Изобретение относится к способу получения ламотриджина и его фармацевтически приемлемых солей, полученных присоединением кислот.
Ламотриджин представляет собой 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин формулы (I)
Это известное соединение описано в патентной заявке EP-A-0021121 и применяется при лечении расстройств центральной нервной системы (ЦНС), в частности эпилепсии. В патентной заявке EP-A-0247892 описан изетионат ламотриджина, являющийся предпочтительной его солью для парентерального введения. В патентной заявке EP-A-0021121 описывается также способ получения ламотриджина, при котором соединение формулы (II)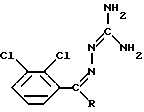
подвергают циклизации, например путем нагревания в сосуде с обратным холодильником в спирте в присутствии основания.
Настоящее изобретение относится к способу получения ламотриджина, при котором соединение формулы (II), в которой R представляет собой CN или CONH2, в органическом растворителе подвергают облучению ультрафиолетовым или видимым светом и, если R представляет собой CN, то нагреванию.
Можно использовать любой органический растворитель, в котором растворяется соединение формулы (II). Предпочтительным примером является C1-C6-алканол.
C1-C6-алканол может представлять собой алканол с нормальной или разветвленной цепью. Это может быть, в частности, C1-C4-алканол, например метанол, этанол, н-пропанол, изопропанол, н-бутанол, втор.-бутанол или трет.-бутанол или их смесь.
Если R в формуле (II) представляет собой CN, то способ по изобретению требует нагревания соединения формулы (II). В этом случае способ предпочтительно осуществляют посредством облучения и нагревания соединения формулы (II) в растворителе. Раствор можно нагревать, например, на паровой бане. Его удобно нагревать до температуры перегонки органического растворителя. Раствор сначала может быть облучен при температуре окружающей среды, а затем последовательно нагрет, например, до начала флегмообразования. Альтернативно раствор может быть сначала нагрет, например, до начала флегмообразовния, а затем последовательно облучен. Во время облучения температуру раствора удобно поддерживать близкой или равной температуре перегонки. Например, раствор можно нагреть до начала флегмообразования, а затем подвергнуть циркуляции через фотохимический реактор с падающей пленкой, в котором он подвергается облучению соответствующей интенсивности и длительности.
Если R в формуле (II) представляет собой CONH2, то способ по изобретению не требует обязательного нагрева. В этом случае раствор соединения формулы (II) в органическом растворителе может быть облучен при температуре окружающей среды, например при температуре от 18oC до 25oC или от 20oC до 25oC, предпочтительно при 25oC. Однако этот раствор может по возможности быть нагрет перед, во время или после облучения, например, как описано выше для соединений формулы (II), в которой R представляет собой CN.
Облучение соединения формулы (II) в органическом растворителе может быть осуществлено путем экспонирования на солнечном свету, под лампой с вольфрамовой нитью, лампой дневного света или ртутной лампой среднего давления. Облучение может также быть осуществлено путем экспонирования как на солнечном свету, так и под лампой с вольфрамовой нитью или как на солнечном свету, так и под лампой среднего давления. Лампа с вольфрамовой нитью представляет собой, как правило, лампу мощностью 150 ватт.
Облучение продолжают в течение периода времени, достаточного для осуществления циклизации соединения формулы (II) в ламотриджин. Продолжительность облучения будет соответственно зависеть помимо прочего от источника или источников облучения, от интенсивности облучения, от характера реакционного сосуда и от температуры и pH облучаемого раствора. Облучение может продолжаться, например, в течение периода времени от 2 часов до 5 дней, в частности от 2 часов до 4 дней или от 2 часов до 2 дней, или 2 часов до 1 дня; или от 4 часов до 4 дней, в частности от 4 часов до 4 дней или от 4 часов до 2 дней, или от 4 часов до 1 дня; или от 2 часов до 24 часов, в частности от 2 часов до 20 часов или от 2 часов до 18 часов; или от 4 часов до 24 часов, в частности от 4 часов до 20 часов, или от 4 часов до 18 часов.
Если R в формуле (II) представляет собой CONH2, то способ по изобретению соответственно осуществляют в присутствии основания. Может быть использовано любое подходящее основание, например гидроксид щелочного металла, такого как гидроксид калия или гидроксид натрия. Гидроксид калия наиболее предпочтителен. Полагают, что щелочное pH, создаваемое путем применения основания, активирует циклизацию соединения формулы (II) в ламотриджин в пользу возможно конкурентных фотохимических процессов. Примером такого конкурентного процесса является E/Z изомеризация соединения формулы (II), продукт которой представляет собой смесь соединения формулы (II) и его Z-изомера, соединения формулы (III)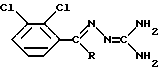
в которой R представляет собой CN или CONH2.
Полагают, что превращение соединения формулы (II) в ламотриджин происходит посредством фотохимической изомеризации в Z-изомер формулы (III), который не выделяют, с последующей циклизацией Z-изомера в сам ламотриджин.
Если R в формуле (II) представляет собой CONH2, то другой фотохимический процесс может, при определенных реакционных условиях, конкурировать с циклизацией в ламотриджин. Этот процесс представляет собой превращение этого соединения в 3-амино-5-гидрокси-6-(2,3- дихлорфенил)-1,2,4-триаэин формулы (IV)
Как указывалось выше, температура и pH, при которых осуществляется способ по изобретению, могут быть существенны при определении хода реакции. Например, облучение раствора соединения II, в котором R представляет собой CONH2, в этаноле при нейтральном pH в течение 18 часов при 80oC приводит к образованию вышеописанного гидрокси-соединения (IV) в качестве основного продукта циклизации, при этом ламотриджин образуется в меньшем количестве. В противоположность этому, нагревание раствора соединения II, в котором R представляет собой CONH2, в этаноле при щелочном pH, например в присутствии KOH, с последующим облучением в течение примерно 3 часов приводит к образованию ламотриджина при фактическом исключении гидрокси-соединения формулы (IV).
Соединение формулы (II), в котором R представляет собой CN, может быть получено дегидратацией аминогуанидоноксима формулы (V)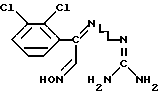
Эту реакцию предпочтительно проводят обработкой соединения формулы (V) подходящим дегидратирующим агентом, например тионилхлорилом, в соответствующем органическом растворителе, например диметилформамиде. Установлено, что в результате дегидратации смеси (E)- и (Z)-изомеров соединения формулы (V) получается по существу только (E)-изомер соединения формулы (II).
Соединение формулы (V) является новым. Поэтому изобретение касается также вышеописанного соединения формулы (V). Соединение формулы (V) может быть получено обработкой 2,3-дихлорфенилглиоксальальдоксима формулы (VI)
аминогуанидином или его солью. Реакцию предпочтительно проводят в подходящем растворителе, таком как диметилсульфоксид или диметилформамид, в присутствии кислоты. Обычно кислота является концентрированной соляной кислотой, например 8N соляной кислотой. Реакция может быть проведена при температуре окружающей среды.
Соединение формулы (VI) может быть получено нитрозированием 2,3-дихлорацетофенона формулы (VII)
соответствующим нитрозирующим агентом, предпочтительно C1-C6-алкилнитритом или азотистой кислотой, более предпочтительно амилнитритом. Реакцию предпочтительно проводят в присутствии кислого или основного катализатора в подходящем органическом растворителе, например в простом эфире или спирте, предпочтительно в диэтиловом эфире или трет.-бутаноле, при температуре в пределах от примерно 15oC до температуры кипения растворителя. Если применяют кислый катализатор, то он предпочтительно является хлороводородом. Если применяют основной катализатор, то он предпочтительно является трет. -бутоксидом калия. Реакция может быть проведена при комнатной температуре.
Соединение формулы (VII) может быть получено обработкой 1,2-дихлорбензола соединением формулы RM1 или RM2X, в котором R представляет собой C1-C6-алкил, M1 представляет собой щелочной металл, M2 представляет собой щелочно-земельный металл, а X представляет собой галоген, после чего следует взаимодействие полученного таким образом соединения с ацетилхлоридом или уксусным ангидридом. RM2X может представлять собой, например, реактив Гриньяра (Grignard), такой как метилмагнийиодид. Предпочтительно применяют соединение формулы RM1, при этом бутиллитий является наиболее предпочтительным, после чего следует реакция с уксусным ангидридом. Эту реакцию предпочтительно проводят при температуре около -70oC.
Соединение формулы (VII) может быть также получено обработкой 2,3-дихлорбензальдегида соединением RM1 или RM2X, где R представляет собой метил, предпочтительно метилмагнийиодидом, и окислением полученного таким образом α-метил-2,3-дихлорбензилового спирта. Приемлемые окисляющие агенты включают, например гипохлорит натрия. Эту реакцию предпочтительно проводят при комнатной температуре.
Соединение формулы (VII) может быть также получено обработкой 2,3-дихлориодбензола магнием и взаимодействием полученного таким образом соединения с ацетилхлоридом в присутствии безводного хлорида железа. Эту реакцию предпочтительно проводят при температуре около -70oC.
Соединение формулы (II), в котором R представляет собой CN, альтернативно может быть получено дегидратацией соответствующего амида, который является соединением формулы (II), где R представляет собой CONH2. Могут быть использованы традиционные дегидратирующие агенты, например пирофосфорилхлорид.
Исходное соединение формулы (II), в которой R представляет собой CONH2, получают способом, который включает обработку 2,3-дихлорфенилглиоксиламида формулы (VIII)
аминогуанидином или его солью.
Если применяют соль аминогуанидина, то она предпочтительно является гидрохлоридом гуанидина.
Реакцию взаимодействия соединения формулы (VIII) с аминогуанидином или его солью предпочтительно проводят в подходящем органическом растворителе, таком как C1-C6-алканол, например этанол, в присутствии кислоты при повышенной температуре. Кислота обычно является концентрированной кислотой, например концентрированной соляной кислотой. Реакцию соответственно проводят при температуре перегонки применяемого органического растворителя.
Соединение формулы (VIII) является новым. Поэтому данное изобретение касается также вышеописанного соединения формулы (VIII).
Соединение формулы (VIII) может быть получено обработкой 1,2-дихлорбензола соединением формулы RM1 или RM2X, в которых R представляет собой C1-C6-алкил, M1 представляет собой щелочной металл, M2 представляет собой щелочноземельный металл, а X представляет собой галоген, с последующей реакцией взаимодействия полученного таким образом соединения с C2-C6-алкилоксаматом. Соединение формулы RM2X может представлять собой, например, реактив Гриньяра, такой как метилмагнийиодид. Предпочтительно используют соединение формулы RM1, наиболее предпочтительным является н-бутиллитий. Алкилоксамат предпочтительно представляет собой этилоксамат.
Реакцию взаимодействия предпочтительно осуществляют обработкой раствора 1,2-дихлорбензола в соответствующем органическом растворителе, например эфире, таком как диэтиловый эфир, диоксан или тетрагидрофуран, раствором н-бутиллития в подходящем органическом растворителе, например гексане, при температуре предпочтительно ниже примерно -60oC. При взаимодействии н-бутилхлорида с литием in situ может образовываться н-бутиллитий.
Соединение формулы (VIII) также может быть получено активацией 2,3-дихлорфенилглиоксиловой кислоты посредством превращения в соответствующий ангидрид или галогенангидрид, а затем обработкой активированного производного аммонием. Альтернативно аммонийная соль 2,3-дихлорфенилглиоксиловой кислоты может быть дегидратирована с получением соединения формулы (VIII). Такие методы являются обычными для превращений кислот в амиды, они традиционны в органическом синтезе.
Соединение формулы (VIII) может быть также получено обработкой этилового эфира 2,3-дихлорфенилглиоксиловой кислоты формулы (IX)
аммонием. Реакцию взаимодействия предпочтительно проводят в соответствующем растворителе, например, C1-C6-алканоле, таком как метанол, этанол, пропан-1-ол или пропан-2-ол, предпочтительно в метаноле.
Этиловый эфир 2,3-дихлорфенилглиоксиловой кислоты является новым. Поэтому данное изобретение касается далее вышеописанного соединения формулы (IX).
Соединение формулы (IX) предпочтительно получают обработкой 1,2-дихлорбензола соединением формулы RM1 или RM2X, в которой R представляет собой C2-C6-алкил, M1 представляет собой щелочной металл, M2 представляет собой щелочноземельный металл, а X представляет собой галоген, с последующей реакцией взаимодействия полученного таким образом соединения с этилоксалилгалогенидом, например этилоксалилхлоридом. RM2X может представлять собой, например, реактив Гриньяра, такой как метилмагнийиодид. Предпочтительно применяют соединение формулы RM1, наиболее предпочтительным является н-бутиллитий.
Данное изобретение касается также способа, который далее включает получение фармацевтически приемлемой соли ламотриджина, образованной присоединением кислоты.
Соответствующие фармацевтически приемлемые соли ламотриджина, полученные присоединением кислоты, включают сульфат, фосфат, метансульфонат, п-толуолсульфонат, бензолсульфонат и изетионат. Изетионат ламотриджина наиболее предпочтителен для парентерального введения, так как он обладает высокой растворимостью в воде.
Изетионат ламотриджина может быть получен взаимодействием ламотриджина с изетионовой кислотой. Предпочтительно молярное соотношение ламотриджина и изетионовой кислоты составляет от 1:3 до 3:1, в частности приблизительно 1: 1.
Изетионовая кислота отсутствует в продаже и поэтому ее обычно получают in situ. Например, изетионат щелочного металла в растворе может быть превращен в изетионовую кислоту, например, посредством пропускания водного раствора изетионата через H+-ионообменную смолу и последующего смешивания триазина с полученным в результате кислым раствором. Обычно растворителем в данной реакции является вода и в этом случае реакцию можно проводить при температурах от 4 до 50oC, лучше при температуре окружающей среды, и без применения каких-либо регуляторов pH или других добавок.
Образовавшийся изетионат может быть перекристаллизован, например, из метилированного технического спирта, с получением кристаллов изетионата ламотриджина, которые легко растворяются в воде.
Альтернативно изетионат ламотриджина может быть получен в результате взаимодействия соли ламотриджина, не являющейся изетионатом, с изетионатным анионом. Предпочтительно, чтобы соотношение соли к аниону составляло от 1:50 до 50: 1. Более предпочтительно соотношение составляет примерно 1:10. Предпочтительно эту реакцию осуществляют путем элюции раствора соли в метаноле через колонку с изетионатной анионообменной смолой. В этом случае соль предпочтительно представляет собой метансульфонат (мезилат) ламотриджина.
Данное изобретение предусматривает также способ, включающий приготовление фармацевтической композиции соединением ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты, с фармацевтически приемлемым разбавителем или носителем.
Ламотриджин должен присутствовать в композициях, полученных в соответствии с изобретением, в форме эффективной единичной дозы, то есть в количестве, эффективном против расстройств ЦНС in vivo.
Фармацевтически приемлемый разбавитель или носитель, присутствующий в композициях, приготовленных согласно изобретению, может представлять собой жидкий или твердый материал, который является инертным или приемлемым с точки зрения медицины и совместим с ламотриджином или его солью.
Фармацевтические композиции могут вводиться перорально или парентерально, применяться в виде суппозиториев или наноситься локально в виде мази, пасты или порошка. Однако предпочтительно пероральное или парентеральное введение состава.
Для перорального введения можно использовать тонкие порошки или гранулы, содержащие разбавляющие, диспергирующие и/или поверхностно-активные вещества, которые могут быть представлены в виде дозы в воде или сиропе, в виде капсул или пастилок в сухом состоянии или в виде неводной суспензии, в которую могут быть включены суспендирующие вещества, и наконец, в виде суспензии в воде или сиропе. Если это желательно или необходимо, могут быть включены ароматизаторы, консерванты, суспендирующие агенты, загустители и эмульгирующие агенты. Если суспензию готовят в воде, то в соответствии с данным изобретением предпочтительно присутствует по меньшей мере одно из этих веществ.
Для парентерального введения активное соединение может быть приготовлено в виде стерильного водного раствора для инъекций, который может содержать антиоксиданты или буферы.
Ламотриджин или его соль могут быть введены в чистой форме, не связанной с другими добавками, и в этом случае предпочтительным носителем является капсула или пастилка.
Альтернативно активное соединение может быть представлено в чистой форме в виде эффективной единичной дозы, например оно может быть спрессованно в виде таблетки и т.п.
Другими веществами, которые могут быть включены в состав препарата, являются, например, инертные с точки зрения медицины ингредиенты, в частности твердые и жидкие разбавители, такие как лактоза, крахмал или фосфат кальция для таблеток или капсул; оливковое масло или этилолеат для мягких капсул; и вода или растительное масло для суспензий или эмульсий; смягчающие агенты, такие как тальк или стеарат магния; желирующие агенты, такие как коллоидные глины; загустители, такие как гуммиарабик или альгинат натрия; а также другие терапевтически приемлемые вспомогательные ингредиенты, такие как увлажнители, консерванты, буферы и антиоксиданты, которые могут быть использованы как носители в таких препаратах.
Таблетки и другие препаративные формы, представленные в виде отдельных единиц, могут содержать количество ламотриджина или его соли, эффективное в этой дозе или в многократных дозах, например единицы, содержащие от 5 мг до 500 мг, предпочтительно примерно от 10 мг до 250 мг, в пересчете на свободное основание.
Водные препараты должны в общем случае содержать фармацевтически приемлемую соль ламотриджина в количестве, эффективном против расстройств ЦНС in vivo и такой препарат может быть представлен в единичной дозировочной форме. В составе водного препарата может находиться до 250 мг/мл соли в пересчете на свободное основание. Однако традиционные концентрации соли в растворе составляют от 10 до 70 мг/мл, предпочтительно, от 10 до 50 мг/мл. Соль для парентерального введения может быть представлена в виде стерильных водных растворов для инъекций, которые могут содержать терапевтически приемлемые вспомогательные ингредиенты, такие как антиоксиданты, буферы и агенты для регулирования осмомолярности раствора. Предпочтительно, чтобы такие анионы, как хлорид и фосфаты, не присутствовали в растворе, так как они имеют тенденцию обмениваться с солью с образованием осадков.
Водный препарат может быть приготовлен растворением соли в водной среде, например в стерильной воде для инъекций. Раствор перед использованием может быть разбавлен до необходимой концентрации.
Нижеследующие примеры иллюстрируют этапы способа по изобретению.
Эталонный пример 1. Получение (E)-2-(2',3'-дихлорфенил)-2-(гуанидинилимино)ацетамида (соединение (II) с R=CONH2
Смесь 2,3-дихлорфенилглиоксиламида (54,5 г, 0,25 моль), гидрохлорида аминогуанидина (33,15 г, 0,30 моль), этанола (1 л) и концентрированной соляной кислоты (4 мл) кипятили с обратным холодильником в течении 6 ч (pH = 1,5). Полученный в результате раствор выпарили досуха, твердый остаток растворили в воде (2 л; полученное pH = 2,5), раствор подщелачивали до pH 13 добавлением 50%-ного (м/о) водного гидроксида натрия (45 мл) при <15o. Смесь фильтровали, темно-желтый твердый остаток промывали раствором 0,88 аммония и сушили до получения сырого продукта (59,5 г, 87%), т.пл 231-3o (эт. 233-4o разлож. ). После перекристаллизации этого продукта (2,2 г) из пропан-1-ола (60 мл) получили чистое вещество (1,83 г, 83%) т.пл. 238-9o (разлож.).
Эталонный пример 2. Получение (E)-2-(2,3-дихлорфенил)-2-(гуанидинилимино)ацетонитрила (соединение (II) с R=CN)
Аминогуанидоноксим формулы (V), определенной выше, растворили в диметилформамиде и обработали тионилхлоридом. Был получен сырой продукт, который содержал несколько второстепенных примесей. Эти примеси легко удалили растиранием с толуолом и получили в результате (E)-2-(2,3-дихлорфенил)-2-гуанидинилиминоацетонитрил с выходом 24%.
Пример 1. Получение ламотриджина
(E)-2-(2', 3'- дихлорфенил)-2-(гуанидинилимино)ацетамид, описанный в эталонном примере 1 (0,3 г), растворили в этаноле (10 мл) и облучили, подвергнув его воздействию солнечного света.
Тонкослойная хроматография (ТСХ) после 4 ч облучения показала, что в растворе присутствует смесь соединения (II) и его Z-изомера, соединения (III), но образования ламотриджина не произошло. Однако через 2 дня ТСХ выявила наличие пятна, соответствующего ламотриджину (Rf = 0,20). Также было обнаружено пятно неидентифицированного вещества на базовой линии. Спустя в общей сложности 4 дня ТСХ анализ раствора показал, что ламотриджин все еще присутствует.
Взвешенное твердое вещество, которое образовалось у насоса, было затем отфильтровано. Установлено, что при ТСХ оно дает пятно около базовой линии. В оставшейся жидкости посредством ТСХ был обнаружен ламотриджин. Поэтому облучение образца жидкости продолжали еще в течение одного дня. ТСХ выявила наличие яркого пятна ламотриджина. Температура плавления ламотриджина = 218oC.
Пример 2. Получение ламотриджина
(E)-2-(2',3'-дихлорфенил)-2-(гуанидинилимино)ацетамид (2,5 г) растворили в горячем этаноле (40 мл). Добавили гидроксид калия (2,0 г, эквивалентно 5% м/о) и облучали полученный раствор, подвергая его воздействию яркого солнечного света и лампы с вольфрамовой нитью (150 Вт).
После 4-дневного облучения с помощью лампы результаты ТСХ показали наличие пятна средней интенсивности, соответствующего ламотриджину (Rf = 0,20).
Затем щелочной этанольный раствор подкислили концентрированной соляной кислотой (3 мл) до pH 6. Добавили силикагель (2,0 г) и упарили весь объем досуха. Полученный тонкий белый порошок ввели в колонку с силикагелем и элюировали (см. табл. 1).
Посредством ТСХ белое твердое вещество было идентифицировано как ламотриджин. Это было подтверждено результатами масс-спектроскопии и измерениями температуры плавления (т.пл. = 218oC).
Пример 3. Получение ламотриджина
(E)-2-(2',3'-дихлорфенил)-2-(гуанидинилимино)ацетамид (2,0 г) растворили в растворе гидроксида калия (5% м/о) в этаноле (40 мл). Полученный раствор облучали при температуре окружающей среды, используя лампу среднего давления в течение полных 12 ч. Раствор оставили стоять, а затем прогревали на паровой бане при 70oC в течение 2 ч.
Спустя 4 ч ТСХ анализ раствора выявил присутствие следового количества ламотриджина (Rf = 0,20) вместе со смесью 50:50 соединения II и его Z изомера, соединения III. ТСХ раствора повторили через 6,5 ч, 9 ч и 12 ч. Каждый раз на пластине обнаруживали слабое пятно, соответствующее ламотриджину, и более яркое пятно, соответствующее E/Z смеси соединений II и III. Эти результаты свидетельствовали о том, что E/Z изомеризация соединения II происходила очень быстро, а циклизация соединения II в ламотриджин в описанных реакционных условиях протекала медленно.
Пример 4. Получение ламотриджина
Способ
Раствор (E)-2-(2',3'-дихлорфенил)-2-(гуанидинилимино)ацетамида (2,2 г) в этаноле (100 мл) нагревали до начала флегмообразовния и подвергали циркуляции через фотохимический реактор с падающей пленкой. Раствор в ходе реакции анализировали посредством ТСХ с интервалами (2,5 ч, 5 ч, 7,5 ч, 10 ч, 13 ч) от начала реакции. После прохождения реакции в течение 10 ч раствор становился темно-желтого цвета. Через 13 ч раствор становился оранжевым и начал выпадать белый осадок. В этот момент реакция была остановлена. Белое твердое вещество отфильтровали и исследовали с помощью ТСХ.
Результат
Через 2,5 ч после начала реакции ТСХ раствора показала, что начинается образование ламотриджина. Непрореагировавшее соединение II также присутствовало. Через 5 ч непрореагировавшее соединение II и ламотриджин присутствовали по-прежнему, но было обнаружено также дополнительное пятно с более высоким значением Rf. В моменты времени, составлявшие 7,5, 10 и 13 ч после начала реакции, проводили такие же наблюдения.
Белое твердое вещество было идентифицировано посредством ТСХ как чистый образец 3-амино-5-гидрокси-6-(2,3-дихлорфенил)-1,2,4-триазина.
Пример 5. Получение ламотриджина
(E)-2-(2',3'-дихлорфенил)-2-(гуанидинилимино)ацетамид (5,0 г) растворили в горячем этаноле (150 мл), содержащем гидроксид калия (2% м/о). Полученный желтый раствор облучали при 25oC, используя лампу среднего давления. ТСХ анализ раствора после облучения в течение 3 ч и 5 ч, соответственно, показал, что ламотриджин в детектируемом количестве не образовался.
Затем раствор нагрели до начала флегмообразования, после чего подвергли облучению, используя лампу среднего давления. После 3 ч облучения анализ с помощью ТСХ выявил присутствие ламотриджина. 3-амино-5-гидрокси-6-(2,3-дихлорфенил)-1,2,4-триазин обнаружен не был.
Перекристаллизация из этанола, содержащего 5 г угля, привела к получению 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин. EtOH (31 г, 56%) в виде очень светлого желтого твердого вещества с т.пл. 219-220oC (эт. 220-222oC). ТСХ в системе силикагель/этилацетат показала присутствие следового количества флуоресцирующей примеси на базовой линии.
Пример 6. Получение ламотриджина
(E)-2-(2,3-дихлорфенил)-2-гуанидинилиминоацетонитрил, описанный в эталонном примере 2, растворили в пропан-1-оле и облучили при кипячении с обратным холодильником, как показано в табл. 2. Выпаривание полученного раствора и перекристаллизация твердого остатка из этанола привели к получению бесцветного гомогенного 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин. EtOH во всех, кроме последнего, случаях (см. табл. 2).
В мелкомасштабных опытах конверсия в 3,5-диамино-6-(2,3-дихлорфенил)- 1,2,4-триаэин была очень чистой (ТСХ), а низкие выходы отражали потери при обработке. В конечном эксперименте с 47 г конверсия в 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин через 8 ч прошла примерно на 50%, но через 32 ч реакционная смесь стала желтой и мутной. Ее профильтровали, выпарили до небольшого объема, охладили и отфильтровали с получением сырого 3,5-диамино-6-(2,3-дихлорфенил)- 1,2,4-триазина (32,2 г, 68%) в виде светло-желтого твердого вещества. Перекристаллизация из этанола, содержащего 5 г угля, привела к получению 3,5-диамино-6-(2,3-дихлорофенил)-1,2,4-триазин.EtOH (31 г, 56%) в виде очень светлого желтого твердого вещества с т.пл. 219-220oC (эт. 220-222oC). ТСХ в системе силикагель/этилацетат показала наличие следового количества флуоресцентной примеси у базовой линии.
Пример 7. Получение аминогуанидоноксима формулы (V)
Альдоксим 2,3-дихлорфенилглиоксаля конденсировали с аминогуанидином в смеси диметилсульфоксида и 8 N соляной кислоты при температуре окружающей среды. Реакционную смесь подщелачивали концентрированным раствором гидроксида аммония и выделяли оксим аминогуанидона с выходом 73% в виде эквимолярной смеси (E)- и (Z)-изомеров.
Эталонный пример 3. Получение 2,3-дихлорфенилглиоксальальдоксима формулы (VI)
Способ А
2,3-дихлорацетофенон (20,00 г, 0,104 моль) растворили в сухом диэтиловом эфире (200 мл) и через раствор в течение получаса барботировали хлороводород. Медленно, по каплям, при перемешивании добавляли амилнитрит (24,36 г, 0,238 моль) так, чтобы поддерживать раствор при мягком кипении с обратным холодильником. После этого раствор мягко кипятили с обратным холодильником в течение 3 ч, продолжая пропускать хлороводород. Раствор оставили стоять при комнатной температуре в течение ночи. Реакционную смесь с исключительной осторожностью вылили в 2 N раствор гидроксида натрия (200 мл) (реакция очень бурная) и разделили. Органическую фазу экстрагировали 2 N раствором гидроксида натрия (2 х 100 мл). Щелочные фазы объединили, вылили на лед (200 мл) и добавили при перемешивании концентрированную соляную кислоту (100 мл). Полученную кислую смесь оставили стоять при комнатной температуре в течение ночи, а образовавшийся осадок отфильтровали на насосе. 2,3-дихлорфенилглиоксальальдоксим получили в виде вещества кремового цвета (10,4 г, 46%-ный выход). ТСХ (SiO2; CHCl3) показала отсутствие примесей, поэтому дальнейшую очистку не проводили.
Метод Б
Трет. -бутоксид калия (239,7 г, 2,136 моль) растворили в трет.-бутаноле (1,5 л) и перемешивали при комнатной температуре в течение 30 мин. В виде разовой порции добавили 2,3-дихлорацетофенон (67,26 г, 0,356 моль), а полученную смесь перемешивали в течение ночи при комнатной температуре. Медленно, по каплям, при перемешивании добавили амилнитрит (83,41 г, 0,712 моль). Когда добавление было завершено, реакционную смесь перемешивали при комнатной температуре в течение 22 ч, а затем нагревали при 50oC в течение 2 ч. Раствору дали остыть и перемешивали при комнатной температуре в течение ночи. Реакционную смесь вылили в воду со льдом (1,5 л), и водный раствор экстрагировали диэтиловым эфиром (3 х 500 мл). Водную фазу подкислили концентрированной соляной кислотой и оставили стоять при комнатной температуре в течение ночи. Твердое вещество, выпавшее в осадок, отфильтровали на насосе, высушили в вакууме над безводной пятиокисью фосфора и получили 2,3-дихлорфенилглиоксальальдоксим (57,3 г, 53,8%) в виде коричневого твердого вещества. ТСХ (SiO2, CHCl3) выявила наличие малого количества примесей, но последующая дегидратация оксима показала, что перекристаллизация была бы желательной. Оксим может быть перекристаллизован из толуола, т.пл. 109oC.
Эталонный пример 4. Получение 2,3-дихлорацетофенона
Способ А
Бутиллитий в гексане (300 мл, 0,474 моль) медленно, по каплям, при перемешивании добавили к 1,2-дихлорбензолу (104,58 г, 0,711 моль), растворенному в сухом тетрагидрофуране (2 л) и поддерживаемому при температуре -70oC в атмосфере азота. Полученный раствор перемешивали при -70oC в течение 1 ч. Этот раствор, по-прежнему при -70oC, в атмосфере азота добавили с помощью иглы с двойным концом к уксусному ангидриду (290,35 г, 2,84 моль), растворенному в сухом тетрагидрофуране (1 л) при -70oC. После завершения добавления полученный раствор перемешивали при -70oC приблизительно в течение 1 ч и давали ему нагреться до комнатной температуры.
Реакционную смесь выливали на лед (5 л) и, после хорошего перемешивания, оставляли стоять в течение ночи при комнатной температуре. Водную смесь экстрагировали эфиром (3 х 1,5 л). Эфирные фазы объединяли, промывали водой (3 х 750 мл), насыщенным раствором бикарбоната натрия (3 х 750 мл) и рассолом (1 х 750 мл). Органическую фазу высушивали над безводным сульфатом магния, фильтровали и выпаривали с получением желтой жидкости. Сырой продукт поместили в высокий вакуум на горячую водяную баню для полного удаления 1,2-дихлорбензола и уксусного ангидрида. Получили 2,3-дихлороацетофенон (67,2 г, выход 73%). ИК, ЯМР и ТСХ (SiO2; CHCl3) показали, что данный продукт содержит мало примесей, поэтому дальнейшую очистку не проводили.
Способ Б
Иодметан (234,89 г, 2,288 моль) добавили по каплям при перемешивании к магниевым стружкам (54,88 г, 2,288 моль) в сухом эфире для получения метилмагнийиодида. 2,3-дихлорбензальдегид (200 г, 1,144 моль), растворенный в смеси бензол/диэтиловый эфир (1 л, 50:50), добавили по каплям при перемешивании к реактиву Гриньяра. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор кипятили с обратным холодильником в течение 2 ч, затем дали ему остыть. Реакционную смесь вылили в насыщенный раствор хлорида аммония (5 л) и органический слой отделили. Водный слой экстрагировали эфиром (3 х 2 л). Органические фазы объединяли, промывали рассолом (1 х 2 л), сушили над безводным сульфатом магния, фильтровали и выпаривали. Получили α- метил-2,3-дихлорбензиловый спирт (196,4 г, выход 90%) в виде желтого масла, которое кристаллизовалось при стоянии с образованием бледно-желтого твердого вещества. ТСХ (SiO2; CHCl3) не выявила наличия примесей, так что дальнейшую очистку не проводили. Однако, если необходимо, спирт может быть перекристаллизован из петролейного эфира при 40-60oC с получением белых призм с температурой плавления 53oC.
α-Метил-2,3-дихлорбензиловый спирт (5,0 г, 0,026 моль) растворили в уксусной кислоте (24 мл) и медленно, по каплям, при перемешивании при температуре 15-25oC добавили 12%-ый (м/о) раствор гипохлорита натрия (23,26 мл, 0,0314 моль). Когда добавление было закончено, реакционную смесь перемешивали при комнатной температуре примерно 1,5 ч, пока проба крахмал/иодид не дала положительный результат. К реакционной смеси добавляли насыщенный раствор бисульфита натрия, пока проба крахмал/иодид не стала отрицательной. Эту смесь вылили в ледяной рассол (100 мл) и экстрагировали диэтиловым эфиром (3 х 75 мл). Эфирные фазы объединили и промывали 2 N раствором гидроксида натрия (3 х 75 мл), пока промывные воды не стали щелочными. Эфирную фазу сушили над безводным сульфатом магния, фильтровали и выпаривали. Был получен 2,3-дихлорацетофенон (3,2 г, выход 65%) в виде бледно-желтого масла. ТСХ (SiO2; CHCl3) и ЯМР показали, что этот материал не содержит примесей.
Способ В
Для получения 2,3-дихлорфенилмагнийиодида 2,3-дихлориодбензол (350 г, 1,282 моль), растворенный в сухом эфире (1250 мл) медленно при перемешивании добавили к магниевым стружкам (30,77 г, 1,282 моль) в сухом диэтиловом эфире (300 мл) в атмосфере азота. По каплям при перемешивании реактив Гриньяра добавили к ацетилхлориду (301,91 г, 3,846 моль), растворенному в сухом диэтиловом эфире (1 л) и безводному хлориду железа (1,925 г, 0,0118 моль) в атмосфере азота при температуре -70oC. Когда добавление было закончено, полученную смесь перемешивали при -70oC в течение еще 5 мин, а затем дали нагреться до комнатной температуры. Реакционную смесь вылили на лед (5 л) и тщательно перемешали. Водную смесь подщелачивали карбонатом натрия и оставили стоять при комнатной температуре в течение ночи. Водный раствор экстрагировали диэтиловым эфиром (3 х 2 л), эфирные фазы объединили, сушили над безводным сульфатом магния, фильтровали и выпаривали. Получили сырой 2,3-дихлорацетофенон (235,7 г) в виде желтой жидкости. Сырое вещество перегнали под вакуумом с получением чистого 2,3-дихлорацетофенона (147,0 г, т.кип. 100oC/2 мм рт. ст. ; выход 60,66%). ЯМР и ТСХ (SiO2; CHCl3) показали, что перегнанный продукт не содержит примесей.
Пример 8. Получение 2,3-дихлорфенилглиоксиламида (соединение VIII)
1. Быстрый лабораторный метод
Перемешиваемый раствор 1,2-дихлорбензола (50,0 г, 0,34 моль) в сухом тетрагидрофуране (500 мл) охладили до -65oC в атмосфере азота и обработали по каплям в течение 1 часа раствором н-бутиллития в гексане (204 мл с концентрацией 1,72 М = 0,35 моль) при температуре, поддерживавшейся ниже -60oC. После перемешивания в течение еще 1 ч бледно-желтый раствор обработали по каплям раствором этилоксамата (19,9 г, 0,17 моль) в теплом тетрагидрофуране (200 мл). В ходе этой процедуры цвет реакционной смеси изменялся от желтого через оранжевый до пурпурно-красного. После перемешивания в течение еще 1 ч при -60oC смесь оставляли нагреваться до 0oC в течение 2 ч, осторожно обработали водой (250 мл) и с помощью соляной кислоты подкислили до pH 6. Желтую органическую фазу отделили, промыли рассолом (2 х 250 мл), высушили (сульфат магния) и выпарили с получением желтого твердого вещества. Его суспендировали в этаноле (300 мл) и отфильтровали с получением сырого 2,3-дихлорфенилглиоксиламида (12,0 г, 32,4%) в виде не совсем белого твердого вещества с т. пл. 213-214oC (эт. т.пл. 216-218oC). В результате концентрирования этанольного фильтрата до 150 мл получили вторую порцию (7,0 г, 18,9%) с т.пл. 205-206oC.
В другом эксперименте сырой 2,3-дихлорфенилглиоксиламид (40 г, т.пл. 199-203oC) был перекристаллизован из уксусной кислоты (240 мл) и воды (180 мл) с получением 33,4 г (83%) очищенного продукта (т.пл. 216-218oC).
2,3-Дихлорфенилглиоксиламид имел следующие характеристики:
Молекулярная масса: 217,9
Температура плавления: 216-218oC
Микроанализ: C; H; N;
Вычислено: 44,07; 2,31; 6,42;
Найдено: 44,79; 2,63; 6,09.
Образование бутиллития in situ
н-Бутилхлорид (55,5 г, 0,6 моль) добавляли по каплям в атмосфере азота к перемешивавшейся суспензии тонкой стружки лития (8,30 г, 1,2 моль) в сухом эфире (300 мл) таким образом, чтобы поддерживать слабое кипение с обратным холодильником. После кипячения с обратным холодильником в течение последующих 3,75 ч смесь охлаждали до комнатной температуры и перемешивали в течение ночи. (Предшествующие анализы показывают, что эта методика дает 66%-ный выход бутиллития ( ≡ 0,4 моль)). Эту смесь затем охлаждали до -60oC и поддерживали при этой температуре во время следующих операций. Добавили сухой тетрагидрофуран (400 мл), затем добавили по каплям раствор 1,2-дихлорбензола (58,8 г, 0,4 моль) в сухом тетрагидрофуране (300 мл) в течение свыше 40 мин. После перемешивания смеси в течение 1 ч был добавлен по каплям в течение свыше 1 ч раствор этилоксамата (23,4 г, 0,2 моль) в теплом тетрагидрофуране (300 мл), и эту смесь перемешивали в течение 1 ч при -60oC. После того, как смеси дали нагреться до 0oC, осторожно добавили воду (300 мл), и смесь подкислили до pH 6 соляной кислотой. Желтую органическую фазу отделили, промыли рассолом (2 х 300), сушили (сульфат магния) и упарили с получением твердого светло-желтого вещества. Его суспендировали в этаноле (250 мл), отфильтровали и высушили с получением сырого 2,3-дихлорфенилглиоксиламида (22,5 г, 52%) с т.пл. 214-215oC (эт. 215-218oC).
Пример 9. Получение этил-2,3-дихлорфенилглиоксилата (соединение IX)
Материалы
- 2,3-дихлориодбензол (0,1М, 27,3 г) в 35 мл сухого эфира;
- магний (2,43 г) в 15 мл сухого эфира;
- хлорид кадмия (0,1М, 9,8 г);
- этилоксалилхлорид (0,079М, 10,9 г, 8,92 мл);
- реакцию проводили в атмосфере азота.
Способ
Реактив Гриньяра был приготовлен обычным способом следующим образом.
Дихлориодбензол добавляли к магнию в течение 1-2 ч и полученный раствор кипятили с обратным холодильником в течение примерно 4 ч. Так как весь Mg не растворился, реактив Гриньяра оставили перемешиваться при комнатной температуре в течение ночи.
Затем колбу охладили льдом и в течение 10 мин добавили порциями сухой хлорид кадмия. Когда был добавлен весь хлорид кадмия, реакционной смеси дали нагреться до комнатной температуры и затем в течение 45 мин кипятили с обратным холодильником.
Эфир выпарили и остаток промыли дважды сухим бензолом, который, в свою очередь, выпарили.
Остаток собрали и обработали этилоксалилхлоридом в 20 мл сухого бензола. Его медленно добавляли из капельной воронки при перемешивании. Эта реакция очень бурная. Когда добавление закончили и спонтанное вскипание утихло, реакционную смесь кипятили с обратным холодильником еще в течение часа.
Затем реакционную смесь охладили на ледяной бане. Осторожно добавили смесь лед/вода. Потом добавили количество 20%-ной H2SO4, достаточное для получения двух четких фаз. Водную фазу отделили и экстрагировали дважды бензолом. Бензольные слои объединили и экстрагировали 1 х водой, 1 х раствором Na2CO3, 1 х водой и 1 х раствором NaCl. Бензольный раствор затем сушили над MgSO4, отфильтровали и выпарили. Получили 17,5 г сырого продукта.
Были проведены ЯМР и ТСХ анализы. ТСХ в SiO2/CHCl3 дала два пятна, одно - соответствующее соединению, указанному в заголовке (продукт), и одно - дихлориодбензолу (исходный материал).
Пример 10. Получение этил-2,3-дихлорфенилглиоксилата (соединение (IX)
Материалы
- 2,3-дихлориодбензол (0,2М, 54,6 г) в 50 мл эфира;
- магниевая стружка (0,2М, 4,86 г) в 50 мл эфира;
- хлорид кадмия (0,2М, 19,6 г);
- этилоксалилхлорид (0,15М, 21,8 г);
- реакцию проводили в атмосфере азота.
Способ
Осуществляли общий способ, описанный в примере 7. Однако реактив Гриньяра не кипятили с обратным холодильником в течение ночи, так как эта реакция протекала очень быстро благодаря большему объему эфира.
После прибавления этилоксалилхлорида реакционную смесь охлаждали льдом и полученную смесь перемешивали при комнатной температуре в течение ночи.
Реакционную смесь разлагали посредством добавления воды со льдом и добавляли количество 20%-ной H2SO4, достаточное для получения двух раздельных слоев.
Смесь разделяли и водные слои промывали дважды бензолом. Бензольные слои объединяли и промывали водой. Никакой щелочной промывки не проводили. Бензольные слои сушили, фильтровали и выпаривали. Получали 39,5 г продукта. ЯМР и ТСХ дали результаты, идентичные полученным в примере 9.
Пример 11. Получение изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина
Раствор изетионата натрия (148 г, 1,0 моль) в воде (4,9 литров) пропустили через колонку с IR 120 (H) ионообменной смолой и элюировали водой. 3,5-Диамино-6-(2,3-дихлорфенил)-1,2,4-триазин (256 г, 1,0 моль) растворили в полученной изетионовой кислоте и этот раствор профильтровали и выпарили в вакууме. Остаток перекристаллизовали из промышленного метилированного спирта с получением изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триаэина.
Выход 273,3 г (72%), т.пл. 242oC.
Пример 12. Получение изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина
50 ммоль Amberlite (торговая марка) IR-45 (OH) смешали с 15 ммоль (10 мл) водной изетионовой кислоты и полученным продуктом заполнили колонку. Затем эту колонку промыли метанолом. 0,7 г (2 ммоль) метанольного раствора мезилата 3,5-диамино-6-(2,3-дихлорфенил)- 1,2,4-триазина элюировали через эту колонку. Элюант выпарили в вакууме и остаток перекристаллизовали из промышленного метилированного спирта и получили изетионат 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина.
Выход 300 мг (40%), т.пл. 242-243oC.
Пример 13
74,625 г (0,195 моль) изетионата 3,5-диамино-6-(2,3-дихлорфенил)- 1,2,4-триазина добавили и растворили примерно в 900 мл воды для инъекций ВР, и разбавили до 1000 мл добавлением еще воды для инъекций ВР, с получением водного раствора, содержавшего изетионат в количестве, эквивалентном 50 мг/мл основания 3,5-диамино-6-(2,3- дихлорфенил)-1,2,4-триазина. Такой раствор был приемлем на основании тонуса.
Пример 14
14,925 г (0,039 моль) изетионата 3,5-диамино-6-(2,3-дихлорфенил)- 1,2,4-триазина добавили к раствору 43,8 г (0,221 моль) моногидрата декстрозы в приблизительно 900 мл воды для инъекций ВР и разбавили до 1000 мл добавлением воды для инъекций ВР с получением водного раствора, содержавшего изетионат в количестве, эквивалентном 10 мг/мл основания 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина. Такой раствор был приемлем на основании тонуса.
Пример 15
Была приготовлена фармацевтическая композиция, включающая следующие ингредиенты.
3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин - 150 мг*
Лактоза - 200 мг*
Кукурузный крахмал - 50 мг*
Поливинилпирролидон - 4 мг*
Стеарат магния - 4 мг*
* - содержание на таблетку
Лекарство смешивали с лактозой и крахмалом и гранулировали с раствором поливинилпирролидона в воде. Полученные гранулы сушили, смешивали со стеаратом магния и прессовали с получением таблеток среднего веса 408 мг.



Описывается способ получения ламотриджина формулы (I), который применяется при лечении расстройств центральной нервной системы, или его фармацевтически приемлемой соли, полученной присоединением кислоты, отличающийся тем, что соединение формулы (II), где R представляет собой CN, подвергают облучению ультрафиолетовым или видимым светом в органическом растворителе при нагревании с последующим, в случае необходимости, переведением целевого продукта в его фармацевтически приемлемую соль обработкой кислотой. Описываются также промежуточные соединения и способ получения фармацевтической композиции. Технический результат - упрощение процесса. 6 с. и 5 з.п. ф-лы, 2 табл.



или его фармацевтически приемлемой соли, полученной присоединением кислоты, отличающийся тем, что соединение формулы II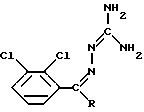
где R представляет собой CN, подвергают облучению ультрафиолетовым или видимым светом в органическом растворителе при нагревании с последующим, в случае необходимости, переведением целевого продукта в его фармацевтически приемлемую соль обработкой кислотой.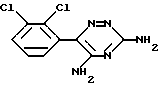
или его фармацевтически приемлемой соли, полученной присоединением кислоты, отличающийся тем, что соединение формулы II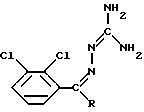
в которой R представляет собой CONH2, в органическом растворителе в присутствии основания подвергают облучению ультрафиолетовым или видимым светом.
5. Способ по п.2 или 3, отличающийся тем, что соединение формулы II, в которой R представляет собой CONH2, получают обработкой 2,3-дихлорфенилглиоксиламида формулы VIII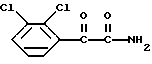
аминогуанидином или его солью.
10. 2,3-Дихлорфенилглиоксиламид формулы VIII
11. Этил-2,3-дихлорфенилглиоксилат формулы IX
| Способ получения амидов -кето-КАРбОНОВыХ КиСлОТ | 1977 |
|
SU799651A3 |
| EP, 0056938 A1, 1982 | |||
| EP, 0150677 A1, 1985 | |||
| US 4992585 A, 1991 | |||
| АЭРОСТАТНО-КОСМИЧЕСКАЯ ЭНЕРГЕТИЧЕСКАЯ СИСТЕМА (АКЭС) | 2019 |
|
RU2733181C1 |
| EP, 0021121 A, 1981 | |||
| EP, 0247892 A1, 1987 | |||
| EP, 0459829 A1, 1991 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1, 2, 4-ТРИАЗИНА ИЛИ 1, 2,4, 5-ТЕТРАЗИНА | 0 |
|
SU310907A1 |

