Настоящее изобретение относится к способу получения ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты, а также к промежуточным соединениям, используемым в этом способе.
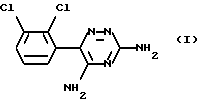
Ламотриджин представляет собой 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин формулы (I)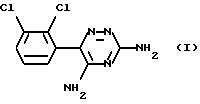
Это известное (EP-A-0021121) соединение, применяемое для лечения расстройств центральной нервной системы (ЦНС), в частности эпилепсии. Известный (EP-A-0247892) изетионат ламотриджина является предпочтительной солью для парентерального введения.
Известен (EP-A-0021121) способ получения ламотриджина, при котором осуществляют циклизацию соединения формулы (I)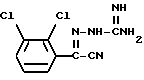
Циклизацию осуществляют путем кипячения с обратным холодильником соединения формулы (I) в алканоле, предпочтительно в C1-4алканоле, таком как метанол или этанол, в присутствии сильного основания, такого как гидроксид калия.
Задачей настоящего изобретения является создание нового способа получения ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты, и использование новых промежуточных соединений при получении ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты.
Таким образом, согласно настоящему изобретению предложен способ получения ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты, при котором 6-(2,3-дихлорфенил)-5-хлор- 3-тиометил-1,2,4-триазин формулы (II)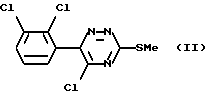
обрабатывают аммиаком.
Взаимодействие соединения формулы (II) с аммиаком можно проводить в органическом растворителе. Подходящие растворители включают C1-6алканолы, такие как метанол, этанол, пропан-1-ол или пропан-2-ол, предпочтительно этанол. Температура реакции может варьировать в пределах от температуры окружающей среды до температуры кипения растворителя. Альтернативно это взаимодействие можно проводить под давлением при температуре выше точки кипения растворителя, например в автоклаве. В этом случае подходящая температура составляет примерно 180oC, а подходящее давление составляет примерно 1930 кПа (280 фунт/кв.дюйм).
Вместо применения аммиака в органическом растворителе можно использовать водный аммиак. Альтернативно соединение формулы (II) можно обработать непосредственно жидким аммиаком под давлением, например в автоклаве.
Соединение формулы (II) является новым.
Соединение формулы (II), 6-(2,3-дихлорфенил)-5-хлор-3-тиометил-1,2,4-триазин, может быть получено обработкой 6-(2,3-дихлорфенил)-5-оксо-3-тиометил-1,2,4-триазина, который имеет формулу (III),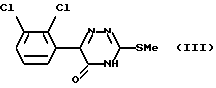
хлорирующим агентом.
Подходящие хлорирующие агенты включают оксихлорид фосфора, тионилхлорид, трихлорид фосфора и пентахлорид фосфора. Оксихлорид фосфора наиболее предпочтителен. Хлорирование является традиционным и проводится в обычных условиях синтеза.
Благодаря кетоенольной таутомерии, соединение формулы (III) может также существовать в виде следующей структуры: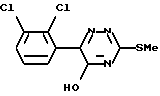
Следует иметь в виду, что все встречающиеся ссылки на соединение формулы (III) относятся к любому из таутомеров или как к кето-, так и к енольному таутомерам.
Соединение формулы (III) является новым. Поэтому в данном изобретении далее предложен 6-(2,3-дихлорфенил)-5-оксо-3-тиометил-1,2,4-триазин.
Соединение формулы (III) можно получить обработкой 6-(2,3-дихлорфенил)-5-оксо-3-тио-1,2,4-триазина формулы (IV)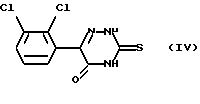
метилирующим агентом.
Подходящие метилирующие агенты включают метилгалогениды, например MeCl, MeBr и MeI, метилсульфонат и метил-п-толуолсульфонат. Наиболее предпочтителен метилиодид. Метилирование проводят в обычных условиях. Например, взаимодействие соединения формулы (IV) с метилиодидом предпочтительно проводят в щелочном водном растворе, например в водном растворе гидроксида щелочного металла, таком как гидроксид натрия или гидроксид калия, при температуре окружающей среды.
Подобно соединению (III) соединение (IV) обладает кетоенольной таутомерией и может соответственно существовать в виде следующей структуры: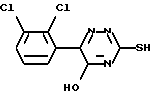
Следует иметь в виду, что все встречающиеся ссылки на соединение формулы (IV) относятся к любому из таутомеров или как к кето-, так и к енольному таутомерам.
Соединение формулы (IV) является новым.
Соединение формулы (IV), 6-(2,3-дихлорфенил)-5-оксо-3-тио-1,2,4-триазин, можно получить обработкой 2,3-дихлорфенилглиоксиловой кислоты формулы (V)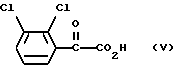
тиосемикарбазидом.
Взаимодействие соединения формулы (V) с тиосемикарбазидом предпочтительно проводят в щелочном водном растворе, например в водном растворе гидроксида натрия или гидроксида калия. Предпочтительно проводить его при температуре, варьирующей от температуры окружающей среды до температуры кипения растворителя, более предпочтительно при кипячении с обратным холодильником.
Соединение формулы (V) можно получить окислением 2,3-дихлорацетофенона формулы (VI)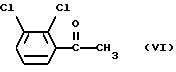
Подходящие окисляющие агенты включают диоксид селена, водный щелочной перманганат калия, дихромат калия и триоксид хрома, все в присутствии пиридина. Окисление осуществляют в обычных условиях.
Соединение формулы (VI) можно получить обработкой 1,2-дихлорбензола соединением формулы RM1 или RM2X, в которой R представляет собой C1-6алкил, М1 представляет собой щелочной металл, М2 представляет собой щелочно-земельный металл и X представляет собой галоген, с последующей реакцией взаимодействия полученного таким образом соединения с ацетилхлоридом или уксусным ангидридом. RM2X может, например, представлять собой реактив Гриньяра (Grignard), такой как метилмагнийиодид. Предпочтительно применяют соединение формулы RM1, причем бутиллитий является наиболее предпочтительным, с последующей реакцией взаимодействия с уксусным ангидридом. Эту реакцию предпочтительно проводят при температуре около -70oC.
Соединение формулы (VI) можно также получить обработкой 2,3-дихлорбензальдегида соединением RM1 или RM2X, где R представляет собой метил, предпочтительно метилмагнийиодидом, и окислением полученного таким образом α- метил-2,3-дихлорбензилового спирта. Подходящие окисляющие агенты включают, например, гипохлорит натрия. Это взаимодействие предпочтительно осуществляют при комнатной температуре.
Соединение формулы (VI) можно также получить обработкой 2,3-дихлориодбензола магнием и обработкой полученного таким образом соединения ацетилхлоридом в присутствии безводного хлорида железа. Реакцию предпочтительно проводят при температуре около -70oC.
В данном изобретении также предложен способ, при котором дополнительно образуют фармацевтически приемлемую соль ламотриджина, полученную присоединением кислоты.
Подходящие фармацевтически приемлемые соли ламотриджина, полученные присоединением кислот, включают сульфат, фосфат, метансульфонат, п-толуолсульфонат, бензолсульфонат и изетионат. Изетионат ламотриджина является наиболее предпочтительным для парентерального введения, так как он обладает высокой растворимостью в воде.
Изетионат ламотриджина можно получить в результате реакции взаимодействия ламотриджина с изетионовой кислотой. Молярное соотношение ламотриджина и изетионовой кислоты предпочтительно составляет от 1:3 до 3:1, в частности примерно 1:1.
Изетионовая кислота отсутствует в продаже и поэтому ее удобно получать in situ. Например, изетионат щелочного металла в растворе можно превратить в изетионовую кислоту, например, путем пропускания водного раствора изетионата через H+ ионообменную смолу с последующим смешиванием полученного раствора с триазином. Реакционный растворитель, как правило, является водой, и в этом случае взаимодействие можно осуществлять при температурах от 4 до 50oC, удобно при температуре окружающей среды, и без необходимости в каких-либо корректорах pH или других добавках.
Образовавшийся изетионат можно перекристаллизовать, например, из технического метилированного спирта с получением кристаллов изетионата ламотриджина, которые легко растворяются в воде.
Альтернативно изетионат ламотриджина можно получить взаимодействием соли ламотриджина, не являющейся изетионатом, с изетионатным анионом. Предпочтительно, если соотношение соли и аниона составляет от 1:50 до 50:1. Более предпочтительно это соотношение составляет примерно 1:10. Предпочтительно взаимодействие осуществляют элюированием раствора данной соли в метаноле через колонку с изетионатанионообменной смолой. В этом случае соль предпочтительно является метансульфонатом (мезилатом) ламотриджина.
В данном изобретении также предложен способ, при котором получают фармацевтическую композицию посредством соединения ламотриджина или его фармацевтически приемлемой соли, полученной присоединением кислоты, и фармацевтически приемлемого разбавителя или носителя.
Ламотриджин должен присутствовать в композициях, полученных в соответствии с данным изобретением, в эффективной единичной дозовой форме, например в количестве, достаточном, чтобы быть эффективным против расстройств ЦНС in vivo.
Фармацевтически приемлемый разбавитель или носитель, присутствующий в композициях, приготовленных в соответствии с данным изобретением, может быть жидким или твердым веществом, которое инертно или приемлемо с медицинской точки зрения и совместимо с ламотриджином или его солью.
Фармацевтические композиции могут вводиться перорально или парентерально, могут применяться в виде суппозитория, или наноситься локально в виде мази, пасты или порошка. Однако предпочтительно пероральное или парентеральное введение композиции.
Для перорального введения можно использовать тонкие порошки или гранулы, содержащие разбавляющие, диспергирующие и/или поверхностно-активные вещества, которые могут быть представлены в виде дозы в воде или сиропе, в виде капсул или пастилок в сухом состоянии или в виде неводной суспензии, в которую могут быть включены суспендирующие вещества, и, наконец, в виде суспензии в воде или сиропе. Если это желательно или необходимо, могут быть включены ароматизаторы, консерванты, суспендирующие агенты, загустители и эмульгирующие агенты. Если суспензию готовят в воде, то в соответствии с данным изобретением предпочтительно присутствует по меньшей мере одно из этих веществ.
Для парентерального введения активное соединение может быть приготовлено в виде стерильного водного раствора для инъекций, который может содержать антиоксиданты или буферы.
Ламотриджин или его соль могут быть введены в чистой форме, не связанной с другими добавками, и в этом случае предпочтительным носителем является капсула или пастилка.
Альтернативно активное соединение может быть представлено в чистой форме в виде эффективной единичной дозы, например оно может быть спрессовано в виде таблетки и т.п.
Другими веществами, которые могут быть включены в состав препарата, являются, например инертные, с точки зрения медицины, ингредиенты, в частности твердые и жидкие разбавители, такие как лактоза, крахмал или фосфат кальция для таблеток или капсул; оливковое масло или этилолеат для мягких капсул; и вода или растительное масло для суспензий или эмульсий; смягчающие агенты, такие как тальк или стеарат магния; желирующие агенты, такие как коллоидные глины; загустители, такие как гуммиарабик или альгинат натрия; а также другие терапевтически приемлемые вспомогательные ингредиенты, такие как увлажнители, консерванты, буферы и антиоксиданты, которые могут быть использованы как носители в таких препаратах.
Таблетки и другие препаративные формы, представленные в виде отдельных единиц, могут содержать количество ламотриджина или его соли, эффективное в этой дозе или в многократных дозах, например единицы, содержащие от 5 мг до 500 мг, предпочтительно примерно от 10 мг до 250 мг, в пересчете на свободное основание.
Водные препараты должны, как правило, содержать фармацевтически приемлемую соль ламотриджина в количестве, эффективном против расстройств ЦНС in vivo, и такой препарат может быть представлен в единичной дозировочной форме. В составе водного препарата может находиться до 250 мг/мл соли, в пересчете на свободное основание. Однако, традиционные концентрации соли в растворе составляют от 10 до 70 мг/мл, предпочтительно от 10 до 50 мг/мл. Соль для парентерального введения может быть представлена в виде стерильных водных растворов для инъекций, которые могут содержать терапевтически приемлемые вспомогательные ингредиенты, такие как антиоксиданты, буферы и агенты для регулирования осмомолярности раствора. Предпочтительно, чтобы такие анионы, как хлоридный и фосфатные, не присутствовали в растворе, так как они имеют тенденцию обмениваться с солью с образованием осадков.
Водный препарат может быть приготовлен растворением соли в водной среде, например в стерильной воде для инъекций. Раствор перед использованием может быть разбавлен до необходимой концентрации.
Нижеследующие Примеры иллюстрируют этапы способа по изобретению.
Пример 1: Получение соединения (VI)
Способ А
В атмосфере азота медленно, по каплям, при перемешивании добавили бутиллитий в гексане (300 мл, 0,474 моль) к 1,2-дихлорбензолу (104,58 г, 0,711 моль), растворенному в сухом тетрагидрофуране (2 л), поддерживавшемуся при температуре -70oC. Полученный раствор перемешивали при -70oC в течение 1 ч. Этот раствор, все еще при -70oC, добавили к уксусному ангидриду (290,35 г, 2,84 моль), растворенному в сухом тетрагидрофуране (1 л) при -70oC, в атмосфере азота через иглу с двойным концом. Когда добавление было закончено, полученный раствор перемешивали при -70oC в течение приблизительно 1 ч и раствору дали нагреться до комнатной температуры.
Реакционную смесь вылили на лед (5 л) и после хорошего перемешивания оставили стоять в течение ночи при комнатной температуре. Водную смесь экстрагировали эфиром (3 х 1,5 л). Эфирные фазы объединили, промыли водой (3 х 750 мл), насыщенным раствором бикабоната натрия (3 х 750 мл) и рассолом (1 х 750 мл). Органическую фазу сушили над безводным сульфатом магния, фильтровали и выпаривали с получением желтой жидкости. Сырой продукт поместили в высокий вакуум на горячую водяную баню для полного удаления остатков 1,2-дихлорбензола и уксусного ангидрида. Получили 2,3-дихлорацетофенон (67,2 г, выход 75%). ИКС, ЯМР и ТСХ (SiO2; CHCl3) показали, что это вещество содержит мало примесей, поэтому дальнейшую очистку не предпринимали.
Способ Б
Иодметан (324,89 г, 2,288 моль) добавляли по каплям при перемешивании к магниевой стружке (54,88 г, 2,288 моль) в сухом эфире (1 л) с образованием метилмагнийиодида. 2,3-Дихлорбензальдегид (200 г, 1,144 моль), растворенный в смеси бензол/диэтиловый эфир (1 л, 50:50), добавили по каплям при перемешивании к реактиву Гриньяра. Реакционную смесь оставили перемешиваться при комнатной температуре в течение ночи. Этот раствор кипятили с обратным холодильником в течение 2 ч и затем дали остыть. Реакционную смесь вылили в насыщенный раствор хлорида аммония (5 л) и отделили органический слой. Водный слой экстрагировали эфиром (3 х 2 л). Органические фазы объединили, промыли рассолом (1 х 2 л), высушили над безводным сульфатом магния, отфильтровали и выпарили. Получили α- метил-2,3-дихлорбензиловый спирт (196,4 г, выход 90%) в виде желтого масла, которое кристаллизуется при стоянии с образованием светло-желтого твердого вещества. ТСХ (SiO2; CHCl3) не выявила наличия примесей, поэтому дальнейшую очистку не проводили. Однако, если необходимо, этот спирт можно перекристаллизовать из петролейного эфира при 40-60oC с получением белых призм с температурой плавления 53oC.
α- Метил-2,3-дихлорбензиловый спирт (5,0 г, 0,026 моль) растворили в уксусной кислоте (24 мл) и прибавили 12%-ый (м/о) раствор гипохлорита натрия (23,26 мл, 0,0314 моль) медленно, по каплям, при перемешивании при температуре 15-25oC. Когда прибавление было закончено, реакционную смесь перемешивали при температуре окружающей среды в течение примерно 1,5 ч, до тех пор, пока проба крахмал/иодид не дала положительный результат.
К реакционной смеси добавляли насыщенный раствор бисульфита натрия, пока проба крахмал/иодид не стала отрицательной. Эту смесь вылили в смесь лед/рассол (100 мл) и экстрагировали диэтиловым эфиром (3 х 75 мл). Эфирные фазы объединили и промывали 2 N раствором гидроксида натрия (3 х 75 мл), пока промывные воды не стали щелочными. Эфирную фазу сушили над безводным сульфатом магния, фильтровали и выпарили. Получили 2,3-дихлорацетофенон (3,2 г, выход 65%) в виде светло-желтого масла. ТСХ (SiO2; CHCl3) и ЯМР показали, что это вещество не содержит примесей.
Способ В
В атмосфере азота 2,3-дихлориодбензол (350 г, 1,282 моль), растворенный в сухом эфире (1250 мл) медленно при перемешивании добавили к магниевым стружкам (30,77 г, 1,282 моль) в сухом диэтиловом эфире (300 мл) для получения 2,3-дихлорфенилмагнийиодида. Этот реактив Гриньяра добавили при перемешивании по каплям, к ацетилхлориду (301,91 г, 3,846 моль), растворенному в сухом диэтиловом эфире (1 л), и безводному хлориду железа (1,925 г, 0,0118 моль), при температуре -70oC в атмосфере азота. Когда прибавление было закончено, полученную смесь перемешивали при -70oC еще в течение 5 мин, а затем дали ей нагреться до комнатной температуры. Затем реакционную смесь вылили на лед (5 л) и тщательно перемешали. Водную смесь подщелочили карбонатом натрия и оставили стоять при комнатной температуре в течение ночи. Этот водный раствор экстрагировали диэтиловым эфиром (3 х 2 л), эфирные фазы объединили, высушили над безводным сульфатом магния, профильтровали и выпарили. Получили сырой 2,3-дихлорацетофенон (235,7 г) в виде желтой жидкости. Сырое вещество перегнали в вакууме с получением чистого 2,3-дихлорацетофенона (147,0 г, т. пл. 100oC/2 мм рт.ст.; выход 60,66%). ЯМР и ТСХ (SiO2; CHCl3) показали, что перегнанный продукт не содержал примесей.
Пример 2: Получение соединения (V)
Раствор KMnO4 (33,33 г, 0,21 М) в воде (1100 мл) по каплям при перемешивании, в течение 2 ч, прибавляли при 10-15oC к 2,3-дихлорацетофенону (12,5 г, 0,066 М), KOH (8,33 г, 0,148 М) в воде (330 мл) и пиридине (833 мл). Полученную смесь перемешивали в течение 1 ч. Избыток перманганата нейтрализовали добавлением метабисульфита натрия и полученный раствор профильтровали через фильтр hyflo. Фильтрат подкислили концентрированной HCl и оставили при комнатной температуре стоять в течение ночи. Кислый раствор экстрагировали эфиром и эфирные фракции объединили, профильтровали и выпарили с получением продукта, который после сушки над P2O5 был идентифицирован как 2,3-дихлорфенилглиоксиловая кислота формулы (V) с помощью ТСХ (SiO2: DI-PE:HCO2H:H2O). Выход сырого продукта = 67,8%
Пример 3: Получение соединения (IV)
2,3-Дихлорфенилглиоксиловую кислоту (12,00 г, 0, 155 М), соединение формулы (V), растворили в теплом 1 N растворе NaOH (165 мл, 0,165 М). Тиосемикарбазид формулы H2N-NH-C(S)-NH2 (10,12 г, 0,11 М) растворили в теплой воде (175 мл). Затем оба раствора смешали и кипятили с обратным холодильником при перемешивании в течение 4 ч. Реакционную смесь оставили охлаждаться в течение ночи, а затем подкислили при 0,5oC концентрированной HCl. Образовавшуюся суспензию перемешивали в течение 30 мин, твердую фазу отфильтровали и дали высохнуть.
Было получено с 48%-ным выходом соединение (IV), 6-(2,3-дихлорфенил)-5-оксо-3-тио-1,2,4-триазин. Его идентичность была подтверждена инфракрасной спектроскопией и ТСХ.
Пример 4: Получение соединения (III)
6-(2,3-дихлорфенил)-5-оксо-3-тио-1,2,4-триазин (16,5 г, 0,06 М), соединение формулы (IV), полученное в примере 3, растворили в 2 N растворе NaOH (33,32 мл). К раствору добавили метилиодид (25,55 г, 0,18 М) в воде (50 мл). Полученный раствор перемешивали в течение ночи при комнатной температуре. Образовавшееся твердое вещество отфильтровали, растворили в 2 N NaOH и подкислили концентрированной HCl с получением осадка, который отфильтровали с помощью насоса и высушили над P2O5 с получением соединения (III), 6-(2,3-дихлорфенил)-5-оксо-3-тиометил-1,2,4-триазина (10,85 г, выход 63%). Температура плавления = 131,6oC.
Пример 5: Получение соединения (II)
6-(2,3-дихлорфенил)-5-оксо-3-тиометил-1,2,4-триазин (2,0 г, 0,0069 М), который представляет собой соединение (III), и оксихлорид фосфора (POCl3, 40 мл) поместили вместе в колбу и кипятили с обратным холодильником в течение 2 ч. Реакционной смеси дали остыть и избыток POCl3 удалили выпариванием. Остаток растворили в CHCl3 и сделали щелочным с помощью NH4OH. После выстаивания в течение 30 мин полученную смесь промывали водой, фильтровали и выпаривали с получением соединения (II), 6-(2,3-дихлорфенил)-5-хлор-3-тиометил-1,2,4-триазина с выходом 80%. Его чистота подтверждена посредством ТСХ.
Пример 6: Получение ламотриджина
6-(2,3-дихлорфенил)-5-хлор-3-тиометил-1,2,4-триазин, полученный в примере 5, растворили в этаноле (100 мл), насытили газообразным аммиаком и нагревали в запаянной стеклянной трубке в автоклаве при 180oC/1930 кПа (280 фунт/дюйм2) в течение 72 ч.
Все содержимое трубки выпарили с получением темно-коричневого сырого продукта. Этот сырой продукт перекристаллизовали из метанола и идентифицировали как соединение (I), (3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин (ламотриджин), с помощью ТСХ (Rf = 0,20). Температура плавления = 218oC.
Пример 7: Получение изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина
Раствор изетионата натрия (148 г, 1,0 моль) в воде (4,9 л) пропустили через колонку с IR 120 (Н) ионообменной смолой и элюировали водой. 3,5-Диамино-6-(2,3-дихлорфенил)-1,2,4-триазин (256 г, 1,0 моль) растворили в полученной изетионовой кислоте, а раствор профильтровали и выпарили в вакууме. Остаток перекристаллизовали из технического метилированного спирта с получением изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина.
Выход 273,3 г (72%), Т.пл. 242oC.
Пример 8: Получение изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина
50 ммоль Amberlite (торговая марка) IR-45 (ОН) смешали с 15 ммоль (10 мл) водной изетионовой кислоты, а полученную смесь поместили в колонку. Эту колонку затем промыли метанолом. Через колонку элюировали 0,7 г (2 ммоль) метанольного раствора мезилата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина. Элюант выпарили в вакууме, остаток перекристаллизовали из технического метилированного спирта и получили изетионат 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина.
Выход 300 мг (40%), т.пл. 242-243oC.
Пример 9:
74,625 г (0,195 моль) изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина добавили и растворили в примерно 900 мл воды для инъекций ВР и разбавили до 1000 мл добавочной порцией воды для инъекций ВР с получением водного раствора, содержавшего изетионат, эквивалентный 50 мг/мл основания 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина. Этот раствор является приемлемым на основании тонуса.
Пример 10:
14,925 г (0,039 моль) изетионата 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина добавили к раствору 43,8 г (0,221 моль) моногидрата декстрозы примерно в 900 мл воды для инъекций ВР и разбавили до 1000 мл добавочной порцией воды для инъекций ВР с получением водного раствора, содержавшего изетионат, эквивалентный 10 мг/мл основания 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазина. Этот раствор приемлем на основании тонуса.
Пример 11:
Была приготовлена фармацевтическая композиция, включающая следующие ингредиенты, содержащиеся в одной таблетке, мг:
3,5-Диамино-6-(2,3-дихлорфенил)-1,2,4-триазин - 150
Лактоза - 200
Кукурузный крахмал - 50
Поливинилпирролидон - 4
Стеарат магния - 4
Лекарство смешивали с лактозой и крахмалом и гранулировали с раствором поливинилпирролидона в воде. Полученные гранулы сушили, смешивали со стеаратом магния и спрессовывали с получением таблеток со средним весом 408 мг.
Описывается способ получения ламотриджина формулы I или его фармацевтически приемлемой соли, полученной присоединением кислоты. Он отличается тем, что 6-(2,3-дихлорфенил)-5-хлор-3-тиометил-1,2,4-триазин формулы II обрабатывают аммиаком. Соединение используют для лечения расстройств центральной нервной системы, в частности эпилепсии. Описывается новое промежуточное соединение, используемое при его получении. Технический результат - упрощение процесса. 2 c. и 8 з.п. ф-лы.
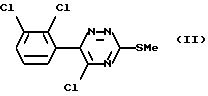

или его фармацевтически приемлемой соли, полученной присоединением кислоты, отличающийся тем, что 6-(2,3-дихлорфенил)-5-хлор-3-тиометил-1,2,4-триазин формулы II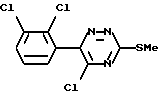
обрабатывают аммиаком.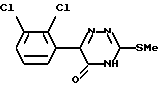
хлорирующим агентом.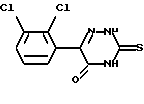
метилирующим агентом.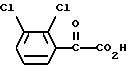
тиосемикарбазидом.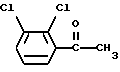
6. Способ по п.5, отличающийся тем, что соединение формулы VI получают обработкой 1,2-дихлорбензола бутиллитием с последующей обработкой полученного таким образом соединения уксусным ангидридом.
| Способ химической регенерации отработанных кобальтовых катализаторов гидрирования нитро- и аминопроизводных углеводородов | 1959 |
|
SU137500A1 |
| GB 759014, 10.10.1956 | |||
| Устройство для передачи подкладных под торфяные кирпичи досок с нижней ветви канатного транспортера на верхнюю его ветвь | 1929 |
|
SU21121A1 |
| СПОСОБ ПРОИЗВОДСТВА КРЕПКИХ АЛКОГОЛЬНЫХ НАПИТКОВ ИЗ ПЛОДОВО-ЯГОДНОГО СЫРЬЯ | 0 |
|
SU247892A1 |
| Устройство для уплотнения подшипниковых узлов во взрывонепроницаемых электрических машинах | 1973 |
|
SU459829A2 |

