Настоящая заявка является частичным продолжением находящейся на рассмотрении заявки на патент США N 07/539763, поданной 18 июня 1990 г. и озаглавленной "Повышенное содержание крахмала в растениях".
Последние успехи генной инженерии позволили создать необходимый инструмент трансформации растений с введением в них чужеродных генов. В настоящее время можно создавать растения, обладающие характеристиками, имеющими важное значение с точки зрения агрономии и обработки урожая. Несомненно, одной из таких важных характеристик является повышенное содержание крахмала и его качество в различных культурных растениях.
Крахмал является полисахаридом, в основном состоящим из звеньев глюкозы, соединенных альфа-1-4- и альфа-1-6-связями. В растительных клетках крахмал находится в виде не растворимых в воде зерен или гранул. В ходе фотосинтеза крахмал образуется и хранится в хлоропластах. Кроме того, крахмал синтезируется в корнях и органах хранения, таких как клубни и семена. В таких не образуемых фотосинтезом тканях крахмал находится в виде пластидов, называемых амилопластами. Как и в хлоропластах, крахмал в амилопластах хранится в виде гранул крахмала. Размер гранул меняется в зависимости от вида растения.
Реально крахмал состоит из амилозы и амилопектина - двух различных полимеров глюкозы. Амилоза в основном состоит из соединенных альфа-1-4-связями линейных цепей молекул глюкозы. В среднем амилоза обладает цепью протяженностью примерно в 1000 молекул глюкозы. Амилопектин характеризуется более короткими цепями, связанными друг с другом альфа-1-6-связями. В среднем амилопектин обладает цепью протяженностью в 20-25 молекул глюкозы.
До недавнего времени в литературе спорным моментом считался субстрат для синтеза крахмала (АДФ-глюкоза или УДФ-глюкоза). Но с выделением мутантов Arabidopsis, не имеющих АДФ-глюкоза-фосфорилазы, признано, что растениями для синтеза крахмала в качестве субстрата используется АДФ-глюкоза. Существуют три этапа синтеза крахмала, и все эти реакции происходят в пределах хлоропластов или амилопластов. На первом этапе из глюкоза-1-фосфата и АТФ под воздействием пирофосфорилазы (EC 2.7.7.27) образуется АДФ-глюкоза. На втором этапе АДФ-глюкоза используется крахмал-синтетазой (EC 2.4.1.21) для образования линейных цепей крахмала, имеющих α- 1-4-связи. На третьем этапе изомеризующий фермент-(ы) (EC 2.4.1.18) вводит альфа-1-6-связи с образованием молекулы амилопектина.
Основной спор возникал вокруг регулирующего этапа синтеза крахмала в растениях. Хотя в качестве регулирующего этапа биосинтеза крахмала предложен синтез АДФ-глюкозы под действием АДФ-глюкоза-пирофосфорилазы, доказано это не было. Так, в заявке на Европейский патент с N публикации 0368506 A2, относящейся к АДФ-глюкоза-пирофосфорилазе, рассматривается роль этого фермента в качестве фактора, лимитирующего скорость биосинтеза крахмала. Доводом против АДФ-глюкоза-пирофосфорилазы в качестве регулирующего фермента могут служить результаты, полученные с мутантом Arabidopsis (Lin, 1988 a, b). Найдено, что данный мутант (TL 46) содержит всего лишь 5% активной АДФ-глюкоза-пирофосфорилазы по сравнению с растениями дикого типа. Однако TL 46 растения продуцируют примерно 40% крахмала от уровня растений дикого типа. Если бы АДФ-глюкоза-пирофосфорилаза была лимитирующим скорость ферментом, в этом случае следовало бы ожидать 95%-ного снижения ферментивной активности для создания понижения в накоплении крахмала в более чем 60%. Аналогично определения in vitro для поддающихся извлечению носителей активности предполагают, что указанный фермент может лимитировать скорость только в случае, если его активность in vivo существенно ингибируется аллостерическими регуляторами ферментивной активности.
Краткое изложение сущности изобретения
Настоящим изобретением даются структуральные ДНК конструкты, кодирующие фермент АДФ-глюкоза-пирофосфорилазу (АДФГПФ) и применимые для создания в растениях повышенного содержания крахмала. Кроме того показано, что ферментивная активность в растительных клетках и тканях является регулирующим фактором биосинтеза крахмала.
В дополнение вышеуказанному одним из аспектов настоящего изобретения дается способ создания генетически трансформированных растений с повышенным содержанием крахмала, включающий стадии:
(a) введения в геном растительной клетки рекомбинантной двуспиральной молекулы ДНК, состоящей из
(I) промотора, действие которого в растениях вызывает продуцирование в целевых растительных тканях РНК последовательности,
(II) структуральной ДНК последовательности, вызывающей продуцирование РНК последовательности, кодирующей слитый полипептид, состоящий из аминоконцевого пластидного транзитного пептида и фермента АДФ-глюкоза-пирофосфорилазы,
(III) 3'-нетрансляционный ДНК последовательности, действие которой в растительных клетках вызывает прекращение транскрипции и присоединение полиаденилированных нуклеотидов к 3'-концу РНК последовательности;
(b) получения трансформированных растительных клеток;
(c) регенерации из трансформированных растительных клеток генетически трансформированных растений с повышенным содержанием крахмала.
Согласно другому аспекту настоящего изобретения дается молекула рекомбинантной двуспиральной ДНК, включающей в своей последовательности:
(a) промотор, действие которого в растениях вызывает продуцирование в целевых тканях растения РНК последовательности;
(b) структурную ДНК последовательность, вызывающую продуцирование РНК последовательности, кодирующей слитый полипептид, состоящий из аминоконцевого пластидного транзитного пептида и фермента АДФ-глюкоза-профосфорилазы;
(c) 3'-нетрансляционной области, действие которой в растительных клетках вызывает прекращение транскрипции и присоединение полиаденилированных нуклеотидов к 3'-концу РНК последовательности, причем промотор гетерологичен по отношению к структуральной ДНК.
Кроме того, согласно еще одному аспекту настоящего изобретения даются бактериальные и трансформированные растительные клетки, содержащие соответственно ДНК, состоящую из вышеупомянутых элементов (a), (b) и (c).
И еще одним аспектом настоящего изобретения даются дифференцированные растения с повышенным содержанием крахмала.
Краткие пояснения к диаграммам
На фиг. 1 приведена нуклеотидная последовательность (Послед. N 1) и соответствующая аминокислотная последовательность (Послед. N 2) для гена АДФ-глюкоза-пирофосфорилазы (gIgC) из E. coli.
На фиг. 2 приведена нуклеотидная последовательность (Послед. N 3) и соответствующая аминокислотная последовательность (Послед. N 4) для мутантного гена АДФ-глюкоза-пирофосфорилазы (gIgC16) из E. coli.
На фиг. 3 приведена нуклеотидная последовательность (Послед. N 5) и соответствующая аминокислотная последовательность (Послед. N 6) для модифицированного хлоропласного транзитного пептида из ssRUBISCO 1A гена из Arabidopsis thaliana.
На фиг. 4 показана плазмидная карта для вектора pMON530 трансформации растений.
На фиг. 5 приведена нуклеотидная последовательность (Послед. N 7) и соответствующая аминокислотная последовательность (Послед. N 8) готовой небольшой субъединицы АДФ-глюкоза-фосфорилазы гена картофеля.
На фиг. 6 приведена почти полная протяженность нуклеотидной последовательности (Послед. N 9) и соответствующей аминокислотной последовательности (Послед. N 10) почти полной большой субъединицы АДФ-глюкоза-пирофосфорилазы гена картофеля.
На фиг. 7 показана плазмидная карта вектора pMON20113 трансформации растений.
На фиг. 8 показана плазмидная карта вектора pMON16938 трансформации растений.
На фиг. 9 показана плазмидная карта вектора pMON977 трансформации растений.
На фиг. 10 показана плазмидная карта вектора pMON6950 трансформации растений.
На фиг. 11 показана плазмидная карта вектора pMON10098 трансформации растений.
Подробное описание изобретения
Экспрессия растительного гена, существующего в виде двуспиральной ДНК, включает транскрипцию матричной РНК (мРНК) из одной из спиралей ДНК при участии фермента РНК-полимеразы и последующую обработку внутри ядра первичного продукта транскрипции мРНК. Такая обработка включает участие 3'-нетрансляционной области, присоединяющей полиаденилированные нуклеотиды к 3'-концу РНК.
Транскрипция ДНК в РНК регулируется областью ДНК, обычно называемой "промотором". Область промотора содержит последовательность оснований, сигнализирующих РНК-полимеразе о связывании с ДНК и об инициации транскрипции мРНК с использованием одной из спиралей ДНК в качестве матрицы для получения соответствующей комплементарной нити РНК.
В литературе описано множество промоторов, проявляющих активность в растительных клетках, в том числе: нопалин-синтетаза (NOS) и октопин-синтетаза (OCS) промотооры (находятся на индуцирующих опухоль плазмидах из Agrobacterium timefaciens, колимовирусные промоторы, такие как: 19S и 35S-промоторы мозаичного вируса цветной капусты (CaMV) и 35S-промоторы мозаичного вируса коричника, световозбуждаемый промотор из малой субъединицы рибулоза-1,5-бисфосфаткарбоксилазы (ssRUBISCO, очень распространенный растительный полипептид), промотор гена a/b связывания белка в хлорофиле и т.д. Все указанные промоторы были использованы для создания ДНК конструктов различного типа, экспрессированных затем в растениях (см., напр., PCT публикацию WO 84/02913 (Rogers и др., Монсанто).
Промоторы, для которых известна или обнаружена способность вызывать транскрипцию РНК в клетках растений, могут быть использованы и в настоящем изобретении. Такие промоторы могут быть получены из различных источников, таких как растения и вирусы растений, в том числе, но без ограничения только ими, усиленный CaMV 35S-промотор и промоторы, в выделенные из генов растения, например генов ssRUBISCO. Как показано ниже, рекомендуется, чтобы конкретный выбранный промотор был способен вызывать достаточную экспрессию с продуцированием в результате эффективного количества фермента АДФ-глюкоза-пирофосфорилазы, вызывающего необходимое повышение содержания крахмала. Кроме того, рекомендуется проводить экспрессию АДФГПФ гена в специфичных тканях растения, таких как листья, корни, клубни, семена, плоды и т.п., и выбранный промотор должен обладать необходимой тканевой и ростовой специфичностью. Для специалиста очевидно, что количество АДФ-глюкоза-пирофосфорилазы, необходимое для индуцирования целевого повышения содержания крахмала, может меняться в зависимости от типа растения, более того, слишком высокая активность АДФ-глюкоза-пирофосфорилазы может оказать нежелательное воздействие на растение. Таким образом, действие промотора следует оптимизировать путем подбора промотора с необходимой экспрессионной способностью в тканях и регулированием силы промотора, а также путем подбора трансформанта, создающего необходимую активность АДФ-глюкоза-пирофосфорилазы в целевых тканях. Подобный подбор в смеси трансформантов, как правило, применяют при экспрессии гетерологичных структуральных генов в растениях, поскольку при этом между трансформантами с одинаковым гетерологичным геном существуют отличия, вызванные различными сайтами внедрения гена в геном растения (обычно называемые "эффектом положения").
Рекомендуется, чтобы промоторы, применяемые в двуспиральной молекуле ДНК настоящего изобретения, обладали сравнительно высокой экспрессирующей способностью в тканях, в которых желателен высокий уровень крахмала, например: клубнях картофеля и плодах томата. Для картофеля особенно рекомендуемым в этом смысле промотором является пататин-промотор, более подробно описанный в прилагаемых примерах. Экспрессия молекулы двуспиральной ДНК настоящего изобретения под действием выбранного промотора во всех или в большинстве тканях растения вряд ли желательна, а в некоторых случаях может оказать нежелательное воздействие на рост растения.
Пататин-промотор класса I, используемый в данном исследовании для экспрессирования E.coli АДФГПФ, как показано, обладает как высокой активностью, так и специфичностью к клубням (Bevan и др., 1986; Jefferson и др., 1990). Известны и другие гены, характеризующиеся специфичностью к клубням или повышенной экспрессирующей способностью, в том числе: гены АДФГПФ клубней картофеля (Muller и др., 1990), сахароза-синтетаза (Salanoubut и Belliard 1987, 1989), основные белки клубней, включая белковые комплексы в 22 к.о. и ингибиторы протеиназы (Hannapel, 1990), и другие палатины класса I и II (Rocha-Sosa и др., 1989; Mignery и др., 1988).
Помимо эндогенных растительных АДФ-глюкоза-пирофосфорилазных промоторов для экспрессирования гена АДФ-глюкоза-пирофосфорилазы в специфичных тканях, таких как: листья, семена или плоды могут быть также использованы и другие промоторы. β-Конглицинин (известные также, как 7 белков) являются одним из основных хранящихся в сое (Glycine max) (Tierney, 1987). Промотор для β-конглицинина может быть использован для сверхэкспрессии E.coli или любого другого АДФ-глюкоза-пирофосфорилазного гена со специфичностью к семенам, что могло бы привести к повышению содержания крахмала в семенах. β-Субъединица β-конглицинина была экспрессирована с помощью его эндогенного промотора в семенах трансгенных петуний и табака, при этом показано, что промотор действует в других растениях специфичных к семенам образом (Bray, 1987).
Зеины образуют группы белков, откладываемых в эндосперме кукурузы. Выделены геномные клоны для генов зеина (Pedersen 1982) и промоторы из этих клонов могут быть также использованы для экспрессирования АДФ-глюкоза-пирофосфорилазного гена в семенах кукурузы и других растений.
Содержание крахмала в плодах томата может быть повышено экспрессированием гена АДФ-глюкоза-пирофосфорилазы и без помощи специфичного к плодам промотора. Промотор из 2А11 геномного клона (Pear, 1989) или Е8-промотор (Deikman, 1988) могли бы экспрессировать АДФ-глюкоза-пирофосфорилазу в плодах томата. Кроме того, выделены новые специфичные к плодам промоторы, характеризующиеся высокой и специфичной экспрессией в ходе созревания плодов томата. Для идентификации приемлемых кДНК клонов, специфично экспрессируемых в зеленых плодах, применен дифференцированный отбор с помощью кДНК библиотеки плодов томата. Использовались кДНК зонды, приготовленные из мРНК, выделенной из плодов на раннем и позднем этапах созревания плодов, из объединенной ткани лист-стебель и из корневой ткани томата. Идентифицированы клоны, обильно экспрессируемые в зеленых плодах и не показавшие поддающейся обнаружению экспрессии в листьях. Геномный саузерн-анализ показал небольшое число (1-2) копий гена. Затем отбором банка геномных клонов томата выделены промоторы для таких кДНК клонов. Характер экспрессии этих промоторов подтвержден слиянием с геном β-глюкоуронидазы (COS) и последующей экспрессией GUS фермента в трансгенных плодах в ходе их созревания. Промоторы, характеризующиеся экспрессией в большей части клеток плодов, затем подвергают слиянию с ХТП-gIgC16 и другими gIgC аллелями или АДФГПФ генами, происходящими либо из водорослей, либо из растений.
Содержание крахмала в корневой ткани может быть повышено экспрессированием гена АДФ-глюкоза-пирофосфорилазы вслед за специфичным к корням промотором. Промотор из гена кислотной хитиназы (Samac и др., 1990) мог бы экспрессировать АДФ-глюкоза-пирофосфорилазу в корневых тканях. Экспрессия в корневых тканях может быть также осуществлена использованием идентифицированных специфичных к корням субдоменов CamV35S-промотора (Benfey и др., 1989). Содержание крахмала в тканях листьев может быть повышено экспрессированием гена АДФ-глюкоза-пирофосфорилазы (напр., gIgC гена) с помощью активного в лиcтьях промотора, такого как ssRUBISCO - промотора или промотора гена а/о связывания белка в хлорофилле.
РНК, продуцируемая ДНК конструктом настоящего изобретения, содержит также 5'-нетрансляционную лидерную последовательность. Такая последовательность может быть получена из промотора, выбранного для экспрессии гена, и может быть специфично модифицирована так, чтобы повысить трансляцию мРНК. 5'-нетрансляционные области могут быть также получены из вирусных РНК, из приемлемых эукариотных генов или из синтетических генных последовательностей. Настоящее изобретение не ограничено конструктами, представленными последующими примерами, в которых нетрансляционная область происходит из 5'-нетрансляционной последовательности, сопутствующей последовательности промотора. Более того, нетрансляционная лидирующая последовательность, как показано выше, может происходить из чужого промотора или кодирующей последовательности.
ДНК конструкты настоящего изобретения, кроме того, содержат структуральную кодирующую последовательность в виде двуспиральной ДНК, кодирующую слитый полипептид, состоящий из аминоконцевого пластидного транзитного пептида и фермента АДФ-глюкоза-пирофосфорилазы. Рекомендуется, чтобы применяемый в настоящем изобретении фермент АДФ-глюкоза-пирофосфорилаза подвергался сниженному аллостерическому контролю в растениях. Такой нерегулируемый фермент АДФ-глюкоза-пирофосфорилаза может быть выбран из известных ферментов, проявляющих нерегулируемую ферментивную активность, или может быть продуцирован в результате мутагенеза нативных ферментов АДФ-глюкоза-пирофосфорилаз из бактерий, водорослей или растений, о чем более подробно речь идет ниже. В отдельных случаях существенные отличия в природе регуляторов, модулирующих активность фермента АДФ-глюкоза-пирофосфорилазы (АДФГПФ) дикого типа, позволяет использовать и сам ген дикого типа; в этих случаях концентрация регуляторов в органеллах растения будет усиливать проявление значительной ферментивной активности АДФГПФ.
Бактериальные АДФ-глюкоза-пирофосфорилазы
АДФ-глюкоза-пирофосфорилаза E. coli хорошо охарактеризована, как четко регулируемый фермент. Активатор-фруктоза-1,6-бисфосфат, как показано, активирует фермент путем повышения его Vmax и повышением сродства фермента к его субстратам (Preiss, 1966 и Gentner, 1967). Кроме того, фруктоза-1,6-бисфосфат (ФБФ) также модулирует чувствительность фермента к таким ингибиторам, как аденозин-5'-монофосфат (АМФ) и неорганический фосфат (Pi) (Gentner, 1968).
В 1981 г. удалось клонировать ген АДФ-глюкоза-пирофосфорилазы вместе с геном для гликоген-синтетазы и геном изомеризующего фермента из E.coli K12 (gIgC), и полученная плазмида была названа pOP12 (Okita, 1981). Ген gIgC, последовательность которого установлена в 1983 г. , содержит 1293 п.о. (Послед. N 1) и кодирует 431 аминокислоту (Послед. N 2) с молекулярной массой 48762, приведенную на фиг. 1 (Baecker, 1983).
Ген gIgC создан в результате химического мутагенеза E.coli К12 штамма PA 601 N-метил-N'-нитрозогуанидином (Cattaneo, 1989 и Creuzet-Sigal, 1972). Синтезирующие гликоген мутанты выявлены при окрашивании йодом мутагенизированных колоний. Найдено, что gIgC16 мутант в ходе стационарной фазы накапливает до 48% гликогена по сравнению с 20% гликогена в родоначальном штамме. При сравнении кинетики gIgC16 АДФ-глюкоза-пирофосфорилазы с исходной обнаружено, что gIgC16 АДФ-глюкоза-пирофосфорилаза отличается более высоким сродством к АДФ-глюкозе в отсутствии активатора-фруктоза-1,6-бисфосфата (ФБФ), и концентрация ФБФ, необходимая для полумаксимальной активации фермента, в gIgC16 снижена. Ингибирование активности АДФ-глюкоза-пирофосфорилазы в gIgC16 под действием 5'-аденозинмонофосфата (АМФ) также уменьшено.
Ген gIg C16 клонирован из E.coli K12 618 (Leung, 1986). Получено два клона с противоположной ориентацией. Эти клоны (pEBL 1 и pEBL 3) содержат оба gIgC16 и gIgB (изомеризующий фермент) гена. Обе плазмиды трансформированы в мутантных штаммах E.coli, не обладающих АДФ-глюкоза-пирофосфорилазной активностью. Штамм E. coli K12 G6MD3 не имеет gIg генов, а E.coli B штамм AC7OR1-504 обладает дефективным геном АДФ-глюкоза-пирофосфорилазы и характеризуется подавленной в пять-семь раз активностью к биосинтезу гликогена. Обе плазмиды (pEBL 1 и pEBL 3) создают в обоих мутантных штаммах АДФ-глюкоза-пирофосфорилазную активность. Клонированная АДФ-глюкоза-пирофосфорилаза была частично очищена из E.coli штамма AC7ORI, трансформированного плазмидой pEBL 3. Осуществлено кинетическое сравнение этого фермента с частично очищенной АДФ-глюкоза-пирофосфорилазой из исходного мутантного штамма (E.coli K-12 618) и с частично очищенной АДФ-глюкоза-пирофосфорилазной из E.coli K-12 штамма 356, являющегося родоначальным штаммом дикого типа штамма 618. Мутантные и дикого типа ферменты сравнивались с точки зрения уровня их активности и ингибирования. АДФ-глюкоза-пирофосфорилаза родоначального штамма 356 активировалась фруктоза-1,6-бисфосфатом примерно в 45 раз. Сигмоидальная кривая активации характеризовалась наклоном Хилла в 1,7 и 50%-ная максимальная активация проявлялась при 62 мкМ ФБФ. АДФ-глюкоза-пирофосфорилаза мутантного штамма 618 более активна в отсутствии ФБФ, и активировалась ФБФ только в 1,8-2 раза. Кривая активации для 618 АДФ-глюкоза-пирофосфорилазы имеет гиперболический характер с наклоном Хилла в 1, и 50%-ная максимальная стимуляция проявляется при 15±3,1 мкМ. Фермент, экспрессированный из pEB13 плазмиды, характеризуется теми же ФБФ кинетическими константами, что и АДФ-глюкоза-пирофосфорилаза из мутантного штамма 618.
В настоящее время ДНК последовательность gIgC16 гена установлена (Послед. N 3) (Kumar, 1989). При рассмотрении фиг. 2 и при сравнении выведенной аминокислотной последовательности gIgC16 (Послед. N4) с неизогенным E. coli K-12 3000 можно отметить два изменения в аминокислотах, а именно: замену Lys 296 на Glu и Gly 336 на Asp.
В E. coli обнаружен и ряд других мутантных АДФ-глюкоза-пирофосфорилаз. Экспрессия любых таких или других бактериальных АДФ-глюкоза-пирофосфорилаз дикого типа или мутантов может быть также использована для повышения продуцирования крахмала в растениях.
E. coli K-12 штамм 6041 (gIgC47) накапливает в ходе стационарной фазы примерно такое же количество гликогена, что и штамм 618 (gIgC16). Штамм 6047, как и штамм 618, отличается более высоким кажущимся сродством к ФБФ и большей активностью в отсутствии ФБФ. Однако фермент из штамма 6047, как сообщается, более чувствителен к АМФ по сравнению с ферментом из штамма 618 (Latil-Damotte, 1977).
E. coli B мутант SG5 при сравнении с его родоначальным штаммом характеризуется более высоким сродством к его аллостерическим активаторам и более низким сродством к его аллостирическим ингибиторам (Govons, 1969; Govons, 1973 и Preiss, 1973). Одни только эти изменения делают фермент более активным в физиологических условиях, и это заставляет бактерию накапливать в два-три раза больше гликогена по сравнению с родоначальным штаммом. Мутантная АДФ-глюкоза-пирофосфорилаза из SG5, как и фермент дикого типа, существует в виде гомотетрамера. В отличие от фермента дикого типа, однако, ФБФ заставляет мутантный фермент образовывать олигомеры большей массы (Carlson, 1976).
АДФ-глюкоза-пирофосфорилаза из E. coli B мутантного штамма GL 1136-504 также характеризуется более высоким кажущимся сродством к активаторам и более низким кажущимся сродством к ингибиторам (KappeL, 1981 и Preiss, 1973). Указанный мутант по сравнению с E.coli дикого типа накапливает в три-четыре раза больше гликогена. В условиях активации очищенный GL 1136-504 фермент и фермент дикого типа (AC70R1) обладают сравнимыми удельными активностями. Однако в отсутствии каких-либо активаторов GL 1136-504 фермент в отличие от фермента дикого типа высоко активен.
Клонированию и секвенс-анализу подвергнут также gIgC из Salmonella typhimurium LT2 (Leung и Preiss, 1987 a). Ген кодирует 431 аминокислоту с соответствующей молекулярной массой 45580. Ген gIgC из Salmonella typhimurium LT2 и тот же ген из E.coli K-12 обладают 90%-ной идентичностью на уровне аминокислот и 80%-ой идентичностью на уровне ДНК. Как и E.coli АДФ-глюкоза-пирофосфорилаза, Salmonella typhimurium LT 2 АДФ-глюкоза-пирофосфорилаза также активируется ФБФ и ингибируется АМФ (Leung и Press, 1987 b). Такая заметная консервативность в аминокислотных последовательностях предполагает, что осуществление мутаций, приводящих к усилению АДФГПФ активности в E.coli, в гене S.typhimurium АДФГПФ должно оказывать аналогичное действие на АДФГПФ фермент этого микроорганизма.
Ряд других бактериальных АДФ-глюкоза-пирофосфорилаз охарактеризован по их реакции на активаторы и ингибиторы (см. обзор Preiss, 1973). Подобно АДФ-глюкоза-пирофосфорилазе из E.coli АДФ-глюкоза-пирофосфорилазы из Aerobacter aerogenes, Aerobacter cloacae, Citrobacter freundii и Escherichia aurescens активируются ФБФ и ингибируются АМФ. Однако АДФ-глюкоза-пирофосфорилаза из Aeromonas formi cans активируется фруктоза-6-фосфатом или ФБФ и ингибируется АДФ. Однако АДФ-глюкоза-пирофосфорилаза из Serratia marcescens не активируется каким-либо испытанным метаболитом. Фотосинтетическая Rhodospirillum rubrum имеет АДФ-глюкоза-пирофосфорилазу, активируемую пируватом, но не одно из испытанных соединений, в том числе: Pi, АМФ и АДФ не ингибируют фермент. Исследовано несколько АДФ-глюкоза-пирофосфорилаз из водорослей, регуляция которых, как найдено, аналогична найденной для растительных АДФ-глюкоза-пирофосфорилаз. Очевидно, АДФ-глюкоза-пирофосфорилазы многих организмов могут быть использованы для повышения биосинтеза крахмала и его накопления в растениях.
Помимо E.coli и растительных АДФГПФ ферментов другие источники, в их числе (но без ограничения только ими): цианобактерии, водоросли и другие прокариотные и эукариотные клетки могут служить источниками АДФГПФ генов. Например, выделение АДФГПФ генов из Synechocystis и Anabaena может быть осуществлено с помощью олигонуклеотидов, соответствующих сайту активатора АДФГПФ в E.coli (аминокислотные остатки 25-42 на фиг. 1), который в высшей степени консервативен в широком спектре разнообразных видов. Олигонуклеотиды, соответствующие этой области, могли бы облегчить выделение гена при их использовании в качестве зондов геномных библиотек. Или же для амплификации сегментов АДФГПФ гена с помощью 5'-праймеров, соответствующих сайту активатора E.coli, и 3'-праймеров, соответствующих каталитическим сайтам E.coli, например сайту связывания АДФ-глюкозы E.coli, может быть использована PCR реакция (описана в примере 1). Продукты PCR реакции могут быть использованы в качестве зондов геномных библиотек для выделения соответствующего гена полной длины.
Растительные АДФ-глюкоза-пирофосфорилазы
Одно время полагали, что УДФ-глюкоза является первичным субстратом биосинтеза крахмала в растениях. Тем не менее обнаружено, что АДФ-глюкоза - лучший субстрат биосинтеза крахмала, нежели УДФ-глюкоза (Recondo), 1961). В том же сообщении утверждается об обнаруженной в растительном материале АДФ-глюкоза-пирофосфорилазной активности.
Из листьев шпината выделена в частично очищенном состоянии АДФ-глюкоза-пирофосфорилаза, которая, как показано, активируется 3-фосфоглиуератом (3-ФГА) и ингибируется неорганическими фосфатами (Ghosh и др., 1966). В работе Ghosh и др. предполагается, что биосинтез крахмала в листьях определяется уровнем АДФ-глюкозы. Активатор (3-ФГА) является первичным продуктом фиксации CO2 в ходе фотосинтеза. При фотосинтезе уровень 3-ФГА должен повышаться с активированием в результате АДФ-глюкоза-пирофосфорилазы. В то же время уровень Pi будет понижаться вследствие фотофосфорилирования с уменьшением ингибирования АДФ-глюкоза-пирофосфорилазы. Указанные изменения будут вызывать рост продуцирования АДФ-глюкозы и биосинтеза крахмала. В темноте уровень 3-ФГА будет понижаться, а уровень Pi - повышаться с уменьшением активности АДФ-глюкоза-пирофосфорилазы и соответственно снижением биосинтеза АДФГ и крахмала (Ghosh, 1966).
Позднее АДФ-глюкоза-пирофосфорилаза из листьев шпината была очищена до однородного состояния, и было показано содержание в ней двух субъединиц в 51 и 54 кДа (Morell, 1987). На основе антител, созданных к двум субъединицам, выяснено, что белок в 51 кДа характеризуется гомологичностью к АДФ-глюкоза-пирофосфорилазе как из эндоспермы кукурузы, так и из клубней картофеля, но не к белку в 54 кДа из листьев шпината.
Имеются сообщения о последовательности клона субъединицы кДНК АДФ-глюкоза-пирофосфорилазы из эндоспермы риса (Anderson, 1989 а). Клон кодирует белок из 483 аминокислот. Сравнение белковых последовательностей АДФ-глюкоза-пирофосфорилазы эндоспермы риса и АДФ-глюкоза-пирофосфорилазы E.coli показало 30%-ную идентичность. В том же 1989 г. установлена последовательность клона кДНК почти полной длины для АДФ-глюкоза-пирофосфорилазы эндоспермы пшеницы (Olive, 1989). Клон АДФ-глюкоза-пирофосфорилазы эндоспермы пшеницы характеризуется примерно 24%-ной идентичностью и белковой последовательностью АДФ-глюкоза-пирофосфорилазы E.coli, в то время как клоны пшеницы и риса характеризуются 40%-ной идентичностью на белковом уровне.
Дополнительные свидетельства о существовании нерегулярных растительных АДФ-глюкоза-пирофосфорилаз дикого типа можно найти в работе Olive и др. (Olive, 1989), в которой утверждается, что АДФ-глюкоза-пирофосфорилазы листьев и эндоспермы пшеницы характеризуются очень разной аллостерической регуляцией. АДФ-глюкоза-пирофосфорилазы эндоспермы не активируется 3-ФГА и требует в десять раз больше ингибитора (ортофосфата) для достижения 50%-ного ингибирования, чем фермент из листьев.
АДФ-глюкоза-пирофосфорилаза эндоспермы кукурузы выделена в чистом виде, и, как показано, ее каталитические и регуляторные свойства аналогичны свойствам других растительных АДФ-глюкоза-пирофосфорилаз (Plaxton, 1987). Молекулярная масса нативного фермента из эндоспермы кукурузы равна 230000, и она состоит из четырех субъединиц одинакового размера.
Молекулярная масса нативной АДФ-глюкоза-пирофосфорилазы из клубней картофеля согласно сообщениям равна 200000, а размер субъединицы - 50000 (Sowokinos, 1982). Активность АДФ-глюкоза-пирофосфорилазы клубней почти целиком зависит от 3-ФГА и, как в случае с другими растительными АДФ-глюкоза-пирофосфорилазами, ингибируется Pi. Показано, что АДФ-глюкоза-пирофосфорилазы клубней и листьев картофеля обладают одинаковыми каталитическими, физическими и аллостерическими свойствами (Anderson, 1989 b).
Продуцирование мутагенезом измененных генов АДФ-глюкоза-пирофосфорилазы
Для специалиста очевидно, что, хотя это и не абсолютно обязательно, повышенные результаты должны быть получены при использовании генов АДФ-глюкоза-пирофосфорилазы, подвергнутых сниженной аллостерической регуляции ("разрегулированных"), но более предпочтительно, не подвергавшихся существенной аллостерической регуляции ("нерегулированных"), с сохранением адекватной каталитической активности. Структуральная кодирующая последовательность для бактериального или растительного фермента - АДФ-глюкоза-пирофосфорилазы может быть подвергнута мутагенезу в E.coli или ином приемлемом хозяине и отобрана на повышенное продуцирование гликогена так, как это описано для gIgC16 гена E.coli. Необходимо указать, что применение гена, кодирующего фермент - АДФ-глюкоза-пирофосфорилазу, на который воздействуют только модуляторы (активаторы/ингибиторы), присутствующие в выбранном растении на уровне, существенно не ингибирующие каталитическую активность, не требует модификации фермента (гена). Указанные "нерегулированные" или "разрегулированные" гены затем могут быть введены в растения описанным здесь способом с получением трансгенных растений, характеризующихся повышенным содержанием крахмала.
К примеру, любой ген АДФ-глюкоза-пирофосфорилазы может быть клонирован в E. coli штамм AC70R1-504 (Leung, 1986). Указанный штамм обладает дефектным геном АДФ-глюкоза-пирофосфорилазы и дерепрессирован в пять-семь раз относительно других ферментов биосинтеза гликогена. Ген/кДНК АДФ-глюкоза-пирофосфорилазы может быть встроен в плазмиду E.coli gIgC промотором или любым другим бактериальным промотором. Такой конструкт затем может быть подвергнут сайтнаправленному или ненаправленному мутагенезу. После мутагенеза клетки следует нанести на пластинки со средой, обогащенной 1% глюкозы. После того, как колонии розовьются, пластинки следует омыть раствором йода (0,2 мас.%/об I2, 0,4 мас.%/об. KI в H2O, Creuzet-Sigal, 1972). Сравнением с идентичными пластинками, содержащими не подвергавшийся мутации E.coli, колонии, продуцирующие больше гликогена, могут быть выявлены по их более темной окраске.
Поскольку процедура мутагенеза способна привести к мутациям промотора, любые предполагаемые мутантные АДФ-глюкоза-пирофосфорилазы первого раунда отбора должны бы обладать геном АДФ-глюкоза-пирофосфорилазы, реклонированным в немутировавший вектор, и полученную плазмиду можно отобрать тем же путем. Мутанты, прошедшие через оба раунда отбора, тем самым пройдут анализ из АДФ-глюкоза-пирофосфорилазную активность в присутствии и в отсутствии активаторов и ингибиторов. Сравнением реакций мутировавших АДФ-глюкоза-пирофосфорилаз на активаторы и ингибиторы с реакциями немутировавших ферментов можно охарактеризовать новый мутант.
В работе Plaxton и Preiss (1987) показано, что АДФ-глюкоза-пирофосфорилаза эндоспермы кукурузы обладает регуляторными свойствами двух растительных АДФ-глюкоза-пирофосфорилаз (Plaxton и Preiss, 1987). Авторами показано, что более ранние сообщения, утверждающие, что АДФ-глюкоза-пирофосфорилаза эндоспермы кукурузы обладает повышенной активностью в отсутствии активатора (3-ФГА) и пониженной чувствительностью к ингибитору (Pi), обусловлены протеолитическим расщеплением фермента в ходе выделения. Изменением гена АДФ-глюкоза-пирофосфорилазы с получением фермента, аналогичного протеолитически расщепленной АДФ-глюкоза-пирофосфорилазе эндоспермы кукурузы, достигается сниженная аллостерическая регуляция.
Для анализа жидкой культуры E. coli на АДФ-глюкоза-пирофосфорилазную активность клетки вращают в центрифуге и вновь суспендируют примерно в 2 мл эктракционного буфера (0,05 М глицилглицина с pH 7, 5 мМ ДТЭ, 1 мМ ЭДТК) на грамм клеточной пасты. Клетки подвергают лизису двукратным пропусканием через французский пресс. Клеточный экстракт центрифугируют в микроцентрифуге 5 минут и надосадочную жидкость обессоливают пропусканием через вращающуюся С-50 колонку.
Анализ фермента на синтез АДФ-глюкозы является модификацией опубликованной методики (Haugen, 1976). Каждые 100 мкл образца содержат: 10 мкмолей Гепес-буфера (pH 7,7), 50 мкг АБС, 0,05 мкмоля /14C/ глюкоза-1-фосфата, 0,15 мкмоля АТФ, 0,5 мкмоля MgCl2, 0,1 мкг кристаллической дрожжевой неорганической пирофосфатазы, 1 мМ молибдата аммония, фермент, активаторы и ингибиторы по желанию и воду. Образец инкубируют 10 минут при 37oC и останавливают кипячением 60 секунд. Образец центрифугируют в микроцентрифуге и 40 мкл надосадочной жидкости инъектируют в Синхром Синхропак АХ-100 анионообменную ВЭЖХ колонку. Образец элюируют 65 мМ KPi (pH 5,5). Непрореагировавший /14C/ глюкоза-1-фосфат элюируется в пределах 7-8 минут, а /14C/ АДФ-глюкоза элюируется примерно через 13 минут. Активность фермента определяют по величине радиоактивности, обнаруживаемой в пике АДФ-глюкозы.
Ферментивная активность растительной АДФГПФ четко регулируется как положительными (3-фосфоглицерат, 3-ФГА), так и отрицательными) эффекторами (неорганический фосфат, Pi) (Ghosh и Preiss, 1981; Morell и др., 1987; Plaxton и Preiss, 1987; Preiss, 1988; Sowokinos и Preiss, 1988; Copeland и Preiss, 1981) и отношение 3-ФГА: Pi играет заметную роль в регулировании биосинтеза крахмала путем модулирования активности АДФГПФ (Santarius и Heben, 1965; Helodt и др. , 1977; Kaiser и Bassham, 1979). Растительные АДФГПФ ферменты являются гетеротетрамерами двух больших /"сжатых" и двух меньших /"хрупких" субъединиц (Morell и др. , 1987; Lin и др., 1988 a, 1988 b; Krishnan и др., 1986; Okita и др., 1990), и есть сильные основания полагать, что гетеротетрамер является наиболее активной формой АДФГПФ. Подтверждением такому предположению служит выделение растительных "безкрахмальных" мутантов с недостатком какой-либо субъединицы (Tsai и Nelson, 1986; Dikinson и Preiss, 1969; Lin и др., 1988 a, 1988 b), а также характеристика состоящей из небольших субъединиц гомотетрамерной "АДФГПФ", для которой обнаружена всего лишь небольшая ферментивная активность (Lin и др., 1988 b). Кроме того, предложенные взаимодействующие остатки эффектора выявлены для обеих субъединиц (Morell и др.., 1988).
Нерегулированные ферментные варианты растительной АДФПГФ, идентифицированные и охарактеризованные путем, аналогичным тому, который привел к выделению E.coli gIg C16 и родственных мутантов. Ряд кДНК растительных АДФГПФ или частей таких кДНК как для больших, так и меньших субъединиц был клонирован как из однодольных, так и двудольных растений (Anderson и др., 1989 a; Olive и др., 1989; Muller и др., 1990; Bhave и др.; du Jardin и Berhin, 1991). Белки, кодируемые растительными кДНК, также как и бактериальными кДНК, отличаются высокой степенью консервативности (Bhave и др., 1990). В частности, идентифицирована высоко консервативная область, содержащая также остатки, обусловливающие ферментативное действие и взаимодействия с эффектором (Morell и др., 1988; du Jardin и Berhin, 1991). Выделены клоны субъединичных генов АДФГПФ клубней картофеля, в том числе: полный небольшой субъединичный ген, завершенный добавлением последовательностей из первого экзона геномного клона с клоном кДНК почти полной длины того же гена, и почти полный ген для большей субъединицы. Нуклеотидная последовательность (Послед. N 7) и аминокислотная последовательность (Послед. N 8) завершенного небольшого субъединичного гена представлена на фиг. 5. Представленная нуклеотидная последовательность отличается от первоначального выделенного гена следующими моментами: BgIII+NcoI сайт введен в АТС кодон для облегчения клонирования гена в E.coli и растительные вектора экспрессии при сайтнаправленном мутагенезе с применением олигонуклеотидной последовательности праймера:
GTTGATAACAAGATCTGTTAACCATGGGGGCTTCC (Послед. N 11),
SacI сайт введен у стоп-кодона с помощью олигонуклеотидной последовательности праймера:
CCAGTTAAAAGGGAGCTCATCAGATGATGATTC (Послед. N 12),
SacI сайт служит 3'-сайтом клонирования. Внутренний BgIII сайт удален с помощью олигонуклеотидной последовательности праймера:
GTGTGAGAACATAAATCTTGGATATGTTAC (Послед. N 13).
Завершенный ген экспрессируют в E.coli под контролем recA промотора в PrecA-ген 10L экспрессионный кассете (Wong и др., 1988) с образованием поддающихся измерению количеств белка. Инициирующий кодон метионина находится и условиях сайтнаправленного мутагенеза в присутствии алигонуклеотидной последовательности праймера:
GAATTCACAGGGCCATGGGTCTAGACCC (Послед. N 14)
с экспрессированием эрелого гена.
Нуклеотидная последовательность (Послед. N 9) и аминокислотная последовательность (Послед. N10) почти полного большого субъединичного гена представлены на фиг. 6. Инициирующий кодон метионина помещен у зрелого N-окончания сайтнаправленным мутагенезом с применением олигонуклеотидной последовательности праймера:
AAGATCAAACCTGCCATGGCTTACTCTGTGATCACTACTG (Послед. N 15).
Назначение инициирующего метионина заключается в облегчении экспрессии данного большого субъединичного гена в E.coli. HindIII сайт расположен в 103 п. о. после стоп-кодона и служит 3'-сайтом клонирования. Полный большой ADPGPP ген выделяют 5'-RACE-методикой (быстрая амплификация концов кДНК; Frohman, 1990; Frohman и др., 1988; Loh и др., 1989). Олигонуклеотидными праймерами для этой методики служат следующие последовательности:
1) GGGAATTCAACCTTCCATCCCGGGCCCCCCCCCCCCCCC (послед. N 16);
2) GGGAATTCAAGCTTGGATCCCGGG (Послед. N 17) и
3) CCTCTAGACAGTCGATCAGGAGCAGATGTACG (Послед. N 18).
Первые две эквиваленты соответственно ANpoIyC и AN праймерам по Loh и др. , (1989), а третья является обратным комплементом последовательности в большом ADPGPP гене, расположенном после Pst I сайта в последовательности на фиг. 6. Продукты PCP 5'-последовательности клонируют в виде EcoRI/HindIII/BamHI-Pst I фрагментов и легко соединяют с существующей частью гена.
Слабо регулируемые мутанты фермента АДФГПФ идентифицируют первоначальным подсчетом колоний из подвергнутой мутагенезу культуры E.coli, характеризующихся повышенным синтезом гликогена, путем окрашивания йодом 24-48-часовых колоний на Лурияагаровых пластинках, содержащих 1% глюкозы, с последующей характеристикой реакций АДФГПФ ферментов из продуктов выделения на положительные и отрицательные эффекторы их активности (Cattaneo и др., 1969; Preiss и др., 1971). Аналогичный подход применим к выделению таких же вариантов растительных АДФГПФ ферментов. При наличии системы экспрессии для каждого субъединичного гена отдельно проводят мутагенез каждого гена любым из известных способов, как химических, так и физических (Miller, 1972) на культуре, содержащей ген или очищенную ДНК. Другой подход заключается в применении PCP методики (Ehrlich, 1989) на полном гене в присутствии ингибирующих ионов Mn ++, т. е. в условиях, приводящих к высокой степени ошибочного введения нуклеотидов. PCR методика может быть использована с праймерами, примыкающими непосредственно к специфичной области гена, и такой мутагенизированный фрагмент затем повторно клонируют в немутагенезированные сегменты гена. Методика неупорядоченного синтеза нуклеотидов может быть также использована для создания короткой области гена с высокой степенью мутагенеза смешиванием нуклеотидов в реакции синтеза с их ошибочным введением в результате во всех положениях этой области. Полученную небольшую область фланкируют с помощью сайтов рестрикции, применяемых для повторной вставки области в остальную часть гена. Полученные культуры или трансформанты отбирают стандартным методом в присутствии йода с выявлением трансформантов, характеризующихся более высоким по сравнению с контролем уровнем гликогена. Отбор рекомендуется проводить со штаммом E.coli с дефицитом только АДФГПФ активности (таким как E.coli LC 618; являющимся спонтанным мутантом штамма IC618 (Cattaneo и др., 1969; Creuzet-Sigal и др., 1972), т.е. фенотипично являющимся гликоген-минус штаммом и дополняемым до гликоген-плюс геном gIgC. Штамм E.coli должен сохранять те другие виды активности, которые необходимы для продуцирования гликогена. Оба гена экспрессируют совместно в одном и том же хозяине E.coli введением генов в совместимые плазмиды с различными подбираемыми маркерными генами, и такие плазмиды, кроме того, отличаются одинаковым числом копий в бактериальном хозяине с целью максимального образования гетеротетрамера. Примеры совместимых плазмид включают серии pBR322/pBR327/pUC (с отбором по ампицилину), основанные на CoIEI репликоне, и плазмиду pAACYC177 (с отбором по канамицину), основанную на p15A репликоне (Change и Cohen, 1978). Применение отдельных плазмид позволяет отобрать мутагенизированные популяции только одного гена или в сочетании со вторым геном с последующей трансформацией в компетентном хозяине, экспрессирующем другой ген, и отбором двух мутагенезированных популяций с последующим их объединением в одном и том же
хозяине. После повторного выделения плазмидной ДНК из колоний с повышенным окрашиванием йодом кодирующие АДФГПФ последовательности вновь клонируют в вектора экспрессии, проверяют фенотип и определяют активность АДФГПФ и его реакцию на молекулу эффектора. Улучшенные варианты отличаются повышенным Vmax, сниженным ингибированием отрицательным эффектором ЭР (Pi) или пониженной зависимостью от активатора Э3-ФГА) с точки зрения максимальной активности. Анализ на такие улучшенные характеристики включает определение активности АДФГПФ в присутствии 0,045 мМ Pi (I0,5 = 0,045 мМ) или в присутствии 0,075 мМ 3-ФГА (A0,5=0,075 мМ). Применимые варианты характеризуются < 40%-ным ингибированием при указанной концентрации Pi или > 50-ной активностью при указанной концентрации 3-ФГА. После выделения улучшенных вариантов и определения ответственной субъединицы или субъединиц мутацию-(и) определяют нуклеотидным секвенированием. Мутацию подтверждают повторным созданием тех же изменений сайтнаправленным мутагенезом и повторным анализом активности АДФГПФ в присутствии активатора и ингибитора. Возникшую мутацию затем переносят в эквивалентный полный АДФГПФ кДНК ген повторным клонированием области, содержащей изменение, из измененной бактериальной экспрессионной формы в растительную форму, имеющую направленную на амилопласт последовательность, или сайтнаправленным мутагенезом полного нативного АДФГПФ растительного гена.
Хлоропласт/амилопласт направленная экспрессия АДФ-глюкоза-пирофосфорилазной активности
Известно, что биосинтез крахмала происходит в хлоропластах и амилопластах растения (называются здесь общим термином "пластиды"). В исследованных растениях АДФ-глюкоза-пирофосфорилаза локализована именно в таких пластидах. В побегах гороха АДФ-глюкоза-пирофосфорилаза ограничена только хлорпластами (Levi, 1978). В листьях шпината вся активность АДФ-глюкоза-пирофосфорилазы вместе с активностью крахмал-синтетазы обнаруживается в хлоропластах (Mares, 1978 и Okita, 1979). Иммуноцитохимическая локализация показывает, что АДФ-глюкоза-пирофосфорилаза клубней картофеля находится исключительно в аминопластах (Kim, 1989). Исследования эндоспермы риса также показывают, что активность АДФ-глюкоза-пирофосфорилазы локализована в амилопластах (Nakamura, 1989).
Многие локализованные к хлоропластах белки экспрессируются генами ядра в качестве предшественников и переправляются в хлоропласт с помощью хлоропластного транзитного пептида (ХТП), удаляемого в ходе этапов импорта. Примеры подобных хлоропластных белков включают: небольшую субъединицу рибулоза-1,5-бисфосфаткарбоксилазы (ssRUBISCO, SSU), 5-енолпируватсгикимат-3-фосфат-синтетазу (EPSPS), феррелоксин-оксидоредуктазу, флуоресцирующий комплексный белок I и белок II и тиоредоксин F. Показано in vivo и in vitro, что нехлоропластные белки могут быть направлены в хлоропласт с помощью белкового слияния с ХТП, и что ХТП-последовательность достаточна для направления белка в хлоропласт. Аналогично локализованные в амилопластах белки экспрессируются генами ядра в качестве предшественников и переправляются в амилопласты с помощью амилопластного транзитного пептида (АТП). Полагают также, что хлоропласт и амилопласт развиваются из общих пропластидов и функционально отличаются только тем, что первый обнаруживается в фотосинтетических клетках, а второй в нефотосинтетических клетках. В самом деле, взаимопревращение двух органелл наблюдалось в таких растениях, как Picea abies (Senser, 1975). Имеются также работы, показывающие, что хлоропластные и амилопластные геномы одного и того же растения неразличимы (Scott, 1984; Macherel, 1985 и Catley, 1987). Кроме того показано, что амилопластный транзитный пептид способен импортировать связанный полипептид в хлоропласты (Klosgen, 1989).
В приведенных в виде примеров воплощениях изобретения применялся специализированный ХТП, происходящий из ssRUBISCO IA гена из Arabidopsis thaliana (SSU IA) (Timko, 1988). Данный ХТП (ХТП1) конструируют комбинацией сайтнаправленных мутагенезов. Кроме того, на фиг. 3 приведены нуклеотидная последовательность ХТПI (Послед. N 5) и соответствующая аминокислотная последовательность (Послед. N 6). ХТПI построен из SSU IA ХТП (аминокислоты 1-55), первых 23 аминокислот зрелого SSU IA белка (56-78), остатка серина (аминокислота 79), нового сегмента, повторяющего аминокислоты 50-56 из ХТП и первые две аминокислоты из зрелого белка (аминокислоты 80-87) и остатков аланина и метионина (аминокислоты 88 и 89). Сайт рестрикции NcoI расположен у 3'-конца (покрывает Met-кодон) и предназначен для облегчения создания точного слияния с 5'-концом гена АДФ-глюкоза-пирофосфорилазы. На более позднем этапе вводят BgIII сайт в восходящем направлении от N-окончании SSUIA последовательностей для облегчения введения продуктов слияния в растительные вектора трансформации. Слияние осуществляют между структуральной ДНК, кодирующей ХТП1, ХТП и gIg C16 геном из E.coli с созданием полной структуральной ДНК последовательности, кодирующей слитный полипетид-пластидный транзитный пептид/АДФ-глюкоза-пирофосфорилаза.
Для специалиста очевидно, независимо от того, будет ли использована в практике настоящего изобретения единственная кДНК растительной АДФ-глюкоза-пирофосфорилазы, кодирующая сжатые и/или хрупкие субъединицы, или же будут использованы обе растительные АДФГПФ кДНК, кодирующие сжатые и хрупкие субъединицы, в том и другом случае с наибольшей легкостью и предпочтительностью могут быть использованы эндогенные ХТП и АТП. Соответственно для целей настоящего изобретения термин "пластидные транзитные пептиды" следует интерпретировать, как включающие хлоропластные транзитные пептиды и амилопластные транзитные пептиды. Для специалиста также очевидно, что могут быть созданы и различные другие, и химерные конструкты, в которых будет использована способность конкретного пластидного транзитного пептида импортировать смежную ферментативную АДФ-глюкоза-пирофосфорилазу в растительный клеточный хлоропласт/аминопласт в зависимости от тканевой специфичности промотора. Действие слитого полипептида может быть подтверждено следующим анализом in vitro.
Анализ на поглощение в пластидах
Из салата (Latica Sativa, сорт лонгифолия) центрифугированием в градиентах перколля/фиколля по модифицированной методике Bartlett и др. (1982) выделяют интактные хлоропласты. Полученный дебрис интактных хлоропластов суспендируют в 0,5 мл стерильного 330 мМ сорбита в 50 мМ Гепес-КОН (pH 7,7), анализируют на хлорофилл (Arnon, 1949) и устанавливают конечную концентрацию хлорофилла в 4 мг/мл (применяют сорбит/гепес). Выход интактных хлоропластов из одного кочана салата составляет 3-6 мг хлорофилла.
Типичный образец для анализа на 300 мкл содержит 5 мМ АТП, 8,8 мМ немеченного метионина, 322 мМ сорбита, 58,3 мМ Гепес-КОН (pH 8), 50 мкл продуктов трансляции лизированных ретикулоцитов и интактные хлоропласты из L.sativa (200 мкг хлорофилла). Анализируемую смесь осторожно вращают при комнатной температуре (в стеклянных пробирочках размером 10 х 75 мм) непосредственно перед волокнооптическим облучателем, настроенным на максимальную интенсивность света (лампа на 150 ватт). Аликвоты анализируемой смеси (50 мкл) отбирают в различные промежутки времени и фракционируют в 100 мкл силиконового масла (в полиэтиленовых пробирочках на 150 мкл) центрифугированием 30 секунд при 11000 х G. В этих условиях интактные хлоропласты образуют дебрис под слоем силиконового масла, а инкубационная среда (содержит лизированные ретикулоциты) всплывает на поверхность. После центрифугирования градиенты силиконового масла сразу же замораживают на сухом льду. Дебрис хлоропластов затем вновь суспендируют в 50-1000 мкл лизисного буфера (10 мМ Гепес-КОН с pH 7,5, 2 мМ PMSF, 1 мМ бензамидина, 5 мМ ε- амино-н-капроновой кислоты и 30 мкг/мл апротинина) и центрифугируют 20 минут при 15000 х G для перевода в дебрис тилакоидных мембран. Прозрачную надосадочную жидкость (стромальные белки) от этого центрифугирования и аликвоту инкубационной среды лизированных ретикулоцитов от каждого опыта на поглощение смешивают с равным объемом 2 х ПаДод 04-ПАГЭ буфера для электрофореза (см. ниже).
НДС-ПАГЭ проводят согласно Laemmli (1970) в 3-17% (мас./об.) акриламидных пластинчатых гелях (60 мм х 1,5 мм) с 3% (мас./об.) концентрирующих гелей (5 мм х 1,5 мм). Гель фиксируют в растворе 40% метанола и 10% уксусной кислоты. Затем гель замачивают в EN3HANCETM(ДюПонт) на 20-30 минут с последующим высушиванием геля в сушилке для гелей. Гель проявляют авторадиографией с применением интенсифицирующего экрана и примерно суточной выдержки с определением того, была ли АДФ-глюкоза-пирофосфорилаза импортирована в выделенные хлоропласты.
Альтернативное средство повышения уровня АДФ-глюкозы в клетках растения заключаются в выделении генов, кодирующих факторы транскрипции, взаимодействующих с восходящими регуляторными элементами гена-(ов) растительной АДФ-глюкоза-пирофосфорилазы. Усиленная экспрессия таких факторов транскрипции в клетках растения может вызвать повышенную экспрессию гена АДФ-глюкоза-пирофосфорилазы. В этих условиях все равно достигается повышенное содержание крахмала за счет повышения ферментативной активности АДФ-глюкоза-пирофосфорилазы, но по другому механизму. Способы выделения факторов транскрипции известны (Katagiri, 1989).
Сигнал полиадениляции
3'-Нетрансляционная область химерного растительного гена содержит сигнал полиадениляции, функция которого в растениях заключается в индуцировании присоединения полиаденилированных нуклеотидов к 3'-концу РНК. Примеры приемлемых 3'-областей включают: (1) 3'-транскрибированные нетрансляционные области, содержащие полиадениляционный сигнал индуцирующих опухоль (Ti) плазмидных генов Agrobacterium, такие как ген нопалин-синтетазы (NOS) и (2) гены растений, подобные генам накапливаемых белков сои и небольшой субъединицы гена рибулоза-1,5-бисфосфат-карбоксилазы (ssRUBISCO). Примером рекомендуемой 3'-области является 3'-область NOS-гена, более подробно описанная в нижеследующих примерах.
Трансформация/регенерация растения
Растения, в которых способом настоящего изобретения может быть повышено содержание крахмала, включают, но без ограничения только ими: кукурузу, пшеницу, рис, морковь, лук, горох, томаты, картофель, сладкий картофель, арахис, канолу/масляничный рапс, ячмень, сорго, кассабу, банан, сою, салат, яблоки и орех.
Молекула двуспиральной ДНК настоящего изобретения, содержащая функциональный растительный ген АДФ-глюкоза-пирофосфорилазы, может быть введена в геном растения любым приемлемым способом. Приемлемые вектора трансформации растения включают вектора, происходящие из T плазмиды Agrobacterium tumefaciens, а также вектора, раскрытые в работах, напр.: Herrera-Estrella (1983), Bevan (1983), KIee (1985) и EPO публикация 120516 (Schilperoort и др.). Помимо растительных векторов трансформации, происходящих из Ti или индуцируемых корнями (Ri) плазмид Agrobacterium, для введения ДНК конструктов настоящего изобретения в клетки растения могут быть использованы альтернативные способы. Подобные способы могут включать, например: применение липосом, электропорацию химикатов, повышающих поглощение свободной ДНК, поставку свободной ДНК путем микропроекционной бомбардировки и трансформацию с помощью вирусов или пыльцы.
Плазмидный вектор экспрессии, пригодный для экспрессии E.coli gIg C16 и других генов АДФГПФ в однодольных растениях, включает следующее: промотор, обладающий специфичностью или повышенной способностью к экспрессии в тканях хранения крахмала однодольных растений, обычно, в эндосперме, такой как промотор для зеин-генов, обнаруживаемых в эндосперме кукурузы (Pedersen и др., 1982); интрон, обеспечивающий сплайс-сайт для облегчения экспрессии гена, такой как ADHI интрон (Callas и др., 1987) и 3'-полиадениляционную последовательность, такую как 3' последовательность нопалин-синтетазы (NOS3'; Fraley и др., 1983). Такая экспрессионная кассета может быть смонтирована на высококопийных репликонах, пригодных для продуцирования больших количеств ДНК.
Особенно привлекательным вектором трансформации растений на основе Agrobacterium, предназначенным для применения в трансформации двудольных растений, является плазмидный вектор pMON530 (Rogers, S.G, 1987). Плазмида pMON530 (см. фиг. 3) является производным плазмиды pMON505, полученной трансфекцией фрагмента StuI - HindIII в 2,3 к.о. плазмиды pMON316 (Rogers, S.G., 1987) в плазмиду pMON526. Плазмида pMON526 является простым производным плазмиды pMON505, в которой гидролизом в присутствии XmaI, обработкой полимеразой Кленова и лигацией удален SmaI сайт. Плазмида pMON530 сохраняет все свойства pMON505 и экспрессионной кассеты CaMV 35S-110 и содержит уникальный сайт расщепления для SmaI между промотором и сигналом полиаденилирования.
Бинарный вектор pMON505 является производным плазмиды pMON200 (Rogers, 1987), в которой область гомологии с Ti плазмидой (LIH) заменена HindIII-SmaI сегментом в 3,8 к.о. мини-RK2 плазмиды (pTIi75) (Schmidhauser и Helinski, 1985). Указанный сегмент содержит RK2 источник репликации (oriV) и источник переноса (ori T) для конъюгации в Agrobacterium с помощью методики трехродственного скрещивания (Horsch и KIee, 1986). Плазмида pMON505 сохраняет все важные признаки pMON200, в том числе синтетический мультилинкер для вставки целевых фрагментов ДНК, химерный NOS/NPTIT/NOS ген для устойчивости к канамицину в клетках растения, детерминанту устойчивости к спектиномицину/стрептомицину для отбора в E.coli и A.tumefaciens, интактный ген нопалин-синтетазы для быстрого подсчета трансформантов и наследственности в потомстве и pBP322 источник репликации для облегчения получения больших количеств вектора в E.coli. Плазмида pMON505 содержит единственную Т-ДНК границу, происходящую из правого конца pTi T37 нопалинового типа Т-ДНК. Саузерн анализ показал, что плазмида pMON505 и любая ДНК, которую она несет, интегрированы в геном растения, т.е. полная плазмида является Т-ДНК, которая вводится в геном растения. Один конец интегрированной ДНК расположен между последовательностью правой границы и геном нопалинсинтетазы, а другой конец находится между граничной последовательностью и pBP322 последовательностями.
Когда будет получено адекватное число клеток (или протопластов), содержащих ген или кДНК АДФ-глюкоза-пирофосфорилазы, клетки (или протопласты) регенерируют во все растение. Выбор методологии для стадии регенерации решающей роли не играет, и приемлемые методики существуют для хозяев из Lequminosae (люцерна, соя, клевер и т.д.), Umbelliferae (морковь, сельдерей, пастернак), Cruciferae (капуста, редис, рапс и т.д.), Cucurbitaceae (дыня и огурец), Cramineae (пшеница, рис, кукуруза и т.д.), Solanaceae (картофель, табак, томаты, перец) и различные культурные растения. См., напр.: Ammirato (1984); Shimamoto (1989); Fronm, (1990); Vasil (1990).
Следующие примеры даются для лучшего разъяснения практики настоящего изобретения, и ни в коей мере не должны рассматриваться, как ограничивающие объем изобретения. Для специалиста очевидно, что в методы и в гены, описываемые здесь, могут быть внесены различные модификации, усечения и т.д. без отхода от духа и объема настоящего изобретения.
Примеры
Пример 1
Для экспрессирования E.coli gIg C16 гена в клетках растения и для направления фермента в пластиды необходимо провести слияние гена с ДНК, кодирующей направляющий в пластиды транзитный пептид (далее обозначаемый как ХТП/АДФ-глюкоза-пирофосфорилазы ген), и соответствующими регуляторными областями растения. Все это осуществляют клонированием gIg C16 гена в ряд плазмидных векторов, содержащих необходимые последовательности.
Плазмиду pLP226, содержащую gIg C16 ген в HindII фрагменте, клонируют в pUC8 вектор у HinCII сайта (Leung и др., 1986). Плазмида pL P226 получена от Др. Jack Preiss, Университет штата Мичиган, и ее трансформируют в замороженные компетентные E. Coli JMI 01 клетки, приготовленные кальцийхлоридным методом (Sambrook и др., 1989). Трансформированные клетки наносят на пластинки 2ХУТ (инфра), содержащие 100 мкг/мл ампицилина. Плазмиду pL P226 очищают методом быстрой щелочной экстракции (БЩЭ) из 5 мл примерно суточной культуры (Birnboim и Doly, 1979).
Для слияния gIg C16 гена с ДНК, кодирующей хлоропластный пептид, у 5'-конца гена необходим NcoI сайт. Кроме того, в нисходящем направлении от терминационного кодона необходим SacI сайт для перемещения гена ХТП/АДФ-глюкоза-пирофосфорилазы в следующий вектор. Для введения указанных сайтов проводят PCR реакцию (#13) использованием примерно 20 нг очищенной быстрой щелочной экстракцией плазмиды pL P226 в качестве матрицы. Реакцию осуществляют согласно рекомендациям изготовителя (Перкин Эльмер Цетус). Использованы праймеры QSP3 и QSP7. Праймер QSP3 создан введением NcoI сайта, который должен включать старт-кондон для gIg C16 гена. Праймер OSP7 гибридизован в нетрансляционной 3'-области gIg C16 гена и добавленном SacI сайте. Термоциклер программируют на 30 циклов по 1 минуте при 94oC на стадию денатурирования, по 2 минуты при 50oC на стадию гибридизации и по 3 минуты при 72oC на стадию удлинения. После каждого цикла время стадии удлинения увеличивают на 15 секунд.
OSP3 праймер:
5' - GGAGTTAGCCATGGTTAGTTTAGAG-3' (Послед. N 19)
QSP7 праймер:
5'- GGCCGAGCTCGTCAACGCCGTCTGCGATTTGTGC-3' (Послед. N 20)
Вектор, в который клонируют продукт PCR, представлен pGEM3zf + (получен от фирмы Промега, Мэдисон, шт. ВИ), гидролизованный в присутствии SacI и HindIII и включающий ДНК для модифицированной небольшой субъединицы ХТП1 из Arabidopsis, легированной у HindIII сайта. ДНК (Послед. N 5) и аминокислотная последовательность (Послед. N 6) этого ХТПI приведены на фиг. 3.
Вектор в линейной форме обрабатывают 30 минут при 56oC щелочной фосфатазой кишечника теленка. Затем и вектор, и PCR # 13 фрагмент, включающий gIg C16 ген с новыми NcoI и SacI сайтами, пропускают через гель агарозы и фрагменты очищают связыванием с DEAE-мембранами. Методика, использованная для очистки фрагмента с помощью DEAE-мембраны, создана Schleicher и Schuell, и носит название "Связывание и выделение ДНК и РНК с помощью S- и S-DEAE-мембраны".
Проводят лигирование gIg C16 гена # 5, слитого с ДНК для модифицированного SSU ХТП из Arabidopsis, с pGEM3zf +. Лигируемая смесь содержит 3 мкл вектора, гидролизованного в присутствии NcoI и SacI, а также 3 мкл продукта PCR # 13, тоже обрезанного в присутствии NcoI и SacI и повторно очищенного на геле, 5 мкл (из полых 20 мкл) продукта лигирования # 5 трансформируют замороженные компетентные клетки JM101 и трансформированные клетки наносят на 2ХУТ пластинки (16 г/л Бактотриптона, 10 г/л дрожжевого экстракта, 10 г/л NaCl, pH 7,3, отверждено 1,5% агара), содержащими ампицилин.
Образец 1 отбирают с пластинки после почти суточного выращивания. Полученным образцом засевают 4 мл 2ХУТ среды и выращивают при 37oC примерно сутки. Плазмиду выделяют методом быстрой щелочной экстракции и ДНК гидролизуют в присутствии EcoRI, NcoI и смеси EcoRI и NcoI. Продукт гидролиза разделяют на геле агарозы с выявлением ожидаемых фрагментов. Выделенная из образца 1 плазмида обозначена pMON20100, и она состоит из pGEM3zf+, ДНК для модифицированного SSU ХТП из Arabidopsis и gIg C16 гена. Продукт слияния имеют ориентацию, допускающую его транскрибирование из SP6 полимеразного промотора.
Для выявления способности конструкта импортировать АДФ-глюкоза-пирофосфорилазу в выделенные хлоропласты салата необходимо слитые ХТП/АДФ-глюкоза-пирофосфорилазу транскрибировать и транслировать с образованием /35S/-меченной АДФ-глюкоза-пирофосфорилазы. Для получения ДНК матрицы для транскрипции с помощью SP6 полимеразы ХТП/АДФ-глюкоза-пирофосфорилазную область pMON 20100 амплифицируют PCR реакцией с образованием большого количества линейной ДНК. Для осуществления этого примерно 0,1 мкл pMON20100, очищенной методом быстрой щелочной экстракции, используют в качестве матрицы в PCR реакции # 80. Примерами служат продажный SP6 промоторный праймер (Промега) и олиго-QSP7. SP6-праймер гибридизован в векторе с SP6 промотором и включает полную последовательность SP6 промотора. Таким образом, PCR-продукт, праймированный олигонуклеотидом, имеет последовательность распознавания SP6 полимеразы. Праймер QSP7 будет гибридизоваться в нетрансляционной 3'-области gIg C16 гена. Это тот же самый праймер, который был использован для введения SacI сайта в нисходящем направлении от терминационного кодона gIg C16 гена. Термоциклер программируют на 30 циклов по 1 минуте на денатурирование при 94oC, по 2 минуты на гибридизацию при 55oC и по 3 минуты на удлинение при 72oC. После каждого цикла стадию удлинения увеличивают на 15 секунд.
SP6 промоторный праймер:
5'-GATTTAGGTGACACTATAG-3' (Послед. N 21).
Продукт PCR реакции #80 (5 мкл) пропускают через гель агарозы и очищают связыванием с DEAE-мембраной. ДНК элюируют и растворяют в 20 мкл ТЕ. Очищенный на геле продукт PCR реакции #80 (2 мкл) используют в in vitro реакции транскрипции SP6 РНК-полимеразы. Условия реакции те, которые предложены поставщиком (Промега) для синтеза больших количеств РНК (100 мкл реакционной смеси). РНК, полученную из ДНК PCR реакции #80, применяют для in vitro трансляции в системе лизата ретикулоцитов кролика (Промега). 35S-Меченый белок, приготовленный из pMON20100 (т. е. в PCR реакции #80), используют в ранее описанном анализе для in vitro импорта в хлоропласт. После обработки образцов, полученных в результате анализа, их подвергают электрофорезу на НДС-ПАГЭ гелях с 3-17% градиентом полиакриламида. Гель фиксируют 20-30 минут в растворе 40% метанола и 10% уксусной кислоты. Затем гель замачивают в EN3HANCETM в течение 20-30 минут с последующим высушиванием геля в сушилке для геля. Гель проявляют авторадиографией с применением интенсифицирующего экрана и примерно суточной выдержки. Полученные результаты показывают, что слитый белок импортируется в выделенные хлоропласты.
Конструкты в pMON20100 затем преобразуют со слиянием с En-CaMV 35S промотором (Kay R. , 1987) и 3'-концом NOS (Bevan M., 1983), выделенным из pMON11999. PCR реакция 114 содержит в качестве матрицы pMON20100, а используемыми праймерами служат QSM11 и QSM10. QSM11 гибридизуют с ДНК для модифицированного SSU ХТП из Arabidopsis и создают BgIII сайт в 7 п.о. в восходящем направлении от АТС старт-кодона. QSM10 гибридизуют с 3'-концом gIg C16 гена и добавляют XbaI сайт сразу же после терминационного кодона, а также добавляют SacI сайт в 5 п.о. после терминационного кодона. SacI сайт, ранее добавленный к gIg C16 гену, находится примерно в 100 п.о. в нисходящем направлении от терминационного кодона. Термоциклер программируют на 25 циклов по 1 минуте на денатурирование при 94oC, по 2 минуты на гибридизацию при 55oC и по 3 минуты на стадию удлинения при 72oC. С каждым циклом к стадии удлинения добавляют 15 секунд.
QSM11 праймер:
5'-AGAGAGATCTAGAACAATGGCTTCCTCTATGCTCTCTTCCGC-3' (Послед. N 22)
QSM10 праймер:
5'-GGCCGAGCTCTAGATTATCGCTCCTGTTTATGCCCTAAC-3' (Послед. 23)
Девяносто пять (95) мкл (из общего объема в 100 мкл) продукта PCR реакции # 114 осаждают этанолом и вновь суспендируют в 20 мкл TE. Пять (5) мкл суспензии гидролизуют примерно сутки при 37oC в присутствии BgIII (4 единицы) и SacI (10 единиц). Пять (5) мкл (5 мкг) вектора pMON999, содержащего En-CaMV35S промотор и 3'-конец NOS, гидролизуют таким же образом. После гидролиза ферментами рестрикции ДНК пропускают через гель агарозы и очищают связыванием с DEAE-мембранами. Каждую ДНК растворяют в 20 мкл ТЕ. Один (1) мкл PCR 114 лигируют с 3 мкл вектора в полном объеме в 20 мкл. Лигируемую смесь инкубируют 7 часов при 14oC. Десятью (10) мкл продукта лигации трансформируют компетентные замороженные ММ294 клетки и наносят на LB пластинки (10 г/л Бактотриптона, 5 г/л дрожжевого экстракта, 10 г/л NaCl и 1,5% агара для отверждения) с добавкой 100 мкг/мл ампицилина. Колонии собирают и ими инокулируют пробирки, содержащие 5 мл LB среды с добавкой 100 мкг/мл ампицилина для примерно суточного выращивания. Пять (5) мл примерно суточной культуры используют для быстрой щелочной экстракции с выделением плазмидных ДНК. ДНК гидролизуют в присутствии EcoRI и отдельные аликвоты гидролизуют в присутствии NotI. После анализа полученных образцов на гелях агарозы подтверждено, что плазмида pMON20102 имеет EcoRI фрагмент в 497 п.о., характерный для gIg C16 гена. Данная плазмида, кроме того, содержит NotI фрагмент в 2,5 к.о., состоящий из En-CaMV 35S промотора, ДНК для модифицированного SSU ХТП из Arabidopsis, gIg C16 гена и 3'-конца NOS.
NotI кассету в 2,5 к.о. затем переносят в вектор трансформации растений - pMON530 (фиг. 4). pMON530 содержит уникальный NotI сайт в РК2 области точно в 600 п.о. после HindIII сайта. Описание конструирования pMON530 можно найти в работе Pogers и др., 1987. Двадцать (20) мкг pMON530 гидролизуют примерно сутки при 37oC в присутствии 40 единиц NotI. Гидролизованный вектор затем дефосфорилируют примерно 1 час при 37oC в присутствии 22 единиц щелочной фосфатазы кишечника теленка. Вектор pMON530 экстрагируют фенол/хлороформом, затем хлороформом и осаждают метанолом. Десять (10) мкг плазмиды pMON20102 также гидролизуют примерно сутки в присутствии 40 единиц NotI. NotI-гидролизованный вектор pMON530 лигируют примерно сутки при 15oC с NotI кассетой из плазмиды pMON20102. Продуктом лигации трансформируют замороженные компонентные JM101 E.coli клетки и трансформированные клетки наносят на IB пластинки, содержащие 75 мкг/мл спектиномицина.
С пластинки с продуктом трансформации собирают девять колоний, которые с целью отбора выращивают в 5 мл LB культур. Плазмиды из 5 мл культуры получают методом быстрой щелочной экстракции. ДНК сначала отбирают гидролизом в присутствии SalI с разделением на 1% геле агарозы. Сравнением полученного образца с продуктом гидролиза в присутствии SalI родоначальной плазмиды (pMON530) отбирают нужный конструкт. Конструкт получил обозначение pMON20104, и его ориентация определена гидролизом в присутствии PstI и двойным гидролизом в присутствии NcoI/BgIII. En-CaMV35S профотор, ведущий ген ХТП/АДФ-глюкоза-пирофосфорилазы, имеет ту же ориентацию, что CaMV35S промотор, уже присутствующий в pMON530.
При подготовке к трансформации клеток табака pMON20104 скрещивают с Agrobacterium ASE трехродственным скрещиванием с плазмидой-хелпером pRK2013. Agrobacterium выращивают 1,5 дня в LB среде при 30oC в присутствии 25 мкг/мл хлорамфеникола и 50 мкг/мл канамицина. Содержащую pRK2013 E.coli выращивают примерно сутки в канамицине (50 мкг/мл). Такую культуру начинают с нескольких колоний, собранных с пластинки. E.coli с pMON20104 выращивают в LB среде в присутствии с 75 мкг/мл спектиномицина. После выращивания всех культур в пробирку добавляют 4 мл LB среды со 100 мкл каждого из: Agrobacterium ASE, pRK2013 и pMON20104. Полученную смесь центрифугируют 5 минут в микроцентрифуге и декантируют. Дебрис вновь суспендируют в оставшейся жидкости и пипеткой наносят в центр LB пластинки. После примерно суточного выращивания при 30oC петельку клеток с пластинки наносят в виде штрихов на LB пластинку в присутствии 75 мкг/мл спектиномицина и 25 мкг/мл хлорамфеникола.
После выдерживания 1-2 дня при 30oC пластинка с трехродственным скрещиванием pMON20104, Agrobacterium ASE и pRK2013 имеет выросшие колонии, в то время как контрольная пластинка от скрещивания pMON20104 и ASE (без pRK2013, необходимой для мобилизации) колоний не имеет. После трехродственного скрещивания с пластинки снимают 2 колонии, высевают в жидкую культуру, содержащую 75 мкг/мл спектиномицина, 25 мкг/мл хлорамфеникола и 50 мкг/мл канамицина, и выращивают при 30oC. Полученные две культуры применяют для трансформации клеток табака.
В методике трансформации табачных листьев в форме диска применяют здоровую ткань листьев в возрасте около 1 месяца. После 15-20-минутной стерилизации поверхности 10% Клорокса плюс поверхностно-активное вещество листья трижды ополаскивают дистиллированной водой. С помощью стерильного бумажного штампа из листьев вырезают диски и помещают в перевернутом виде в MS104 среду (MS соли 4,3 г/л, витамин B5 500X 2 мл/л, НУК 0,1 мг/л и ВА 1 мг/л) для предкультивирования в течение 1 дня.
Затем диски засевают примерно суточной культурой Agrobacterium ASE: pMON20104, разбавленной в отношении 1/5 (т.е. примерно 0,6 OD). Высевание осуществляют помещением дисков в центрифужные пробирки вместе с культурой. Спустя 30-60 секунд жидкость осушают и диски помещают между стерильной фильтровальной бумагой. Затем диски для сокультивирования помещают в перевернутом виде на MS104 питающие пластинки.
После 2-3-дневного сокультивирования диски переносят все еще в перевернутом виде на селекционные пластинки с MS104 средой. Спустя 2-3 недели образуется каллюс и с дисков из листьев отделяют отдельные скопления. Побеги чисто отрезают от каллюса, если они достаточно большие, чтобы отделяться от стержня. Побеги помещают на несодержащую гормонов корневую среду (MSO:MS соли 4,3 г/л, сахароза 30 г/л и витамины B5 500X 2 мл/л) с селекцией. Через 1-2 недели образуются корни. Укоренившиеся побеги помещают в почву и выдерживают в условиях высокой влажности (напр., в пластиковых контейнерах или мешочках). Побеги затвердевают при постепенном воздействии на них условий влажности окружающей среды.
Содержание крахмала в трансформированной ткани каллюса определяют количественно модифицированной методикой 1 и др. (Lin и др., 1988 a). Скопления каллюса удаляют с пластинок, принимая меры для внесения агара. Каллюс помещают в микроцентрифужные пробирки на 1,5 мл и сушат в вакууме в приборе СПИД ВАКR (Савант). После нескольких часов высушивания пробирки вынимают и взвешивают на аналитических весах с точностью до 0,1 мг. Пробирки возвращают еще на несколько часов в СПИД ВАКR и взвешивание повторяют, пока не будет достигнуто устойчивое значение сухой массы. Высушенный каллюс растирают в пробирке и тщательно перемешивают с получением однородного образца. Отбирают аликвоту каждого высушенного образца каллюса и помещают в предварительно взвешенную микроцентрифужную пробирку на 1,5 мл. Пробирки затем снова взвешивают и определяют массу каллюса в них. Масса образцов находилась в интервале 9-34 мг.
В каждую пробирку добавляют примерно 1 мл 80%-ного этанола и пробирки инкубируют 10-20 минут на водяной бане при 70oC. Затем образцы центрифугируют и этанол удаляют. Промывание этанолом проводят еще 2 раза. После последнего промывания этанолом образцы сушат в СПИД BAKR, затем в каждую пробирку добавляют 200 мкл 0,2 н. КОН. Образцы растирают с помощью подвесной мешалки, после чего нагревают 30 минут при 100oC. Перед нагреванием пробирок в пробках иглой делают несколько небольших отверстий. Тем самым исключают выбивание пробок с потерей образца. По окончании стадии нагрева к каждому образцу добавляют 40 мкл 1 н. уксусной кислоты. Добавляют 35 мкл (7,4 единицы) панкреатической альфа-амилазы с последующим инкубированием 30 минут при 37oC. Затем в каждый образец добавляют 5 единиц (в 5 мкл) амилоглюкозидазы (из Aspergillus niger) вместе со 160 мкл 100 мМ ацетата натрия (pH 4,6). Образцы нагревают 1 час при 55oC, кипятят 2-3 минуты и недолго центрифугируют в микронцентрифуге. Затем образцы вновь сушат в СПИД BAKER и снова суспендируют в 1000 мкл 100 мМ Трис-Cl (pH 7,5).
Затем образцы анализируют на глюкозу с помощью аналитического набора на глюкозу (НК) фирмы Сигма (#16-10 по каталогу). Использованием этого анализа глюкоза в образцах (+АТФ) под действием гексокиназы превращается в глюкоза-6-фосфат + АДФ. Глюкоза-6-фосфат (+НАД) превращен в 6-фосфоглюконат + НАДН. Определяют поглощение при 340 нм, вызванное присутствием НАДН, которое прямо пропорционально концентрации глюкозы. Все анализы и расчеты проводят согласно рекомендациям Сигмы. Анализы проводят согласно "Альтернативной методике" фирмы Сигма при комнатной температуре с 10 мкл образца на анализ или 5 мкл образца + 5 мкл 100 мМ Трис-Cl (pH 7,5). Процент крахмала определяют делением количества (массы) глюкозы на сухую массу каллюса.
Для вестерн-блотирования часть высушенного гомогенизированного каллюса от каждого из 12 образцов плюс 2 контрольных образцов вновь суспендируют в 200 мкл экстракционного буфера (100 мМ Трис-Cl с pH 7,1, 1 мМ ЭДТК, 10% глицерина, 5 мМ ДТТ, 1 мМ бензамидина). Каждый образец растирают мешалкой, центрифугируют 5 минут в микроцентрифуге при полной скорости вращения и надосадочные жидкости переносят в новые пробирки. Концентрацию белка в каждом образце определяют БиоРад анализом на белок (Iowry и др., 1951) с АБС в качестве стандарта. Двадцать пять (25) мкг каждого образца наносят на НДС полиакриламидные гели с 7-17%-ным градиентом полиакриламида. Поскольку образцы были нанесены на два геля, на каждый гель наносят и такие же контрольные образцы каллюса. Кроме того, на каждый гель наносят контрольный образец, усиленный 10 нг чистой E.coli АДФ-глюкоза-пирофосфорилазой.
После электрофореза гели в виде пятен наносят на нитроцеллюлозу с помощью аппарата ПолиБлотR фирмы Америкен Бионетикс. Вестерн блотирование осуществляют по методике, предложенной фирмой Промега. Фильтры на 30 минут блокируют 1% АБС в TBST (10 мМ Трис-Cl с pH 8, 150 мМ NaCl и 0,05% Твин 20). Десять (10) мл TBST плюс 1,3 мкл первичных антител кролика к E.coli АДФ-глюкоза-пирофосфорилазы смешивают и фильтры инкубируют с этими первичными антителами 30 минут. Затем фильтры 3 раза промывают TBST по 50 мл и 5 минут на каждую промывку. Затем с фильтрами инкубируют десять (10) мл TBST плюс 1,3 мкл вторичных антител (козьи антикроличьи антитела, конъюгированные с щелочной фосфатазой, Промега) 30 минут с последующим промыванием 3 раза TBST. Сигналы наблюдают визуально использованием реакции щелочной фосфатазы с BCIP и NBT и их количественно определяют лазерным денситометром.
Результаты показаны в табл. 1.
В приведенной табл. 1 показаны результаты количественного определения крахмала и интегрированные площади пиков в вестерн блотировании. Количество крахмала указано в виде процента крахмала относительной сухой массы каллюса. Площадь пика - это интегрированная площадь под пиком, полученным в результате сканирования денситометром соответствующего образца при вестерн блотировании. Образцы 1-6 пропущены через один гель, а образцы 7-12 пропущены через другой гель. Контроль 2 проведен на обоих гелях с добавкой и без 10 нг очищенной E.coli АДФ-глюкоза-пирофосфорилазы. Неусиленные образцы на обоих гелях показали отсутствие перекрывающихся полос. Для усиленных образцов приведены площади пиков. Полученные результаты показывают, что увеличение АДФ-глюкозы приводит к повышению содержания крахмала в клетках растения.
Пример 2
Описанной в примере 1 плазмидой pMON20104 трансформируют картофель сорта Дезири с помощью методики трансформации дисков клубня, опубликованной Sheerman и Bevan (Sheerman и Bevan, 1988). Безвирусные клубни Solanum tuberosum, сорт Дезири очищают от кожуры, недолго промывают дистиллированной водой и стерилизуют с поверхности в течение 15 минут в 10%-ном растворе хлорида натрия, содержащем несколько капель Твин 20. Клубни промывают 6 раз стерилизованной водой и затем погружают в жидкую MS среду. Для получения долек клубня используют корковое сверло диаметром 1 см и полученные дольки нарезают скальпелем в 1-2 мм диски. Диски оставляют плавить в 20 мл MS среды, содержащей Agrobacterium ASE: pMON20104. Культуру Agrobacterium AE: pMON20104 (10 мл) центрифугируют и перед употреблением вновь суспендируют в 20 мл MS среды. Культуру и диски осторожно встряхивают в чашке Петри. Спустя 20 минут диски переносят на табачные питающие пластинки с 3C5ZR средой (MS соли, 1 мг/мл тиамин HCl, 0,05 мг/л никотиновой кислоты, 0,5 мг/л пиридоксин HCl, 3% сахарозы, 5 мкМ зеатинрибозида и 3 мкМ IAA аспарагиновой кислоты, pH 5,9).
Через 48 часов инфицированные диски помещают на новые пластинки с той же средой, но без питающего слоя и 500 мкг/мл карбенициллина и 100 мкг/мл канамицина. Пластинки герметизируют парафином и инкубируют 16 часов светового дня при 25oC. Диски субкультивируют каждые 3 недели на свежих пластинках и концентрацию карбенициллина снижают с 500 до 200 мкг/мл после выдерживания в культуре 4 недели. Развивающиеся побеги удаляют и помещают в большие пробирки, содержащие MS соли и витамины R3 (1 мг/мл тиамин HCl, 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл пиридоксин HCl) плюс 200 мкг/мл карбенициллина и 100 мкг/мл канамицина. После образования корней растения переносят в почву и постепенно закаливают.
Эти предварительные опыты показали, что получение трансгенных растений, экспрессирующих АДФГПФ ген под контролем En-CaMV 35S промотора, проблематично. Получено одно растение картофеля на содержащей сахарозу среде, но при перенесении растения из среды в почву оно погибло. Этот результат не был неожиданным. En-CaM 35S промотор относится к конститутивным промоторам, вызывающим экспрессию АДФГПФ во всех тканях растения. Конститутивная экспрессия АДФГПФ гена наиболее вероятно создает невозможность подачи сахарозы к растущим частям растения вследствие передаваемого АДФГПФ превращения сахарозы в крахмал в экспортирующих сахар клетках и тканях растения. Таким образом, данный пример иллюстрирует экспрессию АДФГПФ в растительных клетках и показывает предпочтительность в большинстве случаев специфичности экспрессии АДФГПФ в намеченной ткани, такой как клубень картофеля или плод томата. Для специалиста будет несложно выбрать из ряда растений, трансформированных En-CaMV 35S промотором, растение, экспрессирующее АДФГПФ в пределах желаемого интервала.
Пример 3
Ткань картофеля была также трансформирована с тем, чтобы экспрессировать ХТП/АДФ-глюкоза-пирофосфорилазный слитый полипептид, ведомый пататин-промотором. Данный конструкт вызывает специфичную экспрессию АДФГПФ в клубнях картофеля с повышением в клубнях содержания крахмала.
Используемый в трансформации картофеля вектор является производным передаваемого Agrobacterium вектора трансформации растений pMON886. Плазмида pMON886 состоит из следующих хорошо охарактеризованных сегментов ДНК. Фрагмента в 0,93 к.о., выделенного из транспозона Т7, кодирующего устойчивость бактерий к спектиномицину/ стрептомицину (Spc/Str) и являющегося детерминантой для отбора в E.coli и Agrobacterium tumefaciens (Fling и др., 1985). Фрагмент присоединения к химерному гену устойчивости к канамицину, созданному для экспрессии в растении с целью селекции трансформированной ткани. Химерный ген состоит из 35 - промотора в 0,35 к.о. мозаичного вируса цветной капсулы (P-35S) (OdeII и др., 1985), неомицин-фосфотрансферазы гена типа II (NPTII) в 0,83 к. о. и 3'-нетрансляционной области в 0,26 к.о. гена нопалин-синтетазы (NOS-3') (Fraley и др., 1983). Следующий сегмент имеет 0,75 к. о. и происходит от репликации из RK2 плазмиды (ori-V) (Stalker и др., 1981). Сегмент присоединен к SalI-PvuI сегменту в 3,1 к.о. плазмиды pBR322, служащему источником репликации для установления в E.coli (ori-322) и bom сайте конъюгационного переноса в клетки Agrobacterium tumefaciens. Затем следуют 0,36 к. о. PvuI фрагмента из pTiT37 плазмиды, содержащей правую граничную область Т-ДНК нопалинового типа (Fraley и др., 1985).
Ген gIg C16 создан для экспрессии в первую очередь в клубнях помещением гена под контроль клубнеспецифичного промотора. Белок G2g C16 направлен на пластиды внутри клетки растения вследствие его синтеза в качестве C-концевого продукта слияния с N-концевой белковой частью, кодирующей направленную на хлоропласт последовательность (ХТП), происходящую из SSU IA гена из Arabidopsis Ihaliana (Timko и др. , 1989). ХТП часть в процессе импорта удаляется с освобождением GIgC16 фермента. Другие растительные сигналы экспрессии также включают 3'-полиадениляционные последовательности, поступающие из NOS-3' последовательностей, расположенных в нисходящем направлении от кодирующей части экспрессионной кассеты. Такую кассету монтируют следующим образом. Пататин-промотор отрезают от pB1241.3 плазмиды в виде HindIII-BamHI фрагмента (pB1241.3 плазмида содержит сегмент пататин-I-промотора, состоящий из ACCI сайта в положении 1323 до DraI сайта в положении 2289 /позиции относятся к последовательности по Bevan и др., 1986/ с HindIII линкером, добавляемым в первом положении, и BamHI линкером, добавляемым во втором положении; Bevan и др., 1986) и лигируют с продуктом слияния ХТПI-gIg C16 (BgIII-SacI фрагмент из pMON20102, см. пример 1) и плазмидным вектором pUC-типа, отрезанным с помощью HindIII и SacI (эти сайты клонирования в векторе фланкированы сайтами распознавания NotI). Затем кассету в виде NotI сайта в pMON886 таким образом, что экспрессия gIg C16 гена происходит с той же ориентацией, что и NPTII (канамицин) гена. Производное обозначается как pMON 20113 и показано на фиг. 7.
Вектор pMON20113 мобилизуют в бесплечевой штамм Agrobacterium tumefaciens с помощью трехродственной системы конъюгации использованием плазмиды-хелпера pRK2013 (Ditta и др., 1980). Использован бесплечевой штамм ABI, несущий Ti плазмиду, переведенную в бесплечевое состояние удалением генов фитогормонов, ответственных за заболевание корневым раком. ABT штаммом служит A208 Agrobacterium tumefaciens, несущий бесплечевую pTiC58 плазмиду pMR90PK (Konez и Schell, 1986). Бесплечевая плазмида Ti обеспечивает функции trfA гена, необходимые для автономной репликации pMON-вектора после конъюгации в ABI штамм. При инкубировании растительной ткани с ABI:: pMON конъюгантом вектор переносится в клетки растений действием vir, кодируемого pMP90RK плазмидой.
pMON20113 конструктом затем трансформируют картофель сорта Руссет Бербанк. Для трансформации картофеля сорта Руссет Бербанк стерильные корневые культуры картофеля помещают в чашки для мороженого, содержащие 8 мл PM среды с добавкой 25 мг/л аскорбиновой кислоты (Murashige и Skoog) (MS) неорганические соли, 30 г/л сахарозы, 0,17 г/л NaH2PO4•H2O, 0,4 мг/л тиамин. HCl и 100 мг/л миоинозита, отвержденные 2 г/л Сельрита при pH 6). Когда длина побегов достигает примерно 5 см, отрезают стеблевые межузелковые сегменты в 3-5 мм и заражают примерно суточной культурой Agrobacterium tumefaciens из 4-дневной выращенной на пластинке культуры, разбавленной в отношении 1:10. Стеблевые экспланты сокультивируют 2 дня при 20oC на стерильной фильтровальной бумаге, помещенной на 1,5 мл питающего слоя табачных клеток, наложенный на 1/10 P среду (MS неорганические соли силой 1/10 и органические добавки без казеина согласно Jarret и др. (1980), /% г/л сахарозы и 8 г/л агара). После сокультивирования экспланты переносят в P-1 среды полной силы для индуцирования каллюса, среда состоит из неорганических М солей, органических добавок согласно Jarret и др. (1980) за исключением казеина, 5 мг/л зеатинрибозида (ZR) и 0,1 мг/л нафталинуксусной кислоты (НУК) (Jarret и др., 1980 a, 1980 b). Для ингибирования роста бактерий добавлен карбенициллин (500 мг/л) и цефтоксим (100 мг/л) и для отбора трансформированных клеток добавлено 100 мг/л канамицина. Трансформированные растения томата, экспрессирующие пататин-промотор-ХТП/АДФ-глюкоза-пирофосфорилаза-NOS ген, характеризуются повышенным содержанием крахмала в клубнях.
Спустя 4 недели экспланты переносят в среду того же состава, но с добавкой 0,3 мг/л гибберелиновой кислоты (ГК3) вместо НУК (Jarret и др., 1981) для промотирования образования побегов. Побеги начинают появляться примерно 2 недели спустя после перенесения в индуцирующую побеги среду. Образовавшиеся побеги обрезают и переносят в сосуды с РМ средой для укоренения. После выдерживания примерно 4 недели в укореняющей среде растения переносят в почву и постепенно закаливают. Побеги испытывают на устойчивость к канамицину, создаваемую ферментом-неомицинфосфотрансферазой II, путем перенесения побегов для укоренения в РМ среду, содержащую 50 мг/л канамицина, с отбором трансформированных клеток.
Регенерированные в культуре растения сорта Руссет Бербанк Уильямс пересаживают в 6-дюймовые горшочки (15,4 см) и выращивают в тепличных условиях до созревания. Клубни собирают и оставляют на два дня при комнатной температуре для заживления. Все клубни длиной более 2 см отбируют и хранят при 9oC и высокой влажности.
Через 3 дня после сбора урожая для 2 или 3 самых больших клубней каждого растения определяют удельный вес (УВ), причем масса каждого клубня в среднем составляет 20-40 грамм. Подсчет удельного веса осуществляют методом вычитания массы на воздухе, где УВ = масса на воздухе/ (масса на воздухе - масса в воде). Расчеты содержания крахмала и сухого вещества на основе УВ
проводят по следующим уравнениям (von Scheelem, 1937):
% крахмала = 17,546 + (199,07ХУВ - 1,0988)
% сухого вещества = 24,182 + (211,04ХУВ - 1,0988).
Вестерн блот-анализ осуществляют для белка, извлеченного из свежей центральной части клубней так, как это описано для тканей листьев томата. Анализ крахмала также проводят на аналогичных свежей центральной части клубней по описанной методике (Lin, 1988 a). Если вкратце, то примерно 300 мг центральной части вырезают, помещают в центрифужные пробирки на 1,5 мл и замораживают в сухом льду. Затем ткань высушивают до постоянной массы в СПИД BAK концентраторе фирмы Савант и определяют конечную сухую массу. Содержание крахмала определяют использованием примерно 60 мг сухого материала из каждого клубня. Первыми удаляют растворимые сахара трехкратным экстрагированием 1 мл 80%-ного этанола при 70oC и 20 минутами на каждую обработку. После конечного инкубирования весь оставшийся этанол удаляют сушкой в СПИД BAK концентраторе. Твердый продукт вновь суспендируют в 400 мкл 0,2 М гидроксида калия, размельчают, после чего выдерживают 30 минут при 100oC для солюбилизации крахмала. Раствор охлаждают и нейтрализуют добавлением 80 мкл 1 н. уксусной кислоты. Крахмал разрушают до глюкозы обработкой 14,8 единицы панкреатической альфа-амилазы (Сигма Кемикал, Сан-Луи) в течение 30 минут при 37oC с последующей обработкой 10 единицами аминоглюкозидазы (Сигма Кемикал, Сан-Луи) в течение 60 минут при 55oC. Образовавшуюся в результате ферментативного гидролиза глюкозу определяют с помощью гексокиназного набора фирмы Сигма Кемикал (Сан-Луи).
Вестерн блотирование и количественный анализ крахмала проводят на вырезанной из клубня центральной части, выращенной в стандартных тепличных условиях. Клубни томатов, экспрессирующие E.coli АДФГПФ, в среднем содержат на 26,4% больше крахмала по сравнению с контрольными растениями. Разброс отдельных точек данных показывает, что две отдельные популяции существуют с учетом содержания крахмала. Одна популяция, представленная контрольными клубнями, имеет содержание крахмала в интервале от 10,2% до 15%, при среднем содержании крахмала в 12,67%. Вторая популяция, представленная экспрессорами E. coli АДФГПФ, имеет интервал содержания крахмала от 12,1% до 19,1%, в среднем 16%. Наблюдаемое повышение содержания крахмала коррелируется с уровнем экспрессии E. coli АДФГПФ с указанием на то, что такая экспрессия приводит к повышению содержания крахмала в клубнях томата.
Удельный вес определяют для 2 или 3 самых больших клубней от каждого из 36 отдельных трансформантов методом вычитания массы в воде из массы на воздухе (KIe кор, 1987). Полученные данные показывают, что клубни, экспрессирующие E.coli АДФГПФ, характеризуются значительным повышением удельного веса по сравнению с контрольными растениями. В среднем удельный вес повышается с 1,068 для контрольных клубней до 1,088 для трансгенных клубней (табл. 2), а для лучших линий удельный вес в среднем равен примерно 1,1. Как и ожидалось, значения удельных весов колеблются и для клубней одного и того растения, также как и между клубнями различных растений. Однако только линии, экспрессирующие E.coli АДФГПФ, дают клубни с повышенным удельным весом, и это повышение грубо коррелируется с уровнем экспрессии gIgC16. Содержание крахмала и сухого вещества в среднем возросло соответственно на 35% и 23,9% в клубнях, экспрессирующих E.coli АДФГПФ, при этом лучшие линии дают повышение соответственно на 59,3% и 40,6%.
Содержание крахмала, определенное глюкозным методом для всех 26 линий картофеля, сравнивалось с содержанием крахмала, подсчитанное для всех тех же самых клубней на основании определения удельных весов. Содержание крахмала, подсчитанное на основании удельного веса, хорошо согласуется с непосредственно определенными величинами (табл. 3). Например, клубни, экспрессирующие E. coli АДФГПФ, содержат 16,01% крахмала при определении количественным анализом против 16,32% при определении на основе удельного веса. При рассмотрении повышения в отдельных линиях экспериментально определенное содержание крахмала отлично коррелируется с ростом сухого вещества (и экспрессией gIgC16 гена). Таким образом, наблюдаемый рост содержания сухого вещества в клубнях, экспрессирующих E.coli АДФГПФ, в сильной степени связан с ростом накопления крахмала.
В скобках указано число исследованных растений, взвешивались два или три клубня одного растения. Стандартное отклонение для образца указано после удельного веса (в скобках). Процент крахмала и сухого вещества подсчитаны на основании удельного веса указанным методом. Контроль представляет собой комбинацию клубней, трансформированных с введением только ДНК вектора без gIgC16 гена, и клубней, являющихся продуктом gIgC16 трансформации, но не экспрессирующих E.coli АДФГПФ.
Средние значения процента крахмала определены экспериментально ферментативным разрушением содержащегося крахмала и рассчитаны на основании определений удельного веса. Стандартные отклонения для образцов указаны в скобках. Разница между E.coli АДФГПФ + и контролем при подсчете на основании удельного веса или ферментативными методами существенна на уровне 0,005 значимых значений в Стъюдент Т-тесте.
Пример 4
Фермент АДФГПФ кодируется единственным геном в E.coli (gIgC), активная форма которого функционирует в виде гетеротетрамера (Preiss, 1984), в то время как растительный фермент является гетеротетрамером, кодируемым по меньшей мере двумя различными генами (Copeland и Preiss, 1981). Как E.coli, так и растительная АДФГПФ являются предметом четкой регуляции, при этом бактериальный фермент активируется фруктоза-1,6-бисфосфатом и ингибируется АМФ (Preiss, 1984), в то время как растительные ферменты активируются 3-фосфоглицератом и ингибируются Pi (Copeland и Preiss, 1981, Preiss, 1984). Охарактеризовано несколько мутантов E. coli, и в табл. 4 суммированы и сравнены кинетические свойства некоторых из них (Romeo T. и Preiss I., 1989).
Показано, что экспрессия gIgC16 варианта, обнаруживаемого в штамме 618 E. coli, приводит к усиленному биосинтезу крахмала в клетках растения. Экспрессия других бактериальных АДФГПФ ферментов в клетках растения также повышает содержание крахмала.
Экспрессия gIgC гена дикого типа также ведет к повышению содержания крахмала. Ген gIgC дикого типа, содержащийся в геномном клоне E.coli, обозначенном pOP12 (Okita и др., 1981), выделен путем, аналогичным способу выделения gIgC16 гена, приведенному в примере 1. Если вкратце, то NcoI сайт вводят у 5'-трансляционного старт-сайта и SacI сайт вводят непосредственно в 3'-конец терминационного кодона PCR реакцией с применением QSP3 и QSM10 праймеров, описанных в примере 1. Полученный NcoI-SacI фрагмент лигируют в вектор pMON20102 (см. пример 1), предварительно гидролизованный в присутствии NcoI и SacI, с получением плазмиды pMON16937. Psu-gIgC химерный ген конструируют лигацией XhoI-BgIII рестрикционного фрагмента, содержащего SsuIA промотор (Timko и др., 1985), BgIII-SacI фрагмента из pMON16937, включающего ХТП1-gIgC ген, и вектора трансформации растений pMON997, гидролизованного в присутствии XhoI и SacI, с образованием pMON16938 (фиг. 8). Плазмида pMON977 содержит следующие хорошо охарактеризованные ДНК сегменты (фиг. 9). Во-первых, фрагмент в 0,93 к.о., выделенный из транспозона Т 7, кодирующего устойчивость бактерий к спектиномицину/стрептомицину (Spc/Str) и являющегося детерминантой для отбора в E.coli и Agrobacterium tumefaciens (Fling и др., 1985). Это фрагмент присоединен к химерному гену устойчивости к канамицину, созданному для экспрессии в растениях с целью отбора трансформированных тканей. Химерный ген состоит из 0,35 к.о. 35 - промотора мозаичного вируса цветной капусты (P-35S) (Odell и др., 1985), 0,83 к.о. гена неомицин-фосфотрансферазы типа II (NPTII) и 0,26 к.о. 3'-нетрансляционной гена нопалин-синтетазы (NOS-3') (Fraley и др., 1983). Следующий сегмент имеет источником 0,75 к.о. из репликационного ori-v плазмиды RK2 (ori-V) (Stalker и др. , 1981). Сегмент присоединен к 33,1 к.о. SaII-PvuI фрагмента из pBR322, обеспечивающего источник репликации для поддержания в E.coli (ori-322) и bom сайте конъюгационного переноса в клетки Agrobacterium tumefaciens. Затем следуют 0,36 к.о. PvuI-BcII фрагмента из pTiT37 плазмиды, содержащего правую граничную область Т-ДНК нопалинового типа (Fraley и др., 1985). Последний сегмент представлен экспрессионной кассетой, состоящей из 0,65 к.о. 35S-промотора мозаичного вируса цветной капусты (CaMV), усиленного удвоением последовательности промотора (P-E35S) (Кау и др., 1987), синтетического мультилинкера с несколькими уникальными сайтами клонирования и 0,7 к.о. 3'-нетрансляционной области rbcs-E9 гена (E9-3') гороха (Coruzzi и др., 1984; Morell и др., 1985). Плазмиду скрещивают со штаммом ABI Agrobacterium tumefaciens с помощью трехродственной системы скрещивания и применяют для трансформации lycopersicon esculentum, сорт UC82B.
Клетки томата трансформируют использованием вышеприведенных штаммов Agrobacterium в целом по методике, описанной McCormick и др. (1986). В частности, семядоли получают из 7-8-дневных ростков. Семена стерилизуют с поверхности в 30%-ном отбеливателе Клорокс и проращивают в коробках для всходов Плантконс на ростковой среде Дэвиса. Ростковая среда Дэвиса состоит из 4,3 г/л MS солей, 20 г/л сахарозы и 10 мл/л витаминов Нитша (pH 5,8). Раствор витамина Нитша состоит из 100 мг/мл миоинозита, 5 мг/л никотиновой кислоты, 0,5 мг/л пиридоксин. HCl, 0,5 мг/л тиамин. HCl, 0,05 мг/л биотина, 2 мг/л глицина. Семена оставляют на проростание на 7-8 дней в ростовой камере при 25oC, влажности 40% и освещении холодным белым светом интенсивностью 80 эйнштейнов м-2 сек-1. Фотопериод состоит из 16 часов освещения и 8 часов темноты.
После появления всходов семядоли экспланируют обрезанием перьевым ножом # 15 верхушечной меристемы и гипокотиля с получением прямоугольного экспланта. Такие обрезы по коротким концам проросшей семядоли увеличивает площадь поверхности для инфекции. Экспланты погружают в стерильную регенерационную жидкость Дэвиса для предотвращения высыхания. Регенерационная среда Дэвиса состоит из IX MS солей, 3% сахарозы, IX витаминов Нитша, 2 мг/л зеатина (pH 5,8). Этот раствор выдерживают в автоклаве с 0,8% агара Нобеля.
Семядоли предкультивируют на "питающих пластинках", состоящих из несодержащей антибиотиков среды Кальгена. Среда Кальгена состоит из 4,3 г/л М солей, 30 г/л сахарозы, 0,1 г/л миоинозита, 0,2 г/л KH2PO4, 1,45 мл раствора 0,9 мг/л тиамин. HCl, 0,2 мл раствора 0,5 мг/мл кинетина и 0,1 мл раствора 0,2 мг/мл 2,4-D, добавлением КОН в растворе устанавливают pH 6. Пластинки покрыты 1,5-2 мл суспензии табачных клеток (TXD) и стерильным Ватмановским фильтром, пропитанным 2С005K средой. 2C005K среда состоит из 4,3 г/л смеси М солей Гибко, 1 мл витамина группы B5 (1000X партию), 30 г/л сахарозы, 2 мл/л PCPA из партии 2 мг/мл, 10 мкл/л кинетина из партии 0,5 мг/мл. Семядоли культивируют 1 день в ростовой камере при 25oC и освещении холодным белым светом интенсивностью 40-50 эйнштейнов м-2 сек-1 с непрерывным световым фотопериодом.
Затем семядоли заражают Iog-фазовым раствором Agrobacterium, содержащей плазмиду pMON1638. Концентрация Agrobacterium примерно 5•108 клеток/мл. Семядоли оставляют на шесть минут пропитываться бактериальным раствором, затем промокают стерильными кружками Ватмановского фильтра с удалением избытка раствора, после чего фильтр заменяют исходной питающей пластинкой, на которой семядоли сокультивируют 2 дня. Спустя два дня семядоли переносят на селекционные пластинки, содержащие регенерационную среду Дэвиса с 2 мг/л зеатин-рибозида, 500 мкг/мл карбенициллина и 100 мкг/мл канамицина. Через 2-3 недели семядоли с каллюсом и/или образовавшимися побегами переносят на свежие пластинки с регенерационной средой Дэвиса, содержащей те же количества карбенициллина и канамицина. В этот момент в опыте подсчитывают число трансформантов. Ткань каллюса регулярно рекультивируют через 3-недельные промежутки и любые ненормальные структуры обрезают так, что развивающиеся почки побегов продолжают регенерировать. Побеги развиваются в пределах 3-4 месяцев.
После того, как побеги развились, их тщательно отрезают от ткани каллюса и высаживают на корневые селекционные пластинки. Такие пластинки содержат 0,5X MSO с 50 мкг/мл канамицина и 500 мкг/мл карбенициллина. В пределах двух недель на селекционной среде побеги образуют корни. Если 2 недели никаких побегов не появляется, тогда побеги обрезают и реплантируют на селекционную среду. Корневые культуры инкубируют в персивалях при температуре 22oC. Побеги с корнями при достижении корнями длины около 2 см высаживают в горшочки. Растения закаливают в ростовой камере при 21oC с фотопериодом в 18 часов света и 6 часов темноты в течение 2-3 недель перед перемещением в теплицу. В теплице растения выращивают при температуре 26oC в течение дня и 21oC в течение ночи. Фотопериод состоит из 13 часов света и 11 часов темноты. В этих условиях и происходит созревание.
Создают растения трансгенного томата, трансформированного вектором pMON16938, и отбирают вестерн блот-анализом на gIgC генный продукт. Для вестерн блот-анализа белки извлекают из ткани листьев или стебля и растирают в отношении (1: 1) со смесью 100 мМ Трис (pH 7,5), 35 мМ KCl, 5 мМ дитиотреитола, 5 мМ аскорбата, 1 мМ ЭДТК, 1 мМ бензамидина и 20% глицерина. Концентрацию белка в экстракте определяют BCA-методом Пирса и белки разделяют ВСА на 3-17% НДС полиакриламидном геле. E.coli АДФГПФ обнаруживают с козьих антител, созданных против очищенной E.coli АДФГПФ и конъюгированных с щелочной фосфатазой кроличьих антикозьих антител (Промега, Мэдисон, ВИ). У большинства растений, экспрессирующих E.coli АДФГПФ дикого типа, уровень E.coli АДФГПФ составлял 0,1% от всего экстрагируемого белка. Для анализа крахмала поздним полднем из 3-4 различных молодых полностью развившихся листьев с выращенного в теплице растения вырезают штампом единственный образец. Вырезанные из листьев образцы с каждого растения объединяют и определяют на аналитических весах Меттлера массу свежих листьев. Общая масса свежих листьев на образец составляла 60-80 мг. Вначале трехкратным экстрагированием при 70oC 1 мл 80%-ным этанолом и 20 минутами на экстрагирование удаляют растворимые сахара. После последнего выдерживания весь оставшийся этанол удаляют сушкой в СПИД ВАК концентраторе. Твердый продукт суспендируют в 400 мкл 0,2 М гидроксида калия, растирают и затем выдерживают 30 минут при 100oC для солюбилизации крахмала. Раствор охлаждают и затем нейтрализуют добавлением 80 мкл 1н уксусной кислоты. Крахмал разрушают до глюкозы обработкой 14,8 единицы панкреатической альфа-амилазы (Сигма Кемикал, Сан-Луи) в течение 30 минут при 37oC с последующей обработкой 60 минут при 55oC 10 единицами амилоглюкозидазы (Сигма Кемикал, Сан-Луи). Образовавшуюся в результате ферментативного гидролиза глюкозу определяют с помощью гексокиназ-набора фирмы Сигма (Сан-Луи) и полученные величины используют для подсчета содержания крахмала.
Листья растений томата, экспрессирующие gIgC ген из Ssu-промотора, в среднем содержат крахмал на 29% больше, чем контрольные растения, а самые лучшие линии дают 107%-ное повышение (табл. 5). В скобках указано число исследованных линий. Таким образом, и другие АДФГПФ с отличающимися кинетическими свойствами также эффективным в повышении содержания крахмала в трансгенных растениях. Необходимо отметить, что высокий уровень экспрессии нерегулированных мутантов АДФГПФ в ткани листьев нежелателен, поскольку может вызвать нежелательное воздействие на рост и развитие растения. В самом деле, применение в вышеприведенных экспериментах gIgC16 гена вместо gIgC не приводит к регенерации трансформантов, экспрессирующих на высоком уровне gIgC16 генный продукт.
Для экспрессирования gIgC из пататин-промотора тот же BgIII-SacI ХТПI-gIgC фрагмент из pMON6937 и HindIII-BamHI фрагмент, содержащий пататин-промотор из плазмиды pB1241.3, лигируют в бинарный вектор pMON10098 (фиг. 11), гидролизованный в присутствии HindIII и SacI, с образованием плазмиды pMON16950 (фиг. 10). Плазмида pB1241.3 содержит сегмент пататин-1-промотора, включающий отрезок от AccI сайта в положении 1323 до DraI сайта в положении 22389/ позиции относятся к последовательности по Bevan и др., 1986/, с HindIII линкером, присоединенным в последнем положении. Плазмида pMON10098 содержит следующие ДНК области, указанные на фиг. 11 по часовой стрелке. 1) Химерный ген устойчивости к канамицину, созданный для экспрессии в растении с целью последующего отбора трансформированной ткани. Химерный ген состоит из 0,35 к. о. 35S-промотора (P-35S) мозаичного вируса цветной капсулы (Odell и др. , 1985) 0,83 к.о. гена неомицин-фосфотрансферазы типа II (KAN) и 0,26 к. о. 3'-нетрансляционной области гена нопалинсинтетазы (NOS-3') (Fraley и др., 1983); 2) фрагмент ClaI-DraI фрагмент в 0,45 к.о. из pTi 15955 октопин-Ti плазмиды, содержащей левую граничную область Т-ДНК (Barker и др., 1983); 3) сегмент в 0,75 к.о., содержащий источник репликации из RK2 плазмиды (ori-V) (Stalker и др. , 1981); 4) сегмент SaII-PstI в 3 к.о. из pBP322, дающего источник репликации для поддержания в E.coli и (ori-322) и bom сайте конъюгационного переноса в клетки Agrobacterium efaciens; 5) фрагмент в 0,93 к. о. , выделенный из транспозона Tn7, кодирующего устойчивость бактерий к спектиномицину/стрептомицину (Spc/Str) (Pling и др., 1985) и являющегося детерминантой для отбора в E.coli и Agrobacterium tumefaciens; 6) фрагмент PvuI-BcII в 0,36 к.о. из pTiT37 плазмиды, содержащей правую граничную область Т-ДНК нопалинового типа (Fraley и др., 1985); и 7) последний сегмент, представленный экспрессионной кассетой, включающей 0,65 к.о. 35S-промотора мозаичного вируса цветной капусты (CaMV), усиленного удвоением последовательности промотора (P-E35S) (Кау и др., 1987), синтетический мультилинкер с несколькими уникальными сайтами клонирования и 0,7 к.о. 3'-нетрансляционной области rbcs-E9 гена (E9-3') гороха (Coruzzi и др., 1984; Morell и др., 1985). Плазмиду скрещивают с ABI штаммом Agrobacterium tumefaciens использованием системы трехродственного скрещивания и применяют для трансформации Руссет Бербанк картофеля линии Вильямс 82. Экспрессия gIgC из пататин-промотора (pMON16950) в картофеле также приводит к повышению содержания крахмала в клубнях.
Способами, аналогичными приведенными для gIgC гена дикого типа и gIgC16 мутантного гена, кроме того, в растениях экспрессирован мутант gIgC-SG5 с повышением в результате содержания крахмала.
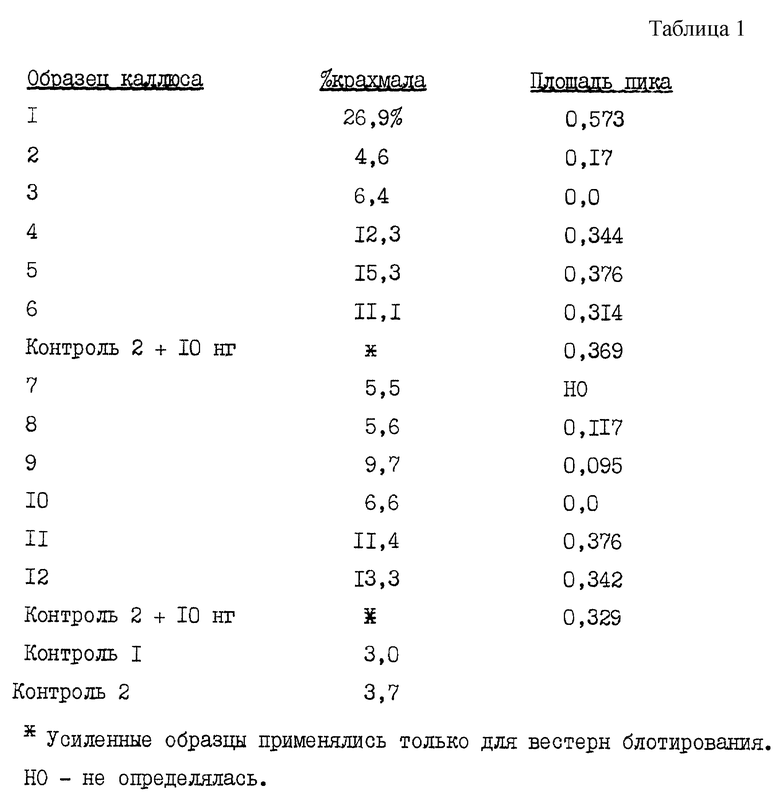









































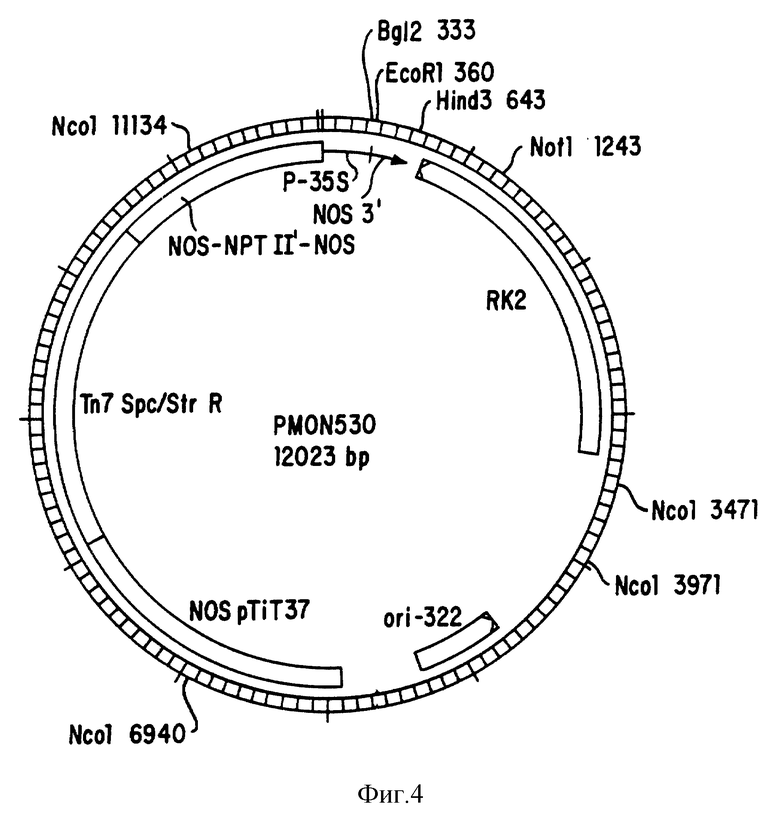





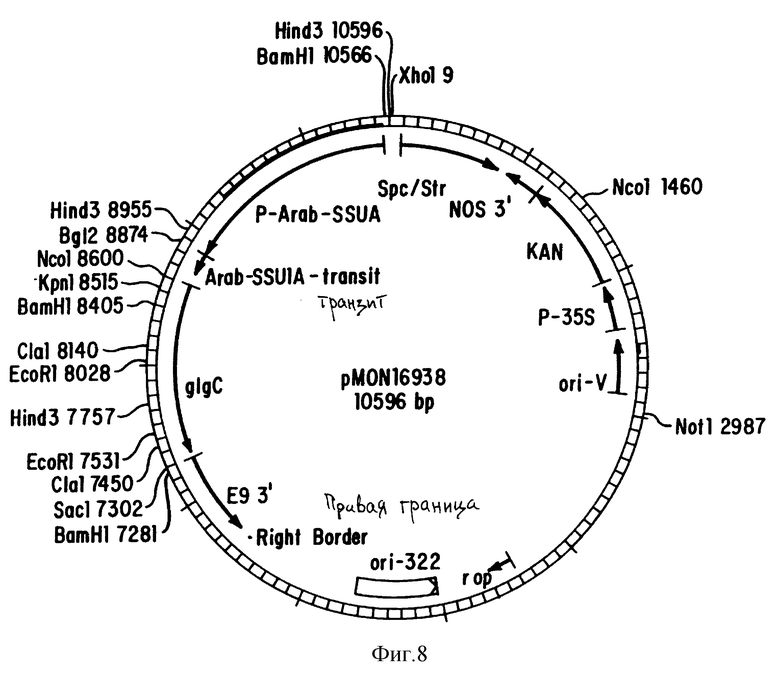
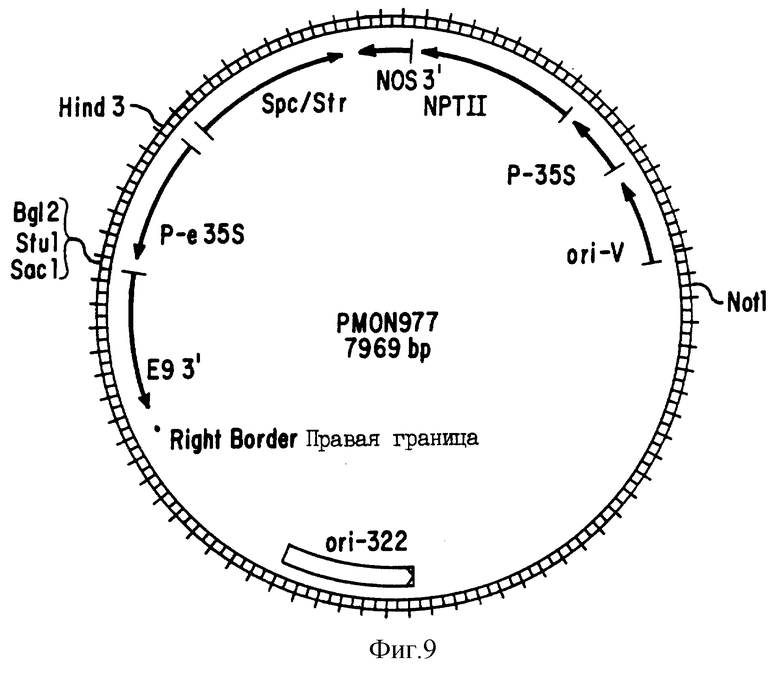
Способ и генный продукт могут быть использованы для селекции растений. За счет введения рекомбинантной двухцепочечной ДНК-молекулы в геном растения в нем продуцируется повышенное количество крахмала. 2 с. и 18 з.п. ф-лы, 11 ил., 5 табл.
Приоритет по пунктам:
18.06.90 - по пп.1-17 и 19-20;
07.06.91 - по п.18.
| УСТРОЙСТВО для ИЗМЕРЕНИЯ ДАВЛЕНИЯ ВЯЗКО-ПЛАСТИЧНЫХ ПРОДУКТОВ | 0 |
|
SU368506A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
| Биохимия растений/ Пер.с англ.Под ред | |||
| В.Л | |||
| Кретовича - М.: Мир, 1968, с.140-154. | |||

