Предметом настоящего изобретения являются производные бензоксазина, их получение и применение в терапии.
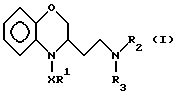
Производные бензоксазина по изобретению отвечают общей формуле (I):
где Y - атом водорода, фтора и хлора или метильная или метоксигруппа,
R1 - либо фенильная группа, замещенная атомом фтора или группой, выбранной из метильной, метокси-, трифторметильной и фенильной групп, либо тиен-2-ильная группа,
R2 - метильная группа,
R3 - либо (C1-C4)-алкильная группа, либо фенил-(C1-C2)-алкильная группа, в которой кольцо, при необходимости, замещено 2-3 метоксигруппами, либо 2-(пиридин-2- ил)-этильная группа, или R2 и R3 образуют вместе с азотом, либо 4-фенил-(пиперидин-1-ильную) группу, либо 4-фенилметил-(пиперидин-1-ильную) группу, либо 1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 6-метокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 5,8- диметокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 2,3,4,5-тетрагидро-1H-3- бензазепин-3-ильную группу, либо 7,8-диметокси-2,3,4,5-тетрагидро- 1H-3-бензазепин-3-ильную группу, а X - либо карбонильная группа, либо сульфонильная группа.
Предпочтительны производные, отвечающие общей формуле (I),
где R1 - фенильная группа, замещенная 3-(трифторметильной) группой,
R2 и R3 образуют вместе с азотом 6,7- диметокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, а
X - карбонильная группа.
Поскольку молекула, представленная общей формулой (I), содержит асимметричный атом углерода, то соединения изобретения могут существовать в виде чистых энантиомеров или смеси энантиомеров. Наконец, соединения изобретения могут быть представлены в виде свободных оснований или солей присоединения фармацевтически приемлемых кислот. Эти различные формы входят в объем изобретения.
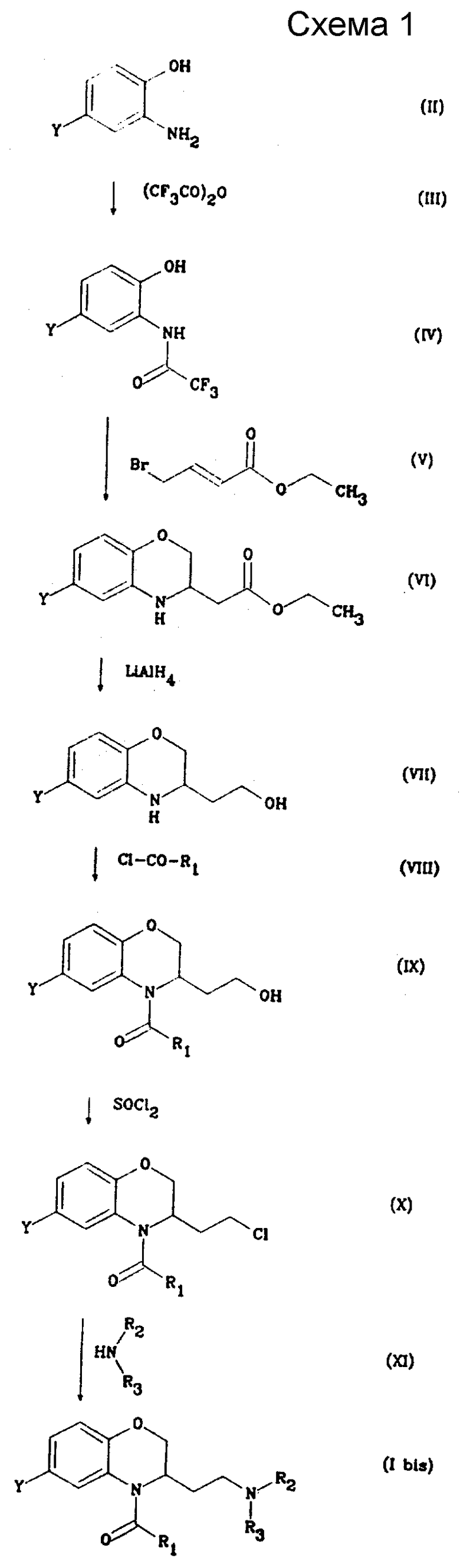
В соответствии с изобретением можно получить соединения, где X является карбонильной группой (схема 1 дана в конце описания).
Проводят реакцию 2-аминофенола общей формулы (II), где Y - определен выше, с трифторуксусным ангидридом формулы (III) в присутствии основания, такого как пиридин, в растворителе, таком как эфир. Получают амид общей формулы (IV), реакцию которого с этил-4-бромобут-2-еноатом формулы (V) проводят в присутствии основания, такого как метилат натрия, в растворителе, таком как этанол, при температуре порядка 80oC. Затем функцию сложного эфира производного 3,4-дигидро-2H-1,4-бензоксазин-3-этилацетата общей формулы (VI) восстанавливают с помощью восстановителя, такого как алюмогидрид лития, для получения производного 3,4- дигидро- 2H-1,4-бензоксазин-3-этанола общей формулы (VII), реакцию которого с хлорангидридом кислоты общей формулы (VIII), где R1 - как определено выше, проводят в растворителе, таком как дихлорметан, для получения спирта общей формулы (IX), который подвергают реакции с тионилхлоридом для получения соединения общей формулы (X). Наконец, проводят реакцию последнего с амином общей формулы (XI), где R2 и R3 - как определено выше, для получения соединения формулы (I bis), которая соответствует общей формуле (I), где X - карбонильная группа.
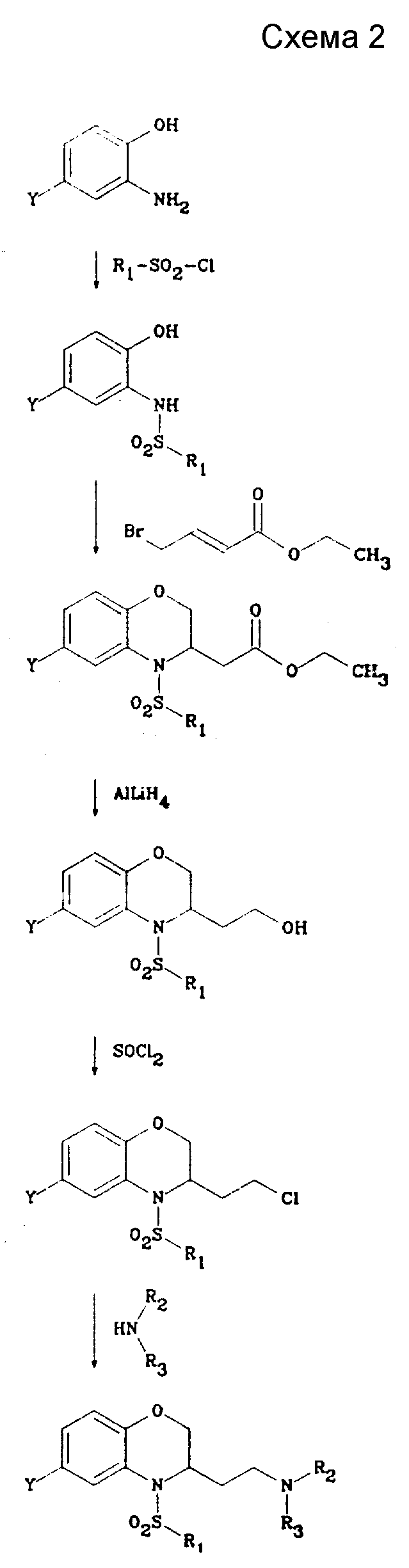
Если X - сульфонильная группа, можно получить соединения общей формулы (I) по схеме 2, приведенной в конце описания.
Проводят реакцию 2-аминофенола общей формулы (II), где Y - как определено выше, с хлоридом общей формулы (III') в присутствии основания, такого как пиридин. Получают соединение общей формулы (IV'), реакцию которого с этил-4- бромобут-2-еноатом формулы (V) проводят в присутствии основания, такого как метилат натрия, в растворителе, таком как этанол, при температуре 80oC. Затем восстанавливают функцию сложного эфира соединения общей формулы (VI') с помощью восстановителя, такого как алюмогидрид лития, для получения соединения общей формулы (VII'), реакцию которого с тионилхлоридом проводят в растворителе, таком как хлороформ, для получения соединения общей формулы (X'), который затем конденсируется с помощью амина общей формулы (XI), где R2 и R3 - как определено выше.
Исходные продукты коммерчески доступны или описаны в литературе, или могут быть синтезированы по описанным или известным специалистам методикам. В частности, 2-амино-4-метоксифенол описан в журнале Y.Am. Chem. Soc. (1949) 71 1265.
При желании, чтобы получить оптически чистое соединение общей формулы (I), можно использовать спирт общей формулы (IX) или (VII'), который изолируют, например, энзиматическим способом.
Основной принцип энзиматического способа состоит в отделении оптически чистого спирта и соответствующего ацетата противоположной конфигурации, например, путем хроматографии на колонке с силикагелем.
По первому варианту, рацемат спирта формулы (IX) или (VII') подвергают химическому ацилированию, например, с помощью уксусного ангидрида, один из двух энантиомеров рацемического ацетата подвергают стереоспецифическому гидролизу в присутствии энзима и отделяют не подвергшийся гидролизу ацетат. Получают оптически чистый спирт и оптически чистый ацетат противоположной конфигурации, который при желании может быть также подвергнут химическому или энзиматическому гидролизу для получения вторичного этантиомера спирта.
В соответствии со вторым вариантом, раценат спирта формулы (IX) или (VII') подвергают стереоспецифическому ацилированию в присутствии энзима, в качестве катализатора этерификации соли энантиомеров, например, с помощью винилацетата. Как описано выше, получают оптически чистый спирт и оптически чистый ацетат противоположной конфигурации, который при желании может также быть подвергнут химическому или энзиматическому гидролизу для получения вторичного энантиомера спирта.
Этими двумя способами можно получить, в зависимости от используемого энзима, левовращающийся или правовращающийся энантиомер спирта (IX) или (VII') и его ацетат противоположной конфигурации. Используются такие энзимы, как, например, липазы Mucor Miehei, Penicillium cyclopim или ростки злаков.
Промежуточные соединения являются новыми и также составляют предмет изобретения. Они отвечают общей формуле (XII)
где Y - как определено выше, а R - либо атом водорода, и R'- 2-гидроксиэтильная группа, либо R-COR1-группа, где R1 - фенильная группа, замещенная атомом фтора или метильной, метокси-, трифторметильной и фенильной группой, или тиен-2-ильная группа, a R' - 2-гидроксиэтильная группа или 2-хлорэтильная группа, либо R -SO2R1 - группа, где R1 - фенильная группа, замещенная атомом фтора или метильной, метокси-, трифторметильной и фенильной группой, или тиен- 2-ильная группа, а R' - 2-гидроксиэтильная или 2- хлорэтильная или этоксикарбонилметильная группа.
Нижеследующие примеры детально иллюстрируют получение некоторых производных по изобретению. Элементарный анализ, инфракрасный спектр и спектр ядерно-магнитного резонанса подтверждают структуру полученных продуктов.
Номера, указанные в скобках при нумерации примеров, соответствуют номерам приведенной ниже таблицы.
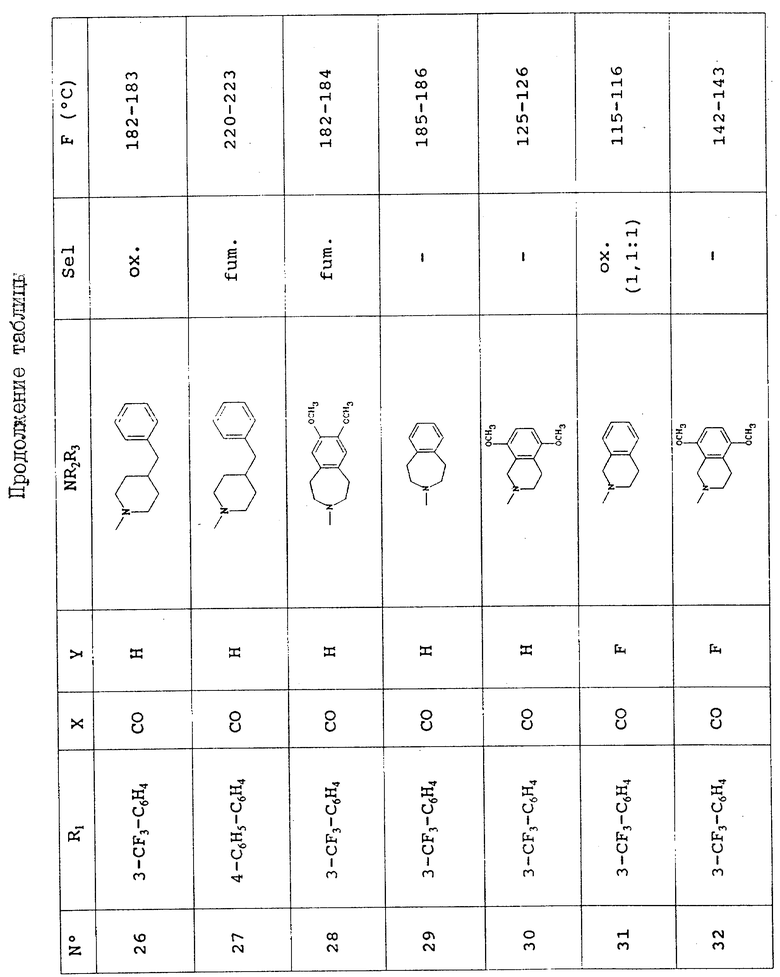
Пример 1 (Соединение N 28).
(±) 3-[2-(7,8-диметокси-2,3,4,5-тетрагидро-1-Н-3-бензазепин-3- ил)этил] -4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин, фумарат.
1.1. N-(2-гидроксифенил)трифторацетамид.
В 4-литровый реактор, при перемешивании магнитной мешалкой, вводят в суспензии в 1,5 л эфира 104 г (0,95 моля) 2-аминофенола, к которым добавляют 77 мл пиридина. Реакционную среду охлаждают смесью льда и этанола. Прикалывают в течение 1 часа 200 г (0,95 моля) трифторуксусного ангидрида. Оставляют до повышения температуры до комнатной, затем продолжают перемешивать в течение 1 часа. Добавляют в реакционную среду ледяную воду, осветляют, органическую фазу промывают последовательно в 1 л 1N раствора соляной кислоты, водой, насыщенным раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия. Высушивают над сульфатом магния и выпаривают досуха.
Получают 170 г продукта, который используют на следующем этапе.
1.2. (±)-3,4-гидро-2Н-1,4-бензоксазин-3-этилацетат.
В 4-литровый реактор, при перемешивании магнитной мешалкой, вводят 165 г (0,8 моля) соединения, полученного, как описано выше, в растворе 2 л этанола. Добавляют последовательно 151 мл 5,3 N раствора метилата натрия и 154,43 г (0,8 моля) этил-4-бромобут-2-еноата. Нагревают до 80oC в течение 1 часа. Выпаривают досуха, снова вводят в 500 мл воды и 200 мл 1N едкого натра, затем экстрагируют с помощью эфира. Органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают досуха. Выделяют 153 г продукта, который очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном.
Получают 55 г продукта.
1.3. (±)-3,4-дигидро-2H-1,4-бензаксазин-3-этанол.
В 2-литровый реактор вводят 300 мл тетрагидрофурана и охлаждают смесью льда и соли. Под струей аргона добавляют 15 г алюмогидрида лития, затем прикапывают 55 г (0,25 моля) соединения, полученного, как описано выше, в растворе в 300 мл тетрагидрофурана. Перемешивают в течение 2 часов. Реактор охлаждают смесью сухого льда и ацетона, затем прикапывают 50 мл воды и 20 мл 1N раствора едкого натра. Перемешивают в течение 2 часов и оставляют на ночь в покое. Осадок отфильтровывают на кизельгуре, промывают последовательно тетрагидрофураном и этилацетатом, концентрируют досуха. Выделяют 40 г неочищенного продукта, который очищают хроматографией на колонке с силикагелем элюируя смесью гексан/этилацетат (50/50).
Получают 35 г продукта.
1.4. (±)-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин-3-этанол.
В 100-миллилитровую колбу помещают 25 мл дихлорметана, 8,96 г (0,08 моля) соединения, полученного по п. 1.3, 7,6 г (0,055 моля) карбоната калия и прикапывают 8,3 мл (0,005 моля) 3- (трифторметил)бензоилхлорида в растворе в 25 мл дихлорметана. Оставляют при комнатной температуре, при перемешивании магнитной мешалкой, в течение 2 часов. Органическую фазу выделяют, промывают последователь 1N раствором едкого натра, водой, затем насыщенным раствором хлорида натрия. Высушивают его над сульфатом магния и концентрируют досуха. Полученное масло очищают хроматографией на колонке с силикагелем, элюируя смесью гексан/этилацетат (50/50). Выделяют 11 г желтого масла, которое кристаллизуется после ночи покоя. Очищают 2 г этого масла хроматографией на колонке с силикагелем, элюируя смесью гексан/этилацетет (3/2).
Получают 1,3 г продукта.
1.5. (±)-3-(2-хлорэтил)-4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4-бензоксазин.
К 3,51 г (0,01 моля) соединения, полученного по п. 1.4, в растворе в 50 мл дихлорметана, добавляют 2,9 мл тионилхлорида и оставляют, перемешивая, при комнатной температуре на 3 часа. Снова добавляют 2,9 мл тионилхлорида и оставляют, перемешивая, при комнатной температуре в течение 2 часов. Концентрируют досуха, извлекают толуолом и снова концентрируют досуха.
Получают 3,2 г продукта.
1.6. 3,4-диметоксибензолацетилхлорид.
В 95 г (0,48 моля) 3,4-диметоксибензолуксусной кислоты в растворе в 200 мл дихлорметана добавляют 103,7 мл (1,42 молей) тионилхлорида. Оставляют при перемешивании при комнатной температуре на 18 часов. Растворители выпаривают. Выделяют 1,4 г неочищенного продукта в виде коричневатого масла.
1.7. N-(2,2-диметоксиэтил)-3,4-диметоксибензолацетамид.
В раствор 52,7 мл (0,48 моля) 2,2-диметоксиэтанамина, охлажденный до 10oC и содержащий 67,5 мл триэтиламина, в 500 мл дихлорметана, прикапывают 104 г (0,48 моля) соединения, полученного по п. 1.6, в растворе в 250 мл дихлорметана. По окончании перемешивания оставляют до восстановления комнатной температуры и оставляют, помешивая, на 1 час. Добавляют 500 мл ледяной воды, а органическую фазу осветляют. Выделяют, промывают насыщенным раствором сульфата магния и концентрируют досуха.
Получают 128 г продукта в виде вязкого масла.
1.8. 7,8-диметокси-1,3-дигидро-2H-3-бензазепин-2-он.
Оставляют, перемешивая, в течение 8 часов при комнатной температуре 128 г (0,45 моля) соединения, полученного по п. 1.7, растворенного в смеси 640 мл концентрированной соляной кислоты и 640 мл уксусной кислоты. Оставляют, помешивая, в течение 3 дней при комнатной температуре. Добавляют 2 кг льда, а полученный продукт, выпавший в осадок, отфильровывают, промывают смесью вода/метанол и высушивают в сушильном шкафу.
Получают 42 г продукта.
Точка плавления: 240-244oC.
1.9. 7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он.
Гидрирование полученного по п. 1.8 соединения проводят в течение 3 часов при давлении 0,42 МПа и температуре 50oC в присутствии 1 г 10% палладия на угле. Концетрируют досуха, фильтруют на кизельгуре и промывают уксусной кислотой. Остаток извлекают дихлорметаном и промывают последовательно насыщенным раствором бикарбоната магния и концетрируют досуха.
Получают 13,2 г продукта.
Точка плавления: 186-190oC.
1.10. 7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин, хлоргидрат.
В суспензию 2,2 г (0,01 моля) соединения, полученного по п. 1.9, в растворе в 25 мл сухого тетрагидрофурана, в атмосфере аргона, прикапывают при комнатной температуре 20 мл 1N раствора диборана в тетрагидрофуране. Нагревают с обратным холодильником в течение 2 часов. Охлаждают смесью льда и спирта и прикапывают 30 мл 6N соляной кислоты. Нагревают в течение 1 часа при 80oC. Подщелачивают 4N раствора едкого натра и экстрагируют этилацетатом. Органическую фазу собирают, промывают насыщенным раствором хлорида натрия, высушивают над сульфатом магния и концентрируют досуха. Остаток снова вводят в 100 мл 1N соляно-кислого пропан-2-ола, а образовавшийся осадок отфильтровывают.
Высушивают и получают 1 г продукта.
Точка плавления: 236oC.
1.11. (±)-3-[2-(7,8-диметокси-2,3,4,5-тетрагидро-IH-3- бензазепин-3-ил)этил] -4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4-бензоксазин, фумарат.
Смешивают 3 г (0,015 моля) соединения, полученного по п. 1.5, 3,56 г (0,0096 моля) соединения, полученного по п. 1.10, 2,66 г карбоната калия, 100 мг иодида калия и 50 мл диметилформамида. Смесь нагревают до 80oC в течение 4 часов, затем выливают на смесь льда и воды. Экстрагируют эфиром, органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом магния и концентрируют досуха. Оставшееся масло очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметан/метанол (99/1), затем смесью дихлорметан/метанол (98/2). Получают 1,8 г основания. Получают фумарат, добавляя эквивалент фумаровой кислоты. Его выделяют и рекристаллизуют в пропан-2-оле.
Точка плавления: 182-184oC.
Пример 2 (Соединение N16).
(±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохи-нолеин-2 -ил)этил] -4-[(4-метилфенил)сульфонил]-3,4-дигидро-2H-1,4- бензоксазин, оксалат.
2.1. N-(2-гидроксифенил)-4-метилбензолсульфонамид.
В 20 г (0,18 моля) 2-аминофенола добавляют 35 г (0,18 моля) тозилхлорида и 60 мл пиридина. Оставляют, перемешивая, при комнатной температуре в течение ночи. Добавляют воду и экстрагируют два раза с помощью эфира. Органические фазы промывают последовательно водой, 1N соляной кислотой и снова 2 раза водой. Высушивают над сульфатом магния и концентрируют досуха.
Получают 44 г продукта.
2.2. (±)-4-[(4-метилфенил)сульфонил] -3,4-дигидро-2H-1,4- бензоксазин-3-этилацетат.
В 5,3 г (0,02 моля) соединения, полученного по п. 2.1, добавляют 5,1 г (0,03 моля) этил-4-бромобут-2-еноата, 3,8 мл 5,3N раствора метилата натрия и 25 мл этанола. Смесь нагревают с обратным холодильником в течение ночи. Выпаривают досуха, остаток извлекают последовательно этилацетатом, водой и, наконец, 1N раствором едкого натра. Органическую фазу собирают, экстрагируют и промывают водой, затем насыщенным раствором хлорида натрия. Высушивают над сульфатом магния, затем выпаривают досуха.
Получают 8 г продукта, который используется в таком виде при последующем этапе.
2.3. (±)-4-[(4-метилфенил)сульфонил] -3,4-дигидро-2H-1,4- бензоксазин-3-этанол. Растворяют в атмосфере аргона 0,23 г (0,006 моля) алюмогидрида лития в 20 мл тетрагидрофурана. Прикапывают 1,5 г (0,004 моля) соединения, полученного по п.2.2, в растворе в 5 мл тетрагидрофурана. По окончании реакции добавляют последовательно 0,7 г воды, 0,3 г 1N раствора едкого натра и снова 0,7 г воды. Смесь фильтруют на кизельгуре, промывают тетрагидрофураном и выпаривают. Остаток извлекают этилацетатом, а органическую фазу промывают водой. Высушивают и выпаривают досуха.
Получают 1,4 г продукта.
2.4. (±)-3-(2-хлорэтил)4-[(4-метилфенил)сульфонил]-3,4-дигидро-2H-1,4-бензоксазин.
Растворяют 2 г (0,006 моля) спирта, полученного по п. 2.3, в 15 мл хлороформа. Прикапывают 2,2 г (0,018 моля) тионилхлорида и каплю диметилформамида. Нагревают с обратным холодильником в течение 5 часов. Выпаривают досуха, остаток извлекают минимальным объемом толуола и снова выпаривают досуха.
Получают 2,1 г продукта.
2.5. (±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил]-4-[(4-метилфенил)сульфонил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
В 1,2 г (0,0034 моля) соединения, полученного в п. 2.4, добавляют 0,65 г (0,0034 моля) 6,7-диметокси-1,2,3,4-тетрагидроизохинолина и 6 мл 3-метилбутанола. Смесь нагревают при 80oC в течение ночи и выпаривают досуха. Остаток снова вводят в разбавленный аммиак и экстрагируют два раза эфиром. Органические фазы промывают водой, высушивают над сульфатом магния и выпаривают досуха. Полученный продукт очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметан/метанол 1%.
Получают 0,8 г продукта.
Оксалат получают, добавляя эквивалент щавелевой кислоты. Рекристаллизуют его в смеси этилацетат/этанол.
Получают 0,6 г оксалата.
Точка плавления: 190-192oC.
Пример 3 (Соединение N18).
(±)-3-[2-(6,7-диметокси-1,2,3,4- тетрагидроизохинолин-2-ил)этил]-4-[3-(трифторметил)бензоил]- 3,4-ди-гидро-2H-1,4 -бензоксазин, оксалат.
3.1. (±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил]-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
В раствор 2,0 г (0,005 моля) соединения, полученного по п. 1.5, в 10 мл диметилформамида, при комнатной температуре, помешивая, в атмосфере аргона, добавляют 1,24 г (0,005 моля) хлоргидрата 6,7-диметокси-1,2,3,4-тетрагидроизохинолина, 4 г карбоната калия и 0,9 г иодида калия. Смесь нагревают при 80oC в течение 4 часов. Охлаждают и добавляют 40 мл воды и 100 мл эфира. Фазы отделяют, а водную фазу экстрагируют два раза 100 мл эфира. Органические фазы объединяют, промывают в 100 мл насыщенного раствора хлорида натрия, высушивают над сульфатом магния, отфильтровывают и выпаривают досуха. Получают 3 г продукта в виде коричневого масла, которое очищают хроматографией на колонке силикагеля, элюируя смесью метанол/дихлорметан (1/9).
Получают 1,89 г основания в виде желтого масла.
Оксалат получают, добавляя эквивалент щавелевой кислоты. Выделяют и рекристаллизуют в виде белых кристаллов в смеси изопропанол/изопропиловый эфир.
Точка плавления: 126-128oC.
Пример 4 (Соединение N 18a).
(±)-3-[2-(6,7-диметокси- , 2,3,4-тетрагидроизохинолеин-2-ил)этил]-4-[3- (трифторметил)бензоил]-3,4-ди-гидро-2H-1,4-бензоксазин, оксалат.
4.1. (±)-4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4- бензоксазин-3-этанол.
Вводят в суспензии 8,7 г (0,025 моля) рацемического спирта, полученного по п. 1.4, в 1,09 л гексана. Добавляют 7,7 мл (0,082 моля) винилацетата и 4,35 г липазы Mucor Miehei. Оставляют на 15 часов при комнатной температуре, смесь отфильтровывают в вакууме. Получают 10,7 г желтого масла, которое содержит смесь правовращающегося спирта и левовращающегося ацетата. Их разделяют хроматографией на колонке силикагеля, элюируя смесью этилацетат/циклогексан (1/1).
Получают 3,62 г химически чистого правовращающегося спирта.
Вращательная способность: [α]
Избыток энантиомеров: ее = 99,7% (хиральная CLHP).
4.2. (±)-3-(2-хлорэтил)-4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4-бензоксазин.
В раствор 4 г (0,011 моля) спирта, полученного по п. 4.1, в 20 мл дихлорметана, при комнатной температуре, помешивая, в атмосфере аргона, добавляют 3 мл (0,041 моля) тионилхлорида. Продолжают перемешивать при комнатной температуре в течение 18 часов. Выпаривают досуха, а полученный продукт используют в таком виде на следующем этапе.
Получают 4,46 г продукта.
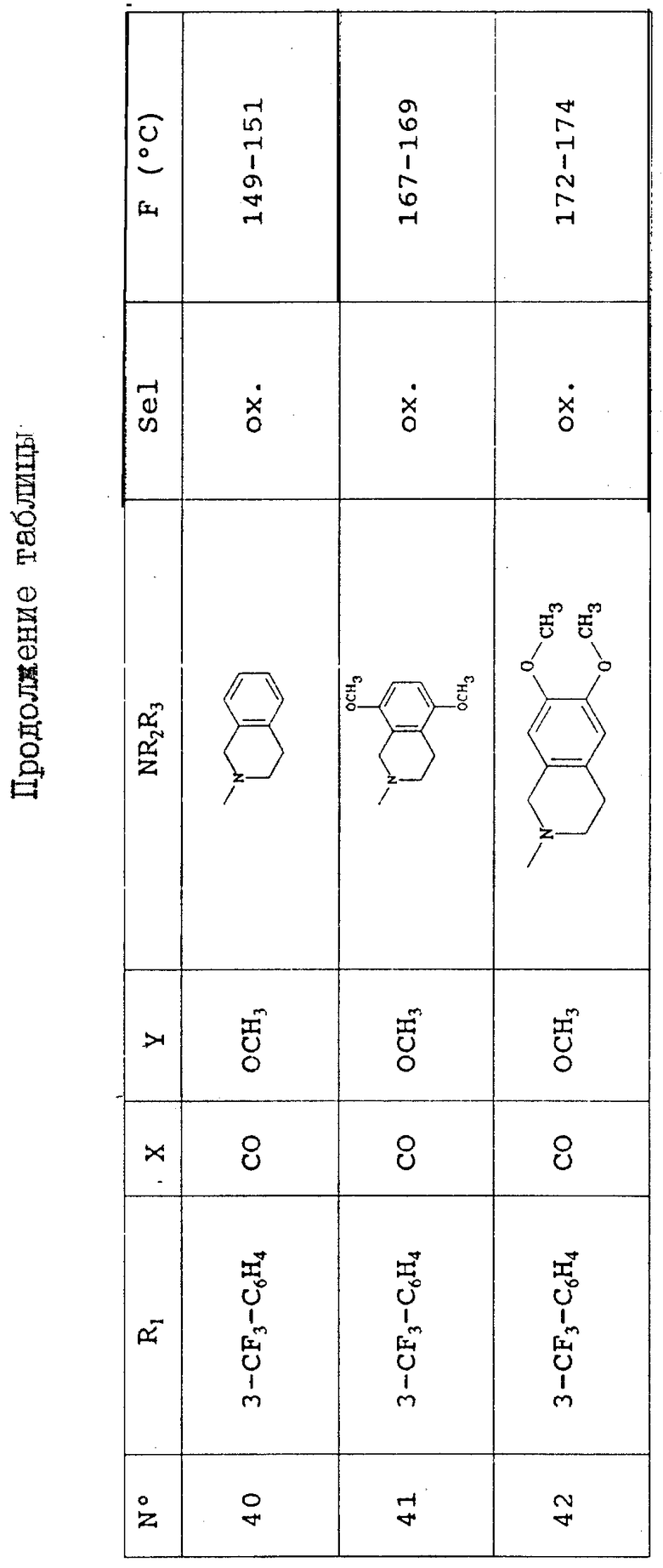
4.3. (±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил]-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
В раствор 4,46 г (0,012 моля) соединения, полученного по п. 4.2, в 40 мл диметилформамида, при комнатной температуре, помешивая, в атмосфере аргона, добавляют 2,31 г (0,012 моля) хлоргидрата 6,7-диметокси-1,2,3,4-тетрагидроизохинолина, 3,31 г (0,024 моля) карбоната калия. Смесь нагревают при 80oC в течение 4 часов, затем охлаждают. Добавляют последовательно 40 мл воды и 100 мл эфира, фазы отделяют, а водную фазу экстрагируют два раза 100 мл эфира. Органические фазы объединяют, промывают в 100 мл насыщенного раствора хлорида натрия. Высушивают над сульфатом магния, отфильтровывают и выпаривают досуха.
Получают 7 г продукта, который очищают хроматографией на колонке с силикагелем, элюируя смесью метанол/дихлорметан (1/9). Получают 1,17 г основания в виде масла.
Оксалат получают, добавляя эквивалент щавелевой кислоты. Выделяют и рекристаллизуют в виде белых кристаллов в смеси этилацетата, изопропилового эфира и ацетона.
Вращательная способность: [α]
Пример 5 (Соединение N 18б).
(-)-3-[2-6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил] -4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
5.1. (-)-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин-З-этанол.
8,7 г (0,025 моля) рацемического спирта, полученного по п. 1.4, вводят в суспензии в 1,09 л гексана. Добавляют 7,6 мл (0,082 моля) винилацетата и 4,35 г липазы Mucor Miehei. Оставляют на 15 часов при комнатной температуре, смесь отфильтровывают в вакууме. Получают 10,7 г желтого масла, которое содержит смесь правовращающегося спирта и левовращающегося ацетата. Их разделяют хроматографией на колонке с силикагелем, элюируя смесью этилацетат/циклогексан (1/1).
Получают 6,14 г левовращающегося ацетата (ее = 70%) в виде масла, которое растирают в порошок в 200 мл гексана. Получают осадок, соответствующий рацемическому ацетату, который фильтруют. Фильтрат выпаривают досуха.
Получают 3,94 г химически чистого левовращающегося ацетата.
Вращательная способность: [α]
Избыток энантиомеров: ее = 99,5% (хиральная CLHP) 3,94 г левовращающегося ацетата растворяют в 40 мл толуола. Добавляют 200 мл 0,01М фосфатного буферного раствора (KH PO/Na PO) при pH 7,2 и 1,2 г липазы Mucor Miehei. Смесь перемешивают в течение ночи при комнатной температуре, поддерживая постоянным pH путем добавления 0,5М водного раствора едкого натра с использованием pH-метра. Добавляют 100 мл этилового эфира, отделяют органическую фазу, а водную фазу экстрагируют два раза 100 мл эфира.
Органические фазы объединяют и промывают насыщенным раствором хлорида натрия. Высушивают над сульфатом магния, фильтруют и выпаривают досуха. Получают 2,8 г химически чистого левовращающегося спирта.
Вращательная способность: [α]
Избыток энантиомеров: ее = 99,5% (хиральная CLHP).
5.2. (-)-3-(2-хлорэтил)-4-[3-(трифторметал)бензоил] -3,4-дигидро-2H-1,4-бензоксазин.
В раствор 2,8 г (0,080 моля) спирта, полученного по п. 5.1, в 20 мл дихлорметана, при комнатной температуре, помешивая, в атмосфере аргона, добавляют 3 мл (0,041 моля) тионилхлорида. Продолжают перемешивать при комнатной температуре в течение 18 часов. Выпаривают досуха, а полученный продукт используют в таком виде на следующем этапе.
Получают 3,04 г продукта.
5.3. (-)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил]-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4- бензоксазин, оксалат.
В раствор 3,04 (0,008 моля) соединения, полученного по п. 5.2, в 40 мл диметилформамида, при комнатной температуре, помешивая, в атмосфере аргона, добавляют 3,17 г (0,016 моля) хлоргидрата 6,7-диметокси-1,2,3,4-тетрагидроизохинолина 2,26 г (0,016 моля) карбоната калия. Смесь нагревают при 80oC в течение 4 часов, затем охлаждают. Добавляют последовательно 40 мл воды и 100 мл эфира, фазы разделяют, а водную фазу экстрагируют два раза 100 мл эфира. Органические фазы объединяют, промывают в 100 мл насыщенного раствора хлорида натрия. Высушивают над сульфатом магния, отфильтровывают и выпаривают досуха. Получают 5 г продукта, который очищают хроматографией на колонке силикагеля, элюируя смесью метанол/дихлорметан (1/9).
Получают 1,30 г основания в виде масла.
Оксалат получают, добавляя эквивалент щавелевой кислоты. Выделяют и рекристаллизуют в виде белых кристаллов в смеси пропан-2-ол, диизопропилового эфира и ацетона.
Точка плавления: 129-130oC.
Вращательная способность: [α]
Пример 6 (Соединение N 38).
(±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2ил)этил] -6-метил-4-[3- (трифторметил)бензоил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
6.1. N-(2-гидроскси-5-метилфенил) трифторацетамид.
В литровом реакторе, при перемешивании магнитной мешалкой, приготавливают суспензию 350 мл диэтилового эфира и 25 г (0,2 моля) 2-амино-4-метилфенола, добавляют 20,5 мл пиридина, реакционную среду охлаждают смесью льда и этанола и прикапывают в течение 1 часа 28 мл (0,2 моля) трифторуксусного ангидрида. Оставляют смесь до повышения температуры до комнатной и продолжают перемешивать в течение 1 часа. Добавляют ледяную воду, органическую фазу отделяют и промывают последовательно 1N соляной кислотой, водой, насыщенным раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают растворитель. Получают 37,07 г продукта, который используют на следующем этапе.
6.2. (±)-6-метил-3,4-дигидро-2H-1,4-бензоксазин-3-этилацетат.
В 3-литровый реактор, охлажденный до 0oC, вводят, при перемешивании магнитной мешалкой, 760 мл этанола и медленно, небольшими частями, добавляют 5,79 г (0,252 моля) натрия, затем прикапывают 37,45 (0,17 моля) N-(2-гидрокси-5-метил-фенил) трифторацетамида и 43,7 г (0,17 моля) 75% чистого этил-4-бромобут-2-еноата, смесь нагревают при 110oC в течение 2 часов. Растворитель выпаривают досуха, снова вводят в 100 мл воды и 40 мл 1N едкого натра, и экстрагируют с помощью диэтилового эфира. Органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают растворитель. Получают 28,58 г продукта, который очищают хроматографией на колонке силикагеля, элюируя смесью циклогексан/изопропиловый эфир (50/50).
Получают 23,48 г продукта.
6.3. (±)-6-метил-3,4-дигидро-2H-1,4-бензоксазин-3-этанол
В литровый реактор вводят 150 мл тетрагидрофурана, охлаждают смесью льда и соли и, в атмосфере аргона, добавляют 6 г (0,158 моля) алюмогидрида лития, затем прикапывают 23,48 г (0,099 моля) (±)-6-метил-3,4-дигидро-2H-1,4-бензоксазин-3-этилацетата в растворе в 150 мл тетрагидрофурана и перемешивают в течение 1,5 часов. Реактор охлаждают смесью сухого льда и ацетона, затем прикапывают 40 мл воды и 20 мл 1N раствора едкого натра и перемешивают в течение 0,5 часа.
Осадок отфильтровывают на кизельгуре, промывают последовательно тетрагидрофураном, затем этилацетатом и выпаривают сольвент. Выделяют 20,29 г неочищенного продукта, который очищают хроматографией на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (50/50).
Получают 22,94 г продукта.
6.4. (±)-6-метил-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-безоксазин-3-этанол.
В литровую колбу помещают 200 мл дихлорметана, 22,87 г (0,118 моля) (±)-6-метил-3,4-дигидро-2H-1,4-бензоксазин-3-этанола, 17,53 г (0,125 моля) карбоната калия и прикапывают 26,19 мл (0,125 моля) 3-(трифторметил) бензоилхлорида в растворе в 200 мл дихлорметана и продолжают перемешивание при комнатной температуре в течение 3 часов. Добавляют 120 мл 1N едкого натра, органическую фазу отделяют, промывают водой, затем насыщенным раствором хлорида натрия, высушивают над сульфатом магния и 46,25 г полученного масла очищают хроматографией на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (6/4).
Получают 19,66 г продукта.
6.5. (±)-3-(2-хлорэтил)-6-метил-4-[3-(трифторметил) бензоил]-3,4-дигидро-2H-1,4-безоксазин.
К 19,66 г (0,054 моля) (±)-6-метил-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4- безоксазин-3-этанола в растворе в 230 мл дихлорметана, добавляют 19,6 мл (0,27 моля) тионилхлорида, и смесь перемешивают при комнатной температуре в течение 3 часов.
Выпаривают растворитель и избыток тионилхлорида, остаток извлекают толуолом и выпаривают, остаток очищают хроматографией на колонке с силикагелем, элюируя смесью циклогексан/изопропиловый эфир (2/1).
Получают 13,83 г продукта.
6.6. (±)-3-[2-(6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ил)этил] -6-метил-4-[3-(трифторметил)бензоил]-3,4- дигидро-2H-1,4-бензоксазин, оксалат
В раствор 2 г (0,005 моля) (±)-3-(2-хлорэтил)-6-метил-4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4-безоксазина в 20 мл N,N-диметилформамида, при комнатной температуре, при перемешивании магнитной мешалкой, в атмосфере аргона, добавляют 1,19 г (0,005 моля) хлоргидрата 6,7-диметокси-1,2,3,4-тетрагидроизохинолина, 1,78 г (0,013 моля) карбоната калия и 0,82 г (0,005 моля) иодида калия и смесь нагревают при 150oC в течение 1 часа. Охлаждают ее, добавляют 55 мл воды и 50 мл диэтилового эфира, фазы разделяют, а водную фазу экстрагируют два раза 50 мл диэтилового эфира, органические фазы объединяют, промывают в 100 мл насыщенного раствора хлорида натрия, высушивают над сульфатом магния, отфильтровывают и выпаривают растворитель.
Получают 3,75 г продукта в виде масла, которое очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметан/метанол (95/5). Получают 0,450 г чистого основания в виде желтого масла.
Оксалат получают, добавляя эквивалент щавелевой кислоты, выделяют его и рекристаллизуют в виде белых кристаллов в пропан-2-оле. Получают 0,180 г оксалана (соотношение кислота:основание = 0,8:1).
Точка плавления: 164-166oC.
Пример 7 (Соединение N35).
(±) 6- хлоро-3-[2-(2,3,4,5-тетрагидро-1H-3-бензазепин-3-ил)этил]-4-[3-(трифторметил) бензоил]-3,4-дигидро-2H-1,4-бензоксазин, оксалат.
7.1. N-(5-хлор-2-гидроксифенил)трифторацетамид.
В литровый реактор, при перемешивании магнитной мешалкой, помещают в суспензии, в 320 мл диэтилового эфира, 25 г (0,174 моля) 2-амино-4-хлорфенола, добавляют 18 мл пиридина, реакционную среду охлаждают смесью льда и этанола и прикапывают в течение 1 часа 24,6 мл (0,174 моля) трифторуксусного ангидрида, смесь оставляют до повышения температуры до комнатной и продолжают перемешивать в течение 1 часа.
Добавляют ледяную воду, осветляют, органическую фазу промывают последовательно 320 мл 1N соляной кислоты, водой, насыщенным раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают растворитель. Получают 40,3 г продукта, который используют при последующем этапе.
7.2. (±) 6-хлор-3,4-дигидро-2H-1,4-бензоксазин-3-этилацетат.
В 3-литровый реактор, охлажденный до 0oC, вводят, при перемешивании магнитной мешалкой, 430 мл этанола и медленно, небольшими частями, добавляют 3,8 г( 0,166 моля) натрия, затем последовательно прикапывают 40 г (0,166 моля) N-(5-хлор-2-гидрокси-фенил)трифторацетамида и 40 г (0,155 моля) 75% чистого 4-бромобут-2-этилэноата, смесь нагревают при 85oC в течение 2 часов. Растворитель выпаривают, снова вводят в 100 мл воды и 40 мл 1N едкого натра, и экстрагируют диэтиловым эфиром.
Органическую фазу отделяют, промывают насыщенным водным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают.
Получают 28,58 г продукта, который очищают хроматографией на колонке с силикагелем, элюируя смесью циклогексан/изопропиловый эфир (50/50).
Получают 23,72 г продукта.
7.3. (±) 6-хлор-4,4-дигидро-2H-1,4-бензоксазин-3-этанол.
В литровый реактор вводят 150 мл тетрагидрофурана, охлаждают смесью льда и соли и, в атмосфере аргона, добавляют 5,92 г (0,156 моля) алюмогидрида лития, затем прикапывают 23,52 г (9,0973 моля)
(±) 6-хлор-3,4-дигидро-2H-1,4-бензоксазин-3-этил-ацетата в растворе в 150 мл тетрагидрофурана и смесь перемешивают в течение 1,5 часов. Реактор охлаждают смесью сухого льда и ацетона, затем прикапывают 40 мл воды и 20 мл 1N раствора едкого натра и перемешивают в течение 0,5 часа, осадок отфильтровывают на кизельгуре, промывают тетрагидрофураном, затем этилацетатом и выпаривают растворитель. Выделяют 27,5 г неочищенного продукта, который очищают хроматографией на колонке силикагеля, элюируя смесью циклогексан/этилацетат (50/50).
Получают 19,67 г продукта.
7.4. (±) 6-хлор-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H- 1,4-безоксазин-3-этанол.
В литровую колбу помещают 100 мл дихлорметана, 19,17 г (0,09 моля) (±)6-хлор-3,4-дигидро-2H-1,4-бензоксазин-3- этанола, 13,3 г (0,096 моля) карбоната калия и прикапывают 20 г (0,096 моля) 3-(трифторметил)бензоилхлорида в растворе в 100 мл дихлорметана и смесь продолжают перемешивать при комнатной температуре в течение 3 часов.
Добавляют 90 мл 1N едкого натра, органическую фазу отделяют, промывают водой, затем насыщенным раствором хлорида натрия, высушивают над сульфатом магния и выпаривают растворитель. Получают 36 г маслянистого продукта, который очищают хломатографией на колонке с силикагелем, элюируя смесью циклогексан/этилацетат (2/1).
Получают 24,21 г продукта.
7.5. (±)6-хлор-3-(2-хлорэтил)-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-безоксазин.
К 24,21 г (±) 6-хлор-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H-1,4-безоксазин-3-этанола в растворе в 260 мл дихлорметана, добавляют 18 мл (0,25 моля) тионилхлорида и смесь перемешивают при комнатной температуре в течение 6 часов.
Выпаривают растворитель, а остаток извлекают толуолом и выпаривают. 25,21 г полученного масла очищают хроматографией на колонке с силикагелем, элюируя смесью циклогексан/изопропиловый эфир (50/50).
Получают 23,73 г продукта.
7.6. (±) 6-хлоро-3-[2-(2,3,4,5-тетрагидро-1H-3- бензазепин-3-ил)этил]-4-[3-(трифторметил)бензоил]-3,4-дигидро-2H- 1,4-бензоксазин, оксалат.
В раствор 2 г (0,005 моля) (±)6-хлор-3-(2-хлорэтил)-4-[3-(трифторметил)бензоил] -3,4-дигидро-2H-1,4-бензоксазина в 20 мл N,N-диметилформамида, при комнатной температуре, при перемешивании в атмосфере аргона, добавляют 0,9 г (0,005 моля) хлоргидрата 2,3,4,5-тетрагидро-1H-3-бензазепина, 1,7 г (0,0124 моля) карбоната калия и 0,82 г (0,005 моля) иодида калия и смесь нагревают при 110oC в течение 1 часа. Охлаждают ее, добавляют 55 мл воды и 50 мл диэтилового эфира, фазы разделяют, а водную фазу отделяют и экстрагируют два раза 50 мл диэтилового эфира. Органические фазы объединяют, промывают в 100 мл насыщенного раствора хлорида натрия, высушивают над сульфатом магния, отфильтровывают и выпаривают растворитель. Получают 2,55 г продукта в виде масла, которое очищают хроматографией на колонке силикагеля, элюируя смесью дихлорметан/метанол (98/2). Получают 1,84 г чистого основания в виде желтого масла, из которого получают оксалат, добавляя эквивалент щавелевой кислоты и рекристаллизуя в этаноле.
Точка плавления: 192-194oC.
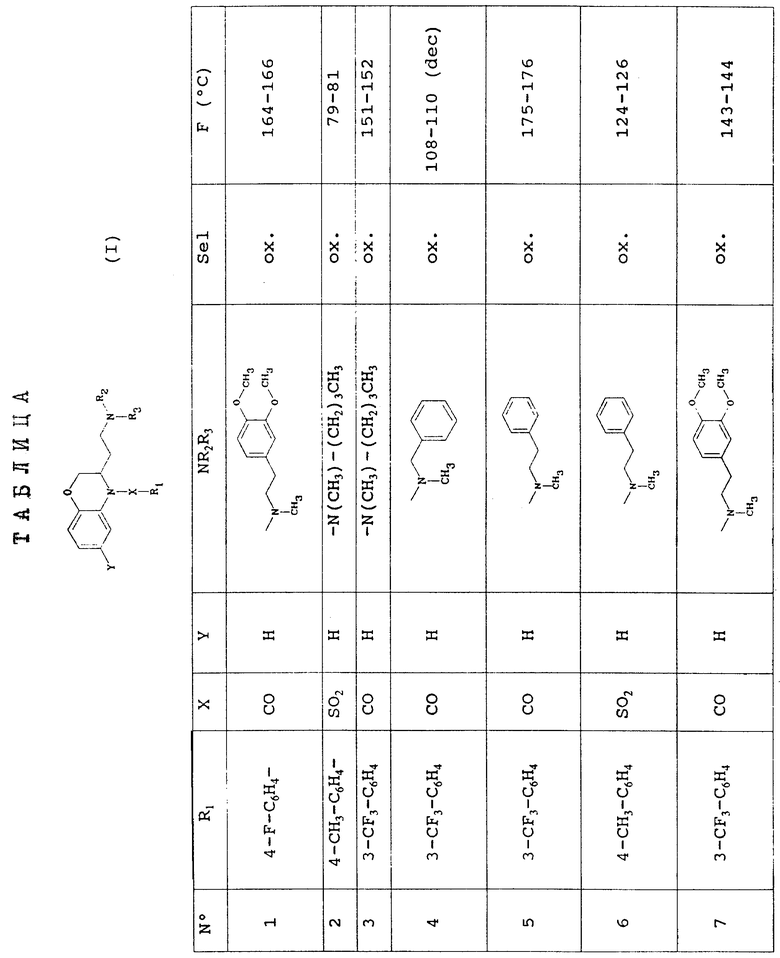
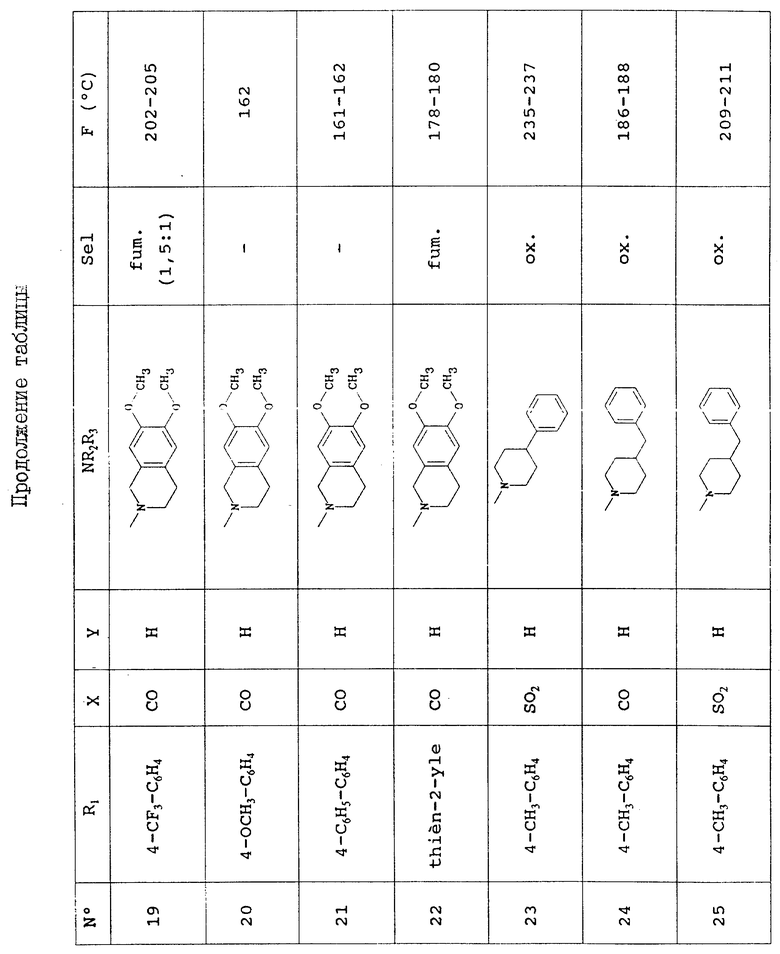
Таблица, приведенная в конце описания, иллюстрирует химическую структуру и физические свойства некоторых соединений по изобретению.
Условные обозначения таблицы.
В колонке "R" "n-A-С6H4" - фенильная группа, замещенная в позиции n кольца группой A.
В колонке "Sel" (соль): "-" обозначает соединение в виде основания, "ox. " означает оксалат, "fum." означает фумарат; если молярное соотношение кислота: основание отлично от 1:1, оно указывается в скобках.
В колонке "F" (oC) (точка плавления) "dec" означает "плавление с разложением"
Соединения по изобретению прошли фармакологические испытания, которые показали их полезные свойства для применения в терапии в качестве активных веществ.
Ингибирование ввода кальция, индуцированного KCl в срезах коры головного мозга неполовозрелых крыс.
Используются крысы Sprague-Dawley, самцы или самки, в возрасте 8 дней. После смещения шейных позвонков вырезают мозг и приготавливают срезы теменной зоны коры головного мозга.
Межклеточная концентрация кальция ([Ca2+]i) измеряется по технологии, описанной в J.Pharm. Exp. Ther. (1992) 261 324-330. Приготовленные таким образом срезы инкубируют в течение 75 мин при 24oC в буферном растворе Кребса, насыщенном O2/CO2 (95%/5%) и содержащем Fura-2 AM в концентрации 7 мкМ, после инкубации, срезы промывают несколько раз тем же буферным раствором и оставляют в этом же буферном растворе до их использования.
Для измерения ([Ca2+]i) срезы помещают, при 30oC, в куб спектрофлуорометра, который перфузируют буферным раствором Кребса с помощью насоса. Деполяризацию срезов осуществляют, перфузируя в течение 3 минут буферный раствор Кребса, содержащий 50 мМ KCl. В перфузионную жидкость вводят тестируемое соединение через 7 минут после первой деполяризации и осуществляют вторую деполяризацию через 7 минут после введения тестируемого соединения. Наблюдают флюоресценцию с двумя длинами волн возбуждения, 340 нм (форма, связанная с кальцием) и 380 нм (свободная форма), причем длину волны эмиссии -510 нм. ([Ca2+]i) рассчитывают по методу, описанному в J.Biol. Chem. (1985) 260 3440-3450. Ингибирующее действие тестируемых соединений рассчитывают в соотношении к повышению ([Ca2+] i), ингибированному 50 мМ KCl, взятым за 100%.
Коэффициент ингибирования ввода Ca2+, индуцированного соединениями по изобретению, является дозо-зависимым и находится в пределах от 10 до 65% для концентраций 10-30 мкМ.
Полная ишемия головного мозга мыши.
Соединения по изобретению прошли испытания на полную ишемию головного мозга мыши. Ишемия была вызвана остановкой сердца, индуцированной путем быстрого внутривенного введения хлорида магния.
При этом испытании измерялось "время выживания", т.е. интервал между моментом введения хлорида магния и последним отмеченным дыхательным движением мыши. Это последнее движение рассматривается как решающий признак функционировании центральной нервной системы.
Остановка дыхания поступает приблизительно через 19 секунд после введения хлорида магния.
Самцов мышей (SWISS OF1 IFFA CREDO) изучали группами по 10. Перед опытами их кормили и поили ad libitum. "Время выживания" измерялось через 10 минут после внутрибрюшинного введения соединений изобретения. Результаты даются в виде разности "времени выживания", измеренного в группе 10 мышей, которым было введено соединение, и "времени выживания", измеренного в группе из 10 мышей, которым была введена жидкость-носитель. Соотношения между "временем выживания" и дозой соединения были отмечены графически на полулогарифмической кривой.
Эта кривая показывает "эффективную 3-секундную дозу" (DE3") кальция, т. е. дозу (в мг/кг), которая вызывает повышение "времени выживания" на 3 секунды по сравнению с контрольной группой из 10 мышей, не подвергнутых обработке. Повышение "времени выживания" на 3 секунды является одновременно статистически достоверным и воспроизводимым.
Дозы DE3'' для соединения изобретения - от 0,2 до 60 мг/кг, внутрибрюшинно.
Изучение потенциал-зависимых ("напряжение-зависимых") бариевых потоков по технологии patch-clamp.
Измерение бариевых потоков по потенциал-зависимым кальциевым каналам осуществлялось на клетках кортекса новорожденных крыс (Sprague-Dawley) в культуре (6-10 дней)
Измерительные камеры объемом 800 мкл, содержащие клетки кортекса крысы, помещают на пластину инвертированного микроскопа Olympus IMT-2TM и наблюдают с увеличением в 400 раз. Камеры подвергаются непрерывной перфузии (4-6 мл/мин) с помощью распределителя растворов, допускающих 9 вводов (смертельный объем < 50 мкл), единственный выход которого в виде полиэтиленовой трубки с отверстием 5 мкм расположен менее, чем в 3 мм от изучаемой клетки. Преимуществом этого устройства является то, что оно обеспечивает быстрый обмен раствора на уровне изучаемых клеток. Используемый метод patch-clapm описан в Pfluegers Archives (1981) 391 85-100. Усилитель Axopatch-1DTM вместе с компьютером типа AT 386-33 МГц с программным обеспечением PCLAMPTM "Axon InstrumentsTM" используются для стимуляции клеток, сбора данных и анализа результатов. Для регистрации бариевых потоков, пипетки из боросиликатного стекла приближают к клеткам с помощью гидравлического микроманипулятора Narishige WR 60.
Кончик пипетки наполняют этанольным межклеточным раствором, имеющим следующий состав (в мМ): CsCl (140), CaCl2 (1), Na2-АТФ, ЭГТК (11, pCa=8), Hepes (10), Tris-OH (pH = 7,2).
При получении полной конфигурации клетки ее подвергают перфузии раствором ТЭА-барий, имеющим следующий состав: ТЭА-C1 (144), BaCl2 (5). MgCl2 (2), CsCl (3), глюкоза (10), Hepes (10), Tris-OH (pH = 7,4).
Этот раствор позволяет измерить кальциевый поток (приравненный к бариевому потоку, проходящему по потенциал зависимым кальциевым каналам), не учитывая натриевых и калиевых потоков.
Общий потенциал-зависимый бариевый поток получают с помощью деполяризующего скачка напряжения продолжительностью 250 мсек при мембранном потенциале от -80 мВ до -16 мВ. Частота стимуляции 0,25 Гц.
Соединения по изобретения вводятся в растворе в среду ТЭА-барий с стабилизированной амплитудой бариевого потока. После получения стабильного ингибирующего эффекта клетка снова перфузируется контрольным ТЭА-бариевым раствором для наблюдения реверсии эффекта.
Полученный эффект сравнивают с эффектом 1 мкМ раствора кадмия. Блокирующий эффект потенциал-зависимых кальциевых каналов изменяется в зависимости от доз рассматриваемых соединений и, для более активных соединений, он составляет порядка 66% при концентрации 1 мкМ и 100% при концентрации 1 мкМ.
Результаты испытаний соединений изобретения показывают, что in vitro они имеют свойства кальций-нейронных антагонистов, a in vivo - нейрозащитные и антиишемические свойства.
В соответствии с этими результатами соединения могут быть использованы для лечения и профилактики церебральных расстройств, например, вызванных приступом ишемии, остановкой сердца или дыхания, тромбозом или закупоркой сосудов головного мозга, для лечения старения головного мозга, слабоумия вследствие многократных инфарктов, старческого слабоумия, например, болезни Альцгеймера или болезни Пика, для лечения оливо-мосто-мозжечковой атрофии и других заболеваний, связанных с перерождением нервных клеток, таких как хорея Huntington, амиотрофический латеральный склероз, для лечения черепных и спинномозговых травм, для профилактики нервных поражений в результате конвульсий, для лечения некоторых видов рака, невралгических изменений при СПИДе и диабетических ретинопатий.
Для этого они могут быть представлены в любой фармацевтически приемлемой форме для энтерального и парэнтерального введения, вместе с соответствующим разбавителем, например, в виде таблеток, драже, гель-капсул, капсул, свечей, растворов и суспензий для питья или впрыскивания; дневная доза от 0,1 до 1000 мг активного вещества.
Примеры фармкомпозиций.
Раствор для инъекций:
Активное вещество - 5 мг
Глюкоза - 250 мг
Вода для инъекций на ампулу объемом 5 мл - до 5 мл
Желатиновые капсулы:
Активное начало - 100 мг
Тальк - 24 мг
Гель диоксида кремния на капсулу 125 мг - 1 мг
Таблетка:
Активное начало - 400 мг
Гель диоксида кремния - 10 мг
Стеариновая кислота - 20 мг
Крахмал зерновых культур на таблетку 475 мг - 45 мг
Сироп:
Активное начале - 5 г
4-гидроксиметилбензоат - 150 мг
Сахароза - 50 г
Дистиллированная вода на флакон объемом 100 мл - до 100 мли



Производные бензоксазина в виде оптически чистого изомера или смеси оптических изомеров формулы 1, где Y - водовод, фтор, хлор, метил или метокси; R1 - фенил, замещенный фтором, метилом, метокси, трифторметилом или фенилом, или тиен-2-ильная группа; R2 - метил; R3 - алкил, фенилалкил, возможно замещенный 2-3 метоксилами, либо 2-(пиридин-2-ил)этил, или R2 и R3 образуют вместе с атомом азота 4-фенил(пиперидин-1-ил), 4-фенилметил-(пиперидин-1-ил), 1,2,3,4-тетрагидроизохинолин -2-ил, 6-метокси-1,2,3,4-тетрагидроизохинолин -2-ил, 5,8-диметокси-1,2,3,4-тетрагидроизохинолин -2-ил, 6,7- диметокси-1,2,3,4- тетрагидроизохинолин -2-ил, 2,3,4,5-тетрагидро-1Н-3-бензазепин-3-ил, 7,8-диметокси- 2,3,4,5-тетрагидро-1Н-3-6ензазепин-3-ил, Х -карбонил или сульфонил; или их соли in vitro проявляют свойства кальций-нейронных антагонистов, a in vivo - нейрозащитные и антиишемические свойства. 3 с. и 2 з.п. ф-лы, 1 табл.
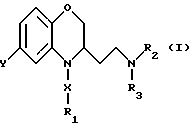
где Y - атом водорода, фтора или хлора или метильная или метокси-группа;
R1 - либо фенильная группа, замещенная атомом фтора или группой, выбранной из метильной, метокси-, трифторметильной и фенильной групп, либо тиен-2-ильная группа;
R2 - метильная группа;
R3 - либо (C1-C4) алкильная группа, либо фенил (C1-C2)алкильная группа, в которой кольцо, при необходимости, замещено 2-3 метокси-группами, либо 2-(пиридин-2-ил) этильная группа;
или R2 и R3 образуют, вместе с азотом, либо 4-фенил(пиперидин-1-ильную) группу, либо 4-фенилметил(пиперидин-1-ильную) группу, либо 1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 6-метокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 5,8-диметокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 6,7-диметокси-1,2,3,4-тетрагидроизохинолин-2-ильную группу, либо 2,3,4,5-тетрагидро-1H-3-бензазепин-3-ильную группу, либо 7,8-диметокси-2,3,4,5-тетрагидро-1H-3-бензазепин-3-ильную группу;
X - либо карбонильная группа, либо сульфонильная группа,
или их соли присоединения фармацевтически приемлемых кислот.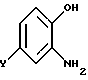
где Y - как определено в п.1,
с трифторуксусным ангидридом для получения амида общей формулы IV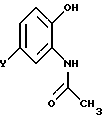
который вводят в реакцию с этил-4-бромобут-2-еноатом, затем восстанавливают функцию сложного эфира в 3,4-дигидро-2H-1,4-бензоксазин-3-этилацетате общей формулы VI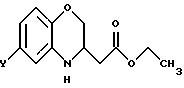
с получением 3,4-дигидро-2H-1,4-бензоксазин-3-этанола общей формулы VII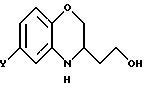
который вводят в реакцию с хлорангидридом кислоты общей формулы VIII
Cl-CO-R1,
где R1 - как определено в п.1,
для получения спирта общей формулы IX
который подвергают реакции с тионилхлоридом для получения соединения общей формулы X
и, наконец, проводят реакцию последнего с амином общей формулы XI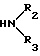
где R2 и R3 - как определено в п.1 или если X - сульфонильная группа,
проводят реакцию 2-аминофенола общей формулы II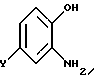
где Y - как определено в п.1, с хлоридом общей формулы III'
R1-SO2 -Cl
для получения соединения общей формулы IV'
которое подвергают реакции с этил-4-бромобут-2-еноатом с получением соединения формулы (VI'), затем восстанавливают функцию сложного эфира полученного соединения общей формулы VI'
с получением соединения формулы VII'
которое подвергают реакции с тионилхлоридом для получения соединения общей формулы X'
которое затем конденсируется с помощью амина общей формулы XI
где R2 и R3 - как определено в п.1.
| Способ получения @ -(2,6-дизамещенных ароматических)- @ -пиридилмочевин или их фармацевтически приемлемых кислотно-аддитивных солей | 1985 |
|
SU1321373A3 |
| Способ получения производных 1,4-бензоксазина | 1975 |
|
SU529162A1 |
| СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ ЛЮМИНОФОРОВ | 0 |
|
SU234412A1 |
| DE 1287584 B, 1969 | |||
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| СПОСОБ УКЛАДКИ ПОД ВОДУ РЕЛЬСОВЫХ ДОРОЖЕК СЛИПОВ И ЭЛЛИНГОВ БЕЗ ПРИМЕНЕНИЯ ОБЩЕЙ МОНТАЖНОЙ РАМЫ | 0 |
|
SU196570A1 |
