Изобретение относится к области органической химии, а именно к способу получения винил-н-бутилового эфира, который используют для получения полимеров, применяемых в медицине (бальзам Шостаковского) и технике (загущающая присадка к маслам "Винипол ВБ-2").
Известные способы получения винил-н-бутилового эфира основаны на реакциях прямого винилирования бутанола ацетиленом (Вацулик П. "Химия мономеров", М. , "ИНЛИТ", т.I, 296, 1960 г, Шильдкнехт К. "Мономеры", М., "ИНЛИТ", 126, 1960 г). Процессы винилирования проводят в присутствии катализатора - 8%-ной спиртовой щелочи при температуре 150oC и давлении до 20 атм, что обеспечивает 87% выхода целевого продукта.
Практическая реализация указанных процессов сопровождается образованием высококипящей примеси, дибутилацетальацетальдегида - продукта присоединения бутанола к винил-н-бутиловому эфиру (Шостаковский М.Ф. и др. "ЖОХ" 18, N 3, 451, 1948 г.), которую для улучшения технико-экономических показателей основного способа перерабатывают в целевой продукт (Лавров В.М. и др., "ЖПХ", 58, N 2, 457, 1985 г., (прототип).
Переработки ацеталей в винил-алкиловые эфиры проводят разложением ацеталей на различных катализаторах (Яновская Л.А. и др., "Химия ацеталей", М., "Наука" 217, 1975 г.). Как правило, активность катализаторов, применяемых в процессах разложения дибутилацеталя, не обеспечивает эффективной переработки ацеталя в целевой продукт, и в сыром эфире, выводимом в ходе реакции из реакционной массы, наряду с бутанолом присутствует в количествах 20% непрореагировавший дибутилацеталь, который после отделения от продуктов разложения возвращают в рецикл на стадию дезалкоксилирования. Наличие в рецикле значительных количеств "возвратного" ацеталя в целом усложняет процессы переработки ацеталя из-за необходимости проведения дополнительных технологических операций.
Известен способ получения винил-н-бутилового эфира дезалкоксилированием дибутилацетальацетальдегида при температуре 300oC на катализаторе палладие, нанесенном на асбест, обеспечивающий 75% выход целевого продукта (Пат, США 1931858, 1933 г.).
Недостаток способа заключается в сложном экспериментальном оформлении процесса дезалкоксилирования из-за необходимости применения высокоэффективной системы улавливания низкокипящего конденсата, смеси винил-н-бутилового эфира, бутанола и непрореагировавшего дибутилацеталя, выводимого в ходе реакции.
Известен способ получения винил-н-бутилового эфира разложением дибутилацеталя при температуре 200oC в присутствии катализатора сульфаниловой кислоты, обеспечивающий 65% выход целевого продукта (Воронков М.Г., "ЖОХ" 20, N 11, 2060, 1950 г.).
Недостаток способа состоит в относительно невысоком выходе винил-н-бутилового эфира.
Наиболее близким к предлагаемому по достигаемому результату является способ получения винил-н-бутилового эфира разложением дибутилацеталя в присутствии катализатора, состоящего из хинолина и ортофосфорной кислоты (Лавров В. М. и др., "ЖПХ", 58, N 2, 457, 1985 г. (прототип). Процесс проводят нагреванием дибутилацеталя и катализатора взятого в количестве 4,3 мас.% от веса ацеталя при температуре до 200oC, удаляя в процессе нагревания легкокипящий конденсат Tкип 100 - 125oC. Фракционированием конденсата отделяют непрореагировавший дибутилацеталь (52% от первоначальной загрузки), который возвращают в рецикл на стадию разложения, а из легкокипящей фракции выделяют целевой продукт известными приемами (Шостаковский М.Ф. "Простые виниловые эфиры", М. АН СССР, 33, 1952 г.) - бутанол отмывают водой, эфирный слой сушат поташом и перегоняют над твердой щелочью. Выход винил-н-бутилового эфира 97,8% на прореагировавший ацеталь.
Недостаток способа состоит в относительно низкой конверсии дибутилацеталя.
В предлагаемом изобретении задача состоит в повышении конверсии дибутилацеталя в процессах дезалкоксилирования.
Это достигается проведением процесса дезалкоксилирования дибутилацеталя в присутствии катализатора трибутилбензиламмонийхлорида, взятого в количествах 2,5 - 5,6 мас.% к массе ацеталя. Разложение дибутилацеталя на указанном катализаторе ведут в температурном интервале 160 - 180oC в течение 7 - 8 часов, удаляя в ходе реакции конденсат Tкип 90 - 106oC состоящий из винил-н-бутилового эфира, бутанола и остаточных количеств 0,9 - 4,2% дибутилацеталя. Завершение реакции контролируют по количеству отогнанного конденсата. По завершении реакции конденсат промывают водой (pH 8), эфирный слой сушат поташом и перегоняют над твердой щелочью. Выход целевого продукта 95,8 - 99%, на исходный дибутилацеталь. Конверсия ацеталя 95,7 - 98,9%.
Ниже приводятся примеры, иллюстрирующие предлагаемое изобретение
Пример 1.
В реактор, снабженный каплеотбойником (насадка с "наколками", длина 15 см), нисходящим холодильником и термометром загружают 26,9 г (0,15 моля) дибутилацетальацетальдегида, 0,19 г (0,0006 моля) трибутилбензиламмонийхлорида и смесь нагревают при температуре 160 - 180oC в течение 11 часов. В ходе разложения ацеталя отгоняют 26,5 г конденсата, Tкип 90 - 110oC. Фракционированием конденсата получают 24,7 г смеси винил-н-бутилового эфира и бутанола, Tкип 90 - 104oC и 1,7 г непрореагировавшего дибутилацеталя, Tкип 90 - 92oC/25 мм рт.ст. η D20 1,4086, d4 20 0,8490, Tкип 90 - 90,5oC/26 мм рт.ст, η D20 1,4031, d4 20 0,8514 (Шостаковский М.Ф. "Простые виниловые эфиры", М. АН СССР, 33, 1952 г.). Легкокипящую фракцию промывают водой (pH 8,5 • 20 мл), эфирный слой сушат поташом и перегоняют над твердой щелочью. Получают 14,4 г винил-н-бутилового эфира, Tкип 93 - 94,5oC, η D20 1,4034, d4 20 0,7809, Tкип 93,7 - 93,8oC, η D20 1,4026, d4 20 0,7792 (Шостаковский М.Ф. "Простые виниловые эфиры", М. АН СССР, 33, 1952 г.). Выход целевого продукта 93,0% на исходный ацеталь. Конверсия ацеталя 93,3%.
Пример 2.
Разложением 30,7 г (0,17 моля) дибутилацеталя в присутствии 0,52 г (0,0016 моля) трибутилбензиламмонийхлорида при температуре 160 - 180oC в течение 8 часов получено 30,6 г конденсата, Tкип 90 - 107oC. Фракционированием конденсата получено 29,1 г смеси винил-н-бутилового эфира и бутанола, Tкип 90 - 104oC и 1,4 г возвратного дибутилацеталя, Tкип 90 - 92oC/25 мм рт. ст. Легковоспламеняющую фракцию промывают водой (5 x 20 мл), эфирный слой сушат поташом и перегонкой над твердой щелочью получают 16,8 г целевого продукта, Tкип 93 - 94,5oC. Выход 95,1% на исходный ацеталь. Конверсий ацеталя 95,1%.
Пример 3.
Разложением 24,5 г (0,14 моля) дибутилацеталя в присутствии 0,62 г при температуре 160 - 180oC в течение 7 часов получено 24,3 г конденсата, Tкип 90 - 106oC. Фракционированием конденсата получено 23,1 г легкокипящей фракции, Tкип 90 - 104oC и 1,0 г непрореагировавшего дибутилацеталя, Tкип 90 - 92,5oC/25 мм рт.ст. Легкокипящую фракцию промывают водой (pH 8), сушат поташом и перегонкой над твердой щелочью получают 13,5 г целевого продукта. Выход 95,8% на исходный ацеталь. Конверсия ацеталя 95,7%.
Пример 4.
Дезалкоксилированием 26,2 г (0,15 моля) дибутилацеталя в присутствии 0,85 г (0,0027 моля) трибутилбензилхлорида при температуре 160 - 180oC в течение 7 часов получено 26,0 г, Tкип 90 - 104oC. Без предварительного фракционирования конденсат промывают водой (5 x 20 мл), эфирный слой сушат поташом и перегонкой над твердой щелочью получают 14,7 г целевого продукта, Tкип 93 - 94oC. Выход 97,5% на исходный ацеталь. Конверсия ацеталя 97,7%.
Пример 5.
К 0,85 г "отработанного" катализатора предыдущего опыта добавляют 21,1 г (0,12 моля) дибутилацеталя и смесь нагревают при температуре 160 - 180oC в течение 7 часов. В процессе нагревания из реакционной массы отгоняют 21,0 г конденсата, Tкип 90 - 104oC. Конденсат промывают водой (pH 8, 5 x 20 мл), сушат поташом и перегонкой получают 11,9 г целевого продукта, Tкип 93 - 94,0oC. Выход 98,3% на исходный ацеталь. Конверсия ацеталя 98,0%.
Пример 6.
Разложением 38,3 г (0,22 моля) дибутилацеталя в присутствии 2,0 г (0,0064 моля) трибутилбензиламмонийхлорида при температуре 160 - 180oC в течение 6 часов получено 38,0 г конденсата, Tкип 90 - 104oC. Конденсат промывают водой (5 x 20 мл), эфирный слой сушат поташом и перегонкой над твердой щелочью получают 21,7 г целевого продукта, Tкип 93 - 94oC. Выход 98,5%. Конверсия дибутилацеталя 98,4%.
Пример 7.
К 2,0 г (0,0064 моля) "отработанного" катализатора предыдущего опыта добавляют 35,8 г (0,20 моля) дибутилацеталя и смесь нагревают при температуре 160-180oC в течение 6 часов. В процессе нагревания из реакционной массы отгоняют 35,7 г конденсата, Tкип 90 - 104oC. Конденсат промывают водой (5 x 20 мл), эфирный слой сушат поташом и перегоняют над твердой щелочью. Получают 20,4 г винил-н-бутилового эфира, Tкип 93 - 94oC. Выход целевого продукта 99,0% на исходный ацеталь. Конверсия ацеталя 98,9%.
Пример 8.
Разложением 40,3 г (0,23 моля) дибутилацеталя в присутствии 3,1 г (0,01 моля) трибутилбензиламмонийхлорида при температуре 160-180oC в течение 6 часов и последующей обработкой полученного конденсата известными приемами получено 22,9 г винил-н-бутилового эфира, Tкип 93 - 94oC. Выход целевого продукта 98,8% на исходный ацеталь. Конверсия ацеталя 98,9%.
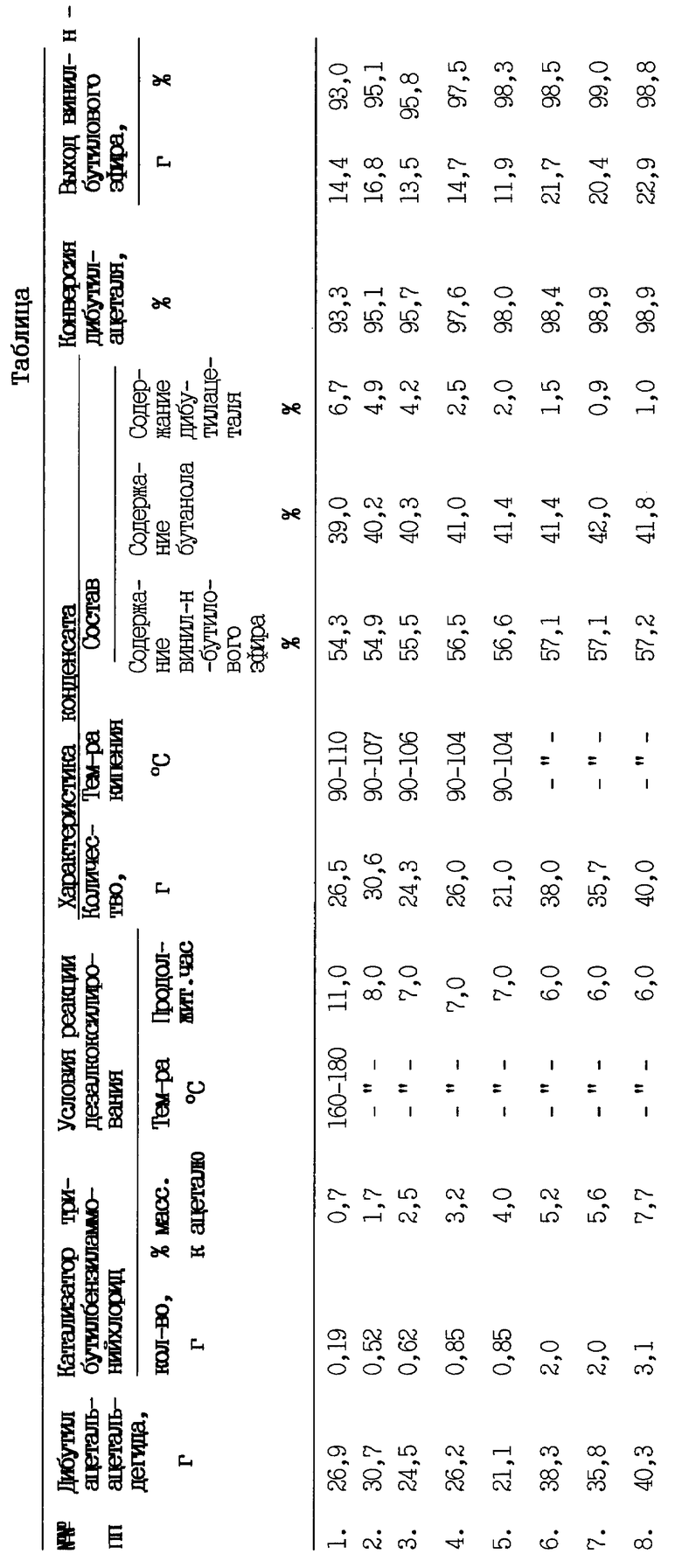
Данные по эффективности применения катализатора трибутилбензиламмонийхлорида в процессах дезалкоксилирования дибутилацетальацетальдегида представлены в таблице.
Из полученных данных следует, что проведение процессов дезалкоксилирования дибутилацеталя в присутствии трибутилбензиламмонийхлорида, взятого в количествах 2,5 - 5,6 мас.% к массе ацеталя обеспечивает 95,8 - 99% выхода винил-н-бутилового эфира и 95,7 - 98,9% конверсию дибутилацеталя (примеры 3-7). Проведение процессов разложения дибутилацеталя с добавлением указанного катализатора в количествах, меньших 2,5 мас.% от массы ацеталя сопровождается увеличением времени завершения реакции до 11 часов и снижением конверсии ацеталя (примеры 1, 2). Разложение дибутилацеталя в присутствии катализатора, взятого в количествах более 5,6 мас.% от массы ацеталя нецелесообразно, так как не дает дополнительных преимуществ по выходу эфира и полноте переработки исходного ацеталя (пример 8). В свою очередь, эффективность процессов дезалкоксилирования дибутилацеталя не снижается при использовании "отработанного" трибутилбензиламмонийхлорида (примеры 5, 7). Проведение процессов дезалкоксилирования дибутилацеталя при температурах выше 180oC нецелесообразно ввиду возможности дополнительного выведения из реакционной массы ацеталя с парами продуктов его разложения. В свою очередь реализация процессов дезалкоксилирования при температуре ниже 160oC сопровождается увеличением времени завершения процессов.
Описанный способ получения винил-н-бутилового эфира каталитическим дезалкоксилированием дибутилацетальацетальдегида с использованием в качестве катализатора трибутилбензиламмонийхлорида в сравнении с известным (прототипом), позволяет при аналогичных выходах эфира увеличить конверсию ацеталя до 95,7 - 98,9% против 48,2% и, как следствие, упрощает процесс получения целевого продукта за счет ликвидации необходимости проведения дополнительных операций по переработке значительных количеств "возвратного" ацеталя.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛ-Н-БУТИЛОВОГО ЭФИРА | 2000 |
|
RU2179967C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛ-Н-БУТИЛОВОГО ЭФИРА | 2022 |
|
RU2798601C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ХЛОРСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 1998 |
|
RU2152381C1 |
| СПОСОБ ПОЛУЧЕНИЯ БУТИЛБЕНЗИЛФТАЛАТА | 1992 |
|
RU2044722C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИСТОГО БЕНЗИЛА | 1997 |
|
RU2125035C1 |
| СПОСОБ ВЫДЕЛЕНИЯ Н-МАСЛЯНОГО АЛЬДЕГИДА ИЗ ПРОДУКТА ГИДРОФОРМИЛИРОВАНИЯ ПРОПИЛЕНА | 1997 |
|
RU2130917C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛКСАНТОГЕНАТОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ | 2002 |
|
RU2211831C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗИЛОВОГО СПИРТА | 1995 |
|
RU2086529C1 |
| СПОСОБ ПОЛУЧЕНИЯ Н-БУТИЛОВОГО КСАНТОГЕНАТА | 1999 |
|
RU2152928C1 |
| Способ получения ацеталей | 1975 |
|
SU697493A1 |
Изобретение относится к винил-н-бутиловому эфиру - полупродукту для получения полимеров, применяемых в медицине и технике. Процесс заключается в дезалкоксилировании дибутилацетальацетальдегида в присутствии катализатора - трибутилбензиламмонийхлорида, взятого в количестве 2,5-5,6 мас.% к дибутилацетальацетальдегиду, и процесс ведут при 160-180°С в течение 6-7 ч. В результате повышается конверсия исходного продукта. 1 табл.
Способ получения винил-н-бутилового эфира каталитическим дезалкоксилированием дибутилацетальацетальдегида при нагревании с последующим выделением целевого продукта известными приемами, отличающийся тем, что в качестве катализатора используют трибутилбензиламмонийхлорид
взятый в количестве 2,5 - 5,6 мас.% к дибутилацеталю и процесс ведут при 160 - 180oC в течение 6 - 7 ч.
| Лавров В.М | |||
| и др | |||
| - Журнал прикладной химии, 1985, т.58, N 2, с.457-459 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТЫХ ВИНИЛОВЫХ ЭФИРОВ | 0 |
|
SU218162A1 |
| US 1931858 A, 1933 | |||
| ПЕРЕХОДНАЯ ПЛОЩАДКА ПАССАЖИРСКОГО ВАГОНА | 1994 |
|
RU2091259C1 |
| МАКСИМАЛЬНЫЙ ЭЛЕКТРОМАГНИТНЫЙ РАСЦЕПИТЕЛЬ ТОКА АВТОМАТИЧЕСКОГО ВЫКЛЮЧАТЕЛЯ | 1997 |
|
RU2117357C1 |
