Изобретение относится к ароматическим амидиновым производным и их солям, которые обладают способностью к сильному антикоагулирующему действию посредством обратимого ингибирования активированного фактора свертывания крови, фактора X (далее обозначаемого "FXA"), и которые могут быть введены перорально. Настоящее изобретение также относится к фармацевтической композиции на их основе, предназначенной для лечения или предупреждения тромбоза или эмболии, и содержащему ароматическое амидиновое производное или его соль в качестве активного ингредиента.
До настоящего времени были предприняты попытки разработать антитромбин в качестве антитромботического средства. Однако известно, что такое антитромботическое средство может вызывать кровотечения и создает трудности для остановки этого кровотечения, поскольку оно ингибирует свертывание крови, а также агрегацию тромбоцитов, индуцированную тромбином. Для решения этой проблемы были предприняты попытки получить антикоагулирующие средства не путем ингибирования тромбина, а посредством другого ингибирующего механизма. В результате проведенных исследований был получен 1,2-бис(5-амидино-2-бензофуранил)-этан (далее обозначаемый "DABE"), который имеет представленную ниже формулу (2), и антикоагулирующее действие которого основано на FXa-ингибировании (Thrombosis Research, vol. 19, pp. 339-34, 1980):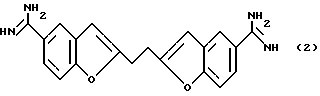
Однако соединение DABE имеет те недостатки, что оно обладает ингибирующими действиями как в отношении FXa, так и в отношении тромбина, которые не могут быть достаточно отделены друг от друга; кроме того, оно обладает очень низкой водорастворимостью и не проявляет антикоагулирующего действия при пероральном введении. Поэтому, для получения антикоагулирующего средства с хорошим клиническим эффектом особые усилия необходимо направить на разработку лекарственного средства, обладающего высокоспецифической и сильной FXa- ингибирующей активностью, хорошей водорастворимостью, и являющегося эффективным при пероральном введении.
Исходя из вышеуказанных соображений, авторами настоящего изобретения были проведены интенсивные исследования, относящиеся к синтезу различных типов ароматических амидовых производных, и к оценке их фармакологических свойств. В результате этих исследований было обнаружено, что ароматическое амидовое производное, представленное общей формулой (I), или его соль, обладает прекрасной водорастворимостью, сильным антикоагулирующим действием, обусловленным его высокоспецифической и обратимой FXa-ингибирующей активностью, даже в случае перорального введения, и поэтому может быть использовано в качестве лекарственного средства для предупреждения и лечения различных заболеваний, связанных с тромбозом и эмболией. На основе вышеуказанных исследований и было разработано настоящее изобретение.
Таким образом, настоящее изобретение относится к ароматическому амидиновому производству, имеющему общую формулу (I):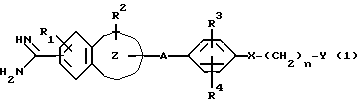
где R1 представляет собой атом водорода или низшую алкоксигруппу;
R2 представляет собой атом водорода, низшую алкильную группу, низшую алкоксигруппу, карбоксильную группу, карбоксиалкильную группу с 2-5 атомами углерода или алкоксикарбонильную группу с 2-5 атомами углерода;
R3 представляет собой атом водорода, карбоксильную группу, карбоксиалкильную группу с 2-5 атомами углерода, алкоксикарбонилалкильную группу с 3-6 атомами углерода, карбоксиалкоксильную группу с 2-5 атомами углерода, алкоксикарбонилалкоксильную группу с 3-6 атомами углерода или алкоксикарбонильную группу с 2-5 атомами углерода;
R4 представляет атом водорода, гидроксильную группу или низшую алкоксигруппу;
n = 0, 1, 2;
A является алкиленовой группой с 1-4 атомами углерода, которая может быть замещена одним или двумя заместителями, выбранными из гидроксиалкильной группы с 1-4 атомами углерода, карбоксила, алкоксикарбонильной группы с 2-5 атомами углерода и карбоксиалкильной группы с 2-5 атомами углерода;
X является простой связью, атомом кислорода или серы, или карбонильной группой;
Y является насыщенным или ненасыщенным 5- или 6-членным гетероциклическим фрагментом, имеющим один или два гетероатома, выбранных из атомов азота, кислорода, серы, или циклическим углеводородным фрагментом, которые могут быть замещены иминогруппой, карбамоилом, моно- или диалкилкарбамоилом, алканоимидоилом, низшим алкилом, низший алканоилом, алкоксикарбонилимином, бензилимидоилом, алканоиламино, алкиламином; где насыщенный или ненасыщенный 5- или 6-членный циклический углеводородный фрагмент также может быть замещен аминоалкильной группой или алканоиламиноалкильной группой; или Y является аминогруппой, которая может быть замещена пирролидинильной или пиридинильной группой;
группа формулы I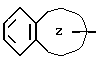
является группой, выбранной из индолила, бензофуранила, бензотиенила, бензотиазолила, нафтила, тетрагидронафтила, бензимидазолила; или к его фармацевтически приемлемой соли.
Настоящее изобретение также относится к фармацевтической композиции, обладающей антикоагулирующим действием, содержащей соединение общей формулы (I) или его соль в качестве активного начала и целевые добавки.
В соединении настоящего изобретения, представленном общей формулой (I), любая прямая, разветвленная или циклическая алкильная группа с 1-6 атомами углерода может быть использована в качестве низшей алкильной группы. Примерами такой алкильной группы могут служить метил, этил, пропил, изопропил, бутил, втор- или трет-бутил, пентил, гексил, циклопропил, циклобутил, циклопентил, циклогексил, и т.п. Низшая алкокси-группа может иметь 1-6 атомов углерода. Примерами такой группы могут служить метокси-, этокси-, пропокси-, изопропокси-, бутокси-, втор- или третбутокси-группа и т.п. Алкоксикарбонильная, алкоксикарбонилалкильная, карбоксиалкоксильная, алкоксикарбонилалкоксильная и гидроксиалкильная группы имеют предпочтительно 1-6 атомов углерода, и более предпочтительно 1-4 атомов углерода. Примерами алкоксикарбонильной группы являются метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил и т.п. Примерами карбоксиалкильной группы являются карбоксиметил, карбоксиэтил, карбоксипропил и т. п. Примерами алкоксикарбонилалкильной группы являются метоксикарбонилметил, этоксикарбонилметил, пропоксикарбонилметил, метоксикарбонилэтил, этоксикарбонилэтил, метоксикарбонилпропил, этоксикарбонилпропил, и т.д. Примерами карбоксиалкокси-группы являются карбоксиметокси-, карбоксиэтокси-, карбоксипропокси-группы и т.п. Примерами алкоксикарбонилалкокси-группы являются метоксикарбонилметокси, этоксикарбонилметокси, пропоксикарбонилметокси, метоксикарбонилэтокси, этоксикарбонилэтоксигруппы и т.п. Примерами гидроксиалкильной группы являются гидроксиметил, гидроксиэтил, гидроксипропил, гидроксибутил и т.п. Примерами алкиленовой группы, имеющей 1-4 атомов углерода и представленной A, являются метилен, этилен, триметилен, тетраметилен и т.п.
Насыщенный или ненасыщенный гетероциклический фрагмент с 5 или 6 членами может содержать, предпочтительно, один или два гетероатома, выбранных из атомов азота, кислорода, серы. Примерами гетероциклических колец такого типа являются пирролидин, пиперидин, имидазолин, пиперазин, тетрагидрофуран, гексагидропиримидин, пиррол, имидазол, пиразин, пирролидинон, пиперидинон, морфолин и т. п. Наиболее предпочтительными являются пирролидин и пиперидин, содержащие в качестве гетероатома один атом азота. Примерами насыщенной или ненасыщенной циклической углеводородной части могут служить циклопентил, циклогексил и т.п. Примерами аминоалкильной группы являются аминометил, аминоэтил, аминопропил и т.п. Примерами заместителей для вышеуказанных гетероциклических колец и циклических углеводородных колец могут служить предпочтительно низший алкил, низший алканоил, карбамоил, моно- или диалкилкарбамоил, алканоимидоил, бензимидоил, алкиламино, алканоиламино, имино, алкоксикарбонилимино, а более предпочтительно, алканоимидоил. Примерами заместителей для вышеуказанных аминогрупп и аминочастей аминоалкильных групп являются пиридинил или пирролидинил.
Соединения формулы (I) настоящего изобретения могут быть оптическими изомерами или стереоизомерами благодаря присутствию асимметрического атома углерода. Объем настоящего изобретения включает в себя оптические изомеры, стереоизомеры соединений настоящего изобретения, и их смеси.
Соли соединения формулы (I) настоящего изобретения могут быть любой природы, с одним лишь ограничением, а именно: они должны быть фармацевтически приемлемыми. Примерами подходящих солей могут служить: соли неорганических кислот, такие, как гидрохлорид, гидробромид, гидроиодид, фосфат, нитрат, сульфат и т.п.; соли органичекой сульфокислоты, такие, как метансульфонат, 2-гидроксиэтансульфонат, p-толуолсульфонат и т.п.; соли органической карбоновой кислоты, такие, как ацетат, пропаноат, оксалат, малоат, сукцинат, глутарат, адипат, тартарат, малеат, малат, манделат, и т.п.
Ниже приводятся примеры наиболее предпочтительных соединений настоящего изобретения, которые могут быть представлены формулой (I):
2-[4[(1-ацетилимидоил-3-пирролидинил)окси]фенил]-3-(7-амидино- 2-нафтил)пропионовая кислота или ее соль;
(+)-2-[4-[[((3S)-1-ацетимидоил-3-пирролидинил)окси] -фенил]- 3-(7-амидино-2-)нафтил)пропионовая кислота или ее соль;
(2S)-2-[4-[(3S)-1-ацетимидоил-3-пирролидинил)окси]-фенил]-3- (7-амидино-2-нафтил)пропионовая кислота или ее соль;
(2R)-2-[4-[((3R)-1-ацетимидоил-3-пирролидинил)окси] -фенил]-3- (7-амидино-2-нафтил)пропионовая кислота или ее соль;
2-[4-[(1-ацетимидоил-2-пирролидинил)метокси] -фенил] -3- (5-амидинобензо[b]тиен-2-ил)пропионовая кислота или ее соль;
(+)-2-[4-[((2S)-1-ацетимидоил-2-пирролидинил)метокси] -фенил]-3- (5-амидинобензо[b]тиен-2-ил)пропионовая кислота или ее соль;
2-[4-[(1-ацетимидоил-4-пиперидинил)окси] -фенил] -3- (7-амидино-2-нафтил)пропионовая кислота или ее соль;
(+)-2-[4-[(1-ацетимидоил-4-пиперидинил)окси]-фенил]-3- (7-амидино-2-нафтил)пропионовая кислота или ее соль;
пентагидрат гидрохлорида (2S)-2-[4-[((3S)-1-ацетимидоил-3- пирролидинил)окси]-фенил]-3-(7-амидино-2-нафтил)пропионовой кислоты.
В основном, соединение формулы (I) настоящего изобретения может быть получено, например, в соответствии с нижеприведенными реакционными формулами. А именно, нитриловое соединение формулы (3) подвергают взаимодействию со спиртом (R5OH) в присутствии галогенводорода. Полученный в результате имидат (4) подвергают реакции с аммиаком с получением ароматического амидинового производного (1a).
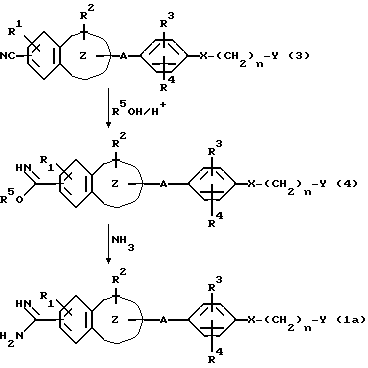
В приведенных выше формулах R1, R2, R3, R4, n, A, X, Y и
являются такими, как они были определены выше, а R5 является низшей алкильной группой.
Ниже приводится подробное описание вышеуказанной реакционной схемы. Реакция нитрилового соединения (3) со спиртом (R5OH) может быть осуществлена, например, посредством взаимодействия нитрила (3) с эквимолярным или избыточным количеством спирта (R5OH), имеющего от 1 до 6 атомов углерода, такого, как метанол, этанол, пропанол, и т.п., в присутствии галогенводорода, такого, как хлороводород, бромоводород и т.п. Если необходимо, может быть использован растворитель, который выбирают, например, из алифатических простых эфиров, таких, как диэтиловый эфир и т.п.; галогенированных углеводородов, таких, как хлороформ, дихлорметан и т.п.; апротонных растворителей, таких, как бензол и т.п., и их смесей. В основном, реакция протекает при температуре от -20 до 60oC в течение от 3 до 220 часов. Предпочтительно, если указанная реакция протекает при температуре от -8 до 30oC в течение периода времени от 10 до 96 часов, в присутствии избыточного количества метанола или этанола, и галогенированного углеводородного растворителя, такого, как хлороформ или дихлорметан.
Реакция полученного таким образом имидата (4) с аммиаком может быть осуществлена посредством взаимодействия указанного имидата (4) с аммиаком в растворителе или в смеси растворителей, выбранных из спиртов, имеющих 1-4 атомов углерода, таких, как этанол, пропанол, и т.п.; алифатических простых эфиров, таких, как диэтиловый эфир и т.п.; галогенированных углеводородов, таких, как хлороформ и т.п.; апротонных растворителей, таких, как бензол и т. п. ; и N, N'-диметилформамида и диметилсульфоксида. Реакция может быть проведена при температуре от -10 до 140oC в течение периода времени от 0,5 до 200 часов, а предпочтительно, при температуре от -8oC до 30oC в течение периода времени от 10 до 96 часов в этаноле.
Если используемое в качестве исходного материала нитриловое соединение (3) имеет карбоксильную группу или алкоксикарбонильную группу, то эту карбоксильную или алкоксикарбонильную группу подвергают эстерификации посредством реакции образования имидата, или посредством переэтерификации при взаимодействии сложного эфира со спиртом (R5OH). В результате этого, поскольку карбоксильная группа в соединении (1a), полученном с помощью этой реакции, является эстерифицированной, то соединение (1a) необходимо гидролизовать, если получают ароматическое амидиновое производное, имеющее свободную гидроксильную группу.
Реакция гидролиза может быть осуществлена путем обработки соединения (1a) в водном растворе неорганической кислоты, такой, как соляная кислота, серная кислота и т. п., или органической кислоты, такой, как толилсульфокислота и т.п., при температуре от -10oC до температуры перегонки, а предпочтительно от -5oC до температуры перегонки, в течение периода времени от 0,5 до 550 часов, а предпочтительно, от 0,5 до 350 часов.
Если соединение (1a) содержит группу, восприимчивую к гидролизу с сильной кислотой, то до проведения реакции гидролиза предпочтительно блокировать амидиногруппу защитной группой, такой, как трет-бутоксикарбонил или аналогичной группой, после чего осуществляют гидролиз сложного эфира в основных условиях с последующим разблокированием. Защита амидино-группы может быть осуществлена с помощью реакции соединения (1a) с 2-(трет-бутоксикарбонилоксиимино)-2-фенил-ацетонитрилом в воде, метаноле, этаноле, тетрагидрофуране, диоксане, ацетоне или в их смеси, в присутствии основания, такого, как 1,8-диазабицикло[5.4.0] -7-ундецен или т.п. Эта реакция может быть осуществлена при температуре от 0oC до 50oC, а предпочтительно от 5oC до 30oC, в течение периода времени от 0,5 до 48 часов, а предпочтительно от 1 до 24 часов.
Сложноэфирный гидролиз защищенного таким образом соединения и последующее разблокирование может быть осуществлено путем обработки защищенного соединения водным раствором гидроксида натрия или гидроксида калия, а затем соляной кислотой, в воде или в водо-содержащем растворителе, таком, как этанол, метанол, тетрагидрофуран, диоксан или т.п. Реакция гидролиза сложного эфира может быть проведена при температуре от 0oC до 50oC, а предпочтительно, от 5oC до 30oC, в течение периода времени от 0,5 до 48 часов, а предпочтительно, от 1 до 24 часов. Реакция разблокирования может быть осуществлена при температуре от 0oC до 60oC, предпочтительно при 25oC, в течение периода времени от 0,5 до 24 часов, а предпочтительно от 1 до 6 часов.
Если в группе A соединения (1a) две алкоксикарбонильные группы связаны с одним атомом углерода, то гидролиз и декарбоксилирование могут быть осуществлены в одно и то же время согласно следующей реакционной схеме: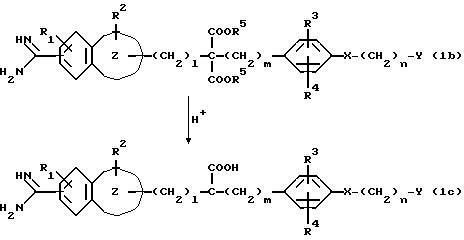
В приведенных выше формулах каждый из l = 1, m = 0 или 1, а R1, R2, R3, R4, R5, n, X, Y и
являются такими, как они были определены выше.
Эта реакция может быть осуществлена в водном растворе неорганической кислоты, такой, как соляная кислота, серная кислота, и т.п., или органической кислоты, такой, как толилсульфокислота или т.п., при температуре от -20oC до температуры перегонки, а предпочтительно от -5oC до температуры перегонки, в течение периода времени от 0,5 до 550 часов, а предпочтительно от 0,5 до 350 часов.
Если в качестве соединения настоящего изобретения формулы (I) получают соединение (1e), имеющее в своей группе имидоильную группу, то оно может быть получено с помощью реакции соединения (1d), имеющего первичную или вторичную аминогруппу в своей группе Y, с имидатом соединения (5) согласно следующей реакционной схеме: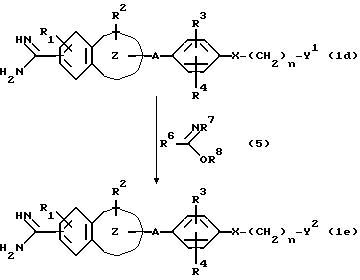
В приведенных выше формулах Y1 представляет собой ряд вышеупомянутых групп Y, имеющих в качестве заместителя первичную или вторичную аминогруппу, Y2 представляет собой другой ряд вышеупомянутых групп Y, имеющих в качестве заместителя имидоильную группу; каждый из R6 и R7 представляет собой атом водорода, низшую алкильную группу или фенильную группу, R8 является низшей алкильной группой или бензильной группой, а R1, R2, R3, R4, n, A, X и
являются такими, как они были определены выше.
Эта реакция может быть осуществлена, например, посредством взаимодействия соединения (1d) с эквимолярным или избыточным количеством имидата (5) в присутствии основания, такого, как триэтиламин, гидроксил натрия, гидроксил калия или т.п., в воде или растворителе или смеси растворителей, выбранных, например, из спиртов, имеющих 1-4 атомов углерода, таких, как этанол, пропанол и т. п., алифатических простых эфиров, таких, как диэтиловый эфир и т.п., галогенированных углеводородов, таких, как хлороформ и т.п., и N, N-диметилформамида и диметилсульфоксида. Эта реакция может быть осуществлена при температуре от -20oC до 70oC в течение периода временит от 1 мин до 168 часов, а предпочтительно при температуре от -10oC до 40oC в течение периода времени от 1 мин до 72 часов.
Когда имидоильная форма (1e) имеет алкоксикарбонильную группу, данная алкоксикарбонильная группа может гидролизоваться в карбоксильную группу.
Реакция гидролиза может проводиться с помощью обработки соединения (1e) в водном растворе неорганической кислоты, такой, как соляная кислота, серная кислота, или т.п., или органической кислоты, такой, как толилсульфокислота или т. п. , при температуре от -10oC до температуры перегонки, а предпочтительно от -5oC до температуры перегонки, в течение периода времени от 0,5 до 550 часов, а предпочтительно, от 0,5 до 350 часов.
В соответствии с настоящим изобретением, если исходное соединение имеет заместитель, такой, как карбоксильная группа, аминогруппа, или т.п., то перед осуществлением нужных реакций предпочтительно защитить такую функциональную группу, после чего защитную группу удаляют. С другой стороны, реакция образованная амидина, реакция образования имидата и аналогичные реакции могут быть проведены без блокирования такой функциональной группы. В этом случае, блокирование первичной или вторичной аминогруппы может быть осуществлено с использованием защитной группы, такой, как трет-бутоксикарбонил, бензилоксикарбонил, р-нитробензилоксикарбонил, трифенилметил, или т.п.
Кроме того, алкоксикарбонил-замещенное соединение может быть получено, например, в соответствии с нижеприведенной реакционной схемой, посредством проведения гидролиза сложного эфира после реакции образования амидина или имидата, с последующей переэстерификацией, если это необходимо: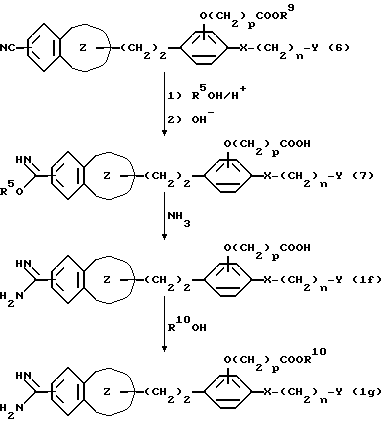
В вышеприведенных формулах R9 является атомом водорода или низший алкильной группой, R10 является низшей алкильной группой, p является целым числом 1 или 2, а R5, n, X, Y
и 
являются такими, как они были определены выше.
То есть, нитриловое соединение формулы (6) подвергают реакции со спиртом (R5OH) в присутствии галогенводорода, и полученное в результате соединение "имидат, сложный эфир" гидролизуют путем обработки основанием, получая производное "имидат • карбоновой кислоты" (7), которое затем подвергают реакции с аммиаком, в результате чего получают амидинозамещенное ароматическое соединение (If). Путем эстерификации соединения (If) получают соединение (Ig).
Реакция нитрилового соединения (6) со спиртом (R5OH) может быть осуществлена посредством взаимодействия указанного нитрилового соединения (6) с эквимолярным или избыточным количеством спирта (R5OH), имеющего 1-6 атомов углерода, такого, как метанол, этанол, пропанол или т.п., в присутствии галогенводорода, такого, как хлороводород, бромоводород и т.п. Если необходимо, может быть использован растворитель или смесь растворителей, которые выбирают, например, из алифатических простых эфиров, таких, как диэтиловый эфир и т.п., галогенированных углеводородов, таких, как хлороформ, дихлорметан и т. п. , апротонных растворителей, таких, как бензол. Эта реакция может быть проведена при температуре от -10oC до 60oC в течение периода времени от 3 до 120 часов. Предпочтительно, если реакция протекает при температуре от -8oC до 30oC в течение периода времени от 10 до 96 часов в галогенированном углеводородном растворителе, таком, как хлороформ или дихлорметан, в присутствии избыточного количества метанола или этанола. После концентрирования и осушки полученной реакционной смеси, твердый остаток обрабатывают концентрированным щелочным раствором для осуществления нейтрализации и сложноэфирного гидролиза, в результате чего получают производное "имидат•карбоновой кислоты" формулы (7). Эта реакция может быть проведена, в основном, при температуре от -10oC до 60oC в течение периода времени от 0,2 до 5 часов, предпочтительно при температуре от 0 до 25oC в течение периода времени от 0,5 до 2 часов, в водном растворе гидроксида натрия или гидроксида калия.
Реакция полученного таким образом производного "имидат•карбоновой кислоты", имеющего формулу (7), с аммиаком может быть осуществлена, например, посредством взаимодействия указанного производного (7) с хлоридом аммония, аммиаком, или с их смесью, в растворителе или в смеси растворителей, которые выбирают, например, из спиртов, имеющих 1-4 атомов углерода, таких, как этанол, пропанол и т.п.; алифатических эфиров, таких, как диэтиловый эфир и т. п. ; галогенированных углеводородов, таких как хлороформ и т.п.; апротонных растворителей, таких, как бензол и т.п., и N,N-диметилформамида и диметилсульфоксида. Эта реакция обычно протекает при температуре от -10oC до 140oC в течение периода времени от 0,5 до 200 часов, а предпочтительно при температуре от -8oC до 30oC в течение периода времени от 10 до 96 часов, в этаноле.
Эстерификация амидино-соединения формулы (1f) может быть осуществлена, например, с помощью реакции соединения (1f) с тионилгалидом, таким как тионилхлорид, тионилбромид и т.п., в спирте с 1-4 атомами углерода, таком, как этанол, пропанол и т.п. Эту реакцию осуществляют, в основном, при температуре от 0oC до температуры перегонки в течение периода времени от 10 мин до 36 часов, предпочтительно при температуре от 10oC до 60oC в течение периода времени от 10 мин до 24 часов.
Кристаллизация соединения формулы (I) настоящего изобретения может быть осуществлена, например, путем обработки реакционного раствора после завершения реакции сильноосновной (OH-типа) ионообменной смолой, или гидроксидом натрия, гидроксидом калия, и т.п., для доведения числа добавленных солей предпочтительно до 1. Полученный раствор обрабатывают при температуре от -10oC до 30oC, а предпочтительно от 0oC до 25oC, в воде или растворителе, таком, как метанол, этанол, изопропанол, ацетон и т.п., или в их смеси, предпочтительно в смеси воды и этанола.
Полученное таким образом ароматическое амидиновое производное формулы (I) или его соль обладает прекрасной и специфической способностью к ингибированию FXa и может быть использовано в качестве антикоагулирующего средства, а также в качестве лекарственного средства для профилактики и лечения тромбоза и эмболии. Поскольку соединение формулы (I) является активным даже при пероральном введении, то оно может быть введено, как перорально, так и парентерально. Соединение настоящего изобретения может быть введено в различных дозах в зависимости от симптомов, возраста, веса пациента и других факторов. В случае перорального введения, обычная доза указанного соединения составляет от 5 до 1000 мг в день для взрослого человека, а предпочтительно от 10 до 500 мг в день для взрослого человека. Примерами стандартных лекарственных форм вводимого средства являются таблетки, капсулы, порошки, гранулы и т. п. , которые могут быть изготовлены с использованием обычно применяемых добавок, таких, как наполнители, замасливатели, связующие вещества и т. п. В случае парентерального введения, соединения настоящего изобретения могут быть введены путем подкожных инъекций, внутривенных инъекций или внутривенных капельных вливаний в дозах, составляющих от 0,1 до 100 мг в день, а предпочтительно от 0,5 до 30 мг в день для взрослого человека.
Поскольку соединение настоящего изобретения обладает высоким антикоагулирующим действием, основанным на его прекрасной FXa-ингибирующей активности, то оно не оказывает влияния на тромбоциты, а может быть использовано для лечения различных заболеваний, вызванных тромбозом или эмболией, таких, как церебральный инфаркт, церебральный тромбоз, церебральная эмболия, преходящее нарушение мозгового кровообращения (TIA), инфаркт миокарда, нестабильная стенокардия, инфаркт легких, эмболия легочной артерии, синдром Бергера, тромбоз глубоких вен, синдром диссеминированного внутрисосудистого свертывания или генерализованный тромбогеморрагический синдром, тромбообразование после хирургической операции на сосудах, введение искусственного клапана, чрезкожная транслюминальная коронарная ангиопластика (PTCA), или чрезкожная транслюминальная коронарная реканализация (PTCP), закупорка после переливания крови, тромбообразование при искусственном кровообращении и т.п.
Для более полной иллюстрации настоящего изобретения ниже приводятся ссылочные примеры, примеры, иллюстрирующие настоящее изобретение, и испытательные примеры. Однако, при этом, необходимо иметь в виду, что указанные примеры представлены лишь с целью иллюстрации настоящего изобретения, но ни в коем случае не ограничивают его объема.
Ссылочный пример 1. Получение (5-циано-3-метил-2-бензофуранил)метилтрифенилфосфонийхлорида.
a) 13,31 г 2-ацетил-4-бромофенола, 11,0 г этилбромоацетона и 9,7 безводного карбоната калия нагревали с обратным холодильником в 70 мл ацетона в течение 2 часов. Нерастворившиеся вещества удаляли путем фильтрации, а полученный фильтрат концентрировали и осушали. Остаток растворяли в хлороформе, промывали водой, а затем осушали для удаления растворителя. Обработанный таким образом остаток промывали смешанным растворителем, состоящим из этанола и н-гексана, а нерастворившиеся кристаллы выделяли путем фильтрации. В результате описанной процедуры получали 16,82 г этил 2-ацетил-4-бромофенил оксиацетата в виде бесцветных пластинчатых кристаллов. Т.пл. 66-68oC.
b) 16,8 г этил (2-ацетил-4-бромофенил)оксиацетата, полученного согласно процедура, описанной в стадии (а), растворяли в 100 мл безводного этанола, в котором предварительно растворяли 1,2 г металлического натрия, и полученный раствор размешивали при комнатной температуре в течение 1,5 часа. Реакционный раствор выливали в воду и экстрагировали этилацетатом, а полученный органический слой промывали водой и затем осушали. После отгонки растворителя, осажденные кристаллы собирали путем фильтрации и промывали этанолом, в результате чего получали 5,3 г этил 5-бромо-3-метил-2-бензофуранкарбоксилата в виде бесцветных тонких игольчатых кристаллов. Т.пл. 96-97oC.
1H-ЯМР (CDCl3) δ: 1,44 (3H, т, J = 8 Гц); 2,54 (3H, c); 4,45 (2H, кв., J = 8 Гц); 7,43 (2H); 7,73 (1H, с).
c) В потоке азота, 4,9 г этил-5-бромо-3-метил-2-бензофуранкарбоксилата, полученного в соответствии с процедурой, описанной в стадии (b), 2,0 г цианида меди и каталитически эффективное количество сульфата меди размешивали в 40 мл N-метил-2-пирролидона в течение 6 часов при 200oC. После охлаждения, реакционный раствор выливали в воду, а нерастворимые вещества удаляли путем фильтрации. Полученный фильтрат экстрагировали этилацетатом, а органический слой промывали водой, концентрировали, осушали и собирали осажденные кристаллы. В результате этой процедуры получали 3,16 г этил-5-циано-3-метил-2-бензофуранкарбоксилата в виде слегка коричневатых кристаллов. Т.пл. 156-158oC.
1H-ЯМР (CDCl3) δ: 1,45 (3H, т, J=8,0 Гц), 2,60 (3H, c); 4,45 (2H, кв., J = 8 Гц); 7,67 (2H), 7,99 (1H, с).
d) 3,1 г этил 5-циано-3-метил-2-бензофуранкарбоксилата, полученного в соответствии с процедурой, описанной в стадии (c), растворяли в 60 мл тетрагидрофурана. К полученному раствору добавляли, охлаждая при этом льдом, 2,1 г иодида кальция (4H2O), 0,63 г борогидрида натрия и каталитически эффективное количество бикарбоната натрия. Полученную смесь размешивали при комнатной температуре в течение 18 часов, затем добавляли еще 2,1 г иодида кальция (4H2O) и 0,63 г борогидрида натрия, и размешивали при комнатной температуре еще 18 часов.
Полученный реакционный раствор разбавляли этилацетатом, промывали водой, а затем осушали путем удаления растворителя. Полученный таким образом остаток подвергали колоночной хроматографии на силикагеле, элюируя хлороформом. В результате этой процедуры получали 1,96 г очищенного 2-гидроксиметил-3-метил-5-бензофуранкарбонитрила.
1H-ЯМР (CDCl3) δ: 1,8 (1H, шир. с); 2,28 (3H, с; 4,78 (2H, с); 7,52 (2H), 7,82 (1H, c).
e) 1,92 г 2-гидроксиметил-3-метил-5-бензофуранкарбонитрила, полученного согласно процедуре, описанной в стадии (d), добавляли к 50 мл диэтилового эфира, а затем добавляли 3 капли пиридина и 1,65 мл тионилхлорида, охлаждая при этом льдом, и полученную смесь размешивали при комнатной температуре в течение 4,5 часов. Реакционный раствор выливали в ледяную воду и экстрагировали хлороформом, а полученный органический слой промывали последовательно водой, насыщенным водным раствором бикарбоната натрия и водой, а затем концентрировали и осушали. В результате этой процедуры получали 1,68 г 2-хлорометил-3-метил-5-бензофуранкарбонитрила.
f) 1,68 г 2-хлорометил-3-метил-5-бензофуранкарбонитрила, полученного согласно процедуре, описанной в стадии (e), и 3 г трифенилфосфина нагревали с обратным холодильником в ксилоле в течение 5 часов. После охлаждения, осажденные кристаллы собирали путем фильтрации, и получали в результате 3,63 г целевого соединения. Т.пл. > 270oC.
1H-ЯМР (CDCl3) δ: 2,0 (1,5H, с); 2,04 (1,5 H, с); 6,09 (2H, д, J = 16 Гц); 7,7 (18H, м).
Ссылочный пример 2. Получение (5-циано-3-бензофуранил)метилтрифенилфосфония бромида
a) 12,15 г этил 5-циано-3-метил-2-бензофуранкарбоксилата, полученного согласно процедуре, описанной в стадии (c), ссылочного примера 1, растворяли в 60 мл этанола, а затем добавляли 5 г гидроксида натрия и 100 мл воды, после чего полученную смесь размешивали при 30-40oC в течение 2 часов. После охлаждения льдом, pH реакционного раствора доводили до 2 с помощью разбавленной HCl, и осажденные таким образом кристаллы собирали путем фильтрации и осушали. В результате этой процедуры получали 10,6 г 5-циано-3-метил-2-бензофуранкарбоновой кислоты в виде бесцветных призмообразных кристаллов. Т. пл.: (сублимация при 275-285oC).
1H-ЯМР (CDCl3) δ: 2,54 (3H, с), 7,88 (2H); 8,44 (1H).
b) 10,64 г 5-циано-3-метил-2-бензофуранкарбоновой кислоты, полученной согласно процедуре, описанной в стадии (a), и 2,5 г медного порошка добавляли к 65 мл хинолина, и полученную смесь размешивали 30 минут при 210oC. После охлаждения ледяной водой и доведения pH до 1 с помощью HCl, реакционную смесь экстрагировали хлороформом, а органический слой осушали при пониженном давлении. Полученный таким образом остаток подвергали хроматографии на колонке с силикагелем, элюируя толуолом. В результате этой процедуры получали 6,89 г очищенного бесцветного 3-метил-5-бензофуранкарбонитрила. Т. пл. 73oC.
1H-ЯМР (CDCl3) δ: 2,26 (3H, д, J = 1,5 Гц), 7,53 (3H); 7,85 (1H, с).
c) 7,28 г 3-метил-5-бензофуранкарбонитрила, полученного согласно описанию в стадии (b), растворяли в 50 мл тетрахлорметана и нагревали с обратным холодильником в условиях светового облучения. К полученному реакционному раствору постепенно добавляли смесь, состоящую из 8,25 г N-бромосукцинимида и 160 г 2,2-азобис-изо-бутилонитрила. После нагревания с обратным холодильником в течение 3 часов, осажденный материал удаляли путем фильтрации, а полученный фильтрат осушали. Осушенный остаток очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента толуол, и получали таким образом 8,65 г смеси (2:5) исходного материала и 3-бромометил-5-бензофуранкарбонитрила. 8,65 г полученного таким образом сырого бромометилового соединения растворяли в ксилоле, затем к полученному раствору добавляли 10 г трифенилфосфина, и полученную смесь нагревали в течение 20 минут. После охлаждения осадок собирали путем фильтрации, в результате чего получали 14,73 г целевого соединения в виде бесцветных кристаллов. Т.пл. > 290oC.
1H-ЯМР (CDCl3) δ: 5,88 (2H, д, J = 16 Гц), 7,0 - 8,0 (19H, м).
Ссылочный пример 3. Получение (5-циано-7-метокси-2-бензофуранил) метилтрифенилфосфонийхлорида.
a) 10,0 г 5-бромо-2-гидрокси-3-метоксибензальдегида растворяли в 39 мл N,N-диметилформамида, а полученный раствор смешивали с 11,9 г безводного карбоната калия и размешивали при комнатной температуре. Затем, при той же температуре, к полученному реакционному раствору по капле добавляли 5,0 г хлорацетона, и размешивали еще один час при повышенной температуре 80oC. Полученный раствор разбавляли этилацетатом и pH доводили по 2 с помощью концентрированной соляной кислоты, а полученный органический слой собирали. Этот органический слой осушали путем отгонки растворителя, а осадок очищали с помощью хроматографии на силикагеле, в результате чего получили 4,0 г 2-ацетил-5-бромо-7-метоксибензофурана. Т.пл. 107-109oC.
1H-ЯМР (CDCl3) δ: 2,62 (3H, c); 3,83 (3H, с); 7,02 (1H), 7,39 (2H).
b) К 107,6 мл 5 н водного раствора гидроксида натрия по капле добавляли 26,8 г брома при температуре -5oC или ниже. Затем, к этому раствору медленно по капле добавляли 100 мл раствора диоксана, содержащего 15,0 г 2-цетил-5-бромо-7-метокси-бензофурана, полученного согласно процедура, описанной в стадии (a). После завершения этого добавления, температуру реакционного раствора постепенно повышали до 60oC, и затем размешивали в течение 30 минут. После охлаждения pH полученного реакционного раствора доводили до 2 с помощью концентрированной соляной кислоты, а затем экстрагировали этилацетатом. Полученный органический слой концентрировали досуха, и осажденные таким образом кристаллы собирали путем фильтрации, в результате чего получали 5-бромо-7-метокси-2-бензофуранкарбоновую кислоту. Эти кристаллы суспендировали в 200 мл этанола, и размешивая при комнатной температуре, к этой суспензии по капле добавляли 10 мл тионилхлорида. Реакционный раствор нагревали с обратным холодильником в течение 2 часов. После охлаждения, этот реакционный раствор нейтрализовали насыщенным водным раствором бикарбоната натрия, затем смешивали с водой, и осажденные кристаллы собирали путем фильтрации. Полученные кристаллы очищали на хроматографической колонке с силикагелем, элюируя хлороформом, и получали в результате 11,33 г этил 5-бромо-7-метокси-2-бензофуранкарбоксилата.
1H-ЯМР (CDCl3) δ: 1,41 (3H, т, J = 7,10 Гц); 4,00 (3H, с); 4,43 (2H, кв. , J = 7 Гц); 7,02 (1H, д); 7,39 (1H, д); 7,42 (1H, с).
c) смесь, состоящая из 2,0 г этил 5-бромо-7-метокси-2-бензофуранкарбоксилата, полученного в соответствии с процедурой, описанной в стадии (b), 1,26 г цианида меди, 100 мл N-метил-2-пирролидона и каталитически эффективного количества сульфата меди, размешивали в течение 2 часа при 180-190oC в потоке аргона. После охлаждения, к реакционному раствору добавляли смесь толуола и этилацетата (1:1) и воду для удаления нерастворившегося материала, а полученный органический слой промывали водой, и затем осушали. После отгонки растворителя, осажденные кристаллы собирали путем фильтрации, промывали этанолом и получали в результате 1,2 г этил 5-циано-7-метокси- бензофуранкарбоксилата.
1H-ЯМР (CDCl3) δ: 1,43 (3H, т, J = 7,0 Гц); 4,06 (3H, с); 4,46 (2H, т, J = 7,0 Гц); 7,10 (1H, д, J = 1,0 Гц); 7,53 (1H, с); 7,64 (1H, д).
d) 8,55 г этил 5-циано-7-метокси-2-бензофуранкарбоксилата, полученного согласно процедуре, описанной в стадии (c), растворяли в 250 мл тетрагидрофурана. При охлаждении в ледяной бане, полученный раствор смешивали с 13,74 г иодида кальция (4H2O), 2,12 г борогидрида натрия и каталитически эффективным количеством бикарбоната натрия, и полученную смесь размешивали при комнатной температуре 1,5 часа, после чего добавляли 13,74 г иодида кальция (4H2O) и 2,12 г борогидрида натрия, и полученную смесь размешивали при комнатной температуре еще один час. При охлаждении в ледяной бане, pH реакционного раствора доводили до 2 с помощью концентрированной соляной кислоты, а растворитель удаляли путем дистилляции. Остаток экстрагировали хлороформом, промывали водой, и осушали путем отгонки растворителя. Полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, используя, в качестве элюента смесь хлороформа и этанола, и получали в результате 1,96 г 2-гидроксиметил-7-метокси-5-бензофуранкарбонитрила.
Т.п. 149-150oC.
1H-ЯМР (CDCl3) δ: 2,17 (1H, т, J = 6,1 Гц); 4,02 (3H, c); 4,80 (2H, д, J = 6,1 Гц); 6,71 (1H, с); 6,99 (1H, д, J = 1,3 Гц); 7,50 (1H, д, J = 1,3 Гц).
e) 5,0 г 2-гидроксиметил-7-метокси-5-бензофуранкарбонитрила, полученного в соответствии с процедурой, описанной в стадии (d), растворяли в 100 мл диэтилового эфира, а затем добавляли несколько капель пиридина. Охлаждая на ледяной бане и размешивая при этом, к полученному раствору по капле добавляли 5,86 г тионилхлорида. После добавления, температуру полученного раствора постепенно повышали до комнатной температуры, и продолжали размешивать при комнатной температуре еще 1 час. При охлаждении на ледяной бане, к реакционному раствору добавляли воду, и образовавшийся органический слой собирали, промывали водой, а затем осушали путем удаления растворителя, в результате чего получали 2-хлорометил-7-метокси-5-бензофуранкарбонитрил. Полученное таким образом хлорометиловое соединение и 9,67 г трифенилфосфина нагревали с обратным холодильником в 50 мл ксилола в течение 18 часов. После охлаждения, осажденные таким образом кристаллы собирали путем фильтрации и получали в результате 10,54 г целевого соединения.
1H-ЯМР (ДМСО-d6) δ: 3,89 (3H, с); 5,6-6,0 (2H, шир. с).
Ссылочный пример 4. Получение (5-цианобензо [b] тиен-2-ил)метилтрифенилфосфония хлорида.
a) 8,13 г 5-бромосалицилальдегида растворяли в 100 мл ацетона, а затем добавляли 6,7 г безводного карбоната калия. Размешивая при комнатной температуре, к полученному раствору добавляли 5,0 г N,N-диаметилтиокарбамоилхлорида, после чего продолжали размешивать еще два часа. Полученный реакционный раствор выливали в ледяную воду, а осажденные таким образом кристаллы собирали путем фильтрации, осушали и получали в результате 9,2 г 5-бромо-2-[N,N-диметилтиокарбамоил)окси]бензальдегида.
Т.пл. 141-143oC.
ИК (KBr): 1690, 1596, 1546, 1470, 1396 см-1
1H-ЯМР (CDCl3) δ: 3,42 (3H, с); 3,47 (3H, с); 7,03 (1H, д, J = 8,3 Гц); 7,72 (1H, дд, J = 8,3 и 2,2 Гц); 8,01 (1H, д, J = 2,2 Гц).
b) 9,0 г 5-бромо-2-[(N,N-диметилтиокарбамоил)окси]-бензальдегида, полученного согласно описанию в стадии a), расплавляли путем его нагревания в течение 10 минут на масляной бане при 210-220oC. Полученный продукт растворяли в 1 мл толуола, а затем добавляли 6 мл метанола. Осажденные таким образом кристаллы собирали путем фильтрации, и получали 4,0 г сырого 5-бромо-2-[(N, N-диметилкарбамоил)тио]бензальдегида.
Т.пл. 118-120oC.
ИК (KBr): 1677, 1365, 1185 см-1
1H-ЯМР (CDCl3) δ: 3,09 (6H, с); 7,31 (1H, д, J = 9,6 Гц); 7,70 (1H, дд, J = 9,6 и 1,8 Гц); 8,14 (1H, д, J = 1,8 Гц), 10,25 (1H, с).
c) 21,0 г 5-бромо-2-[(N,N-диметилкарбамоил)тио]бензальдегида растворяли в 50 мл метилортоформата. Полученный раствор смешивали с 1,0 г p-толуолсульфоната, а затем нагревали с обратным холодильником 50 минут. После охлаждения, реакционный раствор выливали в насыщенный раствор бикарбоната натрия и экстрагировали бензолом. Полученный органический слой осушали путем удаления растворителя. Полученный осадок растворяли в 100 мл метанола, после чего добавляли 37 мл 2н гидроксида натрия и нагревали с обратным холодильником в потоке азота в течение 1 часа. После охлаждения, полученный реакционный раствор корректировали до получения pH 1 с помощью концентрированной соляной кислоты, экстрагировали бензолом, а затем осушали с удалением растворителя. Осадок растворяли в 20 мл ацетона, и по капле добавляли при комнатной температуре, к размешанной смеси, состоящей из 6,74 г хлороацетона, 22,1 г безводного карбоната калия, и 150 мл ацетона. После 30-минутного размешивания, реакционную смесь нагревали с обратным холодильником в течение 30 минут. После охлаждения нерастворившиеся материалы удаляли путем фильтрации, и полученный фильтрат концентрировали досуха. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя толуолом, и полученный продукт перекристаллизовывали из этанола, в результате чего получали 7,5 г 2-ацетил-5-бромобензо о тиофена. Т. пл. 120-121oC. ИК (KBr): 1668, 1512, 1326, 1266 см-1.
1H-ЯМР (CDCl3) δ: 2,67 (3H, с); 7,54 (1H, дд, J = 8,8 и 1,8 Гц); 7,75 (1H, д, J = 8,8 Гц); 7,85 (1H, с); 8,03 (1H, д, J = 1,8 Гц).
d) Размешивая, 5,4 мл брома по капле добавляли к 5 н водному раствору гидроксида натрия, который был охлажден до -5-0oC. К этому раствору, при температуре -5oC или ниже, по капле добавляли 50 мл диоксанового раствора 2-ацетил-5-бромобензо /b/ тиофена, полученного согласно описанию в стадии (c). Полученную смесь размешивали в течение 30 минут при комнатной температуре, а затем в течение 30 минут при 50oC. При охлаждении льдом, pH реакционного раствора доводили до 2 с помощью концентрированной соляной кислоты, а осажденные кристаллы собирали путем фильтрации и промывали водой. Полученные таким образом кристаллы растворяли в этилацетате, и раствор осушали и концентрировали. Осажденные кристаллы собирали путем фильтрации и промывали толуолом, в результате чего получали 6,6 г 5-бромобензо[b]тиофен-2-карбоновой кислоты. Т.пл. 238-241oC.
ИК (KBr): 1671, 1554, 1518, 1443 см-1
1H-ЯМР (CDCl3) δ: 7,57 (1H, дд, J = 8,6 и 1,8 Гц); 7,82 (1H, д, J = 8,6 Гц); 8,00 (1H, с); 8,07 (1H, д, J = 1,8 Гц).
e) 6,4 г 5-бромобензо[b]тиофен-2-карбоновой кислоты, полученной в стадии (d), суспендировали в 250 мл этанола. Охлаждая на ледяной бане и размешивая, к полученной суспензии по капле добавляли 4,45 г тионилхлорида, а затем нагревали с обратным холодильником в течение 1 часа. После чего к полученной смеси, охлаждая при этом льдом, по капле добавляли 8,15 г тионилхлорида, а затем нагревали с обратным холодильником в течение 2 часов. Полученный реакционный раствор концентрировали, а pH доводили до 9 с помощью насыщенного водного раствора бикарбоната натрия. Осажденные кристаллы собирали путем фильтрации, осушали и получали 7,0 г этил-5-бромобензо о тиофен-2-карбоксилата. Часть полученного таким образом соединения перекристаллизовывали из метанола и получали игольчатые кристаллы. Т.пл. 94-95oC.
1H-ЯМР (CDCl3) δ: 1,42 (3H, т, J = 7,0 Гц); 4,41 (2H, кв., J = 7,0 Гц); 7,54 (1H, дд, J = 8,8 и 1,8 Гц); 7,73 (1H, д, J = 8,8 Гц); 7,96 (1H, с); 8,01 (1H, д).
f) 7,0 г этил-5-бромобензо[b]тиофен-2-карбоксилата, полученного в стадии (a), и 5,4 г цианида меди суспендировали в 70 мл N-метил-2-пирролидона, и полученную суспензию размешивали в течение 2 часов при нагревании при температуре 200oC в потоке азота. После охлаждения, реакционную смесь разбавляли этилацетатом, нерастворившиеся вещества удаляли путем фильтрации, и полученный фильтрат промывали водой и осушали. После отгонки растворителя, осажденные кристаллы собирали путем фильтрации и промывали этанолом, в результате чего получали 5,02 г 5-дианобензо о тиофен-2-карбоксилата в виде кристаллов. Т.пл. 138-139oC.
ИК (KBr): 2232, 1728, 1262 см-1
1H-ЯМР (CDCl3) δ: 1,43 (3H, т, J = 7,0 Гц); 4,45 (2H, кв., J = 7,0 Гц); 7,70 (1H, дд, J = 9,0 и 1,8 Гц), 8,04 (1H, д, J = 9,0 Гц); 8,08 (1H), 8,20 (1H)
g) К 150 мл тетрагидрофурана добавляли 4,92 г этил 5-цианобензо[b]тиофен-2-карбоксилата, полученного в стадии (f), а затем добавляли 3,33 г иодида кальция (4H2O). Охлаждая льдом и размешивая, к полученной смеси добавляли 1,0 г борогидрида натрия и каталитически эффективное количество бикарбоната натрия, после чего смесь размешивали при комнатной температуре в течение 1 часа. Затем добавляли еще 3,33 г иодида кальция (4H2O), и к полученной смеси, которую охлаждали на ледяной бане, размешивая при этом, добавляли 1,0 г борогидрида натрия, после чего смесь размешивали при комнатной температуре. После размешивания в течение 1 часа, к смеси опять добавляли 3,33 г иодида кальция (4H2O), а к полученной смеси, которую размешивая, охлаждали на ледяной бане, добавляли еще 1,0 г борогидрида натрия, после чего смесь размешивали при комнатной температуре еще 1 час. Полученный таким образом реакционный раствор разбавляли водой, экстрагировали этилацетатом и осушали для удаления растворителя. Затем осажденные кристаллы собирали путем фильтрации и промывали смесью бензола и н-гексана, в результате чего получали 4,0 г 2-гидроксиметилбензо[b]тиофен-5-карбонитрил. Т.пл. 78-79oC.
ИК (KBr): 3496, 2236, 1026 см-1
1H-ЯМР (CDCl3) δ: 4,97 (2H, с); 7,26 (1H), 7,51 (1H, дд, J = 8,3 и 1,8 Гц); 7,90 (1H, д, J = 8,3 Гц); 8,03 (1H).
h) 4,0 г 2-гидроксиметилбензо[b] тиофен-5-карбонитрила, полученного в стадии (g), растворяли в 100 мл диэтилового эфира с последующим добавлением 0,1 мл пиридина. При охлаждении льдом и размешивании, к полученному раствору добавляли 5 мл диэтилэфирного раствора 5,5 тионилхлорида, и эту смесь размешивали при комнатной температуре в течение 2 часов. Полученный реакционный раствор выливали в ледяную воду и экстрагировали бензолом. Органический слой промывали насыщенным водным раствором бикарбоната натрия и концентрировали досуха. Полученный таким образом осадок растворяли в 100 мл ксилола, и раствор смешивали с 7,2 г трифенилфосфина, и нагревали с обратным холодильником в течение 10 часов. После этого осажденные кристаллы собирали путем фильтрации и получали 6,3 г целевого соединения. Т.пл. 271-274oC (разлож.)
1H-ЯМР (CDCl3) δ: 6,70 (2H, д, J = 15,1 Гц); 7,30 - 8,10 (19H, м).
Ссылочный пример 5. Получение (7-циано-2-нафтил)метилтрифенилфосфония бромида
a) 11,0 г 7-метил-2-нафталинкарбоновой кислоты, полученной в соответствии с процедурой, описанной в Australian Journal of Chemistry (т. 18, стр. 1351-1364, 1965), смешивали с 70 мл тионилхлорида и нагревали с обратным холодильником в течение 4 часов. Реакционный раствор концентрировали досуха. К остатку добавляли 300 мл концентрированного водного аммиака, охлаждая при этом. Смесь размешивали 3 часа при комнатной температуре, а затем экстрагировали этилацетатом. Органический слой промывали водой, а затем насыщенным водным раствором хлорида натрия, после чего этот слой осушали и растворитель удаляли. В результате этой процедуры получали 8,5 г 7-метил-2-нафталинкарбоксамида в виде бесцветных игольчатых кристаллов. Т. пл. 210 - 212oC.
1H-ЯМР (ДМСО-d6) δ: 2,50 (3H, с.); 7,4 - 8,5 (6H, м).
b) 8,0 г 7-метил-2-нафталинкарбоксамида, полученного в стадии (a), суспендировали в 200 мл тетрагидрофурана, к которому затем добавляли при комнатной температуре 100 мл раствора тетрахлорметана, содержащего 22,66 г трифенилфосфина. Полученную смесь размешивали при комнатной температуре в течение 30 минут, а затем при 60oC в течение 40 часов. После охлаждения до комнатной температуры, нерастворившиеся материалы удаляли путем фильтрации, а полученный фильтрат концентрировали при пониженном давлении. 28,35 г полученного остатка хроматографировали на колонке с силикагелем, элюируя смесью н-гексана и этилацетата, и получали в результате 5,73 г 7-метил-2-нафталинкарбонитрила в виде бесцветных кристаллов. Т. пл. 134 - 136oC.
1H-ЯМР (CDCl3) δ: 2,54 (4H, с); 7,4 - 8,2 (6H, м).
c) 5,7 г 7-метил-2-нафталинкарбонитрила, полученного в стадии (b), суспендировали в 100 мл тетрахлорметана. К этой суспензии добавляли 6,37 г N-бромосукцинимида и 30 мг 2,2-азобис-изо-бутилонитрила. После 2-х часового нагревания с обратным холодильником, реакционный раствор разбавляли дихлорметаном, промывали водой, а затем насыщенным водным раствором хлорида натрия, и осушали. После отгонки растворителя, получали 8,34 г 7-бромометил-2-нафталинкарбонитрила в виде бледно-желтых игольчатых кристаллов. Т. пл. 110 - 116oC.
1H-ЯМР (CDCl3) δ: 4,65 (2H, с); 7,55 - 8,25 (6H, м).
d) 8,34 г 7-бромометил-2-нафталинкарбонитрила, полученного в стадии (c), растворяли в 200 мл ксилола, затем раствор смешивали с 11,6 г трифенилфосфина, и полученную смесь нагревали с обратным холодильником в течение 16 часов. К реакционному раствору добавляли диэтиловый эфир, и осажденные кристаллы собирали фильтрацией, высушивали, и получали в результате 12,10 г целевого соединения.
1H-ЯМР (CDCl3) δ: 5,96 (3H, д, J = 15,3 Гц); 7,1 - 8,0 (2H, м).
Ссылочный пример 6. Получение (6-циано-1-метил-2- индолил)метилтрифенилфосфония бромида.
a) 1,5 г метил 6-циано-2-индолкарбоксилата, полученного в соответствии с процедурой, описанной в Liebigs Annalen der Chemie (1986, стр. 438 - 455), растворяли в 20 мл N-диметилформамида. 320 мг 60% гидрида натрия добавляли к полученному раствору, охлаждая при этом льдом и размешивая, и полученную смесь размешивали при комнатной температуре в течение 10 минут. К этой смеси, затем, добавляли 0,47 мл метилиодида, и размешивали при комнатной температуре в течение 2 часов. К полученному реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и осажденные таким образом кристаллы собирали путем фильтрации, и промывали метанолом. Промытые кристаллы перекристаллизовывали из смеси дихлорметана и метанола и получали в результате 1,4 г метил 6-циано-1-метил-2-индолкарбоксилата.
1H-ЯМР (CDCl3) δ: 3,92 (3H, с.); 4,10 (3H, с.); 7,42 (1H, с); 7,52 (1H, дд); 7,98 (1H, дд); 8,38 (1H, шир.).
b) 5,7 г метил 6-циано-1-метил-2-индолкарбоксилата, полученного в стадии (a), растворяли в 120 мл тетрагидрофурана. Охлаждая на ледяной бане и размешивая при этом, к полученному раствору добавляли каталитически эффективное количество бикарбоната натрия, 5,6 г иодида кальция и 1,8 г борогидрида натрия, после чего смесь размешивали 5 часов. Реакционный раствор смешивали с ледяной водой и уксусной кислотой, затем из смеси отгоняли тетрагидрофуран, и полученный водный раствор экстрагировали этилацетатом, а затем осушали. После отгонки растворителя, остаток растворяли в 50 мл дихлорометана. При охлаждении льдом и размешивании, к реакционному раствору по капле добавляли 10 мл раствора дихлорметана, содержащего 1 мл трибромида фосфора, и полученную смесь размешивали при той же температуре 2 часа, а затем еще 2 часа при комнатной температуре. Обработанный таким образом реакционный раствор смешивали с ледяной водой, промывали водным раствором карбоната натрия и осушали. Органический слой концентрировали при пониженном давлении до уменьшения объема в 2 раза, затем смешивали с 15 г трифенилфосфина и нагревали с обратным холодильником 12 часов. После этого осадок собирали путем фильтрации и получали 10,5 г целевого соединения.
1H-ЯМР (ДМСО-d6) δ:/ (3H, с); 5,55 (2H, д); 6,26 (1H, с); 7,20 - 8,10 (18H, м).
Соединения ссылочных примеров 7 и 8 получали в соответствии с процедурой, описанной в Сравнительном примере 6.
Ссылочный пример 7. (6-циано-1-этил-2- индолил)метилтрифенилфосфония бромид
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т); 3,83 (2H); 5,57 (2H, д); 6,26 (1H, с); 7,39 (1H, д); 7,59 (1H, д); 7,70 - 8,00 (16H, м).
Ссылочный пример 8. [1-(2-хлорометил)-6-циано-2-индолил] метилтрифенилфосфония бромид.
1H-ЯМР (ДМСО-d6) δ: 3,40 - 3,80 (2H); 4,30 - 4,60 (2H); 5,60 (2H, д); 6,25 (1H, с); 7,10 - 8,00 (18H, м).
Ссылочный пример 9. Получение 2-бромометил-5-бензотиазолкарбонитрила
a) 28,0 г 5-бромо-2-метилбензотиазола растворяли в 200 мл N-метил-2-пирролидона, и полученный раствор смешивали с 13,8 г цианида меди и каталитически эффективным количеством сульфата меди, после чего смесь размешивали 4 часа с нагреванием при температуре 180 - 190oC в потоке азота. Полученный реакционный раствор выливали в воду, и нерастворившиеся материалы собирали путем фильтрации. Этот нерастворившийся материал смешивали со смесью, содержащей 22 мл этилендиамина и 50 мл воды, и полученную смесь тщательно перемешивали. После экстрагирования бензолом органический слой промывали водой, и осушали путем отгонки бензола. Затем образовавшийся остаток промывали этанолом и получали 10,22 г 2-метил-5-бензотиазолкарбонитрила в виде слегка коричневатых кристаллов, Т. пл. 158 - 160oC.
1H-ЯМР (CDCl3) δ: 2,90 (3H, с); 7,60 (1H, дд); 7,95 (1H, д); 8,25 (1H, д).
b) 7,46 г 2-метил-5-бензотиазолкарбонитрила, полученного в стадии (a), растворяли в 250 мл тетрахлорметана, и раствор нагревали с обратным холодильником в условиях светового облучения. К полученному реакционному раствору постепенно добавляли смесь, содержащую 7,62 г N-бромосукцимида и 150 мг 2,2-азобис-изобутилонитрила, а затем нагревали с обратным холодильником 20 часов. После охлаждения, нерастворившиеся материалы удаляли путем фильтрации, а растворитель отгоняли. Образовавшийся таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя толуолом, и получали в результате 2,18 г целевого соединения в виде светло-желтых призмообразных кристаллов. Т. пл. 185 - 186oC.
1H-ЯМР (CDCl3) δ: 4,83 (2H, с); 7,67 (1H, дд); 8,02 (1H, д); 8,34 (1H, д).
Ссылочный пример 10. Получение (6-циано-1,2,3,4-тетрагидро- 2-нафтил)метилтрифенилфосфония р-толуолсульфоната.
a) 10,0 г метил 6-гидроксиметил-5,6,7,8-тетрагидро-2- нафталинкарбоксилата добавляли к 3,82 г 2,3-дигидропирана. Затем добавляли 5 капель концентрированной серной кислоты и полученную смесь размешивали в течение 1 часа. К этой смеси добавляли 1,00 г 2,3-дигидропирана и 3 капли концентрированной серной кислоты, после чего смесь размешивали 5 часов. Полученную реакционную смесь смешивали с 100 мл диэтилового эфира, и смесь промывали насыщенным водным раствором бикарбоната натрия, затем водой, а затем насыщенным водным раствором хлорида натрия, после чего смесь осушали. После отгонки растворителя, получали 13,72 г метил 6-[(2-тетрагидропиранил)оксиметил]-5,6,7,8-тетрагидро-2- нафталинкарбоксилата в виде желтого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,50 - 3,00 (13H, м); 3,30 - 4,10 (4H, м); 3,86 (3H, с); 4,60 (1H, шир.); 7,10 (1H, д); 7,80 - 7,90 (2H, м).
b) 13,72 г метил 6-[(2-тетрагидрофуранил)оксиметил]-5,6,7,8- тетрагидро-2-нафталинкарбоксилата, полученного в стадии (a), растворяли в 180 мл метанола. После добавления раствора 2,96 г гидроксида натрия в 60 мл воды, полученную смесь нагревали с обратным холодильником в течение 3 часов. После охлаждения, реакционный раствор концентрировали при пониженном давлении, затем смешивали с хлороформом и водой и нейтрализовали уксусной кислотой. Полученный органический слой промывали водой, а затем насыщенным водным раствором хлорида натрия, и осушали. После отгонки растворителя, полученный остаток кристаллизовали в изопропиловом эфире и получали в результате 10,51 г 6-[(2-тетрагидропиранил)оксиметил] -5,6,7,8- тетрагидро-2-нафталинкарбоновой кислоты.
1H-ЯМР (CDCl3) δ: 1,50 - 3,00 (13H, м); 3,30 - 4,00 (4H, м); 4,60 (1H, шир.); 7,16 (1H, д); 7,80 - 7,90 (2H, м).
c) 12,0 г 6-[(2-тетрагидропиранил)оксиметил]-5,6,7,8- тетрагидро-2-нафталинкарбоновой кислоты, полученной в стадии (b), и 4,1 г триэтиламина растворяли в 100 мл тетрагидрофурана, и полученный раствор охлаждали до -15oC. Затем к этому раствору добавляли, размешивая при этом, 5,64 г изобутилового эфира хлормуравьиной кислоты. Полученный реакционный раствор размешивали 20 минут при той же температуре, а затем выливали в 200 мл охлажденного льдом этанола, содержащего 14% (масс./об.) аммиака. После удаления нерастворившихся материалов путем фильтрации, полученный фильтрат осушали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью н-гексана и этилацетата, и очищенный продукт кристаллизовали в изопропиловом эфире, в результате чего получали 7,20 г 6-[(2-тетрагидропиранил)оксиметил]-5,6,7,8-тетрагидро-2- нафталинкарбоксамида.
1H-ЯМР (CDCl3) δ: 1,40 - 3,00 (13H, м); 3,30 - 4,00 (4H, м); 4,60 (1H, шир.); 6,10 (2H, шир.); 7,20 (1H, д); 7,50 - 7,70 (2H, м).
b) 15,0 г 6-[(2-тетрагидропиранил)оксиметил] -5,6,7,8- тетрагидро-2-нафталинкарбоксамида, полученного в стадии (c), суспендировали в 60 мл диоксана. После добавления 8,35 мл пиридина, суспензию охлаждали до -8oC - 0oC.
Затем, к этой суспензии, размешивая при этом, по капле добавляли 7,89 мл безводного трифторацетата. Полученный реакционный раствор размешивали 30 минут при -5oC, затем 2 часа при комнатной температуре. Обработанный таким образом реакционный раствор разбавляли хлороформом и последовательно промывали водой и насыщенным водным раствором хлорида натрия. Полученный органический слой осушали, растворитель отгоняли, и получали в результате 9,78 г 6-[(2-тетрагидропиранил)оксиметил-5,6,7,8-тетрагидро-2- нафталинкарбонитрила в виде маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,50-3,00 (13H, м), 3,30-4,40 (4H, м); 4,61 (1H, шир); 7,05 - 7,50 (3H, м).
е) 9,78 г 6-[(2-тетрагидропиранил)оксиметил] -5,6,7,8-тетрагидро- 2-нафталинкарбонитрила растворяли в 100 мл этанола. После добавления 100 мг р-толуолсульфоновой кислоты, полученную смесь размешивали 15 часов при комнатной температуре. Затем полученный реакционный раствор нейтрализовали с помощью насыщенного водного раствора бикарбоната натрия, с последующим удалением растворителя путем дистилляции. Полученный таким образом остаток растворяли в хлороформе, а раствор промывали водой, а затем насыщенным водным раствором хлорида натрия. Органический слой осушали, а растворитель отгоняли. После кристаллизации из изопропанола получали 5,26 г 6-гидроксиметил-5,6,7,8-тетрагидро-2-нафталинкарбонитрила в виде бесцветных кристаллов. Т.пл. 83-85oC.
1H-ЯМР (CDCl3) δ: 1,30 - 3,00 (7H, м); 3,64 (2H, д, J = 6,0 Гц); 7,05 - 7,50 (3H, м).
f) 15,0 г 6-гидроксиметил-5,6,7,8-тетрагидро-2-нафталинкарбонитрила, полученного в стадии (e), и 3,50 г р-толуолсульфонилхлорида растворяли в 150 мл пиридина, и полученный раствор размешивали при комнатной температуре 15 часов. Полученный реакционный раствор выливали в ледяную воду, и осажденные кристаллы собирали путем фильтрации, промывали водой, а затем изопропанолом, и осушали. В результате этой процедуры получали 24,72 г бесцветного 5,6,7,8- тетрагидро-6-[(p-толуолсульфонил)оксиметил]-2-нафталинкарбонитрила.
Т.пл. 100- 102oC.
1H-ЯМР (CDCl3) δ: 1,20 - 3,80 (7H, м); 2,47 (3H, с); 4,00 (2H, д, J = 6,0 Гц), 7,10 (1H, д, J = 9,0 Гц), 7,30 - 7,50 (4H, м); 7,80 (2H, д).
q) 24,00 г 5,6,7,8-тетрагидро-6-[(p-толуолсульфонил)-оксиметил]-2-нафталинкарбонитрила и 18,38 г трифенилфосфина смешивали, а затем нагревали 15 часов при температуре 130-140oC в герметично закрытом сосуде. Полученный реакционный продукт кристаллизовали из смеси ацетона и н-гексана, в результате чего получали 23,3 г целевого соединения в виде светло-желтого порошка.
1H-ЯМР (CDCl3) δ: 1,40 - 2,90 (7H, м); 2,27 (3H, с); 3,60 - 3,90 (2H, м); 6,80 - 7,30 (5H, м); 7,40 - 8,00 (17H, м).
Ссылочный пример 11. Получение (6-циано-2-нафтил)метилтрифенилфосфония бромида
а) 6,11 г 6-метил-2-нафталинкарбонитрила растворяли в 100 мл тетрахлорметана, и полученный раствор смешивали с 6,63 г N-бромсукцинимида и 30 г 2,2-азобис-изо-бутилонитрила. После 4-х часового нагревания с обратным холодильником, реакционный раствор смешивали с хлороформом, промывали водой, а затем осушали. После отгонки растворителя получали 7,07 г бесцветного 6-бромометил-2-нафталинкарбонитрила.
Т.пл.134-137oC.
1H-ЯМР (CDCl3) δ: 4,65 (2H, с); 7,60 - 7,80 (2H, м); 7,80 - 8,00 (3H, м); 8,22 (1H, с).
b) 2,0 г 6-бромометил-2-нафталинкарбонитрила, полученного в стадии (a), и 2,77 г трифенилфосфина растворяли в 50 мл ксилола. После 18-часового нагревания с обратным холодильником, осажденные кристаллы собирали путем фильтрации и получали 3,31 г целевого соединения. Т.пл. > 270oC.
1H-ЯМР (CDCL3) δ: 5,93 (2H, д, J = 15,2 Гц); 7,40 - 8,00 (21H, м).
Ссылочный пример 12. Получение (S)-(+)-3-гидрокситетрагидрофурана.
0,23 г р-толуолсульфоновой кислоты добавляли к 25 г (S)-(-)-1,2,4-бутантриола, и полученную смесь размешивали при 100oC 5 минут, а затем при 180-200oC 10 минут. Эту реакционную смесь подвергали дистилляции, и собирали фракцию при 95-100 C/300 мм рт.ст., получая тем самым 16,2 г целевого соединения в виде маслянистого материала.
1H-ЯМР (CDCl3) δ: 1,80 - 2,20 (2H, м); 3,76 (2H, д); 3,70 - 4,10 (2H, м); 4,40 - 4,60 (1H, м).
Ссылочный пример 13. Получение этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил окси]фенил]-2-оксоацетата.
В 40 мл тетрагидрофурана растворяли 1,8 г этил 2-(4-гидроксифенил)-2-оксоацетата, 1,74 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина и 2,92 г трифенилфосфина. К этому раствору, при комнатной температуре, добавляли 1,94 г диэтилазокарбоксилата, и полученную смесь размешивали 18 часов. После отгонки растворителя, остаток растворяли в этилацетате, и раствор промывали водой, а затем осушали. После этого растворитель отгоняли, а полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью толуола и хлороформа, в результате чего получали 2,53 г целевого соединения в виде вязкого желтого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,41 (3H, т, J = 7,0 Гц), 1,46 (9H, с); 2,00 - 2,40 (2H, м); 3,00 - 3,75 (4H, м); 4,43 (2H, кв., J = 7,0 Гц); 5,00 (1H, шир.); 6,93 (2H, д, J = 9,0 Гц); 8,00 (2H, д, J = 9,0 Гц).
Соединения, представленные в ссылочных примерах 14-25, были получены в соответствии с процедурой, описанной в ссылочном примере 13.
Ссылочный пример 14. Метил-2-[4-[((3S)-трет-бутоксикарбонил-3-пирролидинил)окси]-фенил]-2- оксоацетат, вязкое желтое маслянистое вещество.
Ссылочный пример 15. Этил-2-[4-[((3R)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил]-2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCL3) δ: 1,40 (3H, т, J = 7,0 Гц); 1,46 (9H, с); 2,00 -2,35 (2H, м); 3,45 - 3,75 (4H, м); 4,90 (2H, кв., J=7,0 Гц); 4,9 - 5,1 (1H, шир. ); 4,9 - 5,1 (1H, шир.); 6,95 (2H, д, J = 9,0 Гц); 8,00 (2H, д, J = 9,0 Гц).
Ссылочный пример 16. Этил 2-[4-[((2S-1-трет-бутоксикарбонил-2-пирролидинил))-метокси]фенил]-2- оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,41 (3H, т); 1,47 (9H, с); 2,0 (4H, шир.), 3,37 (2H, шир.); 4,20 (3H, шир.); 4,43 (2H, кв.), 7,0 (2H, д), 7,95 (2H, д).
Ссылочный пример 17. Этил 2-[4-[((2S, 4S)-1-трет-бутоксикарбонил-2-карбамоил-4-пирролидинил)окси] фенил] -2- оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,42 (3H, т, J = 7,0 Гц); 1,48 (9H, с); 2,20 - 2,90 (2H, шир.); 3,64 - 3,90 (2H, шир.); 4,30 - 4,60 (1H, шир.); 4,42 (2H, кв., J = 7,0 Гц); 5,60 (1H, шир.); 6,97 (2H, д, J = 9,0 Гц); 8,07 (2H, д, J = 9,0 Гц).
Ссылочный пример 18. Этил-2-[4-[((2S, 4S)-1-трет-бутоксикарбонил-2-диметилкарбамоил-4-пирролидинил)окси] фенил] -2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,37 - 1,50 (12H, м); 1,96 - 2,30 (1H, м); 2,50 - 2,82 (1H, м); 2,90 - 3,15 (6H, шир.); 3,70 (1H, дд, J = 10,8 и 5,1 Гц); 3,90 - 4,16 (1H, м); 4,46 (2H, кв., J = 7,0 Гц); 4,60 - 5,14 (2H, м); 7,00 (2H, д, J = 9,4 Гц); 8,08 (2H, д, J = 9,4 Гц).
Ссылочный пример 19. Этил 2-[4-[2-(трет-бутоксикарбонилоамино)-1-(трет-бутоксикарбониламинометил) этокси] фенил]-2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,00 - 1,70 (21H, шир.); 2,80 - 3,80 (4H, м); 4,20 - 4,60 (3H, м); 7,10 (2H, д, J=8,3 Гц); 7,98 (2H, д, J=8,3 Гц).
Ссылочный пример 20. Этил 2-[4-[(1-трет-бутоксикарбонил-4- пиперидинил)окси]фенил]-2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,35 (3H, т, J=6 Гц); 1,49 (9H, с); 1,8 - 2,0 (4H, м), 3,2 - 4,0 (4H, м); 4,46 (2H, кв., J=6 Гц); 4,6 - 4,8 (1H, м); 7,01 (2H, д, J=9 Гц); 8,04 (2H, д, J=9 Гц).
Ссылочный пример 21. Этил 2-[4-(2-трет-бутоксикарбониламиноэтокси)фенил] -2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,42 (3H, т, J=7,0 Гц); 1,46 (9H, с); 3,56 (2H, кв., J=5,4 Гц); 4,12 (2H, квинтет, J=5,4 Гц); 4,44 (2H, кв., J=7,0 Гц); 5,04 (1H, шир.); 6,98 (2H, д, J=9,0 Гц); 8,00 (2H, д, J=9,0 Гц).
Ссылочный пример 22. Этил 2-[4-[(1-трет-бутоксикарбонил-4-пиперидинил)метокси]фенил]-2- оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,2 - 1,3 (2H, м); 1,42 (3H, т, J=7,1 Гц); 1,47 (9H, с); 1,65 - 1,80 (2H, м); 3,89 (2H, д); 4,10 - 4,25 (2H, м); 4,43 (2H, кв., J=7,1 Гц); 6,95 (2H, д, J=8,8 Гц); 7,99 (2H, д, J=8,8 Гц).
Ссылочный пример 23. Этил 2-[4-[((2S)-1-трет-бутоксикарбонил-5-оксо-2-оксо-2-пирролидин) метокси] фенил] -2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,35 (3H, т); 1,41 (9H); 1,80 - 2,20 (2H, м); 2,47 (2H, т); 4,05 (2H, шир.); 4,41 (2H, кв.); 4,70 - 5,00 (1H, м); 6,98 (2H, д); 8,00 (2H, д).
Ссылочный пример 24. Этил 2-[4-[(2R, 4S)-1-трет-бутоксикарбонил-2-метил-4-пирролидинил)окси] фенил] - 2-оксоацетат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,20 - 1,42 (6H, м); 1,47 (9H, с); 2,90 - 2,60 (1H, м); 3,50 - 3,80 (2H, м); 3,90 - 4,22 (1H, м); 4,42 (2H, кв.); 4,90 - 5,10 (1H, м); 6,95 (2H, д); 8,00 (2H, д).
Ссылочный пример 25. Метил 2-оксо-2-[4-[((3R)-тетрагидро-3- фуранил)окси]фенил]ацетат, вязкое желтое маслянистое вещество.
Ссылочный пример 26. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил оксифенил]-2-этоксикарбонилацетат.
a) 27,7 г этил 4-метоксифенилацетат и 34 мл диэтилкарбоната растворяли в 150 мл N,N-диметилформамида, и полученный раствор нагревали с обратным холодильником, постепенно, в течение 1 часа добавляя 6,5 г гидрида натрия. Затем продолжали нагревать с обратным холодильником еще 2 часа, после чего реакционный раствор выливали в смесь ледяной воды и соляной кислоты и экстрагировали этилацетатом. Полученный органический слой промывали водой, осушали с отгонкой растворителя. Полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя толуолом, в результате чего получали 26,7 г этил 2-этоксикарбонил-2-4-метоксифенил ацетата в виде светло-желтого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,25 (6H, т, J=7,0 Гц); 3,79 (3H, с); 4,20 (4H, кв., J=7,0 Гц); 4,55 (1H, с); 6,88 (2H, д, J=8,0 Гц); 7,32 (2H, д, J=8,0 Гц).
b) 5,8 г этил 2-этоксикарбонил-2-4-метоксифенил ацетата, полученного в стадии (a), растворяли в 70 мл дихлорметана, и раствор охлаждали до -40oC. К этому раствору, размешивая, при этом, по каплям добавляли 6,2 мл трибромида бора, растворенного в 5 мл дихлорметана. После завершения добавления, раствор нагревали до комнатной температуры и размешивали 30 минут. Полученный реакционный раствор выливали в смесь ледяной воды и соляной кислоты, после чего экстрагировали хлороформом. Полученный органический слой осушали путем отгонки растворителя, а остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 4,7 г этил 2-этоксикарбонил-2-4-гидроксифенил ацетата в виде бесцветного маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,27 (6H, т, J=7,0 Гц); 4,22 (4H, кв., J=7,0 Гц); 4,55 (1H, с); 5,66 (1H, шир.); 6,76 (2H, д, J=8,0 Гц); 7,25 ((2H, д, J=8,0 Гц).
c) В 150 мл тетрагидрофурана растворяли 4,7 г этил-2-этоксикарбонил-2-(4-гидроксифенил)ацетата, полученного в стадии (b); 6,58 г трифенилфосфина и 4,7 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина. К полученному таким образом раствору, размешивая при этом, добавляли 4,37 г диэтилазодикарбоксилата, и продолжали размешивать в течение 18 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью толуола и этилацетата, в результате чего получали 4,0 г целевого соединения в виде бесцветного маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,25 (6H, т, J=7,0 Гц); 1,46 (9H, с); 2,1 (2H, шир.); 3,55 (4H, шир. ); 4,20 (4H, кв., J=7,0 Гц); 4,52 (1H, с); 4,82 (1H, шир.); 6,82 (2H, д, J=8,0 Гц); 7,28 (2H, д, J=8,0 Гц).
Ссылочный пример 27. Этил 2-[4-[(2R)-1-трет-бутоксикарбонил-2-пирролидинил)-метокси]фенил]-2-этоксикарбонилацетат.
Это соединение было получено согласно процедуре, описанной в ссылочном примере 26, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,25 (6H, т, J=7,0 Гц); 1,47 (9H, с); 2,0 (4H, шир.); 3,40 (2H, шир.); 3,9 (1H); 4,20 (6H); 4,54 (1H, с); 6,82 (2H, д, J=8,0 Гц); 7,28 (2H, д, J=8,0 Гц).
Ссылочный пример 28. Этил 2-этоксикарбонил-2-[4-[(2- имидазолин-2-ил)метокси]фенилацетат.
a) к 150 мл ацетона добавляли 14,58 г этилэтоксикарбонил-2-4-гидроксифенил ацетата, 8,8 г бромоацетонитрила и 9,6 г безводного карбоната калия. После 5-часового нагревания с обратным холодильником нерастворившиеся материалы
удаляли путем фильтрации, а полученный фильтрат концентрировали досуха. Остаток очищали с помощью колоночной хроматографии, элюируя толуолом, и получали в результате 14,2 г этил-2-[4-(цианометокси)фенил]-2-этоксикарбонилацетата в виде бесцветного маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,26 ((6H, т, J=8,0 Гц); 4,22 (4H, кв., J=8,0 Гц); 4,58 (1H, с); 4,75 (2H, с); 7,02 (2H, д, J=9,0 Гц); 7,36 (2H, д, J=9,0 Гц).
b) 14,2 г этил 2-4-цианометоксифенил-2-этоксикарбонилацетата, полученного в стадии (a), растворяли в смеси, состоящей из 20 мл этанола и 150 мл диэтилового эфира. Полученный раствор размешивали при комнатной температуре в течение 18 часов, охлаждая при этом льдом, в потоке хлорводорода. После отгонки растворителя, получали 16,9 г этил-2-этоксикарбонил-2-[4-(2-этокси-2-иминоэтокси)фенил]ацетата гидрохлорида в виде твердого вещества.
c) При охлаждении льдом и размешивании, 40 мл этанолового раствора, содержащего 3,6 г этил 2-этоксикарбонил-2-[4-(2-этокси-2-иминоэтокси]фенил] ацетата, полученного в стадии (b), по капле добавляли к 10 мл этанолового раствора, содержащего 0,6 г этилендиамина, и полученную смесь размешивали при комнатной температуре в течение 1,5 часов, после чего смесь нагревали с обратным холодильником в течение 0,5 часов. После охлаждения, pH полученного реакционного раствора доводили по кислотного значения с помощью этанола, содержащего 13% (мас. /об.) соляную кислоту, а затем смесь концентрировали досуха. Остаток растворяли в воде и промывали диэтиловым эфиром. После этого pH полученного водного слоя доводили до 9 - 10 с помощью разбавленного водного раствора гидроксида натрия, а осажденные кристаллы собирали путем фильтрации. В результате этой процедуры получали 1,83 г целевого соединения в виде бесцветных кристаллов.
Т.пл. 72 - 110oC постепенное смачивание
FAB-MC (масс-спектроскопия путем бомбардировки быстрыми атомами) (m/z): 335 (M++1).
1H-ЯМР (CDCl3) δ: 1,23 (6H, т, J=8,0 Гц); 3,62 (4H, с); 4,10 (4H, кв., J= 8,0 Гц); 5,52 (1H, с); 4,68 (2H, с); 6,94 (2H, д, J=10 Гц); 7,26 (2H, д, J=10 Гц).
Ссылочный пример 29. Получение этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(5- циано-2-бензофуранил)пропионата
a) 3,12 г этил 2-[4-[((3S)-1-третбутоксикарбонил-3- пирролидинил)окси] фенил]-2-оксоацетата растворяли в 100 мл тетрагидрофурана, а затем добавляли 4,65 г 5-циано-2-бензофуранил метилтрифенилфосфония хлорида. К полученному раствору добавляли 400 мг 60% гидрида натрия. К этой смеси, размешивая, по капле добавляли 3 мл этанола, и полученную смесь размешивали один час при комнатной температуре. Реакционный раствор нейтрализовали с помощью 10% раствором лимонной кислоты, экстрагировали этилацетатом, и осушали путем отгонки растворителя. Полученный остаток хроматографировали на колонке с силикагелем, элюируя смесью толуола и этилацетата, в результате чего получали 3,1 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3-5- циано-2-бензофуранил акрилата в виде вязкого маслянистого продукта, представляющего собой смесь E- и Z-форм. Часть полученного таким образом соединения разделяли на E- и Z- формы.
E-форма (менее полярная):
1H-ЯМР (CDCl3) δ: 1,32 (3H, т, J=7,6 Гц); 1,49 (9H, с); 1,70-2,40 (2H, м); 3,30-3,80 (4H, м); 4,30 (2H, кв., J= 7,6 Гц); 4,92 (1H, шир.); 6,62 (1H, с); 6,94 (2H, д, J=9,0 Гц); 7,24 (2H, д, J=9,0 Гц); 7,38 (1H, д, J=8,6 Гц); 7,56 (1H, д, J=8,6 Гц); 7,74 (1H, с); 7,77 (1H, с);
Z-форма:
1H-ЯМР (CDCl3) δ: 1,10-1,60 (12H, м); 2,00-2,30 (2H, м); 3,30-3,80 (4H, м); 4,50 (3H, кв., J=7,2 Гц); 4,92 (1H, шир.), 6,76 (1H, с); 6,81 (1H, с); 6,88 (2H, д, J=8,75 Гц); 7,88 (2H, д, J=8,75 Гц); 7,31-7,60 (2H), 7,85 (1H, с).
b) 3,1 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3- 5-циано-2-бензофуранил] акрилата, полученного в стадии (a), растворяли в смеси, состоящей из 100 мл тетрагидрофурана и 100 мл этанола. К полученному раствору добавляли 700 мг окиси палладия 1H2O сульфата бария, полученного согласно процедуре, описанной в Angewandte Chemie (vol.67, стр. 785, 1955). После 6-часового каталитического гидрирования при нормальном давлении, катализатор удаляли путем фильтрации, а полученный фильтрат концентрировали. После этого, остаток хроматографировали на колонке с силикагелем, элюируя смесью толуола и этилацетата, в результате чего получали 1,9 г целевого соединения в виде вязкого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,00-1,40 (3H, м); 1,46 (9H, с); 2,00-2,30 (2H, м); 3,16 (1H, дд, J=14,4 и 7,2 Гц); 3,40-3,80 (5H, м); 3,90-4,30 (3H, м); 4,94 (1H, шир. ); 6,40 (1H, с); 6,80 (2H, д, J=8,7 Гц); 7,25 (2H, д, J=8,7 Гц); 7,46 (2H, с); 7,76 (1H, с).
Ссылочный пример 30. Получение этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]- 3-(7-циано-2-нафтил)пропионата.
a) 8,40 г (7-циано-2-нафтил)метилтрифенилфосфония бромида и 5,0 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил]-2-оксоацетата и суспендировали в смеси 100 мл тетрагидрофурана и 100 мл этанола. К полученной суспензии добавляли, размешивая при этом, 2,51 г 1,8-диазобицикло[5.4.0]-7-ундецена, а затем перемешивали 3 часа при комнатной температуре. После отгонки растворителя, остаток хроматографировали на колонке с силикагелем, элюируя смесью н-гексана и этилацетата, в результате чего получали 6,06 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]- фенил] -3-(7-циано-2-нафтил)акрилата в виде смеси E- и Z-форм. Часть полученного таким образом соединения разделяли на E- и Z-форм.
E-форма: т.пл. 104-106oC кристаллизация в этаноле
1H-ЯМР (CDCl3) δ: 1,35 (3H, т, J=7,3 Гц); 1,48 (9H, с); 2,05-2,30 (2H, м); 3,45-3,70 (4H, м); 4,31 (2H, кв., J=7,3 Гц); 4,92 (1H, шир.); 6,86 (2H, д, J= 8,8 Гц); 7,16 (2H, д, J=8,8 Гц); 7,20 (1H, дд, J=8,8 и 1,5 Гц); 7,56 (1H, дд, J=8,3 и 1,5 Гц); 7,62 (1H, д, J=8,8 Гц); 7,73 (1H, с); 7,80 (1H, д, J=8,3 Гц); 7,93 (1H, с); 8,07 (1H, с);
Z-форма:
1H-ЯМР (CDCl3) δ: 1,19 (3H, т, J=7,3 Гц); 1,48 (9H, с); 2,05-2,30 (2H, м); 3,45-3,70 (4H, м); 4,29 (2H, кв., J=7,3 Гц); 4,93 (1H, шир.); 6,90 (2H, д, J= 8,8 Гц); 7,09 (1H, с); 7,44 (2H, д); 7,60 (1H, дд, J=8,3 и 1,5 Гц); 7,63 (1H, с); 7,90 (1H, д, J=8,3 Гц); 8,18 (1H, с).
b) 6,06 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил]- 3-(7-циано-2-нафтил)акрилата, полученного в виде смеси E- и Z-форм в стадии (a) (см. выше), растворяли в смеси, состоящей из 80 мл тетрагидрофурана и 80 мл этанола. К полученному раствору добавляли 2,0 г окиси палладия • 1H2O сульфата бария. После каталитической гидрогенизации при нормальном давлении в течение 3,5 часов, катализатор удаляли путем фильтрации, а растворитель отгоняли. Полученный остаток подвергали колоночной хроматографии на силикагеле, используя в качестве элюента смесь н-гексана и этилацетата, в результате чего получали 6,24 г целевого соединения в частично отвержденной форме.
1H-ЯМР (CDCl3) δ: 1,11 (3H, т, J=7,3 Гц); 1,47 (9H, с); 2,00-2,33 (2H, м); 3,18 (1H, дд, J=14,2 и 6,8 Гц); 3,40-3,65 (5H, м); 3,88 (1H, т, J=7,5 Гц); 4,06 (2H, кв., J=7,3 Гц); 4,85 (1H, шир.); 6,80 (2H, д, J=8,8 Гц); 7,24 (2H, д); 7,42 (1H, дд, J=8,8 и 1,5 Гц); 7,54 (1H, дд, J=8,3 и 1,5 Гц); 7,62 (1H, c); 7,77 (1H, д, J=8,8 Гц); 7,85 (1H, д, J=8,3 Гц); 8,13 (1H, с).
Соединения, представленные в ссылочных примерах 31-39, получали в соответствии с процедурой, описанной в ссылочном примере 30.
Ссылочный пример 31. Этил 2-[4-[((3R)-1-трет-бутоксикарбонил-3-пирролидинил)оксифенил]-3- (7-циано-2-нафтил)пропионат.
1H-ЯМР (CDCl3) δ: 1,11 (3H, т, J=7,3 Гц); 1,47 (9H, с); 2,00-2,35 (2H, м); 3,18 (1H, дд, J=14,2 и 6,8 Гц); 3,40-3,70 (5H, м); 3,38 (1H, шир.); 4,06 (2H, кв. , J= 7,3 Гц); 4,85 (1H, шир.); 6,80 (2H, д, J=8,8 Гц); 7,24 (2H); 7,42 (1H, дд, J=8,8 Гц и 1,5 Гц); 7,54 (1H, дд, J=8,3 и 1,5 Гц); 7,62 (1H, с); 7,77 (1H, д, J=8,8 Гц), 7,84 (1H, д, J=8,3 Гц); 8,11 (1H, с).
Ссылочный пример 32. Этил 2-[4-[(1-трет-бутоксикарбонил-4-пиперидинил)окси]фенил- 3-(7-циано-2-нафтил)пропионат.
1H-ЯМР (CDCl3) δ: 1,11 (3H, т); 1,49 (9H, с); 1,70-2,00 (4H, м); 3,00-4,10 (9H, м); 4,45 (1H, шир.); 6,80-8,10 (10H, м);
FAB-MC (m/z): 418 (M++1).
Ссылочный пример 33. Этил 2-[4-[((2S, 4S)-1-трет-бутоксикарбонил-2-карбамоил-4-пирролидинил)окси] фенил] - 3-(5-циано-2-бензофуранил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J=7,0 Гц); 1,47 (9H, с); 2,10-2,80 (2H, шир. ); 3,16 (1H, дд, J=14,4 и 7,2 Гц); 3,40-4,50 (6H, м); 5,08 (1H, шир.); 6,39 (1H, с); 6,76 (2H, д, J=8,35 Гц); 7,26 (2H, д, J=8,35 Гц); 7,50 (2H, с); 7,80 (1H).
Ссылочный пример 34. Этил 2-[4-[((2S, 4S)-1-трет-бутоксикарбонил-2-диметилкарбамоил-4-пирролидинил)окси] фенил]-3-5-циано-2-бензофуранил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,23 (3H, т); 1,44 (9H, с); 1,90-2,30 (1H, шир.); 2,40-2,80 (1H, шир.); 2,98 (1H, с); 3,10-4,23 (7H, м); 4,40-5,00 (2H, шир.); 6,38 (1H, с); 6,90 (2H, д, J=8,3 Гц); 7,20 (2H, д, J=8,35 Гц); 7,45 (2H, с); 7,76 (1H, с).
Ссылочный пример 35. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси]фенил]-3-(5-циано-3-метил-2- бензофуранил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т); 1,47 (9H, с); 2,02 (3H, с); 2,1 (2H, шир. ); 3,1 (1H, шир.); 3,6 (5H, шир.); 4,1 (3H, м); 4,85 (1H, шир.); 6,83 (2H, д); 7,15 (2H, д); 7,46 (2H); 7,7 (1H, с).
Ссылочный пример 36. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)оксифенил] -3-(5- циано-7-метокси-2-бензофуранил)пропионат, вязкое желтое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J=7 Гц); 1,46 (9H, с); 2,00-2,30 (2H, м); 3,16 (1H, дд, J=14,5 и 7,4 Гц); 3,40-3,76 (5H, м); 3,80-4,30 (3H, м); 4,02 (3H, с); 4,70-5,00 (1H, шир.); 6,37 (1H, с); 6,80 (2H, д, J=8,75 Гц); 6,95 (1H, д, J=1,3 Гц); 7,23 (2H, д, J=8,75 Гц); 7,41 (1H, д, J=1,3 Гц).
Ссылочный пример 37. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)оксифенил] -3- (5-циано-3-бензофуранил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,14 (3H, т); 1,45 (9H, с); 2,12 (2H, шир.); 2,90-4,00 (7H, м); 4,08 (2H, кв.); 4,84 (1H, шир.); 6,85 (2H, д); 7,2 (2H, д); 7,41 (1H, с); 7,50 (2H); 7,72 (1H).
Ссылочный пример 38. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)оксифенил]-3-(6-циано-2-нафтил)пропионат.
1H-ЯМР (CDCl3) δ: 1,25 (3H, т, J = 7,0 Гц); 1,46 (9H, с); 2,00-2,20 (2H, м); 3,00-4,00 (7H, м); 4,08 (2H, кв.); 4,85 (1H, шир.); 6,80-8,20 (10H, м).
Ссылочный пример 39. Этил 2-[4-[(1-трет-бутоксикарбонил-4- пиперидинил)метокси]фенил]-3-(7-циано-2-нафтил)пропионат.
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,1 Гц); 1,1-1,2 (2H, м); 1,39 (9H, с); 1,68-1,76 (2H, м); 2,65-2,75 (2H, м); 3,78 (2H, д); 3,9-4,1 (5H, м); 4,55-4,65 (1H, м); 6,85 (2H, д, J = 8,3 Гц); 7,25 (2H, д, J = 8,3 Гц); 7,55-7,65 (1H, м); 7,68-7,73 (1H, м); 7,82 (1H, с); 7,90-7,95 (1H, м); 8,03 (1H, д, J = 8,8 Гц); 8,44 (1H, с).
Ссылочный пример 40. Получение этил (+)-2-[4-[((3S)-1- третбутоксикарбонил-3-пирролидинил)окси] фенил]-3-(7-циано-2- нафтил)пропионата и этил (-)-2-[4-[((3S)-1-третбутоксикарбонил-3- пирролидинил)-окси]фенил]-3-(7-циано-2-нафтил)пропионата.
2,0 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси] фенил] -3-(7-циано-2-нафтил)пропионата растворяли в 10 мл этанола, нагревая при этом. После охлаждения до комнатной температуры, осажденные кристаллы собирали путем фильтрации, а затем перекристаллизовывали дважды из этанола, в результате чего получали 640 мг этил (+)-2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси] фенил] -3-(7-циано-2-нафтил)пропионата. Т.пл. 132-133,5oC.
[α]
1H-ЯМР (CDCl3) δ: 1,11 (3H, т; J = 7,3 Гц); 1,47 (9H, с ); 2,00-2,30 (2H, м); 3,18 (1H, дд, J = 14,2 и 6,8 Гц); 3,40-3,70 (5H, м); 3,87 (1H, т, J = 7,6 Гц); 4,00-4,10 (2H, м); 4,85 (1H, шир.); 6,80 (2H, д, J = 8,8 Гц); 7,20-7,30 (2H, м); 7,42 (1H, д, J = 8,3 Гц); 7,55 (1H, д, J = 8,3 Гц); 7,63 (1H, с); 7,77 (1H, д, J = 8,3 Гц); 7,85 (1H, д, J = 8,3 Гц); 8,12 (1H, с).
ВЭЖХ: Колонка: колонка с амилозой для разделения оптических изомеров (CHIPALPAK AD, 4,60 ⊘ х 250 мм, Diacel Chemical Industries, Ltd.)
Растворитель: изо-пропанол:н-гексан = 15:85 (об./об.)
Скорость потока: 1 мл/мин.
Температура колонки: 25oC
Время удерживания: 31,37 минут
Фильтрат концентрировали досуха и кристаллизовали из смеси н-гексана и этанола. Собранные кристаллы перекристаллизовывали из той же смеси, и получали в результате 80 мг этил (-)-2-[4-[((3S)-1- трет-бутоксикарбонил-3-пирролидинил)-окси]фенил]-3-(7-циано-2- нафтил)пропионата. Т.пл. 82,5-85,0oC
[α]
1H-ЯМР (CDCl3) δ: 1,11 (3H, т, J = 7,3 Гц); 1,47 (9H, с); 2,00-2,30 (2H, м); 3,18 (1H, дд, J = 14,2 и 6,8 Гц); 3,40-3,66 (5H, м); 3,87 (1H, т, J = 7,6 Гц); 4,00-4,10 (2H, м); 4,85 (1H, шир.); 6,80 (2H, д, J = 8,8 Гц); 7,20-7,30 (2H, м); 7,42 (1H, д, J = 8,3 Гц); 7,56 (1H, д, J = 8,3 Гц); 7,62 (1H, с); 7,77 (1H, д, J = 8,3 Гц); 7,85 (1H, д, J = 8,3 Гц); 8,12 (1H, с).
ВЭЖХ: Колонка: колонка с амилозой для разделения оптических изомеров (CHIPALPAK AD, 4,6 ⊘ х 250 нм, Diacel Chemical Industries, Ltd.)
Растворитель: изопропанол:н-гексан = 15:85 об./об.
Скорость потока: 1 мл/мин.
Температура колонки: 25oC
Время удерживания: 23,22 мин.
Ссылочный пример 41. Получение этил 3-(5-циано-2-бензофуранил)- 2-[4-[((3S)-1-метил-3-пирролидинил)окси]фенил]пропионата.
1,8 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3-(5- циано-2-бензофуранил)пропионата растворяли в 28 мл муравьиной кислоты, и полученный раствор размешивали при 70oC в течение 1 часа. Полученный реакционный раствор концентрировали досуха, и остаток растворяли в 8 мл муравьиной кислоты. К этому раствору добавляли 0,29 мл 37% формальдегида, а затем нагревали с обратным холодильником в течение 4 часов. После охлаждения, реакционный раствор смешивали с хлороформом, а pH смеси доводили до 10-11 с помощью водного аммиака, затем полученный органический слой собирали и осушали. После отгонки растворителя, остаток очищали с помощью хроматографии на силикагеле, используя в качестве элюента смесь хлороформа и метанола. В результате описанной процедуры получали 1,07 г целевого соединения в виде маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J = 7,2 Гц); 1,60-2,30 (2H, м); 2,38 (3H, с); 2,00-4,00 (7H, м); 4,11 (2H, кв., J = 7,2 Гц), 4,60-4,90 (1H, шир.); 6,39 (1H, с); 6,78 (2H, д, J = 8,8 Гц); 7,21 (2H, д, J = 8,8 Гц); 7,47 (2H, с); 7,77 (1H, с).
Ссылочный пример 42. Получение этил 2-[4-[((3S)-1-ацетил-3- пирролидинил)окси]фенил]-3-(5-циано-2-бензуфуранил)пропионата.
2,3 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси]фенил]-3-(5-циано-2-бензофуранил)пропионата растворяли в 3 мл анизола. К полученному раствору добавляли 25 мл трифторуксусной кислоты, охлаждая при этом льдом, и полученную смесь размешивали при комнатной температуре в течение 1 часа. После отгонки трифтороуксусной кислоты при пониженном давлении, pH остатка доводили до 10-11 с помощью насыщенного водного раствора бикарбоната натрия, затем экстрагировали хлороформом и осушали. При комнатной температуре органический слой смешивали с 2 мл триэтиламина, а затем с 555 мл ацетилхлорида, после чего перемешивали при той же температуре в течение 0,5 часа. После отгонки растворителя, остаток хроматографировали на колонке с силикагелем, элюируя смесью хлороформа и этанола, в результате чего получали 1,8 г целевого соединения.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J = 7,0 Гц); 2,04 (1,5H); 2,08 (1,5H); 3,14 (1H, дд, J = 15,1 и 3,6 Гц); 3,40-4,30 (8H, м); 4,70-5,04 (1H, шир.); 6,40 (1H, с); 6,60-6,92 (2H, м); 7,10 (2H, м); 7,47 (2H, с ); 7,77 (1H, с).
Ссылочный пример 43. Получение этил 3-(5-циано-2-бензофуранил)- 2-[4-[((3S)-1-диметилкарбамолил- 3-пирролидинил)окси]фенил}пропионата.
2,3 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси} фенил] -3-(5-циано-2-бензофуранил)пропионата растворяли в 3 мл анизола. К полученному раствору добавляли 25 мл трифторуксусной кислоты, охлаждая при этом льдом, после чего смесь размешивали 1 час при комнатной температуре. После отгонки трифторуксусной кислоты, pH остатка доводили до 10-11 с помощью насыщенного водного раствора бикарбоната натрия, а затем экстрагировали хлороформом и осушали. При комнатной температуре, органический слой смешивали с 2 мл триэтиламина, а затем с 760 мг N,N-диметилкарбамоилхлорида, после чего смесь размешивали 1 час при той же температуре. После отгонки растворителя, остаток хроматографировали на колонке с силикагелем, элюируя смесью хлороформа и этанола, в результате чего получали 1,7 г целевого соединения.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J = 7,0 Гц); 1,9-2,20 (2H, м); 2,86 (6H, с); 3,14 (1H, дд, J = 16,0 и 7,2 Гц); 3,30-4,50 (8H, м); 4,72-4,96 (1H, шир. ); 6,41 (1H, с); 6,83 (2H, д, J = 8,7 Гц); 7,25 (2H, д, J = 8,7 Гц); 7,49 (2H, с); 7,78 (1H, с).
Ссылочный пример 44. Получение этил 2-(4-ацетоксифенил)-2- оксоацетата.
7,25 г этил 2-(4-гидроксифенил)-2-оксоацетата растворяли в 15 мл пиридина, затем добавляли 4 мл уксусного ангидрида, и размешивали при комнатной температуре в течение 1 часа. Полученный раствор выливали в воду, и экстрагировали диэтиловым эфиром. Органический слой промывали водой и концентрировали досуха. Затем остаток растворяли в бензоле, концентрировали, и получали в результате 8,3 г целевого соединения в виде маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,41 (3H, т); 2,32 (3H, с); 4,43 (2H, кв.); 7,29 (2H, д); 8,01 (2H, д).
Ссылочный пример 45. Получение этил 3-(5-циано-2-бензофуранил)-2-(4-гидроксифенил)пропионата.
a) 15,93 г 5-циано-2-бензофуранил метилтрифенилфосфония хлорида и 8,29 г этил 2-(4-ацетоксифенил)-2-оксоацетата растворяли в смеси, состоящей из 80 мл тетрагидрофурана и 80 мл этанола. При комнатной температуре, к этому раствору добавляли 5,34 г 1,8-диазабицикло[5.4.0]-7-ундецена, и полученную смесь размешивали при этой температуре 18 часов. Затем реакционный раствор концентрировали досуха, в остаток очищали на хроматографической колонке с силикагелем, используя в качестве элюента смесь толуола и этилацетата, в результате чего получали 11,28 г этил 2-4-ацетоксифенил-3-5-циано-2-бензофуранил акрилата в виде светло-желтых кристаллов, представляющих собой смесь E- и Z-форм.
1H-ЯМР (CDCl3) δ: 1,32 (3H, т); 2,36 (3H, с); 4,30 (2H, кв.); 6,30 (1H, с); 7,2-7,8 (8H, м).
b) 3,8 г этил-2-4-ацетоксифенил-3-5-циано-2-бензофуранил акрилата, полученного в стадии (a), растворяли в смеси растворителей: этанола и тетрагидрофурана. Полученный раствор смешивали с 750 мг оксида палладия • 1H2O сульфата бария, и подвергали каталитической гидрогенизации при нормальном давлении. После удаления растворителя путем фильтрации, фильтрат концентрировали досуха и получали в результате 3,8 г этил 2-(4-ацетоксифенил)-3-(5-циано-2-бензофуранил)-пропионата.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J = 7,2 Гц); 2,25 (1H, с); 3,20 (1H, дд, J= 16,2 и 7,0 Гц); 3,40-4,30 (4H, м); 6,50 (1H, с); 7,10 (2H, д, J=8,3 Гц); 7,40 (2H, д, J=8,3 Гц); 7,56 (2H, с); 7,86 (1H, с).
с) 8,1 г этил 2-(4-ацетоксифенил)-3(5-циано-2-бензофуранил) пропионата, полученного в стадии (b), растворяли в 100 мл этанолового раствора, содержащего 15% аммиак, и полученный раствор выдерживали при комнатной температуре в течение 18 часов. Затем реакционный раствор концентрировали досуха, а остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом. В результате этой процедуры получали 5,62 г целевого соединения в виде бесцветных кристаллов. Т. пл. 140-142oC.
1H-ЯМР(CDCl3) δ: 1,15 (3H, т); 3,0-4,0 (3H, м); 2H, кв); 4,98 (1H, с); 6,39 (1H, с); 6,76 (2H, д); 7,15 (2H, д), 7,45 (2H); 7,75 (1H).
Ссылочный пример 46. Получение этил 3-(5-циано-2-бензофуранил)-3-(4-гидроксифенил)пропионата.
а) В 150 мл ацетона суспендировали 20 г 5-бромосалицилальдегида, 22,9 г 2-бромо-4-метоксиацетофенона и 27,6 г безводного карбоната калия. Затем перемешивали при комнатной температуре в течение 4 часов, реакционный раствор концентрировали досуха, а затем смешивали с водой и осажденные кристаллы собирали путем фильтрации. После промывания водой и последующей перекристаллизации из этанола, получали 14,02 г 5-бромо-2-(4-метоксибензоил)бензофурана в виде бесцветных призмообразных кристаллов. Т. пл. 143-146oC.
ИК (KBr): 1644, 1605, 1257 см-1
1H-ЯМР (ДМСО-d6) δ: 3,35 (3H, с); 7,15 (2H, д, J=9 Гц); 7,72 (3H, м); 8,0-8,2 (3H)
b) 15,0 г 5-бромо-2-(4-метоксибензоил)бензофурана, полученного в стадии (а), и 6,09 г цианида меди суспендировали в 75 мл N-метил-2-пирролидона, и полученную суспензию размешивали в потоке азота в течение 5 часов при 200-220oC. После охлаждения, реакционный раствор разбавляли хлороформом, нерастворившиеся материалы удаляли путем фильтрации, а полученный фильтрат промывали разбавленной соляной кислотой. После осушки органического слоя с последующим концентрированием при пониженном давлении, получали 6,60 г 2-(4-метоксибензоил)-5-бензофуранкарбонитрила в виде коричневого порошка.
ИК (KBr): 2224, 1644 см-1
1H-ЯМР (ДМСО-d6) δ: 3,30 (3H, с); 7,15 (2H, д, J = 9 Гц); 7,83 (1H, с); 8,00 (2H, д); 8,07 (2H, д, J= 9 Гц); 8,42 (1H, с).
c) 1,85 г этилдиэтилфосфоноацетата растворяли в 20 мл тетерагидрофурана. Размешивая при комнатной температуре, к полученному раствору добавляли 320 мг 60% гидрида натрия, после чего продолжали размешивать до получения прозрачного реакционного раствора. Через 10 минут к этому реакционному раствору добавляли 1,75 г 2-(4-метоксибензоил)-5-бензофуранкарбонитрила, полученного в стадии в), и смесь нагревали с обратным холодильником в течение 30 минут. После охлаждения, реакционную смесь концентрировали досуха. Остаток смешивали с разбавленной соляной кислотой, экстрагировали дихлорметаном, промывали насыщенным водным раствором хлорида натрия и концентрировали досуха. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью н-гексана дихлорметана, в результате чего получали 1,78 г этил 3-(5-циано-2-бензофуранил)-3-(4-метоксифенил)акрилата в виде смеси E- и Z-изомеров.
1H-ЯМР (CDCl3) δ: 1,20 (3H, т; J= 7 Гц); 3,84 (3H, с); 4,18 (2H, кв., J= 7 Гц); 6,32 (1H, с); 6,8-7,4 (5H, м); 7,56 (2H, с); 7,93 (1H, шир).
d) 1,78 г E/Z - смеси этил 3-(5-циано-2-бензофуранил)-3-(4-метоксифенил)акрилата, полученного в стадии (с), растворяли в смеси растворителей, состоящей из 6 мл тетрагидрофурана и 20 мл этанола. К полученному раствору добавляли 200 мг 5% палладированного угля, и эту смесь подвергали каталитической гидрогенизации при нормальном давлении в течение 1,5 часов. После удаления катализатора путем фильтрации, полученный фильтрат концентрировали досуха, в результате чего получали 1,79 г 3-(5-циано-2-бензофуранил)-3-(4-метоксифенил)пропионата.
1H-ЯМР (CDCl3) δ: 1,29 (3H, т, J=7 Гц); 2,9-3,1 (2H, м); 3,78 (3H, с); 4,09 (2H, кв. , J= 7 Гц); 4,5-4,7 (1H, м); 6,47 (1H, с); 6,88 (2H, д, J= 9 Гц); 7,47 (2H, с); 7,80 (1H, с).
e) 1,79 г этил 3-(5-циано-2-бензофуранил)-3-(4-метоксифенил)пропионата, полученного в стадии (d), растворяли в 20 мл безводного дихлорметана, и полученный раствор охлаждали до -50oC. К этому раствору по капле добавляли 10 мл раствора дихлорметана, содержащего 1,36 мл трибромида бора. Полученную смесь постепенно нагревали, и размешивали 3 часа при комнатной температуре. Затем, реакционный раствор разбавляли дихлорметаном, и полученный органический слой промывали разбавленной соляной кислотой, а затем насыщенным водным раствором хлорида натрия, после чего концентрировали досуха. В результате этой процедуры получали 1,34 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,15 (3H, т, J=7 Гц); 2,9-3,3 (2H, м); 4,09 (2H, кв., J = 7 Гц); 4,5-4,7 (1H, м); 6,15 (1H, шир.); 6,46 (1H, с); 6,80 (2H, д, J = 9 Гц); 7,15 (2H, д, J = 9 Гц); 7,42 (2H, с); 7,76 (1H, с).
Ссылочный пример 47. Получение этил-2-(2-(5-циано-2-бензофуранил)этил)-5-гидроксибензоата.
а) 4,87 г 2-формил-5-метоксибензойной кислоты растворяли в 30 мл хлороформа. Размешивая при комнатной температуре, раствор смеси бензола и н-гексана (1: 1), содержащего дифенилдиазометан, полученный в соответствии с процедурой, описанной в Journal of Chemical Society (Parkin 1, стр. 2030-2033, 1975), добавляли к полученному раствору до тех пор, пока реакционный раствор не станет пурпурно-красного цвета. Этот реакционный раствор подвергали очистке с помощью колоночной хроматографии на силикагеле, элюируя смесью толуола и этилацетата, и получали в результате 8,2 г дифенилметил-2-формил-5-метоксибензоата в виде вязкого маслянистого вещества.
1H- ЯМР (CDCl3) δ: 3,87 (3H, с); 7,13 (1H, дд, J = 11,5 и 2,9 Гц); 7,20 (1H, с); 7,24 (11H, м); 7,97 (1H, д, J = 11,5 Гц); 10,45 (1H, с).
b) 6,0 г дифенил 2-формил-5-метоксибензоата, полученного в стадии (a), и 8,1 г (5-циано-2-бензофуранил)метилтрифенилфосфонийхлорида растворяли в смеси, содержащей 70 мл тетрагидрофурана и 70 мл метанола. Размешивая при комнатной температуре, 2,91 г 1,8-диазабицикло [5.4.0]-7-ундецена добавляли к полученному раствору, и эту смесь размешивали 2 часа при той же температуре. После отгонки растворителя, полученный остаток очищали с помощью хроматографии на силикагеле, элюируя смесью толуола и хлороформа, в результате чего получали 8,2 г дифенилметил 2-[2-(5-циано-2-бензофуранил)винил]-5-метоксибензоата в виде смеси E- и Z-изомеров.
1H-ЯМР (CDCl3) δ: 3,84 (1H, с); 3,88 (3H, с); 6,20-8,28 (19H, м).
c) 8,2 г E/Z - смеси дифенилметил 2-[2-(5-циано-2-бензофуранил)винил]-5-метоксибензоата, полученного в стадии b), растворяли в смеси растворителей, состоящей из 60 мл тетрагидрофурана и 60 мл этанола. К этому раствору добавляли 2,0 г окиси палладия• 1H2O сульфата бария, и полученную смесь подвергали каталитической гидрогенизации при нормальном давлении. После удаления катализаторов путем фильтрации полученный фильтрат концентрировали, осажденные кристаллы собирали путем фильтрации, и получали в результате 4,45 г 2-[2-(5-циано-2-бензофуранил)этил] -5-метоксибензойной кислоты. Т. пл. 179-182oC.
1H-ЯМР (CDCl3) δ: 2,90-3,42 (4H, м); 3,75 (3H, с); 6,67 (1H, с); 7,01 (1H, дд, J = 8,7 и 2,2 Гц); 7,24 (1H, д, J = 8,7 Гц), 7,34 (1H, д, J=2,2 Гц); 7,69 (2H, с), 8,06 (1H, с); 12,98 (1H, шир.).
FD-МС m/Z: 321 (M+), 311, 283.
d) 4,45 г 2-[2-(5-циано-2-бензофуранил)этил]-5- метоксибензойной кислоты, полученной в стадии (с), растворяли в 200 мл этанола, и полученный раствор смешивали с 4 мл концентрированной серной кислоты, а затем смесь нагревали с обратным холодильником в течение 16 часов. После охлаждения, реакционный раствор нейтрализовали с помощью насыщенного водного раствора бикарбоната натрия, а этанол удаляли путем дистилляции. Полученный остаток экстрагировали этилацетатом и осушали путем отгонки растворителя. Остаток очищали с помощью хроматографии на силикагеле, а затем перекристаллизовывали из н-гексана, в результате чего получали 4,11 г этил 2-[2-(5-циано-2-бензофуранил)этил]5-метоксибензоата в виде бесцветных игольчатых кристаллов. Т. пл. 92-93oC.
1H-ЯМР (CDCl3) δ: 1,38 (3H, т, J= 7,0 Гц); 2,90-3,48 (4H, м); 3,82 (3H, с); 4,34 (2H, кв., J = 7,0 Гц); 6,41 (1H, с); 6,96 (1H, дд, J = 8,7 и 2,6 Гц); 7,10 (1H, д, J=8,7 Гц); 7,46 (1H, д, J = 2,6 Гц); 7,48 (2H, с); 7,79 (1H, с).
e) 4,11 г этил 2-[2-(5-циано-2-бензофуранил)этил]-5-метоксибензоата, полученного в стадии (d), растворяли в 40 мл дихлорметана, и полученный раствор охлаждали до -78oC. Затем, при той же температуре, к этому раствору по капле добавляли 8,85 г трибромида бора, и полученную смесь постепенно нагревали до -5oC - 0oC, после чего перемешивали в течение 1 часа. Реакционный раствор выливали в ледяную воду и экстрагировали этилацетатом. Полученный органический слой промывали 4 н соляной кислотой, а затем водой, после чего осушали, а растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле, получая в результате 2,80 г целевого соединения в виде призмообразных кристаллов.
Т.пл. 133-135oC.
1H ЯМР (CDCl3) δ: 1,40 (3H, т, J = 7,0 Гц); 2,96 - 3,50 (4H, м); 4,36 (2H, кв. , J = 7,0 Гц); 6,45 (1H, с); 6,93 (1H, дд, J = 8,7 и 2,9 Гц); 7,13 (1H, д, J = 8,7 Гц); 7,50 (1H, д, J = 2,9 Гц); 7,56 (2H, с); 7,84 (1H, с).
Ссылочный пример 48. Получение этил 2-[2-[2-(5-циано-2-бензофуранил)этил]-5-гидроксифенил]ацетата.
a) 2,0 г 2-[2-(5-циано-2-бензофуранил)этил]-5-метоксибензойной кислоты суспендировали в 10 мл бензола, и полученную суспензию смешивали с 1 мл тионилхлорида. После 1-часового нагревания полученной смеси с обратным холодильником и концентрирования этой смеси досуха, получали сырой хлорангидрид.
Смешанный раствор, состоящий из 10 мл н-гексана, содержащего 10% (мас. /об.) триметилсилилдиазометана, 1,3 мл триэтиламина, 10 мл ацетонитрила и 10 тетрагидрофурана охлаждали до -5oC. Затем к этому раствору, размешивая, по капле добавляли 5 мл ацетонитрилового раствора полученного выше хлорангидрида. Этот реакционный раствор размешивали 48 часов при 0oC, после чего растворитель отгоняли при пониженном давлении и низкой температуре. Полученный таким образом остаток растворяли в смеси, состоящей из 4 мл коллидина и 4 мл бензилового спирта, и этот раствор размешивали 7 минут при 180oC в потоке азота. Полученный реакционный раствор растворяли в бензоле, промывали 10% уксусной кислоты, а затем осушали. После отгонки растворителя, остаток хроматографировали на колонке с силикагелем, элюируя смесью толуола и этилацетата, в результате чего получали 830 мг бензил 2-[2-[2-(5-циано-2-бензофуранил)этил]-5-метоксифенил]-ацетата. Т. пл. 127-128oC.
1H-ЯМР (CDCl3) δ: 3,00 (4H), 3,68 (2H, с); 3,76 (3H, с); 5,13 (2H, с); 6,32 (1H, c); 6,76 (1H, дд, J = 7,9 и 1,3 Гц); 6,80 (1H, c); 7,08 (1H, д, J = 7,9 Гц); 7,30 (5H, с); 7,48 (1H, д, J = 1,3 Гц); 7,77 (1H, c).
b) 855 мг бензил 2-[2-[2-(5-циано-2-бензофуранил)-этил]-5-метоксифенил] ацетата, полученного в стадии a), растворяли в 20 мл дихлорметана, и полученный раствор охлаждали до -50oC. К этому раствору по капле добавляли 5 мл дихлорметанового раствора, содержащего 1,75 г трибромида бора, а затем постепенно повышали температуру до 15oC и при этой температуре размешивали 20 минут. Реакционный раствор экстрагировали этилацетатом, промывали разбавленной соляной кислотой, а затем осушали. После концентрирования досуха, остаток растворяли в 30 мл этанола, смешивали с 2 мл тионилхлорида и нагревали с обратным холодильником в течение 1 часа. После охлаждения, реакционный раствор разбавляли этилацетатом, а полученный органический слой промывали водой и осушали путем отгонки растворителя. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом. В результате этой процедуры получали 680 мг целевого соединения в виде порошка. Т.пл. 84-86oC.
1H-ЯМР (CDCl3) δ: 1,25 (3H, т, J = 7,0 Гц); 3,02 (4H), 3,59 (2H, c); 4,57 (2H, кв., J = 7,0 Гц); 6,19 (1H, с); 6,41 (1H, с); 6,55-6,84 (2H, м); 6,99 (1H, д, J = 7,9 Гц); 7,48 (2H, с); 7,77 (1H, с).
Ссылочный пример 49. Получение этил 5-циано-2-[2-(4-гидроксифенил)этил] -3-бензофуранкарбоксилата.
a) 91,5 г (5-бромо-2-бензофуранил)метилтрифенилфосфония хлорида и 25 г р-анизальдегида растворяли в смеси, состоящей из 180 мл тетрагидрофурана и 180 мл этанола. К этому раствору, размешивая при комнатной температуре, добавляли 27,58 г 1,8-диазабицикло[3,4,0] -7-ундецена, и полученную смесь размешивали 18 часов. После этого, реакционный раствор концентрировали, осажденные кристаллы собирали фильтрацией, и получали в результате 32,8 г 5-бромо-2-[2-(4-метоксифенил)-винил] бензофурана в виде одного из стереоизомеров.
Т.пл.190-194oC.
1H-ЯМР (CDCl3) δ: 3,83 (3H, с); 6,54 (1H, с); 6,9 (3H); 7,25 (1H, д, J = 17 Гц); 7,31 (2H); 7,45 (2H, д); 7,62 (1H).
Полученный фильтрат концентрировали досуха, а осадок очищали с помощью колоночной хроматографии на силикагеле, элюируя толуолом, в результате чего получали 22 г 5-бромо-2-[2-(4-метоксифенил)винил] бензофурана в виде другого стереоизомера.
1H-ЯМР (CDCl3) δ: 3,84 (3H, с); 6,35 (1H, д, J = 14 Гц); 6,53 (1H, с); 6,62 (1H, д, J = 14 Гц); 6,9 (2H, д); 7,24 (2H); 7,3 (2H, д); 7,38 (1H).
b) 84 г смеси двух стереоизомеров 5-бромо-2-[2-(4-метоксифенил)винил] бензофурана, полученного в стадии (a), растворяли в 600 мл дихлорметана. К этому раствору, охлаждая льдом и размешивая при этом, добавляли 18,5 мл ацетилхлорида, а затем по капле добавляли 28,9 мл тетрахлорида титана. Полученный реакционный раствор выливали в ледяную воду, экстрагировали хлороформом, а полученный органический слой промывали разбавленной соляной кислотой, а затем водой, а затем осушали путем отгонки растворителя. После этого остаток суспендировали в эфире, нерастворившиеся кристаллы собирали путем фильтрации, и получали в результате 76 г 3-ацетил-5-бромо-2-[2-(4-метоксифенил)винил]бензофурана в виде тонких желтых игольчатых кристаллов, один и тот же изомер был образован из E- и из Z-форм. Т.пл. 163-165oC.
1H- ЯМР (CDCl3) δ: 2,69 (3H, с); 3,85 (3H, с); 6,95 (2H, д, J = 10 Гц); 7,4 (2H, м); 7,6 (2H, д, J = 10 Гц); 7,65 (2H, с); 8,08 (1H).
c) Смесь 20,7 г 3-ацетил-5-бромо-2-[2-(4-метоксифенил)-винил]бензофурана, полученного в стадии (b), 6 г цианида меди и 800 мл N-метил-2-пирролидона размешивали в потоке азота в течение 8,5 часов при 210-220oC. Реакционный раствор выливали в ледяную воду, осажденные материалы удаляли путем фильтрации, а полученный фильтрат экстрагировали этилацетатом. После удаления нерастворившихся материалов путем фильтрации, полученный органический слой промывали водой и осушали путем отгонки растворителя. Полученный таким образом остаток подвергали колоночной хроматографии на силикагеле, элюируя толуолом, после чего полученный продукт промывали метанолом. В результате этой процедуры получали 7,82 г 3-ацетил-2-[2-(4-метоксифенил)винил]-5-бензофуранкарбонитрила в виде тонких кристаллов. Т.пл. 190-191oC.
1H-ЯМР (CDCl3) δ: 2,69 (3H, с); 3,85 (3H, с); 6,98 (2H, д, J = 10 Гц); 7,50 -7,80 (6H, м); 8,36 (1H).
d) 7,8 г 3-ацетил-2-[2-(4-метоксифенил)винил]-5-бензофуранкарбонитрила, полученного в стадии c), растворяли в смеси растворителей, состоящей из 600 мл тетрагидрофурана и 500 мл этанола. К этому раствору добавляли 900 мг оксида палладия • 1H2O сульфата бария, и полученную смесь подвергали каталитической гидрогенизации при нормальном давлении в течение 3,5 часов. После удаления катализатора путем фильтрации, фильтрат концентрировали досуха. Полученный остаток экстрагировали этилацетатом, а органический слой промывали водой и осушали путем отгонки растворителя. После этого осадок промывали метанолом, осажденные кристаллы собирали фильтрацией, и получали в результате 5,47 г 3-ацетил-2-[2-(4-метоксифенил)этил]-5-бензофуранкарбонитрила в виде бесцветных призмообразных кристаллов. Т.пл. 130-131oC.
1H-ЯМР (CDCl3) δ: 2,54 (3H, с); 3,04 (2H, м); 3,4 (2H, м); 3,77 (3H, с); 6,85 (2H, д, J = 10 Гц); 7,05 (2H, д); 7,57 (2H, с); 8,33 (1H).
е) 5,2 г гидроксида натрия растворяли в 30 мл воды, и полученный раствор охлаждали до температуры 0oC или ниже. К этому раствору, размешивая при этом, по капле добавляли 2,7 мл брома, а затем 40 мл диоксанового раствора, содержащего 4,14 г 3-ацетил-2-[2-(4-метоксифенил)этил] 5-бензофуранкарбонитрила, полученного в стадии (d). Полученную смесь размешивали 45 минут при 0oC, а затем еще 1 час с охлаждением льдом. Реакционный раствор смешивали с водой, и pH смеси доводили до 2 с помощью концентрированной соляной кислоты, после чего смесь экстрагировали хлороформом. Полученный органический слой промывали водой и осушали путем отгонки растворителя. Затем, остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 1,44 г 5-циано-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоновой кислоты.
Т.пл. 205-208oC (после перекристаллизации из метанола, с получением тонких призмообразных кристаллов).
1H-ЯМР (CDCl3) δ: 3,13 (2H, м); 3,5 (2H, м); 3,78 (3H, с); 6,83 (2H, д); 7,07 (2H, д); 7,56 (2H, с); 8,34 (1H).
f) 1,81 г 5-циано-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоновой кислоты, полученной в стадии е), добавляли к 5 мл тионилхлорида, и полученную смесь нагревали с обратным холодильником в течение 1 часа. Полученный раствор концентрировали досуха, а остаток смешивали с этанолом и размешивали 30 минут при 50oC. Осажденные кристаллы собирали путем фильтрации, и получали в результате 1,82 г этил 5-циано-2-(2-(4-метоксифенил)этил)-3-бензофуранкарбоксилата.
Т.пл. 135-139oC (тонкие призмообразные кристаллы)
ИК (KBr): 224, 1695, 1614, 1587, 1515 см-1
g) 1,78 г этил 5-циано-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоксилата, полученного в стадии f), обрабатывали в соответствии с процедурой, описанной в стадии e) сравнительного примера 46, и получали в результате 2,27 г целевого соединения в виде тонких игольчатых кристаллов.
Т.пл. 182-183oC.
1H-ЯМР (CDCl3) δ: 1,45 (3H, т, J = 8,0 Гц); 3,0 (2H, м); 3,4 (2H, м); 4,4 (2H, кв., J = 8,0 Гц); 6,7 (2H, д); 7,1 (2H, д); 7,55 (2H); 8,29 (1H).
Ссылочный пример 50. Получение этил[5-циано-2-(2-(4-гидроксифенил)этил] -3-бензофуранил)ацетата.
a) 128 г 5-бромо-2-[2-(4-метоксифенил)винил] бензофурана в виде смеси двух стереоизомеров растворяли в смеси, состоящей из 1,3 л тетрагидрофурана и 0,7 л этанола. Полученный раствор смешивали с 3,0 г диоксида платины, и в течение 4 часов подвергали каталитической гидрогенизации при нормальном давлении. Затем, катализатор удаляли путем фильтрации, фильтрат концентрировали, а осажденные кристаллы собирали путем фильтрации и промывали этанолом. В результате описанной процедуры получали 97,08 г 5-бромо-2-[2-(4-метоксифенил)этил]бензофурана. Т.пл. 109-111oC.
1H-ЯМР (CDCl3) δ: 3,00 (4H, с); 3,77 (3H, с); 6,28 (1H, с); 6,88 (2H, д, J = 9,0 Гц); 7,08 (2H, д, J = 9,0 Гц); 7,32 (2H); 7,60 (1H).
b) 97 г 5-бромо-2-[2-(4-метоксифенил)-этил]бензофурана, полученного в стадии a), обрабатывали в соответствии с процедурой, описанной в стадии b) ссылочного примера 49, в результате чего получали 79,9 г 3-ацетил-5-бромо-2-[2-(4-метоксифенил)этил]бензофурана. Т. пл. 100-101oC.
1H-ЯМР (CDCl3) δ: 2,52 (3H, с); 3,05 (2H, м); 3,35 (2H, м); 3,76 (3H, с); 6,80 (2H, д, J = 9,0 Гц); 7,10 (2H, д, J = 9,0 Гц); 7,35 (2H, м); 8,05 (1H).
c) 79,9 г 3-ацетил-5-бромо-2-[2-(4-метоксифенил)этил]-бензофурана, полученного в стадии b), обрабатывали в соответствии с процедурой, описанной в стадии e) ссылочного примера 49, в результате чего получали 64,2 г 5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоновой кислоты.
1H-ЯМР (ДМСО-d6) δ: 3,00 (2H, м); 3,35 (2H, м); 3,69 (3H, с); 6,80 (2H, д, J = 8,0 Гц); 7,07 (2H, д, J = 8,0 Гц); 7,50 (1H, дд); 7,55 (1H, д); 8,00 (1H, д).
d) 64,2 г 5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоновой кислоты, полученной в стадии c), суспендировали в 900 мл этанола. 30 мл тионилхлорида по капле добавляли к полученной суспензии, охлаждая при этом льдом. После 5-часового нагревания с обратным холодильником, к реакционной смеси по капле добавляли еще 50 мл тионилхлорида, а затем полученную смесь нагревали с обратным холодильником еще 3 часа. Реакционный раствор концентрировали досуха, а остаток смешивали с водой и нерастворившиеся материалы собирали путем фильтрации. Эти нерастворимые материалы растворяли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия, затем водой, а после этого насыщенным водным раствором хлорида натрия, а растворитель отгоняли.
Полученный остаток суспендировали в этаноле, фильтровали и получали 59,23 г этил 5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоксилата. Т. пл. 73-75oC.
1H-ЯМР (CDCl3) δ: 1,43 (3H, т, J = 8,9 Гц); 3,10 (2H, м); 3,40 (2H, м); 3,77 (3H, с); 4,40 (2H, кв., J = 8,9 Гц); 6,80 (2H, д); 7,2 (2H, д); 7,33 (2H, м); 8,10 (1H).
e) 35,5 г этил 5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранкарбоксилата, полученного в стадии d), растворяли в 400 мл тетрагидрофурана с последующим постепенным добавлением 3,5 г алюмогидрида лития, а затем полученную смесь размешивали при комнатной температуре в течение 1 часа. Реакционный раствор выливали в воду, pH раствора доводили до 2 соляной кислотой, и полученный раствор экстрагировали бензолом. Затем органический слой промывали водой, и концентрировали досуха, в результате чего получали 30 г 5-бромо-3-гидроксиметил-2-[2-(4-метоксифенил)этил] бензофурана в виде кристаллов. Т. пл. 65-75oC.
1H-ЯМР (CDCl3) δ: 2,95 (4H, с); 3,69 (3H, м); 4,33 (2H, с); 6,77 (2H, д); 6,90 (2H, д); 7,26 (2H, м); 7,65 (1H).
f) 30 г 5-бромо-3-гидроксиметил-2-[2-(4-метоксифенил)-этил]бензофурана, полученного в стадии e), суспендировали в 150 мл диэтилового эфира. К этой суспензии добавляли 12 капель пиридина. Затем, охлаждая льдом, по капле добавляли 12 мл тионилхлорида. Реакционную смесь размешивали при комнатной температуре в течение 1 часа. Затем полученный реакционный раствор выливали в ледяную воду, и экстрагировали диэтиловым эфиром. После этого органический слой промывали водой, а затем насыщенным водным раствором бикарбоната натрия, и концентрировали досуха, в результате чего получали 28,3 г 5-бромо-3-хлорометил-2-[2-(4-метоксифенил)этил]бензофурана. Т.пл. 70-75oC.
1H-ЯМР (CDCl3) δ: 3,00 (4H, с); 3,76 (3H, с); 4,38 (2H, с); 6,82 (2H, д); 6,97 (2H, д); 7,31 (2H); 7,68 (1H).
g) К 75 мл ацетонитрила добавляли 10,82 г 5-бромо-3-хлорометил-2-[2-(4-метоксифенил)этил] бензофурана, полученного в стадии f), 3,7 г цианида калия и 0,6 г 18-кроун-6-эфира. Полученную смесь нагревали с обратным холодильником в течение 2,5 часов. Затем реакционный раствор смешивали с водой и экстрагировали бензолом, а полученный органический слой промывали водой и осушали путем отгонки растворителя. Полученный остаток очищали путем хроматографии на колонке с силикагелем, используя в качестве элюента смесь толуола и н-гексана, в результате чего получали 9,17 г 5-бромо-3-цианометил-2-[2-(4-метоксифенил)этил]бензофурана.
1H-ЯМР (CDCl3) δ: 2,95 (4H, с); 3,20 (2H, с); 3,73 (3H, с); 6,80 (2H, д); 6,90 (2H, д); 7,33 (2H); 7,61 (1H).
h) 9,17 г 5-бромо-3-цианометил-2-[2-(4-метоксифенил)этил]бензофурана, полученного в стадии (g), добавляли к смешанному раствору, состоящему из 100 мл этанола и 5 мл концентрированной серной кислоты, и полученную смесь нагревали с обратным холодильником в течение 18 часов. Реакционный раствор выливали в воду и экстрагировали этилацетатом. Затем органический слой промывали водой, затем насыщенным водным раствором бикарбоната натрия и снова водой, после чего этот слой осушали путем отгонки растворителя. В результате этой процедуры получали 8,96 г этил 5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранил ацетата.
1H-ЯМР (CDCl3) δ: 1,21 (3H, т, J = 7,0 Гц); 2,96 (4H, с); 3,34 (2H, с); 3,74 (3H, с); 4,10 (2H, кв., J = 7,0 Гц); 6,80 (2H, д, J = 9 ГЦ); 7,00 (2H, д, J = 7,0 Гц); 7,28 (2H); 7,59 (1H).
i) 8,2 г этил [5-бромо-2-[2-(4-метоксифенил)этил]-3-бензофуранил]ацетата, полученного в стадии h), обрабатывали в соответствии с процедурой, описанной в стадии c) ссылочного примера 49, в результате чего получали 4,5 г этил [5-циано-2-[2-(4-метоксифенил)этил] -3-бензофуран ацетата в виде бесцветных игольчатых кристаллов.
Т.пл. 85-86oC
1H-ЯМР (CDCl3) δ: 1,23 (3H, т, J = 7,0 Гц); 3,01 (4H, с); 3,40 (2H, с); 3,75 (3H, с); 4,11 (2H, кв., J = 7,0 Гц); 6,80 (2H, д, J = 9 Гц); 7,00 (2H, д, J = 7,0 Гц); 7,47 (2H); 7,81 (1H).
j) 4,45 г этил[5-циано-2-[2-(4-метоксифенил)этил]-3-бензофуранил]ацетата, полученного в стадии i), обрабатывали в соответствии с процедурой, описанной в стадии e) сравнительного примера 46, в результате чего получали 2,98 г целевого соединения в виде бесцветных кристаллов.
Т.пл. 134-136oC
1H-ЯМР (CDCl3) δ: 1,22 (3H, т, J = 7,0 Гц); 2,98 (4H, с); 3,39 (2H, с); 4,12 (2H, кв. , J = 7,0 Гц); 6,74 (2H, кв, J = 9 Гц); 6,91 (2H, д, J = 7,0 Гц); 7,48 (2H); 7,80 (1H).
Ссылочный пример 51. Получение этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-4-этокси-5- гидроксиэтил]пропионата.
а) 20,0 г феруловой кислоты растворяли в 250 мл метанола и полученный раствор подвергали каталитическому восстановлению в течение 3 часов при нормальном давлении в присутствии 10% палладированного угля (катализатор, на 50% мокрого типа). После удаления катализатора путем фильтрации, полученный фильтрат концентрировали, а осажденные кристаллы собирали фильтрацией, в результате чего получали 19,3 г 3-(4-гидрокси-3-метоксифенил)пропионовой кислоты.
Т. пл. 87 - 89oC.
1H-ЯМР (CDCl3) δ: 2,5 - 3,0 (4H, м); 3,85 (3H, с); 6,5 - 6,9 (3H, м).
b) 19,3 г 3-(4-гидрокси-3-метоксифенил)пропионовой кислоты, полученной в стадии а), растворяли в 300 мл этанола. После добавления 2,0 мл концентрированной серной кислоты, полученную смесь нагревали с обратным холодильником в течение 2 часов. Затем реакционный раствор концентрировали при пониженном давлении, экстрагировали хлороформом, промывали водой, а затем осушали. После отгонки растворителя получали 23,0 г этил 3-(4-гидрокси-3-метоксифенил)пропионата в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,23 (3H, т, J = 7,2 Гц); 2,4 - 3,0 (4H, м); 3,85 (3H, с); 4,12 (2H, кв., J = 7,12 Гц); 6,6 - 6,9 (3H, м).
c) 10,0 г этил 3-(4-гидрокси-3-метоксифенил)пропионата полученного в стадии b), растворяли в 300 мл тетрагидрофурана. Затем добавляли 1,96 г 60% гидрида натрия. Полученную таким образом смесь размешивали в течение 30 минут при 50oC. После этого к смеси по капле добавляли 7,17 г этилбромида. После 6-часового нагревания с обратным холодильником реакционный раствор выливали в воду, экстрагировали хлороформом, промывали водой, а затем концентрировали при пониженном давлении. После этого, полученный остаток подвергали колоночной хроматографии на силикагеле, элюируя хлороформом, и получали в результате 5,6 г маслянистого этил 3-(4-этокси-3-метоксифенил)пропионата.
1H-ЯМР (CDCl3) δ: 1,23 (3H, и, J = 7,2 Гц); 1,43 (3H, т, J = 7,1 Гц); 2,4 - 3,0 (4H<м); 3,85 (3H, с); 4,06 (2H, кв., J = 7,1 Гц); 4,11 (2H, кв., J = 7,12 Гц); 6,7 - 6,9 (3H, м).
d) 9,3 г этил 3-(4-этокси-3-метоксифенил)пропионата, полученного в стадии c), растворяли в 10 мл уксусной кислоты, с последующим добавлением 7,4 г хлорметилового метилэфира, а затем размешивали 22 часа при комнатной температуре. Реакционный раствор выливали в ледяную воду, экстрагировали этилацетатом, промывали водой и осушали путем отгонки растворителя. Полученный таким образом остаток растворяли в 10 мл ксилола, и раствор смешивали с 8,54 г трифенилфосфина. Полученную смесь размешивали при комнатной температуре 18 часов, а затем 5 часов при 70 - 80oC. После охлаждения ксилол удаляли путем декантации, а оставшуюся часть отверждали путем добавления н-гексана, в результате чего получали 6,0 г сырого [5-этокси-2-(2-этоксикарбонилэтил)-4-метоксифенил]-метилтрифенилфосфония хлорида.
e) В раствор, состоящий из 50 мл тетрагидрофурана и 50 мл этанола, растворяли 1,5 г 5-цианобензо[b]тиофен-2-карбальдегида и 6,34 г сырого [5-этокси-2-(2-этоксикарбонилэтил)-4-метоксифенил] метилтрифенилфосфония хлорида, полученного в стадии d). К полученному раствору добавляли 1,83 г 1,8-диазабицикло[5.4.0] -7-ундецена, и размешивали 18 часов при комнатной температуре. Полученный реакционный раствор концентрировали при пониженном давлении, а остаток растворяли в смеси, состоящей из 20 мл тетрагидрофурана и 20 мл этанола. Полученный таким образом раствор смешивали с 1,70 г 10% палладированного угля (катализатор на 50% мокрого типа), и подвергали каталитической гидрогенизации при нормальном давлении до тех пор, пока не завершится абсорбция водорода. После удаления катализатора путем фильтрации, растворитель отгоняли. После хроматографии полученного остатка на колонке с силикагелем, получали 1,2 г маслянистого этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-4-этокси- 5-метоксифенил]пропионата.
1H-ЯМР (CDCl3) δ: 1,24 (3H, т, J = 7,1 Гц); 1,36 (3H, т, J = 7,0 Гц); 2,4 - 3,3 (8H, м); 3,84 (3H, с); 3,98 (2H, кв., J = 7,0 Гц); 4,13 (2H, кв., J = 7,1 Гц); 6,64 (1H, с); 6,70 (1H, с); 7,04 (1H, с); 7,46 (1H, дд, J = 8,4 и 1,5 Гц); 7,83 (1H, д, J = 8,4 Гц); 7,96 (1H, с).
f) 2,1 г этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-4- этокси-5-метоксифенил]пропионата, полученного в стадии e), растворяли в 20 мл ν-коллидина, с последующим добавлением 7,94 г иодида лития, а затем нагревали с обратным холодильником в течение 18 часов. Реакционный раствор выливали в воду, экстрагировали хлороформом, промывали водой, а затем осушали. После отгонки растворителя, полученный остаток растворяли в 100 мл этанола, смешивали с 0,3 мл концентрированной серной кислоты, а затем нагревали с обратным холодильником в течение 1 часа. После отгонки растворителя при пониженном давлении, полученный реакционный раствор разбавляли хлороформом, промывали водой, а затем осушали путем отгонки растворителя. После этого полученный остаток очищали с помощью хроматографии на колонке с силикагелем, элюируя хлороформом. В результате описанной процедуры получали 2,0 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,24 (3H, т, J = 7,1 Гц); 1,35 (3H, т, J = 7,0 Гц); 2,4 - 3,3 (8H, м); 3,98 (2H, кв., J = 7,0 Гц); 4,13 (2H, кв., J = 7,1 Гц); 6,60 (1H, с); 7,44 (1H, дд, J = 8,4 и 1,5 Гц); 7,82 (1H, д, J = 8,4 Гц); 7,96 (1H, с); 7,94 (1H, с).
Ссылочный пример 52. Получение этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-5-гидроксифенил пропионата.
a) 33,5 г 6-метокси-2-тетралона растворяли в 27,6 мл этанола, а затем добавляли 37,8 мл этилортоформата и одну каплю концентрированной серной кислоты. Полученную смесь размешивали 4 часа при 100oC. После отгонки растворителя при пониженном давлении, полученный остаток подвергали колоночной хроматографии на силикагеле, элюируя хлороформом. Нужные фракции объединяли, концентрировали, и осажденные кристаллы собирали, в результате чего получали 5,82 г 3,4-дигидро-2-этокси-6-метоксинафталена.
1H-ЯМР (CDCl3) δ: 1,37 (3H, т, J = 7,0 Гц); 2,20 - 3,00 (4H, м); 3,79 (3H, с); 3,84 (2H, кв., J = 7,0 Гц); 5,48 (1H, с); 6,60 - 7,00 (3H, м).
b) 5,8 г 3,4-дигидро-2-этокси-6-метоксинафталина, полученного в стадии (a), растворяли в смеси, состоящей из 90 мл этанола и 10 мл дихлорметана. Затем, размешивая при температуре -20oC, через полученный раствор в целях окисления барботировали озоном. К полученному реакционному раствору при той же температуре, постепенно, по капле добавляли 10 мл диметилсульфида, после чего смесь размешивали 30 мин при комнатной температуре. После отгонки растворителя при пониженном давлении, остаток растворяли в 100 мл смеси тетрагидрофурана и этанола (1:1). К полученному раствору добавляли 12,5 г (5-цианобензо[b] тиен-2-ил)метилтрифенилфосфония хлорида, а затем 4,46 мл 1,8-диазабицикло[5.4.0] -7-ундецена, после чего смесь размешивали 5 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом. Очищенный продукт растворяли в 60 мл смеси этанола и тетрагидрофурана (1:1), и полученный раствор смешивали с 3,9 г 10% палладированного угля (катализатор на 50% мокрого типа). После 3-часового каталитического гидрирования смеси при нормальном давлении, получали 2,75 г этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-5-метоксифенилпропионата.
1H-ЯМР (CDCl3) δ: 1,22 (3H, т, J = 7,2 Гц); 2,2 - 3,4 (8H, м); 3,76 (3H, с); 4,16 (2H, кв., J = 7,2 Гц); 6,60 - 7,30 (4H, м); 7,48 (1H, д, J = 8,2 Гц); 7,85 (1H, д, J = 8,2 Гц); 7,99 (1H, с).
c) 2,75 г этил 3-[2-[2-(5-цианобензо[b]тиен-2-ил)этил]-5- метоксифенил пропионата, полученного в стадии [b] , обрабатывали согласно процедуре, описанной в стадии (e) ссылочного примера 46, в результате чего получали 2,3 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,23 (3H, т, J = 7,1 Гц); 2,4 - 3,34 (8H, м); 4,13 (2H, кв., J = 7,2 Гц); 5,60 (1H, с); 6,50 - 7,20 (3H, м); 7,25 (1H, с); 7,44 (1H, д, J = 8,3 Гц); 7,82 (1H, д, J = 8,3 Гц), 7,92 (1H, с).
Ссылочный пример 53. Получение этил 2-(5-цианобензо[b]тиен-2-ил)-3-(4-гидроксифенил)пропионата.
a) 0,5 г 5-бромо-2-гидроксиметилбензо[b] тиофена растворяли в 20 мл дихлорметана, а затем добавляли 230 мг трибромида фосфора. После 1-часового размешивания при комнатной температуре, реакционный раствор смешивали с водой, промывали насыщенным водным раствором бикарбоната натрия, а затем осушали путем отгонки растворителя. Полученный остаток растворяли в смеси, состоящей из 10 мл ацетонитрила и 3 мл диметилсульфоксида, затем добавляли 300 мг цианида меди, и нагревали с обратным холодильником в течение 2 часов. После охлаждения, к реакционному раствору добавляли толуол, нерастворившиеся материалы удаляли путем фильтрации, а фильтрат промывали водой, осушали и концентрировали. После сбора осажденных кристаллов путем фильтрации, получали 200 мг 5-бромо-2-цианометилбензо о тиофена. Т. пл. 94 - 96oC.
1H-ЯМР (CDCl3) δ: 3,98 (2H,с); 7,25 (1H, с); 7,42 1H, дд, J = 8,5 и 1,8 Гц); 7,65 (1H, д, J = 8,5 Гц); 7,90 (1H, д, J = 1,8 Гц).
b) 12,0 г 5-бромо-2-цианометилбензо[b]тиофена, полученного в стадии (a), растворяли в 80 мл этанола, а затем добавляли 1,0 мл воды и 7 мл концентрированной серной кислоты. После 7-часового нагревания с обратным холодильником, реакционный раствор смешивали с 40 мл этанола, 15 мл концентрированной серной кислоты и 0,5 мл воды, и полученную смесь нагревали с обратным холодильником в течение 2 часов. После охлаждения, реакционный раствор смешивали с водой и экстрагировали равным объемом смеси толуола и этилацетата. Полученный органический слой промывали водой и насыщенным водным раствором бикарбоната натрия, а затем осушали путем отгонки растворителя. После этого остаток подвергали хроматографии на силикагеле и получали 8,0 г этил 2-(5-бромобензо[b]тиен-2-ил)ацетата. Т. пл. 56 - 57oC.
1H-ЯМР (CDCl3) δ: 1,28 (3H, т, J = 7,0 Гц); 3,88 (2H, с); 4,23 (2H, кв., J = 7,0 Гц); 7,11 (1H, с); 7,38 (1H, дд, J = 8,3 и 1,8 Гц); 7,68 (1H, д, J = 8,3 Гц); 7,82 (1H, д, J = 1,8 Гц).
c) 800 мг этил 2-(5-бромобензо[b]тиен-2-ил)ацетата, полученного в стадии b), и 965 мг диэтилкарбоната растворяли в 4 мл N,N-диметилформамида. К полученному раствору, нагревая в масляной бане при 120 - 130oC, добавляли 162 мг гидрида натрия (60%). После 10-минутного размешивания при той же температуре, к реакционному раствору добавляли 30 мг гидрида натрия (60%), и размешивание продолжали еще 10 минут. Полученный раствор разбавляли равным объемом смеси толуола и этилацетата, промывали последовательно разбавленной соляной кислотой и водой, а затем осушали. После отгонки растворителя, полученный остаток очищали с помощью хроматографии на колонке с силикагелем, элюируя смесью толуола и этилацетата. В результате этой процедуры, получали 600 мг этил 2-(5-бромобензо[b]тиен-2-ил)-2-этоксикарбонилацетата.
1H-ЯМР (CDCl3) δ: 1,28 (6H, т, J = 7,0 Гц); 4,25 (4H, кв., J = 7,0 Гц); 4,95 (1H, с); 7,17 (1H, с); 7,35 (1H, дд, J = 8,3 и 2,1 Гц); 7,62 (1H, д, J = 8,3 Гц); 7,83 (1H, д, J = 2,1 Гц).
d) 6,2 г этил 2-(5-бромобензо[b] тиен-2-ил)-2-этоксикарбонилацетата, полученного в стадии c), и 5,2 г 4-метоксибензилхлорида растворяли в 30 мл N, N-диметилформамида. К полученному раствору, при комнатной температуре, добавляли 1,34 г гидрида натрия (60%), и смесь размешивали в течение 3 часов. Охлаждая льдом, реакционный раствор смешивали с 10% водным раствором лимонной кислоты, экстрагировали толуолом, промывали водой, а затем осушали. После отгонки растворителя, полученный остаток хроматографировали на колонке с силикагелем, элюируя толуолом, в результате чего получали 8,2 г этил 2-(5-бромобензо[b] тиен-2-ил)-2-этоксикарбонил-3-(4-метоксифенил) пропионата. Т. пл. 58 - 60oC.
1H-ЯМР (CDCl3) δ: 1,22 (6H, т, J = 7,0 Гц); 3,65 (5H, с); 4,30 (4H,. кв. , J = 7,0 Гц); 6,60 (2H, д, J = 8,5 Гц); 6,79 (2H, д, J = 8,5 Гц); 7,31 (1H, с); 7,35 (1H, дд, J = 8,8 и 1,8 Гц); 7,60 (1H, д, J = 8,8 Гц); 7,82 (1H, д, J = 1,8 Гц).
e) 3,0 г этил 2-(5-бромобензо[b]тиен-2-ил)-2-этоксикарбонил-3- (4-метоксифенил)пропионата, полученного в стадии (d), растворяли в 25 мл этанола. К полученному раствору добавляли 0,91 г гидроксида калия, растворенного в 2,5 мл воды, и эту смесь размешивали при комнатной температуре в течение 4 дней. Охлаждая льдом, полученный реакционный раствор смешивали разбавленной соляной кислотой и экстрагировали этилацетатом, а органический слой промывали водой, и осушали путем отгонки растворителя. Полученный остаток растворяли в 60 мл этанола, смешивали с 4 мл концентрированной серной кислоты, а затем нагревали с обратным холодильником в течение 1 часа. После охлаждения льдом, реакционный раствор промывали насыщенным водным раствором бикарбоната натрия, а затем водой, после чего растворитель отгоняли. Остаток очищали с помощью хроматографии на колонке с силикагелем, и получали 1,6 г этил 2-(5-бромобензо[b]тиен-2-ил)-3-(4-метоксифенил)пропионата. Т.пл. 62 - 65oC.
1H-ЯМР (CDCl3) δ: 1,15 (3H, т, J = 7,0 Гц); 3,08 (1H, дд. J = 13,5 и 7,3 Гц); 3,37 (1H, дд, J = 13,5 и 7,3 Гц); 3,71 (3H, с); 4,10 (2H, кв., J = 7,0 Гц); 4,14 (1H, т, J = 7,3 Гц); 6,75 (2H, д, J = 8,7 Гц); 6,83 (1H, с); 7,05 (2H, д, J = 8,7 Гц); 7,30 (1H, дд, J = 8,8 и 2,3 Гц); 7,57 (1H, д, J = 8,8 Гц); 7,74 (1H, д, J = 2,3 Гц).
f) 1,6 г этил 2-(5-бромобензо[b]тиен-2-ил)-3-(4-метоксифенил) пропионата, полученного в стадии e), обрабатывали в соответствии с процедурой, описанной в ссылочном примере 49, в стадии c), в результате чего получали 1,0 г этил 2-(5-цианобензо[b]тиен-2-ил)-3-(4-метоксифенил)пропионата. Т. пл. 93 - 96oC.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J = 7,0 Гц); 3,09 (1H, дд, J = 14,0 и 8,0 Гц); 3,39 (1H, дд, J = 14,0 и 8,0 Гц); 3,73 (3H, с); 4,12 (2H, кв., J = 7,0 Гц); 4,16 (1H, т); 6,75 (2H, д, J = 8,8 Гц); 7,05 (2H, д, J = 8,8 Гц); 7,13 (1H, с); 7,43 (1H, дд, J = 8,3 и 1,3 Гц); 7,81 (1H, д, J = 8,3 Гц); 7,92 (1H, шир.).
g) 3,3 г этил-2-(5-цианобензо[b]тиен-2-ил)-3-(4-метоксифенил) пропионата, полученного в стадии f), обрабатывали в соответствии с процедурой, описанной в стадии e) ссылочного примера 46, в результате чего получали 2,8 г целевого соединения. Т. пл. 146 - 147oC.
1H-ЯМР (CDCl3) δ: 1,19 (3H, т, J = 7,0 Гц); 3,09 (1H, дд, J = 13,5 и 7,5 Гц); 3,38 (1H, дд, J = 13,5 и 7,5 Гц); 4,13 (2H, кв., J = 7,0 Гц); 4,18 (1H, т); 6,70 (2H, д, J = 8,5 Гц); 7,00 (2H, д, J = 8,5 Гц); 7,15 (1H, с); 7,47 (1H, дд, J = 8,3 и 1,3 Гц); 7,85 (1H, д, J = 8,3 Гц); 7,95 (1H, шир.).
Ссылочный пример 54. Получение этил 2-[4-[((2S)-1-трет-бутоксикарбонил-2-пирролидинил)метокси]фенил]-3- (5-циано-2-бензофуранил)пропионата.
1,04 г диэтилазодикарбоксилата добавляли к 300 мл раствора тетрагидрофурана, содержащего 1 г этил 3-(5-циано-2-бензофуранил)-2- (4-гидроксифенил)пропионата, 1,2 г (2S)-1-трет-бутоксикарбонил-2-пирролидинметанола и 1,56 г трифенилфосфина, и полученную смесь размешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли 0,6 г (2S)-1-трет-бутоксикарбонил-2-пирролидинметанола, 0,78 г трифенилфосфина и 0,52 г диэтилазодикарбоксилата, а затем размешивали 18 часов при комнатной температуре. Затем, реакционный раствор концентрировали досуха, а остаток очищали с помощью хроматографии на колонке с силикагелем, элюируя смесью толуола и этилацетата. В результате описанной процедуры получали 790 мг целевого соединения в виде бесцветного маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,15 (3H, т); 1,46 (9H, с); 1,98 (4H, шир.), 3,0 - 4,2 (8H, м); 4,1 (2H), 6,37 (1H, с); 6,9 (2H, д); 7,2 (2H, д); 7,45 (2H); 7,76 (1H, с).
Соединения ссылочных примеров 55 - 61 были получены в соответствии с процедурой, описанной в ссылочном примере 54.
Ссылочный пример 55. Этил 3-[4-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)окси]фенил]-3-(5-циано-2-бензофуранил)пропионат,
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J = 7 Гц); 1,46 (9H, с); 2,0 - 2,2 (2H, м); 2,8 - 3,2 (2H, м); 3,5 - 3,7 (4H, м); 4,10 (2H, кв., J = 7 Гц); 4,5 - 4,7 (1H, м); 4,9 (1H, м); 6,49 (1H, с); 6,82 (2H, д, J = 9 Гц); 7,23 (2H, д, J = 9 Гц); 7,47 (2H, с); 7,80 (1H, с).
Ссылочный пример 56. Этил 5-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]-2-2-[2-(5- циано-2-бензофуранил)этил]бензоат.
1H-ЯМР (CDCl3) δ: 1,38 (3H, т, J = 7,0 Гц); 1,48 (9H, с); 2,00 - 2,30 (2H, м); 2,96 - 3,76 (8H, м); 4,36 (2H, кв.); 4,90 (1H, шир.); 6,44 (1H, с); 6,93 (1H, дд, J = 8,8 и 2,7 Гц); 7,15 (1H, д, J = 8,8 Гц); 7,48 (1H, д, J = 2,7 Гц); 7,52 (2H, с); 7,80 (1H, с)
Ссылочный пример 57. Этил 2-[5-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)окси]-
-2-[2-(5-циано-2-бензофуранил)этил]фенил]ацетат
1H-ЯМР (CDCl3) δ: 1,25 (3H, т. J = 7,0 Гц); 1,47 (9H, с); 1,90 - 2,30 (2H, м); 3,04 (4H, с); 3,36 - 3,70 (6H, м); 4,16 (2H, кв., J = 7,0 Гц); 4,90 - 5,12 (2H, шир.); 6,42 (1H, с); 6,60 - 6,80 (2H, м); 7,08 (1H, д, J = 7,6 Гц); 7,43 (2H, с); 7,77 (1H, д, J = 0,87 Гц).
Ссылочный пример 58. Этил 2-[2-[4-((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси]фенил]этил]-5-циано-3-бензофуранкарбоксилат.
1H-ЯМР (CDCl3) δ: 1,46 (12H, м); 2,05 (2H, м); 2,95 (2H, м); 3,5 (6H, м); 4,4 (2H, кв.); 4,80 (1H, шир.); 6,82 (2H, д); 7,08 (2H, д); 7,55 (2H); 8,30 (1H).
Ссылочный пример 59. Этил 3-[5-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси]-2-[2-(5-цианобензо[b]тиен-2-ил)этил]-4- этоксифенил]пропионат.
1H-ЯМР (CDCl3) δ: 1,23 (3H, т, J = 7,2 Гц); 1,33 (3H, т, J = 7,0 Гц); 1,47 (9H, с); 2,00 - 3,30 (10H, м); 3,4 - 3,7 (4H, м); 3,94 (2H, кв., J = 7,0 Гц); 4,12 (2H, кв., J = 7,2 Гц); 4,70 - 5,00 (1H, шир.); 6,67 (1H, с); 6,72 (1H, с); 7,05 (1H, с); 7,46 (1H, дд, J = 8,4 и 1,6 Гц); 7,84 (1H, дд, J = 8,4 Гц); 7,95 (1H, д, J = 1,6 Гц).
Ссылочный пример 60. Этил 3-[5-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси-2-[2-(5-цианобензо[b]тиен-2-ил)этил]фенил]пропионат.
Т.пл. 117 - 119oC
1H-ЯМР (CDCl3) δ: 1,24 (3H, т. J = 7,1 Гц); 1,49 (9H, с); 1,6 - 3,6 (14H, м); 4,13 (2H, кв., J = 7,1 Гц); 4,6 - 4,9 (1H, м); 6,50 - 7,20 (4H, м); 7,45 (1H, д, J = 8,5 Гц); 7,83 (1H, д, J = 8,5 Гц); 7,95 (1H, с).
Ссылочный пример 61. Этил-3-[4-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси]фенил]-2-(5-цианобензо[b]тиен-2-ил)пропионат.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J = 7,0 Гц); 1,45 (9H, с); 3,08 (1H, дд, J = 13,7 и 7,4 Гц); 3,30 - 3,70 (5H, м); 4,11 (2H, кв., J = 7,0 Гц); 4,00 - 4,30 (1H); 6,72 (2H, д, J = 8,3, Гц); 7,07 (2H, д, J = 8,3, Гц); 7,10 (1H, с); 7,40 (1H, дд, J = 8,3 и 1,3 Гц); 7,77 (1H, д, J = 8,3 Гц); 7,89 (1H, шир.).
Ссылочный пример 62. Получение метил 3-(5-циано-2-бензофуранил)-2-[4-[(тетрагидро-3-фуранил)окси] фенил]пропионата.
В 30 мл тетрагидрофурана растворяли 3 г 3-гидрокситетрагидрофурана, 6,5 г метил 2-(4-гидроксифенил)-2-оксоацетата и 9 г трифенилфосфина. Полученный раствор смешивали с 6,5 г диэтилазодикарбоксилата, и эту смесь размешивали в течение 2 часов. После отгонки растворителя, полученный остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, элюируя дихлорметаном, в результате чего получали 7,5 г метил 2-[4-[(тетрагидро-3-фуранил)окси]фенил]-2-оксоацетата в виде маслянистого вещества.
В смеси растворителей, состоящей из 30 мл тетрагидрофурана и 50 мл метанола, растворяли 2,2 г метил 2-[4-[(тетрагидро-3-фуранил) окси)фенил]-2-оксоацетата, полученного выше, и 3,6 г (5-циано-2-бензофуранил)метилтрифенилфосфония хлорида. К этому раствору, охлаждая льдом, добавляли 1,5 мл 1,8-диазабицикло[5.4.0]-7-ундецена. После 18-часового размешивания при комнатной температуре, реакционный раствор концентрировали досуха, остаток очищали с помощью хроматографии на силикагеле, элюируя смесью хлороформа и ацетона, в результате чего получали метил 3-(5-циано-2-бензофуранил)окси] фенил] акрилат в виде смеси E- и Z-форм. Полученное таким образом производное акриловой кислоты растворяли в 80 мл метанола, смешивали с 4 г окиси палладия • 1H2O • сульфата бария, и подвергали каталитической гидрогенизации при нормальном давлении. Затем, катализатор удаляли путем фильтрации, полученный фильтрат концентрировали досуха, а остаток очищали в помощью колоночной хроматографии на силикагеле, элюируя смесью хлороформа и ацетона. В результате этой процедуры, получали 2,5 г целевого соединения.
1H-ЯМР (CDCl3) δ: 2,0 - 2,3 (2H, м); 3,2 (1H, дд); 3,6 (1H, дд); 3,65 (3H, с); 3,97 (2H, д); 3,8 - 4,2 (1H, м); 4,8 - 5,0 (1H, м); 6,40 (1H, м); 6,8 (2H, д); 7,25 (1H, д); 7,5 (2H, с); 7,79 (1H, с).
Ссылочный пример 63. Получение метил 3-(5-циано-2-индолил)-2-[4-[((3R)-тетрагидро-3-фуранил)окси] фенил]пропионата.
В 30 мл тетрагидрофурана растворяли 3,0 г (S)-(+)-3- гидрокситетрагидрофурана, 6,6 г метил 2-(4-гидроксифенил)-2-оксоацетата и 8,90 г трифенилфосфина. Полученный раствор смешивали с 6,0 г диэтилазодикарбоксилата, и эту смесь размешивали в течение 2 часов. После отгонки растворителя, остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 4,60 г метил-2- [4-[((3R)-тетрагидро-3-фуранил)окси]фенил]-2-оксоацетата в виде маслянистого продукта.
В смеси растворителей, состоящей из 30 мл метанола, растворяли 1,70 г метил 2-[4-[((3R)-тетрагидро-3-фуранил)окси)фенил] -2- оксоацетата, полученного выше, и 3,0 г (5-циано-2-индол)метил трифенилфосфония бромида. К полученному раствору добавляли 2,1 мл 1,8-диазабицикло[5.4.0]-7-ундецена, размешивая при охлаждении льдом, а затем смесь размешивали при комнатной температуре в течение 2 часов. После отгонки растворителя, остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью хлороформа и ацетона, в результате чего получали метил 3-(5-циано-2-индолил)-2- [4-[((3R)-тетрагидро-3-фуранил)окси] фенил] акрилат в виде смеси E- и Z-форм. Полученную таким образом E/Z-смесь растворяли в 50 мл метанола, смешивали с 4,0 г окиси палладия • 1H2O • сульфата бария, а затем подвергали каталитической гидрогенизации при нормальном давлении в течение 3 часов. После этого катализатор удаляли путем фильтрации, растворитель в полученном фильтрате отгоняли, а остаток очищали с помощью хроматографии на силикагеле с использованием в качестве элюента смеси хлороформа и ацетона. В результате описанной процедуры получали 1,50 г целевого соединения в виде вязкого маслянистого материала.
1H-ЯМР (CDCl3) δ: 3,10 (1H, дд); 3,60 (3H, с); 3,78 - 4,10 (5H, м); 4,75 - 5,00 (1H, м); 6,25 (1H, шир.); 6,80 (2H, д); 7,20 (2H, д); 7,30 - 7,90 (3H, м); 10,00 (1H, с).
Ссылочный пример 64. Получение метил 3-(5-циано-2-индолил)-2-[4-[((3S)-тетрагидро-3-фуранил)окси] фенил]пропионата.
a) В 80 мл тетрагидрофурана растворяли 5,0 г (S)-(+)-3- гидрокситетрагидрофурана, 3,3 г муравьиной кислоты и 17,0 г трифенилфосфина. К этому раствору, размешивая и охлаждая льдом, по капле добавляли 12,0 г диэтилазодикарбоксилата. После 2-часового размешивания при комнатной температуре, растворитель отгоняли, а полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали (S)-(+)-тетрагидро-3-фуранилформат, который затем растворяли в 50 мл этанола. К этому эфирному раствору, размешивая, добавляли 5,0 г гидроокиси натрия, растворенной в 5 мл воды, а затем размешивали в течение 3 часов. После растворения в диэтиловом эфире, нерастворившиеся материалы удаляли путем фильтрации. После отгонки растворителя, получали 4,50 г сырого (R)-(-)-3-гидрокситетрагидрофурана.
b) Сырой (R)-(-)-3-гидрокситетрагидрофурана, полученный в стадии (a), обрабатывали согласно процедуре, описанной в ссылочном примере 63, в результате чего получали 1,50 г целевого соединения в виде вязкого маслянистого материала.
1H-ЯМР (CDCl3) δ: 3,15 (1H, дд); 3,65 (3H, с); 3,30 - 4,20 (5H, м); 4,80 - 5,05 (1H, м); 6,30 (1H, шир.); 6,82 (2H, д); 7,22 (2H, д); 7,30 - 7,90 (3H, м); 9,30 (1H, шир.).
Ссылочный пример 65. Получение метил 3-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-2-(5-циано-2-бензофуранил)пропионата.
a) 21,0 г 2-ацетил-5-бензофуранкарбонитрила растворяли в 300 мл дихлорметана. К этому раствору, размешивая при температуре -10oC, по капле добавляли 30 мл дихлорметанового раствора, содержащего 18,2 г брома. После постепенного нагревания до температуры охлаждения льдом, полученный реакционный раствор смешивали с хлороформом, и промывали 10% водным раствором тиосульфата натрия. Затем органический слой осушали и концентрировали досуха, а остаток перекристаллизовывали из смеси бензола и н-гексана, в результате чего получали 21,0 г 2-(2-бромо-1-оксоэтил)-5-бензофуранкарбонитрила в виде бесцветных кристаллов. Т.пл. 156 - 158oC.
ИК (KBr): 2228, 1696, 1616, 1564, 1290, 1166, 1122 см-1.
1H-ЯМР (CDCl3) δ: 4,44 (2H, с); 7,60 - 7,90 (3H, м); 8,11 (1H, с).
FD-MC (m/z): 263 (M+), 265 (M+)
b) 444 мг диоксида селена растворяли в 10 мл сухого метанола, нагревая при этом, а затем добавляли 1,056 г 2-(2-бромо-1-оксоэтил)-5-бензофуранкарбонитрила, полученного в стадии (a). Полученную таким образом смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения, нерастворившиеся материалы удаляли путем фильтрации, а полученный фильтрат концентрировали досуха. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью толуола и этилацетата, в результате чего получали 129 мг метил 2-(5-циано-2-бензофуранил)-2- оксоацетата в виде бесцветных игольчатых кристаллов. Т. пл. 196 - 199oC.
ИК (KBr): 1740, 1674, 1614, 1552 см-1.
1H-ЯМР (CDCl3) δ: 4,03 (3H, с); 7,66 - 7,96 2H, м); 8,17 (2H, с).
FD-MC (m/z): 321 (M+ + 92), 229 (M+).
c) 3,1 г метил 2-(5-циано-2-бензофуранил)-2-оксоацетата, полученный в стадии b), и 6,2 г (4-метоксифенил)метилтрифенилфосфония хлорида растворяли в смеси растворителей, состоящей из 100 мл тетрагидрофурана и 100 мл метанола. К этому раствору, размешивая при комнатной температуре, добавляли 2,19 г 1,8-диазабицикло[5.4.0]-ундецена, и продолжали размешивать в течение 1 часа. Затем к этой смеси добавляли 1,3 г (4-метоксифенил)метилтрифенилфосфония хлорида и 0,65 г 1,8-диазабицикло[5.4.0]-ундецена. После 1-часового размешивания с последующей отгонкой растворителя, полученный остаток очищали с помощью колоночной хроматографии, элюируя хлороформом, в результате чего получали вязкое маслянистое олефиновое соединение в виде смеси E- и Z-форм.
1H-ЯМР (CDCl3) δ: 3,78 (1,5H, с); 3,84 (3H, с); 3,87 (1,5H, с); 6,60 (9H, м).
Полученное олефиновое соединение растворяли в смеси растворителей, состоящей из 100 мл метанола и 100 мл тетрагидрофурана, с последующим добавлением 1,1 г окиси палладия • 1H2O • сульфата бария, а затем подвергали каталитической гидрогенизации при нормальном давлении в течение 3 часов. После удаления катализатора путем фильтрации и отгонки растворителя, остаток очищали с помощью хроматографии на силикагеле, в результате чего получали 4,2 г вязкого и маслянистого метил 2-(5-циано-2-бензофуранил)-2-(4-метоксифенил)пропионата.
1H-ЯМР (CDCl3) δ: 3,20 (1H, дд, J=14,4 и 7,8 Гц); 3,41 (1H, дд, J=14,4 и 7,4 Гц); 3,69 (3H, с); 3,75 (3H, с); 4,10 (1H, дд, J=7,8 и 7,4 Гц); 6,60 (1H, с); 6,76 (2H, д, J=8,8 Гц); 7,05 (2H, д, J=8,8 Гц); 7,53 (2H); 7,82 (1H, с).
d) 4,2 г метил 2-(5-циано-2-бензофуранил)-3-(4-метоксифенил) пропионата, полученного в стадии (c), растворяли в 150 мл дихлорметана, и полученный раствор охлаждали до -50oC. Затем к этому раствору, размешивая, по капле добавляли 30 мл дихлорметанового раствора, содержащего 9,97 г трибромида бора. Затем температуру реакционного раствора постепенно повышали до 15oC. После 30-минутного размешивания при этой температуре, реакционный раствор разбавляли хлороформом, промывали разбавленной соляной кислотой, а затем осушали путем отгонки растворителя. Полученный остаток хроматографировали на колонке с силикагелем, элюируя смесью хлороформа и этанола, и полученные объединенные нужные фракции концентрировали с осаждением кристаллов. Эти кристаллы промывали бензолом, и собирали путем фильтрации, в результате чего получали 3,1 г метил 2-(5-циано-2-бензофуранил)-3-(4-гидроксифенил) пропионата в виде бесцветных кристаллов.
Т.пл. 110 - 111oC.
ИК (KBr): 2228, 1722, 1594, 1518, 1272 см-1
1H-ЯМР (CDCl3) δ: 3,18 (1H, дд, J=14,4 и 7,8 Гц); 3,36 (1H, дд, J=14,4 и 7,4 Гц); 3,69 (3H, с); 4,09 (1H, дд, J=7,8 и 7,4 Гц); 6,60 (1H, с); 6,69 (2H, д, J=8,4 Гц); 7,00 (2H, д, J=8,4 Гц); 7,53 (2H, с); 7,83 (1H, с).
e) В 150 мл сухого тетрагидрофурана растворяли 3,0 г метил-2-(5-циано-2-бензофуранил)-3-(4-гидроксифенил)-пропионата, полученного в стадии d), 1,92 г (3R)-1-трет-бутоксикарбонил-3- гидроксипирролидина и 2,69 г трифенилфосфина. К полученному раствору, размешивая при комнатной температуре, добавляли 1,79 г диэтилазодикарбоксилата, и продолжали размешивать в течение 1 часа. После отгонки растворителя, остаток подвергали колоночной хроматографии на силикагеле, элюируя смесью толуола и этилацетата, в результате чего получали смесь, содержащую исходное соединение метил 2-(5-циано-2-бензофуранил)-3-(4-гидроксифенил)пропионата и целевое соединение
Полученную таким образом смесь растворяли в 100 мл тетрагидрофурана. К этому раствору добавляли последовательно 0,95 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина, 1,35 г трифенилфосфина и 0,85 г диэтилазодикарбоксилата. Полученную смесь размешивали при комнатной температуре в течение 16 часов. Затем реакционный раствор обрабатывали и очищали в соответствии с процедурой, описанной выше, в результате чего получали 2,02 г целевого соединения в виде вязкого и маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,46 (9H, с); 1,88 - 2,24 (2H, м); 3,10 - 3,60 (6H, м); 3,69 (3H, с); 4,10 (1H, т); 4,81 (1H, шир.); 6,1 (1H, с); 6,73 (2H, д, J=8,3 Гц); 7,04 (2H, д, J=8,3 Гц); 7,54 (2H, с); 7,83 (1H, с).
FD-MC (m/z): 321 (M+).
Ссылочный пример 66. Получение этил 3-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-4- (5-цианобензо[b]тиен-2-ил)бутирата.
a) 14,2 г этил 2-этоксикарбонил-2-4-метоксифенил ацетата растворяли в 150 мл тетрагидрофурана. К этому раствору, размешивая при этом с охлаждением льдом, добавляли 2,6 г гидрида натрия (маслянистого типа, 60%), а затем продолжали размешивать в течение 2 минут. После этого, к смеси добавляли 17,2 г 5-бромо-2- бромометил-бензо[b]тиофена. После 18-часового размешивания при комнатной температуре, реакционный раствор смешивали с водным раствором хлорида аммония, а затем экстрагировали этилацетатом. После отгонки растворителя, остаток хроматографировали на колонке с силикагелем, элюируя хлороформом, в результате чего получали 24,2 г этил-3-(5-бромобензо[b]тиен-1-ил)-2-этоксикарбонил-2-(4-метоксифенил) пропионата.
1H-ЯМР (CDCl3) δ: 1,2 (6H, т); 3,78 (3H, с); 3,85 (2H, с); 6,76 - 7,0 (3H, м); 7,2 - 7,8 (5H, м).
b) Раствор 7,3 г гидроксида калия, растворенных в 20 мл воды, добавляли к 200 мл этанолового раствора, содержащего 24,2 г 3-(5-бромобензо[b]тиен-2-ил)-2-этоксикарбонил-2-(4-метоксифенил) пропионата, полученного в стадии (a), и полученную смесь размешивали в течение 4 дней. Затем реакционный раствор выливали в охлажденную разбавленную соляную кислоту, а осажденные кристаллы собирали путем фильтрации. Эти кристаллы растворяли в этилацетате, а затем осушали. После отгонки растворителя, осадок растворяли в 200 мл этанола, смешивали в 3 мл концентрированной серной кислоты и нагревали с обратным холодильником в течение 2 часов. После охлаждения, реакционный раствор концентрировали, смешивали с хлороформом, промывали водой и осушали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 20 г этил 3-(5-бромобензо[b]тиен-2-ил)-2-(4- метоксифенил)пропионата.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т); 3,2 (1H, дд); 3,55 (1H, дд); 3,77 (3H, с); 3,81 (1H, дд); 4,10 (2H, кв.); 6,82 (2H, д); 7,2 - 7,8 (6H, м).
c) 20 г этил 3-(5-бромобензо[b]тиен-2-ил)-2-(4-метоксифенил) пропионата, полученного в стадии b), растворяли в 200 мл тетрагидрофурана, а затем добавляли 12 г борогидрида натрия. К этой смеси, охлаждая при этом льдом, по капле добавляли 80 мл метанола. После 3-часового размешивания, реакционный раствор доводили до pH 6 с помощью концентрированной соляной кислоты, а затем экстрагировали этилацетатом. Полученный органический слой осушали для отгонки растворителя, а остаток хроматографировали на колонке с силикагелем, элюируя смесью хлороформа и метанола, в результате чего получали 16 г 3-(5-бромобензо[b]тиен-2-ил)-2-(4-метоксифенил)-1-пропанола.
1H-ЯМР (CDCl3) δ: 2,9 - 3,4 (3H, м); 3,73 (3H, с); 3,62 - 3,90 (2H, шир. ); 6,70 - 7,80 (8H, м).
d) 16 г 3-(5-бромобензо[b]тиен-2-ил)-2-(4-метоксифенил)-1- пропанола, полученного в стадии c), растворяли в 40 мл дихлорметана. К этой смеси, размешивая при охлаждении льдом, добавляли 6,3 мл триэтиламина и 4 мл метансульфонилхлорида. После 2-часового размешивания при той же температуре, реакционный раствор смешивали с дихлорметаном, промывали с водой и осушали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 18,5 г 3-(5-бромобензо[b]тиен-2-ил)-2-(4-метоксифенил)пропилметансульфоната.
1H-ЯМР (CDCl3) δ: 3,78 (3H, с); 3,9 - 4,5 (3H, м); 3,70 (3H, с), 4,3 (2H, м); 6,70 - 7,80 (8H, м).
e) 1,2 г цианида натрия растворяли в 30 мл диметилсульфоксида при температуре 90oC. К этому раствору постепенно добавляли 18,5 г 3-(5-бромбензо[b] тиен-2-ил-2-(4-метоксифенил)пропилметансульфоната, а затем размешивали в течение 1 час при 80oC. Реакционный раствор смешивали со смесью этилацетата и толуола, промывали водой, а затем осушали с отгонкой растворителя. Осажденные кристаллы промывали эталоном и осушали, в результате чего получали 5 г 3-(5-бромобензо[b] тиен-2-ил)-2-ил-(4-метоксифенил)бутиронитрила. То же самое соединение получали также с выходом 2 г путем концентрирования этанолового раствора, полученного в результате промывки кристаллов, и хроматографирования полученного концентрата на колонке с силикагелем, элюируя хлороформом.
1H-ЯМР (CDCl3) δ: 2,5 - 2,7 (2H, шир.); 3,2 - 3,4 (3H, шир.); 3,76 (3H, с); 6,70 - 7,80 (8H, м).
MC m/z: 386, 388.
f) 7 г 3-(5-бромобензо[b]тиен-2-ил)-2-(4-метоксифенил) бутиронитрила, полученного в стадии e), суспендировали в 80 мл этанола, а затем добавляли 5 мл концентрированной серной кислоты и несколько капель воды. Полученную смесь нагревали с обратным холодильником в течение 7 часов. После отгонки растворителя, реакционный раствор смешивали с хлороформом и водой, а полученный органический слой осушали путем отгонки растворителя. Затем остаток хроматографировали на колонке с силикагелем, элюируя хлороформом, и получали 6,3 г этил 4-(5-бромобензо[b]тиен-2-ил)-3-(4-метоксифенил)бутирата.
1H-ЯМР (CDCl3) δ: 1,12 (3H, т); 2,65 (2H, дд); 3,10 - 3,80 (3H, м); 3,76 (3H, с); 4,01 (2H, кв. ); 6,70 - 6,95 (3H, м); 7,10 (2H, д); 7,20 - 7,40 (1H); 7,55 (1H, д); 7,72 (1H, д);
FAB-MC (m/z): 433, 435.
g) 6,0 г этил 4-(5-бромобензо[b]тиен-2-ил)-3-(4-метоксифенил) бутирата, полученного в стадии f, растворяли в 50 мл N-метил-2-пирролидона, с последующим добавлением 1,6 г цианида меди и каталитически эффективного количества сульфата меди. Полученную смесь размешивали при температуре 190 - 200oC в потоке аргона. После охлаждения, реакционный раствор смешивали с этилацетатом и толуолом, а затем промывали водой, с последующей отгонкой растворителя. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя смесью хлороформа и ацетона, в результате чего получали 4,5 г этил 4-(5-цианобензо[b]тиен-2-ил)-3-(4-метоксифенил)- бутирата
1H-ЯМР (CDCl3) δ: 1,18 (3H, т); 2,70 (2H, дд); 3,16 - 3,70 (3H, м); 3,78 (3H, с); 4,02 (2H, кв.); 6,85 (2H, д); 6,98 (1H, с); 7,18 (2H, д); 7,5 (1H, дд); 7,8 (1H, д); 7,96 (1H, д).
h) 4,5 г этил 4-(5-цианобензо[b]тиен-2-ил)-3-(4-метоксифенил) бутирата, полученного в стадии g), растворяли в 20 мл дихлорметана. К полученному раствору, охлажденному до -70oC, добавляли 3,4 мл трибромида бора. Полученную таким образом смесь нагревали до комнатной температуры и размешивали в течение 1 часа. К реакционному раствору добавляли измельченный лед, и собирали слой дихлорметана, который затем осушали для отгонки растворителя. Полученный остаток растворяли в 50 мл тетрагидрофурана. К смеси, размешивая в потоке аргона и при охлаждении льдом, добавляли 1,9 г (3R)-1-третбутоксикарбонил-3-гидроксипирролидина, 3,2 г трифенилфосфина и 2,3 г диэтилазодикарбоксилата. Полученную смесь размешивали 18 часов при комнатной температуре. После отгонки растворителя, остаток очищали с помощью колоночной хроматографии, элюируя смесью н-гексана и этилацетата. В результате этой процедуры, получали 4 г целевого соединения.
1H-ЯМР (CDCl3) δ: 1,48 (9H, с); 1,95 - 2,20 (2H, м); 2,65 (2H, дд); 3,15 - 3,70 (7H, м); 4,78 - 5,00 (1H, м); 6,80 (2H, д); 6,98 (1H, с); 7,17 (2H, д); 7,5 (1H, дд); 7,82 (1H, д); 7,98 (1H, д).
Ссылочный пример 67. Получение этил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)тио] фенил] -3-(5-цианобензо[b] тиено- 2-ил)-2-этоксикарбонилпропионата.
a) 20,2 г этил 4-меркаптофенилацетата растворяли в 450 мл тетрагидрофурана. К этому раствору, размешивая и охлаждая льдом, добавляли 21,0 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина, 29,4 г трифенилфосфина и 19,5 г диэтилазодикарбоксилата. Полученную смесь размешивали при комнатной температуре в течение 18 часов. После отгонки растворителя, остаток очищали с помощью хроматографии на силикагеле, элюируя смесью н-гексана и этилацетата, в результате чего получали 7,0 г этил этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)тио]фенил]ацетата.
1H-ЯМР (CDCl3) δ: 1,25 (3H, т, J = 7,2 Гц), 1,45 (9H, с); 1,7 - 2,4 (2H, м); 3,2 - 4,4 (5H, м); 3,58 (2H, с); 4,15 (2H, кв., J = 7,2 Гц); 7,0 - 7,6 (4H, м).
b) 4,0 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) тио] фенил] ацетата, полученного в стадии (a), растворяли в 21 мл N,N-диметилформамида, а затем добавляли 4,02 мл диэтилкарбоната. К полученному раствору, перемешивая при температуре 130oC, добавляли 530 мг (60%) гидрида натрия, после чего смесь размешивали 10 минут, затем добавляли 106 мг гидрида натрия и опять размешивали 10 минут. Реакционный раствор выливали в ледяную воду, нейтрализовали разбавленной соляной кислотой, экстрагировали этилацетатом, а затем осушали. После отгонки растворителя, остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью н-гексана и этилацетата, в результате чего получали 1,74 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) тио)фенил] -2-этоксикарбонилацетата в виде маслянистого продукта.
1H-ЯМР (CDCl3) δ: 1,27 (6H, т, J = 7,2 Гц); 1,46 (9H, с); 1,4 - 2,4 (2H, м); 3,0 - 4,0 (5H, м); 4,22 (4H, кв., J = 7,2 Гц); 4,58 (1H, с); 7,2 - 7,5 (4H, м).
c) 1,7 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)тио]фенил] -2-этоксикарбонилацетата, полученного в стадии (b), растворяли в смеси растворителей, состоящей из 20 мл тетрагидрофурана и 1 мл N,N-диметилформамида, после чего добавляли 155 мг гидрида натрия (60%), и полученную смесь размешивали 20 минут. К реакционному раствору добавляли 980 мг 2-бромометилбензо[b] тиофен-5-карбонитрила, а затем размешивали в течение 24 часов. Полученный реакционный раствор выливали в ледяную воду, экстрагировали этилацетатом, а затем осушали. После отгонки растворителя, остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью н-гексана и этилацетата, в результате чего получали 2,05 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,23 (6H, т, J = 7,2 Гц), 1,46 (9H, с); 1,50 - 2,50 (2H, м); 3,2 - 4,4 (5H, м); 3,89 (2H, с); 4,25 (4H, кв., J = 7,2 Гц); 7,28 (4H, с); 7,44 (1H, дд, J = 8,4 и 1,5 Гц); 7,78 (1H, д, J = 8,4 Гц); 7,91 (1H, дд).
Ссылочный пример 68. Получение этил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(5-цианобензо[b] тиен-2-ил)-2-пропионата.
В смеси, состоящей из 50 мл тетрагидрофурана и 50 мл этанола, растворяли 3,0 г (5-цианобензо[b]тиен-2-ил-метилтрифенилфосфония хлорида и 2,55 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)окси] фенил] -2-оксоацетата. К полученному раствору, размешивая при комнатной температуре, добавляли 1,07 г 1,8-диазабицкло[5.4.0]-7-ундецена, и эту смесь размешивали при комнатной температуре в течение 1 часа. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью толуола и этилацетата, в результате чего получали этил-2-[4-[((3S)-1-третбутоксикарбонил-3-пирролидинил] окси] фенил]-3-(5-цианобензо[b]тиен-2-ил)акрилат в виде смеси E- и Z-форм. Полученное таким образом соединение растворяли в смеси, состоящей из 50 мл тетрагидрофурана и 50 мл этанола, и этот раствор смешивали с 0,5 г 10% палладированного угля (катализатор на 50% мокрого типа), а затем подвергали каталитической гидрогенизации при нормальном давлении. После удаления катализатора фильтрацией и отгонки растворителя, полученный остаток очищали с помощью хроматографии на силикагеле, элюируя смесью толуола и этилацетата, в результате чего получали 2,2 г целевого соединения в виде вязкого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J = 7,0 Гц); 1,47 (9H, с); 1,90 - 2,20 (2H, м); 3,10 - 3,95 (7H, м); 4,10 (2H, кв., J = 7,0 Гц); 4,84 (1H, шир.); 6,81 (2H, д, J = 9,0 Гц); 7,20 (1H, с); 7,25 (2H, д, J = 9,0 Гц); 7,44 (1H, дд, J = 9,0 и 1,6 Гц); 7,81 (1H, дд, J = 9,0 и 1,6 Гц); 7,94 (1H, с).
Соединения ссылочных примеров 69-75 получали в соответствии с процедурой, описанной в ссылочном примере 68.
Ссылочный пример 69. Этил 2-[4-[2-(трет-бутоксикарбониламино)- 1-(трет-бутоксикарбониламиноэтил)этокси] фенил] -3-(5-цианобензо[b] тиен-2-ил)пропионат, вязкое маслянистое вещество
1H-ЯМР (CDCl3) δ: 1,25 (3H, т, J = 7,0 Гц); 1,45 (18H, с); 2,90 - 4,50 (8H, м); 6,80 - 7,35 (5H); 7,45 (1H, дд, J = 8,3 и 1,3 Гц); 7,80 (1H, д, J = 8,3 Гц); 7,93 (1H).
Ссылочный пример 70. Этил 2-[4-[((2S)-1- бутоксикарбонил-2-пирролидинил)-метокси] фенил]-3-(5-цианобензо[b]тиен-2-ил)пропионат.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т); 1,47 (9H, с); 2,00 (4H, шир.с.); 3,40 (2H, шир.с.); 3,60 - 4,30 (6H); 6,90 (2H, д, J = 10 Гц); 7,25 (2H, д, J = 10 Гц); 7,00 - 8,00 (4H, м).
Ссылочный пример 71. Этил 2-[4-[(1-трет-бутоксикарбонил-4- пиперидинил)окси]фенил- -3-(5-цианобензо[b]тиен-2-ил)пропионат твердое вещество
1H-ЯМР (CDCl3) δ: 1,10 (3H, т, J = 6,0 Гц); 1,50 (9H, с); 1,70 - 2,00 (4H, м); 3,20 - 4,00 (4H, м); 4,15 (2H, кв.); 4,30 - 4,60 (1H, шир.с); 6,80 - 8,10 (8H).
Ссылочный пример 72. Этил 2-[4-(2-трет- бутоксикарбониламиноэтокси)фенил] -3-(5-цианобензо[b] тиен-2-ил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,16 (3H, т, J = 7,0 Гц); 1,45 (9H, с); 3,05 - 4,40 (9H); 5,12 (1H, шир.с.); 6,84 (2H, д, J = 8,3 Гц); 7,01 (1H, с); 7,25 (2H, д, J = 8,3 Гц); 7,41 (1H, дд, J = 8,3 и 1,2 Гц); 7,77 (1H, дд, J = 8,3 Гц); 7,89 (1H, с).
Ссылочный пример 73. Этил 2-[4-[((2S)-1-трет- бутоксикарбонил-5-оксо-2-пирролидинил)метокси]фенил]- 3-(5-цианобензо[b]тиен-2-ил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т); 1,42 (9H, с); 1,80 - 2,25 (2H, м); 2,30 - 2,60 (2H, м); 3,20 (1H, дд); 3,37 (1H, дд); 3,50 - 3,82 (1H, дд); 3,82 - 4,50 (4H, м); 4,80 - 5,10 (1H, м); 6,75 - 8,10 (8H, м).
Ссылочный пример 74. Этил 2-[4-[((2S,4S)-1-трет- бутоксикарбонил-2-метил-4-пирролидин)окси]фенил]-3-(5-цианобензо[b]тиен-2-ил)пропионат.
1H-ЯМР (CDCl3) δ: 1,15 - 1,50 (6H, м); 1,50 (9H, с); 1,80 - 2,60 (2H, м); 3,00 - 4,50 (8H, м); 4,80 - 5,10 (1H, м); 6,80 - 8,20 (8H, м).
Ссылочный пример 75. Этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси]фенил]- 3-(6-цианобензо[b]тиен-2-ил)пропионат.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т); 1,46 (9H, с); 2,10 (2H, м); 3,60 (6H, м); 3,83 (1H, м); 4,10 (2H); 4,85 (1H, шир. с); 6,86 (2H, д); 7,04 (1H, с); 7,25 (2H); 7,55 (1H, дд); 7,65 (1H, д); 8,04 (1H).
Ссылочный пример 76. Получение этил (+)-2-[4-[((2S)-1-трет-бутоксикарбонил-2-пирролидинил)метокси] фенил]-3- (5-цианобензо[b]тиен-2-ил)пропионата и этил (-)-2-[4-[((2S)-1-трет-бутоксикрабонил-2-пирролидинил)метокси]фенил]- 3-(5-цианобензо[b]тиен-2-ил)пропионата.
5,0 г этил 2-[4-[((2S)-1-трет-бутоксикарбонил-2-пирролидинил)метокси] фенил] -3- (5-цианобензо[b]тиен-2-ил)пропионата разделяли на (+) и (-) формы с использованием колонки для разделения оптических изомеров, в результате чего получали 2,5 г (+) и 1,7 (-) целевого соединения.
Изомер (-): т.пл. 102-104oC
[α]
1H-ЯМР (CDCl3) δ: 1,13-1,22 (3H, м); 1,47 (8H, с); 1,80 - 2,10 (4H, м); 3,25 - 3,50 (4H, м); 3,64 - 3,75 (1H, м); 3,70 - 3,90 (1H, шир. с); 3,90 (1H, шир. с); 4,05 - 4,20 (4H, м); 6,88 (2H, д, J = 8,3 Гц); 7,02 (1H, с); 7,23 (2H, д, J = 8,3 Гц); 7,45 (1H, д, J = 8,3 Гц); 7,80 (1H, д, J = 8,3 Гц), 7,94 (1H, с).
ВЭЖХ: Колонка; колонка на амилозе для использования в разведении оптических изомеров (CHIPALPAK AD, 20 ⊘ х 250 мм, Daicel Chemical Industries, Ltd).
Растворитель: н-гексан: изо-пропанол = 70:30.
Скорость потока: 4 мл/мин
Время удерживания: 20-23 минуты
Изомер (+): т.пл. 111-112oC
[α]
1H-ЯМР (CDCl3) δ: 1,13 - 1,22 (3H, м); 1,47 (9H, с); 1,80 - 2,10 (4H, м); 3,25 - 3,50 (4Н, м); 3,64 - 3,75 (1H, м); 3,70 - 3,90 (1H, шир. с); 3,90 (1H, шир. с); 4,05 - 4,20 (4H, м); 6,88 (2H, д, J = 8,3 Гц); 7,02 (1H, с); 7,23 (2H, д, J = 8,3 Гц); 7,45 (1H, д, J = 8,3 Гц); 7,80 (1H, д, J = 8,3 Гц); 7,94 (1H, с).
ВЭЖХ: Колонка, колонка на амилозе для использования в разделении оптических изомеров (CHAPALPAK AD, 20 ⊘ х 250 мм, Daicel Chemical Industries, Ltd).
Растворитель: н-гексан: изо-пропанол = 70:30
Скорость потока: 4 мл/мин
Время удерживания: 23-27 минут
Ссылочный пример 77. Получение этил (-)-2-[4-[(1-трет-бутоксикарбонил-3-пиперидинил)окси] фенил]-3- (7-циано-2-нафтил)пропионата и этил (+)-2-[4-[(1-трет-бутоксикарбонил-4-пиперидинил)-окси] фенил] -3-(7-циано-2- нафтил)пропионата.
Эти соединения получали тем же способом, описанным в ссылочном примере 76.
Изомер (+):
[α]
1H-ЯМР (CDCl3) δ: 1,11 (3H, т, J = 6,9 Гц); 1,47 (9H, с); 1,70 - 1,80 (2H, м); 1,85 - 1,95 (2H, м); 3,15 - 3,20 (1H, м); 3,30 - 3,40 (2H, м); 3,50 - 3,60 (1H, м); 3,65 - 3,75 (2H, м); 3,85 - 3,90 (1H, шир. с.); 4,0 - 4,1 (2H, м); 4,40 - 4,45 (1H, м); 6,85 (2H, н, J = 8,3 Гц); 7,23 (2H, д); 7,40 - 7,45 (1H, м); 7,53 - 7,58 (1H, м); 7,62 (1H, с); 7,77 (2H, д); 7,85 (1H, д, J = 8,3 Гц); 8,12 (1H, с).
ВЭЖХ: Колонка: колонка на амилозе для разделения оптических изомеров (CHIPALPAK, AD, 4,6 ⊘ х 250 мм, Diacel Chemical Industries, Ltd).
Растворитель: н-гексан:изо-пропанол = 90:10
Скорость потока: 1 мл/мин
Время удерживания: 26,9 минут
Изомер (+):
[α]
1H-ЯМР (CDCl3) δ: 1,11 (3H, т, J = 7,3 Гц); 1,65 - 1,70 (2H, м); 1,85 - 2,00 (2H, м); 3,15 - 3,20 (2H, м); 3,30 - 3,35 (2H, м); 3,50 - 3,60 (1H, м); 3,65 - 3,75 (2H, м); 3,85 - 3,90 (1H, шир. с); 4,0 - 4,1 (2H, м); 4,40 - 4,45 (1H, м); 6,85 (2H, д, J = 8,8 Гц); 7,23 (2H, д, J = 8,3 Гц); 7,40 - 7,45 (м, 1H, Ar-H); 7,52 - 7,57 (1H, м); 7,62 (1H, с); 7,77 (1H, д, J = 8,3 Гц); 7,85 (1H, д, J = 8,3 Гц); 8,11 (1H, с).
ВЭХЖ: Колонка; колонка на амилозе для разделения оптических изомеров (CHIRALPAK AD, 4,6 ⊘ х 250 мм, Daicel Chemical Industries, Ltd).
Растворитель: н-гексан: изо-пропанол = 90:10
Скорость потока: 1 мл/мин
Время удерживания: 31 минута
Ссылочный пример 78. Получение этил (+)-2-[4-[((2S)-1-трет- бутоксикарбонил-2-пирролидинил)метокси] фенил] -3-(5-цианобензо[b] -тиен-2-ил) пропионата.
54,0 г 2-[4-[((2S)-1-трет-бутоксикарбонил-2-пирролидинил)-метокси]фенил] -3-5-цианобензо[b] тиен-2-ил-пропионата, растворяли в 400 мл сухого этанола во время нагревания, и к полученному раствору добавляли 800 мл сухого н-гексана. К полученной таким образом смеси добавляли 100 мг гидрида натрия и затравочные кристаллы этил (+)-2-[4-[((2S)-1-трет-бутоксикарбонил-2-пирролидинил)-метокси] фенил] - 3-(5-цианобензо[b]тиен-2-ил)пропионата. После перемешивания при комнатной температуре в течение 4 часов, полученную таким образом смесь смешивали со 100 мг гидрида натрия, и продолжали перемешивать еще 18 часов при комнатной температуре, и собирали осажденные кристаллы путем фильтрации. Собранные таким образом кристаллы перекристаллизовывали из 22 объемов (м. /об.) смеси этанола и н-гексана (30:70, м/об.). Процесс перекристаллизации повторяли 3 раза, и получали 37,0 г целевого соединения с чистотой диастереоизомера 99,5% или более.
Ссылочный пример 79. Получение этил (+)-2-[4-[((3S)-трет-бутоксикарбонил-3-пирролидин)окси]фенил]-3-(7-циано- 2-нафтил)пропионата.
Это соединение было получено в соответствии с процедурой, описанной в ссылочном примере 78.
Ссылочный пример 80. Получение этил 2-[4-[((2R)-1-трет-бутоксикарбонил-2- пирролидинил)метокси]фенил]-3-(5-дианобензо[b]тиен-2-ил)- 2-этокси-карбонилпропионата.
4,1 г этил-2-[4-[((2R)-1-трет-бутоксикарбонил-2-пирролидинил)метокси] фенил] -2- этоксикарбонилацетата растворяли в 100 мл тетрагидрофурана. Полученный таким образом раствор при комнатной температуре смешивали с 0,38 г 60% гидрида натрия и перемешивали в течение 30 минут. Перемешивая при комнатной температуре, к полученному реакционному раствору по капле добавляли 10 мл раствора тетрагидрофурана, содержащего 2,1 г 2-бромометилбензо[b]-тиофен-5-карбонитрила. После концентрирования реакционного раствора досуха, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смесь толуола и хлороформа, в результате чего получали 4,34 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,21 (6H); 1,46 (9H, с); 2,0 (4H, шир. с); 3,40 (2H, шир. с); 3,88 (3H); 4,22 (6H); 6,90 (3H); 7,20 (2H, д); 7,50 (1H); 7,78 (1H, д); 7,93 (1H, д).
В соответствии с процедурой, описанной в ссылочном примере 80, получали соединения ссылочных примеров 81 и 82.
Ссылочный пример 81. Этил 3-(5-цианобензо[b]-тие-2-ил)-2-этоксикарбонил-2-[4-[(2-имидазолин-2-ил) метокси] фенил] пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,22 (6H, т); 3,63 (4H, с); 3,89 (2H, с); 4,24 (4H); 4,69 (2H, с); 6,86 (3H); 7,27 (2H); 7,42 (1H); 7,76 (1H); 7,88 (1H).
Ссылочный пример 82. Этил-2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси] фенил] -3- (5-циано-2-бензотиазолил)-2-этоксикарбонил-пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,22 (6H, т); 1,46 (9H, с); 2,09 (2H, шир.с); 3,56 (4H, шир.с); 4,13 (2H); 4,28 (4H, д); 4,85 (1H, шир. с); 6,82 (2H, д); 7,26 (2H, д); 7,63 (1H, дд); 7,95 (1H, д); 8,25 (1H, д).
Ссылочный пример 83. Получение этил 3-(5-цианобензо[b]тиен-2-ил)-2-[4-[[2-(этоксикарбонилимино) гексагидропиримидин-5-ил]окси]фенил]пропионата
1,0 г этил 2-[4-[2-(трет-бутоксикарбониламино)-1-(трет- бутоксикарбониламинометил)-этокси] фенил] -3-(5-цианобензо[b]тиен-2-ил) пропионата растворяли в 2 мл анизола. К вышеуказанному раствору добавляли 10 мл трифтороуксусной кислоты, размешивая при охлаждении льдом, и полученную таким образом смесь перемешивали при комнатной температуре в течение 1 часа. Полученный реакционной раствор концентрировали при пониженном давлении, и полученный таким образом остаток растворяли в воде и промывали н-гексаном. Полученный водный слой доводили до pH 9-10 с использованием концентрированного водного раствора аммиака и затем экстрагировали хлороформом. Полученный органический слой концентрировали досуха и полученный таким образом остаток растворяли в 20 мл сухого этанола. К полученному таким образом раствору добавляли 300 мг этил N-(этокси(метилтио)метилен)карбамата, который был синтезирован в соответствии с процедурой, описанной в Journal of the Chemical Society, Parkin 1, 1973, pp. 2644-2646. После этого полученную таким образом смесь перемешивали 20 часов, а осадок собирали и в результате чего получали 560 мг целевого соединения.
Т.пл. 179-182oC.
ИК (KBr): 2230, 1725, 1638, 1512, 1337 см-1
1H-ЯМР (CDCl3) δ: 1,13 (3H, т, J = 7,0 Гц); 1,17 (3H, т, J = 7,0 Гц), 3,10 - 4,30 (11H, м); 4,50 - 4,80 (1H, м); 6,89 (2H, д, J = 8,75 Гц); 7,03 (1H, с); 7,26 (2H, д, J = 8,75 Гц); 7,46 (1H, дд, J = 8,31 и 1,75 Гц); 7,82 (1H, д, J = 8,31 Гц); 7,95 (1H, д); 8,70 - 9,50 (2H, шир. с).
Ссылочный пример 84. Получение этил 3-(5-цианобензо[b]тиен-2-ил)-2-[4-[[2-(имино)-гексагидропиримидин-5-ил]окси]фенил] пропионата гидрохлорида.
a) 2,9 г тиоцианата калия растворяли в 150 мл сухого ацетона. Размешивая при охлаждении льдом, к вышеуказанному раствору добавляли по капле 6,8 г p-нитробензилхлороформата, который был растворен в 20 мл ацетона. Полученную таким образом смесь перемешивали в течение 2 часов, охлаждая льдом, и смешивали с 1,15 г метанола, а затем перемешивали при комнатной температуре в течение 20 часов. После этого, осажденные таким образом кристаллы собирали путем фильтрации и промывали с использованием хлороформа, в результате чего получали 2,88 г p-нитробензилметокси(тиокарбамоил)карбамата в виде порошка.
1H-ЯМР (CDCl3) δ: 4,03 (3H, с; 5,33 (2H, с); 7,70 (2H, д, J = 9,0 Гц); 8,80 (2H, д, J = 9,0 Гц).
b) 3,5 г p-нитробензилметокси(тиокарбамоил)карбамата, полученного в вышеуказанной стадии (a), и 1,79 г безводного карбоната калия растворяли в смеси раствора 40 мл воды и 40 мл диоксана. К полученному таким образом раствору постепенно до капле добавляли 1,72 г диметилсульфата, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К полученному реакционному раствору снова добавляли 300 мг безводного карбоната калия, добавляли при этом по капле 300 мг диметилсульфата. Полученный таким образом реакционный раствор разбавляли этилацетатом, промывали водой и последовательно насыщали водным раствором хлорида натрия, а затем концентрировали. После этого осажденные таким образом кристаллы собирали путем фильтрации и тщательно промывали н-пентаном, в результате чего получали 3,23 г p-нитробензил N-(метокси метилтио)метилен)карбамата.
1H-ЯМР (CDCl3) δ: 2,40 (3H, с); 4,00 (3H, с); 5,28 (2H, с); 7,56 (2H, д, J = 9,0 Гц); 8,22 (2H, д, J = 9,0 Гц).
c) В части в 10 мл анизола растворяли 2,0 г этил 2-[4-[2-(трет-бутоксикарбониламино)-1-(трет-бутоксикарбониламино-метил) этокси]фенил]-3-(5-цианобензо[b] тиен-2-ил)пропионата. К вышеуказанному раствору добавляли 30 мл трифторуксусной кислоты, перемешивая при охлаждении льдом, и смесь перемешивали при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и полученный таким образом остаток растворяли в воде и промывали н-гексаном. Полученный водный слой доводили до pH 10 с использованием концентрированного водного раствора аммиака и затем экстрагировали хлороформом. Полученный органический слой концентрировали досуха, и полученный таким образом остаток растворяли в 50 мл тетрагидрофурана. К полученному таким образом раствору добавляли 921 мг p-нитробензил N-(метокси-(метилтио) метилен)карбамата, полученного в вышеуказанной стадии b), перемешивая при этом 18 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смесь хлороформа и этанола, в результате чего получали 1,5 г этил 3-(5-цианобензо[b] тиен-2-ил)-2-[4-[[2-(p-нитробензилоксикарбонилимино) гексагидропиримидин-5-ил]окси]фенил]пропионата в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,17 (3H, т, J = 7,0 Гц); 3,00 - 4,30 (1H, м); 4,40 - 4,70 (1H, м); 5,08 (2H, c); 6,81 (2H, д, J = 8,3 Гц); 7,03 (1H, с); 7,10 - 7,56 (5H, м); 7,81 (1H, д, J = 9,3 Гц); 7,94 (1H, с); 8,10 (2H, д, J = 8,75 Гц); 8,70 - 9,40 (2H, шир. с.).
d) В 100 мл этанола растворяли 1,5 г этил 3-(5-циано-бензо[b]тиен-2-ил)-2-[4-[[2-(p-нитробензилоксикарбонилимино)- гексагидропиримидин-5-ил] окси] фенил]пропионата, полученного в вышеуказанной стадии c). К полученному таким образом раствору добавляли 0,5 г хлорида аммония и 0,5 г 10% палладированного угля (катализатор 50% мокрого типа). Полученную смесь подвергали 2-часовой каталитической гидрогенизации при нормальном давлении. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смеси хлороформа и этанола, в результате чего получали 1,0 г целевого соединения.
1H-ЯМР (CDCl3) δ: 1,21 (3H, т, J = 7,0 Гц); 3,00 - 3,90 (7H, м); 4,17 (2H, кв., J = 7,0 Гц); 4,50 - 4,80 (1H, шир. с); 6,87 (2H, д, J = 8,75 Гц); 7,01 (1H, с); 7,06 - 7,36 (5H); 7,44 (1H, дд, J = 7,0 и 1,3 Гц); 7,81 (1H, с); 8,07 (2H, с).
Ссылочный пример 85. Получение этил 3-(5-цианобензо[b]тиен-2-ил)-2-[4-[2-(1-пирролин-2-ил)аминоэтокси]фенил] пропионата гидрохлорида.
Перемешивая, 1,3 г этил 2-[4-[2-трет-бутоксикарбонил-аминоэтокси)фенил] -3-(5-цианобензо[b] тиен-2-ил) пропионата растворяли в 50 мл этанола и затем перемешивали. К этому раствору добавляли 25 мл этанола, содержащего 13% (м. /об. ) соляной кислоты. Полученную таким образом смесь перемешивали при 50oC в течение 30 минут. После отгонки растворителя, полученный остаток растворяли в 50 мл этанола и затем перемешивали. Полученный таким образом раствор смешивали с 78,2 мг 2-этокси-1-пирролина, и смесь нагревали с обратным холодильником в течение 1,5 часа. После охлаждения, осажденные кристаллы собирали путем фильтрации, в результате чего получали 1,1 г целевого соединения.
т.пл.: 212-215oC
1H-ЯМР (CDCl3) δ: 1,14 (1,5H, т, J = 7,0 Гц); 1,16 (1,5H, т, J = 7,0 Гц); 2,16 (2H, т, J = 7,5 Гц); 2,87 (2H, т, J = 8,0 Гц); 3,20 - 4,40 (9H); 6,87 (2H, д, J = 8,3 Гц); 7,13 (1H, с); 7,27 (2H, д, J = 8,3 Гц); 7,48 (1H, дд, J = 8,3 и 1,3 Гц); 7,92 (1H, д, J = 8,3 Гц); 8,04 (1H, с); 10,04 (1H, шир. с.); 10,40 (1H, шир.с.).
Ссылочный пример 86. Получение метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(5-циано-2- индолил)пропионата.
В смеси растворителей, состоящих из 50 мл тетрагидрофурана и 50 мл метанола, растворяли 5,0 г (5-циано-2-индолил)-метилтрифенилфосфония бромида и 3,6 г метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси]фенил] -2- оксоацетата. К полученному таким образом раствору, перемешивая при комнатной температуре, добавляли 1,07 г 1,8-диазабицикло[5.4.0]-7-ундецена, и смесь перемешивали при той же температуре в течение 2 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь дихлорметана и ацетона, в результате чего получали метил 2-[4-[((3S)-1-трет-бутокси-карбонил-3- пирролидинил)окси] фенил] -3-(5-циано-2-индолил)акрилат в виде смеси E- и Z-изомеров. Полученное таким образом соединение растворяли в смеси растворителей, состоящих из 50 мл тетрагидрофурана и 50 мл метанола, и полученный раствор смешивали с 5,0 г оксида палладия • 1H2O сульфата бария и затем подвергали каталитической гидрогенизации при нормальном давлении. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента смеси дихлорметана и ацетона. В результате этой процедуры получали 3,5 г целевого соединения в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,46 (9H, с), 2,00 - 2,20 (2H, м); 2,95 - 4,22 (7H, м); 4,75 - 4,90 (1H, шир.с); 6,23 (1H, д); 6,80 (2H, д); 7,18 (2H, д); 7,20 - 7,40 (2H, м); 7,80 (1H, с); 8,80 (1H, шир. с).
Соединения ссылочных примеров 87-92 получали в соответствии с процедурой, описанной в ссылочном примере 86.
Ссылочный пример 87. Метил-2-[4-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)-окси]фенил]-3-(6-циано-2-индолил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,50 (9H, с); 2,00 - 2,25 (2H, шир.с); 3,13 (1H, дд); 3,37 - 3,75 (3H, м); 3,97 (1H, дд); 4,70 - 4,90 (1H, шир. с); 6,37 (1H, с); 6,80 (2H, д); 7,10 - 7,70 (5H, м); 9,25 (1H).
Ссылочный пример 88. Метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси] фенил] -3- (6-циано-1-метил-2-индолил)пропионат, вязкое маслянистое вещество,
1H-ЯМР (CDCl3) δ: 1,45 (9H, с); 2,00 - 2,25 (2H, шир.с); 3,13 (1H, дд); 3,60 (3H, с); 3,62 (3H, с); 3,90 - 4,10 (1H, дд); 4,75 - 4,90 (1H, шир.с.); 6,30 (1H, с); 6,80 (2H, д); 7,10 - 7,70 (5H, м); 9,25 (1H).
Ссылочный пример 89. Метил 2-[4-[((3R)-1-трет- бутоксикарбонил-3-пирролидинил)-окси]фенил]-3-(6-циано-1-этил-2- индолил)пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,34 (3H, т, J = 7,2 Гц); 1,47 (9H, с); 2,00 - 2,30 (2H, м); 2,90 - 3,30 (3H, м); 3,40 - 3,80 (4H, м); 3,66 (3H, м); 4,15 (2H, кв. , J = 7,2 Гц); 4,70 - 5,00 (1H, шир.с.); 6,30 (1H, с); 6,84 (2H, д, J = 8,8 Гц); 7,27 (2H, д, J = 8,8 Гц); 7,26 (1H, д, J = 7,0 Гц); 7,54 (1H, д, J = 7,0 Гц).
Ссылочный пример 90. Метил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)-окси]фенил]-3-(6-циано-1-этил-2- индолил)пропионат, вязкое маслянистое вещество
1H-ЯМР (CDCl3) δ: 1,32 (3H, т, J = 7,2 Гц); 1,48 (9H, с); 2,00 - 2,30 (2H, м); 2,90 - 3,30 (3H, м); 3,40 - 3,80 (4H, м); 3,64 (3H, c); 4,15 (2H, кв. , J = 7,2 Гц); 4,70 - 5,00 (1H, шир.с.); 6,30 (1H, с); 6,84 (2H, д, J = 8,8 Гц); 7,26 (2H, д, J = 8,8 Гц); 7,26 (1H, д, J = 7,0 Гц); 7,54 (1H, д, J = 7,0 Гц).
Ссылочный пример 91. Метил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)-окси] фенил]-3- [1-(2-хлороэтил)-6-циано-2-индолил]пропионат, вязкое маслянистое вещество.
1H-ЯМР (CDCl3) δ: 1,47 (9H, с); 2,00 - 2,30 (2H, м); 3,00 - 4,20 (9H, м); 3,66 (3H, с); 4,20 - 4,60 (2H, м); 4,80 - 5,00 (1H, м); 6,37 (1H, с); 6,84 (2H, д); 7,20 - 7,80 (5H, м).
Ссылочный пример 92. Метил 2-[4-[(1-трет- бутоксикарбонил-4-пиперидинил)окси]фенил]-3-(6-циано-1-этил-2- индолил)пропионат.
1H-ЯМР (CDCl3) δ: 1,34 (3H, т); 1,60 - 2,00 (4H, м); 3,10 (1H, дд); 3,30 - 3,40 (2H, м); 3,57 (1H, дд); 3,62 - 3,75 (2H, м); 3,90 - 4,30 (3H, м); 4,35 (1H, м); 6,30 (1H, с); 6,90 (2H, д); 7,30 (3H, м); 7,54 (1H, д); 7,58 (1H, с).
Ссылочный пример 93. Получение метил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(5-циано-1-метил- 2-индолил)пропионата.
3,0 г метил 2-[4-[(3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]фенил] -3-(5-циано-2-индолил) пропионата растворяли в 30 мл N,N-диметилформамида, и затем перемешивали, охлаждая льдом. К вышеуказанному раствору добавляли 270 мг 60% гидрида натрия, и перемешивали в течение 10 минут. Полученный реакционный раствор смешивали с 0,4 мл метилиодида, а смесь перемешивали при комнатной температуре в течение 1 часа. Обработанный таким образом реакционный раствор разбавляли смесью толуола и этилацетата, а затем промывали водным раствором хлорида аммония. После осушки органического слоя с отгонкой растворителя, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смесь дихлорметана и ацетона. В результате этой процедуры получали 2,0 г целевого соединения в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,45 (9H, с); 2,00 - 2,22 (2H, м); 3,05 (1H, дд); 3,35 - 3,80 (5H, м); 3,63 (3H, с); 4,00 (1H, дд), 4,75 - 5,00 (1H, шир.с.); 6,25 (1H, д); 6,85 (2H, д); 7,20 - 7,50 (2H, м); 7,90 (1H, с).
Ссылочный пример 94. Получение этил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)-3-(6-циано-1,2,3,4-тетрагидро-2-нафтил)пропионата.
9,0 г (6-циано-1,2,3,4-тетрагидро-2-нафтил) метилтрифенилфосфония р-толуолсульфоната суспендировали в 150 мл тетрагидрофурана, постепенно добавляя 600 мг 60% гидрида натрия. Полученную таким образом смесь нагревали с обратным холодильником в течение 20 минут. После охлаждения до комнатной температуры, к полученному реакционному раствору добавляли 10 мл раствора тетрагидрофурана, содержащего 4,16 г этил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси] фенил] -2-оксоацетата. Полученную реакционную смесь перемешивали в течение 10 минут, а затем нагревали с обратным холодильником 2 часа. После охлаждения до комнатной температуры, полученный таким образом реакционный продукт растворяли в этилацетате и промывали последовательно водой и насыщенным водным раствором хлорида натрия. После осушки полученного органического слоя с отгонкой растворителя, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смеси н-гексана и этилацетата, в результате чего получали 3,90 г этил 2-[4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(6-циано-1,2,3,4- тетрагидро-2-нафтил) акрилата в виде желтого маслянистого вещества смеси E- и Z-формах. 2,58 г полученной таким образом смеси E/Z растворяли в 40 мл этанола, и полученный раствор смешивали с 650 мг оксида палладия • 1H2O • сульфата бария, и подвергали каталитической гидрогенизации при нормальном давлении около 5 часов. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь н-гексана и этилацетата. В результате этой процедуры получали 1,69 г целевого соединения в виде желтого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,20 (3H, т, J = 7,0 Гц); 1,45 (9H, с); 1,50 - 3,90 (16H, м); 4,10 (2H, кв.); 4,82 (1H, м); 6,81 (2H, кв., J = 9,0 Гц); 7,00 - 7,40 (5H, м).
Ссылочный пример 95. Получение этил 2-[4-[((2S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(5-циано-2- бензимидазолил)пропионата.
a) 3,42 г 3,4-диаминобензонитрила и 4,06 г этилхлороацетоимидата гидрохлорида растворяли в 100 мл этанола, и раствор нагревали с обратным холодильником в течение 3 часов. После охлаждения и отгонки растворителя, полученный остаток растворяли в этилацатате, промывали водой, а затем высушивали. После отгонки растворителя, осажденные таким образом кристаллы собирали путем фильтрации, в результате чего получали 2,7 г 2-хлорометил-5-бензимидазолкарбонитрила.
т.пл.: 144-146oC
1H-ЯМР (CDCl3) δ: 4,83 (2H, c); 7,48 (1H, д, J = 7,1 Гц); 7,57 (1H, д, J = 7,1 Гц); 7,95 (1H, с).
b) 1,0 г 2-хлорометил-5-бензимидазолкарбонитрила, полученного в вышеуказанной стадии a), и 2,19 г трифенилфосфина растворяли в 30 мл 1,2-дихлороэтана, а полученный раствор нагревали при температуре 140oC в течение 1 часа. После охлаждения и отгонки растворителя, полученный таким образом остаток и 2,03 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил] -2- оксоацетата растворяли в смеси растворителей, состоящих из 20 мл тетрагидрофурана и 20 мл этанола. Перемешивая при комнатной температуре, к полученному таким образом раствору добавляли 1,1 г 1,8-диазацибикло[5.4.0] -7-ундецена, и смесь перемешивали при той же температуре 72 часа. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь хлороформа и этанола, в результате чего получали 1,5 г маслянистого этил-2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3-(5-циано-2-бензимидазолил)акрилата в виде смеси E- и Z-форм. Полученную таким образом смесь E/Z растворяли в смеси растворителей, состоящих из 50 мл тетрагидрофурана и 50 мл этанола, и полученный раствор смешивали с 1,5 г оксида палладия • 1H2O • сульфата бария, и затем повергали каталитической гидрогенизации при нормальном давлении. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смеси хлороформа и этанола. В результате этой процедуры получали целевое соединение в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,14 (3H, т, J = 7,0 Гц); 1,48 (9H, с); 1,90 - 2,30 (2H, шир. с.); 3,05 - 3,90 (6H, м); 4,12 (2H, кв., J = 7,0 Гц); 4,00 - 4,30 (1H); 4,70 - 4,95 (1H, шир.с.); 6,79 (2H, д, J = 8,8 Гц); 7,19 (2H, д, J = 8,8 Гц); 7,35 - 8,10 (3H, м).
FD-MC (m/z): 504 (M+); 505 (M+ + 1).
Ссылочный пример 96. Получение (+)-((2S)-1-р-толуолсульфонил-2- пирролидинил метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) окси]фенил] -3-(6-циано-2-индолил)пропионата и (-)-((2S)-1-р- толуолсульфонил-2-пирролидинил метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) окси]фенил] -3-(6-циано-2-индолил)пропионата.
a) водный раствор, содержащий 3 г гидроксида натрия, растворенного в 10 мл воды, добавляли к 100 мл раствора метанола, содержащего 22 г метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) окси]фенил]-3-(6-циано-2-индолил)пропионата, и полученную смесь перемешивали при комнатной температуре в течение 24 часов. После отгонки растворителя, pH оставшейся части доводили до значения 4-5 с помощью лимонной кислоты, а затем экстрагировали этилацетатом. Путем осушки экстракта с отгонкой растворителя, получали 20 г 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) окси]фенил]-3-(6-циано-2-индолил)пропионовой кислоты.
ИК (KBr): 3352, 2218, 1710, 1677 см-1
b) В 300 мл диоксана растворяли 20 г 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил] -3- (6-циано-2-индолил)пропионовой кислоты, полученной в вышеуказанной части (a) и 11,9 г ((2S)-1-п-толуолсульфонил-2-пирролидинил)-метанола. Полученный таким образом раствор смешивали с каталитически эффективным количеством 4-диметиламсинопиридина и 9 г 1,3-дициклогексилкарбодиимида, перемешивая при охлаждении льдом, и смесь перемешивали при комнатной температуре 24 часа. После удаления осажденных материалов путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смеси хлороформа и ацетона, в результате чего получали 10,5 г (+)-((2S)-1-р-толуолсульфонил-2-пирролидинил)метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил] -3- (6-циано-2-индолил)пропионата.
1H-ЯМР (CDCl3) δ: 1,00 - 1,80 (4H, м); 1,46 (9H, с); 2,00 - 2,30 (2H, м); 2,43 (3H, с); 3,00 - 4,40 (12H, м); 4,75 - 5,00 (1H, м); 6,30 (1H, с); 6,82 (2H, д); 7,10 - 7,90 (9H, м); 9,00 (1H, с).
После повторного элюирования колонки той же системой растворителей, получали 9,5 г ((-)-((2S)-1-п-толуолсульфонил-2-пирролидинил метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-3- (6-циано-2-индолил)пропионата.
1H-ЯМР (CDCl3) δ: 1,00 - 2,00 (4H, м); 1,44 (9H, с); 2,00 - 2,25 (2H, м); 2,41 (3H, с); 2,95 - 4,10 (10H, м); 4,20 (2H, д); 4,70 - 4,90 (1H, м); 6,25 (1H, с); 6,80 (2H, д); 7,10 - 7,80 (9H, м); 9,20 (1H, с).
Ссылочный пример 97. Получение метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-3-(6-циано-1- этоксикарбонилметил-2-индолил)пропионата.
3,0 г метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3- -3-(6-циано-2-индолил)пропионата растворяли в 30 мл N,N-диметилформамида. Полученный таким образом раствор смешивали с 280 мг 60% гидрида натрия, перемешивая при охлаждении льдом, и еще перемешивали в течение 20 минут при той же температуре. Полученный реакционный раствор смешивали с 0,7 мл бромоацетата и смесь перемешивали в течение 1 часа. Обработанный таким образом раствор смешивали с разбавленной соляной кислотой, экстрагировали смесью этилацетата и толуола, промывали водой и затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси дихлорметана и ацетона в качестве элюирующего растворителя. В результате этой процедуры получали 3,2 г целевого соединения в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl) δ: 1,26 (3H, т); 1,46 (9H, с); 3,02 (1H, дд); 3,30 - 3,70 (5H, м); 3,66 (3H, с); 4,00 (1H, дд); 4,20 (2H, кв.); 4,80 (2H, с); 4,78 - 4,90 (1H, м); 6,40 (1H, с); 6,90 (2H, д); 7,20 - 7,70 (5H, м).
Ссылочный пример 98. Получение 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]- -3-(6-циано-2-индолил)пропанола.
2,7 г метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3-(6-циано-2-индолил)пропионата растворяли в 30 мл тетрагидрофурана, добавляя при этом 660 мг борогидрида натрия. К полученному таким образом раствору добавляли по капле 12 мл метанола, перемешивая при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Полученный реакционный раствор смешивали с 10% водным раствором лимонной кислоты, экстрагировали дихлорметаном и затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя смеси дихлорметана и метанола. В результате этой процедуры получали 2,2 г целевого соединения в виде вязкого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,48 (9H, с); 1,95 - 2,25 (2H, м); 2,48 (1H, т); 3,00 - 3,22 (2H, м); 3,40 - 3,69 (6H, м); 3,70 - 3,90 (1H, м); 4,70 - 4,90 (1H, м); 6,21 (1H, с); 6,80 (2H, д); 7,00 - 7,65 (5H, м); 9,20 (1H, с).
Ссылочный пример 99. Получение этил 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-3- гидроксипропил]-6-циано-1-индолацетата.
2,0 г 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-3-(6-циано-2-индолил)пропанола растворяли в 30 мл N,N-диметилформамида. К вышеуказанному раствору добавляли 280 мг 60% гидрида натрия, перемешивая при охлаждении льдом, и затем перемешивали 20 минут при комнатной температуре. Полученный реакционный раствор смешивали с 0,5 мл этилбромацетата, и смесь перемешивали в течение 1 часа. Обработанный таким образом раствор смешивали с водным раствором хлорида аммония, экстрагировали смесью толуола и этилацетата, промывали водой, а затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь дихлорметана и ацетона. В результате этой процедуры получали 1,5 г целевого соединения в виде вязкого маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,23 (3H, т); 1,45 (9H, с); 1,90 - 2,20 (2H, с); 4,20 (2H, кв.); 4,50 - 4,90 (3H); 6,20 (1H, с); 6,78 (2H, д).
Ссылочный пример 100. Получение 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] этил]-6-индолкарбонитрила.
a) В 40 мл тетрагидрофурана растворяли 1,31 г р-гидроксибензальдегида, 1,87 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина и 2,88 г трифенилфосфина. Перемешивая при комнатной температуре, к полученному таким образом раствору добавляли 1,91 г диэтилазодикарбоксилата, и смесь перемешивали в течение 45 минут. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь бензола и этилацетата. В результате этой процедуры получали 2,9 г [4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси] бензальдегида в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,48 (9H, с); 2,00 - 2,40 (2H, с); 3,30 - 3,80 (4H, м); 4,90 - 5,10 (1H, м); 6,98 (2H, д, J = 9,0 Гц); 7,84 (2H, д, J = 9,0 Гц); 9,89 (1H, с).
b) 0,93 г [4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)-окси] бензальдегида, полученного в вышеуказанной части a), и 1,6 г (6-циано-2-индолил)метилтрифенилфосфония бромида растворяли в смеси растворителей, состоящих из 20 мл метанола и 20 мл тетрагидрофурана. В полученном таким образом растворе растворяли 490 мг 1,8-диазабицикло[5.4.0]-7-ундецена, перемешивали, охлаждая при этом льдом, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь хлороформа и метанола, в результате чего получали 700 мг 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил] винил]-6-индолкарбонитрила в виде смеси E- и Z-форм.
1H-ЯМР (CDCl3) δ: 1,43 (9H, с); 1,90 - 2,23 (2H, м); 3,30 - 3,70 (4H, м); 4,75 - 4,95 (1H, м); 8,65 (1H, шир.с.).
c) 700 мг 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] винил]-6-индолкарбонитрила, полученного в вышеуказанной части b), растворяли в смеси растворителей, состоящей из 20 мл метанола и 40 мл тетрагидрофурана. К полученному раствору добавляли 70 мг оксида палладия • 1H2O • сульфата бария, и смесь подвергали каталитической гидрогенизации при нормальном давлении в течение 3 часов. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь хлороформа и метанола. В результате этой процедуры получали 650 мг целевого соединения в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,50 (9H, с); 1,95 - 2,20 (2H, м); 4,70 - 4,90 (1H, м); 6,30 (1H, с); 6,75 (2H, д); 7,10 (2H, д); 7,10 - 7,65 (3H, м); 9,46 (1H, шир.с.).
Ссылочный пример 101. Получение этил 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] этил]-6-циано-1-индолацетата.
2,4 г 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] этил] -6-индолкарбонитрила растворяли в 50 мл N,N-диметилформамида. К полученному таким образом раствору добавляли 300 мг 60% гидрида натрия и перемешивали, охлаждая при этом льдом, а затем полученную смесь нагревали до комнатной температуры и перемешивали при той же температуре в течение 20 минут. К вышеуказанной смеси добавляли 0,76 мл этилбромоацетата и перемешивали, охлаждая льдом, а затем перемешивали еще 1 час. Полученный реакционный раствор смешивали с водным раствором хлорида аммония и экстрагировали смесью толуола и этилацетата, и полученный органический слой промывали водой и высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь дихлорметана и ацетона. В результате этой процедуры получали целевое соединение 2,3 г в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,2 (3H, т); 2,00 - 2,20 (2H, м); 2,95 (4H, с); 3,30 - 3,60 (4H, м); 4,18 (2H, кв.); 4,70 (2H, с); 6,36 (1H, с); 6,75 (2H, д); 7,00 - 7,60 (5H, м).
Ссылочный пример 102. Получение 2-[4-[(1-трет-бутоксикарбонил-4-пиперидинил)окси-фенил]метил]-5- бензофуранкарбонитрила.
a) 5,07 г гидроксида калия добавляли к 30 мл диэтиленгликоля, и смесь перемешивали при комнатной температуре с добавлением 5,5 г 80% гидразина дигидрата 2H2O и 5,0 г 5-бромо-2-(4-метоксибензоил)бензофурана. Полученную таким образом смесь нагревали с обратным холодильником. После охлаждения, полученный реакционный раствор доводили до pH 4-5, экстрагировали бензолом, а затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, с использованием в качестве элюирующего растворителя смесь н-гексана и изопропанола, в результате чего получали 3,95 г 5-бромо-2-(4-метоксибензил)бензофурана в виде коричневого маслянистого продукта.
1H-ЯМР (CDCl3) δ: 3,80 (3H, с); 4,02 (2H, с); 6,23 (1H, с); 6,90 (2H, д, J = 9,0 Гц); 7,20 - 7,40 (4H, м); 7,57 (1H, м).
b) 3,95 г 5-бромо-2-(4-метоксибензоил)бензофурана, полученного в вышеуказанной стадии a), и 1,67 г цианида меди суспендировали в 20 мл N-метил-2-пирролидона, а суспензию нагревали при температуре 200-220oC в потоке азота. После охлаждения, полученный реакционный продукт растворяли в хлороформе, и нерастворимые вещества удаляли с помощью фильтрования. Полученный органический слой промывался водой и концентрировался досуха. После этого, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь н-гексана и изопропилового эфира, в результате чего получали 3,10 г 2-(4-метоксибензил)-5-бензофуранкарбонитрила, т.пл. 78-80oC.
1H-ЯМР (CDCl3) δ: 3,80 (3H, с); 4,10 (2H, с); 6,39 (1H, с); 6,90 (2H, д, J = 9,0 Гц); 7,22 (2H, д); 7,46 (2H); 7,78 (1H, с).
c) 3,0 г 2-(4-метоксибензил)-5-бензофуранкарбонитрила, полученного в вышеуказанной части (b), растворяли в 30 мл дихлорметана, и раствор охлаждали до -50oC. К раствору при перемешивании добавляли по капле 20 мл раствора дихлорметана, содержащего 2,23 мл трибромида бора. После постепенного нагревания смеси до комнатной температуры, ее перемешивали в течение 1 часа. Полученный реакционный раствор разбавляли хлороформом, промывали разбавленной соляной кислотой и затем высушивали. После отгонки растворителя, осажденные таким образом кристаллы собирали путем фильтрации, в результате чего получали 2,48 г 2-(4-гидроксибензил)-5-бензофуранкарбонитрила в виде желтых призмообразных кристаллов.
1H-ЯМР (CDCl3) δ: 4,02 (2H, с); 6,45 (1H, с); 6,77 (2H, д, J = 9,0 Гц); 7,10 (2H, д); 7,48 (2H); 7,81 (1H, с).
d) В 50 мл тетрагидрофурана растворяли 1,50 г 2-(4-гидроксибензоил)-5-бензофуранкарбонитрила, полученного в вышеуказанной части c), 2,37 г трифенилфосфина и 1,21 г 1-трет-бутоксикарбонил-4-гидроксипиперидина. Перемешивая при комнатной температуре, полученный таким образом раствор смешивали с 1,57 г диэтилазодикарбоксилата, и перемешивали еще в течение 40 часов. После отгонки растворителя полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя, смесь н-гексана и этилацетата. В результате этой процедуры, получали 1,30 г целевого соединения в виде бесцветных игольчатых кристаллов. Т.пл. 144-146oC.
1H-ЯМР (CDCl3) δ: 1,47 (9H, с); 1,60 - 2,00 (4H, м); 3,20 - 3,90 (4H, м); 4,05 (2H, с); 4,44 (1H, м); 6,41 (1H, с); 6,87 (2H, д, J = 9,0 Гц); 7,20 (2H, д, J = 9,0 Гц); 7,47 (2H); 7,79 (1H, с).
Ссылочный пример 103. Получение 3-[3-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] пропил]-5-бензофуранкарбонитрила.
a) 2,14 г (5-циано-3-бензофуранил)метилтрифенилфосфония хлорида и 0,7 г 4-метоксифенилацетальдегида растворяли в смеси растворителей, содержащей 100 мл тетрагидрофурана и 100 мл этанола. Полученный таким образом раствор смешивали с 0,71 г 1,8-диазабицикло[5.4,0]-7-ундецена и перемешивали в течение 24 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя толуол, в результате чего получали 0,86 г желтого и маслянистого 3-[3-(4-метоксифенил)аллил] -5-бензофуранкарбонитрила в виде смеси E- и Z-форм. Полученную таким образом смесь E/Z растворяли в 100 мл этанола, а раствор подвергали каталитической гидрогенизации при нормальном давлении в присутствии 370 мг 5% палладированного угля. После этого катализатор удаляли путем фильтрации, а растворитель отгоняли, в результате чего получали 0,6 г 3-[3-(4-метоксифенил)пропил] -5-бензофуранкарбонитрила. Полученное таким образом метокси-соединение растворяли в 20 мл дихлорметана. Перемешивая при -40oC, к полученному раствору добавляли по капле 10 мл раствора дихлорметана, содержащего 0,4 мл трибромида бора. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный реакционный раствор выливали в холодную разбавленную соляную кислоту и экстрагировали хлороформом. После осушки полученного органического слоя и отгонки растворителя, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюирующего растворителя толуола. В результате этой процедуры получали 280 мг 3-[3-(4-гидроксифенил)пропил]-5-бензофуранкарбонитрила.
1H-ЯМР (CDCl3) δ: 2,0 (2H, м); 2,64 (4H, м); 6,80 (2H, д); 7,16 (2H, д); 7,52 (3H, м); 7,79 (1H).
b) В 20 мл тетрагидрофурана растворяли 280 мг 3-[3-(4-гидроксифенил)пропил]-5-бензофуранкарбонитрила, полученного в вышеуказанной части (a), 280 мг (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина и 400 мг трифенилфосфина. Перемешивая при комнатной температуре, полученный таким образом раствор смешивали с 265 мг диэтилазодикарбоксилата, и продолжали перемешивать в течение 24 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь н-гексана и этилацетата. В результате этой процедуры получали 400 мг целевого соединения в виде желтого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,46 (9H, с); 2,05 (4H, м); 2,66 (4H, м); 3,60 (4H, шир. с); 4,85 (1H, шир. с); 6,85 (4H, д); 7,05 (2H, д); 7,53 (3H, м); 7,83 (1H).
Ссылочный пример 104. Получение 4-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси]-3-метоксибензальдегида.
В 50 мл тетрагидрофурана растворяли 3,04 г ванилина, 3,74 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина и 5,24 г трифенилфосфина. Полученный таким образом раствор смешивали с 4,00 г диэтилазодикарбоксилата, и смесь перемешивали при комнатной температуре 18 часов. После концентрации реакционного раствора, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя смесь этанола и хлороформа. В результате этой процедуры, получали 5,0 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,47 (9H, с); 2,00 - 2,40 (2H, м); 3,50 - 3,80 (4H, м); 3,90 (3H, с); 5,02 (1H, шир.с.); 6,80 - 7,60 (3H, м); 9,86 (1H, с).
Ссылочный пример 105. Получение 4-[[4-(N-ацетил) аминометилциклогексил] метокси]бензальдегида.
Получали целевое соединение в соответствии с процедурой, описанной в ссылочном примере 104.
1H-ЯМР (CDCl3) δ: 0,80 - 2,10 (10H, м); 3,13 (2H, дд, J = 6,1 Гц и 6,1 Гц); 3,84 (2H, д, J = 6,1 Гц); 5,56 (1H, шир.с.); 6,97 (2H, д, J = 8,7 Гц); 7,82 (2H, д, J = 8,7 Гц); 9,88 (1H, с).
Ссылочный пример 106. Получение 3-ацетокси-4-[((3S)-1- ацетил-3-пирролидинил)окси]-бензальдегида.
a) 5,5 г 4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси]-3-метоксибензальдегида растворяли в 20 мл дихлорметана с последующим добавлением 30 мл муравьиной кислоты. Полученную таким образом смесь перемешивали при комнатной температуре 1 час и затем еще 1 час при 50oC. Растворитель и муравьиная кислота отгонялись при пониженном давлении, и полученный остаток растворяли в 100 мл тетрагидрофурана. Полученный таким образом раствор смешивали с 2,68 г ацетилхлорида, и 8,63 г триэтиламина по капле добавляли к этой смеси и перемешивали, охлаждая при этом льдом. После этого растворитель отгоняли и полученный остаток растворяли в хлороформе, и промывали водой, и осушали полученный органический слой. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в качестве элюента используя толуол, в результате чего получали 4,3 г 4-[((3S)-1-ацетил-3-пирролидинил)окси] -3-метоксибензальдегида в виде масляной вязкой жидкости.
1H-ЯМР (CDCl3) δ: 2,04 (5H, м); 3,50 - 4,00 (4H, м); 3,90 (3H, с); 5,10 (1H, шир.с.); 6,80 - 7,60 (3H, м); 9,86 (1H, с).
b) 4,3 г 4-[((3S)-1-ацетил-3-пирролидинил)окси]-3-метоксибензальдегида, полученного в вышеуказанной части (a), растворяли в 40 мл дихлорметана, и раствор охлаждали до -70oC. К вышеуказанному раствору при перемешивании добавляли по капле 12,6 г трибромида бора. Полученный таким образом реакционный раствор нагревали до 0oC и затем выливали в ледяную воду, и экстрагировали хлороформом. Полученный органический слой концентрировали досуха, и полученный таким образом остаток кристаллизовали из системы растворителей хлороформа и н-гексана, в результате чего получали 2,0 г 4-[((3S)-1-ацетил-3-пирролидинил) окси]-3-гидроксибензальдегида. Полученное таким образом соединение использовали в последующей реакции без дополнительной очистки.
1H-ЯМР (CDCl3 : ДМСО-d6 = 9:1) δ: 2,00 - 2,50 (5H, м); 3,50 - 4,00 (4H, м); 5,32 (1H, шир.с); 6,96 (1H, д, J = 8,0 Гц); 7,22 - 7,58 (2H, м); 9,81 (1H, с).
c) 1,1 г 4-[((3S)-1-ацетил-3-пирролидинил)окси]-3- гидробензальдегида, полученного в вышеуказанной части b), суспендировали в 5 мл пиридина, с последующим добавлением 0,86 г уксусного ангидрида. Полученную таким образом смесь перемешивали при комнатной температуре в течение 1 часа. После осушки реакционного раствора при пониженном давлении, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этанола и хлороформа. В результате этой процедуры получали 1,30 г целевого соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3) δ: 2,06 (3H, с); 2,28 (3H, с); 2,00 - 2,40 (2H, м); 3,40 - 3,90 (4H, м); 5,60 (1H, шир.с); 6,90 - 7,90 (3H, м); 9,90 (1H, с).
Ссылочный пример 107. Получение 4-[(1-тритил-4-имидазолил) метокси] бензальдегида.
1,22 г р-гидроксибензальдегида и 4,08 г 4-хлорметил-1-тритилимидазола растворяли в 40 мл N,N-диметилформамида, с последующим добавлением 1,66 г безводного карбоната калия и затем перемешивали при комнатной температуре 40 часов. Полученный таким образом реакционный раствор распределяли между водой и бензолом, а органический слой концентрировали досуха. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, и очищенный продукт кристаллизовали из системы растворителей н-гексана и бензола. В результате этой процедуры получали целевое соединение (2,3 г).
Т.пл. 181-182oC.
1H-ЯМР (CDCl3) δ: 5,09 (2H, c), 6,90 (1H, д, J = 1,1 Гц); 7,00 - 7,40 (17H, м); 7,47 (1H, д, J = 1,1 Гц); 7,81 (2H, д, J = 8,8 Гц); 9,88 (1H, c).
Ссылочный пример 108. Получение 2-[2-[4-[(3S)-1-ацетил-3-пирролидинил)окси]-3-гидроксифенил]этил]-5- бензофуранкарбонитрила.
a) 1,3 г 3-ацетилокси-4-[((3S)-1-ацетил-3-пирролидинилокси] бензальдегида и 2,22 г (5-циано-2-бензофуранил)метилтрифенил-фосфония хлорида растворяли в смеси растворителей, состоящей из 10 мл тетрагидрофурана и 10 мл этанола, с последующим добавлением 0,943 г 1,8-диазабицикло[5.4.0]-7-ундецена и затем перемешивали при комнатной температуре в течение 2 часов. К полученной смеси затем добавляли 1,88 г 1,8-диазабицикло [5.4.0]-7-ундецена и 3 мл воды, с последующим перемешиванием при комнатной температуре в течение 2 часов. Полученный таким образом реакционный раствор доводили до pH 4-5 с 10% водным раствором лимонной кислоты и концентрировали при пониженном давлении, а полученный остаток экстрагировали хлороформом и затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью хлороформа и этанола, в результате чего получали 1,8 г 2-[2-[4-[((3S)-пирролидинил)окси]-3-гидроксифенил] винил] -5- бензофуранкарбонитрила в виде порошкообразной смеси E- и Z-изомеров.
1H-ЯМР (CDCl3) δ: 2,00 - 2,50 (5H, м); 3,40 - 4,00 (4H, м); 5,00 (1H, шир. с); 6,30 - 7,90 (9H, м).
b) 1,8 г 2-[2-[4-[((3S)-1-ацетил-3-пирролидинил)окси]-3-гидроксифенил] винил] -4- бензофуранкарбонитрила, полученного в вышеуказанной части (a), растворяли в смеси растворителей, состоящей из 300 мл тетрагидрофурана и 300 мл этанола, и раствор подвергали каталитическому восстановлению при нормальном давлении в течение 5 часов в присутствии 340 мг оксида палладия• 1H2O•сульфата бария. После удаления катализатора путем фильтрации и концентрирования полученного фильтрата, осажденные таким образом кристаллы собирали путем фильтрации, в результате чего получали 1,6 г целевого соединения в виде бесцветных кристаллов. Т.пл. 191-193oC.
ИК (KBr): 2224, 1644, 1512 см-1
1H-ЯМР (CDCl3) δ: 2,09 (3H, c); 2,00 - 2,50 (2H, м); 3,04 (4H, c); 3,50 - 3,90 (4H, м); 4,96 (1H, шир.с); 6,40 (1H, с); 6,50 - 6,90 (3H, м); 7,50 (2H, с); 7,80 (1H, с).
Последующие соединения ссылочных примеров 109 и 110 получали в соответствии с процедурой, описанной в ссылочном примере 108.
Ссылочный пример 109. 2-[2-[4-[[4-(N-Ацетил)аминометилциклогексил]метокси]фенил]этил]- 5-бензофуранкарбонитрил.
Т.пл.: 159-161oC
1H- ЯМР (CDCl3) δ: 0,80 - 2,10 (10H, м); 1,99 (3H, с); 3,04 (4H, с); 3,19 (2H, дд, J = 6,1 и 6,1 Гц); 3,63 (2H, д, J = 6,1 Гц); 5,50 (1H, шир.с. ); 6,40 (1H, с); 6,79 (2H, д, J = 8,3 Гц); 7,08 (2H, д, J = 8,3 Гц); 7,49 (2H); 7,49 (1H).
Ссылочный пример 110. 2-[2-[4-[(1-Тритил-4-имидазолил)метокси]фенил] этил]-5- бензофуранкарбонитрил.
Т.пл. 165-167oC
1H-ЯМР (CDCl3) δ: 3,03 (4H, c); 4,97 (2H, с); 6,38 (1H, c); 6,90 - 7,70 (22H, м); 7,78 (1H, с).
Ссылочный пример 111. Получение 2-[2-[4-[(1-имидазолил)метил)фенил]этил] -5-бензофуранкарбонитрила.
a) 911 мг 4-гидроксиметилбензальдегида и 2,0 г (5-циано-2-бензофуранил)-метилтрифенилфосфония хлорида растворяли в смеси растворителей, состоящей из 10 мл тетрагидрофурана и 10 мл этанола, добавляли 845 мг 1,8-диазабицикло[5.4.0]-7-ундецена и перемешивали при комнатной температуре в течение 1 часов. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 2,3 г 2-[2-(4-гидроксиметилфенил)винил] -5-бензофуранкарбонитрила в виде смеси E и Z изомеров. 2,3 г полученной таким образом смеси E/Z смеси растворяли в смеси растворителей, состоящей из 10 мл тетрагидрофурана и 10 мл этанола, а раствор подвергали каталитической гидрогенизации при нормальном давлении в течение 7 часов в присутствии 600 мг оксида палладия • 1H2O • сульфата бария. После удаления катализатора путем фильтрации и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом, в результате чего получали 835 мг 2-[2-(4-гидроксиметилфенил)этил]-5-бензофуранкарбонитрила в виде кристаллов.
т.пл.: 123-124oC.
1H-ЯМР (CDCl3) δ: 1,60 (1H, с); 3,10 (4H, с); 4,67 (2H, с); 6,41 (1H, с); 7,20 (2H, д, J = 8,2 Гц); 7,34 (2H, д, J = 8,2 Гц); 7,52 (2H, с); 7,82 (1H, с).
b) 835 мг 2-[2-(4-гидроксиметилфенил)этил]-5-бензофуран карбонитрила, полученного в вышеуказанной части (a), растворяли в 15 мл тионилхлорида, и раствор перемешивали при комнатной температуре в течение 1 часа. После этого тионилхлорид отгоняли, и полученный остаток растворяли в 30 мл ацетонитрила, вместе с 550 мг N-ацетилимидазола и 600 мг иодида натрия. Полученный таким образом раствор нагревали с обратным холодильником 3 часа. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя хлороформом. В результате этой процедуры получали 300 мг целевого соединения в виде коричневых кристаллов.
Т.пл.: 72-73oC.
1H-ЯМР (CDCl3) δ: 3,08 (4H, с); 5,09 (2H, с); 6,40 (1H, с); 6,89 (1H, с); 7,00 - 7,18 (5H, м); 7,49 (2H, с); 7,56 (1H, с); 7,79 (1H, с).
Ссылочный пример 112. Получение 4-[2-(5-циано-2-бензофуранил)этил]бензойной кислоты
a) 5,17 г метил 4-формилбензоата и 13,97 г (5-циано-2-бензофуранил)метилтрифенилфосфония хлорида растворяли в смеси растворителей, состоящей из 50 мл тетрагидрофурана и 50 мл метанола. 5,02 г 1,8-диазабицикло[5.4.0] -7-ундецена добавляли к полученному таким образом раствору и перемешивали, охлаждая льдом, а затем перемешивали при комнатной температуре 2 часа. Посредством сбора осажденных кристаллов через фильтр, получали метил 4-[2-(5-циано-2-бензофуранил)винил] бензоат в виде смеси E- и Z-форм. Таким образом полученные кристаллы растворяли в смеси растворителей, состоящей из 300 мл тетрагидрофурана и 100 мл этанола, а раствор подвергали каталитической гидрогенизации при нормальном давлении в течение 2 часов в присутствии 2,0 г оксида палладия • 1H2O • сульфата бария. После удаления катализатора путем фильтрации и концентрирования полученного фильтрата, полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя бензолом, в результате чего получали 8,1 г метил 4-[2-(5- циано-2-бензофуранил)этил] бензоата в виде призмообразных кристаллов, т.пл.: 114-115oC.
1H-ЯМР (CDCl3) δ: 3,13 (4H, с); 3,90 (3H, с); 6,31 (1H, с); 7,26 (2H, д, J = 8,5 Гц); 7,50 (2H, с); 7,80 (1H, с); 7,98 (2H, д, J = 8,5 Гц).
b) 1,5 г метил 4-[2-(5-циано-2-бензофуранил)этил]бензоата, полученного в вышеуказанной части (a), растворяли в смеси растворителей, состоящей из 20 мл тетрагидрофурана и 20 мл этанола, и затем добавляли 11 мл 1 н. водного раствора гидроксида натрия и еще перемешивали при комнатной температуре в течение 14 часов. После этого, полученный реакционный раствор доводили до pH 2 концентрированной соляной кислотой, и осажденные таким образом кристаллы собирали путем фильтрации, промывали водой, а затем высушивали. В результате этой процедуры, получали 1,41 г целевого соединения, т.пл. 234-235oC.
1H-ЯМР (ДМСО-d6) δ: 3,13 (4H, с); 6,70 (1H, с); 7,44 (2H, д, J = 8,0 Гц); 7,69 (2H, с); 7,88 (2H, д, J = 8,0 Гц); 8,06 (1H, с).
Ссылочный пример 113. Получение 2-[2-[4-[(4-метил-1-пиперазинил) карбонил]фенил]этил]-5-бензофуранкарбонитрила.
1,35 г 4-[2-(5-циано-2-бензофуранил)этил]бензойной кислоты нагревали с обратным холодильником в течение 2 часов в 15 мл тионилхлорида. После этого тионилхлорид отгоняли, а полученный таким образом остаток растворяли в 10 мл тетрагидрофурана. Полученный таим образом раствор добавляли по капле к 20 мл раствора тетрагидрофурана, содержащего 1,0 г 1-метиленпиперазина, перемешанного при охлаждении льдом. После перемешивания при комнатной температуре в течение 1 часа и последующего удаления растворителя путем дистилляции, полученный остаток растворяли в хлороформе и промывали насыщенным водным раствором бикарбоната натрия. После осушки полученного органического слоя и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью хлороформа в этаноле. В результате этой процедуры получали 1,35 г целевого соединения в виде кристаллов, т.пл. 115-116oC.
1H-ЯМР (ДМСО-d6) δ: 2,34 (3H, с); 2,43 (4H, шир. с); 3,11 (4H, с); 3,64 (4H, шир. с.); 6,42 (1H, с); 7,26 (2H, д, J = 9,0 Гц); 7,40 (2H, д, J = 9,0 Гц); 7,50 (2H, с); 7,80 (1H, с).
Ссылочный пример 114. Получение 2-[2-[4-[[(2-пиразинил)амино]карбонил] фенил]этил]-5- бензофуранкарбонитрила.
a) 1 г 4-[2-(5-циано-2-бензофуранил)этил]бензойной кислоты и 573 мг 1-гидроксибензотриазола, при комнатной температуре растворили в 100 мл дихлорметана с последующим добавлением 780 мг 1,3-дициклогексилкарбодиимида и затем перемешивали при той же температуре 3 часа. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью хлороформа и этанола, а очищенный таким образом продукт кристаллизовали из смеси бензола и н-гексана. В результате этой процедуры получали 1,2 г 2-[2-[4-[[(1-бензотриазолил)окси] карбонил]фенил]-этил]-5-бензофуранкарбонитрила в виде порошкообразного вещества, т.пл.: 171-172oC.
1H-ЯМР (CDCl3) δ: 3,14 (4H, с); 6,48 (1H, с); 7,30-8,30 (11H, м);
MC (m/z): 409 (M++1).
b) 100 мг 2-[2-[4-[[(1-бензотриазолил)окси] карбонил]фенил] этил]-5-бензофуранкарбонитрила, полученного в вышеуказанной части a), и 23,3 мг аминопиразина растворяли в 2 мл N,N-диметилформамида. Полученный таким образом раствор смешивали с 13,0 мг (60%) гидрида натрия и перемешивали при комнатной температуре в течение 2 часов. Полученный реакционный раствор разбавляли этилацетатом, промывали водой, а затем высушивали. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 55 мг целевого соединения в виде порошкообразного вещества, т.пл.: 183-185oC.
1H-ЯМР (CDCl3) δ: 3,17 (4H, с); 6,42 (1H, с); 7,34 (2H, д, J=9,0 Гц); 7,59 (2H, с); 7,81 (1H, с); 7,90 (2H, д, J=9,0 Гц); 8,26 (1H, дд, J=3,0 и 1,6 Гц); 8,40 (1H, дд, J=3,0 Гц); 8,65 (1H, шир.с.); 9,74 (1H, д, J=1,6 Гц).
Ссылочный пример 115. Получение метил[5-[((3S)-1-трет- бутоксикарбонил-3-пирролидинил)окси] -2-[2-(5-циано-2- бензофуранил)этил]фенил]оксоацетата.
a) В 40 мл тетрагидрофурана растворяли 1,38 г 2,4-дигидроксибензальдегида, 1,87 г (3R)-1-трет-бутоксикарбонил-3-гидроксипирролидина и 2,88 г трифенилфосфина. Полученный таким образом раствор смешивали с 1,91 г диэтилазодикарбоксилата и перемешивали при комнатной температуре 1 час. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 1,2 г 4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил] -окси] -2- гидроксибензальдегида в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,47 (9H, с); 2,00-2,36 (2H, м); 3,30-3,75 (4H, м); 4,94 (1H, кв.); 6,38 (1H, д, J=2,1 Гц); 6,52 (1H, дд, J=8,0 и 2,1 Гц); 7,44 (1H, д, J=8,0 Гц); 9,72 (1H, с); 11,45 (1H, с).
b) 1,27 г 4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]- 2-гидроксибензальдегида, полученного в вышеуказанной части a), и 0,884 г этилбромоацетата растворяли в 100 мл ацетона. Полученный таким образом раствор смешивали с 1,12 г безводного карбоната калия, и смесь нагревали с обратным холодильником в течение 1,5 часа. После охлаждения, нерастворившиеся вещества удаляли путем фильтрации, а растворитель отгоняли. После этого, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 1,44 г этил[2-формил-5-[((3S)-1-трет-бутоксикарбонил- 3-пирролидинил)окси] фенил]оксиацетата в виде вязкого и маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,30 (3H, т, J=7,0 Гц); 1,47 (9H, с); 2,00-2,28 (2H, м); 3,36-3,70 (4H, м); 4,28 (2H, кв.); 4,71 (2H, с); 4,94 (1H, квинт); 6,31 (1H, д, J=2,2 Гц); 6,53 (1H, дд, J=8,8 и 2,2 Гц); 7,84 (1H, с); 10,39 (1H, с).
c) 1,44 г этил[2-формил-5-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)окси] фенил] оксоацетата, полученного в вышеуказанной части b), и 1,71 г (5-циано-2-бензофуранил)метилтрифенилфосфония хлорида растворяли в смеси растворителей, состоящей из 10 мл тетрагидрофурана и 10 мл метанола. Полученный таким образом раствор смешивали с 0,664 г 1,8-диазабицикло[5.4.0]-7-ундецена, и смесь перемешивали в течение 1 часа. После отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью бензола и этилацетата, в результате чего получали 1,8 г олефинового соединения в виде смеси E- и Z-форм. 1,8 г полученного таким образом олефинового соединения растворяли в смеси растворителей, состоящей из 40 мл тетрагидрофурана и 40 мл этанола, и полученный раствор подвергали каталитической гидрогенизации при нормальном давлении в присутствии 0,22 г оксида палладия • сульфата бария • 1H2O. После удаления катализатора путем фильтрации, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью бензола и этилацетата. В результате этой процедуры получали 1,6 г целевого соединения в виде вязкого маслянистого вещества. Во время реакции осуществляли переэтерификацию.
1H-ЯМР (CDCl3) δ: 1,46 (9H, с); 1,96-2,08 (2H, м); 3,08 (4H, с); 3,36-3,68 (4H, м); 3,80 (3H, м); 4,62 (2H, с); 4,80 (1H, шир. с); 6,30 (1H, с); 6,36 (1H, д, J=8,0 Гц); 6,43 (1H, с); 7,02 (1H, д, J=8,0 Гц); 7,46 (2H, с); 7,77 (1H, с).
Иллюстративный пример 1 (изобретения). Этил 3-(5-амидино-2-бензофуранил)-2-[4-[((3)-3-пирролидинил)окси]фенил] пропионата дигидрохлорид.
1,96 г этил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -3- (5-циано-2-бензофуранил)пропионата растворяли в 150 мл этанола. При охлаждении льдом и перемешивании, хлороводород барботировали в полученный таким образом раствор до насыщенного уровня, и затем оставляли на 18 часов. Полученный реакционный раствор концентрировали досуха при пониженном давлении, и полученный таким образом остаток растворяли в 300 мл раствора этанола, содержащего 15% (м./об.) аммиака и затем раствор оставляли еще на 18 часов. После отгонки растворителя, полученный остаток подвергали колоночной хроматографии, используя колонку, заполненную высокопористым полимером (синтетическим адсорбентом; полимер стирол-дивинилбензола Diaion HP-20), используя в качестве элюента смесь воды и ацетонитрила. Полученные таким образом нужные фракции подвергали обращенно-фазовой жидкостной хроматографии высокого разрешения (ЖХВР), с использованием колонки, заполненной октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого элюированные таким образом фракции собирали, смешивали с разбавленной соляной кислотой, и затем концентрировали досуха. В результате этой процедуры получали 610 мг целевого соединения в виде твердого вещества.
1H-ЯМР (DMCO-d6) δ: 1,08 (3H, т, J=7,0 Гц); 1,90-2,30 (2H, м); 3,00-3,80 (6H, м); 3,80-4,30 (3H, м); 5,08 (1H, шир.с.); 6,73 (1H, с); 6,93 (2H, д, J= 8,3 Гц); 7,33 (2H, д, J=8,3 Гц); 7,73 (2H, с); 8,08 (1H, с); 9,25 (2H, шир. с); 9,40 (2H, шир.с); 9,50-10,00 (2H, шир.с.).
Последующие соединения иллюстративных примеров 2-17 получали в соответствии с процедурой, описанной в иллюстративном примере 1.
Иллюстративный пример 2. Этил 3-(5-амидино-2-бензофуранил)- 2-[4-[((2S, 4S)-2-карбамоил-4-пирролидинил)окси] фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 1,10 (3H, т, J=7,0 Гц); 1,70-3,20 (2H, м); 3,00-4,50 (8H, м); 5,00-5,30 (1H, шир.с.); 6,71 (1H, с); 6,87 (2H, д, J=8,3 Гц); 7,30 (2H, д, J=8,3 Гц); 7,72 (2H, с); 8,10 (1H, с); 9,00-10,00 (6H).
Иллюстративный пример 3. Этил 3-(5-амидино-2-бензофуранил)-2-[4-[((2S, 4S)-2-диметилкарбамоил- 4-пирролидинил)окси] фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO6) δ: 1,11 (3H, т, J=7,0 Гц); 1,70-3,30 (2H, м); 2,91 (3H, с); 2,96 (3H, с); 3,00-4,20 (7H, м); 4,70 (1H, шир.с.); 5,10 (1H, шир.с.); 6,69 (1H, с); 6,86 (2H, д, J=8,7 Гц); 7,29 (2H, д, J=8,7 Гц), 7,69 (2H, с); 8,07 (1H, с); 8,80 (1H, шир.с.); 9,10 (2H, шир.с.); 9,34 (2H, шир.с.); 10,08 (1H, шир.с.).
Иллюстративный пример 4. Этил 2-(5-амидино-2-бензофуранил)-3-[4- [((3S)-3-пирролидинил)окси] фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,08 (3H, т, J = 7,0 Гц); 1,80-2,30 (2H, шир. с); 2,70-3,70 (6H, м); 4,08 (2H, кв., J = 7,0 Гц); 4,35 (1H, т, J = 7,9 Гц); 5,08 (1H, шир. с); 6,84 (2H, д, J = 8,3 Гц); 6,9 (1H, c); 7,17 (2H, д, J = 8,3 Гц); 7,79 (2H, с); 8,12 (1H, с); 9,33 (2H, шир. с); 9,51 (2H, шир. с); 9,80 (2H, шир. с).
Иллюстративный пример 5. Этил 3-(5-амидино-2-бензофуранил)-3- [4-[((3S)-3-пирролидинил-окси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,17 (3H, т, J = 7 Гц); 2,0-2,2 (2H, м); 3,0-3,8 (6H, м); 4,07 (2H, кв.); 4,5-4,7 (1H, м); 5,13 (1H, м); 6,94 (1H, с); 6,94 (2H, д, J = 9 Гц); 7,32 (2H, д, J = 9 Гц); 7,73 (2H, с); 8,13 (1H, с); 9,21 (2H, шир. с); 9,40 (2H, шир. с); 9,4-10,0 (2H, шир. с).
Иллюстративный пример 6. Этил 3-(5-амидино-3-бензофуранил)- 2-[4-[((3S)-3-пирролидинил)окси] фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,06 (3H, т); 2,10 (2H, шир.); 3,0-3,7 (7H); 4,05 (2H, кв. ); 5,09 (1H, шир. с); 6,95 (2H, д); 7,28 (2H, д); 7,77 (3H); 8,21 (1H, с); 9,2-9,8 (6H).
Иллюстративный пример 7. Этил 2-[2-(5-амидино-2-бензофуранил)- этил]-5-[((3S)-3-пирролидинил)окси]бензоата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,29 (3H, т, J = 7,0 Гц); 2,00-2,35 (2H, м); 2,90-3,60 (8H, м); 4,18 (2H, кв., J = 7,0 Гц); 5,20 (1H, шир. с); 6,75 (1H, c); 7,20 (1H, дд, J = 7,9 и 2,8 Гц); 7,39 (1H, д, J = 7,9 Гц); 7,41 (1H, д, J = 2,8 Гц); 7,74 (2H, с); 8,09 (1H, с); 9,23 (2H, шир. с); 9,40 (2H, шир. с.); 9,50-10,20 (2H, шир. с).
Иллюстративный пример 8. Этил[2-[2-(5-амидино-2-бензофуранил)- этил]-5-[((3S)-3-пирролидинил-окси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,18 (3H, т, J = 7,0 Гц); 2,0-2,30 (2H, м); 3,02 (4H, c); 3,00-4,00 (6H, м); 4,90 (2H, кв., J = 7,0 Гц); 5,12 (1H, шир. с); 6,80-7,00 (3H, м); 7,24 (1H, д, J = 8,64 Гц); 7,76 (2H, с); 8,12 (1H, с); 9,29 (1H, с); 9,45 (2H, шир. с); 9,40-10,10 (2H, шир. с).
Иллюстративный пример 9. Этил 5-амидино-2-[2-[4-[((3S)-3- пирролидинил)окси] фенил]этил]-3-бензофуранкарбоксилата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,36 (3H, т); 2,08 (2H, м); 4,30 (2H); 5,00 (1H, шир. с); 6,90 (2H, д); 7,10 (2H, д); 7,83 (2H); 8,34 (1H, с); 9,27 (2H, шир. с); 9,51 (4H, шир. с).
Иллюстративный пример 10. Этил 3-(5-амидинобензо[b]тиен-2-ил)- 2-[4-[((3S)-3-пирролидинил)окси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,19 (3H, т, J = 7,0 Гц); 2,00-2,40 (2H, м); 3,00-4,20 (9H, м); 5,08 (1H, шир. с); 6,92 (2H, д, J = 8,75 Гц); 7,27 (1H, с); 7,31 (2H, д, J = 8,75 Гц); 7,68 (1H, д, J = 8,3 и 1,5 Гц); 8,10 (1H, д, J = 8,3 Гц)); 8,27 (1H, д, J = 1,5 Гц); 9,19 (2H, шир. с.); 9,42 (2H, шир. с); 9,10-10,00 (2H, шир. с).
Иллюстративный пример 11. Этил 3-(6-амидинобензо[b]тиен-2- ил)-2-[4-[((3S)-3-пирролидинил-окси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,05 (3H, т); 2,10 (2H, м); 3,30 (7H, м); 4,0 (2H, м); 5,05 (1H, шир. с); 6,90 (2H, д); 7,22 (3H, м); 7,60-7,90 (2H, м); 8,38 (1H, с); 9,10 (2H, шир. с); 9,35 (2H, шир. с); 9,40 (2H, шир. с).
Иллюстративный пример 12. Этил 3-(6-амидино-1-этил-2-индолил)- 2-[4-[(4-пиперидинил)окси]фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,05 (3H, т); 1,28 (3H); 1,83 (2H шир. с); 2,08 (2H, шир. с); 2,90-3,20 (5H, шир. с); 3,90-4,40 (3H, м); 4,62 (1H, шир. с); 6,34 (1H, с); 6,97 (2H, д); 7,34 (2H); 7,47 (1H, д); 7,58 (1H, д); 8,13 (1H, с); 8,90-9,40 (6H, шир. с).
Иллюстративный пример 13. Этил 3-(6-амидино-1-этил-2-индолил)- 2-4-[((3R)-3-пирролидинил)окси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,08 (3H, т, J = 7,0 Гц); 1,31 (3H, т, J = 7,0 Гц); 2,10-2,30 (2H, шир. с); 3,17 (1H, дд); 3,20-3,40 (2H, м); 3,90-4,40 (5H, м); 5,14 (1H, шир. с); 6,37 (1H, с); 6,97 (2H, д, J = 8,8 Гц); 7,38 (2H, д, J = 8,8 Гц); 7,49 (1H, д, J = 8,3 Гц); 7,62 (1H, д, J = 8,3 Гц); 8,14 (1H, с); 8,99 (2H, шир. с.); 9,32 (2H, шир. с); 9,50-9,70 (2H, шир. с).
Иллюстративный пример 14. Этил 3-(6-амидино-1-метил-2-индолил)- 2-4-[((3S)-3-пирролидинил)окси] фенил]пропионата дигидрохлорид (переэтерификация с использованием этанолового растворителя) (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,05 (3H, т); 1,95-2,30 (2H, м); 3,76 (3H, с); 4,02 (2H, кв.); 4,00-4,30 (1H, м); 5,00-5,20 (1H, м); 6,38 (1H, с); 7,00 (2H, д); 7,40 (2H, д); 7,50-7,70 (2H, м); 8,25 (1H, с); 9,30-10,10 (6H).
Иллюстративный пример 15. Этил 3-(6-амидино-2-нафтил)-2- [4-((3S)-3-пирролидинил)-окси]-фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,06 (3H, т, J = 7,0 Гц); 2,00-2,20 (2H, м); 3,00-4,00 (7H, м); 3,99 (2H, кв., J = 7,0 Гц); 5,11 (1H, м); 6,92 (2H, д, J = 9,0 Гц); 7,31 (2H, д, J = 9,0 Гц); 7,55 (1H, д, J = 8,0 Гц); 7,80-8,10 (4H, м); 8,51 (1H, с); 9,40 (2H, шир. с); 9,58 (2H, шир. с); 9,50-10,00 (2H, шир. с).
Иллюстративный пример 16. Этил 3-(7-амидино-2-нафтил)-2- [4-[((3S)-3-пирролидинил)-окси]фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,0 Гц); 2,00-2,20 (2H, м); 3,10-3,80 (7H, м); 3,98 (2H, кв., J = 7,0 Гц); 5,10 (1H, м); 6,93 (2H, д, J = 9,0 Гц); 7,32 (2H, д, J = 9,0 Гц); 7,50-8,10 (5H, м); 8,44 (1H, м); 9,41 (2H, шир. с); 9,59 (2H, шир. с); 9,30-10,00 (2H, шир. с).
Иллюстративный пример 17. Этил 3-(7-амидино-2-нафтил)-2- [4-[(4-пиперидинил-метокси]фенил]пропионата дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,1 Гц); 1,45-1,55 (2H, м); 1,85-1,95 (2H, м); 2,80-2,95 (2H, м); 3,15-3,50 (5H, м); 3,81 (2H, д); 3,95-4,05 (2H, м); 4,05-4,15 (1H, м); 6,87 (2H, д, J = 8,3 Гц); 7,27 (2H, д, J = 8,3 Гц); 7,58-7,63 (1H, м); 7,75-7,80 (1H, м); 7,84 (1H, с); 7,95 (1H, д, J = 8,8 Гц); 8,07 (1H, д, J = 8,8 Гц); 8,40 (1H, с); 9,29 (2H); 9,53 (2H).
Иллюстративный пример 18. 3-(7-Амидино-2-нафтил)-2-[4- [(4-пиперидинил)-метокси]фенил]-пропионовой кислоты гидрохлорида моногидрат.
1,51 г этил 3-(7-амидино-2-нафтил)-2-[4-[(4-пиперидинил)- метокси]фенил] пропионата дигидрохлорида растворяли в 50 мл концентрированной соляной кислоты, а раствор оставляли в герметично закрытом сосуде при комнатной температуре в течение 62 часов. После осушки полученного реакционного раствора при пониженном давлении, полученный таким образом остаток очищали путем нанесения его на колонку, упакованную высокопористым полимером (синтетический адсорбент; полимер стирола и дивинилбензола HP-20). После этого, элюированные таким образом фракции собирали и смешивали с небольшим количеством этанола, а затем осажденные кристаллы собирали путем фильтрации. В результате этой процедуры получали 0,79 г целевого соединения в виде кристаллов. Т.пл.: 285 - 287oC (с разл.).
Поскольку растворимость таким образом полученного продукта в любом растворителе очень низка, то соединение обрабатывали дигидрохлоридом с соляной кислотой и затем до проведения измерения ЯМР осушали.
1H-ЯМР (ДМСО-d6) δ: 1,45 - 1,60 (2H, м); 1,85 - 1,95 (2H, м); 1,95 - 2,05 (1H, шир. с); 2,8 - 2,9 (2H, м); 3,1 - 3,2 (1H, м); 3,2 - 3,3 (2H, м); 3,4 - 3,5 (1H, м); 3,80 (2H, д, J = 6,4 Гц); 3,9 - 4,0 (1H, м); 6,87 (2H, д, J = 8,8 Гц); 7,27 (2H, д, J = 8,8 Гц); 7,60 (1H, д, J = 8,8 Гц); 7,75 - 7,80 (1H, м); 7,83 (1H, с); 7,94 (1H, д, J = 8,8 Гц); 8,07 (1H, д, J = 8,3 Гц); 8,40 (1H, с); 8,8 - 8,9 (1H, шир.с.); 9,33 (2H); 9,54 (2H).
Иллюстративный пример 19. 3-(5-Амидино-2-бензофуранил)-2- [4-[((3S)-3-пирролидинил)окси]фенил]пропионовой кислоты дигидрохлорид.
3,2 г этил 3-(5-амидино-2-бензофуранил)-2- [4-[((3S)-3-пирролидинил)окси] фенил] пропионата дигидрохлорида растворяли в 80 мл 2 н. соляной кислоты, а раствор нагревали с обратным холодильником в течение 30 минут. После охлаждения и отгонки растворителя, полученный остаток очищали с помощью колоночной хроматографии с использованием колонки, упакованной высокопористым полимером (синтетический адсорбент; полимер стирола-дивинилбензола Diaion HP-20), а в качестве элюента использовали 5 - 10% ацетонитрила. После этого, элюированные таким образом фракции собирали, и доводили до значения pH 2 - 3 разбавленной соляной кислотой, а затем концентрировали досуха. В результате этой процедуры получали 1,25 г целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,30 (2H, м); 3,00 - 3,80 (6H, м); 4,10 (1H, т, J = 7,2 Гц); 5,10 (1H, шир. с); 6,74 (1H, с); 6,94 (2H, д, J = 8,3 Гц); 7,40 (2H, д, J = 8,3 Гц); 7,74 (2H, с); 8,09 (2H, с); 9,22 (2H, шир. с); 9,40 (2H, шир. с); 9,10 (2H, шир. с).
Последующие соединения иллюстративных примеров 20 - 26 получали в соответствии с процедурой, описанной в иллюстративном примере 19.
Иллюстративный пример 20. 3-(5-Амидино-2-бензофуранил)-2- [4-[((2S, 4S)-2-диметилкарбамоил-4-пирролидинил)окси] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,80 - 3,00 (2H, м); 2,89 (3H, с); 2,95 (3H, с); 3,00 - 3,70 (4H, м); 4,09 (1H, т, J = 7,9 Гц); 4,70 (1H, шир. с); 5,12 (1H, шир. с); 6,71 (1H, с); 6,86 (2H, д, J = 8,3 Гц); 7,30 (2H, д, J = 8,3 Гц); 7,73 (2H, с); 8,10 (1H, с); 8,76 (1H, шир. с); 9,30 (2H, шир. с); 9,46 (2H, шир. с); 10,80 (1H, шир. с).
Иллюстративный пример 21. 2-(5-Амидино-2-бензофуранил)-3- [4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90 - 2,30 (2H, м); 2,90 - 3,70 (6H, м); 4,26 (1H, т, J = 7,9 Гц); 5,06 (1H, шир. с); 6,83 (2H, д, J = 8,3 Гц); 6,93 (1H, с); 7,17 (2H, д, J = 8,3 Гц); 7,78 (2H, с); 8,14 (1H, с); 9,30 (2H, шир. с); 9,47 (2H, шир. с); 9,80 (2H, шир. с).
Иллюстративный пример 22. 3-(5-Амидино-2-бензофуранил)-3- [4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,0 - 2,2 (2H, м); 3,0 - 4,0 (6H, м); 4,6 (1H, м); 5,10 (1H, м); 6,92 (1H, с); 6,92 (2H, д, J = 9,0 Гц); 7,32 (2H, д, J = 9,0 Гц); 7,73 (2H, с); 8,16 (1H, с); 9,30 (2H, шир. с); 9,46 (2H, шир. с); 9,6 - 10,0 (2H, шир. с).
Иллюстративный пример 23. 2-[2-(5-Амидино-2-бензофуранил) этил] -5-[((3S))-3-пирролидинил)окси] бензойной кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90 - 2,30 (2H, м); 2,90 - 3,70 (3H, м); 4,96 (1H, шир. с); 6,75 (1H, с); 7,08 (1H, дд, J = 7,9 и 2,8 Гц); 7,28 (1H, д, J = 7,9 Гц); 7,41 (1H, д, J = 2,8 Гц); 7,75 (2H, с); 8,09 (1H, с); 9,25 (2H, шир. с); 9,42 (2H, шир. с); 9,50 - 10,00 (2H, шир. с).
Иллюстративный пример 24. [2-[2-(5-Амидино-2-бензофуранил) этил]-5-[((3S))-3-пирролидинил)окси] фенил] уксусной кислоты дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 1,95 - 2,30 (2H, м); 3,03 (4H, с); 5,04 (1H, шир. с); 6,68 - 6,90 (3H, м); 7,14 (1H, д, J = 8,3 Гц); 7,74 (2H, с); 8,10 (1H, с); 9,38 (2H, шир. с); 9,66 (2H, шир. с); 9,00 - 10,00 (2H, шир. с).
Иллюстративный пример 25. 3-(5-Амидинобензо[b]тиен-2-ил)- 2-[4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90 - 2,40 (2H, м); 3,00 - 4,10 (7H, м); 5,14 (1H, шир. с); 6,93 (2H, д, J = 8,2 Гц); 7,28 (1H, с); 7,33 (2H, д, J = 8,2 Гц); 7,70 (1H, д, J = 8,8 Гц); 8,09 (1H, д, J = 8,8 Гц); 8,26 (1H, с); 9,24 (2H, шир. с); 9,47 (2H, шир. с); 9,00 - 10,20 (2H, шир. с).
Иллюстративный пример 26. 3-(7-Амидино-2-нафтил)-2- [4-[((3S)-3-пирролидинил)окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,20 (2H, м); 3,00 - 3,70 (6H, м); 5,01 (1H, м); 5,11 (1H, м); 6,92 (2H, д, J = 9,0 Гц); 7,33 (2H, д, J = 9,0 Гц); 7,50 - 8,20 (5H, м); 8,43 (1H, с); 9,00 - 10,50 (6H).
Иллюстративный пример 27. Этил (+)-3-(7-амидино-2-нафтил)-2-[4-[((3S))-3-пирролидинил)окси]фенил]пропионата дигидрохлорид.
123,1 г этил (+)-2-[4-[((3S)-1-трет-бутоксикарбонил-3- пирролидинил)окси]фенил]-3-(7-циано-2-нафтил)пропионата растворяли в смеси растворителей, состоящей из 480 мл дихлорметана и 1286 мл этанола. Перемешивая при -10oC, хлороводород барботировали в полученный таким образом до уровня насыщения, полученный раствор оставляли еще на 26 часов при температуре от -8 до -5oC. После этого полученный реакционный раствор концентрировали при пониженном давлении при температуре от 10oC и ниже, в результате чего получали 154 г маслянистого продукта. Полученный таким образом маслянистый продукт растворяли в 1480 мл этанола и, поддерживая внутреннюю температуру при -10oC или ниже, вводили аммиачный газ до тех пор, пока концентрация его не станет 21% (об. /об. ) или больше. После выдерживания при температуре от -8oC до 5oC в течение 107 часов, полученный реакционный раствор концентрировали при пониженном давлении при температуре от 10oC или ниже для отгонки растворителя, и полученный таким образом остаток растворяли в 200 мл воды. После доведения до значения pH 3 - 5 разбавленной соляной кислотой, полученный раствор очищали с помощью колоночной хроматографии с использованием колонки, упакованной высокопористым полимером (синтетический адсорбент; полимер стирол-дивинилбензола Diaion HP-20) и использованием в качестве элюента смеси воды и ацетонитрила. После этого элюированные таким образом фракции собирали, смешивали с небольшим количеством соляной кислоты и концентрировали досуха. В результате этой процедуры получали 107 г целевого соединения в виде бесцветного твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,2 Гц); 2,00 - 2,30 (2H, м); 3,1 - 3,6 (6H, м); 3,90 - 4,05 (2H, м); 4,05 - 4,15 (1H, м); 5,10 (1H, шир. с); 6,93 (2H, д, J = 8,8 Гц); 7,32 (2H); 7,60 (1H, д, J = 8,3 Гц); 7,78 (1H, д, J = 8,3 Гц); 7,85 (1H, с); 7,96 (1H, д, J = 8,3 Гц); 8,08 (1H, д, J = 8,3 Гц); 8,41 (1H, с); 9,20 - 9,30 (2H, шир. с); 9,40 - 9,70 (4H, шир. с).
Последующие соединения иллюстративных примеров 28 - 32 получали в соответствии с процедурой, описанной в иллюстративном примере 27.
Иллюстративный пример 28. Этил (-)-3-(7-амидино-2-нафтил)-2- [4-[((3S))-3-пирролидинил)окси] фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,02 (3H, т, J = 7,2 Гц); 2,00 - 2,30 (2H, м); 3,1 - 3,6 (6H, м); 3,90 - 4,05 (2H, м); 4,05 - 4,15 (1H, м); 5,10 (1H, шир. с); 6,94 (2H, д, J = 8,8 Гц); 7,32 (2H); 7,60 (1H, д, J = 8,3 Гц); 7,78 (1H, д, J = 8,3 Гц); 7,86 (1H, с); 7,96 (1H, д, J = 8,3 Гц); 8,08 (1H, д, J = 8,3 Гц); 8,42 (1H, с); 9,20 - 9,30 (2H, шир. с); 9,40 - 9,70 (4H, шир. с).
Иллюстративный пример 29. Этил (+)-3-(5-амидинобензо[b]тиен-2 ил)-2-4-[((2S)-2-пирролидинил)метокси] фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,10 (3H, т, J = 7,3 Гц); 1,73 (1H, д. кв. J = 12,3 и 8,3 Гц); 1,84 - 2,05 (2H, м); 2,06 - 2,16 (1H, м); 3,12 - 3,27 (2H, шир. с); 3,39 (1H, дд, J = 15,0 и 7,8 Гц); 3,64 (1H, дд, J = 15,0 и 7,8 Гц); 3,80 - 3,93 (1H, шир. с); 4,00 - 4,24 (5H, м); 6,93 (2H, д, J = 8,3 Гц); 7,30 (1H, с); 7,31 (2H, д); 7,67 (1H, д, J = 8,3 Гц); 8,11 (1H, д, J = 8,3 Гц); 8,23 (1H, с); 9,12 - 9,30 (3H); 9,45 (2H, с); 9,43 (2H, с); 9,74 - 9,94 (1H, шир. с).
Иллюстративный пример 30. Этил (-)-3-(5-амидинобензо[b]тиен-2- ил)-2-[4-[((2S)-2-пирролидинил)метокси] фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,09 (3H, т, J = 7,3 Гц); 1,72 (1H, д. кв., J = 12,1 и 8,3 Гц); 1,82 - 2,03 (2H, м); 2,06 - 2,16 (1H, м); 3,12 - 3,27 (2H, шир. с); 3,39 (1H, дд, J = 15,0 и 7,8 Гц); 3,64 (1H, дд, J = 15,0 и 7,8 Гц); 3,80 - 3,93 (1H, шир. с); 4,00 - 4,24 (5H, м); 6,93 (2H, д, J = 8,8 Гц, 2 • AH); 7,30 (1H, с); 7,31 (2H, д, J = 8,8 Гц); 7,67 (1H, дд, J = 8,3 и 1,5 Гц); 8,11 (1H, д, J = 8,3 Гц); 8,23 (1H, д, J = 1,5 Гц); 9,10 - 9,25 (1H, шир. с); 9,21 (2H, с); 9,43 (2H, с); 9,74 - 9,84 (1H, шир. с).
Иллюстративный пример 31. Этил (+)-3-(7-амидино-2-нафтил)-2-[4-[(4-пиперидинил)оксо]-фенил]пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,1 Гц); 1,75 - 1,85 (2H, м); 2,05 - 2,15 (2H, м); 3,0 - 3,1 (2H, м); 3,1 - 3,2 (3H, м); 3,9 - 4,0 (2H, м); 4,0 - 4,1 (1H, шир. с); 4,1 - 4,2 (1H, м); 4,61 (1H, шир. с); 6,95 (2H, д, J = 8,8 Гц); 7,29 (2H, д, J = 8,8 Гц); 7,61 (1H, д, J = 8,3 Гц); 7,78 (1H, д, J = 8,3 Гц); 7,84 (1H, с); 7,95 (1H, д, J = 8,3 Гц); 8,07 (1H, д, J = 8,8 Гц); 8,38 (1H, с); 8,9 - 9,1 (2H, шир. с); 9,20 (2H, шир. с); 9,49 (2H, шир. с).
Иллюстративный пример 32. Этил (-)-3-(7-амидино-2-нафтил)-2-[4-[(4-пиперидинил)окси]-фенил] пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 7,1 Гц); 1,75 - 1,80 (2H, м), 2,05 - 2,15 (2H, м); 3,0 - 3,1 (2H, м); 3,1 - 3,3 (3H, м); 3,50 - 3,60 (1H, м); 3,65 - 3,75 (2H, м); 3,9 - 4,0 (2H, м); 4,0 - 4,1 (1H, шир. с); 4,1 - 4,2 (1H, м); 4,61 (1H, шир. с); 6,95 (2H, д, J = 8,3 Гц); 7,29 (2H, д, J = 8,3 Гц); 7,61 (1H, д, J = 8,8 Гц); 7,78 (1H, д, J = 8,3 Гц); 7,84 (1H, с); 7,95 (1H, д, J = 8,3 Гц); 8,07 (1H, д, J = 8,8 Гц); 8,39 (1H, с); 8,9 - 9,1 (2H, шир. с); 9,23 (2H, шир. с); 9,50 (2H, шир. с).
Иллюстративный пример 33. Этил (+)-2-[4-[((3S)-1-ацетимидоил-3-пирролидинил)окси]фенил]-3- (7-амидино-2-нафтил)пропионата дигидрохлорид.
В 1,000 мл этанола растворяли 105,3 г этил (+)-3-(7-амидино-2-нафтил)-2-[4-[((3S)-3-пирролидинид)окси] фенил] пропионата дигидрохлорида. Перемешивая при комнатной температуре, полученный таким образом раствор смешивали с 51,5 г этилацетимидата гидрохлорида. Перемешивая и охлаждая льдом, к вышеуказанному раствору по капле добавляли 89 мл триэтиламина, поддерживая при внутренней температуре от 3 - 5oC, и перемешивали в течение 2,5 часов, поддерживая при температуре 5oC или ниже. После отгонки растворителя при пониженном давлении и низкой температуре, полученный реакционный раствор доводили до значения pH 4-5 разбавленной соляной кислотой, затем продолжали дистилляцию при пониженном давлении для удаления растворителя. Полученный остаток очищали с помощью колоночной хроматографии с использованием колонки, упакованной высокопористым полимером (синтетический адсорбент; полимер стирола-дивинилбензола Diaion HP-20) и в качестве элюента использовали смесь воды и ацетонитрила. После этого, элюированные фракции собирали, смешивали с небольшим количеством разбавленной соляной кислоты, а затем концентрировали досуха. В результате этой процедуры получали 110,1 г целевого соединения в виде бесцветного твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,02 (3H, м); 2,10 - 2,35 (2H, м); 2,26 (1,5H, с); 2,30 (1,5H, с); 3,19 (1H, м); 3,40 - 3,85 (5H, м); 3,90 - 4,05 (2H, м); 4,05 - 4,15 (1H, м); 5,13 (0,5H, шир. с); 5,20 (0,5H, шир. с); 6,90 - 6,97 (2H, м); 7,32 (2H, м); 7,61 (1H, д, J = 8,3 Гц); 7,80 (1H, дд, J = 8,3 и 1,5 Гц); 7,84 (1H, с); 7,96 (1H, д. J = 8,3 Гц); 8,08 (1H, д, J = 8,3 Гц); 8,43 (1H, с); 8,52 (0,5H, шир. с); 8,61 (0,5H, шир. с); 9,28 - 9,40 (3H, шир. с); 9,50 - 9,60 (2H, шир. с).
Последующие соединения иллюстративных примеров 34 - 38 получали в соответствии с процедурой, описанной в иллюстративном примере 33.
Иллюстративный пример 34. Этил (-)-2-[4-[((3S)-1-ацетимидоил-3-пирролидинил)окси]фенил]-3- (7-амидино-2-нафтил)пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,02 (3H, м); 2,10 - 2,35 (2H, м); 2,26 (1,5H, с); 2,30 (1,5H, с); 3,19 (1H, м); 3,40 - 3,85 (5H, м); 3,90 - 4,05 (2H, м); 4,05 - 4,15 (1H, м); 5,13 (0,5H, шир. с); 5,20 (0,5H, шир. с); 6,90 - 6,97 (2H, м); 7,32 (2H, м); 7,61 (1H, д, J = 8,3 Гц); 7,80 (1H, дд, J = 8,3 и 1,5 Гц); 7,84 (1H, с); 7,96 (1H, д, J = 8,3 Гц); 8,08 (1H, д, J = 8,3 Гц); 8,42 (1H, с); 8,52 (0,5H, шир. с); 8,61 (0,5H, шир. с); 9,28 - 9,40 (3H, шир. с); 9,50 - 9,60 (2H, шир. с).
Иллюстративный пример 35. Этил (+)-2-[4-[((2S)-1-ацетимидоил-3-пирролидинил)метокси] фенил] -3- (5-амидинобензо[b]тиен-2-ил)пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,09 (3H, т, J = 7,3 Гц); 1,95 - 2,60 (4H, м); 2,26 (1H, с); 2,47 (2H, с); 3,30 - 3,70 (4H, м); 3,90 - 4,10 (5H, м); 4,40 - 4,60 (1H, м); 6,85 - 6,95 (2H, м); 7,28 - 7,33 (3H, м); 7,67 (1H, д, J = 8,3 Гц); 8,11 (1H, д, J = 8,3 Гц); 8,23 (1H, с); 8,54 (2/3H, с); 8,69 (1/3H, с); 9,23 (2H, с); 9,35 - 9,50 (3H, м).
Иллюстративный пример 36. Этил (-)-2-[4-[((2S)-1-ацетимидоил-3- пирролидинил)метокси] фенил] -3-(5-амидинобензо[b] тиен-2-ил)пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,09 (3H, т, J = 7,3 Гц); 1,95 - 2,60 (4H, м); 2,26 (1H, с); 2,47 (2H, с); 3,30 - 3,70 (4H, м); 3,90 - 4,10 (5H, м); 4,40 - 4,60 (1H, м); 6,85 - 6,95 (2H, м); 7,28 - 7,33 (3H, м); 7,67 (1H, д, J = 8,3 Гц); 8,10 (1H, д, J = 8,3 Гц); 8,23 (1H, с); 8,51 (2/3H, с); 8,66 (1/3H, с); 9,16 (2H, с); 9,30 - 9,48 (3H, м).
Иллюстративный пример 37 Этил (+)-2-[4-[(1-ацетимидоил-4- пиперидинил)окси]фенил]-3-(7-амидино-2-нафтил)пропионата дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,01 (3H, т, J = 6,9 Гц); 1,65 - 1,80 (2H, м); 2,0 - 2,1 (2H, м); 2,30 (3H, с); 3,1 - 3,2 (1H, м); 3,2 - 3,8 (5H, м); 3,9 - 4,0 (2H, м); 4,0 - 4,1 (1H, шир. с); 4,67 (1H, шир. с); 6,95 (2H, д, J = 8,8 Гц); 7,30 (2H, д, J = 8,8 Гц); 7,61 (1H, д, J = 8,3 Гц); 7,78 - 7,84 (1H, м); 7,84 (1H, с); 7,95 (1H, д, J = 8,3 Гц); 8,08 (1H, д, J = 8,3 Гц); 8,40 (1H, с); 8,80 - 9,55 (6H).
Иллюстративный пример 38. Этил (-)-2-[4-[(1-ацетимидоил-4-пиперидинил)-окси] фенил] -3- (7-амидино-2-нафтил)пропионата дигидрохлорид (твердое вещество)
[α]D= -67,69o (с = 0,585, H2O)
Иллюстративный пример 39. (+)-2-[4-[((3S)-1-Ацетимидоил-3-пирролидинил)-окси]фенил]-3- (7-амидино-2-нафтил)пропионовой кислоты дигидрохлорид.
При поддерживании внутренней температуры при -5oC или ниже, 110,1 г этил (+)-2-[4-[((3S)-1-ацетимидоил-3-пирролидинил)окси] фенил]-3-(7-амидино-2-нафтил)пропионата дигидрохлорида растворяли в 3 300 мл концентрированной соляной кислоты, а полученный раствор оставляли еще на 232 часа при 5oC. Полученный реакционный раствор концентрировали путем отгонки соляной кислоты и воды при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии, на колонке, наполненной синтетическим адсорбентом типа высокопористого полимера (полимер стирола-дивинилбензола Diaion HP-20), элюируя смесью воды и ацетонитрила. После этого, элюированные фракции собирали, смешивали с небольшим количеством соляной кислоты, и затем концентрировали досуха. В результате этой процедуры, получали 103,6 г целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,10 - 2,4 (2H, м); 2,28 (1,5H, с); 2,31 (1,5H, с); 3,10 - 3,30 (1H, м); 3,40 - 4,10 (6H, с); 5,14 (0,5H, шир. с); 5,20 (0,5H, шир. с); 6,90 - 7,00 (2H, м); 7,35 - 7,40 (2H, м); 7,60 (1H, д, J = 8,3 Гц); 7,80 (1H, д, J = 8,3 Гц); 7,84 (1H, с); 7,94 (1H, д, J = 8,3 Гц); 8,06 (1H, д, J = 8,3 Гц); 8,42 (1H, с); 8,55 (0,5H, шир. с); 8,65 (0,5H, шир. с); 9,30 - 9,70 (5H).
ЖХВР: Колонка: лиганд-обменного типа с D-пеницилламином в качестве оптически активного центра (SUMICHIPA OA-5000, 4,6 ⊘ • 150 мм, Sumika Analysis Center
Растворитель: 2 мМ водного раствора сульфата меди: ацетонитрил = 85:15 (об./об.)
Скорость потока: 1 мл/мин
Колоночная температура: 60oC
Время удержания: 43,60 минут
Последующие соединения иллюстративных примеров 40 - 44 получали в соответствии с процедурой, описанной в иллюстративном примере 39.
Иллюстративный пример 40. (-)-2-[4-[((3S)-1-Ацетимидоил-3- пирролидинил)окси] фенил] -3-(7-амидино-2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,05 - 2,4 (2H, м); 2,28 (1,5H, с); 2,31 (1,5H, с); 3,10 - 3,30 (1H, м); 3,40 - 4,10 (6H, м); 5,13 (0,5H, шир. с); 5,20 (0,5H, шир. с); 6,90 - 7,00 (2H, м); 3,5 - 7,40 (2H, м); 7,60 (1H, д, J = 8,3 Гц); 7,81 (1H, д, J = 8,3 Гц); 7,84 (1H, с); 7,94 (1H, д, J = 8,3 Гц); 8,06 (1H, д, J = 8,3 Гц); 8,42 (1H, с); 8,55 (0,5H, шир. с.); 8,64 (0,5H, /ир. с.); 9,30 - 9,70 (5H).
ЖХВР: Колонка: лиганд-обменного типа с D-пеницилламином в качестве оптически активного центра (SUMICHIPA OA-5000, 4,6 ⊘ x 150 мм, Sumika Analysis Center)
Растворитель: 2 мМ водного раствора сульфата меди: ацетонитрил = 85:15 (об./об.)
Скорость потока: 1 мл/мин
Колоночная температура: 60oC
Время удерживания: 38, 14 минут.
Иллюстративный пример 41. (+)-2-[4-[((2S)-1-Ацетимидоил-2- пирролидинил)метокси] фенил]-3-(5-амидинобензо[b]тиен-2-ил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,95 - 2,60 (4H, м); 2,25 (1H, с); 2,44 (2H, с); 3,15 - 3,80 (5H, м); 4,40 - 4,60 (1H, м); 6,83 - 6,95 (2H, м); 7,26 (1H, с); 7,32 (2H, д, J = 8,8 Гц); 7,61 (1H, д, J = 8,5 Гц); 8,04 (1H, д, J = 8,50 Гц); 8,21 (1H, с); 8,40 - 10,90 (6H).
Иллюстративный пример 42. (-)-2-[4-[((2S)-1-Ацетимидоил-2- пирролидинил)метокси] фенил]-3-(5-амидинобензо[b]тиен-2-ил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,95 - 2,60 (4H, м); 2,25 (1H, с); 2,44 (2H, с); 3,15 - 3,75 (4H, м); 3,82 (1H, т, J = 7,5 Гц); 4,40 - 4,60 (1H, м); 6,83 - 6,95 (2H, м); 7,27 (1H, м); 7,31 (2H, д, J = 8,8 Гц); 7,64 (1H, д, J = 8,8 Гц); 8,06 (1H, д, J = 8,8 Гц); 8,21 (1H, с); 8,40 - 10,40 (6H).
Иллюстративный пример 43. (+)-2-[4-[(1-Ацетимидоил-4- пиперидинил)окси] фенил]-3-(7-амидино-2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,65 - 1,80 (2H, м); 1,95 - 2,05 (2H, м); 2,31 (3H, с); 3,1 - 3,2 (1H, м); 3,3 - 3,9 (5H, м); 3,95 - 4,05 (1H, м); 4,66 (1H, шир. с.); 6,95 (2H, д, J = 8,8 Гц); 7,29 (2H, д, J = 8,3 Гц); 7,60 (1H, д, J = 8,8 Гц); 7,81 (1H, д, J = 8,8 Гц); 7,84 (1H, с); 7,95 (1H, д, J = 8,8 Гц); 8,07 (1H, д, J = 8,8 Гц); 8,43 (1H, с); 8,80 - 9,65 (6H).
Иллюстративный пример 44. (-)-2-[4-[(1-Ацетимидоил-4- пиперидинил)окси] фенил]-3-(7-амидино-2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,65 - 1,80 (2H, м); 2,00 - 2,10 (2H, м); 2,30 (3H, с); 3,1 - 3,2 (1H, м); 3,3 - 3,85 (5H, м); 3,95 - 4,05 (1H, м); 4,66 (1H, м); 6,95 (2H, д, J = 8,8 Гц); 7,30 (2H, д, J = 8,8 Гц); 7,61 (1H, д, J = 8,8 Гц); 7,78 (1H, д, J = 8,8 Гц); 7,85 (1H, с); 7,95 (1H, д, J = 8,8 Гц); 8,08 (1H, д, J = 8,8 Гц); 8,40 (1H, с); 8,60 - 9,65 (6H).
Иллюстративный пример 45. (+)-2-[4-[((3S)-1-Ацетимидоил-3- пирролидинил)окси] фенил]-3-(7-амидино-2-нафтил)пропионовой кислоты гидрохлорида пентагидрат.
102,6 г (+)-2-[4-[((3S)-1-ацетимидоил-3-пирролидинил)окси] фенил] -3-(7-амидино-2-нафтил)пропионовой кислоты дигидрохлорида растворяли в 1000 мл воды. При перемешивании, полученный таким образом раствор доводили до значения pH 4,8, постепенно добавляя высокоосновную ионно-обменную смолу OH-типа (Amberlite IA-410). После этого смолу удаляли путем фильтрации, а полученный фильтрат концентрировали досуха. Таким образом полученный остаток (94,6 г) растворяли в 142 мл воды, и раствор смешивали с 1570 мл этанола, и перемешивали при комнатной температуре в течение 1 часа. После удаления кристаллов путем фильтрации, в полученный маточный раствор вносили затравочные кристаллы и перемешивали при 8oC в течение 40 часов. После этого, осажденные таким образом кристаллы собирали путем вакуумной фильтрации, промывали этанолом, а затем высушивали воздухом в течение 6,5 часов при нормальном давлении и относительной влажности от 60 до 70%. В результате этой процедуры, получали 70,3 г целевого соединения в виде бесцветных призмообразных кристаллов.
[α]
1H-ЯМР (ДМСО-d6) δ: 2,20 - 2,35 (2H, м); 2,29 (1,5H, с); 2,32 (1,5H, с); 2,80 - 2,95 (1H, м); 3,30 - 4,00 (6H, м); 5,16 (0,5H, шир. с); 5,22 (0,5H, шир. с); 6,90 - 7,00 (2H, м); 7,45 - 7,51 (2H, м); 7,57 (1H, д, J = 8,3 Гц); 7,66 (1H, д, J = 8,0 Гц); 7,93 (1H, д, J = 8,3 Гц); 7,97 (1H, д, J = 8,3 Гц); 8,11 (1H, с); 8,68 (1H, шир. с); 8,70 - 9,30 (шир. с.); 11,50 - 12,20 (шир. с).
Элементарный анализ для C26H28N4O3 • HCl • 5H2O:
Вычислено: C 54,68; H 6,88; N 9,80; Cl 6,21
Найдено: C 54,77; H 6,76; N 9,68; Cl 6,42
Результаты кристаллографического рентгеновского анализа подтвердили, что полученное целевое соединение является (2S)-2-[4-[((3S)-1-ацетимидоил-3-пирролидинил)окси]фенил]-3-(7- амидино-2-нафтил)пропионовой кислотой.
Иллюстративный пример 46. Метил (+)-3-(6-амидино-2-индолил)-2-[4-[((3S)-3-пирролидинил)окси]фенил] пропионата дигидрохлорид.
При охлаждении льдом, хлороводород барботировали в смеси растворителей, состоящей из 10 мл дихлорметана и 20 мл метанола. К насыщенному таким образом раствору добавляли 10 мл раствора дихлорметана, содержащего 450 мг (+)-((2S)-1-р-толуолсульфонил-2-пирролидинил)метил 2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси] фенил]- 3-(6-циано-2-индолил)-пропионата. Полученную таким образом смесь оставляли еще на 72 часа при 5oC. После концентрирования досуха при пониженном давлении при температуре от 40oC или ниже, полученный остаток растворяли в 20 мл растворе этанола, содержащем 14% (м. /об. ) аммиака, и перемешивали при комнатной температуре в течение 24 часов. После отгонки растворителя, полученный таким образом остаток подвергали обращенно-фазовой ЖХВР с использованием колонки, заполненной октадецилсвязанным силикагелем и элюируя смесью воды и ацетонитрила. После этого элюированные фракции собирали, смешивали с разбавленной соляной кислотой и затем концентрировали досуха. В результате этой процедуры получали целевое соединение (95 мг) в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,30 (2H, м); 3,30 (1H, дд); 3,50 - 3,60 (5H, м); 3,70 (3H, с); 4,20 (1H, т); 5,20 (1H, м); 6,33 (1H, с); 6,96 (2H, д); 7,33 (2H, д); 7,40 (1H, д); 7,64 (1H, д); 7,80 (1H, с); 9,30 - 9,80 (6H, м).
Иллюстративный пример 47. Метил (-)-3-(6-амидино-2-индолил)-2- [4-[((3S)-3-пирролидинил)окси]фенил]пропионата дигидрохлорид.
Это соединение получали в соответствии с процедурой, описанной в иллюстративном примере 46 (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,30 (2H, м); 3,30 (1H, дд); 3,50 - 3,60 (5H, м); 3,70 (3H, с); 4,20 (1H, т); 5,20 (1H, м); 6,33 (1H, с); 6,96 (2H, д); 7,33 (2H, д); 7,40 (1H, д); 7,64 (1H, д); 7,80 (1H, с); 9,30 - 9,80 (6H, м).
Иллюстративный пример 48. (+)-3-(6-Амидино-2-индолил)-2- [4-[((3S)-3-пирролидинил)-окси]фенил]пропионовой кислоты дигидрохлорид.
1,8 г метил (+)-3-(6-амидино-2-индолил)-2-[4-[((3S)-3- пирролидинил)окси] фенил] пропионата дигидрохлорида растворяли в 60 мл концентрированной соляной кислоты, и раствор перемешивали при 5oC в течение 7 дней. После концентрирования реакционного раствора досуха, при пониженном давлении при температуре от 50oC или ниже, полученный остаток подвергали обращенно-фазовой ЖХВР с использованием колонки, заполненной октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого, элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой, а затем концентрировали досуха. В результате описанной процедуры, получали 1,3 г целевого соединения в виде твердого вещества.
ИК (KBr): 3600 - 3300, 1730, 1680 см-1
Иллюстративный пример 49. (-)-3-(6-Амидино-2-индолил)-2-[4-[((3S)-3-пирролидинил)окси] фенил]пропионовой кислоты дигидрохлорид.
Это соединение получали в соответствии с процедурой, описанной в иллюстративном примере 48 (твердое вещество)
ИК (KBr): 3600 - 3300, 1730, 1680 см-1.
Иллюстративный пример 50. 3-(5-Амидино-2-бензофуранил)-2-[4-[((3S)-1-метил-3-пирролидинил)окси]фенил] пропионовой кислоты дигидрохлорид.
1,0 г Этил 3-(5-циано-2-бензофуранил)-2-[4-[((3S)-1-метил-3-пирролидинил)окси]фенил] пропионата растворяли в 70 мл эталона. Размешивая и охлаждая льдом, в полученный раствор барботировали хлороводород до уровня насыщения. Затем насыщенный раствор оставляли на 20 часов при 25oC. После отгонки растворителя, остаток растворяли в 50 мл этанола, содержащего 14% масс./об. аммиака, и полученный раствор оставляли еще на 20 часов при 25oC. Затем, растворитель удаляли путем дистилляции и получали в результате этил 3-(5-амидино-2-бензофуранил)-2[4-[((3S)-1-метил-3-пирролидинил)окси]фенил] пропионат-дигидрохлорид. Полученное сложноэфирное соединение растворяли в 50 мл 2 н. соляной кислоты, и нагревали с обратным холодильником в течение 30 минут. После отгонки растворителя при пониженном давлении, полученный остаток подвергали колоночной хроматографии, используя колонку, заполненную синтетическим адсорбентом типа высокопористого полимера (полимер стирола и дивинилбензола, Diaion HP-20), и элюируя смесью воды и ацетонитрила. Нужные фракции объединяли и подвергали обращенно-фазовой ЖХВР, используя колонку, заполненную октадецилсвязанным силикагелем, и элюируя смесью воды и ацетонитрила. После этого элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 200 мг целевого соединения в виде твердого вещества.
1 H-ЯМР (DMCO-d6) δ: 2,40-3,40 (6H,м); 2,92 (3H, м); 5,10-5,40 (1H, шир. с): 6,82 (1H, с); 7,01 (2H, д, J= 8,4 Гц); 7,43 (2H, д, J = 8,4 Гц): 7,82 (2H, с); 8,17 (1H, с); 9,34 (2H, шир, с); 9,23 (2H, шир. с).
Соединения, представленные иллюстративными примерами 51-82, были получены в соответствии с процедурой, описанной в иллюстративном примере 50.
Иллюстративный пример 51. 2-[4-[((3S)-1-Ацетил-3-пирролидинил)окси]фенил]-3-(5-амидино-2-бензофуранил) пропионовой кислоты дигидрохлорид (твердое вещество)
1H-ЯМР (DMCO-d6) δ: 1,90-2,38 (5H, м); 3,00-3,90 (6H, м); 4,06 (1H, т, J = 7,2 Гц); 4,88 (1H, шир. с), 6,67 (1H, с); 6,87 (2H, д, J = 8,3 Гц); 7,29 (2H, д, J = 8,3 Гц); 7,70 (2H, с); 8,08 (1H, с); 9,20 (2H, шир, с); 9,41 (2H, шир, с.).
Иллюстративный пример 52. 3-(5-Амидино-2-бензофуранил)-2-[4-[((3S)-1-диметилкарбамоил-3-пирролидинил) окси]фенил]пропионовой кислоты гидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 1,84-2,20 (2H, м); 2,75 (6H, с); 3,00-3,90 (6H, м); 4,09 (1H, т, J= 7,2 Гц); 4,87-5,10 (1H, шир.); 6,68 (1H, с); 6,87 (2H, д, J= 8,75 Гц); 7,29 (2H, д, J = 8,75 Гц); 7,70 (2H, с); 8,07 (1H, с); 9,23 (2H, шир, с); 9,39 (2H, шир, с.).
Иллюстративный пример 53. 3-(5-Амидино-2-бензофуранил)-2-[4-[((2S)-2-пирролидинил-метокси] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,95 (4H, шир, с); 6,71 (1H, с); 6,97 (2H, д); 7,27 (2H, д); 7,71 (2H, с); 8,06 (1H, с); 9,15-9,35 (5H); 9,7 (1H).
Иллюстративный пример 54. 3-(5-Амидино-2-бензофуранил)-2-[4-[(тетрагидро-3-фуранил)-окси]фенил] пропионовой кислоты гидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,6-2,4 (2H, м); 3,0-3,9 (6H, м); 4,0 (1H, дд); 4,8-5,1 (1H, м); 6,75 (1H, с); 6,9 (2H, д); 7,32 (2H); 7,77 (2H, с); 8,1 (1H, с); 9,37 (4H, д).
Иллюстративный пример 55. 3-(5-Амидино-3-метил-2-бензофуранил)-2-[4-[((3S)-3-пирролидинил)окси]фенил] пропионовой кислоты дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 2,06(5H, м); 5,05 (1H, шир.); 6,94 (2H, д); 7,22 (2H, д); 7,70 (2H, с); 8,08 (1H, с); 9,10-9,50 (5H, м).
Иллюстративный пример 56. 3-(5-Амидино-7-метокси-2-бензофуранил)-2-[4-[((3S)-3-пирролидинил)окси]фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90-2,40 (2H, м); 2,90-3,80 (6H, м); 4,03 (3H, с); 5,00-5,20 (1H, шир.); 6,65 (1H, с); 6,91 (2H, д, J = 8,3 Гц); 7,31 (2H, д, J = 8,3 Гц); 7,32 (1H, с); 7,68 (1H, с); 9,16 (2H, шир.); 9,40 (2H, шир); 9,20-10,0 (2H, шир).
Иллюстративный пример 57. 3-(5-Амидино-3-бензофуранил)-2-[4-[((3S)-3-пирролидинил)-окси] -фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,1 (2H, шир.); 3,00-4,00 (7H, м)ж 5,08 (1H, шир); 6,90 (2H, д, J = 8 Гц); 7,30 (2H, д, J = 8 Гц); 7,77 (3H); 8,22 (1H, с); 9,0-10,00 (6H).
Иллюстративный пример 58. 5-Амидино-2-[2-[4-[((3S)-3-пирролидинил)окси] фенил] этил] -3- бензофуранкарбоновой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,07 (2H, м); 3,00-3,50 (8H), 5,05 (1H, шир.); 6,85 (2H, ш, J = 8,0 Гц); 7,15 (1H, д, J = 8,0 Гц); 7,82 (2H, с); 8,35 (1H, с); 9,30 (1H, с); 9,50 (2H, шир.).
Иллюстративный пример 59. 3-[2-[2-(5-Амидинобензо[b] тиен-2-ил этил]-4-этокси-5-[((3S)-3-пирролидинил)окси] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,30 (3H, т, J = 7,0 Гц); 2,00-3,90 (14H, м); 4,01 (2H, кв. , J = 7,0 Гц); 6,94 (1H, с); 6,96 (1H, с); 7,39 (1H, с); 7,79 (1H, д, J = 9,0 Гц); 8,20 (1H, д, J = 9,0 Гц); 8,37 (1H, с); 9,41 (2H, шир.); 9,59 (2H, шир.); 9,0 - 10,0 (2H, шир.).
Иллюстративный пример 60. 3-[2-[2-(5-Амидинобензо[b]тиен-2-ил)этил]-5-[((3S)-3-пирролидинил)окси]фенил] пропионовой кислоты дигидрохлорид (твердое вещество),
1H-ЯМР (ДМСО-d6) δ: 2,00 - 4,80 (14H, м); 5,08 (1H, м); 6,77 (1H, д, J = 8,5 Гц); 6,82 (1H, с); 7,18 (1H, д, J = 8,5 Гц); 7,35 (1H, с); 7,72 (1H, д, J = 8,7 Гц); 8,16 (1H, д, J = 8,7 Гц); 8,23 (1H, с); 9,31 (2H, шир.); 9,51 (2H, шир.); 9,3-9,8 (2H, шир.).
Иллюстративный пример 61. 4-(5-Амидинобензо[b]тиен-2-ил)-3-[4-[((3S)-3-пирролидинил)-окси] фенил] масляной кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00-2,30 (2H, м); 5,00-5,20 (1H, м); 6,85 (2H, д); 7,20 (1H, с); 7,25 (2H, д); 7,65 (1H, дд); 8,05 - 8,25 (2H, м);
Иллюстративный пример 62. 3-(5-Амидинобензо[b]тиен-2-ил)2-[4-[(2-этоксикарбонилимино)- гексагидропиримидин-5-ил] окси] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,27 (3H, т, J=7,0 Гц); 3,00-4,04 (шир.); 4,24 (2H, кв. , J = 7,0 Гц); 4,90-5,10 (1H, шир); 6,99 (2H, д, J = 8,3 Гц); 7,34 (2H, д, J = 8,3 Гц); 7,39 (1H, с); 7,68 (1H, дд, J= 9,0 и 1,8 Гц); 8,10 (1H, д, J = 9,0 Гц); 8,24 (1H, д, J= 1,8 Гц); 8,98 (2H, шир.); 9,23 (2H, шир.); 9,44 (2H, шир.); 11,65 (1H, с).
Иллюстративный пример 63. 3-(5-Амидинобензо[b] тиен-2-ил)-2-[4-[(2-(имино-)гексагидропиримидин-5-ил] оксо] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 3,20 - 4,20 (3H, м); 3,44 (2H); 4,80-5,00 (1H, шир. ); 6,98 (2H, д, J = 8,31 Гц); 7,17 (2H, с); 7,29 (1H, с); 7,34 (2H, д, J= 8,31 Гц); 7,70 (1H, дд, J = 8,2 и 2,0 Гц); 8,06 (2H, с); 8,12 (1H, д, J = 8,2 Гц); 8,25 (1H, с); 9,46 (2H, шир.); 9,57 (2H, шир.).
Иллюстративный пример 64. 3-(5-Амидинобензо[b]тиен-2-ил)-2- [4-[((2S)-2-пирролидинилметокси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,95 (4H, м); 3,00 - 4,20 (8H, м); 6,95 (2H, д, J= 8,0 Гц); 7,28 (3H); 7,70 (1H, д, J=8,0 Гц); 8,06 (1H, д, J=8,0 Гц); 8,23 (1H, с); 9,20 - 9,50 (6H).
Иллюстративный пример 65. 3-(5-Амидинобензо[b]тиен-2-ил)-2-[4- [(4-пиперидинил)окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (CDCl3) δ: 1,80 - 2,15 (4H, м); 3,00 - 3,25 (4H, м); 3,30 - 4,00 (3H, м); 4,60 - 4,70 (1H, м); 6,97 (2H, д, J=8,3 Гц); 7,31 (3H, м); 7,69 (1H, дд); 8,13 (1H, д, J=8,8 Гц); 8,26 (1H, с); 9,31 (2H, шир.); 9,50 (2H, шир.); 9,00 - 10,00 (2H, шир.).
Иллюстративный пример 66. 3-(5-Амидинобензо[b]тиен-2-ил)-2- [4-[(2-аминоэтил)-окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 3,00 - 4,40 (7H, м); 6,93 (2H, д, J=8,3 Гц); 7,29 (1H, с); 7,32 (2H, д, J=8,3 Гц); 7,67 (1H, дд, J=9,0 и 1,0 Гц); 8,20 (1H, д, J= 9,0 Гц); 8,32 (1H, с); 8,10 - 8,60 (3H, шир.); 9,24 (2H, шир.); 9,46 (2H, шир.).
Иллюстративный пример 67. 3-(5-Амидинобензо[b]тиен-2-ил) -2-[4-(2-(1-пирролин-2-ил)-аминоэтокси] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,88 - 2,30 (2H, м); 2,60 - 3,00 (2H, м); 3,00 - 4,30 (9H, м); 6,90 (2H, д, J=8,3 Гц); 7,30 (1H, с); 7,31 (2H, д, J=8,3 Гц); 7,70 (1H, дд, J=8,50 и 1,00 Гц); 8,11 (1H, д, J=8,50 Гц); 8,25 (1H, с); 9,28 (2H, шир.); 9,48 (2H, шир.); 10,00 (1H, шир.); 10,19 (1H, шир.).
Иллюстративный пример 68. 3-(5-Амидино-2-индолил)-2-[4-[((3S) -3-пирролидинил)окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,35 (2H, м); 4,00 - 4,30 (1H, м); 5,00 - 5,30 (1H, шир. ); 6,37 (1H, с); 7,00 (2H, д); 7,40 (2H, д); 7,60 (2H, д); 8,10 (1H, с); 11,60 (1H, с).
Иллюстративный пример 69. 3-(6-Амидино-2-индолил)-2-[4-[((3S) -3-пирролидинил)окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,20 (2H, м); 3,40 (4H, м); 5,16 (1H, шир.); 6,36 (1H, с); 7,00 (2H, д); 7,27 (2H, д); 7,36 - 7,96 (3H, м); 9,20 - 9,50 (6H, м); 11,80 (1H, с).
Иллюстративный пример 70. 3-(5-Амидино-2-индолил)-2-[4-[((3R) тетрагидро -3-фуранил]окси]фенил]пропионовой кислоты гидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 5,00 (1H, шир.); 6,28 (1H, с); 6,82 (2H, д); 7,30 (2H, д); 7,58 (2H, д); 8,00 (1H, с); 9,10 (4H); 11,8 (1H, с).
Иллюстративный пример 71. 3-(5-Амидино-2-индолил)-2-[4- [((3S)-тетрагидро-3-фуранил] -окси] фенил] пропионовой кислоты гидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 5,10 (1H, шир.); 6,27 (2H, с); 6,82 (2H, д); 7,29 (2H, д); 7,58 (2H, с); 8,00 (1H, с); 9,12 (4H); 11,8 (1H, с).
Иллюстративный пример 72. 3-(5-Амидино-1-метил-2-индолил)-2-[4- [((3S)-3-пирролидинил)-окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90 - 2,25 (2H, м); 3,73 (3H, с); 5,00 - 5,20 (1H, шир.); 6,40 (1H, д); 6,95 (2H, с); 7,40 (2H, д); 7,62 (2H, с); 8,10 (1H, с); 9,00 - 9,80 (6H, шир.).
Иллюстративный пример 73. 3-(6-Амидино-1-этил-2-индолил)-2-[4- [((3S)-3-пирролидинил)-окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,30 (3H, т); 1,95 - 2,30 (2H, м); 5,10 (1H, м); 6,37 (1H, с); 6,92 (2H, д); 7,30 - 7,70 (4H, м); 8,10 (1H, с); 9,30 - 9,90 (6H, шир.).
Иллюстративный пример 74. 3-(6-Амино-1-этил-2-индолил)-2-[4- [((3S)-3-пирролидинил)-окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,31 (3H, т); 2,00 - 4,50 (11H); 5,11 (1H, шир.); 6,38 (1H, с); 6,96 (2H, д, J=8,4 Гц); 7,30 - 7,70 (4H, м); 8,17 (1H, с); 9,07 (2H, шир.); 9,34 (2H, шир.); 9,30 - 10,00 (2H, шир.).
Иллюстративный пример 75. 3-[6-Амидино-1-(2-хлороэтил)-2- индолил]-2-[4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 - 5,00 (13H); 5,13 (1H, шир.); 6,42 (1H, с); 6,97 (2H, д, J=8,4 Гц); 7,50 - 7,70 (2H, м); 8,22 (1H, с); 9,13 (2H, шир.); 9,39 (2H, шир.); 9,50 - 10,00 (2H, шир.).
Иллюстративный пример 76. 3-[6-Амидино-1,2,3,4-тетрагидро-2- нафтил]-2-[4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,30 - 4,00 (16H, м); 5,10 (1H, м); 6,94 (2H, д, J= 9,0 Гц); 7,20 - 7,70 (5H, м); 9,18 (2H, шир.); 9,34 (2H, шир.); 9,50 - 10,00 (2H, шир.).
Иллюстративный пример 77. 3-[5-Амидино-2-бензимидазолил-2- [4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,98 - 2,28 (2H, шир.); 3,00 - 4,80 (7H, м); 5,00 - 5,20 (1H, шир.); 6,93 (2H, д, J=9,0 Гц); 7,34 (2H, д, J=9,0 Гц); 7,91 (2H, с); 8,28 (1H, с); 9,36 (2H, шир.); 9,61 (2H, шир.); 9,40 - 10,10 (2H, шир.).
Иллюстративный пример 78. 3-(7-Амидино-2-нафтил)-2-[4-[(3R)-3- пирролидинил)окси]фенил]- пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,20 (2H, м); 3,00 - 4,20 (7H, м); 5,10 (1H, шир. ); 6,92 (2H, д, J=9,0 Гц); 7,33 (2H, д, J=9,0 Гц); 7,50 - 8,20 (5H, м); 8,43 (1H, с); 9,39 (2H, шир.); 9,60 (2H, шир.); 9,50 - 10,00 (2H, шир.).
Иллюстративный пример 79. 3-(7-Амидино-2-нафтил)-2-[4-[(4- пиперидинил)окси]фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,70 - 2,20 (4H, м); 2,80 - 4,10 (7H, м); 4,50 - 4,80 (1H, м); 6,95 (2H, д, J=8,0 Гц); 7,30 (2H, д, J=8,0 Гц); 7,60 - 8,50 (6H, м); 9,35 (2H, шир.); 9,57 (2H, шир.); 9,10 - 9,80 (2H, шир.).
Иллюстративный пример 80. 3-(6-Амидино-1-карбоксиметил-2-индолил)-2-[4-[((3S)-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,93 - 2,30 (2H, м); 2,80 - 3,80 (6H, м); 3,90 - 4,25 (1H, т); 5,00 - 5,50 (3H, шир. с); 6,41 (1H, с); 7,00 (2H, д); 7,42 (2H, д); 7,60 - 7,90 (2H, м); 8,30 (1H, с); 9,10 - 10,00 (6H, шир.).
Иллюстративный пример 81. 6-Амидино-2-[3-гидрокси-2- [4-[((3S)-3-пирролидинил)окси] фенил]пропил]-1-индолуксусной кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,95 - 2,20 (2H, м); 4,90 - 5,15 (3H, м); 6,20 (1H, с); 6,90 (2H, д); 7,25 (2H, д); 7,57 (2H, м); 8,20 (1H, с); 9,20 - 9,90 (6H, шир.)
Иллюстративный пример 82. 6-Амидино-2-[2-[4-[((3S)-3-пирролидинил) окси] фенил]этил]-1-индолоуксусной кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,90-2,30 (2H, м); 3,10 - 3,50 (4H, м); 4,80-5,30 (3H, шир. ); 6,42 (1H, с); 6,90 (2H, д); 7,25 (2H, д); 7,60 (3H, м); 8,25 (1H, с); 9,20-10,00 (6H, шир.).
Иллюстративный пример 83. Этил 3-(5-амидинобензо[b]тиен-2-ил)-2-этоксикарбонил-2-[4-[((2R)-2-пирролидинил) метокси] фенил] пропионата дигидрохлорид.
4,34 г этил 2-[4-[((2R)-1-трет-бутоксикарбонил-2-пирролидинил)метокси] фенил] -3-(5- цианобензо [b]тиен-2-ил)-этоксикарбонилпропионата растворяли в 150 мл этанола. Хлороводород барботировали в полученный раствор при охлаждении льдом и перемешивании до насыщенного уровня. Насыщенный раствор оставляли при комнатной температуре в течение 18 часов. После отгонки растворителя при пониженном давлении, остаток растворяли в 100 мл этанолового раствора, содержащего 13% (м./об.) аммиака, и полученный раствор оставляли еще на 24 часа. После отгонки растворителя, полученный остаток хроматографировали на колонке с синтетическим адсорбентом типа высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20) и элюируя смесью воды и ацетонитрила. Объединенные нужные фракции подвергали обращено-фазовой ЖХВР на колонке с октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и затем концентрировали досуха. В результате описанной процедуры получали 1,0 г целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,15 (6H, т, J = 7,0 Гц); 2,0 (4H, шир.); 3,00-4,00 (3H); 3,95 (2H); 4,2 (4H); 7,00 (2H, д); 7,16 (1H); 7,31 (2H, д); 7,70 (2H, дд); 8,10 (1H, д); 8,26 (1H, д); 9,20-9,60 (5H); 9,9 (1H).
Иллюстративный пример 84. 3-5-Амидинобензо [b]тиен-2-ил-2-[4-[(2-имидазолин-2-ил)метокси]фенил]пропионовой кислоты дигидрохлорид.
1,6 г этил 3-(5-цианобензо[b]тиен-2-ил)-2-этоксикарбонил-2-[4-[(2-имидазолин-2-ил)- метокси] фенил] пропионата растворяли в 100 мл этанола. Охлаждая льдом и перемешивая, хлороводород барботировали в полученный раствор до уровня насыщения. Насыщенный раствор оставляли на 18 часов при 5oC. После отгонки растворителя при пониженном давлении, полученный остаток растворяли в 100 мл этанолового раствора, содержащего 13% (м./об.) аммиака, и раствор отставляли при комнатной температуре еще на 24 часа. После этого растворитель отгоняли до получения этил 3-(5-аминобензо [b]тиен-2-ил)-2-этоксикарбонил-2-[4-[(2-имидазолин-2-ил)метокси]фенил] пропионата дигидрохлорида. Полученное сложноэфирное соединение растворяли в 50 мл 5 н. соляной кислоты, и раствор нагревали с обратным холодильником в течение 1 часа. После отгонки растворителя, остаток хроматографировали на колонке с синтетическим адсорбентом типа высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20) и элюируя смесью воды и ацетонитрила. Объединенные нужные фракции подвергали обращенно-фазовой ЖХВР на колонке с октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и затем концентрировали досуха. В результате описанной процедуры получали 200 мг целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 3,35 (1H, дд); 3,65 (1H, дд); 3,89 (4H, с); 3,99 (1H, т); 5,07 (2H, с); 6,98 (2H, д); 7,32 (1H, с); 7,37 (2H, д); 7,66 (1H, д); 8,12 (1H, д); 8,21 (1H, с); 9,10 (2H); 9,39 (2H); 10,38 (2H).
Последующие соединения иллюстративных примеров 85 и 86 получали в соответствии с процедурой, описанной в иллюстративном примере 82.
Иллюстративный пример 85. 3-(5-Амидинобензо[b]тиен-2-ил)-2-[4-[((3S) -3-пирролидинил)-тио] фенил]пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,5-4,5 (10H, м); 6,95 (2H, д); 7,32 (1H, с); 7,40 (2H, д); 7,71 (1H, д); 8,13 (1H, д); 8,28 (1H, с); 9,3 (2H, шир.); 9,5 (2H, шир.); 9,8 (2H, шир.).
Иллюстративный пример 86. 3-(5-Амидино-2-бензотиазолил)-2-[4-[((3S)-3-пирролидинил)-окси] -фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,08 (2H, шир.); 3,00-4,25 (7H); 5,10 (1H, шир.); 6,95 (2H, д); 7,34 (2H, д); 7,82 (1H, дд); 8,29 (1H, д); 8,41 (1H, д); 9,20 - 9,60 (6H).
FAB MC (m/Z): 411 (M++1).
Иллюстративный пример 87. 2-[4-((3S)-1-Ацетимидоил-3-пирролидинил)окси] фенил]-3- (5-амидино-2-бензофуранил)пропионовой кислоты дигидрохлорид.
1,1 г (3-(5-амидино-2-бензофуранил)-2-[4-[((3)-3-пирролидинил)окси]фенил] пропионовой кислоты дигидрохлорида растворяли в 20 мл воды. Перемешивая и охлаждая льдом, 1,4 г этилацетамида гидрохлорида постепенно добавляли к полученному раствору, при этом pH раствора доводили до значения 8,5 с помощью 1 н. водного раствора гидроксида натрия. Полученную смесь перемешивали 15 минут, охлаждая льдом, и затем доводили значение pH до 2,0 разбавленной соляной кислотой. После концентрации досуха полученного реакционного раствора остаток подвергали обращенно-фазовой ЖХВР на колонке с октадецилсвязанным силикагелем, элюируя смесью воды и ацетонитрила. После этого, элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и затем концентрировали досуха. В результате описанной процедуры получали 780 мг целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,90-2,40 (5H, м); 2,90-4,30 (7H, шир.); 4,96 (1H, шир. ); 6,73 (1H, с); 6,93 (2H, д, J = 8,8 Гц); 7,33 (2H, д, J = 8,8 Гц); 7,73 (2H, с); 8,10 (1H, с); 8,50-8,80 (1H, шир.); 9,33 (2H, шир.); 9,46 (3H, шир.).
Последующие соединения иллюстративных примеров 88-91 были получены в соответствии с процедурой, описанной в иллюстративном примере 87.
Иллюстративный пример 88. 2-[4-[((3S)-1-Ацетимидоил-3-пирролидинил)окси] фенил]-3-5-аминобензо[b]тиен-2-ил)пропионовой кислоты дигидрохлорид (твердое вещество)
1H-ЯМР (ДМСО-d6) δ: 2,00-2,50 (5H, м); 3,10-4,20 (7H, м); 4,96 (1H, шир. ); 6,93 (2H, д, J = 7,9 Гц); 7,29 (1H, с); 7,34 (2H, д, J = 7,9 Гц); 7,73 (1H, д, J = 8,3 Гц); 8,10 (1H, д, J = 8,3 Гц); 8,30 (1H, с); 8,50-9,30 (1H, шир.); 9,37 (2H, шир.); 9,54 (2H, шир.).
Иллюстративный пример 89. 2-[4[((3S)-1-Ацетимидоил-3-пирролидинил)окси] фенил] -3-(5-амидино-2 -бензотиазолил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 (2H, шир.); 2,26 (1,5H); 2,30 (1,5H); 3,00-4,25 (7H); 5,17 (1H, шир.); 6,99 (2H, д); 7,31 (2H, д); 7,88 (1H, д); 8,25 (1H, д); 8,44 (1H, д); 8,50 (1H); 9,33 (2H, шир.); 9,55 (2H, шир.).
Иллюстративный пример 90. 2-[4-[((3S)-1-Ацетимидоил-3-пирролидинил)окси] фенил] -3-(6-амидино-1-этил-2 -индолил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,31 (3H, т, J = 7,0 Гц); 2,15-2,40 (2H, м); 2,28 (1,5 H); 2,31 (1,5 H); 3,15 (1H, дд); 3,40-4,05 (5H); 4,10 (1H, т); 4,28 (2H, м); 5,16 (0,5H, шир.); 5,22 (0,5H, шир.); 6,40 (1H, с); 6,97 (2H, дд); 7,40 (2H); 7,48 (1H, д, J = 8,3 Гц); 7,62 (1H, д, J = 8,3 Гц); 8,13 (1H, с); 8,50 (0,5 H, с); 8,59 (0,5H, с); 8,98 (2H, с); 9,32 (2H, с); 9,25-9,35 (1H).
Иллюстративный пример 91. 2-[4-[((3S)-1-Ацетимидоил-3-пирролидинил)окси] фенил] -3-(7-амидино- 2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00-2,40 (5H, м); 2,90 - 4,10 (7H, м); 5,15 (1H, шир. ); 6,93 (2H, д, J = 8,0 Гц); 7,33 (2H, д, J = 8,0 Гц); 7,50 - 8,40 (6H, м); 8,50 - 8,70 (1H); 9,30 (3H, шир.); 9,55 (2H, шир.).
Иллюстративный пример 92. 2-[4-[(2R)-1-Ацетимидоил-2-пирролидинил)метокси] фенил] -3-(5-амидинобензо [b]тиен-2-ил)пропионовой кислоты гидрохлорид.
1,0 г этил 3-(5-амидинобензо[b]тиен-2-ил)-2-этоксикарбонил-2-[4-[(2R)-2-пирролидинил) метокси] фенил]пропионата дигидрохлорида растворяли в 20 мл этанола с последующим добавлением 0,42 г этилацетамидата гидрохлорида. К полученному раствору, размешивая при охлаждении льдом, добавляли 0,51 г триэтиламина, и полученную смесь нагревали до комнатной температуры и размешивали в течение 18 часов. После отгонки растворителя получали этил 2-[4-[((2R)-1-ацетимидоил-2-пирролидин) метокси] фенил]-2-этоксикарбонил-3-(5-амидино-бензо[b]тиен-2-ил) пропионата дигидрохлорид. Полученное таким образом сложноэфирное соединение растворяли в 50 мл 5 н. соляной кислоты, а затем нагревали с обратным холодильником в течение 60 минут. После отгонки растворителя, остаток хроматографировали на колонке, упакованной высокопористым полимером в качестве синтетического абсорбента (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. Нужные фракции объединяли и концентрировали, а полученный остаток подвергали обращенно-фазовой жидкой хроматографии высокого разрешения (ЖХВР) на колонке, наполненной октадецил-связанным силикагелем, и используя в качестве элюента смесь воды и ацетонитрила. Затем, нужные элюированные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 360 мг целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,00 (4H, шир.); 2,24 - 2,43 (3H); 3,00 - 4,00 (5H); 4,00 (2H); 4,50 (1H, шир.); 6,90 (2H, д); 7,30 (3H); 7,70 (1H, д); 8,10 (1H, д); 8,23 (1H, с); 9,20 - 9,60 (5H, м).
Иллюстративный пример 93. 3-(5-Амидинобензо[b]тиен-2-ил)-2-[4-[((3S)-1-бензимидоил-3- пирролидинил)окси]фенил]пропионовой кислоты дигидрохлорид.
1,0 г этил 3-(5-амидинобензо[b] тиен-2-ил)-2-[4-[((3S)- 3-пирролидинил)окси]фенил]пропионата дигидрохлорида растворяли в 15 мл этанола. К этому раствору добавляли 773 мг этил бензимидатгидрохлорида, который был получен с помощью реакции бензонитрила с этанолом в присутствии хлорводорода. К полученному раствору, размешивая при охлаждении льдом, добавляли 631 мг триэтиламина, и смесь нагревали до комнатной температуры, а затем размешивали в течение 18 часов. После отгонки растворителя, получали этил 3-(5-амидинобензо [b] тиен-2-ил)-2-[4-[((3S)-1-бензимидоил-3-пирролидинил)окси] фенил] - пропионата гидрохлорид. Полученное таким образом сложноэфирное соединение растворяли в 60 мл 3н. соляной кислоты и нагревали с обратным холодильником в течение 30 минут. После отгонки растворителя, остаток подвергали колоночной хроматографии на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. Нужные фракции объединяли и концентрировали, а полученный остаток подвергали обращенно-фазовой ЖХВР, используя колонку, наполненную октадецил-связанным силикагелем, и элюируя смесью воды и ацетонитрила. Нужные элюированные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 350 мг целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,80 (2H, м); 3,00 - 3,30 (7H, м); 4,04 (0,5H, шир.); 4,30 (0,5H, шир.); 6,80 - 7,90 (11H, м); 8,12 (1H, д, J = 8,3 Гц); 8,30 (1H, с); 9,20 - 9,70 (6H, м).
Соединения, представленные в иллюстративных примерах 94 - 100, были получены в соответствии с процедурой, описанной в иллюстративном примере 93.
Иллюстративный пример 94. 3-(5-Амидинобензо [b]тиен-2-ил)-2-[4-[((3S)-1-н-гексан-имидоил-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 0,80 - 0,95 (3H, м); 1,20 - 1,40 (4H, м); 1,45 - 1,70 (2H, м); 2,15 - 2,40 (2H, м); 2,45 - 2,60 (2H, м); 3,25 - 3,90 (6H, м); 3,96 (1H, т, J = 7,5 Гц); 6,85 - 7,00 (2H, м); 7,25 - 7,40 (3H, м); 7,69 (1H, дд, J = 8,3 и 1,5 Гц); 8,11 (1H, д, J = 8,3 Гц); 8,25 (1H, с); 8,58 (0,5 H, с); 8,66 (0,5H, с); 9,20 - 9,30 (3H, шир.); 9,47 (2H, шир.).
Иллюстративный пример 95. 3-(5-Амидинобензо [b] тиен-2-ил)-2- [4-[((3S)-1-цикло-пропанкарбоксимидоил-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 0,90 - 1,30 (3H, м); 1,80 - 4,10 (10H, м); 5,10 - 5,30 (1H, м); 6,96 (2H, д, J = 8,4 Гц); 7,32 (1H, с); 7,36 (2H, д, J = 8,4 Гц); 7,71 (1H, д, J = 7,4 Гц); 8,14 (1H, д, J = 7,4 Гц); 8,29 (1H, с); 8,40 - 8,70 (2H, м); 9,36 (2H, шир.); 9,52 (2H, шир.).
Иллюстративный пример 96. 2-[4-[((2S)-1-Ацетимидоил-2-пирролидинил)метокси] фенил] -3- (5-амидинобензо[b]тиен-2-ил) пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,05 (4H, шир.); 2,25 - 2,43 (3H), 3,00 - 4,50 (8H); 6,95 (2H); 7,30 (3H); 7,70 (1H, д); 8,10 (1H, д); 8,25 (1H, с); 8,60 (1H, с); 9,20 - 9,60 (5H, м).
Иллюстративный пример 97. 2-[4-[(1-Ацетимидоил-4-пиперидинил)окси]фенил] -3-5-амидинобензо[b] тиен-2-ил пропионовой кислоты дигидрохлорид.
1H-ЯМР (ДМСО-d6) δ: 1,65 - 2,10 (4H, м); 2,32 (3H, с); 3,20 - 4,00 (7H, м); 4,60 - 4,70 (1H, м); 6,96 (2H, д, J = 8,3 Гц); 7,30 (3H, м); 7,69 (1H, д, J = 8,3 Гц); 8,10 (1H, д, J = 8,3 Гц); 8,26 (1H, с); 8,95 (1H, с); 9,32 (2H, шир.); 9,52 (2H, шир.).
Иллюстративный пример 98. 2-[4-[(1-Ацетимидоил-4-пиперидинил)окси]фенил] -3-(6-амидино -1-этил-2-индоил)пропионовой кислоты дигидрохлорид.
1H-ЯМР (ДМСО-d6) δ: 1,30 (3H, т, J = 7,0 Гц); 1,73 - 2,10 (4H, м); 2,31 (3H, с); 3,10 (1H, м); 3,30 - 3,30 (5H); 4,05 (1H, т), 4,30 (2H, м); 4,70 (1H, шир. ); 6,38 (1H, c); 6,97 (2H, д, J = 8,5 Гц); 7,37 (2H, д, J = 8,5 Гц); 7,48 (1H, д, J = 8,3 Гц); 7,61 (1H, д, J = 8,3 Гц); 8,14 (1H, с); 8,86 (1H, шир.); 9,15 - 9,50 (5H, м).
Иллюстративный пример 99. 3-(7-Амидино-2-нафтил)-2-[4-[((3S)-1- бутанимидоил-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 0,60 - 4,00 (16H); 5,00 (1H, шир.); 6,79 (2H, д, J = 8,0 Гц); 7,21 (2H, д, J = 8,0 Гц); 7,30 - 8,10 (5H, м); 8,34 (1H, с); 8,30 (1H, с); 8,40 - 8,70 (1H); 9,00 - 10,00 (5H).
Иллюстративный пример 100. 3-(7-Амидино-2-нафтил)-2-[4-[((3S)-1- бензомидоил-3-пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,00 - 4,10 (9H); 4,90 (0,5 H, шир.); 5,20 (0,5 H, шир.); 6,70 - 8,10 (1H, м); 8,32 (1H, с); 9,10 - 9,50 (4H).
Иллюстративный пример 101. 3-(5-Амидинобензо[b] тиен-2-ил)- 2-[4-[((3S)-1-(N-метилацетимидоил)-3-пирролидинил]окси]фенил] пропионовой кислоты дигидрохлорид
2,0 г 3-(5-амидинобензо[b] тиен-2-ил)-2-4-[((3S)-3- пирролидинил)окси] фенил] пропионовой кислоты дигидрохлорид растворяли в смеси, состоящей из 10 мл воды и 10 мл ацетонитрила. К полученному раствору, размешивая, постепенно добавляли 20 г этил (N-метил)ацетимидата гидрохлорида, полученного в соответствии с процедурой, описанной в The Journal of Organic Chemistry vol. 33, pp. 1679 - 1681, 1968), поддерживая при этом pH раствора при 8,5 с помощью 2 н. водного раствора гидроксида натрия. После отгонки растворителя, остаток промывали дихлорметаном и подвергали хроматографии на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), используя в качестве элюента смесь воды и ацетонитрила, осуществляя тем самым обессоливание. Нужные фракции объединяли и подвергали обращенно-фазовой ЖХВР, используя колонку, наполненную октадецил-связанным силикагелем. Затем элюированные нужные фракции объединяли и концентрировали досуха, пропуская через сильноосновную ионообменную смолу CI-типа (Diaion SA-10, Nippon Rensui Co., Ltd.). В результате описанной процедуры получали 370 мг целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,00 - 2,44 (2H, м); 2,30 (1,5 H); 2,33 (1,5 H); 2,98 (3H); 3,06 - 4,20 (7H, м); 5,00 - 5,40 (1H, шир.); 6,92 (2H, д, J = 8,3 Гц); 7,28 (1H, с); 7,33 (2H, д, J = 8,3 Гц); 7,72 (1H, д. J = 9,0 Гц); 8,06 (1H, д, J = 9,0 Гц); 8,28 (1H, с); 8,8 - 9,20 (1H, шир.); 9,23 (2H, шир. с); 9,50 (2H, шир.).
Иллюстративный пример 102. 3-(5-Амидино-2-бензофуранил)-2- [4-[((2R)-2-амино-1-бутил)окси]фунил]пропионовой кислоты дигидрохлорид.
В 300 мл тетрагидрофурана растворяли 1,1 г этил 3-(5-циано-2-бензофуранил)-2-(4-гидроксифенил)пропионата, 1,24 г (2R)-2-трет-бутоксикарбонил-амино-1-бутанола, и 1,72 г трифенилфосфина. Полученный таким образом раствор смешивали с 1,14 г диэтилазодикарбоксилата и размешивали при комнатной температуре в течение 18 часов. Затем полученный раствор смешивали с 0,83 г (2R)-2-трет-бутоксикарбониламино-1-бутанола, 1,2 г трифенилфосфина и 0,76 г диэтилазодикарбоксилата, и полученную смесь размешивали при комнатной температуре в течение 18 часов. После концентрирования реакционного раствора досуха, остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве проявляющего растворителя смесь толуола и этилацетата, в результате чего получали 660 мг этил 2-[4-[((2R)-2-трет-бутоксикарбониламино-1-бутил)окси] фенил] -3-(5-циано-2-бензофуранил)пропионата в виде бесцветного маслянистого вещества.
Затем, 660 мг этил 2-[4-[((2R)-2-трет-бутоксикарбониламино- 1-бутил)окси] фенил]-3-(5-циано-2-бензофуранил)пропионата, полученного выше, обрабатывали и очищали в соответствии с процедурой, описанной в иллюстративном примере 50, в результате чего получали 78 мг целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,04 (3H, т); 1,70 (2H); 3,0 - 4,2 (6H); 6,41 (1H, с); 6,99 (2H, д); 7,27 (2H, д); 7,72 (2H, с); 8,07 (1H, с); 8,3 (3H, шир.); 9,34 - 9,40 (4H);
Иллюстративный пример 103. 3-(5-Амидино-2-бензофуранил)-2- [4-[((2S)-2-амино-1-бутил)окси]фенил]пропионовой кислоты дигидрохлорид.
Повторяли процедуру, описанную в иллюстративном примере 102, за исключением того, что вместо (2R)-2-трет-бутоксикарбониламино-1- бутанола использовали (2S)-2-трет-бутоксикарбониламино-1-бутанол, в результате чего получали 620 мг целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,04 (3H, т); 1,70 (2H); 3,0 - 4,2 (6H); 6,68 (1H, с); 6,96 (2H, д); 7,24 (2H, д); 7,70 (2H, с); 8,05 (1H, с); 8,4 (3H, шир.); 9,40 (4H, шир.).
Иллюстративный пример 104. 3-[4-[((3S)-1-Ацетимидоил-3- пирролидинил]окси]фенил]-4-(5-амидинобензо[b]тиен-2-ил)масляной кислоты дигидрохлорид.
1 мл тионилхлорида по капле добавляли к 50 мл этанола. К этому раствору, размешивая при комнатной температуре, добавляли 1,0 г 4-(5-амидинобензо[b] тиен-2-ил)-3-[4-[((3S)-3-пирролидинил)окси] фенил] масляной кислоты дигидрохлорида, с последующим нагреванием с обратным холодильником в течение 1 часа. После охлаждения и последующего удаления растворителя путем дистилляции, полученный реакционный раствор тщательно осушали при пониженном давлении, в результате чего получали этил 4-(5-амидинобензо[b]тиен-2-ил)-3- [4-[((3S)-3-пирролидинил)окси] фенил]бутират-дигидрохлорид. Полученное таким образом сложноэфирное соединение растворяли в 20 мл тетрагидрофурана. Размешивая при охлаждении льдом, полученный раствор смешивали с триэтиламином, а затем с 360 мг этилацетамидатгидрохлорида, и эту смесь размешивали в течение 1 часа. После отгонки растворителя, полученный остаток хроматографировали на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. Нужные фракции собирали и концентрировали досуха, а остаток растворяли в 50 мл 2 н. соляной кислоты и нагревали с обратным холодильником в течение 1 часа. После отгонки растворителя, остаток хроматографировали на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), используя в качестве элюента смесь воды и ацетонитрила. Нужные фракции объединяли и подвергали обращенно-фазовой ЖХВР на колонке, наполненной октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 850 мг целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 2,0 - 2,45 (2H, м); 2,32 (3H, д); 2,5 - 2,9 (2H, м); 3,1 - 4,0 (7H, м); 5,1 - 5,35 (1H, м); 6,92 (2H, д); 7,30 (2H, д); 7,8 (1H, д); 8,20 (1H, д); 8,37 (1H, с); 8,6 - 8,9 (1H, м); 9,30 - 9,80 (5H).
FAB -MC (m/z): 465.
Иллюстративный пример 105. 2-[4-[((3S)-1-(Ацетимидоил-3- пирролидинил] окси]фенил]-3-(6-амидино-1-этил-2-индолил)пропионовой кислоты дигидрохлорид.
Это соединение было получено в соответствии с процедурой, описанной в иллюстративном примере 104 (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,35 (3H, т); 2,37 (3H); 4,10 - 4,40 (1H, м); 6,42 (1H, с); 7,00 (2H, д); 7,45 (2H, д); 7,60 (2H, м); 8,30 (1H, с); 8,70 (1H, шир.); 9,25 - 9,80 (5H).
Иллюстративный пример 106. 3-[4-[((3S)-1-(Ацетимидоил-3- пирролидинил] окси]фенил]-2-(5-амидинобензо[b]тиен-2-ил)пропионовой кислоты дигидрохлорид.
2,0 г этил 3-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил) окси]фенил]-2-(5-цианобензо[b]тиен-2-ил)пропионата растворяли в 100 мл этанола. Размешивая при охлаждении льдом, в полученный раствор барботировали хлороводород до уровня насыщения. Полученный раствор оставляли на 18 часов при комнатной температуре. После отгонки растворителя, полученный остаток растворяли в 100 мл этанолового раствора, содержащего 13% аммиака, и этот раствор оставляли еще на 18 часов. После отгонки растворителя, остаток хроматографировали на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила, в результате чего получали 1,1 г этил 2-(5-амидинобензо[b] тиен-2-ил)-3-[4-[((3S)-3-пирролидинил)окси] фенил] пропионатгидрохлорида. 1,1 г полученного соединения растворяли в 15 мл этанола, и этот раствор смешивали с 566 мг этилацетамидгидрохлорида, а затем с 694 мг триэтиламина, после чего полученную смесь размешивали при комнатной температуре в течение 18 часов. После отгонки растворителя, полученный остаток растворяли в 50 мл 2 н. соляной кислоты, а затем нагревали с обратным холодильником в течение 30 минут. После охлаждения и последующей отгонки растворителя, полученный остаток хроматографировали на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюирую смесью воды и ацетонитрила. Собранные нужные фракции подвергали обращенно-фазовой ЖХВР, используя колонку, наполненную октадецил-связанным силикагелем, и элюируя смесью воды и ацетонитрила. Затем элюированные нужные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 490 мг целевого соединения в виде твердого продукта.
1H-ЯМР (DMCO-d6) δ: 2,1-2,4 (2H, м); 2,22 (1,5H); 2,29 (1,5H); 3,08 (1H, дд, J= 13,7 и 7,8 Гц); 3,30-4,00 (5H, м); 4,36 (1H); 5,00-5,20 (1H); 6,80-6,90 (2H, м); 7,15-7,25 (2H, м); 7,44 (1H, с); 7,72 (1H, д, J=8,3 Гц); 8,15 (1H, д, J=8,3 Гц); 8,32 (1H, с); 8,58 (0,5 H); 8,66 (0,5 H); 9,32 (2H, шир. ); 9,38 (0,5 H); 9,45 (0,5 H); 9,50 (2H, шир.).
Соединения настоящего изобретения, представленные в иллюстративных примерах 107-110, были получены в соответствии с процедурой, описанной в иллюстративном примере 106.
Иллюстративный пример 107. 2-[4-[((3R)-1-Ацетимидоил-3- пирролидинил)окси] фенил] -3-(7-амидино-2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 2,00-2,40 (5H, м); 2,90-4,10 (7H, м); 5,20 (1H, шир. ); 6,93 (2H, д, J=8,0 Гц); 7,33 (2H, д, J=8,0 Гц); 7,56 (1H, д); 7,70-8,20 (4H, м); 8,45 (1H, с); 8,50-8,80 (1H); 9,45 (3H, шир.); 9,63 (2H, шир.).
Иллюстративный пример 108. 2-[4-[(1-Ацетимидоил-4-пиперидинил)окси]фенил] -3-(7-амидино- 2-нафтил)пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 1,50-2,10 (4H, м); 2,31 (3H, с); 3,00-4,20 (7H, м); 4,60-4,80 (1H, м); 6,95 (2H, д, J=9,0 Гц); 7,31 (2H, д, J=9,0 Гц); 7,50-8,50 (6H, м); 8,93 (1H); 9,45 (3H, шир.); 9,62 (2H, шир.).
Иллюстративный пример 109. 3-(7-Амидино-2-нафтил)- 2-[4-[(1-бутанимидоил-4-пиперидинил)-окси] фенил] пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 0,96 (3H, т, J=7,0 Гц); 1,57 (2H, м); 1,71 (2H, шир. (2,03 (2H, шир.), 2,52 (2H, т); 3,18 (1H, м); 3,40-3,90 (5H); 4,00 (1H, т); 4,68 (1H, шир.); 6,96 (2H, д, J=8,0 Гц); 7,30 (2H, д, J=8,0 Гц); 7,60-8,40 (6H, м); 8,86 (1H); 9,32 (3H, шир.); 9,58 (2H, шир.).
Иллюстративный пример 110. 2-[4-[((2R, 4S)-1-Ацетимидоил-2- метил-4-пирролидинил)окси] фенил] -3-(5-амидинобензо [b] тиен-2-ил) пропионовой кислоты дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 1,85 (3H, м); 2,27 (1,5H, с); 2,37 (1,5H, с); 5,10-5,50 (1H, шир.); 7,00 (2H, д); 7,10-8,70 (6H, м); 9,10-9,60 (6H, шир.).
FAB-MC (m/z): 465 (M++1).
Иллюстративный пример 111. 3-(5-Аминобензо [b] тиен-2-ил)-2-[4-[((2S)-5-оксо-2-пирролидинил) метокси]фенил]пропионовой кислоты гидрохлорид
a) 3,2 г этил 2-[4-[((2S)-1-трет-бутоксикарбонил-5-оксо-2-пирролинидил)метокси] фенил] -3-(5-цианобензо [b] тиен-2-ил пропионата растворяли в смеси, состоящей из 50 мл этанола и 50 мл дихлорметана. Затем, через этот раствор, размешивая при охлаждении льдом, барботировали хлороводород до уровня насыщения. Полученную реакционную смесь оставляли на 48 часов при 5oC. После отгонки растворителя, остаток растворяли в 100 мл этанолового раствора, содержащего 13% (масс./об.) аммиака, и полученный раствор выдерживали при комнатной температуре 24 часа. После этого, растворитель удаляли путем дистилляции и получали этил 3-(5-амидинобензо[b]тиен-2-ил)-2-[4-[((2S)-5-оксо-2-пирролидинил) метокси]фенил]пропионатгидрохлорид. Полученное таким образом сложноэфирное соединение растворяли в смеси из 30 мл тетрагидрофурана и 30 мл воды. К полученной смеси добавляли 1,6 г 2-(трет-бутоксикарбонилимино)-2-фенилацетонитрила и 2 мл 1,8-дизабицикло[5.4.0]-7-ундецена. После 24-часового размешивания при комнатной температуре, реакционный раствор экстрагировали этилацетатом и осушали. После отгонки растворителя, остаток очищали с помощью хроматографии на колонке с силикагелем, элюируя смесью хлороформа и метанола, в результате чего получали 2,4 г этил 3-[5-(N-трет-бутоксикарбонил)аминоиминометилбензо[b] тиен-2-ил] - 2-[4-[((2S)-5-оксо-2-пирролидинил)метокси] фенил] пропионата в виде твердого вещества.
1H-ЯМР (CDCl3) δ: 1,18 (3H, т); 1,58 (9H, с); 1,80-2,50 (4H, м); 3,28 (1H, дд); 3,70 (1H, дд); 3,80-4,50 (6H, м).
b) 2,4 г этил 3-[5-(N-трет-бутоксикарбонил аминоиминометилбензо[b]тиен-2-ил-2-[4-[((2S)-5-оксо-2-пирролидинил)- метокси] фенил]пропионат, полученного в стадии a), растворяли в 30 мл тетрагидрофурана. Полученный раствор смешивали с водным раствором 200 мг гидроксида натрия, растворенного в 5 мл воды, и эту смесь размешивали в течение 72 часов. После отгонки растворителя, полученный остаток растворяли в 10 мл концентрированной соляной кислоты, и этот раствор размешивали при комнатной температуре в течение 1 часа. После отгонки растворителя, остаток подвергали хроматографии на колонке, наполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. Затем элюированные нужные фракции объединяли, смешивали с соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 1,1 г целевого соединения.
1H-ЯМР (DMCO-d6) δ: 1,70-2,30 (4H, м); 6,90 (2H, д); 7,29 (2H, д); 7,30 (1H, с).
FAB-MC (m/z): 438 (M++1).
Иллюстративный пример 112. 2-[2-[4-[(4-Имидазолил)метокси]фенил]этил]-5-бензофуран-карбоксамидина дигидрохлорид.
3,53 г части 2-[2-[4-[(1-тритил-4-имидазолил)метокси]-фенил] этил]-5-бензофуранкарбонитрила растворяли в смеси, состоящей из 150 мл этанола и 100 мл дихлорметана. Затем, размешивая при охлаждении льдом, через этот раствор барботировали газообразный хлороводород, после чего этот раствор выдерживали 24 часа при комнатной температуре. После отгонки растворителя, остаток растворяли в этанольном растворе, содержащем 15% (мас./об.) аммиака, и полученный раствор размешивали при комнатной температуре в течение 80 часов. После отгонки растворителя, остаток растворяли в смеси 100 мл муравьиной кислоты и 2 мл концентрированной соляной кислоты, и полученный раствор размешивали в течение 6 часов. После удаления муравьиной кислоты путем дистилляции, полученный остаток растворяли в горячей воде, а нерастворившиеся материалы удаляли путем фильтрации. После концентрирования полученного фильтрата досуха, остаток хроматографировали на колонке, заполненной синтетическим адсорбентом в виде высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. Нужные фракции объединяли и концентрировали, а остаток подвергали обращенной фазовой ЖХВР на колонке, заполненной октадецил-связанным силикагелем, элюируя смесью воды и ацетонитрила. После этого нужные элюированные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 730 мг целевого соединения в виде светло-желтого твердого вещества.
1H-ЯМР (DMCO-d6) δ: 2,95 (4H, м); 5,12 (2H, с); 6,72 (1H, с); 6,94 (2H, д, J=8,7 Гц); 7,20 (2H, д, J=8,7 Гц); 7,72 (2H, с); 7,75 (1H, с); 8,08 (1H, с); 9,13 (3H, шир.); 9,38 (2H, шир.).
Иллюстративный пример 113. 2-[2-[4-[((3S)-3-Пирролидинил)окси] фенил] этил]-6-индолкарбоксамидина дигидрохлорид
650 мг 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил] -6-индолкарбонитрила растворяли в смеси, состоящей из 100 мл этанола и 30 мл дихлорметана. Размешивая при охлаждении льдом, в полученный раствор барботировали хлороводород до уровня насыщения. Полученный раствор оставляли на 24 часа при комнатной температуре. После отгонки растворителя, остаток растворяли в этаноловом растворе, содержащем 11% (мас./об.) аммиака, и полученный раствор размешивали 24 часа при комнатной температуре. После отгонки растворителя, остаток подвергали обращенно-фазовой ЖХВР, используя колонку, заполненную октадецил-связанным силикагелем, и элюируя смесью воды и ацетонитрила. После этого, нужные элюированные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 90 мг целевого соединения в виде кристаллов. Т.пл. 229-233oC.
1H-ЯМР (DMCO-d6) δ: 1,95-2,35 (2H, м); 5,00-5,30 (1H, шир.); 6,36 (1H, с); 6,80-7,80 (7H, м); 8,00 (1H, с); 9,30-9,60 (6H, шир.).
Соединения, представленные в проиллюстрированных примерах 114-121, были получены в соответствии с процедурой, описанной в иллюстрированном примере 113.
Иллюстрированный пример 114. 2-[3-Гидрокси-2-[4-[((3S)-3-пирролидинил)окси]фенил]пропил-6- индолкарбоксамида дигидрохлорид (твердое вещество).
1H-ЯМР (DMCO-d6) δ: 1,97-2,30 (2H, м); 2,90-4,60 (9H, м); 5,00-5,20 (1H, шир. с); 6,22 (1H, с); 6,90 (2H, д); 7,18-7,70 (2H, м); 7,96 (1H, с); 9,10-9,90 (6H, шир. с); 11,05 (1H, с).
Иллюстративный пример 115. 2-[2-[4-[[(2-Пиразинил)амино]- карбонил]фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 3,20 (4H, с); 6,78 (1H, с); 7,08 (1H, шир. с); 7,48 (2H, д, J = 7,9 Гц); 7,80 (2H, с); 8,03 (2H, д, J = 7,9 Гц); 8,12 (1H, с); 8,40-8,60 (2H, д); 9,25 (2H, шир.с); 9,39 (3H, шир. с); 11,05 (1H, с).
Иллюстративный пример 116. 2-[2-[4-[(1-Имидазолил)метил]- фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид (твердое вещество )
1H-ЯМР (ДМСО-d6) δ: 3,10 (4H, с); 5,45 (2H, с); 6,75 (1H, с); 7,24 (2H, д, J = 8,3 Гц); 7,40 (2H, д, J = 8,3 Гц); 7,66 (1H, с); 7,72 (2H, с); 7,80 (1H, с); 8,12 (1H, с); 9,30 (2H, шир. с.); 9,45 (3H, шир. с).
Иллюстративный пример 117. 2-[2-[4-[(4-Метил-1-пиперазинил)- карбонил] фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид.
1H-ЯМР (ДМСО-d6) δ: 2,80 (3H, с); 4,09 (4H, с); 3,10 (4H, шир. с); 4,00 (4H, шир. с); 6,74 (1H, с); 7,36 (4H, с); 7,74 (2H, с); 8,12 (1H, с); 9,28 (2H, шир. с); 9,48 (2H, шир. с).
Иллюстративный пример 118. 3-[3-[4-[((3S)-3-Пирролидинил)окси]- фенил] пропил]-5-бензофуранкарбоксамидина дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 2,10 (4H, м); 2,70 (4H, м); 3,30 (4H); 5,07 (1H, шир. с); 6,90 (2H, д); 7,15 (2H, д); 7,79 (2H, с); 8,00 (1H, с); 8,23 (1H, с); 9,20-9,80 (6H, шир.).
Иллюстративный пример 119. 2-[[4-[(4-Пиперадинил)окси]фенил]- метил]-5-бензофуранкарбоксамидина дигидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 1,70-2,20 (2H, м); 2,70-3,30 (4H, м); 4,14 (2H, с); 4,60-4,80 (1H, м); 6,79 (1H, с); 6,97 (2H, д, J = 9,0 Гц); 7,26 (2H, д, J = 9,0 Гц); 7,74 (2H, с); 8,13 (1H, с); 9,30 (2H, шир.); 9,44 (2H, шир); 9,00-9,60 (2H, шир.).
Иллюстративный пример 120. 2-[2-[4-[(3S)-1-Ацетил-3- пирролидинил)окси] -3-гидроксифенил]-этил]-5-бензофуранкарбоксамидина гидрохлорид.
Т.пл. 175-176oC
1H-ЯМР (ДМСО-d6) δ: 1,80-2,20 (5H, м); 2,70-3,80 (8H, м); 4,88 (1H, шир. ); 6,60 (1H, д, J = 8,3 Гц); 6,77 (1H, с); 6,82 (1H, д, J = 8,3 Гц); 7,72 (1H, д, J = 8,1 Гц); 7,80 (1H, д, J = 8, 1 Гц); 8,08 (1H, с); 8,90-8,98 (1H); 9,23 (2H, шир.); 9,40 (2H, шир.).
Иллюстративный пример 121. 2-[2-[4-(N-Ацетил)аминометилциклогексил метокси фенил этил-5- бензофуранкарбоксамидина гидрохлорид (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 0,70-2,00 (10H, м); 1,83 (3H, с); 2,75-3,20 (6H, м); 3,70 (2H, д, J = 5,7 Гц); 6,64 (1H, с); 6,77 (2H, д, J = 8,8 Гц); 7,15 (2H, д, J = 8,8 Гц); 7,65-7,68 (3H); 8,04 (1H, с); 9,00 (2H, шир.); 9,31 (2H, шир.).
Иллюстративный пример 122. 2-[2-[4-[((3S)-1-Формимидоил-3- пирролидинил)-окси]фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид.
a) Повторяли процедуру иллюстративного примера 100, и получали 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]фенил]-этил]-5-бензофуранкарбонитрил
1H-ЯМР (CDCl3) δ: 1,66 (9H, с); 3,04 (4H, с); 3,30-3,70 (4H, шир.); 4,85 (1H); 6,40 (1H, с); 6,80 (2H, д); 7,12 (2H, д); 7,52 (2H, с); 7,82 (1H, с).
b) 1,66 г 2-[2-[4-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)- окси] фенил] этил]-5-бензофуранкарбонитрила, полученного в стадии a), обрабатывали и очищали в соответствии с процедурой, описанной в иллюстративном примере 113, в результате чего получали 800 мг 2-[2-[4-[((3S)-3-пирролидинил)окси] фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорида.
1H-ЯМР (ДМСО-d6) δ: 1,90-2,30 (2H, м); 3,06 (4H, шир.); 3,00-3,80 (4H, шир. ); 5,08 (1H, шир.); 6,73 (1H, с); 6,88 (2H, д); 7,19 (2H, д); 7,74 (2H, с); 8,11 (1H, с); 9,26 (2H, шир.); 9,47 (2H, шир.).
c) 1,0 г 2-[2-[4-[((3S)-3-пирролидинил)окси]фенил]этил]-5- бензофуранкарбоксамидингидрохлорида, полученного в стадии b), растворяли в 15 мл воды. Размешивая и охлаждая льдом, к полученному раствору добавляли 1,83 г бензилметанимидата
гидрохлорида, поддерживая при этом pH реакционного раствора при значении 8 с помощью 1 н. водного раствора гидроксида натрия. Полученную смесь размешивали 20 минут на ледяной бане. Затем pH реакционного раствора доводили до 2,0 с помощью разбавленной соляной кислоты, промывали диэтиловым эфиром и концентрировали досуха. Полученный остаток подвергали хроматографии на колонке, наполненной синтетическим адсорбентом типа высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. В результате описанной процедуры получали целевое соединение в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,80-2,60 (2H, м); 3,08 (4H, шир.); 3,20-4,00 (4H, шир. ); 5,14 (1H, шир.); 6,80 (1H, с); 6,92 (2H, д); 7,25 (2H, д); 7,76 (1H, д); 7,86 (1H, д); 8,21 (1H, с); 8,40 (1H, шир.); 9,08 (1H, шир.); 9,18 (2H, шир.); 9,57 (3H, шир.).
Иллюстративный пример 123. 2-[2-[3-Гидрокси-4-[((3S)-3- пирролидинил)окси]фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид.
1,0 г 2-[2-[4-[((3S)-1-ацетил-3-пирролидинил)окси]-3- гидроксифенил]-5-бензофуранкарбоксамидина гидрохлорида растворяли в 30 мл 6 н. соляной кислоты, и полученный раствор нагревали с обратным холодильником в течение 4 часов. После отгонки растворителя, полученный остаток подвергали колоночной хроматографии на колонке с синтетическим адсорбентом типа высокопористого полимера (полимер стирола и дивинилбензола: Diaion HP-20), элюируя смесью воды и ацетонитрила. В результате описанной процедуры получали 320 мг целевого соединения в виде твердого продукта.
1H-ЯМР (ДМСО-d6) δ: 1,90-2,20 (2H, м); 2,70-3,50 (8H, м); 5,08 (1H, шир. ); 6,66 (1H, дд, J = 9,0 и 1,8 Гц); 6,80 (2H, с); 6,94 (1H, с); 7,76 (2H, с); 8,12 (2H, с); 9,26 (2H, шир); 9,44 (2H, шир.); 9,24 (2H, шир.).
Иллюстративный пример 124. 2-[2-[4-[(4-Аминометилциклогексил)- метокси] фенил]этил]-5-бензофуранкарбоксамидина дигидрохлорид.
Это соединение было получено в соответствии с процедурой, описанной в иллюстративном примере 123 (твердое вещество).
1H-ЯМР (ДМСО-d6) δ: 0,80-2,00 (10H, м); 2,90-3,20 (4H, м); 3,73 (4H, м); 6,72 (1H, с); 6,79 (2H, д, J = 8,5 Гц); 7,14 (2H, д, J = 8,5 Гц); 7,73 (2H, с); 8,07 (3H, шир.); 9,18 (2H, шир); 9,38 (2H, шир.).
Иллюстративный пример 125. [2-[2-(5-Амидино-2-бензофуранил)- этил]-5-[((3S)-3-пирролидинил)окси]фенил]оксиуксусной кислоты дигидрохлорид.
1,6 г метил [5-[((3S)-1-трет-бутоксикарбонил-3-пирролидинил)окси]- 2-[2-(5-циано-2-бензофуранил)этил] фенил] оксиацетата растворяли в 100 мл этанола. Размешивая и охлаждая льдом, через полученный раствор барботировали хлороводород до уровня насыщения. Обработанный таким образом раствор нагревали до комнатной температуры, и выдерживали при этой температуре 18 часов. После отгонки растворителя, полученный остаток растворяли в 20 мл этанола, и этот раствор по капле добавляли, размешивая при этом, к водному раствору 5 н. гидроксида натрия. После 10-минутного размешивания, полученный раствор насыщали хлоридом натрия, экстрагировали хлороформом три раза 200 мл, 100 мл и 100 мл, последовательно, а затем осушали в присутствии смеси карбоната калия и сульфата магния. После отгонки растворителя, остаток растворяли в 50 мл этанола. Полученный раствор смешивали с 1,0 г хлорида аммония, а затем с 200 мл этанолового раствора, содержащего 12% аммиак, и полученную смесь размешивали в течение 96 часов. После отгонки растворителя, остаток подвергали обращенно-фазовой ЖХВР, используя колонку с октадецил-связанным силикагелем, и элюируя смесью воды и ацетонитрила. После этого, нужные элюированные фракции объединяли, смешивали с разбавленной соляной кислотой и концентрировали досуха. В результате описанной процедуры получали 0,7 г целевого соединения в виде порошка.
1H-ЯМР (ДМСО-d6) δ: 1,96-2,32 (2H, шир.); 2,80-3,60 (8H, шир); 4,75 (2H, с); 5,10 (1H, шир.); 6,48 (1H, д, J = 8,8 Гц); 6,51 (1H, с); 6,76 (1H, с); 7,54 (1H, д, J = 8,8 Гц); 7,74 (2H, с); 8,10 (1H, с); 9,27 (2H, шир.); 9,42 (2H, шир.); 9,00-10,20 (2H, шир.).
Иллюстративный пример 126. Этил [2-[2-(5-амидино-2-бензофуранил)- этил] -5-[(3S)-3-пирролидинил)окси]фенил]оксиацетата дигидрохлорид.
0,65 г [2-[2-(5-амидино-2-бензофуранил)этил-5-[((3S)-3- пирролидинил)окси]фенил]оксиуксусной кислоты дигидрохлорида растворяли в 50 мл этанола. К этому раствору, охлаждая льдом, добавляли 0,2 мл тионилхлорида, и полученную смесь размешивали при комнатной температуре 18 часов. Затем реакционный раствор концентрировали досуха, растворяли в воде и снова концентрировали досуха. В результате описанной процедуры получали 0,65 г целевого соединения в виде твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,20 (3H, т); 2,11 (2H, шир.); 3,30 (4H, шир.); 4,20 (2H, кв. ); 4,84 (2H, с); 5,08 (1H, шир.); 6,53 (2H, с); 6,75 (1H, сж; 7,10 (1H, д); 7,72 (2H, с); 8,07 (1H, с); 9,18 (2H); 9,38 (2H); 9,66 (2H, шир.).
Иллюстративный пример 127. Пентагидрат гидрохлорида (2R)-2-[4-[[(3R)-1-ацетимидоил-3-пирролидинил] окси] фенил]-3- (7-амидино-2-нафтил)пропановой кислоты.
Структурная формула: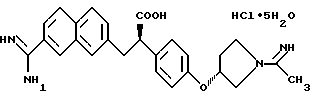
Молекулярная формула (М. в. ): C26H21N4O3 • 1HCl • 5H2O (571.071) N 1730801
Свойства: Бесцветные призматические кристаллы
[α]
1H-ЯМР (ДМСО-d6) δ: 2,20 - 2,40 (м., 2H, пирролидин C4-H1), 2,28 (c., 1,5H, 1/2 x-CH2), 2,31 (c., 1,5H, 1/2 x-CH2), 2,83-2,93 (м., 1H, -C(COOH)C  H-), 3,30 - 4,03 (м. , 6H, пирролидин C2-H1 C5-H1, -C(COOH)C
H-), 3,30 - 4,03 (м. , 6H, пирролидин C2-H1 C5-H1, -C(COOH)C  и -C
и -C 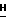 (COOH)CHH-), 5,18 (шир. , 0,5H, пирролидин 1/2 х C3-H), 5,24 (шир., 0,5H, пирролидин 1/2 x C3-H), 6,86 - 6,95 (д., 2H, J = 7,8 Гц, ArH), 7,50 (д., 2H, J = 7,8 Гц, ArH), 7,56 (д., 1H, J = 8,3 Гц), 7,65 (д., 1H, J = 8,3 Гц), 7,93 (д. , 1H, J = 8,8 Гц, ArH), 7,96 (д., 1H, J = 8,8 Гц, ArH), 8,13 (с., 1H, ArH), 8,65 (c., 1H, ArH), 8,70 - 9,00 (шир., 1H).
(COOH)CHH-), 5,18 (шир. , 0,5H, пирролидин 1/2 х C3-H), 5,24 (шир., 0,5H, пирролидин 1/2 x C3-H), 6,86 - 6,95 (д., 2H, J = 7,8 Гц, ArH), 7,50 (д., 2H, J = 7,8 Гц, ArH), 7,56 (д., 1H, J = 8,3 Гц), 7,65 (д., 1H, J = 8,3 Гц), 7,93 (д. , 1H, J = 8,8 Гц, ArH), 7,96 (д., 1H, J = 8,8 Гц, ArH), 8,13 (с., 1H, ArH), 8,65 (c., 1H, ArH), 8,70 - 9,00 (шир., 1H).
MS (FAB): m/z 445 (M+H)+.
ИК (KBr): 1682, 1634, 1610, 1504, 1384, 1242 см-1.
Анализ: Вычислено C26H21N4O3 • 1HCl • 5H2O: C 54,68; H 6,88; N 9,81; Cl 6,21. Найдено: C 54,60; H 6,84; N 9,74; Cl 6,38
ВЭЖХ: колонка: SUMICHIRAL OA-5000L 4,6 ⊘ x 150 мм
растворитель: 2 мМ CuSO4 : MeOH = 83:17
скорость потока: 1 мл/мин.
температура: 50oC
детекция: УФ 254 нм
время удерживания: 55,28 мин.
Испытательный пример 1. Измерение растворимости в воде.
К различным количествам каждого образца добавляли фиксированное количество воды, и полученную смесь размешивали на вибраторе в течение 10 минут при 25oC. Полученные результаты представлены в таблице 1.
Указанные результаты свидетельствуют о том, что соединения настоящего изобретения являются более растворимыми в воде, чем соединение прототипа.
Испытательный пример 2. Измерение антикоагулирующей активности.
С использованием центрифуги получали образец плазмы крови человека. 100 мкл от полученного препарата плазмы крови добавляли к 100 мкл физиологического раствора, содержащего или не содержащего испытуемое соединение, и полученную смесь оставляли на 2 минуты при 37oC. Затем к этой смеси добавляли 100 мкл 0,02 М раствора хлорида кальция, предварительно инкубированного при 37oC. После этого, с помощью CLOTEC (Sanko Iunyaku, Co., Ltd) измеряли время коагуляции. Время коагуляции, измеренное без добавления испытуемого соединения, рассматривали как контрольное, и исходя из этого, рассчитывали концентрацию испытуемого соединения, которая давала время коагуляции крови, в два раза превышающее контрольное время, и эту концентрацию принимали за показатель антикоагулирующей активности (обозначаемый далее "СТ2"). Полученные результаты показаны в таблице 2.
Испытательный пример 3. Измерение способности к ингибированию активированного фактора свертывания крови X (FXa).
180 мкл физиологического раствора, содержащего испытуемое соединение, смешивали с 200 мкл Трис-HCl-буфера (pH 8,4) и с 100 мкл водного раствора 1 мМ S-2222 (Kabivitrum AB), и полученную смесь инкубировали при 37oC. Затем к этой смеси добавляли 20 мкл Трис-HCl-забуференного физиологического раствора (pH 7,45), содержащего 0,6 ед./мл FXa человека. После 15-минутного инкубирования при той же температуре, реакцию останавливали путем добавления 100 мкл 60% уксусной кислоты, и измеряли оптическую плотность реакционной смеси. Смесь, полученную без добавления испытуемого соединения, использовали в качестве "пустышки", а другую реакционную смесь, где 60% уксусную кислоту добавляли перед добавлением FXa, использовали в качестве контроля. В качестве показателя FXa-ингибирующей активности рассчитывали концентрацию каждого испытуемого соединения, которое на 50% ингибирует FXa-активность (эту концентрацию далее обозначали "IC50"). Характерные примеры полученных результатов представлены в таблице 2.
Испытательный пример 4. Измерение тромбин-ингибирующей активности.
100 мкл Трис-HCl-забуференного физиологического раствора (pH 7,45) (TBS), содержащего 6 мг/мл фибриногена (тип 1, Daiichi Pure Chemicals Co., Ltd), смешивали с 100 мкл физиологического раствора. При 37oC, 100 мкл трис-HCl-забуференного физиологического раствора (pH 7,45) (TBS), содержащего различные количества тромбина (для наружного использования, Sankyo Co., Ltd), добавляли к полученной выше смеси, и измеряли время свертывания крови, используя при этом CLOTEC (Sanko Junyak Co., Ltd), после чего строили калибровочную кривую. Степень ингибирования (%) каждого испытуемого соединения определяли путем измерения времени коагуляции с использованием 100 мкл физиологического раствора, к которому добавляли каждое из указанных соединений. Концентрацию каждого соединения, которая на 50% ингибирует активность тромбина, рассчитывали исходя из процента ингибирования (далее эту концентрацию обозначали "IC50"), и концентрацию этого соединения определяли как показатель антитромбиновой активности. Характерные примеры полученных результатов представлены в таблице 2.
Как видно из результатов, представленных в таблице 2, каждое соединение настоящего изобретения обладает сильной антикоагулирующей активностью, обусловленной его специфической активностью против FXa (по сравнению с соединением DABE, которое известно специалистам как антикоагулирующий агент).
Испытательный пример 5. Измерение антикоагулирующей активности при пероральном введении
Водный раствор каждого испытуемого соединения вводили перорально каждой крысе отдельно (самцы SID: Wistar) под анестезией, в дозе 10 мл/кг тела. Периодически собирали пробы крови, и препараты плазмы крови, полученные из этих проб, измеряли для определения времени образования и активности тромбопластина (APTT). Аналогичным образом, APTT определяли при введении крысам чистой воды, а эти измерения использовали в качестве контроля. Затем вычисляли отношение испытат. /контроль APTT, и это отношение использовали в качестве показателя антикоагулирующей активности. Характерные примеры полученных результатов представлены в таблице 3.
Как видно из таблицы 3, при пероральном введении каждого соединения настоящего изобретения ясно наблюдается пролонгированное действие этих соединений на свертывание плазмы крови.
Испытательный пример 6. Испытание на токсичность путем однократного введения крысам пероральной дозы.
Соединение иллюстративного примера 45 растворяли в дистиллированной воде и перорально вводили каждой из двух 6-недельных крыс (самцы, SLc: CD) в дозе 2000 мг/кг веса тела. В течение времени наблюдения 14 дней, ни одного смертельного случая зарегистрировано не было.
Испытательный пример 7. Испытание на токсичность путем многократного введения крысам пероральной дозы.
Каждое из соединений настоящего изобретения растворяли в дистиллированной воде и вводили перорально каждой из пяти 5-недельных крыс (самцы, Slc: SD) в дозе 800 мг на кг веса тела. Это пероральное введение проводили один раз в день и повторяли в последующие 10 дней в целях определения возможной смертности. Результаты наблюдений представлены в таблице 4.
Испытательный пример 8. Определение антитромбиновой активности путем перорального введения соединения крысам в модели артериовенозного анастомаза.
С помощью слегка модифицированной процедуры, описанной в Thrombosis Research, vol. 54, pp. 399-410, 1989, определяли антитромбиновое действие соединений настоящего изобретения при их пероральном введении.
Заранее определенное количество испытуемого соединения растворяли в очищенной воде и вводили перорально самцам крыс (STD : Wistar) и этих крыс анестезировали через 15 минут после введения соединения. Затем, возле сонной артерии анастезированных крыс налагали артериальный зажим для остановки кровообращения, и вводили, фиксируя при этом, один конец шунта, наполненного физиологическим раствором, а другой конец шунта вводили, фиксируя, в яремную вену. В настоящих испытаниях, шунт состоял из медной проволоки длиной 20 см и диаметром 0,17 мм, находящейся в полиэтиленовой трубке (Hibiki N 5), с внешним диаметром 5/3 мм и длиной 21 см), где каждый конец этой трубки соединяли с полиэтиленовой трубкой (Hibiki N 3; с внешним диаметром 11 мм и длиной 3 см), используя 3-миллиметровую силиконовую трубку. Через 30 минут после введения, артериальный зажим удаляли для того, чтобы кровь поступала в шунт. Через 7 минут после восстановления кровообращения, медную проволоку вынимали вместе с образовавшимися тромбами и промывали 10 мл физиологического раствора. После этого, в соответствии с процедурой, описанной в Journal of Biological Chemistry, vol, 193, pp. 265-275, 1951, определяли количество тромбов, образовавшихся на мелкой проволоке, в виде белка. При расчете степени ингибирования тромбоза после введения соединения, использовали контрольный вариант, где вместо испытуемого соединения вводили воду. Полученные результаты представлены в таблице 5.
Как видно из таблицы 5, при пероральном введении наблюдалось значительное ингибирование тромбообразования.
Хотя настоящее изобретение описано на предпочтительных примерах его осуществления, однако, любому специалисту ясно, что в него могут быть внесены различные изменения и модификации, не выходящие за рамки существа и объема настоящего изобретения.
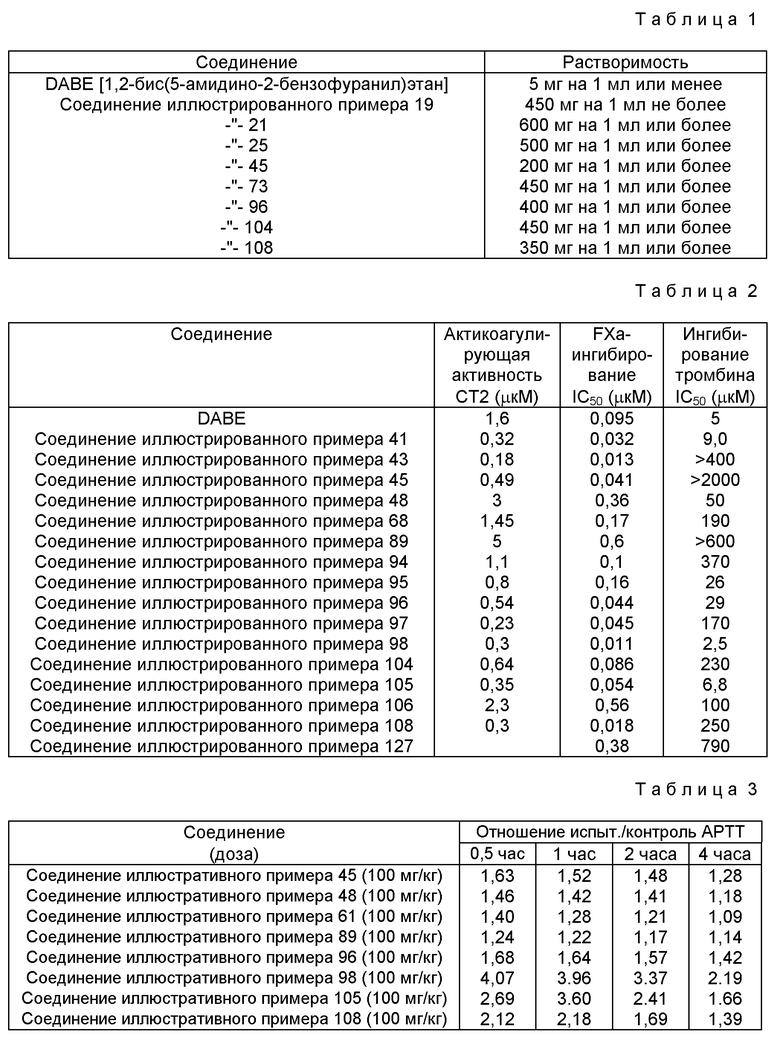
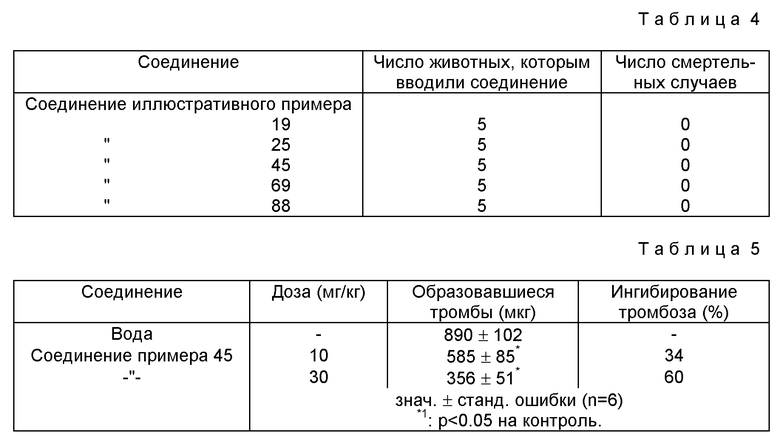
Описываются новые ароматические амидиновые производные общей формулы I, где R1 - атом водорода или низшая алкоксигруппа; R2 - атом водорода, низшая алкильная группа, низшая алкоксигруппа, карбоксильная группа, карбоксиалкильная группа с 2 - 5 атомами углерода или алкоксикарбонильная группа с 2 - 5 атомами углерода; R3 - атом водорода, карбоксильная группа, карбоксиалкильная группа с 2 - 5 атомами углерода, алкоксикарбонилалкильная группа с 3 - 6 атомами углерода, карбоксиалкоксильная группа с 2 - 5 атомами углерода и алкоксикарбонилалкоксильная группа с 3 - 6 атомами углерода; R4 - атом водорода, гидроксильная группа или низшая алкоксигруппа; n = 0, 1, 2; А - алкиленовая группа с 1 - 4 атомами углерода, которая может быть замещена одним или двумя заместителями, выбранными из гидроксиалкильной группы с 1 - 4 атомами углерода, карбоксила, алкоксикарбонильной группы с 2 - 5 атомами углерода и карбоксиалкильной группы с 2 - 5 атомами углерода; Х - простая связь, атом кислорода или серы или карбонильная группа; Y - насыщенный или ненасыщенный 5- или 6-членный гетероциклический фрагмент, имеющий один или два гетероатома, выбранных из атомов азота, кислорода, серы, или циклический углеводородный фрагмент, которые могут быть замещены иминогруппой, карбамоилом, моно- или диалкилкарбамоилом, алканоимидоилом, низшим алкилом, низшим алканоилом, алкоксикарбонилимином, бензилимидоилом, алканоиламино, алкиламином; насыщенный или ненасыщенный 5- или 6-членный циклический углеводородный фрагмент, которые могут быть замещены аминоалкильной группой или алканоиламиноалкильной группой, или Y является аминогруппой, которая может быть замещена пирролидинильной или пиридинильной группой; и группа формулы II является группой, выбранной из индолила, бензофуранила, бензотиенила, бензотиазолила, нафтила, тетрагидронафтила, бензимидазолила, или его фармацевтически приемлемая соль. Целевые продукты обладают способностью к сильному антикоагулирующему действию посредством обратимого ингибирования активированного фактора свертывания крови ("FХА"). Описывается также антикоагулирующая композиция на основе соединений формулы (I). 11 с. и 1 з.п.ф-лы, 5 табл.
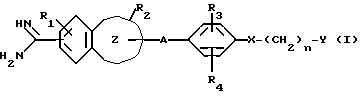
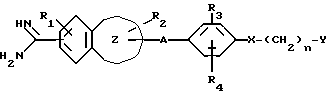
где R1 - атом водорода или низшая алкоксигруппа;
R2 - атом водорода, низшая алкильная группа, низшая алкоксигруппа, карбоксильная группа, карбоксиалкильная группа с 2-5 атомами углерода или алкоксикарбонильная группа с 2-5 атомами углерода;
R3 - атом водорода, карбоксильная группа, карбоксиалкильная группа с 2-5 атомами углерода, алкоксикарбонилалкильная группа с 3-6 атомами углерода, карбоксиалкоксильная группа с 2-5 атомами углерода, алкоксикарбонилалкоксильная группа с 3-6 атомами углерода или алкоксикарбонильная группа 2-5 атомами углерода;
R4 - атом водорода, гидроксильная группа или низшая алкоксигруппа;
n = 0, 1, 2;
A - алкиленовая группа с 1-4 атомами углерода, которая может быть замещена одним или двумя заместителями, выбранными из гидроксиалкильной группы с 1-4 атомами углерода, карбоксила, алкоксикарбонильной группы с 2-5 атомами углерода и карбоксиалкильной группы с 2-5 атомами углерода;
X - простая связь, атом кислорода или серы или карбонильная группа;
Y - насыщенный или ненасыщенный 5- или 6-членный гетероциклический фрагмент, имеющий один или два гетероатома, выбранных из атомов азота, кислорода, серы, или циклический углеводородный фрагмент, которые могут быть замещены иминогруппой, карбамоилом, моно- или диалкилкарбамоилом, алканоимидоилом, низшим алкилом, низшим алканоилом, алкоксикарбонилимино, бензилимидоилом, алканоиламино или алкиламиногруппой; где насыщенный или ненасыщенный 5- или 6-членный циклический углеводородный фрагмент может быть также замещен аминоалкильной группой или алканоиламиноалкильной группой, или Y является аминогруппой, которая может быть замещена пирролидинильной или пиразинильной группой;
группа формулы
является группой, выбранной из индолила, бензофуранила, бензотиенила, бензотиазолила, нафтила, тетрагидронафтила, бензимидазолила;
или его фармацевтически приемлемая соль.
| Способ получения амидиновых производных | 1981 |
|
SU1176832A3 |
| Лопата для очистки покрытий крыш из рулонных кровельных материалов | 1935 |
|
SU48433A1 |
| DE 3402628 А, 02.08.84 | |||
| Thrombosis Research | |||
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Ручной ткацкий станок | 1922 |
|
SU339A1 |

