Настоящее изобретение относится к новым замещенным 1-(7-хлорхинолин-4-ил)-пиразол-3-карбоксамид-N-оксидам, имеющим большое сродство к рецептору нейротензина, к способу их получения и к фармацевтическим композициям, содержащим их в качестве активного начала.
Первые синтетические непептидные лекарственные средства, способные связываться с рецептором нейротензина, описаны в европейской заявке EP-0477049. Речь идет об амидах пиразол-3-карбоновой кислоты, различно замещенных аминокислотами, которые смещают иодированный нейротензин с его рецептора при дозах ниже микромоля на мембранах мозга человека. Из этой серии соединений следует отметить одно соединение, 2-{[(7-хлорхинолин-4-ил)-5-(2,6- диметоксифенил)пиразол-3-ил] -карбониламино} адамантан-2-карбоновую кислоту, называемую далее SR48692, которая обладает мощной и селективной активностью антагониста нейротензина /D. Gully et al., Proc. Natl. Acad. Sci. USA, 1993, 90, 65-69/.
Характерным признаком этой группы соединений, описанных в EP-0477049, является наличие в положении 1 пиразольного цикла, фенильной, нафтильной, хинолин-4-ильной группы, возможно замещенной. Более конкретно SR48692 имеет в положении 1 пиразола 7-хлорхинолин-4-ильную группу.
Теперь обнаружено, что при окислении в мягких условиях азота 7-хлорхинолин-4-ильной группы в производных пиразол-3-карбоксамидов получают молекулы, которые обладают по сравнению с их не N-оксидными предшественниками по крайней мере такой же активностью по отношению к рецепторам нейротензина и, кроме того, обладают лучшей растворимостью, в частности в воде.
Так, эти новые соединения согласно изобретению являются особенно интересными, потому что они позволяют получать растворы для инъекций.
Кроме того, они совместимы с многочисленными лечебными препаратами, вводимыми оральным путем, потому что они обладают лучшей биодоступностью.
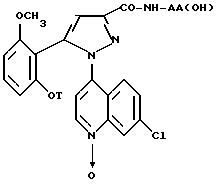
Следовательно, согласно одному из аспектов настоящее изобретение относится к замещенным 1-(7-хлорхинолин-4-ил)-пиразол-3-карбоксамид- N-оксидам формулы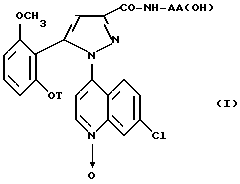
в которой T является водородом, C1-C4-алкилом, C3-C8-циклоалкилом, (C3-C8)-циклоалкилметилом, метоксиэтилом,
группа -NH-AA(OH) является остатком аминокислоты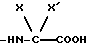
где X является водородом, C1-C5-алкилом или карбоциклическим радикалом, неароматическим, содержащим 3 - 15 атомов углерода, и X' является водородом, или же X и X', взятые вместе с атомом углерода, к которому они присоединены, образуют неароматический C3-C15-карбоцикл, и их солям.
Под C1-C4-алкилом или C1-C5-алкилом понимают прямые или разветвленные алкилы.
Неароматические C3-C15-карбоциклические радикалы представляют собой моно- или полициклические, конденсированные или мостиковые, насыщенные или ненасыщенные, возможно терпеновые радикалы. Эти радикалы могут быть замещены одним или несколькими C1-C4-алкилами.
Моноциклические радикалы включают C3-C12-циклоалкилы, например циклопропил, циклопентил, циклогексил, циклогептил, циклооктил, циклододецил.
В приведенном выше аминокислотном остатке, когда X и X' вместе с атомом углерода, к которому они присоединены, образуют неароматический C3-C15-карбоцикл, указанный карбоцикл является таким, как определено для соответствующих приведенных выше радикалов.
Среди неароматических полициклических карбоциклов предпочтительным является адамантан. Так, когда X' является водородом, X является 1-адамантильной или 2-адамантильной группой, и когда -C(XX') вместе образуют карбоцикл, этот радикал представляет собой 2-адамантилиденовый радикал.
Среди неароматических карбоциклов особенно предпочтительными являются циклопентан и циклогексан.
N-оксиды предпочтительных замещенных хинолинилпиразолов согласно настоящему изобретению являются N-оксидами формулы /I/, в которой:
T является метильной или циклопропилметильной группой, а группа -NH-AA(OH) является остатком 2-аминоадамантан-2-карбоновой кислоты или (S)-2-амино-2-циклогексилуксусной кислоты или их солей.
Соли являются солями щелочных металлов, предпочтительно натрия или калия, щелочноземельных металлов, предпочтительно кальция, и органических оснований, например диэтиламина, трометамина, меглумина (N-метил-D-глюкамина), лизина, аргинина, гистидина или диэтаноламина.
Соли соединений формулы I согласно настоящему изобретению также являются солями с минеральными или органическими кислотами, которые позволяют осуществить удобное разделение или кристаллизацию соединений формулы I, с такими как пикриновая кислота, щавелевая кислота или оптически активная кислота, например, миндальная кислота или камфосульфоновая кислота, или с кислотами, которые образуют фармацевтически приемлемые соли, такие как хлоргидрат, кислый сульфат, первичный фосфат, метансульфонат, малеат, фумарат, 2-нафталин-2-сульфонат, изетионат.
Когда соединения /I/ содержат асимметричный атом углерода, энантиомеры являются частью изобретения.
Когда группа -NH/AA/OH является остатком циклоалифатической аминокислоты, соединения формулы /I/ также включают соединения, у которых аминная функция находится в эндо-положении по отношению к циклической алифатической системе, а также те, у которых аминная функция находится в экзо-положении по отношению к алифатической циклической системе.
Согласно другому из аспектов изобретения настоящее изобретение относится к способу получения замещенных 1-(7-хлорхинолин-4-ил)пиразол-3-карбоксамид-N-оксидов формулы /I/ и их солей с минеральными или органическими основаниями, отличающемуся тем, что обрабатывают производные формулы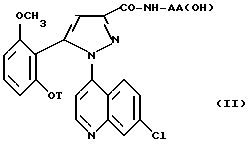
в которой T и AA/OH/ имеют значения, указанные выше для соединения формулы /I/, окисляющим агентом при комнатной температуре в апротонном растворителе, чтобы получить соединения /I/ или одну из их солей.
Используемые окисляющие агенты являются хорошо известными специалистам и выбраны, например, среди:
гексагидрата моноопероксифталата магния /или ГМПФ/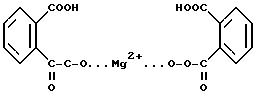
пербензойной кислоты,
метахлорпербензойной кислоты /или ХПБК/,
перфталевой кислоты,
пермуравьиной кислоты,
перуксусной кислоты.
Также можно использовать другие перкислоты.
Используемыми растворителями являются растворители, обычно применяемые специалистами в реакциях окисления, например диполярные апротонные растворители, такие как диметилформамид, или хлорированные растворители, такие как дихлорметан или хлороформ.
Предпочтительным окисляющим агентом является метахлорпербензойная кислота, которая приводит к хорошим выходам в условиях мягкоокисления. Температура реакции предпочтительно является комнатной температурой, что позволяет избежать образования продуктов разложения или гидроксилирования.
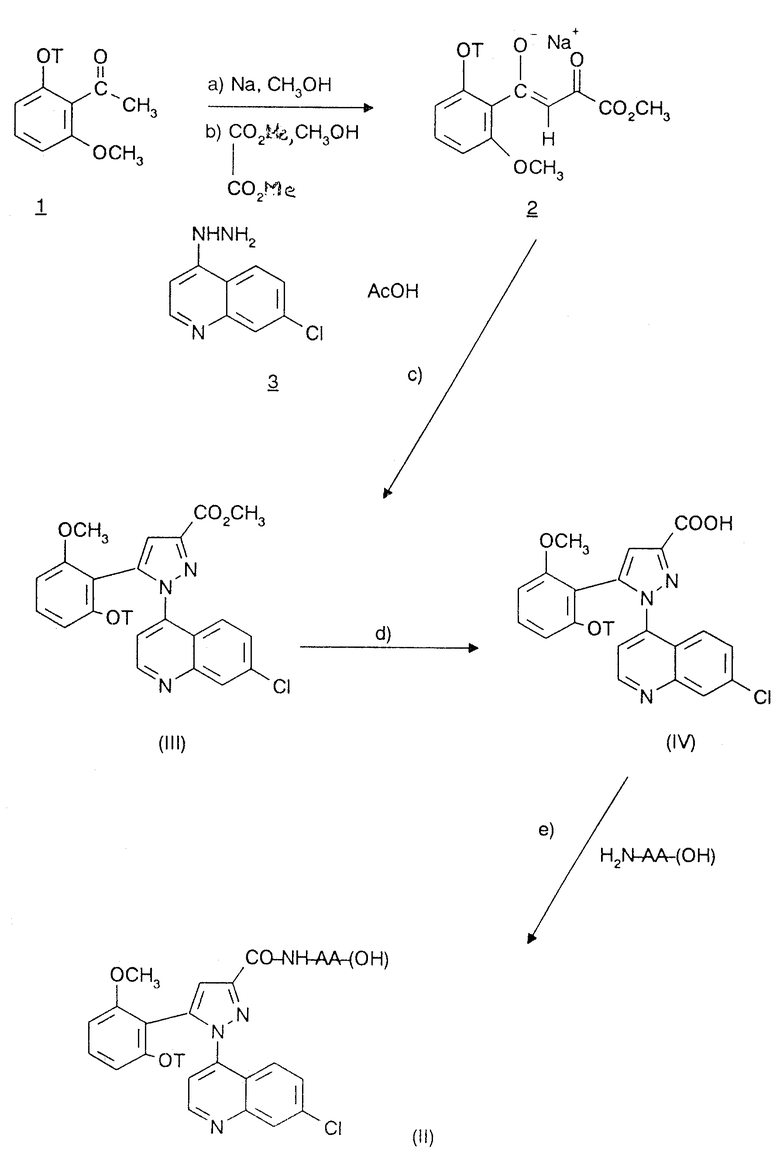
Соединения формулы /II/ получают согласно EP-0477049 по схеме 1, приведенной в конце текста.
На первой стадии a/ проводят реакцию сильного основания, такого как метилат натрия, с кетоном формулы I, в которой T имеет указанные выше значения, потом проводят реакцию /стадия b/ с эквимолекулярным количеством метилоксалата в спирте, например, метаноле, согласно L.Claisen, Ber., 1909, 42, 59. После осаждения простым эфиром, таким как диэтиловый или диизопропиловый эфир, еноляты натрия 2 отделяют фильтрованием. Можно также получать енолят лития по W.V. Murray et al., J. Heterocyclic Chem., 1989, 26, 1389.
Полученный таким образом енолят металла 2 и избыток производного 7-хлор-4-(гидразин-1-ил)-хинолина 3 или одной из его солей затем кипятят с обратным холодильником с уксусной кислотой /стадия c//, чтобы получить сложные эфиры /III/.
При омылении сложных эфиров /III/ под действием щелочного агента, например, гидроксида калия или гидроксида натрия с последующим подкислением получают кислоты /IV/ (этап d).
В качестве функционального производного замещенной (7-хлорхинолин-4-ил)пиразол-3-карбоновой кислоты формулы /IV/ можно использовать хлорангидрид кислоты, ее ангидрид, смешанный ангидрид, C1-C4-сложный алкиловый эфир, активированный сложный эфир, например п-нитрофениловый, или своевременно активированную свободную кислоту, например, N,N-дициклогексилкарбодиимидом или гексафторфосфатом бензотриазол-N-окситрис-/диметиламино/-фосфония /BOP/.
Аминокислоты формулы NH2-AA-/OH/ могут быть использованы или как таковые, или после предварительной защиты обычными защищающими группами, используемыми в пептидном синтезе.
Так, на стадии e/ способа можно провести взаимодействие хлорангидрида 1-(7-хлорхинолин-4-ил)пиразол-3-карбоновой кислоты, полученного при взаимодействии тионилхлорида с кислотой формулы /IV/, с аминокислотой, в таком растворителе, как ацетонитрил, ТГФ, ДМФ или дихлорметан, в инертной атмосфере при комнатной температуре в течение времени от нескольких часов до нескольких дней в присутствии основания, такого как пиридин, гидроксид натрия или триэтиламин.
Вариант стадии e/ заключается в получении хлорангидрида кислоты или смешанного ангидрида (7-хлорхинолин-4-ил)пиразол-3-карбоновой кислоты при взаимодействии изобутилхлорформиата или этилхлорформиата с кислотой формулы /IV/ и с последующей его реакцией с N,O-бис-триметилсилильным производным аминокислоты, полученным в соответствии с методикой, описанной в публикации M. T. Nagasawa и сотр. J. Med. Chem., 1975, 18, 8, 826-830, при взаимодействии бис/триметилсилил/ацетамида или 1,3-бис/триметилсилил/мочевины, или бис/трифторметил/ацетамида с аминокислотой формулы NH2-AA-/OH/ в таких растворителях, как ацетонитрил, дихлорметан, в инертной атмосфере при комнатной температуре или при температуре кипения растворителя в течение времени от нескольких часов до одного дня.
В другом варианте осуществления стадии e/ проводят взаимодействие смешанного ангидрида пиразол-3-карбоновой кислоты с аминокислотой формулы NH2-AA-/OH/ в таком растворителе, как дихлорметан, в инертной атмосфере при комнатной температуре в течение времени от одного до нескольких дней в присутствии основания, такого как триэтиламин.
(7-Хлорхинолин-4-ил)-4-пиразол-3-карбоновые кислоты формулы /IV/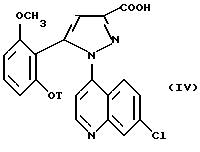
в которой T является C3-C8-циклоалкилом, C3-C8-циклоалкилметилом или метоксиэтилом, а также функциональные производные по кислотной функции являются новыми и как ключевые промежуточные продукты для получения соединения /I/ составляют последующий аспект изобретения
Соединения формулы /II/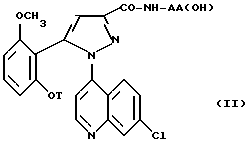
в которой T является C3-C8-циклоалкилом, C3-C8-циклоалкилметилом или метоксиэтилом, также являются новыми и составляют другой аспект настоящего изобретения.
Когда продукт формулы /II/ получают в виде кислоты, он может быть превращен в металлическую соль, например в щелочную, такую как натриевая соль, или соль щелочноземельного металла, такую как соль кальция, по классическим методикам.
Некоммерческие аминокислоты могут быть получены по методике Strecker, Ann. 1850, 75, 27 или по методике H.T.Bucherer и сотр., J.Proct. Chem., 1934, 141, 5, с последующим гидролизом, приводящим к аминокислотам; например, 2-аминоадамантан-2-карбоновую кислоту получают согласно H.T.Nagasawa и сотр., J. Med. Chem., 1973, 16 /7/, 823 или согласно M.Paventi и сотр., Can. J. Chem., 1987, 65, 2114.
α - Амино-1-адамантилуксусную и α -амино-2-адамантилуксусную кислоты получают согласно B. Gaspern и сотр., Croatino Chemica Acta, 1976, 48 /2/, 169-178.
2-Аминонорборнан-2-карбоновую кислоту получают согласно H.S. Tager и сотр., J. Am. Chem. Soc., 1972, 94, 968.
α -Аминоциклоалкилкарбоновые кислоты получают согласно J.W. Tsang и сотр., J. Med. Chem., 1984, 27, 1663.
R- и S-циклопентилглицины получают в соответствии с европейской заявкой на патент EP-477049.
R- и S-циклогексилглицины получают согласно Rudman и сотр., J. Am. Chem. Soc., 1952, 74, 551.
R- и S-циклогексилглицины можно получать также каталитическим гидрированием R- и S-фенилглицинов.
α -Аминоциклоалкилкарбоновые кислоты R- или S-конфигурации также можно получить ферментативным гидролизом, стереоспецифическим, соответствующих рацемических N-ацетильных производных согласно J.Hill и сотр., J. Org. Chem. , 1965, 1321.
Соединения формулы /I/ также охватывают соединения, в которых один или несколько атомов водорода или углерода заменены их радиоактивным изотопом, например, тритием или углеродом-14. Такие меченые соединения полезны для исследовательских работ по метаболизму или фармацеи, в биохимических опытах в качестве лиганд рецепторов.
Соединения формулы /I/ и их соли с минеральными или органическими основаниями обладают очень большим сродством к человеческим рецепторам нейротензина, в тестах, описанных в публикации D. Gully и сотр., приведенной выше. Более конкретно, по сравнению с 1-нафтил- и 4-хлор-1-нафтильными производными, описанными в EP-0477049, которые имеют ИК50, равную или больше 100 нМ, соединения изобретения обладают значительно меньшей ИК50, доходящей от нескольких нМ до 50 нМ. Особенно интересными являются продукты формулы /I/, в которой T является метилом или циклопропилметилом, более конкретно амид 2-амино-2-адамантанкарбоновой кислоты, который имеет ИК50 порядка 2 нМ.
Следовательно, это соединение является даже более активным, чем SR48692, что является неожиданным, если сослаться на уже очень высокую активность соединений, описанных в EP-0477049.
Кроме того, N-оксидные соединения, в частности описанное в примере 2 ниже, 2-{[(1-(1-оксид-7-хлорхинолин-4-ил)-5-(2,6- диметоксифенил)-пиразол-2-ил]карбониламино}адамантан-2-карбоновая кислота являются объектом изучения растворимости в сравнении с SR48692.
Исследованными средами являлись вода, этанол и смесь вода/полиэтиленгликоль 400 /70/30 объем/объем/.
Измерения растворимости проводили при 25oC после перемешивания в течение 3 или 5 часов при насыщении.
После помещения в условия, в которых солюбилизированные в воде продукты не адсорбируются на некоторых фильтрах, растворы фильтруют, потом анализируют жидкостной хроматографией /колонка μ Бондапак C18, элюент ацетонитрил-трифторуксусная кислота, определение при 254 нМ, расход 1 мл/мин/.
Получают следующие результаты (см. таблицу в конце описания).
Это изучение показало более высокую и неожиданную растворимость соединения примера 2, в частности, в воде и в смеси вода/полиэтиленгликоль, которые являются растворителями, позволяющими готовить формы для инъекций. В качестве дополнительного примера приведена растворимость соединений в этаноле.
Соединения настоящего изобретения являются малотоксичными, например, их острая токсичность совместима с их использованием в качестве лекарственного средства. Для такого использования млекопитающим вводят эффективное количество соединения /I/ или одной из его фармацевтически приемлемых солей для лечения патологических состояний, связанных с дисфункцией допаминэргетических систем, например, в качестве антипсихотических средств /D.R. Handrich и сотр. Brain Research, 1982, 231, 216-221 и C.B. Nemeroff, Biological Psychiatry, 1980, 15, /2/, 283-302/, при расстройствах сердечно-сосудистых или желудочно-кишечных систем.
Так объектом настоящего изобретения согласно другому из его аспектов являются фармацевтические композиции, содержащие в качестве активного начала соединения формулы /I/ и его возможные фармацевтически приемлемые соли.
В фармацевтических композициях настоящего изобретения для введения оральным, сублингвальным, внутримышечным, трансдермальным или ректальным путями активные начала могут быть введены в виде единичных доз введения или в смеси, или с классическими фармацевтическими носителями животным или людям. Подходящие единичные формы введения представляют собой формы для орального введения, такие как таблетки, капсулы, порошки, гранулы и растворы или суспензии для орального введения, формы для сублингвального или буккального введения, формы для подкожного введения, внутримышечного или внутривенного введения и формы для ректального введения.
Наконец для получения желаемого эффекта доза активного начала может варьировать между 0,5 и 1000 мг в день, предпочтительно между 2 и 500 мг.
Каждая единичная доза может содержать от 0,5 до 250 мг активного начала, предпочтительно 1 - 125 мг, в сочетании с фармацевтически приемлемым носителем. Эта единичная доза может быть введена 1 - 4 раза в день.
Когда готовят твердую композицию в виде таблеток, смешивают активное начало с фармацевтически приемлемым носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или их аналоги. Таблетки можно покрывать оболочкой из сахарозы или других подходящих материалов или их можно обработать таким образом, чтобы они обладали пролонгированной или замедленной активностью и что они будут высвобождать непрерывно заранее определенное количество активного начала.
Препарат в капсулах получают при смешивании активного начала с разбавителем и выливании полученной смеси в мягкие или твердые капсулы.
Препарат в виде сиропа или элексира может содержать активное начало совместно с подслащивателем, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептика, а также агентом, придающим вкус, и подходящим красителем.
Порошки или гранулы, диспергирующиеся в воде, могут содержать активное начало в смеси с диспергаторами, смачивателями или суспендирующими агентами, такими как поливинилпирролидон и ему подобные, а также с подслащивателями или корректорами вкуса.
Активное начало также может находиться в виде комплекса с циклодекстрином, например, α,β,γ-декстрином, 2-оксипропил-β-циклодекстрином, метил-β-циклодекстрином.
Для ректального введения прибегают к суппозиториям, которые готовят со связующими, плавящимися при ректальной температуре, например маслом какао или полиэтиленгликолями.
Активное начало может быть сформулировано также в виде микрокапсул, возможно с одним или несколькими носителями.
Следующие примеры, приведенные в качестве неограничивающих, иллюстрируют изобретение. В приготовлениях, приведенных ниже, описаны методики синтеза различных промежуточных продуктов, позволяющих получать соединения изобретения.
Точки плавления были измерены на нагревательном столике Кофлера.
Соединения согласно изобретению были подвергнуты анализу с точностью до сотых согласно теории.
Спектры ядерного магнитного резонанса и масс-спектры также подтверждают структуру соединений, описанных в примерах ниже.
Приготовление I.
Растворяют 5 г 2-окси-6-метоксиацетофенона в 100 мл изопропанола в присутствии 1,2 эквивалента /6,3 мл/ гидроксида цезия концентрацией 50% в воде. Смесь перемешивают в течение 10 минут, потом концентрируют в вакууме и растворяют в изопропаноле и концентрируют в вакууме. Остаток поглощают в 30 мл диметилформамида, потом прибавляют раствор 1,2 эквивалента /3,5 мл/ циклопропилметилбромида и нагревают реакционную смесь при 80oC в течение 4 часов. Концентрируют в вакууме, растворяют остаток в этилацетате, последовательно промывают насыщенным раствором хлорида натрия, водой, декантируют органическую фазу, сушат и концентрируют в вакууме, чтобы получить 5,1 г целевого 2-циклопропилметокси-6-метоксиацетофенона.
Приготовление II
Растворяют 0,53 г натрия в 15 мл метанола, потом прибавляют раствор 5,1 г соединения, полученного выше, и 1 эквивалент /3,4 г/ диэтилоксалата в 25 мл метанола. Реакционную смесь кипятят с обратным холодильником в течение 6 часов, потом охлаждают. Прибавляют диизопропиловый эфир до осаждения и отфильтровывают осадок, чтобы получить 5,3 г натриевой соли метилового эфира 4-/2-циклопропилметокси-6-метоксифенил/-2,4-диоксобутановой кислоты.
Приготовление III
Суспендируют 1 г натриевой соли, полученной выше, и 1,1 эквивалента /0,65 г/ (7-хлор-4-гидразин-4-ил)-хинолина в 10 мл уксусной кислоты. Реакционную смесь нагревают при 100oC в течение 5 часов, потом выливают в 150 мл ледяной воды и отфильтровывают осадок, чтобы получить 0,74 г метилового эфира 5-/2-циклопропилметокси-6-метоксифенил/-1-/7-хлорхинолин-4-ил/- пиразол-3-карбоновой кислоты.
Приготовление IV
Растворяют 2,7 г полученного выше сложного эфира в смеси 25 мл метанола и 25 мл воды в присутствии 2,5 эквивалента /0,815 г/ гидроксида калия. Реакционную смесь 2 часа кипятят с обратным холодильником, потом выливают в ледяную воду. Экстрагируют диэтиловым эфиром, потом подкисляют эфирную фазу раствором соляной кислоты /pH 2/, отделяют осадок фильтрованием и промывают водой, чтобы получить 2,5 г целевой 5-/2-циклопропилметокси-6-метоксифенил/-1-/7- хлорхинолин-4-ил/-пиразол-3-карбоновой кислоты.
Приготовление V
К 39 г 2-аминоадамантан-2-карбоновой кислоты в 680 мл ацетонитрила прибавляют при комнатной температуре 2,1 эквивалента /42,7 г/ бис-триметилсилилацетамида, потом реакционную смесь кипятят с обратным холодильником в течение 2 часов. Раствор становится прозрачным и содержит N,O-бис-триметилсилильное производное 2-амино-2-адамантанкарбоновой кислоты.
Приготовление VI
Растворяют 1,35 г полученной в приготовлении IV пиразолкарбоновой кислоты в 30 мл толуола в присутствии 2,25 мл тионилхлорида. Реакционную смесь кипятят с обратным холодильником в течение 5 часов, потом концентрируют в вакууме. Прибавляют полученный таким образом хлорангидрид кислоты к раствору, полученному в приготовлении V и содержащему эквивалент 0,59 мг адамантанкарбоновой кислоты. Эту реакционную смесь затем кипятят с обратным холодильником в течение 3 часов, потом концентрируют растворитель в вакууме. Остаток поглощают смесью 12 мл метанола и 2 мл воды, потом перемешивают 1 час при комнатной температуре, в течение этого времени осаждается целевой продукт. Завершают осаждение в конце гидролиза при добавлении 10 мл воды. Перемешивают еще полчаса, потом отфильтровывают осадок и последовательно промывают водой, пентаном, потом диэтиловым эфиром, чтобы получить после сушки 1,8 г 2-{[(7-хлорхинолин-4-ил)-5-(2-циклопропилметокси-6- метоксифенил)-пиразол-3-ил]карбониламино}адамантан-2-карбоновой кислоты; т.пл. = 200oC.
Пример 1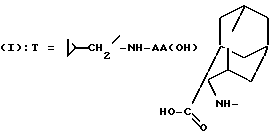
Растворяют 0,626 г кислоты, полученной в приготовлении VI, в 100 мл хлороформа в присутствии 0,294 мг метахлорпербензойной кисоты и перемешивают смесь 24 часа при комнатной температуре. Реакционную смесь концентрируют в вакууме, потом поглащают 10 мл дихлорметана. Отделяют фильтрованием полученный осадок, потом промывают дихлорметаном, получают 0,24 г 2-{[(1-(1-оксид-7-хлорхинолин-4-ил)-5-(2- циклопропилметокси-6-метоксифенил)-пиразол-3-ил]карбониламино}адамантан- 2-карбоновой кислоты, т.пл. = 190oC.
Пример 2
Получают 2-{[(1-7-хлорхинолин-4-ил)-5-(2,6-диметоксифенил)- пиразол-3-ил] карбониламино}адамантан-2-карбоновую кислоту /SR48692/, работая по методикам, описанным в приготовлениях I - VI выше, но используя метилбромид вместо циклопропилметилбромида. Растворяют 0,5 г этой кислоты в 80 мл диметилформамида, потом прибавляют 4,15 г гексагидрата монопероксифталата магния. Реакционную смесь перемешивают и выдерживают при комнатной температуре в течение 24 часов. Затем прибавляют 250 мл 0,1%-ного раствора трифторуксусной кислоты в воде и экстрагируют последовательно с помощью 150 мл, потом 100 мл, потом 50 мл дихлорметана. Объединяют экстракты и промывают 2 раза 250 мл дистиллированной воды. Экстракт, полученный после промывки, концентрируют в вакууме при 40oC, затем очищают остаток препаративной высокоэффективной жидкостной хроматографией на оксиде кремния. Для этого его растворяют в 4,5 мл элюента, состоящего из смеси дихлорметана/изопропанола (97/3 /объем/объем) и элюируют на фазе Кромасил 100 -10 μ при 40 бар. Фракции собирают каждые 0,2 минуты, объем фракции 25 мл. Концентрирование фракцией с чистым продуктом дает 0,080 г белых кристаллов, которые промывают дихлорметаном, чтобы получить в результате 0,050 г 2-{[(1-(1-оксид-7-хлорхинолин-4-ил)-5-(2,6-диметоксифенил)-3- пиразолил]карбониламино}адамантан-2-карбоновой кислоты, т.пл. = 205oC.
-10 μ при 40 бар. Фракции собирают каждые 0,2 минуты, объем фракции 25 мл. Концентрирование фракцией с чистым продуктом дает 0,080 г белых кристаллов, которые промывают дихлорметаном, чтобы получить в результате 0,050 г 2-{[(1-(1-оксид-7-хлорхинолин-4-ил)-5-(2,6-диметоксифенил)-3- пиразолил]карбониламино}адамантан-2-карбоновой кислоты, т.пл. = 205oC.
Пример на фармацевтическую композицию:
Ингредиенты - Количество
активное начало(a) - 75,0
полиэтиленгликоль 400 - 969,0
гидроксид натрия - 5,77
дистиллированная вода - 90,4
(a)активным началом является соединение из примера 1 или 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ 1-НАФТИЛПИРАЗОЛ-3-КАРБОКСАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ СИНТЕЗА | 1994 |
|
RU2140912C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ СИНТЕЗА | 1993 |
|
RU2119917C1 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ VLA-4 | 2002 |
|
RU2318815C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
| ПРОИЗВОДНЫЕ ГЛИЦИНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2130923C1 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| КОНЪЮГАТ, СОДЕРЖАЩИЙ ЛИГАНД РЕЦЕПТОРОВ НЕЙРОТЕНЗИНА, И ЕГО ПРИМЕНЕНИЕ | 2015 |
|
RU2743781C2 |

Описываются новые производные замещенных 1-(7-хлорхинолин-4-ил)-пиразол-3-карбоксамид-N-оксидов формулы I, где Т является водородом, C1-C4-алкилом, С3-С8-циклоалкилом, (С3-С8)-циклоалкилметилом, метоксиэтилом, группа -NН-АА(ОН) является остатком аминокислоты формулы
где Х является водородом, С1-С5-алкилом или C3-C15 неароматическим карбоциклическим радикалом, а Х' является водородом, или же Х и X' вместе с атомом углерода, к которому они присоединены, образуют С3-С15 неароматический карбоцикл, и их соли. Соединения имеют большое сродство к рецептору нейротензина. Описывается также способ их получения, промежуточные соединения и фармацевтическая композиция на основе соединений общей формулы I. 5 с. и 2 з.п. ф-лы, 1 табл. 
в которой T является водородом, C1 - C4-алкилом, C3 - C8-циклоалкилом, (C3 - C8)-циклоалкилметилом, метоксиэтилом;
группа - NH - AA(OH) является остатком аминокислоты формулы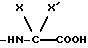
где X является водородом, C1 - C5-алкилом или C3 - C15 неароматическим карбоциклическим радикалом;
X' является водородом, или же X и X' вместе с атомом углерода, к которому они присоединены, образуют C3 - C15 неароматический карбоцикл,
и их соли.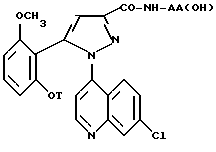
в которой T и -NH - AA(OH) имеют значения, указанные в п.1,
окисляющим агентом при комнатной температуре в апротонном растворителе, и выделяют соединение I или его соль.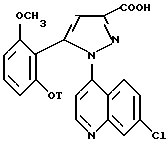
в которой T является C3 - C8-циклоалкилом, C3 - C8-циклоалкилметилом или метоксиэтилом.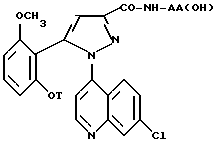
в которой T является C3 - C8-циклоалкилом, C3 - C8-циклоалкилметилом или метоксиэтилом;
NH - AA(OH) имеет значения, указанные в п.1.
| Способ получения 2-гетерилзамещенных бензимидазола | 1975 |
|
SU541846A1 |
| EP 322126 A, 1988 | |||
| СПОСОБ СВАРКИ ДЕТАЛЕЙ КОСВЕННОЙ ДУГОЙ | 0 |
|
SU289879A1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-АЛКИЛ-2-МЕТИЛ(ФЕНИЛ)-3-АЦЕТИЛ- | 0 |
|
SU277794A1 |
| Спасательный плот | 1972 |
|
SU477049A1 |
