Область техники
Изобретение относится к новым производным пиридонкарбоновой кислоты, противоопухолевым средствам, содержащим их в качестве эффективных ингредиентов, к способам получения новых производных пиридонкарбоновой кислоты и т. д.
Предпосылки создания изобретения
По существу являются известными как различные производные пиридонкарбоновой кислоты, которые содержат 2-тиазопильные группы, так и тот факт, что эти производные пиридонкарбоновой кислоты показывают антимикробную активность. Так например, в примере 24 заявки на патент Японии (Кокаи) N 152682/1986, выложенной для ознакомления, представлено следующее соединение А (в дальнейшем ссылка 1):
Соединение А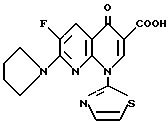
В примере 5 ссылки 1 показано следующее соединение:
Соединение В: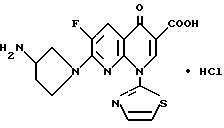
Кроме того, в примере 12 ссылки 1 раскрыто следующее Соединение С:
Соединение С: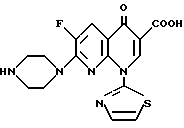
Вышеприведенные соединения В и С представлены также в Таблице 1 заявки на патент Японии (Кокаи) N 33176/1987, выложенной для ознакомления (в дальнейшем ссылка 2).
Кроме того, в примере 24-4 заявки на патент Японии (Кокаи) N 56959/1985, выложенной для ознакомления (соответствующей заявке на Европейский патент N 131839, выложенной для ознакомления, и патенту США N 4730000; в дальнейшем они будут упомянуты вместе как ссылка 3), и в примере 15 заявки на патент Японии (Кокаи) N 251667/1986, выложенной для ознакомления (в дальнейшем ссылка 4), упомянуто следующее соединение D:
Соединение D: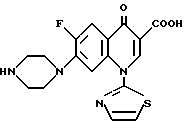
В примере 28-16 заявки на патент Японии (Кокаи) N 163866/1985, выложенной для ознакомления (соответствующей заявке на Европейский патент N 154780, выложенной для ознакомления, и патенту США N 4774246; в дальнейшем они будут упомянуты вместе как ссылка 5), упомянуто следующее Соединение E:
Соединение E: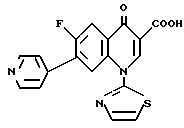
Далее, в примере 8 заявки на патент Японии (Кокаи) N 85255/1990, выложенной для ознакомления (в дальнейшем ссылка 6), показано следующее Соединение F.
Соединение F: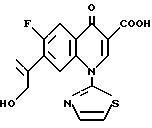
Однако химическая структура этих соединений отличается от структуры Соединения (1) этого изобретения следующими особенностями (1) и (2).
(1) 6-Положение Соединений A-F всегда замещено атомом фтора.
(2) Заместитель в 7-положении Соединений А, С, D, E и F не является замещенной 1-пирролидинильной группой. Кроме того, в ссылках 1-6 лишь упоминается о том, что Соединения A-F показывают антимикробную активность, и в них ничего не упоминается о противоопухолевой и противораковой активности.
Является известным, что определенные виды производных пиридонкарбоновой кислоты показывают противоопухолевую активность. Так, например, в источнике: Cancer Research 52, 2818(1992) сообщается, что следующее соединение G обладает противоопухолевой активностью:
Соединение G:
В этом научном труде сообщается о том, что на противоопухолевую активность исследовалось 90 видов производных пиридонкарбоновой кислоты и о том, что большая часть упомянутых производных не обладает противоопухолевой активностью, за исключением лишь нескольких типов производных. Кроме того, в этом труде упоминается о том, что главную роль для проявления противоопухолевой активности играют циклопропильная группа, которая замещена в 1-положении, и два атома галогена, которые замещены в 6 и 8-положениях, и о том, что производная пиридонкарбоновой кислоты, которая не содержит таких заместителей, не показывает противоопухолевую активность.
Краткое описание чертежей.
Фиг. 1-9 показывают изменение степени ингибирования роста опухоли (IR) во времени в том случае, когда соединение этого изобретения вводили оголенной мыши, которой трансплантировали различные раковые клетки человека.
Фиг. 10-14 показывают изменение веса опухоли с течением времени в вышеприведенном эксперименте.
Такие цифры на каждой фиг., как 1-1(50)[91], означают в указанной последовательности "Номер соединения (введенное количество мг/кг), [IR,% Степень ингибирования роста опухоли в последний день наблюдений].
Фиг. 15-19 показывают схемы реакций, которые упомянуты в примере Серии А [Получение промежуточных Соединений этого изобретения], в примере Серии В [Получение исходных материалов этого изобретения] и в примере Серии С [Получение Соединений этого изобретения], которые будут упомянуты позднее.
Раскрытие изобретения
После интенсивного поиска соединений, обладающих противоопухолевой активностью, настоящие изобретатели в итоге обнаружили, что новые производные пиридонкарбоновой кислоты, имеющие 2-тиазолильные группы, которые могут быть замещены, показывают высокую противоопухолевую активность.
Это изобретение относится к производным пиридонкарбоновой кислоты, которые имеют 2-тиазолильные группы и следующую общую формулу (I), и к их солям: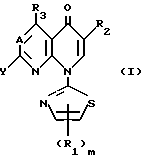
в которой R1 является атомом водорода, низшей алкоксильной группой, атомом галогена, низшей алкильной группой, которая может быть замещена атомом галогена, или фенильной группой, которая может быть замещена атомом галогена;
R2 является карбоксильной группой или группой, способной превращаться в карбоксильную группу;
R3 является атомом водорода, аминогруппой, которая может быть защищена атомом галогена или низшей алкильной группой, которая может быть замещена атомом галогена;
А представляет атом азота или CH;
m является целым числом, равным 1 или 2;
Y представляет удаляемую группу или группу, имеющую следующую формулу Y':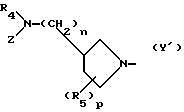
в которой R4 является атомом водорода или низшей алкильной группой;
Z является атомом водорода, низшей алкильной группой или группой, способной превращаться в атом водорода;
R5 является атомом водорода, атомом галогена, низшей алкоксильной группой, низшей алкилтиогруппой или низшей алкильной группой, которая может быть замещена атомом галогена;
n является целым числом, равным 0 или 1;
р является целым числом, равным 1, 2, 3 или 4.
Соединения этого изобретения включают, конечно, их стереоизомеры, оптические изомеры, гидраты, сольваты и т.д.
Соединения (1) этого изобретения подразделяются в соответствии с характером заместителей на две категории.
Одна из категорий включает соединения формулы (I), в которой Y представляет "удаляемую группу", и эти соединения являются пригодными в качестве непосредственных промежуточных соединений для соединений, в которых Y представляет вышеуказанный Y'. Таким образом, одна из целей этого изобретения состоит в обеспечении промежуточных соединений для производных пиридонкарбоновой кислоты, которые являются пригодными в качестве противоопухолевых средств.
В качестве "удаляемой группы", включенной при определении заместителя Y, может быть использована любая группа до тех пор, пока она может быть замещена далее описанной производной пирролидина (III), и вследствие этого удалена, и примеры такой группы включают атом галогена, низшую алкоксильную группу, низшую алкилтиогруппу, низшую алкилсульфинильную группу, низшую алкилсульфонильную группу, арилсульфонильную группу, низшую алкилсульфонилоксигруппу, арилсульфонилоксигруппу и т.д. Среди этих групп предпочтительными являются атомы галогена, например атом фтора и атом хлора. Что касается других заместителей или солей, то конкретные их примеры можно увидеть ниже при объяснении Соединения (I-а) этого изобретения.
Соединениями этого изобретения другой категории являются соединения вышеприведенной формулы (I), в которой заместитель Y является Y', и они являются пригодными в качестве превосходных противоопухолевых или противораковых средств. Таким образом, это изобретение обеспечивает производные пиридонкарбоновой кислоты, имеющие следующую формулу (I-а), их физиологически приемлемые соли, способы получения таких производных и солей, и противоопухолевые средства, которые содержат такие производные или соли в качестве эффективных ингредиентов: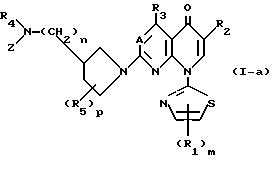
в которой A, R1, R2, R3, R4, R5, Z, m, n и p являются такими, как они определены выше. Соли соединений, имеющих формулу (I-а), включают как соли, произведенные от части карбоксильной группы, которая содержалась в определении R2 формулы (I-а), так и кислые соли присоединения, произведенные от части основной замещающей группы, которая связана с 3-положением 1-пирролидинильной группы.
Примеры таких солей, произведенных от части карбоксильной группы, включают соли металлов, например натрия, калия, магния, цинка, серебра, алюминия и платины, и соли органических оснований, например таких, как диметиламиноэтанол, метиламиноэтанол, триэтаноламин и гуанидин.
В качестве примеров кислых солей присоединения в основной замещающей группе, которая связана с 3-положением 1- пирролидинильной группы формулы (I-а), можно рассмотреть соли неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота и фосфорная кислота, и соли органических кислот, таких как щавелевая кислота, малеиновая кислота, фумаровая кислота, малоновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, бензойная кислота, метансульфоновая кислота, п-толуолсульфокислота, аскорбиновая кислота, глюкуроновая кислота, 2-окси- этансульфокислота, лактобионовая кислота и глюкогептоновая кислота.
Каждый из вышеприведенных заместителей объясняется следующим образом:
В этом описании "низшая алкильная группа" означает алкильную группу с прямой или разветвленной цепью, имеющую от 1 до 5 атомов углерода, и примеры такой группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, трет-бутильную группу и т. д. "Низшая алкоксильная группа" означает алкоксильную группу, имеющую от 1 до 5 атомов углерода, при этом предпочтительными примерами являются метоксильная группа и этоксильная группа.
Примеры "атома галогена" включают атом хлора, атом фтора и атом брома.
Заместитель R1 в формуле (I-а) расположен в 4- и/или 5-положении 2-тиазолильной группы, и предпочтительные примеры включают атом водорода, атом галогена, например атом фтора, атом хлора и атом брома, низшую алкоксильную группу, такую как метоксильная группа и этоксильная группа, низшую алкильную группу, такую как метильная группа и этильная группа, низшую алкильную группу, замещенную атомом галогена, такую как трифторметильная группа, и фенильную группу, которая может быть замещена атомом галогена, такую как 3,4-дифторфенильная группа.
Что касается "группы, способной превращаться в карбоксильную группу", содержащейся при определении заместителя R2, то может быть использована любая группа до тех пор, пока она является способной превращаться в карбоксильную группу посредством химических или ферментологических средств, и предпочтительные примеры такой группы включают оксиметильную группу, формильную группу, сложноэфирную форму и физиологически приемлемые соли карбоксильной группы.
Примеры сложноэфирных форм, которые являются способными превращаться в карбоксильные группы посредством, главным образом, химических средств, включают низшие алкиловые сложные эфиры, такие как сложный метиловый и сложный этиловый эфир.
Примеры сложноэфирных форм, которые являются способными превращаться в карбоксильную группу не только посредством химических средств, но также посредством ферментологических средств, включают низшие алканоилоксинизшие алкиловые сложные эфиры, такие как ацетоксиметиловый эфир, 1-ацетоксиэтиловый эфир и пивалоилоксиметиловый эфир: низшие алкоксикарбонилокси- низшие алкиловые сложные эфиры, такие как 1- этоксикарбонилоксиэтиловый эфир; ди-низший алкиламино-низшие алкиловые сложные эфиры, такие как 2-диметиламиноэтиловый эфир; 2- (1-пиперидинил) этиловый эфир; 3-бутиролактониловый эфир; холиновый эфир; флалидиловый эфир; (5-метил-2-оксо-1,3-диоксол-4-ил) метиловый эфир и т.д.
Предпочтительные примеры заместителя R3 включают атом водорода, атом галогена, например атом фтора и атом хлора, аминогруппу, аминогруппу, защищенную посредством защитной группы для амино, и низшую алкильную группу, замещенную атомом галогена, такую как трифторметильную группу. В качестве упомянутой защитной группы для амино может быть использована любая защитная группа до тех пор, пока она легко устранима посредством обычной реакции удаления защиты, такой как гидролиз или гидрогенолиз, без оказания существенного воздействия на другие структурные части. Более конкретно, упомянутая защитная группа является по существу той же самой, что и "группа, способная обращаться в атом водорода" при определении заместителя Z, который будет объяснен позднее. Предпочтительные примеры защитной группы для амино включают бензильную группу и третильную группу.
Заместитель R4 представляет атом водорода или низшую алкильную группу, и предпочтительно применяют атом водорода, метильную группу и этильную группу. В данном случае 3-положение 1- пирролидинильной группы, с которой связана основная замещающая группа, обладающая R4 и Z, занято асимметрическим атомом углерода, что может привести к наличию оптических изомеров.
Предпочтительные примеры заместителя Z включают атом водорода, низшую алкильную группу, такую как метильная группа и этильная группа, или "группу, способную превращаться в атом водорода". Что касается "группы, способной превращаться в атом водорода", то может быть использована любая группа до тех пор, пока она способна превращаться в атом водорода посредством химических средств, таких как гидролиз или гидрогенолиз, или посредством ферментологических средств.
Примеры Z в качестве "группы, способной превращаться в атом водорода", включают, во-первых, гидролизуемые группы. Конкретные примеры гидролизуемых групп включают ацильные группы, группы, имеющие оксикарбонильную группу, аминокислотные остатки и пептидные остатки, и кроме того, например, O-нитрофенилсульфенил, триметилсилил, тетрагидропиранил, дифенилфосфинил и т.д. В общем, Соединения (I-а) этого изобретения, в которых Z в качестве группы, обратимой в атом водорода, является аминокислотным остатком или пептидным остатком, имеют превосходную растворимость по сравнению с такими соединениями, где Z не является ни аминокислотным, ни пептидным остатком и их выгодно используют в форме жидких веществ, например в виде инъекционного раствора.
Примеры вышеупомянутых ацильных групп включают формил, ацетил, трифторацетил и т.д.
Примеры вышеупомянутых групп, имеющих оксикарбонильную группу, включают, кроме того, этоксикарбонил, трет-бутоксикарбонил- [(CH3)3C-OCО], бензилоксикарбонил, п-метоксибензилоксикарбонил, вилилоксикарбонил, β- (п-толуолсульфонил) этоксикарбонил и т.д.
Далее, примеры аминокислотных остатков включают аминокислотные остатки сами по себе и такие аминокислотные остатки, которые защищены защитной группой, которую обычно применяют в синтезе пептида. Примеры защитных групп для аминогруппы, обычно используемых в синтезе пептида, включают ацильные группы, такие как формильные и ацетильные, арилметилоксикарбонильные группы, такие как бензилоксикарбонильные и п-нитробензилоксикарбонильные, трет-бутоксикарбонильную группу [(CH3)3C-OCO] и т.д.
Что касается остатков аминокислоты, то может быть использован любой остаток, например, аланиновый остаток [(CH3CH(NH2)CO-] и лейциновый остаток [(CH3)2CHCH2CH(NH2)CO-].
В общем эти аминокислоты представлены рядом из трех английских букв, и этот принцип поддерживается также в настоящем описании. Кроме того, за счет добавления символа "L", "D" или "DL" впереди этих трех букв можно различить L-формы, D-формы или их смеси. Эти символы не включают, когда эти изомеры относят к целому.
Конкретные примеры аминокислотных остатков включают остатки таких аминокислот, как Gly (глицин), Ala (аланил), Arg (аргинил), Asn (аспарагин), Asp (аспарагиновая кислота, Cys (цистеин), Glu (глутаминовая кислота), His (гистидин), Ile (изолейцин), Leu (лейцин), Lys (лизин), Met (метионин), Phe (фенилаланин), Pro (пролин), Ser (серин), Thr (треонин), Trp (триптофан), Tyr (тирозин), Val (валин), Nva (норвалин), Hse (гомосерин), 4-Нур (4-оксипролин), 5-Hyl (5-оксилизин), Orn (орнитин) и β-Ala.
От двух до пяти, предпочтительно от двух до трех из вышеупомянутых кислот, образуют пептидные остатки. В качестве примеров таких пептидных остатков можно принять остатки таких пептидов, как: Ala-Ala [CH3CH(NH2)CO-NHCH(CH3)CO-] , Gly-Phe, Nva-Nva, Ala-Phe, Gly-Gly, Gly-Gly -Gly, Ala-Met, Met-Met, Leu-Met и Ala-Leu.
Остатки этих аминокислот или пептидов могут принимать стереохимическую конфигурацию D-формы, L-формы или их смеси, но предпочтительной является L-форма. Кроме того, когда Z является остатком аминокислоты или пептида, такой остаток может также иметь при данных обстоятельствах асимметрический атом углерода. Примеры аминокислотных остатков, имеющих асимметрический атом углерода, включают остатки таких аминокислот, как Ala, Leu, Phe, Trp, Nva, Val, Met, Ser, Lys, Thr и Tyr, и в качестве примеров пептидных остатков, которые имеют асимметрический атом углерода, здесь можно упомянуть такие, которые имеют эти аминокислотные остатки, имеющие асимметрический атом углерода, в качестве составляющего ингредиента.
Кроме того, группа Z, "способная превращаться в атом водорода", может представлять элиминируемую гидрогенолизируемую восстановлением группу, и примеры такой группы включают арилсульфонильные группы, такие как о-толуолсульфонильные; метильные группы, замещенные фенилом или бензилокси-, такие как бензильные, тритильные и бензилоксиметильные; арилметоксикарбонильные, такие как бензилоксикарбонильные и о- метоксибензилоксикарбонильные; и галогенэтоксикарбонильные группы, такие как β,β,β- трихлорэтоксикарбонильные и β- иодэтоксикарбонильные и т.д.
Заместитель "(R5)p -" 1-пирролидинильной группы формулы Y' связан с пирролидиновым кольцом упомянутой 1- пирролидинильной группы. Знак "p" означает целое число от 1 до 4. Когда p равно от 2 до 4, R5 могут быть одинаковыми или разными. R5 может быть связан с любым положением 1-пирролидинильной группы, но предпочтительно или с положением, с которым связан основной заместитель, содержащий R4 и Z (это положение в дальнейшем будет названо 3-положением 1-пирролидинильной группы) или с положением, являющимся, к тому же, соседним (это положение в дальнейшем будет названо 2- и/или 4-положением 1-пирролидинильной группы). Когда R5 связан с положением, иным чем 3-положение 1-пирролидинильной группы, и когда R5 не является атомом водорода, следует, что упомянутая 1-пирролидиниль-ная группа имеет, по крайней мере, два асимметрических атома углерода, при этом в результате Соединения (I-а) того изобретения могут существовать в виде стереоизомера (цис-форма или транс-форма) и оптического изомера. Предпочтительные примеры заместителя R5 включают атом водорода, низшие алкильные группы, например метильную группу и этильную группу, низшие алкильные группы, замещенные атомом галогена, например фторметильную группу и трифторметильную группу, низшую алкоксильную группу, например метоксильную и этоксильную группу, низшие алкилтиогруппы, например метилтиогруппу, и атом галогена, например атом хлора и атом фтора.
Соединения (I-а) этого изобретения, имеющие такие заместители, которые подробно описаны выше, и их физиологически приемлемые соли являются новыми и обладающими превосходной противоопухолевой активностью.
У соединений этого изобретения, имеющих общую формулу (I-а), ядерные соединения, которые показывают противоопухолевую активность, имеют следующую формулу (I-b):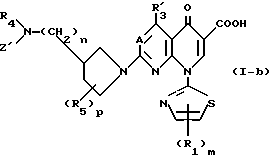
в которой R1, m, R4, n, R5, p и A являются такими, как они определены в формуле (I-а); R3 является атомом водорода, аминогруппой, атомом галогена или низшей алкильной группой, которая может быть замещена атомом галогена; и Z' является атомом водорода или низшей алкильной группой.
В последующем соединения, представленные вышеприведенной формулой (I-b), будут иногда сокращенно представлены как "активные соединения". Когда соединение, которое не является активным соединением, вводят в живой организм, упомянутое соединение при таких обстоятельствах превращается в живом организме в активное соединение. В таком случае соединение, которое не является активным соединением, иногда сокращенно называют "пролекарство". В этом изобретении примеры такого пролекарства включают соединения формулы (I-а), в которых является аминокислотным остатком или пептидным остатком, или в которых R2 является формильной группой или сложноэфирной формой, такой как ацетоксиметоксикарбонил.
Кроме того, при рассмотрении настоящего описания и формулы изобретения, можно видеть, что соединения, которые являются способными превращаться в активные соединения посредством определенных средств, например химических средств или ферментологических средств, относят обычно к "способным превращаться соединениям".
Структурные характеристики соединений (I-а) этого изобретения основаны на том, что упомянутые соединения имеют следующую структуру:
(1) соединения в качестве основы имеют пиридонкарбоновую кислоту, представленную следующей формулой 
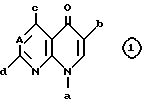
в которой А является такой, как она определена выше, и a, b, c и d означают положения, в которых связывают заместители,
(2) 2-тиазолильнуго группу, которая может иметь заместитель, связывают в положении "а",
(3) карбоксильную группу или группу, способную превращаться в карбоксильную группу, связывают в положении "b",
(4) положение "с" является незамещенными, или в таком положении связывают группу, такую как аминогруппа,
(5) А является атомом азота или атомом углерода, который не замещен атомом галогена, например атомом фтора, и
(6) положение "d" замещено определенной 1- пирролидинильной группой, которая имеет, по крайней мере, заместитель, представленный следующей формулой 
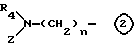
в которой R4, Z и n являются такими, как они определены выше.
Соединения (I-а) этого изобретения являются новыми соединениями, которые структурно характеризуются, в частности, комбинацией заместителей, которые связаны в положениях "а" и "d", и тем фактом, что А не содержит атом фтора.
Все соединения этого изобретения, включенные в формулу (I-а), и их физиологически приемлемые соли являются превосходными противоопухолевыми или противораковыми средствами. В частности, соединения, в которых А является CH, являются предпочтительными в качестве противоопухолевых средств. Наиболее предпочтительными являются соединения, в которых А является CH, m и p равны 1, и n равно 0.
В особенности предпочтительными являются соединения, в которых А являются CH, m и p равны 1, n равно 0, R1 является атомом водорода, R2 является карбоксильной группой, R3 является атомом водорода, R4 является атомом водорода или низшей алкильной группой, и R5 является атомом водорода, низшей алкильной группой или низшей алкоксильной группой. Примеры таких соединений включают 1,4-дигидро-7-(3-метокси-4-метиламино-1-пирролидинил)-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновую кислоту (смотри Соединение 1-1 и т.д. в далее представленной Таблице 1) и соединения способные в нее превращаться.
Хотя конкретные примеры предпочтительных соединений, включенных в формулу (I-а), упомянуты позднее в примерах, в качестве предпочтительных примеров соединений 1,8-нафтиридинового типа формулы (I-а) этого изобретения могут быть, далее рассмотрены следующие соединения и, кроме того, соединения, способные в них превращаться:
7-(3-амино-4-фтор-1-пирролидинил) -1,4-дигидро-4-оксо- 1-(2-тиазолил) -1,8-нафтиридин-3-карбоновая кислота;
7-(3-амино-4-метокси-3-метил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил) -1, 8-нафтиридин-3-карбоновая кислота;
7-(3-амино-4-метокси-4-метил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2- тиазолил) -1, 8-нафтиридин-3-карбоновая кислота;
7-(3-амино-3-фторметил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновая кислота;
7- (3-амино-4-фторметил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновая кислота;
7- (3-амино-4-трифторметил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновая кислота;
7- (3-амино-1-пирролидинил) -1- (4-хлор-2-тиазолил) - 1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота;
7- (3-амино-1-пирролидинил) -1- (4,5-дифтор-2-тиазолил) -1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота;
5-амино-7- (3-амино-1-пирролидинил) -1- (4-фтор-2-тиазолил)-1,4- дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота;
7-(3-амино-1-пирролидинил) -1,4-дигидро-4-оксо-1-(4- трифторметил-2-тиазолил) -1,8-нафтиридин-3-карбоновая кислота;
7-(3-амино-1-пирролидинил) -1-[4-(3,4-дифторфенил) -2-тиазолил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота;
7-(3-амино-1-пирролидинил) -1-(5-бром-2-тиазолил) -1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновая кислота.
В качестве примеров соединений пиридопиримидинового типа формулы (I-а) этого изобретения могут быть рассмотрены следующие соединения и соединения, способные в них превращаться:
5,8-дигидро-2-(транс-3-метокси-4-метиламино-1-пирролидинил) -5-оксо-8-(2-тиазолил) пиридо[2,3-d]-пиримидин-6-карбоновая кислота;
8-(4-фтор-2-тиазолил)-5,8-дигидро-2-(транс-3- метокси-4-метиламино-1-пирролидинил)-5-оксо-пиридо[2,3-d] пиримидин-6-карбоновая кислота.
Фармакологические испытания.
Примеры испытаний.
Противоопухолевая активность Соединений (I-а) этого изобретения описана в дальнейшем. В качестве контрольных групп были приняты как Соединение А, которое раскрыто в заявке на патент Японии N 152682/1986, выложенной для ознакомления (Ссылка 1), на которую ссылались в начале этого описания, так и Этопозид, т.е. коммерчески пригодное противораковое средство, которое имеет далее описанную структурную формулу.
Пример 1 испытания.
Противоопухолевая активность in vitro (С50: мкг/мл) против мышиных Р388 лимфоцитарных лейкозных клеток
Исследуемые соединения испытывания на противоопухолевую активность в соответствии с МТТ [3-(4,5-диметилтиазол-2-ил)-2,5- дифенилтетразолийбромидным] методом, используя мышиные Р388 лимфоцитарные лейкозные клетки.
В каждую ячейку чашки, содержащей 96 ячеек, поместили питательную среду, содержащую от 1.000 до 2.000 мышиных Р388 лимфоцитарных лейкозных клеток, и испытуемое соединение с заданной концентрацией в количестве 0.1 мл, и клетки культивировали в течение 72 часов при 37oC и содержании газа диоксида углерода в воздухе 5%. После культивирования добавили раствор МТТ (5 мг/мл) в количестве 0.02 мл в каждую ячейку, и затем клетки культивировали в течение еще 4 часов. Питательную среду центрифугировали (4oC, 2000 оборотов в минуту, в течение 20 минут), и полученные надосадочные жидкости удалили путем всасывания. Затем в каждую ячейку для растворения образованного формазона поместили 0.1 мл диметилсульфоксида и затем добавили еще 0.1 мл диметилсульфоксида.
Затем с помощью метода многократного двухцветного сканирования измерили поглощательную способность (O. D).) каждого из полученных растворов (длина основной волны 570 нм, длина подволны - 690 нм). Предположив, что поглощательная способность необработанных клеток (контрольных) составляет 100%, методом наименьших квадратов вычислили дозу, угнетающую возрастание клеток на 50% (концентрация лекарственного вещества, дающая 50% ингибирование: IC50: мкг/мл). Результаты показаны в таблице 1.
Соединение H: Этопозид.

Как показано в Таблице 1, противоопухолевые активности соединений (IC50) этого изобретения in vitro против мышиных P 388 лимфоцитарных лейкозных клеток в 2-22 раза меньше, чем противоопухолевая активность Соединения А.
Пример 2.
Противоопухолевые активности in vitro против линий раковых клеток человека.
В каждую ячейку чашки, содержащей 96 ячеек, поместили питательную среду в количестве 0.1 мл, содержащую от 500 до 2000 раковых клеток человека, и клетки культивировали в течение 20 часов при 37oC и содержании диоксида углерода в воздухе 5%. После культивирования добавили раствор испытуемых соединений заданной концентрации, и клетки культивировали в течение еще 72 часов. После культивирования в соответствующие ячейки добавили раствор МТТ (5 мг/мл) в количестве 0.01 мл, и клетки культивировали в течение 4 часов. Надосадочные жидкости питательной среды удалили путем всасывания, и затем в соответствующую ячейку для растворения образованного формазана поместили 0.1 мл диметилсульфоксида и, кроме того, добавили еще 0.1 мл диметилсульфоксида. Тем же самым способом, что и в примере 1 испытаний, вычислили дозу, угнетающую разрастание клеток на 50%.
Результаты показаны в таблице 2.
Как показано в Таблице 2, Соединения этого изобретения показывают превосходные противоопухолевые активности in vitro (IC50) против раковых клеток человека. С другой стороны, активность Соединения A, т.е. контрольного, составляет только от 1/2 до 1/50 от активности соединений этого изобретения.
Пример 3 испытаний.
Увеличение продолжительности жизни мышей, которым имплантировали мышиные P388 лимфоцитарные лейкозные клетки.
Каждой мыши SLC:BDF 1 (возрастом от 8 до 10 недель, самки, 7 животных на 1 группу) внутрибрюшинно имплантировали 1•106 мышиных P388 лимфоциторных лейкозных клеток. Испытуемое соединение (лекарственное вещество) или растворяли в 0.1 N NaOH, или суспендировали в 0.4% растворе карбоксиметилцеллюлозы, и полученный раствор или суспензию разбавили дистиллированной водой или 0.4% раствором карбоксиметилцеллюлозы для того, чтобы получить заданные концентрации для введения.
Полученный раствор ввели дважды внутрибрюшинно (ip), а именно в первый день следующий за имплантацией (первый день) и в пятый день после имплантации, каждый раз по 0.2 мл. В течение 30 дней наблюдали за состоянием (жизнью и смертью) мышей и для каждой группы определили среднее время выживания (в дальнейшем MST) и таким образом, в соответствии с последующим уравнением, вычислили увеличение продолжительности жизни (ILS; %):
ILS(%) = [{(MST испытуемой группы/
(MST контрольной группы)} -1] х 100
Действие лекарственного средства оценили в соответствии с критерием Национального института рака США (NCI):
ILS = 75% или выше: ++ (в высшей степени эффективное);
от 20 до 74%: + (эффективное);
19% или ниже: - (неэффективное).
Результаты показаны в Таблице 3.
Как показано в Таблице 3, воздействие соединений этого изобретения на увеличение продолжительности жизни мышей, которым имплантировали мышиные P388 лимфоцитарные лейкозные клетки, намного превосходит воздействие соединения А, являющегося контрольным.
Пример 4.
Эффект ингибирования роста опухоли у мышей, которым имплантировали мышиные опухолевые клетки толстой кишки 26.
2% brei мышиных опухолевых клеток толстой кишки 26 в количестве 0.1 мл имплантировали в отдел брюшной полости мыши-самки SLC:CDF1 (возрастом от 7 до 9 недель, 7 животных на одну группу). Затем испытуемое соединение (лекарственное вещество) растворили в 0.1 N NaOH и разбавили дистиллированной водой для получения заданных концентраций для введения. Полученные растворы вводили внутрибрюшинно один раз в день по 0.2 мл, начиная с первого дня после имплантации и до 9 дня. На 21 или 22 день после имплантации оценили вес опухоли, исходя из диаметра опухоли, и таким образом в соответствии с последующим уравнением вычислили степень ингибирования роста опухоли в группе, которой осуществляли введение лекарства (IR%) по сравнению с контрольной группой:
IR(%) = [1-{ (MTW в группе, подвергнутой лечению)/(MTW в контрольной группе)}] х 100
MTW: средний вес опухоли
Результаты показаны в Таблице 4.
Как показано в Таблице 4, степень ингибирования роста опухоли (IR) соединениями этого изобретения в мышиных опухолевых клетках толстой кишки 26, имплантированных мышам, является превосходной. С другой стороны, воздействие Соединения А является явно более худшим по сравнению с соединениями этого изобретения как с точки зрения доз, так и с точки зрения степеней ингибирования.
В последующих примерах 5-9 испытаний раковые клетки человека имплантировали оголенным мышам, которым вводили испытуемые соединения, растворенные в водном растворе, содержащем NaOH, и таким образом следили за степенью ингибирования роста раковых клеток.
Результаты примеров 5-9 испытаний показаны на фиг. 1-14.
Фиг. 1-9 показывают изменения IR с течением времени.
Фиг. 10-14 показывают соотношение между весом опухоли и временем в том случае, когда в примерах 5-9 испытаний использовали Соединение 1-1. Вертикальная ось соответствует весу опухоли, а горизонтальная ось соответствует количеству дней, прошедших после начала введения. Вес опухоли оценивали, исходя из диаметра опухоли. Термин "Контроль" на фиг. 10-14 означает изменение веса опухоли во времени у оголенных мышей, которым хотя и трансплантировали раковые клетки, но не вводили испытуемые соединения.
Каждая из фигур показывает количество каждого введенного соединения (мг/кг/день) и IR(%) в последний день наблюдений, как объяснено в примере 4 испытаний.
В дальнейшем будет разъяснен режим введения испытуемых соединений оголенным мышам. После истечения "x" количества дней со времени имплантации оголенным мышам раковых клеток человека, в течение "y" дней внутрибрюшинно (ip) вводили испытуемые соединения, после чего в течение "z" дней следовали периоды вымывания. Затем испытуемые соединения вводили опять в течение "y" дней.
В этом случае цикл, состоящий из введения ("y" дней) и прекращения введения ("z" дней), называется "курс". Такой режим обозначен посредством знаков следующим образом:
[(х)(y)(z) (курс) (ip)=внутрибрюшинно)]
Эти знаки объяснены ниже с помощью примера. Знак  означает день, в который вводили испытуемое соединение.
означает день, в который вводили испытуемое соединение.
Пример: испытуемые соединения вводили (ip) внутрибрюшинно через 25 дней после имплантации, и затем введение временно прекратили на 6 дней, после чего введение осуществляли опять. Эту операцию повторили пять раз (см. табл. 5).
Пример 5 испытаний.
Противоопухолевое действие на раковые клетки носоглотки человека KB, имплантированные оголенным мышам.
Опыт проводили при следующих условиях:
Условия:
Использованные животные: оголенные мыши-самки BALB/c и AnNCrj-nu/nu (возрастом 9 и 14 недель, 6 животных на одну группу).
Использованные раковые клетки: раковые клетки носоглотки человека КВ.
Имплантация раковых клеток:
2.5 х 106 раковых клеток имплантировали внутрикожно в отдел брюшной полости оголенных мышей.
Режим введения:
[(5) (1) (6) (6 курсов) внутрибрюшинно)].
Результаты: показаны на фиг. 1-2 и 10.
Пример 6 испытаний.
Противоопухолевое действие на раковые клетки молочной железы человека МХ-1, имплантированные оголенным мышам.
Опыт проводили при следующих условиях:
Условия:
Использованные животные: оголенные мыши-самки BALB/c AnNCrj-nu/nu (возрастом 9 недель, 5-6 животных на одну группу).
Использованные раковые клетки: раковые клетки молочной железы человека МХ-1.
Имплантация раковых клеток:
В спинку оголенных мышей внутрикожно имплантировали кусочек раковой ткани размером 2х3 мм.
Режим введения:
[(16 и 23) (1) (6) (6 курсов) внутрибрюшинно)]
Результаты: показаны на фиг. 3-5 и 11
Пример 7 испытания.
Противоопухолевое действие на раковые клетки прямой кишки человека WiDr, имплантированные оголенным мышам.
Опыт проводили при следующих условиях:
Условия:
Использованные животные: оголенные мыши самки BALB/cAnNCrj-nu/nu (возрастом 9 недель, 6 животных на одну группу).
Использованные раковые клетки: раковые клетки прямой кишки человека WiDr.
Имплантация раковых клеток:
2.5х106 раковых клеток имплантировали внутрикожно в отдел брюшной полости оголенных мышей.
Режим введения:
[(9) (1) (6) (6 курсов) внутрибрюшинно)].
Результаты: показаны на фиг. 6 и 12.
Пример 8 испытания.
Противоопухолевое действие на клетки меланомы человека HMV-2, имплантированные оголенным мышам.
Опыт проводили при следующих условиях:
Условия:
Использованные животные: оголенные мыши-самки BALB/c AnNCrj-nu/nu и BALB/с nu/nu (возрастом 11-15 недель, 6-7 животных на одну группу).
Использованные раковые клетки:
Раковые клетки меланомы человека HMV-2
Имплантация раковых клеток:
4.4х106 раковых клеток имплантировали внутрикожно в отдел брюшной полости оголенных мышей.
Режим введения:
[(8-9) (1) (6) (9 курсов) внутрибрюшинно]
Результаты: показаны на фиг. 7-8 и 13.
Пример 9 испытаний
Противоопухолевое действие на раковые клетки легкого человека LX-1, имплантированные оголенным мышам.
Опыт проводили при следующих условиях:
Условия:
Использованные животные: оголенные мыши-самки BAlB/c AnNCrj-nu/nu (возрастом 13 недель, 6 животных на одну группу).
Использованные раковые клетки: раковые клетки легкого человека LX-I.
Имплантация раковых клеток:
В спину оголенных мышей внутрикожно имплантировали кусочек раковой ткани размером 2х3 мм.
Режим введения:
[(19 и 26) (1) (6) (5-6 курсов) внутрибрюшинно)].
Результаты: показаны на фиг. 8 и 14.
Как видно из упомянутых фиг. 1-14, соединения этого изобретения замечательно ингибируют рост раковых клеток человека, имплантированных оголенным мышам.
Пример 10 испытаний.
Острая токсичность.
Растворы испытуемых соединений с заданными концентрациями вводили мышам-самкам BALB/с CrSl с (возрастом 10 недель, от 5 до 10 животных на одну группу (0.1 мл/10 г массы тела) и исходя из смертности мышей на 14 день после введения вычислили значения LD50. Результаты показаны в табл. 6.
Как показано в табл. 6, острая токсичность соединений этого изобретения почти равна острой токсичности Соединения H (Этопозида), являющегося коммерчески пригодным противораковым средством.
Пример 11 испытаний.
Растворимость.
Измерили растворимости Соединения 1-1, 1-1-3, 3 и 3-1 в 0.1М фосфатном буферном растворе (pH 7.2) и в дистиллированной воде и получили следующие результаты.
Соединение 1-1-3 является L-Ala производной Соединения 1-1 и его получили в соответствии с примером С-1 (5), в то время как Соединение 3-1 является L-Ala производной Соединения 3 и его получили в соответствии с примером С-6 (4) (См. табл. 7)
Как показано в табл. 7, Соединения 1-1-3 и 3-1, Z которых является аминокислотным остатком, показывают растворимость, в 10 или более раз превышающую растворимость Соединения 1-1 и 3, Z которых является атомом водорода. Кроме того, растворимость Соединения 1-1 в нейтральном состоянии является настолько высокой, что оно является подходящим в качестве жидкого средства, например инъекционного раствора.
Как показано в вышеприведенных результатах испытаний, соединения этого изобретения показывают замечательную противоопухолевую активность не только против неплотных опухолей, таких как лимфоцитарная лейкозная опухоль, но также против различных плотных опухолей, которые встречаются в тканях, например, легкого, молочной железы, желудка, матки, кожи, кишечника, мочевого пузыря и носоглотки. Кроме того, соединения этого изобретения обладают сравнительно высокой безвредностью. Поэтому эти соединения пригодны в качестве средств для лечения или профилактики опухолей человека.
Соединения этого изобретения вводят в таком количестве, чтобы ингибировать опухоль, это количество изменяется в зависимости от фармакодинамических свойств соединений, способа введения, симптомов и возраста, цели введения (профилактика или лечение) и т.д. Однако обычно соединения вводят в количестве от около 0.25 мг до около 50 мг, предпочтительно от 0.5 мг до около 20 мг, в течение одного дня и на мг массы тела. Так например, пациенту, имеющему массу тела примерно 50 кг, вводят в день от около 13 мг до около 2.5 г, предпочтительно в целом от 25 мг до 1 г активного ингредиента. Вышеприведенная суточная доза может быть разделена на 2-4 дозы и введена отдельно.
Способ применения лекарственного средства может быть или пероральным, или парентеральным, но при этом рекомендуется парентеральное введение.
Соединение этого изобретения обычно вводят в форме лекарственных препаратов. Эти препараты можно получить путем смешивания соединений этого изобретения с лекарственными носителями. Так например, лекарственный носитель для жидких средств, используемых в качестве лекарственных препаратов для парентерального введения, содержит в качестве необходимого ингредиента растворитель и, если необходимо, вспомогательные средства, например тонизирующие вещества, солюбилизаторы, успокаивающие средства, регуляторы pH, буферы и консерванты.
Что касается растворителя, то обычно применяют воду, органические растворители, такие как пропиленгликоль, или смесь воды с органическим растворителем.
Примеры тонизирующих веществ включают сахара, например сорбит и маннит, и хлорид натрия, но более предпочтительными являются сахара.
Что касается регулятора pH, то могут быть использованы основания, например гидроксид натрия, и кислоты, например хлористоводородная кислота и фосфорная кислота.
Примеры солюбилизатора включают поверхностно-активные вещества, такие как Polysorbate 80 и Pluronic F 68, и органические кислоты, такие как молочная кислота и метансульфоновая кислота, которые могут образовывать вместе с соединениями этого изобретения кислую соль присоединения.
Что касается успокаивающих средств, то здесь можно использовать гидрохлорид лидокаина и гидрохлорид прокаина. Что касается консервантов, то можно применять бензиловый спирт, а в качестве примеров стабилизатора здесь можно рассмотреть антиоксидант, такой как аскорбиновая кислота. Что касается буферного раствора, то можно применять соли кислот, например фосфорной кислоты, лимонной кислоты и молочной кислоты.
Жидкие средства, например инъекционные растворы и настои, можно получить путем растворения или суспендирования, предпочтительно растворения, соединения этого изобретения в растворителе и, если необходимо, смешивания с другими вспомогательными средствами перед растворением или суспендированием или после растворения или суспендирования. Лиофилизованные лекарственные препараты можно получить путем сушки вымораживанием этих жидкостей. При введении лиофилизованные лекарственные препараты повторно растворяют или повторно суспендируют.
Что касается носителей в твердых лекарственных препаратах, таких как таблетки, капсулы, гранулы, тонкоизмельченные гранулы и порошки, то они могут быть использованы до тех пор, пока они не провзаимодействуют с соединениями этого изобретения, и до тех пор, пока они будут использоваться в данной области. Конкретные примеры таких носителей включают крахмалы, маннит, кристаллическую целлюлозу, карбоксиметилцеллюлозу и т.д.
Эти лекарственные препараты могут, кроме того, содержать ингредиенты, пригодные для консервативного лечения, иного, чем соединения этого изобретения.
Способы приготовления.
Способы приготовления соединений этого изобретения являются следующими. Соединения (I) этого изобретения и их соли могут быть получены в соответствии с (a) реакцией замещения пирролидина, (b) реакцией замыкания кольца, (c) реакцией окисления и т.д.
(a) Реакция замещения пирролидина.
Среди соединений этого изобретения формулы (I) и соединения (I-а), у которого Y является 1-пирролидинильной группой (Y'), имеющей заместитель, и его соль могут быть получены путем побуждения соединения, имеющего следующую форму (II), или его соли;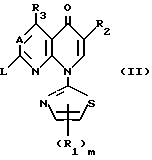
где L является элиминируемой группой, и A1, R1, R2, R3 и m являются такими, как они определены выше,
к взаимодействию с производной пирролидина следующей формулы (III):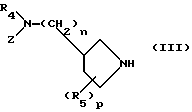
в которой R4, R5, Z, n и p являются такими, как они определены выше.
Что касается элиминируемой группы (L) в формуле (II), то здесь можно упомянуть те же самые группы, которые упоминались в случае формулы (I), где Y является элиминируемой группой, и предпочтительные примеры группы (L) включают атом галогена, низшую алкоксильную группу, низшую алкилтиогруппу, низшую алкилсульфинильную группу, низшую алкилсульфонильную группу, арилсульфонильную группу, низшую алкилсульфонилоксигруппу, арилсульфонилоксигруппу и т.д.
Вышеприведенную реакцию можно проводить или без растворителя или в соответствующем растворителе, и предпочтительно в присутствии основания при температуре в диапазоне от 10 до 150oC. Что касается растворителя, то можно использовать ацетонитрил, воду, этанол, пиридин, диметилсульфоксид, 1-метил-2-пирролидон и т. д. Что касается основания, то здесь используют, например такое, которое функционирует как акцептор кислоты, и конкретные примеры такого основания включают триэтиламин, 1,8-диазабицикло[5,4,0]-7- ундецен и карбонаты, например карбонат натрия и бикарбонат натрия. Соединение (III) может быть использовано в избытке для того, чтобы оно могло действовать как акцептор кислоты.
Соединения (II), которые используют в качестве исходного материала, являются также новыми и могут быть получены, например, посредством следующей реакции замыкания кольца.
(b) Реакция замыкания кольца.
Соединения (I) этого изобретения и его соли могут быть получены путем подвержения соединения, представленного следующей формулой (IV):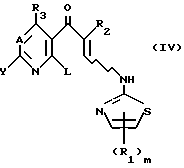
где L является элиминируемой группой, и R1, R2, R3, A, Y и m являются такими, как они определены выше,
реакции замыкания кольца.
Что касается элиминируемой группы L, то могут быть использованы те же самые группы, которые включены при определении Y и объяснены выше в отношении формулы (II).
Реакцию замыкания кольца можно проводить путем перемешивания смеси Соединения (IV) с растворителем в присутствии основания, например карбоната калия, карбоната натрия, гидрида натрия, калий-трет-бутоксида или фторида калия, количество которого в молях в 1-3 раза превышает количество Соединения (IV), при температуре в диапазоне от 30 до 150oC, предпочтительно от 30oC до 100oC в течение времени от 1 до 6 часов. Примеры подходящих растворителей включают этанол, диоксан, тетрагидрофуран, диметилформамид, диметилсульфоксид и т.д.
Соединения (IV), которые используют в качестве исходных материалов, являются также новыми и могут быть получены в соответствии с процессами, приведенными в примерах, которые будут описаны позже.
(с) Реакция окисления.
Соединения этого изобретения, представленные формулой (I), и их соли могут быть получены путем подвержения соединения, представленного следующей формулой (V);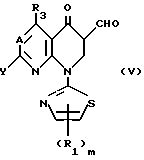
где R1, R3, A, Y и m являются такими, как они определены выше,
реакции окисления.
Реакцию окисления осуществляют путем смешивания вышеприведенного Соединения (V) с окислителем в растворителе и перемешивания полученной смеси в течение нескольких часов при температуре 100oC или ниже, предпочтительно до 50oC. Примеры окислителя включают 2,3-дихлор-5,6-дицианбензохинон, тетрахлор-1,4-бензохинон, тетрацианэтилен, палладий на углероде, N-бромсукцинимид и диоксид марганца. Что касается примеров растворителя, то могут быть рассмотрены 1,4-диоксан, толуол, ксилол, этанол, трет-бутанол, этилацетат, диметилформамид и т.д.
Соединения (I) этого изобретения и их соли могут быть также получены путем побуждения Соединения (I) этого изобретения, в котором Z является атомом водорода, к взаимодействию с аминокислотой или пептидом в соответствии с обычным методом, или путем аминирования Соединения (I), в котором R3 является атомом галогена, для того, чтобы превратить упомянутое соединение в соединение, в котором R3 является аминогруппой.
Когда соединения этого изобретения, полученные вышеупомянутым способом, являются сложноэфирными формами, упомянутые соединения можно превратить в формы карбоновой кислоты посредством гидролиза сложноэфирных частей с помощью обычного метода. Карбоновокислотные части Соединений (I) можно, кроме того, также этерифицировать посредством общепринятого способа. Далее, когда Z соединений этого изобретения является аминокислотным остатком или пептидным остатком, защищенным защитной группой, посредством обычного метода упомянутая защитная группа может быть элиминирована.
Соединения этого изобретения, полученные таким путем, могут быть выделены и очищены обычными способами. В соответствии с условиями выделения и очистки эти соединения получают в форме солей, свободных карбоновых кислот или свободных аминов, которые затем превращают из одной формы в другую в соответствии с назначениями, вследствие чего могут быть получены соединения желательных форм.
Когда соединения этого изобретения являются рецемическими, их можно, если это является необходимым, посредством известных методов разделить на соответствующие оптические изомеры. Стереоизомеры (цис-форма и транс-форма) соединений этого изобретения, если это является необходимым, можно отделить друг от друга в соответствии с общепринятым методом, например таким, как метод фракционированной кристаллизации или метод хроматографии.
В качестве исходного материала можно, конечно, использовать оптический изомер или стереоизомер и довести его до желательного соответствующего вещества, и такой метол является обычно выгодным.
Примеры.
Это изобретение будет подробно объяснено с помощью нижеприведенных рабочих примеров.
В дальнейшем Примеры Серии А включают конкретные примеры способов получения промежуточных соединений (II) [общей формулы (II)]. Примеры Серии В включают конкретные примеры способов получения исходных материалов (III) [общей формулы (III)]. Примеры Серии С включают конкретные примеры способов получения целевых веществ (I-а), и Примеры Серии D включают конкретные примеры способов получения лекарственных препаратов.
Кроме того, приложенные фиг. 15-19 показывают схемы реакций, которые представлены в последующих Примерах Серии А, Серии В и Серии С - 14.
1. Серия А.
Пример А-1. Получение промежуточного соединения (II) этилового эфира 7-хлор-1,4-дигидро- 4-оксо-1-(2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты.
(1)-1. В тетрагидрофуране (200 мл) растворили 2,6-дихлорпиридин (20 г) и к полученному раствору по каплям добавили раствор н-бутиллития (1.6 М) в н-гексане (84.5 мл) в потоке газа аргона при температуре -78oC в течение 30 минут. Затем полученный раствор перемешали в течение одного часа при этой же температуре и добавили значительный избыток диоксида углерода (твердого). После перемешивания в течение одного часа температуру подняли до -10oC и затем для установления pH раствора, равного 1-2, добавили воду и соляную кислоту, после чего полученный раствор экстрагировали этилацетатом. Затем полученный экстракт сушили над безводным сульфатом натрия и под пониженным давлением досуха отогнали растворитель.
К полученному твердому добавили тионилхлорид (40 мл) и полученную смесь нагревали в колбе с обратным холодильником в течение трех часов. Под пониженным давлением отогнали избыток тионилхлорида и затем под пониженным давлением перегнали сырой продукт для образования 2,6-дихлорникотиноилхлорида (19.8 г).
Температура кипения: 97-99oC/1мм Hg.
ИК (чистый) см-1: 1784.
(1)-2. Смесь 2,6-дихлорникотиновой кислоты (50.6 г) с тионилхлоридом (100 мл) нагрели в колбе с обратным холодильником в течение 2 часов. Под пониженным давлением отогнали избыток тионилхлорида и под пониженным давлением перегнали сырой продукт, получив при этом 2,6-дихлорникотиноилхлорид (44.9 г).
Температура кипения: 115-120oC/3 мм Hg.
ИК (чистый) см-1: 1784.
(2) Диэтиловый эфир этоксимагниймалоновой кислоты, полученный из металлического магния (5.36 г), диэтилового эфира малоновой кислоты (35.4 г) и этанола (27 мл), растворили в смешанном растворе, состоящем из тетрагидрофурана (35 мл) и толуола (140 мл). Полученный раствор охладили льдом, и к этому раствору при перемешивании по каплям добавили смешанный раствор, состоящий из тетрагидрофурана (19 мл) и толуола (32 мл), содержащий хлорид кислоты (44.9 г), полученный в (1)-1 или (1)-2. После завершения добавления по каплям полученную смесь перемешали в течение ночи при комнатной температуре. Смесь концентрировали под пониженным давлением, к остатку добавили водный раствор хлористоводородной кислоты и затем смесь экстрагировали этилацетатом. Полученный экстракт сушили над безводным сульфатом магния, под пониженным давлением отогнали растворитель и, таким образом, получили маслянистое вещество. К маслянистому веществу добавили воду (190 мл) и п-толуолсульфоновую кислоту (0.1 г), и смесь нагрели в колбе с обратным холодильником в течение 2 часов. После охлаждения смесь экстрагировали хлороформом и сушили над безводным сульфатом натрия, затем под пониженным давлением отогнали растворитель, получив при этом маслянистое вещество. Полученный сырой продукт отогнали под пониженным давлением до получения этилового эфира 2,6-дихлорникотиноилуксусной кислоты (45.2 г).
Температура кипения: 135-140oC/2мм Hg.
(3) Смесь из соединения (44.9 г), полученного выше в (2), уксусного ангидрида (43.8 г) и этилортоформиата (37.7 г) нагрели в колбе с обратным холодильником в течение одного часа. Смесь досуха концентрировали под пониженным давлением, при охлаждении льдом добавили диизопропиловый эфир (500 мл) и 2-аминотиазол (20 г) и затем полученную смесь перемешали при комнатной температуре в течение 5 часов. После фильтрации получили кристаллы этилового эфира 2-(2,6-дихлорникотиноил)-3-(2- тиазолиламино)акриловой кислоты (52.8 г).
Температура плавления: 119-122oC (перекристаллизовали из диизопропилового эфира).
ИК (KBr) см-1: 1700.
(4) Соединение (51.7 г), полученное выше в (3), растворили в диоксане (310 мл), затем добавили карбонат калия (21.4 г) и полученную смесь перемешали при 60oC в течение одного часа. После этого добавили смесь воды со льдом, полученную смесь нейтрализовали 10% водным раствором хлористоводородной кислоты и посредством фильтрации получили кристаллы. Кристаллы перекристаллизовали из смешанного раствора, состоящего из хлороформа и диизопропилового эфира, получив при этом этиловый эфир 7-хлор-1,4-дигидро-4-оксо-1-(2-тиазолил)-1,8- нафтиридин-3-карбоновой кислоты (44.6 г).
Температура плавления: 176-177oC.
ИК (KBr) см-1: 1724.
ЯМР (CDCl3) δ: 1.43 (t, 3H, J = 6.5 Hz), 4.45 (q, 2H, J=6.5 Hz), 7.38 (d, 1H, J = 3.5 Hz), 7.52 (d, 1H, J= 8.5 Hz), 7.75 (d, 1H, J = 3.5 Hz), 8.78 (d, 1Н, J = 8.5 Hz), 10.0 (s, 1H)
Пример A-2. Получение промежуточного соединения (II) этилового эфира 7-хлор-1-(4-фтор-2-тиазолил)-1,4- дигидро-4-оксо-1,8 -нафтиридин-3 -карбоновой кислоты.
Смесь, состоящую из сложного эфира (250 мг), полученного в Примере 1 (4), N-фтор-2,6-дихлорпиридинийтетрафторбората (240 мг) и 1,2- дихлорэтана (10 мл), нагрели в колбе с обратным холодильником в течение 2 дней.
К раствору добавили воду и полученный раствор экстрагировали хлороформом. После этого полученный экстракт сушили над безводным сульфатом натрия и под пониженным давлением отогнали растворитель. Полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ) и перекристаллизацией из этилацетата получили вышеуказанное соединение (40 мг).
Температура плавления: 174-175oC/
ИК(KBr)см-1: 1700/
ЯМР (CDCl3) δ: 1.42 (t, 3H, J=6.5 Hz), 4.45 (q, 2H,J= 6.5 Hz), 7.33 (d, 1H, J= 3.5 Hz), 7.51 (d, 1H, J= 8.5 Hz), 8.78 (d, 1H, J = 8.5 Hz), 9.85 (s, 1H).
Пример A-3. Получение промежуточного соединения (II) 5,7- дихлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3- карбоновой кислоты.
(1) К смеси, состоящей из 4-амино-2,6-дихлорпиридина (5,5 г), хлорида одновалентной меди (4,4 г) и концентрированной хлористоводородной кислоты (50 мл), при охлаждении льдом постепенно добавили нитрит натрия (3.5 г) и хлорид натрия. После этого полученную смесь перемешали в течение одного часа при этой же температуре и в течение полутора часов при комнатной температуре, добавили воду и смесь экстрагировали хлороформом. Затем полученный экстракт сушили над безводным сульфатом натрия, под пониженным давлением отогнали растворитель, получив при этом 2,4,6-трихлорпиридин (5,5 г).
ИК (чистый) см-1: 1563, 1357, 1155, 851, 823.
ЯМР (CDCl3) δ: 7,31 (s, 2H).
(2) К смеси, состоящей из соединения (5,5 г), полученного выше в (1), и тетрагидрофурана (55 мл), по каплям добавили раствор н-бутиллития (1,6 М) в гексане (20 мл) при температуре -78oC. Затем полученный раствор перемешали в течение одного часа при этой же температуре и добавили значительный избыток диоксида углерода (твердого). После перемешивания в течение одного часа температуру подняли до 0oC и затем добавили водный раствор хлористоводородной кислоты с тем, чтобы раствор был кислым, после чего полученный раствор экстрагировали этилацетатом. После этого полученный экстракт сушили над безводным сульфатом натрия и под пониженным давлением отогнали растворитель. К полученному остатку по каплям добавили диизопропиловый эфир, и посредством фильтрации получили 2,4,6- трихлорникотиновую кислоту (6.5 г).
Температура плавления: 138-141oC.
BK (KBr) см-1: 1715.
(3) Смесь соединения (6.5 г), полученного выше в (2), с тионилхлоридом (25 мл) нагревали в колбе с обратным холодильником в течение трех часов. Под пониженным давлением отогнали избыток тионилхлорида и под пониженным давлением перегнали сырой продукт, получив при этом 2,4,6-трифторникотиноил-хлорид (6.6 г).
Температура кипения: 93-95oC/1 мм Hg
ИК (чистый) см-1: 1791
(4) К смеси, состоящей из моноэтилового эфира малоновой кислоты (3.6 г) и тетрагидрофурана (30 мл), при температуре 0oС по каплям добавили раствор метилмагнийбромида (3М), растворенного в простом эфире (19 мл). После этого полученный раствор перемешали в течение одного часа при комнатной температуре и по каплям добавили смесь, состоящую из соединения (6.6 г), полученного выше в (3), и тетрагидрофурана (30 мл), и полученный раствор нагрели в течение полутора часов при 60oC в колбе с обратным холодильником. Под пониженным давлением отогнали растворитель, к полученному раствору добавили водный раствор хлористоводородной кислоты и затем смесь экстрагировали хлороформом. Полученный экстракт сушили над безводным сульфатом магния, под пониженным давлением отогнали растворитель и затем полученный сырой продукт перегнали под пониженным давлением, при этом получили этиловый эфир 2,4,6- трифторникотиноилуксусной кислоты (4.8 г).
Температура кипения: 160-162oC/2 мм Hg.
ИК (чистый) см-1: 1746.
(5) Смесь соединения (4.8 г), полученного выше в (4), уксусного ангидрида (4.2 г) и этилортоформиата (3.6 г) в течение полутора часов нагрели в колбе с обратным холодильником. Смесь досуха концентрировали под пониженным давлением, при охлаждении льдом добавили диизопропиловый эфир (100 мл) и 2-аминотиазол (1.6 г) и затем полученную смесь перемешали при комнатной температуре в течение трех часов. Под пониженным давлением отогнали растворитель и полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ) и перекристаллизацией из этилацетата получили этиловый эфир 2-(2,4,6-никотиноил)-3-(2- тиазолиламино)акриловой кислоты (4.0 г).
Температура плавления: 126-127oC.
ИК (KBr) см-1: 1691.
(6) Смесь, состоящую из соединения (4.0 г), полученного выше в (5), карбоната калия (1.5 г) и этилацетата (40 мл), нагрели при 60oC в течение одного часа. Под пониженным давлением отогнали растворитель, к полученному остатку добавили воду и затем образованную смесь экстрагировали хлороформом. Полученный экстракт сушили над безводным сульфатом натрия, под пониженным давлением отогнали растворитель и затем полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ), и перекристаллизацией из хлороформа получили этиловый эфир 5,7-дихлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты.
Температура плавления: 226-227oC.
ИК (KBr) см-1: 1737, 1692.
(7) Смесь сложного эфира (1.8 г), полученного выше в (6), с 20% водным раствором хлористоводородной кислоты (60 мл) нагрели в колбе с обратным холодильником в течение пяти часов. После охлаждения к смеси добавили воду, при фильтрации получили кристаллы, затем кристаллы промыли водой, получив при этом 5,7-дихлор-1,4- дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновую кислоту (1,4 г).
Температура плавления: 264-266oC.
ИК (KBr) см-1:1729.
Пример А-4. Получение промежуточного соединения (II) 5-амино-7- хлор-1,4-дигидро-4-оксо-1-(2-тиазолил)-1,8-нафтиридин-3- карбоновой кислоты.
(1) Смесь из соединения (500 мг), полученного в примере А-3 (6), бензиламина (140 мг), триэтиламина (280 мг) и толуола (15 мл) нагрели в колбе с обратным холодильником в течение 30 минут. Под пониженным давлением отогнали растворитель, к полученному остатку добавили воду и затем образованную смесь экстрагировали хлороформом. Полученный экстракт сушили безводным сульфатом натрия и под пониженным давлением отогнали растворитель. Полученный остаток подвергли перекристаллизации из этилацетата и получили этиловый эфир 5-бензиламино-7-хлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (510 мг).
Температура плавления: 141-143oC.
ИК (KBr) см-1: 1733  4,41 (q, 2H, J=7 Hz),
4,41 (q, 2H, J=7 Hz),
ЯМР (CDCl3) δ: 1.42 (t, 3H, J= 7Hz),  4.49 (q, 2H, J = 6.5 Hz), 6.47 (s, 1H), 7.31 (d, 1H, J= 3.5 Hz), 7.32-7.40 (m, 5H), 7.70 (d, 1H, J=3.5 Hz), 9.87 (s, 1H), 11.2-11.7 (m, 1H).
4.49 (q, 2H, J = 6.5 Hz), 6.47 (s, 1H), 7.31 (d, 1H, J= 3.5 Hz), 7.32-7.40 (m, 5H), 7.70 (d, 1H, J=3.5 Hz), 9.87 (s, 1H), 11.2-11.7 (m, 1H).
(2) Смесь, состоящую из сложного эфира (1.0 г), полученного выше в (1), концентрированной серной кислоты (2 мл) и уксусной кислоты (8 мл), перемешали в течение пяти часов при 110oC. После охлаждения добавили 8 мл воды и смесь перемешали при 110oC в течение одного часа. При фильтрации получили кристаллы, которые промыли водой, получив при этом 740 мл вышеназванного соединения.
Температура плавления: 264-265oC.
ИК (KB) см-1: 1727.
Пример А-5. Получение промежуточного соединения (II) этилового эфира 7-хлор-1,4-дигидро-4-оксо-1-(2-тиазолил)-5-трифтор-метил-1,8-нафтиридин-3-карбоновой кислоты.
(1) Смесь 2,6-дихлор-4-трифторметилпиридина (5 г) с тетрагидрофураном (50 мл) охладили до -78oC и к смеси по каплям добавили раствор н-бутиллития (1.6 М) в н-гексане (16 мл), которую затем перемешали в течение 30 минут. Затем к этой смеси добавили значительный избыток диоксида углерода (твердого) и затем перемешали в течение одного часа. После того как температура поднялась до 0oC, смесь экстрагировали этилацетатом и разбавленной хлористоводородной кислотой и полученный органический слой сушили над сульфатом натрия. Под пониженным давлением отогнали растворитель, к полученному остатку добавили тионилхлорид (20 мл) и затем полученную смесь нагрели в колбе с обратным холодильником в течение шести часов. Под пониженным давлением отогнали избыточный тионилхлорид, полученный остаток перегнали под пониженным давлением, получив при этом 2,6-дихлор-4- трифтор-метилникотиноилхлорид (3.8 г).
Температура кипения: 77-78oC/2 мм Hg.
ИК (чистый) см-1: 1797.
(2) К смеси магния (0.36 г) с этанолом (1.5 г) добавили каплю тетрахлорида углерода и затем к полученной смеси по каплям добавили смесь, состоящую из диэтилмалоната (2.4 г), этанола (1.5 мл) и толуола (10 мл), и затем перемешали в течение двух часов. После охлаждения смеси льдом к смеси по каплям добавили смесь, состоящую из соединения (3.8 г), полученного выше в (1), и тетрагидрофурана (10 мл) и перемешали в течение трех часов при комнатной температуре. Затем смесь экстрагировали этилацетатом и разбавленной хлористоводородной кислотой и полученный органический слой сушили над сульфатом натрия. Под пониженным давлением отогнали растворитель, к полученному остатку добавили воду (20 мл) и п-толуолсульфоновую кислоту (50 мг) и затем полученную смесь нагрели в колбе с обратным холодильником в течение трех часов. Затем смесь экстрагировали хлороформом и водой и полученный органический слой сушили над сульфатом натрия. Под пониженным давлением отогнали растворитель, получив при этом этиловый эфир 2,6-дихлор-4-трифторметилникотиноилуксусной кислоты (0.9 г).
ИК (чистый) см-1: 1744, 1721.
MS (m/z): 330 (МН+).
(3) Смесь, состоящую из соединения (0.9 г), полученного выше в (2), этилортоформиата (0.6 г) и уксусного ангидрида (0.7 г), нагрели в колбе с обратным холодильником при 140oC в течение 1.5 часов и затем концентрировали досуха под пониженным давлением. К полученному остатку добавили изопропиловый эфир (20 мл) и затем при охлаждении льдом добавили 2-аминотиазол (0.3 г). После перемешивания в течение 3 часов при комнатной температуре отогнали растворитель. К этому остатку для экстракции добавили хлороформ и воду, и полученный органический слой сушили над сульфатом натрия. Под пониженным давлением отогнали растворитель и полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ), получив при этом этиловый эфир 2-(2,6-дихлор-4- трифторметилникотиноил)-3-(2-тиазолиламино) акриловой кислоты (0.37 г).
ИК (чистый) см-1: 1713.
MS (m/z): 440 (МН+).
(4) Смесь, состоящую из соединения (0.37 г), полученного выше в (3), карбоната калия (0.13 г) и этилацетата (10 мл), нагрели в колбе с обратным холодильником в течение 15 минут. К смеси для экстракции добавили этилацетат и воду, и полученный органический слой сушили над сульфатом натрия. Под пониженным давлением отогнали растворитель и полученный остаток подвергли перекристаллизации из этилацетата, получив при этом вышеуказанный этиловый эфир 7-хлор- 1,4-дигидро-4-оксо-1-(2-тиазолил) -5-трифторметил-1,8- нафтиридин-3-карбоновой кислоты (0.27г).
Температура плавления: 184-185oC
ИК (KBr)см-1: 1736, 1703
Пример А-6. Получение промежуточного соединения (II) этилового эфира 5,8-дигидро-2- метансульфонил-5-оксо-8-(2-тиазолил) пиридо [2,3-d] пиримидин-6-карбоновой кислоты.
(1) Раствор моноэтилового эфира малоновой кислоты (12.3 г), растворенного в тетрагидрофуране (80 мл), охладили льдом и затем к полученному раствору по каплям добавили раствор метилмагнийбромида (3М) в простом эфире (64 мл). После перемешивания полученной смеси в течение 20 минут к смеси по каплям добавили раствор 2-метилтио-4-хлорпиримидин-5- карбонилхлорида (8.6 г), растворенного в тетрагидрофуране (100 мл), и затем перемешали в течение двух часов при комнатной температуре. Эту реакционную смесь влили в смесь воды со льдом и затем к полученному раствору для установления pH 5-6 добавили концентрированную хлористоводородную кислоту и раствор экстрагировали этилацетатом. Полученный экстракт сушили над безводным сульфатом натрия и под пониженным давлением отогнали растворитель. Затем посредством силикагельной колоночной хроматографии осуществили очистку (элюент: хлороформ) и таким образом получили этиловый эфир 3-(2-метилтио-4-хлорпиримидин-5- ил)-3-оксопропионовой кислоты (8.0 г).
ИК (чистый) см-1: 1743.
MS (m/z): 275 (МН+).
(2) Смесь, состоящую из соединения (7.95 г), полученного выше в (1), ортоэтилформиата (6.80 г) и уксусного ангидрида (7.76 г), нагрели в колбе с обратным холодильником при 130oC в течение одного часа и затем концентрировали под пониженным давлением. К смеси при охлаждении добавили диизопропиловый эфир (100 мл) и 2-аминотиазол (3.28 г) и перемешивали всю ночь при комнатной температуре. При фильтрации получили кристаллы, которые затем промыли диизопропиловым эфиром. Эти кристаллы растворили в 1,4-диоксане (70 мл), к полученному раствору при охлаждении добавили карбонат калия (2.72 г) и полученную смесь перемешали при комнатной температуре в течение пяти часов. Эту смесь охладили льдом, к ней добавили смесь воды со льдом (200 мл) и затем нейтрализовали 10% водным раствором хлористоводородной кислоты. При фильтрации получили кристаллы, которые последовательно промыли водой, 1,4- диоксаном и диизопропиловым эфиром и таким образом получили этиловый эфир 5,8-дигидро-2-метилтио-5-оксо-8-(2-тиазолил)- пиридо[2,3-d] -пиримидин-6-карбоновой кислоты (6.0 г).
Температура плавления: 183-184oC.
ИК (KBr) см-1: 1736.
(3) Раствор соединения (5.99 г), полученного выше в (2), растворенного в метиленхлориде (450 мл), охладили льдом и к полученной смеси постепенно добавили 80% м-хлорпербензойную кислоту (9.30 г), и полученную смесь перемешивали всю ночь при комнатной температуре. Затем смесь последовательно промыли водным раствором тиосульфата натрия, водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. После этого смесь сушили над сульфатом натрия и под пониженным давлением отогнали растворитель. Затем перекристаллизацией из смешанного раствора, состоящего из этилацетата и диизопропилового эфира, получили вышеуказанный этиловый эфир 5,8-дигидро-2-метансульфонил-5-оксо-8-(2-тиазолил) пиридо[2, З-d] пиримидин-6-карбоновой кислоты (4,48 г).
Температура плавления: 185-187oC.
ИК (KBr) см-1: 1741.
2. Серия В.
Пример В-1. Получение транс-3-(N-трет-бутоксикарбонилметиламино)-4- метилпирролидина, являющегося исходным материалом, представленным соединением формулы (III).
(1) Транс-3-амино-1-бензил-4-метилпирролидин (19 г) растворили в метиленхлориде (200 мл) и к полученному раствору при охлаждении льдом добавили раствор ди-трет-бутилкарбоната (22.9 г) в метиленхлориде (20 мл). Полученную смесь перемешали при комнатной температуре в течение одного часа. Затем этот реакционный раствор концентрировали под пониженным давлением, при этом получили транс-1-бензил-3-(трет- бутоксикарбониламино)-4-метилпирролидин (28.2 г).
Температура плавления: 138-140oC (перекристаллизовали из этилацетата-н-гексана).
ИК (KBr) см-1: 3198, 1706.
MS (m/z): 291 (МН+).
(2) 70% толуоловый раствор (40 мл) натрийбис(2-метокси) алюминий-гидрида растворили в 150 мл толуола и к полученному раствору при охлаждении льдом постепенно добавили соединение, полученное выше в (1), (10 г). Полученную реакционную смесь нагрели в колбе с обратным холодильником в течение одного часа и после охлаждения льдом избыток реагента разложили водой. Затем нерастворимое вещество отделили фильтрацией, полученный фильтрат сушили над безводным сульфатом магния и под пониженным давлением отогнали растворитель. Полученный остаток растворили в метиленхлориде (100 мл) и к полученному раствору при охлаждении льдом добавили раствор ди-третбутилкарбоната (7.5 г) в метиленхлориде (10 мл). Полученную смесь перемешали при комнатной температуре в течение 3.5 часов и затем концентрировали под пониженным давлением. Образованный остаток очистили силикагельной колоночной хроматографией (элюент : н-гексан : этилацетат = 5:1), при этом получили транс-1-бензил-3-(N-трет-бутоксикарбонилметиламино) -4- метилпирролидин (9.9 г)
ИК (чистый вес) см-1: 1694.
MS (m/z): 305 (МН+).
(3) Соединение (1.52 г), полученное выше в (2), растворили в этаноле (50 мл), к полученному раствору добавили 10% палладий на углероде (200 мг) и при 50oC поглотили теоретическое количество водорода. После этого фильтрацией отделили катализатор, под пониженным давлением отогнали растворитель и таким образом получили транс-3-(N-трет-бутоксикарбонил-метиламино)-4- метилпирролидин (950 мг).
ИК (чистый) см-1: 3337, 1685.
MS (m/z): 215 (МН+).
Пример В-2. Получение (+) -Транс-3- (N-трет-бутоксикарбонил-метиламино)-4-метоксипирролидина, являющегося исходным материалом, представленным соединением формулы (III).
(1) Транс-3-амино-1-бензил-4-метоксипирролидин (рацемический: 22,4 г), раскрытый в опубликованном патенте Японии, выложенном для ознакомления, N 69474/1990, и 19.6 г L-винной кислоты растворили в метаноле (350 мл), и полученный раствор оставили в этом состоянии на 7 часов. Фильтрацией получили осажденный L-тартрат и затем его подвергли перекристаллизации с помощью метанола и воды, таким образом получили транс-3-амино-1-бензил-4- метоксипирролидин-L-тартрат (14.1 г), имеющий далее указанные физические свойства. Затем весь маточный раствор объединили и под пониженным давлением отогнали растворитель, затем добавили насыщенный рассол и потом карбонат кальция для того, чтобы pH полученной смеси стал основным. Затем смесь экстрагировали этилацетатом. Полученный экстракт промыли насыщенным рассолом, затем сушили над безводным сульфатом натрия и под пониженным давлением отогнали растворитель. Полученный остаток и D-винную кислоту (6.73 г) растворили в метаноле (180 мл) на 7 часов. Фильтрацией получили осажденный D-тартрат, который подвергли перекристаллизации метанолом и водой, вследствие чего получили транс-3-амино-1-бензил-4-метоксипирролидин-D-тартрат (9.9 г), имеющий далее указанные физические свойства.
L-тартрат
Температура плавления 206-208oC (разложенный).
[α]
Элементарный анализ (%): в виде С12H18N2O • 3/2 C4Н6O6
Вычисленное значение: С 50.11; H 6.31; N 6.49;
Найденное значение: С 49.85; H 6.26; N 6.27.
D-тартрат
Температура плавления: 207-209oC (разл.).
[α]
Элементарный анализ (%): в виде С12H18N2O • 3/2 C4H6O6.
Вычисленное значение: С 50.11; H 6.31; N 6.49.
Найденное значение: С 50.35; H 6.32; N 6.47.
(2) К L-тартрату (3.65 г), полученному выше в (1), добавили ненасыщенный рассол, полученную смесь нейтрализовали карбонатом калия и затем экстрагировали этилацетатом. Полученный экстракт промыли насыщенным рассолом и затем сушили над безводным сульфатом натрия. Затем под пониженным давлением отогнали растворитель, и таким образом получили (+)- транс-3-амино-1-бензил-4-метоксипирролидин (1.23 г).
[α]
(3) Соединение (5.74 г), полученное выше в (2), растворили в метаноле (65 мл) и при охлаждении льдом к полученному раствору добавили ди-третбутилкарбонат (7.29 г), затем перемешивали при этой же температуре в течение 30 минут и при комнатной температуре в течение 4 часов. Под пониженным давлением отогнали растворитель и полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол = 50:1), таким образом получили (+)-транс-1-бензил-3- (трет- бутоксикарбониламино) -4-метоксипирролидин (8.55 г).
Температура плавления: 44-45oC.
[α]
(4) Алюмогидрид лития (3.43 г) суспендировали в безводном тетрагидрофуране (150 мл), и к полученной суспензии по каплям добавили безводный тетрагидрофурановый раствор (50 мл) соединения (8.4 г), полученного выше в (3), и образованную смесь перемешали при комнатной температуре в течение одного часа. После этого смесь нагрели в колбе с обратным холодильником в течение 5 часов, при охлаждении льдом разложили избыток реагента с помощью воды и затем нерастворимое вещество удалили фильтрацией. Затем полученный фильтрат экстрагировали этилацетатом. Образованный экстракт промыли насыщенным рассолом и после этого сушили над безводным сульфатом натрия. Под пониженным давлением отогнали растворитель и полученный остаток растворили в метиленхлориде (180 мл), к полученному раствору при охлаждении льдом добавили ди-трет- бутилбикарбонат (6.3 г). Образованную смесь перемешали при этой же температуре в течение 30 минут и при комнатной температуре в течение 2 часов и затем под пониженным давлением отогнали растворитель. Образованный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ: метанол = 50: 1), таким образом получили (+)-транс-1-бензил-3-(N-трет- бутоксикарбонилметиламино)-4-метоксипирролидин (8.26 г).
[α]
(5) Желательный (+)-транс-3-(N-трет- бутоксикарбонилметиламино)-4-метоксипирролидин (5.59 г) получили из соединения (8.15 г), полученного выше в (4), тем же самым способом, что и в примере В-1 (3).
[α]
ИК (чистый) см-1: 3318, 1693.
МS (m/z): 231 (MH+).
Пример В-3. Получение (-)-транс-3-(N-трет-бутоксикарбонилметиламино)-4- метоксипирролидина, являющегося исходным материалом, представленным соединением формулы (III).
(1) (-) -Транс-3-амино-1- бензил-4-метоксипирролидин (1.01 г) получили из D-тартрата (2.57 г), полученного в примере В-2(1), тем же самым способом, что и в примере В-2(2).
[α]
(2) (-) -Транс-1-бензил-3-(трет-бутоксикарбониламино) -4- метоксипирролидин (4.5 г) получили из соединения (3.03 г), полученного выше в (1), тем же самым способом, что и в примере В-2 (3).
Температура плавления: 44-45oC.
[α]
(3) (-) -Транс-1-бензил-1-3-(N-трет-бутоксикарбонилметиламино)-4- метоксипирролидин (4.25 г) получили из соединения (4.25 г), полученного выше в (2), тем же самым способом, что и в примере В-2 (4).
[α]
(4) Желательный (-)-Транс-3-(N-трет-бутоксикарбонилметиламино)-4- метоксипирролидин (2.81 г) получили из соединения (4.25 г), полученного выше в (3), тем же самым способом, что и в примере В-2 (5).
[α]
ИК (чистый) см-1: 3318, 1693
M/S (m/z): 231 (MH+).
Пример В-4. Получение 3-(N-трет- бутоксикарбонилметиламино) -3-метилпирролидина.
(1) 1-Бензил-3-(N-трет-бутоксикарбонилметиламино) -3-метилпирролидин (28.3 г) получили из 3-амино-1-бензил-3-метилпирролидина (20 г), тем же самым способом, что и в примере В-1 (1).
ИК (чистый) см-1: 3356, 1716, 1697.
MS (m/z): 291 (MH+).
(2) 1-Бензил-3- (N-трет-бутоксикарбонилметиламино)-3-метилпирролидин (7.5 г) получили из соединения (11.4 г), полученного выше в (1), тем же самым способом, что и в примере В-1 (2).
ИК (чистый) см-1: 1697.
MS (m/z): 305 (MH+).
(3) Желательный 3-(N-трет-бутоксикарбонилметиламино)-3- метилпирролидин (5.5 г) получили из соединения (7.5 г), полученного выше в (2), тем же самым способом, что и в примере В-1 (3).
ИК (чистый) см-1: 3337, 1682.
MS (m/z): 215 (МН+).
3. Серия С.
Пример С-1. Получение желательного продукта (I)
1,4-Дигидро-7-(транс-3-метокси-4-метиламино-1-пирролидинил) -4-оксо-(2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты, ее соли и ее -А а производной.
(1) К суспензии, состоящей из этилового эфира 7-хлор- 1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3- карбоновой кислоты (7.1 г), полученного в примере А-1 (4), транс-3-метокси-4-метиламинопирролидиндихлорида (6.0 г) и ацетонитрила (150 мл), добавили триэтиламин (18 мл). Полученную реакционную смесь перемешали при комнатной температуре в течение 5 часов и затем концентрировали под пониженным давлением. После этого добавили водный раствор бикарбоната натрия и полученную смесь экстрагировали хлороформом. Затем полученный экстракт сушили над безводным сульфатом натрия, под пониженным давлением отогнали растворитель и образованный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол = 6:1), таким образом получили этиловый эфир 1,4-дигидро-7-(транс-3- метокси-4-метиламино-1-пирролидинил) -4-оксо-1- (2-тиазолил) -1, 8-нафиридин-3-карбоновой кислоты (6.1 г).
Температура плавления 72-76oC.
(2) Раствор, состоящий из вышеприведенного сложного эфира (6.0 г) и 18% водного раствора хлористоводородной кислоты (100 мл), перемешали при 100oC в течение 28 часов. При фильтрации получили кристаллы, которые промыли смешанным раствором, состоящим из этанола и диизопропилового эфира, при этом получили гидрохлорид 1,4-дигидро-7-(транс-3-метокси-4-метиламино-1-пирролидинил) -4- оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (Соединение I-1) (4.45 г).
Температура плавления 270-273oC.
(3) Раствор, состоящий из гидрохлорида (51.5 г), полученного выше в (2), воды (500 мл) и водного раствора аммиака (40 мл), перемешивали всю ночь при 50oC. К этому раствору добавили ацетонитрил и концентрировали под пониженным давлением, фильтрацией получили кристаллы. Кристаллы промыли водой и ацетонитрилом, при этом получили 1,4-дигидро-7- (транс-3-метокси-4-метиламино-1-пирролидинил) -4-оксо-1- (2- тиазолил)-1,8-нафтиридин-3-карбоновую кислоту (Соединение I-1-1) (32.6 г).
Температура плавления: 290-292oC (разложенная).
(3) Раствор, состоящий из гидрохлорида (51.5 г), полученного выше в (2), воды (500 мл) и водного раствора аммиака (40 мл), перемешивали всю ночь при 50oC. К этому раствору добавили ацетонитрил и концентрировали под пониженным давлением, фильтрацией получили кристаллы. Кристаллы промыли водой и ацетонитрилом, при этом получили 1,4-дигидро-7-(транс-3-метокси-4- метиламино-1-пирролидинил) -4-оксо-1- (2-тиазолил) -1,8- нафтиридин-3-карбоновуго кислоту (Соединение I-1-1) (32.6 г).
Температура плавления: 290-292oС (разложенный).
(4) Смесь, состоящую из соединения (2.0 г), полученного выше в (3), молочной кислоты (3.1 г) и дистиллированной воды (4 мл), нагрели при 60oC для растворения. После этого полученный раствор охладили до комнатной температуры, добавили этанол (70 мл), фильтрацией получили кристаллы, промыли их этанолом, получив при этом 2 г лактата (Соединение I-1-2).
Температура плавления: 288-291oC (разложенный).
(5) К смеси, состоящей из сложного эфира (1.89 г), полученного выше в (1), N-трет-бутоксикарбонил-L-аланина (1.26 г) и метиленхлорида (80 мл), добавили 1.27 г гидрохлорида 1- этил-3-(3-диметил-аминопропил)карбодиимида (WSC), и полученную смесь перемешали при комнатной температуре в течение 3 часов. После промывки водой смесь сушили над безводным сульфатом натрия и затем концентрировали под пониженным давлением. Образованный концентрат очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол = 50: 1) до получения этилового эфира 7-{транс-3-[N-(N-трет-бутоксикарбонил-L- аланил)] метиламино-4-метокси-1-пирролидинил} -1,4-дигидро-4-оксо-1- (2-тиазолил) -1, 8-нафтиридин-3-карбоновой кислоты (2.15 г).
Температура плавления: 120-123oC.
[α]
Смесь, состоящую из этого этилового эфира (1.71 г), 0.5 N хлористоводородной кислоты (42 мл) и этанола (22 мл), перемешивали при нагревали (80oC) в течение 17.5 часов. Полученный раствор концентрировали под пониженным давлением, фильтрацией получили кристаллы, которые затем промывали 10% хлористоводородной кислотой и этанолом, при этом получили 1.16 г гидрохлорида 7- [транс-3-(N-L-аланилметиламино)-4-метокси-1-пирролидинил]-1,4- дигидро-4-оксо-1- (2-тиазолил) -1, 8-нафиридин-3-карбоновой кислоты (Соединение I-1-3).
Температура плавления 230-233oC.
[α]
Пример С-2. Получение желательного соединения (I)
(+) -1,4- дигидро-7- (транс-3-метокси-4-метиламино-1-пирролидинил) -4-оксо- 1- (2-тиазолил) -1,8-нафтиридин-З-карбоновой кислоты (Соединение I-2-1) и ее гидрохлорида (Соединение I-2).
(1) При использовании этилового эфира 7-хлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8- нафтиридин-3-карбоновой кислоты, полученного в примере А-1 (4), и (+)-транс-3-(N-трет-бутоксикарбонилметиламино)-4- метоксипирролидина, полученного в примере В-2, получили этиловый эфир (аморфный) (+)-7-[транс-3-(N- трет-бутоксикарбонилметиламино)-4- метокси-1-пирролидинил]- 1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3- карбоновой кислоты, тем же самым способом, что и в примере С-1(1).
[α]
(2) Из этилового эфира, полученного выше в (1), в соответствии со способом, описанным в примере С-1 (2), получили вышеназванный гидрохлорид (Соединение I-2).
Температура плавления (разложенный) 278-282oC.
[α]
(3) Раствор, состоящий из гидрохлорида (28.1 г), полученного выше в (2), воды и водного раствора аммиака (25 мл), перемешивали всю ночь при 50oC и затем концентрировали под пониженным давлением. Кристаллы промывали водой и метанолом, при этом получили желательную карбоновую кислоту. (Соединение I-2-1) (19.5 г).
Температура плавления: 268- 271oC.
[α]
Пример C-3. Получение желательного соединения (I) гидрохлорида (-)-1,4-Дигидро-7- (транс-3-метокси-4-метиламино-1-пирролидинил) -4-оксо-1- (2- тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (Соединение I-3).
(1) При использовании этилового эфира 7-хлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1, 8-нафтиридин-3-карбоновой кислоты, полученного в примере А-1 (4), и (-)-транс-3-(N-трет-бутоксикарбонилметиламино)-4-метоксипирролидина, полученного в примере В-3, получили этиловый эфир (аморфный) (-)-7-[транс-3-(N-трет- бутоксикарбонилметиламино) -4-метокси-1-пирролидинил] -1,4-дигидро- 4-оксо-1-(2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты, тем же самым способом, что и в примере С-1 (1).
[α]
(2) Указанное в заголовке соединение получили из сложного этилового эфира, полученного выше в (1), в соответствии с методикой, описанной в примере С-1 (2).
Температура плавления: 278-282oC (разложенный).
[α]
Примеры С-4 и C-5. Получение желательного соединения (I).
В соответствии с почти той же самой методикой, которая описана в примере С-1, получили соединения, приведенные в табл. 8.
Пример С-6. Получение желательного продукта (I) гидрохлорида 7- (3-Амино-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты (Соединение 3) и его L-Ala производной (Соединение 3-1).
(1) К суспензии, состоящей из сложного эфира (2.0 г), полученного в Примере А-1 (4), и ацетонитрила (80 мл), по каплям добавили ацетонитрил (10 мл), содержащий З-аминопирролидин (1.6 г). Образованную смесь перемешали при комнатной температуре в течение полутора часов и фильтрацией получили кристаллы. Путем перекристаллизации из смешанного раствора, состоящего из хлороформа, метанола и диизопропилового эфира, получили этиловый эфир (1.89 г) 7-(3- амино-1-пирролидинил)-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8- нафтиридин-3-карбоновой кислоты.
Температура плавления: 219-221oC.
(2) Суспензию, состоящую из вышеприведенного сложного эфира (1.0 г) и 10% водного раствора хлористоводородной кислоты (15 мл), перемешали при 95oC в течение 3.5 часов. Фильтрацией получили кристаллы, которые промыли смешанным раствором, состоящим из хлороформа и метанола, при этом получили гидрохлорид 7- (3-амино- 1-пирролидинил) -1,4-дигидро-4-оксо-1-(2-тиазолил)-1,8- нафтиридин-3-карбоновой кислоты (Соединение 3) (0.93 г).
Температура плавления: 267oC (разложенный).
(3) К смеси, состоящей из сложного этилового эфира (2.1 г), полученного выше в (1), N-трет-бутоксикарбонил-L-ананила (1.5 г) и метиленхлорида (80 мл), добавили 1.6 г гидрохлорида 1-этил-3-(3-диметиламинопропил)- карбодиимида (WSC), и полученную смесь перемешали при комнатной температуре в течение двух часов. После промывки водным раствором бикарбоната натрия смесь сушили над безводным сульфатом натрия, концентрировали под пониженным давлением и затем очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол = 20:1), получив при этом этиловый эфир 7-[3-(N-третбутоксикарбонил- L-аланил) амино-1-пирролидинил]-1,4-дигидро-4- оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (2.9 г).
Температура плавления: 138-140oC.
[α]
(4) Смесь, состоящую из сложного эфира (1.4 г), полученного выше в (3), 0.5 N хлористоводородной кислоты (30 мл) и этанола (16 мл), перемешали при 70oC в течение двух дней. Под пониженным давлением концентрировали растворитель и перекристаллизацией из 10% хлористоводородной кислоты и этанола получили гидрохлорид 7-(3-L- аланиламино-1-пирролидинил)-1,4-дигидро-4-оксо-1- (2-тиазолил) - 1,8-нафтиридин-3-карбоновой кислоты (Соединение 3-1) (0.77 г).
Температура плавления: 229-231oC (разложенная).
[α]
Примеры С-7 - С-10. Получение желательного продукта (I).
Почти тем же самым способом, который упомянут в Примере С-1, получили соединения, приведенные в табл. 9.
Пример С-11. Получение желательного продукта (I) гидрохлорида 7- (3-Амино-1-пирролидинил) -1- (4-фтор-2-тиазолил) - 1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты (Соединение 8).
(1) Смесь, состоящую из этилового эфира 7-хлор-1-(4-фтор-2- тиазолил) -1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты (30 мг), полученного в примере А-2, 3-(трет-бутоксикарбониламино)- пирролидина (19 мг), триэтиламина (26 мг) и ацетонитрила (10 мл), перемешали при комнатной температуре в течение 30 минут. Под пониженным давлением отогнали растворитель и добавили воду, затем полученную смесь экстрагировали хлороформом. Образованный экстракт сушили над безводным сульфатом натрия и под пониженным давлением отогнали растворитель. Образованный остаток подвергли перекристаллизации с использованием этилацетата и таким образом получили этиловый эфир 7-(3-трет-бутоксикарбониламино-1- пирролидинил)-1-(4-фтор-2 -тиазолил) -1, 4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты (40 мг). Т.пл. 233-234 oС.
(2) Раствор, состоящий из вышеприведенного сложного эфира (40 мг) и 20% хлористоводородной кислоты (2 мл), нагрели в колбе с обратным холодильником в течение 1.5 часа. После охлаждения кристаллизацией получили кристаллы, промыли их разбавленным водным раствором хлористоводородной кислоты и таким образом получили названное в заголовке соединение 8 (32 мг).
Температура плавления: 283-284oC (разложенная).
Пример С-12. Получение желательного продукта (I) Гидрохлорида 5-Амино-7-(3-амино-1-пирролидинил) -1,4-дигидро-4- оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (Соединение 9).
(1) 5-Амино-7-хлор-1,4-дигидро-4-оксо-1- (2- тиазолил) -1,8-нафтиридин-3-карбоновая кислота (1.0 г), полученная в примере А-4 (2), и аминопирролидин (800 мг) провзаимодействовали почти тем же самым путем, как это описано в Примере С-1, таким образом получили названное в заголовке Соединение 9 (610 мг).
Температура плавления: 261-263oC.
Пример С-13. Получение желательного продукта (I).
Почти тем же самым способом, который упомянут в Примере С-1, получили соединения, приведенные в табл. 10
Пример С-14. Получение желательного продукта (I) гидрохлорида 3-формил-1,4-дигидро-7-(транс-3-метокси-4- метиламино-1-пирролидинил) -4-оксо-1-(2-тиазолил) -1,8- нафтиридина (Соединение II).
(1) К суспензии, состоящей из этилового эфира (8.49 г) 7-хлор-1,4-дигидро-4-оксо-1- (2- тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (8.49 г), полученного в Примере А-1 (4), дигидрохлорида транс-3-метокси-4- метиламинопирролидина (6.5 г) и ацетонитрила (400 мл), добавили триэтиламин (13.4 мл). После перемешивания в течение всей ночи при комнатной температуре полученную реакционную смесь концентрировали под пониженным давлением. Затем добавили водный раствор бикарбоната натрия и полученную смесь экстрагировали хлороформом. Полученный экстракт промыли насыщенным рассолом, затем сушили над безводным сульфатом натрия и после этого под пониженным давлением отогнали растворитель. К полученному остатку добавили метиленхлорид (500 мл) и при охлаждении льдом к полученной смеси добавили ди-трет-бутилбикарбонат (6.4 г). После перемешивания в течение всей ночи при комнатной температуре образованную реакционную смесь концентрировали под пониженным давлением и полученный остаток очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол = 100:1), при этом получили этиловый эфир 7-[транс-3-(N-трет-бутоксикарбонилметиламино) -4-метокси-1-пирролидинил] -1,4-ди-гидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (9.1 г).
ИК (KBr) см-1: 1735, 1695
MS (m/z): 530 (MH+)
(2) К смеси, состоящей из соединения (8.95 г), полученного выше в (1), и этанола (150 мл), добавили 1N водный раствор гидроксида натрия (150 мл), и полученную смесь перемешали при этой же температуре в течение 30 минут и в течение ночи при комнатной температуре. Затем с помощью водного раствора уксусной кислоты смесь сделали кислой и экстрагировали хлороформом. Полученный экстракт промыли насыщенным рассолом и затем сушили над безводным сульфатом натрия. Под пониженным давлением отогнали растворитель и таким образом получили 7-[транс-3-(N-трет- бутоксикарбонилметиламино) -4-метокси-1-пирролидинил] -1,4- дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновую кислоту (7.19 г).
Температура плавления: 185-188oC.
ИК (KBr) см-1: 1715, 1690.
MS (m/z): 502 (МН+).
(3) К смеси, состоящей из соединения (7.15 г), полученного выше в (2), и метанола (400 мл), при охлаждении льдом добавили 2.16 г боргидрида натрия, и полученную смесь перемешали в течение 15 минут при этой же температуре и затем перемешивали всю ночь при комнатной температуре. Под пониженным давлением отогнали растворитель и полученный остаток экстрагировали хлороформом. Полученный экстракт промыли насыщенным рассолом и затем сушили над безводным сульфатом натрия. Под пониженным давлением отогнали растворитель и полученный остаток затем очистили силикагельной колоночной хрома- тографией (элюент: хлороформ : метанол = 100:1), при этом получили 7-[транс-3-(N-трет-бутоксикарбонилметиламино) -4-метокси-1- пирролидинил]-1,2,3,4-тетрагидро-4-оксо-1- (2-тиазолил) -1,8- нафтиридин (3.9 г).
ИК (чистый) см-1: 1690.
MS (m/z): 460 (MH+).
(4) Соединение (3.8 г), полученное выше в (3), растворили в тетрагидрофуране (500 мл) и к полученному раствору по каплям добавили 6.1 мл раствора н-бутиллития (1.6 М) в н-гексане при температуре -78oC. После этого полученный раствор перемешали в течение 30 минут при этой же температуре, добавили этилформиал (1.34 мл), затем медленно подняли температуру и полученную смесь перемешивали всю ночь. Под пониженным давлением отогнали растворитель, к полученному остатку добавили водный раствор уксусной кислоты и затем образованную смесь экстрагировали хлороформом. Полученный экстракт промыли насыщенным рассолом и затем сушили над безводным сульфатом натрия. Под пониженным давлением отогнали растворитель и таким образом получили 7- [транс-3-(N-трет-бутоксикарбонил-метиламино) -4-метокси-1- пирролидинил]-3-формил-1,2,3,4-тетрагидро-4-оксо-1-(2-тиазолил)- 1,8-нафтиридин (3.87 г).
ИК (чистый) см-1: 1690, 1615.
MS (m/z): 488 (МН+).
(5) Соединение (3.6 г), полученное выше в (4), растворили в 1,4-диоксане (160 мл) и к образованному раствору постепенно добавили 2,3-дихлор-5,6-дицианбензохинон (2.51 г). После этого полученную смесь перемешали в течение 2.5 часов, под пониженным давлением отогнали растворитель, к полученному остатку добавили водный раствор гидроксида натрия и образованную смесь экстрагировали хлороформом. Полученный экстракт промыли насыщенным рассолом и затем сушили над безводным сульфатом натрия. Под пониженным давлением отогнали растворитель и полученный остаток затем очистили силикагельной колоночной хроматографией (элюент: хлороформ : метанол =100:1), получив при этом 7-[транс-3-(N-трет- бутоксикарбонилметиламино) -4-метокси-1-пирролидинил]-3-формил- 1,4-дигидро-4-оксо-1-(2-тиазолил)-1,8-тиазолил)-1,8-нафтиридин (1.73 г).
Температура плавления: 130-132oC.
ИК (KBr) см-1: 1695, 1645, 1615.
MS (m/z) : 486 (MH+).
(6) Смесь, состоящую из соединения (1.65 г), полученного выше в (5), 10% водного раствора хлористоводородной кислоты и этанола (40 мл), нагрели в течение 7 часов при 50-60oC. Фильтрацией получили кристаллы, промыли их этанолом и диизопропиловым эфиром, при этом получили гидрохлорид 3-формил-1,4-дигидро-7- (транс-3-метокси-4-метиламино-1-пирролидинил) -4-оксо-1- (2-тиазолил) -1,8-нафтиридина (Соединение II) (1.05 г).
Температура плавления: 255-263oC (разложенный).
ИК (KBr) см-1: 3460, 1695, 1645.
MS (m/z): 386 (MH+).
Примеры С-15 - С-27 Получение желательного продукта (I)
В соответствии со способом, упомянутым в примере С-1, получили соединения, приведенные в табл. 11.
Пример С-28. Получение желательного продукта (I) Гидрохлорида 1,4-Дигидро-7- (транс-3-метиламино-4-метилтио-1- пирролидинил) -4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (Соединение 25).
(1) При использовании этилового эфира, 7-хлор-1,4-дигидро-4-оксо-1- (2-тиазолил) -1,8- нафтиридин-3-карбоновой кислоты, полученного в Примере А-1 (4), и транс-3-метиламино-4-метилтиопирролидина получили этиловый эфир 1,4-дигидро-7-(транс-3-метиламино-4-метилтио-1-пирролидинил) -4- оксо-1-(2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты тем же самым способом, что и в Примере С-1 (1).
Температура плавления: 164-165oC.
(2) Указанное в заголовке соединение получили из сложного этилового эфира, полученного выше в (1), в соответствии со способом, упомянутым в Примере С-1 (2).
Температура плавления: 271-272oC.
Пример C-29
Гидрохлорид 7-(3 -амино-1 -пирролидинил)-1,4-дигидро-1 -(5- метил-2-тиазолил)-4-оксо-1,8-нафтиридин-3-карбоновой кислоты
1. Смесь, состоящую из этил 2,6-дихлорникотиноилацетата (3,0 г), триэтилового эфира ортомуравьиной кислоты (2,5 г) и уксусного ангидрида (2,9 г), кипятили с обратным холодильником в течение 2,5 часов при 140oC, при этом образующийся этилацетат отгоняли под атмосферным давлением. После концентрирования под пониженным давлением полученный осадок разбавляли этилацетатом (20 мл) и добавляли 2-амино-5-метилтиазол (1,6 г). После перемешивания при комнатной температуре в течение 3,5 часов к этому раствору добавляли карбонат калия (1,8 г). Полученную смесь нагревали при 60oC в течение 30 минут и разбавляли ледяной водой (50 мл). Полученные осадки собирали на фильтре, промывали этилацетатом, водой и затем высушивали с получением этилового эфира 7-хлор-1,4- дигидро-1- (5-метил-2-тиазолил) -4-оксо-1,8-нафтиридин-3- карбоновой кислоты (2,6 г).
Температура плавления: 193-195oC.
ИК (KBr) см-1: 1706, 1665.
МС (m/z): 350 (МН+).
2. К раствору, содержащему 3-аминопирролидин (0,35 г), триэтиламин (1,5 мл) в ацетонитриле (25 мл) добавляли соединение (0,98 г), полученное выше (1). Образовавшуюся смесь перемешивали при комнатной температуре в течение 1 дня. Полученные осадки собирали на фильтре, промывали ацетонитрилом и затем высушивали с получением 1,2 г 0,8 N гидрохлорида этилового эфира 7- (3-амино-1-пирролидинил-1,4-дигидро-1-(5-метил-2-тиазолил) -4- оксо-1,8 -нафтипиридин-3-карбоновой кислоты.
Температура плавления: 215-218oC (разл.).
ИК (KBr) см-1: 1719, 1628.
МС (m/z): 400 (MH+).
3. Суспензию, состоящую из соединения (0,61 г), полученного выше (2), 0,5N водного раствора гидроокиси натрия (3,6 мл) и этанола (1 мл), перемешивали при комнатной температуре в течение 1 дня, а затем нагревали при 60oC в течение 30 минут и охлаждали. К этому раствору добавляли 10% водный раствор соляной кислоты (1 мл) и нерастворенное вещество удаляли фильтрованием, а фильтрат концентрировали под пониженным давлением. Осадок переносили в смешанный раствор воды и этанола и промывали 10% водным раствором соляной кислоты, этанолом и затем высушивали с получением гидрохлорида 7-(3-амино-1-пирролидинил) -1,4-дигидро-1- (5-метил-2-тиазолил-4-оксо-1,8-нафтиридин-3-карбоновой кислоты (0,41 г).
Температура плавления: 275-277oC (разл.).
ИК (KBr) см-1: 1718, 1627.
МС (m/z): 372 (MH+).
Пример C-30
Гидрохлорид 7-(3-амино-1 -пирролидинил)-1,4-дигидро-4-оксо-1 - (4-фенил-2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты
1. Смесь, состоящую из этил 2,6-дихлорникотиноилацетата (3,0 г), триэтилового эфира ортомуравьиной кислоты (2,5 г) и уксусного ангидрида (2,9 г), кипятили с обратным холодильником в течение 2,5 часов при 140oC, при этом образующийся этилацетат отгоняли под атмосферным давлением. После концентрирования под пониженным давлением полученный осадок разбавляли этилацетатом (20 мл) и добавляли 2-амино-4-фенилтиазол (2,4 г). После перемешивания при комнатной температуре в течение 2,5 часов к этому раствору добавляли карбонат калия (1,8 г). Полученную смесь нагревали при 60oC в течение 30 минут и разбавляли ледяной водой (50 мл). Образовавшиеся осадки собирали на фильтре, промывали этилацетатом, водой и затем высушивали с получением этилового эфира 7-хлор-1,4- дигидро-4-оксо-1- (4-фенил-2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (3,2 г).
Температура плавления: 240-241oC.
ИК (KBr) см-1: 1713, 1651.
МС (m/z): 412 (МН+).
2. К раствору, содержащему 3-аминопирролидин (0,33 г), триэтиламин (1,5 мл) в ацетонитриле (25 мл), добавляли соединение (1,2 г), полученное выше (1). Образовавшуюся смесь перемешивали при комнатной температуре в течение 1 дня. Полученные осадки собирали на фильтре, промывали ацетонитрилом и затем высушивали с получением 1,4 г 0,8 N гидрохлорида этилового эфира 7-(3-амино-1-пирролидинил)-1,4-дигидро-4-оксо-1- (4-фенил-2- тиазолил) -1,8-нафтиридин-3-карбоновой кислоты.
Температура плавления: 285-288oC (разл.).
ИК (KBr) см-1: 1714, 1629.
МС (m/z): 462 (MH+).
3. Суспензию, состоящую из соединения (0,83 г), полученного выше (2), 0,5N водного раствора гидроокиси натрия (5,6 мл) и этанола (4 мл), перемешивали при комнатной температуре в течение 1,5 часов, затем нагревали при 60oC в течение 2 часов и охлаждали. К этому раствору добавляли 10% водный раствор соляной кислоты (4 мл). Полученные осадки собирали на фильтре и промывали водой, этанолом и затем высушивали с получением гидрохлорида 7- (3-амино-1-пирролидинил) -1,4-дигидро-4-оксо-1-(4-фенил-2- тиазолил)-1,8-нафтиридин-3-карбоновой кислоты (0,58 г).
Температура плавления: 272-274oC (разл.).
ИК (KBr)см-1: 1717, 1623.
МС (m/z): 434 (MH+).
Пример C-31
Гидрохлорид 7-(3-амино-1 -пирролидинил)-1 -[4-(3,4- дифторфенил)-2-тиазолил]- 1,4-дигидро-4-оксо-1,8 нафтиридин-3- карбоновой кислоты
1. Смесь, состоящую из ацетата этил 2,6-дихлорникотиноила (1,2 г), триэтилового спирта ортомуравьиной кислоты (1,0 г) и уксусного ангидрида (1,2 г), кипятили с обратным холодильником в течение 1,5 часов при 140oC, при этом образующийся этилацетат отгоняли под атмосферным давлением. После концентрирования под пониженным давлением полученный остаток разбавляли этилацетатом (20 мл) и добавляли 2-амино-4-(3,4-дифторфенил)тиазол (1,0 г). После перемешивания при комнатной температуре в течение 4,5 часов к этому раствору добавляли карбонат калия (0,7 г). Полученную смесь нагревали при 60oC в течение 30 минут и разбавляли ледяной водой (50 мл). Полученные осадки собирали на фильтре, промывали этилацетатом, водой, а затем высушивали с получением 1,4 г этилового эфира 7-хлор-1-[4- (3,4-дифторфенил) - 2-тиазолил]-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.
Температура плавления: 273-276oC (разл.).
ИК (KBr) см-1: 1722, 1632.
МС (m/z): 448 (МН+).
2. К раствору 3-аминопирролидина (0,27 г), триэтиламина (1,1 мл) в ацетонитриле (25 мл) добавляли соединение (1,0 г), полученное выше (1). Образовавшуюся смесь перемешивали при комнатной температуре в течение 1 дня. Полученные осадки собирали на фильтре, промывали ацетонитрилом и затем высушивали с получением 1,1 г этилового эфира 7-(3-амино-1-пирролидинил) -1-[4- (3,4-дифторфенил) -2-тиазолил] - 1,4-дигидро-4- оксо-1,8-нафтиридин-3-карбоновой кислоты.
Температура плавления: 270-272oC.
ИК (KBr) см-1: 1724, 1631.
МС (m/z): 498 (MH+).
3. Суспензию, состоящую из соединения (0,50 г), полученного выше (2), 0,5N водного раствора гидроокиси натрия (4,0 мл) и этанола (1 мл), перемешивали при комнатной температуре в течение 2 часов и затем нагревали при 60oC в течение 2 дней и охлаждали. К этому раствору добавляли 10% водный раствор соляной кислоты (1,4 мл). Образовавшиеся осадки собирали на фильтре, промывали водой, этанолом и затем высушивали с получением 0,43 г гидрохлорида 7-(3- амино-1-пирролидинил)-1-[4- (3,4-дифторфенил) -2-тиазолил]-1,4- дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.
Температура плавления: 279-281oC (разл.).
ИК (KBr) см-1: 1717, 1624.
МС (m/z): 470 (MH+).
Пример C-32
Гидрохлорид 7-(транс-3-амино-4-хлорметил-1-пирролидинил)-1,4- дигидро- 4-оксо-1 -(2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты
1. Смесь, состоящую из 3-(бензиламино)пропионитрила (50 г), карбоната калия (86 г) и эпибромгидрина (27 мл) в N,N- диметилформамиде (400 мл), нагревали при 85-90oC в течение 1 часа, в течение этого периода дважды добавляли эпибромгидрин (27 мл после 20 мин и 30 мл после 40 мин). После охлаждения нерастворимое вещество отделяли фильтрованием, а фильтрат концентрировали под пониженным давлением. Остаток растворяли в толуоле (500 мл) и диметилсульфоксиде (200 мл) и раствор нагревали при 65oC. К раствору добавляли амид натрия (25 г), и образовавшуюся смесь перемешивали при той же температуре в течение 10 мин. Добавляли ледяную воду, разделяли смесь, а водный слой экстрагировали толуолом. Объединенные органические фазы промывали дважды водой и экстрагировали четыре раза 3% водным раствором уксусной кислоты. Водную фазу подщелачивали водным раствором гидроокиси натрия и экстрагировали хлороформом. После высушивания над безводным сульфатом магния растворитель отгоняли под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле с хлороформом в качестве элюента, получали транс-1-бензил-3-циан-4-гидроксиметилпирролидин (9 г).
ИК (чист.) см-1: 3417, 2241.
2. Смесь, состоящую из соединения (9 г), полученного выше (1), тионил хлорида (7,6 мл) и хлороформа (120 мл), кипятили с обратным холодильником в течение 4 часов. Добавляли ледяную воду, смесь подщелачивали 20% водным раствором гидроокиси натрия и экстрагировали хлороформом. Органическую фазу промывали насыщенным рассолом и затем высушивали над безводным сульфатом магния, растворитель отгоняли под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: смесь н-гексан : этилацетат 3: 1), при этом получали транс- 1-бензил-3-хлорметил-4-цианпирролидин (8 г).
ИК (чист) см-1: 2250.
МС (m/z): 236 (М++2), 234 (М+), 199 (M+-C1), 91 (основание).
3. К раствору концентрированной серной кислоты (18 мл) добавляли полученное выше (2) соединение (8 г), растворенное в толуоле. Полученную смесь перемешивали при комнатной температуре в течение 40 мин и затем нагревали при 60 oС в течение 30 мин. На ледяной бане раствор подщелачивали 50% водным раствором гидроокиси натрия и экстрагировали этилацетатом. Органическую фазу промывали насыщенным рассолом и затем высушивали над безводным сульфатом магния, растворитель отгоняли под пониженным давлением. Остаток перекристаллизовывали из смеси этилацетат - п-гексан и получали транс-1-бензил-4-хлорметилпирролидин-3-карбоксамид (7,5 г).
Температура плавления: 98oC.
ИК (KBr) см-1: 3400, 3200, 1650, 1620.
MC (m/z): 254 (M++2), 252 (М+), 217 (М+-C1), 91 (основание).
4. К метанольному раствору (150 мл) соединения (7 г), полученного выше (3), добавляли по каплям 8,5% раствор гипохлорита натрия (26,7 г) на ледяной бане. Смесь перемешивали при комнатной температуре в течение 30 мин и затем нагревали при 65oC в течение 1,5 часов. Растворитель отгоняли под пониженным давлением и остаток экстрагировали хлороформом. Органическую фазу высушивали над безводным сульфатом магния и затем растворитель отгоняли под пониженным давлением, получали транс-1-бензил-3- хлорметил-4-метоксикарбониламинопирролидин (8 г).
ИК (чист.) см-1: 3310, 1720.
MC (m/z): 284 (M++2), 282 (М+), 91 (основание).
5. Смесь, состоящую из соединения (8 г), полученного выше (4), и концентрированной соляной кислоты (50 мл), кипятили с обратным холодильником в течение 8 часов. Раствор охлаждали и разбавляли водой (100 мл), а к полученному раствору добавляли активированный уголь. После фильтрования фильтрат подщелачивали 50% водным раствором гидроокиси натрия и экстрагировали этилацетатом. Органическую фазу промывали насыщенным рассолом и затем высушивали над безводным сульфатом магния. Растворитель отгоняли под пониженным давлением, полученный остаток растворяли в хлороформе (50 мл) и к образовавшемуся раствору добавляли раствор ди-трет-бутил дикарбоната (4,4 г) в хлороформе (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель отгоняли под пониженным давлением и затем остаток очищали с помощью колоночной хроматографии на силикагеле (элюент хлороформ) с получением транс-1-бензил-3-трет-бутокси-карбониламино-4-хлорметилпирролидина (4,8 г).
Температура плавления: 87-88oC.
ИК (KBr) см-1: 3210, 1694.
МС (m/z): 326 (М++2), 324 (М+), 91 (основание).
6. К раствору, состоящему из соединения (1,0 г), полученного выше (5), уксусной кислоты (0,7 мл) и этанола (30 мл), добавляли 5% палладированный уголь (0,24 г) и муравьинокислый аммоний (0,97 г) на ледяной бане. Полученную смесь перемешивали при комнатной температуре в течение 45 мин. Катализатор удаляли фильтрованием, а фильтрат отгоняли под пониженным давлением и получали неочищенное вещество ацетат транс-3-трет-бутоксикарбониламино-4-хлорметилпирролидина (1,7 г).
7. К суспензии, состоящей из вещества (1,7 г), полученного выше (6), триэтиламина (2,2 мл) и ацетонитрила (35 мл), добавляли этиловый эфир 7-хлор-1,4-дигидро-4-оксо-1-(2-тиазолил)- -1,8-нафтиридин-карбоновой кислоты (0,77 г). Полученную смесь перемешивали при комнатной температуре в течение 1 дня. Образовавшиеся осадки собирали на фильтре, промывали ацетонитрилом и затем высушивали с получением этилового эфира 7- (транс-3-трет- бутоксикарбониламино-4-хлорметил-1-пирролидинил) -1,4-дигидро-4- оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (1,2 г).
Температура плавления: 223-225oC.
ИК (KBr)см-1: 3340, 1717, 1676.
МС (m/z): 534 (MH+).
8. Суспензию, состоящую из соединения (0,20 г), полученного выше (7), 20% соляной кислоты (10 мл) и этанола (3 мл), нагревали при 100oC в течение 1 дня и затем охлаждали. Полученные осадки собирали на фильтре, промывали 10% соляной кислотой и этанолом, а затем высушивали с получением гидрохлорида 7- (транс-3-амино-4-хлорметил-1-пирролидинил) -1,4- дигидро-4-оксо-1-(2-тиазолил)-1,8-нафтиридин-3-карбоновой кислоты (0,16 г).
Температура плавления: 255-258oC (разл.).
ИК (KBr) см-1: 1734, 1636.
МС (m/z): 406 (MH+).
Пример C-33
Гидрохлорид 7-(транс-3-амино-4-трифторметил-1 - пирролидинил)-1,4-дигидро-4- оксо-1 -(2-тиазолил)-1,8-нафтиридин- 3-карбоновой кислоты
1. К суспензии, состоящей из ацетата 3-трет-бутоксикарбониламино- 4-трифторметилпирролидина, который получали согласно "Bioorg. Med.Chem.Lett., 1998, 2833", триэтиламина (1,0 мл) и ацетонитрила (20 мл), добавляли этиловый эфир 7-хлор-1,4- дигидро-4-оксо-1- (2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (0,46 г). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем нагревали при 60oC в течение 1 дня. Образовавшиеся осадки собирали на фильтре, промывали ацетонитрилом и затем высушивали с получением этилового эфира 7-(транс-3-трет-бутоксикарбониламино-4- трифторметил-1-пирролидинил) -1,4-дигидро-4-оксо-1- (2-тиазолил)- 1,8-нафтиридин-3-карбоновой кислоты (0,43 г).
Температура плавления: 226-228oC.
ИК (KBr) см-1: 3361, 1687, 1618.
МС (m/z): 554 (MH+).
2. Суспензию, состоящую из соединения (0,20 г), полученного выше (1), 20% соляной кислоты (10 мл) и этанола (3 мл), нагревали при 100oC в течение 2 часов и затем охлаждали. Полученные осадки собирали на фильтре и промывали 10% соляной кислотой, этанолом и затем высушивали с получением гидрохлорида 7- (транс-3-амино-4-трифторметил-1-пирролидинил) -1,4-дигидро-4-оксо-1-(2-тиазолил) -1,8-нафтиридин-3-карбоновой кислоты (0,16 г).
Температура плавления: 265-268oC.
ИК (KBr) см-1: 1734, 1635.
МС (m/z): 426 (MH+).
Соединения по примерам C-29 - C-33 приведены в конце описания
Серия D.
Пример D-1. Получение жидкого средства.
Рецепт
Соединение 1-1 - 2 г
Сорбит - 50 г
Гидроксид натрия - Соответствующее количество
Дистиллированная вода для инъекции - Соответствующее количество - 1000 мл
Способ получения:
Соединение 1-1 и сорбит растворили в части дистиллированной воды для инъекции и добавили остаток дистиллированной воды для установления pH образованного раствора, равного 4.0. Этот раствор отфильтровали с помощью мембранного фильтра (0.22 м), при этом получили жидкое средство для инъекции.
Пример D-2. Получение средства, высушенного вымораживанием.
Рецепт
Соединение 1-1 - 1 г
Маннит - 5 г
Гидроксид натрия - Соответствующее количество
Дистиллированная вода для инъекции - Соответствующее количество - 1000 мл
Способ получения.
Соединение 1-1 и маннит растворили в части дистиллированной воды для инъекции и к полученному раствору добавили остаток дистиллированной воды для установления pH образованного раствора, равного 5.0. Этот раствор отфильтровали с помощью мембранного фильтра (0.22 м), и полученный фильтрат подвергли сушке вымораживанием для получения порошка для инъекций.
Промышленная применимость.
Соединение этого изобретения является пригодным в качестве лекарственного средства, в частности, в качестве противоопухолевого средства для млекопитающих, включая людей.
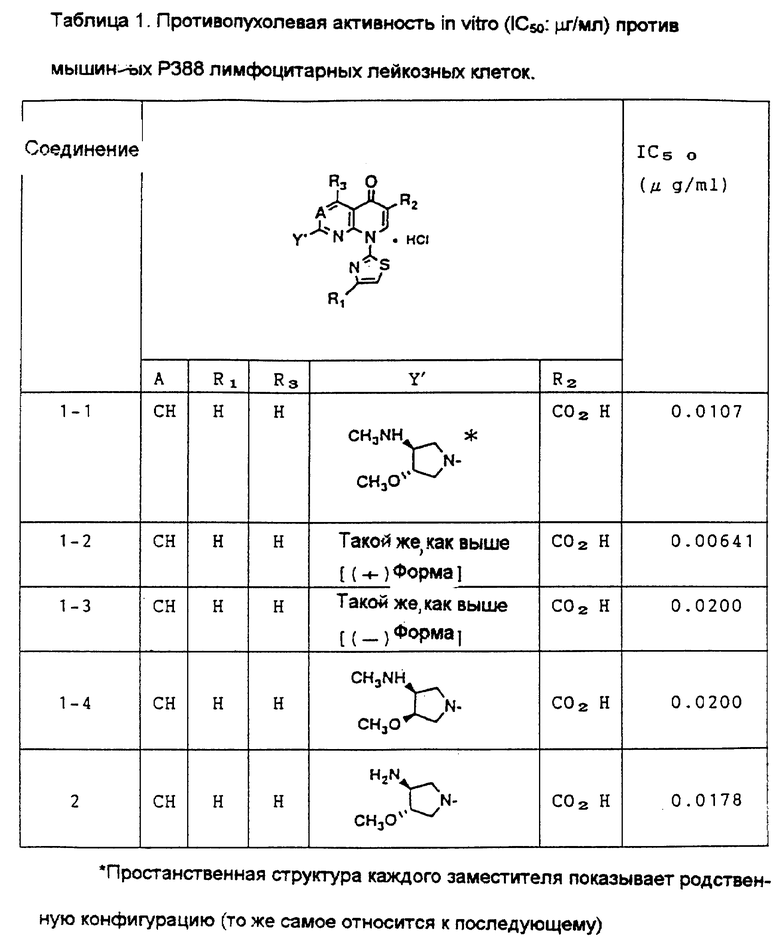
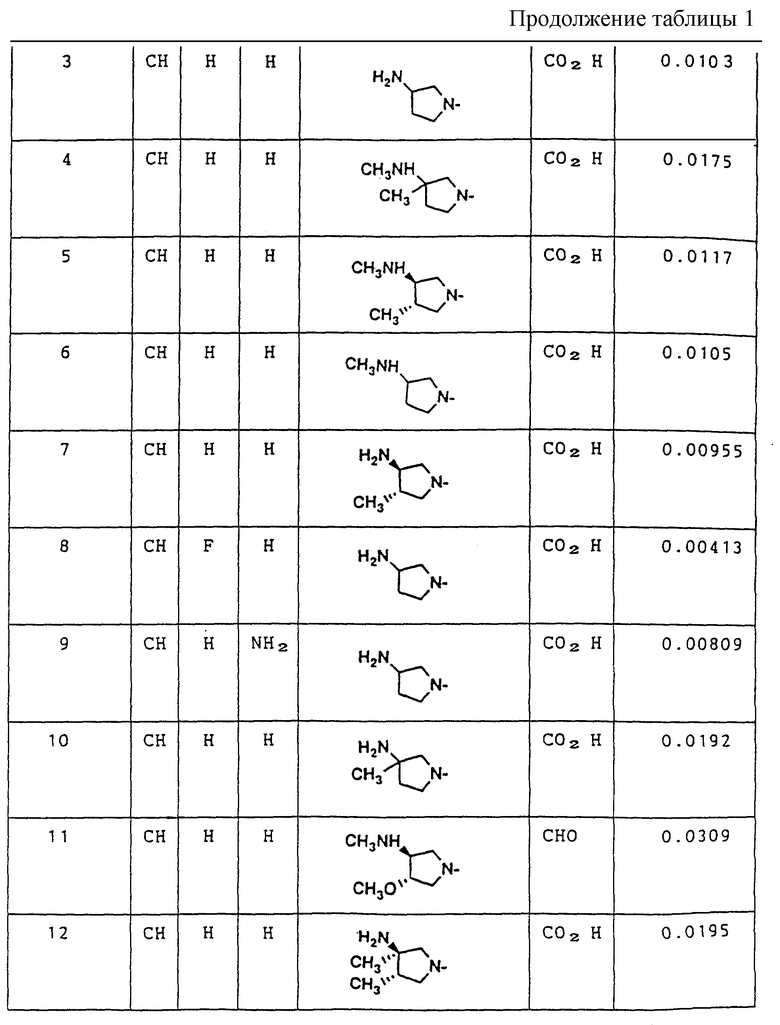
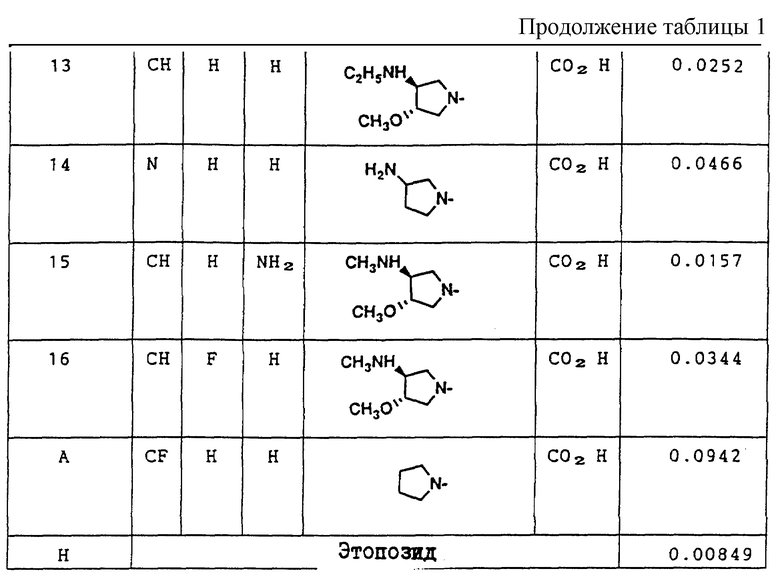
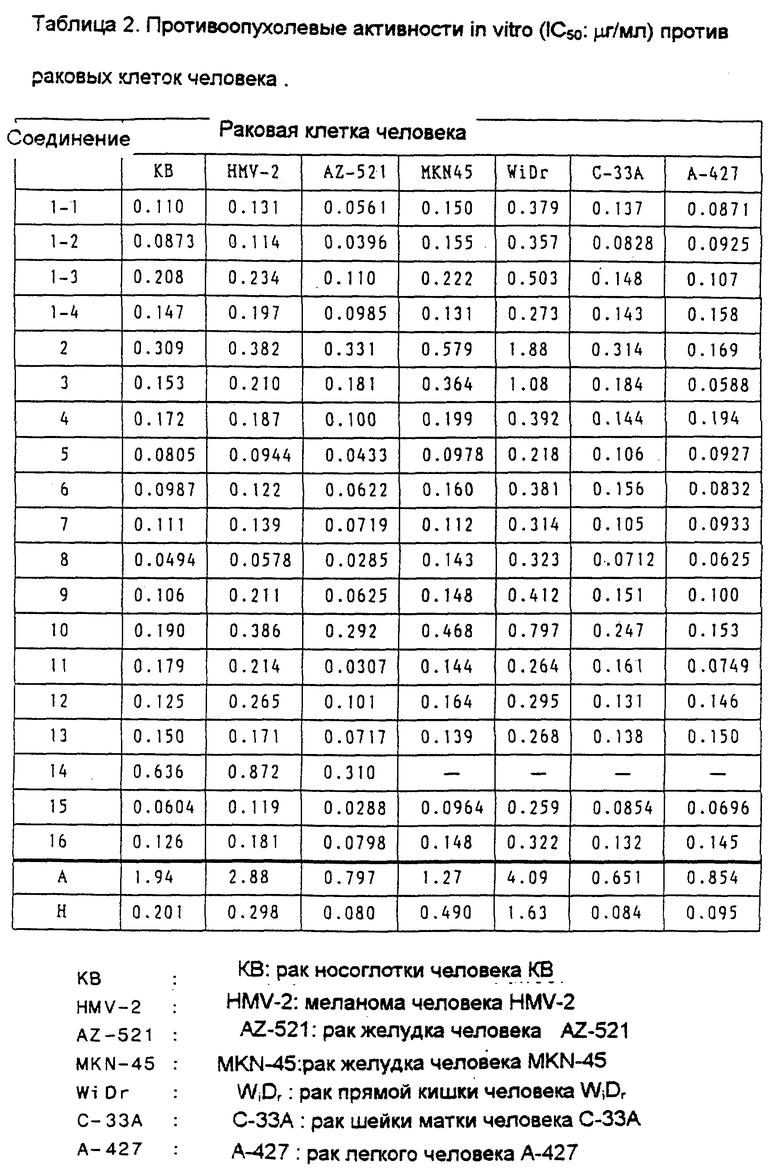
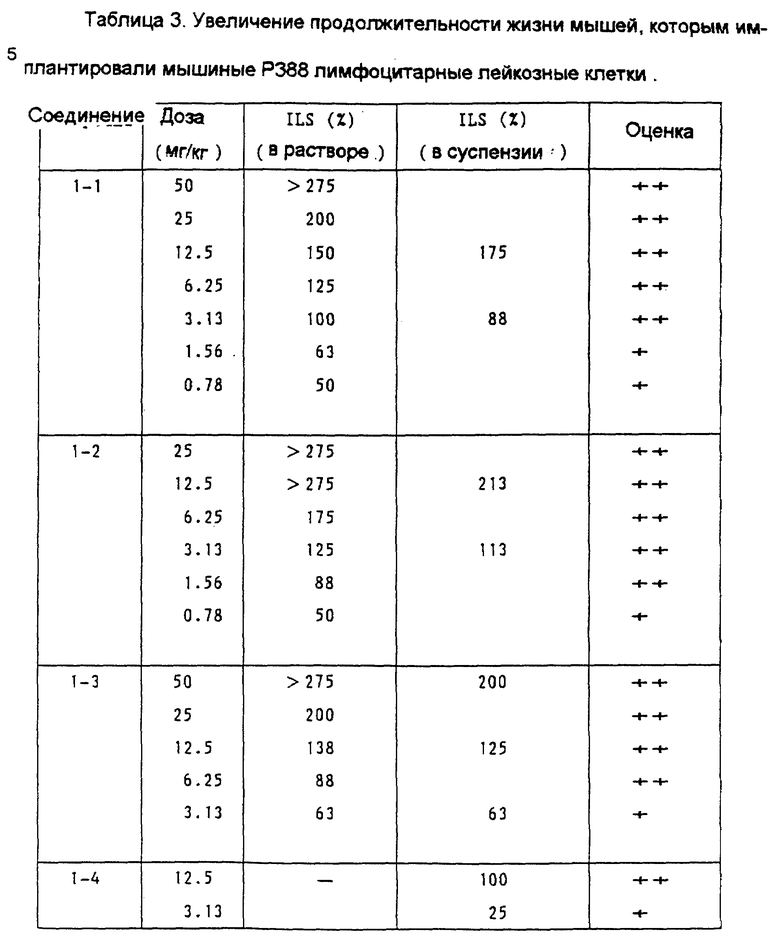
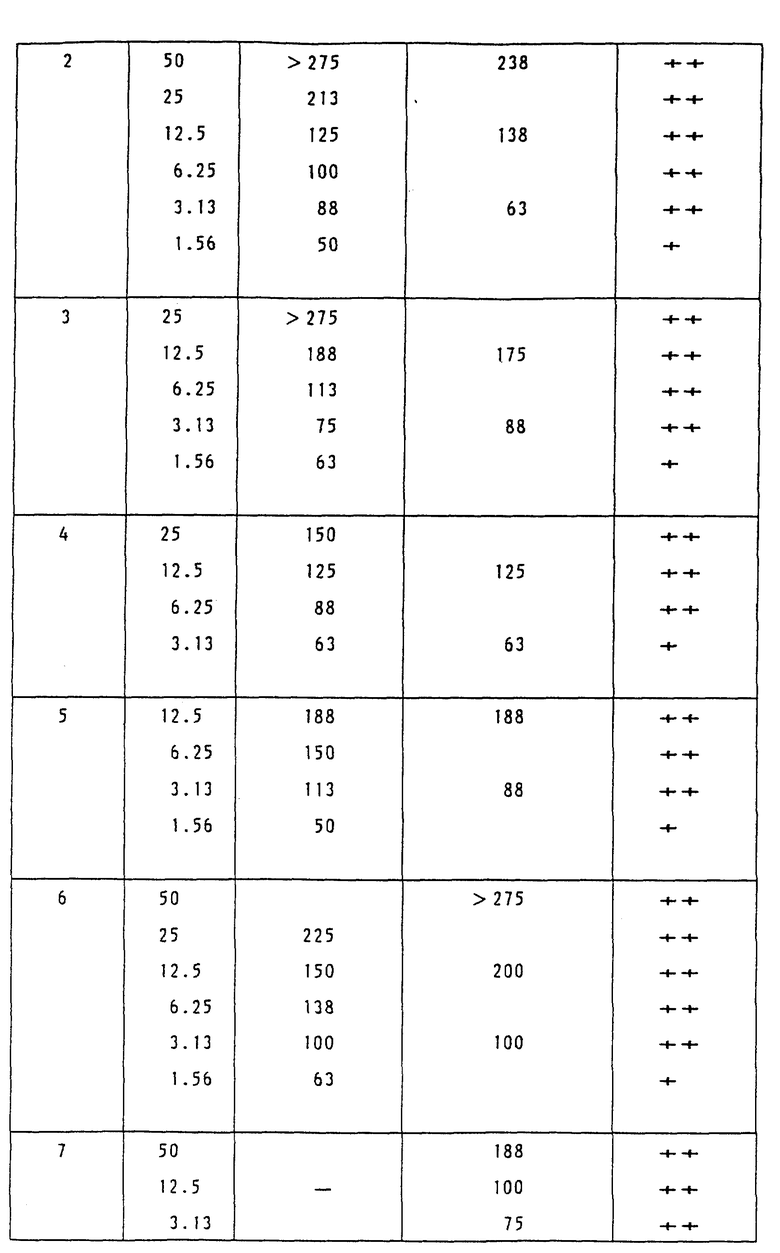
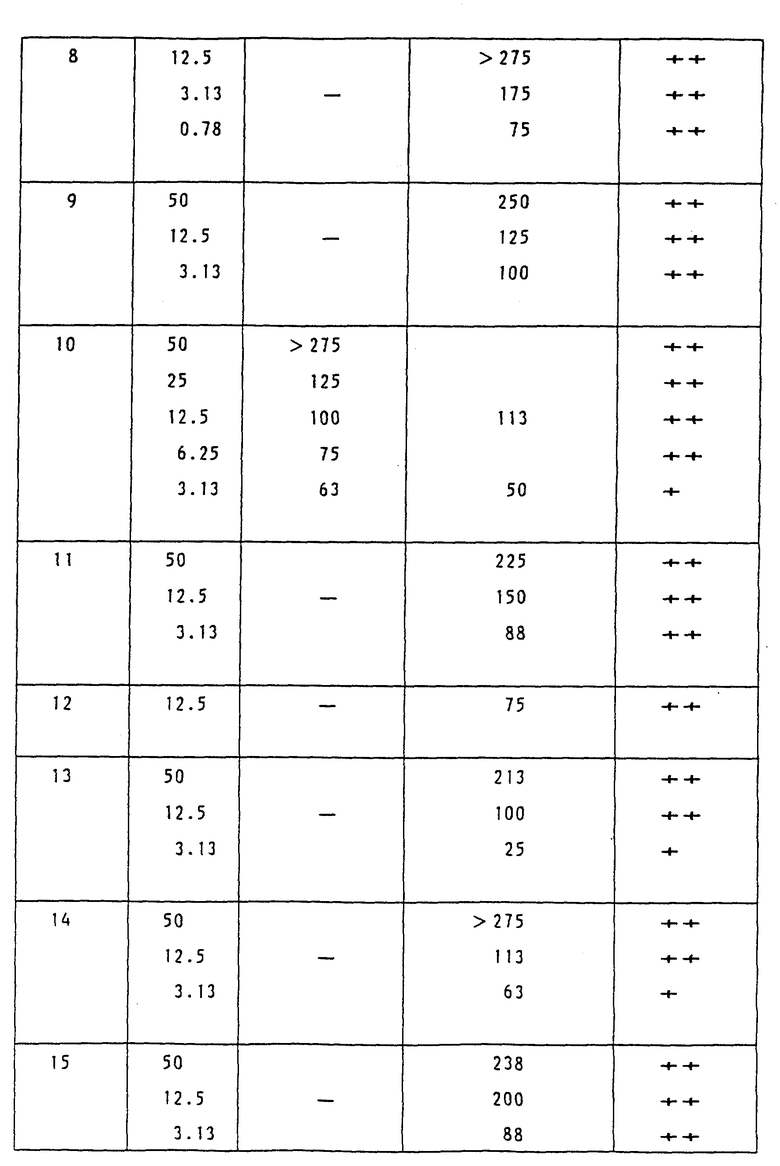
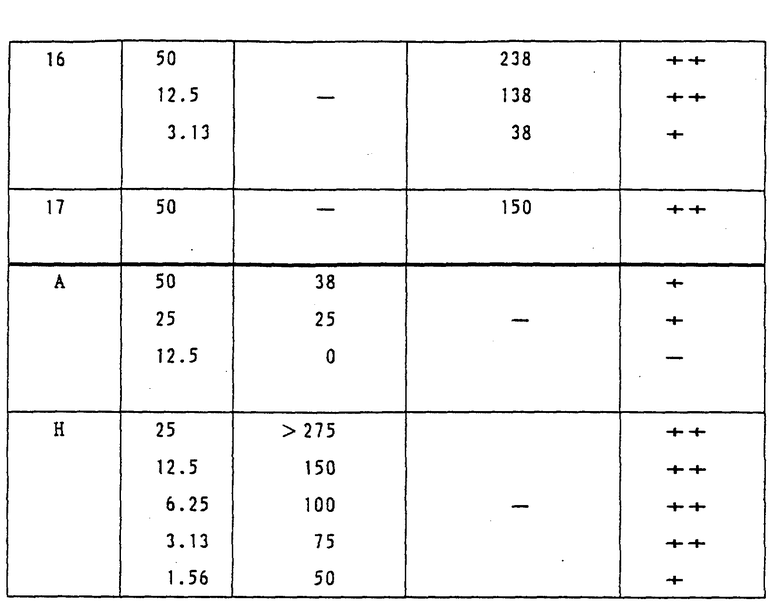
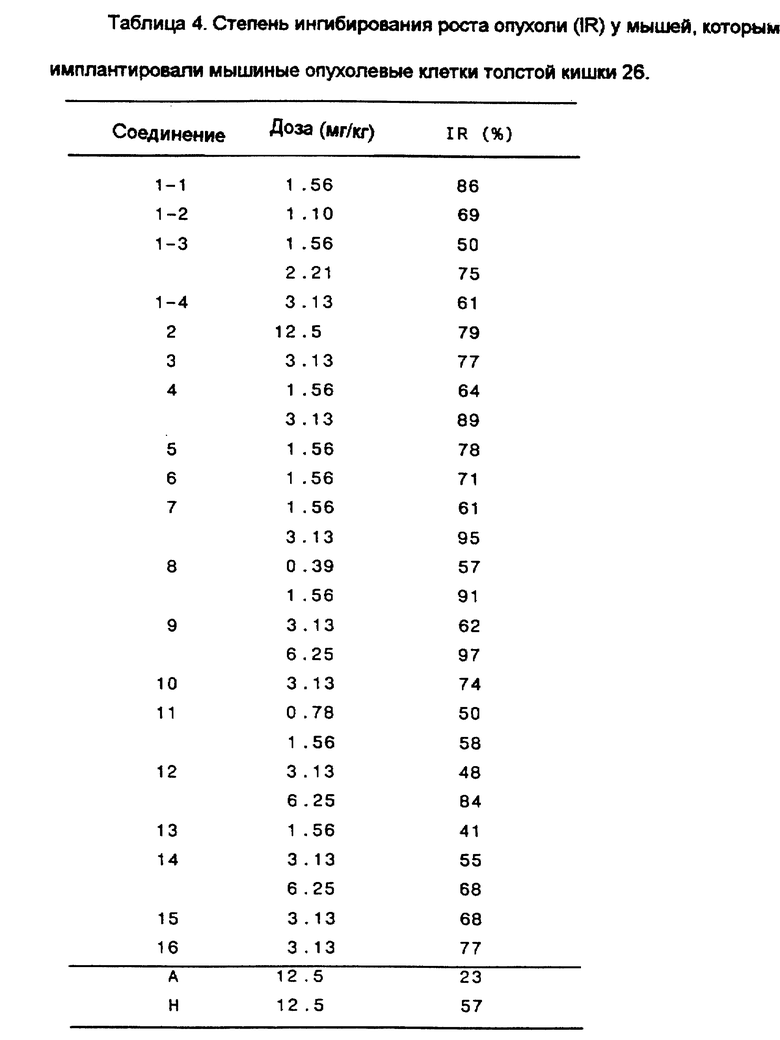
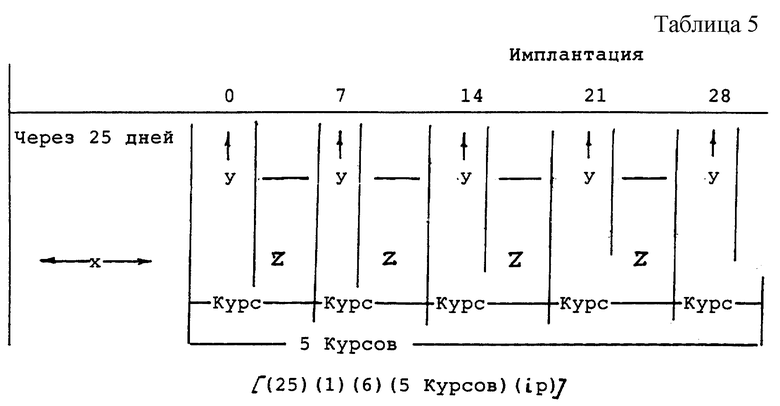
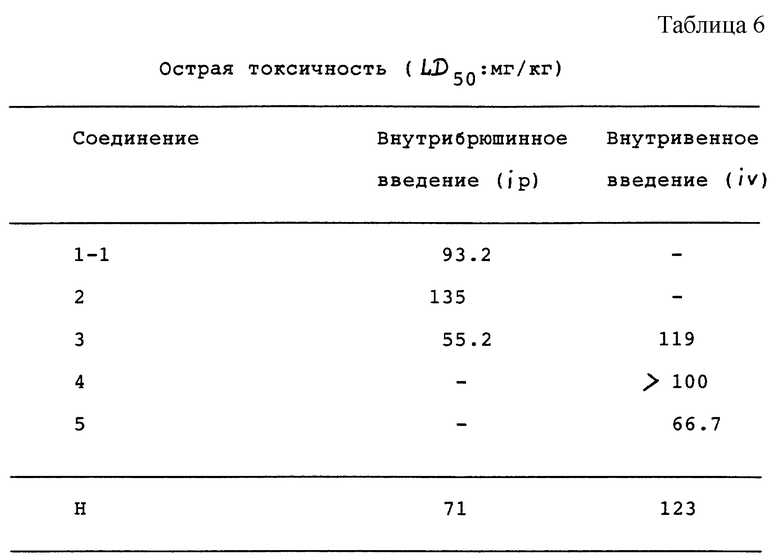
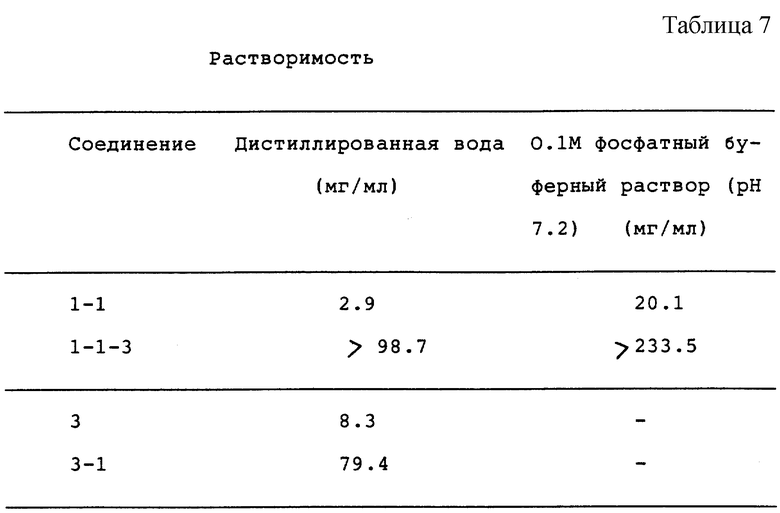
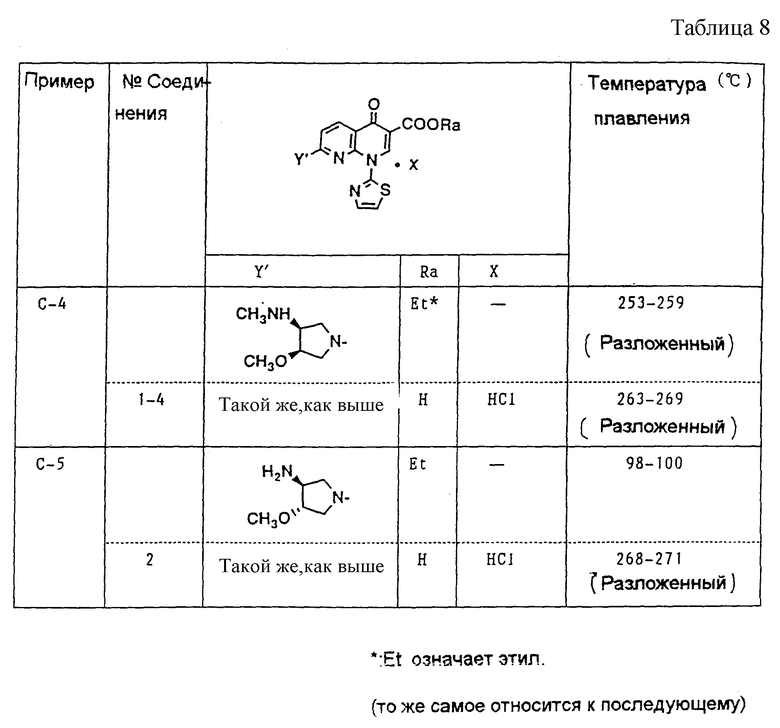
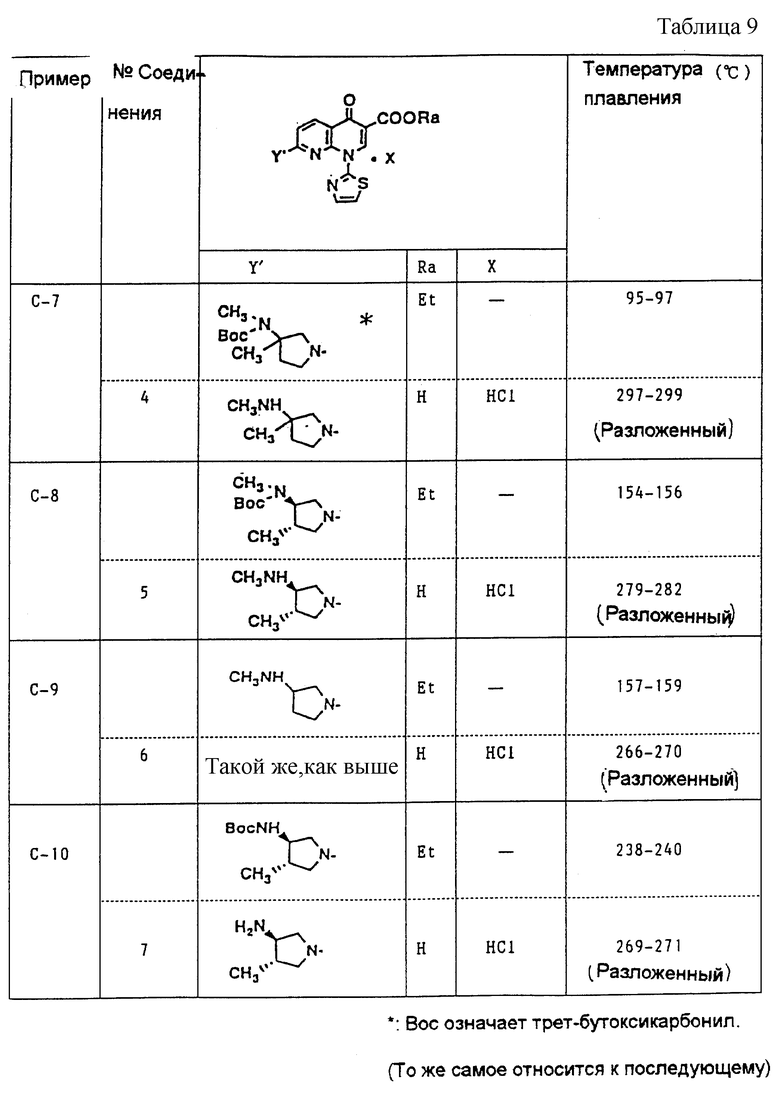
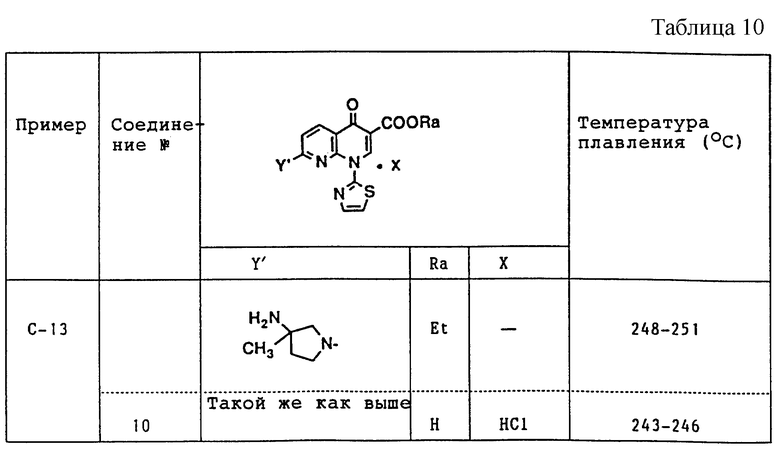
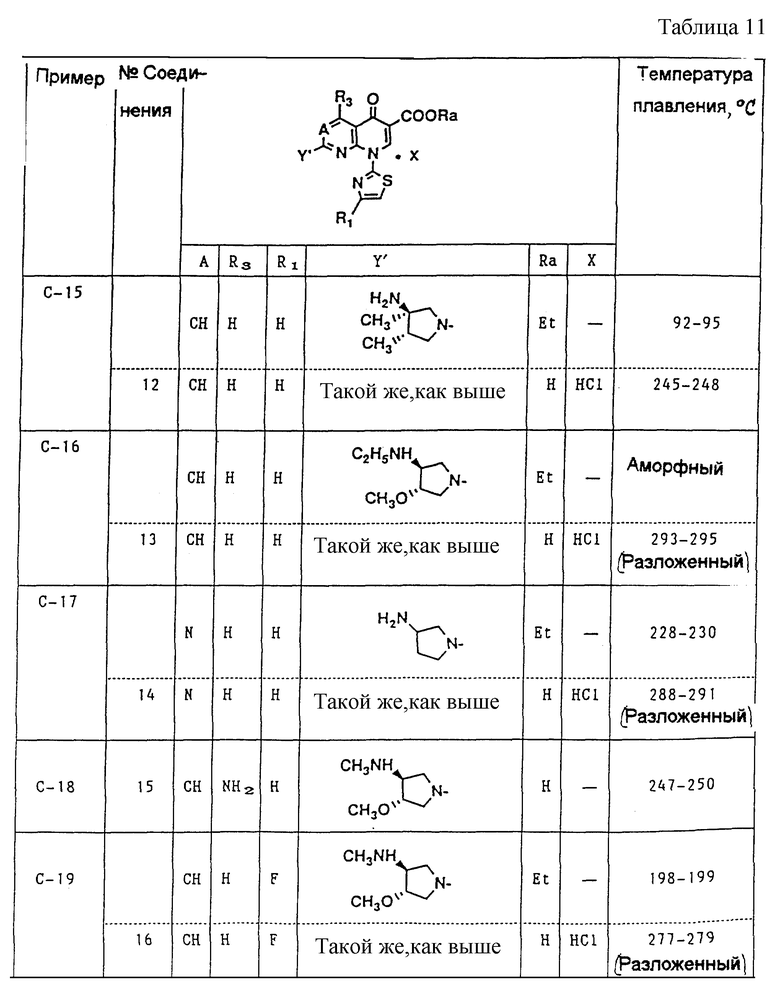
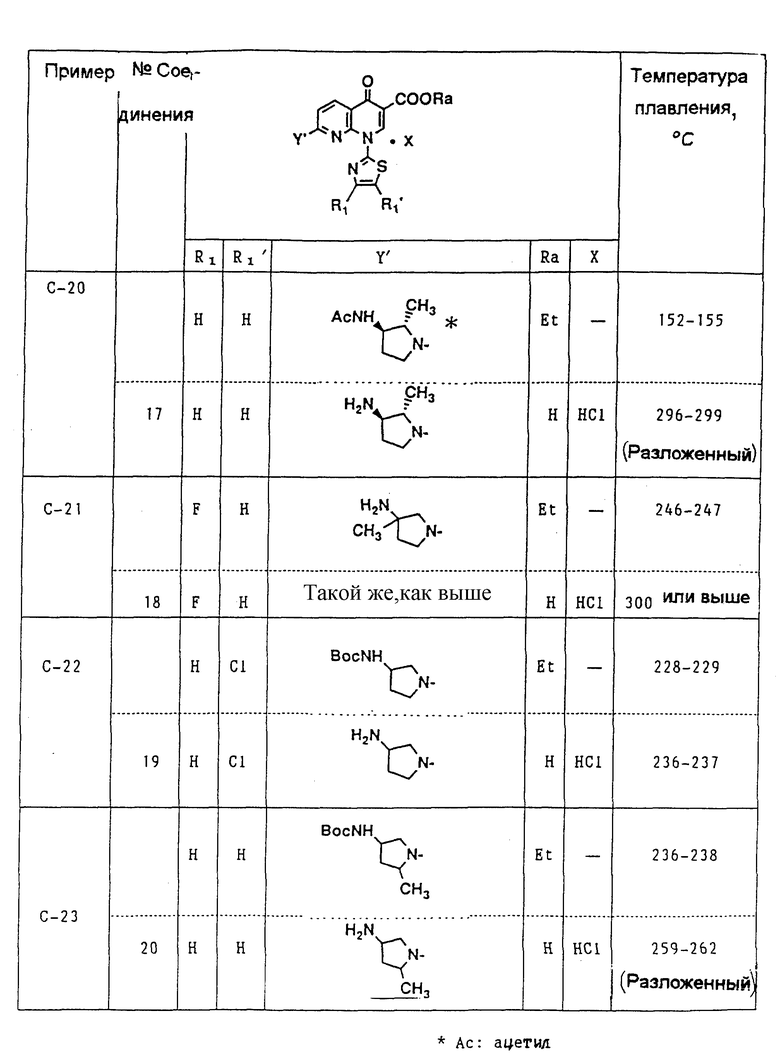
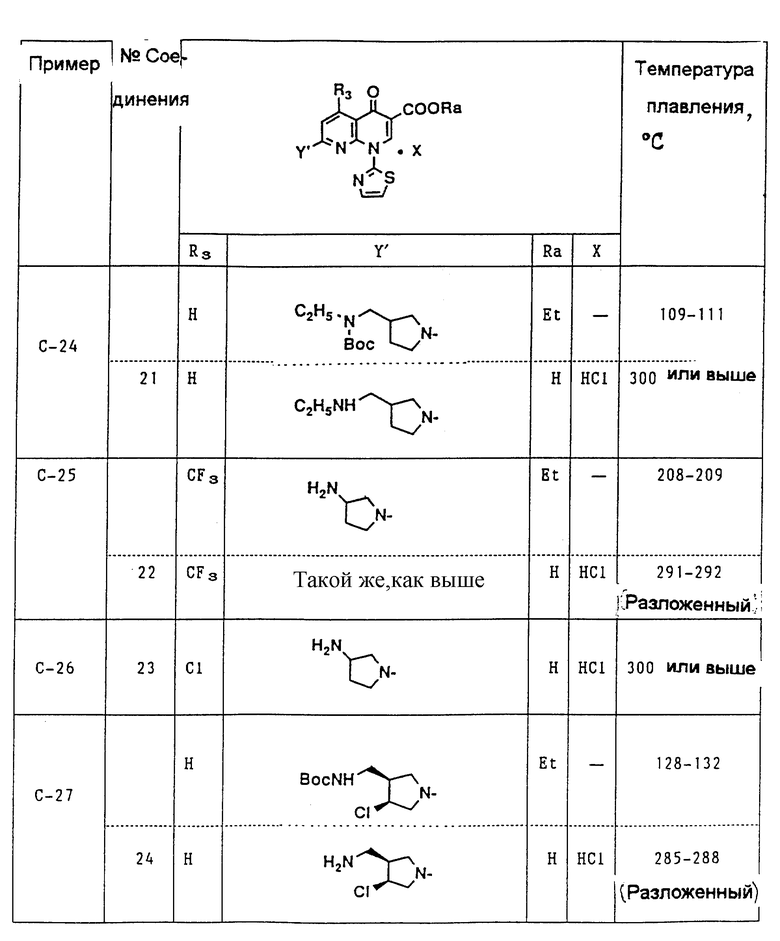
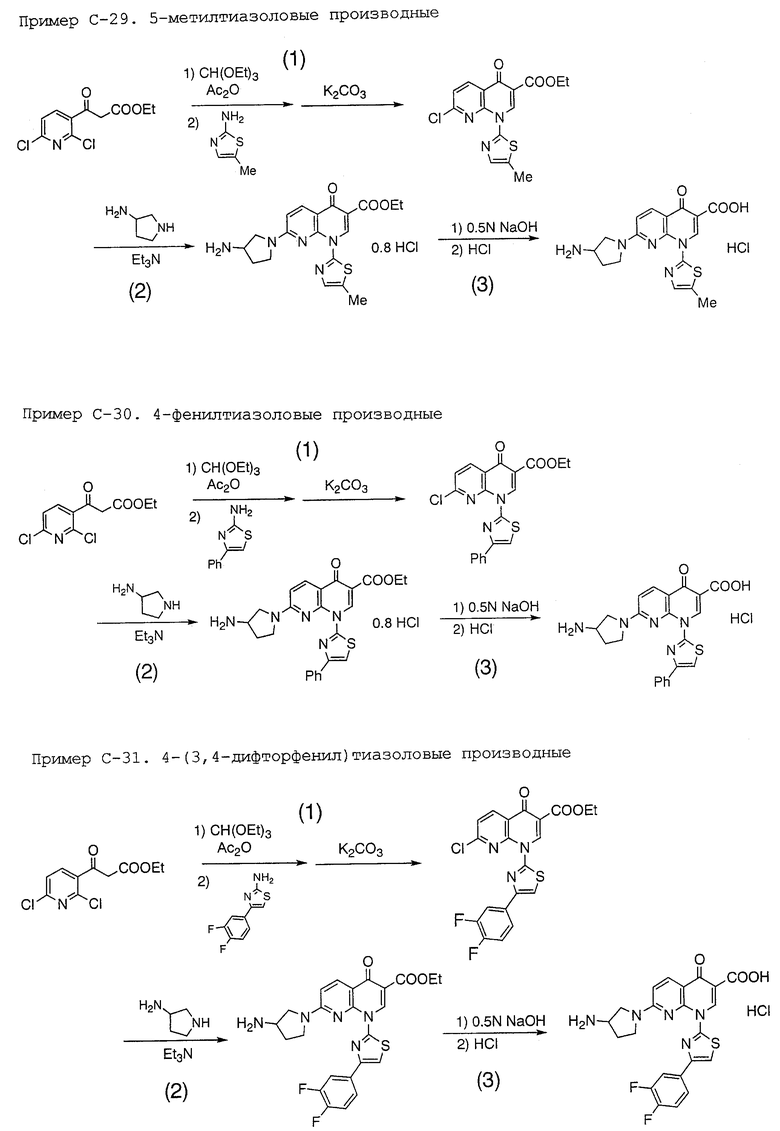
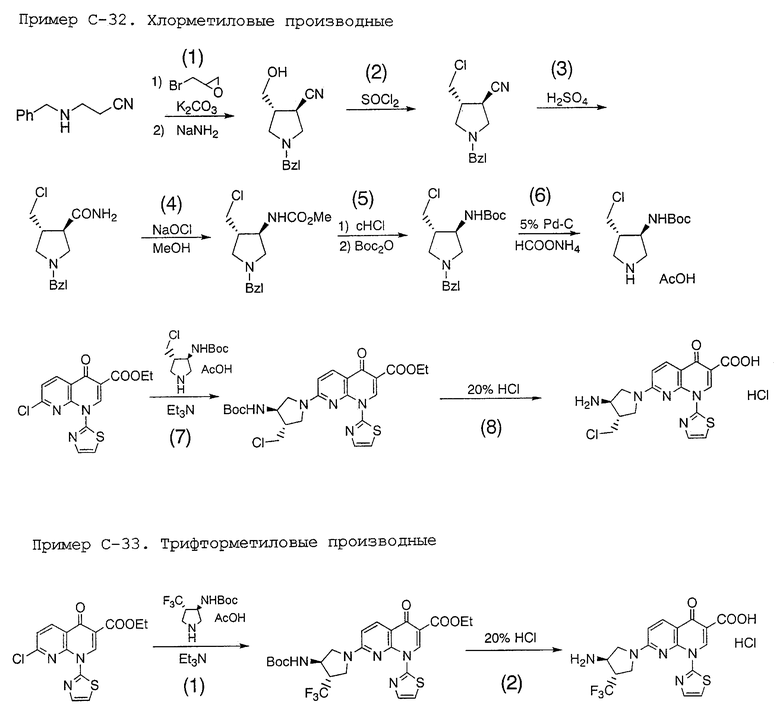
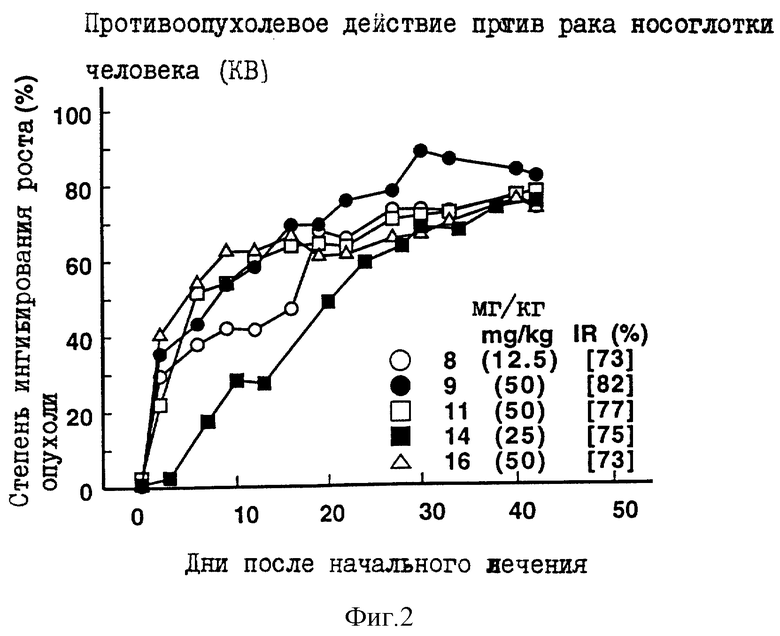
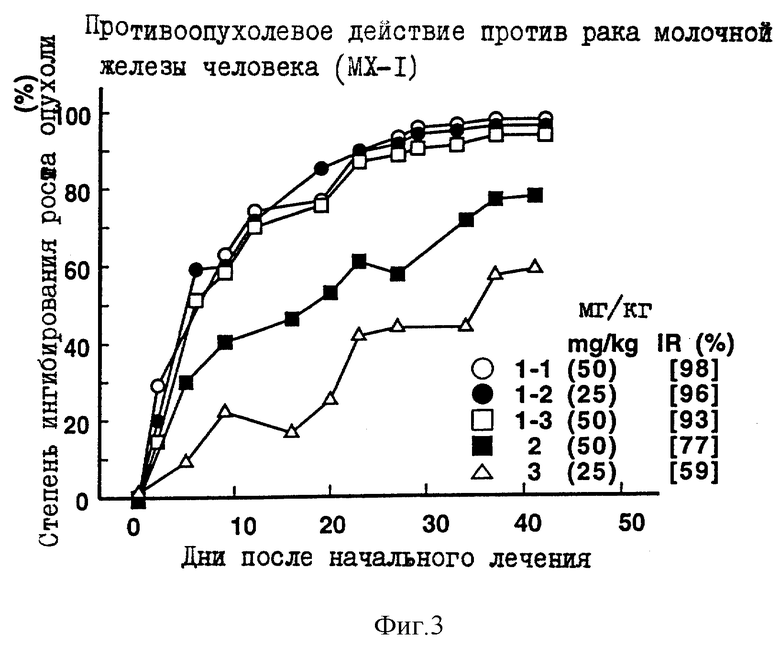
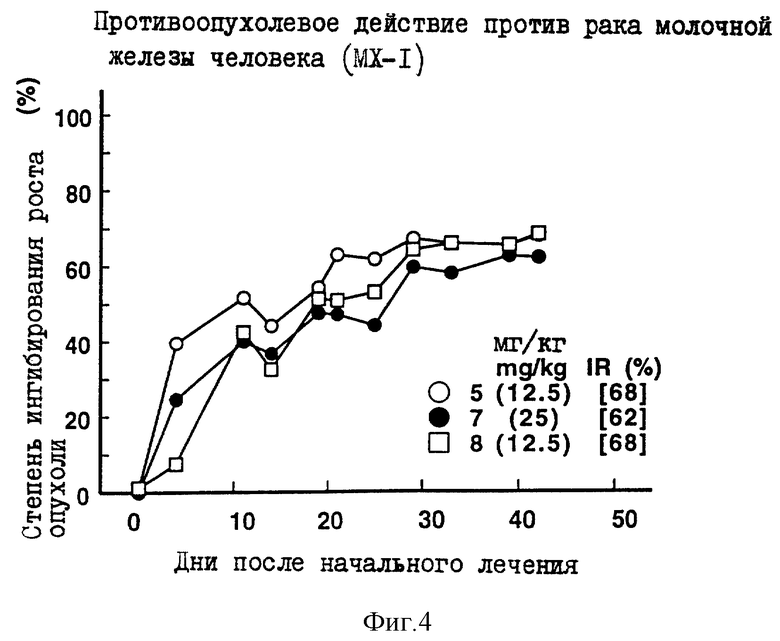
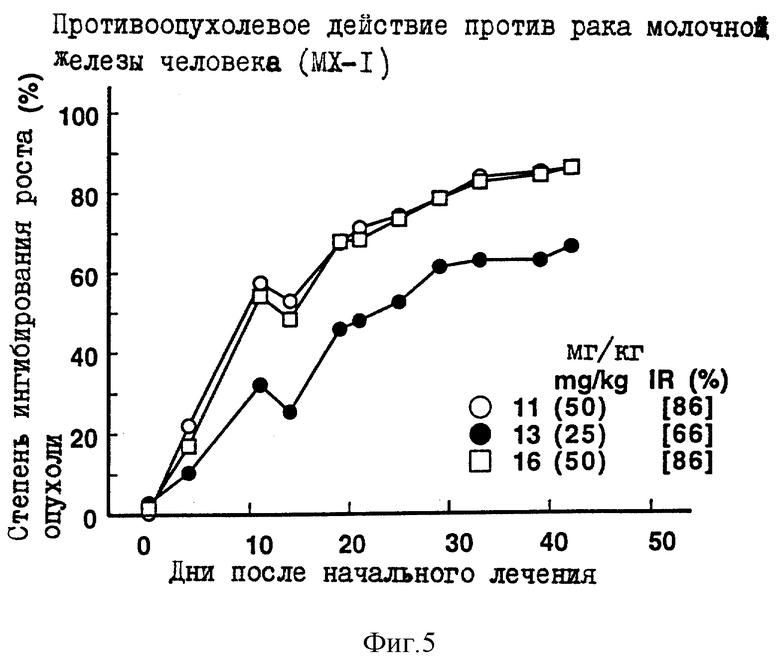
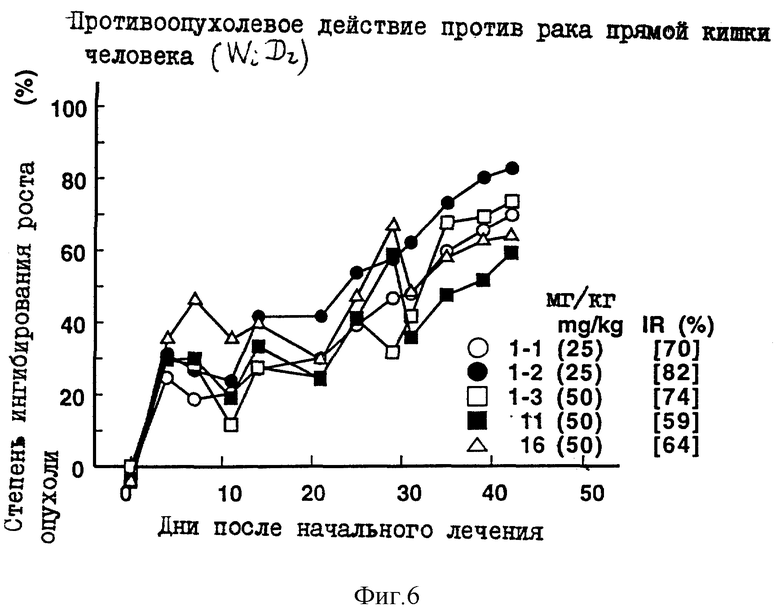
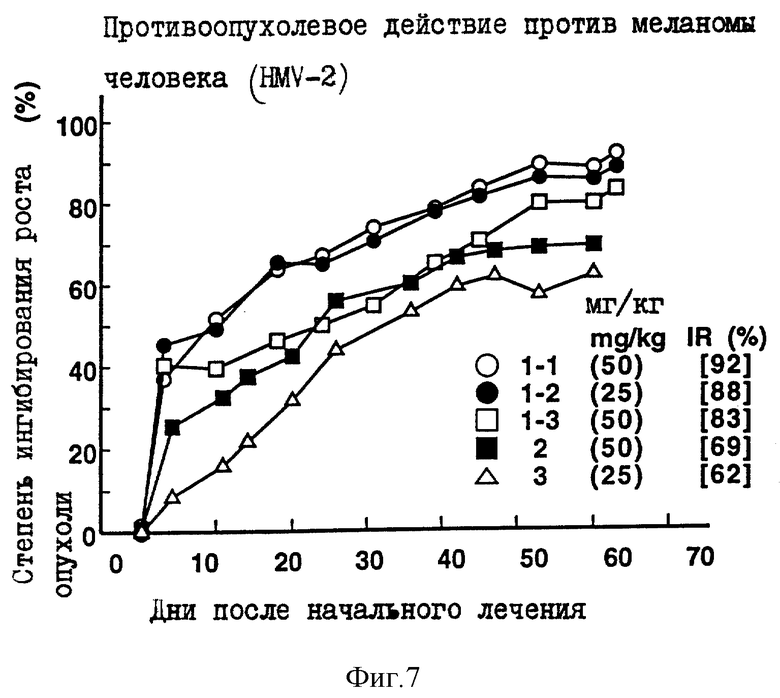
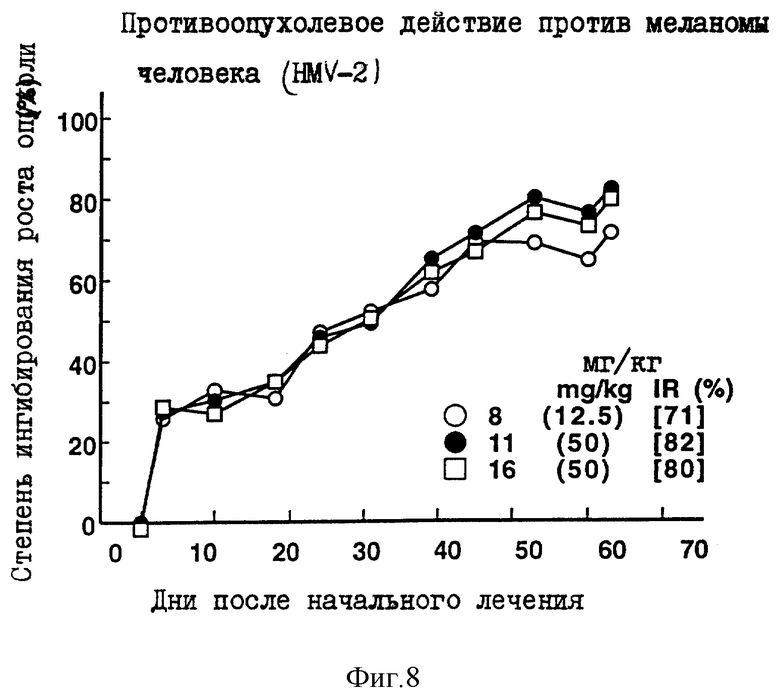
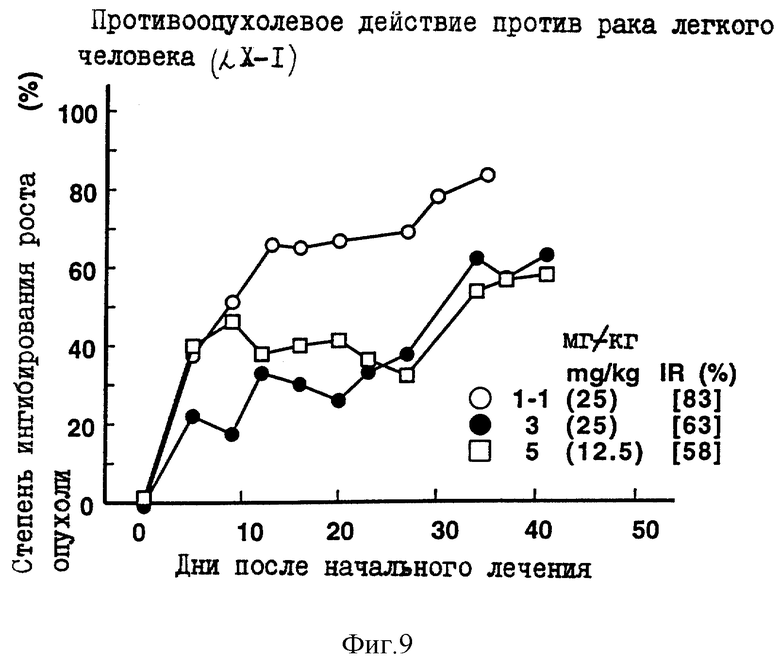
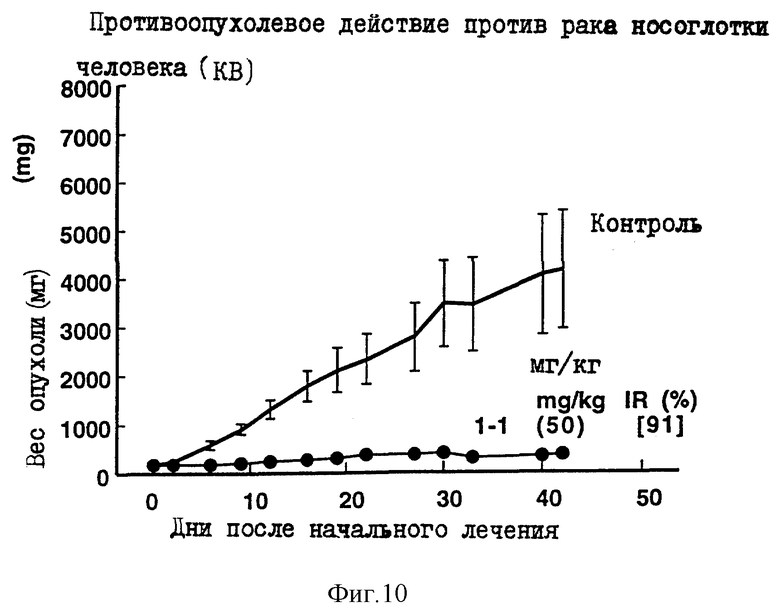
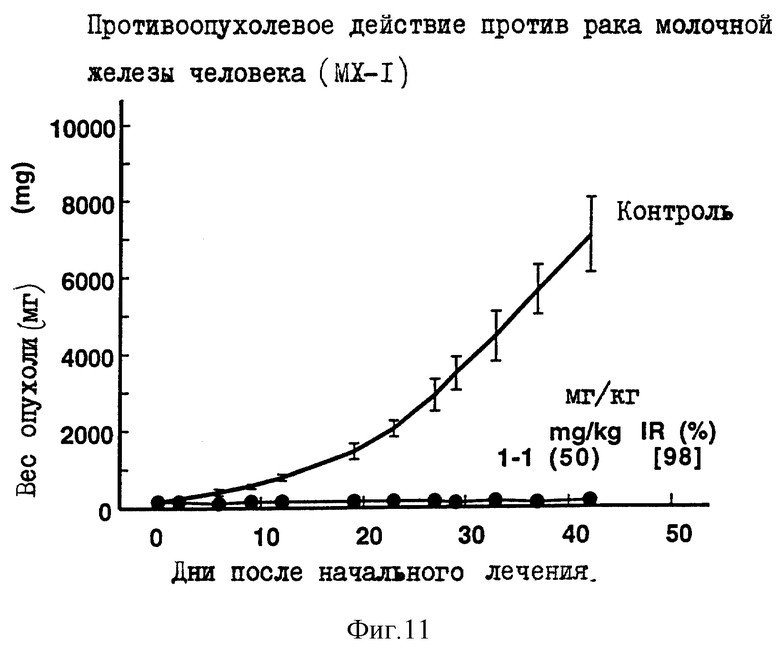
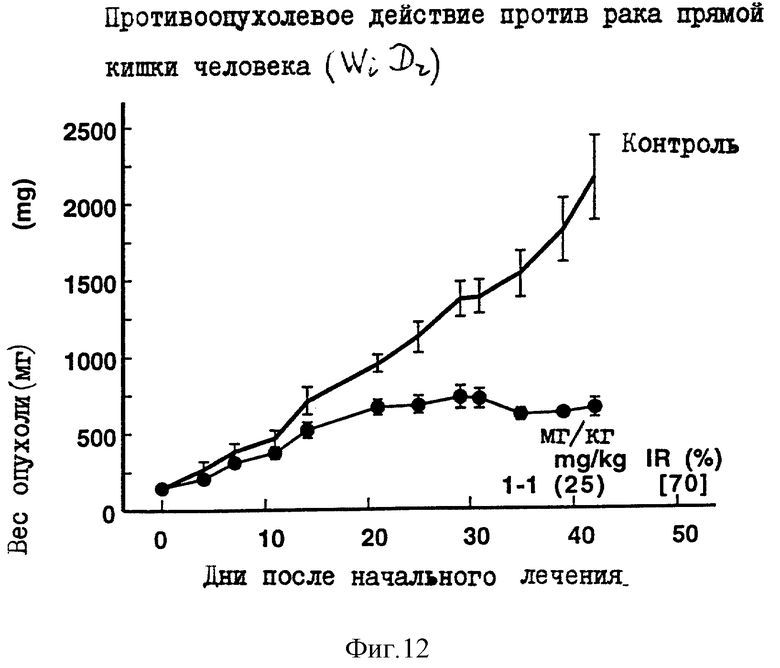
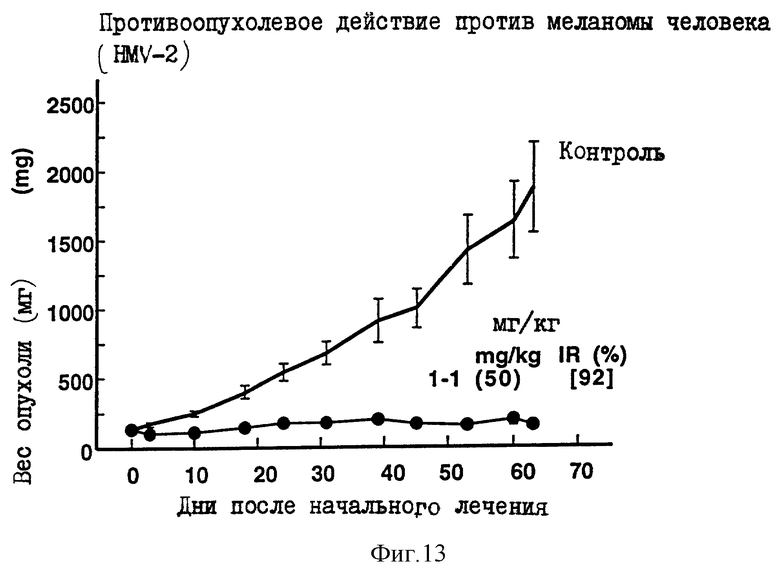
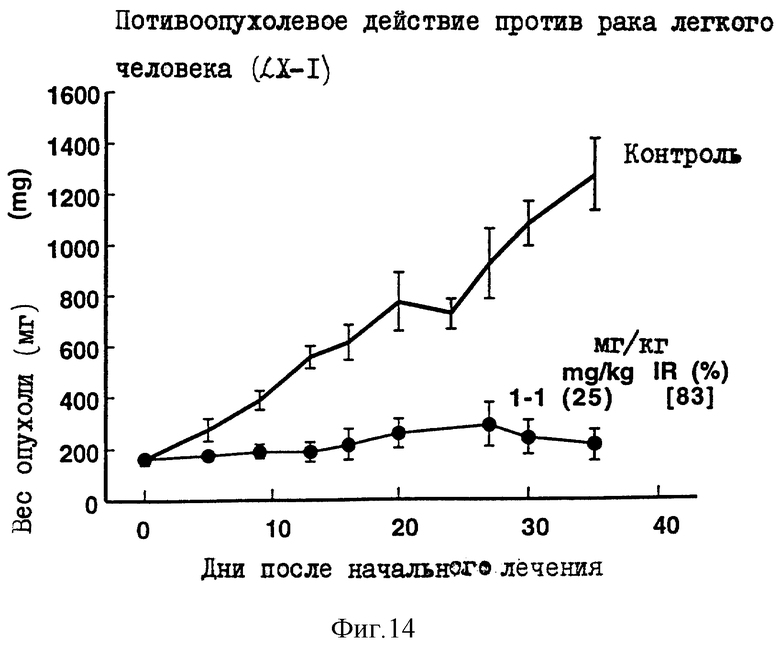
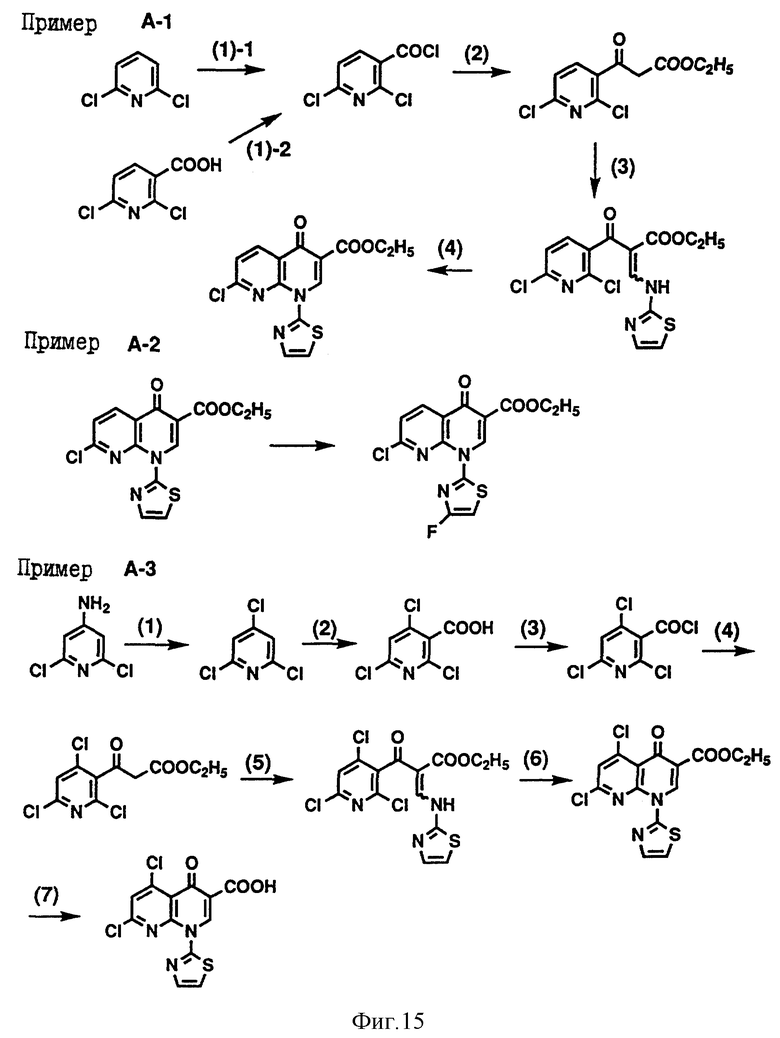
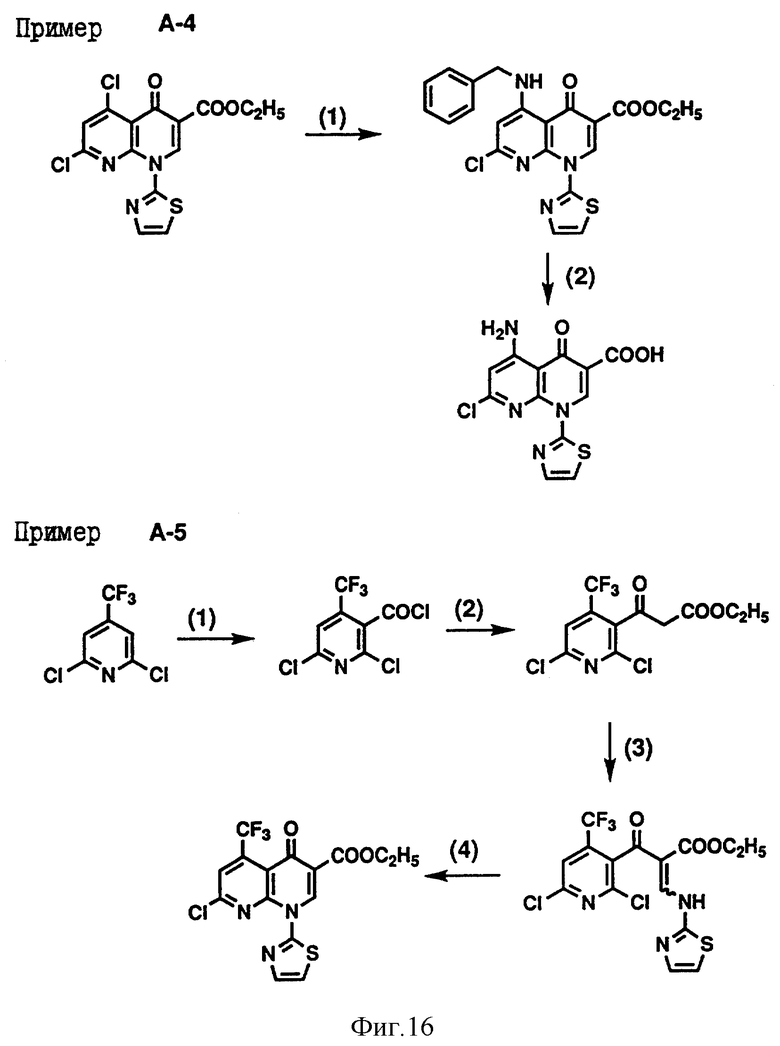
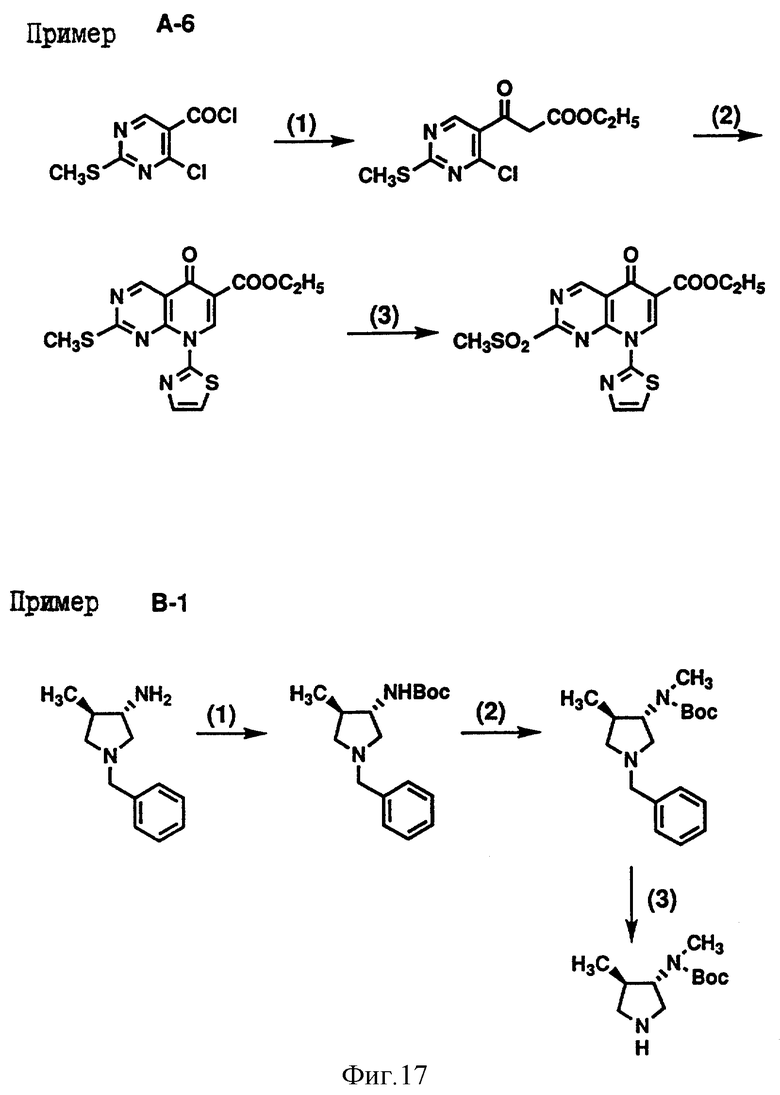
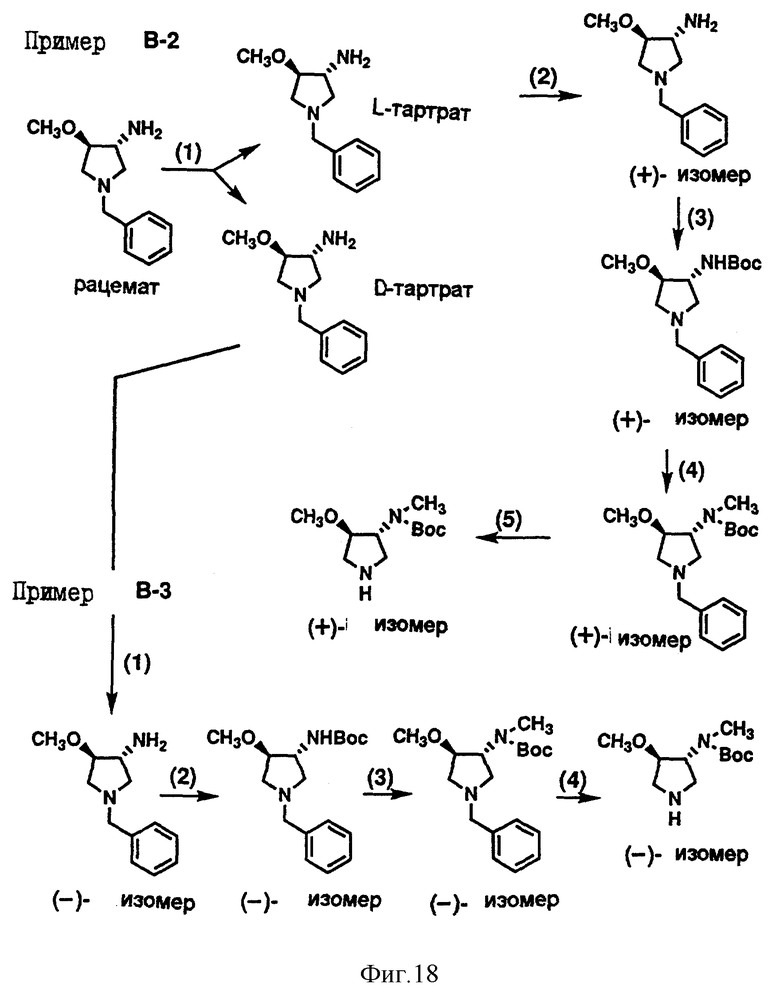
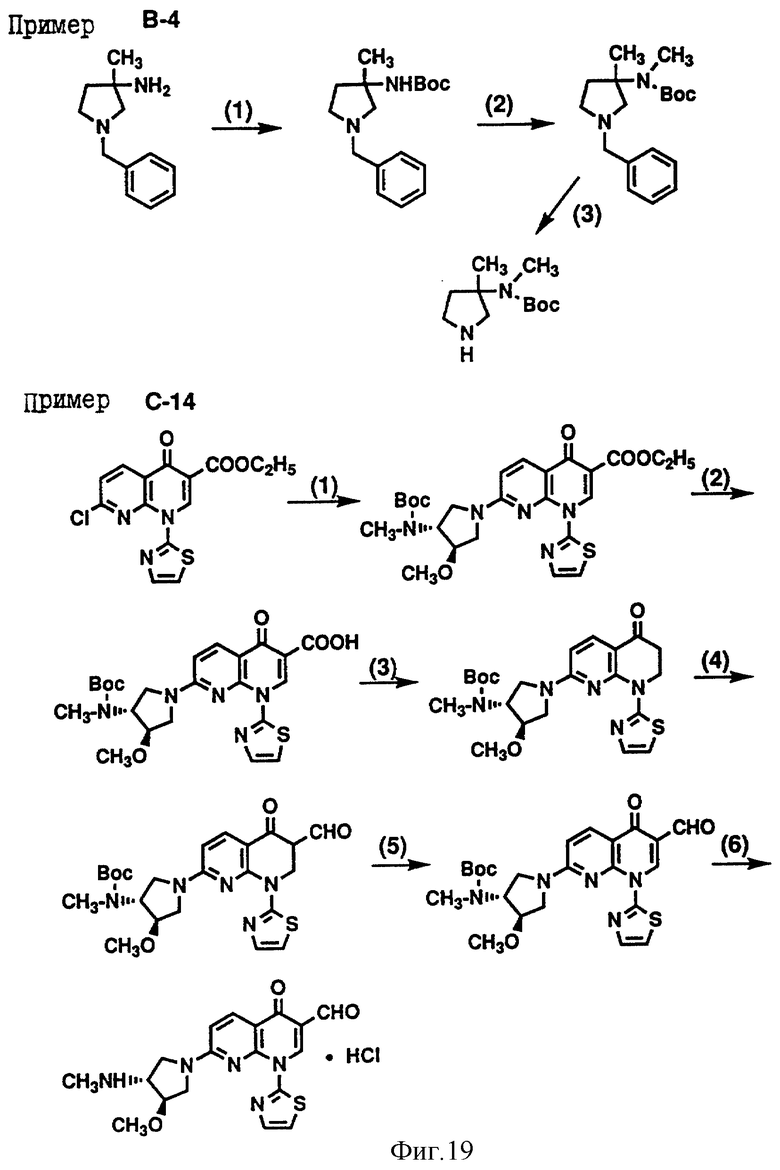
Производные пиридонкарбоновой кислоты формулы I, где R1 - водород, галоген, низший алкил или фенил, замещенный галогеном; R2 - карбоксильная группа или группа, способная превращаться в карбоксильную группу; R3 - водород, аминогруппа, галоген или низший алкил, возможно замещенный галогеном; A - CH; m = 1 или 2; Y - удаляемая группа или группа, имеющая формулу Y'; Z - H, низший алкил или группа, способная превращаться в атом водорода; R5 - Y, галоген, низший алкокси, низший алкилтиогруппа или низший алкил, возможно замещенный галогеном; n = 0 или 1; p = 1 или 2. Соединения формулы (I) могут быть использованы в качестве эффективных ингредиентов противоопухолевых и противораковых средств. 8 с. и 16 з.п. ф-лы, 11 табл., 19 ил.
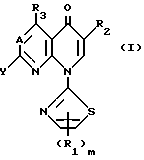
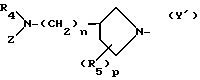
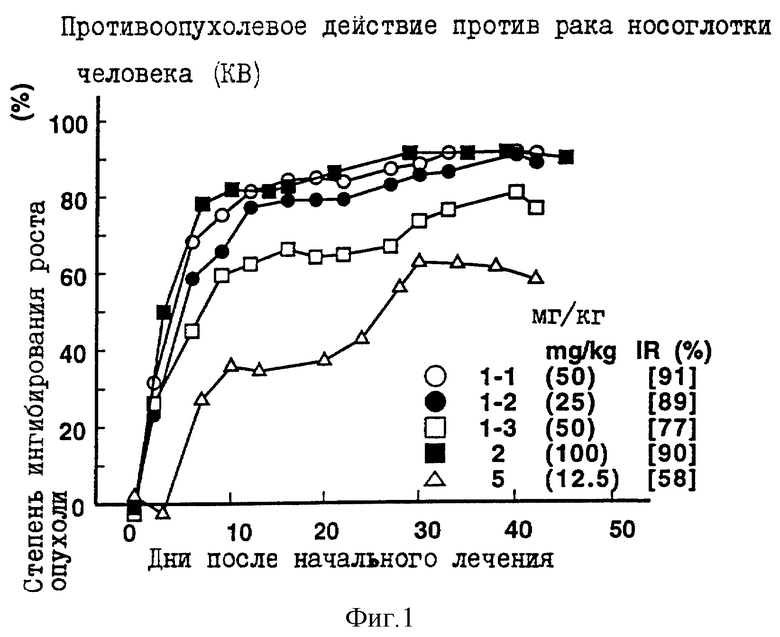
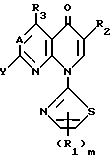
в которой R1 является атомом водорода, атомом галогена, низшей алкильной группой или фенильной группой, которая может быть замещена атомом галогена;
R2 является карбоксильной группой или группой, способной превращаться в карбоксильную группу;
R3 является атомом водорода, аминогруппой, которая может быть защищена, атомом галогена или низшей алкильной группой, которая может быть замещена атомом галогена;
A представляет CH;
m является целым числом, равным 1 или 2;
Y представляет удаляемую группу или группу, имеющую следующую формулу V':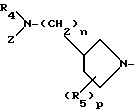
в которой R4 является атомом водорода или низшей алкильной группой;
Z является атомом водорода, низшей алкильной группой или группой, способной превращаться в атом водорода;
R5 является атомом водорода, атомом галогена, низшей алкоксигруппой, низшей алкилтиогруппой или низшей алкильной группой, которая может быть замещена атомом галогена;
n является целым числом, равным 0 или 1;
p является целым числом, равным 1 или 2.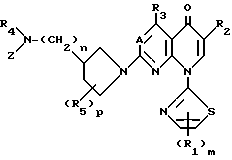
в которой R1 является атомом водорода, атомом галогена, низшей алкильной группой или фенильной группой, которая может быть замещена атомом галогена;
R2 является карбоксильной группой или группой, способной превращаться в карбоксильную группу;
R3 является атомом водорода, аминогруппой, которая может быть защищена, атомом галогена или низшей алкильной группой, которая может быть замещена атомом галогена;
R4 является атомом водорода или низшей алкильной группой;
Z является атомом водорода, низшей алкильной группой или группой, способной превращаться в атом водорода;
R5 является атомом водорода, атомом галогена, низшей алкокси группой, низшей алкилтиогруппой или низшей алкильной группой, которая может быть замещена атомом галогена;
A является CH;
m является целым числом, равным 1 или 2;
p является целым числом, равным 1 или 2;
n является целым числом, равным 0 или 1.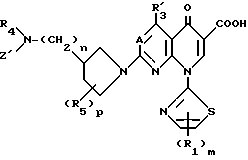
в которой R1 является атомом водорода, атомом галогена, низшей алкильной группой или фенильной группой, которая может быть замещена атомом галогена;
R'3 является атомом водорода, аминогруппой, атомом галогена или низшей алкильной группой, которая может быть замещена атомом галогена;
R4 является атомом водорода или низшей алкильной группой;
Z' является атомом водорода или низший алкильной группой;
R5 является атомом водорода, атомом галогена, низшей алкокси группой, низшей алкилтиогруппой или низшей алкильной группой, которая может быть замещена атомом галогена;
A представляет CH;
m является целым числом, равным 1 или 2;
n является целым числом, равным 0 или 1;
p является целым числом, равным 1 или 2.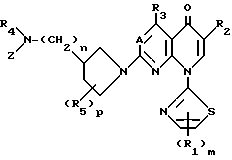
где A, R1, R2, R3, R4, R5, z, m, n и p являются такими, как они определены в п.1,
отличающийся тем, что соединение, имеющее следующую общую формулу II, или его соли: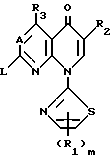
где L является удаляемой группой и A, R1, R2, R3 и m являются такими, как они определены в п.1,
подвергают реакции с циклическим амином, имеющим общую формулу III: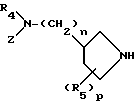
где R4, R5, n и p являются такими, как они определены в п.1.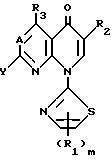
где A, Y, R1, R2, R3 и m являются такими, как они определены в п.1,
отличающийся тем, что соединение общей формулы IV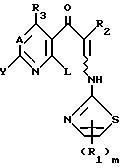
где L является удаляемой группой и A, R1, R2, R3 Y и m являются такими, как они определены в п.1,
подвергают реакции замыкания кольца.
Приоритет по пунктам:
13.03.95 - по пп.1 - 24;
14.06.94, 28.07.94, 28.12.1994, 13.03.1995 - по п.10;
28.07.1994, 15.11.1994 - по пп.11 и 12.
| Способ диагностики плацентарной недостаточности | 1986 |
|
SU1627987A1 |
| EP 154780 A1, 18.09.1985 | |||
| Способ получения замещенных тиазолидиниловых эфиров серной кислоты или их солей | 1982 |
|
SU1181545A3 |
| Генератор с переменным числом оборотов и, примерно, постоянным напряжением | 1933 |
|
SU37175A1 |
| US 3988463, 26.10.76 | |||
| СПОСОБ НЕПРЕРЫВНОГО СУЛЬФИРОВАНИЯ И/ИЛИ СУЛЬФАТИРОВАНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1993 |
|
RU2039736C1 |

