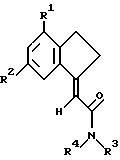
Настоящее изобретение касается группы замещенных карбоциклических амидов, фармацевтических составов, которые их содержат, способов получения этих соединений и их использования в терапии, в частности, при лечении воспалительных состояний.
Задача настоящего изобретения заключалась в создании новых соединений, обладающих полезными противовоспалительными и анальгезирующими свойствами и не проявляющих других фармакологических свойств, которые могут быть использованы для лечения воспалительных состояний и снятия боли, в создании фармацевтического состава, содержащего предложенные соединения и проявляющего противовоспалительную и анальгезирующую активность, и в разработке способа изготовления такого лекарственного средства, а также в разработке способа лечения указанных состояний с использованием этих соединений. В задачу изобретения также входила разработка эффективных способов получения заявленных соединений с использованием новых промежуточных соединений.
Поставленная задача решается тем, что предложены замещенные карбоциклические амиды формулы I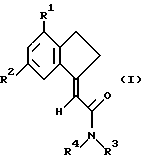
где R1 и R2 независимо выбраны из хлора, фтора, брома, C1-C6 алкила, C1-C6 алкокси или C1-C6 галоалкила при условии, что оба R1 и R2 не являются фтором;
R3 и R4 независимо выбраны из водорода и C1-C6 алкила.
Замещенные карбоциклические амиды формулы I могут представлять собой фармацевтически приемлемую соль, сольват или физиологически функциональное производное.
Предпочтительно, когда по меньшей мере один из R1 и R2 является хлором.
Наиболее предпочтительными соединениями формулы I по изобретению являются:
(E)-2-(4-хлор-6-фтор-1-инданилиден)-N-метилацетамид,
(E)-2-(4-хлор-6-фтор-1-инданилиден)ацетамид,
(E)-2-(4,6-дихлор-1-инданилиден)ацетамид,
(E)-2-(6-фтор-4-метил-1-инданилиден)ацетамид,
(E)-2-(6-фтор-4-метил-1-инданилиден)-N-метилацетамид,
(E)-2-(6-хлор-4-фтор-1-инданилиден)ацетамид,
(E)-2-(4-бром-6-фтор-1-инданилиден)ацетамид или
(E)-2-(4-хлор-6-метил-1-инданилиден) ацетамид.
Предпочтительной группой соединений формулы I являются замещенные карбоциклические амиды формулы IA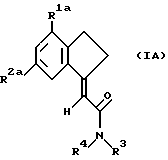
где R1a является хлором,
R2a является хлором, фтором, бромом или метилом, а
R3 и R4 независимо выбраны из водорода, метила или этила.
Соединения формулы I обладают противовоспалительной и анальгезирующей активностью.
Задача изобретения решается также тем, что предложен фармацевтический состав, проявляющий противовоспалительную и анальгезирующую активность, включающий активный ингредиент и один или несколько фармацевтически приемлемых носителей, отличающийся тем, что в качестве активного ингредиента содержит соединение формулы I или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное в эффективном количестве.
Для решения поставленной задачи предложен способ профилактики или лечения состояний, связанных с воспалением, артритом или болью у млекопитающего, при котором указанному млекопитающему вводят терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного.
Поставленная задача решается также тем, что предложен способ изготовления лекарственного средства для профилактики или лечения состояний, связанных с воспалением, артритом или болью, включающий в себя смешивание активного ингредиента с одним или более чем одним носителем, отличающийся тем, что в качестве активного ингредиента используют соединение формулы I или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное.
В рамках решения вышеуказанной задачи предложен способ получения соединений формулы I и их солей, сольватов и физиологически функциональных производных, при котором соединение формулы II
подвергают взаимодействию с амином NHR3R4, где R1-R4 такие, как определено ранее, и X является отщепляемой группой, после чего возможно превращают полученное таким образом соединение формулы I в его соль или физиологически функциональное производное.
Предложен также способ получения соединений формулы I и их солей, сольватов и физиологически функциональных производных, при котором соединение формулы III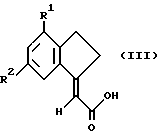
подвергают взаимодействию с соответствующим реагентом сочетания, с последующим взаимодействием с амином HNR3R4, где R1-R4 такие, как определено ранее, после чего возможно превращают полученное таким образом соединение формулы I в его соль или физиологически функциональное производное.
Еще один аспект изобретения в рамках решения поставленной задачи составляют производное индана формулы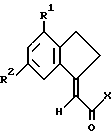
где R1 и R2 такие, как определено ранее, и Х является отщепляемой группой или ОН, и
производное индана формулы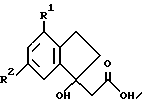
где R1 и R2 такие, как определено ранее, представляющие собой новые промежуточные соединения, которые являются особенно ценными для получения некоторых соединений формулы I.
Производные индана, которые являются особенно ценными, включают в себя:
2-(4-хлор-6-фтор-1-гидрокси-1-инданил) уксусную кислоту,
(E)-2-(4-хлор-6-фтор-1-инданилиден) уксусную кислоту,
(E)-2-(4-хлор-6-фтор-1-инданилиден) ацетилхлорид,
2-(4,6-дихлор-1-гидрокси-инданил) уксусную кислоту,
(E)-(4,6-дихлор-1-инданилиден) уксусную кислоту,
(E)-2-(4,6-дихлор-1-инданилиден) ацетилхлорид,
2-(6-фтор-1-гидрокси-4-метил-1-инданил) уксусную кислоту,
(E)-2-(6-фтор-4-метил-1-инданилиден) уксусную кислоту,
(E)-2-(6-фтор-4-метил-1-инданилиден) ацетилхлорид,
2-(6-хлор-4-фтор-1-гидрокси-1-инданил) уксусную кислоту,
(E)-2-(6-хлор-4-фтор-1-инданилиден) уксусную кислоту,
(E)-2-(6-хлор-4-фтор-1-инданилиден) ацетилхлорид,
2-(4-бром-6-фтор-1-гидрокси-1-инданилиден) уксусную кислоту,
(E)-2-(4-бром-6-фтор-1-инданилиден) уксусную кислоту,
(E)-2-(4-бром-6-фтор-1-инданил) ацетилхлорид,
2-(4-xлop-6-мeтил-1-гидpoкcи-1-индaнил) уксусную кислоту,
(E)-2-(4-хлор-6-метил-1-инданилиден) уксусную кислоту,
(E)-2-(4-хлор-6-метил-1-инданилиден) ацетилхлорид.
В качестве использованных здесь терминов
а) "C1-C6алкил" означает алкильную группу, имеющую от 1 до 6 углеродных атомов, включая алкильные группы с нормальной, разветвленной цепью или циклические. Такие алкильные группы предпочтительно имеют от 1 до 3 углеродных атомов, и более предпочтительны метил, этил, пропил, проп-2-ил, бутил, бут-2-ил, 2-метилпроп-2-ил, циклопропил или циклобутил. Наиболее предпочтительными алкильными группами являются метил, этил или циклопропил.
б) "C1-C6алкокси" как группа или часть группы означает моновалентный радикал с нормальной или разветвленной цепью, имеющей от 1 до 6 углеродных атомов, присоединенный к основной части через атом кислорода. Такие алкоксигруппы предпочтительно имеют от 1 до 4 углеродных атомов и более предпочтительно представляют собой метокси или этокси, наиболее предпочтительно метокси.
в) "галоалкил" означает алкил, замещенный атомами фтора или хлора в количестве от 1 до 4, предпочтительно атомами фтора.
г) "физиологически функциональные производные" означают любое другое физиологически приемлемое производное какого-либо соединения по настоящему изобретению, например сложный эфир, который при введении реципиенту, такому как человек, способен дать (прямо или косвенно) указанное соединение или его активный метаболит или остаток.
д) "соль" означает основные соли, как дополнительно определено далее.
е) "сольват" означает комбинацию в определенных соотношениях соединения, предусмотренного настоящим изобретением, с растворителем.
Следует учитывать, что соединения формулы I могут существовать в различных геоизомерных формах и в виде их смесей в любых соотношениях. Настоящее изобретение включает также геоизомерные формы или смеси геоизомеров, включая индивидуальные Е и Z изомеры соединений формулы I и смеси таких изомеров в любых соотношениях. Предпочтительными соединениями формулы I являются те, в которых группа, соседняя с экзодвойной связью, и карбонильная группа находятся на противоположных сторонах экзодвойной связи. Соединения формулы I могут существовать в формах, в которых один или более углеродных центров является/являются хиральными.
В объем настоящего изобретения входит каждый возможный оптический изомер по существу свободный, т.е. ассоциированный с менее чем 5% любого другого оптического изомера (изомеров), так же как смеси одного или нескольких оптических изомеров в любых пропорциях, включая их рацемические смеси.
Фармацевтически приемлемые соли включены в объем данного изобретения и являются особенно подходящими для медицинских применений из-за своей относительно большей водорастворимости сравнительно с исходными (т.е. основными) соединениями. Такие соли должны несомненно иметь фармацевтически приемлемый анион или катион. Подходящие фармацевтически приемлемые основные соли включают соли аммония, соли щелочных металлов, такие как соли натрия и калия, и соли щелочноземельных металлов, такие как соли магния и кальция.
Соли, имеющие фармацевтически неприемлемый анион, также включены в сферу данного изобретения как полезные промежуточные продукты для приготовления или очистки фармацевтически приемлемых солей и/или для использования в нетерапевтических, например in vitro, применениях.
Соединения формулы I могут также быть использованы при лечении воспалительных и артритных состояний, включая ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит, так же как несуставных воспалительных состояний, включая синдром грыжи/перфорации/выпадения межпозвоночного диска, синдром бурсита, тендинита, теносиновита, фибромиалгии и других воспалительных состояний, связанных со связочным растяжением и местным скелетно-мышечным растяжением сухожилий. Особенно отмечают, что соединения формулы I проявляют пониженную склонность к образованию язвы по сравнению с другими противовоспалительными средствами, такими как ибупрофен, напроксен или аспирин.
Анальгезирующая активность соединений формулы I делает их полезными для сдерживания боли, например боли, связанной с воспалением и/или травмой. Соответственно, соединения по изобретению используют как мягкие и сильные анальгетики.
Соединения в соответствии с изобретением могут быть использованы в сочетании с другими терапевтическими агентами для лечения состояний, связанных с воспалением, артритом и/или болью. Примеры таких других терапевтических агентов включают анальгетики, такие как кодеин, оксикодон, ацетаминофен, фенацетин или ибупрофен; противоартритные средства, такие как метотрексат или азатиоприн; и противозастойные средства, такие как эфедрин или псевдоэфедрин.
Фармацевтические составы по настоящему изобретению включают в себя активный ингредиент, представляющий собой соединение формулы I, как было определено ранее, вместе с одним или несколькими приемлемыми носителями для него и, возможно, другими терапевтическими агентами. Каждый носитель должен быть "приемлемым" в смысле совместимости с другими ингредиентами состава и ненанесения вреда реципиенту.
Составы должны быть удобны для перорального, ректального, назального, местного (включая трансбуккальный и подъязычный), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения. Эти составы могут быть удобно представлены в виде единичной дозовой формы.
Составы в соответствии с данным изобретением, пригодные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, облатки или таблетки, каждая из которых содержит предусмотренное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии масло в воде или жидкой эмульсии вода в масле. Активный ингредиент также может быть представлен в виде большой пилюли, лекарственной кашки или пасты.
Составы, пригодные для перорального использования как описано ранее, могут также включать буферные агенты, предназначенные для нейтрализации желудочной кислотности. Такие буферы могут быть выбраны из разнообразных органических и неорганических агентов, таких как слабые кислоты или основания, смешанные с соответствующими им солями.
Составы, пригодные для местного введения в рот, включают лепешки, содержащие активный ингредиент в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик; и полоскания для рта, содержащие активный ингредиент в подходящем жидком носителе.
Составы для ректального введения могут быть представлены в виде суппозитория с подходящей основой, включающей, например, масло какао или салицилат.
Составы, пригодные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пенок и аэрозолей, содержащих вдобавок к активному ингредиенту такие носители, которые, как известно специалистам, будут подходящими для этих случаев.
Составы, пригодные для парентерального введения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические факторы и растворы, которые делают составы изотоническими с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители, такие как липосомы или другие системы из микрочастиц, которые предназначены для доставки этих соединений к компонентам крови или одному или более органам. Данные составы могут быть представлены в виде однодозовых или многодозовых герметичных контейнеров, например ампул или флаконов, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требуя только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Предназначенные для немедленного использования растворы для инъекций и суспензии можно приготовлять из стерильных порошков, гранул и таблеток ранее описанного вида.
Составы, пригодные для введения через кожу, могут быть представлены в виде отдельных кусочков пластыря, приспособленных оставаться в непосредственном контакте с эпидермой реципиента в течение продолжительного времени. Такие кусочки пластыря соответственно содержат активное соединение в виде возможно забуференного водного раствора с концентрацией, например, от 0,1 до 0,2 М по отношению к указанному соединению. В качестве одной специфической возможности активное соединение может высвобождаться из кусочка пластыря путем электрофореза как в общем описано (Pharmaceutical Research, 3(6), 318 (1986)).
Предпочтительными составами единичной дозы являются те, которые содержат суточную дозу или единицу, суточную субдозу, как здесь ранее изложено, или соответствующую их долю активного ингредиента.
Следует понимать, что в дополнение к ингредиентам, в частности, упомянутым ранее, составы в соответствии с этим изобретением могут включать другие традиционные агенты, имеющие отношение к типу рассматриваемого состава, например составы, пригодные для перорального введения, могут включать такие дополнительные агенты, как подслащивающие вещества, загустители и корригенты.
Следует учитывать, что предпочтительный способ введения фармацевтических составов по изобретению будет меняться с состоянием и возрастом реципиента, природой заболевания и выбранным активным ингредиентом.
При реализации способа профилактики или лечения состояний, связанных с воспалением, артритом или болью, требуемое количество индивидуального активного ингредиента будет, конечно, зависеть от большого числа факторов, включая тяжесть состояний, которые нужно лечить, и индивидуальности реципиента, и будет в конечном счете оставаться на усмотрение лечащего врача.
В общем случае для упомянутых состояний подходящая доза соединения формулы I или его солей, сольватов или физиологически функциональных производных (рассматриваемых как исходное соединение) находится в пределах от 0,05 до 100 мг на кг массы тела реципиента в день, предпочтительно от 0,1 до 50 мг на кг массы тела в день, наиболее предпочтительно в пределах от 0,5 до 20 мг на кг массы тела в день и оптимально от 1 до 10 мг на кг веса тела в день. Желаемая доза предпочтительно представлена как две, три, четыре, пять, шесть и более субдоз, вводимых через определенные интервалы в течение дня. Эти субдозы могут быть введены в виде единичных дозовых форм, например, содержащих от 1 до 1500 мг, предпочтительно от 5 до 1000 мг и наиболее предпочтительно от 10 до 700 мг активного ингредиента на единичную дозовую форму.
При реализации способа изготовления лекарственного средства для профилактики или лечения состояний, связанных с воспалением, артритом или болью, это средство может быть приготовлено любыми способами, хорошо известными в фармации. Такие способы включают стадию смешения активного ингредиента с носителем, который составляет один или несколько дополнительных ингредиентов. В общем, эти составы готовят равномерным и однородным смешением активного ингредиента с жидкими носителями или тонко диспергированными твердыми носителями, или обоими, и затем, если необходимо, формированием продукта.
Например, таблетку можно изготовить прессованием или формованием по желанию с одним или несколькими дополнительными ингредиентами. Прессованную таблетку можно изготовить прессованием в соответствующем приспособлении активного ингредиента в свободно-текучей форме, такой как порошок или гранулы, по желанию смешанные со связующим веществом (например, повидоном, желатином, гидроксипропилметилцеллюлозой), смазывающим веществом, инертным разбавителем, консервантом, разрыхлителем (например, натрий крахмалгликолятом, поперечно-сшитым повидоном, поперечно-сшитой натрий-карбоксиметилцеллюлозой), поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием в соответствующем приспособлении смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Эти таблетки по желанию могут быть покрыты оболочкой или на них могут быть сделаны метки, или они могут быть изготовлены так, чтобы обеспечить медленное или контролируемое высвобождение из них активного ингредиента, используя, например, гидроксипропилметилцеллюлозу в разных соотношениях для достижения желаемого профиля высвобождения. Таблетки могут быть покрыты энтеросолюбильными оболочками для обеспечения высвобождения в областях пищеварительного тракта, иных, чем желудок.
При реализации первого их указанных выше способов получения соединений формулы I и их солей, сольватов и физиологически функциональных производных удобно осуществлять реакцию в инертном растворителе, например галогенированном алкане, таком как дихлорметан, при неэкстремальной температуре, например от -20oC до 120oC; и предпочтительно 0oC - 30oC.
Удобные отщепляемые группы включают атомы галогена, такие как хлор или бром, активированные сложные эфиры (например, N-гидроксисукцинимид, пентафторфенил, нитрофенил, 1-гидрокси-бензотриазол), смешанные ангидриды (например, этоксикарбонилокси) или C1-C6 алкокси (например, этокси) группы.
В случае, когда R3 и R4 представляют собой водород, соединение HNR3R4, т. е. NH3, предпочтительно используют в гидратированной форме в виде гидроксида аммония и Х является атомом галогена.
Соединения формулы II, где Х является атомом галогена, могут быть получены из соединений формулы III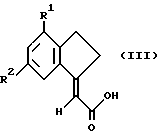
(где R1, R2 и R5 такие, как определено ранее) посредством взаимодействия с галогенирующим агентом (например, оксалилхлоридом или тионилхлоридом) в подходящем органическом растворителе (например, бензоле, толуоле, дихлорметане), возможно, в присутствии катализатора (например, ДМФ) при температуре от около -20oC до температуры кипения с обратным холодильником.
Соединения формулы II, где Х представляет собой алкокси (например, этокси), могут быть получены из соединений формулы III взаимодействием с соответствующим полярным растворителем (например, органическим спиртом, таким как этанол), возможно, в присутствии каталитического количества кислоты (например, п-толуолсульфокислоты) при температуре от примерно 0oC до температуры кипения с обратным холодильником.
Соединения формулы II, где Х представляет собой активированный сложный эфир (как описано ранее), могут быть получены из соединений формулы III взаимодействием с фенолом или N-гидроксисоединением и карбодиимидом (например, дициклогексилкарбодиимидом или 1-(3-иметиламинопропил)-3-этилкарбодиимидом) в растворителе, таком как диметилформамид (ДМФ) или дихлорметан, при температуре от 0oC до 50oC.
Соединения формулы II, где Х представляет собой активированный сложный эфир, могут быть получены из соединений формулы III взаимодействием с алкилгалоэфиром, например этилхлорформиатом, в соответствующем растворителе, таком как тетрагидрофуран, при соответствующей температуре, например от 0oC до комнатной температуры.
При реализации следующего заявленного способа получения соединений формулы (I) или их солей, сольватов и физиологически функциональных производных их получают непосредственно из соединений формулы III взаимодействием с подходящим сочетающим реагентом (например, дициклогексилкарбодиимидом (ДЦК) или этилхлорформиатом) с последующим взаимодействием полученного таким образом активированного сложного эфира (без выделения его) с соответствующим амином, HNR3R4.
Соединения формулы III могут быть получены дегидратацией соединений формулы IV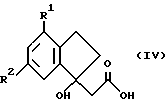
(где R1 и R2 такие, как определено ранее) взаимодействием с соответствующим дегидратирующим агентом (например, кислотой, такой как трифторуксусная кислота) в соответствующем органическом растворителе (например, дихлорметане) при температуре от -20oC до температуры кипения с обратным холодильником.
Соединения формулы IV могут быть получены омылением соответствующего C1-C6 алкилового сложного эфира, например этилового, основанием (например, гидроксидом натрия) в подходящем полярном растворителе (например, этаноле) при температуре от около 0oC до температуры кипения с обратным холодильником или водной кислотой (например, соляной кислотой) при температуре от примерно 0oC до температуры кипения с обратным холодильником.
Алкиловые сложные эфиры соединений формулы IV могут быть получены из соединений формулы V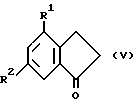
(где R1 и R2 такие, как определено ранее) взаимодействием с X1CH2C(O)OR5, где X1 является атомом галогена, таким как хлор, бром или иод (предпочтительно бром), R5 представляет собой C1-C6алкил, предпочтительно этил, в присутствии металла (например, цинка, предпочтительно активированного цинка) и каталитического количества галогена (например, иода) в соответствующем органическом растворителе (например, этиловом эфире, бензоле) при температуре от примерно 0oC до температуры кипения с обратным холодильником или взаимодействием с литиевой солью этилацетата в подходящем растворителе (например, тетрагидрофуране) при температуре от -100oC до комнатной температуры (например, -78oC).
Соединения формулы V могут быть получены из соединений формулы VI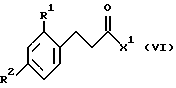
где R1, R2 и Х1 такие, как определено ранее, циклизацией в присутствии кислоты Льюиса (например, хлорида алюминия) в подходящем растворителе (например, дихлорметане) при температуре от примерно 0oC до температуры кипения с обратным холодильником.
Соединения формулы VI могут быть получены из соответствующей кислоты взаимодействием с галогенирующим агентом (например, оксалилхлоридом или тионилхлоридом) либо с чистым, либо находящимся в подходящем органическом растворителе (например, метиленхлориде или N,N-диметилформамиде) при температуре от примерно 0oC до температуры кипения с обратным холодильником.
Такие кислоты могут быть получены
(1) омылением соответствующих C1-C6 алкиловых сложных эфиров основанием (например, гидроксидом натрия) в соответствующем полярном растворителе (например, в воде или этаноле) при температуре от примерно 0oC до температуры кипения с обратным холодильником или водной кислотой (например, соляной кислотой) при температуре от приблизительно 0oC до температуры кипения с обратным холодильником.
C1-C6 алкиловые сложные эфиры могут быть получены каталитическим гидрированием соответствующих C1-C6 алкил-акриловых сложных эфиров в подходящем растворителе (например, 95%-ном этаноле) с катализатором (например, оксидом платины) при комнатной температуре и давлении водорода от 0,0981 МПа до 0,3924 МПа.
Акриловые сложные эфиры могут быть получены взаимодействием соединения формулы VII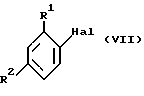
где R1 и R2 такие, как определены ранее, a Hal является отщепляемой группой (например, Br, I или OSO2CF3), с C1-C6 сложными эфирами акриловой кислоты (предпочтительно этилакрилатом) в подходящем растворителе (например, ацетонитриле или диметилформамиде) в присутствии катализатора (например, ацетат палладия (II)/три-о-толил фосфина или бис(трифенилфосфин) палладий (II) хлорида) и третичного амина (например, триэтиламина).
Соединения формулы VII, где Hal является Br или I, коммерчески доступны или могут быть получены способами, хорошо известными специалистам или описанными в литературе.
Соединения формулы VII, где Hal является OSO2CF3, могут быть получены из соответствующего фенола взаимодействием с трифторметансульфоновым ангидридом в подходящем растворителе (например, дихлорметане) в присутствии основания (например, пиридина). Фенолы доступны коммерчески или могут быть получены способами, хорошо известными специалистам или описанными в литераторе.
(2) каталитическим гидрированием соответствующих акриловых кислот в подходящем растворителе (например, 95%-ном этаноле) с катализатором (например, оксидом платины) при комнатной температуре и давлении водорода от 0,0981 МПа до 0,3924 МПа.
Акриловые кислоты могут быть получены из соответствующих альдегидов взаимодействием с малоновой кислотой в соответствующем растворителе (например, пиридине) в присутствии соответствующего основания (например, пиперидина).
Такие альдегиды доступны коммерчески или могут быть получены способами, хорошо известными специалистам или описанными в химический литературе.
(3) из соединений формулы VIII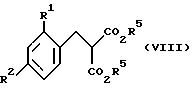
где R1, R2 и R5 такие, как определено ранее, взаимодействием с сильным основанием (например, водным гидроксидом калия) при температуре кипения с обратным холодильником, с последующей обработкой сильной кислотой (например, H2SO4) при температуре кипения с обратным холодильником.
Соединения формулы VIII могут быть получены взаимодействием соединения формулы IX с соединением формулы VII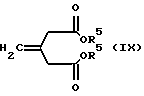
где R1, R2 и R5 такие, как определено ранее, в органическом растворителе (например, безводном диэтиловом эфире) и, возможно, в присутствии галогенида меди (например, иодида меди (I)) при температуре между -50oC и температурой кипения с обратным холодильником.
Соединения формулы IX могут быть получены взаимодействием соединения формулы Х с формальдегидом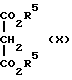
где R5 такой, как определено ранее, в органическом растворителе (например, этиловом эфире или дихлорметане) при температуре между комнатной и температурой кипения с обратным холодильником.
Соединения формулы Х доступны коммерчески или могут быть получены способами, известными специалистам или хорошо описанными в химической литературе.
Как уже упоминалось выше, промежуточные соединения формул II, III и IV, представляющие собой производные индана, составляют еще один аспект изобретения.
Альтернативно, соединения формулы I могут быть получены взаимодействием R3R4NC(O)CH2PO(OR6)2 (R3 и R4 такие, как определено ранее, и R6 представляет собой C1-C6алкил) с основанием (например, NaH) в подходящем органическом растворителе (например, ТГФ или ДМСО) и взаимодействием образовавшихся анионных структур с соединением формулы V при температуре от примерно 0oС до температуры кипения с обратным холодильником. Добавление анионного стабилизирующего реагента (например, гексаметилдисилизана калия) или краун-эфира (например, 15-краун-5) может способствовать этой реакции.
Соединение R3R4NC(O)CH2PO(OR6)2 может быть в зависимости от R3 и R4 доступным коммерчески или быть получено способами, хорошо известными специалисту или описанными в химической литературе. Альтернативно, эти соединения могут быть получены взаимодействием соответствующего R3R4NC(O)CH2X (где X является атомом галогена) с соответствующим P(OR6)3 в подходящем органическом растворителе (например, ТГФ) при температуре от примерно 0oC до 50oC.
Соединение R3R4NC(O)CH2X может быть получено взаимодействием соответствующего амина R3R4NH с XCH2С(O)X в подходящем органическом растворителе (например, диэтиловом эфире) при температуре от приблизительно 0oC до температуры кипения с обратным холодильником.
Соединение XCH2C(O)X доступно коммерчески или может быть получено способами, хорошо известными специалистам в приготовлении таких соединений или хорошо описанными в химической литературы.
Альтернативно, соединения формулы I могут быть получены взаимодействием R3R4NC(O)CH2P(+)Ph3Cl(-) (где R3 и R4 такие, как определено ранее, и Ph означает фенил) с подходящим основанием (например, NaH) в соответствующем органическом растворителе (например, диметоксиэтане) при температуре от приблизительно 0oC до 50oC и взаимодействием полученных в результате анионных структур с соединением формулы V соответственно при температуре от приблизительно 0oC до температуры кипения с обратным холодильником.
Соединение R3R4NC(О)CH2P(+)(Ph)3Cl(-) может быть получено взаимодействием R3R4NC(O)CH2X с приблизительно 50%-ным молярным избытком P(Ph)3 (трифенилфосфин) в подходящем органическом растворителе (например, ТГФ) при температуре от приблизительно 20oC до температуры кипения с обратным холодильником.
Соединения формулы I, так же как и любые из промежуточных соединений, используемых при получении этих соединений, могут участвовать в одном или более следующих необязательных превращений:
(1) превращение соединения формулы I или полученных промежуточных соединений в основную соль или другие физиологически функциональные их производные;
(2) в том случае, когда образована основная соль или другое физиологически функциональное производное соединения формулы I или его промежуточного соединения, превращение указанной соли или производного в соединение формулы I или его промежуточное соединение или другое его производное.
Следующие примеры иллюстрируют настоящее изобретение, но не должны истолковываться как ограничение его объема.
Пример 1
Получение (E)-2-(4-хлор-6-фтор-1-инданилиден)-N-метилацетамида.
а) Получение 2-хлор-4-фторкоричной кислоты
К смеси 2-хлор-4-фторбензальдегида (20,0 г, 0,13 моль, Aldrich) и малоновой кислоты (26,2 г, 0,25 моль, Aldrich) в пиридине (100 мл) при 50oC по каплям добавили пиперидин (10 мл). Через 24 ч при 70oC смесь вылили в ледяной раствор концентрированной HCl (120 мл) и воды (1,5 л). Образовавшийся твердый осадок был отфильтрован и промыт несколько раз водой и представлял собой 24,4 г (96%) 2- хлор-4-фторкоричной кислоты в виде белого твердого вещества. Перекристаллизация 1,5 г из смесей ацетон-вода дала 1,1 г 2-хлор-4-фторкоричной кислоты в виде белого твердого вещества с т.пл. 243-245oC.
б) Получение 3-(2-хлор-4-фторфенил)пропановой кислоты
Смесь 2-хлор-4-фторкоричной кислоты (22,9 г, 0,11 моль) и гидрата оксида платины (0,5 г, EM Scientific) в 95%-ном спирте (140 мл) помещают в прибор Парра для гидрирования. После поглощения определенного количества водорода катализатор отделяют фильтрованием, и смесь концентрируют в вакууме с получением 22,6 г (98%) 3-(2-хлор-4-фторфенил)пропановой кислоты в виде твердого вещества пурпурного цвета. Этот материал использовали без дальнейшей очистки.
3а) 3-(2,4-дихлорфенил)пропановая кислота была получена способом, подобным тому, который описан в примере 1б, из 2,4-дихлоркоричной кислоты (25,0 г, 0,12 моль, Aldrich). Этот материал использовали без дальнейшей очистки.
в) Получение 4-хлор-6-фтор-1-инданона
К смеси 3-(2-хлор-4-фторфенил)пропановой кислоты (21,6 г, 0,11 моль) и дихлорметана при комнатной температуре по каплям добавили оксалилхлорид (19,2 мл). Смесь перемешивали при комнатной температуре до прекращения газовыделения. Избыток оксалилхлорида удалили отгонкой с получением 3-(2-хлор-4-фторфенил)пропионил хлорида. Раствор 3-(2-хлор-4-фторфенил)пропионил хлорида в дихлорметане (100 мл) по каплям добавили к смеси хлорида алюминия (17,3 г, 0,13 моль, Aldrich) в дихлорметане (100 мл) при комнатной температуре. После того, как добавление было закончено, смесь кипятили с обратным холодильником в течение 2,5 ч. Реакционную смесь вылили в ледяную воду (1,5 л). Разделили две фазы и дихлорметановую фазу промыли 0,1 н. водным раствором гидроксида натрия, сушили (Na2SO4) и сконцентрировали с получением сырого 4-хлор-6-фтор-1-инданона. Хроматография на силикагеле со смесью гексан: дихлорметан (1:1) в качестве элюента дала 11,1 г (55%) 4-хлор-6-фтор-1-инданона в виде белого твердого вещества с т.пл. 94-96oС.
3б) 4,6-дихлор-1-инданон был получен способом, подобным описанному в примере 1в, из 3-(2,4-дихлорфенил)пропановой кислоты (24,3 г, 0,11 моль). Хроматография на силикагеле со смесью гексан:дихлорметан (1:1) в качестве элюента дала 12,5 г (55%) 4,6-дихлор-1-инданона в виде белого твердого вещества. Перекристаллизация 1,0 г из гексана дала 0,7 г 4,6-дихлор-1-инданона в виде белого твердого вещества с т.пл. 118-120oC.
г) Получение этил 2-(4-хлор-6-фтор-1-гидрокси-1-инданил) ацетата
Этилацетат (5 г, 0,07 моль) добавили по каплям к перемешиваемому, охлаждаемому (баня - сухой лед с ацетоном) раствору диизопропиламида, полученному при добавлении по каплям 2,5 М раствора н-бутиллития (26,8 мл, 0,07 моль) в гексане к охлаждаемому (баня - сухой лед с ацетоном) раствору диизопропиламина (6,8 г, 0,07 моль) в тетрагидрофуране (35 мл). Через 30 мин добавили по каплям раствор 4-хлор-6-фтор-1-инданона (12,4 г, 0,07 моль) в тетрагидрофуране (100 мл), и смесь перемешивали в течение 1 ч (баня - сухой лед с ацетоном). Затем добавили раствор хлорида аммония (10,6 г, 0,2 моль) в воде (80 мл), и этой смеси дали нагреться до комнатной температуры. Водную фазу отделили и экстрагировали диэтиловым эфиром. Объединенную органическую фазу высушили (сульфат натрия), профильтровали и сконцентрировали в вакууме с получением 19,5 г сырого этил 2-(4-хлор-6-фтор-1-гидрокси-1-инданил)ацетата. Хроматография на силикагеле со смесью гексан:этилацетат (8:2) в качестве элюента дала 15,2 г (83%) желтого масла; ЯМР (ДМСО-d6): δ 7,13-7,28. (m, 2H), 5,55 (s, 1H), 3,98 (m, 2H), 2,79 (2m's, 4H), 2,50 (m, 1H), 2,11 (m, 1H), 1,08 (t, 3H).
3в) Этил 2-(4,6-дихлор-1-гидрокси-1-инданил)ацетат получали способом, подобным описанному в примере 1г, из 4,6-дихлор-1-инданона. Хроматография на силикагеле со смесью гексан:этилацетат (8:2) в качестве элюента дала 10,6 г (65%) желтого масла; ЯМР (CDCl3): δ 7,22-7,27 (m, 2H), 4,28 (br, 1H), 4,21 (m, 2H), 3,03 (m, 1H), 2,75 (m, 3H), 2,30 (m, 2H), 1,28 (t, 3H).
д) Получение 2-(4-хлор-6-фтор-1-гидрокси-1-инданил) уксусной кислоты
Смесь этил 2-(4-хлор-6-фтор-1-гидрокси-1-инданил) ацетата (14,5 r, 0,05 моль), 1 н. раствор гидроксида натрия (52 мл) и абсолютный этанол (100 мл) перемешивали в течение 18 ч при комнатной температуре. Смесь концентрировали в вакууме, разбавили водой и промыли диэтиловым эфиром. Водную фазу нейтрализовали 1,0 н. соляной кислотой (52 мл) и экстрагировали диэтиловым эфиром. Экстракты диэтилового эфира сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 12,5 (96%) сырого 2-(4-хлор-6-фтор-1-гидрокси-1-инданил) уксусной кислоты. Этот материал был использован немедленно без дальнейшей очистки.
3г) 2-(4,6-дихлор-1-гидрокси-1-инданил)уксусную кислоту получали способом, подобным описанному в примере 1д, из этил 2-(4,6-дихлор-1-гидрокси-1-инданил)ацетата (9,9 г, 0,03 моль). Этот материал использовали немедленно без дальнейшей очистки.
е) Приготовление (E)-2-(4-хлор-6-фтор-1-инданилиден) уксусной кислоты
Трифторуксусную кислоту (27,4 мл) добавили к перемешиваемому холодному (баня - лед с ацетоном) раствору 2-(4-хлор-6-фтор-1-гидрокси-1-инданил) уксусной кислоты (12,5 г, 0,05 моль) в дихлорметане (200 мл). Через 1,5 ч концентрировали в вакууме с получением 10,6 г сырого (E)-2-(4-хлор-6-фтор-1-инданилиден) уксусной кислоты. Хроматография 1,0 г образца на силикагеле со смесью гексан:этилацетат (1:1) в качестве элюента дала 0,32 г (E)-2-(4-хлор-6-фтор-1-инданилиден) уксусной кислоты в виде белого твердого вещества с т.пл. 229-230oC.
3д) (E)-2-(4,6-дихлор-1-инданилиден) уксусная кислота может быть получена способом, подобным описанному в примере 1е, из 2-(4,6-дихлор-1-гидрокси-1-инданил) уксусной кислоты (8,6 г, 0,03 моль). 1,0 г образца перекристаллизовали из смесей изопропанол:вода с получением 0,6 г (E)-2-(4,6-дихлор-1-инданилиден) уксусной кислоты в виде белого твердого вещества с т.пл. 245- 247oC.
ж) Получение (E)-2-(4-хлор-6-фтор-1-инданилиден) ацетил хлорида
Суспензию (E)-2-(4-хлор-6-фтор-1-инданилиден) уксусной кислоты (9,6 г, 0,04 моль) в дихлорметане (100 мл) обработали оксалилхлоридом (10,7 г, 0,08 моль) и перемешивали при комнатной температуре в течение 3 ч. Образовавшийся раствор концентрировали в вакууме, и этот остаток использовали для дальнейшей очистки.
3е) (E)-2-(4,6-дихлор-1-инданилиден) ацетил хлорид был получен способом, подобным описанному в примере 1ж, из (E)-2-(4,6-дихлор-1-инданилиден) уксусной кислоты (5,3 г, 0,02 моль). Полученный осадок был использован для дальнейшей очистки.
з) Получение (E)-2-(4-хлор-6-фтор-1-инданилиден)-N-метилацетамида
Раствор (E)-2-(4-хлор-6-фтор-1-инданилиден) ацетил хлорида (4,0 г, 0,015 моль) в дихлорметане (35 мл) по каплям добавили к ледяной смеси 40%-ного водного метиламина (2,6 мл, 0,03 моль) и дихлорметана (100 мл), и смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь сконцентрировали в вакууме, и остаток распределился между 5%-ным водным раствором бикарбоната натрия и этилацетатом. Раствор в этилацетате сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Этот остаток был подвергнут очистке колоночной хроматографией на силикагеле, используя смесь этилацетат: гексан (1:1) в качестве элюента с получением 1,59 г (44%) (E)-2-(4-хлор-6-фтор-1-инданилиден)-N-метилацетамида в виде белого твердого вещества с т.пл. 173-175oC; ЯМР (CDCl3): δ 7,10-7,30 (m, 2H), 6,16 (s, 1H), 5,64 (br, 1H), 3,42-3,48 (m, 2H), 3,01-3,07 (m, 2H), 2,95 (s, 3H).
Пример 2
(E)-2-(4-хлор-6-фтор-1-инданилиден)ацетамид
Раствор (E)-2-(4-хлор-6-фтор-1-инданилиден) ацетилхлорида (4,0 г, 0,015 моль) [такой как получен в примере 1ж] в дихлорметане (36 мл) добавили по каплям к ледяной смеси 30%-ного водного гидроксида аммония (2,0 мл, 0,03 моль) и дихлорметана (100 мл), и эту смесь перемешивали при комнатной температуре в течение 18 ч. Смесь концентрировали в вакууме, и остаток распределялся между 5%-ным водным раствором бикарбоната натрия и этилацетатом. Раствор в этилацетате сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Этот остаток очищали колоночной хроматографией на силикагеле со смесью этилацетат:гексан (2:1) в качестве элюента. Растирание получившегося твердого вещества с пентаном дало 1,47 г (43%) (E)-2-(4-хлор-6-фтор-1-инданилиден) ацетамида в виде белого твердого вещества с т.пл. 182-184oC.
Пример 3
Получение (Е)-2-(4,6-дихлор-1-инданилиден)ацетамида
(E)-2-(4,6-дихлор-1-инданилиден)ацетамид был получен из промежуточного соединения 3е способом, аналогичным описанному в примере 2.
Этот остаток был очищен колоночной хроматографией со смесью этилацетат: гексан (3:2) в качестве элюента. Растирание полученного твердого вещества с пентаном дало 1,01 г (52%) (E)-2-(4,6-дихлор-1-инданилиден)ацетамида в виде белого твердого вещества с т.пл. 210-212oC.
Пример 4
Получение (E)-2-(6-фтор-4-метил-1-инданилиден)ацетамида
а) Получение (E)-этил 3-(4-фтор-2-метилфенил)акрилата
Смесь 2-бром-5-фенилтолуола (17,6 г, 0,09 моль, Aldrich), этилакрилата (9,3 г, 0,09 моль), триэтиламина (9,4 г, 0,09 моль), ацетата палладия (II) (2,7 г, 0,01 моль) и три-о-толилфосфина (7,3 г, 0,02 моль) в ацетонитриле (60 мл) поместили в бомбу Парра и нагревали при 110oC в течение 12 ч. После охлаждения до комнатной температуры смесь разбавили диэтиловым эфиром и профильтровали. Фильтрат был сконцентрирован в вакууме с получением 33,0 г оранжевого масла. Хроматография на силикагеле с использованием вначале смеси гексан:дихлорметан (8:2) и затем гексан:дихлорметан (6:4) в качестве элюента привела к получению 18,0 г (93%) (E)-этил 3-(4-фтор-2-метилфенил)акрилата в виде бледно-желтого твердого вещества: ЯМР (ДМСО-d6): δ 7,78 (d, 1H, CH=, J= 16 Hz), 7,78 (m, 1H, ArH), 7,02-7,15 (m, 2H, Ar), 6,48 (d, 1H, CH=, J=16 Hz), 4,17 (q, 2H, CH2), 2,38 (s, 3H, CH3), 1,24 (t, 3H, CH4).
б) Получение этил 3-(4-фтор-3-метилфенил)пропионата
Смесь (E)-этил 3-(4-фтор-2-метилфенил)акрилата (32,9 г, 0,16 моль) и гидрата оксида платины (0,5 г, EM Scientific) в 95%-ном этаноле (125 мл) поместили в прибор Парра. После поглощения соответствующего количества водорода катализатор отфильтровали, и фильтрат концентрировали в вакууме с получением 33,7 г этил 3-(4-фтор-3-метилфенил)пропионата. 1,0 г образца очистили хроматографией на силикагеле со смесью гексан:дихлорметан в качестве элюента с получением 0,92 г этил 3-(4-фтор-3-метилфенил)пропионата в виде бесцветного масла: ЯМР (CDCl3): δ 6,78-7,26 (m, 3H, Ar), 4,13 (q, 2H, CH2), 2,90 (t, 2H, CH2), 2,54 (t, 2H, CH2), 2,31 (s, 3H, CH3), 1,24 (t, 3H, CH3).
в) Получение 3-(4-фтор-2-метилфенил)пропионовой кислоты
К смеси этил 3-(4-фтор-3-метилфенил)пропионата (32,7 г, 0,16 моль) в этаноле (150 мл), охлажденной до температуры ледяной бани, добавили в одной порции 1,0 н. раствор гидроксида натрия (156 мл), и смесь перемешивали в течение 18 ч при комнатной температуре. Эту смесь сконцентрировали в вакууме, остаток растворили в воде, и водную фазу промыли диэтиловым эфиром. Водную фазу охладили в ледяной бане и подкислили добавлением 1,0 н. раствора соляной кислоты (160 мл). Отфильтровывание полученного твердого вещества дало 25,8 г (91%) 3-(4-фтор-2-метилфенил)пропионовой кислоты. Перекристаллизовали 0,5 г образца из воды с получением 0,16 г 3-(4-фтор-2-метилфенил)пропионовой кислоты в виде белого твердого вещества с т.пл. 112-113oC.
г) Получение (E)-2-(6-фтор-4-метил-1-инданилиден)ацетамида
Получали из (E)-2-(6-фтор-4-метил-1-инданилиден)ацетил хлорида (4,0 г, 0,018 моль) согласно способу, описанному в примерах 1в-1ж и 2, через следующие промежуточные соединения:
(1) 6-фтор-4-метил-1-инданон, белое твердое вещество с т.пл. 90-92oC.
(2) этил 2-(6-фтор-1-гидрокси-4-метил-1- инданил)ацетат, светло-желтое масло, ЯМР (CDCl3): 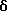 6,76-6,88 (m, 2H, Ar), 4,22 (q, 2H, CH2CH3), 2,79 (2m's, 4H, CH2•s), 2,30 (m, 2H, CH2), 2,24 (s, 3H, CH3), 1,28 (t, 3H, CH3).
6,76-6,88 (m, 2H, Ar), 4,22 (q, 2H, CH2CH3), 2,79 (2m's, 4H, CH2•s), 2,30 (m, 2H, CH2), 2,24 (s, 3H, CH3), 1,28 (t, 3H, CH3).
(3) 2-(6-фтор-1-гидрокси-4-метил-1-инданил)уксусная кислота, используемая немедленно без дальнейшей очистки.
(4) (E)-2-(6-фтор-4-метил-1-инданилиден)уксусная кислота. Перекристаллизация 0,5 г из 2-пропанола дала 0,23 г (E)-2-(6-фтор-4-метил-1-инданилидан)уксусной кислоты в виде белого твердого вещества с т.пл. 243-246oC.
(5) (E)-2-(6-фтор-4-метил-1-инданилиден)ацетилхлорид, используемый без дальнейшей очистки.
Хроматография на силикагеле со смесью этилацетат:гексан (6:4) в качестве элюента и растирание полученного в результате твердого вещества в пентане дало 1,8 г (49%) (E)-2-(6-фтор-4-метил-1-инданилиден)ацетамида в виде не совсем белого твердого вещества: т.пл. 178-180oC; ЯМР (ДМСО-d6): δ 7,25 (br s, 1H, NH2), 7,07-7,11 (m, 1H, Ar), 6,99-7,03 (m, 1H, Ar), 6,84 (br s, 1H, NH2), 6,34 (t, 1H, =CH), 3,15-3,20, 2,80-2,84 (2m's, 4H, 2XCH2), 2,22 (s, 3H, CH3).
Пример 5
Получение (E)-2-(6-фтор-4-метил-1-инданилиден)-N-метилацетамида
Вышеуказанное соединение получали из (E)-2-(6-фтор-4-метил-1-инданилиден) ацетилхлорида (4,0 г, 0,018 моль) способом, аналогичным описанному в примере 1ж. Ацетилхлорид получали как описано в примере 1ж. Хроматография на силикагеле со смесью этилацетат:гексан (6:4) в качестве элюента и растирание полученного в результате твердого вещества в пентане дали 1,71 г (43%) (E)-2-(6-фтор-1-метил-1-инданилиден)-N-метилацетамида в виде не совсем белого твердого вещества с т.пл. 202-204oC; ЯМР (ДМСО-d6): δ 7,78 (br d, 1H, NH), 7,07-7,11 (m, 1H, Ar), 6,99-7,02 (m, 1H, Ar), 6,31 (t, 1H, =CH), 3,17-3,22, 2,80-2,84 (2m's, 4H, 2XCH2), 2,64 (d, 3H, CH3), 2,22 (s, 3H, CH3).
Пример 6
Получение (E)-2-(6-хлор-4-фтор-1-инданилиден)ацетамида
а) Получение 4-хлор-2-фторфенилтрифторметансульфоната
Смесь 4-хлор-2-фторфенола (25,0 г, 0,17 моль, Aldrich) и пиридина (13,5 г, 0,17 моль, Aldrich) в дихлорметане (120 мл) по каплям добавили к раствору трифторметансульфонового ангидрида (50,0 г, 0,18 моль, Aldrich) в дихлорметане (120 мл) при температуре ледяной бани. После перемешивания при комнатной температуре в течение 60 ч реакционную смесь промыли водой и высушили над сульфатом натрия, профильтровали и сконцентрировали в вакууме с получением 45 г сырого 4-хлор-2-фторфенилтрифторметансульфоната. Хроматография на силикагеле с гексаномм в качестве элюента дала 32,2 г (68%) 4-хлор-2-фторфенилтрифторметансульфоната в виде бесцветного масла: ЯМР (CDCl3):  7,20-7,33 (m, 3H, Ar).
7,20-7,33 (m, 3H, Ar).
б) Получение (E)-этил 3-(4- хлор-2-фторфенил)акрилата
Смесь 4-хлор-2-фторфенилтрифторметансульфоната (5,0 г, 0,02 моль), этилакрилата (1,8 г, 0,02 моль, Aldrich), триэтиламина (1,8 г, 0,02 моль) и хлорида бис(трифенилфосфин) палладия (II) (1,4 г, 0,02 моль, Aldrich) в диметилформамиде (20 мл) поместили в бомбу Парра и нагревали при 110oC в течение 12 ч. После охлаждения до комнатной температуры смесь разбавили диэтиловым эфиром и профильтровали. Фильтрат промыли водой, профильтровали и сконцентрировали в вакууме с получением 6,6 г оранжевого масла. Хроматография на силикагеле с использованием первоначально смеси гексан:дихлорметан (7: 3) в качестве элюента дала (а) 1,57 г чистого (E)-этил 3-(4-хлор-2-фторфенил)акрилата в виде зеленого масла, которое твердеет при стоянии, и (б) 0,93 г (E)-этил 3-(4-хлор-2-фторфенил)акрилата, содержащего незначительную примесь. Перекристаллизация (а) из водно-ацетоновых смесей дала 0,82 г (E)-этил 3-(4-хлор-2-фторфенил)акрилата в виде твердого белого вещества с т. пл. 38-40oC.
в) Получение 3-(4-хлор-2-фторфенил)пропионовой кислоты Вышеуказанное соединение получали из (E)-этил 3-(4-хлор-2-фторфенил)акрилата (37,9 г, 0,17 моль) согласно способам, описанным в примерах 4б и 4в, через следующее промежуточное соединение:
(1) Этил 3-(4-хлор-2-фторфенил)пропионат. 1,0 г образца был очищен хроматографически на силикагеле со смесью гексан:этилацетат (98:2) в качестве элюента с получением 0,38 г
этил 3-(4-хлор-2-фторфенил)пропионата в виде бесцветного масла. ЯМР (CDCl3): δ 7,03-7,18 (m, 3H, Ar), 4,13 (q, 2H, CH2), 2,93 (t, 2H, CH2), 2,60 (t, 2H, CH2), 1,23 (t, 3H, CH3).
0,5 г образца перекристаллизовали из воды и получили 0,18 г 3-(4-хлор-2-фторфенил)пропионовой кислоты в виде белого твердого вещества с т.пл. 83-85oC.
г) Получение 6-хлор-4-фтор-1-инданона
Вышеуказанное соединение получали из 3-(4-хлор-2-фторфенил)пропионовой кислоты (8,4 г, 0,04 моль) способом, аналогичным описанному в примере 1в. Хроматография на силикагеле с гексан:метиленхлорид (7:3) в качестве элюента дала 4,1 г (54%) 6-хлор-1-фтор-1-инданона в виде белого твердого вещества с т.пл. 105-107oC.
д) Получение этил 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)ацетата
Раствор этилацетата (8,3 г, 0,09 моль) в тетрагидрофуране (10 мл) по каплям добавили к раствору диизопропиламида лития (62,7 мл 1,5 М раствора в циклогексане, 10,1 г, 0,09 моль, Aidrich) в тетрагидрофуране (100 мл) при -78oC в атмосфере азота. Через 30 мин по каплям добавили раствор 6-хлор-4-фтор-1-инданона в тетрагидрофуране (175 мл), и смесь перемешивали при -78oC в течение 70 мин. Реакцию обрывали раствором хлорида аммония (15,1 г, 0,27 моль) в воде (100 мл), и реакционную смесь оставили на ночь для доведения до комнатной температуры. Фазы разделили, и водную фазу экстрагировали диэтиловым эфиром. Объединенную органическую фазу сушили (сульфат натрия), фильтровали и концентрировали в вакууме с получением 24,4 г сырого этил 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)ацетата. Хроматография на силикагеле, используя смесь гексан:этилацетат (9:1) в качестве элюента, дала 14,7 г (57%) этил 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)ацетата в виде желтого масла. Повторная хроматография 0,5 г образца на силикагеле с использованием дихлорметана в качестве элюента дала 0,27 г этил 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)ацетата в виде бесцветного масла; ЯМР (CDCl3): δ 6,96-7,12 (m, 2H, Ar), 4,35 (br s, 1H, OH), 4,22 (q, 2H, CH2CH3), 3,04 (m, 1H, CH2), 2,75 (2m's, 3H, CH2•s), 2,32 (m, 2H, CH2), 1,28 (t, 3H, CH3).
e) Получение (E)-2-(6-хлор- 4-фтор-1-инданилиден)ацетамида
Вышеуказанное соединение получали из этил 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)ацетата (16,0 г, 0,06 моль) способами, аналогичными описанным в примерах 1е-1ж и 2, через следующие промежуточные соединения:
(1) 2-(6-хлор-4-фтор-1-гидрокси-1-инданил)уксусная кислота. Использовали без дальнейшей очистки.
(2) (E)-2-(6-хлор-4-фтор-1-инданилиден)уксусная кислота. Очищали 1,0 г образца с помощью колоночной хроматографии на силикагеле с гексан:этилацетат (1:1) в качестве элюента, затем перекристаллизацией из 2-пропанола с получением 0,21 (E)-2-(6-хлор-4-фтор-1-инданилиден)уксусной кислоты в виде белого твердого вещества с т.пл. 254-256oC.
(3) (E)-2-(6-хлор-4-фтор-1-инданилиден)ацетил хлорид, использован без дальнейшей очистки.
Хроматография на силикагеле со смесью этилацетат:гексан (7:3) в качестве элюента, и растирание образовавшегося в результате твердого вещества в пентане дало 1,77 г (49%) (E)-2-(6-хлор-4-фтор-1-инданилиден)ацетамида в виде белого твердого вещества с т.пл. 171-173oC, ЯМР (ДМСО-d6): δ 7,43 (d, 1H, Ar), 7,37 (dd, 1H, Ar), 7,31 (br s, 1H, NH2), 6,99 (br s, 1H, NH2), 6,46 (t, 1H, =CH), 3,17-3,22, 2,92-2,97 (2m's, 4H, 2XCH2).
Пример 7
Получение (E)-2-(4-бром-6-фтор-1-инданилиден)ацетамида
а) Получение 2-бром-1-(бромметил)-4-фторбензола
Смесь 2-бром-4-фтортолуола (46,6 г, 0,25 моль, Aldrich), N-бромсукцинимида (46,3 г, 0,25 моль, Aldrich) и перекиси бензоила (0,5 г, 0,002 моль, Aldrich) в четыреххлористом углероде (500 мл) кипятили с обратным холодильником и освещали (250 Вт, инфракрасная лампа) в течение 18 ч. После охлаждения до комнатной температуры сукцинимид фильтровали, и фильтрат концентрировали в вакууме. Хроматография на силикагеле с гексаном в качестве элюента дала 41,8 г (62%) 2-бром-1-(бромметил)-4-фторбензола в виде белого твердого вещества с т.пл. 47-49oC.
б) Получение диэтил 2-(бром-4-фторбензил)малоната
Раствор диэтилмалоната (25,9 г, 0,16 моль) в диметоксиэтане (10 мл) по каплям добавили к суспензии гидрида натрия (6,0 г 60%-ной дисперсии в минеральном масле, 0,15 моль, Aldrich) в диметоксиэтане (25 мл) при комнатной температуре. Через 1 ч добавили по каплям раствор 2-бром-1-(бромметил)-4-фторбензола (40,8 г, 0,15 моль) в диметоксиэтане (125 мл), и смесь кипятили с обратным холодильником в течение 1,5 ч. Реакционную смесь охладили до комнатной температуры и концентрировали в вакууме. Остаток распределяется между дихлорметаном и водой. Экстракты дихлорметана сушили (сульфат натрия) и концентрировали в вакууме с получением 63,4 г желтого масла. Хроматография на силикагеле со смесью дихлорметан:гексан (3:2) дала 21,3 г (40%) диэтил 2-(2-бром-4-фторбензил)малоната в виде бесцветного масла (была получена вторая фракция, 11,5 г, которая содержала незначительную примесь и могла быть использована без дальнейшей очистки); ЯМР (CDCl3): δ 7,21-7,31 (m, 2H, Ar), 6,90-6,97 (m, 1H, Ar), 4,11-4,21 (m, 4H, 2XCH2), 3,77 (t, 1H, CH), 3,30 (d, 2H, CH2), 1,22 (t, 6H, 2XCH3).
в) Получение 3-(2-бром-4-фторфенил)пропионовой кислоты
Смесь диэтил 2-(бром-4-фторбензил)малоната (31,8 г, 0,09 моль) и гидроксида калия (10,3 г, 0,18 моль) в воде (200 мл) кипятили с обратным холодильником в течение 4,5 ч. Смесь концентрировали в вакууме для удаления этанола. К полученному в результате раствору добавили концентрированную серную кислоту (15,7 мл, 0,29 моль), и смесь кипятили с обратным холодильником в течение 18 ч. Реакционную смесь охладили в ледяной бане, и полученное в результате твердое вещество отфильтровали, промыли водой и высушили на воздухе с получением 20,6 г (31%) неочищенной 3-(2-бром-4-фторфенил)пропионовой кислоты. Этот материал был использован без дальнейшей очистки.
г) Получение (E)-2-(4-бром-6-фтор-1-инданилиден)ацетамида
Вышеуказанное соединение получали из 3-(2-бром-4-фторфенил)пропионовой кислоты (19,6 г, 0,08 моль) способами, аналогичными указанным в примерах 1в-1ж и 2, через следующие промежуточные соединения:
(1) 4-бром-6-фтор-1-инданон. Перекристаллизация 0,8 г образца из гексана дала 0,54 г 4-бром-6-фтор-1-инданона в виде белого твердого вещества с т.пл. 129-131oC.
(2) Этил 2-(4-бром-6-фтор-1-гидрокси-1-инданил)ацетат.
Хроматография на силикагеле с использованием смеси гексан:этилацетат (4: 1) дала 14,1 г (70%) этил 2-(4-бром-6-фтор-1-гидрокси-1-инданил)ацетата в виде бледно-желтого масла; ЯМР (CDHCl3): δ 6,98-7,19 (m, 2H, Ar), 4,21 (q, 2H, CH2CH3), 3,04 (m, 1H, CH2), 2,74 (m, 3H, CH2•s), 2,31 (m, 2H, CH2), 1,28 (t, 3H, CH3).
(3) 2-(4-бром-6-фтор-1-гидрокси-1-инданил)уксусная кислота. Использовали немедленно без дальнейшей очистки.
(4) (E)-2-(4-бром-6-фтор-1-инданилиден)уксусная кислота. Использовали без дальнейшей очистки.
(5) (E)-2-(4-бром-6-фтор-1-инданилиден)ацетил хлорид. Использовали без дальнейшей очистки.
Хроматография конечного продукта на силикагеле со смесью этилацетат: гексан (7: 3) в качестве элюента с растиранием образовавшегося в результате твердого вещества в пентане дала 1,4 г (47%) (E)-2-(4-бром-6-фтор-1-инданилиден)ацетамида в виде белого твердого вещества с т.пл. 183-185oC; ЯМР (ДМСО-d6): δ 7,54 (dd, 1H, Ar), 7,37 (m, 2H, Ar и NH2), 6,98 (br s, 1H, NH2), 6,39 (t, 1H, =CH), 3,17-3,22, 2,86-3,00 (2m's, 4H, 2XCH2).
Пример 8
Получение (E)-2-(4-хлор-6-метил-1-инданилиден)ацетамида
Вышеуказанное соединение было получено из 2-хлор-4-метилфенола (50,0 г, 0,35 моль, Aldrich) способами, аналогичными описанным в примерах 6а, 6б, 4б, 4в, 1в-1ж и 2, через следующие промежуточные соединения:
(1) 2-хлор-4-метилфенилтрифторметансульфонат. Хроматография на силикагеле с гексаном в качестве элюента дала 58,2 г (61%) 2-хлор-4-метилфенилтрифторметансульфоната в виде бесцветного масла. ЯМР (ДМСО-d6): δ 7,6 (s, 1H, Ar), 7,5 (d, 1H, J=8,95 Hz, Ar), 7,3 (d, 1H, J=8,95 Hz, Ar), 3,95 (s, 3H).
(2) (E)-этил 3-(2-хлор-4-метилфенил)акрилат. Хроматография на силикагеле с использованием смеси гексан: этилацетат (95:5) как элюента дала 24,1 г (выход 70%) чистого (E)-этил 3-(2-хлор-4-метилфенил)акрилата. ЯМР (ДМСО-d6): δ 7,89 (d, 1H, CH=, J=16,0 Hz), 7,85 (d, 1H, J=8,2 Hz, ArH), 7,35 (s, 1H, Ar), 7,21 (d, 1H, J=8,2 Hz), 6,67 (d, CH=, J=16,0 Hz), 4,21 (q, 2H, J=7,1 Hz), 2,32 (s, 3H), 1,27 (t, 3H, J=7 Hz).
(3) Этил 3-(2-хлор-4-метилфенил)пропионат. Хроматография на силикагеле со смесью гексан:этилацетат (98:2) в качестве элюента с получением 29,10 г этил 3-(2-хлор-4-метилфенил)пропионата в виде бесцветного масла; ЯМР (CDCl3): δ 7,03-7,24 (m, 3H, Ar), 4,13 (q, 2H, CH2), 2,93 (t, 2H, CH2), 2,60 (t, 2H, CH2), 2,27 (s, 3H, CH3), 1,17 (t, 3H, CH3).
(4) 3-(2-хлор-1-метилфенил)пропионовая кислота. Использовали без дальнейшей очистки. ЯМР (ДМСО-d6): δ 12,2 (s, 1H), 7,05-7,25 (m, 3H, Ar), 2,89 (t, 2H, CH2), 2,75 (t, 2H, CH2), 2,27 (s, 1H).
(5) 4-хлор-6-метил-1-инданон. Хроматография на силикагеле со смесью гексан: этилацетат (градиент 99: 1 - 99:5) в качестве элюента дала 14,12 г (64%) 4-хлор-6-метил-1-инданона. ЯМР (ДМСО-d6): δ 7,56 (s, 1H, Ar), 7,37 (s, 1H, Ar), 2,96 (m, 2H, CH2), 2,64 (m, 2H, CH2), 2,35 (s, 3H).
(6) Получение этил 2-(4-хлор-6-метил-1-гидрокси-1-инданил) ацетата. Хроматография на силикагеле с использованием смеси гексан:этилацетат (8:2) как элюента дала 6,36 г этил 2-(4-хлор-6-метил-1-гидрокси-1-инданил)ацетата в виде желтого масла и 6,54 г возвращенного исходного материала. Этот возвращенный исходный материал был вновь обработан в идентичных условиях. Обработка и хроматография как ранее дали 6,4 г этил 2-(4-хлор-6-метил-1-гидрокси-1-инданил)ацетата с суммарным выходом 12,7 г (86%) в виде бесцветного масла; ЯМР (ДМСО): δ 7,09 (s, 2H, Ar), 5,37 (br s, 1H, OH), 4,0 (m, 2H, CH2CH3), 2,6-2,95 (m, 4H), 4-2,6 (m, 1H), 2,28 (s, 3H), 2,0-2,2 (m, 1H), 1,08 (t, 3H, CH3).
(7) 2-(4-хлор-6-метил-1-гидрокси-1-инданил)уксусная кислота. Использована немедленно без дальнейшей очистки.
(8) (E)-2-(4-хлор-6-метил-1-инданилиден)уксусная кислота. Использована без дальнейшей очистки.
(9) (E)-2-(4-хлор-6-метил-1-инданилиден)ацетилхлорид. Использован без дальнейшей очистки.
Хроматография конечного продукта на силикагеле со смесью этилацетат:гексан (градиент 1:1 - 3:1) в качестве элюента дала 2,01 г (69%) (E)-2-(4-хлор-6-метил-1-инданилиден)ацетамида в виде белого твердого вещества с т.пл. 213-215oC; ЯМР (ДМСО-d6): δ 7,35 (br s, 1H, NH), 7,33 (s, 1H, Ar), 7,29 (s, 1H, Ar), 6,93 (br s, 1H, NH2), 6,41 (t, 1H, =CH, J=2,6 Hz), 3,17-3,22, 2,90-2,97 (2m's, 4H, 2XCH2), 2,35 (s, 1H, CH3).
Фармацевтические составы и примеры с 9 по 17 смотри после формулы изобретения.









Описываются соединения формулы (1), в которой R1 и R2 независимо выбраны из хлора, фтора, брома, С1 - C6алкила, C1 - С6 алкоксила или C1 - C6галоалкила при условии, что оба R1 и R2 не являются фтором, R3 и R4 независимо выбраны из водорода и С1 - C6алкила, и его фармацевтически приемлемые соли, сольваты или физиологически функциональные производные, обладающие противовоспалительной или анальгезирующей активностью, их использование в медицине, особенно для профилактики или лечения состояний, связанных с воспалением, артритом или болью, фармацевтические составы, включающие их, и способы их получения. 8 с. и 6 з.п.ф-лы.
где R1 и R2 независимо выбраны их хлора, фтора, брома, C1 - C6-алкила, C1 - C6-алкокси или C1 - C6-галоалкила, при условии, что оба R1 и R2 не являются фтором;
R3 и R4 независимо выбраны из водорода и C1 - C6-алкила,
или их фармацевтически приемлемая соль, или сольват.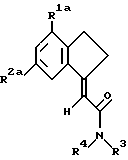
где R1а является хлором;
R2а является хлором, фтором, бромом или метилом;
R3 и R4 независимо выбраны из водорода, метила или этила.
где R1, R2, R3 и R4 такие, как определено в п.1,
для производства лекарства для профилактики или лечения состояний, связанных с воспалением, артритом или болью.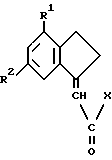
подвергают взаимодействию с амином NHR3R4,
где R1-R4 такие, как определено ранее;
X является отщепляемой группой,
после чего возможно превращают полученное таким образом соединение формулы I в его соль.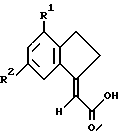
подвергают взаимодействию с соответствующим реагентом сочетания, с последующим взаимодействием с амином HNR3R4,
где R1-R4 такие, как определено ранее,
после чего возможно превращают полученное таким образом соединение формулы I в его соль.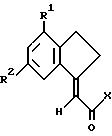
где R1 и R2 такие, как определено ранее;
X является отщепляемой группой или OH.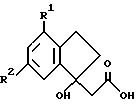
где R1 и R2 такие, как определено ранее.
| Способ получения амидов ненасыщенных кислот | 1987 |
|
SU1695825A3 |
| Способ получения (4,2,0)-бициклооктановых производных или их фармацевтически приемлемых нетоксичных солей | 1987 |
|
SU1588275A3 |
| Приспособление к свеклокомбайну для уборки маточной сахарной свеклы | 1973 |
|
SU476846A1 |

