Данное изобретение относится к новым калиевым солям производных бифенилметана, обладающим сильной активностью антагониста ангиотензина II и гипотензивной активностью, а также фармацевтическим препаратам, содержащим данные соли.
Ангиотензин II является активным центром системы ренин-ангитензин и обладает сильным сосудосуживающим действием и стимулирующим действием на синтез и секрецию альдостерона в корковом веществе надпочечника. Известно также, что он является веществом, вызывающим гипертензию. Полагают, что его действие вызывается посредством специфического рецептора на разных органах-мишенях, например корковом веществе надпочечников, почках, артериолах и периферии симпатических нервах.
Известные обычные примеры веществ, которые проявляют гипотензивное действие путем фармакологического ингибирования системы ренин-ангиотензин, включают ингибиторы фермента, превращающего ангиотензин, например каптоприил и энараприл, антагонисты ангиотензина II и ингибиторы ренина. В качестве антагониста ангиотензина II, не относящегося к этим веществам, уже известны саралазин ([Sar', Ala8] AG11), пептид типа ангиотензина II, и непептидные производные, например производные имидазола (выложенные патентные заявки Японии NN 7103/1981 и 71074/1981 и выложенные публикации на японском языке N 501020/1991), производные пиразола (выложенная патентная заявка Японии N 218371/1991) и производные аминоазола (WO93/17681).
При клинических применениях производных пептидов, однако, возникают трудности из-за их короткого in vivo периода жизнедеятельности, отсутствия эффективности при пероральном введении и существенной агонистической активности. Среди непептидных производных также ни один еще не применяли клинически в качестве лекарственного средства.
Цель данного изобретения заключается в том, что предлагается лекарственное средство, которое можно применять клинически и которое обладает описанной выше активностью.
С целью разработки клинически превосходного лекарственного средства в таких обстоятельствах авторы данного изобретения проводили экстенсивное исследование. В результате этого они нашли, что производное бифенилметана, представленное следующей формулой (1), или его соль обладает превосходной активностью антагониста ангиотензина II и полезно в качестве терапевтического средства для болезней, протекающих с расстройством кровообращения, болезней сердца и церебральной апоплексии, и подали после этого заявку на PCT (PCT/JP93/01134).
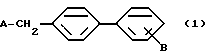
где А представляет собой группу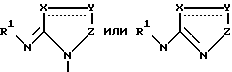
где R1 представляет собой атом водорода, низшую алкильную группу, низшую циклоалкильную группу, замещенную или незамещенную фенильную группу, замещенную или незамещенную аралкильную группу, замещенную или незамещенную ацильную группу или аминокислотный остаток; X представляет собой атом кислорода, атом серы или группу =CH-, Y представляет собой атом азота или группу =CR2-, Z представляет собой атом кислорода, атом азота или группу =CR3-, причем эти Y и Z не могут быть гетероатомами одновременно, каждый из R2 и R3 представляет собой атом водорода, атом галогена, замещенную или незамещенную низшую алкильную группу, защищенную или незащищенную карбоксильную группу, низшую циклоалкильную группу, низшую алкенильную группу, низшую алкоксильную группу, низшую алкилтиогруппу или арильную группу, или R2 и R3 могут образовать вместе с соседними атомами углерода замещенное или незамещенное бензольное кольцо. B представляет собой цианогруппу, защищенную или незащищенную карбоксильную группу, или защищенную, или незащищенную тетразолил-5-ильную группу и  обозначает двойную связь или одинарную связь.
обозначает двойную связь или одинарную связь.
В результате дальнейшего исследования авторы настоящего изобретения нашли, что среди описанных выше производных дифенилметана (1) моно- и дикалий 2-[[5-этил-3-[2'-(1H-тетразол-5-ил) бифенил-4-ил]метил-1,3,4-тиадиазолин-2-илиден] аминокарбонил]-1-циклопентенкарбоксилаты обладают особенно сильной активностью антагониста ангиотензина II и гипотензивной активностью и обладают также очень высокой биологической доступностью при пероральном введении, что приводит к завершению данного изобретения.
Данное изобретение, следовательно, предлагает моно- или дикалиевую соль 2-[[5-этил-3-[2'-(1H- тeтpaзoл-5-ил)бифeнил-4-ил] метил-1,3,4-тиадиазолин-2-илиде] аминокарбонил]1-цикло-пентенкарбоксилата, представленного следующей формулой (2):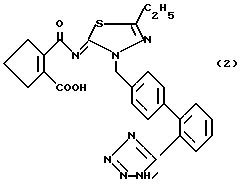
Данное изобретение предлагает также фармацевтическую композицию, которая содержит моно- или дикалиевую соль соединения (2) и фармацевтический носитель.
Данное изобретение предлагает также применение моно- или дикалиевой соли соединения (2) в качестве фармацевтического средства.
Кроме того, данное изобретение предлагает также способ терапии болезней, протекающих с нарушением кровообращения, который содержит введение эффективного количества моно- или дикалиевой соли соединения (2).
Из моно- и дикалиевой солей соединения (2), причем эти соли относятся к данному изобретению, дикалиевая соль особенно предпочтительна.
Моно- или дикалиевая соль соединения (2), к которой данное изобретение относится, может находиться в виде различных стереоизомеров (цис-формы, транс-формы). Эти стереоизомеры также включаются в данное изобретение. Соединения данного изобретения могут также присутствовать в виде сольватированных продуктов, например гидратов, которые также включаются в данное изобретение.
Моно- или дикалиевую соль соединения (2) можно получить различными способами. Предпочтительным является способ получения, указанный ниже: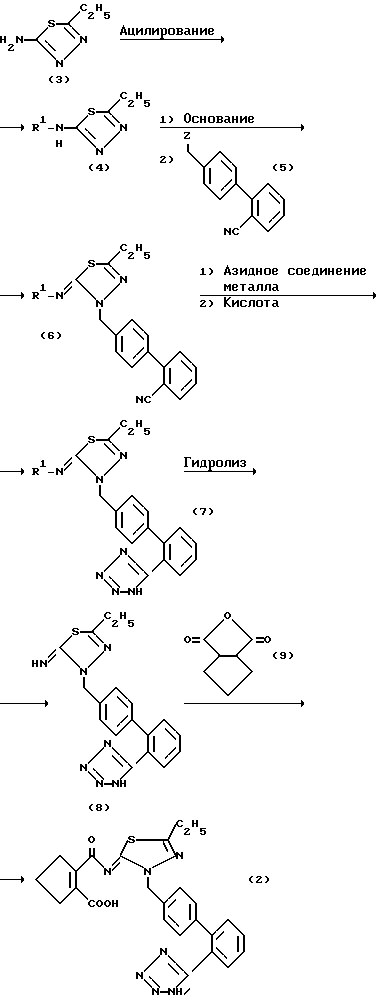
где R1 представляет собой замещенную или незамещенную ацильную группу и Z представляет собой атом галогена или сульфонилоксигруппу.
А именно, 2-амино-5-этил-1,3,4-тиадиазол (3) подвергают ацилированию для получения соединения (4). Соединение (4) конденсируют с соединением (5) в присутствии основания, получая соединение (6). Соединение (6) затем тетразолилируют для получения соединения (7), которое затем гидролизуют для удаления ацильной группы, посредством чего получают соединение (8). Затем реакцией соединения (8) с соединением (9) получают соединение (2). Полученное таким образом соединение (2) превращают в его моно- или дикалиевую соль способом, известным per se в настоящей области. Соединение данного изобретения можно получить таким способом. Соединение (7) можно также получить реакцией соединения (4) с 4- галогенметил-2-(1H-тетразол-5-ил)бифенилом.
Указанные выше стадии реакции затем будут описаны соответственно подробно.
Примеры ацилирующего агента, пригодного для ацилирования соединения (3), включают уксусную кислоту, трифторуксусную кислоту или подобные агенты. Ацилирование можно проводить при помощи любой желаемой реакции, обычно применяемой для ацилирования аминогруппы. Описанное, в частности, ацилирование можно проводить реакцией соединения (3) с ацилхлоридом или ангидридом, который соответствует желаемой ацильной группе, в апротонном полярном растворителе, таком как галогенированный углеводород, например хлористый метилен, хлороформ, четыреххлористый углерод или хлорбензол; ароматический углеводород, например бензол или толуол: простой эфир, например тетрагидрофуран или диоксан; ацетонитрил или N,N-диметилформамид, при температурах от 0oC до комнатной температуры в присутствии или в отсутствие основания, например пиридина, пиколина, N,N-диметиланилина, N-метилморфолина, диметиламина, триэтиламина; карбоната натрия или карбоната калия при температуре от -70 до 100oC; или реакцией его с кислотой, например уксусной кислотой или ангидридом ее, при температурах от комнатной температуры до 150oC.
Примеры основания, пригодного в указанной выше конденсации соединения (4) и соединения (5), включают гидрид натрия, гидрид лития, карбонат калия, карбонат натрия, алкоголяты натрия, трет-бутоксид калия, едкий натр, едкий кали, триэтиламин и диизопропилэтиламин. Здесь можно применять любой растворитель, если он не оказывает действия на реакцию. Примеры пригодных растворителей включают апротонные полярные растворители, например N,N-диметилформамид и диметилсульфоксид, простые эфиры, например диэтиловый эфир, тетрагидрофуран, диоксан, моноглимы (1,2-диметоксиэтаны) и диглимы (диметиловые эфиры диэтиленгликолей); галогенированные углеводороды, например хлористый метилен, хлороформ и четыреххлористый углерод; и спирты, например метанол, этанол и пропанол.
В качестве ускорителя реакции можно добавлять межфазный катализатор. Примеры межфазного катализатора включают четвертичные аммониевые соли, например хлорид тетраметиламмония, хлорид тетраоктиламмония и бромид тетрабутиламмония; пиридиниевые соли, например хлорид N-неопентил-4-(N',N'-диметиламино)-пиридиния и хлорид N-(2-этилгексил)-4-(N', N'-диметиламино)пиридиния; и четвертичные фосфониевые соли, например бромид тетрабутилфосфония и бромид тетрафенилфосфония.
Конденсацию можно обычно проводить при температурах от 30 до 150oC, предпочтительно от 10 до 100oC. Время реакции может обычно быть от 10 мин до 24 ч, предпочтительно от 1 до 10 ч.
Особенно предпочтительным примером конденсации является пример, в котором соль металла соединения (4) получают в апротонном полярном растворителе, например N, N-диметилформамиде, при применении гидрида натрия или карбоната калия в качестве основания и полученную в результате соль металла обрабатывают соединением (5) при температурах от 0oC до комнатной температуры.
Примеры атома галогена, представленного Z в соединении (5), включают атом фтора, атом хлора, атом бромa, атом иода и т.д. Иллюстративные примеры сульфонилоксигруппы включают алкилсульфонилоксигруппы, например метансульфонилокси, этансульфонилокси и трифторметансульфонилоксигруппу, и арилсульфонилоксигруппы, например бензолсульфонилокси и n-толуолсульфонилоксигруппу
Тетразолилирование соединения (6) можно проводить в соответствии с известным способом (выложенная патентная заявка Японии N 23868/1988), а именно, присоединением 1,3-диполярного кольца к соединению (6) с применением азидного соединения металла, например азида три-C1-C1-8-алкилолова, три-C1-C18-алкилсилилазида или азида натрия. В частности, тетразол-3-ил производное (7) можно получить добавлением азидного соединения металла к соединению (6) в растворителе, например бензоле или толуоле, реакцией их при нагревании и затем обработкой продукта реакции соляной кислотой или подобным реагентом.
Соединение (5) можно получить способом, известным на сегодняшний день (выложенная патентная заявка Японии N 23868/1988, 27362/1991 или 74369/1991; J.Org.Chem. 56, 2395-2400 (1991) или т.п.).
Для деацилирования (удаления защитной группы) соединения (7) можно применять любую желаемую известную реакцию. Например, удаление защитной группы можно осуществить реакцией соединения (7) в водном щелочном растворе, например водном растворе едкого натра, водном растворе едкого кали или водном растворе карбоната натрия, или в кислотном растворе, например соляной кислоте или уксусной кислоте, при температуре от комнатной температуры до 100oC, с применением растворителя, смешиваемого с водой, например этанола, метанола, тетрагидрофурана или N,N-диметилформамида, или без растворителя.
Реакцию между соединением (8) и соединением (9) можно проводить в апротонном полярном растворителе, таком как галогенированный углеводород, например хлористый метилен, хлороформ, четыреххлористый углерод или хлорбензол, ароматический углеводород, например бензол или толуол, простой эфир, например тетрагидрофуран или диоксан; ацетонитрил или N,N-диметилформамид, при температурах от 0oC до комнатной температуры в присутствии или в отсутствие основания, например пиридина, пиколина, N,N-диметиланилина, N-метилморфолина, диметиламина, триэтиламина, карбоната натрия или карбоната калия, при температурах от -70 до 100oC.
В указанном способе получения, когда тетразол-5-ильная группа имеет защитную группу, если нужно, защитную группу можно удалить.
Для указанного выше удаления защитной группы желательно проводить удаление защитной группы реакцией соединения в содержащем воду спирте или простом эфире, например диоксане или тетрагидрофуране, который содержит соляную кислоту, уксусную кислоту или подобную кислоту, при комнатной температуре или около этого в течение приблизительно 1-10 ч.
Полученное таким образом соединение (2) можно превратить в его моно- или дикалиевую соль способом, известным per se в настоящей области. В частности, необходимо только растворить соединение (2) в растворе едкого натра и затем осадить в виде соли. Предпочтительно применение в качестве раствора едкого кали раствора едкого кали в воде, метаноле, этаноле, н-пропиловом спирте, изопропиловом спирте, ацетоне и подобном растворителе, применяемом в количестве, которое по меньшей мере эквивалентно количеству соединения (2). Соединение (2) растворяют в полученном таким образом растворе едкого кали. Температуру растворения можно определить между комнатной температурой и желаемой температурой нагревания, которая зависит от соединения (2). Кроме этого, можно выбрать желаемый способ осаждения соли, поскольку некоторые соли осаждаются, когда растворы просто оставляют, но некоторые соли не осаждаются до тех пор, пока не будет до некоторой степени удален растворитель.
Полученную таким образом моно- или дикалиевую соль соединения (2), если нужно, можно очищать способом, известным per se в настоящей области, например растворением соли в одном или нескольких растворителях, выбранных из воды, метанола, этанола, н-пропилового спирта, изопропилового спирта или ацетона, с последующей перекристаллизацией ее из раствора.
Когда моно- и дикалиевую соль соединения (2), которые относятся к данному изобретению, применяют в качестве терапевтических средств для болезней, протекающих с нарушением кровообращения, их можно вводить в состав композиции вместе с фармацевтически приемлемым носителем для парентерального введения, например инъекционного или ректального введения, или для перорального введения в твердой или жидкой форме.
Композиции данного изобретения для применения в качестве инъекций могут быть в форме фармацевтически приемлемых, не содержащих патогенные микроорганизмы водных, неводных растворов, суспензий или эмульсий. Примеры пригодных неводных носителей, разбавителей, растворителей и наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла, например оливковое масло, инъецируемые органические сложные эфиры, например этилолеат. Эти препараты могут содержать одно или несколько вспомогательных средств, например антисептики, увлажняющие средства, эмульгаторы и диспергаторы. Эти готовые препаративные формы можно стерилизовать, например, фильтрованием их через бактериальный фильтр или смешиванием непосредственно перед применением со стерилизующим средством в форме не содержащей патогенные микроорганизмы твердой композиции, растворимой в стерилизованной воде или одной из некоторых других сред, которые можно стерилизовать и инъецировать.
Примеры твердых препаратов для перорального введения включают капсулы, таблетки, пилюли, порошки, гранулы и т.д. При изготовлении этих твердых препаратов соединения в соответствии с данным изобретением обычно смешивают по меньшей мере с одним инертным наполнителем, например сахарозой, лактозой или крахмалом. При изготовлении препаратов обычным способом в них можно также ввести один или несколько материалов, не являющихся инертными наполнителями, например смазывающее вещество, такое как стеарат магния. В случае капсул, таблеток и пилюль можно также ввести буфер. Таблетки и пилюли можно покрыть энтеросолюбильной оболочкой.
Иллюстративные жидкие препараты для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, которые содержат инертный разбавитель, применяемый обычно специалистами настоящей области, например воду. Кроме такого инертного разбавителя в жидкие препараты можно также добавлять одно или несколько вспомогательных средств, например увлажняющие средства, эмульгаторы, суспендирующие средства, подслащивающие вещества, кондиционеры и отдушки. Предпочтительны препараты для ректального введения, которые содержат наполнители, например какао-масло или воск суппозиторий, кроме соединения данного изобретения.
Доза моно- или дикалиевой соли соединения (2) в соответствии с настоящим изобретением зависит от свойств соединения, которое вводят, способа введения, желаемого времени лечения и других факторов. Она обычно варьирует от около 0,1 мг/кг до 100 мг/кг в сутки, особенно предпочтительно около 0,5-50 мг/кг в сутки. При желании эту суточную дозу можно вводить в виде 2-4 порций.
Данное изобретение будет в дальнейшем описываться более конкретно примерами. Однако следует учитывать, что данное изобретение никоим образом не ограничивается до этих примеров или этими примерами.
Пример 1 синтеза.
1) Синтез 2-трифторацетамидо-5-этил-1,3,4-тиадиазола.
В суспензию 200 г 2-амино-5-этил-1,3,4-тиадиазола в 3 л толуола добавляли 260 мл триэтиламина при комнатной температуре, затем добавляли 265 мл трифторуксусного ангидрида при охлаждении льдом. Смесь перемешивали 1 ч при комнатной температуре. В реакционную смесь добавляли воду и осажденные кристаллы отделяли фильтрованием. В фильтрат добавляли этилацетат. Органический слой отделяли и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме, получая 237,5 (68%) заглавного соединения.
2) Синтез 4-бромметил-2'-цианобифенила.
В 110 мл четыреххлористого углерода добавляли 10,5 г 4- метил-2'-цианобифенила, 9,79 г N-бромсукцинимида и 120 мг 2,2'-азобисизобутиронитрила. Полученную смесь кипятили с обратным холодильником в течение 2 ч. Нерастворимый материал удаляли фильтрованием при нагревании. Фильтрат охлаждали при стоянии. Осажденные кристаллы отделяли фильтрованием, в результате чего получали 6,6 г (44%) заглавного соединения.
3) Синтез 2-трифторацетиламино-5-этил-3-(2'-цианобифенил-4-ил)-метил-1,3,4-тиадиазолина.
В суспензию 124,1 г гидрида натрия (55% в масле) в 1,5 л N,N-диметилформамида добавляли 533,7 г 2-трифтор-ацетамидо-5-этил-1,3,4-тиадиазола, полученного в (1), при охлаждении льдом. После окончания выделения водорода в реакционную смесь по каплям добавляли раствор 643,6 г 4-бромметил-2'-цианобифенила, полученного в (2), в 3 л N,N-диметилформамида. Реакционную смесь перемешивали 1 ч при комнатной температуре и затем перемешивали 5 ч при 80oC. Растворитель выпаривали в вакууме и в остаток добавляли воду и этилацетат. Органический слой промывали водой и сушили над безводным сульфатом магния. После выпаривания растворителя в вакууме остаток очищали хроматографией на колонке с силикагелем (элюент:смесь н-гексана и этилацетат 3:1), получая неочищенные (сырые) кристаллы. Эти кристаллы перекристаллизовали из этанола, получая 490,8 г (50%) заглавного соединения.
4) Синтез 2-трифторацетилимино-5-этил-3-(2'-цианобифенил-4-ил)-метил-1,3,4-тиадиазолина.
В 18 л N,N-диметилформамида добавляли 2,44 кг (10,8 моль) 2-трифторацетамидо-5-этил-1,3,4-тиадиазола и 2,80 кг (10,3 моль) 4-бромметил-2'-цианобифенила, затем добавляли 894 г (6,5 моль) безводного карбоната калия и 60 г иодида калия. Реакционную смесь перемешивали 41 ч при комнатной температуре. Нерастворимый материал удаляли фильтрованием и фильтрат концентрировали в вакууме. В остаток добавляли 45 л воды и 18 л этилацетата. Органический слой отделяли и сушили над безводным сульфатом магния и концентрировали до половины первоначального его объема. Осажденные кристаллы отделяли фильтрованием и промывали последовательно этанолом и диизопропиловым эфиром. Кристаллы (чистота: 95,8%) перекристаллизовывали из 5 л этанола, получая 2,70 кг кристаллов (чистота 94%). Эти кристаллы (чистота 94%) очищали колоночной хроматографией на силикагеле (10 кг силикагеля, элюент: смесь н-гексана и этилацетата, 3: 1 - 2:1), получая 2,64 кг (чистота: 99%) заглавного соединения. Все фильтраты объединяли и растворитель выпаривали. Остаток растворяли в 3 л горячего этанола и в раствор добавляли активированный уголь. Смесь перемешивали в течение 30 мин при 80oC и фильтровали через целит. Фильтрат оставляли на ночь при комнатной температуре. Осажденные кристаллы отделяли фильтрованием. Кристаллы обрабатывали снова активированным углем, как описано выше, посредством чего получали 562 г (чистота: 98%) заглавного соединения. Фильтрат выпаривали и остаток очищали колоночной хроматографией на силикагеле (4 кг силикагеля, элюент: смесь н-гексана и этилацетат, 5:1), получая 371 г заглавного соединения. Всего получали 3,57 г заглавного соединения.
Свойства: бесцветные кристаллы в форме призм.
Температура плавления: 106-107oC.
1H ЯМР-спектр (δ м.д., в CDCl3): 7,43-7,79 (8H, м), 5,60 (2H, с), 2,95 (2H, к), 1,39 (3H, т).
5) Синтез 2-трифторацетилимино-5-этил-3- [2'-(1H-тетра-зол-5-ил)бифенил-4-ил]метил-1,3,4-тиадиазолина
В 1 л толуола добавляли 490,8 г 2-трифторацетилимино-5-этил-3-(2'-цианобифенил-4-ил)метил-1,3,4-тиадиазолина, полученного в (4), и 485,5 азида триметилолова. Смесь кипятили с обратным холодильником в течение 40 ч. В реакционную смесь добавляли 200 мл концентрированной соляной кислоты и затем перемешивали в течение 10 мин. Реакционную смесь экстрагировали 5 л этилацетата. Органический слой промывали водой и сушили над безводным сульфатом магния. Растворитель удаляли в вакууме, получая заглавное соединение, которое не очищали для применения в следующей стадии.
6) Синтез гидрохлорида 2-имино-5-этил-3-[2'-(1H-тетразол-5-ил) бифенил-4-ил]метил-1,3,4-тиадиазолина.
В смесь 4 л тетрагидрофурана и 200 мл воды и 2-трифторацетиламино-5-этил-3-2'-(1H-тетразол-5-ил)биферил-4-ил метил-1,3,4-тиадиазолина, полученного в (5), добавляли 94,4 г едкого натра и смесь кипятили с обратным холодильником в течение 7 ч. Реакционную смесь концентрировали в вакууме. В остаток добавляли воду и этилацетат. Водный слой отделяли и подкисляли соляной кислотой. Осажденные кристаллы отделяли фильтрованием и получали 168 г заглавного соединения. Из слоя этилацетата осаждали кристаллы и отделяли их фильтрованием, посредством чего получали дополнительно 80 г заглавного соединения.
Свойства: бесцветный порошок.
Температура плавления: 205-206oC.
1H ЯМР-спектр (δ м.д., в DMSO-d6): 7,53-7,73 (4H, м), 7,28 (2H, д), 7,14 (2H, д) 5,43 (2H, с), 2,89 (2H, к), 1,22 (3H, т).
7) Синтез 2-[[5-этил-3-[2'-(1H-тетразол-5-ил)бифенил-4- ил] метил-1,3,4-тиадиазолин-2-илиден]аминокарбонил]-1- циклопентенкарбоновой кислоты.
В 2 мл N,N-диметилформамида добавляли 200 мг гидрохлорида 5-этил-2-имино-3-[2'-(1H-тетразол-5-ил)бифенил-4-ил] метил-1,3,4-тиадиазолина и 76 мг ангидрида 1,2-цикло-пентендикарбоновой кислоты. Реакционную смесь перемешивали 2 ч при комнатной температуре и выливали в воду. Осажденные кристаллы отделяли фильтрованием, промывали водой и этанолом и затем сушили, посредством чего получали 170 мг заглавного соединения.
Свойства: бесцветные кристаллы.
Температура плавления: 234-235oC.
1H ЯМР-спектр (δ м.д., в CDCl3 + CD3OD): 7,76 (1H, д), 7,35-7,65 (ЗH, м), 7,29 (2H, д), 7,11 (2H, д), 5,52 (2H, с), 3,03 (4H, т), 2,94 (2H, к), 1,89 (2H, м), 1,39 (3H, т).
Пример 1.
Синтез 2-[[5-этил-3-[2'-(1H-тетразол-5-ил) бифенил-4-ил] метил-1,3,4-тиадиазолин-2-илиден] аминокарбонил] - 1-циклопентенкарбоксилата дикалия (соединение A).
В смесь 400 мл 0,1 N раствора (спиртового) едкого кали и 600 мл этанола добавляли 10 г 2-[[5-этил-3-[2'-(1H-тетразол-5-ил)бифенил- 4-ил] метил-1,3,4-тиадиазолин-2-илиден] аминокарбонил]-1- циклопентенкарбоновой кислоты. После того, как смесь полностью растворилась при нагревании на водяной бане, растворитель выпаривали в вакууме до тех пор, пока объем раствора не уменьшился всего до около 200 мл. Осажденные кристаллы отделяли фильтрованием, промывали этанолом и затем сушили в вакууме, посредством чего получали 11 г заглавного соединения.
Температура плавления: > 280oC.
ИК-спектр (KBr), см-1 1642-(COOK), 1570(=N-CO-).
1H ЯМР-спектр (δ м.д., в D2O): 7,51-7,62 (ЗH, м), 7,37-7,39 (1H, м), 7,31 (2H, д), 7,04 (2H, д), 5,49(2H, с), 2,66- 2,86 (6H, м), 1,95-2,00 (2H, м), 1,23 (3H, т)
Пример 2 Синтез 2-[[5-этил-3-[2'-(1H-тетразол-5-ил бифенил-4-ил]метил-1,3,4-тиадиазолин-2-илиден] аминокарбонил] -1- циклопентенкарбоксилата монокалия (соединение B).
В смесь 8,3 мл 0,05 N раствора (спиртового) едкого кали и 50 мл этанола добавляли 206 мг 2-[[5-этил-3-[2'-(1H-тетразол-5-ил)бифенил-4-ил]метил-1, 3,4-тиадиазолин-2-илиден] аминокарбонил] -1- циклопентенкарбоновой кислоты. После того, как смесь полностью растворилась при нагревании на водяной бане, растворитель выпаривали в вакууме. В остаток добавляли этанол. Осажденную часть отделяли фильтрованием и сушили в вакууме, посредством чего получали 180 мг заглавного соединения.
ИК-спектр (KBr): 1680 (-COOH), 1570(=N-CO-).
Испытание 1.
Гипотензивное действие на крыс с почечной гипертензией (инвазивное).
Каждую из крыс с почечной гипертензией получали сжатием левой почечной артерии самца крысы SD (возраст: 6 недель, масса тела: 190-220 г) серебряным зажимом (внутренний диаметр: 0,4318 мм) при анестезии. Гипотензивное действие изучали, применяя крыс, среднее кровяное давление которых поднималось до 150 мм Hg или выше за 4-8 недель после сжатия почечной артерии. За один день до испытания в бедренную артерию каждой из крыс с почечной гипертензией вставляли канюлю для измерения кровяного давления. Крысам предоставляли сколько угодно (ad libitum) корма и воды непосредственно до начала испытания. Вставленную таким образом канюлю соединяли с датчиком кровяного давления и среднее значение кровяного давления регистрировали на полиграфе. После того, как кровяное давление становилось стабильным, каждой из крыс перорально вводили 0,3 мг/кг испытуемого соединения, которое суспендировали в 0,5%-ной карбоксиметилцеллюлозе. В соответствии с уравнением, приведенным в табл. 1, степень понижения кровяного давления (%) рассчитывали из величин кровяного давления до и после введения испытуемого соединения. Результаты приводятся в табл. 1.
Испытание 2.
Ингибирующее действие испытуемых соединений, перорально введенныx крысам с нормальным кровяным давлением, но кровяное давление которых преднамеренно повышено ангиотензином II (внутривенной инъекцией 0,1 мкг/кг) (инвазивное).
За один день до испытания в правую бедренную артерию и правую бедренную вену каждого самца крыс SD (масса тела: 200-350 г) при анестезии вставляли канюлю для измерения кровяного давления и другую канюлю для введения раствора ангиотензина II (AII) в физиологическом растворе (внутривенное введение 0,1 мкг/кг) соответственно. Крысам предоставляли сколько угодно (ad libitum) корма и воды непосредственно до начала испытания. В день испытания первую указанную канюлю, вставленную в артерию, соединяли с датчиком кровяного давления и среднее значение кровяного давления регистрировали на полиграфе. Каждой из крыс для повышения давления внутривенно вводили раствор AII в физиологическом растворе. Реакцию повышения давления повторяли несколько раз. После того, как было подтверждено, что реакция стала стабильной, перорально вводили крысе 1,0 мг/кг испытуемого соединения, которое суспендировали в 0,5%-ной карбоксиметилцеллюлозе. В соответствии с уравнением, приведенным в табл. 2, степень ингибирования (%) повышения кровяного давления рассчитывали из данных повышения кровяного давления до и после введения испытуемого соединения. Результаты приводятся в табл.2.
Испытание 3.
Измерение биологической доступности испытуемого соединения у крыс.
Самцов крыс SD возраста 6 недель (5 крыс в группе) держали голодными в течение ночи. Затем каждой крысе вводили 3 мг/кг испытуемого соединения внутривенно после растворения в физиологическом растворе и перорально после суспендирования в 0,5%-ной карбоксиметилцеллюлозе соответственно. После введения периодически отбирали кровь. Из отобранной крови плазму отделяли центрифугированием. Испытуемое соединение в плазме определяли жидкостной хроматографией высокого разрешения и на основании этого определяли площади с концентрацией (AUC) при внутривенном введении и пероральном введении соответственно. Биологическую доступность (BA) испытуемого соединения для крыс рассчитывали в соответствии с уравнением, приведенным в табл.3. Результаты испытания приводятся в табл.3.
Как очевидно из результатов табл. 1,2,3, моно- или дикалиевая соль соединения (2) по сравнению с соединением (2) и его натриевой солью обладает сильной активностью антагониста ангиотензина II и сильным гипотензивным действием и обладает также высокой биологической доступностью при пероральном введении.
Моно- или дикалиевая соль производного бифенилметана в соответствии с данным изобретением обладает сильной активностью антагониста ангиотензина II и высокой биологической доступностью по сравнению со свободными производными бифенилметана или их солями, другими чем калиевые соли производных бифенилметана, так что она не только полезна в качестве терапевтического средства для болезней, протекающих с расстройством кровообращения, например гипертензии, болезней сердца и церебральной апоплексии, но также стабильна к действию света и тепла. Следовательно, она является очень полезным лекарственным средством при клиническом применении.



Изобретение относится к новым калиевым солям производных бифенилметана - моно- или дикалий 2-[[5-этил-3-[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил-1,3,4-тиадиазолин-2-илиден] аминокарбонил] -1-циклопентенкарбоксилату, обладающим гипотензивной активностью. 3 с.п. ф-лы, 3 табл.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Способ получения производных тиазолидиндиона-2,4 или их фармацевтически приемлемых солей с щелочными металлами | 1989 |
|
SU1766260A3 |
| US 5283252 A, 1994. | |||
