Настоящее изобретение относится к новым производным 1-[ω--(3,4-дигидро-2-нафталинил)алкил] циклического амина, оказывающим ингибирующее действие на рефлекс мочеиспускания и др., способу их получения и содержащей их фармацевтической композиции.
Предпосылки изобретения
К настоящему времени существует много сообщений по производным 1-[ω--(3,4-дигидро-2-нафталинил)алкил]циклического амина.
Однако насколько известно авторам настоящей заявки, только одно указанное далее сообщение относится к производному 1-[ω--(3,4-дигидро-2-нафталинил)алкил] циклического амина, в котором алкильная часть является этилом. Эта работа, Pharmazie, 42,369 (1987), раскрывает, что 1-[2-(3,4-дигидро-2-нафталинил)этил] пиперидингидрохлорид проявляет очень слабое родство с рецептором допамина in vitro.
Известныследующиесоединения-производные1-[ω--(3,4-дигидро-2-нафталинил)алкил]циклического амина, где алкильная часть является метилом.
Патент США 4,022,791 раскрывает, что соединения указанной далее формулы полезны в качестве аналгетиков и транквилизаторов, а в журнале J.Med.Chem., 21, 257 (1978) приведена работа почти аналогичного содержания.
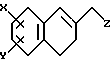
где X и Y независимо являются H, F, Cl, Br и алкильной группой, имеющей от 1 до 4 атомов углерода, или алкоксигруппой, имеющей от 1 до 4 атомов углерода, и Z является вторичной или третичной аминогруппой, при условии, что X и Y одновременно не могут быть H.
Кроме того, в Chem.Pharm.Bull., 31, 2006 (1983) описано, как на собаках и крысах с самопроизвольно повышающимся давлением исследовали сосудорасширяющее действие и активность по снижению артериального давления 1-[(3,4-дигидро-6-морфолино-2-нафталинил)метил] пиперазина и 4-бензил-1-[(3,4-дигидро-6-морфолино-2-нафталинил)метил]пиперидина.
Вместе со старением общества год от года увеличивается количество пациентов, страдающих от частого мочеиспускания и недержания мочи. В настоящее время клиническое применение для лечения этих симптомов находят три лекарственных препарата, а именно, флавоксат, оксибутинин и пропиверин, которые отличаются от медикаментов для лечения частого мочеиспускания и недержания мочи в сочетании с гипертрофией простаты. Фармакологическая активность (увеличение объемных возможностей мочевого пузыря) всех этих лекарственных препаратов основана на расслаблении гладкой мышцы мочевого пузыря, и что касается побочных эффектов, они не обязательно являются удовлетворительными в смысле меньшей эффективности и сложности использования в случае частого мочеиспускания и недержания мочи в сочетании с закупоркой мочевых путей (сложность мочеиспускания).
В таких обстоятельствах стараются развивать лекарственные препараты для лечения частого мочеиспускания и недержания мочи через центральный механизм, который отличается от механизма действия существующих медикаментов. Например сообщают, что 1-(4-этилфенил)-2-метил-3-(1-пирролидинил)-1-пропанонгидрохлорид (общее название: инаперизон гидрохлорид), который является мышечным релаксантом центрального действия (сравните, Druqs Fut.,. 18, 375 (1993)), эффективен при таких симптомах, как нейрогенный мочевой пузырь, нестабильный мочевой пузырь и поллакиурия (сравните, Nishinihon J.Urol., 54, 1472 и 1820 (1992)). Однако необязательно в достаточной степени повышена эффективность и исправлены побочные воздействия.
Авторы настоящего изобретения интенсивно исследовали производное 1-[ω--(3,4-дигидро-2-нафталинил)алкил] циклического амина приведенной далее формулы (I) и обнаружили, что оно оказывает сильное ингибиторное действие на рефлекс мочеиспускания, опосредованный главным образом - через центральный механизм, без таких побочных эффектов как депрессия центральных нервов и ингибирование спинального рефлекса.
Описание изобретения
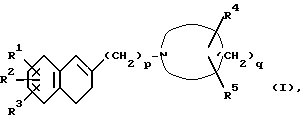
Настоящееизобретениеобеспечиваетпроизводное1-[ω--(3,4-дигидро-2-нафталинил)алкил]циклического амина формулы (I):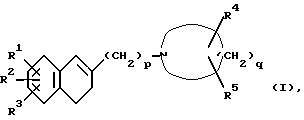
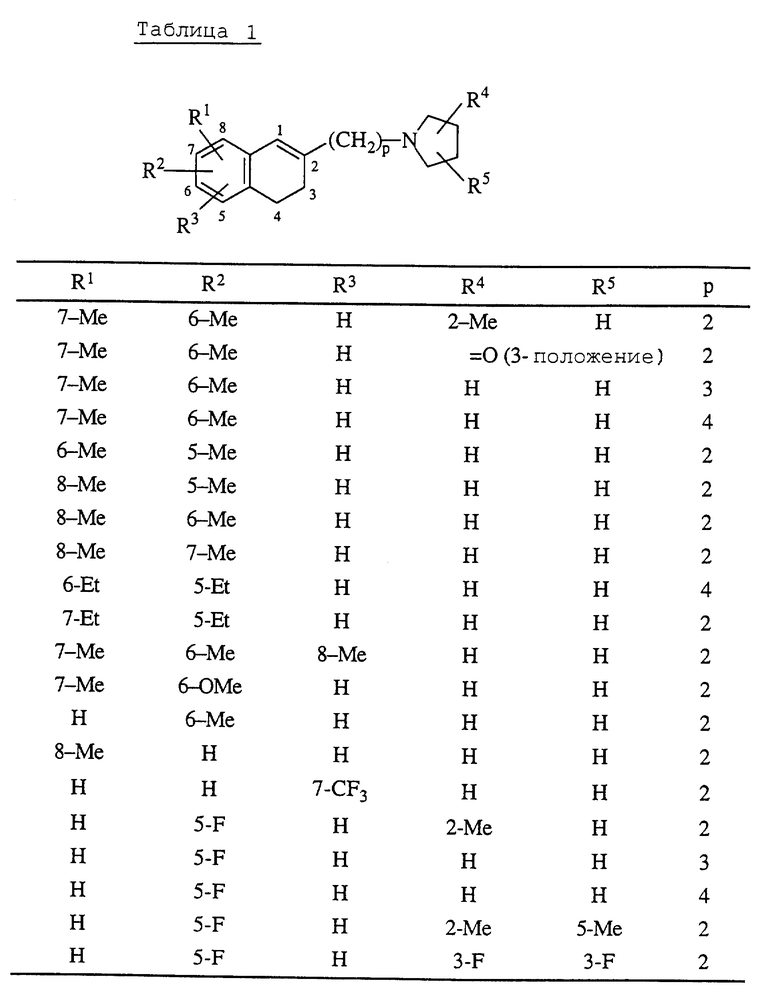
где R1 и R2 являются одинаковыми или разными и каждый представляет собой атом водорода, атом галогена, гидроксильную группу, C1-C5-алкильную группу, C1-C3-алкоксигруппу, гидроксиметильную группу, формильную группу, карбоксильную группу или C1-C3-алкоксикарбонильную группу, или если R1 и R2 соединены с соседними атомами углерода, то R1 и R2 могут вместе образовать метилендиоксигруппу, этиленоксигруппу (-CH2CH2O-), триметиленовую группу или тетраметиленовую группу; R3 является атомом водорода, атомом галогена, трифторметильной группой, C1-C5-алкильной группой, C1-C3-алкоксигруппой или фенильной группой; R4 является атомом водорода, атомом галогена, гидроксильной группой, C1-C3-алкильной группой или (C1-C2-алкокси)метильной группой; R5 является атомом водорода, атомом галогена, C1-C3-алкильной группой или (C1-C2 алкокси)метильной группой; или когда R4 и R5 присоединены к атомам углерода, не соседним с атомом азота, то R4 и R5 могут вместе образовывать оксогруппу; p - целое число от 2 до 6; q - целое число от 3 до 7; при условии, что когда p=2 и q=5, то R1, R2, R3, R4 и R5 одновременно не являются атомами водорода, его фармацевтически пригодную соль присоединения кислоты и его N-оксидное производное, способ их получения и содержащую их фармацевтическую композицию.
Фармацевтически пригодные соли присоединения кислот соединения формулы (I) включают такие соли с неорганическими кислотами как гидрохлорид, гидробромид, гидроиодид, сульфат или фосфат, или соли с органическими кислотами, такие как оксалат, малонат, сукцинат, малеат, фумарат, лактат, малат, цитрат, тартрат, бензоат, метансульфонат или паратолуолсульфонат. Соединение формулы (I), его соль и его N-оксидное производное могут существовать в виде гидратов или сольватов, и настоящее изобретение включает также эти гидраты и сольваты.
Соединение формулы (I) может иметь один или более асимметрических атомов углерода и/или проявлять геометрическую изомерию. В соответствии с этим соединение формулы (I) может существовать в виде различных стереоизомеров. Настоящее изобретение включает также эти стереоизомеры, их смесь и их рацемическую смесь.
Ниже приведено объяснение терминов, использованных в настоящем описании и формуле изобретения.
Обозначение "алкильная группа" и "алкильная часть" подразумевает либо линейные, либо разветвленные цепи. Атом галогена включает фтор, хлор, бром и йод, предпочтительными являются фтор и хлор, и наиболее предпочтителен фтор. Предпочтительными группами для R1 и R2 являются атом водорода, атом галогена (особенно фтор), гидроксильная группа, метильная группа, этильная группа, пропильная группа, метоксигруппа, этоксигруппа, гидроксиметильная группа, карбоксильная группа, метоксикарбонильная группа и этоксикарбонильная группа. Более предпочтительными примерами R1 и R2 являются метильная группа, этильная группа, метоксигруппа, этоксигруппа и гидроксиметильная группа, и особо предпочтительно для этих групп присоединение по 6-ой и 7-ой позиции. Кроме того, более предпочтительно, чтобы R1 представлял собой атом водорода или атом галогена (особенно фтора), а среди этих соединений особо предпочтительны 6,7- дигалоген - или 5-галоген-замещенные.
Предпочтительной группой для R3 является атом водорода, а предпочтительными группами для R4 являются атом водорода, атом галогена (особенно фтора), гидроксильная группа и метильная группа, а среди них более предпочтительны атом водорода и метильная группа.
Предпочтительными группами для R5 являются атом водорода и метильная группа. Кроме того, предпочтительно, чтобы R4 и R5 вместе образовывали оксогруппу. Предпочтительными значениями p являются целые числа от 2 до 5, особо предпочтительны 2, 3 или 4, и наиболее предпочтительно 4.
Предпочтительными соединениями настоящего изобретения являются соединения формулы (I), где R1 и R2 являются одинаковыми или разными, и каждый представляет, собой атом водорода, атом галогена, гидроксильную группу, метильную группу, этильную группу, пропильную группу, метоксигруппу, этоксигруппу, гидроксиметильную группу, карбоксильную группу, метоксикарбонильную группу или этоксикарбонильную группу, R3 является атомом водорода, a R4, R5, p и q такие, как определено выше, их фармацевтически пригодные соли присоединения кислот и их N-оксидные производные.
Более предпочтительными соединениями настоящего изобретения являются соединения формулы (I), где R1 и R2 присоединены по 7-ой и 6-ой позиции, соответственно, и являются одинаковыми или разными, и каждый представляет собой метильную группу, этильную группу, метоксигруппу, этоксигруппу или гидроксиметильную группу, R3 является атомом водорода, R4 и R5 являются одинаковыми или разными, и каждый представляет собой атом водорода или метильную группу,
p является целым числом от 2 до 5, и q является целым числом от 3 до 7, и их фармацевтически пригодные соли присоединения кислот.
Другими более предпочтительными соединениями являются соединения формулы (I), где R1 является атомом водорода, R2 является атомом галогена в 5-ой позиции или R1 и R2 каждый представляет собой атом галогена в 7-ой и 6-ой позиции, соответственно, и R3 является атомом водорода, R4 и R5 являются одинаковыми или разными, и каждый представляет собой атом водорода или метильную группу,
p является целым числом от 2 до 5, и q является целым числом от 3 до 7, и их фармацевтически пригодные соли присоединения кислот.
Наиболее предпочтительными соединениями являются соединения формулы (I), где R1 и R2 присоединены по 7-ой и 6-ой позиции, соответственно, являются одинаковыми или разными, и каждый представляет собой метильную группу, этильную группу, метоксигруппу, этоксигруппу и гидроксиметильную группу, все R3, R4 и R5 представляют собой атомы водорода,
p равно 2, 3 или 4, и q равно 4 или 5, и их фармацевтически пригодные соли присоединения кислот.
Другими наиболее предпочтительными соединениями являются соединения формулы (I), где R1 является атомом водорода, R2 является атомом фтора в 5-ой позиции, или R1 и R2 являются атомами фтора в 7-ой и 6-ой позиции, соответственно, все R3, R4 и R5 представляют собой атомы водорода, p равно 2, 3 или 4, и q равно 4 или 5, и их фармацевтически пригодные соли присоединения кислот.
Особо предпочтительными соединениями являются соединения формулы (Ia):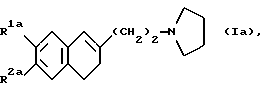
где R1a и R2a одинаковые или разные, и каждый представляет собой метильную группу, этильную группу, метоксигруппу, этоксигруппу или гидроксиметильную группу, и их фармацевтически пригодные соли присоединения кислот.
Подходящими примерами наиболее предпочтительных соединений настоящего изобретения являются приведенные далее соединения и их фармацевтически пригодные соли присоединения кислот, среди которых первые четыре соединения являются самыми предпочтительными и особо предпочтительны из них - первые два.
1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил)этил]пирролидин,
1-[2-(5-фтор-3,4-дигидро-2-нафталинил)этил]пирролидин,
1-[2-(3,4-дигидро-6,7-диметокси-2-нафталинил)этил] пирролидин,
1-[2-(3,4-дигидро-7-метокси-6-метил-2-нафталинил)этил]пирролидин,
1-[2-(3,4-дигидро-6-гидроксиметил-7-метил-2- нафталинил)этил]пирролидин,
1-[2-(6,7-диэтил-3,4-дигидро-2-нафталинил)этил]пирролидин,
1-[2-(6,7-дифтор-3,4-дигидро-2-нафталинил)этил]пирролидин и
1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил) этил]пиперидин.
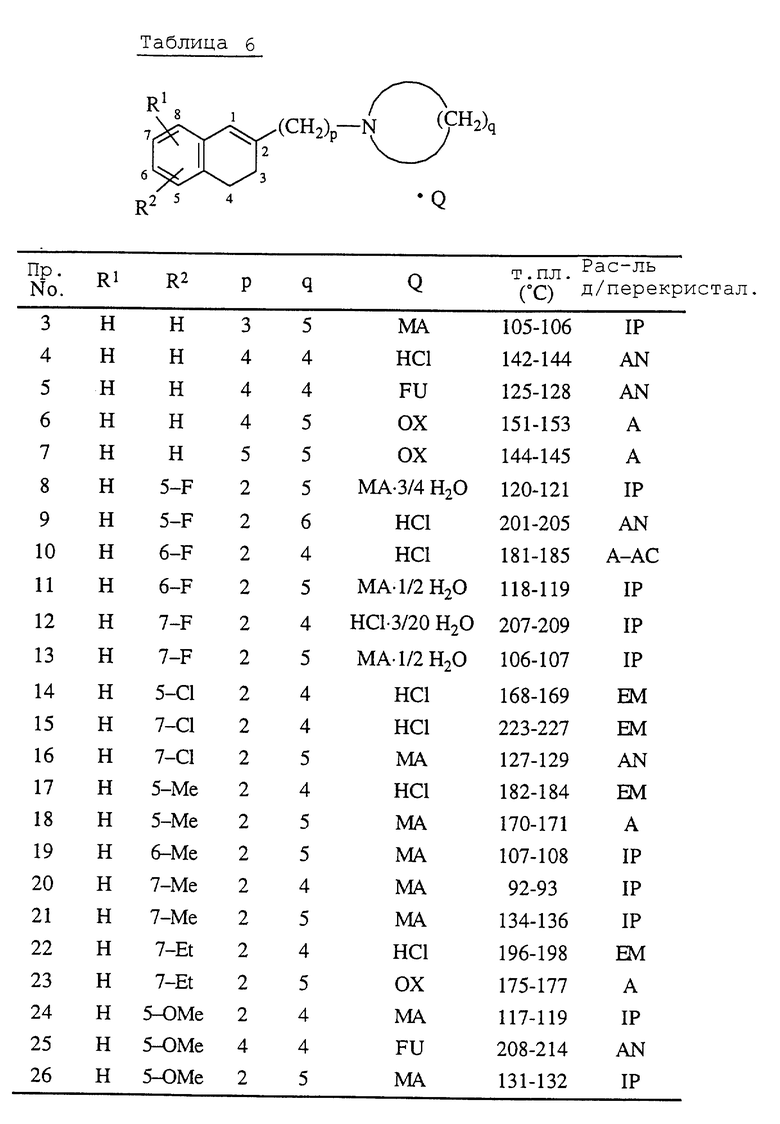
Данные соединения являются типичными представителями настоящего изобретения в дополнение к соединениям примеров, описанных здесь далее, соединениям приведенных в конце описания табл. 1 и 2, и их фармацевтически пригодным солям присоединения кислот и N-оксидным производным. В таблицах Me обозначает метильную группу, Et обозначает этильную группу.
Соединения настоящего изобретения можно получить, например следующими способами:
Способ (a)
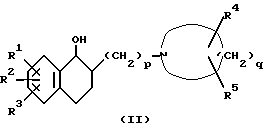
Соединение формулы (I) получают посредством дегидратации соединения формулы (II):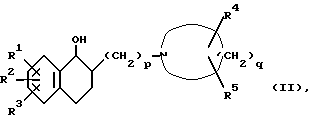
где R1, R2, R3, R4, R5, p и q такие, как определено выше.
Реакцию дегидратации проводят в условиях, подходящих для реакции дегидратации спиртов до олефинов. Например соединение формулы (II) реагирует с агентом дегидратации в подходящем растворителе или без растворителя. Агент дегидратации включает, например неорганическую кислоту (например, соляную, бромистоводородную, серную, фосфорную или борную кислоту), органическую кислоту (например, оксалиновую, муравьиную или трифторуксусную кислоту), ароматическую сульфоновую кислоту (например, паратолуолсульфоновую кислоту), ангидрид органической кислоты (например, уксусный ангидрид), ортосульфобензойный ангидрид, безводную неорганическую соль (например, гидросульфат калия), хлорангидрид неорганической кислоты (например, тионилхлорид или фосфорный оксихлорид), хлорангидрид органической кислоты (например, ацетилхлорид), сульфоновой кислоты (например, паратолуолсульфонилхлорид или метансульфонилхлорид); кислоту Льюиса (например, комплекс бор фторид-диэтиловый эфир или хлорид цинка), йод, оксид алюминия и силикагель. Растворитель необходимо выбирать в соответствии, например с типом используемого агента дегидратации, растворитель включает, например ароматические углеводороды (например, бензол, толуол и ксилол), эфиры (например, диэтиловый эфир, тетрагидрофуран или диоксан), кетоны (например, ацетон или этилметилкетон), ацетонитрил, спирты (например, метанол, этанол или изопропиловый спирт), этиленгликоль, органические кислоты (например, муравьиную кислоту, уксусную кислоту или пропионовую кислоту), пиридин, диметилсульфоксид и воду. Эти растворители можно использовать по одному или в смеси двух или более растворителей. Температуру реакции можно варьировать в зависимости, например от типа агента дегидратации, но обычно она составляет величину в диапазоне примерно от -20 до 200oC. Кроме того, используемое в реакции дегидратации соединение формулы (II) может быть в виде комплекса с борсодержащим восстановителем, таким как бораны или продукты их разложения, который можно перевести в соединение формулы (I), используя кислотный агент дегидратации, такой как неорганическая кислота или органическая кислота.
Исходное соединение формулы (II) можно получить способом в соответствии со следующей схемой реакции.
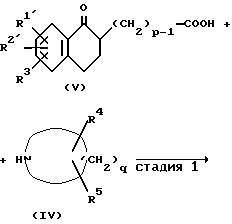
где R3, R4, R5, p и q такие, как определено выше, R1', R2' - такие же группы, как R1 и R2 за исключением того, что в формильную группу и карбоксильную группу введена защита.
В приведенной выше реакционной схеме защищенная формильная группа для R1' и R2' включает, например ацетали (например, диметоксиметил, диэтоксиметил или этилендиоксиметил), оксимы (например, гидроксииминометилен), и защищенная карбоксильная группа включает, например низшую алкоксикарбонильную группу (например, метоксикарбонил или этоксикарбонил), аралкилоксикарбонильную группу (например, бензилоксикарбонил). Далее объяснена каждая стадия приведенной выше реакционной схемы.
Стадия 1
Данную стадию осуществляют, проводя реакцию соединения (V) или его реакционноспособного производного с соединением (IV) в тех же условиях, что и обычную реакцию амидирования.
Реакционноспособное производное соединения (V) включает, например сложные эфиры с низшими алкилами (главным образом, метиловые эфиры), активированные сложные эфиры, ангидриды кислот и галогенангидриды кислот (главным образом, хлориды). Активированные сложные эфиры включают, например паранитрофениловый эфир, 2,4,5-трихлорфениловый эфир и N- гидроксисукцинимидный эфир. Ангидриды кислот включают симметричные ангидриды кислот и смешанные ангидриды кислот; смешанные ангидриды кислот включают, например смешанный ангидрид кислоты с таким алкилхлоркарбонатом, как этилхлоркарбонат или изобутилхлоркарбонат, смешанный ангидрид кислоты с таким аралкилхлоркарбонатом как бензилхлоркарбонат, смешанный ангидрид кислоты с таким арилхлоркарбонатом как фенилхлоркарбонат и смешанный ангидрид кислоты с такой алкановой кислотой как изовалериановая кислота или пивалиновая кислота.
Если на этой стадии используют соединение (V) само по себе, реакцию предпочтительно проводить в присутствии конденсирующего агента, такого как N, N'-дициклогексилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, N, N'-карбонилдиимидазол, N,N'-карбонилдисукцинимид, 1-этоксикарбонил-2-этокси-1,2-дигидрохинолин, дифенилфосфорилазид, пропанфосфоновый ангидрид или гексафторфосфат бензотриазол-1-илокси-трис-(диметиламино)фосфония.
Реакцию соединения (V) или его реакционноспособного производного с соединением (IV) проводят в растворителе или без растворителя. Растворитель, который необходимо выбирать в соответствии, например с типом используемого исходного соединения, включает, например ароматические углеводороды (например, бензол, толуол и ксилол), простые эфиры (например, диэтиловый эфир, тетрагидрофуран и диоксан), галогенированные углеводороды (например, дихлорметан и хлороформ), спирты (например, этанол и изопропиловый спирт), этилацетат, ацетон, ацетонитрил, диметилформамид, 1,3-диметил-2-имидазолидинон, диметилсульфоксид, этиленгликоль и воду. Эти растворители можно использовать по одному или в виде смеси двух или более растворителей. Обычно, если необходимо, реакцию проводят в присутствии основания, эти основания включают, например гидроксид щелочного металла (например, гидроксид натрия или гидроксид калия), карбонат щелочного металла (например, карбонат натрия или карбонат калия), гидрокарбонат щелочного металла (например, гидрокарбонат натрия или гидрокарбонат калия), или органические основания (например, триэтиламин, трибутиламин, диизопропилэтиламин или N-метилморфолин), но избыточное количество соединения (IV) может действовать вместо основания. Температуру реакции можно варьировать в зависимости, например от типа используемых исходных соединений, но обычно она составляет величину в диапазоне примерно от -30 до 200oC, предпочтительно в диапазоне от -10 до 150oC.
Исходное соединение (V) данной стадии можно получить из 3,4-дигидро-1(2H)-нафталинонов по существу известным способом по методу, раскрытому в J. Med. Chem., 17, 273 (1974); первой публикации патента Японии (Kokai) N 54-24861 (Chem. Abstr., 91, 56702b (1979)); Pharmazie, 41, 835 (1986); Yakugaku Zasshi, 110, 561 и 922 (1990); Heterocycles, 34, 1303 (1992); Tetra hedron, 48, 4027 (1992), или способом, раскрытым в справочных примерах с 1 по 3, или модифицированным способом.
С другой стороны, исходные 3,4-дигидро-1-(2H)-нафталиноны для получения соединения (V) могут быть коммерчески доступными соединениями или их можно получить по существу известным способом, например по методу, раскрытому в J. Chem. , Soc. , 1961, 4425; J. Org. Chem., 26, 1109 (1961); J. Heterocycl. Chem. , 10, 31 (1973); патенте США 46022791; Chem. Pharm. Bull, 25, 632 (1977); там же, 31, 2006 (1983); J. Med. Chem., 17, 273 (1974); там же, 50, 4933 (1985); первой публикации патента Японии (Kokai) N 6-87746; Synth. Commun., 21, 981 (1991); или Tetrahedron Lett., 33, 5499 (1992), или модифицированным способом.
Другое исходное соединение (IV) этой стадии может быть коммерчески доступным соединением или его можно получить по существу известным способом, например по методу, раскрытому в Synlett, 1995, 55, или модифицированным способом.
Соединение формулы (VI), в котором p равно 2, также получают способами параграфов с (1) по (3) справочных примеров 9-12, описанных здесь далее, или модифицированными способами.
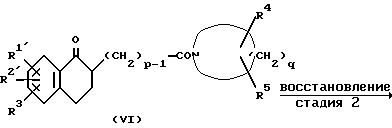
Стадия 2
Данную стадию осуществляют, проводя обработку соединения (VI) восстановителем, подходящим для восстановления карбонильной группы кетона в спиртовую гидроксильную группу и восстановления карбонильной группы амида в метиленовую группу, в подходящем растворителе. Восстановитель включает, например гидрид или гидридный комплекс алюминия, такой как литийалюминийгидрид, диизобутилалюминийгидрид, натрий-бис-(2-метоксиэтокси)алюминийгидрид или гидрид алюминия, боргидрид натрия или комбинацию боргидрида натрия с кислотой Льюиса, такой как безводный хлорид алюминия, хлорид кобальта (П) или комплекс фторида бора-диэтилового эфира, водородный комплекс бора, такой как ацетоксиборгидрид натрия или трифторацетоксиборгидрид натрия, бораны, такие как диборан, и хлорид триэтилсиланцинка. Растворитель, который необходимо выбирать в соответствии, например с типом используемого восстановителя, включает, например простые эфиры (например, диэтиловый эфир, тетрагидрофуран, диметоксиэтан, диоксан и диглим), ароматические углеводороды (например, бензол и толуол), галогенированные углеводороды (например, дихлорметан и хлороформ), спирты (например, метанол и этанол), уксусную кислоту и пиридин. Эти растворители можно использовать по одному или в виде смеси двух или более растворителей. Температуру реакции можно варьировать в зависимости, например от типа используемого восстановителя, но обычно она составляет величину в диапазоне примерно от -10 до 130oC.
Реакцию восстановления можно проводить в инертной атмосфере, например под азотом или аргоном. Если на этой стадии используют соединение (VI), где R4 и R5 вместе образуют оксогруппу, то предпочтительно до проведения реакции защитить ее обычной защитной группой, такой как ацеталь (например, диметилацеталь, диэтилацеталь или этиленацеталь) или оксим. Если используют соединение (VI), где R1' и/или R2' являются защищенной формильной группой и/или защищенной карбоксильной группой, то после восстановления защитные группы в продукте удаляют обычным способом, получая соединение (II). Полученное на этой стадии соединение (II) можно, не выделяя и не чистя, использовать в следующей реакции дегидратации, как указано выше. Если в качестве восстановителя используют борсодержащий агент, такой как бораны, то соединение (II) получают в виде комплекса с борсодержащим восстановителем или продуктом его разложения, который сам по себе также можно применять в реакции дегидратации.
Способ (в)
Соединение формулы (I) получают также посредством реакции соединения формулы (III):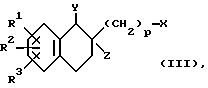
где R1, R2, R3 и p такие, как определено выше, X и Y одинаковые или разные, и каждый представляет собой реакционноспособный эфирный остаток от спирта, и Z является атомом водорода, или Y и Z могут вместе образовывать связь, с соединением формулы (IV);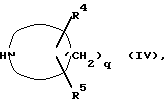
где R4, R5 и q такие, как определено выше.
Реакционноспособный эфирный остаток от спирта X и Y формулы (III) включает, например атом галогена (например, хлор, бром или йод), низшую алкилсульфонилоксигруппу (например, метансульфонилокси- или этансульфонилоксигруппу) и арилсульфонилоксигруппу (например, бензолсульфонилокси-, паратолуолсульфонилокси- или метанитробензолсульфонилоксигруппу).
Реакцию соединения (III) и соединения (IV) обычно проводят в присутствии основания в подходящем растворителе. Основание включает, например карбонат щелочного металла (например, карбонат натрия или карбонат калия), органическое основание (например, триэтиламин, трибутиламин, диизопропилэтиламин или N-метилморфолин), алкоголят щелочного металла (например, метилат натрия или этилат натрия) и гидрид щелочного металла (например, гидрид натрия или гидрид калия), но избыточное количество соединения (IV) может действовать вместо основания. Растворитель, который необходимо выбирать в соответствии, например с типом используемых исходных соединений, включает, например ароматические углеводороды (например, бензол и толуол), простые эфиры (например, тетрагидрофуран, диоксан и диглим), галогенированные углеводороды (например, дихлорметан и хлороформ), кетоны (например, ацетон и этилметилкетон), ацетонитрил, спирты (например, метанол, этанол и изопропиловый спирт), диметилформамид и 1,3-диметил-2-имидазолидинон. Эти растворители можно использовать по одному или в виде смеси двух или более растворителей. Температуру реакции можно варьировать в зависимости, например от типа используемых исходных соединений, и обычно она составляет величину в диапазоне примерно от 30 до 150oC, предпочтительно в диапазоне от 80 до 120oC.
Исходное соединение (III) получает по существу известным способом, например по методу справочных примеров 10 и 11, описанных здесь далее, или модифицированным способом.
Когда соединение (I), в котором R1 и/или R2 является C1- C3-алкоксикарбонильной группой, получают описанными выше способами (а) или (в), указанное соединение (I) можно превратить в соединение (I), в котором R1 и/или R2 является формильной группой или гидроксиметильной группой, посредством восстановления обычным образом, или в соединение (I), в котором R1 и/или R2 является карбоксильной группой, посредством обычного гидролиза. Конверсия в гидроксиметильную группу проиллюстрирована в приведенном далее примере 71. Конверсия в карбоксильную группу проиллюстрирована в приведенном далее Примере 73. Конверсию в формильную группу проводят путем восстановления при температуре примерно от -78oC до -50oC с использованием в качестве восстановителя гидрида или водородного комплекса алюминия (например, литийалюминийгидрида, изобутилалюминийгидрида, диизобутилалюминийгидрида или натрий-бис-(2-метоксиэтокси)алюминийгидрида).
Полученное в описанных выше процессах соединение (I) можно выделить и очистить такими обычными способами как хроматография, перекристаллизация или переосаждение. Соединение (I) получают либо в виде свободного основания, либо в виде соли присоединения кислоты, в соответствии с типом исходных соединений, условий реакции и др. Соль присоединения кислоты переводят в форму свободного основания обычными способами, например, обрабатывая ее основанием (например, карбонатом щелочного металла или гидроксидом щелочного металла). С другой стороны, свободное основание можно превратить в соль присоединения кислоты обычным образом посредством обработки различными кислотами.
Кроме того, соединение (I) можно превратить в N-оксидное производное по его циклической аминной части посредством окисления в условиях, обычных для N-окисления. Реакцию N-окисления осуществляют посредством взаимодействия соединения (I) с окислителем в подходящем растворителе; окислитель включает, например перекись водорода и органические надкислоты, такие как надуксусная кислота, надбензойная кислота, метахлорнадбензойная кислота и мононадфталевая кислота. Окислитель обычно используют в количестве примерно от 0,9 до 2 эквивалентов относительно количества соединения (I). Растворитель, который необходимо выбирать в соответствии, например с типом используемого окислителя, включает, например воду, уксусную кислоту, спирты (например, метанол и этанол), кетоны (например, ацетон), эфиры (например, диэтиловый эфир и диоксан) и галогенированные углеводороды (например, дихлорметан и хлороформ). Температуру реакции можно варьировать в зависимости, например от типов используемых окислителей, но обычно она составляет величину в диапазоне примерно от -30 до 100oC, предпочтительно в диапазоне примерно от -20 до 30oC.
В следующих далее фармакологических экспериментах на примерах соединений настоящего изобретения и инаперизон гидрохлорида (inaperisone hydrochloride, здесь далее обычно обозначают как "Соединение A"), который, как известно, является агентом центрального действия для лечения частого мочеиспускания и недержания мочи, проиллюстрирована фармакологическая активность соединений настоящего изобретения.
Эксперимент 1. Ингибирующее действие на периодические сокращения мочевого пузыря (рефлекс мочеиспускания)
Эксперимент проводят согласно способу Maggi, C.A. и Meli, A. (J. Pharmacol. Methods, 10, 79 (1983)). Обычно принимают, что периодические сокращения мочевого пузыря, вызываемые введением в мочевой пузырь соляного раствора, опосредованы через проводящий путь рефлекса мочеиспускания подобно естественному мочеиспусканию.
Для эксперимента используют самок крыс Std-Wistar весом 160-190 г в группах по 3-4 животных. Под уретановой анестезией (1 г/кг, подкожно) после серединной лапаротомии перевязывают оба мочеточника и отрезают со стороны почки. В мочевой пузырь через внешнее отверстие мочеиспускательного канала вставляют канюлю, соединенную со шприцем и датчиком давления, и перевязывают вокруг проксимальной уретры. Животное после операции оставляют приблизительно на 30 минут. При помощи шприца медленно заполняют мочевой пузырь теплым соляным раствором (0,4-1 мл) до тех пор, пока не индуцируют периодические сокращения. Изменение внутрипузырного давления регистрируют на самописце при помощи датчика давления. После того, как периодические сокращения станут постоянными, через канюлю, предназначенную для интрадуоденального введения, вводят исследуемое соединение, растворенное или суспендированное в 0.5% водном растворе трагаканта. Действие каждого соединения на частоту сокращений проверяют каждые 15 минут в течение 2 часов после введения.
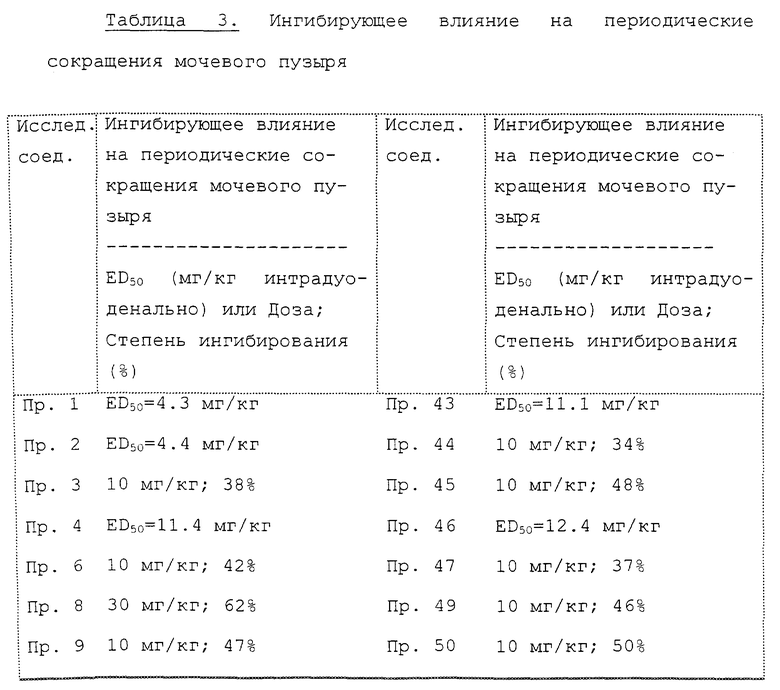
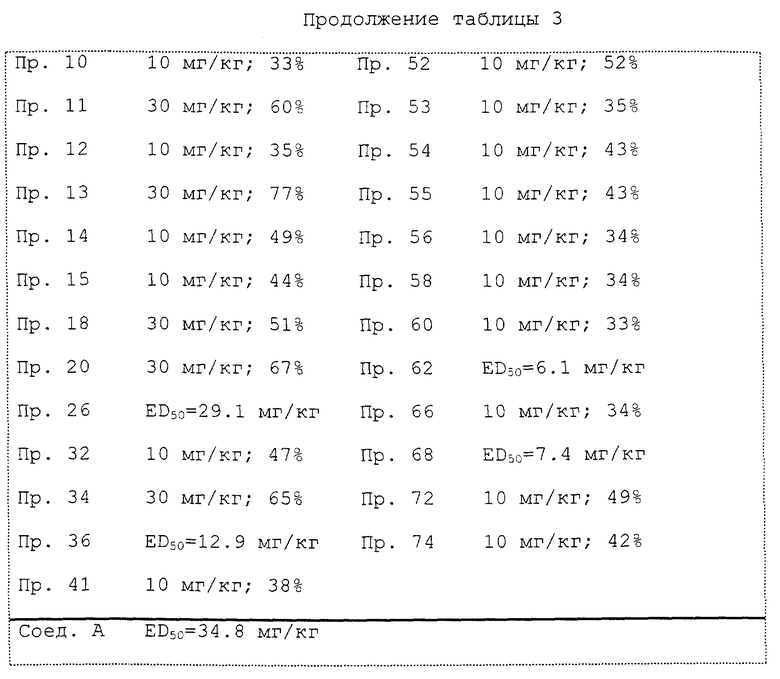
В табл. 3 показано ингибирующее действие на частоту сокращений (два часа) при указанной дозе каждого исследуемого соединения или доза исследуемого соединения, необходимая для 50% ингибирования частоты сокращений (величина ED50). Степень ингибирования частоты сокращений (два часа) выражают средней величиной степени ингибирования при измерениях через каждые 15 минут в течение двух часов после введения. Величину ED50 определяет способом Litchfield-Wilcoxon [J. Pharmacol. Exp. Ther, 96, 99 (1949)] на основании степени ингибирования частоты сокращений (два часа) при каждой дозе.
Как ясно из табл. 3, все исследованные в данном эксперименте соединения настоящего изобретения демонстрируют заметное ингибирующее действие на частоту периодических сокращений мочевого пузыря. Особенно существенно действие соединений примеров 1 и 2, примерно в 8 раз больше, чем действие инаперизон гидрохлорида (Соединения A). Кроме того действие соединений примеров 62, 68, 4, 36 и 43 приблизительно в 5.7, 4.7, 3.3 и 3 раза, соответственно, больше по сравнению с действием инаперизон гидрохлорида.
Кроме этого соединения примеров 2, 3, 4, 12, 20, 26, 36, 41, 43-47, 49, 50, 56, 62, 68 и 72 демонстрируют не только ингибирование частоты сокращений, но также подавляют амплитуду сокращений.
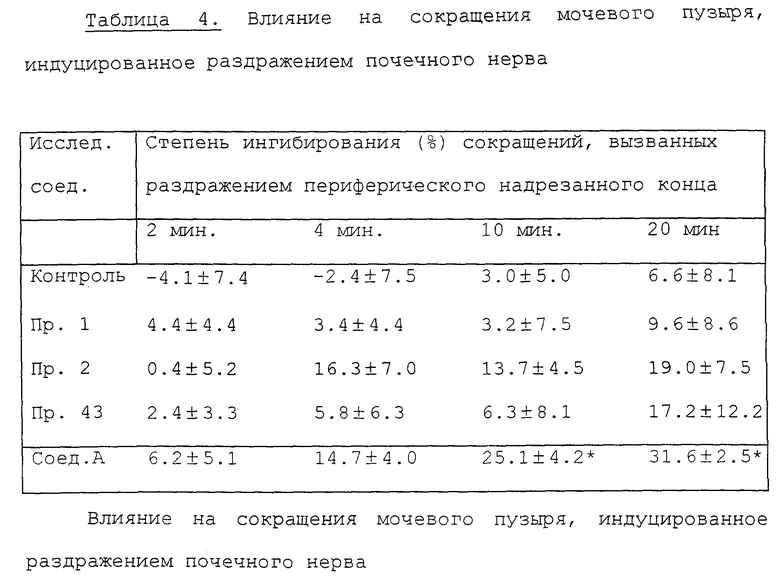
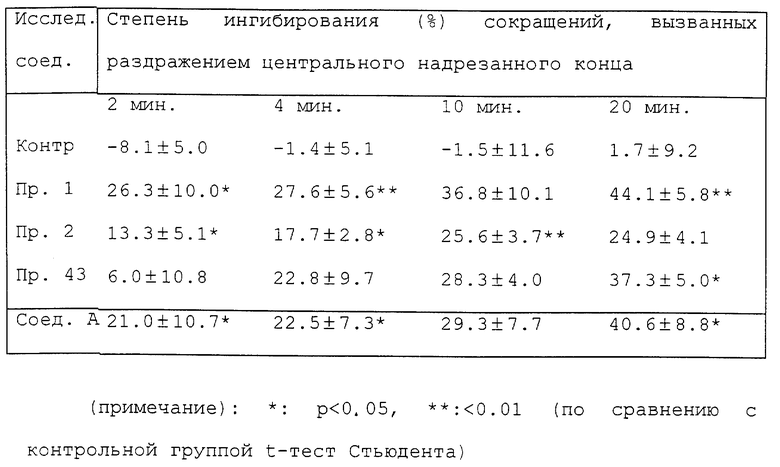
Эксперимент 2. Действие на сокращения мочевого пузыря, вызванные стимулированием почечного нерва (pelvic nerve).
Для эксперимента используют самцов крыс Std-Wistar весом 250-350 г. Под уретановой анестезией (1 г/кг, внутрибрюшинно) после серединной лапаротомии перевязывают оба мочеточника и отрезают со стороны почки. Раздражая периферический конец почечного нерва, разрезают его с одной стороны. Раздражая центральный разрезанный конец этого нерва, разрезают одну сторону почечного нерва, как при раздражении периферического конца после того, как отрезаны оба подчревных нерва. Затем выделяют мочевой пузырь, в него через небольшой разрез на верхушке свода вставляют канюлю, соединенную со шприцем и датчиком давления, и после этого перевязывают проксимальную уретру. Через пятнадцать минут после операции при помощи шприца в мочевой пузырь вводят теплый соляной раствор в количестве (0.1-0.2 мл), достаточно малом, чтобы не вызвать периодическое сокращение. Изменение внутрипузырного давления регистрируют на самописце при помощи датчика давления. Почечный нерв раздражают со стороны надрезанного периферического или центрального конца при помощи платиновой электродной пары импульсом продолжительностью 1 мсек, с напряжением 3 В и частотой 10 Гц (для периферического) или напряжением 5 В и частотой 20 Гц (для центрального) в течение 5 секунд каждые 2 минуты. Растворенные в дистиллированной воде исследуемые соединения вводят через канюлю, вставленную в шейную вену при дозе 5 мг/кг.
В табл. 4 показана степень ингибирования сокращений при использовании исследуемых соединений через 2, 4, 10 и 20 минут после их введения, которую рассчитывают, сравнивая с контрольной реакцией до введения исследуемого соединения, и сравнивают с соответствующей величиной для группы животных, обработанных наполнителем - дистиллированной водой (непарный t-тест). Каждое значение в таблице соответствует среднему значению ± стандартная ошибка средних величин для 3-4 животных.
Как ясно из табл. 4, влияние соединений примеров 1, 2 и 43 на сокращения мочевого пузыря, вызванные раздражением центрального надрезного конца является более сильным, чем влияние на сокращения, вызванные раздражением периферического надрезанного конца. Результат предполагает, что влияние соединений данного изобретения может быть опосредовано главным образом через центральную нервную систему.
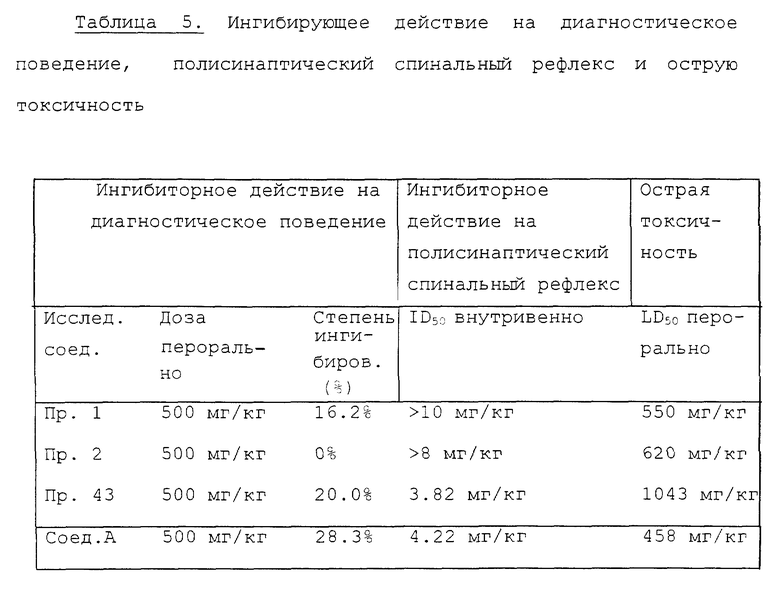
Эксперимент 3. Ингибиторное действие на диагностическое поведение
Для эксперимента используют самцов крыс Std-Wistar весом 150-200 г в группах по 5 животных. Через час после орального введения исследуемых соединений, суспендированных в 0.5% водном растворе трагаканта, животных индивидуально помещают в клетку для исследования (23x35x30 см) на приборе "Animex activity meter" (Farad Co., Sweden). Сразу после этого начинают подсчет двигательной активности и продолжают в течение трех минут. Определяют среднее значение диагностического повеления (подсчет/3 минуты) в группах, обработанных исследуемыми соединениями, и затем рассчитывают ингибиторное действие каждого исследованного соединения по сравнению с контрольной группой (группа, обработанная 0,5% водным раствором трагаканта). Результаты показаны в табл. 5 вместе с результатами экспериментов 4 и 5.
Эксперимент 4. Ингибиторное действие на полисинаптический спинальный рефлекс.
Эксперимент проводят согласно способу ltoh и др. [Japan (period) J. Pharmacol, 32, 1125 (1982)] используют самцов крыс Std-Wistar весом 250-350 г в группах по 4-6 животных. Под анестезией комбинированной внутрибрюшинной инъекции уретана (400 мг/кг) и альфа-хлоралозы (50 мг/кг) крысу, фиксированную в стереотаксическом аппарате, зажимают в позвоночнике и большеберцовой кости. В левую икроножную мышцу вводят концентрический игловой электрод и центральный конец ипсилатерального общего перонеального нерва с той же стороны предельно (supramaximally) раздражают при помощи электрического стимулятора (прямоугольный импульс, 0.1 мсек, 0.1 Гц). Исследуемые соединения, растворенные в дистиллированной воде, вводят через канюлю, вставленную в правую бедренную вену, и периодически снимают вызванную электромиограмму (2, 5, 10, 20 и 30 минут после инъекции). Ингибирующее влияние исследуемых соединений на амплитуду вызванной электромиограммы выражают в процентах от амплитуды до введения и затем рассчитывают величину ID50 (доза, необходимая для 50% ингибирования амплитуды) согласно способу Litchfield-Wilcoxon на основании максимальной степени ингибирования при каждой дозе. Результаты представлены в табл. 5 вместе с результатами экспериментов 3 и 5.
Эксперимент 5. Острая токсичность.
Используют самцов мышей ddY весом 18-25 г в группах по 5-15 животных.
Исследуемые соединения, суспендированные в 0.5% водном растворе трагаканта, вводят орально и наблюдают смертность в течение 7 дней после введения. Рассчитывают LD50 (50% летальная доза) согласно способу Litchfield-Wilcoxon. Результаты представлены в табл. 5 вместе с результатами экспериментов 3 и 4.
Как ясно из табл. 5, ингибиторные эффекты соединений примеров 1 и 2 на диагностическое поведение менее сильны, чем эффект Соединения А, а ингибиторный эффект соединения примера 43 на диагностическое поведение почти равен эффекту Соединения A. В эксперименте по изучению спинального рефлекса ингибиторный эффект соединения примера 43 почти равен эффекту Соединения A, а эффекты соединений примеров 1 и 2 значительно слабее, чем эффект Соединения A. Острая токсичность соединения примера 43 примерно в два раза ниже, чем соединения A, а острая токсичность соединений примеров 1 и 2 также ниже, чем у Соединения A.
Судя по всем этим результатам и результатам эксперимента 1, ингибиторные эффекты соединений Примеров 1, 2 и 43 на периодические сокращения мочевого пузыря (рефлекс мочеиспускания) предельно освобождены от таких побочных воздействий как центральная депрессия, ингибирование спинального рефлекса и токсичность по сравнению с соответствующими характеристиками Соединения A.
Как ясно из приведенных выше результатов, соединение формулы (I), его фармацевтически пригодная соль присоединения кислоты и N-оксидное производное (здесь далее обозначены как "соединение настоящего изобретения") демонстрируют сильный ингибиторный эффект на рефлекс мочеиспускания и проявляют низкую токсичность, а следовательно, эти соединения являются полезными в качестве агентов для лечения частого мочеиспускания и недержания мочи, особенно для лекарственных средств, предназначенных для лечения различных заболеваний, связанных со снижением объемной способности мочевого пузыря (например, нестабильный мочевой пузырь, нейрогенный мочевой пузырь, хронический цистит, хронический простатит и нервная поллакиурия), вызванным различными факторами.
Соединения настоящего изобретения можно вводить орально, парентерально или ректально, но предпочтителен оральный прием. Дозу соединений настоящего изобретения можно варьировать в соответствии, например с типами соединений, способом приема, а также симптомами и возрастом пациентов, но обычно доза составляет количество в диапазоне 0.1-20 мг/кг/день, предпочтительно 0.4-10 мг/кг/день, при приеме один раз в день или несколькими порциями.
Соединения настоящего изобретения обычно вводят в виде фармацевтических композиций, которые готовят, смешивая активные соединения с фармацевтически пригодным носителем или разбавителем. Фармацевтически пригодным носителем или разбавителем может быть любое общеизвестное вещество, которое обычно используют в фармацевтике и которое не реагирует с соединениями настоящего изобретения. Подходящими примерами фармацевтически пригодных носителей или разбавителей являются, например лактоза, инозит, глюкоза, маннит, декстран, крахмал, частично желатинированный крахмал, сахароза, алюмосиликат магния, синтетический силикат алюминия, кристаллическая целлюлоза, натрийкарбоксиметилцеллюлоза, гидроксипропилкрахмал, кальцийкарбоксиметилцеллюлоза, ионообменная смола, метилцеллюлоза, желатин, гуммиарабик, гидроксипропилцеллюлоза, слабозамещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон, поливиниловый спирт, альгиновая кислота, альгинат натрия, легкая безводная кремниевая кислота, стеарат магния, тальк, карбоксивиниловый полимер, оксид титана, эфиры сорбитановой жирной кислоты, лаурилсульфат натрия, глицерин, эфиры глицериновой жирной кислоты, очищенный ланолин, глицерожелатин, полисорбат, макрогол, растительные масла, воск, жидкий парафин, белый вазелин, нейонные поверхностно-активные вещества, пропиленгликоль и вода.
Фармацевтические композиции находятся, например в виде таблеток, капсул, гранул, порошков, сиропов, суспензий, суппозиториев, припарок и препаратов для инъекции. Эти препаративные формы можно приготовить общеизвестным способом. При получении жидкостей для медицинского применения соединение настоявшего изобретения можно растворить или суспендировать в воде или другом подходящем растворителе. На таблетки и гранулы можно нанести покрытие, используя общеизвестный способ.
Эти фармацевтические композиции могут содержать соединение настоящего изобретения в количестве более 0.1%, предпочтительно 1-70%. Эти фармацевтические композиции могут также содержать другие терапевтически эффективные соединения.
Лучший способ осуществления данного изобретения
Настоящее изобретение более подробно проиллюстрировано следующими примерами и справочными примерами, которые нельзя считать ограничительными.
Идентификацию соединений проводили посредством элементного анализа, масс-спектроскопии, ИК-спектроскопии, ЯМР-спектроскопии и др.
Для облегчения описания в приведенных далее примерах и справочных примерах могут быть использованы следующие сокращения:
Me: Метильная группа
Et: Этильная группа
Ph: Фенильная группа
Fu: Фумаровая кислота
Ma: Малеиновая кислота
OX: Щавелевая кислота
A: Этанол
AC: Ацетон
AN: Ацетонитрил
DE: Диэтиловый эфир
EM: Этилметилкетон
Ip: Изопропиловый спирт
Пример 1. Получение 1-[2-(5-фтор-3,4-дигидро-2- нафталинил)этил]пирролидина.
К раствору 5-фтор-1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил]-1-нафталинола (20.0 г) в толуоле (250 мл) добавляют моногидрат паратолуолсульфокислоты (16.2 г) и смесь кипятят с обратным холодильником в течение ночи. После охлаждения реакционный раствор последовательно промывают 1N водным раствором гидроксида натрия, водой, насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол:этилацетат = 5:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (12,6 г) в виде маслянистого продукта.
Полученное описанным выше способом свободное основание растворяют в 30% растворе хлористого водорода в этаноле (100 мл) и смесь концентрируют при пониженном давлении, удаляя этанол. К остатку добавляют диэтиловый эфир, а осажденные кристаллы собирают фильтрацией и перекристаллизовывают из ацетонитрила, получая гидрохлорид требуемого соединения (4,5 г), т.пл. 194-198oC.
Пример 2. Получение 1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил)этил]пирролидина.
1,2,3,4-тетрагидро-6,7-диметил-2-[2-(1-пирролидинил)этил] -1-нафталинол (24,0 г) растворяют в 30% растворе хлористого водорода в этаноле (240 мл) и смесь кипятят с обратным холодильником в течение одного часа. Реакционный раствор концентрируют при пониженном давлении и остаток растворяют в воде. Смесь подщелачивают карбонатом калия и экстрагируют этилацетатом, экстракт промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ: метанол = 10:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (14.5 г) в виде маслянистого продукта.
Полученное описанным выше способом свободное основание обрабатывают 30% раствором хлористого водорода в этаноле таким же образом, как описано в примере 1, получая его гидрохлорид, который затем перекристаллизовывают из этанола, получая гидрохлорид требуемого соединения, т.пл. 212-214oC.
Спектр 1H-ЯМР (200 МГц, (CD3)2SO, δ м.д.): 1.75-2.08 (м, 4H), 2.10-2.27 (м, 2H), 2.14 (с, 6H), 2.53-2.77 (м, 4H), 2.83-3.15 (широкий с, 2H), 3.15-3.39 (м, 2H), 3.39-3.68 (широкий с, 2H), 6.24 (с, 1H), 6.79 (с, 1H), 6.88 (с, 1H), 10.85 (широкий с, 1H).
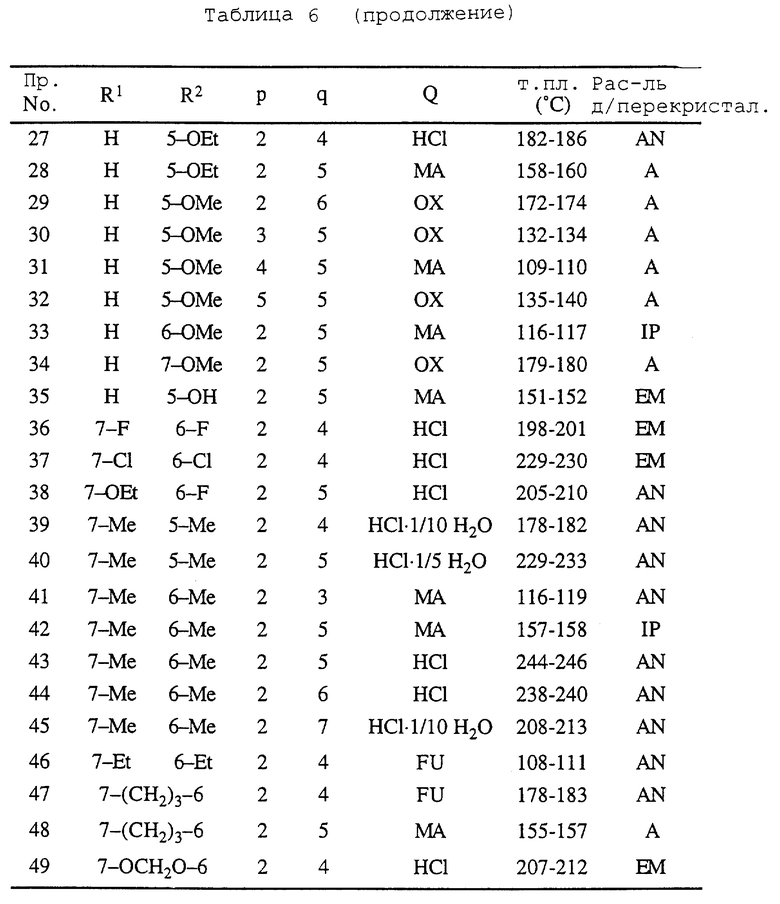
Примеры 3-41. Соответствующие 1,2,3,4-тетрагидро-2-[ω--(1-циклический амино)алкил] -1-нафталинолы обрабатывают таким же образом, как описано в примере 1 или 2, получая соединения, перечисленные в табл. 6. 1,2,3,4-Тетрагидро-2-[ω-(1-циклический амино)алкил]-1-нафталинолы получают, обрабатывая соответствующие 1-[ω-(1,2,3,4-тетрагидро-1-оксо-2-нафталинил)алканоил] циклические амины, которые получают в справочных примерах 4-7, таким же образом, как описано в справочном примере 8.
Пример 50. Получение 1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил)этил]-2,5-диметилпирролидина.
1-[(1,2,3,4-Тетрагидро-6,7-диметил-1-оксо-2-нафталинил)ацетил]-2,5-диметилпирролидин обрабатывают таким же образом, как описано в справочном примере 8, получая 1,2,3,4-тетрагидро-6,7-диметил-2-[2-(2,5-диметил-1-пирролидинил)этил] -1-нафталинол в виде маслянистого продукта, который затем обрабатывают таким же образом, как в примере 2, получая гидрохлорид требуемого соединения, т.пл. 227-229oC (перекристаллизован из ацетонитрила).
Пример 51. Получение 1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил)этил]-2-(метоксиметил)пирролидина.
1-[(1,2,3,4-Тетрагидро-6,7-диметил-1-оксо-2-нафталинил)ацетил] -2-(метоксиметил)пирролидин обрабатывают таким же образом, как в справочном примере 8, получая 1,2,3,4-тетрагидро-6,7-диметил-2-[2-(2-(метоксиметил)-1-пирролидинил)этил] -1-нафталинол в виде маслянистого продукта, который затем обрабатывают таким же образом, как в примере 1, получая фумарат требуемого соединения, т.пл. 166-168oC (перекристаллизован из ацетонитрила).
Пример 52. Получение 1-[2-(2-(5,7-дифтор-3,4-дигидро-2-нафталинил)этил] -2-(метоксиметил)пирролидина.
К комплексу борана с 5,7-дифтор-1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил]-1-нафталинолом, получение которого описано в следующем далее справочном примере 9, добавляют моногидрат паратолуолсульфокислоты (4.1 г) и толуол (54 мл) и смесь кипятят с обратным холодильником в течение трех часов. После охлаждения реакционный раствор промывают 10% водным раствором гидроксида натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ:метанол = 50:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (2.0 г) в виде маслянистого продукта. Полученный таким образом продукт обрабатывают обычным образом щавелевой кислотой в этаноле, получая соответствующий оксалат, который затем перекристаллизовывают из ацетонитрила, получая оксалат требуемого соединения, т.пл. 146-149oC.
Комплекс борана с 5-фтор-1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил] -1-нафталинолом, получение которого описано в следующем далее справочном примере 12, обрабатывают таким же образом, как в приведенном выше примере, получая соединение примера 1.
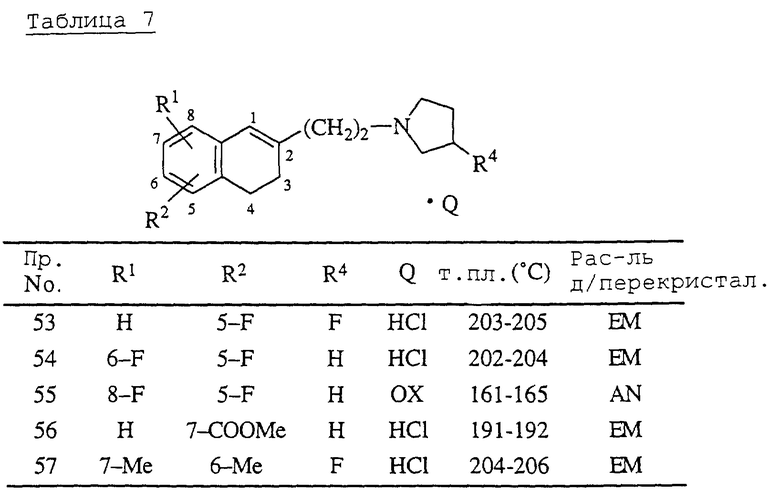
Примеры 53-57.
Комплексы борана с соответствующими 1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил] -1-нафталинолами, которые получают таким же способом, как в описанном ниже справочном примере 9, обрабатывают таким же образом, как в примере 52, получая соединения, перечисленные в табл. 7.
Пример 58. Получение 1-[2-(3,4-дигидро-7-метокси-2-нафталинил)этил] пирролидина.
К 3,4-дигидро-7-метокси-2-нафталинэтил метансульфонату, получение которого описано в следующем ниже справочном примере 10, добавляют ацетонитрил (70 мл) и пирролидин (3.5 г) и смесь кипятят с обратным холодильником в течение шести часов. Смесь концентрируют при пониженном давлении, удаляя растворитель, и остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ:метанол - 50:1). Фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (2.5 г) в виде маслянистого продукта.
Полученное таким образом свободное основание обрабатывают 30% раствором хлористого водорода в этаноле, таким же образом, как в примере 1, получая соответствующий гидрохлорид, который затем перекристаллизовывают из этилметилкетона, получая гидрохлорид требуемого соединения, т.пл. 161-163oC.
Примеры 59-61.
Соответствующие 3,4-дигидро-2-нафталинэтил метансульфонаты, которые получают таким же способом, как в описанном ниже справочном примере 10, обрабатывают таким же образом, как в примере 58, получая следующие соединения.
(Пример 59).
Гидрохлорид 1-[2-(3,4-дигидро-2-нафталинил)этил] пирролидина, т. пл. 205-207oC (перекристаллизован из смеси этанолдиэтиловый эфир).
(Пример 60). Гидрохлорид 1-[2-(3,4-дигидро-2-нафталинил)этил] -3-гидроксипирролидина, т.пл. 126-128oC (перекристаллизован из этилметилкетона).
(Пример 61).
Гидрохлорид 3-фтор-1-[2-(3,4-дигидро-6,7-диметокси-2-нафталинил)этил] пирролидина, т.пл. 202-205oC (перекристаллизован из этилметилкетона).
Пример 62. Получение 1-[2-(3,4-дигидро-6,7-диметокси-2-нафталинил)этил] пирролидина.
К 1,2,3, 4-тетрагидро-2-(2-метансульфонилоксиэтил)-6,7-диметокси-1-нафталинил метансульфонату, получение которого описано в следующем ниже справочном примере 11, добавляют ацетонитрил (72 мл) и пирролидин (4.5 г) и смесь кипятят с обратным холодильником в течение шести часов. Смесь концентрируют при пониженном давлении, удаляя растворитель, и остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ:метанол = 50:1). Фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (3.5) г в виде маслянистого продукта.
Полученное таким образом свободное основание обрабатывают 30% раствором хлористого водорода в этаноле, таким же образом, как в примере 1, получая соответствующий гидрохлорид, который затем перекристаллизовывают из этилметилкетона, получая гидрохлорид требуемого соединения, т.пл. 204-206oC.
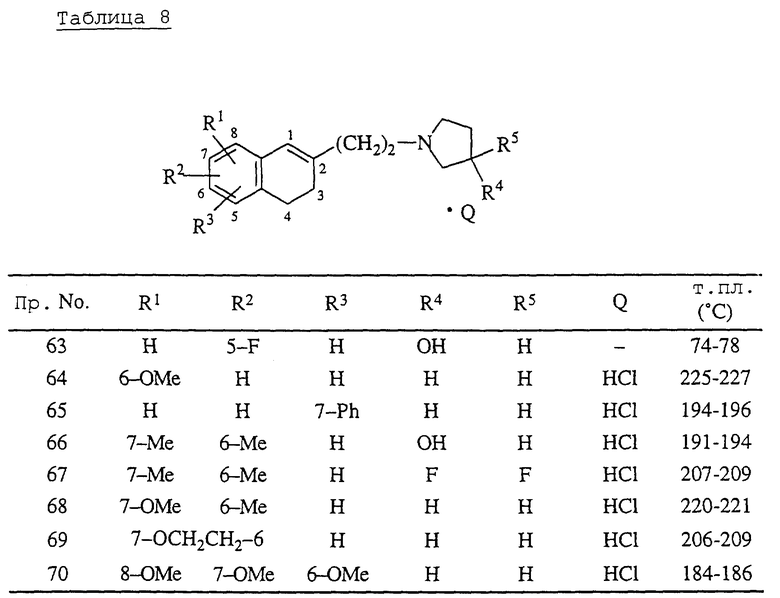
Примеры 63-70.
Соответствующие 1,2,3,4-тетрагидро-2-(2-метансульфонилоксиэтил)-1-нафталинил метансульфонаты, которые получают таким же способом, как в описанном ниже справочном примере 11, обрабатывают таким же образом, как в примере 62, получая соединения, перечисленные в табл. 8. Растворителем для перекристаллизации соединения примера 63 является диэтиловый эфир, а для соединений всех остальных примеров этилметилкетон.
Пример 71. Получение 1-[2-(3,4-дигидро-7-гидроксиметил-2-нафталинил)этил]пирролидина.
1-[2-(3,4-Дигидро-7-гидроксиметил-2-нафталинил)этил] пирролидин (1.0 г) растворяют в тетрагидрофуране (20 мл) и при охлаждении на льду добавляют туда по капле 1.5 М раствор диизобутилалюминийгидрида в толуоле (7.0 мл), смесь перемешивают в течение одного часа. К реакционному раствору добавляют по капле воду для разложения избыточного количества восстановителя и смесь экстрагируют этилацетатом. Экстракт сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ: метанол = 30: 1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (0.8 г) в виде маслянистого продукта.
Полученное описанным выше способом свободное основание обрабатывают 30% раствором хлористого водорода в этаноле таким же образом, как в примере 1, получая соответствующий гидрохлорид, который затем перекристаллизовывают из этилметилкетона, получая гидрохлорид требуемого соединения, т.пл. 193-194oC.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.72 (м. 1H), 1.95-2.20 (м. 4H), 2.27 (т, 2H, J= 8.5), 2.72-2.91 (м, 6H), 3.15-3.29 (м, 2H), 3.75-3.94 (м, 2H), 4.55 (д, 2H, J=5), 6.28 (с, 1H), 7.01 (с, 1H), 7.05-7.17 (м, 2H), 12.65 (м, 1H).
Пример 72. Получение 1-[2-(3,4-дигидро-6-гидроксиметил-7-метил-2-нафталинил)этил]пирролидина.
1-[2-(3,4-Дигидро-6-метоксикарбонил-7-метил-2-нафталинил)этил]пирролилин обрабатывают таким же образом, как в примере 71, получая требуемое соединение, т.пл. 76-77oC (перекристаллизован из смеси диэтиловый эфир-гексан).
Пример 73. Получение 1-[2-(7-карбокси-3,4-дигидро-2-нафталинил)этил]пирролидина.
1-[2-(3,4-Дигидро-7-метоксикарбонил-2-нафталинил)этил]пирролидин (1.0 г) растворяют в этаноле (5 мл) и добавляют туда 1N водный раствор гидроксида натрия (5.2 мл), смесь перемешивают при 25oC в течение шести часов. Смесь концентрируют при пониженном давлении, удаляя растворитель, и остаток подкисляют 10% соляной кислотой. Осажденные кристаллы собирают фильтрацией и проводят обессоливание, используя СНР-20Р [производства Mitsubishi Kasei Corporation, высокопористая полистирольная смола, 75-150 μM] (элюент: вода, затем ацетонитрил). Элюаты концентрируют при пониженном давлении и остаток перекристаллизовывают из смеси этанол - этилметилкетон, получая требуемое соединение, т.пл. 202-203oC.
Спектр 1H-ЯМР (200 МГц, (CD3)2SO, δ м.д.): 1.80-2.10 (м, 4H), 2.28 (т, 2H, J=7), 2.62 (т, 2H, J=8), 2.85 (т, 2H, J=8), 2.90-3.12 (м, 2H), 3,32 (т, 2H, J= 7), 3.41-3.65 (м, 2H), 6.44 (с, 1H), 7.25 (д, 1H, J=8), 7.60 (д, 1H, J=l), 7.70 (дд, 1H, J-8,1), 10.56 (м, 1H), 12.80 (м, 1H).
Пример 74. Получение 1-[2-(6-карбокси-3,4-дигидро-7-метил-2-нафталинил)этил]пирролидина.
1-[2-(3,4-Дигидро-6-метоксикарбонил-7-метил-2-нафталинил)этил)пирролидин обрабатывают таким же образом, как в примере 73, получая гидрохлорид требуемого соединения, т. пл. 258-259oC (перекристаллизован из смеси этанол - этилметилкетон).
Применяемые в приведенных выше примерах исходные соединения получают следующим образом.
Справочный пример 1. Получение 1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2-нафтилуксусной кислоты.
(1) Смесь 37% формалина (18 г) и диметиламингидрохлорида (18 г) перемешивают при 25oC в течение 30 минут и при 70oC в течение 30 минут. Температуру реакции повышают до 80oC и по капле добавляют к реакционной смеси уксусный ангидрид (80 мл). Реакционную смесь перемешивают при 80oC в течение одного часа и добавляют туда 3,4-дигидро-6,7-диметил-1(2H)-нафталинон (26 г). Температуру реакции повышают до 90oC и реакционную смесь перемешивают в течение 6 часов. Эту смесь концентрируют при пониженном давлении, удаляя растворитель, и к остатку добавляют ацетон. Осажденные кристаллы собирают фильтрацией и промывают ацетоном, получая гидрохлорид 2-диметиламинометил-3,4-дигидро-6,7-диметил-1-(2H)нафталинона (39 г).
(2) Полученный выше гидрохлорид основания Манниха (39 г) растворяют в ледяной воде, смесь подщелачивают водным аммиаком и экстрагируют дихлорметаном. Слой дихлорметана промывают водой и сушат над безводным сульфатом магния, полученную смесь концентрируют при пониженном давлении при температуре ниже 40oC, удаляя растворитель. Остаток растворяют в ацетоне и добавляют туда по капле метилиодид (10.8 мл) при перемешивании и охлаждении на льду. Смесь перемешивают еще 30 минут при охлаждении на льду и затем нагревают до 25oC. Смесь перемешивают в течение двух часов. Кристаллы собирают фильтрованием и промывают ацетоном, получая иодид 1,2,3,4-тетрагидро-N,N,N,6,7-пентаметил-1-оксо-2-нафтилметанаминия (46 г).
(3) Полученную выше четвертичную соль (46 г) растворяют в метаноле (300 мл), добавляют туда раствор цианида калия (9,6 г) в воде (80 мл) и перемешивают смесь при 25oC в течение трех часов. Реакционную смесь концентрируют при пониженном давлении и экстрагируют остаток этилацетатом. Этилацетатный слой промывают водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом магния. Полученную смесь концентрируют при пониженном давлении, удаляя растворитель и получая 1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2-нафтилацетонитрил (22 г) в виде кристаллов.
(4) Полученное выше ацетонитрильное соединение (22 г) растворяют в смеси концентрированной соляной кислоты (200 мл) и ледяной уксусной кислоты (200 мл), и смесь кипятят с обратным холодильником в течение 6 часов. К реакционному раствору добавляют воду и осажденные кристаллы собирают фильтрованием, промывают водой и перекристаллизовывают из этилметилкетона, получая требуемое соединение (16.5 г), т.пл. 193-194oC.
ИК-спектр (KBr, см-1): 1707, 1676
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.81-2.24 (м, 2H), 2.27 (с, 3H), 2.29 (с, 3H), 2.38-2.56 (м, 1H), 2.89-3.15 (м, 4H), 7.02 (с, 1H), 7.80 (с, 1H), 10, 50 (широкий с, 1H).
Справочный пример 2. Получение 1,2,3,4-тетрагидро-1-оксо-2-нафтилпропионовой кислоты.
(1) К раствору диизопропиламина (16 мл) в тетрагидрофуране (350 мл) по капле добавляют 1.6 М раствор бутиллития в гексане (71 мл) при охлаждении до -78oC и смесь перемешивают в течение 30 минут. К реакционной смеси по капле добавляют раствор 3,4-дигидро-1(2H)-нафталинона (16.5 г) в тетрагидрофуране (60 мл) в течение периода примерно 20 минут при охлаждении до -78oC, смесь перемешивают в течение 30 минут. К смеси добавляют по капле раствор этил-3-бромпропаноата (20.5 г) в тетрагидрофуране (60 мл) в течение периода примерно 20 минут и затем перемешивают еще 30 минут. Реакционную смесь перемешивают в течение ночи при 20oC и разбавляют диэтиловым эфиром. Смесь последовательно промывают водой, 5% водным раствором гидрокарбоната натрия и 5% соляной кислотой и сушат над безводным сульфатом магния. Остаток концентрируют при пониженном давлении, удаляя растворитель и получая этил-1,2,3,4-тетрагидро-1-оксо-2-нафтилпропаноат (10.1 г) в виде маслянистого продукта.
(2) Полученный выше этиловый эфир (10.1 г) растворяют в этаноле (200 мл) и добавляют туда 2N водный раствор гидроксида натрия (170 мл), и смесь кипятят с обратным холодильником в течение трех часов. Смесь концентрируют при пониженном давлении, удаляя этанол, подкисляют водный слой концентрированной соляной кислотой и экстрагируют дихлорметаном. Слой дихлорметана промывают водой, сушат над безводным сульфатом магния и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: хлороформ) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (6,3 г) в виде маслянистого продукта.
Соответствующие исходные вещества обрабатывают таким же образом, как в описанном выше справочном примере, получая 1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафтилпропионовую кислоту в виде маслянистого продукта.
Справочный пример 3. Получение 1,2,3,4-тетрагидро-1-оксо-2-нафтилбутановой кислоты.
(1) К раствору диэтилкарбоната (200 г) в толуоле (800 мл) добавляют 60% гидрид натрия (27 4 г), смесь перемешивают при 50oC в течение 30 минут и добавляют туда по капле раствор 3,4-дигидро-1(2H)нафталинона (50 г) в толуоле (200 мл). Смесь кипятят с обратным холодильником в течение одного часа, выливают в ледяную воду и нейтрализуют уксусной кислотой. Слой толуола собирают, последовательно промывают водным раствором карбоната калия и водой, сушат над безводным сульфатом магния и концентрируют при пониженном давлении. Остаток чистят перегонкой при пониженном давлении, получая этил-1,2,3,4-тетрагидро-1-оксо-2-нафтилкарбоксилат (47 г) в виде маслянистого продукта, т.к. 135-145oC/2 мм рт.ст.
(2) Полученное выше этилкарбоксилатное соединение (32 г) растворяют в трет-бутаноле (120 мл) и добавляют туда трет-бутилат калия (25 г), и смесь кипятят с обратным холодильником в течение 30 минут. Смесь оставляют стоять для охлаждения до 25oC и добавляют туда этил-4-бромбутаноат (34 г). Смесь кипятят с обратным холодильником в течение ночи и концентрируют при пониженном давлении. К остатку добавляют воду и смесь экстрагируют диэтиловым эфиром. Эфирный слой промывают водой, сушат над безводным сульфатом магния и концентрируют при пониженном давлении, получая этил-2- этоксикарбонил-1,2,3,4-тетрагидро-1-оксо-2-нафтилбутоноат (50 г) в виде маслянистого продукта.
(3) Полученное выше соединение - этилбутоноат растворяют в этаноле (500 мл) и добавляют туда 30% водный раствор гидроксида калия (300 мл) и смесь перемешивают при кипячении с обратным холодильником в течение ночи. Эту смесь концентрируют при пониженном давлении, удаляя этанол, полученный остаток подкисляют концентрированной соляной кислотой и экстрагируют дихлорметаном. Слой дихлорметана промывают водой, сушат над безводным сульфатом магния и концентрируют при пониженном давлении, удаляя растворитель и получая требуемое соединение (23 г) в виде маслянистого продукта.
Соответствующие исходные соединения обрабатывают таким же образом, как в описанном выше справочном примере, получая соединения в виде маслянистых продуктов:
1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафтилбутановую кислоту,
1,2,3,4-тетрагидро-1-оксо-2-нафтилпентановую кислоту и
1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафтилпентановую кислоту.
Справочный пример 4. Получение 1-[(1,2,3,4-тетрагидро-6-метил-1-оксо-2-нафталинил)ацетил]пиперидина.
1,2,3,4-Тетрагидро-6-метил-1-оксо-2-нафтилуксусную кислоту (7.0 г) и тионилхлорид (7 мл) растворяют в хлороформе (150 мл) и смесь кипятят с обратным холодильником в течение одного часа. После охлаждения смесь концентрируют при пониженном давлении, удаляя растворитель, остаток растворяют в толуоле (110 мл) и туда по капле добавляют пиперидин (8.2 г) при охлаждении на льду. Смесь перемешивают при 25oC, в течение одного часа, последовательно промывают разбавленной соляной кислотой и водой, сушат над безводным сульфатом магния и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол: этилацетат = 10:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (4.4) г в виде маслянистого продукта.
Соответствующие исходные вещества обрабатывают таким же образом, как в описанном выше справочном примере, получая 1-[(1,2,3,4-тетрагидро-7-метил-1-оксо-2-нафталинил)ацетил]пиперидин в виде маслянистого продукта.
Справочный пример 5. Получение 1-[1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2-нафталинил)ацетил]пирролидина.
К раствору 1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2-нафтилуксусной кислоты (8.0 г), пирролидина (3.8 г) и гексафторфосфата бензотриазол-1-илокси-трис-(диметиламино)фосфония (реагент ВОР, 16.7) в дихлорметане (80 мл) по капле добавляют триэтиламин (3.8 г) при 25oC и смесь перемешивают в течение трех часов. Смесь концентрируют при пониженном давлении, удаляя растворитель, и к остатку добавляют воду и толуол, нерастворимые материалы удаляют фильтрованием. Слой толуола собирают, последовательно промывают 1N водным раствором гидроксида натрия, водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом натрия. Остаток концентрируют при пониженном давлении, удаляя растворитель, чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол:этилацетат = 10:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (9.5 г) в виде маслянистого продукта.
ИК-спектр (KBr, см-1): 1672, 1638
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.76-2.09 (м, 5H), 2.26 (с, 3H), 2.28 (с, 3H), 2.17-2.40 (м, 2H), 2.76-3.28 (м, 4H), 3.36-3.66 (м, 4H), 7.00 (с, 1H), 7.78 (с, 1H).
1,2,3,4-Тетрагидро-1-оксо-нафтилуксусную кислоту, полученную таким же способом, как описано в справочном примере 1, и циклические аминные соединения обрабатывают так же, как в справочном примере 5, получая соединения, перечисленные в табл. 9.
Справочный пример 6.
Соответствующие исходные вещества обрабатывают таким же образом, как в описанном выше справочном примере 5, получая следующие соединения в виде маслянистых продуктов:
1-[(1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2-нафталинил)ацетил]-2,5-диметилпирролидин и 1-[(1,2,3,4- тетрагидро-6,7-диметил-1-оксо-2-нафталинил)ацетил]-2-метоксиметил)пирролидин.
Справочный пример 7. Получение 1-[3-(1,2,3,4-тетрагидро-1- оксо-2-нафталинил)пропаноил]пиперидина.
К раствору 1,2,3,4-тетрагидро-1-оксо-2-нафтилпропановой кислоты (6.3 г), пиперидина (3.7 г) и ВОР реагента (15.4 г) в дихлорметане (150 мл) по капле добавляют триэтиламин (3.5 г) при 25oC и смесь кипятят с обратным холодильником в течение трех часов. Реакционную смесь последовательно промывают водой и 10% соляной кислотой и сушат над безводным сульфатом магния. Полученный остаток концентрируют при пониженном давлении, удаляя растворитель, чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол: этилацетат = 10:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (7.1 г) в виде маслянистого продукта.
Соответствующие исходные вещества обрабатывают таким же образом, как в описанном выше справочном примере, получая следующие соединения:
1-[3-(1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафталинил)пропаноил] пиперидин,
1-[4-(1,2,3,4-тетрагидро-1-оксо-2-нафталинил)бутаноил]пирролидин,
1-[4-(1,2,3,4-тетрагидро-1-оксо-2-нафталинил)бутаноил]пиперидин,
1-[4-(1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафталинил)бутаноил] пирролидин,
1-[4-(1,2,3,4-тетрагидро-5-метокси-1-оксо-2-нафталинил)бутаноил] пиперидин,
1-[5-(1,2,3, 4-тетрагидро-1-оксо-2-нафталинил)пентаноил]пиперидин и
1-[5-(1, 2, 3, 4-тетрагидро-5-метокси-1-оксо-2-нафталинил)пентаноил]пиперидин.
Справочный пример 8. Получение 1,2,3,4-тетрагидро-6,7-диметил-2-[2-(1-пирролидинил)этил]-1-нафталинола.
К раствору 1-[(1,2,3,4-тетрагидро-6,7-диметил-1-оксо-2- нафталинил)ацетил] пирролидина (9.5 г), в толуоле (100 мл) по капле при перемешивании добавляют 70% раствор бис (2-метоксиэтокси)алюминийгидрида натрия в толуоле (29.0 г) при охлаждении на льду, смесь перемешивают при той же температуре в течение одного часа и при температуре 25oC в течение трех часов. Реакционную смесь снова охлаждают льдом, добавляют туда по капле насыщенный водный раствор тартрата натрия-калия (30 мл) и смесь перемешивают при той же температуре в течение 30 минут для разложения избыточного количества восстановителя. Органический слой отделяют, последовательно промывают водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол: этилацетат = 10:1) и фракции, содержащие требуемое соединение, объединяют и концентрируют при пониженном давлении, получая требуемое соединение (8.5 г) в виде маслянистого продукта.
ИК-спектр (в чистом виде, см-1): 3360, 3120
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.46-1.95 (м, 9H), 2.20 (с, 3H), 2.23 (с, 3H), 2.44-2.94 (м, 9H), 4.31-4.41 (м, 1H), 6.81 (с, 1H), 7,42 (с, 1H).
Справочный пример 9. Получение 5,7-дифтор-1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил]-1-нафталинола.
(1) Смесь 5,7-дифтор-3,4-дигидро-1(2H)-нафталинона (3.2 г), моногидрата глиоксиловой кислоты (1.6 г) и 85% фосфорной кислоты (3 мл) нагревают при перемешивании при 90oC в течение четырех часов. После охлаждения к реакционному раствору добавляют воду, а выпавшие кристаллы собирают фильтрованием, промывают водой и сушат, получая (5,7-дифтор-3,4-дигидро-1-оксо-2-(1H)-нафталинилиден)уксусную кислоту (4.2 г).
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 3.00 (т, 2H, J=7), 3.40-3.60 (м, 2H), 6.91 (т, 1H, J=1), 7.00-7.13 (м, 1H), 7.58-7.66 (м, 1H).
(2) К раствору полученного производного уксусной кислоты (4.2 г), пирролидина (1.9 г) и ВОР реагента (8.6 г) в дихлорметане (40 мл) по капле при перемешивании добавляют триэтиламин (2.0 г) при 25oC и смесь перемешивают в течение трех часов. Смесь концентрируют при пониженном давлении, удаляя растворитель, и к остатку добавляют воду и толуол. Нерастворимые материалы удаляют фильтрованием, толуольный слой отделяют, последовательно промывают 1N водным раствором гидроксида натрия, водой и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом натрия. Полученную смесь концентрируют при пониженном давлении, удаляя растворитель, остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь толуол:этилацетат = 2:1), а фракции, содержащие 1-[(5,7-дифтор-1,2,3,4-тетрагидро-1-оксо-2-нафталинилиден)ацетил] пирролидин, объединяют и концентрируют при пониженном давлении, получая указанное амидное соединение (2.6 г) в виде маслянистого продукта.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.85-2.06 (м, 4H), 2.96 (т, 2H, J= 7), 3.27-3.38 (м, 2H), 3.48-3.67 (м, 4H), 6.97-7.09 (м, 1H), 7.16 (т, 1H, J=1), 7.56-7.64 (м, 1H).
(3) Полученное выше амидное соединение (2.6 г) растворяют в этаноле (130 мл) и пропускают через смесь газообразный водород при температуре 25oC и перемешивании с применением в качестве катализатора 10% палладий-углерод (0.3 г). После абсорбции теоретически рассчитанного количества газообразного водорода катализатор удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении, удаляя растворитель и получая 1-[(5,7-дифтор-1,2,3,4-тетрагидро-1-оксо-2-нафталинил)ацетил]пирролидин (2.3 г) в виде маслянистого продукта.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.60 (с, 2H), 1.80-2.08 (м, 4H), 2.28-2.41 (м, 1H), 2.74-3.26 (м, 4H), 3.40-3.58 (м, 4H), 6.90-7.04 (м, 1H), 7.48-7.57 (м, 1H).
(4) Полученный выше продукт (2.3 г) растворяют в тетрагидрофуране (23 мл) и по капле добавляют туда 1М раствор комплекса боран-тетрагидрофуран в тетрагидрофуране (26 мл) при охлаждении льдом, смесь перемешивают при 25oC в течение ночи. К реакционному раствору добавляют по каплям метанол (26 мл) при охлаждении льдом и разлагают избыточное количество восстановителя. Смесь концентрируют при пониженном давлении, удаляя растворитель и получая требуемое соединение в виде комплекса с бораном.
Справочный пример 10. Получение метансульфоната 3,4-дигидро-7-метокси-2-нафтилэтила.
(1) Смесь 3,4-дигидро-7-метокси-2(1H)-нафталинона (5.0 г), бензойной кислоты (0.69 г), этил(трифенилфосфоранилиден)ацетата (14.9 г) и толуола (20 мл) кипятят с обратным холодильником в течение ночи. После охлаждения нерастворимые материалы удаляют фильтрованием, а фильтрат концентрируют при пониженном давлении. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь гексан:этилацетат = 10:1), а фракции, содержащие этил-3,4-дигидро-7-метокси-2-нафтилацетата, объединяют и концентрируют при пониженном давлении, получая указанный эфир (6.2 г) в виде маслянистого продукта.
(2) Полученный выше эфир (4.9 г) растворяют в безводном тетрагидрофуране (50 мл) и туда по капле добавляют 1М раствор диизобутилалюминийгидрида в толуоле (48 мл) при -10oC, смесь перемешивают при 0oC в течение одного часа. К реакционному раствору добавляют по капле воду и разлагают избыточное количество восстановителя. Нерастворимые материалы удаляют фильтрованием, а фильтрат экстрагируют этилацетатом. Органический слой сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель и получая 3,4-дигидро-7-метокси-2-нафтилэтанол (3.5 г) в виде маслянистого продукта.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.65 (широкий с, 1H), 2.25 (т, 2H, J= 8.5), 2.48 (т, 2H, J=7.5), 2.75 (т, 2H, J=8.5), 3.78 (с, 3H), 3.75-3.84 (м, 2H), 6.29 (с, 1H), 6.59 (д, 1H, J=3), 6.66 (дд, 1H, J=8,3), 7.01 (д, 1H, J=8).
(3) Полученное выше производное этанола (3.5 г) и триэтиламин (2.8 г) растворяют в дихлорметане (70 мл) и добавляют туда по капле метансульфонилхлорид (2.2 г) при 0oC. Смесь перемешивают при той же температуре в течение одного часа и концентрируют при пониженном давлении, удаляя растворитель и получая требуемое соединение в виде маслянистого продукта.
Справочный пример 11. Получение метансульфоната 1,2,3,4-тетрагидро-2-(2-метансульфонилоксиэтил)-6,7-диметокси-1-нафталинила.
(1) 3,4-Дигидро-6,7-диметокси-1-(2H)-нафталинон (10.0 г), растворяют в тетрагидрофуране (200 мл) и добавляют туда по капле 2М раствор диизопропиламида лития в тетрагидрофуране (37 мл) при -70oC. Смесь перемешивают в течение 30 минут и добавляют туда по капле этилбромацетат (10.5 г) при -70oC. Смесь перемешивают при той же температуре в течение двух часов и затем перемешивают при 20oC в течение ночи. Реакционную смесь выливают в ледяную воду и экстрагируют диэтиловым эфиром. Эфирный слой промывают последовательно водой, 10% соляной кислотой и 10% водным раствором гидрокарбоната натрия и сушат над безводным сульфатом натрия. Остаток концентрируют при пониженном давлении, удаляя растворитель и получая этил-1,2,3,4-тетрагидро-6,7-диметокси-1-оксо-2-нафтилацетат (12.0 г) в виде маслянистого продукта.
(2) Полученный выше эфир (12.0 г) растворяют в толуоле (100 мл) и туда добавляют 70% раствор бис-(2-метоксиэтокси)алюминийгидрида натрия (24 г) при охлаждении на льду и смесь перемешивают при 25oC в течение шести часов. К реакционному раствору добавляют насыщенный водный раствор тартрата натрия-калия при охлаждении на льду и смесь перемешивают в течение одного часа. Органический слой отделяют, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, удаляя растворитель. Остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь хлороформ:метанол = 20: 1), а фракции, содержащие 1,2,3,4-тетрагидро-2-(2-гидроксиэтил)-6,7-диметокси-1-нафталинол, объединяют и концентрируют при пониженном давлении, получая указанный диол (3.6 г) в виде маслянистого продукта.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.52-2.02 (м, 5H), 2.45-2.90 (м, 4H), 3.71-3.95 (м, 2H), 3.84 (с, 3H), 3.89 (с, 3H), 4.44 (д, 1H, J=7), 6.55 (с, 1H), 7.05 (с, 1H).
(3) Полученный выше диол (3.6 г) и триэтиламин (4.3 г) растворяют в дихлорметане (72 мл) и добавляют туда по капле метансульфонилхлорид (3.8 г) при охлаждении на льду. Смесь перемешивают в течение одного часа и концентрируют при пониженном давлении, удаляя растворитель и получая требуемое соединение в виде маслянистого продукта.
Справочный пример 12. Получение 5-фтор-1,2,3,4-тетрагидро-2-[2-(1-пирролидинил)этил]-1-нафталинола.
(1) Смесь 5-фтор-3,4-дигидро-1(2H)-нафталинона (10 г), моногидрата глиоксиловой кислоты (6.2 г) и 85% фосфорной кислоты (10 мл) перемешивают при 90oC в течение трех часов. После охлаждения к реакционному раствору добавляют воду, а выпавшие кристаллы собирают фильтрованием, промывают водой и сушат, получая (5-фтор-3,4-дигидро-1-оксо-2(1H)-нафталинилиден) уксусную кислоту (13 г).
(2) К раствору полученного производного уксусной кислоты (20 г) и ВОР реагента (45 г) в дихлорметане (200 мл) добавляют при перемешивании пирролидин (10 г), затем по капле добавляют триэтиламин (10 г) при охлаждении льдом и смесь перемешивают в течение трех часов. Эту смесь последовательно промывают водой, 10% соляной кислотой, 10% водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом натрия. Полученную смесь концентрируют при пониженном давлении, удаляя растворитель, и остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь гексан:этилацетат = 2:1), получая 5-фтор-1,2,3,4-тетрагидро-1-оксо-2-нафталинилиден)ацетил] пирролидин (15 г) в виде маслянистого продукта.
(3) Полученное выше амидное соединение (15.4 г) суспендируют в этаноле (300 мл) и пропускают через смесь газообразный водород при температуре 25oC и перемешивании с применением в качестве катализатора 10% палладий-углерод (1.5 г). После абсорбции теоретически рассчитанного количества газообразного водорода катализатор удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении, удаляя растворитель, а остаток чистят способом колоночной хроматографии на силикагеле (элюент: смесь гексан:этилацетат = 2:1), получая 5-фтор-1,2,3,4-тетрагидро-1-оксо-2-нафталинил)ацетил] пирролидин (9.5 г) в виде маслянистого продукта.
Спектр 1H-ЯМР (200 МГц, CDCl3, δ м.д.): 1.80-2.10 (м, 5H), 2.24-2.45 (м, 2H), 2.80-3.03 (м, 2H), 3,08-3.30 (м, 2H), 3.38-3.54 (м, 4H), 7,15-7.34 (м, 2H), 7.78-7,85 (м, 1H).
(4) Полученный выше продукт (9.5 г) растворяют в тетрагидрофуране (100 мл) и по капле добавляют туда 1М раствор комплекса боран-тетрагидрофуран в тетрагидрофуране (138 мл) при охлаждении льдом, смесь перемешивают при 20oC в течение ночи. К реакционной смеси добавляют по каплям метанол (100 мл) при охлаждении льдом и разлагают избыточное количество восстановителя. Смесь концентрируют при пониженном давлении, удаляя растворитель и получая требуемое соединение в виде комплекса с бораном.
Пример 75. Приготовление таблеток, г:
1-[2-(5-Фтор-3,4-дигидро-2-нафталинил)этил]пирролидингидрохлорид - 15
Кукурузный крахмал - 30
Лактоза - 68
Кристаллическая целлюлоза - 30
Гидроксипропилцеллюлоза - 5
Легкая безводная кремневая кислота - 1
Стеарат магния - 1
Указанные выше компоненты соединяют и замешивают обычным способом, смесь гранулируют и прессуют на 1000 ядер для таблеток (каждая по 150 мг). Затем на каждое ядро наносят покрытие обычным образом, используя гидроксипропилметилцеллюлозу, макрогол, оксид титана, тальк и легкую безводную кремневую кислоту, получая таблетки в оболочке.
Пример 76. Приготовление порошка, г:
1-[2-(3,4-дигидро-6,7-диметил-2-нафталинил)этил] пирролидингидрохлорид - 20
D-маннит - 935
Гидроксипропилцеллюлоза - 30
Стеарат магния - 10
Легкая безводная кремневая кислота - 5
Указанные выше компоненты смешивают и гранулируют, получая 2% порошкообразный препарат.
Возможность промышленного применения
Как объяснено выше, производные 1-[ω-(3,4-дигидро-2-нафталинил)алкил]циклического амина формулы (I), их фармацевтически пригодные соли присоединения кислот и N-оксидные производные оказывают сильное ингибирующее действие на рефлекс мочеиспускания и проявляют низкую токсичность, а следовательно, эти соединения являются полезными в качестве агентов для лечения частого мочеиспускания и недержания мочи, или средств для лечения различных заболеваний, вызванных уменьшением объемных возможностей мочевого пузыря, что связано с различными факторами.



Описывается новое производное 1-[1-[ω-(3,4--(3,4-дигидро-2-нафталинил)алкил]циклического амина формулы I, где значения R1 - R5, p, q указаны в п.1 формулы изобретения. Соединения настоящего изобретения оказывают сильное ингибиторное действие на рефлекс мочеиспускания и являются полезными агентами для лечения частого мочеиспускания и недержания мочи. Описывается также фармацевтическая композиция на основе соединений формулы I. 2 с. и 9 з.п. ф-лы, 9 табл.
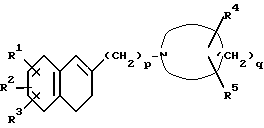
где R1 и R2 являются одинаковыми или разными и каждый представляет собой атом водорода, атом галогена, гидроксильную группу, C1-C5-алкильную группу, C1-C3-алкоксигруппу, гидроксиметильную группу, формильную группу, карбоксильную группу или C1-C3-алкоксикарбонильную группу, или, если R1 и R2 соединены с соседними атомами углерода, то R1 и R2 могут вместе образовывать метилендиоксигруппу, этилендиоксигруппу (-CH2CH2O-), триметиленовую группу или тетраметиленовую группу;
R3 является атомом водорода, атомом галогена, трифторметильной группой, C1-C5-алкильной группой, C1-C3 алкоксигруппой или фенильной группой;
R4 является атомом водорода, атомом галогена, гидроксильной группой, C1-C3-алкильной группой или (C1-C2-алкокси) метильной группой;
R5 является атомом водорода, атомом галогена, C1-C3-алкильной группой или (C1-C2-алкокси) метильной группой;
p является целым числом от 2 до 6;
q является целым числом от 3 до 7,
при условии, что если p = 2 и q = 5, то R1, R2, R3, R4 и R5 одновременно не являются атомами водорода;
его фармацевтически пригодная соль присоединения кислоты или N-оксидное производное.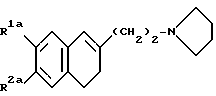
где R1a и R2a являются одинаковыми или разными и каждый представляет собой метильную группу, этильную группу, метоксигруппу, этоксигруппу или гидроксиметильную группу;
или его фармацевтически пригодная соль присоединения кислоты.
| СПОСОБ ПОЛУЧЕНИЯ 1- | 0 |
|
SU166350A1 |
| ЯДТЕНТНО- ТЕХНИЧЕСКАЯ БИБЛИОТЕКА | 0 |
|
SU240564A1 |
| Оплеточная машина | 1975 |
|
SU583485A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
