Протеинкиназа C (РКС) представляет собой семейство близкородственных ферментов, обладающих функциями серин/треонинкиназы. Протеинкиназа C играет важную роль в передаче сигнала от клетки к клетке, в экспрессии генов, а также в регулировании дифференцировки и роста клеток. В настоящее время известно по крайней мере 10 изозимов протеинкиназы C (РКС), которые отличаются между собой по своему распределению в тканях, регулированию и по своей ферментной специфичности. Nishizuka Y. Annu, Rev. Biochem. 58: 31-44 (1989); Nishizuka Y. Science 258: 607-614 (1992).
Изозимы протеинкиназы C представляют собой одноцепочечные полипептиды длиной от 592 до 737 аминокислот. Эти изозимы содержат регуляторный домен и каталитический домен, связанные линкерным пептидом. Регуляторный и каталитический домены могут также подразделяться на константные и вариабельные области. Каталитические домены протеинкиназы C и других протеинкиназ очень схожи между собой, тогда, как регуляторный домен является уникальным для каждого изозима РКС. Аминокислотные последовательности изозимов РКС обнаруживают 40 - 80% гомологии внутри своей группы. Однако, гомология различных видов одного изозима составляет обычно более чем 97%.
Протеинкиназа C представляет собой мембрано-ассоциированный фермент, который аллостерически регулируется рядом факторов, включая мембранные фосфолипиды, кальций и некоторые мембранные липиды, такие, как диацилглицерины, высвобождаемые в ответ на активацию фосфолипаз Bell R.M. и Burns D.J. J.Biol. Chem, 266: 4661 - 4664 (1991); Nishizuka Y. Science 258: 607 - 614 (1992). Изозимы протеинкиназы C, альфа, бета-1, бета-2 и гамма, для полной своей активации требуют присутствия мембранного фосфолипида, кальция и диацилглицерин/форболовых сложных эфиров. Дельта-, эпсилон-, эта- и тета-формы РКС, по способу своей активации, являются кальций-независимыми. Дзета- и лямбда-формы РКС являются кальций- и диацилглицерин-независимыми и для своей активации, очевидно, требуют лишь мембранного фосфолипида.
Лишь один или два изозима протеинкиназы C могут быть ассоциированы с каким-либо определенным заболеванием. Например, повышенные уровни глюкозы в крови, обнаруживаемые при диабете, приводят к изозим-специфическому повышению уровня изозима бета-2 в сосудистых тканях. Inoguchi и др., Proc. Natl. Acad. Sci. USA 89: 11059 - 11065 (1992). Связанное с диабетом повышение бета-изозима в тромбоцитах человека коррелирует с изменением его ответной реакции на агонистов. Bastyr 111, E.J. и L и J.Diabetes 42: (Suppl. 1) 97A (1993). Было показано, что рецептор витамина D человека селективно фосфорилируется бета-изозимом протеинкиназы C. Это фосфорилирование связано с изменениями функции рецептора. Hsiel и др., Proc. Natl. Acad. Sci USA 88: 9315 - 9319 (1991); Hsiel и др., J. Biol. Chem. 268: 15118 - 15126 (1993). Кроме того, недавно было показано, что изозим бета-2 является ответственным за пролиферацию клеток эритролейкоза, а альфа-изозим участвует в дифференцировке мегакариоцитов в этих же самых клетках. Murray и др., J. Biol. Chem. 268: 15847-15853 (1993).
Повсеместная распространенность изозимов протеинкиназы C и их важная роль в физиологии побуждает исследователей разрабатывать высокоселективные ингибиторы РКС. В случае, если имеются данные, свидетельствующие о связи некоторых изозимов с определенными заболеваниями, есть все основания предполагать, что наилучшими терапевтическими средствами являются ингибирующие соединения, селективные к одному или двум изозимам протеинкиназы C в сравнении с другими изозимами протеинкиназы C или другими протеинкиназами. Благодаря своей специфичности такие соединения должны обладать большей эффективностью и меньшей токсичностью.
Микробный индолокарбазол, стауроспорин, является сильным ингибитором протеинкиназы C, который взаимодействует с ее каталитическим доменом. Tamaoki и др., Biochem. Biophys. Rec. Commun. 135: 397 - 402 (1986); Gross и др. , Biochem. Pharmacol. 40: 343 - 350 (1990). Однако, терапевтический эффект этой молекулы и близкородственных соединений ограничен отсутствием специфичности к протеинкиназе C по сравнению с другими протеинкиназами. Reugg U. T. и Burgess G.M., Trend Pharmacol. Sci. 10: 218 - 220 (1989). Это отсутствие селективности в указанном классе молекул приводит к их неприемлемой токсичности.
В последних работах был рассмотрен еще один класс соединений, родственных стауроспорину, а именно, бисиндолмалеимиды. Davis и др. FEBS Lett. 259: 61 - 63 (1989); Twoemy и др., Biochem. Biophys. Res. Commun 171: 1087 - 1092 (1990); Toullec и др., J. Biol. Chem. 266: 15771 - 15781 (1991); Davis и др., J. Med. Chem. 35: 994 - 1001 (1992); Bit и др., J. Med. Chem. 36: 21 - 29 (1993). Некоторые из этих соединений обнаруживали селективность к протеинкиназе C.
Хотя ранее были обнаружены соединения, обладающие специфичностью к протеинкиназе C, однако, мало что известно о их селективности к изозимам. Например, анализ стауроспорина на изозимную селективность показал очень низкую селективность к изозимам за исключением лишь слабого ингибирования дзета-изозима по сравнению с другими изозимами. McGlynn и др., J. Cell. Biochem. 49: 239 - 250 (1992); Ward N.E. и O'Brian C.A. Molec. Pharmacol. 41: 387 - 392 (1992). Исследование РКС-селективного соединения 3-[1-(3-диметиламинопропил)-индол-3-ил] -4-(1H-индол-3-ил)-1H-пиррол-2,5- диона обнаружило слабую селективность к кальций-зависимым изозимам. Toullec и др.., J. Biol. Chem. 266: 15771 - 15781 (1991). Последующие исследования этого соединения не обнаружили какого-либо отличия в селективности (либо существует очень слабая селективность) к альфа-изозиму по сравнению с селективностью к бета-1 и бета-2-изозимам. Martiny-Baron и др. , J. Biol. Chem. 268: 9194 - 9197 (1993); Wilkinson и др., Biochem. J. 294: 335 - 337 (1993). Поэтому, несмотря на годы, посвященные исследованиям и идентификации классов соединений, ингибирующих преимущественно протеинкиназу C, а не другие протеинкиназы, потребность в получении терапевтически эффективных изозим-селективных ингибиторов пока еще остается актуальной.
Настоящее изобретение относится к новым и сильным ингибиторам протеинкиназы C. Соединения настоящего изобретения являются селективными по отношению к протеинкиназе C, но не по отношению к другим киназам, и что, совершенно неожиданно, эти соединения обладают высокой изозим-селективностью. Как селективные ингибиторы, эти соединения могут быть использованы для лечения заболеваний, связанных с сахарным диабетом и его осложнениями, а также таких заболеваний, как ишемия, воспалительные заболевания, нарушения центральной нервной системы, сердечно-сосудистые заболевания, кожные заболевания и рак.
Настоящее изобретение относится к соединениям формулы I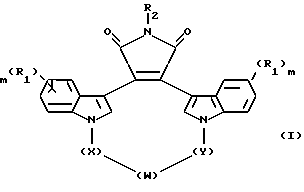
где W - -O-, -S-, -SO-, -SO2-, -CO-, C2-C6-алкилен, замещенный алкилен, C2-C6-алкенилен, -арил-, -арил(CH2)m-O-, гетероцикл, -гетероцикл-(CH2)m-O-,
конденсированный бицикл-, -конденсированный бицикл-(CH2)mO-, NR3-, -NOR3-, -CONH или -NHCO-;
X и Y - независимо - C1-C4-алкилен, замещенный алкилен, либо X, Y и W, взятые вместе, образуют -(CH2)n-AA-;
R1 независимо - водород, галоген, C1-C4-алкил, гидрокси, C2-C4-алкокси, галогеноалкил, нитро, NR4R5 или -NHCO(C1-C4-алкил);
R2 - водород, CH3CO-, NH2 или гидрокси;
R3 - водород, (CH2)m арил, C1-C4 - алкил, -COO(C1-C4-алкил), -CONR4R5, -(C=NH)NH2, -SO(C1-C4-алкил), -SO2(NR4R5) или -SO2(C1-C4-алкил);
R4 и R5 независимо - водород, C1-C4-алкил, фенил, бензил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо;
AA - аминокислотный остаток;
m независимо является 0, 1, 2 или 3; и
n независимо является 2, 3, 4 или 5.
Настоящее изобретение также относится к новым промежуточным соединениям, предназначенным для получения вышеописанных соединений. Такими промежуточными соединениями являются соединения формулы II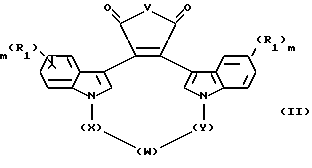
где V - -O- или N-CH3;
W - -O-, -S-, -SO-, -SO2-, -CO-, C2-C6 -алкилен, замещенный алкилен, C2-C6- алкенилен, арил, - арил(CH2)mO -, гетероцикл, - гетероцикл-(CH2)mO - конденсированный бицикл-, -конденсированный бицикл-(CH2)mO-, -NR3-, -NOR3-, -CONH- или -NHCO-;
X и Y независимо - C1-C4-алкилен, замещенный алкилен, либо X, Y и W, взятые вместе, образуют -(CH2)n-AA-;
R1 независимо - водород, галоген, C1-C4-алкил, гидрокси, C1-C4-алкокси, галогеноалкил, нитро, NR4R5 или -NHCO(C1-C4-алкил);
R3 - водород, (CH2)m-арил, C1-C4-алкил, -COO (C1-C4-алкил), -CONR4R5, -(C=NH)NH2, - SO(C1-C4-алкил), -SO2(NR4R5) или -NO2(C1-C4-алкил);
R4 и R5 независимо - водород, C1-C4 - алкил, фенил, бензил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо;
AA - аминокислотный остаток;
m независимо является 0, 1, 2 или 3;
n независимо является 2, 3, 4 или 5.
В другом своем варианте, настоящее изобретение относится к способу получения соединений формулы II, заключающемуся в том, что смесь соединения, имеющего концентрацию от около 1,5 М до 0,001 М, и формулу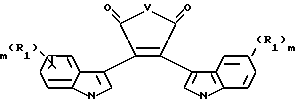
где V - O или N-CH3;
R1 независимо - водород, галогено, C1-C4-алкил, гидрокси, C1-C4-алкокси, галогеноалкил, нитро, NR4R5 или -NHCO(C1-C4-алкил);
m независимо является 0, 1, 2 или 3;
и алкилирующего агента, имеющего концентрацию от около 1,5 M до около 0,001 М, и формулу: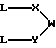
где L является уходящей группой;
W - -O-, -S-, -SO-, -SO2-, -CO-, C2-C6-алкилен, замещенный алкилен, C2-C6-алкенилен, -арил-, -арил(CH2)m-O-, -гетероцикл-, -гетероцикл-(CH2)mO-, конденсированный бицикл-, -конденсированный бицикл-(CH2)mO-, -NR3-, -NOR3-, -CONH- или -NHCO-;
X и Y независимо - C1-C4-алкилен или замещенный алкилен;
R3 - водород, (CH2)m-арил, C1-C4-алкил, -COO(C1-C4-алкил), - CONR4R5, -(C=NH)NH2, -SO(C1-C4-алкил), -SO2(NR4R5) или -SO2(C1-C4-алкил);
R4 и R5 независимо - водород, C1-C4 -алкил, фенил, бензил, либо R4 и R5, взятые вместе с азотом, с которым они связаны, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо;
m независимо является 0, 1, 2 или 3;
объединяют примерно с 0,5 - 10 эквивалентами Cs2CO3 при дозе от около 0,1 мл/ч до около 2,0 мл/ч в полярном апротонном растворителе.
Еще один способ получения соединений формулы II заключается в том, что соединение в концентрации от около 3 М до около 0,001 М, имеющее формулу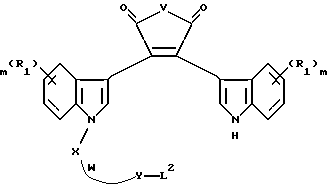
где L2 независимо является уходящей группой;
V - -O- или N-CH3;
W - -O-, -S-, -SO-, -SO2-, -CO-, C2-C6-алкилен, замещенный алкилен, C2-C6 - алкенилен, -арил-, -арил (CH2)mO-, -гетероцикл-, -гетероцикл-(CH2)mO-, конденсированный бицикл, - конденсированный бицикл-(CH2)mO-, -NR3-, -NOR3-, -CONH- или NHCO-;
X и Y независимо - C1-C4-алкилен или замещенный алкилен;
R1 независимо - водород, галоген, C1-C4-алкил, гидрокси, C1-C4-алкокси, галогеноалкил, нитро, NR4R5 или -NHCO(C1-C4-алкил);
R3 - водород, (CH2)m-арил, C1-C4- алкил, -COO(C1-C4-алкил), -CONR4R5, -(C=NH)NH2, -SO(C1-C4-алкил), -SO2(NR4R5) или -SO2(C1-C4-алкил);
R4 и R5 независимо - водород, C1-C4-алкил, фенил, бензил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо;
m независимо является 0, 1, 2 или 3;
объединяют приблизительно с 0,5-10 эквивалентами Cs2CO3 при дозе от около 0,1 мл/ч до около 2,0 мл/ч в полярном апротонном растворителе.
В другом своем варианте, настоящее изобретение относится к способу ингибирования протеинкиназы C, заключающемуся в том, что млекопитающему, нуждающемуся в таком лечении, вводят фармацевтически эффективное количество соединения формулы I. Настоящее изобретение также относится к способу селективного ингибирования бета-1 и бета-2-изозимов протеинкиназы C, заключающемуся в том, что млекопитающему, нуждающемуся в таком ингибировании, вводят фармацевтически эффективное количество соединения формулы I.
Кроме того, настоящее изобретение относится к способам лечения состояний, о которых известно, что определенную роль в их патологии играет протеинкиназа C, например, таких состояний, как ишемия, воспалительные реакции, нарушения центральной нервной системы, сердечно-сосудистые заболевания, кожные заболевания и рак; причем, указанные способы заключаются в том, что млекопитающему, нуждающемуся в таком лечении, вводят фармацевтически эффективное количество соединения формулы I.
Настоящее изобретение особенно предпочтительно использовать для лечения осложнений, связанных с диабетом. Поэтому, настоящее изобретение также относится к способу лечения сахарного диабета, заключающемуся в том, что млекопитающему, нуждающемуся в таком лечении, вводят фармацевтически эффективное количество соединения формулы I.
И наконец, в еще одном своем варианте, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями.
Как указывалось выше, настоящее изобретение относится к соединениям формулы I, которые способны к селективному ингибированию протеинкиназы C. Предпочтительными соединениями настоящего изобретения являются соединения формулы I, где части -X-W-Y- содержат 4-8 атомов, которые могут быть замещенными или незамещенными. Наиболее предпочтительно, если части -X-W-Y- содержат 6 атомов.
Другими предпочтительными соединениями настоящего изобретения являются соединения формулы I, где R1 и R2 - водород; а W - замещенный алкилен, -O-, -S-, -CONH-, -NHCO- или -NR3-.
Особенно предпочтительными соединениями являются соединения формулы Ia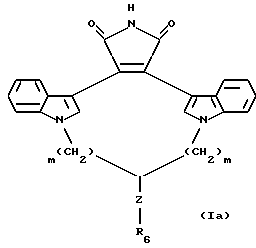
где Z - -(CH2)p- или -(CH2)p-O-(CH2)p-;
R6 - гидрокси, -SH, C1-C4-алкил, (CH2)m-арил, -NH(арил) или -NR4R5;
R4 - водород или C1-C4-алкил;
R5 - водород или C1-C4-алкил или бензил;
p = 0, 1 или 2;
m = 2 или 3, независимо.
Наиболее предпочтительными соединениями формулы Ia являются соединения, в которых Z - CH2, R6 - -NH2 или N(CH3)2.
Другими предпочтительными соединениями являются соединения, в которых W - -O-; Y - замещенный алкилен; а X - алкилен. Эти соединения могут быть представлены формулой I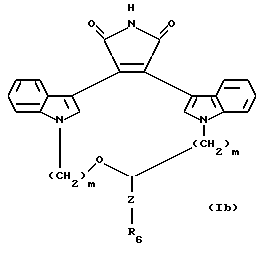
где Z - -(CH2)p-;
R6 - NR4R5;
R4 и R5 независимо - H или C1-C4-алкил;
p = 0, 1 или 2;
m = 2 или 3, независимо.
Наиболее предпочтительными соединениями формулы Ib являются соединения, в которых p = 1, а R4 и R5 - метил.
Термин "галогено" означает фтор, хлор, бром и йод.
Термин "C1-C4-алкил" означает алкильную группу с циклической, прямой или разветвленной цепью, имеющую от 1 до 4 атомов углерода, например, такую, как метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, т-бутил и т.п.
Термин "галогеноалкил" означает одну из указанных алкильных групп, замещенную одним или несколькими атомами галогена, предпочтительно 1 - 3 атомами. Примером галогеноалкила является трифторметил.
"C1-C4-алкокси" означает C1-C4-алкильную группу, ковалентно связанную посредством -O-связи.
Термин "C1-C4-алкилен" означает прямую и содержащую 1 - 4 атомов углерода алкиленовую часть формулы -(CH2)r, где r = 1 - 4. Примерами C1-C4-алкилена являются метилен, этилен, триметилен, метилэтилен, тетраметилен и т.п.
Аналогично, термин "C2-C6-алкилен" означает алкиленовую часть, содержащую 2 - 6 атомов углерода. Предпочтительно, если C2-C6-алкилен является алкиленом, имеющим 2 - 4 атома углерода.
Термин "C2-C6-алкенилен" означает прямой или разветвленный углеводород, имеющий 2 - 6 атомов углерода и одну или несколько двойных связей, предпочтительно одну или две двойных связей. Примерами C2-C6-алкенилена являются этенилен, пропенилен, 1,3-бутадиенил и 1,3,5-гексатриенил.
Термин "арил" означает замещенный или незамещенный фенил или нафтил. Арил может быть необязательно замещенным одной или двумя группами, независимо выбранными из гидрокси, карбокси, C1-C4-алкокси, C1-C4-алкила, галогеноалкила, нитро, - NR4R5, -NHCO (C1-C4-алкила), -NHCO(бензила), -NHCO (фенила), SH, S(C1-C4-алкила), -SO2(NR4R5), -SO2(C1-C4-алкила), -SO2 (фенила) или галогена.
Термин "(CH2)m-арил" предпочтительно является бензилом или фенилом.
Термин "замещенный алкилен" означает часть формулы: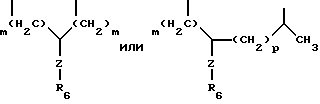
где Z - -(CH2)p или -(CH2)p-O-(CH2)p-;
R6 - C1-C4-алкил, C1-C4-алкокси, (CH2)m-арил, (CH2)m-арилокси, гидрокси, карбокси, -COO- (C1-C4-алкил), -COO(CH2)m-арил, -CO(C1-C4-алкил), -NR4R5,
-N(R4R5)(OR5), -NH(CH2)m-арил, -NH(CH2)m-пиридил, -CONH(CH2)m-арил,
-CONH(C1-C4-алкил), -NHCO- (C1-C4-алкил), -NHCO(CH2)m-арил, -OCONH (C1-C4-алкил), -OCONH(CH2)m-арил, -NHCOO(алкил), -NHCOO(бензил), -NHSO2(C1-C4-алкил), -NHSO2(CH2)m-арил, -CN, -SH, -S(C1-C4-алкил), S(арил), -SO2(NR4R5), -SO2 (C1-C4-алкил), -SO(C1-C4-алкил), гликозил или гетероцикл; R4 и R5 независимо - C1-C4-алкил, фенил, бензил, либо R4 и R5, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо;
p независимо равно 0, 1 или 2;
m независимо равно 0, 1, 2 или 3.
Предпочтительно, если Z - -CH2-, а R6 - C1-C4-алкил, арил или -NR4R5.
Термин "гетероцикл" означает стабильное замещенное или незамещенное, насыщенное или ненасыщенное 5- или 6-членное кольцо, имеющее 1 - 4 гетероатома, которые могут быть одинаковыми или различными и представляют собой атомы серы, кислорода и азота; а в случае, если гетероцикл содержит два смежных атома углерода, то соседние атомы углерода могут быть структурированы с образованием группы формулы -CH=CH-; при условии, что (1) если гетероциклическое кольцо имеет 5 членов, то гетероатомы могут содержать более чем два атома серы или два атома кислорода, но не одновременно; и (2) если гетероциклическое кольцо имеет 6 членов и является ароматическим, то атомы серы и кислорода отсутствуют. Гетероцикл может быть присоединен у любого атома углерода или азота, который делает структуру стабильной. Гетероцикл может быть замещен одной или двумя группами, независимо выбранными из C1-C4-алкила, C1-C4-алкокси, гидрокси, ацетила, карбокси, галогеноалкила, нитро, -NR4R5, -NHCO (C1-C4-алкила), -NHCO(бензила), -NHCO(фенила), SH, S (C1-C4-алкила), -OCO(C1-C4-алкила), -SO2(NR4R5), -SO2 (C1-C4-алкила),
-SO2(фенила) или галогена. Примерами гетероцикла могут служить пиразол, пиразолин, имидазол, ацетилимидазол, изоксазол, триазол, тетразол, оксазол, 1,3-диоксолон, тиазол, оксадиазол, тиадиазол, пиридин, дипиридил, пиримидин, пиперазин, морфолин, пиразин, пирролидин, пиперидин, пиперазин, оксазолидинон, имидазолидинон и аминопиридин.
Термин "гликозил" означает сахар с 5 или 6 атомами углерода, выбранный из группы, включающей в себя аллозил, альтрозил, глюкозил, минозил, гулозил, галактозил талозил, арабинозил, ксилозил, ликсозил, рамнозил, рибозил, дезоксифуранозил, дезоксипиранозил и дезоксирибозил. Гликоза может быть замещенной, O-ацетилированной, O-метилированной, аминозамещенной, монозамещенной, ди-алкиламино-замещенной или ациламинозамещенной.
Термин "конденсированный бицикл" означает стабильную конденсированную бициклическую кольцевую систему формулы: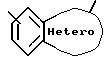
где Hetero - замещенное или незамещенное, насыщенное или ненасыщенное 5- или 6-членное кольцо, имеющее 1- 3 гетероатома, которые могут быть одинаковыми или различными и представляют собой атомы серы, кислорода и азота; а в случае, если Hetero содержит два смежных атома углерода, то соседние атомы углерода могут быть структурированы с образованием группы формулы -CH=CH-; при условии, что (1) если Hetero-кольцо имеет 5 членов, то гетероатомы могут включать в свое число не более чем два атома серы или два атома кислорода, но не одновременно; и (2) если Hetero-кольцо имеет 6 членов и является ароматическим, то атомы серы и кислорода отсутствуют. Конденсированный бицикл может быть присоединен у любого атома углерода или азота, обеспечивающего стабильность структуры. Конденсированный бицикл может быть замещен одной или двумя группами, независимо выбранными из следующих групп: C1-C4-алкил, C1-C4-алкокси, гидрокси, карбокси, галогеноалкил, нитро, -NR4R5, -NHCO(C1-C4-алкил), -NHCO(бензил), -NHCO-(фенил), SH, S(C1-C4-алкил), -OCO (C1-C4-алкил), -SO2- (NR4R5), -SO2(C1-C4-алкил), -SO2(фенил) или галоген.
Примерами конденсированного бицикла являются индол, имидазо(1,2-a)пиридин, бензотриазол, бензимидазол, бензотриазол, бензоксазол, бензоксатиазол, хинолин, изохинолин, фталазин, хиназолин, хиназолинон, хиноксалин и аминоизохинолин.
Термин "аминокислотный остаток" относится к части, имеющей формулу: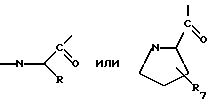
где R - вариабельная боковая цепь аминокислоты;
R7 - водород или гидрокси.
Вариабельная боковая цепь аминокислоты является атомом или группой, связанной с атомом α- углерода, который также является связанным с карбоксильной или аминогруппой. Примерами вариабельной области природных аминокислот являются следующие структуры: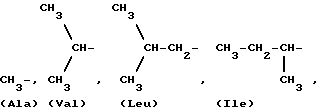
CH3-S-CH2-CH2-,
(Met)
H-,
(Gly)
HO-CH2,
(Ser)
H2N-CH2-CH2-CH2-CH2-
(Lys)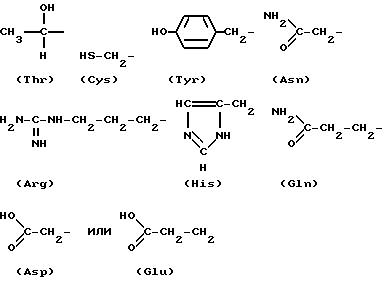
Помимо природных аминокислот, термин "аминокислотный остаток" также включает в себя изомеры положения и варианты. Примерами изомеров положения и вариантов могут служить следующие аминокислотные остатки: 2-аминоадипиновая кислота (Aad), 3-аминоадипиновая кислота (bAad), β-аланин (bAla), 2-аминомасляная кислота (Abu), 4-аминомасляная кислота (4Abu), 6-аминокапроновая кислота (Acp), 2-аминогептановая кислота (Ahe), 2-аминоизомасляная кислота (Aib), 3-аминоизомасляная кислота (bAib), 2-аминопимелиновая кислота (Apm), 2,4-диаминомасляная кислота (Dbu), десмозин (Des), 2,2'-диаминопимелиновая кислота (Dpm), 2,3-диаминопропионовая кислота (Dpr), N-этилглицин (EtGly), N-этиласпарагин (EtAsn), гидроксизин (Hyl), аллогидроксилизин (aHyl), 3-гидроксипролин (3Hyp), 4-гидроксипролин (4Hyp), изодесмозин (Ide), алло-изолейцин (alle), нафтилглицин, N-метилглицин (MeGly), N-метилизолейцин (Melle), N-метиллизин (MeLys), норвалин (Nva), норлейцин (Nle), орнитин (Orn), фенилглицин, цианоаланин (CA), γ-карбоксиглутамат, O-фосфосерин, α-нафтилаланин (NA), β-нафтилаланин (bNA), S-галактозил, цистеин, глицинамид, N-формилметионин, тирозин-O-сульфат и т.п. Эти аминокислотные остатки могут присутствовать либо в D-, либо в L-конфигурации. Если это не указано особо, упоминаемые аминокислоты имеют L-конфигурацию.
Термин "уходящая группа", используемый в настоящем описании, имеет свое обычное значение и хорошо известен специалистам. В основном, уходящей группой может быть любая группа или атом, который способствует повышению электрофильности атома, с которым он связан для замещения. Предпочтительными уходящими группами являются трифталат, мезилат, тозилат, имидат, хлорид, бромид и иодид. Если алкилирующий агент содержит аминокислотный остаток (т.е., X, W, и Y, взятые вместе, образуют -(CH2)n-AA-), то уходящей группой, связанной с карбоксигруппой, является предпочтительно, пентафторфениловый эфир или паранитрофениловый эфир.
Термин "карбоксизащитная группа", используемый в настоящем описании, означает одно из сложноэфирных производных карбоновокислотной группы, обычно используемых для блокирования или защиты карбоновокислотной группы, в том случае, когда реакцию осуществляют на других функциональных группах данного соединения. Конкретно выбранный тип карбоксизащитной группы не имеет решающего значения, поскольку дериватизированная карбоновая кислота является стабильной в условиях последующей реакции и может быть удалена в любой подходящей стадии этой реакции, не оказывая неблагоприятного воздействия на остальную часть молекулы. T.W.Greene и P.Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, N.Y., 1991, гл. 5 (где приводится список обычно используемых защитных групп). См. также, E.Haslam, Protective Groups in Organic Chemistry, J. G.W.McOmie, Plenum Press, New York, N.Y. 1973.
Термин "защищенная крбоксигруппа" соответствует термину "карбоксизащитная группа".
Термин "гидроксизамещенная группа", используемый в настоящем описании, относится к одному из эфирных или сложноэфирных производных гидроксигруппы, обычно используемых для блокирования или защиты в том случае, когда реакцию осуществляют на других функциональных группах данного соединения. Конкретно выбранный тип карбоксизащитной группы не имеет решающего значения, поскольку дериватизированная гидроксигруппа является стабильной в условиях последующей реакции и может быть удалена в любой подходящей стадии этой реакции, не оказывая неблагоприятного воздействия на остальную часть молекулы. T.W.Green и P.Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, N. Y., 1991 (где приводится список обычно используемых защитных групп). Предпочтительными гидроксизащитными группами являются трет-бутилдифенилсилилокси (TBDPS), трет-бутилдиметилсилилокси (TBDMS), трифенилметил (тритил), метокситритил или алкиловый или ариловый сложный эфир.
Термин "защищенная гидроксигруппа" соответствует термину "гидроксизащитная группа".
Термин "аминозащитная группа", используемый в настоящем описании, относится к заместителям аминогруппы, обычно используемым для блокирования или защиты функциональных аминогрупп в том случае, когда реакцию осуществляют на других функциональных группах данного соединения. Конкретно выбранный тип аминозащитной группы не имеет решающего значения, поскольку дериватизированная аминогруппа является стабильной в условиях последующей реакции и может быть удалена в любой подходящей стадии этой реакции, не оказывая неблагоприятного воздействия на остальную часть молекулы. T.W.Green и P.Wuts, Protective Groups in Organic Synthesis, гл. 7 (где приводится список обычно используемых защитных групп). См. также J.W.Barton, Protective Groups in Organic Chemistry, гл. 2. Предпочтительными аминозащитными группами являются т-бутоксикарбонил, фталимид, циклический алкил и бензилоксикарбонил.
Термин "защищенная амино" означает аминогруппу, замещенную аминозащитной группой, определенной выше.
Термин "-NH-защитная группа", используемый в настоящем описании, относится к подклассу аминозащитных групп, обычно используемых для блокирования или защиты -NH-функциональной группы, в том случае, когда реакцию осуществляют на других функциональных группах данного соединения. Конкретно выбранный тип защитной группы не имеет решающего значения, поскольку дериватизированная аминогруппа является стабильной в условиях последующей реакции и может быть удалена в любой подходящей стадии этой реакции, не оказывая неблагоприятного воздействия на остальную часть молекулы. T.W.Greene и P.Wuts, Proptective Groups in Organic Synthesis, гл. 7 (где приводится список обычно используемых защитных групп). Предпочтительными -NH-защитными группами является карбамат, амид, алкил-, или арилсульфонамид.
Термин "защищенная-NH" означает группу, замещенную -NH-защитной группой, определенной выше.
Термин "фармацевтически эффективное количество", используемый в настоящем описании, означает количество соединения настоящего изобретения, способное ингибировать РКС-активность у млекопитающих. Конкретная доза соединения, вводимого в соответствии с настоящим изобретением, может быть определена в каждом конкретном случае и зависит от вводимого соединения, способа введения, конкретного заболевания и т.п. Указанные соединения могут быть введены различными способами, например, путем перорального, ректального, чрескожного, подкожного, локального, внутривенного, внутримышечного или интраназального введения. В основном, обычная суточная доза может содержать от около 0,01 мг/кг до около 20 мг/кг активного соединения настоящего изобретения. Предпочтительная дневная доза может составлять от около 0,05 до около 10 мг/кг, а наиболее предпочтительно от около 0,1 до около 5 мг/кг. Однако, для наружного применения, типичная доза составляет от около 1 до около 500 мкг соединения на см2 пораженной ткани. Предпочтительно, количество локально наносимого соединения составляет от около 30 до около 300 мкг/см2, более предпочтительно от около 50 до около 200 мкг/см2 и наиболее предпочтительно, от около 60 до около 100 мкг/см2.
Термин "лечение", используемый в настоящем описании, означает помощь пациенту в целях устранения заболевания, состояния или нарушения путем введения соединения настоящего изобретения для предупреждения возникновения симптомов или осложнений, ослабления симптомов или осложнений или устранения заболевания, состояния или расстройства.
Термин "изозим-селективный" относится к предпочтительному ингибированию бета-1 или бета-2-изозимов протеинкиназы C по сравнению с другими изозимами протеинкиназы C (т.е. альфа, гамма, дельта, эпсилон, дзета и эта). В основном, как было определено в РКС-анализе, доза соединения настоящего изобретения, необходимая для ингибирования бета-1- или бета-2-изозима РКС, как минимум, в 8 раз ниже (а предпочтительно в 10 раз) дозы, требующейся для равного ингибирования альфа-изозима протеинкиназы C. Указанные соединения обнаруживают также различия в диапазонах ингибирования при IC50, т.е., 50%-ингибирования. Так, например, изозим-селективные соединения ингибируют бета-1- и бета-2-изозимы протеинкиназы C при гораздо меньших концентрациях с гораздо меньшей токсичностью, чем при их минимальном ингибировании других изозимов РКС.
Благодаря своим кислотным группам, соединения формулы I могут существовать в виде их фармацевтически приемлемых основных аддитивных солей. Эти соли могут быть получены с использованием неорганических оснований, таких как гидроксиды, карбонаты, бикарбонаты аммония, щелочных металлов и щелочноземельных металлов и т.п., а также с использованием основных органических аминов, таких как алифатические и ароматические амины, алифатические диамины, гидроксиалкамины и т.п. Примерами оснований, которые могут быть использованы для получения вышеуказанных солей, являются гидроксид аммония, карбонат калия, бикарбонат натрия, гидроксид кальция, метиламин, диэтиламин, этилендиамин, циклогексиламин, этаноламин и т.п.
Благодаря своим основным группам, соединения формулы I могут также существовать в виде фармацевтически приемлемых кислых аддитивных солей. Кислотами, обычно используемыми для получения таких солей, являются хлористоводородная, бромистоводородная, иодистоводородная, серная и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфоновая, метансульфоновая, щавелевая, парабромофенилсульфоновая, угольная, янтарная, лимонная, бензойная, уксусная и родственные органические и неорганические кислоты. Указанными фармацевтически приемлемыми солями могут быть сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, монобифосфат, дибифосфат, метафосфат, пирофосфат, хлорид, иодид, ацетат, пропионат, деканоат, каприлат, формат, изобутират, гептаноат, оксалат, малонат, сукцинат, суберат, себацат, фумарат, малеат, 2-бутин-1,4-диоат, 3-гексин-2,5-диоат, бензоат, хлорбензоат, гидроксибензоат, метоксибензоат, фталат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гиппурат, β-гидроксибутират, гликолат, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и др. соли.
Помимо фармацевтически приемлемых солей, настоящее изобретение относится и к другим солям. Эти соли могут служить в качестве промежуточных соединений для очистки целевых соединений, для получения других солей или для идентификации и характеризации соединений или промежуточных соединений.
Фармацевтически приемлемые соли соединений формулы I могут также существовать в виде различных сольватов, например, с водой, метанолом, этанолом, диметилформамидом, этилацетатом и т.п. Кроме того, могут быть получены смеси таких сольватов. Источником образования такого сольвата может служить растворитель кристаллизации, специально используемый в таких целях, либо любой другой обычный растворитель. Указанные сольваты входят в объем настоящего изобретения.
Следует отметить, что соединение формулы I может существовать в виде различных стереоизомерных форм, например, W может содержать хиральный атом углерода в замещенной алкиленовой части. В основном, указанные соединения получают в виде рацематов, которые могут быть использованы как таковые, но, если необходимо, могут быть получены и отдельные энантиомеры путем их разделения или синтеза с использованием традиционной техники. Указанные рацематы, отдельные энантиомеры, а также их смеси являются частью настоящего изобретения.
Настоящее изобретение также относится к фармацевтически приемлемым предшественникам соединений формулы I. Такие предшественники или пролекарства представляют собой лекарственные средства, которые были химически модифицированы и могут быть биологически неактивными в области их действия, но которые могут разлагаться или модифицироваться под действием одного или нескольких ферментов, или под действием других in vivo-процессов с образованием родительской биоактивной формы. Указанное пролекарство обычно имеет другой фармакокинетический профиль, чем его родительская форма, что способствует его лучшей абсорбции эпителием слизистой оболочки, лучшему солеобразованию или растворимости, и/или повышению системной стабильности (например, повышению времени полужизни в плазме крови). Указанными химическими модификациями являются обычно следующие формы:
1) сложноэфирные или амидные производные, которые могут расщепляться эстеразами или липазами;
2) пептиды, которые могут распознаваться специфическими или неспецифическими протеазами; или
3) производные, которые аккумулируются в областях их действия посредством мембранной селекции пролекарственной формы или модифицированной пролекарственной формы;
либо любые комбинации из вышеуказанных форм (1) - (3). Стандартные процедуры, используемые для селекции и получения соответствующих пролекарственных производных, описаны, например, в работе H.Bundgaard, Design of Prodrugs, (1985).
Синтез некоторых бис-индол-N-малеимидных производных описан Davis и др. в патенте США N 5057614, который вводится в настоящее описание посредством ссылки. В основном, соединения настоящего изобретения могут быть получены следующим образом:
Схема 1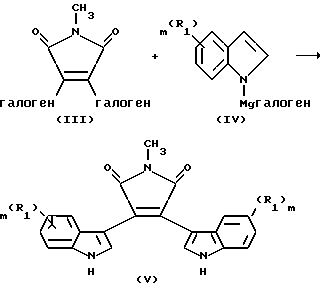
R1, m, "галоген" являются такими, как они были определены выше. "Галогено" являются предпочтительно хлоро-, бромо- или иодо-. Соединение формулы III, предпочтительно, представляет собой 2,3-дихлоро-N-метилмалеимид.
Реакция между соединением III и индолом (Соединение IV) известна как реакция Гриньяра. Эту реакцию осуществляют в инертном органическом растворителе, таком как толуол, при температуре в диапазоне от комнатной температуры до температуры перегонки реакционной смеси. Важно отметить, что реакция, проиллюстрированная на схеме 1, зависит от выбранного растворителя. При использовании системы растворителей: толуол: ТГФ: простой эфир, соединение V получают с выходом более чем 80% и с чистотой более чем 95%. Полученный продукт осаждают из реакционной смеси с помощью хлорида аммония (H4Cl). Полученное промежуточное соединение V может быть выделено стандартными методами.
После этого соединение V (бис-3,4-(3'-индолил)-1N-метилпиррол-2,5-дион) может быть, путем щелочного гидролиза, превращено в соответствующий ангидрид формулы VI с использованием стандартной техники, хорошо известной специалистам и описанной Breenner и др., Tetrahedron 44: 2887 - 2892 (1988). Затем, предпочтительно, соединение V подвергают реакции с 5 н. KOH в этаноле при температуре в диапазоне от 25oC до температуры перегонки.
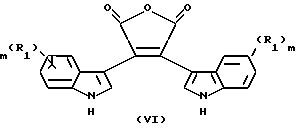
Соединения формулы V являются обычно более стабильными, чем соединения формулы VI. Поэтому, предпочтительно, чтобы реакция соединения V протекала в соответствии с нижеприведенной схемой 2. В результате этой реакции образуется соединение формулы I. Однако, любому специалисту известно, что соединение формулы VI может быть также подвергнуто реакции в соответствии со схемой 2.
Схема 2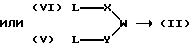
где X, Y и W являются такими, как они были определены выше. L является хорошей уходящей группой, такой, как хлоро, бромо, иодо, мезил, тозил и т.п. L может быть также гидроксигруппой или другим предшественником, который может быть легко превращен в хорошую уходящую группу стандартными методами. Например, гидроксигруппа может быть легко превращена в сульфоновую сложноэфирную группу, такую как мезил, посредством реакции гидрокси с метансульфонилхлоридом с образованием мезилатной уходящей группы.
Реакция, показанная на схеме 2, может быть осуществлена любыми традиционными методами, которые обычно используются для получения N-замещенных индолов. В этой реакции обычно используются примерно эквимолярные количества двух реагентов, хотя возможны и другие соотношения, например, такие, где алкилирующий реагент используется в избыточном количестве. Предпочтительно, если данная реакция протекает в полярном апротонном растворителе в присутствии соли щелочного металла, или в других алкилирующих условиях, хорошо известных специалистам. Если уходящей группой является бромо или хлоро, то для ускорения реакции может быть добавлено каталитическое количество иодидной соли, такой как иодид калия. Реакционные условия представляют собой: гексаметилдисилазид калия в диметилформамиде или в тетрагидрофуране, гидрид натрия в диметилформамиде.
Предпочтительно, если реакцию осуществляют в условиях медленного обратного добавления карбоната цезия в ацетонитриле, диметилформамиде (ДМФ) или в тетрагидрофуране (ТГФ). В основном, реакция протекает при температуре в диапазоне примерно от комнатной температуры до температуры перегонки реакционной смеси.
Следует отметить, что реакция, описанная в схеме 2, может быть осуществлена с использованием соединений формулы VIIa:
L - Y'
L - X' (VIIa)
где X' и Y' являются защищенной карбокси-группой, защищенной гидрокси-группой или защищенным амином. После алкилирования, проведенного по схеме 2, X' и Y' могут быть превращены в части, способные к присоединению с образованием W. Этот способ является предпочтительным способом для получения соединений формулы I, где W представляет собой -S-, -O- или NR3. Реакция взаимодействия X' и Y' с образованием различных эфирных, тиоэфирных или аминоэфирных производных является известной и описана, например, Ito и др., Chem. Pharm. Bull. 41(6): 1066 - 1073 (1993); Kato и др., J.Chem. Pharm. Bull. 34: 486 (1986); Goodrow и др., Synthesis 1981: 457; Harpp и др., J. Am. Chem. Soc. 93: 2437 (1971); и Evans и др., J. Org. Chem. 50: 1830 (1985).
Следует также отметить, что соединения формулы V могут быть также превращены в соединения формулы II посредством двухстадийного синтеза, проиллюстрированного на схеме 3.
Схема 3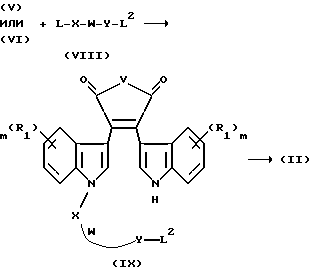
R1, X, W, Y, V и L являются такими, как они были определены выше. L2 является защищенной гидркоси-группой или другой группой, которая может быть легко превращена в хорошую уходящую группу стандартными способами. Взаимодействие между соединением V или VI и соединением VII является реакцией алкилирования, описанной выше. Моноалкилированное промежуточное соединение IX подвергается разблокированию, а L2 превращается в уходящую группу. Например, если гидрокси-группа защищена т-бутилдиметилсилилом (TBDMS), то TBDMS может быть селективно удален с использованием подкисленного метанола. Затем, полученную свободную гидроксигруппу превращают в уходящую группу, такую как алкилгалогенид, а предпочтительно, алкилиодид или бромид (CBr4 в трифенилфосфине) или сульфонат (мезилхлорид и триэтиламине). Затем образуют макролид (соединение с макролитическим лактонным кольцом) с помощью алкилирования путем медленного обратного добавления основания к раствору, такого, как гексаметилдисилазид калия или гидрид натрия, но предпочтительно Cs2CO3, в полярном апротонном растворителе, таком как ацетонитрил, ДМФ, ТГФ, при температуре в диапазоне от комнатной температуры до температуры перегонки.
Схемы 2 и 3 иллюстрируют способ настоящего изобретения. Наиболее неожиданным является тот факт, что соединения формулы II могут быть получены со значительно более высоким выходом при условии медленного обратного добавления к Cs2CO3 в полярном апротонном растворителе. Под медленным обратным добавлением подразумевается объединение смеси соединения и алкилирующего агента (схема 2) или соединения (схема 3) с основанием со скоростью от около 0,1 мл/ч до около 2,0 мг/ч. Концентрация каждого реагента в смеси составляла от около 1,5 М до около 0,001 М. Если в реакции использовали моноалкилированное соединение (схема 3), то его концентрация составляла от около 3 М до около 0,001 М. Медленное добавление приводило к концентрации реагентов в реакционном сосуде от около 0,01 мкМ до 1,5 М. При этом, следует отметить, что может быть использована более высокая скорость добавления при более низкой концентрации реагентов. Аналогично, при более низкой скорости добавления может быть использована более высокая концентрация реагентов. Предпочтительно, если соединение добавляют со скоростью около 0,14 мл/ч, при концентрации соединения и алкилирующего агента 0,37 М. Предпочтительно также, если Cs2CO3 добавляют в избыточном количестве, а более предпочтительно, если отношение количества Cs2CO3 к количеству алкилирующего агента составляет 4 : 1. Предпочтительными полярными апротонными растворителями являются ацетонитрил, диметилформамид (ДМФ), ацетон, диметилсульфоксид (ДМСО), диоксан, метиловый эфир диэтиленгликоля (диглим), тетрагидрофуран (ТГФ) или другие полярные апротонные растворители, в которых данные реагенты являются растворимыми. Реакцию осуществляли при температуре от 0oC до температуры перегонки. Следует отметить, что соотношение соединения и алкилирующего агента в смеси не является решающим параметром. Однако, предпочтительно, чтобы указанные реагенты были смешаны друг с другом в соотношении 0,5:3. Наиболее предпочтительно, чтобы это соотношение составляло 1 : 1.
Если V является N-CH3, то соединение II может быть превращено в соответствующий ангидрид (V является O) с помощью щелочного гидролиза. Щелочной гидролиз представляет собой реакцию соединения с основанием, например гидроксидом натрия или калия, в C1-C4-спирте (предпочтительно в этаноле), либо в смеси ДМСО/вода, диоксан/вода или ацетонитрил/вода, при температуре в диапазоне от около 25oC и предпочтительно примерно до температуры перегонки. Концентрация реагентов не имеет решающего значения.
Ангидрид (V является O) превращают в малеимид формулы I путем аммонолиза. Аммонолиз представляет собой реакцию ангидрида с избыточным количеством гексаметилдисилазана или аммониевой соли (ацетат, бромид или хлорид аммония) и C1-C4-спирта (предпочтительно метанола) в полярном апротонном растворителе, таком как ДМФ, при комнатной температуре. В этой реакции, предпочтительно, чтобы отношение количества гексаметилдисилизана или аммониевой соли к количеству ангидрида составляло более чем 5 : 1.
Еще один способ получения соединений формулы I проиллюстрирован на схеме 4. Этот способ может быть, в частности, использован, если W является -NH, а X или Y является замещенным алкиленом.
Схема 4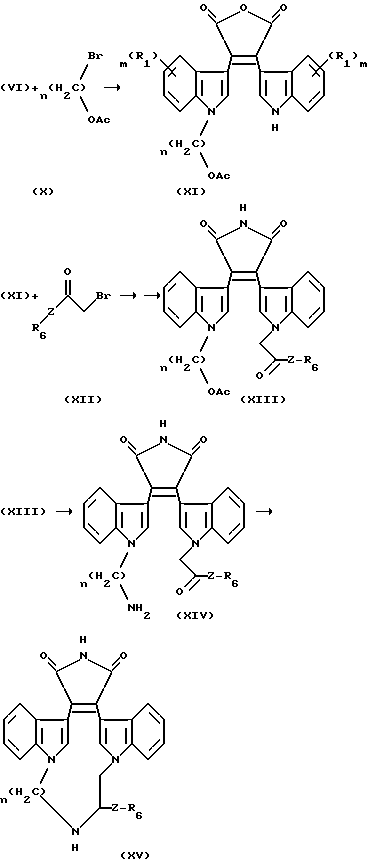
Ac является ацетилом, а R1, R6, Z, n и m являются такими, как они были определены выше. Реакцию алкилирования соединения VI с X проводили в условиях, описанных выше и известных специалистам. Аналогично, реакцию алкилирования соединения XI с α-галогенокетоном (соединение XII) осуществляют в условиях, обсуждаемых выше. Реакцию превращения ангидрида в малеимид (соединение XV) осуществляют как описано ранее. Например, ангидрид может быть превращен в бис-индолмалеимид с помощью реакции ангидрида с гексаметилдисилизаном и метанолом в инертном органическом растворителе, таком как ДМФ, при комнатной температуре.
Защищенная гидрокси-группа, представленная OAc, может быть легко гидролизована с образованием спирта (например, K2CO3 в водном метаноле и ТГФ). Полученный спирт превращают в уходящую группу известными методами, например, путем реакции спирта с мезилхлоридом в триэтиламине при 0oC. Уходящую группу замещают азидом, таким как NaN3 в ДМФ при 50oC. Полученный азид восстанавливают с использованием катализатора Lindlar в присутствии H2, в результате чего получали амин. Макроцикл замыкали посредством внутримолекулярного основания Шиффа. Основание Шиффа восстанавливали в стандартных условиях, например, с использованием NaCNBH3 или другого восстановительного агента, в результате чего получали макроциклы формулы I.
На нижеприведенной схеме 5 проиллюстрирован еще один способ получения соединений формулы I. Этот способ может быть, в частности, использован в том случае, если X, W и Y, взятые вместе, образуют -(CH2)n-AA-.
Схема 5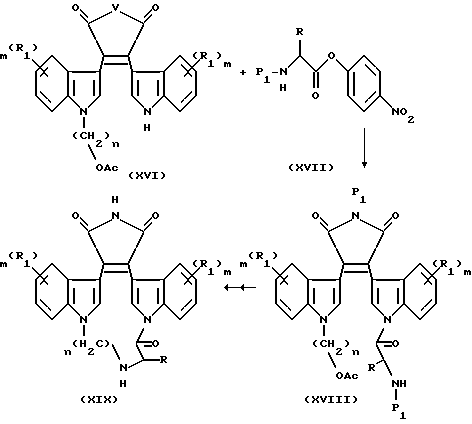
R1, Ac, V, m и n являются такими, как они были определены выше. R1 представляет собой амино-защитную группу, R представляет собой вариабельную боковую цепь аминокислоты. Реакцию ацилирования соединения XVI с активированной аминокислотой (такой, как пара-нитрофениловый сложный эфир; показано) осуществляли с использованием 18-краун-6- и KF в ТГФ, ДМФ или диметоксиэтане при комнатной температуре, как описано Klaunser и др., J.Chem. Soc. PERKIN I 607 - 631 (1977); и Nakagawa и др., J.Am. Chem. Soc. 105: 3709 - 3710 (1983). Замыкание макроцикла с образованием соединения XIX осуществляли посредством образования внутримолекулярного основания Шиффа, как описано на схеме 4.
На схеме 6, представленной ниже, показан другой метод получения соединений формулы I, который предпочтительно использовать в том случае, если W является -CONH- или -NHCO-.
Схема 6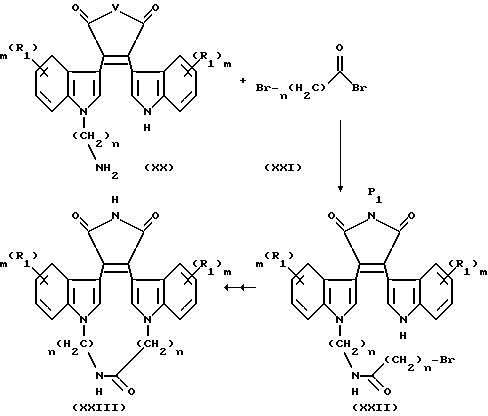
R1, Ac, V, P1, m и n являются такими, как они были определены ранее. Реакция между соединением XX и соединением XXI протекает в присутствии этилдиизопропиламина в метиленхлориде при 0oC. Макоцикл образуется в результате внутримолекулярного алкилирования свободного индолового азота и α- галогенкарбонильного конца в условиях алкилирования, описанных выше. Защищенный малеимид разблокировали, как обсуждалось ранее, в результате чего получали соединение XXIII.
Альтернативный метод получения промежуточных соединений XI и XX проиллюстрирован на схеме 7.
Схема 7.
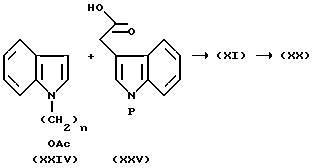
Ac является таким, как он был определен выше; P представляет собой индолзащитную группу, такую, как т-бутоксикарбонил, либо какую-нибудь другую индолзащитную группу, известную специалистам. T.W.Greene и P.Wuts. Protecting Groups in Organic Synthesis, гл. 7, стр. 385. Реакция, проиллюстрированная на схеме 7, известна как конденсация Перкина. Эта реакция описана Hill и др. , J.Med. Chem. 36: 21 - 29 (1993). К безводному раствору соединения XXIV в инертном органическом растворителе, таком как галогенированный углеводород, например, метиленхлорид, добавляли оксалилхлорид при температуре в диапазоне от -78oC до температуры перегонки (предпочтительно, при 0oC). Примерно, через 1 - 3 ч летучие вещества удаляли. Полученный твердый материал растворяли в безводном галогенированном алифатическом углеводородном растворителе, например, метиленхлориде, и добавляли к соединению XXV в присутствии акцептора кислоты, предпочтительно, третичного амина, такого, как триэтиламин, при комнатной температуре.
Полученный ангидрид, соединение XI, подвергали реакции в соответствии со схемами 4 или 5, либо превращали в малеимид или защищенный малеимид, как описано выше.
Защищенная гидрокси-группа (предпочтительно OAc, как показано) соединения XI может быть превращена в спирт с помощью известной техники, например, соединение XI подвергают реакции с NH4OH или водным аммиаком в ДМФ при повышенных температурах, например, 140oC. Полученный спирт превращают в амин (соединение XX) известными способами. Например, спирт в дихлорэтане и коллидине в атмосфере азота подвергают реакции с ангидридом трифторуксусной кислоты в дихлорметане. Приблизительно через два часа, смесь обрабатывают водным аммиаком. Полученный амин (соединение XX) затем подвергали реакции в соответствии со схемой 6.
Промежуточные соединения настоящего изобретения могут быть получены в соответствии с нижеприведенной схемой 8. Эту схему предпочтительно использовать для получения соединений, где W является -O-, Y - замещенным алкиленом, а X - алкиленом.
Схема 8.
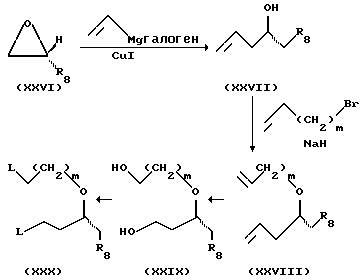
R8 представляет собой N3, NH-защитную группу, аминозащитную группу или гидроксизащитную группу; m независимо равно 0, 1, 2 или 3; а L является хорошей уходящей группой, такой, как хлоро, бромо, йодо, мезил, тозил и т.п. Предпочтительно L является мезилом. R8 является предпочтительно защищенной гидрокси-группой, а наиболее предпочтительно, -O-тритилом.
На схеме 8 проиллюстрирован стереоселективный синтез линкерной части (-X-W-Y-) макроцикла. S-энантиомер показан выше; однако, следует отметить, что аналогичным образом могут быть получены комплементарный энантиомер или смесь энантиомеров. Кроме того, следует отметить, что аналогичная реакция с метил-замещенным эпоксидом или реагентом Гриньяра может быть использована для получения различных линкеров (-X-W-Y-), содержащих метил-замещенный алкилен.
В вышеуказанной реакции эпоксид (соединение XXVI) подвергают реакции размыкания кольца с использованием реагента Гриньяра. Эту реакцию осуществляют в присутствии агента, образующего комплекс с медью; однако, могут быть использованы и другие условия алкилирования. Реакцию проводят в инертном растворителе при температуре в диапазоне от -30oC до температуры перегонки реакционной смеси. В результате этой реакции получают соединение XXVII, которое может быть подвергнуто последующей реакции без дополнительной очистки. Соединение XXVII подвергают аллилированию в условиях, которые обычно используются для получения простых эфиров. Реакция, проиллюстрированная на схеме 8, известна как синтез Вильямсона. После образования алкоксида натрия с использованием NaH, NaOH или KOH с последующим аллилированием аллилбромидом получают диен (соединение XXVIII). Соединение XXVIII превращают в спирт (соединение XXIX) стандартными способами. Например, соединение XXVIII может быть превращено в озонид путем обработки озоном при низкой температуре. Затем озонид восстанавливают с использованием NaBH4, LiAlH4, BH3, либо с помощью каталитического гидрирования с избыточным количеством H2, в результате чего получают спирт (соединение XXIX). Гидрокси-части соединения XXIX превращают в уходящую группу L стандартными способами, например, посредством реакции спирта с мезилхлоридом в триэтиламине.
Во всех схемах, приведенных выше, предпочтительно, чтобы реакции проводились с использованием соответствующих защитных групп. В частности, предпочтительно, чтобы в процессе реакции алкилирования и/или ацилирования, R1 был защищен, а затем разблокирован. Аналогично, если R6 является -NR4R5, то реакцию предпочтительно проводить с использованием аминозащитной группы. При этом, следует отметить, что многие из этих реакций могут быть осуществлены без использования защитных групп, и если это позволяют реакционные условия, с использованием блокирующих реагентов или т.п. Если W содержит гидрокси-часть, то предпочтительно, чтобы в процессе алкилирования или ацилирования индола, эта гидрокси-часть была защищена как трет-бутилфенилсилилокси (TBDPS) или трифенилметил (тритил). Полученные соединения формулы I могут быть выделены и очищены стандартными методами.
Соединения III, IV, V, VII, VIIa, VIII, X, XII, XVII, XXI, XXIV, XXV, XXVI и любые другие реагенты, необходимые для вышеуказанных реакций, являются либо коммерчески доступными, либо они могут быть получены стандартными способами. Например, соединение III может быть получено способом, описанным Edge и др., Chem. and. Ind. 130 (1991); соединение IV получают предпочтительно in situ с помощью реакции соответствующим образом замещенного индола с галидом алкилмагния, таким как бромид этилмагния, с использованием стандартной техники.
Для более подробного описания настоящего изобретения ниже приводятся Получения и Примеры. Однако, указанные примеры не должны рассматриваться как некое ограничение объема настоящего изобретения. Для наглядности, ниже приводится структура характерного соединения в соответствии с номенклатурой, принятой в настоящем описании: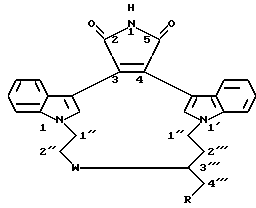
В нижеприведенных Примерах и Получениях были использованы следующие обозначения:
точка плавления - т.пл.,
ядерный магнитный резонанс - ЯМР,
масс-спектроскопия - МС,
жидкостная хроматография высокого давления на силикагеле - ЖХВД,
N,N-диметилформамид - ДМФ,
палладированный уголь - Pdc,
тетрагидрофуран - ТГФ,
этилацетат - EtOAc.
Термины "ЯМР" и "МС" указывают на то, что полученные спектры соответствуют нужной структуре.
Получение 1
2,3-бис-(3'-Индолил)-фуран-1,4-дион
К раствору, содержащему 2,3-дихлоромалеиновый ангидрид (5,56 г, 33,3 мМ) и метиламингидрохлорид (3,50 г, 55,0 мМ) в 40 мл уксусной кислоты, добавляли этоксид натрия (3,56 г, 50 мМ). Затем смесь перемешивали в пробирке в присутствии CaCl2 в качестве осушающего вещества при 25oC и в течение 16 ч, после чего нагревали с обратным холодильником в течение 4 ч. Охлажденную смесь выливали в воду (350 мл) и экстрагировали этилацетатом (3 х 75 мл). Объединенные органические экстракты промывали 100 мл-порциями насыщенного водного раствора гидрокарбоната натрия, водой и солевым раствором, а затем осушали сульфатом магния. После этого растворитель выпаривали при пониженном давлении. Образовавшийся остаток перекристаллизовывали из этанола и получали 3,82 г (64%) 2,3-дихлоро-N-метилмалеимида в виде белых кристаллов. После концентрирования маточного раствора, остаток подвергали радиальной препаративной тонкослойной хроматографии (Chromatotron, Harrison Research) и получали еще 0,81 г 2,3-дихлоро-N-метилмалеимида (выход 77%).
Раствор индола (10,5 г, 90 мМ) в 175 мл сухого толуола по капле в течение часа и в атмосфере азота обрабатывали раствором этилмагнийбромида (1,0 М в ТГФ, 90 мл, 90 мМ). После завершения добавления, светло-зеленый раствор нагревали при 40oC в течение 30 мин, а затем охлаждали до 25oC. Затем в течение 30 минут добавляли раствор 2,3-дихлоро-N-метилмалеимида (3,8 г, 21 мМ) в 50 мл толуола. Полученную реакционную смесь нагревали 3 ч при 100oC, охлаждали до 25oC и гасили 100 мл 20%-ной водной лимонной кислоты. После этого слои отделяли. Водную фазу экстрагировали этилацетатом (50 мл). Объединенные органические слои осушали безводным сульфатом магния. Растворитель выпаривали при пониженном давлении. Образовавшийся остаток растворяли в 30 мл ацетона и оставляли на 40 ч при 5oC. Твердые вещества собирали и промывали льдом/холодным эфиром, в результате чего получали 5,25 г (73%) 3,4-бис-(3'-индолил)-1-метил-пиррол-2,5-диона в виде красного твердого продукта с т. пл. 276 - 278oC.
К раствору 3,4-бис-(3'-индолил)-1-метил-пиррол-2,5-диона в 150 мл этанола добавляли 5 н. KOH (50 мл). Полученную смесь перемешивали в течение 4 ч при 25oC и разводили 150 мл воды. Большую часть этанола выпаривали при пониженном давлении. После этого смесь подкисляли до pH 1. Осажденный продукт фильтровали и промывали водой. Образовавшийся неочищенный продукт растворяли в минимальном количестве метиленхлорида и медленно фильтровали через 2-дюймовую колонку с силикагелем, элюируя 50%-этилацетатом в гексане. В результате этой процедуры получали целевое соединение (3,10 г, 77%) в виде красного твердого вещества, т.пл. 225 - 228oC.
Получение 2
бис-2,6-Дибромометилпиридин
К смеси, содержащей 2,6-пиридиндиметанол (375 мг, 5,28 мМ) и трифенилфосфин (3,20 г, 12,2 мМ) в 35 мл сухого метиленхлорида, при 0oC и в атмосфере азота порциями в течение 10 мин добавляли N-бромосукцинимид (2,16 г, 12,2 мМ). Затем смесь перемешивали в течение часа при 0oC, после чего оставляли на 16 ч при 5oC. Большую часть растворителя удаляли при пониженном давлении. К образовавшемуся остатку добавляли 100 мл эфира. Затем эфирный слой декантировали и концентрировали до объема 20 мл, после чего разводили гексаном/этилацетатом (3 : 1) (50 мл). Образовавшийся мутноватый раствор помещали на ночь в холодильник. После выпаривания растворителей в вакууме, неочищенный продукт перекристаллизовывали из гексана, в результате чего получали 766 мг (55%) бис-2,6-дибромометилпиридина в виде белого кристаллического твердого вещества. МС.
Получение 3
(±)-3-(Бензилокси)метилен-1,6-дибромогексан
Раствор т-бутоксида калия (1,0 М в ТГФ, 8,27 мл, 8,27 мМ) при 25oC и в атмосфере азота по капле добавляли к раствору (±)-3-циклогексан-1-метанола (853 мг, 7,60 мМ) в ТГФ (35 мл). Полученную смесь перемешивали при 25oC в течение 30 мин. После этого по капле добавляли бензилбромид (1,0 мл, 8,37 мМ). Реакционную смесь оставляли для перемешивания при комнатной температуре на 16 ч, а затем обрабатывали 5 мл насыщенного водного раствора NH4Cl и концентрировали. Образовавшийся остаток растворяли в эфире (50 мл), промывали водой (20 мл) и солевым раствором (20 мл), а затем осушали сульфатом магния. Растворитель выпаривали при пониженном давлении. Остаток подвергали радиальной хроматографии на силикагеле, элюируя 5% EtOAc в гексане, в результате чего получали (±)-3-(бензилокси)метил-1-циклогексен (1,42 г, 92%) в виде бесцветного маслообразного вещества. ЯМР-анализ.
Озон барботировали через раствор (±)-3-бензилоксиметилен-1-циклогексена (1,35 г, 6,70 мМ) в метиленхлориде (65 мл) при -78oC до тех пор, пока наблюдалась синяя окраска непрореагировавшего озона. Реакционную смесь оставляли для нагревания до комнатной температуры, и в это время через смесь барботировали сухой азот. Затем в течение нескольких минут с помощью шприца добавляли боран-диметилсульфидный комплекс (10,0 М в ТГФ, 2,7 мл, 27,8 мМ) и реакционную смесь оставляли при комнатной температуре на 24 ч. После этого, реакционную смесь обрабатывали 5%-ным водным раствором соляной кислоты (1 мл) и энергично перемешивали в течение часа. Затем добавляли твердый гидрокарбонат натрия до тех пор, пока смесь не становилась полностью протестированной с помощью лакмусовой бумаги. Эту смесь осушали безводным сульфатом магния. Реакционную смесь фильтровали и концентрировали с получением неочищенного (±)-3-(бензилокси)метил-1,6-гександиола (1,49 г, прибл. 100%) в виде маслообразного вещества. Этот материал, который обнаруживал, в основном, одно пятно, о чем свидетельствовал ТСХ-анализ (Rf = 0,25, 25% EtOAc в гексане), использовали в последующей стадии без дополнительной очистки.
К перемешанной смеси (±)-3-(бензилокси)метил-1,6-гексендиола (1,45 г, 6,10 мМ) и трифенилфосфина (3,67 г, 14,0 мМ) в сухом метиленхлориде (50 мл), при 0oC и в атмосфере азота добавляли N-бромосукцинимид (2,49 г, 14,0 мМ). Через 12 ч реакционную смесь концентрировали и к остатку добавляли эфир (100 мл). Эту смесь перемешивали в течение 15 мин и эфирный слой декантировали из твердых остатков. Эту процедуру повторяли с 50 мл эфира. Объединенные эфирные экстракты концентрировали до объема 50 мл, а затем разводили 100 мл гексана. После отстаивания при 5oC в течение ночи, раствор декантировали из осажденных твердых остатков и концентрировали, в результате чего получали дибромид (±)-3-(бензилокси)метил-1,6-дибромогексана (1,09 г, 49%) в виде светло-желтого маслообразного вещества. Это вещество являлось, в основном, гомогенным, о чем свидетельствовала ТСХ, Rf = 0,75 (10% EtOAc в гексане). ЯМР-анализ.
Получение 4
1-(трет-Бутилдиметилсилилокси)-4-(трет-бутилдифенилсилилокси) бутан-3-ол
К безводному метиленхлоридному (110 мл) раствору 3-бутен-1-ола (15 г, 0,21 М) добавляли имидазол (28,6 г, 0,42 М 2 экв.), а затем трет-бутилдиметилсилилхлорид (32 г, 0,22 М). Через 90 мин реакция была полностью завершена, на что указывала ТСХ (10% EtOAc в гексане). Метиленхлоридный раствор переносили в делительную воронку, разводили метиленхлоридом (110 мл), промывали водой (200 мл) и солевым раствором (200 мл). Органический слой собирали, осушали сульфатом магния, фильтровали, а растворитель выпаривали, в результате чего получали маслообразное вещество (1-(O-TBDMS)-3-бутен), которое использовали для последующей реакции. МС.
Вышеописанное маслообразное вещество растворяли в смеси ацетона (400 мл) и воды (50 мл). После этого добавляли N-метилморфолин-N-оксид (85,2 г, 0,63 М, 3 эквивалента). Полученную суспензию охлаждали до 0oC и через 10 мин добавляли 0,3 г каталитического количества OsO4. Эту суспензию оставляли для перемешивания в течение ночи, постепенно нагревая до комнатной температуры. ТСХ (25% EtOAc/гексан) указывала на полное завершение реакции. После этого реакционную смесь гасили бисульфитом натрия, разводили эфиром (1 л), а затем промывали водой (400 мл) и солевым раствором (400 мл). Органический слой собирали, а водный слой экстрагировали эфиром (2 х 500 мл). Объединенные органические слои осушали, фильтровали и концентрировали с получением 4-(O-TBDMS)-1,2-бутендиола в виде маслообразного вещества, которое использовали в последующей реакции.
Вышеописанное маслообразное вещество растворяли в безводном метиленхлориде (250 мл). К раствору добавляли имидазол (30 г, 0,44 М, 2,5 экв.), перемешивая при этом. Полученный раствор охлаждали до 0oC. После 15-минутного охлаждения, по капле в течение 45 мин добавляли метиленхлоридный (50 мл) раствор трет-бутилдифенилсилилхлорида (50 г, 0,18 М, 1 экв.). После завершения добавления, перемешивание продолжали в течение 2,5 ч при 0oC. Затем раствор переносили в делительную воронку, разводили метиленхлоридом (250 мл), промывали водой, солевым раствором и осушали сульфатом магния, после чего фильтровали. Растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт в виде маслообразного вещества. Неочищенный продукт очищали путем элюирования (10% EtOAc/гексан) через узкую колонку с силикагелем. Элюирующий растворитель удаляли в вакууме и получали целевое промежуточное соединение в виде вязкого маслообразного вещества (78,1 г, полный выход 93%). МС.
Получение 5
1-(трет-Бутилдиметилсилилокси)-3-(3-иодопропилокси)-4- (трет-бутилдифенилсилилокси)-бутан
К метиленхлоридному (20 мл)/циклогексановому (100 мл) раствору спирта Получения 4, в атмосфере азота (баллонного) добавляли аллилтрихлорацетимидат (17,82 г, 88 мМ, 2,2 экв.), а затем трифторметансульфоновую кислоту (50 мкл/г исходного материала, 0,92 мл). Через 20 ч раствор фильтровали и фильтрат промывали насыщенным водным раствором гидрокарбоната натрия, водой, а затем солевым раствором. Органический слой собирали и осушали сульфатом магния. Растворитель удаляли с получением маслообразного вещества, которое очищали с помощью флеш-хроматографии на силикагеле (элюент: гексан) и повышали полярность подвижной фазы, используя свыше нескольких литров 5%-ного этилацетата в гексане. В результате этой процедуры получали 19,27 г аллилового эфира, 1-(трет-бутилдиметилсилилокси)-3-(пропенокси)- 4-(трет-бутилдифенилсилилокси)-бутана в виде светло-коричневого маслообразного вещества (выход 97%). МС.
К ТГФ-раствору (60 мл) вышеописанного аллилового эфира (14,16 г, 28,38 мМ, 1 экв.), по капле в атмосфере азота добавляли 9-BBN (9-борабицикло 3.3.1 нонан, 0,5 М-раствор в ТГФ, 60 мл, 30 мМ, 1,1 экв.). Через 3 ч ТСХ (10% EtOAc в гексане) реакционной смеси указывала на полное израсходование исходного материала. К раствору добавляли 3 М-водный NaOH (10,41 мл, 31,22 мМ 1,1 экв.), а затем медленно (в течение 1,5 ч) по капле добавляли 30%-ный пероксид водорода (10,3 мл, 90,82 мМ, 3,2 экв.). Реакционную температуру в течение добавления поддерживали ниже 50oC (с использованием ледяной бани).
Через 30 мин к раствору добавляли хлорид натрия до тех пор, пока раствор не становился насыщенным. Органический слой удаляли, водный слой экстрагировали эфиром, объединенные органические слои осушали и фильтровали, а фильтрат концентрировали с получением маслообразного вещества. Неочищенное маслообразное вещество очищали с помощью флешхроматографии на силикагеле, элюируя 10% EtOAc/гексаном и повышая полярность, используя около 1,5 л 20% EtOAc/гексана, получали 9,53 г светло-желтого маслообразного вещества (выход 65%). МС.
К безводному (0oC) эфирному (150 мл) раствору вышеописанного спирта добавляли триэтиламин (2,93 г, 28,91 мМ, 1,5 экв.), а затем по капле добавляли мезилхлорид (3,31 г, 28,91 мМ, 1,5 экв.), энергично перемешивая при этом. После выдерживания смеси при 0oC в течение 3 ч ТСХ (10% EtOAc/гексан) указывала на израсходование исходного материала. Полученную реакционную смесь разводили эфиром, промывали водой, солевым раствором, осушали сульфатом магния и растворитель удаляли. Полученное маслообразное вещество пропускали через слой двуокиси кремния, элюируя 25% EtOAc-гексаном, после чего элюент концентрировали. К ацетоновому (200 мл) раствору полученного маслообразного вещества добавляли гидрокарбонат натрия (0,17 г, 1,93 мМ, 0,1 экв.) и NaI (28,88 г, 192,7 мМ, 10 экв.). После 30-минутного перемешивания при комнатной температуре в атмосфере азота реакционную смесь нагревали до 50oC с использованием водяной бани. Через 2,5 ч ТСХ (10% EtOAc в гексане) указывала на полное израсходование мезилата. Полученную реакционную смесь разводили эфиром (500 мл), а затем промывали холодным насыщенным водным раствором Na2SO3, водой, солевым раствором и осушали сульфатом магния. Растворитель удаляли. Полученное маслообразное вещество пропускали через слой двуокиси кремния (элюент: 5% EtOAc в гексане) и получали очищенное целевое соединение в виде бесцветного маслообразного вещества (выход 85%, 10,3 г).
Получение 6
3-Бромопропилацетат
3-Бромопропан-1-ол (0,54 М, 75 г) в метиленхлориде (500 мл) при 0oC в атмосфере азота обрабатывали ацетилхлоридом (0,5 М, 40,2 мл). Затем к этому раствору медленно, с помощью шприца, порциями (5 мл) добавляли триэтиламин (0,54 М, 75 мл). Реакционную смесь оставляли на 12 ч для постепенного нагревания до комнатной температуры. Осадок отфильтровывали и фильтр промывали метиленхлоридом. Фильтрат промывали дважды водой, а затем дважды промывали солевым раствором и осушали сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и получали целевое соединение (ацетат) в виде маслообразного вещества (91 г, выход 93%). МС.
Получение 7
N-(3-Ацетоксипропил)-индол
К перемешанной (0oC) диметилформамидной (400 мл) суспензии NaH (60% в минеральном масле, 0,705 М, 28,2 г, 1,5 экв.) в трехгорловой колбе, снабженной обратным холодильником и капельной воронкой, добавляли диметилформамидный (150 мл) раствор индола (55 г, 0,47 М). Через 30 - 60 мин диметилформамидный (50 мл) раствор алкилгалида добавляли к раствору индола, а затем добавляли 3-бромопропилацетат (170 г, 0,94 М). Реакционную смесь нагревали 6 ч при 50oC, а затем оставляли для перемешивания при комнатной температуре на 5 - 15 ч.
Растворитель удаляли в вакууме. Образовавшийся остаток распределяли между метиленхлоридом и водой. Органический слой промывали 3 раза 1 н. соляной кислотой, водой и солевым раствором, а затем осушали сульфатом натрия и фильтровали. Фильтрат концентрировали и получали целевой алкилиндол (102 г) в виде маслянистого вещества, которое медленно кристаллизовали. МС.
Получение 8
N-(трет-Бутоксикарбонил)-индол-3-ил-уксусная кислота
К перемешанному ацетоновому (800 мл) раствору индол-3-уксусной кислоты (26,25 г, 0,15 М) добавляли карбонат цезия (48,9 г, 0,15 М), а затем аллилбромид (15 мл, 0,17 М, 1,16 экв.). Через 12 ч растворитель удаляли. Остаток распределяли между водой и CHCl3. Органический слой промывали солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали и получали 27,9 г аллилового сложного эфира в виде маслообразного вещества (выход 74%).
К ацетонитриловому (500 мл) раствору аллилового сложного эфира (27,9 г) добавляли ди-трет-бутилдикарбонат (29,1 г, 0,133 М, 1,2 экв.) и 4-диметиламинопиридин (1,36 г, 0,011 М, 0,1 экв.). Через 15 мин реакционную смесь разводили этилацетатом (1,2 л), а затем промывали 0,1 н. соляной кислотой, водой (2 раза) и солевым раствором (2 раза). Органической слой осушали сульфатом натрия, фильтровали и концентрировали с получением ВОС-защищенного сложного эфира (32,9 г, 94%) в виде маслообразного вещества, которое медленно кристаллизовали.
К метиленхлоридному/этилацетатному (10 : 3) (325 мл) раствору ВОС-защищенного сложного эфира добавляли 2-этилгексаноат натрия (17,3 г, 0,104 М), трифенилфосфин (4,93 г, 18,8 мМ, 0,18 экв.) и Pd (PPh3)4 (4,56 г, 3,95 М, 0,04 экв.). Через час растворитель удаляли. Остаток распределяли между этилацетатом и водой. Основный водный слой подвергали обратному экстрагированию этилацетатом, а затем эфиром, после чего осторожно подкисляли 0,10 н. соляной кислотой. Подкисленный водный слой экстрагировали этилацетатом. Органический слой промывали водой, солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали и получали ВОС-защищенную кислоту (21,8 г, выход 77%) в виде маслообразного вещества, которое медленно кристаллизовали. Выход этого целевого соединения из трех стадий составлял свыше 53%. МС.
Получение 9
(±)-3,4-[(N, N'-1,1'-(3''-3-трет-Бутилдифенилсилилоксиметилен) гексан)-бис-(3,3'-индолил)]-1-(метил)-пиррол-2,5-дион
Диметилформамидный (50 мл) раствор бис-(3,3'-индолил)]-1-(метил)-пиррол-2,5-диона (3,41 г, 10,0 мМ), содержащий 3-трет-бутилдифенилсилилоксиметилен-1,6-дибромогександибромид (5,64 г, 11 мМ, полученный способом, аналогичным описанному в Получении 2 для получения бензоилового производного), с помощью шприца, в течение 15 мин и при 60oC добавляли к диметилформамидной (350 мл) суспензии Cs2CO3 (11,2 г, 34,3 мМ). Через 4 ч после завершения добавления, реакционную смесь охлаждали до комнатной температуры, выливали в воду (1,5 л) и экстрагировали метиленхлоридом (3 х 300 мл). Органическую фазу промывали водой, осушали, фильтровали и концентрировали. Концентрат очищали с помощью флеш-хроматографии, элюируя 10 - 25% этилацетатом/гексаном, и получали 2,95 г макроциклического 3,4-[(N,N'-1,1'-(3''-3-трет- бутилдифенилсилилоксиметилен)гексан)-бис-(3,3'-индолил)] -1-(метил)-пиррол- 2,5-диона (выход 43%) в виде красного маслообразного вещества. МС.
Получение 10
(S)-Метил 4-трет-бутилдифенилсилилокси-3-(аллилокси)-бутират
К циклогексановому (400 мл) раствору (S)-метил 4-трет-бутилдифенилсилилокси-3-(гидрокси)бутирата (20,0 г, 53,7 мМ) добавляли аллилтрихлороацетимидат (21,74 г, 107,4 мМ), а затем трифторометансульфоновую кислоту (1 мл, 50 мл/г спирта) добавляли к этому раствору пятью порциями в течение 30 минут, перемешивая при этом в атмосфере азота. Через 70 часов твердые вещества отфильтровывали, осадок на фильтре промывали циклогексаном, а летучие вещества удаляли в вакууме. Полученное маслообразное вещество фильтровали через слой двуокиси кремния, промывали гексаном и продукт элюировали 10% этилацетатом/гексаном. ЯМР-анализ указывал на присутствие остаточного имидата (прибл. 10%); однако, материал использовали без дополнительной очистки. В результате этой процедуры получали 24,76 г материала (выход 100%). МС.
Получение 11
(S)-4-трет-Бутилдифенилсилилокси-3-(2-иодоэтокси)-1-иодобутан
К раствору (S)-метил 4-трет-бутилдифенилсилилокси-3-(аллилокси)-бутирата (23,8 г, 57 мМ), растворенного в безводном ТГФ (1,0 л), по капле, при -75oC, в атмосфере азота и в течение 40 мин добавляли DIBAL-H (231 мл, 1,0 М в толуоле, 231 мМ). После перемешивали в течение 1,5 часа, смесь оставляли для нагревания до -10oC и гасили 5% водой в метаноле и большим количеством целита. Полученную реакционную смесь фильтровали через слой целита, а фильтрат концентрировали и распределяли между эфиром и 20% лимонной кислотой. Эфирный слой осушали и концентрировали в вакууме. Остаточное маслообразное вещество пропускали через слой двуокиси кремния, элюируя хлороформом, в результате чего получали 20,6 г (93%) (S)-4-трет-бутилдифенилсилилокси-3-аллилокси-бутан-1-ола.
К метаноловому (500 мл) раствору (S)-4-трет-бутилдифенилсилилокси-3-аллилоксибутан-1-она (20,6 г, 53,6 мМ), при -78oC и приблизительно в течение 12 минут добавляли озон. После появления слабого синего оттенка реакционной смеси, к содержимому реакционного сосуда добавляли NaBH4 (12,2 г, 321 мМ, 6 экв. ). Полученную реакционную смесь оставляли для нагревания до комнатной температуры. Летучие вещества удаляли в вакууме. Образовавшийся остаток пропускали через слой силикагеля, элюируя этилацетатом, в результате чего получали 16,4 г (79%) (S) 4-трет-бутилдифенилсилилокси-3-(2-гидрокси)этокси)-бутан-1-ола в виде бесцветного маслообразного вещества.
К эфирному (600 мл) раствору (S) 4-трет-бутилдифенилсилилокси-3-(2-гидрокси-этокси)-бутан-1-ола (15,7 г, 40,4 мМ) при 0oC и в атмосфере азота добавляли триэтиламин (16,8 мл, 121 мМ), а затем мезилхлорид (9,38 мл, 121 мМ). Через 3 часа раствор фильтровали, фильтрат промывали водой (2 раза), солевым раствором (2 раза), осушали сульфатом натрия и концентрировали в вакууме. В результате этой процедуры получали 21,9 г (> 99%) бисмезилата в виде желтого маслообразного вещества, которое использовали сразу в последующей реакции. Этот бисмезилат растворяли в ацетоне (1,4 л), который дистиллировали из карбоната калия. К полученному раствору добавляли NaI (90,4 г, 603 мМ) и 0,05 экв. NaHCO3 (170 мг, 2 мМ). Реакционную смесь поддерживали при 56oC в течение 24 ч и фильтровали. Фильтрат концентрировали в вакууме. Образовавшийся остаток распределяли между эфиром и 10% Na2CO3, эфирный слой промывали солевым раствором, осушали сульфатом натрия и концентрировали с получением 17,9 г (73,2%) (S)-4-трет-бутилдифенилсилилокси-3-(2-иодоэтокси)-1-иодобутана в виде бесцветного маслообразного вещества. Полный выход составлял 54%, МС: MW = 608,39; Найдено: 559 (М-трет-бутил; FD, CHCl3).
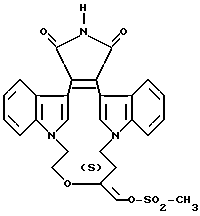
Получение 12
(S)-3,4-[(N, N'-1,1')-((2''-Этокси)-3'''-(O)-4''- (метансульфонилокси)-бутан)-(бис)-(3-индолил)]-1H-пиррол-2,5-дион
3,4-(бис)-(3-Индолил)-1H-пиррол-2,5-дион (10,04 г, 29,4 мМ) и (S)-4-(трет-бутилдифенилсилилокси)-3-(2-иодоэтокси)-1-(иодо)бутан (17,9 г, 29,4 мМ) объединяли и растворяли в безводном ДМФ (80 мл). Затем этот раствор с помощью шприца, в течение 72 ч, при 50oC и в атмосфере азота добавляли к суспензии карбоната цезия (38,3 г, 118 мМ) в безводном диметилформамиде (1,7 л). После этого ДМФ удаляли в вакууме. Остаток распределяли между CHCl3 и 1 н. соляной кислотой. Кислотный слой подвергали обратной экстракции хлороформом и этилацетатом. Объединенные органические слои промывали 1 н. соляной кислотой, 2 раза промывали водой, 2 раза солевым раствором, а затем осушали сульфатом натрия и концентрировали с получением твердого фуксина. Эту неочищенную реакционную смесь использовали без дополнительной очистки.
Неочищенную реакционную смесь суспендировали в этаноле (700 мл) и обрабатывали 5 н. раствором КОН (800 мл). Реакционную температуру повышали до 80oC. Через 72 ч этанол удаляли в вакууме; водную суспензию охлаждали до 0oC и подкисляли 5 н. соляной кислотой. Фиолетовый осадок собирали и пропускали через слой двуокиси кремния, элюируя этилацетатом. Элюент концентрировали и получали 8,7 г частично силилированного малеимида в виде пурпурного твердого красителя, который использовали в последующей стадии без дополнительной очистки.
К диметилформамидному (1 л) раствору вышеописанного ангидрида (8,7 г, 19,7 мМ) при комнатной температуре и в атмосфере азота добавляли 1,1,1,3,3,3-гексаметилдисилазан (41,6 мл, 197 мМ) и метанол (4 мл, 98,5 мМ). Через 40 часов реакционную смесь концентрировали в вакууме, а затем добавляли раствор MeCN/1 н. соляной кислоты (2 : 1, об/об) (100 мл). Образовавшийся остаток перемешивали в течение часа. Органический растворитель удаляли, а водную суспензию экстрагировали этилацетатом. После удаления растворителей получали 8,9 г малеимида, который использовали без дополнительной очистки.
К метиленхлоридной (800 мл) суспензии вышеописанного малеимида (8,9 г, 20 мМ), в атмосфере азота и при комнатной температуре добавляли пиридин (4,85 мл, 60 мМ) и избыточное количество метансульфонового ангидрида (4,21 г, 24 мМ). Через 16 часов реакционную смесь промывали 0,1 н. соляной кислотой, солевым раствором и органический слой концентрировали. Остаток пропускали через слой двуокиси кремния, элюируя градиентом 0 - 10% MeCN в CH2Cl2. Элюентную фракцию, содержащую нужный мезилат, концентрировали и получали 2,8 г целевого соединения в виде твердого пурпурного красителя. Полный выход по отношению к дииодиду составлял 18%. МС: MW = 520; найдено: 520 (FD, CHCl3).
Получение 13
3-(трет-Бутилдифенилсилилоксиметилен)-1-циклогексен
К смеси 3-циклогексен-1-метанола (Aldrich, 13,0 мл, 0,11 М), N,N-диизопропилэтиламина (43 мл, 0,244 М) и 4-диметиламинопиридина (2,70 г, 0,022 М) в 375 мл безводного метиленхлорида, в атмосфере азота и при 25oC добавляли трет-бутилдифенилхлоросилан (32 мл, 0,123 М). После этого смесь перемешивали при 25oC в течение 48 ч. Полученную реакционную смесь промывали 150 мл-порциями 1 н. соляной кислоты, а затем водой и солевым раствором, после чего осушали безводным сульфатом магния. Растворитель выпаривали. Остаток загружали в колонку (4'' • 4'') с двуокисью кремния и медленно элюировали гексаном. Таким образом получали 33,6 г (86%) 3-(трет-бутилдифенилсилилоксиметилен)-1-циклогексена в виде бесцветного маслообразного вещества, которое являлось гомогенным, о чем свидетельствовал ТСХ-анализ (Rf = 0,4, гексан).
Элементный анализ для C23H30OSi (0,3 H2O):
Вычислено: C 77,6; H 8,67.
Найдено: C 77,38; H 8,72.
Получение 14
3-(трет-Бутилдифенилсилилоксиметилен)-1,6-гександиол
Озон барботировали через тщательно перемешанный раствор 3-(трет-бутилдифенилсилилоксиметилен)-1-циклогексена (18,0 г, 51,3 мМ) в метиленхлориде (550 мл) при -78oC до тех пор, пока наблюдался синий оттенок непрореагировавшего озона. Полученную реакционную смесь оставляли для нагревания до 25oC. Затем в течение 30 минут через раствор барботировали сухой азот. После этого к раствору по капле в течение 10 минут добавляли боран-диметилсульфидный комплекс (10,0 М, 23 мл, 0,23 М). Смесь медленно перемешивали в атмосфере азота при 25oC в течение 24 ч. После добавления 5%-ной соляной кислоты (15 мл), реакционную смесь перемешивали в течение часа. К смеси добавляли твердый гидрокарбонат натрия до тех пор, пока эта смесь не становилась основной, о чем свидетельствовала индикаторная бумага для определения pH (с влажной поверхностью). После фильтрования фильтрат промывали 200 мл-порциями 5%-ного гидрокарбоната натрия, а затем водой, после чего осушали безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении, неочищенный продукт очищали с помощью хроматографии через слой (4'' • 4'') двуокиси кремния, элюируя этилацетатом. В результате этих процедур получали 17,8 г (90%) 3-трет-(бутилдифенилсилилокси)метилен)-1,6-гександиола в виде бесцветного вязкого маслообразного вещества, которое являлось гомогенным, на что указывал ТСХ-анализ (Rf = 0,5, эфир).
Аналитический анализ для C23H34O3Si (0,2 H2O):
Вычислено: C 70,80; H 8,88.
Найдено: C 70,72; H 8,86.
Получение 15
3-трет-бутилдифенилсилилоксиметилен-1,6-дибромогексан
При 0oC и в атмосфере азота к перемешанному раствору, содержащему 3-(трет-бутилдифенилсилилоксиметилен)-1,6-гександиол (17,5 г, 45,2 мМ) и трифенилфосфин (28,6 г, 109 мМ) в безводном метиленхлориде (550 мл), порциями в течение 5 мин добавляли N-бромосукцинимид (19,3 г, 109 мМ). Полученную реакционную смесь перемешивали 5 ч при 0oC, а затем помещали в холодильник (при 5oC) на 16 ч. После удаления большей части растворителя, к остатку медленно добавляли сухой эфир (300 мл). Эфирный слой декантировали из осажденных твердых веществ. Твердые вещества промывали еще 200 мл свежеприготовленного эфира. Объединенный эфирный слой концентрировали (100 мл), растирали с 300 мл гексана, а затем декантировали из осажденных твердых веществ. После этого твердые вещества промывали 25%-ным эфиром в гексане, а объединенные органические слои осушали сульфатом магния (безводным) и концентрировали. Неочищенный продукт помещали на колонку (4'' • 4'') с силикагелем и элюировали 25%-ным эфиром в гексане, в результате чего получали 3-трет-бутилдифенилсилилоксиметилен-1,6-дибромогексана (20,1 г, 86%) в виде бесцветного маслообразного вещества, которое являлось гомогенным, о чем свидетельствовала ТСХ (Rf = 0,75, 10%-ный EtOAc в гексане).
1H-ЯМР (300 МГц, CDCl3): δ 1,06 (с, 9H); 1,35 - 2,10 (м, 7H); 3,55 (м, 4H); 3,56 (прбл. д, 2H, J = 4 Гц), 7,40 и 7,64 (м, 10H).
13C-ЯМР (75 МГц, CDCl3): 19,2, 26,9, 29,3, 30,0, 31,9, 33,8, 34,7, 38,5, 65,0, 127,7, 129,7, 133,4, 135,5.
Получение 16
(S)-(-)-3-Циклогексен-1-метанол
К охлажденному раствору сложного эфира (Ireland и др., J. Org. Chem. 1992, 57(19), 5071 - 5073 и их ссылки), а именно (S)-(-)-3-циклогексен-1-метокси-(S)-N-метил-2-гидроксисукцинимида (8,20 г, 34,5 мМ) в ТГФ (90 мл), по капле в течение 15 мин добавляли раствор LiAlH4 (1,0 М в ТГФ, 75,8 мл, 75,8 мМ). Полученную реакционную смесь оставляли для нагревания до комнатной температуры, а затем перемешивали в течение 2 ч при 25oC, охлаждали и гасили водой и 1 н. раствором NaOH. После этого смесь фильтровали через целит. Твердые вещества промывали тетрагидрофураном (100 мл). После выпаривания фильтрата при пониженном давлении, остаток растворяли в 150 мл эфира, а затем промывали водой (2 х 50 мл), солевым раствором (50 мл) и осушали безводным сульфатом магния. После выпаривания растворителя получали 3,24 г (83%) (S)-(-)-3-циклогексен-1-метанола в виде прозрачного маслообразного вещества. [α]D = -90:3 (C = 1, CH3OH). ТСХ-свойства и 1H-ЯМР-спектр этого материала показали полную идентичность с рацемическим материалом (Aldrich).
1H-ЯМР (300 МГц, CDCl3): δ 1,21 - 1,42 (м, 2H); 1,68 - 1,88 (м, 3H); 2,04 - 2,21 (м, 3H); 3,54 (шир.с, 2H); 5,69 (с, 2H).
Получение 17
(S)-(-)-3-Тетрабутилдифенилсилилоксиметилен-1-циклогексен
(S)-(-)-3-Циклогексен-1-метанол (3,17 г, 28,3 мМ) обрабатывали трет-бутилдифенилхлоросиланом (8,15 мл, 31,1 мМ), N,N-диизопропилэтиламином (10,9 мл, 62,3 мМ) и диметиламинопиридином (1,03 г, 8,5 мМ) в метиленхлориде (100 мл), в результате чего после обработки и хроматографии получали 8,73 г (88%) силилового сложного эфира, (S)-(-)-3-третбутилдифенилсилилоксиметилен-1-циклогексена в виде прозрачного маслообразного вещества. ТСХ-свойства и 1H-ЯМР-спектр этого материала показал полную идентичность с рацемическим силиловым эфиром, 3-трет-бутилдифенилсилилоксиметилен)-1-циклогексеном.
1H-ЯМР (300 МГц, CDCl3): δ 1,05 (с, 9H); 1,29 (м, 1H); 1,71 - 2,18 (м, 4H), 3,54 (д, 2H, J = 6 Гц), 5,66 (шир.с, 2H), 7,38 и 7,66 (м, 10H).
Получение 18
(S)-(-)-3-трет-Бутилдифенилсилилоксиметилен)-1,6-гексендиол
В соответствии с аналогичной процедурой, описанной для получения рацемического диола, а именно 3-(трет-бутилдифенилсилилоксиметилен)-1,6-гексендиола, силиловый эфир, а именно (S)-(-)-3-трет-бутилдифенилсилилоксиметилен)-1-циклогексен (8,35 г, 23,9 мМ) подвергали озонированию, после чего восстанавливали путем обработки смесью BH3 и Me2S, в результате чего получали (S)-(-)-3-(трет-бутилдифенилсилилоксиметилен)-1,6-гександиол (прибл. 5,01 г, 55%) в виде бесцветного вязкого маслообразного вещества, которое являлось гомогенным, о чем свидетельствовала ТСХ (Rf = 0,4, EtOAc).
1H-ЯМР (300 МГц, CDCl3): 1,05 (с, 9H), 1,21 - 1,81 (м, 7H), 2,32 (шир.с, 2H), 3,50 - 3,75 (м, 6H), 7,32 и 7,70 (м, 10H).
Получение 19
(S)-3-(трет-Бутилдифенилсилилоксиметилен)-1,6-дибромогексан
В соответствии с процедурой, описанной для получения рацемического дибромида, а именно 3-(трет-бутилдифенилсилилоксиметил)-1,6-дибромогексана, (S)-(-)-3-(трет-бутилдифенилсилилоксиметил)-1,6-гександиол (4,85 г, 12,53 мМ) подвергали реакции с N-бромосукцинимидом (5,35 г, 30,1 мМ) и трифенилфосфином (7,87 г, 30,1 мМ) в метиленхлориде (150 мл) при 0oC. Таким образом было получено соединение (S)-(-)-3-(трет-бутилдифенилсилилоксиметилен)-1,6-дибромогексан (4,81 г, 75%) в виде прозрачного бесцветного маслообразного вещества, которое являлось гомогенным, о чем свидетельствовала ТСХ (Rf = 0,8, 10% EtOAc в гексане). ТСХ-свойства и 1H-ЯМР-спектр этого соединения показали полную идентичность с рацемическим изомером. МС.
1H-ЯМР (300 МГц, CDCl3): 1,06 (с, 9H), 1,35 - 2,10 (м, 7H), 3,55 (м, 4H), 3,56 (соотв.д, 2H, J = 4 Гц), 7,40 и 7,64 (м, 10H).
Получение 20
(R)-3-(трет-Бутилдифенилсилилоксиметилен)-1,6-дибромогексан
В соответствии с процедурой, описанной для получения (S)-(-)-3-(трет-бутил-дифенилсилилоксиметилен)-1,6-дибромогексана, (S)-(-)-3-(трет-бутилдифенилсилилоксиметилен)-1,6-гександиол (5,05 г, 13,04 мМ) подвергали реакции с N-бромосукцинимидом (5,75 г, 31,32 мМ) и трифенилфосфином (8,21 г, 31,32 мМ) в метиленхлориде (160 мл) при 0oC. Таким образом получали хиральный дибромид, (R)-3-(трет-бутилдифенилсилилоксиметилен)-1,6-дибромогексан (5,85 г, 87%) в виде прозрачного бесцветного маслообразного вещества, которое являлось гомогенным, на что указывала ТСХ (Rf = 0,8, 10% EtOAc в гексане). МС.
1H-ЯМР (300 МГц, CDCl3): 1,06 (с, 9H), 1,35 - 2,10 (м, 7H), 3,55 (м, 4H), 3,56 (соотв.д, 2H, J = 4 Гц), 7,40 и 7,64 (м, 10H).
Получение 21
2-Аллил-4-пентеновая кислота
К перемешанной суспензии метоксида натрия (59,4 г, 1,1 М) в сухом метаноле (1 л), при 0oC, по капле и в атмосфере азота добавляли диметилмалонат (57 мл, 0,5 М). Через 30 минут одной порцией добавляли аллилбромид (95 мл, 1,1 мМ). После отстаивания в течение 14 ч при комнатной температуре, реакционную смесь концентрировали в вакууме. Остаток растворяли в метаноле (0,5 л) и обрабатывали 500 мл 5 н. NaOH. После 20-часового перемешивания метанол удаляли в вакууме, а основный водный слой два раза промывали этилацетатом. После этого водный слой подкисляли путем добавления 0,5 л 5 н. соляной кислоты, а затем экстрагировали этилацетатом. Органический экстракт два раза промывали водой, затем солевым раствором, после чего осушали сульфатом натрия и концентрировали в вакууме с получением белого твердого вещества. Полученное твердое вещество растирали с пентаном и осушали при атмосферном давлении. Таким образом получали 51,4 г (выход 57%) двухосновной кислоты. Эту кислоту (50 г, 274 мМ) нагревали при 150oC до тех пор, пока не прекращалось выделение CO2 (около 2 ч). Остаточное коричневое маслообразное вещество элюировали этилацетатом через небольшой слой двуокиси кремния, в результате чего получали 32,8 г (85%) целевого соединения в виде золотистого маслообразного вещества. Полный выход из трех стадий составлял 48%.
1H-ЯМР (CD3CN) δ: 2,4 (м, 4H), 2,5 (м, 1H), 5,05 (дд, 2H), 5,15 (дд, 2H), 5,9 (м, 2H), 12,8 (шир.с, 1H). МС.
Получение 22
3-(трет-Бутилдифенилсилилоксиметилен)-пентан-1,5-диол
К перемешанной при 0oC суспензии NaH (4,33 г, 114 мМ) в безводном эфире (125 мл), по капле в атмосфере азота добавляли 2-аллил-4-пентеновую кислоту (16,0 г, 114 мМ). Полученную реакционную смесь оставляли для нагревания до комнатной температуре. Затем реакцию гасили путем добавления этанола (25 мл) и 40 мл 4 н. соляной кислоты, после чего дважды экстрагировали эфиром, осушали и концентрировали в вакууме с получением спирта, 2-аллил-4-пентен-1-ола в виде бесцветного маслообразного вещества (11,7 г, 82%), которое использовали в последующей стадии без дополнительной очистки.
К сухому метиленхлоридному (0,5 мл) раствору 2-аллил-4-пентен-1-ола (11,7 г, 93 мМ) добавляли имидазол (12,6 г, 185 мМ), а затем хлоро-трет-бутилдифенилсилан (25,48 г, 93 мМ) и смесь перемешивали 16 ч. Реакционную смесь фильтровали, фильтрат промывали водой и солевым раствором, а затем осушали и концентрировали в вакууме с получением силилового эфира, 3-(трет-бутилдифенилсилилоксиметилен)-пент-1,4-ена (32,5 г, 96%) в виде маслообразного вещества, которое использовали без дополнительной очистки.
При -78oC, через метаноловый (500 мл) раствор 3-(трет-бутилдифенилсилилоксиметилен)-1,5-пентадиола (17 г, 477 мМ) барботировали озон до тех пор, пока наблюдался синий оттенок (30 мин). После этого реакционную смесь продували азотом (20 мин) и добавляли NaBH4 (17,6 г, 47 мМ). Холодную баню удаляли и реакционную смесь доводили до комнатной температуры. После этого реакционную смесь концентрировали в вакууме, а остаток распределяли между эфиром и солевым раствором. Эфирный слой концентрировали и образовавшийся остаток элюировали через слой двуокиси кремния, используя в качестве элюента смесь 0 - 50% этилацетата/гексана. Минорный компонент собирали и концентрировали с получением диола, 3-(трет-бутилдифенилсилилоксиметилен)-пентан-1,5-диола (3,8 г, 22%) в виде бесцветного маслообразного вещества. Полный выход из трех стадий составлял 17%. МС.
1H-ЯМР: δ 1,17 (с, 9H), 1,6 (дт, 4H), 1,83 (м, 1H), 2,14 (с, 2H), 3,6 (м, 6H), 7,41 (т, 4H), 7,45 (т, 2H), 7,66 (д, 4H).
Получение 23
1,5-Дииодо-3-(трет-бутилдифенилсилилоксиметилен)-пентан
К эфирному (0oC) (300 мл) раствору 3-(трет-бутилдифенилсилилоксиметилен)-пентан-1,5-диола (6,9 г, 19 мМ) добавляли метансульфонилхлорид (4,3 мл, 56 мМ), а затем Et3N (7,7 мл, 56 мМ). Через 3 - 16 ч реакционную смесь медленно нагревали до комнатной температуры, а затем промывали водой и солевым раствором, после чего осушали сульфатом магния и концентрировали, в результате чего получали 8,5 г (90%) 1,5-бис(метансульфонилокси)-3-(трет-бутилдифенилсилилоксиметилен)- пентана в виде бесцветного маслообразного вещества, которое использовали без дополнительной очистки.
К свежедистиллированному ацетоновому (500 мл) раствору бис-мезилата, а именно 1,5-бис(метансульфонилокси)-3-(трет-бутилдифенилсилилоксиметилен)- пентана (8,5 г, 16 мМ) добавляли избыточное количество NaI (36,1 г, 241 мМ) и NaHCO3 (67 мг, 0,8 мМ). Реакционную смесь нагревали с обратным холодильником при 57oC в течение 72 ч, охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме. Остаток разводили эфиром, промывали 10%-ным Na2SO3, осушали и концентрировали, в результате чего получали 7,4 г (выход 78%) бесцветного маслообразного вещества. Полный выход целевого соединения из двух стадий составлял 70%. МС.
1H-ЯМР-анализ: (ДМСО-d6) δ: 1,06 (с, 9H), 1,78 (м, 1H), 1,8 - 2,06 (м, 4H), 3,13 (м, 4H), 3,57 (д, 2H), 7,38 - 7,46 (м, 3H), 7,64 (д, 2H).
Получение 24
2-(2'-Бромоэтокси)-бензилбромид
Озон барботировали через сухой метаноловый раствор 2-(аллилокси)бензилового спирта (LaChapelle и др., Tetrahedron 44(16), 5033 - 5044 (1988)) (7,0 г, 43 мМ) при -78oC в течение 13 мин, контролируя ТСХ-профиль реакции каждые 2 мин на полное исчезновение исходного олефина (Rf = 0,8, 75% EtOAc/гексан). Полученную реакционную смесь продували азотом, добавляли NaBH4 (9,7 г, 0,25 М) и реакционную температуру повышали до 0oC. Через 30 мин реакционную смесь нагревали до комнатной температуры, концентрировали, разводили эфиром, промывали водой и солевым раствором, а затем осушали и концентрировали с получением остатка. Этот остаток элюировали через слой двуокиси кремния (градиентом 25% EtOAc /75% гексана). После выпаривания элюирующего растворителя был получен диол, 2-(2'-гидроксиэтокси)-бензиловый спирт (4,8 г, 67%) в виде маслообразного вещества. МС: MW = 168; найдено: 168, FD, CHCl3.
При 0oC к сухому метиленхлоридному (250 мл) раствору диола, 2-(2'-гидроксиэтокси)-бензилового спирта (4,38 г, 26 мМ) добавляли трифенилфосфин (15,8 г, 60 мМ) и N-бромосукцинимид (10,7 г, 60 мМ). После выдерживания в течение 2 часов при 0oC, реакция была завершена, о чем свидетельствовал ТСХ-анализ (20% EtOAc/CH2Cl2), и полученную реакционную смесь концентрировали в вакууме. Концентрат элюировали градиентом гексана/15% EtOAc в гексане, через слой силикагеля. После концентрирования элюирующих фракций получали дибромид, 2-(2'-бромоэтокси)-бензилбромид (6,91 г, выход 90%) в виде бесцветного твердого вещества. МС.
13C-ЯМР (CHCl3, 75,4 МГц): δ 28,7, 29,1; 68,2; 112,3, 121,6, 126,8, 130,2, 131,1, 156,0.
1H-ЯМР (CHCl3, 200 МГц): δ 3,72 (2H, т, J = 5 Гц), 4,34 (2H, т, J = 5 Гц), 4,59 (2H, с), 6,84 (H, д, J = 7 Гц), 6,95 (H, т, J = 7 Гц), 7,25 - 7,38 (2H).
Получение 25
1-(трет-Бутилдиметилсилилокси)-3-(2-иодоэтокси)-4-(трет- бутилдифенил)-бутан
Аллиловый эфир, а именно, 1-(трет-бутилдиметилсилилокси)-3-(аллилокси)-4-(трет-бутилдифенил)- бутан (21,6 г, 43,4 мМ) растворяли в метаноле (500 мл) и охлаждали до -78oC в атмосфере азота. Через реакционную смесь барботировали озон, и через 11 мин завершение реакции контролировали с помощью ТСХ (9% гексан/1% этилацетат). После этого добавляли борогидрид натрия (9,9 г, 6 экв.) и через 5 мин реакционную смесь оставляли для нагревания до комнатной температуры. Метанол удаляли в вакууме. Образовавшийся остаток суспендировали в эфире (800 мл). Эфир промывали водой, а водный слой подвергали обратному экстрагированию эфиром. Объединенные органические слои промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали в вакууме с получением маслообразного вещества. Это вещество пропускали через слой двуокиси кремния (элюент: 5% этилацетат/гексан и затем 25% этилацетат/гексан), в результате чего получали 11,0 г (50%) спирта, 1-(трет-бутилдиметилсилилокси)-3-(2-(гидрокси)этокси)-4-(трет- бутилдифенил)-бутана в виде светло-желтого маслообразного вещества. МС. ЯМР.
К безводному эфирному (200 мл) раствору спирта, 1-(трет-бутилдиметилсилилокси)-3-(2-(гидрокси)этокси)-4-(трет- бутилдифенил)-бутана (11,0 г, 21,9 мМ), в атмосфере азота и при 5oC добавляли триэтиламин (4,6 мл, 1,5 экв. ) и метансульфонилхлорид (2,5 мл, 1,5 экв.). Через 1,5 ч реакцию завершали, о чем свидетельствовала ТСХ (5% этилацетат/дихлорметан). Реакционную смесь разводили эфиром (250 мл), промывали водой (дважды), а затем солевым раствором (дважды). После осушки сульфатом натрия, фильтрования и концентрирования в вакууме получали маслообразное вещество. Это вещество пропускали через слой двуокиси кремния (элюент: 5% этилацетат/гексан, а затем 25% этилацетат/гексан), в результате чего получали 11,6 г (выход 91%) мезилата, 1-(трет-бутилдиметилсилилокси)-3-(2-(метансульфонилокси) этокси)-4-(трет-бутилдифенил)-бутана в виде маслообразного вещества. МС. ЯМР.
К ацетоновому (300 мл) раствору мезилата, 1-(трет-бутилдиметилсилилокси)-3-(2-метансульфонилокси) этокси)-4-(трет-бутилдифенил)-бутана (11,6 г, 20 мМ), в атмосфере азота добавляли иодид натрия (44 г, 15 экв.) и бикарбонат натрия (170 мг, 0,1 экв.). Полученную смесь нагревали с обратным холодильником в течение 18 ч с последующим удалением ацетона в вакууме. Образовавшийся остаток суспендировали в эфире, дважды промывали водой и водный слой промывали эфиром. Объединенные эфирные фракции промывали 10%-ным раствором сульфита натрия, а затем дважды промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 10,7 г (выход 87%) целевого иодида в виде маслообразного вещества, которое использовали без дополнительной очистки. МС. ЯМР.
Получение 26
1-(2-(Метилсульфонилокси)-этокси)-2-((метилсульфонилокси)этил)- 3-(трет-бутилдифенилсилилокси)-пропан
К перемешанному раствору диметилаллилмалоната (34 г, 0,2 М) в т-бутиловом спирте (0,5 л) добавляли твердый борогидрид натрия (19 г, 0,5 М). Полученную реакционную смесь нагревали при 70oC, а затем по капле в течение часа добавляли метанол (162 мл). Затем смесь перемешивали в течение ночи при комнатной температуре. Для разложения избыточного количества борогидрида натрия добавляли 20 мл воды. Полученную смесь фильтровали через целит. Фильтрат концентрировали (100 мл) и экстрагировали этилацетатом (20 мл х 4). Объединенные экстракты осушали сульфатом магния и концентрировали при пониженном давлении, в результате чего получали относительно чистый диол, 2-аллилпропан-1,3-диол (19 г, выход 83%), который использовали в последующей стадии без дополнительной очистки.
К перемешанному раствору диола, 2-(2-пропен-1-ил)-пропан-1,3-диола (23,2 г, 0,19 М) в толуоле (1 л) добавляли анизальдегид (27,3 г, 0,20 М) и PPTS-кислоту (4 г, 10 М). Реакционную смесь загружали в колбу, снабженную ловушкой Дина-Старка, а затем реакционную смесь нагревали с обратным холодильником. Через 5 ч реакционную смесь охлаждали до комнатной температуры, разводили эфиром (1 л), промывали насыщенным гидрокарбонатом натрия (50 мл х 3), а затем водой (50 мл х 3) и солевым раствором (50 мл). Органический слой осушали сульфатом магния и концентрировали при пониженном давлении с получением остатка. Этот остаток элюировали через узкую колонку с силикагелем (элюент: 10% этилацетат в гексане). После выпаривания элюирующего растворителя получали анизилиден, 1,3-O-анизилиден-2-(2-пропен-1-ил)пропан (40 г, 89%) с Rf = 0,62 (25% этилацетат в гексане).
К перемешанному раствору анизилидена, 1,3-O-анизилиден-2-(2-пропен-1-ил)пропана (20,0 г, 85,3 мМ) в CH2Cl2 (500 мл) и буфере (pH 7,0; 25 мл), при 0oC добавляли DDQ (38,7 г, 170,4 мМ). Полученную реакционную смесь тщательно перемешивали и оставляли для нагревания до комнатной температуры. Через 12 ч реакционную смесь разводили эфиром (1 л), промывали водным насыщенным гидрокарбонатом натрия (200 мл х 2) и 10%-ным водным Na2SO3 (200 мл х 3), а затем осушали и концентрировали при пониженном давлении с получением остатка. Этот остаток элюировали через колонку с силикагелем (10 - 25% этилацетата в гексане). После выпаривания элюирующего растворителя получали анизоат, содержащий спирт, 3-O-(4-метоксибензоат)-2-(2-пропен-1-ил)пропан-1-ол (12,7 г, 61%). Rf = 0,14 (25% этилацетата в гексане). ЯМР.
К перемешанному раствору спирта, 3-O-(4-метоксибензоат)-2-(2-пропен-1-ил)-пропан-1-ола (16,58 г, 66,32 мМ) в метиленхлориде (250 мл) добавляли трихлороаллилимидат (24,80 г, 132,64 мМ) в циклогексане (500 мл). К этой смеси в атмосфере азота добавляли 1 мл трифтороуксусной кислоты. Через 12 ч образовывался белый осадок. Полученную реакционную смесь фильтровали. Фильтрат разводили эфиром (500 мл), промывали водой (100 мл х 3) и солевым раствором (100 мл), а затем осушали и концентрировали при пониженном давлении с получением остатка. Этот остаток элюировали через колонку с силикагелем, элюируя этилацетатом/гексаном (градиент 0 - 25% этилацетата). Полученный диен, 1-(2-пропен-1-окси)-2- (2-пропен-1-ил)-3-O-(4-метоксибензоат)-пропан (24 г), содержащий некоторое количество ацетамида, использовали в последующей стадии без дополнительной очистки. (Rf = 0,38, 25% этилацетат в гексане).
Сложный эфир, 1-(2-пропен-1-окси)-2-(2-пропен-1-ил)-3-O-(4-метоксибензоат)-пропан (24 г) растворяли в ТГФ (60 мл) и добавляли метанол (100 мл) и 1 н. водный NaOH (40 мл). Полученную смесь перемешивали в течение ночи, а метанол и ТГФ удаляли при пониженном давлении. Концентрированную реакционную смесь разводили эфиром (250 мл), экстрагировали эфиром (100 мл х 3), осушали и концентрировали при пониженном давлении с получением остатка. Этот остаток элюировали через колонку с силикагелем (10% этилацетат/гексан), а элюирующий растворитель выпаривали, в результате чего получали спирт, 1-(2-пропен-1-окси)-2-(2-пропен-1-ил)-пропен-3-ол (4,10 г, 30% из 2 стадий). ЯМР: Rf = 0,23 (25% этилацетат в гексане).
К перемешанному метиленхлоридному (150 мл) раствору спирта, 1-(2-пропен-1-окси)-2-(2-пропен-1-ил)-пропен-3-ола (4,10 г, 26,2 мМ), в атмосфере азота добавляли имидазол (2,70 г, 39,7 мМ). После растворения имидазола, в течение 10 мин добавляли трет-бутилхлородифенилсилан (8,24 г, 29,97 М) в метиленхлориде (50 мл). После 12-часового перемешивания, реакционную смесь разводили эфиром (100 мл), а затем гасили водой (100 мл) и экстрагировали эфиром (100 мл х 3). Объединенную органическую фазу промывали солевым раствором (100 мл), осушали и концентрировали при пониженном давлении с получением остатка. Этот остаток элюировали через узкую колонку с силикагелем (0 - 25% этилацетат/гексан). После выпаривания элюирующего растворителя получали силиловый эфир, 1-(2-пропен-1-окси)-2-(2-пропен-1-ил)-3-(трет- бутилдифенилсилилокси)-пропан (7,61 г, выход 72%). Rf = 0,76 (25% этилацетат в гексане).
Озон барботировали через метаноловый (-78oC, 500 мл) раствор диена, 1-(2-пропен-1-окси)-2-(2-пропен-1-ил)-3-(трет- бутилдифенилсилилокси)-пропана (7,41 г, 18,80 мМ). После исчезновения исходного материала (ТСХ: 25% этилацетат/гексан), реакционную смесь продували азотом и добавляли борогидрид натрия (2,13 г, 56,30 мМ). Реакционную смесь нагревали до комнатной температуры. Через 12 ч реакционную смесь концентрировали. Белый остаток гасили путем добавления воды, а затем экстрагировали этилацетатом (100 мл х 3). Объединенную органическую фазу промывали солевым раствором, осушали и концентрировали при пониженном давлении с образованием остатка. Остаток элюировали через узкую колонку с силикагелем (градиент 10 - 50% этилацетата/гексана). После выпаривания элюирующего растворителя получали 1,7-диол, 1-(2-гидроксиэтокси)-2-(2-гидроксиэтил)-3-(трет-бутилдифенилсилилокси)- пропан (5,48 г, выход 72%). Rf = 0,21 (50% этилацетат в гексане). ЯМР.
К перемешанному метиленхлоридному (400 мл) раствору диола, 1-(2-гидроксиэтокси)-2-(2-гидроксиэтил)-3-(трет-бутилдифенилсилилокси)- пропана (5,48 г, 13,6 мМ), в атмосфере азота добавляли ТЕА (11,2 мл, 78 мМ), а затем по капле в течение 30 мин добавляли метансулфонилхлорид (3 мл, 39,00 мМ) в метиленхлориде (100 мл). Через 12 ч реакционную смесь разводили эфиром (500 мл), после чего промывали водой (100 х 3) и солевым раствором (100 мл), а затем осушали и концентрировали при пониженном давлении с получением остатка. Остаток элюировали через узкую колонку с силикагелем (градиент 10 - 50% этилацетата/гексана). Элюирующий растворитель выпаривали и получали бисмезилат, 1-(2-метилсульфонилокси)-этокси)-2-((метилсульфонилокси)этил)-3- (трет-бутилдифенилсилилокси)-пропан (7,40 г, 97%). Rf = 0,55 (50% этилацетат в гексане). ЯМР.
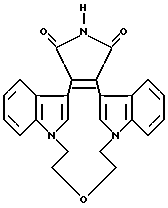
Пример 1
3,4-[(N,N'-Этоксиэтил)-бис-(3,3'-индолил)]-1H-пиррол-2,5-дион
В атмосфере азота, к раствору 3,4-бис(3'-индолил)-фуран-2,5-диона (337 мг, 1,02 мМ) в 5 мл сухого ДМФ, порциями в течение 15 мин добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 113 мг, 2,82 мМ). Полученную смесь перемешивали в течение 1,5 ч, а затем разводили 5 мл ДМФ. К образовавшемуся зеленому раствору по капле добавляли бис 2,2'-дибромо-этиловый эфир (0,14 мл, 1,13 мМ). Полученную реакционную смесь перемешивали в течение 30 мин при 25oC, а затем нагревали в течение ночи при 50oC. Охлажденную смесь выливали в разбавленную водную лимонную кислоту (75 мл) и экстрагировали этилацетатом (2 х 40 мл). Объединенные органические экстракты промывали водой (3 х 20 мл) и солевым раствором (20 мл), после чего осушали сульфатом магния. Растворитель выпаривали при пониженном давлении. Остаток пропускали через узкую колонку с силикагелем (50% EtOAc/гексан), а затем подвергали радиальной препаративной тонкослойной хроматографии (Chromatotron), элюируя 50% этилацетатом/гексаном. В результате этой процедуры получали 82 мг (20%) 2,3-[(N, N'-1,1'-этоксиэтил)-бис-(3,3'-индолил)] -1H-фуран-2,5-дион в виде твердого вещества цвета бордо, т.пл. > 320oC.
Раствор 2,3-[(N, N'-1,1'-этоксиэтил)-бис(3,3'- индолил)]-1H-фуран-2,5-диона (58 мг, 0,15 мМ) в ДМФ (1,5 мл), в атмосфере азота обрабатывали смесью 1,1,1,3,3,3-гексаметилдисилазана (0,33 мл, 1,45 мМ) и CH3OH (23 мг, 0,73 мМ) (предварительно смешанного в течение 10 мин). После перемешивания в течение 16 ч при комнатной температуре смесь выливали в воду (20 мл) и экстрагировали этилацетатом (3 х 5 мл). Объединенные органические экстракты несколько раз промывали водой, осушали сульфатом магния и концентрировали. Образовавшийся остаток очищали с помощью радиальной хроматографии (элюент: 3% CH3OH в CHCl3) и получали 3,4-[(N,N'-1,1'-этоксиэтил)-бис-(3,3'-индолил)]-1H-пиррол-2,5-дион (41,5 мг, 72%) в виде фиолетового твердого вещества, т.пл. > 320oC. МС: Анализ для C24H19N3O3: вычислено: 3971426; найдено: 3971438.
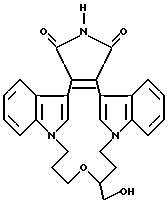
Пример 2.
3,4-[(N, N'-1,1')-((3''-пропокси)-3'''(O)-4''' (гидрокси)бутан)-(3,3'-индолил)]-1(H)-пиррол-2,5-дион
К перемешанному диметилформамидному (125 мл) раствору бис-(3, 3'-индолил)] -1-(метил)-пиррол-2,5-диона (4,35 г, 12,8 мМ), содержащего карбонат цезия (8,31 г, 25,5 мМ), по капле в течение 15 мин и в атмосфере азота добавляли диметилформамидный раствор (20 мл) 1-(трет-бутилдиметилсилилокси)-3-(3-иодопропилокси)-4-(трет- бутилдифенилсилилокси)-бутана (4,0 г, 6,4 мМ). Через 3 ч ТСХ (1:1 этилацетат/гексан) указывала на израсходование исходного иодида. Полученную реакционную смесь разводили этилацетатом (200 мл) и промывали водой. Водный слой экстрагировали этилацетатом (200 мл), а объединенные органические слои осушали и концентрировали. Концентрат осушали с помощью флэш-хроматографии (элюент: 10-25% этилацетат/гексан) и получали нужный моноалкилированный продукт, а именно, 3-[(N-1-(3-пропокси-3(O)- 4-трет-бутилдифенилсилилокси-1-трет-бутилдиметилсилилокси)-бутан] -4- (3'-индолил)-1-(метил)-пиррол-2,5-дион (3,94 г, выход 69%) в виде красного маслообразного вещества. МС.
К метаноловому (100 мл) раствору вышеописанного алкилированного продукта (3,14 г, 3,74 мМ) добавляли толуолсульфоновую кислоту (60 мг, 2%). Через 2 ч ТСХ (50% этилацетат/гексан) указывала на израсходование исходного материала. Полученную реакционную смесь концентрировали до половины объема, разводили этилацетатом (300 мл), промывали 1 н. гидроксидом натрия, а затем солевым раствором, после чего осушали и концентрировали. Концентрат очищали путем элюирования через слой двуокиси кремния (50% этилацетат/гексан), в результате чего получали нужный спирт, 3-[(N-1-(3-пропокси-O-4-трет-бутилдифенилсилилоксибутан-1-ол] -4-(3'- индолил)-1-(метил)-пиррол-2,5-дион (1,76 г, выход 65%) в виде красного пенообразного вещества. МС.
К эфирному (0oC) (200 мл) раствору вышеописанного спирта 3-[(N'-1-(3-пропокси-O-4-трет-бутилдифенилсилилокси-бутан-1-ол] -4- (3'-индолил)-1-(метил)-пиррол-2,5-диона (1,76 г, 2,4 мМ) добавляли триэтиламин (0,5 мл, 1,5 экв. ), а затем мезилхлорид (0,28 мл, 1,5 экв.). Реакционную смесь доводили до комнатной температуры и завершали через 1 ч. После этого реакционную смесь разводили эфиром (200 мл), а затем промывали водой и солевым раствором. После осушки и концентрирования, концентрат пропускали через слой двуокиси кремния (элюент: 50% этилацетат/гексан), в результате чего получали мезилатный продукт, который использовали непосредственно в последующей стадии.
К ацетоновому (250 мл) раствору вышеописанного мезилата добавляли иодид натрия (3,6 г, 10 экв.) и NaHCO3 (20 мг). После 4-часового перемешивания ТСХ обнаруживала присутствие исходного материала (50% этилацетат/гексан) и к реакционной смеси добавляли дополнительное количество иодида натрия (10 экв.), а затем смесь нагревали при 60oC. Через 4 ч ТСХ (50% этилацетат/гексан) указывала на израсходование исходного материала. Эту реакционную смесь концентрировали, разводили этилацетатом (250 мл), промывали водой и 10%-ным сульфитом натрия, а затем осушали и концентрировали. Концентрат очищали путем элюирования через слой силикагеля (50% этилацетат/гексан), в результате чего получали нужный иодид в виде маслообразного вещества, а именно, 3-[(N-1-(3-пропокси-3(O)-4-трет-бутилдифенилсилилокси-1-иодобутан] - 4-(3'-индолил)-1-(метил)-пиррол-2,5-дион (1,71 г, выход 85%). МС.
Диметилформамидный (10 мл) раствор вышеописанного иодида, 3-[(N-1-(3-пропокси-3(O)-4-трет-бутилдифенилсилилокси-1-иодобутан] -4- (3'-индолил)-1-(метил)-пиррол-2,5-диона (2,0 г, 2,4 мМ), медленно, с помощью шприца в течение 80 ч добавляли к диметилформамидному (400 мл) раствору карбоната цезия (3,12 г, 9,6 мМ). Через 3 ч после завершения добавления ТСХ (50% этилацетат/гексан) указывала на израсходование исходного материала. Реакционную смесь разводили этилацетатом (1 л), а затем промывали водой и солевым раствором. Водную часть раствора экстрагировали 500 мл этилацетата. Объединенные органические слои концентрировали и полученный концентрат очищали на колонках с двуокисью кремния (элюент: 50% этилацетат/гексан). После концентрирования элюента получали нужный макроциклический 3,4-[(N,N'-1,1')-(3''-пропокси)-3'''(O)-4'''(гидрокси)бутан)-бис-(3,3-индолил)] -1-(метил)пиррол-2,5-дион (1,65 г, выход 97%). МС.
К этаноловому (100 мл) раствору вышеописанного N-метилмалеимида, 3,4-[(N,N'-1,1')-((3'''-пропокси)-3'''(O)-4'''-(гидрокси)бутан)-бис) 3,3-индолил)] -1-(метил)-пиррол-2,5-диона (1,7 г, 2,4 мМ) добавляли 50 мл 5 н. KOH. Через 12 ч реакционную смесь нагревали 2 ч при 50oC. После этого реакционную смесь охлаждали до комнатной температуры, концентрировали, разводили этилацетатом и промывали водой. Органическую фазу осушали и концентрировали, в результате чего получали нужный ангидрид, 2,3-[(N,N'-1,1'-(3''-пропокси-3'''-(O)-4'''-гидроксибутан)-бис-(3,3'- индолил)] -фуран-1,4-дион (1,37 г, выход 83%) в виде красного маслообразного твердого вещества. МС.
К диметилформамидному (100 мл) раствору вышеописанного ангидрида, 2,3-[(N, N'-1,1'-((3''-пропокси)-3'''-(O)-4'''-(гидрокси)-бутан)-бис- (3,3'-индолил)] -фуран-1,4-диона (1,37 г, 3 мМ) добавляли 1,1,1,3,3,3-гексаметилдисилазан (12,6 мл, 20 экв.) и метанол (1,21 мл, 10 экв.). Через 24 ч ТСХ (50% этилацетат/гексан) указывала на полное израсходование исходного материала. Реакционную смесь разводили этилацетатом, промывали 1 н. соляной кислотой и водой, а затем осушали и концентрировали. Концентрат перемешивали в 1 н. соляной кислоте или с фторидом цезия для удаления остаточной TMS-группы. После этого, реакционную смесь разводили этилацетатом, промывали водой, а затем осушали и концентрировали, в результате чего получали 1,02 г (выход 75%) нужного малеимида, 3,4-[(N, N'-1,1')-((3''-пропокси)-3'''(O)-4'''- (гидрокси)-бутан)-бис-(3,3'- индолил)]-1(H)-пиррол-2,5-диона в виде красного твердого вещества, МС.
1H-ЯМР: (300 МГц в ДМСО-d6): 2,1 (м, 4H), 2,4 (м, 2H), 3,28 (шир.с,м), 3,4 (м, 4H), 4,5 (т. J= 6 Гц, 1H), 7,0-7,9 (м, 10H), 11,0 (с, 1H).
13C-ЯМР (75 МГц в ДМСО-d6): 20,9, 28,9, 30,3, 30,9, 34,3, 40,4, 41,6, 42,4, 62,4, 65,9, 78,1, 104,0, 104,1, 110,0, 110,1, 119,6, 119,7, 121,4, 121,8, 24,8, 126,5, 126,6, 127,9, 131,5, 131,6, 131,7, 135,8, 135,9, 139,1, 151,4, 172,2.
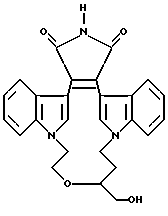
Пример 3
3,4-[(N, N'-1,1'-(2''-этокси)-3'''(O)-4'''-(гидрокси)-бутан)-бис- (3,3'-индолил)]-1(H)-пиррол-2-,5-дион
К диметилформамидному (250 мл) раствору бис-(3,3'-индолил)-1-(метил)-пиррол-2,5-диона (17,9 г, 52,5 мМ, 3 экв.), в атмосфере азота добавляли карбонат цезия (68,4 г, 4 экв.). К полученной суспензии добавляли иодид, 1-(трет-бутилдиметилсилилокси)- 3-(2-иодоэтокси)-4-(трет-бутилдифенилсилилокси)-бутан (10,7 г, 17,5 мМ). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. ТСХ (5% этилацетат/гексан) указывала на исчезновение иодида. После этого, реакционную смесь выливали в этилацетат (1200 мл) и промывали 400 мл 1 н. соляной кислоты, а затем подвергали обратному промыванию этилацетатом (дважды). Объединенные этилацетатные части промывали насыщенным раствором бикарбоната натрия, дважды промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме. Диметилформамид удаляли путем азеотропной дистилляции с использованием ксилена. Полученную красную камедь суспендировали в дихлорметане и ацетонитриле, в результате чего получали твердую суспензию. После концентрирования, добавляли еще дихлорметан, суспензию охлаждали и фильтровали с получением красного твердого вещества. Некоторую часть нужного продукта экстрагировали из полученного твердого вещества путем повторного растирания в дихлорметане, а затем в этилацетате. Фильтраты концентрировали в вакууме и полученный остаток абсорбировали на двуокиси кремния, а затем помещали в большую флеш-колонку. Диалкилированные побочные продукты удаляли путем элюирования 5%-ным гексаном/1%-ным этилацетатом, а затем путем элюирования 3% гексаном/1% этилацетатом, в результате чего получали 8,2 г (57%) моноалкилированного продукта, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''-(трет- бутилдиметилсилилокси)-бутан))-индол-3-ил] -4-[индол-3-ил]1N(метил)-пиррол-2,5- диона, МС, ЯМР.
К метаноловому (450 мл) раствору трет-бутилдиметилсилилового эфира, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''- (трет-бутилдиметилсилилокси)-бутан))-индол-3-ил]-4-[индол-3-ил]-1N- (метил)-пиррол-2,5-диона (8,2 г, 9,9 мМ), в атмосфере азота и при 5oC добавляли моногидрат толуолсульфоновой кислоты (0,16 г, 0,085 экв.). Через 2 ч ТСХ (50% этилацетат/гексан) указывала на полное завершение реакции. После этого реакционную смесь гасили путем добавления твердого бикарбоната натрия (0,14 г). Метанол удаляли в вакууме. Полученный остаток растворяли в этилацетате, промывали 0,1 н. гидроксидом натрия, а затем дважды промывали солевым раствором. После осушки сульфатом магния, фильтрования и концентрирования в вакууме, получали красное пенообразное вещество. Это вещество абсорбировали на двуокиси кремния и помещали на слой двуокиси кремния. Остаточный исходный материал элюировали 2%-ным гексаном/1% этилацетатом, а затем 1% гексаном/1% этилацетатом. После этого остаточный исходный материал удаляли и получали 6,4 г (91%) спирта, 3-[(N-1-(2-этокси-(3'''(O)-4'''-(трет-бутилдифенилсилилокси)-1-(гидрокси)- бутан))-индол-3-ил] -4-[индол-3-ил] -1N (метил)-пиррол-2,5-диона, МС, ЯМР.
К безводному эфирному (500 мл) раствору спирта, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''-(гидрокси)- бутан))-индол-3-ил]-4-[индол-3-ил]-1N (метил)-пиррол-2,5-диона (6,36 г, 8,9 мМ), в атмосфере азота и при 5oC добавляли триэтиламин (1,9 мл, 1,5 экв.) и метансульфонилхлорид (1,0 мл, 1,5 экв.). Через 3 часа добавляли еще 1,25 мл триэтиламина (1,0 экв) и 0,7 мл (1,0 экв.) метансульфонилхлорида. Через час реакция была полностью завершена, на что указывала ТСХ (50% этилацетат/гексан). Полученную реакционную смесь разводили эфиром (250 мл), промывали водой, 0,1 н. соляной кислотой и два раза солевым раствором. Эфирный слой осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 7,0 г мезилата, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''- (метансульфонилокси)-бутан))-индол-3-ил] -4-[индол-3-ил]-1N (метил)-пиррол-2,5-диона, МС.
К ацетоновому (200 мл) раствору мезилата, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''- (метансульфонилокси)-бутан))-индол-3-ил] -4-[индол-3-ил] -1N-(метил)-пиррол-2,5- диона (7,0 г, 8,9 мМ), в атмосфере азота добавляли иодид натрия (13,3 г, 10 экв.) и бикарбонат натрия (75 мг, 0,1 экв.). После этого, смесь перемешивали при 50oC в течение 13 ч. Реакционную смесь концентрировали в вакууме, а остаток растворяли в эфире и промывали 10% раствором сульфита натрия. После этого слои отделяли, а эфирную часть промывали 10%-ным раствором сульфата натрия, водой, а затем дважды промывали солевым раствором. После осушки и концентрирования в вакууме, остаток пропускали через слой двуокиси кремния, элюируя 1% гексаном/1% этилацетатом и 1% гексаном/2% этилацетатом. В результате этих процедур получали 7,6 г иодида, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''-(иодо)- бутан))-индол-3-ил] -4-[индол-3-ил] -1N (метил)-пиррол-2,5-диона в виде красного твердого вещества (количественный выход из двух стадий), МС, ЯМР.
К диметилформамидной (1 л) суспензии карбоната цезия (12,0 г, 4 экв.), в атмосфере азота, с помощью шприца и в течение 65 ч добавляли иодид, 3-[(N-1-(2-этокси-(3'''-(O)-4'''-(трет-бутилдифенилсилилокси)-1'''- (иодо)-бутан))-индол-3-ил] -4-[индол-3-ил]-1N (метил)пиррол-2,5-дион (7,6 г, 9,2 мМ), растворенный в 25 мл диметилформамида. Через 3 ч после завершения добавления, реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате (700 мл), промывали водой (2х300 мл), а водный слой подвергали обратному промыванию этилацетатом (2х200 мл). Объединенные этилацетатные части промывали солевым раствором (2х200 мл), осушали сульфатом магния, фильтровали и концентрировали в вакууме с получением остатка пурпурного цвета. Этот остаток абсорбировали на двуокись кремния и загружали в флеш-колонку. После элюирования 3% гексаном/1% этилацетатом, а затем 1% этилацетатом/1% гексаном получали 5,2 г (82%) макроциклического 3,4-[(N,N'-1,1'-((2'''-этокси)-3'''-(O)-4'''-(трет-бутилдифенилсилилокси)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона, МС, ЯМР.
Суспензию N-метилмалеимида, 3,4-[(N,N'-1,1'-[(2-этокси)-3'''(O)-4'''-(трет-бутилдифенилсилилокси)-бутан)-бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона в 5 н. KOH (150 мл) и этаноле (300 мл) перемешивали при комнатной температуре в течение 65 ч, а затем 1 ч при 60oC. Реакционную смесь концентрировали в вакууме до объема 150 мл и образовавшийся остаток суспендировали в воде, охлаждали до 5oC, после чего подкисляли до pH 3 путем добавления концентрированной соляной кислоты. Полученную красную водную суспензию экстрагировали этилацетатом (4х200 мл), осушали и концентрировали в вакууме, в результате чего получали 3,3 г неочищенного ангидридного спирта, 2,3-[(N, N'-1,1'-((2-этокси)-3'''(O)-4'''-(гидрокси)-бутан)-бис-(3,3- индолил))-фуран-1,4-диона в виде пурпурного твердого вещества, MC.
К диметилформамидному (250 мл) раствору ангидрида, 2,3-[(N,N'-1,1'-(2'''-этокси)-3'''-(O)-4'''-(гидрокси)бутан)-бис-(3,3- индолил)] -фуран-1,4-диона (3,3 г, 7,5 мМ), в атмосфере азота добавляли 1,1,1,3,3,3-гексаметилдисилазан (32 мл, 2 экв.) и метанол (3 мл, 10 экв.). Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем нагревали при 60oC в течение 2 ч. Диметилформамид удаляли в вакууме и полученный остаток растворяли в ацетонитриле (250 мл), после чего добавляли 1 н. соляную кислоту (50 мл). Реакционную смесь перемешивали в течение 15 мин, после чего концентрировали и распределяли между этилацетатом и водой (250 мл). Твердый продукт осаждали и получали спиртовой малеимид, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''(O)-4'''-(гидрокси)-бутан-бис-(3,3'- индолил)] -1(H)-пиррол-2,5-дион (0,92 мл, 28%). Небольшое количество (50 мл) этого продукта абсорбировали на двуокиси кремния и загружали во флеш-колонку. После элюирования дихлорметаном, 5% ацетонитрилом-дихлорметаном, а затем 10% ацетонитрилом/дихлорметаном получали 38 мг аналитически чистого материала. Этилацетат концентрировали и хроматографировали, в результате чего получали дополнительное количество 8%-ного неочищенного продукта. МС.
1H-ЯМH (ДМСО-d6): δ 1,96 (1H, м), 2,09 (1H, м), 3,31 (1H, м), 3,40 (1H, м), 3,51 (1H, м), 3,62 (1H, м), 3,89 (1H, м), 4,18 (3H, м), 4,35 (1H, м), 4,68 (1H, т, J = 2 Гц), 7,11 (2H, м), 7,19 (2H, м), 7,44 (1H, с), 7,46 (1H, д, J = 9 Гц), 7,51 (1H, с), 7,53 (1H, д, J = 9 Гц), 7,79 (1H, д, J = 8 Гц), 7,83 (1H, д, J = 8 Гц), 10,91 (1H, с).
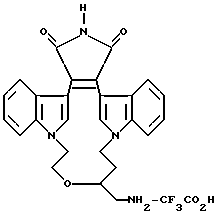
Пример 4
3,4-[(N, N'-1,1'-((2''-Этокси)-3'''(O)-4'''-(амино)-бутан)-бис- (3,3'-индолил)]-1(H)-пиррол-2,5-диона трифторацетатная соль
К безводному тетрагидрофурановому (15 мл) раствору спирта, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''(O)-4'''(гидрокси)-бутан)-бис- (3,3'-индолил)] -1(H)-пиррол-2,5-диона (155 мг, 0,35 мМ), в атмосфере азота добавляли 2,4,6-коллидин (280 мкл, 3 экв.). Полученный раствор охлаждали до -78oC и обрабатывали ангидридом трифторметансульфоновой кислоты (118 мкл, 2 экв.). После выдерживали в течение 1,5 ч при -78oC, добавляли избыточное количество концентрированного гидроксида аммония (2 мл). Через 10 мин реакционную смесь нагревали до -42oC в бане из сухого льда/ацетонитрила, после чего перемешивали в течение 18 ч, нагревали при этом до комнатной температуры. Реакционную смесь концентрировали в вакууме. Полученный остаток растворяли в этилацетате (400 мл), а затем промывали водой и солевым раствором, после чего осушали и концентрировали в вакууме, в результате чего получали неочищенный первичный амин. Этот амин абсорбировали на двуокиси кремния и загружали во флеш-колонку, после чего последовательно элюировали 1% этилацетатом/1% гексаном, этилацетатом, этилацетатом/5% метанолом и, наконец, 50% этилацетатом/45% ацетонитрилом/4% метанолом/2% изопропиламином, в результате чего получали амин, а именно, 3,4-[(N,N'-1,1'-(2''-этокси)-3'''(O)-4'''-(амино)-бутан)-бис-(3,3' -индолил)] -1(H)-пиррол-2,5-дион (38 мг). Также выделялся исходный спирт (104 мг, 67%). Этот продукт очищали с помощью обращенно-фазовой хроматографии высокого разрешения (элюент: 85% ацетонитрил/0,01% TFA-вода). Объединенные фракции подвергали азеотропной дистилляции с использованием этилацетата, в результате чего получали 23 мг (12%) TFA-соли в виде порошка, МС.
1H-ЯМР (d6-ДМСО): δ 1,99 (1H, м), 2,08 (1H, м), 2,82 (1H, м), 3,18 (1H, м), 3,57 (2H, м), 3,75 (1H, м), 4,13 (2H, м), 4,29 (1H, м), 4,44 (1H, м), 7,09 (2H, т, J = 7 Гц), 7,18 (2H, т, J = 7 Гц), 7,47 (4H, м), 7,70 (3H, шир. с), 7,78 (2H, м).
Аналогичным способом были получены S-энантиомер (4s) в качестве гидрохлоридной соли и R-энантиомер (4r) в качестве гидрохлоридной соли.
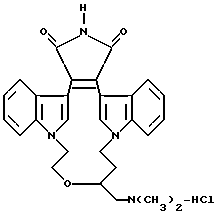
Пример 5
3,4-[(N, N-1,1'-((2''-Этокси)-3'''(O)-4'''-(N, N-диметиламино) бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
К безводной дихлорметановой (140 мл) суспензии спирта, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(гидрокси)-бутан)-бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона (472 мг, 1,07 мМ), в атмосфере азота добавляли пиридин (260 мкл, 3 экв.) и метансульфоновый ангидрид (242 мг, 1,3 экв.). Через 4 ч реакционную смесь разводили дихлорметаном, промывали дважды 1 н. соляной кислотой и фильтровали для удаления исходного материала (54 мг). Дихлорметановую часть промывали солевым раствором (дважды), осушали и концентрировали с получением неочищенного мезилата в виде пурпурного твердого вещества. Это вещество абсорбировали на двуокиси кремния и загружали во флеш-колонку, после чего элюировали дихлорметанам, 5% ацетонитрилом/дихлорметаном и 10% ацетонитрилом/дихлорметаном, в результате чего получали 288 мг (выход 52%) мезилата, 3,4-[(N, N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(метан-сульфонилокси)-бутан)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона, МС, ЯМР.
К тетрагидрофурановому (20 мл) раствору мезилата, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''(O)-4'''-(метансульфонилокси)-бутан)- бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона (304 мг, 0,59 мМ) добавляли 8,9 М-раствор диметиламина в тетрагидрофуране (7 мл, 100 экв.). После нагревания при 65oC в течение 24 ч в герметично закрытом сосуде, реакционную смесь разводили этилацетатом (200 мл), дважды промывали солевым раствором, осушали и концентрировали с получением неочищенного диметиламинового производного в виде твердого веществ. Это вещество абсорбировали на двуокиси кремния и загружали во флеш-колонку, после чего элюировали 3% этилацетатом/1% гексаном, этилацетатом и 2% изопропиламином/этилацетатом, в результате чего получали 193 мг (выход 70%) диметиламинового производного, которое имело 90% чистоту (ЖХВР-анализ). Это диметиламиновое производное, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(N, N'-диметиламино)- бутан)-бис-(3,3'-индолил)] -1(H)-пиррол-2,5-дион очищали более чем на 95%. Это вещество было получено в качестве трихлорацетатной соли, которую подвергали обращенно-фазовой эксклюзионной ЖХВР, элюируя 85% ацетонитрилом/15% смесью TFA/воды.
Трифтороацетатную соль 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(N, N'-диметиламино)-бутан)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона превращали в гидрохлоридную соль путем суспендирования соли в этилацетате и осторожного промывания 0,1 н. гидроксидом натрия (5х50 мл). Этилацетатную часть дважды промывали солевым раствором, осушали и концентрировали с получением свободного основания, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(N,N'-диметиламино)-бутан)- бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона. К безводной метаноловой суспензии (50 мл) свободного основания, 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(N, N'-диметиламино)-бутан)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона, добавляли 1 н. соляную кислоту в безводном эфире (13 мл, 50 экв.). После выпаривания эфира, остаток осушали в вакууме и получали 143 мг (выход 52%) гидрохлоридной соли 3,4-[(N,N'-1,1'-((2''-этокси)-3'''-(O)-4'''-(N, N-диметиламино)-бутан)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона в виде красного твердого вещества, МС.
1H-ЯМР (d6-ДМСО): δ 2,03 (1H, м), 2,26 (1H, м), 2,68 (6H, т, J = 5 Гц), 3,24 (1H, м), 3,28 (1H, м, после встряхивания с D2O), 3,64 (1H, м), 3,77 (2H, м), 4,07 - 4,38 (4H, м), 7,08 (2H, м), 7,17 (2H, м), 7,43 (3H, м), 7,52 (1H, д, J = 8 Гц), 7,79 (2H, м), 10,33 (1H, шир. с.), 10,92 (1H, с).
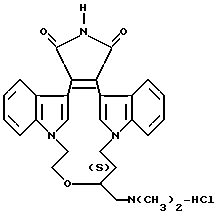
Пример 5s
(S)-3,4-[(N, N'-1,1'-((2''-Этокси)-3'''-(O)-4'''-(N, N-диметиламино)- бутан)-бис-(3,3'-индол)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
К тетрагидрофурановому (300 мл) раствору мезилата, (S)-3,4-[(N,N'-1,1')-((2''-этокси)-3''-(O)-4'''-(метансульфонилокси)- бутан-(бис)-(3-индолил)] -1H-пиррол-2,5-диона (2,8 г, 5,30 мМ), в герметично закрытом сосуде добавляли диметиламин (100 мл, 40% в воде). После нагревания при 50oC в течение 24 ч реакционную смесь концентрировали. Образовавшийся остаток пропускали через слой двуокиси кремния, элюируя этилацетатом, а затем 10% триэтиламином/этилацетатом, в результате чего получали нужное (S)-диметиламиновое производное. Элюент концентрировали с получением 1,7 г (выход 67%) свободного основания, а именно, (S)-3,4-[(N,N-1,1'-((2''-этокси)-3'''-(O)-4'''-(N, N-диметиламино)-бутан)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона в виде фиолетового твердого веществ. Это свободное основание превращали в гидрохлоридную соль путем суспендирования свободного основания, а именно, 3,4-[(N, N'-1,1')-(4''-N, N-диметиламино-3-(S)-''этокси-бутан)] -(бис)- (3-индолил)-1H-пиррол-2,5-диона (1,7 г, 3,6 мМ) в метаноле (300 мл) и добавления 1,0 н. безводной соляной кислоты в эфире (10 мл, 10 мМ). После отстаивания в течение 0,5 ч при комнатной температуре, ярко-оранжевый осадок собирали, промывали эфиром, а затем осушали в вакууме, в результате чего получали 1,4 г (выход 77%) гидрохлоридной соли 3,4-[(N,N'-1,1')-(4''-N,N-диметиламино-3-(S)-''-этокси-бутан)]-(бис)- (3-индолил)-1H-пиррол-2,5-диона, МС.
1H-ЯМР (d6-ДМСО): δ 2,1 (м, 1H), 2,35 (м, 1H), 2,68 (с, 6H), 3,2 (м, 1H), 3,33 (м, 1H), 3,66 (шир.т., 1H), 3,8 (шир. т, 1H), 3,85 (м, 1H), 4,17 (м, 1H), 4,2 - 4,4 (м, 3H), 7,1 (д, 1H), 7,13 (д, 1H), 7,2 (м, 2H), 7,44 (с, 1H), 7,48 (с, 1H), 7,5 (д, 1H), 7,56 (д, 1H), 7,82 (шир.т, 2H), 10,59 (шир. т, 1H), 10,96 (с, 1H),.
Пример 5r
(R)-3,4-[(N,N'-1,1-'-((2''-Этокси)-3'''-(O)-4'''-(N,N-диметиламино)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
(R)-Энантиомер получали способом, аналогичным способу получения (S)-энантиомера, за исключением того, что в качестве исходного материала использовали (R)-4-трет-бутилдифенилсилилокси-3-(2-иодоэтокси)- 1-иодобутан, МС, ЯМР.
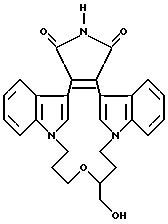
Пример 6
3,4-[(N, N'-1,1'-(2''-Этокси-(3'''(O)-метилен)-4'''-(гидрокси)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-дион
Сухой диметилформамидный (100 мл) раствор (бис)мезилата, 1-(2-метилсульфонилокси)-этокси)-2-((метилсульфонилокси)этил)-3- (трет-бутилдифенилсилилокси)-пропана (7,4 г, 13,30 мМ) и бис-(3,3'-индолил)]-1(метил)-пиррол-2,5-диона (4,43 г, 13,30 мМ), в течение 16 ч при 50oC добавляли к перемешанной суспензии Cs2CO3 (25,4 г, 78 мМ) в ДМФ (400 мл). Через 8 часов реакционную смесь концентрировали при пониженном давлении (80oC) и получали остаток. Этот остаток разводили этилацетатом (200 мл), а затем промывали водой (50 мл). Органический слой отделяли, а водный слой экстрагировали этилацетатом (50 мл x 3). Объединенные органические части осушали и концентрировали с получением остатка. Остаток элюировали на колонке с силикагелем (25% этилацетат в гексане, а затем 5% метанол в метиленхлориде), в результате чего получали три преобладающих продукта: силилэфирный макроциклический продукт, 2,3-[(N, N'-1,1'-(4'''-этокси-1'-ил-(3'''-(трет- бутилдифенилсилилокси)метилен)бутан-1-ил)- бис-(3,3'-индолил)] -1-(метил)-пиррол-1,4-дион (2,35 г); МС: Анализ для C44H45N3O4Si: Мол. масса: 707,31, найдено: 708; Rf = 0,84 (50% этилацетат в гексане) и десилилированный спирт (макроциклический продукт; 600 мг), МС.
К перемешанному EtOH (500 мл)-раствору N-метилмакроциклического продукта, 2,3-[(N, N'-1,1'-(-4'''-этокси-1'-ил-(3'''-(трет- бутилдифенилсилилокси)метилен)бутан-1-ил)- бис-(3,3'-индолил)] -1(метил)-пиррол-1,4-дион (1,65 г, 2,33 мМ) добавляли 100 мл 5 н. КОН. После выдерживания в течение 12 ч при 50oC, реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением остатка. Полученный остаток подкисляли путем добавления концентрированной соляной кислоты до pH 1, а затем экстрагировали этилацетатом (200 мл x 5). Объединенные органические фазы осушали, концентрировали при пониженном давлении, а затем элюировали через узкую колонку с двуокисью кремния (5% метанол в дихлорметане). После выпаривания элюирующего растворителя получали остаток, содержащий ангидрид, а именно, 2,3-[(N,N'-1,1'-(4'''-этокси-(1'-ил-(3'''-(трет- бутилдифенилсилилокси)метилен)-бутан-1-ил)-бис-(3,3'-индолил)] - фуран-1,4-дион, который использовали в последующей стадии.
К безводному диметилформамидному (250 мл) раствору ангидрида, 2,3-[(N, N'-(4'''-этокси-1'-ил-(3'''-(трет- бутилдифенилсилилокси)метилен)-бутан-1-ил)-бис-(3,3'- индолил)] -фуран-1,4-диона (600 мг, 1,3 мМ) добавляли HMDS (2,1 г, 13 мМ), а затем метанол (209 мг, 6,5 мМ). Через 48 ч реакционную смесь концентрировали и полученный остаток растворяли в этилацетате (100 мл), а затем промывали 25 мл 1 н. соляной кислоты, водой (25 мл) и солевым раствором (25 мл). Полученную органическую фазу осушали и концентрировали в вакууме с получением остатка. Полученный остаток элюировали на колонке с силикагелем (0 - 5% метанол/CH2Cl2). После выпаривания элюирующего растворителя получали 300 мг (выход 50%) имида, 3,4-[(N, N'-1,1'-(2''-этокси-(3'''(O)-метилен)-4'''-(гидрокси)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона в виде твердого вещества, МС.
1H-ЯМР (CDCl3) δ: 9,65 (с, 1H), 7,79 (т, J = 7,65 Гц), 7,61 (с, 1H), 7,54 (с, 1H), 7,46 - 7,40 (м, 2H), 7,24 - 7,08 (м, 2H), 7,07 - 7,02 (м, 2H), 4,43 - 4,33 (м, 2H), 4,30 - 4,21 (м, 1H), 4,14 - 4,06 (м, 1H), 3,64 (т, J = 4,64 Гц), 3,58 - 3,38 (м, 5H), 3,71 (т, J = 8,64 Гц, 1H), 1,89 - 1,85 (м, 1H).
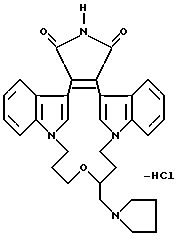
Пример 7
3,4-[(N, N'-1,1'-(2''-Этокси-(3'''((O)-метилен)-4'''-(N-пирролидино)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
К безводному метиленхлоридному (50 мл) раствору имидного спирта, 3,4-[(N, N'-1,1'-(2''-этокси-(3'''(O)-метилен)-4'''-(гидрокси)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона (140 мг, 0,30 мМ), содержащего пиридин (120 мг, 1,5 мМ) в атмосфере азота добавляли метансульфоновый ангидрид (106 мг, 0,61 мМ). Через 12 ч реакцию гасили водой (25 мл), разводили метиленхлоридом (50 мл), а затем промывали 0,2 н. соляной кислотой (20 мл x 2), водным раствором бикарбоната натрия (20 мл), снова водой (20 мл) и солевым раствором (20 мл), после чего осушали и концентрировали с получением остатка. Этот остаток элюировали через узкую колонку с силикагелем (5% метанол в дихлорметане) и после выпаривания элюирующего растворителя получали мезилат, 3,4-[(N,N'-1,1'-(2''-этокси-(3'''(O)-метилен)-4'''-(метансульфонилокси)- бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-дион, который использовали в последующей стадии.
В герметично закрытом сосуде, к тетрагидрофурановому (20 мл) раствору мезилата, 3,4-[(N,N'-1,1'-(2-этокси-(3'''(O)-метилен)-4'''-(метансульфонилокси)-бутан)-бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона (157 мг, 0,29 мМ) добавляли пирролиден (203 мг, 2,90 мМ). После нагревания при 50oC в течение 12 ч реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении, растворяли в метиленхлориде (50 мл), промывали водой (20 мл x 2) и солевым раствором (20 мл), после чего осушали и концентрировали при пониженном давлении с получением остатка. Остаток элюировали через узкую колонку с силикагелем (5% метанол в дихлорметане), после чего элюирующий растворитель выпаривали и получали пирролидин, 3,4-[(N,N'-1,1'-(2'-этокси-(3'''(O)-метилен)-4'''-(N-пирролидино)- бутан)-бис-(3,3'-индолил)] -1(H)-пиррол-2,5-дион. Масс-спектроскопия для C31H32N4O3: Молярная масса: 508,62; найдено: 508, Rf = 0,14 (5% метанол в дихлорметане, следовые количества триэтиламина). Пирролидин, кроме того, очищали с помощью обращенно-фазовой гель-хроматографии, в результате чего получали пирроловый макроциклический продукт в качестве трифтороуксусной соли (50 мг, выход 37%). Соль трифтороуксусной кислоты пиррола превращали в гидрохлоридное целевое соединение путем экстрагирования 1 н. гидроксида натрия (5 мл) с суспензией соли трифтороуксусной кислоты (55 мг) и смесью этилацетата (25 мл) и метанола (2 мл). Полученный экстракт осушали и концентрировали, в результате чего образовывался остаток. Этот остаток суспендировали в эфире/метаноле (10 : 1), а затем добавляли гидрохлоридный раствор эфира. Через 30 мин суспензию концентрировали и осушали в вакууме, в результате чего получали целевое соединение (48 мг, выход 88%), МС.
1H-ЯМР: δ 10,98 (с, 1H), 7,90 (с, 1H), 7,82 (с, 1H), 7,70 - 7,62 (м, 3H), 7,56 - 7,50 (м, 1H), 7,24 - 7,02 (м, 4H), 4,50 - 4,20 (м, 4H), 2,82 - 2,44 (м, 4H), 2,26 - 2,24 (м, 1H), 1,82 - 1,60 (м, 6H), 1,26 - 1,02 (м, 2H).
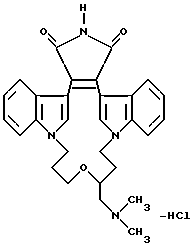
Пример 8
3,4-[(N, N'-1,1'-(2''-Этокси-(3'''(O)-метилен)-4'''- (N, N-диметиламино)-бутан)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
Целевой третичный амин получали путем замены мезилата на диметиламин (58 мг, выход 75%). МС.
1H-ЯМР (CDCl3): δ 10,93 (с, 1H), 7,84 (с, 1H), 7,77 (с, 1H), 7,69 - 7,64 (м, 3H), 7,47 (д, J = 7,97 Гц, 1H), 7,13 - 7,02 (м, 4H), 4,40 - 4,11 (м, 4H), 3,73 - 3,20 (м, 4H), 2,50 (с, 3H), 2,33 (с, 1H), 2,13 - 1,96 (м, 2H), 1,86 - 1,70 (м, 1H), 1,21 - 1,10 (м, 2H).
Следующие соединения были получены способом, аналогичным способу, описанному в Примерах настоящей заявки, и, кроме того, проиллюстрированы соединения настоящего изобретения. В последующих примерах структуры указанных соединений были подтверждены ЯМР-анализом, МС и/или элементным анализом. В процессе синтеза R представляет собой защищенную гидрокси-группу, предпочтительно, силилэфирную группу, а более предпочтительно, трет-бутилдифенилсилилокси-группу (TBDPS). Силиловый эфир может быть превращен в уходящую группу и замещен для получения следующих соединений (см. примеры 9 - 25r в конце описания).
Пример 26
3,4-[(N, N'-1,1'-(2-Метилен-6-метиленпиридин)-бис-(3,3'- индолил)-1H-пиррол-2,5-дион
В соответствии с аналогичной процедурой, описанной в примере 1, 2,3-бис-индолмалеиновый ангидрид (287 мг, 0,88 мМ) в 5 мл диметилформамида обрабатывали гидридом натрия (60% в минеральном масле, 88 мг, 2,19 мМ) в течение полутора часов, после чего разводили диметилформамидом до объема 11 л и обрабатывали бис-2,6-дибромометилпиридином (245 мг, 0,93 мМ). После перемешивания в течение ночи при 50oC реакционную смесь обрабатывали этилацетатом и фильтровали через узкую пробку из двуокиси кремния (50% EtOAc в гексане). Таким образом получали N,N-(2,6-пиридин-мостиковая связь)-бис-индолмалеиновый ангидрид (142 мг, 37%) в виде темно-красного твердого вещества, которое показало, в основном, одно пятно (ТСХ-анализ) и было использовано непосредственно в последующей стадии без дополнительной очистки.
3,4-[N,N'-1,1'-(2-Метилен-6-метиленпиридин)-бис-(3,3'- индолил)-1H-фуран-2,5-дион (140 мг, 0,32 мМ) в 2 мл ДМФ обрабатывали смесью 1,1,1,3,3,3-гексаметилдисилазана (0,72 мл, 3,2 мМ) и CH3OH (0,063 мл, 1,6 мМ), в результате чего после обработки и очистки с помощью радиальной хроматографии на силикагеле получали 42 мг целевого соединения, N,N'-(2,6-пиридин-мостиковая связь)-бис- индолмалеимида в виде бордового твердого вещества. Это вещество являлось гомогенным, на что указывала ТСХ (Rf = 0,35, 3% CH3OH в CHCl3).
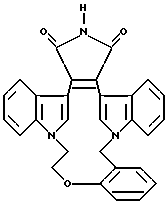
Пример 27
Гидрохлорид 3,4-[(N,N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'- индолил)]-1(H)-пиррол-2,5-диона
Сухой диметилформамидный (45 мл) раствор дибромида, 2-(2''-бромоэтокси)-бензилбромида (2,0 г, 6,8 мМ) и бис-(3,3'-индолил)]-1(метил)-пиррол-2,5-диона (2,3 г, 6,8 мМ), с помощью шприца и в течение 20 ч добавляли к суспензии Cs2CO3 (8,9 г, 27 мМ) в сухом ДМФ (550 мл), тщательно перемешивая при этом в атмосфере азота. Через 2 ч реакционную смесь концентрировали в вакууме, а остаток растворяли в метиленхлориде, промывали 1 н. соляной кислотой и солевым раствором, после чего осушали и концентрировали в вакууме, в результате чего получали фиолетовое маслообразное вещество. Это маслообразное вещество пропускали через узкую пробку из двуокиси кремния, элюируя гексаном/этилацетатом (1:1). Элюент концентрировали и получали макроциклический 3,4-[(N, N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'-индолил)]-1-(метил)- пиррол-2,5-дион (2,76 г, выход 71%) в виде пурпурного твердого красителя. После перекристаллизации из изопропанола/метиленхлорида получали аналитически чистый материал.
МС: MW = 473; найдено: 473 (FD, CHCl3). EA: Вычислено: C 76,09 (75,86); H 4,90 (4,93); N 8,87 (8,79).
К этаноловому (100 мл) раствору макроциклического 3,4-[(N,N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'-индолил)] -1-(метил)- пиррол-2,5-диона (710 мг, 15 мМ), содержащего ТГФ (20 мл), добавляли 5 н. KOH (80 мл). Реакционную смесь нагревали при 55oC в течение 70 ч, перемешивали при этом, после чего охлаждали до комнатной температуры, а этанол удаляли в вакууме. Концентрат подкисляли путем добавления 5 н. соляной кислоты до pH 1 (325 мл), экстрагировали этилацетатом, промывали дважды солевым раствором, осушали и концентрировали в вакууме, в результате чего получали ангидрид, 3,4-[(N,N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'-индолил)] -фуран- 2,5-дион (700 мг, количественное превращение) в качестве остатка.
К сухому диметилформамидному (500 мл) раствору ангидрида, 3,4-[(N,N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'-индолил)] -фуран- 2,5-диона (760 мг, 17 мМ) добавляли метанол (0,34 мл, 8,3 мМ) и 1,1,1,3,3,3-гексаметилдисилазан (3,5 мл, 17 мМ). После 22-часового нагревания при 55oC реакционную смесь концентрировали в вакууме, разводили этилацетатом и промывали 0,1 н. соляной кислоты. Объединенный органический слой осушали и концентрировали с получением фиолетового остатка. Этот остаток пропускали через узкую пробку с силикагелем и элюировали CH2Cl2/гексаном (градиент 0 - 100% CH2Cl2). После выпаривания элюирующего растворителя получали H-малеимид, 3,4-[(N,N'-1,1'-(2''-этокси)-бензил)-бис-(3,3'-индолил)] -1(H)- пиррол-2,5-дион (483 мг, выход 70%) в виде пурпурного твердого вещества. Полученное целевое соединение кристаллизовали из CH2Cl2/гексана. МС.
1H-ЯМР: (ДМСО-d6): δ 4,29 (2H, шир.с), 4,59 (2H, шир.с), 5,23 (2H, шир. с), 6,90 - 6,99 (2H), 7,01 - 7,18 (3H), 7,20 - 7,27 (2H), 7,59 - 7,68 (2H), 7,71 - 7,80 (5H), 10,92 (H, с).
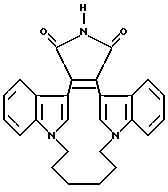
Пример 28
3,4-[(N,N'-1,1'-Гексан)-бис-(3,3'-индолил)]-1H-пиррол-2,5-дион
К раствору 3,4-бис-(3-индолил)]-1-метил-пиррол-2,5-диона (499 мг, 1,46 мМ) в 10 мл ДМФ, в атмосфере азота, порциями в течение 30 мин добавляли гидрид натрия (60% в масле, 146 мг, 3,65 мМ). Полученный зеленый раствор перемешивали в течение часа. После этого смесь разводили 10 мл ДМФ, а затем по капле обрабатывали 1,6-дибромогексаном (0,24 мл, 1,57 мМ). Реакционную смесь перемешивали 30 мин при комнатной температуре, а затем нагревали 16 ч при 45oC. Охлажденную смесь выливали в разбавленный водный NH4Cl (125 мл) и экстрагировали этилацетатом (3 х 40 мл). Объединенные органические экстракты промывали водой и осушали сульфатом магния. После удаления растворителя в вакууме, остаток очищали с помощью флеш-хроматографии на силикагеле (элюент: градиент CH2Cl2/гексан, 1 : 1 - 3 : 1), в результате чего получали целевое соединение, а именно, 3,4-[(N,N'-1,1'-гексан)-бис-(3,3'-индолил)]-1-метил-пиррол-2,5-дион (137 мг, 22%) в виде пурпурного твердого вещества, т.пл. > 320oC.
Смесь, содержащую 3,4-[(N,N'-1,1'-гексан)-бис-(3,3'-индолил)]-1-метил-пиррол-2,5-дион (137 мг, 322 мМ), этанол (15 мл), 5 н. KOH (5 мл) и ТГФ (2 мл), перемешивали в течение 4 ч при комнатной температуре. В это время ТСХ-анализ указал на израсходование исходного материала. Полученную смесь разводили водой (15 мл) и концентрировали на роторном испарителе. Смесь охлаждали, подкисляли 3 н. соляной кислотой до pH 1 и экстрагировали метиленхлоридом (3 х 10 мл). Объединенные органические экстракты тщательно промывали водой, осушали безводным сульфатом магния и концентрировали. Полученное пурпурное твердое вещество (116 мг), идентифицированное с помощью ЯМР-анализа, представляло собой смесь (4 : 1) целевого ангидрида и исходного материала. Эту смесь использовали непосредственно в последующей стадии без дополнительной очистки.
В соответствии с процедурой, описанной в примере 1, раствор 3,4-[(N, N'-1,1'-гексан)-бис-(3,3'-индолил)-фуран-2,5-диона (108 мг, 0,263 мМ) в ДМФ (1,5 мл), в течение ночи и в атмосфере азота обрабатывали смесью 1,1,1,3,3,3-гексаметилдисилазана (0,59 мл, 2,62 мМ) и CH3OH (0,05 мл, 1,31 мМ). После обработки этилацетатом, неочищенный продукт подвергали флеш-хроматографии на силикагеле (элюируя градиентом CH2Cl2/EtOAc, 10 : 1 - 5 : 1), в результате чего получали две окрашенные фракции. Первая окрашенная проэлюированная фракция содержала 3,4-[(N,N'-1,1'-гексан)-бис-(3,3'-индолил)-1-метил-пиррол-2,5-дион в виде примеси, выходящей из предыдущих фракций. Вторая окрашенная фракция содержала нужный продукт, 3,4-[(N,N'-1,1'-гексан)-бис-(3,3'-индолил)-1H-пиррол-2,5-дион (56 мг). Т.пл. > 320oC. МС.
Анализ для C26H32N3O2 (0,3 H2O):
Вычислено: C 76,26, H 5,66, N 10,26.
Найдено: C 75,21, H 5,65, N 10,05.
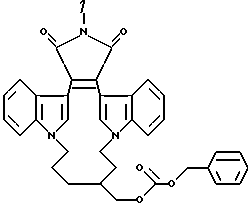
Пример 29
3,4-[(N,N'-1,1'-(3''-Бензилкарбонат)метилен)гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-дион
При 0oC к дихлорметановому раствору 3,4-[(N,N'-1,1'-(3''-(гидрокси)метилен)гексан)-бис-(3,3'-индолил)] -1H- пиррол-2,5-диона (24 мг, 0,054 мМ) добавляли диизопропилэтиламин (10,6 мг, 0,081 мМ), а затем бензилхлороформат (13,8 мг, 0,081 мМ). Через 72 ч реакционную смесь гасили 2,5 н. бикарбонатом натрия, органический слой удаляли, а водный слой экстрагировали дихлорметаном. Объединенные органические слои объединяли, промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали с получением маслообразного вещества. Это вещество очищали с помощью обращенно-фазовой ЖХВР (5% ацетонитрил в воде/0,1% TFA в 100% ацетонитриле) на C18-колонках, в результате чего получали 6 мг целевого соединения. МС.
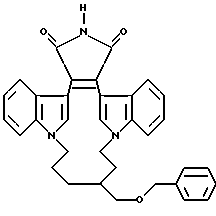
Пример 30
(±)-3,4-[(N, N'-1,1'-(3''-Бензилоксиметилен)гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-дион
В соответствии с аналогичной процедурой, описанной в предыдущих примерах, 3,4-бис-(3'-индолил)]-1-метил-пиррол-2,5-дион (400 мг, 1,17 мМ) в 8 мл ДМФ обрабатывали гидридом натрия (60% в масле, 117 мг, 2,93 мМ), а затем (±)-3-бензилоксиметилен-1,6-дибромогексаном в 7 мл ДМФ. После нагревания при 50oC в течение ночи неочищенный продукт обрабатывали и очищали с помощью флеш-хроматографии на силикагеле, элюируя CH2Cl2/гексаном (1 : 1 - 2 : 1), и получали чистый (±)-3,4-[(N,N'-(3''-(бензилоксиметилен)гексан)-бис- (3,3'- индолил)] -1-метил-пиррол-2,5-дион (149 мг, 23%) в виде фиолетового твердого вещества.
Смесь, содержащую (±)-3,4-[(N,N'-1,1'-(3''-бензилоксиметилен)гексан)-бис- (3,3'-индолил)]-1-метил-пиррол-2,5-дион (141 мг, 0,259 мМ), этанол (15 мл) и 5 н. KOH (5 мл) перемешивали при комнатной температуре в течение 3 ч. ТСХ-анализ указывал на израсходование исходного материала. После подкисления и экстрагирования метиленхлоридом, неочищенный продукт (101 мг) показал два пятна (ТСХ-анализ) (метиленхлорид), соответствующих исходному материалу и нужному ангидриду, (±)-3,4-[(N,N'-1,1'-(3''-бензилоксиметилен)гексан)-бис-(3,3'-индолил)] -фуран-2,5-диону. По грубой оценке, ЯМР-анализ указал на присутствие смеси (4 : 1) ангидрида и исходного материала. Этот материал использовали непосредственно в последующей стадии без дополнительной очистки.
(±)-3,4-[(N, N'-1,1'-(3''-Бензилоксиметилен)гексан)-бис- (3,3'-индолил)] -фуран-2,5-дион (98 мг, 0,180 мМ) в 1 мл ДМФ обрабатывали смесью 1,1,1,3,3,3-гексаметилдисилазана (0,41 мл, 1,80 мМ) и CH3OH (0,036 мл, 0,90 мМ) в течение ночи при 25oC. После этого смесь обрабатывали этилацетатом и подвергали флеш-хроматографии на силикагеле (элюируя градиентом CH2Cl2, а затем CH2Cl2 - EtOAc, 10 : 1), в результате чего получали 30 мг очищенного (±)-3,4-[(N, N'-1,1'-(3''-(бензилокси)метилен)гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-диона, Т.пл. 171 - 173oC. МС.
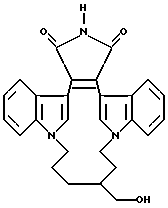
Пример 31
3,4-[(N, N'-1,1'-(3''-(гидрокси)метилен)гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-дион
Смесь, содержащую бис-(3,3'-индолил)]-1-(метил)-пиррол-2,5-дион (3,41 г, 10,0 мМ) и 3-трет-бутилдифенилсилилоксиметилен-1,6-дибромогексан (5,64 г, 11,0 мМ) в 50 мл ДМФ, в атмосфере азота с помощью шприца в течение 30 мин и при 55oC добавляли к тщательно перемешанному раствору карбоната церия (11,2 г, 34,3 мМ) в ДМФ (350 мл). После завершения добавления, реакционную смесь нагревали при 55oC еще 16 ч. Затем охлажденную смесь выливали в 1,2 л воды, содержащей 20 мл 3 н. соляной кислоты, после чего экстрагировали 300-миллилитровыми порциями метиленхлорида. Объединенные органические экстракты промывали водой и солевым раствором, а затем осушали сульфатом магния и концентрировали. Остаток пропускали через колонку (3'' • 3'') с силикагелем, элюируя хлороформом. Полученный таким образом неочищенный продукт очищали с помощью флеш-хроматографии на силикагеле (элюент: CHCl3), в результате чего получали 2,87 г (41%) 3,4-[(N,N'-(3''-трет-бутилдифенилсилилоксиметилен)-гексан)-бис- (3,3'-индолил)] -1-(метил)-пиррол-2,5-диона в виде пурпурного твердого вещества, т.пл. 220 - 224oC.
ВРМС для C44H45N3SiO [M + 1]:
Вычислено: 6923307.
Найдено: 6923299.
Смесь, содержащую 3,4-[(N,N'-1,1'-(3''-трет-бутилдифенилсилилоксиметилен)-гексан)-бис- (3,3'-индолил)] -1(H)-метил-пиррол-2,5-дион (1,55 г, 2,22 мМ), 4 н. KOH (100 мл), ТГФ (10 мл) и 95% EtOH (200 мл), нагревали в течение 16 ч при 90oC. После удаления большей части EtOH на роторном испарителе, смесь подкисляли до pH 1 путем добавления 6 н. соляной кислоты, а затем экстрагировали метиленхлоридом (3 х 75 мл). Объединенные органические экстракты промывали водой и солевым раствором, после чего осушали безводным сульфатом натрия. После удаления растворителя в вакууме, образовавшийся остаток растворяли в минимальном количестве 5%-ного метанола в CHCl3 и помещали на 3'' • 3''-колонку с силикагелем. После элюирования хлороформом, а затем 100% метанолом в хлороформе получали две фракции. После выпаривания второй фракции получали ангидридный спирт в виде пурпурного твердого вещества, которое являлось гомогенным, на что указывала ТСХ (Rf = 0,5; 10% метанол в CHCl3). Это вещество использовали непосредственно в последующей стадии без дополнительной очистки.
К раствору вышеописанного ангидрида (510 мг, 1,15 мМ) в ДМФ (11 мл) добавляли предварительно перемешанный раствор, содержащий 1,1,1,3,3,3-гексаметилдисилазан (5,14 мл, 23 мМ) и CH3OH (0,45 мл, 11,5 мМ). Полученную смесь нагревали в течение 24 ч (при 50oC) в атмосфере азота. Охлажденную реакционную смесь выливали в 100 мл воды. Осажденный продукт промывали водой и осушали в течение ночи, в результате чего получали 409 мг 3,4-[(N,N'-1,1'-(3''-гидроксиметилен)-гексан)-бис-(3,3'-индолил)] -1H- пиррол-2,5-диона в виде рыжевато пурпурного твердого вещества. В соответствии с обращенно-фазовым ЖХВР-анализом, это вещество имело 93% чистоту с примесью неидентифицированного соединения с аналогичным Rf.
ВРМС для C27H25N3O3:
Вычислено: 4391896.
Найдено: 4391911.
Пример 31r
(R)-3,4-[(N, N'-1,1'-(3''-(Гидроксиметилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион
В соответствии с процедурой, описанной выше в примере 31, (R)-3,4-[(N, N'-1,1'-(3''-(гидроксиметилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-дион был получен с полным 25% выходом из дибромида, (R)-3-(трет-бутилдифенилсилилоксиметилен)-1,6- дибромогексана, путем диалкилирования бис-(3,3'-индолил)] -1-(метил)-пиррол-2,5-диона, с последующим гидролизом и образованием 1-H-пиррол-2,5-диона, т.пл. > 300oC.
1H-ЯМР (300 МГц, ДМСО-d6): 1,05 - 2,25 (м, 7H), 4,04 - 4,45 (м, 6H), (м, 8H), 7,08 - 7,88 (м, 10H).
Пример 31s
(S)-3,4-[(N, N'-1,1'-(3''-(Гидроксиметилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион
В соответствии с аналогичной процедурой, описанной выше в примере 31, (S)-3,4-[(N, N'-1,1'-(3''-(гидроксиметилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион был получен (4,5 г) с полным 39% выходом из дибромида, (S)-3-(трет-бутилдифенилсилилоксиметилен)-1,6- дибромогексана, путем диалкилирования бис-(3,3'-индолил)] -1-(метил)-пиррол-2,5-диона, с последующим гидролизом и образованием 1-H-пиррол-2,5-диона. МС.
1H-ЯМР (d6-ДМСО): δ 1,05 - 2,25 (2H), 1,23 - 1,24 (1H), 1,50 - 1,52 (1H), 1,71 (1H), 1,94 (1H), 2,07 - 2,12 (1H), 4,05 - 4,4 (м, 6H), 7,09 - 7,21 (м, 4H), 7,35 (д, J = 15 Гц, 2H), 7,49 (д, J = 9 Гц, 2H), 7,8 (д, J = 9 Гц, 2H), 10,93 (с, 1H).
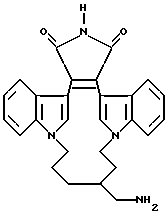
Примеры 32 и 33
3,4-[(N, N'-1,1'-(3''-Аминометилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион примера 32 в качестве TFA-соли и примера 33 в качестве HCl-соли
К перемешанному раствору ангидридного спирта, 2,3-[(N,N'-1,1'-(3''-(гидроксиметилен)-гексан)-бис-(3,3'- индолил)]-фуран-1,4-диона (0,18 г, 0,41 мМ) в безводном дихлорметане (10 мл), в атмосфере азота добавляли триэтиламин (0,10 г, 1,06 мМ) и метансульфонилхлорид (0,11 г, 0,98 мМ). Полученный раствор перемешивали 30 мин при комнатной температуре. Растворитель удаляли в вакууме. Образовавшийся остаток растворяли в 10 мл безводного диметилформамида, а затем добавляли азид натрия (0,26 г, 4,1 мМ). Реакционную смесь нагревали в атмосфере азота при 50oC в течение 1,5 ч. После этого охлажденную реакционную смесь распределяли между 0,2 н. соляной кислотой и этилацетатом. Объединенный органический экстракт осушали сульфатом магния, фильтровали и выпаривали, в результате чего получали 185 мг азида, который использовали непосредственно в следующей реакции. Этот неочищенный азид растворяли в диметилформамиде (3 мл) и в атмосфере азота добавляли 1,1,1,3,3,3-гексаметилдисилазан (1,25 г, 7,75 мМ) и метанол (0,12 г, 3,87 мМ). Полученную реакционную смесь нагревали при 50oC. Через 12 ч реакционную смесь охлаждали, разводили этилацетатом, промывали водой, а затем 2 н. соляной кислотой. Водные промывки подвергали обратному экстрагированию этилацетатом (3 х 50 мл). Объединенные органические слои осушали сульфатом магния, фильтровали и выпаривали, в результате чего получали азид-имид, 3,4-[(N,N'-1,1'-(3''-азидометилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион (175 мг) в виде пурпурного твердого вещества. Это вещество хроматографировали на C18-колонке (Rainin Dynamex R-60 (21,4 х 250 см) с использованием линейного градиента от 80% A (0,1% TFA и 5% ацетонитрил в воде) до 100% B (чистый ацетонитрил) в течение 60 мин при 15 мл/мин. Таким образом получали очищенный 3,4-[(N, N'-1,1'-(3''-азидометилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-дион с полным 57% выходом. МС. ЯМР.
К раствору азида, 3,4-[(N,N'-1,1'-(3''-азидометилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-диона (0,1 г, 0,21 мМ) в этилацетате (15 мл) и этаноле (5 мл) добавляли 0,1 г катализатора Lindlar. После этого реакционную смесь перемешивали под давлением водорода в 1 атмосферу при комнатной температуре. Через 12 ч катализатор удаляли путем фильтрации и концентрировали в вакууме. После очистки с помощью препаративной обращенно-фазовой ЖХВР на C18-колонках (Rainin Dynamex R-60) (21,4 х 250 мм) с использованием линейного градиента от 80% A (0,1% TFA и 5% ацетонитрил в воде) до 100% B (чистый ацетонитрил), в течение 60 мин и при скорости потока 15 мл/мин, получали первичный амин в виде TFA-соли, а именно, трифтороуксуснокислотной соли 3,4-[(N, N'-1,1'-(3''-аминометилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-диона в виде твердого продукта (80 мг, выход 63%). МС.
1H-ЯМР (d6-ацетон): δ 0,77 - 0,78 (м, 1H), 1,0 - 1,1 (м, 1H), 1,27 - 1,34 (м, 1H), 1,43 (м, 1H), 1,52 - 1,65 (м, 4H), 1,60 - 1,68 (м, 1H), 1,90 - 1,94 (м, 1H), 3,17 - 3,21 (м, 1H), 3,35, 3,38 (м, 1H), 3,64 - 3,67 (м, 1H), 3,75 - 3,82 (м, 2H), 6,61 - 6,72 (м, 4H), 6,824 (д, J = 16 Гц, 2H), 6,936 (т, J = 8,31 Гц, 2H), 7,397 (т, J = 7,83 Гц, 2H), 9,3 (с, 1H).
13C-ЯМР (d6-ацетон): δ 26,0, 28,0, 32,1, 35,4, 40,8, 41,0, 41,1, 45,1, 45,8, 50,9, 105,1, 105,2, 110,8, 111,0, 121,24, 121,29, 122,7, 122,9, 123,0, 128,4, 128,6, 131,5, 132,0, 134,0, 134,1, 136,8, 137,1, 172,6, 172,7, 192,5.
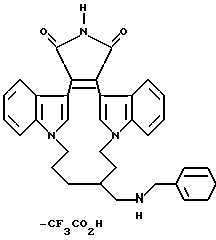
Пример 34
Трифтороацетатная соль 3,4-[(N, N'-1,1'-(3''-N-бензиламино) метилен)-гексан)-бис-(3,3'-индолил)]-1H-пиррол-2,5-диона
В атмосфере азота к перемешанному раствору первичного амина, 3,4-[(N, N'-1,1'-(3''-аминометилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-диона (40 мг, 0,05 мМ) в безводном ТГФ добавляли бензальдегид (9,39 мг, 0,08 мМ). Через 30 мин к смеси добавляли триацетоксиборогидрид натрия (18,75 мг, 0,08 мМ). После часового перемешивания реакционную смесь разводили водой и экстрагировали этилацетатом (3 х 25 мл). Объединенные органические экстракты осушали сульфатом магния, фильтровали и концентрировали в вакууме. После очистки с помощью обращенно-фазовой ЖХВР на C18-колонках (Rainin Dynamex R-60) (21,4 х 250 мм) с использованием линейного градиента от 80% A (0,1% TFA и 5% ацетонитрил в воде) до 100% B (чистый ацетонитрил) в течение 60 мин и при 15 мл/мин, получали две различные фракции монобензилового соединения: (I) 3,4-[(N, N'-1,1'-(3''-(N-бензиламино)метилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-диона (16 мг, выход 66%) и (II) дибензиламиновое соединение, а именно, 3,4-[(N, N'-1,1'-(3''-(N,N-дибензиламино)метилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-дион (7 мг, выход 20%). МС.
1H-ЯМР (d6-ацетон): δ 1,1 - 1,3 (м, 1H), 1,5 - 1,6 (м, 1H), 1,71 - 1,77 (м, 1H), 1,93 - 2,10 (м, 3H), 2,5 (м, 1H), 3,1 - 3,2 (м, 1H), 3,37 - 3,41 (м, 1H), 3,37 - 3,41 (м, 1H), 4,13 (т, J = 5,1 Гц, 2H), 4,28 (т, J = 5,1 Гц, 2H), 4,36 (д, J = 3,6 Гц, 2H), 7,13 - 7,24 (м, 4H), 7,33 (д, J = 25 Гц, 2H), 7,39 - 7,51 (м, 7H), 7,89 - 7,96 (м, 2H), 9,76 (с, 1H).
13C-ЯМР (d6-ацетон): δ 25,6, 27,3, 32,1, 32,9, 44,7, 45,4, 50,1, 52,2, 105,0, 105,2, 110,8, 111,1, 121,8, 121,3, 122,8, 122,9, 123,1, 128,5, 129,8, 130,3, 131,2, 131,3, 132,0, 132,4, 133,7, 134,0, 136,8, 137,0, 172,5, 172,6.
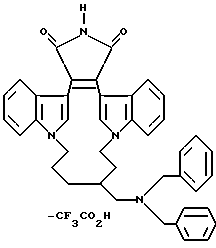
Пример 35
3,4-[(N, N'-1,1'-(3''-(N, N-Дибензиламино)метилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-диона трифтороацетатная соль
В соответствии со способом, описаным в примере 34, был получен 3,4-[(N, N'-1,1'-(3''-(N,N-дибензиламино)метилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-дион. МС.
1H-ЯМР (d6-ацетон): δ 0,2 - 0,3 (м, 1H), 0,6 - 0,9 (м, 4H), 1,2 - 1,3 (м, 1H), 1,50 (д, J = 5,4 Гц, 2H), 2,27 (м, 1H), 3,3 - 3,8 (м, 8H), 6,6 - 6,9 (м, 18H), 7,35 (дд, J = 7,5 Гц, J = 24,9 Гц, 2H), 9,1 (с, 1H).
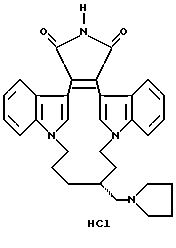
Пример 36r
(R)-3,4-[(N, N'-1,1'-(3''-(N-Пирролидино)метилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-диона гидрохлоридная соль
Перемешанную смесь мезилата, (R)-3,4-[(N,N'-1,1'-(3''-(метансульфонилокси)метилен)-гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-диона (202 мг) и пирролидина (1,5 мл) в ТГФ (15 мл) нагревали при 50oC до тех пор, пока ТСХ не обнаруживала израсходование исходного материала (16 ч). После этого добавляли EtOAc (30 мл). Органическую фазу промывали 10-миллилитровыми порциями 5%-ного гидрокарбоната натрия, водой и солевым раствором. В результате концентрирования получали темно-красный остаток, который подвергали препаративной ЖХВР (обращенно-фазовые колонки Waters, 0,1% TFA и 5% CH3CN в воде/100% CH3CN), в результате чего получали чистый (R)-3,4-[(N, N'-1,1'-(3''-(N-пирролидинометилен)-гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-дион в качестве его TFA-соли. Аналогично в результате превращения TFA-соли в HCl-соль, получали (R)-3,4-[(N,N'-1,1'-(3''-(N-пирролидинометилен)-гексан)-бис- (3,6'-индолил)] -1H-пиррол-2,5-диона гидрохлоридную соль (42 мг) в виде светло-красного твердого продукта, т.пл. 220oC (с разлож.).
ВРМС для C31H33N4O2 [M + 1]:
Вычислено: 4932604.
Найдено: 4932605.
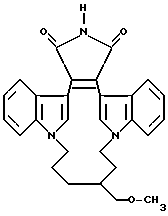
Пример 37
3,4-[(N, N'-1,1'-(3''-Метоксиметилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-дион
Раствор 3,4-[(N, N'-1,1'-(3''-трет-бутилдифенилсилилоксиметилен)-гексан)-бис- (3,3'-индолил)]-1(метил)-пиррол-2,5-диона (1,25 г, 1,81 мМ) в ТГФ (20 мл) обрабатывали раствором фторида тетра-н-бутиламмония в ТГФ (1 М, 2,0 мл, 2,0 мМ). Полученную смесь перемешивали в течение часа при 25oC. После этого реакционную смесь гасили 5 мл 1 н. соляной кислоты и разводили этилацетатом (75 мл). После промывания водой и солевым раствором, органический слой осушали сульфатом магния и концентрировали. Образовавшийся остаток очищали с помощью флеш-хроматографии на силикагеле, элюируя 3 - 5% метанолом в смеси ТГФ/гексане (1 : 1), в результате чего получали спирт, 3,4-[(N,N'-1,1'-(3''-гидроксиметилен)-гексан)-бис- (3,3'-индолил)] -1(метил)-пиррол-2,5-дион (509 мг, 62%) в виде пурпурного твердого вещества. Это вещество использовали непосредственно в следующей стадии. К перемешанному раствору, содержащему вышеописанный спирт (285 мг, 0,63 мМ) и 47% водную трифтороборную кислоту (170 мг, 0,95 мМ) в метиленхлориде (6 мл), при 0oC, по капле в течение 5 мин добавляли раствор триметилсилилдиазометана (Aldrich, 2,0 М гексан, 0,47 мл, 0,95 мМ). Полученную смесь перемешивали при 0oC в течение 2 ч, а затем при 25oC в течение 4 ч. ТСХ-анализ реакционной смеси указывал на присутствие большого количества непрореагировавшего исходного материала. После этого смесь охлаждали и добавляли дополнительное количество (1 экв.) триметилсилилдиазометантетрафтороборной кислоты. Затем эту смесь перемешивали в течение 2 ч при 0oC, после чего перемешивали 6 ч при 25oC. Полученную смесь разводили метиленхлоридом (20 мл), после чего промывали водой (10 мл) и 2 н. соляной кислотой (10 мл). Органический слой осушали сульфатом магния и концентрировали. Остаток загружали в 3'' • 3''-колонку с силикагелем и элюировали метиленхлоридом, в результате чего получали метиловый эфир, 3,4-[(N, N'-1,1'-(3''-метоксиметилен)-гексан)-бис- (3,3'-индолил)] -1(метил)-пиррол-2,5-дион (114 мг, 39%) в виде рыжевато-пурпурного твердого вещества, т.пл. 234 - 236oC, ЯМР.
ВРМС для C29H29N3O3:
Вычислено: 4672208.
Найдено: 4672210.
Смесь 3,4-[(N,N'-1,1'-(3''-метоксиметилен)-гексан)-бис- (3,3'-индолил)] -1(метил)-пиррол-2,5-диона (110 мг, 0,243 мМ) и 5 н. KOH (8 мл) в 15 мл EtOH, содержащего 1 мл ТГФ, нагревали при 90oC в течение 24 ч. После удаления большей части этанола при пониженном давлении, смесь подкисляли 6 н. соляной кислотой до pH 1, а затем экстрагировали этилацетатом (3 х 15 мл). Объединенные органические экстракты промывали разбавленным водным раствором гидрокарбоната натрия и водой, после чего осушали безводным сульфатом магния. После удаления растворителей в вакууме, неочищенный продукт загружали на 2'' • 2''-колонку с силикагелем (элюируя метиленхлоридом), в результате чего получали ангидрид, который использовали непосредственно в последующей стадии.
К раствору полученного ангидрида (76 мг, 0,17 мМ) в ДМФ (1,5 мл) добавляли предварительно перемешанный в течение 5 мин раствор 1,1,1,3,3,3-гексаметилдисилазана (0,75 мл, 3,34 мМ) и CH3OH (0,07 мл, 1,67 мМ). Полученную реакционную смесь перемешивали 1 ч при 25oC, после чего нагревали в течение 20 ч при 50oC. ТСХ-анализ указал на полное завершение реакции. Охлажденную реакционную смесь обрабатывали этилацетатом. Неочищенный продукт очищали с помощью флеш-хроматографии на силикагеле (элюируя градиентом метиленхлорида - 4% этилацетата в метиленхлориде), в результате чего получали 42 мг (55%) 3,4-[(N, N'-1,1'-(3''-метоксиметилен)-гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-диона в виде темно-красного твердого вещества. После перекристаллизации из ацетона/воды получали 28 мг аналитически чистого 3,4-[(N,N'-1,1'-(3''-метоксиметилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-диона в виде красновато-фиолетового твердого вещества, т.пл. 272 - 274oC.
Аналитический элементный анализ для C28H27N3O3 (0,1 • H2O):
Вычислено: C 73,86; H 6,02; N 9,23.
Найдено: C 73,51; H 5,92; N 8,99.
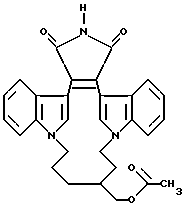
Пример 38
3,4-[(N, N'-1,1'-(3''-(Ацетокси)метилен)-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-дион
К перемешанной смеси ангидрида, 2,3-[(N,N'-1,1'-(3''-(гидроксиметилен-гексан)-бис-(3,3'-индолил)] - фуран-1,4-диона (1,49 мг, 0,34 мМ), 4-диметиламинопиридина (27 мг, 0,22 мМ), пиридина (0,75 мл) и ТГФ (1,5 мл) добавляли уксусный ангидрид (0,064 мл, 0,68 мМ). Полученную реакционную смесь перемешивали в течение 16 ч атмосфере азота и при 25oC. Затем эту смесь разводили 20 мл EtOAc промывали 2 н. соляной кислотой (2 х 10 мл) и водой (2 х 10 мл), после чего осушали безводным сульфатом магния. После выпаривания растворителей при пониженном давлении, неочищенный продукт очищали с помощью хроматографии на узкой колонке с силикагелем, элюируя метиленхлоридом, в результате чего получали О-ацетатный ангидрид 2,3-[(N,N'-1,1'-(3''-(ацетоксиметилен)-гексан)- бис-(3,3'-индолил)]-фуран-1,4-дион (111 мг, 68%) в виде пурпурного твердого вещества, т.пл. 252 - 254oC.
К перемешанному раствору О-ацетатного ангидрида, 2,3-[(N,N'-1,1'-(3''-(ацетоксиметилен)-гексан)-бис-(3,3'-индолил)]- фуран-1,4-диона (103 мг, 0,22 мМ) в ДМФ (2 мл) добавляли предварительно перемешанный в течение 5 мин раствор, содержащий 1,1,1,3,3,3-гексаметилдисилазан (0,48 мл, 2,2 мМ) и CH3OH (0,043 мл, 1,1 мМ). Полученную реакционную смесь обрабатывали этилацетатом и неочищенный продукт очищали с помощью флеш-хроматографии на силикагеле, элюируя градиентом CH2Cl2 - 5% EtAOc в CH2Cl2, в результате чего получали О-ацетилмалеимид, 3,4-[(N,N'-1,1'-(3''-(Ацетокси)метилен-гексан)-бис-(3,3'-индолил)] - 1H-пиррол-2,5-дион (74 мг, 72%) в виде темно-красного твердого вещества, которое являлось гомогенным, о чем свидетельствовала ТСХ (метиленхлорид). После перекристаллизации из ацетона/воды получали целевое соединение в виде красного твердого вещества, т.пл. 250 - 252oC.
Элементный анализ для C29H27N3O4 (0,1 • H2O)
Вычислено: C 72,06; H 5,67; N 8,69.
Найдено: C 71,72; H 5,67; N 8,29.
Следующие соединения были получены способом, аналогичным способу, описанному в Примерах настоящей заявки, и, кроме того, проиллюстрированы соединения настоящего изобретения. В следующих примерах структуры данных соединений подтверждены ЯМР-анализом, МС и/или элементным анализом (примеры 39 и 40 см. в конце описания).
Пример 40r
(R)-3,4-[(N, N'-1,1'-(3''-(N,N-Диметиламино)метилен-гексан)- бис-(3,3'-индолил)]-1H-пиррол-2,5-диона гидрохлоридная соль
При 0oC к перемешанному раствору хирального спирта, (R)-3,4-[(N,N'-1,1'-(3''-(гидрокси)метилен-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-диона (200 мг, 0,45 мМ) и пиридина (0,11 мл, 1,35 мМ) в метиленхлориде (5 мл), в течение 10 мин добавляли метансульфоновый ангидрид (84 мг, 0,54 мМ). Полученную реакционную смесь перемешивали при 25oC в течение 4 ч. Затем добавляли метиленхлорид (20 мл), и смесь промывали 10-мл порциями 3% соляной кислоты, водой и солевым раствором. После сушки безводным сульфатом магния и удаления растворителя в вакууме оставлялся неочищенный мезилат (205 мг), который являлся гомогенным, о чем свидетельствовала ТСХ (1% метанола в CHCl3). Этот материал использовали непосредственно в последующей стадии.
К раствору полученного мезилата (205 мг) в 10 мл ТГФ добавляли 40%-ный водный раствор диметиламина (2 мл) и реакционную смесь нагревали при 50oC в течение 36 ч. После удаления ТГФ при пониженном давлении, к остатку добавляли 20 мл метиленхлорида. После этого смесь промывали 5%-ным водным раствором гидрокарбоната натрия, водой и солевым раствором, а затем осушали безводным сульфатом магния. После концентрирования получали неочищенный (R)-3,4-[(N, N'-1,1'-(3''-(N, N-диметиламин)метилен-гексан)-бис-(3,3'- индолил)]-1H-пиррол-2,5-дион (158 мг) в виде красного твердого вещества, которое очищали с помощью препаративной ЖХВР (обращенно-фазовые колонки Waters), элюируя градиентом 0,1% TFA и 5% CH3CN в воде - 100% CH3CN, и получали амин - TFA-соль. Этот материал растворяли в метиленхлориде и превращали в свободное основание с использованием разбавленного водного KOH. После осушки органической фазы сульфатом магния (15 мин), растворитель выпаривали, а свободный амин (60 мг) растворяли в смеси метанола/ТГФ (5 мл) (1:1), охлаждали до 0oC в атмосфере азота и медленно подкисляли путем добавления 1 н. соляной кислоты в эфире до pH 4 - 5, с использованием индикаторной бумаги для определения pH (с увлажненной поверхностью). Осажденную соль фильтровали и промывали сухим эфиром в потоке азота, после чего осушали в вакууме в течение ночи для разложения CaSO4. Таким образом получали диметиламингидрохлоридную соль, (R)-3,4-[(N,N'-1,1'-(3''-(N,N-диметиламин)метилен-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион гидрохлоридную соль (43 мг) в виде светло-красного твердого вещества, т.пл. 230oC (при разложении). МС.
1H-ЯМР (300 МГц, ацетон-d6): 0,9 - 3,5 (м, 7H), 3,20- 3,42 (м, 8H), 4,05 - 4,18 (м, 4H), 7,02 - 7,80 (м, 10H), 10,94 (с, 1H).
Пример 40s
(S)-3,4-[(N, N'-1,1'-(3''-(N,N-Диметиламин)метилен)-гексан)-бис- (3,3'-индолил)]-1H-пиррол-2,5-диона гидрохлоридная соль
В соответствии с аналогичной процедурой, описанной в Примере 40r, (S)-3,4-[(N, N'-1,1'-(3''-(N, N-диметиламин)метилен-гексан)-бис- (3,3'-индолил)] -1H-пиррол-2,5-диона гидрохлоридную соль (90 мг) получали с полным 27% выходом из спирта, (S)-3,4-[(N,N'-1,1'-(3''-гидроксиметилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-диона путем образования мезилата, с последующей заменой его на диметиламин. МС.
1H-ЯМР (d6-ДМСО) δ 0,92 (широкий синглет, 1H) 1,35 (шир. с., 1H), 1,60 (шир. с. , 2H), 1,85 (шир.с., 1H) 2,37 - 2,42 (м, 2H), 2,91 - 3,05 (м, 2H), 4,13 (шир. с., 2H), 4,23 (шир.с., 2H), 7,11 - 7,23 (м, 4H), 7,34 (д, J = 20 Гц, 2H), 7,50 (дд, J = 8,1 Гц, J = 12,6 Гц, 2H), 7,79 (д, J = 8 Гц, 2H), 9,92 (шир.с., 1H), 10,98 (1H, с).
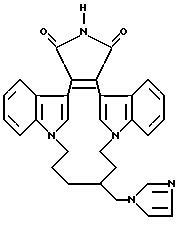
Пример 41
3,4-[(N, N'-1,1'-(3''-(N-имидазол)метилен)-гексан)-бис-(3,3'- индолил)] -1H-пиррол-2,5-дион
К перемешанному раствору, содержащему 3,4-[(N,N'-1,1'-(3''- (гидрокси)метилен)-гексан)-бис-(3,3'-индолил)] -1H-пиррол-2,5-диона (100 мг, 0,23 мМ) и триэтиламина (0,05 мл, 0,36 мМ) в безводном CHCl3, в атмосфере азота, при 25oC по капле добавляли метансульфонилхлорид (0,025 мл, 0,32 мМ). После 20-минутного перемешивания реакционную смесь разводили 15 мл CHCl3, промывали водой и солевым раствором, а затем осушали, фильтровали и концентрировали. Образовавшийся остаток очищали с помощью хроматографии на узкой колонке с силикагелем, элюируя хлороформом, а затем 10% этилацетатом в хлороформе. Таким образом получали 3,4-[(N,N'-1,1'-(3''-метансульфонилоксиметилен)-гексан)- бис-(3,3'-индолил)] -1H-пиррол-2,5-дион (53 мг) в виде красного твердого вещества, которое являлось гомогенным, на что указывала ТСХ (5% EtOAc в CH2Cl2).
К перемешиваемому раствору 3,4-[(N,N'-1,1'-(3''-(метансульфонилокси)метилен)-гексан)- бис-(3,3'-индолил)] -1H-пиррол-2,5-диона (49 мг, 0,095 мМ) в ДМФ (0,75 мл), в атмосфере азота по капле добавляли раствор натриевой соли имидазола в диметилформамиде (полученный путем добавления 60% NaH (8,7 мг, 0,22 мМ) к раствору имидазола (16 мг, 0,24 мМ) в ДМФ (0,5 мл)). Полученную реакционную смесь перемешивали 15 мин при 25oC, после чего нагревали при 50oC в течение 30 мин. После этого реакционную смесь разводили 25 мл метиленхлорида, содержащего 3% метанол. Затем смесь промывали 10-миллилитровыми порциями воды и солевым раствором, после чего осушали безводным сульфатом натрия. После выпаривания растворителей при пониженном давлении, неочищенный продукт загружали на 3'' x 3''-колонку с силикагелем, элюируя метиленхлоридом, а затем 5% метанолом в метиленхлориде, содержащем 1% триэтиламин. В результате этой процедуры получали 3,4-[(N, N'-1,1'-(3''-(N-имидазол)метилен)- гексан)-бис-(3,3'-индолил)] -1H-пиррол-2,5-дион (21,5 мг, 46%) в виде красного твердого вещества. Это вещество подвергали обращенно-фазовой ЖХВР (градиент 5% CH2CN в воде, содержащей 0,1% TFA/CH3CN), в результате чего получали аналитически чистый 3,4-[(N,N'-1,1'-(3''-(N-имидазол)метилен)-гексан)- бис-(3,3'-индолил)]-1H-пиррол-2,5-дион (12,4 мг) в виде красного твердого вещества, т.пл. 261 - 266oC. ЯМР.
ВРМС для C30H27N5O2 [M + 1]:
Вычислено: 4902244.
Найдено: 4902242.
Следующие соединения были получены способом, аналогичным способу, описанному в Примерах настоящей заявки, и, кроме того, проиллюстрированы соединения настоящего изобретения. В следующих примерах структура данных соединений подтверждена ЯМР-анализом, МС и/или элементным анализом (примеры 42 - 49 см. в конце описания).
Пример 50
3,4-[(N, N'-1,1'-(Пропилтиопропил))-бис-(3,3'-индолил)]-1H-пиррол- 2,5-дион
К перемешанному безводному метиленхлоридному (1,0 л) раствору N-(3-ацетоксипропил)-индола (102 г, 0,47 М), при 0oC по капле добавляли оксалилхлорид (43,04 мл, 0,494 М, 1,05 экв.). Через 15 мин ледяную баню удаляли. Полученную реакционную смесь оставляли для нагревания до комнатной температуры, перемешивая при этом в течение 3 ч. Летучие вещества удаляли в вакууме, в результате чего получали твердый пурпурный краситель, который снова растворяли в сухом метиленхлориде (1,0 л) в атмосфере азота. При энергичном перемешивании к раствору добавляли N-трет-бутоксикарбонил-индол-3-уксусную кислоту (129,25 г, 0,47 М), после чего быстро добавляли триэтиламин (130,6 мл, 0,94 М, 2 экв.). Через 16 ч реакционную смесь концентрировали и очищали с помощью флеш-хроматографии на силикагеле (элюент: гексан/этилацетат, 3:1). Мажорную окрашенную фракцию концентрировали и получали ангидрид, 2-[1-(3-ацетоксипропил)-3-индолил] -3-[1-трет-бутоксикарбонил-3- индолил] -фуран-1,4-дион (101 г, 40% выход) в виде кристаллического красного твердого вещества. МС.
К ВОС-защищенному ангидриду (7,4 г, 14 мМ) при перемешивании добавляли трифторуксусную кислоту (27 мл, 350 мМ), содержащую этантиол (1 мл, 14 мМ). Через час реакционную смесь распределяли между метиленхлоридом и насыщенным гидрокарбонатом натрия. Органический слой промывали солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали и получали неочищенный разблокированный ангидрид в виде красного наполовину твердого вещества. Это вещество пропускали через узкий слой двуокиси кремния, промывая гексаном, а затем метиленхлоридом. Окрашенную фракцию элюировали через слой двуокиси кремния (этилацетат) и осушали в вакууме, в результате чего получали очищенный разблокированный ангидрид, 2-[1-(ацетоксипропил)-3-индолил] - 3-(3-индолил)-фуран-1,4-дион (5,7 г, 95% выход) в виде красного твердого вещества. МС.
К перемешанному безводному диметилформамидному (125 мл) раствору разблокированного ангидрида (3,0 г, 7 мМ), при комнатной температуре добавляли NaH (420 мг, 10,5 мМ, 60% в минеральном масле). При этом наблюдалось изменение ярко-оранжевого оттенка на фиолетовый. Через 30 мин быстро добавляли 3 экв. 3-бромопропилацетата. Полученную реакционную смесь нагревали до 75oC, что приводило к постепенному появлению оранжевого оттенка. Через 6 ч диметилформамид удаляли в вакууме. Остаток загружали во флеш-колонку с двуокисью кремния, элюируя гексаном/этилацетатом (3:2). Мажорный красный слой собирали, а растворитель выпаривали, в результате чего получали алкилированный ангидрид, 2,3-бис[1-(3-ацетоксипропил)-3-индолил] -фуран- 1,4-дион (1,36 г, 36%) в виде красного твердого вещества. МС.
2,3-Бис[1-(3-ацетоксипропил)-3-индолил]-фуран-1,4-дион (1,32 г, 2,52 мМ) суспендировали в абсолютном этаноле (125 мл), перемешивая при этом, а затем обрабатывали 125 мл 5 н. KOH. После 16-часового перемешивания, реакционную смесь концентрировали до объема 126 мл. Образовавшийся остаток медленно подкисляли 5 н. соляной кислотой до образования красного твердого осадка. Это осажденное вещество фильтровали и осушали в вакуумной печи при 60oC, в результате чего получали спиртовый ангидрид (1,1 г, 99%) в виде красного порошкообразного вещества.
В атмосфере азота спиртовый ангидрид (1,1 г, 2,47 мМ) растворяли в безводном ДМФ (30 мл), перемешивая при этом. После этого добавляли предварительно перемешанный раствор 1,1,1,3,3,3-гексаметилдисилазана (5,22 мл, 24,7 мМ, 10 экв.) и метанола (0,50 мл, 12,4 мМ, 5 экв.). Полученную реакционную смесь оставляли для перемешивания при комнатной температуре на 16 ч. Диметилформамид удаляли в вакууме. К образовавшемуся остатку добавляли 100 мл ацетона и приблизительно 500 мг избыточного количества CsF. После 4-часового перемешивания реакционную смесь концентрировали. Образовавшийся остаток распределяли между этилацетатом и водой. Органический слой промывали 5 раз 1 н. соляной кислотой и 2 раза солевым раствором, после чего осушали сульфатом натрия и фильтровали. Фильтрат концентрировали с получением бисиндолилмалеимида, 3,4-бис[1-(3-гидроксипропил)-3-индолил]-1H-пиррол-2,5-диона (1,0 г, выход 91%) в виде красного порошкообразного вещества. Полный выход из 2 стадий составлял 90%. МС.
В атмосфере азота, 3,4-бис[1-(3-гидроксипропил)-3-индолил]-1H-пиррол-2,5-дион (1,0 г, 2,25 мМ) растворяли в безводном метиленхлориде (250 мл) при комнатной температуре. В реакционный сосуд одновременно добавляли CBr4 (2,09 г, 6,3 мМ, 2,8 экв.) и трифенилфосфин (2,83 г, 10,8 мМ, 4,8 экв.). Полученную смесь оставляли на 16 ч при перемешивании. Неочищенную реакционную смесь концентрировали и очищали с помощью флеш-хроматографии на колонках с силикагелем (элюент: гексан/этилацетат, 7:3). Таким образом получали проэлюированный мажорный красный слой. После удаления растворителей из этого слоя получали дибромовое соединение 3,4-бис[1-(3-бромопропил)-3-индолил]-1H-пиррол-2,5-дион (876 мг, выход 68%) в виде красного порошкообразного вещества.
Дибромовое соединение (47,8 мг, 0,084 мМ) растворяли в ацетоне при комнатной температуре, перемешивая при этом. После этого к смеси добавляли избыточное количество нонагидрата сульфида натрия (229 мг, 0,95 мМ, 11,3 экв.). Образовавшуюся смесь перемешивали в течение ночи, после чего ацетон удаляли в вакууме. Остаток распределяли между водой и метиленхлоридом. Органический слой промывали солевым раствором, осушали сульфатом натрия и концентрировали, в результате чего получали 35,5 мг (выход 94%) целевого продукта в виде красно-оранжевого твердого вещества. МС.
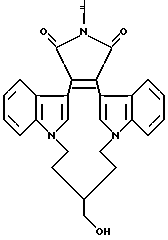
Пример 51
3,4-[(N, N'-1,1'-(3''-(Гидрокси)метилен)пентан)-бис-(3,3'-индолил)] - 1(H)-пиррол-2,5-дион
При 55oC и в атмосфере азота безводный диметилформамидный (35 мл) раствор 1,5-дийодо-3-(трет-бутилдифенилсилилоксиметилен)-пентана (7,3 г, 12 мМ) и бис-(3,3'-индолил)] -1(H)-пиррол-2,5-диона (4,21 г, 12 мМ) с помощью шприца в течение 48 ч добавляли к суспензии Cs2CO3 (16,06 г, 49,3 мМ) в безводном диметилформамиде (1 л), энергично перемешивая при этом. Еще через 2 ч реакционную смесь концентрировали в вакууме с получением остатка. Это остаток растворяли в метиленхлориде, после чего промывали 1 н. соляной кислотой, солевым раствором, а затем осушали и концентрировали в вакууме с получением фиолетового маслообразного вещества. Полученное маслообразное вещество пропускали через пробку из двуокиси кремния (элюент: гексан/этилацетат, 4: 1). После концентрирования элюента получали макроциклический продукт, 3,4-[(N, N'-1,1'-(3''-(трет-бутилдифенилсилилоксиметилен) пентанил)-бис-(3,3'-индолил)] -1(метил)-пиррол-2,5-дион (4,5 г, 55% выход) в виде твердого пурпурного красителя.
К этаноловой (300 мл) суспензии 3,4-[(N,N'-1,1'-(3''-(трет-бутилдифенилсилилоксиметилен)пентанил)- бис-(3,3'-индолил)]-1(метил)-пиррол-2,5-дион (4,2 г, 6,2 мМ) добавляли 300 мл 5 н. KOH. Полученную реакционную смесь нагревали с обратным холодильником (86oC) в течение 48 ч. После перемешивания, охлаждения до комнатной температуры и удаления этанола в вакууме получали концентрат. Этот концентрат подкисляли до pH 1 путем добавления 325 мл 5 н. соляной кислоты, а затем экстрагировали этилацетатом, промывали солевым раствором (дважды), осушали и концентрировали с получением остаточного ангидрида, 3,4-[(N, N'-1,1'-(3''-(гидроксиметилен)пентан)- бис-(3,3'-индолил)]-фуран-2,5-диона (2,6 г, 100% выход).
К безводному диметилформамидному (500 мл) раствору ангидрида 3,4-[(N, N'-1,1'-(3''-(гидроксиметилен)пентанил)- бис-(3,3'-индолил)]-фуран-2,5-диона (2,6 г, 6,2 мМ) добавляли раствор метанола (1,21 мл, 31 мМ) и 1,1,1,3,3,3-гексаметилдисилазана (13,1 мл, 62 мМ). После 36-часового нагревания при 55oC реакционную смесь концентрировали в вакууме, разводили этилацетатом и промывали 1 н. соляной кислотой. Кислотную промывку, содержащую некоторое количество твердых веществ, подвергали обратному экстрагированию хлороформом. Объединенные органические слои осушали и концентрировали с получением фиолетового остатка. Этот остаток загружали на узкий слой двуокиси кремния (элюент: 2 - 10% MeCN/CH2Cl2). После элюирования, фракцию, содержащую мажорный продукт, концентрировали в вакууме, в результате чего получали целевой спирт, 3,4-[(N,N'-1,1'-(3''-(гидроксиметилен)пентанил)- бис-(3,3'-индолил)] -1(H)-пиррол-2,5-дион 650 мг, 25%) в виде твердого пурпурного красителя. МС.
1H-ЯМР (ДМСО-d6): δ 0,7 (м, 1H), 1,48 (м, 2H), 1,82 (м, 2H), 3,19 (дд, 2H), 4,16 (м, 4H), 4,4 (т, 1H), 7,05 (т, 2H), 7,16 (т, 2H), 7,46 (д, 2H), 7,17 (с, 2H), 7,65 (д, 2H), 10,96 (с, 1H).
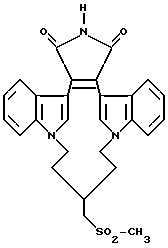
Пример 52
3,4-[(N, N'-1,1'-(3''-(Метансульфонилокси)метилен)-пентан-1'',5''- ил)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-дион
К сухому метиленхлоридному (80 мл) раствору 3,4-[(N,N'-1,1'-(3''-(гидроксиметилен)пентан-1'', 5''-ил)-бис- (3,3'-индолил)]-1(H)-пиррол-2,5-диона (334120) (650 мг, 1,5 мМ) добавляли метансульфоновый ангидрид (400 мг, 2,29 мМ), а затем избыточное количество пиридина (370 мл, 4,58 мМ). После 16-часового отстаивания при комнатной температуре реакционную смесь помещали на узкую пробку с двуокисью кремния и элюировали со смесью 0 - 7% MeCN/CH2Cl2. Окрашенную фракцию концентрировали в вакууме и получали мезилат, 3,4-[(N,N'-1,1'-(3''-(метансульфонилокси)метилен)пентан-1'',5''-ил)-бис- (3,3'-индолил)]-1(H)-пиррол-2,5-дион (501 мг, выход 67%) в виде фиолетового твердого вещества. МС.
1H-ЯМР (ДМСО-d6): δ 0,89 (м, 1H), 1,61 м, 2H), 1,82 (м, 2H), 2,99 (с, 3H), 4,02 (д, 2H), 4,22 (м, 4H), 7,06 (т, 2H), 7,17 (т, 2H), 7,17 (с, 2H), 7,54 (д, 2H), 7,63 (д, 2H), 10,98 (с, 1H).
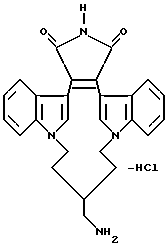
Пример 53
3,4-[(N, N'-1,1'-(3''-(Аминометилен)пентан-1'', 5''-ил)- бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлоридная соль
В герметично закрывающийся реакционный сосуд, содержащий тетрагидрофурановый (20 мл) раствор мезилата, 3,4-[(N,N'-(метансульфонилокси)метилен)пентан-1'', 5''-ил)-бис- (3,3'-индолил)]-1(H)-пиррол-2,5-диона (250 мг, 0,5 мМ), добавляли водный 33%-ный раствор NH4OH (10 мл), реакционный сосуд герметично закрывали, а затем нагревали при 60oC. Через 48 ч реакционную смесь охлаждали и элюировали через пробку из силикагеля (этилацетат/ацетон). Затем ацетоновую фракцию концентрировали в вакууме, в результате чего получали рыжеватый твердый остаток. Часть этого остатка очищали с помощью обращенно-фазовой гель-проникающей ЖХВР (85% MeCN/вода, 0,01% TFA). Чистые фракции собирали и концентрировали с получением красного твердого вещества. Затем это твердое вещество распределяли между этилацетатом и 0,1 н. NaOH. Органический слой концентрировали с получением свободного основания в качестве остатка. Образовавшийся остаток растворяли в метаноле (2 мл) и в течение часа обрабатывали соляной кислотой (2 мл, 1,0 М в эфире). Реакционную смесь концентрировали в вакууме и получали 28,5 мг (выход 13%) целевого соединения в виде твердого пурпурного красителя, которое имело менее 95%-чистоту (ЖХВР-анализ). МС.
1H-ЯМР (ДМСО-d6): δ 1,17 (м, 1H), 1,5- 1,63 (м, 2H), 1,8 - 1,95 (м, 2H), 2,73 (м, 2H), 4,18 (м, 4H), 7,12 (т, 2H), 7,15 (с, 2H), 7,23 (т, 2H), 7,56 (д, 2H), 7,75 (д, 2H), 7,8 (шир., 3H), 11,01 (с, 1H).
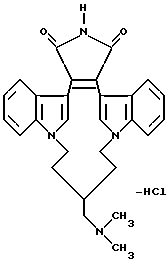
Пример 54
3,4-[(N, N'-1,1'-(3''-(N, N-(Диметиламино)метилен)-пентанил)-бис-(3,3'-индолил)]-1(H)-пиррол-2,5-диона гидрохлорид
Целевое соединение получали в виде гидрохлоридной соли, используя диметиламин (40% водный раствор; 5 мл) вместо мезилата, 3,4-[(N,N'-1,1'-(3''-(метансульфонилокси)метилен)пентанил)-бис- (3,3'-индолил)] -1(H)-пиррол-2,5-диона (110 мг, 0,2 мМ), с последующим превращением в гидрохлоридную соль в качестве целевого соединения (28 мг, выход 26%). МС.
1H-ЯМР (ДМСО-d6): δ 1,17 (м, 1H), 1,5 - 1,63 (м, 2H), 1,8 - 1,95 (м, 2H), 2,73 (м, 2H), 4,18 (м, 4H), 7,12 (т, 2H), 7,15 (с, 2H), 7,23 (т, 2H), 7,56 (д, 2H), 7,75 (д, 2H), 7,8 (шир, с, 3H), 11,01 (с, 1H).
Аналогичным образом были получены следующие соединения, и, кроме того, проиллюстрированы следующие соединения настоящего изобретения (примеры 55-113 см. в конце описания).
Как указывалось выше, соединения настоящего изобретения являются сильными ингибиторами протеинкиназы C. Эти соединения являются селективными в отношении протеинкиназы C.
Способность соединения настоящего изобретения к селективному ингибированию протеинкиназы C определяли с помощью анализа на кальций-кальмодулинзависимую протеинкиназу, анализа на казеиновую протеинкиназу II, анализа на каталитическую субъединицу сАМР-зависимой протеинкиназы и анализа на протеин-тирозин-киназу.
Анализ на кальций-кальмодулинзависимую протеинкиназу (CaM) (см. табл.I).
Анализ на кальций-кальмодулинзависимую протеинкиназу описан в Journal of Neuorcience, 3: 818-831 (1983). Компонентами анализа (в полном объеме 250 мкл) являются: 55 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота), pH 7,5; 2,75 мМ дитиотреитол; 2,2 мМ EGTA (этиленбис(оксиэтиленнитрило) тетрауксусная кислота, используемая в буфере-"пустышке"); 1,1 мМ хлорид кальция (Sigma, St.Louis, Missouri) (используемый в контрольном буфере); 10 мМ хлорид магния (Sigma, St.Louis, Missouri); 200 мкг/мл гистона типа HL (Worthington); 10 мкл ДМСО или ДМСО/ингибитор и 30 мкМ (гамма-32P) АТР (DuPont). Реакцию инициировали путем добавления кальций-кальмодулинзависимой протеинкиназы (выделенной из гомогената головного мозга крысы), затем инкубировали 10 мин при комнатной температуре и прекращали реакцию путем добавления 0,5 мл охлажденной льдом трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл альбумина бычьей сыворотки (Sigma, St. Louis, Missouri). Осадок собирали путем вакуумной фильтрации на стекловолоконных фильтрах, а затем оценивали с помощью бета-сцинтилляционного счетчика.
Компоненты анализа: 165 мкл буфера; 25 мкл кальмодулина (250 мкг/мл); 10 мкл ДМСО или ДМСО/ингибитора; 25 мкл киназы и 25 мкл АТ32Р.
Анализ на казеиновую протеинкиназу II (CK-II)
Анализ на казеиновую протеинкиназу II описан в Neuochem Res. 13: 829 - 836 (1988). Компонентами анализа (в полном объеме 250 мкл) являются: 20 мМ Трис-HCl, pH 7,5, 5 мМ фторида натрия, 50 мг/мл казеина (Sigma, St.Louis, Missouri), 10 мкл ДМСО или ДМСО/ингибитора и 30 мкм (гамма-32Р) АТР (DuPont). Реакцию инициировали путем добавления казеиновой протеинкиназы II (выделенной из гомогената головного мозга крысы), инкубировали 10 мин при комнатной температуре и реакцию прекращали путем добавления 0,5 мл охлажденной льдом трихлоруксусной кислоты (Amresco), с последующим добавлением 100 мкл 1 мг/мл альбумина бычьей сыворотки (Sigma, St.Louis, Missouri). Осадок собирали путем вакуумной фильтрации на стекловолоконном фильтре, а затем оценивали с помощью бета-сцинтилляционного счетчика.
Компоненты анализа для добавления: 175 мкл буфера; 10 мкл ДМСО или ДМСО/ингибитора; 25 мкл АТ32Р в 300 мкМ хлорида магния; 40 мкл фермента (неразбавленного).
Буфер получали следующим образом:
(Конечный объем = 3,5 мл; количество 20 анализов)
500 мкл каждого: 200 мМ Трис-HCl pH 7,5
50 мМ фторида натрия
50 мг/мл казеина
+ 2 мл деионизованной воды
Полный объем: 3,5 мл
Анализ на каталитическую субъединицу сАМР-зависимой протеинкиназы (РКА)
Компонентами анализа (в полном объеме 250 мкл) являются: 20 мМ HERES (Sigma, St. Louis, Missouri) буфера pH 7,5, 200 мкг/мл гистона типа HL (Worthington), 10 мМ хлорида магния (Sigma, St.Louis, Missouri), 10 мкл ДМСО или ДМСО/ингибитора и 30 мкМ (гамма 32Р) АТР (DuPont). Реакцию инициировали путем добавления каталитической субъединицы сАМР-зависимой киназы, выделенной из бычьего сердца (Sigma, St.Louis, Missouri), инкубировали 10 мин при 30oC и реакцию прекращали путем добавления 0,5 мл охлажденной льдом трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл альбумина бычьей сыворотки (Sigma). Осадок собирали с помощью вакуумной фильтрации на стекловолоконных фильтрах, а затем оценивали с помощью бета-сцинтилляционного счетчика. Этот анализ идентичен анализу на протеинкиназу C (РКС), за исключением того, что в этом анализе не использовали фосфолипидов или диацилглицерина, а гистоновый субстрат был специфическим к каталитической субъединице сАМР-зависимого фермента.
Анализ на протеин-тирозин-киназу (src)
Этот анализ имел следующие компоненты: 10 мкл Раитида (Paytide); 10 мкл киназы; 4 мкл ДМСО или ДМСО/ингибитора; 6 мкл 200 мМ HEPES, pH 7,5; 10 мкл АТ32Р.
Этот анализ описан в Onogene Science, Inc.Cat. # РК02 и РК03 (1990).
Неожиданно было обнаружено, что соединения настоящего изобретения являются также изозим-селективными ингибиторами, то есть, эти соединения селективно ингибируют бета-1 и бета-2-изозимы протеинкиназы C. В анализе на РКС-фермент определяли селективность по отношению к этим изозимам.
Анализ РКС-фермента
РКС-ферменты = альфа, бета I, бета II, гамма, дельта, эпсилон, эта и дзета.
В данном анализе были использованы следующие компоненты (в полном объеме 250 мкл):
Везикулы, состоящие из 120 мкг/мл фосфатидилсерина (Avanti Polar Lipids) и достаточного количества диацилглицерина (Avanti Polar Dupnot) для активации фермента до максимальной активности в 20 мМ HEPES-буфера (Sigma, St. Louis, Missouri), pH 7,5, 940 мкМ хлорида кальция (Sigma, St. Louis, Missouri) для анализа только альфа-, бета-1-, бета-2- и гамма-ферментов; 1 мМ EGTA для всех ферментов, 10 мМ хлорида магния (Sigma, St. Louis, Missouri) и 30 мкМ (гамма-32Р) АТР (DuPont). Для всех ферментов, в качестве субстрата использовали либо гистон типа HL (Worthington), либо основный белок миелин. Реакцию инициировали путем добавления протеинкиназы C, инкубировали 10 мин при 30oC, и после этого реакцию прекращали путем добавления 0,5 мл холодной трифторуксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл альбумина бычьей сыворотки (Sigma, St.Louis, Missouri). Осадок собирали путем вакуумной фильтрации на стекловолоконных фильтрах с использованием системы фильтрации TOMTECTM и оценивали путем подсчета на бета-сцинтилляционном счетчике.
В Таблице 1 проиллюстрированы РКС-селективность характерных соединений, используемых в вышеописанном анализе.
Соединения настоящего изобретения ингибируют протеинкиазу C при IC50-величине, составляющей менее 100 мкм. Кроме того, соединения настоящего изобретения селективно ингибируют бета-1- и бета-2-изозимы протеинкиназы C и имеют значения IC50 по отношению к этим изозимам, составляющие менее чем 10 мкм.
Как ингибиторы протеинкиназы C, соединения настоящего изобретения могут быть использованы для лечения заболеваний, патологии которых в той или иной степени связаны с протеинкиназой C. Такими заболеваниями являются: сахарный диабет и его осложнения, ишемия, воспалительные заболевания, нарушения центральной нервной системы, сердечно-сосудистые заболевания, болезнь Альцгеймера, кожные заболевания и рак.
Было показано, что ингибиторы протеинкиназы C блокируют воспалительные реакции, такие как нейтрофильный кислородный "взрыв", C 3-супрессирующее действие Т-лимфоцитов и форболиндуцированный отек конечностей. Twoemy B. и др. , Biochem. Biophys, Res. Commun. 171: 1087-1092 (1990); Mulgueen, M.J. и др. Agents Actions 37: 85-98 (1992). В соответствии с этим, соединения настоящего изобретения как ингибиторы РКС могут быть использованы для лечения воспалительных заболеваний.
Активность протеинкиназы C играет существенную роль в функционировании центральной нервной системы. Huang K.P. Trends Neurosci 12: 425-432 (1989). Кроме того, было показано, что ингибиторы протеинкиназы C предупреждают нарушения, связанные с местной или центральной ишемией головного мозга, и отек мозга. Hara H. и др., J.Cereb Blood Flow Metab. 10: 646-653 (1990); Shibata S. и др., Brain Res 594: 290-294 (1992). Недавно было установлено, что протеинкиназа C вызывает осложнения при болезни Альцгеймера. Shimohama S. и др. , Neurology 43: 1407-1413 (1993). Поэтому, соединения настоящего изобретения могут быть использованы для лечения болезни Альцгеймера и ишемии головного мозга.
Долгое время активность протеинкиназы C связывали с клеточным ростом, стимулированием опухолевого роста и рака. Rotenberg S.A и Weinstein U.B. Biochem. Mol. Aspects Sel. Cancer 1: 25 - 73 (1991). Ahmad и др., Molecular Pharmacology 43: 858-862 (1993). Известно, что ингибиторы протеинкиназы C являются эффективными средствами для предупреждения роста опухолей у животных. Meyer T. и др., Int, J.Cancer 43: 851-856 (1989); Akinagaka S. и др., Cancer Res. 51: 488-4892 (1991). Соединения настоящего изобретения могут также действовать как множественные агенты-реактиваторы, повышающие эффективность химиотерапевтических средств при их введении совместно с этими соединениями.
Активность протеинкиназы C играет также важную роль в сердечно-сосудистых заболеваниях. Было показано, что повышенная активность протеинкиназы C в сосудистой сети приводит к сужению кровеносных сосудов и гипертензии. Известный ингибитор протеинкиназы C способствует предупреждению повышения этой активности. Bilder G.E. и др., J. Pharmacol. Exp. Ther. 252: 526-530 (1990). Поскольку ингибиторы протеинкиназы C способствуют подавлению нейтрофильного кислородного "взрыва", то ингибиторы протеинкиназы C могут быть также использованы для лечения ишемии сердца и сосудов и последующего улучшения сердечной деятельности. Muid R. E. и др. FEBS Lett. 293: 169-172 (1990); Sonoki H. и др. , Kokyu-To Junkan 37: 669-674 (1989). Была также исследована роль потеинкиназы C в функции тромбоцитов, в результате чего было показано, что повышение уровней протеинкиназы C коррелирует с повышенной реакцией в ответ на введение агонистов. Bastyr III, E, J. и Lu, J. Diabetes 42: (Suppl.I) 97А (1993). Было показано, что РКС участвует в биохимическом пути модуляции микрососудистой проницаемости фактором активации тромбоцитов. Kooayashi и др., Amer. Phys. Soc. H1214-H1220 (1994). Было проиллюстрировано, что сильные ингибиторы протеинкиназы C воздействуют на агонист-индуцированную агрегацию тромбоцитов. Toullec D. и др., J. Biol, Chem. 266: 15771 - 15781 (1991). Ингибиторы протеинкиназы C также блокируют агонист-индуцированную пролиферацию клеток гладких мышц. Matsumoto H. и Saaki Y. Biochem. Biophys. Res. Commun. 158: 105-109 (1989). Поэтому, соединения настоящего изобретения могут быть использованы для лечения сердечно-сосудистых заболеваний, атеросклероза и рестеноза.
Кроме того, кожные заболевания, такие как псориаз, также ассоциируются с аномальной активностью протеинкиназы C. Horn F. и др., J. Invest. Dermatol, 88: 220-222 (1987); Paynaud F. и Evain-Brion, D.Br. J.Dermatol. 124: 542-546 (1991). Псориаз характеризуется патологической пролиферацией кератиноцитов. Было показано, что известные ингибиторы протеинкиназы C ингибируют пролиферацию кератиноцитов с эффективностью, зависящей от потенции данных РКС-ингибиторов. Hegemann L. и др., Arch. Dermatol. Res. 283: 456-460 (1991); Bollag W.B.. и др. J. Invest. Dermatol. 100: 240-246 (1993). В соответствии с этим, соединения настоящего изобретения как ингибиторы РКС могут быть использованы для лечения псориаза.
Было показано, что некоторые виды диабета связаны с активностью протеинкиназы C. Чрезмерная активность того фермента связана с нарушением инсулинового "сигнала", а поэтому, приводит к возникновению инсулин-независимого диабета Типа П. Kasasaki A. и др., J.Biol. Chem. 265: 10226-10231 (1990); Chen K.S. и др., Trans.Assoc. Am. Physician 104: 206-212 (1991); Chin J.E. и др. , J. Biol. Chem 268: 6338-6347 (1993). Кроме того, исследования показали заметное увеличение РКС-активности в тканях, о которых известно, что они являются восприимчивыми к осложнениям диабета (благодаря испытаниям этих тканей в гипергликемических условиях). Lee T.-S. и др., J.Clin, Invest. 83: 90-94 (1989); Lee T. -S. и др., Proc. Natl. Acad. Sci. USA 86: 5141-5145 (1989); Craven P.A. и DeRubertis F.R. J. Clin. Inve. 83: 1667-1675 (1989); Wolf B. A. и др., J.Clin. Invest 87: 31-38 (1991); Tesfamariam B. и др., J. Clin. Invest. 87: 1643-1648 (1991).
Соединения настоящего изобретения являются также изозим-селективными. Эти соединения ингибируют преимущественно бета-1- и бета-2-изозимы протеинкиназы C, по отношению к другим изозимам протеинкиназы C, т.е., альфа-, гамма-, дельта-, эпсилон-, дзета- и эта-изозимам. В основном, для ингибирования бета-1- и бета-2-изозимов РКС, соединения настоящего изобретения требуют, как минимум, в 10 раз меньшую дозу, чем это потребовалось бы для равного ингибирования альфа-изозима РКС, как это показано в РКС-анализе. В соответствии с этим, соединения настоящего изобретения ингибируют бета-1- и бета-2-изозимы протеинкиназы C при концентрациях намного меньших, чем это понадобилось бы для ингибирования других изозимов РКС. Такая изозим-селективность была проиллюстрирована в табл. 2 для наиболее характерного соединения настоящего изобретения.
Благодаря своей селективности, соединения настоящего изобретения могут быть с особой эффективностью использованы для лечения заболеваний, связанных с активностью бета-1- или бета-2-изозима протеинкиназы C. Например, повышение уровней глюкозы в крови, наблюдаемое при диабете, приводит к изозим-специфическому повышению уровня бета-2-изозима в тканях сосудов. Inoguchi и др. , Proc. Natl. Acad. Sci. USA, 89: 11059 - 11065 (1992). Было показано, что диабет-ассоциированное повышение уровня бета-изозима в тромбоцитах человека коррелирует с изменением их реакции в ответ на введение агонистов. Bastyr III, E.J. и Lu. J. Diabetes 42: (Suppl.I) 97A (1993). Кроме того, было показано, что рецептор витамина D человека селективно фосфорилируется бета-изозимом протеинкиназы. Это фосфорилирование связано с изменениями функции рецептора. Hsieh и др., Proc. Natl. Acad. Sci. USA 88: 9315 - 9319 (1991); Hsieh и др., J. Biol. Chem. 268: 15118 - 15126 (1993). Кроме того, было недавно показано, что изозим бета-2 является ответственным за пролиферацию клеток эритролейкоза, а альфа-изозим участвует в дифференцировке мегакариоцитов в этих же самых клетках. Murrey и др., J. Biol. Chem. 168: 15847 - 15853 (1993).
Предпочтительно, чтобы перед введением Соединений формулы I, они были изготовлены в виде композиций. Поэтому, в еще одном варианте своего осуществления, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей.
Фармацевтические композиции настоящего изобретения изготавливают с использованием традиционной техники и доступных ингредиентов. При изготовлении композиций настоящего изобретения, активный ингредиент обычно смешивают с носителем или разбавляют носителем, либо включают в носитель, который может быть в форме капсулы, сашэ, бумаги или другой емкости. Если носитель служит в качестве разбавителя, то таким носителем может быть твердый, полутвердый или жидкий материал, который служит в качестве наполнителя или среды для активного ингредиента. Так, например, композиции настоящего изобретения могут быть изготовлены в форме таблеток, пилюль, порошков, драже, сашэ, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как в твердой, так и в жидкой среде), мягких и твердых желатиновых капсул, суппозиториев, стерильных инъецируемых растворов и стерильных порошковых упаковок.
Некоторыми примерами подходящих носителей, наполнителей и разбавителей является лактоза, декстроза, сахароза, сорбит, маннит, крахмал, аравийская камедь, фосфат кальция, альгинаты, трагакантовая камедь, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, водный сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. В указанные композиции могут быть также введены замасливающие агенты, смачивающие агенты, эмульгирующие и суспендирующие агенты, консерванты, подслащивающие агенты или ароматизирующие агенты. Композиции настоящего изобретения могут быть изготовлены таким образом, чтобы обеспечивалось быстрое или замедленное высвобождение активного ингредиента после введения этой композиции пациенту. Предпочтительно, если композиция будет изготовлена в виде разовой лекарственной формы, каждая из которых содержит от около 1 до около 500 мг, а предпочтительно от около 5 до около 300 мг активного ингредиента. При этом, следует отметить, что доза введения данного терапевтического препарата может быть определена лечащим врачом, исходя из конкретных условий, включая заболевание, на которое направлено лечение, тип вводимого соединения, способ введения и т.п. В соответствии с этим, вышеуказанные диапазоны доз являются иллюстративными и не ограничивают объема настоящего изобретения. Термин "разовая лекарственная форма" означает физически дискретную лекарственную единицу с унифицированной дозой, предназначенной для введения человеку и другим млекопитающим и содержащей заранее определенное количество активного материала, рассчитанное на нужный терапевтический эффект, в сочетании с подходящим фармацевтическим носителем.
Кроме того, соединения настоящего изобретения могут быть использованы для наружного применения. Композиции, предназначенные для этих целей, изготавливают в виде мазей, кремов и гелей.
Мази обычно изготавливают с использованием либо (1) масляной основы, т. е. , основы, состоящей из жирных масел или углеводородов, таких как нефтяное или минеральное масло; либо (2) абсорбирующей основы, т.е., основы, состоящей из безводного вещества или вещества, способного абсорбировать воду, например, безводного ланолина. Количество активного ингредиента (соединения) добавляется в композицию в соответствии с масляной или абсорбирующей основой, используемой в данной композиции.
Кремы представляют собой эмульсии "масло в воде". Эти эмульсии состоят из масляной фазы (дисперсная фаза), обычно содержащей жирные масла, углеводороды и т.п., такие, как воски, нефтяное, минеральное масло и т.п.; и водной фазы (непрерывная фаза), содержащей воду и любые водорастворимые вещества, например, аддитивные соли. Эти две фазы могут быть стабилизированы путем использования эмульгирующего агента, например, поверхностно-активного вещества, такого как лаурилсульфат натрия, гидрофильные коллоиды, например, аравийская камедь, коллоидальные глины, пчелиный воск и т.п. При составлении эмульсий, активный ингредиент обычно добавляют в количестве, необходимом для получения нужной концентрации.
Гели изготавливают обычно на масляной, водной или эмульсионно-суспензионной основе. К этой основе добавляют гелеобразующий агент, который формирует матрицу, увеличивая вязкость композиции. Примерами гелеобразующих агентов являются гидроксипропилцеллюлоза; полимеры акриловой кислоты и т.п. Обычно, активное соединение добавляют к композиции в нужной концентрации перед добавлением гелеобразующего агента.
Количество соединения, вводимое в композицию для наружного применения, не является критическим; это количество должно быть лишь достаточным для свободного нанесения указанной композиции на пораженные участки ткани и для обеспечения нужного терапевтического эффекта.
Количество композиции для наружного применения зависит от размеров пораженной ткани и от концентрации активного соединения в данной композиции. В основном, композиция для наружного применения может быть нанесена в количестве от около 1 до около 500 мкг/см2 пораженной ткани, предпочтительно от около 30 до около 300 мкг на см2 пораженной ткани, более предпочтительно от 50 до 200 мкг/см2 и наиболее предпочтительно от около 60 до около 100 мкг/см2.
Ниже представлены примеры, иллюстрирующие осуществление настоящего изобретения, и не должны рассматриваться как некое ограничение его объема.
Композиция 1
Жесткие желатиновые капсулы получали с использованием следующих ингредиентов, (мг/капсула)
Активный агент - 250
Крахмал, осушенный - 200
Стеарат магния - 10
Всего - 460
Вышеуказанные ингредиенты смешивали и наполняли ими жесткие желатиновые капсулы в количестве 460 мг.
Композиция 2
Таблетку получали с использованием следующих ингредиентов, (мг/капсула)
Активный агент - 250
Микрокристаллическая целлюлоза - 400
Коллоидальный диоксид кремния - 10
Стеариновая кислота - 5
Всего - 65
Эти компоненты смешивали и спрессовывали в таблетки весом по 665 мг каждая.
Композиция 3
Аэрозольный раствор получали с использованием следующих ингредиентов, (мг/капсула)
Активный агент - 0,25
Этанол - 29,75
Пропеллент 22 (хлородифторометан) - 70,0
Всего - 100,00
Активное соединение смешивали с этанолом. Полученную смесь добавляли к порции пропеллента 22, охлаждали до -30oC и переносили в устройство для заполнения. Затем требуемое количество соединения помещали в контейнер из нержавеющей стали и разводили остатком распыляющего вещества. После этого к контейнеру подсоединяли клапанное устройство.
Композиция 4
Таблетки, содержащие по 60 мг активного ингредиента, получали следующим образом, (мг/капсула)
Активный агент - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (10% раствор в воде) - 4
Натрийсодержащий карбоксиметилкрахмал - 4,5
Стеарат магния - 0,5
Тальк - 1
Всего - 150
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 18 меш, США, и энергично перемешивали. Раствор поливинилпирролидона перемешивали с полученными поршкообразными веществами, которые затем пропускали через сито N 14, меш, США. Полученные таким образом гранулы осушали при 50oC и пропускали через сито N 18, меш, США. Натрийсодержащий карбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш, США, добавляли к этим гранулам, которые после перемешивания спрессовывали в таблетировочной машине и получали таблетки массой по 150 мг каждая.
Композиция 5
Капсулы, содержащие по 80 мг лекарственного средства каждая, получали следующим образом, (мг/капсула)
Активный агент - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния перемешивали, пропускали через сито N 45 меш, США, а затем наполняли ими жесткие желатиновые капсулы в количестве 200 мг.
Композиция 6
Суппозитории, содержащие 225 мг активного ингредиента каждый, получали следующим образом, (мг/капсула)
Активный агент - 225
Глицериды насыщенных жирных кислот - 2000
Всего - 2225
Активный ингредиент пропускали через сито N 60 меш, США, и суспендировали в предварительно расплавленных глицеридах насыщенных жирных кислот, с использованием минимальной требуемой температуры. После этого смесь выливали в массу для суппозиториев с номинальной емкостью 2 г и оставляли для охлаждения.
Композиция 7
Суспензии, содержащие по 50 мг лекарственного средства каждая на 5 мл-дозу, изготавливали следующим образом: (мг/капсула)
Активный агент, мг - 50
Натрийсодержащая карбоксиметилцеллюлоза, мг - 50
Сироп, мл - 1,25
Раствор бензойной кислоты, мл - 0,10
Отдушка - Дост. кол-во
Краситель - Дост. кол-во
Очищенная вода, мл - 5
Лекарственное средство пропускали через сито N 45 меш, США, а затем смешивали с натрийсодержащей карбоксиметилцеллюлозой и сиропом для образования зубной пасты. После этого, при перемешивании добавляли раствор бензойной кислоты, отдушку и краситель, разбавленный некоторым количеством воды. Для получения требуемого объема добавляли достаточное количество воды.
Композиция 8
Внутривенная композиция может быть получена следующим образом, мг/капсула:
Активный агент - 250
Изотонический физиологический раствор - 1000
Раствор из вышеописанных ингредиентов вводят внутривенно в дозе 1 мл/1 мин пациенту, нуждающемуся в таком лечении.
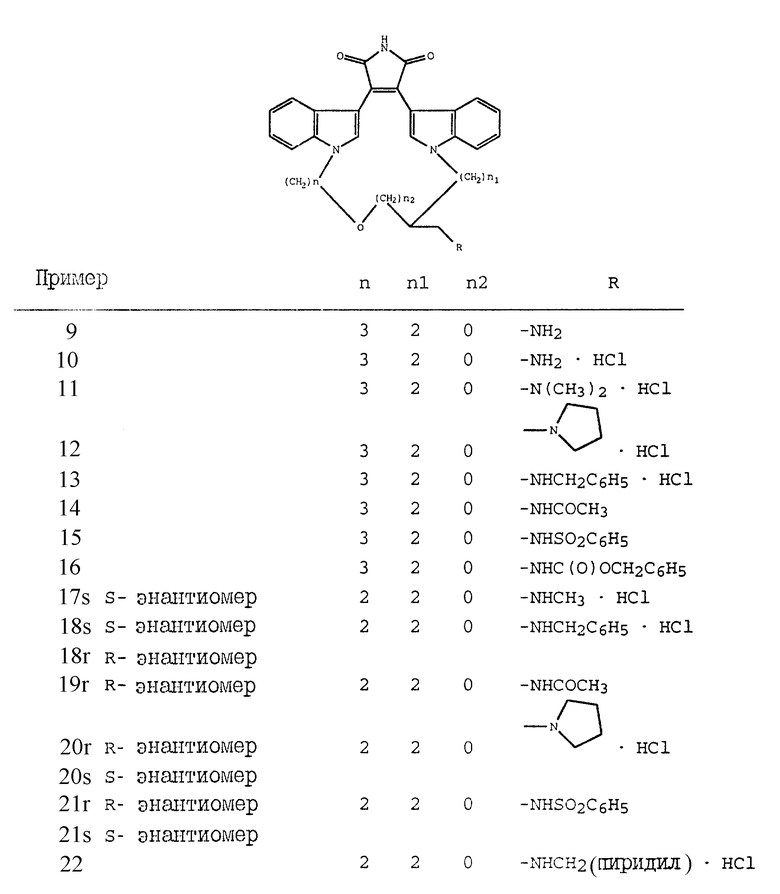
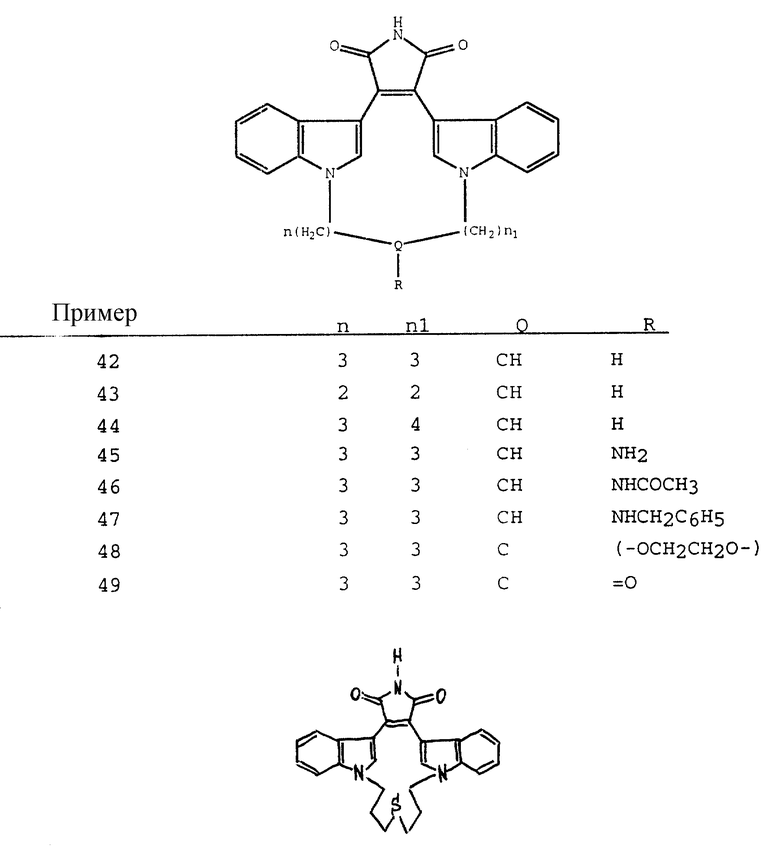
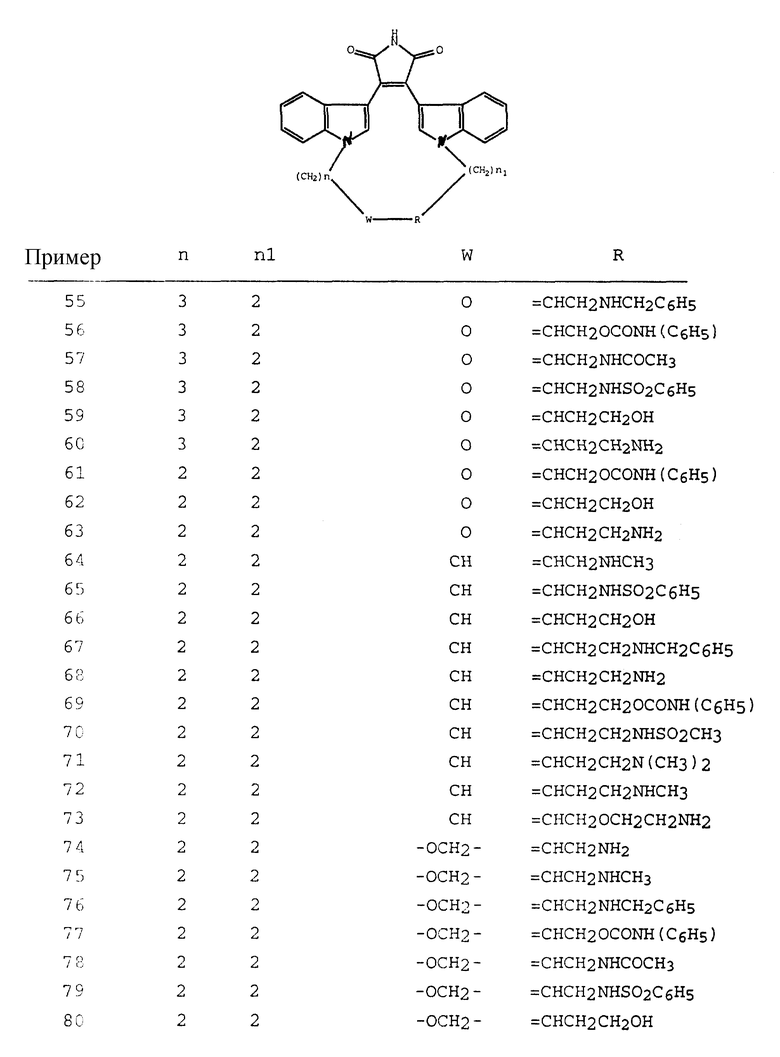
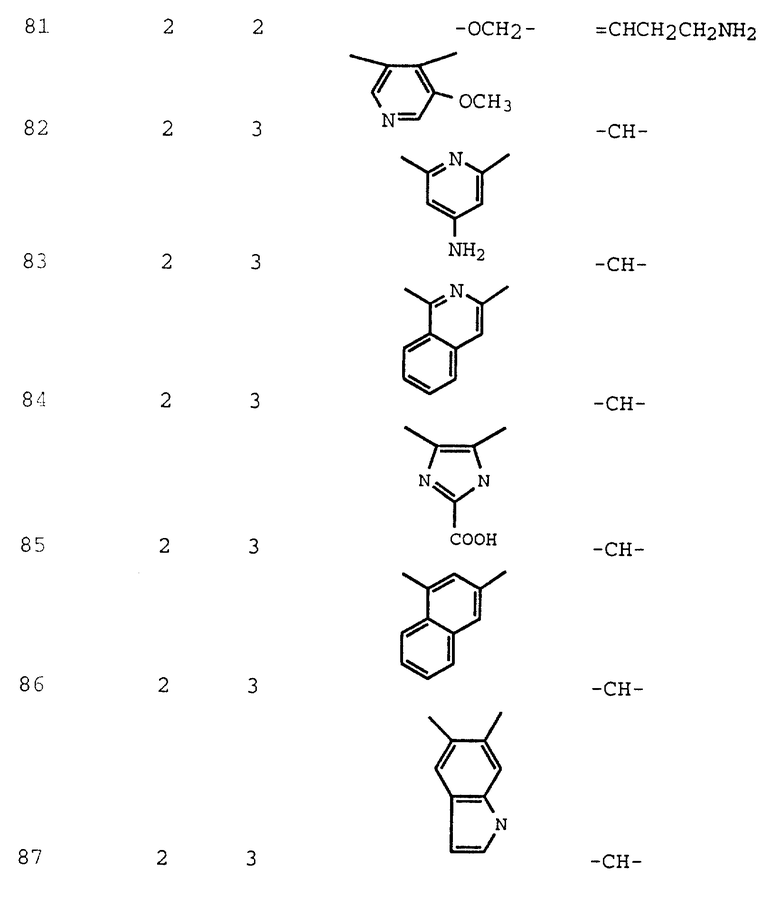
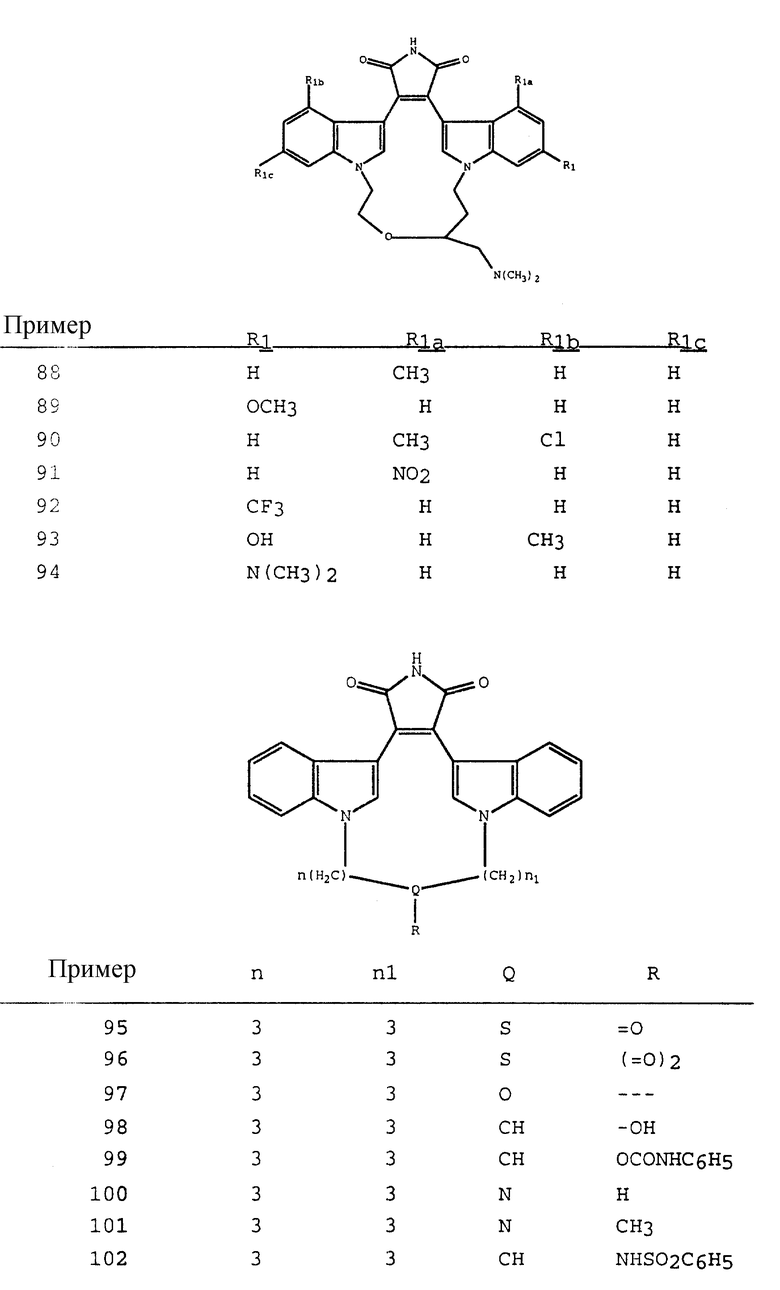
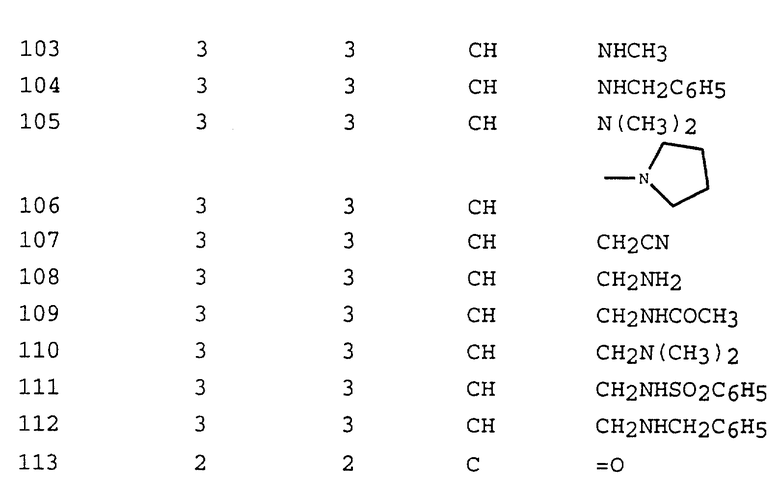

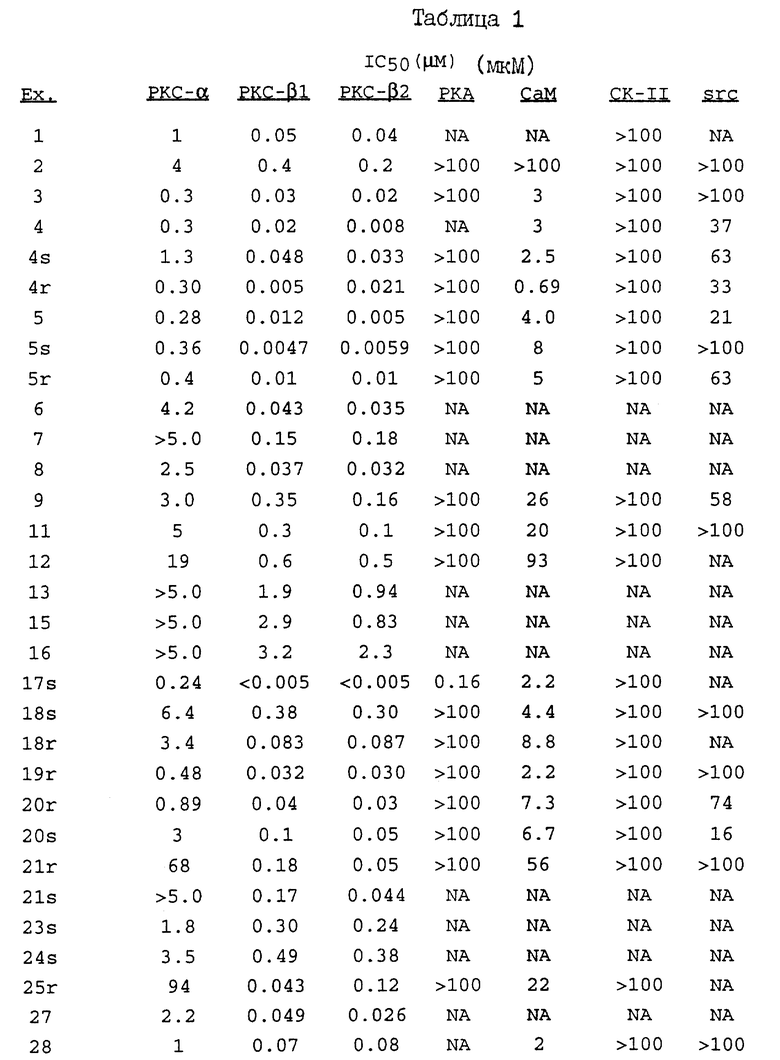
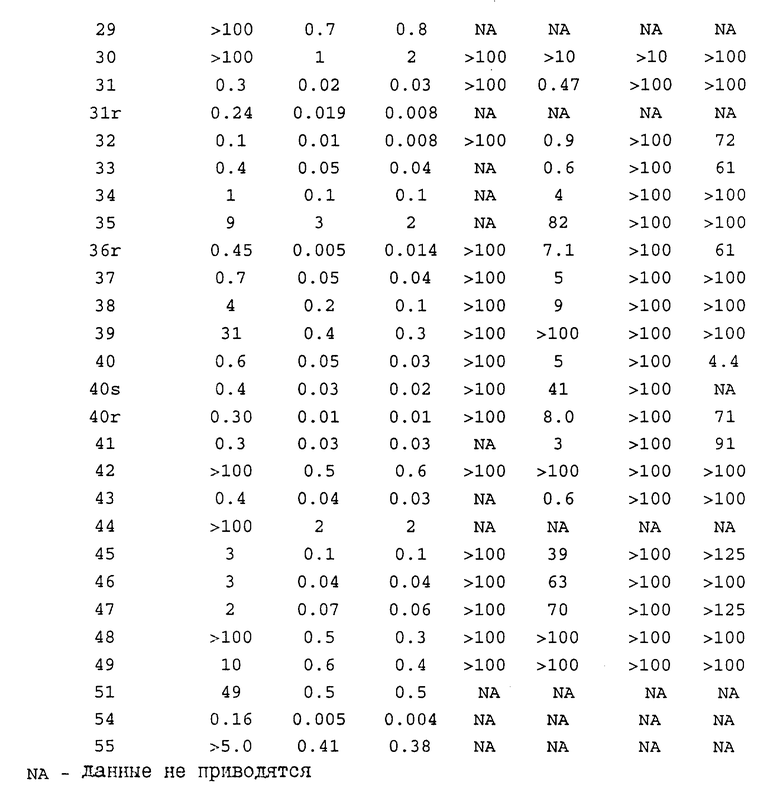

Описываются новые бис-индолмалеимидные макроциклические производные формулы (I), где значения R1, R2, X, Y, W, m указаны в п.1 формулы. Они относятся к новым и сильным ингибиторам протеинкиназы С и обладают высокой изозим-селективностью. Они могут найти применение при лечении заболеваний, связанных с сахарным диабетом, а также таких заболеваний, как ишемия, кожные заболевания и рак. Описывается также способ их получения (варианты) и фармацевтическая композиция на основе соединений формулы (I). 5 с. и 8 з.п. ф-лы, 3 табл.
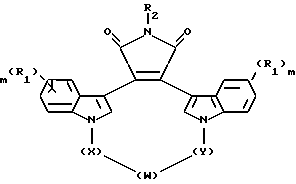
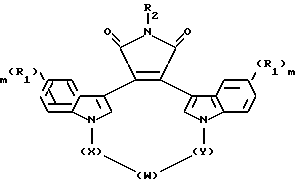
где W - -O-, -S-, -SO-, -SO2-, -CO-, -NR3-, -гетероцикл-, выбранный из группы, включающей 5-6-членные гетероциклы, содержащие один или два атома азота; -конденсированный бицикл-, представляющий собой 5-6-членный азотсодержащий гетероцикл, сконденсированный с фенильным кольцом, или нафтилдиил, C2 - C6-алкилен, замещенный алкилен формулы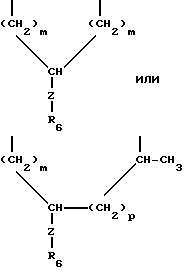
где Z - -(CH2)p- или -(CH2)p-O-(CH2)p-, когда R6 представляет -NH2; R6 - C1 - C4-алкил, C1 - C4-алкокси, (CH2)m-арил, (CH2)m-арилокси, гидрокси, карбокси, -COO(C1 - C3-алкил), -COO(CH2)m-арил, -NR4R5, -NH(CH2)m-арил, -NH(CH2)m-пиридил, -CONH(CH2)m-арил, -NHCO(C1 - C4-алкил), -OCONH(CH2)m-арил, -NHCOO(бензил), -NHSO2(C1 - C4-алкил), -NHSO2(CH2)m-арил, -CN, -SO2(C1 - C4-алкил), гетероцикл, выбранный из группы, включающей 5-6-членный азотсодержащий гетероцикл или морфолин; арил-фенил;
X и Y независимо представляют собой C1 - C4-алкилен, возможно замещенный группой Z-R6;
R1 независимо - водород, галоген, C1 - C4-алкил, гидрокси, C1 - C4-алкокси, галогеноалкил, нитро, -NR4R5;
R2 - водород;
R3 - водород, C1 - C4-алкил;
R4 и R5 независимо - водород, C1 - C4-алкил, фенил, бензил;
p = 0, 1 или 2, независимо;
m = 0, 1, 2 или 3, независимо,
или их фармацевтически приемлемая соль или сольват.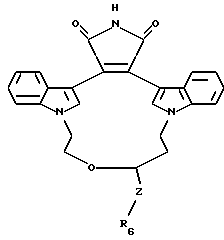
где Z - (CH2)p-;
R6 - гидрокси, C1 - C4-алкил, (CH2)m-арил, -NH(арил), -NHSO2(C1 - C4-алкил), -NHSO2(CH2)m-арил или -NR4R5; арил-фенил;
R4 - водород или C1 - C4-алкил;
R5 - водород, C1 - C4-алкил, бензил;
p = 0, 1 или 2;
m = 0, 1, 2 или 3.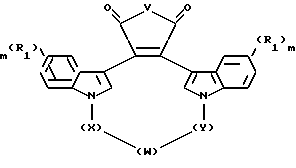
где V - -O- или N-CH3 ;
W - -O-, -S-, -SO-, -SO2-, -CO-, -NR3-, C2 - C6-алкилен, -гетероцикл-, выбранный из группы, включающей 5-6-членные гетероциклы, содержащие один или два атома азота; -конденсированный бицикл-, представляющий собой 5-6-членный азотсодержащий гетероцикл, сконденсированный с фенильным кольцом, или нафтилдиил, замещенный алкилен формулы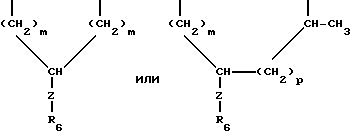
где Z - -(CH2)p- или -(CH2)p-O-(CH2)p-, когда R6 представляет -NH2;
R6 - C1 - C4-алкил, C1 - C4-алкокси, (CH2)m-арил, (CH2)m-арилокси, гидрокси, карбокси, -COO(C1 - C4-алкил), -COO(CH2)m-арил, -NR4R5, -NH(CH2)m-арил, -NH(CH2)m-пиридил, -CONH(CH2)m-арил, -NHCO(C1 - C4-алкил), -OCONH(CH2)m-арил, -NHCOO(бензил), -NHSO2(C1 - C4-алкил), -NHSO2(CH2)m-арил, -CN, SO2(C1 - C4-алкил), гетероцикл, выбранный из группы, включающей 5-6-членный азотсодержащий гетероцикл или морфолин; арил-фенил;
X и Y независимо - C1 - C4-алкилен;
R1 независимо - водород, галоген, C1 - C4-алкил, гидрокси, C1 - C4-алкокси, галогеноалкил;
R3 - водород, C1 - C4-алкил;
R4 и R5 независимо - водород, C1 - C4-алкил, фенил, бензил;
m = 0, 1, 2 или 3, независимо,
p = 0, 1 или 2, независимо.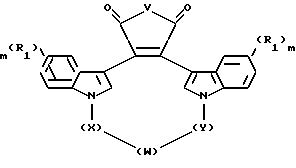
где значения R1, X, W, V и m указаны в п.7,
заключающийся в том, что смесь соединения, имеющего концентрацию от около 1,5 М до около 0,001 М, формулы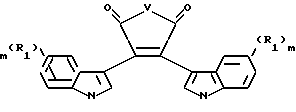
где значения R1, m, V приведены в п.7,
и алкилирующего агента, имеющего концентрацию от около 1,5 М до около 0,001 М, формулы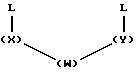
где значения X, W, V определены в п.7,
L является удаляемой группой,
в полярном апротонном растворителе прибавляют к суспензии приблизительно 0,5 - 10 эквивалентов Cs2CO3 в полярном апротонном растворителе, со скоростью от около 0,1 мл/ч до около 2,0 мл/ч.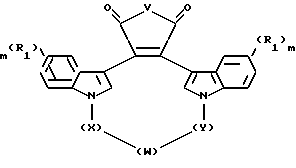
где значения R1, X, W, V и m определены в п.7,
заключающийся в том, что смесь соединение, имеющее концентрацию от около 3 М до около 0,001 М, формулы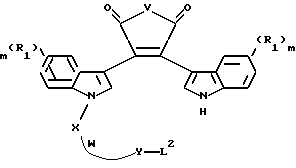
где L2 независимо является удаляемой группой;
R1, X, W, V и m определены в п.7,
объединяют приблизительно с 0,5 - 10,0 эквивалентами Cs2CO3 со скоростью от около 0,1 мл/ч до около 2,0 мл/ч в полярном апротонном растворителе.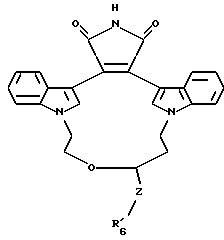
где Z - (CH2)p-;
R6' представляет собой защищенную гидроксигруппу или защищенный амин;
p = 0, 1 или 2;
и b) необязательно замещают гидроксигруппу или аминогруппу для получения соединения по любому из пп.3 - 6, где R6 представляет собой гидроксигруппу, C1 - C4-алкил, (CH2)m-арил, -NH(арил), -NHSO2(C1 - C4-алкил), -NHSO2(CH2)m-арил или -NR4R5, R4 - водород или C1 - C4-алкил; R5 - водород, C1 - C4-алкил, бензил, арил-фенил.
Приоритет по признакам:
07.12.93 - все значения радикалов за исключением R4 - фенил по пп.1 и 7.
| УСТРОЙСТВО ДЛЯ ФОРМОВАНИЯ ПОКРЫШЕК ПНЕВМАТИЧЕСКИХ ШИН | 0 |
|
SU328000A1 |
| SU 1179932 A, 1985 | |||
| US 4886802 A, 1989. | |||

