин-1-ил;
R12 и R13 - каждый, независимо друг от друга, означают атом водорода или алкил (С1-C4);
n = 2, 3, 4 или 5;
р = 2 или 3;
Z - метил или галоген;
Y-метиленовая группа или атом кислорода; при условии, что в формуле (I) любой один из заместителей R1, R3 или R4 означает группу -COR8 и что: если R1 = -COR8, то R3 означает группу -CH2CHR10NR6R11; или группу -(CH2)2NR'6R'11, а R4 имеет одно из значений, указанных для R5; если R3 = -COR8, то R1 означает группу, выбранную из групп -CH2CHR10NR6R11, -CHR9CH2NR'6R'11, -(CH2)2NR'6R'11 и -(CH2)nZ, R4 имеет одно из значений R5 и по меньшей мере один из радикалов R4, R5 и R7 является водородом; если R4 = -COR8, то R1 означает группу, выбранную из групп: -CH2CHR10NR6R11, -CHR9CH2NR'6R'11, -(CH2)2NR'6R'11 и -(CH2)nZ и R3 - водород или алкил (С1-C4),
а также их фармацевтически приемлемые соли.
в которой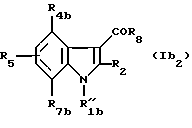
R'' 1b означает группу -(CH2)nZ,
R4b - обозначает атом водорода,
R7b обозначает (С1-C4)алкокси или атом галогена,
R2 обозначает (С1-C4)алкил,
R5 обозначает атом водорода,
R8 обозначает нафт-1-ил, незамещенный или замещенный 1 или 2 заместителями, выбранными из галогена,
n означает 2, 3, 4 или 5,
Z обозначает группу метил или атом галогена,
и их фармацевтически приемлемые соли.
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1993, ч.2, с | |||
| Вагонный распределитель для воздушных тормозов | 1921 |
|
SU192A1 |
| US 5013837, 07.05.1991 | |||
| ЕР 0444451 А2, 04.09.1991. | |||

