Данная заявка является частичным продолжением находящейся на рассмотрении заявки N 07/543236, поданной 25 июня 1990 г.
Последние достижения генной инженерии обеспечили специалистов необходимым инструментом трансформации растений с введением в них чужеродных генов. В настоящее время можно создавать растения с уникальными с точки зрения агрономии показателями. Несомненно, среди достигаемых при этом преимуществ: более экономически эффективная, экологически совместимая борьба с сорняками путем создания толерантности к гербицидам. Толерантные к гербицидам растения могут уменьшить необходимость в обработке почвы с эффективным снижением тем самим эрозии почвы.
Одним из гербицидов, подвергавшимся в этой связи тщательному изучению, является N-фосфонометилглицин, обычно называемый глифозатом. Глифозат ингибирует участие шикимовой кислоты в биосинтезе ароматических соединений, в том числе аминокислоты и витамины. Более конкретно, глифозат ингибирует превращение фосфоенолпировиноградной кислоты и 3-фосфошикимовой кислоты в 5-енолпирувил-3-фосфошикимовую кислоту путем ингибирования фермента 5-енолпирувил-3-фосфошикимовой кислоты-синтетазы (ЕПФШ-синтетаза или ЕПФШС).
Показано, что толерантные к глифозату растения могут быть созданы введением в геном растения способности продуцировать на высоком уровне ЕПФШ-синтетазу, при этом рекомендуется, чтобы фермент был толерантным по отношению к глифозату. (Shah и др., 1986). Введение в растения гена-(ов) разрушения глифозата могло бы послужить способом придания растениям толерантности к глифозату и/или повышения толерантности трансгенного растения, уже экспрессирующего толерантную к глифозату ЕПФШ-синтетазу, в зависимости от физиологического действия продуктов разрушения.
Метаболизм (разрушение) исследован на самых различных растениях, и в большинстве таких исследований установлена небольшая степень такого разрушения. В тех случаях, когда разрушение происходит, начальным продуктом разрушения является аминометилфосфонат (АМФК) (Coupland, 1985; Marshall и др., 1987). И в таких случаях неясно, метаболизируется ли глифозат растением или же посторонними бактериями на поверхности листьев, на которые наносят глифозат. Согласно сообщениям, АМФК для большинства видов растений менее фитотоксичен, чем глифозат (Franz, 1985), но не для всех видов растений (Maier, 1983; Tanaka и др., 1988). В почве разрушение глифозата происходит более интенсивно и быстро (Torstensson, 1985). Основным идентифицированным продуктом разрушения является АМФК (Rueppel и др., 1977; Nomura и Hilton, 1977) - фосфонат, который может быть метаболизирован самыми различными микроорганизмами (Zeleznick и др., 1963; Masfalerz и др., 1965; Cook и др., 1978; Daughton и др., 1979a, 1979b; Wackett и др., 1987a). Идентифицирован целый ряд чистых бактериальных культур, разрушающих глифозат по одному из двух известных путей (Moore и др., 1983; Talbot и др., 1984; Shinabarger и Braymer, 1986; Ballthazor и Hallas, 1986; Kishore и Jacob, 1987; Wackett и др., 1987a; Pipke и др., 1987a; Pipke и др., 1987; Hallas и др., 1988; Jacob и др. , 1985 и 1988; Pipke и Amrhein, 1988; Quinn и др., 1988 и 1989; Lerbs и др. , 1990; Schowanek и Verstraete, 1990; Weidhase и др., 1990; Liu и др., 1991). Путь с участием "C-P-лиазы", разрушающей глифозат до саркозина и неорганического фосфата (Pi), указан для вида Pseudomonas (Shinabarger и Braymer, 1986; Kishore и Jacob, 1987) и вида Arthrobacler (Pipke и др., 1987b). Чистые культуры, способные разрушать глифозат до АМФК, указаны для вида Flavobacterium (Balthazor и Hallas, 1986), для вида Pseudomonas (Jacob и др. , 1988) и для Arthrobacter atrocyaneus (Pipke и Amrhein, 1988). Кроме того, большое число изолятов, превращающих глифозат в АМФК, идентифицирован в промышленных активированных илах, которыми обрабатывают отходы производства глифозата (Hallas и др., 1988). Однако, число и природа бактериальных генов, ответственных за указанные разрушения, до настоящего времени не были определены, как не был выделен и ген-(ы).
Соответственно, в одном из его аспектов целью изобретения является создание новых генов, кодирующих фермент метаболизма глифозата, превращающий глифозат в аминометилфосфонат и глиоксилат.
Другой целью изобретения является повышение активности фермента метаболизма глифозата по отношению к глифозату путем замены специфичных аминокислотных остатков.
Еще одна цель настоящего изобретения заключается в создании генетически модифицированных растений, экспрессирующих ген, кодирующий фермент метаболизма глифозата и отличающийся повышенной толерантностью к гербицидному глифозату.
И еще одна цель изобретения заключается в том, чтобы показать, что фермент метаболизма может быть направлен с помощью хлоропластного транзитного пептида в пластиды и что направленный в пластиды фермент придает им толерантность к глифозату на высоком уровне.
И наконец, целью изобретения является создание метода отбора трансформированной растительной ткани использованием фермента метаболизма глифозата в качестве поддающегося отбору маркера в присутствии ингибирующих концентраций глифозата.
Эти и другие цели, аспекты и признаки настоящего изобретения для специалиста станут очевидными из нижеследующего описания и рабочих примеров.
Настоящим изобретением даются структуральные ДНК конструкты, кодирующие фермент глифозат-оксидоредуктазу и применимые в создании способности разрушать глифозат в гетерологичных микроорганизмах (напр.: бактериях и растениях), а также в создании толерантных к глифозату растений.
В соответствии с вышеизложенным согласно с одним из аспектов настоящего изобретения дается способ создания генетически трансформированных растений, толерантных по отношению к гербициду глифозату, включающий стадии:
(a) внедрения в геном клетки растения рекомбинантной двунитевой молекулы ДНК, состоящей из:
(I) промотора, действие которого в клетках растения вызывает продуцирование РНК последовательности,
(II) структуральной ДНК последовательности, вызывающей продуцирование РНК последовательности, кодирующей фермент глифозат-оксидоредуктазу,
(III) 3'-нетрансляционной ДНК последовательности, действие которой в клетках растения вызывает присоединение полиаденилированных нуклеотидов к 3'-концу РНК последовательности;
причем промотор гетерологичен по отношению к кодирующей последовательности и способен вызывать достаточную экспрессию указанного фермента в растительных тканях, включая меристематическую ткань, с повышением устойчивости клетки растения, трансформированной указанным геном, к глифозату;
(b) получения трансформированной клетки растения и
(c) регенерирования из трансформированной клетки растения генетически трансформированного растения с повышенной толерантностью к гербициду глифозату.
Согласно другому аспекту настоящего изобретения дается рекомбинантная двунитевая молекула ДНК, состоящая последовательно из:
(a) промотора, действие которого в клетках растения вызывает продуцирование РНК последовательности;
(b) структуральной ДНК последовательности, вызывающей продуцирование РНК последовательности, кодирующей фермент глифозат-оксидоредуктазу; и
(c) 3'-нетрансляционной области, действие которой в растении вызывает присоединение полиаденилированных нуклеотидов к 3'-концу РНК последовательности.
Согласно еще одному аспекту изобретения даются бактериальные и трансформированные растительные клетки, содержащие, соответственно, ДНК, состоящую из вышеупомянутых элементов (a), (b) и (c).
Согласно еще одному аспекту настоящего изобретения даются дифференцированные растения, включающие трансформированные растительные клетки, охарактеризованные выше, и отличающиеся толерантностью к гербициду глифозату.
В соответствии с еще одним аспектом настоящего изобретения дается способ избирательной борьбы с сорняками на полях с культурными растениями, на которых высажены семена или рассада культурных растений, способ включает стадии:
(a) высаживания семян культурных растений или самих растений, отличающихся толерантностью к глифозату в результате введения в семена или растения рекомбинантной двунитевой молекулы ДНК, состоящей из:
(I) последовательности промотора, действие которой в растениях вызывает продуцирование РНК последовательности,
(II) структуральной ДНК последовательности, вызывающей продуцирование РНК, кодирующей фермент глифозат-оксидоредуктазу,
(III) 3'-нетрансляционной области, кодирующей сигнал полиаденилирования, действие которого в растениях вызывает присоединение полиаденилированных нуклеотидов к 3'-концу РНК последовательности. Причем промотор гетерологичен по отношению к кодирующей последовательности и способен вызывать достаточную экспрессию указанного фермента в растительной ткани, включая меристематическую ткань, с повышением толерантности клетки растения, трансформированной указанным геном; и
(b) нанесения на растения и семена на полях достаточного количества для борьбы с сорняками гербицида глифозата без существенного действия на урожай.
В особенно рекомендуемом воплощении изобретения двунитевая молекула ДНК включает ген для экспрессии в растении, представляющий собой структуральную ДНК, кодирующую слитый полипептид, содержащий аминоконцевой хлоропластный транзитный пептид, способный вызывать импортацию карбоксиконцевого фермента глифозат-оксидоредуктазу в хлоропласт клетки растения, экспрессирующей указанный ген.
Другое воплощение настоящего изобретения заключается в применении гена глифозат-оксидоредуктазы в качестве поддающегося отбору маркера для отбора и идентификации трансформированной растительной ткани.
На фиг. 1 приведена ДНК последовательность для полной длины промотора мозаичного вируса норичника (FMV).
На фиг. 2 приведена структуральная ДНК последовательность для гена глифозат-оксидоредуктазы из бактериального изолята LBAA.
На фиг. 3 приведено сравнение подвергнутого обработке структурального гена глифозат-оксидоредуктазы с модифицированным геном глифозат-оксидоредуктазы, предназначенным для повышенной экспрессии в растениях. Подвергнутый обработке ген глифозат-оксидоредуктазы показан в виде верхней ДНК последовательности. Единственные изменения, осуществленные в модифицированном гене, показаны в нижерасположенной нити последовательностей.
На фиг. 4 приведено сравнение подвергнутого обработке структурального гена глифозат-оксидоредуктазы с синтетическим геном глифозат-оксидоредуктазы, предназначенным для повышенной экспрессии в растениях. Подвергнутый обработке ген глифозат-оксидоредуктазы показан в виде верхней ДНК последовательности.
На фиг. 5 показано строение pMON17032 - pMON1886 вектора, содержащего модифицированный ген глифозат-оксидоредуктазы, внедренный в виде En-CaMV35S -модифицированной глифозат-оксидоредуктазы-NOS-3'-кассеты в NotI сайт вектора. Описание вектора pMON886 можно найти в тексте.
На фиг. 6 приведена нуклеотидная последовательность ХТП1 хлоропластного транзитного пептида, происходящего из A.thaliana SSUIA гена.
На фиг. 7 приведена генетическая/структуральная карта плазмиды pMON17066 - вектора pMON979 типа, содержащего слитый полипептид ХТП/синтетическая глифозат-оксидоредуктаза. Родственными pMON979 типу производными являются pMON17065 и pMON17073.
На фиг. 8 приведена генетическая/структуральная карта плазмиды pMON17138, являющейся примером вектора pMON981 типа, содержащего ген, кодирующий слитый полипептид ХТП/синтетическая глифозат-оксидоредуктаза. В данном примере ген ХТП1-синтетической глифозат-оксидоредуктазы клонирован в pMON979 в виде XbaI-BamHI фрагмента.
На фиг. 9 приведена нуклеотидная последовательность ХТП2 хлоропластного транзитного пептида, происходящего из A.thaliana EPSPS гена.
На фиг. 10 приведена структуральная карта плазмиды pMON17159.
На фиг. 11 приведена структуральная карта плазмиды pMON17226.
На фиг. 12 приведена структуральная карта плазмиды pMON17164.
Экспрессия растительного гена, существующего в форме двунитевой ДНК, включает синтез матричной РНК (мРНК) из одной из нитей ДНК при участии фермента РНК-полимеразы и последующую обработку внутри ядра первичного транскрипта мРНК. Такая обработка включает участие 3'-сигнальной области, облегчающей присоединение полиаденилированных нуклеотидов к 3'-концу РНК.
Транскрипция ДНК в мРНК регулируется областью ДНК, обычно называемой "промотором". Область промотора содержит последовательность оснований, подающих сигналы РНК-полимеразе связываться с ДНК и инициировать транскрипцию в мРНК использованием в качестве матрицы одной из нитей ДНК с образованием соответствующей комплементарной нити РНК.
В литературе описан целый ряд промоторов, проявляющих активность в клетках растений, в их числе; нопалин-синтетаза (NOS) и октопин-синтетеза (OCS) промоторы (находящиеся на индуцирующих опухоль плазмидах из Agrobacterium tumefaciens), каульмовирусные промоторы, такие как 19S-35S-промоторы мозаичного вируса цветной капусты (CaMV) и 35S-промотор мозаичного вируса норичника (FMV), светочувствительный промотор из малой субъединицы рибулозабисфосфаткарбоксилазы (SSRUBISCO, очень распространенного растительного полипептида). Все перечисленные промоторы были использованы для создания различного типа ДНК конструктов, экспрессированных в растениях (см., напр., PCT публикацию WO 84/02913 (Pogers и др., Монсанто)).
Промоторы, для которых известна или обнаружена способность вызывать транскрипцию в клетках растения, могут быть использованы и в настоящем изобретении. Подобные промоторы могут быть получены из различных источников, таких как: растения и растительные ДНК вирусы, в том числе, но без ограничения только ими: CaMV 35S и FMV35S-промоторы и промоторы, выделенные из растительных генов, таких как: гены SSRUBISCO или белки a/b связывания хлорофила. Как показано ниже, рекомендуется, чтобы конкретный выбранный промотор был способен вызывать достаточную экспрессию с продуцированием в результате глифозат-оксидоредуктазы в количестве, эффективном для придания растениям заметной толерантности к глифозатным гербицидам. Количество глифозат-оксидоредуктазы, необходимое для создания целевой толерантности, может меняться в зависимости от вида растения.
Рекомендуется, чтобы используемый промотор характеризовался сравнительно высокой экспрессией во всех меристематических тканях помимо других тканей, поскольку сейчас известно, что глифозат перемещается и накапливается в растительной ткани этого типа. Или же может быть использована комбинация химерных генов с целью кумулятивного создания необходимого общего уровня экспрессии фермента глифозат-оксидоредуктазы с получением в результате толерантного к глифозату генотипа.
мРНК, продуцируемая ДНК конструктом настоящего изобретения, содержит также 5'-нетрансляционную лидер-последовательность. Такая последовательность может происходить из промотора, выбранного для экспрессирования гена, и может быть специально модифицирована с тем, чтобы повысить трансляцию мРНК. 5'-Нетрансляционные области могут быть также получены из вирусных РНК, из приемлемых эукариотных генов или из синтетических генных последовательностей. Настоящее изобретение не ограничено конструктами, представленными в нижеследующих примерах, в которых нетрансляционная область происходит как из 5'-нетрансляционной последовательности, сопутствующей последовательности промотора, так и из части 5'-нетрансляционной области гена белка оболочки вируса. Желательно, чтобы нетрансляционная лидер-последовательность могла происходить из неродственного промотора или кодирующей последовательности, о чем речь шла выше.
Рекомендуемым для использования в настоящем изобретении промотором является полной длины транскриптный (35S) промотор мозаичного вируса норичника (FMV), действующий как сильный и однородный промотор для химерных генов, вводимых в растения, в частности, двудольные растения. В целом, полученные трансгенные растения экспрессируют белок, кодируемый введенным геном на более высоком и равномерном уровне во всех тканях и клетках, чем тот же самый ген, ведомый усиленным CaCV 35S-промотором. ДНК последовательность промотора (см. фиг. 1) расположена между нуклеотидами 6368 и 6930 (ПОСЛЕД. N 1) FMV генома. Рекомендуется, чтобы с промотором сочеталась 5'-нетрансляционная лидер-последовательность, и пример лидер-последовательности приведен на фиг. 1 (ПОСЛЕД. N 2). Лидер-последовательность может происходить из того же FMV генома или же может происходить из источника, отличного от FMV.
3'-нетрансляционная область химерного растительного гена содержит сигнал полиаденилирования, действие которого в растениях вызывает присоединение полиаденилированных нуклеотидов к 3'-концу РНК. Примеры приемлемых 3'-областей включают: (1) 3'-транскрибированные, нетрансляционные области, содержащие сигнал полиаденилирования из Agrobacterium индуцирующих опухоль (Ti) плазмидных генов, таких как ген нопалин-синтетазы (NOS), и (2) растительные гены, такие как гены белков хранения сои и ген малой субъединицы рибулоза-1,5-бисфосфаткарбоксилазы (ssRUBISCO). Пример рекомендуемой 3'-области представлен областью из ssRUBISCO гена гороха (E9), более подробно раскрытая в нижеследующих примерах.
ДНК конструкты настоящего изобретения кроме того содержат структуральную кодирующую последовательность в форме двунитевой ДНК, кодирующую фермент глифозат-оксидоредуктазы, превращающий глифозат в аминометилфосфонат и глиоксилат.
Краткие сведения о реакциях глифозат-оксидоредуктазы
Фермент глифозат-оксидоредуктаза катализирует расщепление C-N связи глифозата с образованием в качестве продуктов реакции аминометилфосфоната (АМФК) и глиоксилата. В аэробных условиях кислород участвует в реакции в качестве сосубстрата. Другие доноры электронов, такие как: метосульфат феназина и убихинон в аэробных условиях стимулируют реакцию. В отсутствие кислорода указанные соединения действуют, как акцепторы электронов.
Анализ на ферментативную реакцию может быть выполнен по поглощению кислорода с помощью кислородного электрода. Глифозат-оксидоредуктаза из LBAA не образует перекиси водорода в качестве продукта восстановления кислорода. Данный фермент требует стехиометрически двух молей окисленного глифозата на моль потребленного кислорода с образованием в качестве продуктов реакции по два моля каждого АМФК и глиоксилата.
Альтернативный метод анализа глифозат-оксидоредуктазы включает реакцию образца с 2,4-динитрофенилгидразином и определение количества глиоксилат-2,4-динитрофенилгидразона с помощью ВЭЖХ-анализа, более подробно описанного в последующем разделе.
Третий метод анализа глифозат-оксидоредуктазы заключается в использовании /3-14C/-глифозата в качестве субстрата, при этом образуемый ферментом радиоактивный АМФК отделяют от субстрата с помощью ВЭЖХ на анионообменной колонке по нижеприведенной методике. Связанная с АМФК радиоактивность является мерой глубины реакции глифозат-оксидоредуктазы.
Глифозат-оксидоредуктаза из LBAA относится к флавопротеинам с участием ФАД в качестве софактора. Один из механизмов, предложенных нами для реакции, катализируемой данным ферментом, включает восстановление ФАД по активному сайту фермента глифозатом. Это приводит к образованию восстановленного ФАД и основания Шиффа аминометилфосфоната с глиоксилатом. Основание Шиффа гидратируется водой и гидролизуется до его компонентов: АМФК и глиоксилата. Восстановленный флавин вновь окисляется молекулярным кислородом. Мы полагаем, что в процессе повторного окисления восстановленного ФАД в качестве промежуточного продукта образуется окисленный флавин. Промежуточный флавин может катализировать окисление глифозата с образованием АМФК и глиоксилата. Данная гипотеза согласуется с наблюдаемой стехиометрией и невозможностью обнаружить нами в реакционной смеси перекись водорода.
Помимо глифозата глифозат-оксидоредуктаза из LBAA окисляет иминодиуксусную кислоту (ИДУ) до глицина и глиоксилата. Скорость реакции с ИДУ значительно быстрее, чем с глифозатом.
Выделение бактерий, эффективно разлагающих глифозат до АМФК
Бактерии, способные разлагать глифозат, известны (Hallas и др., 1988; Malik и др., 1988). Ряд таких бактерий отобран для быстрого разрушения глифозата следующим путем. Двадцать три бактериальных изолята перенесены с TSA-пластинок (Триптиказный соевый агар, BBL) в среду A, состоящую из среды с солями Дворкина-Фостера, содержащей глюкозу, глюконат и цитрат (каждый в концентрации 0,1%) в качестве источника углерода и содержащей глифозат в качестве источника фосфора (концентрация глифозата 0,1 мМ).
Минимальную среду Дворкина-Фостера готовят смешиванием в 1 л обработанной в автоклаве воды по 1 мл каждого из компонентов A, B и C и 10 мл компонента D, тиамин•HCl (5 мг), C-источников до конечной концентрации в 0,1% каждого и P-источника (глифозат или иной фосфонат, или Pi) до необходимой концентрации.
A. Соли Д-Ф (1000Х партия, на 100 мл, с обработкой в автоклаве):
H3BO3 - 1 мг
MnSO4•7H2O - 1 мг
ZnSO4•7H2O - 12,5 мг
CuSO4•5H2O - 8 мг
NaMoO3•3H2O - 1,7 мг
B. FeSO4•7H2O (1000Х партия, на 100 мл, с обработкой в автоклаве) 0,1 г;
C. MgSO4•7H2O (1000Х партия, на 100 мл, с обработкой в автоклаве) 0,1 г;
D. (NH4)2SO4 (1000Х партия, на 100 мл, с обработкой в автоклаве) 20 г.
Дрожжевой экстракт (УЕ, Дифко) добавляют до конечной концентрации 0,01-0,001%.
Кроме того, каждый 1 мл культурной среды содержит примерно 200000 cpm /3-14C/-глифозата (Амерсхам, CFA-745). Культуры инкубируют с встряхиванием при 30oC. Изоляты LBAA показали значительный рост в первый день, в то время как другие испытуемые культуры показали незначительный рост до третьего дня. Определение радиоактивности (сцинтилляционный счетчик) в культуре, клеточных дебрис и надосадочной жидкости культуры (на четвертый день) показали общее снижение 14C-радиоактивности и распределение остаточной радиоактивности в отношении 1:1 в надосадочной жидкости и дебрис, что указывает на произошедший значительный метаболизм глифозата с его поглощением (см. табл. I).
На пятый день 75 мкл надосадочной жидкости всех испытуемых культур анализируют с помощью ВЭЖХ следующим образом. Применяют анионообменную колонку СИНХРОПАКR AX100 (P. J.Cobert), подвижная фаза состоит из 65 мМ KH2PO4 (pH 5,5 добавлением NaOH, в зависимости от требований эксперимента для изменения времен удерживания материала концентрацию фосфатного буфера меняют в пределах 50-75 мМ), режим изократный и элюируемый продукт регистрируют непрерывно с помощью детектора радиоактивности. Данным анализом выявлено, особенно в одном из изолятов (LBAA) полное отсутствие пика глифозата (Время удерживания /ВУ/ = 7 мин в данном анализе) и появление нового пика радиоактивности с тем же временем удерживания, что у метиламина или N-ацетилметиламина (ВУ = 3,5 мин). Охарактеризована коллекция бактерий, в которую входит и штамм LBAA, как разрушающих глифозат до АМФК (Hallas и др., 1988), обнаружение метиламина или N-ацетилметиламина предполагает, что АМФК или N-ацетил АМФК были метаболизированы за счет активности LBAA "C-P-лиазы" с выделением фосфата, необходимого в данном эксперименте для роста культуры. Штамм LBAA исследован более подробно.
Превращение глифозата в АМФК в микробиальных изолятах
Для четкости и краткости описана нижеследующая методика выделения генов, кодирующих фермент глифозат-оксидоредуктазу, дается для выделения подобного гена из бактериального изолята (LBAA). Для специалиста очевидно, что такая же или аналогичная стратегия может быть использована для выделения таких генов из других микробиальных изолятов.
Путь разрушения глифозата охарактеризован на покоящихся клетках выращенного в присутствии глифозата штамма LBAA следующим образом. Клетки из культуры LBAA (100 мл) выращивают в DF среде с глюкозой, глюконатом и цитратом в качестве источников углерода, с тиамином и дрожжевым экстрактом (0,01%) для поступления следовых добавок (=DF3S среде) и с 0,2 мМ глифозата в качестве источника фосфора, собирают при значении Klett = 200, промывают дважды 20 мл DF3S среды и эквивалент в 20 мл клеток повторно суспендируют в 100 ul той же среды, содержащей /3-14C/-глифозат (2,5 ul с радиоактивностью 52 мCi/ммоль). Клеточную смесь инкубируют со встряхиванием при 30oC и через промежутки времени отбирают образцы (20 мл). Образцы центрифугируют и как надосадочную жидкость, так и клеточные дебрис анализируют с помощью ВЭЖХ (дебрис вновь суспендируют в 100 ul кислой DF3S среды /=DF3S, 0,65 н. HCl/, кипятят 5 мин, недолго центрифугируют и надосадочную жидкость анализируют; подкисленный контроль с глифозатом также анализируют). За 2 ч количество радиоактивности в пике глифозата (ВУ = 7,8 мин) из надосадочной жидкости уменьшилось до -33% от исходного уровня, примерно 3% глифозата обнаружено в клетках. Продукт, элюирущийся совместно с метиламином в качестве стандарта, составил -5% от исходных показаний счетчика для надосадочной жидкости и -1,5% для дебрис. Новый пик, составивший -1,5% от исходной радиоактивности при ВУ = 7,7 мин (ВУ глифозата = 8,9 мин при подкислении в данном эксперименте), идентифицирован в клеточном содержимом. Большое уменьшение общей радиоактивности также предполагает, что в данном эксперименте глифозат подвергается интенсивному метаболизму. Такой путь метаболизма получил дополнительное освещение в опыте, в котором метаболизм /14C/-АМФК сравнивался с метаболизмом /3-14C/-глифозата (см. выше) в покоящихся клетках, собранных при значении Klett = 165 и вновь суспендированных при эквиваленте в 15 мл клеток на 100 ul DF3S среды. Образцы, анализируемые с помощью ВЭЖХ, состоят из полных культур, подкисленных и обработанных вышеописанным способом. В первые 2 ч опыта с глифозатом 25% радиоактивности обнаруживаются в пике метиламин/N-ацетилметиламин (БУ = 4,8 мин), 12,5% в виде пика АМФК (ВУ = 6,4 мин), 30% в виде пика, указанного выше (ВУ = 9,4 мин) и 30% в виде пика глифозата (ВУ = 11,8 мин). В опыте с АМФК 15% радиоактивности обнаружено в виде N-ацетилметиламина/метиламина, 59% в виде АМФК и 18% в виде пика с ВУ = 9,4 мин. Модифицированная форма АМФК идентифицирована, как N-ацетил-АМФК. Вывод об аналогичной стадии ацетилирования сделан на основе продуктов, идентифицированных в E.coli, выращенной в аминометилфосфонатах в качестве единственного источника P (Avita и др., 1987). Полученные данные указывают на следующий путь разложения глифозата в LBAA: глифозат ---> АМФК (---> метиламин)---> N-ацетилАМФК ---> N-ацетилметиламин.
Клонирование гена-(ов) глифозат-оксидоредуктазы в E.coli
После установления превращения глифозата в АМФК в штамме LBAA был изучен общий подход клонирования в E.coli гена-(ов), участвующего в подобном превращении. Клонирование и генные технологии, если нет особых указаний, соответствуют в целом ранее опубликованным (Maniatis и др., 1982). Стратегия клонирования заключалась в следующем. Введение космидного банка штамма LBAA в E. coli и отбор на ген-(ы) превращения глифозата в АМФК определялись ростом на глифозате в качестве источника фосфора (P). Отбор основывался на применении АМФК, образованным ферментом метаболиза глифозата, в качестве источника P с последующим выделением из АМФК под действием E.coli "C-P-лиазы" неорганических фосфатов (Pi). Большинство штаммов E.coli неспособны усваивать фосфонаты в качестве источника P в исходном состоянии, однако, такие штаммы быстро адаптируются (независимо от RecA) и усваивают фосфонаты (становятся Mpu+) (Wackett и др. 1987b). E.coli Mpu+ выделен из E.coli SR200 (Leu-, Pro-, recA, hsdR, supE, Smr, tonA) следующим образом. Аликвоты свежей культуры E.coli SR200 в 1-бульоне наносят на MOPS (Neidhardt и др., 1974) полный агар (т. е. содержит L-лейцин и L-пролин в концентрации 25 ug/мл и витамин B1 /тиамин/ в концентрации 10 ug/мл; агар - ДИФКО "Очищенный"), содержащий в качестве источника P аминометилфосфонат (АМФК, 0,2 мМ, Сигма),
MOPS среда включает:
10 мл 10X MOPS солей
2 мл 0,5 мг/мл Тиамин•HCl
1 мл 20% глюкозы
10X MOPS соли включает:
на 100 мл
40 мл 1М MOPS, pH 7,4
4 мл 1М Трицина, pH 7,4
1 мл 0,01М FeSO4•7H2O
5 мл 1,9M NH4Cl
1 мл 0,276 М K2SO4
1 мл 0,5 мМ CaCl2
1 мл 0,528 М MgCl2
10 мл 5 М NaCl
1 мл 0,5% L-Метионина
1 мл питательных микродобавок
В число микродобавок входят:
3•10-9 М (NH4)6Mn7O24
4•10-7 М H3BO4
3•10-8 М CoCl2
1,6•10-8 М CuSO4
8•10-8 М MnCl2
1•10-8 М ZnSO4
Шесть отдельных колоний собирают с пластинки с агаром после трехдневного инкубирования при 37oC и наносят в виде штрихов на MOPS полный агар, содержащий в качестве источника P либо АМФК, либо метилфосфонат (Альфа). Одна колония, обозначенная как E.coli SR200 Mpu+, выбрана из тех колоний, которые в равной степени и однородно росли на обоих фосфонатных средах.
Хромосомную ДНК получают из штамма LBAA следующим путем. Дебрис из 100 мл поздней log-фазы культуры LBAA в L-бульоне (Miller, 1972) вновь суспендируют в 10 мл Раствора 1 (Birnboim и Doly, 1979). Добавляют НДС до конечной концентрации в 1% и суспензию подвергают трем циклам замораживания-оттаивания, каждый из которых заключается в погружении на 15 мин в сухой лед и в воду на 10 мин при 70oC. Лизат затем экстрагируют четыре раза равными объемами смеси фенол-хлороформ (1:1, фенол насыщен TE) (TE = 10 мМ Трис с pH 8; 1 мМ ЭДТК) и фазы разделяют центрифугированием (15000 G, 10 мин). Осаждаемый этанолом продукт получают из надосадочной жидкости в виде дебрис непродолжительным центрифугированием (8000 G, 5 мин) после добавления двух объемов этанола. Дебрис вновь суспендируют в 5 мл TE и диализуют 16 ч при 4oC в 2 л TE. В результате получено 6 мл раствора ДНК в концентрация 150 мкг/мл.
Частично рестриктированную ДНК получают следующим образом. Три аликвотных образца по 100 мкг LBAA ДНК обрабатывают 1 ч при 37oC эндонуклеазой рестрикции HindIII в количестве соответственно 4,2 и 1 ферментных единиц/мкг ДНК. Образцы ДНК объединяют, добавляют ЭДТК до концентрации 0,25 мМ и экстрагируют равным объемом фенол-хлороформ. После добавления NaАцетата и этанола ДНК осаждается двумя объемами этанола и отделяется в виде дебрис центрифугированием (12000 G, 10 мин). Высушенные дебрис ДНК вновь суспендируют в 500 мкл TE и расслаивают на 10-40% градиенты сахарозы (5% приращение по 5,5 каждый раз) в 0,5 М NaCl, 50 мМ Трис с pH 8, 0,5 мМ ЭДТК. После центрифугирования в течение 20 ч со скоростью 2600 об/мин в SW28 роторе пробирки пунктируют и отбирают фракции по 1 мл. Пятнадцать мкл образца из каждой третьей фракции пропускают на 0,8% геле агарозы и размер ДНК определяют сравнением с линейной лямбда-ДНК и HindIII-гидролизованной лямбда-ДНК в качестве стандартов. Фракции, содержащие ДНК в виде фрагментов в 25-35 к.о., объединяют, обессоливают на колонках АМИКОН10 (7000 об./мин, 20oC, 45 мин) и концентрируют осаждением. По этой методике получено 50 ug LBAA ДНК необходимого размера.
Плазмидную pHC79 (Hohn и Collins, 1980) ДНК и вектор, обработанный HindIII-фосфатазой, получают по литературной методике (Maniatis и др., 1982). Лигация проводилась в следующих условиях:
Векторная ДНК (обработана HindIII и щелочной фосфатазой теленка) - 1,6 мкг
Фракционированные по размеру LBAA HindIII фрагменты - 3,75 мкг
10X буфер лигации - 2,2 мкл
250 мМ Трис-HCl, pH 8
100 мМ MgCl2
100 мМ дитиотреитола
2 мМ спермидина
T4 ДНК-лигаза (Боэрингер-Маннхэйм) (400 ед./ul) - 1 мкл
H2O - До 22 ul
18 ч при 16oC.
Подвергнутую лигации ДНК (4 мкл) упаковывают в частицы лямбда-фага (Стратаген, Гигпак Гоулд) использованием методики изготовителя.
E. coli SR200 Mpu+, выращиваемую около суток в L-бульоне (с 0,2% мальтозы), заражают 50 мкл упакованной ДНК. Трансформанты отбирают на MOPS полном агаре плюс ампициллин и с глифозатом (0,2 мМ) в качестве источника P.
Для титрования упакованных космид аликвотные образцы также наносят на MOPS (Neidhardt и др., 1974) полный агар плюс ампициллин, содержащий Pi (1 мМ). Космидные трансформанты выделяют спустя 2 дня при 37oC на той же среде при отношении - 10-5 на мкг/LBAA HindIII ДНК. Колонии возникают на глифозат-агаре в интервале от дня 3 до дня 10 с конечным отношением 1 на 200-300 космид. Плазмидную ДНК получают из двадцати одного космидного трансформанта, отобранных с глифозатных пластинок. Данные космиды, основываясь на характере HindIII рестрикции плазмидной ДНК, можно разделить по меньшей мере на два класса. Общими для космид класса I являются клонированные фрагменты HindIII рестрикции в 6,4 и 4,2 к.о., для класса II общим является фрагмент в 23 к.п. о. Десять космид, представляющие отклонения от клонированных фрагментов, вновь трансформируют с E.coli SR200 Mpu+ и способность усваивать глифозат проверяют отбором путем выращивания на пластинках с MOPS полным агаром плюс ампициллин плюс глифозат. Кроме того определяют конечную плотность клеток, достигаемую культивированием в присутствии глифозата (0,2 мМ в MOPS среде) в качестве источника P, при этом между различными трансформантами удалось обнаружить лишь небольшие отличия. Трансформантами также инокулируют MOPS полный бульон с АМФК в концентрации 0,1 мМ в качестве источника P (для подтверждения наличия активности "C-P-лиазы) и после выдерживания 24 ч при 37oC разбавляют 100-кратно MOPS полной средой с глифозатом в концентрации 0,1 мМ и /3-14C/-глифозатом (40000 cpm/мл). Все содержащие космиду клетки разрушают глифозат и образуют N-ацетилАМФК и N-ацетилметиламин без заметной разницы в скорости. В данных испытаниях N-ацетилАМФК обнаружен в надосадочной жидкости культуры. Одна космида класса I, идентифицированная как pMON7468, выбрана для дальнейшего исследования. Второй ген глифозат-оксидоредуктазы идентифицирован в космидном клоне класса II.
Бесклеточные лизаты E. coli SR200 Mpu+/pMON17468 получены из клеток, выращенных на MOPS полной среде с глифозатом в концентрации 1 мМ (и с добавками L-фенилаланина, L-тирозина и L-триптофана, концентрация каждого 100 мкг/мл, а также п-аминобензойной кислоты, п-гидроксибензойной кислоты и 2,3-дигидроксибензойной кислоты, концентрация каждой 5 мкг/мл для сведения к минимуму ингибирующего действия E.coli ЕПФП-синтетазы). Дебрис (масса примерно 0,5 г во влажном состоянии) вновь суспендируют 1 мл лизисного буфера (40 мМ MOPS с pH 7,4, 4 мМ трицина с pH 7,4, 10% глицерина, 1 мМ ДТТ) и дважды пропускают через французский пресс. Клеточные дебрис удаляют центрифугированием 10 мин при 15000 об./мин. Надосадочную жидкость после добавления MgCl2 до концентрации 10 мМ анализируют на разрушение радиомеченного глифозата. Глифозат в качестве субстрата поставляется в виде /3-14С/-глифозата (конечная концентрация = 17 мкМ). Выявленными продуктами являются преимущественно АМФК и некоторое количество N-ацетилАМФК; образование АМФК указывает на клонирование ферментативной активности из штамма LBAA, однако образование N-ацетилАМФК может быть вызвано эндогенной E.coli активностью (Avila и др., 1987). Удельная активность для образования в этих условиях АМФК составляет 13,3 пмоля АМФК/мин•мг белка.
Характеристика гена превращения глифозата в АМФК
Затем в космиде локализуют клонированную область, ответственную за ферментативную активность глифозат-оксидоредуктазы. Выделяют делеции pMON7468 преимущественно в пределах клонированной области использованием ферментов рестрикции, обрезающих внутри вставки случайным образом, применением следующей методики. Образцы плазмидной ДНК по 0,5-2 мкг полностью гидролизуют следующими эндонуклеазами рестрикции; NotI, SacI, BglII или BamHI, экстрагируют смесью фенол-хлороформ, осаждают этанолом, вновь суспендируют в TE буфере и подвергают лигации 2-4 ч при комнатной температуре (или 18 ч при 16oC) в конечном объеме 50 мкл с буфером лигации и T4 ДНК-лигазой. Трансформанты отбирают в E.coli SR200 Mpu+ и эти делеции исследуют на потерю или сохранение фенотипа усвоения глифозата. Эти данные в сочетании с рестрикционным картированием клонов использованы для локализации активной области, оказавшейся вблизи центральной части вставки в pMON7468, включающей два общих HindIII фрагмента (6,4 и 4,2 к.о.). HindIII рестрикционные фрагменты из этой области затем субклонируют в pBIueScript (Стратаген) и их глифозатный фенотип определяют в E.coli JMIOI Mpu+ (Mpu+ производное JM101 выделено по методике выделения SR200 Mpu+). Клоны, содержащие HindIII фрагмент в 6,4 к.о., в любой ориентации дают усвоение глифозата. После рестрикционного картирования данного HindIII фрагмента из двух HindIII клонов в 6,4 к.о. использованием ферментов, обрезающих вставку, а также область полилинкера случайным образом, выделен ряд клонов делеции. Кроме того субклонировано несколько рестрикционных фрагментов, внутренних по отношению к HindIII фрагменту. PstI (3,5 к. о. ) и BglII (2,5 к.о.) фрагменты в любой ориентации оказались положительными на усвоение глифозата. Эти данные в сочетании с данными делеции использованы для локализации активной области, оказавшейся BglII-Xhol фрагментом примерно в 1,8 к.о. Кроме того, делеции, выделенные из HindIII фрагмента в 6,4 к.о., указывают на минимальный размер кодирующей области около 0,7 к.о., при этом E.coRI и SacI сайты, вероятно, расположены в пределах кодирующих последовательностей.
Направление транскрипции/экспрессии локуса, ответственного за ферментативную активность превращения глифозата в АМФК, определяют следующим образом. E. coli JMIOI Mpu+ трансформанты из pMON7469 #1 и #4 (клоны 2,5 к.о. BglII фрагмента в BamHI сайте pUC118 с противоположной ориентацией) выращивают в M9-глюкоза-тиамин-ампициллин бульоне в присутствии и отсутствие Plac индуктора IPTG, собирают в поздней log-фазе (Klett = 109-220), по вышеприведенной методике получают бесклеточные лизаты четырех культур и анализируют на активность превращения глифозата в АМФК при концентрации глифозата 1,7 мкМ. Наибольшая ферментативная активность получена для pMON7468 #1 плюс IPTG, где XhoI сайт доставлен относительно Plac, что предполагает экспрессирование гена-(ов) в направлении от BglII к XhoI (см. табл. II).
Единственным выявленным продуктом был АМФК, что предполагает индуцирование в E.coli трансформантах, выращенных на глифозате в качестве источника P, ранее описанной АМФК ацетилирующей активности.
В более позднем эксперименте клеточные лизаты из pMON7468 #1 и pMON7470 (BglII-XhoI 1,8 к.о. в pUC118, образован из pMON7469 #1 делецией ~700 п.о. XhoI-SaII фрагмента) на активность по превращению глифозата в АМФК при концентрации глифозата 2 мМ /уд. акт. /3-14C/-глифозата = 3,7 мCi/ммоль, 0,2 мкCi/реакцию, культуры выращивают в присутствии в среде ИПТГ, при этом зарегистрирована более высокая ферментативная активность, что отражает улучшенные условия анализа (см. табл. III).
Белки, кодируемые BglII фрагментом, определяют in vivo использованием T7 экспрессионной системы (Tabor и Richardson, 1985) после клонирования указанного фрагмента в BamHI сайт вектора pBlue Script (+) (pMON7471 #1, #2, ориентации противоположны). Испытуемые и контрольные плазмиды трансформируют в E.coli K38, содержащем pGPI-2 (Tabor и Richardson, 1985) и выращивают при 30oC в L-бульоне (2 мл) в присутствии ампициллина и канамицида (100 и 50 мкг/мл соответственно) до значения Klett ~50. Отбирают аликвоту, клетки отделяют центрифугированием, промывают M9 солями (Miller, 1972) и вновь суспендируют в M9 среде (1 мл), содержащей 0,2% глюкозы, 20 мкг/мл тиамина и 18 аминокислот в концентрации 0,01% (минус цистеин и метионин). После инкубирования 90 мин при 30oC культуры переносят в нагретую до 42oC водяную баню и выдерживают 15 мин. Добавляют рифампицин (Сигма) до концентрации 200 мг/мл, культуры выдерживают еще 10 мин при 42oC и затем 20 мин при 30oC. Образцы пульс обрабатывают 5 мин при 30oC 35S-метионином (10 мк Ci), клетки отделяют центрифугированием и суспендируют в 60-120 мкл крекинг-буфера (60 мМ Трис-HCl с pH 6,8/1% НДС/1% 2-меркаптоэтанола/10% глицерина/0,01% бромфенол голубого). Аликвоты образцов подвергают электрофорезу на 12,5% НДС-ПАГЭ и после вымачивания 60 мин в 10 объемах смеси уксусная кислота-метанол-вода (10: 30: 60) гель вымачивают в ЭНЛАЙТНИНГR (ДЮПОНТ) согласно указаниям изготовителя, сушат и экспонируют на рентгеновской пленке при -70oC. Белки, меченные 35S-метионином, обнаруживаются только для направления от BglII к XhoI, и самый крупный из них имеет размер около 45 кД. Когда BglII-XhoI фрагмент исследуют после клонирования в BamHI-XhoI сайты pBIue Script (с образованием pMON7472), то и в этом случае такой белок в ~45 кД тоже экспрессируется.
Влияние экспрессии активности по превращению глифозата в АМФК на глифозатную толерантность E.coli вначале определяют изучением роста рекомбинантов в среде, содержащей ингибирующие концентрации глифозата. В испытании сравнивают рост E.coli
JM101, содержащего контрольный вектор (pUC118; Viera, и Nessing, 1987) или pUC118 клоны BglII фрагмента в 2,5 к.о. (pMON7469 #1, #4). Наблюдается очень четкая корреляция между способностью усваивать глифозат и толерантность к глифозату. Такой генотип толерантности (устойчивость к 1,5 мМ глифозата) затем используют в качестве сита для быстрого определения фенотипа клонов делеции, таких как pMON7470 (BglII-XhoI 1,8 к.о. в pUC118, образован из pMON7469 #1 делецией ~700 п.о. XhoI- SaII фрагмента) и последующих клонов.
Нуклеотидная последовательность структурального гена глифозат-оксидоредуктазы
Нуклеотидную последовательность BqlII-XhoI фрагмента (ПОСЛЕД. N 3) определяют использованием однонитевых ДНК матриц (созданы использованием фагемидных клонов и "хелпера" M13 фага R408) и продажного набора СЕКВЕНАЗАR (Интернейшнл Биотехнолоджис. Инк.). Компьютерный анализ последовательности (ПОСЛЕД. N 3) выявил единственную большую открытую рамку считывания (ОРС) в направлении от BglII к XhoI (см. фиг. 2), включающую месторасположение отдельных соответствующих сайтов рестрикции. Предполагаемый стоп-кодон (UAA) расположен в 2 п.о. 5' от ScaI сайта рестрикционного обрезания. Данные, подтверждающие то, что UAA-кодон является терминационным колоном в ~ 45 кД ОРС, получены на основании следующего. Ранее на основании фенотипа усвоения глифозата определено, что 3'-границы расположены между SacI сайтом (95 п.о. в восходящем направлении от ScaI сайта) и XhoI сайтом. При клонировании BglII-ScaI фрагмента в BamHI-SmaI сайты pBlue Script и экспрессировании белков in vivo все равно получают белок размером ~45 кД. BgIII-ScaI фрагмент затем повторно клонируют из такого pBlue Script клона в виде XbaI-HindIII в вектор pUC118 XbaI-HindIII, что, как найдено, придает устойчивость к 15 мМ глифозата трансформантам E. coli JM101. На основании этих данных месторасположение C-окончания белка размером ~45 кД находится между Sacl и Scal сайтами. Единственным стоп-кодоном в любой рамке считывания, который находится между указанными сайтами, может быть кодон, находящийся непосредственно в восходящем направлении от ScaI сайта.
Имеются два метиониновых кодона (AUG, расположены в положениях 120 и 186), которые при использовании как fMet должны были бы привести к белкам размерами 46, 140 и 44,002 кД соответственно, но ни одному из них не предшествовала четко распознаваемая последовательность Шайн-Дельгарно.
Начало белка более точно очерчено следующим образом. Последовательности, распознающие сайт рестрикции BgIII, введены в положениях, находящихся в восходящем направлении от потенциальных старт-кодонов, сайт-направленным мутагенезом pMON7470 замещением AGATCT на последовательности AGACTG ("Bg120") и GTATCG ("Bg186) в 21 и 9 п.о. в восходящем направлении от AUG120 и AUG186 соответственно. За исключением особо отмеченных случаев олигонуклеотидные примеры для мутагенеза представляют собой последовательности, подлежащие фланкированным изменениям 8-10 гомологичными основаниями с каждой стороны. Толерантность к глифозату определена для мутированных клонов. Введение BglII сайта в восходящем направлении от AUG120 не оказало воздействия на толерантность к глифозату, но была ликвидирована мутагенезом с введением BglII сайта в восходящем направлении от AUG186. Действие обоих случаев мутагенеза на белок размером ~45 кД определено субклонированием мутированных последовательностей в T7 экспрессионные вектора использованием в полилинкере плазмиды pMON7470 сайта KpnI, находящегося сразу же в восходящем направлении от исходного BglII сайта и нисходящего HindIII сайта. Этот полный фрагмент реклонирован в p18UT3T7 (ФАРМАЦИЯ) KpnI - HindIII и испытан in vivo по вышеприведенной методике. И в этом случае белок размером ~ 45 кД экспрессировался и в сравнимых количествах из обоих "BglII" мутагенизированных последовательностей. При использовании новых BglII сайтов в виде 5'-концов (и нисходящего HindIII сайта) для клонирования в pBIue Script BamHI-HindIII сайты белок размером ~ 45 кД экспрессировался, если новый BglII сайт в восходящем направлении от AUG120 служил в качестве 5'-конца, но не в том случае, когда тот же сайт был расположен в восходящем направлении от AUG186 и являлся 5'-концом. Эти данные служат очень сильным основанием того, что AUG120 (или какой-то кодон, расположенный очень близко к нему) является N-окончанием белка глифозат-оксидоредуктазы. BglII сайт, введенный в восходящем направлении от AUG186, не приводит к преждевременно терминированному или неустойчивого белка, на основании чего можно предположить, что предсказания в результате такого мутагенеза в кодирующей последовательности (Val18-Cys19--->Arg18-Ala19) оказали тяжелые последствия на активность фермента.
Дополнительные данные, подтверждающие расположение N-окончания, получены введением по отдельности последовательности распознавания NcoI сайта рестрикции (CCATGG на CTATGT; замена во втором кодоне серина на аланин) или NdeI последовательности (CATATG на GCTATG) у AUC120 и экспрессированием этой ОРС с помощью эффективных E.coli векторов экспрессии. Экспрессия NdeI варианта более подробно излагается ниже. NdeI-HindIII фрагмент, начинающийся у предполагаемого AUG кодона, клонируют в pMON2123 (NdeI-HindIII) с заменой ompF-IGF-I слитого фрагмента (Wong и др., 1988). Полученный клон вводят в E. coli JM101 и клетки индуцируют 2 ч налидиксиновой кислотой по описанной методике (Wong и др., 1988). Полученный не различим на ДС-ПАГЭ по размеру от белка размером ~ 45 кД, а клеточный лизат из индуцированной культуры обладает удельной активностью глифозат-оксидоредуктазы в 12,8 ммоля АМФК/мин•мг. При сравнении в отдельном опыте никакой разницы в глифозат-оксидоредуктазной активности не наблюдалось, если вторым кодоном был аланин, а не серин. Структуральная ДНК последовательность для фермента глифозат-оксидоредуктазы (ПОСЛЕД. N 4) начинается у нуклеотида 120 и кончается у нуклеотида 1415 BglII-XhoI фрагмента (см. фиг. 2) и фермент глифозат-оксидоредуктаза состоит из 431 аминокислоты (ПОСЛЕД. N 5).
Конструирование векторов трансформации растительного гена глифозат-оксидоредуктазы
Для облегчения манипуляций со структуральным геном глифозат-оксидоредуктазы внутренние последовательности распознования сайтов EcoRI и NcoI удаляют сайт-направленным мутагенезом с замещением последовательности GAATTT на GAATTC и CCACGG на CCATGG соответственно. Кодирующую последовательность глифозат-оксидоредуктазы, пригодную для введения и экспрессирования в векторах трансформации растений, конструируют следующим образом. NcoI ("Met-Ala") N-окончание связывают с NcoI- и EcoRI-делетированными кодирующими последовательностями и C-окончания подвергают делеции до ScaI сайта в серии этапов клонирования использованием внутренних SpnI и EcoRV сайтов рестрикции. На этих этапах BglII расположен непосредственно за NotI сайтом в восходящем направлении, а EcoRI и HindIII сайты располагаются в нисходящем направлении сразу же за стоп-кодоном. Последовательность обработанного таким образом гена глифозат-оксидоредуктазы (ПОСЛЕД. N 6) приведена на фиг. 3. Обработанный ген глифозат-оксидоредуктазы тем не менее кодирует белок глифозат-оксидоредуктазу дикого типа. Проведенные манипуляции не меняют аминокислотной последовательности глифозат-оксидоредуктазы. Структуральная последовательность глифозат-оксидоредуктазы (ПОСЛЕД. N 6) в виде BglII/NcoI-EcoRI/HindIII фрагмента в 1321 п.о. легко клонируется в соответствующую растительную экспрессионную кассету. Такой ген глифозат-оксидоредуктазы (ПОСЛЕД. N 6) клонируют в виде BglII-EcoRI фрагмента в растительный вектор трансформации и экспрессии pMON979 с образованием PMON17073.
Модификация и повторный синтез генной последовательности глифозат-оксидоредуктазы
Ген глифозат-оксидоредуктазы из LBAA содержит последовательности, которые могут оказаться несовместимыми с высокой экспрессией гена в растениях. Такие последовательности включают потенциальные сайты полиаденилирования, которые часто A+T-обогащены, характеризуются более высоким G+C% по сравнению с теми же показателями, обычными для растительных генов (56% против ~ 50%), отличаются концентрированными участками G и C остатков и кодонами, редко используемыми в растительных генах. Высокий G+C% в гене глифозат-оксидоредуктазы ведет к ряду потенциальных последствий, в том числе: более высокая используемость G или C в третьем положении в кодонах по сравнению с растительными генами и потенциальная способность образовывать сильные шпилечные структуры, которые могут повлиять на экспрессию или стабильность РНК. Снижение содержания G+C в гене глифозат-оксидоредуктазы, разрушение скоплений G и C, удаление потенциальных последовательностей полиаденилирования и улучшения в используемости кодонов с приближением к более часто встречающемуся в растительных генах может привести к более высокой экспрессии в растениях глифозат-оксидоредуктазы.
В первой фазе данного эксперимента сайт-направленным мутагенезом модифицируют выбранные области гена. Эти модификации направлены в первую очередь (но не исключительно) на снижение G+C% и на разрушение G+C кластеров. Обработанный ген глифозат-оксидоредуктазы вначале реклонируют в фрагмидный вектор pMON7258 в виде NcoI-HindIII фрагмента с образованием pMON17014. Однонитевую ДНК получают из dut und E.coli штамма. Семь областей гена модифицируют сайт-направленным мутагенезом использованием перечисленных в Таблице IV праймеров и набора для мутагенеза ВиоРад (Каталожный N 170-3576) по методике, прилагаемой к набору.
Для ясности приведены обратные комплементы настоящих праймеров. Положения оснований в последовательностях, представленных на фиг. 2 и 3, соответствующие праймерам, указаны соответственно первым и вторым рядом цифр.
Строение полученного гена (ПОСЛЕД. N 7) подтверждено секвес-анализом и его способностью создавать толерантность к глифозату на уровне, сравнимом с контрольным, подвергнутым обработке геном глифозат-оксидоредуктазы. Такой модифицированный ген (ПОСЛЕД. N 7) далее называется "модифицированной глифозат-оксидоредуктазой". G+C% гена глифозат-оксидоредуктазы (ПОСЛЕД. N 6) снижен от ~ 56% в обработанном варианте до ~ 57% в модифицированном варианте (ПОСЛЕД. N 7). Сравнение обработанного и модифицированного генов глифозат-оксидоредуктазы приведен на фиг. 3, где обработанный вариант показан сверху, а изменения, введенные для получения модифицированного варианта, показаны снизу. Такой модифицированный ген глифозат-оксидоредуктазы в виде BglII-EcoRI фрагмента клонируют в растительную экспрессионную кассету, содержащую En-CaMV35S промотор и NOS-3'-последовательности. Такую кассету затем клонируют в виде NotI фрагмента в pMON886 вектор с образованием pMON17032 (фиг. 5).
Синтетический ген глифозат-оксидоредуктазы (ПОСЛЕД. N 8) сконструирован с изменением возможно большего числа несовместимых последовательностей, о которых речь шла выше. Короче, генная последовательность пересмотрена с изъятием возможно большего числа последовательностей или признаков последовательностей (избегая при этом введения ненужных сайтов рестрикции): скоплений G или C числом 5 или более, A+T-обогащенных областей (преимущественно), которые могут действовать, как сайты полиаденилирования или как потенциальные области дестабилизации РНК, или кодоны, редко встречающиеся в растительных генах. Сравнение обработанного (ПОСЛЕД. N 6) и синтетического (ПОСЛЕД. N 8) генов глифозат-оксидоредуктазы приведено на фиг. 4, где обработанный ген (ПОСЛЕД. N 6) показан сверху, а изменения, внесенные в синтетический ген (ПОСЛЕД. N 8), показаны снизу. G+C% для синтетического гена глифозат-оксидоредуктазы ~ 51%, а потенциал образования коротких шпилечных структур высокой энергии снижен. Такой синтетический ген клонируют в виде BglII-EcoRI фрагмента в pMON979 с образованием pMON1765, предназначенного для введения в растения.
Экспрессия хлоропласт-направленной глифозат-оксидоредуктазы
Мишенью для глифозата в растениях является фермент 5-енолпирувилшикимат-3-фосфат-синтетаза (ЕПШФС), располагающийся в хлоропласте. Хотя глифозат-оксидоредуктазная активность, связанная с цитоплазмой, снижает/предотвращает достижения глифозатом хлоропласта в трансгенном растении, направление фермента глифозат-оксидоредуктазы в хлоропласт, как найдено, еще больше уменьшает действие глифозат на ЕПШФ-синтетазу. Многие локализованные в хлоропласте белки экспрессируются генами ядра в виде предшественников и направляются в хлоропласт хлоропластным транзитным пептидом (ХТП), который удаляется на этапах импорта. Примеры подобных хлоропластных белков включают: малую субъединицу (SSU) рибулоза-1,5-бисфосфаткарбоксилазы (RUBIS CO), 5-енолпирувилшикимат-3-фосфат-синтетазу (ЕПШФС), ферредоксин, ферредоксин-оксидоредуктазу, светоулавливающий комплексный белок I и белок II и тиоредоксин F. Показано in vivo и in vitro, что нехлоропластные белки могут быть направлены в хлоропласт использованием слитых белков с ХТП и что ХТП-последовательность достаточна для направления белков в хлоропласт (della-Gioppa и др., 1987).
Белок глифозат-оксидоредуктаза направлялся в хлоропласт созданием слияния между C-окончанием ХТП и N-окончанием глифозат-оксидоредуктазы. В первом примере использован специализированный ХТП, происходящий из SSU1A гена из Arabidopsis thaliana (Timko и др., 1988). Такой ХТП (обозначен как ХТП1) конструируют сочетанием сайт-направленных мутагенезов. Структура ХТП1 (ПОСЛЕД. N 9) (фиг. 6) включает SSU1A ХТП (аминокислоты 1-55), первые 23 аминокислоты зрелого SSU1A белка (аминокислоты 56-78), остаток серина (аминокислота 79), новый сегмент, повторяющий аминокислоты 50-56 из SSU1A ХТП и первые две аминокислоты зрелого белка (аминокислоты 80-87), а также остатки аланина и метионина (аминокислоты (88 и 89). NotI сайт рестрикции расположен у 3'-конца (перекрывает Met кодон), что облегчает создание точного слияния с 5'-концом глифозат-оксидоредуктазы гена или другого гена. На последнем этапе вводят BglII сайт в восходящем направлении от N-окончания SSU1A последовательности для облегчения введения продукта слияния в векторы трансформации растений. Слитый белок встраивают между ХТП1 (ПОСЛЕД. N 9) и обработанной глифозат-оксидоредуктазой (ПОСЛЕД. N 6) (через NcoI сайт) в pGEM3 ϶ f (+) вектор с образованием pMON17034. Такой вектор может быть транскрибирован in vitro с помощью SP6-полимеразы и транскрибирован и трансляцией РНК с 35S-метионином с получением продукта, который может быть исследован на импорт в хлоропласты, выделенные из Lactuca sativa с помощью методов, описанных ниже (della-Cioppa и др., 1986). Продукт слияния ХТП1-глифозат-оксидоредуктаза затем соединяют с синтетическим геном глифозат-оксидоредуктазы (ПОСЛЕД. N 8) и полученный продукт вводят в виде BglII-EcoRI фрагмента в растительный вектор pMON979 с образованием pMON17138 (фиг. 7). После промежуточного этапа клонирования для создания большего числа сайтов клонирования такой продукт слияния ХТП1-глифозат-оксидоредуктаза клонируют также в виде XbaI-BamHI сайта в pMON981 с образованием pMON17138 (фиг. 8).
Во втором примере продукт слияния ХТП-глифозат-оксидоредуктаза встраивают между Arabidopsis thaliana ЕПШФС (Klee и др., 1987) ХТП и синтетическими кодирующими последовательностями глифозат-оксидоредуктазы. Arabidopsis ХТП сначала подвергают сайт-направленному мутагенезу с введением SphI сайта рестрикции у сайта обработки ХТП. В результате мутагенеза происходит замена Glu-Lys в указанном месторасположении на Cys-Met. Последовательность такого ХТП, названного ХТП2 (ПОСЛЕД. N 10), приведена на фиг. 9. NcoI сайт синтетического гена глифозат-оксидоредуктазы (ПОСЛЕД. N 8) заменяют SphI сайтом, перекрывающим Met кодон. На этом же этапе второй кодон превращают в кодон для лейцина. Такое изменение не оказывает видимого влияния на in vivo активность глифозат-оксидоредуктазы в E.coli. Продукт слияния ХТП2-синтетическая глифозат-оксидоредуктаза затем клонируют в pBIue Script KS (+) и полученную матрицу транскрибируют in vitro с помощью Т7-полимеразы, и меченный 35S-метионином продукт, как показано, импортируется с эффективностью, сравнимой с эффективностью импорта продукта слияния ХТП1-глифозат-оксидоредуктаза. Продукт слияния ХТП2-синтетическая глифозат-оксидоредуктаза затем клонируют в виде XbaI-BamI фрагмента в растительный вектор экспрессии с образованием pMON17164. Структуральная карта этой плазмиды представлена на фиг. 12.
Часть растительного вектора pMON17164 (фиг. 12) состоит из следующих сегментов. Химерного гена устойчивости к канамицину, созданного для экспрессии в растении для возможности отбора трансформированной ткани. Химерный ген состоит из 35S-промотора мозаичного вируса цветной капусты в 0,35 к.о. (P-35S) (Odell и др., 1985), 0,83 к.о. гена неомицин-фосфотрансферазы типа II (KAN) и 0,26 к. о. 3'-нетрансляционной области гена нопалин-синтетазы (NOS3') (Fraley и др., 1983). ClaI-DraI фрагмента в 0,45 к.о. из pT 15955 октопин Ti плазмиды, содержащего Т-ДНК левую граничную область (Barker и др. , 1983). Сегмента в 0,75 к.о., содержащего источник репликации из RK2 плазмиды (ori-V ) (Stalker и др., 1981). SalI-PstI сегмента в 3 к.о. из pBR322, обеспечивающего источник репликации в E.coli (ori-322) и дающего bom сайт для конъюгационного переноса в клетки Agrobacterium tumefaciens фрагмента в 0,93 к.о., выделенного из транспозона T7 и кодирующего устойчивость бактерий к спектиномицину/стрептомицину (Scp/Str) (Fling и др., 1985), который является детерминантной для отбора в E. coli и Agrobacterium tumefaciens. PvuI-bclI фрагмента в 0,36 к.о. из pTiT37 плазмиды, содержащего Т-ДНК правую граничную область нопалинового типа (Fraley и др., 1985). Экспрессионной кассеты, состоящей из 0,6 к.о. 35S-промотора из мозаичного вируса норичника (P-FMV) (Cowda и др., 1989), нескольких уникальных сайтов клонирования и 0,7 к.о. 3'-нетрансляционной области rbcS-E9 гена гороха (E9 3') (Coruzzi и др., 1984 и Morelli и др., 1985). Продукт слияния ХТП2-синтетическая глифозат-оксидоредуктаза клонируют в указанную экспрессионную кассету. Введение этой плазмиды в Agrobacterium и последующая трансформация растений раскрывается в нижеследующих примерах.
Для специалиста очевидно, что могут быть созданы разнообразные химерные конструкты, использующие способность конкретного ХТП импортировать соответствующий фермент глифозат-оксидоредуктазу в хлоропласт растительной клетки. Импорт глифозат-оксидоредуктазы в хлоропласт может быть определен с помощью следующего анализа.
Анализ на поглощение хлоропластом
Центрифугированием из салата (Lactuca sativa, сорт лонгифолия) в градиента Перколь/фиколь выделяют интактные хлоропласты по видоизмененной методике Bartlett и др., (1982). Полученные дебрис интактных хлоропластов суспендируют в 0,5 мл стерильного 330 мМ сорбита в 50 мМ Гепес-КОН (pH 7,7), проверенного на хлорофил (Arnon, 1949), и устанавливают конечную концентрацию хлорофила в 4 мг/мл (использованием сорбита/Гепеса). Выход интактных хлоропластов из одного кочна салата составляет 3-6 мг хлорофила.
В типичном эксперименте на поглощение с 300 мкл используют 5 мМ АТФ, 8,3 мМ немеченного метионина, 322 мМ сорбита, 58,3 мМ Гепес-KOH (pH 8), 50 мкл продукта трансляции лизата ретикулоцитов и интактные хлоропласты из L. sativa (200 мкг хлорофила). Полученную смесь осторожно вращают при комнатной температуре (в пробирках из стекла размером 10 х 75 мм) непосредственно перед волоконнооптическим излучателем, настроенным на максимальную интенсивность света (лампа на 150 Вт). Аликвоты образцов смеси (примерно 50 мкл) отбирают в различные промежутки времени и фракционируют в градиентах силиконового масла (в полиэтиленовых пробирках на 150 мкл) центрифугированием 30 с при 11000 X G. В этих условиях интактные хлоропласты образуют под слоем силиконового масла дебрис, а инкубационная среда (содержащая лизат ретикулоцитов) всплывает на поверхность. После центрифугирования градиенты силиконового масла сразу же замораживают в сухом льду. Хлоропластные дебрис затем вновь суспендируют в 50-100 мкл лизисного буфера (10 мМ Гепес-KOH с pH 7,5, 1 мМ PMSF, 1 мМ бензамидина, 5 мМ ∈ амино-н-капроновой кислоты и 30 мкг/мл апротинина) и центрифугируют 20 мин при 15000 Х G для превращения в дебрис тилакоидных мембран. Прозрачную надосадочную жидкость (стромальные белки), полученную центрифугированием, и аликвоту инкубационной среды с лизатом ретикулоцитов от каждого эксперимента на поглощение смешивают с равным объемом 2X НДС-АГЭ образца буфера для электрофореза (см. ниже).
НДС-ПАГЭ проводят по методике Laemmli (1970) в 3-17% (мас./об.) акриламидных пластинках геля (60 мм х 1,5 мм) с 3% (мас./об.) акриламидного концентрирующего геля (5 мм х 1,5 мм). Гель фиксируют 20-30 мин в растворе 40% метанола и 10% уксусной кислоты. Затем гель замачивают 20-30 мин в EN3HANCER (ДюПонт) с последующим высушиванием геля в сушилке для гелей. Гель проявляют авторадиографией использованием интенсифицирующего экрана и примерно суточной выдержки для определения того, была ли глифозат-оксидоредуктаза импортирована в выделенные хлоропласты.
Альтернативная методика выделения других структуральных генов глифозат-оксидоредуктазы
Ряд других генов глифозат-оксидоредуктазы идентифицирован и клонирован, в том числе второй LBAA ген глифозат-оксидоредуктазы из космиды pMON7477 класса II. Ген расположен (Саузерн-гибридизация) на HindIII фрагменте в ~ 23 к. о., охарактеризованном в вышеприведенном разделе о клонировании, с использованием первого гена глифозат-оксидоредуктазы в качестве зонда. Саузерн-анализ показал также PstI и BglII гибридизационные полосы в ~ 3,5 и ~ 2,5 к. о. соответственно. BglII фрагмент из pMON7477 субклонируют в BamHI сайт pBlue Script вектора. Клон в Е.coli JM101 (pMON7482), в котором клонированный фрагмент ориентирован относительно Iac-промотора так же, как в pMON17469 # 1, индуцируют ИПТГ и анализируют на глифозат-оксидоредуктазную активность. В данном эксперименте достигнута уд.акт. ~93 нмоль/мин•мг. В последующем эксперименте также выделены космиды класса I и класса II после инфицирования E. coli JM101 препаратом упакованной космиды и отбора непосредственно на толерантность к глифозату при концентрации глифозата 3-5 мМ на М9 среде.
Ген глифозат-оксидоредуктазы также был субклонирован из другого микробиального изолята, первоначально идентифицированного по его способности усваивать глифозат в качестве источника фосфора, и для которого позднее показано содержание предполагаемого гена глифозат-оксидоредуктазы при гибридизации с LBAA глифозат-оксидоредуктазным генным зондом. Этот вначале был клонирован в T7 промоторную космиду отбором на толерантность к глифозату в E.coli HB101/pGPI-2 (Boyer и Rolland-Dussoix, 1969; Tabor и Richardson, 1985) на М9 среде, содержащей 3 мМ глифозата. На присутствие гена глифозат-оксидоредуктазы вначале указал положительный сигнал гибридизации с LBAA геном, а также его положение на BglII фрагменте в 2,5 к.о. Этот BglII фрагмент был клонирован в BamHI сайт в pBlue Script (pMON17183) и экспрессирован из Iac-промотора добавлением ИПТГ. В этом опыте получена глифозат-оксидоредуктаза с удельной активностью 53 нмоль/мин•мг, подтверждая выделение гена при такой стратегии. Следующие признаки обычно обнаруживаются у этих генов глифозат-оксидоредуктазы: гены определяются (Саузерн-гибридизацией применением генных зондов глифозат-оксидоредуктазы полной длины) на BqlII фрагментах ~ 2,5 к. о., на PstI фрагментах ~3,5 к.о., содержат один EcoRI сайт в пределах гена и гены не содержат HindIII сайта. Схематичная диаграмма (см. фиг. 13) иллюстрирует некоторые общие признаки этих генов.
Большая схожесть генов глифозат-оксидоредуктазы предполагает кроме того и иной путь, которым могут быть клонированы глифозат-оксидоредуктазы. Кажущаяся консервативность областей, фланкирующих гены, и отсутствие определенных сайтов рестрикции предполагает применение однонитевых олигонуклеотидных зондов к фланкирующим областям, содержащим сайты рестрикции для BglII, HindIII, PstI, BamHI, NdeI или других приемлемых сайтов клонирования ЦРП (цепная реакция полимеразы, все подробности о ЦРП и ее применении см. Erlich, 1989) для амплификации приемлемого для клонирования фрагмента гена глифозат-оксидоредуктазы. Фланкирующие последовательности для 119 п.о. в восходящем направлении (ПОСЛЕД. N 11) гена глифозат-оксидоредуктазы дикого типа (LBAA изолят) и для ~290 п.о. (ПОСЛЕД. N 12) в нисходящем направлении гена представлены на фиг. 2.
С помощью ЦРП подхода выделены гены глифозат-оксидоредуктазы из ряда источников. Наличие глифозат-оксидоредуктазной активности подтверждено клонированием гена глифозат-оксидоредуктазы из хромосомной ДНК, полученной от вида Pseudomanas, штамм LBr (Jacob и др., 1988), и использованием праймеров, гомологичных N- и C-окончаниям LBAA гена глифозат-оксидоредуктазы и содержащих следующие приемлемые рестрикционные сайты клонирования:
5'-GAGAGACTGT CGACTCCGCG GGAGCATCAT ATG-3' (ПОСЛЕД. N 13)
и 5'-GAACGAATCC AAGCTTCTCA CGACCGCGTA AGTAC-3'(ПОСЛЕД. N 14)
Для этих ЦРП реакций используют следующие циклотермические параметры:
Денатурирование при 94oC 1 мин;
Гибридизация при 60oC 2 мин;
Полимеризация при 72oC 3 мин
30 циклов без автоусиления, связывание инкубированием при 4oC.
Образован ожидаемый ЦРП продукт в ~1,3 к.о. и после гидролиза в присутствии NdeI и HindIII фрагмент клонирован в pMON2123 для экспрессии кодированного фермента. Глифозат-оксидоредуктазную активность определяют вышеприведенным методом, и Km для глифозата аналогично вышеприведенным значениям для ферментов из LBAA.
Источник гена глифозат-оксидоредуктазы Km (глифозат, мМ) Вид Pseudomonas, штамм LBr
Бактерии, выделенные из вспомогательных потоков обработки отходов производства глифозата, также могут оказаться способными превращать глифозат в АМФК. Штаммы Pseudomonas LBAA и LBr являются примерами таких бактерий. Такие бактерии могут бить выделены de novo из таких продуктов обработки отходов.
Популяцию бактерий выделяют с колонки с фиксированным слоем иммобилизованных клеток, в которой используются шарики из Маннвиль R-635 диатомовой земли с нанесением на триптоновый соевый агар (Дифко), содержащий 100 ug/мл циклогексимида, и инкубированием при 28oC. Колонка работает три месяца на отработанных водах в качестве сырья, поступающих с завода по производству глифозата филиала Монсанто Компани, шт. МС. Колонка содержит 50 мг/мл глифозата и NH3 в виде NH4Cl. Общее содержание органического углерода 300 мг/мл и ПБК (Потребность в биологическом кислороде - мера доступности "мягкого" углерода) менее 30 мг/мл. Такая колонка для обработки описана в работе Heitkamp и др. (1990). Одним из преобладающих членов этой популяции, идентифицированным, как штамм Т10 вида Agrobacterium, растет так же, как обнаружено, в минимальном бульоне, в котором единственным источником углерода является глифозат в концентрации 10 мМ (такой бульон готовят так же, как DF среду, но с заменой глюкозы, глюконата и цитрата глифозатом). Из этого изолята получают хромосомную ДНК и подвергают той же ЦРП процедуре и с теми же праймерами, что и описанные выше для штамма LBr. Образован фрагмент нужного размера, который клонируют в E.coli экспрессионный вектор. Проводят анализ на глифозат-оксидоредуктазную активность и кроме того определяют K для глифозата:
Источник гена - К (глифозат, мМ)
Вид Agrobacterium, штамм T10 - 28
Превращение глифозата в АМФК установлено для многих различных почв (обзор см. : Malik и др., 1989), и существует ряд методик извлечения общей ДНК из смешанных образцов окружающей среды, например, почвы (Holben. и др., 1988; Steffan и Atlas 1988; Tsai и Olson, 1991), что указывает на возможность клонирования генов глифозат-оксидоредуктазы без необходимости получать вначале изолят, такой как разрушенные микроорганизмы. Разумеется, методика, раскрытая для клонирования генов глифозат-оксидоредуктазы и основанная на придании E. coli способности усваивать глифозат или толерантности к глифозату, предлагает схему, по которой могут быть клонированы другие гены глифозат-оксидоредуктазы и другие гены метаболиза глифозата, не полагаясь на гомологию, определенную для гена глифозат-оксидоредуктазы, раскрытого здесь. Возможно также обогащение разрушающими глифозат бактериями, например, неоднократным внесением глифозата в участок почвы (Quinn и др., 1988; Talbot и др. , 1984). Такая стадия обогащения может быть использована для большей простоты выделения из почвы или иных образцов окружающей среды генов глифозат-оксидоредуктазы.
Свидетельства присутствия гена глифозат-оксидоредуктазы в почвенных бактериях и методика выделения таких генов представлены ниже. Популяцию соответствующих бактерий обогащают с целью селекции бактерий, способных расти в жидкой среде с глифозатом (10 мМ) в качестве источника углерода (такую среду готовят по методике приготовления среды Дворкина-Фостера, но с исключением источников углерода и использованием P в качестве источника P). Инокулум создают экстрагированием почвы (с полей в Джерсейвилле с недавно убранной соей, Иллинойс) и отбором популяции последовательным культивированием при 28oC в вышеописанной среде (для предотвращения роста грибков добавлено 100 мкг/мл циклогексимида). При нанесении на пластинки с L-агаром в качестве среды идентифицированы колонии 5 типов. Хромосомную ДНК получают из 2 мл культуры этих изолятов в 1-бульоне и присутствие гена глифозат-оксидоредуктазы зондируют с помощью ЦРП отбора. Использованием праймеров: GCCGAGATGACCGTGGCCGAAAGC (ПОСЛЕД. N 15) и GGGAATGCCGCATGCTTCAACGGC (ПОСЛЕД. N 16) получен фрагмент ДНК предсказанного размера с хромосомной ДНК от одного из изолятов (обозначен, как 3). Условия ЦРП следующие: 1 мин при 94oC; 2 мин при 40oC; 3 мин при 72oC, 35 циклов. ДНК фрагмент, полученный таким путем, применяют в качестве зонда (после радиомечения) для выделения 3 кандидата в гены глифозат-оксидоредуктазы из космидного банка, сконструированного так, как описано для LBAA ДНК, что сильно ускоряет выделение других генов глифозат-оксидоредуктазы. Применяемые праймеры гомологичны внутренним последовательностям в LBAA гене глифозат-оксидоредуктазы. Используемые условия ЦРП допускают значительную степень несоответствия между праймерами, а полученный результат предполагает, что ген глифозат-оксидоредуктазы из 3 может и не быть и не столь близок к другим генам глифозат-оксидоредуктазы, последовательно выделенным с помощью праймеров к N- и C-окончанию LBAA гена.
Существуют самые различные методики выделения генов. Некоторые из этих методик основаны на знании назначения гена, что позволяет создавать фенотипные средства отбора, помогающие выделению. Другие методики основаны на информации о по меньшей мере части ДНК последовательности, что позволяет применять зонды или праймеры с частичной или полной гомологией, или же методики основаны на применении антител, с помощью которых обнаруживают генный продукт. Все эти возможности могут быть применены для клонирования генов глифозат-оксидоредуктазы.
Улучшение кинетических свойств глифозат-оксидоредуктазы
Прежние примеры создания гербицидной устойчивости ферментативной дезактивацией гербицида основывались на применении ферментов со способностью связывать и подвергать метаболизму гербициды с гораздо большей эффективностью по сравнению с метаболизмом глифозата под действием глифозат-оксидоредуктазы. Фермент глифозат-оксидоредуктаза характеризуется значением K к глифозату в 20-30 мМ, поэтому скорость реакции разрушения глифозата может быть доведена до оптимального уровня в трансгенных растениях либо понижением Km, либо повышением Vmax.
Технология случайного мутагенеза в сочетании с соответствующей селекцией и/или отбором являются мощным инструментом, успешно используемым для создания большого числа мутагенизированных генных последовательностей и потенциальных вариантов. Этот же подход может быть использован для выделения и идентификации вариантов глифозат-оксидоредуктазы с улучшенной эффективностью в разрушении глифозата. Технология мутагенеза, которая может быть использована, включает химический мутагенез бактериальных культур, содержащих представляющий интерес ген, или очищенной ДНК, содержащей этот ген, а также ЦРП методы, применяемые для создания копий гена (или его частей) в условиях, благоприятствующих ошибочному введению нуклеотидов (ошибок) в новую нить. Например, этому способствует проведение ЦРП в присутствии Mn++.
Приемлемые средства отбора in vivo улучшенных в результате мутагенеза вариантов могут заключаться в улучшении толерантности к глифозату в E.coli или в возрастании роста на глифозате в Mpu+ штаммах. Для отбора ген глифозат-оксидоредуктазы клонируют в вектор, содержащий слабый бактериальный промотор и/или репликон с небольшим числом копий. Фенотипы толерантности к глифозату, как показано, меняются в интервале концентрация глифозата и коррелируются с уровнем экспрессии глифозат-оксидоредуктазы. Например, в неиндуцируемых условиях векторы Plac-глифозат-оксидоредуктазы экспрессируют меньше глифозат-оксидоредуктазы, чем PrecA-глифозат-оксидоредуктазные векторы, а также характеризуются меньшей толерантностью к глифозату. Мутагенизированный генный фрагмент клонируют в наиболее приемлемый вектор и затем отбирают полученную библиотеку. Варианты отбирают по их способности расти при концентрациях глифозата, ингибирующих рост контрольного штамма, содержащего родственный клон глифозат-оксидоредуктазы. Глифозат-оксидоредуктазная активность придает E.coli способность превращать глифозат в АМФК, и в приемлемых штаммах E.coli эта АМФК может служить источником фосфата после расщепления C-P-связи C-P-лиазой. К приемлемым штаммам E.coli относятся B штаммы или Mpu+ производные K штаммов. Ген глифозат-оксидоредуктазы придает штамму E. coli JM101 Mpu+ (= GB993) минимальный рост на глифозате в качестве единственного источника фосфора. Показано, что скорость роста на глифозате также коррелируется с уровнем экспрессии глифозат-оксидоредуктазы. Мутагенизированный ген глифозат-оксидоредуктазы клонируют в приемлемый вектор и библиотеку вариантов отбирают по различным скоростям роста на пластинках или культивированием в среде, содержащей глифозат в качестве единственного источника фосфора. Клоны, показавшие более быстрый рост на пластинках по сравнению с контрольным штаммом, затем повторно отбирают анализом кривой роста.
Варианты глифозат-оксидоредуктазы, идентифицированные при каждой селекции/отборе, клонируют в вектор для экспрессии на высоком уровне и подвергают ферментативному анализу с определением Km и Vmax значений для глифозата. Наилучшие варианты глифозат-оксидоредуктазы очищают для полной кинетической характеристики. Варианты глифозат-оксидоредуктазы, для которых определены более низкие Km значения и аналогичные или более высокие Vmax значения по сравнению со значениями для фермента дикого типа, подвергают секвенс-анализу нуклеиновых кислот с определением мутации-(ий). Цель выделения вариантов будет заключаться в повышении kcat/Km отношения для катализируемого глифозат-оксидоредуктазой разрушения глифозата.
Выделен вариант с подобными улучшениями. Используемая методика мутагенеза заключалась в применении ЦРП с отравлением Mn++, а матрицей служила переведенная в линейную форму генная плазмида глифозат-оксидоредуктазы, содержащая синтетический ген глифозат-оксидоредуктазы (ПОСЛЕД. N 8). Применяемые олигонуклеотидные праймеры были гомологичны к областям вектора и фланкируют ген глифозат-оксидоредуктазы. Применялись следующие условия ЦРП: 1 мин при 94oC, 2 мин при 55oC, 3 мин при 72oC, 35 циклов. Применяют 5:1 отношение dCTP+dCTP+TTP к dATP. Реакционная смесь содержит MnCl2 в концентрациях 125, 250, 375 или 500 мкМ. После реакции амплифицированный продукт реклонируют в вектор, содержащий слабый E. coli промотор. Этим вектором является pBR327 производное, содержащее araBAD-промотор и приемлемые сайты клонирования. Сто колоний с этого этапа клонирования затем отбирают в E.coli GB993 на улучшенную толерантность к глифозату и фенотипы усвоения в среде, представляющей собой MOPS минимальную среду с глифозатом и Pi или только с глифозатом соответственно. Скорости роста определяют измерением A550 в течение 96 ч. Идентифицировано три клона, характеризующихся при таком отборе более быстрыми скоростями роста. Эти трансформанты отличаются в 1,5-2 раза более быстрым фенотипом усвоения. Ген глифозат-оксидоредуктазы реклонируют в часть вектора экспрессии с проверкой этого генотипа. Весь кинетический анализ проведен на сырых E. coli лизатах. Предполагаемые варианты белков глифозат-оксидоредуктазы были сверхэкспрессированы после субклонирования NcoI/HindIII варианта гена глифозат-оксидоредуктазы в PrecA-генIOL вектор экспрессии. Для сверхэкспрессий в PrecA-генIOL конструктах содержащие вектор CB993 клетки индуцируют при значении Klett = 110-120 в М9 минимальной среде с 50 мкг/мл налидиксиновой кислоты и оставляют расти 2,5 ч при 37oC и интенсивном встряхивании. Клетки собируют центрифугированием 5 мин при 4000G и 4oC и вновь суспендируют в 100 мМ Трис-HCl (pH 7,1), 1 мМ ЭДТК, 35 мМ KCl, 20% глицерина и 1 мМ бензамидина при 3 мл/г дебрис. Лизаты получают разрушением клеток во Французском прессе дважды при 1000 psi (68 атм). Нерастворимые дебрис удаляют центрифугированием 15 мин при 12000G и 4oC, а надосадочную жидкость обессоливают пропусканием через PD-10 колонку (Сефадекс G-25, Фармация). Фракцию свободного объема используют в качестве источника фермента в кинетическом анализе. Концентрации белка определяют с помощью анализа на связывания белка с красителем фирмы БиоРад. Для определения линейных интервалов строят кривые времени и концентрации фермента. Ферментативный анализ проводят следующим образом. Лизат и глифозат-оксидоредуктазную смесь (конечная концентрация = 0,1 М MOPS, 0,01 М трицина с pH 7,4, 0,01 мМ ФАД, 10 мМ MgCl2) в 100 мкл реакционной среды прединкубируют 2 мин при 30oC перед добавлением глифозата (чистый для анализа образец получен из воды с установленным pH 7 добавлением NaOH). Определено, что оптимальное время ферментативного анализа при использовании 10 мкг лизата - 10 мин. После выдерживания 10 мин при 30oC и встряхивания добавляют 0,25 мл реактива динитрофенилгидразина (ДНФГ; 0,5 мг/мл в 0,5 М HCl) и реакционную смесь оставляют еще на 5 мин при 30oC и встряхивании. Затем к анализируемой смеси добавляют раствор (400 мкл) 1,5 М NaOH и реакцию продолжают 5 мин при 30oC и встряхивании. Ферментативную активность определяют по количеству образовавшегося аддукта глиоксилат-ДНФГ измерением A520 относительно стандарта глифозата. Ферментативный анализ осуществляют в дублях на по меньшей мере двух изолятах колоний предполагаемого варианта глифозат-оксидоредуктазы. Для определения Km и Vmax ферментативный анализ осуществляют в (0,2-2)хK интервале концентраций глифозата. Значения Km и Vmax определяют на основании кривых линейного построения по Burk, Eadie-Hofstee и гиберболических кинетических кривых. Значение Vmax получают после определения в лизате количества иммунореактивного белка глифозат-оксидоредуктазы с помощью иммуноблот-анализа, описанного ниже. Иммуноблот-анализ проводят после НДС-ПАГЭ и переноса белка с геля на нитроцеллюлозу при 500 мА в аппарате для переноса Hoeffer в 25 мМ Трис-HCl, 192 мМ глицина, содержащих 0,1% НДС и 25% метанола, в течение 1-2 ч. После переноса нитроцеллюлозу инкубируют при комнатной температуре и встряхивании по меньшей мере 30 мин с 50 мМ Трис-HCl (pH 7,5), 0,9% NaCl, 0,01% Твин 20, 0,02% NaN3, содержащих 2% бычьего сывороточного альбумина. После блокирования добавляют тот же буфер, содержащий в разбавлении 1:25000 козью анти-глифозат-оксидоредуктазную антисыворотку, и фильтр встряхивают 45 мин при комнатной температуре. После инкубирования с первичными глифозат-оксидоредуктазными антителами фильтр промывают 45 мин в буфере без антител, затем добавляют в разбавлении 1:5000 вторые антитела кролика к козьим антителам, связанным с щелочной фосфатазой (от Pierce), и фильтр инкубируют 45 мин при комнатной температуре и встряхивании. Затем фильтр промывают 30 мин в буфере без антител перед добавлением NBT и BCIP (Промега) для появления окрашивания. Иммунореактивный белок глифозат-оксидоредуктазу кроме того определяют количественно дот-блотированием лизата на нитроцеллюлозе с последующей обработкой фильтра вышеприведенным методом за исключением того, что для детектирования применяют 125I-белок G. Количество белка глифозат-оксидоредуктазы в лизате определяют подсчетом пятен и сравнением уровня радиоактивности со стандартом белка глифозат-оксидоредуктазой. Один из вариантов (V.247) показал в 3-4 раза более высокую удельную активность для глифозат-оксидоредуктазы при 25 мМ глифозата, и иммуноблот-анализ показал, что это не вызвано повышенным уровнем белка глифозат-оксидоредуктазы. Последующими анализами установлено, что этот вариант обладает в 10 раз более низким значением Km для глифозата по сравнению с дикого типа глифозат-оксидоредуктазой. Аналогичным путем определено значение Km для ИДУ, и полученные данные представлены в табл. А.
Ген глифозат-оксидоредуктазы подвергнут секвенс-анализу (ПОСЛЕД. N 17), при этом обнаружено пять изменений в нуклеотидах. Эти изменения указаны ниже в их связи с кодонами: GCT на GCC (кодон 43), без изменения аминокислоты; AGC на GGC (кодон 84), Ser на Gly; AAG на AGG (кодон 153), Lys на Arg; GAC на CGC (кодон 334), His на Arg и CCA на CCG (кодон 362), без изменения аминокислоты. Аминокислотная последовательность гена глифозат-оксидоредуктазы из V.247 представлена ПОСЛЕД. N 18. Важность этих различных изменений аминокислот первоначально определена реклонированием измененных областей в глифозат-оксидоредуктазу дикого типа с последующим выявлением влияния изменений на активность и кинетику глифозат-оксидоредуктазы. Эта операция сопровождалась реклонированием NcoI-NheI фрагмента (содержит кодон 84), NheI-ApaLI фрагмента (содержит кодон 153) и ApaLI-HindIII фрагмента (содержит кодон 334) по отдельности в ген дикого типа. Эти гены глифозат-оксидоредуктазы затем экспрессируют с последующим проведением кинетического анализа. Полученные данные, представленные в табл. Б, показывают, что только изменения, произошедшие в ApaLI-HindIII фрагменте (содержит кодон 334), ответственны за изменение фермента.
Полученные результаты подтверждены и расширены повторением замены His на Arg у кодона 334 и введением других специфичных изменений у этого остатка сайт-направленным мутагенезом. Ниже перечислены использованные праймеры:
Arg - CGTTCTCTAC ACTCGTGCTC GTAAGTTGC (ПОСЛЕД. N 19);
Lys - CGTTCTCTAC ACTAAGGCTC GTAAGTTGC (ПОСЛЕД. N 20);
Gln - CGTTCTCTAC ACTCAAGCTC GTAAGTTGC (ПОСЛЕД. N 21);
Ala - CGTTCTCTAC ACTGCTGCTC GTAAGTTGC (ПОСЛЕД. N 22).
(приведенные последовательности противоположны реально использованным последовательностям). Наличие указанных изменений подтверждено проведением секвенс-анализа мутагенизированных генов глифозат-оксидоредуктазы и кинетического анализа ферментов глифозат-оксидоредуктазы. Полученные данные, представленные в табл. В, показывают, что в этом положении возможен целый ряд замен, которые приводят к ферменту с измененными кинетическими свойствами.
Для замены H 334 остатка другими аминокислотами осуществлен дополнительный мутагенез. Использованные при этом праймеры и новые кодоны перечислены ниже.
Trp - CTCTACACTTGGGCTCGTAAGCTTCTTCCAGC (ПОСЛЕД. N 23);
Ile - CTCTACACTATCGCTCGTAAGCTTCTTCCAGC (ПОСЛЕД. N 24);
Leu - CTCTACACTCTGGCTCCTAAGCTTCTTCCAGC (ПОСЛЕД. N 25);
Glu - CTCTACACTGAAGCTCGTAAGCTTCTTCCAGC (ПОСЛЕД. N 26)
(приведенные последовательности противоположны реально использованным последовательностям, кроме того эти праймеры добавляют "тихий" HindIII сайт, ускоряющий мутагенизированное потомство данной популяции). GLU 334 вариант сохраняет значительную глифозат-оксидоредуктазную активность, в то время как TRP334, ILE334 и LEU334 варианты сохраняют гораздо меньше активности.
Из вариантов первого поколения те, которые отличаются наиболее высоким kcat/Km отношением, рекомендуют подвергать второму раунду мутагенеза с последующим отбором и анализом. Альтернативный подход заключается в конструировании второго поколения вариантов глифозат-оксидоредуктазы сочетанием одноточечного мутагенеза, идентифицированного в вариантах первого поколения.
ТРАНСФОРМАЦИЯ РАСТЕНИЙ
Растения, которым практикой настоящего изобретения может быть придана толерантность к глифозату, включают, но без ограничения только ими: сою, хлопок, кукурузу, брюкву, масличный рапс, лен, сахарную свеклу, подсолнечник, картофель, табак, томаты, пшеницу, рис, люцерну, салат, яблони, тополь и сосну.
Молекула двунитевой ДНК настоящего изобретения ("химерный ген") может быть введена в геном растения любым приемлемым способом. Приемлемые вектора трансформации растений включают вектора, происходящие из Ti плазмиды Agrobacterium tumefaciens, а также вектора, списанные в работах, напр.: Herrera-Estrella (1983), Bevan (1984), Klee (1985) и в EPO-публикации 120516 (Schilperoort и др.). Помимо векторов трансформации растений, происходящих из Ti или индуцируемых корнями (Ri) плазмид Agrobactеrium, в альтернативных методах в клетки растения могут быть введены ДНК конструкты настоящего изобретения. Подобные могут включать, например: применение липосом, электропорацию, применение химикатов, повышающих поглощение свободной ДНК, выделение свободной ДНК использованием микросфокусированной бомбардировки, а также трансформацию с помощью вирусом или пыльцы.
Вектор трансформации растений pMON979 происходит из pMON886 (описана ниже) при замене гена неомицин-фосфотрансферазы типа II (KAN) в pMON886 фрагментом в 0,89 к. о. , содержащим бактериальный ген гентамицин-3-N-ацетил-трансферазы типа III (AAC(3)-III) (Hayford и др. , 1988). Химерный P-35S/AA(3)-III/NOS3' ген кодирует устойчивость к гентамицину, что позволяет отбирать трансформированные клетки растения. pMON979 кроме того содержит экспрессионную кассету в 0,95 к.о., состоящую из усиленного CaMV35S-промотора (Kay и др., 1987), нескольких уникальных сайтов рестрикции и NOS3' конец (P-En-CaMV35S/NOS 3'). Остальные сегменты ДНК в pMON979 точно такие же, как в pMON886.
Плазмиду pMON886 конструируют из следующих сегментов ДНК. Первым является фрагмент в 0,93 к. о. от AvaI до созданного EcoRV, выделенный из транспозона Tn7, кодирующего бактериальную устойчивость к cпектиномицину/стрептомицину (Spc/Str), что является детерминантной для отбора в E.coli и Agrobacterium tumefaciens. Этот фрагмент соединен с сегментом ДНК в 1,61 к.о., кодирующим химерный ген устойчивости к канамицину, позволяющий отбирать трансформированные клетки растения. Химерный ген (P-35S/KAN/NOS 3') состоит из 35S-промотора мозаичного вируса цветной капусты (CaMV), гена неомицин-фосфотрансферазы типа II (KAN) и 3'-нетрансляционной области гена нопалин-синтетазы (NOS 3') (Fraley и др., 1983). Следующий сегмент представлен 0,75 к.о. oriV, содержащий источник репликации из RK2 плазмиды. Сегмент присоединен к SaII-PvuI сегменту в 3,1 к.о. плазмиды pBR322 (ori 322), обеспечивающему источник репликации для установления в E.coli и bom сайте конъюгационного переноса в клетки Agrobacterium tumefaciens. Следующий представлен 0,36 к.о. PvuI-BclI из pTiT37, несущими правую граничную область Т-ДНК нопалинового тина (Fraley и др., 1985).
Плазмида pMON981 содержит следующие ДНК сегменты: 0,93 к.о. фрагмента, выделенного из транспозона Tn7, кодирующего бактериальную устойчивость к спектиномицину/стрептомицину /Spc/Str детерминанта для отбора в E.coli и Agrobacterium tumefaciens (Fling и др., 1985)/; химерный ген устойчивости к канамицину, созданный для экспрессии в растениях с целью отбора трансформированной ткани и состоящей из 0,35 к.о. 35S-промотора мозаичного вируса цветной капусты (P-35S) (Odell и др., 1985), 0,83 к.о. гена неомицин-фосфотрансферазы типа II (KAN) и 0,26 к.о. 3'-нетрансляционной области гена нопалин-синтетазы (NOS 3') (Fraley и др., 1983); 0,75 к.о. источника репликации из RK2 плазмиды (oriV) (Stalker и др., 1981); 3,1 к.о. SaII-PvuI сегмента из pBR322, обеспечивающего источник репликации для установления в E. coli (ori-322) и bom сайте конъюгационного переноса в клетки Agrobacterium tumefaciens, и 0,36 к.о. PvuI-BclI фрагмента из pTiT37, плазмиды, содержащего правую граничную область Т-ДНК нопалинового типа (Fraley и др., 1985). Экспрессионная кассета состоит из 0,6 к.о. 35S-промотора из мозаичного вируса норичника (P-FMV) (Cowada и др., 1989) и 0,7 к.о. 3'-нетрансляционной области rbc S-E9 гена гороха (E9 3') (Coruzzi и др., 1984 и Morelli и др., 1985), SspI фрагмент в 0,6 к.о., содержащий FMV35S-промотор (фиг. 1), создают для внедрения приемлемых сайтов клонирования в нисходящем направлении от сайта начала транскрипции.
Растительный вектор мобилизуют в ABI штамм Agrobacterium tumefaciens. ABI штамм является A208 Agrobacterium tumefaciens, несущей бесплечевую Ti плазмиду pTiC58 (pMP90RK) (Koncz u Schell, 1986). Ti плазмида не несет генов T-ДНК фитогормонов, вследствие чего штамм не способен вызывать заболевание корончатым галлом. Скрещивание растительного вектора в ABI проводят в системе трехродственной конъюгации использованием плазмиды-хельпера pRK2013 (Ditta и др., 1980). При инкубировании растительной ткани с конъюгатом ABI: растительный вектор переносится в клетки растения с помощью vir-функций, кодируемых бесплечевой pTiC58 плазмидой. Вектор раскрывается у правой граничной области Т-ДНК, и полная последовательность растительного вектора может быть внедрена в хромосому растений-хозяина. Плазмида pTiC58 не переносится в клетку растения, а остается в Agrobacterium.
РЕГЕНЕРАЦИЯ РАСТЕНИЙ
При достижении в трансформированных клетках (или протопластах) адекватного продуцирования глифозат-оксидоредуктазной активности клетки (или протопласты) регенерируют в целое растение. Выбор методологии этапа регенерации решающей роли не играет, и существуют приемлемые методики для хозяев из Leguminosae (Люцерна, соя, клевер и т.д.), Umbelliferae (морковь, сельдерей, пастернак), Cruciferae (капуста, редис, рапс и т.д.), Cucurbitaceae (дыня и огурец), Gramineae (пшеница, рис, кукуруза и т.д.), Solanaceae (картофель, табак, томаты, перец) и различных цветочных культур. См. напр.: Ammirato, 1984, Shimamoto , 1989; Fromm 1990; Vasil 1990.
Нижеследующие примеры даются для лучшего освещения практики настоящего изобретения и ни в коей мере не должны рассматриваться как ограничивающие объем настоящего изобретения. Для специалиста очевидно, что в раскрываемые здесь методы и гены могут быть внесены различные модификации, усечения и т. д. без отхода от духа и объема настоящего изобретения.
ПРИМЕРЫ
Экспрессия, активность и фенотип глифозат-оксидоредуктазы в трансформированных растениях
В следующих, представленных в виде примеров воплощения изобретения описаны трансформация, экспрессия, активность глифозат-оксидоредуктазы и фенотип толерантности к глифозату, придаваемый растениям генами глифозат-оксидоредуктазы, вводимыми в Nicoliana tabacum, сорт "Самсун" и/или brassica napus, сорт Вестар с помощью векторов pMON17073, pMON17032, pMON17065, pMON17066, pMON17138 и pMON17164. Исходные данные для табака по экспрессии обработанного гена глифозат-оксидоредуктазы (ПОСЛЕД. N 6) под контролем En-CaMV35S-промотора (см. данные для pMON17073 в таблицах VIII и IX, например) указывают на очень низкий уровень экспрессии глифозат-оксидоредуктазы. Транскрипция гена подтверждена в случае 3-4 растений Нозерн и S1 анализом, но никакого белка глифозат-оксидоредуктазу обнаружить не удалось (предел обнаружения в данном анализе ~0,01% от уровня экспрессии). Анализ R0 растений после опрыскивания 0,4 фунт/га (примерно 0,448 кг/га) глифозата также выявил очень низкий уровень толерантности. Модификация генной последовательности (см. выше) привела к улучшению экспрессии в табаке, как и применение FMV-промотора и применение ХТП слияния с геном глифозат-оксидоредуктазы. По этой причине большинство представленных данных относится к трансгенным растениям, полученным с помощью векторов, содержащих эти улучшенные конструкты глифозат-оксидоредуктазы. Один ряд экспериментов с модифицированным вектором глифозат-оксидоредуктазы (pMON17032) представлен в примере 1, а исследование обработанной глифозат-оксидоредуктазы, синтетической глифозат-оксидоредуктазы и ХТП1-синтетической глифозат-оксидоредуктазы представлено в примере 2. Трансформация и экспрессия глифозат-оксидоредуктазы в каноле раскрываются примером 3.
Пример 1
Методика трансформации на кружках табачных листьев требует здоровых листьев возрастом около 1 месяца. После стерилизации поверхности 15-20 мин 10% Хлорокса плюс поверхностно-активное вещество листья ополаскивают 3 раза стерильной водой. С помощью стерильного бумажного штампа вырубают из листьев кружочки, которые помещают внутренней поверхностью вверх на MS104 среду (MS соли 4,3 г/л, сахароза 30 г/л, B5 витамины 500X 2 мл/л, HYK 0,1 мг/л, БА 1 мг/л) для предкультивирования в течение 1 дня.
Затем кружочки инокулируют примерно суточной культурой бесплечевого штамма Agrobacterium ABI, содержащего целевой вектор, разбавленный в отношении 1/5 (т.е. примерно 0,6 ОП). Инокуляцию проводят помещением кружков в центрифужные пробирки с культурой. Спустя 30-60 с жидкость сливают и кружки помещают между стерильной фильтровальной бумагой. Затем кружки помещают внутренней поверхностью вверх на питательную среду MS104 вместе с кружками из фильтровальной бумаги для сокультивирования.
После 2-3 дней сокультивирования кружки переносят опять же внутренней поверхностью вверх на селекционные пластинки с MS104 средой. Спустя 2-3 недели образуется каллюс, и с кружков из листьев удаляют отдельные побеги. Побеги чисто отрезают от каллюса, когда они становятся достаточно большими, чтобы отличить их от стебля. Побеги помещают на не содержащую гормонов среду укоренения (MSO: MS соли 4,3 г/л, сахароза 30 г/л и B5 витамины 500X 2 мл/л) с селекцией. За 1-2 недели образуются корни. Любой анализ каллюса рекомендуют проводить с укоренившимися побегами и в стерильном состоянии. Укоренившиеся побеги помещают в почву и выдерживают в условиях высокой влажности (напр., в пластиковых контейнерах или мешках). Побеги закаливают постепенным воздействием обычно влажности.
Всего исследовано 45 устойчивых к канамицину pMON17032 табачных линий (таблица V).
Повторное каллюсообразование листьями на культурной среде растительной ткани указывает на низкий уровень толерантности к глифозату (оценена, как +/- фенотип) для по меньшей мере 11 из этих линий. По меньшей мере 24 из этих линий экспрессируют глифозат-оксидоредуктазу на поддающемся обнаружению уровне в интервале 0,5-2 нг на 50 мкг извлекаемого белка. Толерантность к глифозату, проявившаяся в анализе с повторным каллюсообразованием листьями, и более высокий уровень экспрессии глифозат-оксидоредуктазы указывают на то, что изменения, проведенные в кодирующих глифозат-оксидоредуктазу последовательностях для получения модифицированного гена глифозат-оксидоредуктазы (ПОСЛЕД. N 7), оказывают заметное влияние на способность этого гена экспрессироваться в растениях. Того же эффекта можно также затем добиться экспрессированием обработанного гена глифозат-оксидоредуктазы (ПОСЛЕД. N 6) использованием более сильного растительного промотора, использованием лучших сигнальных последовательностей 3'-полиаденилирования, оптимизацией последовательностей возле кодона инициации для рибосомной загрузки и инициации трансляции или сочетанием этих и других последовательностей или факторов экспрессии или регуляции. R1 потомство ряда этих линий с наиболее высоким уровнем экспрессии глифозат-оксидоредуктазы (NN 18854 и 18848) опрыскивают глифозатом в дозировке (0,4 и 1 фунт/ар (0,448 и 1,12 кг/га соответственно) и в течение четырех недель оценивают вегетативное поведение (таблица VI).
После начального промежутка времени и особенно для растений, экспрессирующих глифозат-оксидоредуктазу на самом высоком уровне, указанные линии растений показали вегетативную толерантность к глифозату (которая улучшалась со временем). Глифозат-оксидоредуктазная ферментативная активность определена для двух pMON17032 линий (NN 18858 и 18881). Собирают ткань листьев (1 г), замораживают в жидком N2 и хранят при -80oC перед экстрагированием. Для экстрагирования ткань листьев размельчают в ступке с пестиком и жидким N2. К размельченной в порошок ткани листьев затем добавляют 1 мл экстракционного буфера (100 мМ Трис Cl с pH 7,4, 1 мМ ЭДТК, 20% глицерина, 35 мМ KCl, 1 мМ бензамидин•HCl, 5 мМ аскорбата Na, 5 мМ дитиотреитола и 1 мг/мл бычьего сывороточного альбумина, 4oC) и образец растирают еще 1 мин. Полученную смесь 5 мин центрифугируют (высокоскоростная центрифуга Эппендорфа) и обработкой надосадочной жидкости насыщенным раствором сульфата аммония достигают 70%-ного конечного насыщения (2,33 мл насыщенного раствора/мл экстракта). Осадившийся белок отделяют центрифугированием в вышеуказанной центрифуге и дебрис вновь суспендируют в 0,4 мл экстракционного буфера. После повторного центрифугирования для удаления веществ в виде частиц образец обессоливают на Сефадексе G50, загруженном в шприц на 1 мл и уравновешанном экстракционным буфером по методике Penefsky (1976). Обессоленные растительные экстракты хранят на льду и концентрацию белка определяют методом Bradford (1976). Реакции глифозат-оксидоредуктазы проводят в дублях 60 мин при 30oC в смеси для анализа, состоящей из 0,1 MOPS/0,01 трицинового буфера (pH 7,4), содержащих 10 мМ MgCl2, 0,01 мМ флавинадениндинуулеотида (ФАД, Сигма) и 1 мМ убихинона Qo (Сигма). Растительные экстракты (75 мкл) прединкубируют 2 мин в смеси для анализа и реакцию инициируют добавлением в качестве субстрата иминодиуксусиой кислоты (ИДУ, 20 мкл) до конечной концентрации 50 мМ (полный объем анализируемой смеси 0,2 мл). Реакционную смесь нейтрализуют и дериватизируют (см. ниже). Кроме того, проведены контрольные реакции без ИДУ и без растительного экстракта. Детектирование глиоксилата осуществляют применением 2,4-динитрофенилгидразина (2,4-ДНФГ) для дериватизации и высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенными фазами по модифицированной методике Qureshi и др. (1982). Реакционную смесь глифозат-оксидоредуктазы (0,2 мл) нейтрализуют 0,25 мл реактива ДНФГ (0,5 мг/мл ДНФГ /Олдрич/ в 0,5 M HCl) и оставляют на 5 мин при 25oC для дириватизации. Затем образцы экстрагируют этилацетатом (2х0,3 мл) и объединенные этилацетатные экстракты экстрагируют 10% Na2CO3 (0,3 мл). Фазу Na2CO3 промывают один раз этилацетатом (0,2 мл) и инъектируют (100 мкл) в Бекман Ультрасфер C18 IP ВЭЖХ колонку (5 мкм, 4,6 мм х 25 см) использованием ЛКБ GTi бинарной ВЭЖХ системы с Вотерс 990 УФ/ВИЗ ВЭЖХ детектором фотодиодной конструкции и Вотерс WISP ВЭЖХ автовпрыскивателем. Изократной подвижной фазой служит смесь метанол-вода-уксусная кислота (60: 38,5: 1,5) с 5 мМ тетрабутиламмонийфосфата (Пирс). ДНФГ-глиоксилатный пик (время удерживания = 6,7 мин) детектируется при 365 нм и сравнивается со стандартом глиоксилата (Сигма, 20 мкМ в 0,2 мл), дериватизированным таким же образом (табл. VII).
Пpимеp 2
С помощью "изогенных" векторов глифозат-оксидоредуктазы pMON17073 (обработанная глифозат-оксидоредуктаза) (ПОСЛЕД. N 6), pMON17065 (синтетическая глифозат-оксидоредуктаза) (ПОСЛЕД. N 8) и pMON17066 (ХТП1-синтетическая глифозат-оксидоредуктаза) создан ряд трансформированных линий табака. Вестерн-анализом (см. таблицу VIII) для некоторых из таких линий обнаружено, что растения с обработанной глифозат-оксидоредуктазой экспрессируют вплоть до ~0,5 нг глифозат-оксидоредуктазы на 50 мкг растительного белка, растения с синтетической глифозат-оксидоредуктазой - на уровне ~0,5-2 нг на 50 мкг и растения с ХТП1-синтетической глифозат-оксидоредуктазой - на уровне ~2-20 нг на 50 мкг.
Ряд первичных трансформантов R0 линий, экспрессирующих обработанную или синтетическую глифозат-оксидоредуктазу, или ХТП1-синтетическую глифозат-оксидоредуктазу опрыскивают глифозатом в дозировке 0,4 фунт/ар (0,448 кг/га) и оценивают по прежней шкале (см. табл. IX).
Линия синтетической глифозат-оксидоредуктазы характеризуется реакцией, аналогичной той, которая отмечена для R1 растений с модифицированной глифозат-оксидоредуктазой, т. е. немедленным действием глифозата, исчезающим со временем в результате метаболизма гербицида под действием глифозат-оксидоредуктазы до производных: АМФК и глиоксилата. Поскольку мишень для глифозата (ЕПШФ-синтетаза) расположена в хлоропласте, активность глифозат-оксидоредуктазы должна снижать уровень глифозата в этой органеле удалением гербицида прежде, чем тот достигнет хлоропласта. Растения с ХТП1-синтетической глифозат-оксидоредуктазой характеризуются более высокой толерантностью к глифозату, проявляющейся в том, что на эти растения глифозат не оказывает сильного, если вообще оказывает, немедленного воздействия при указанной дозировке. В целом, обработанные толерантные растения также отличаются нормальным развитием, цветением и плодоношением.
Растения с ХТП1-синтетической глифозат-оксидоредуктазой характеризуются заметно более высоким уровнем экспрессии глифозат-оксидоредуктазы по сравнению с другими конструктами глифозат-оксидоредуктазы. Повышенный уровень глифозат-оксидоредуктазы может быть связан с усиленной трансляцией продукта слияния или с секвестированием глифозат-оксидоредуктазы в хлоропласте, что увеличивает период полураспада белка. Более высокий уровень глифозат-оксидоредуктазы и/или ее расположение в хлоропласте может привести к более высокому уровню толерантности за счет быстрой детоксификации глифозата в хлоропласте. Присутствие глифозат-оксидоредуктазы в хлоропласте подтверждено. Пять листьев от каждого из четырех растений (NN 22844, 22854, 22886, 22887), для которых доказана Вестерн-положительность, гомогенизируют в смесителе Варинга в 0,9 л GR+буфер (Bartlet и др., 1982) в течение 3 х 3 с при высокой скорости. Гомогенизат фильтруют через 4 слоя Мираклоз и центрифугируют при 6000 об. /мин в GS-3 роторе. Дебрис вновь суспендируют в GR+буфере (всего 4 мл) и помещают в верхнюю часть 40/80% ступенчатого градиента Перколя и вращают 10 мин при 9500 об./мин. Интактные хлоропласты (нижняя полоса) промывают один раз GR-буфером (Bartlet и др., 1982) и центрифугируют (вплоть до 6000 об. /мин без торможения). Дебрис снова суспендируют в 300 мкл 50 мМ Гепес (pH 7,7), 330 мМ сорбита и лизируют на льду с помощью ультразвука (небольшая проба, 30%-3 отстоя в микронаконечнике х 10 с). Дебрис осаждают в надосадочную жидкость пропускают через колонку с Сефадексом G50 в 50 мМ Гепеса (pH 7,5). Концентрация растворимого белка 2,4 нг/мл. Анализ фермента проводят вышеописанными методами с применением в качестве субстратов 50 мМ ИДУ и 50 мМ глифозата (30 мин на анализ), но без добавления 1 мМ убихинона (табл. IX').
Пример 3
Ряд трансформированных линий брюквы создают с векторами pMON17138 (ХТП1-синтетическая глифозат-оксидоредуктаза) и pMON17164 (ХТП2-синтетическая глифозат-оксидоредуктаза) следующим образом.
Растительный материал
Рассаду Brassica napus, сорт Вистар высаживают в 2-х-дюймовые (~5 см) горшочки, содержащие Метро Микс 350. Рассаду выращивают в ростовой камере при 24oC, фотопериоде 16/8 ч, интенсивности света 400 yEм-2 с-1 (HID лампы). Удобряют удобрением марки Питерс 20-10-20 Специальное, общего назначения. Через 2,5 недели рассаду пересаживают в 6-дюймовые (~15 см) горшки и выращивают в ростовой камере при температуре 15/10oC (день/ночь), фотопериоде 16/8 ч, интенсивности света 800 yEм-2 с-1 HID лампы. Удобряют удобрениями марки Питерс 15-30-15 Специальное Хай-Фос.
Трансформация/селекция/регенерация
Четыре концевых междоузлий от растений непосредственно перед стрелкованием или в процессе стрелкования, но перед цветением удаляют и стерилизуют с поверхности в 70%-ном (об./об.) этаноле в течение 1 мин, 2%-ном (мас./об.) гипохлорите натрия в течение 20 мин и ополаскивают 3 раза стерильной деионизированной водой. Стебли с листьями могут быть охлаждены в увлажненных пластиковых мешках в течение до 72 ч перед стерилизацией. Шесть-семь сегментов стебля нарезают на кружочки в 5 мм с помощью Редко растительного резака 200 с установлением ориентации базального конца.
Agrobacterium выращивают примерно сутки в ротаторе при 24oC в 2 мл бульона Луриа, содержащего 50 мг/л канамицина, 24 мг/л хлорамфеникола и 100 мг/л спектиномицина. Готовят 1:10 разбавление в MS (Murashige и Skoog) среде, что дает примерно 9•108 клеток на мл. Это подтверждено показателем оптической плотности при 660 mu. Кружки стебля (экспланты) инокулируют 1 мл Agrobacterium, избыток с эксплантов отсасывают.
Экспланты помещают базальной стороной вниз на пластинки Петри, содержащие 1/10X стандартных MS солей, B5 витамины, 3% сахарозы, 0,8% агара (pH 5,7), 1 мг/л 6-бензиладенина (БА). На пластинки наносят слой 1,5 мл среды, содержащей MS соли, B5 витамины, 3% сахарозы (pH 5,7), 4 мг/л п-хлорфеноксиуксусной кислоты, 0,005 мг/л кинетина и покрывают стерильной фильтровальной бумагой.
После 2-3 дней сокультивирования экспланты переносят в глубокие плоские чашки Пэтри, содержащие MS соли, B5 витамины, 3% сахарозы, 0,8% агара (pH 5,7), 1 мг/л БА, 500 мг/л карбенициллина, 50 мг/л цефотаксима, 200 мг/л канамицина или 175 мг/л гентамицина для селекции. На каждую пластинку помещают семь эксплантов. Спустя 3 недели экспланты переносят в свежую среду по 5 эксплантов на пластинку. Экспланты культивируют в ростовой комнате при 25oC и непрерывном освещении (холодный белый свет).
Анализ экспрессии
Через 3 недели с эксплантов срезают побеги. Для подтверждения модификации R0 побегов проводят анализ на повторное каллюсообразование в листьях. Три крохотных кусочка ткани листьев помещают на среду повторного каллюсообразования, содержащую MS соли, B5 витамины, 3% сахарозы, 0,8% агара (pH 5,7), 0,5 мг/л нафталинуксусной кислоты (НУК), 500 мг/мл карбенициллина, 50 мг/л цефтотксима и 200 мг/л канамицина или гентамицина, или глифозата. Анализируемые листья инкубируют в ростовой комнате в тех же условиях, что культура эксплантов. Спустя 3 недели проводят оценку анализа на повторное каллюсообразование с позицией толерантности к гербициду (каллюс или ткань зеленого листа) или чувствительности (отбеливание).
Трансплантация
В момент обрезки стебли побегов погружают в РутонR и помещают в 2-х дюймовые (~ 5 см) горшочки, содержащие Метро-Микс 350, и горшочки помещают в замкнутое увлажненное окружение. Затем горшочки помещают в ростовую камеру при 24oC, фтопериоде 16/8 ч, 480 yEм-1 с-2 (HID лампы на 3 недели периода закаливания).
Семена, собранные с R0 растений, являются R1 семенами, из которых выращивают R1 растения. Для выявления толерантности R0 растения к глифозату исследуют его потомство. Поскольку считается, что R0 растение гемизиготно у каждого местоположения вставки, "свое" приводит к максимуму генотипной сегрегации в R1. Поскольку каждая вставка действует, как доминантный аллель, в отсутствие связи, и приняв, что только одна гемизиготная вставка необходима для экспрессии толерантности, одна вставка будет сегрегировать в отношении 3:1, две вставки - 15:1, три вставки - 63:1 и т/д. Поэтому необходимо вырастить сравнительно мало растений для обнаружения по меньшей мере одного фенотипа устойчивости.
Семена R0 растения собирают, обмолачивают и сушат перед высаживанием в испытании на опрыскивание глифозатом. Применяют самые различные технологии для выращивания R1 растений для последующего испытания с опрыскиванием. Испытания проводят как в теплицах, так и в ростовых камерах. Применяют две системы высаживания: горшочки ~10 см или подносы для высаживания с 32 или 36 ячейками. Применяемой для высаживания почвой служит либо Метро 350 плюс три типа удобрений постепенного действия, либо Метро 350 для рассады. Поливка либо поверхностная в теплицах, либо применяют субирригацию в ростовых камерах. Удобрения вносят вместе с водой для поливки. Поддерживают приемлемый для брюквы температурный режим. Устанавливают фотопериод в 16 ч. В начале цветения растения пересаживают в горшочки ~15 см для образования семян.
Опрыскиваемая "партия" состоит из нескольких групп R1 потомства, которые опрыскивают в один и тот же день. Некоторые партии могут кроме того включать потомство отличных от R1 растений. Каждая партия включает опрысканные и неопрысканные нетрансгенные генотипы, представляющие в конкретной партии генотипы, которые предположительно были трансформированы. Кроме того в партию включают один или несколько несегрегированных трансформированных генотипов, для которых ранее выявлена некоторая устойчивость.
Двадцать шесть растений от каждого отдельного R0 потомства не опрыскивают и используют в качестве контроля для сравнения и оценки толерантности к глифозату, а также для оценки любых отклонений, не связанных с глифозатом. Когда другие растения достигают стадии 2-4 листьев (обычно 10-20 дней после посева), наносят глифозат в дозировке 0,28-1,12 кг/га в зависимости от целей исследования. Лабораторный трековый опрыскиватель калибруют для поступления дозировки, эквивалентной полевым условиям.
Для оценки вегетативной устойчивости опрысканных растений применяют шкалу в 0-10 баллов. Шкала соотнесена с неопрысканными растениями того же R0 семейства. 0 означает гибель растения, а 10 означает отсутствие видимых отличий от неопрысканного растения. Более высокое число в интервале 0-10 представляет прогрессивно меньшие повреждения по сравнению с неопрысканным растением. Оценку растений проводят на 7, 14 и 28 день после обработки (ДПО) или до стрелкования, и линии дается средняя оценка для опрысканных растений в пределах R0 растительного семейства.
Используют шесть цифровых показателей для качественной оценки степени репродуктивного урона от глифозата:
0 - отсутствие развития цветочных почек,
2 - цветочные почки присутствуют, но отпадают до раскрытия,
4 - цветки раскрываются, но пыльники отсутствуют или пыльники не способны экструдировать за лепестки,
6 - стерильные пыльники,
8 - частично стерильные пыльники,
10 - полностью плодоносные цветы.
Растения оценивают с помощью этой шкалы в момент или сразу же после начала цветения в зависимости от скорости развития цветочных структур.
В нижеследующих таблицах X и XI сведены вегетативные и репродуктивные оценки растений брюквы, трансформированных pMON17138 (опрыскивание в дозировке 0,56 кг/га) и pMON17164 (опрыскивание в дозировке 0,84 кг/га) соответственно. Нижепредставленные результаты иллюстрируют толерантность к глифозату, придаваемые растениям брюквы в результате экспрессии в растениях гена глифозат-оксидоредуктазы.
Пример 4
Ген глифозат-оксидоредуктазы кроме того введен и экспрессирован в сое с приданием этому растению толерантности к глифозату. Ген слияния ХТП2-синтетическая глифозат-оксидоредуктаза (см. выше) вводят в сою под контролем FMV-промотора и с NOS3'-последовательностями в pMON17159, карта которой представлена на фиг. 10. Этот вектор состоит из следующих элементов помимо генных последовательностей глифозат-оксидоредуктазы: pUC - источник репликации, NPT11 - бактериальный ген селекционного маркера (канамиция) и гена бета-глюкоронидазы (GUS : Jefferson, и др., 1986) под контролем E35S-промотора с E9 3'-последовательностями. С последним геном дается поддающийся оценке маркер, облегчающий идентификацию трансформированного растительного материала.
Растения сои трансформируют pMON17159 методом микрофокусированной инъекции при использовании технологии с впрыскивателем частиц по методике Christou и др. (1988). Семена, собранные с R0 растений, являются R1 семенами, из которых вырастают R1 растения. Для выявления толерантности R0 растений к глифозату исследуют потомство этого растения. Поскольку предполагается, что R0 растение гемизиготно у каждого местоположения вставки, "свое" приводит к максимуму генотипной сегрегации в R1. Поскольку каждая вставка действует в отсутствие связи, как доминантный аллель, и полагая, что для экспрессирования толерантности необходима только одна гемизиготная вставка, которая будет сегрегировать в отношении 3:1, две вставки - 15:1, три вставки - 63:1 и т.д. Таким образом, для выявления по меньшей мере одного фенотипа устойчивости необходимо вырастить сравнительно небольшое число R1 растений.
Семена R0 растения сои собирают и сушат перед высевом для испытаний с опрыскиванием глифозатом. Семена высевают в 4-х дюймовые (~5 см) квадратные горшочки, содержащие Метро 350. Двадцати ростков от каждого R0 растения считают достаточными для испытания. Растения выдерживают и выращивают в тепличных условиях. Устанавливают фотопериод в 12,5-14 ч и температуру 30oC днем и 24oC ночью. По мере необходимости вносят водорастворимые удобрения марки Питерс Пит Лайт.
"Партия" для опрыскивания состоит из нескольких групп R1 потомства, которые все опрыскивают в один и тот же день. Некоторые партии могут также включать потомство отличных от R1 растений. Каждая партия кроме того включает опрысканные и неопрысканные нетрансгенные генотипы, представляющие в конкретной партии генотипы, предположительно трансформированные. В партию включены также один или несколько несегрегированных генотипов, для которых ранее выявлена некоторая устойчивость.
Одно-два растения из каждого отдельного R0 потомства не опрыскивают и используют в качестве контроля для сравнения и оценки толерантности к глифозату, а также для оценки любых отклонений, не связанных с глифозатом. Когда другие растения достигают первой стадии трех листьев (обычно 2-3 недели после высева), глифозат наносят в дозировке, эквивалентной 128 унций/ар (8,895 кг/га) РаундапR. Лабораторный трековый опрыскиватель калибруют для опрыскивания в дозировке, эквивалентной указанным условиям.
Применяют вегетативную шкалу оценок в 0-10 баллов. Оценка соотносится с неопрысканным потомством того же R0 растения. 0 означает гибель растения. 10 означает отсутствие видимой разницы с неопрысканным растением. Более высокое числовое значение в интервале 0-10 представляет прогрессивно меньшее повреждение по сравнению с неопрысканным растением. Оценку растений проводят на 7, 14 и 28 день после обработки (ДПО) (см. табл. XII).
Пример 5
Ген глифозат-оксидоредуктазы кроме того введен в клетки черной мексиканской сладкой (ЧМС) кукурузы с экспрессией белка, обнаруживаемого в каллюсе.
Для введения гена глифозат-оксидоредуктазы в клетки кукурузы применяют плазмиду pMON19632. Скелет такой плазмиды конструируют вставкой 0,6 к.о. 35S-РНК-промотора (E35S) мозаичного вируса цветной капусты (CaMV), содержащего удвоение области от -90 до -300 (Kay и др., 1987), 0,58 к.о. оснований фрагмента, содержащего первый интрон гена спиртовой гидрогеназы кукурузы (Callis и др., 1987) и 3'-терминационные области из гена нопалин-синтетазы (NOS) (Fraley и др., 1983) в pUC119 (Yanisch-Perron и др., 1985). pMON19632 образуют вставкой 1,7 к.о. BglII/EcoRI фрагмента из pNON17064, содержащего Arabidopsis SSU ХТП, слитый с кодирующей последовательностью синтетической глифозат-оксидоредуктазы (ПОСЛЕД. N 8).
Плазмиду pMON19632 вводят в клетки ЧМС кукурузы совместной бомбардировкой с ЕС9 - плазмидой, содержащей сульфонилмочевиноустойчивую форму гена ацетолактат-синтетазы кукурузы. Каждой плазмидой (2,5 мкг) покрывают поверхность частиц вольфрама и вводят с помощью PDS - 1000 инъектора в ЧМС клетки в Iog-фазе, по существу, по методике Klein и др. (1989). Трансформанты отбирают на М среде, содержащей 20 ч/блн хлорсульфурона. После первоначальной селекции на сульфурон каллюсы анализируют вестерн-блотированием глифозат-оксидоредуктазы.
Каллюс ЧМС (3 г влажной массы) сушат на фильтровальной бумаге (Ватман N 1) под вакуумом, перевешивают, после чего добавляют экстракционный буфер (500 мкл/г сухой массы; 100 мМ Трис, 1 мМ ЭДТК, 10% глицерина). Ткань гомогенизируют с помощью подвесной мешалки Вийтона в течение 30 с с интервалами в 2,8 с. После центрифугирования (3 мин, микроцентрифуга Эппендорфа) надосадочную жидкость удаляют и белок определяют количественно (анализ белка набором БиоРад). Образцы (50 мкг/ячейку) наносят на НДС-ПАГЭ гель (JuIe 3-17%) вместе со стандартом глифозат-оксидоредуктазы (10 нг), подвергают электрофорезу и переносят на нитроцеллюлозу аналогично ранее описанному методу (Padgette 1987). Нитроцеллюлозный блот зондируют козьим анти-глифозат-оксидоредуктазным ИгС и проявляют 125I-белком G. Радиоактивный блот делают видимым с помощью авторадиографии. Результаты получают количественно денситометрией
на ЛКБ УльтраСкан XI лазерном денситометре (см. таблицу XIII).
Данные таблицы XIII показывают, что глифозат-оксидоредуктаза может экспрессироваться и детектироваться в однодольных растениях, таких как кукуруза.
Пример 6
Ген глифозат-оксидоредуктазы может быть использован в качестве селекционного маркера трансформации растений непосредственно на среде, содержащий глифозат. Способность отбирать и идентифицировать трансформированный растительный материал зависит в большинстве случаев от применения доминантного селекционного маркерного гена, способствующего ткани в присутствии обычно ингибирующего вещества. Гены устойчивости к антибиотику и толерантности к гербициду использовались почти исключительно в качестве таких доминантных селекционных маркерных генов в присутствии соответствующего антибиотика или гербицида. Схема селекции nptIII/канамицин, вероятно, используется наиболее часто. Показано, что глифозат-оксидоредуктаза также применима и, возможно, с большим успехом в схеме селекционный маркер/селекция для продуцирования и идентификации трансформированных растений.
Вектором трансформации растений, который может быть использован в такой схеме, является pMON17226 (фиг. 11). Данная плазмида сходна со многими другими уже описанными плазмидами и, по-существу, состоит из ранее раскрытой бактериальной репликоновой системы, позволяющей этой плазмиде реплицироваться в E.coli, а также быть введенной и реплицироваться в Agrobacterium, бактериального селекционного маркерного гена (Spc/Str), а между правой границей Т-ДНК и левой границей расположен синтетический ген глифозат-оксидоредуктазы в FMS-промотор-E9 3'-кассете. Данная плазмида обладает также единственными сайтами для ряда ферментов рестрикции, расположенными в пределах границ и вне экспрессионной кассеты. Это делает возможным легко добавлять другие гены и генетические элементы к вектору для введения в растения.
Методика прямой селекции трансформированных растений на глифозате приведена для табака. Экспланты готовят для предкультивирования по стандартной методике, приведенной в примере 1. Поверхность листьев растения табака в возрасте 1 месяца стерилизуют (15 минут в 10%-ном хлороксе + поверхностно-активное вещество; промывка 3X дH2O); экспланты нарезают квадратиками размером 0,5 х 0,5 см с удалением краев листьев, средней жилки листа, кончика и черешка для получения однородного тканевого типа; экспланты помещают внутренней поверхностью вверх одним слоем на MS104 пластинки + 2 мл 4C005K среда для увлажнения поверхности; предкультивируют 1-2 дня. Экспланты инокулируют с помощью примерно суточной культурой Agrobacterium, содержащей плазмиду трансформации растений, в которой устанавливают титр 1,2 • 109 бактерий/мл с 4C005K средой. Экспланты помещают в центрифужную пробирку, добавляют суспензию Agrobacterium и смесь бактерий и эксплантов интенсивно вращают 25 секунд при максимальных оборотах с гарантированием равномерного проникновения бактерий. Бактерии сливают, а экспланты помещают между слоями сухой стерильной фильтровальной бумаги для удаления избытка бактерий. Высушенные экспланты помещают внутренней поверхностью вверх на MS104 пластинки + 2 мл 4C005K среды + кружок фильтра. Сокультивируют 2-3 дня. Экспланты переносят на MS104 + 1000 мг/л карбенициллина + 100 мг/л цефотаксима на 3 дня (фаза выдерживания). Затем экспланты переносят M104 + 0,05 мМ глифозата + 1000 мг/л карбенициллина + 100 мг/л цефотаксима для фазы селекции. В течение 3-5 дней образуются корни и в этот момент кусочки листа могут быть отобраны из укоренившихся пластинок для подтверждения толерантности к глифозату и того, что материал трансформирован.
Присутствие белка глифозат-оксидоредуктаза в этих трансформированных тканях подтверждено иммуноблот-анализом кружков из листьев. Ниже представлены данные одного из экспериментов с pMON17226. Побеги (25 штук), образовавшиеся на глифозате из 100 эксплантов, инокулируют Agrobacterium ABI/ pMON17226; 15 из них оказались положительными на повторное каллюсообразование на глифозате и 19 из них оказались положительными для белка глифозат-оксидоредуктазы при детектировании иммуноблотированием. Полученные данные указывают на степень трансформации 15-19 на 100 эксплантов, что характеризует такую процедуру трансформации растений, как высокоэффективную и времясберегающую. Аналогичная частотность трансформации достигнута и с производным pMON17226 (pMON17241), содержащим ген для глифозат-оксидоредуктазы V. 247 (ПОСЛЕД. N 17). Показано, что ген глифозат-оксидоредуктазы позволяет также проводить прямую селекцию трансформантов в других видах растений, в том числе: Arabidopsis, картофеле и сахарной свекле.
Из вышеизложенного можно видеть, что настоящее изобретение отвечает поставленным целям и обладает преимуществами, которые очевидны и неотделимы от изобретения.
Необходимо указать, что определенные признаки и подкомбинации могут быть использованы без ссылок на другие признаки и подкомбинации. Это определяется и охватывается объемом формулы изобретения.
Поскольку возможно осуществление многих воплощений изобретения без отхода от его объема, необходимо указать, что весь изложенный материал или показанный в диаграммах следует рассматривать, как иллюстративный, но не как ограничительный.
Список литературы
Ammirato, P. V. , et al. Handbook of Plant Cell Culture - Crop Species. Macmillan Publ. Co. (1984).
Avila, L.Z., Loo, S.H. and Frost, J.W. (1987) Chemical and mutagenic analysis of aminomethylphosphonate biodegradation. J. Amer. Chem. Soc. 109: 6758-6764.
Balthazor, T.M. and Hallas, L.E. (1986) Glyphosate-degrading microorganisms from industrial activated sludge. Appl. Environ. Microbiol. 51:432-434.
Bartlett, S. G. , Grossman, A.R., and Chua, N.H. (1982) in Methods in Chloroplast Molecular Biology, pp. 1081-1091. M. Edelman, R.B., Hallick, and Chua, N.H., eds.
Bevan, M. (1984) Nucleic Acids Res 12 (22):8711-8721.
Birnboim, H.C. and Doly, J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl. Acids. Res. 7:1513-1525.
Boyer, H.W. and Rolland-Dussoix, D. (1969) A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459.
Bradford, M. Anal. Biochem. 72, 248 (1976).
Callis, J. , Fromm, M. , and Walbot, V. (1987) Introns increase gene expression in cultured maize cells. Genes and Dev. 1:1183-1200.
Christou, P. , D.E. McCabe, and W.F. Swain (1988) Stable transformation of Soybean Callus by DNA-Coated Gold Particles. Plant Physiol. 87:671-674.
Cook, A.M., Daughton, C.G. and Alexander, M. (1978) Phosphonate utilization by bacteria. J. Bacteriol. 133:85-90.
Coruzzi, G. , Broglie, R., Edwards, C., and Chua, N.H. (1984). Tissue-specific and light-regulated expression of a pea nuclear gene encoding the small subunit of ribulose-1,5-bisphosphate carboxylase. EMBO J 3, 1671-1679.
Coupland, D, (1985) Metabolism of glyphosate in plants, in The Herbicide Glyphosate. eds. E. Grossbard and D. Atkinson. Butterworths. pp. 25-34.
Daughton, C. G., Cook, A.M. and Alexander, M. (1979a) Bacterial conversion of alkylphosphonates to natural products via carbon-phosphorus bond cleavage. J. Agric. Food Chem. 27: 1375-1382.
Daughton, C. G., Cook, A.M. and Alexander, M. (1979b) Biodegradation of phosphonate toxicants yields methane or ethane on cleavage of the C-P bond. FEMS Microbiol. Lett. 5:91-93.
Daughton, C.G., Cook, A.M. and Alexander, M. (1979c) Phosphate and soil binding: factors limiting bacterial degradation of ionic phosphorous-containing pesticide metabolites. Appl. Environ. Microbiol. 37:605-609.
della-Cioppa, G., Bauer, S.C., Klein, B.K., Shah, D.M., Fraley, R.T. and Kishore G. K. (1986) Translocation of the precursor of 5-enolpyruvylshikimate-3-phosphate synthase into chloroplasts of higher plants in vitro. Proc, Natl. Acad Sci. USA 83:6873-6877.
della-Cioppa, G., Bauer, S.C., Taylor, M.T., Rochester, D.E., Klein, B. K. , Shah, D.M., Fraley, R.T. and Kishore G.M. (1987) Targeting a herbicide-resistant enzyme from Escherichia coli to chloroplasts of higher plants. Bio/Technology 5:579-584.
Ditta, G. , Stanfield, S., Corbin, D., and Helinski, D.R. (1980). Broad host range DNA cloning system for Gram-Negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc Natl Acad Sci USA 77, 7347-7351.
Erlich, H.A. (1989) Ed. PCR Technology - Principles and Applications for DNA Amplification. Stockton Press, New York.
Fling, M. E., Kopf, J., and Richards, C. (1985). Nucleotide sequence of the transposon Tn7 gene encoding an aminoglycoside-modifying enzyme, 3''(9)-O-nucleotidyltransferase. Nucleic Acids Research 13 no.19, 7095-7106.
Fraley, R. T. , Rogers, S.G., Horsch, R.B., Sanders, P.R., Flick, J.S., Adams, S.P., Bittner, M.L., Brand, L.A., Fink, C.L., Fry, J.S., Galluppi, G. R. , Goldberg, S.B., Hoffmann, N.L. and Woo, S.C. (1983) Expression of bacterial genes in plant cells. Proc. Natl. Acad. Sci. USA 80:4803-4807.
Fraley, R. T., Rogers, S.G., Horsch, R.B., Eichholtz D.A., Flick, J.S., Fink, C. L. , Hoffmann, N.L. and Sanders, P.R. (1985) The SEV system: a new disarmed Ti plasmid vector system for plant transformation.
Franz, J. E. (1985) Discovery, development and chemistry of glyphosate, in The Herbicide Glyphosate. eds, E. Grossbard and D. Atkinson. Butterworths. pp. 3-17.
Fromm, M. , (1990) UCLA Symposium on Molecular Strategies for Crop Improvement, April 16-22, 1990. Keystone, CO.
Gowda, S., Wu, F.C., and Shepard, R.J. (1989). Identification of promoter sequences for the major RNA transcripts of figwort mosaic and peanut chlorotic streak viruses (caulimovirus group). Journal of Cellular Biochemistry supplement 13D, 301. (Abstract).
Hallas, L. E., Hahn, E.M. and Korndorfer, C. (1988) Characterization of microbial traits associated with glyphosate biodegradation in industrial activated sludge. J. Industrial Microbiol. 3:377-385.
Hayford, M.B., Medford, J.I., Hoffmann, N.L., Rogers, S.G. and Klee, H. J. (1988) Development of a plant transformation selection system based on expression of genes encoding gentamicin acetyltransferases. Plant Physiol. 86:1216-1222.
Heitkamp, M.A., Hallas, L. and Adams, W.J. (1990) Biotreatment of industrial wastewater with immobilized microorganisms - Presented in Session 11, Paper S40, Society for Industrial Microbiology Annual Meeting, Orlando, Florida, July 29 - August 3, 1990.
Herrera-Estrella, L. , et al. (1983) Nature 303:209 Horsch, R.B. and H. Klee. (1986) Proc. Natl. Acad. Sci. U.S.A. 83:4428-32.
Holben, W. E. , Jannson, J.K., Chelm, B.K. and Teidje, J.M. (1988) DNA probe method for the detection of specific microorganisms in the soil bacterial community. Appl. Environ. Microbiol. 54:703-711.
Hohn, B. and Collins J. (1980) A small cosmid for efficient cloning of large DNA fragments. Gene 11:291-298.
Jacob, G. S. , Schaefer, J., Stejskal, E.O. and McKay, R.A. (1985) Solid-state NMR determination of glyphosate metabolism in a Pseudomonas sp. J. Biol. Chem. 260:5899-5905.
Jacob, G. S. , Garbow, J., Hallas, L.E., Kishore, G.M. and Schaefer, J. (1988) Metabolism of glyphosate in Pseudomonas sp. strain LBr. Appl. Environ. Microbiol. 54:2953-2958.
Jefferson, R.A., T.A.. Kavanaugh, M.W. Bevan (1987) GUS fusions: β -glucuronidase as a sensitive and versatile gene fusion marker in higher plants. The EMBO Journal vol. 6, N 13. pp 3901-3907.
Kay, R., Chan, A., Daly, M. and McPherson, J. (1987) Duplication of the CaMV 35S promoter sequences creates a strong enhancer for plant genes. Science 236:1299-1302.
Kishore, G. M. and Jacob G.S. (1987) Degradation of glyphosate by Pseudomonas sp. PG2928 via a sarcosine intermediate. J. Biol. Chem. 262:12164-12168.
Klee, H.J., et al. (1985) Bio/Technology 3:637-42.
Klee, H. J., Muskopf, Y.M. and Gasser, C.S. (1987) Cloning of an Arabidopsis thaliana gene encoding 5-enolpyruvylshikimate-3-phosphate synthase: sequence analysis and manipulation to obtain glyphosate tolerant plants. Mol. Gen. Genet. 210:437-442.
Klein, T.M., Kornstein, L., Sanford, J.C., and Fromm, M.E. (1989) Genetic transformation of maize cells by particle bombardment Plant Phys. 91: 440-444.
Koncz, C. and Schell, J. (1986) The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimeric genes carried by a novel type of Agrobacterium binary vector. Mol. Gen. Genet 204:383-396.
Lerbs, W. , Stock, M. and Parthier, B. (1990) Physiological aspects of glyphosate degradation in Alcaligenes sp. strain GL. Arch. Microbiol. (1990) 153:146-150.
Liu, C. -M., McLean, P.A., Sookdeo, C.C. and Cannon, F.C. (1991) Degradation of the herbicide glyphosate by members of the family Rhixobiaceae. Appl. Environ. Microbiol. 57:1799-1804.
Maier, L. (1983) Phosphorus Sulfur 14:295.
Malik, J. , Barry, G. and Kishore, G. (1989) The herbicide glyphosate. BioFactors 2:17-25.
Maniatis, T., Fritsch, E.F. and Sambrook, J. (1982) Molecular Cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
Marshall, G. , Kirkwood, R.C. and Martin, D.J. (1987) Pestic. Sci. 18: 65-77.
Mastalerz, P., Wieczorek, Z. and Kochman, M (1965) Utilization of carbon-bound phosphorus by microorganisms. Acta Biochim. Pol. 12:151-156.
Miller, J.H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
Moore, J.K., Braymer, H.D. and Larson, A.D. (1983) Isolation of a Pseudomonas sp. which utilizes the phosphonate herbicide glyphosate. Appl. Environ. Microbiol. 46:316-320.
Morelli, G. , Nagy, F. , Fraley, R.T., Rogers, S.G., and Chua, N.H. (1985). A short conserved sequence is involved in the light-inducibility of a gene encoding ribulose 1,5-bisphosphate carboxylase small subunit of pea. Nature 315, 200-204.
Neidhardt, F. C. , Bloch, P.L. and Smith, D.F. (1974) Culture media for enterobacteria. J. Bacteriol. 119: 736-747.
Nomura, N. S. and Hilton, H.W. (1977) The adsorption and degradation of glyphosate in five Hawaiian sugarcane soils. Weed Res. 17:113-121.
Odell, J. T. , Nagy, F., and Chua, N.H. (1985). Identification of DNA sequences required for activity of the cauliflower mosaic virus 35S promoter. Nature 313, 810-812.
Padgette, S., et al. (1987) Bacterial Expression and Isolation of Petunia hybrida EPSP Synthase. Arch. Biochem. Biophys. 258:564-573.
Penefsky, H.S., Meth. Enzymol. 56, 527-530 (1979).
Pipke, R., Schulz, A. and Amrhein, N. (1987b) Uptake of glyphosate by an Arthrobacter sp. Appl. Environ. Microbiol. 53: 974-978.
Pipke, R. and Amrhein, N. (1988) Degradation of the phosphonate herbicide glyphosate by Arthrobacter atrocyaneus ATCC 13752. Appl. Environ. Microbiol. 54: 1293-1296.
Pipke, R., Amrhein, N., Jacob, G.S. and Kishore, G.M. (1987a) Metabolism of glyphosate by an Arthrobacter sp. GLP-1. Eur. J. Biochem. 165:267.273.
Quinn, J.P., Peden, J.M.M. and Dick, E. (1988) Glyphosate tolerance and utilization by the microflora of soils treated with the herbicide. Appl. Microbiol. Biotechnol. 29: 511-516.
Quinn, J. P. , Peden, J.M.M. and Dick, R.E. (1989) Appl. Microbiol. and Biotechnology 31:283-287.
Qureshi, A.A., Elson, C.E., and Lebeck, L.A., J. Chromotog. 249,333-345 (1982).
Rueppel, M. L. , Brightwell, B.B., Schaefer, J. and Marvel, J.T. (1977) Metabolism and degradation of glyphosate in soil and water. J. Agric. Food Chem. 25: 517-528.
Schowanek, D. and Verstraete, W. (1990) Phosphonate utilization by bacterial cultures and enrichments from environmental samples. Appl. Environ. Microbiol. 56: 895-903.
Shah, D. , Horsch, R. , Klee, H., Kishore, G., Winter, J., Tumer, N., Hironaka, C. , Sanders, P., Gasser, C., Aykent, S., Siegal, N., Rogers, S., and Fraley, R. (1986). Engineering herbicide tolerance in transgenic plants. Science 233,478-481.
Shimamoto, K. et al. (1989) Nature 338:274-276.
Shinabarger, D. L. and Braymer, H.D. (1986) Glyphosate catabolism by Pseudomonas sp. strain PG2982. J. Bacteriol. 168: 702-707.
Stalker, D. M., Thomas, C.M., and Helinski, D.R. (1981). Nucleotide sequence of the region of the origin of replication of the broad host range plasmid RK2. Mol Gen Genet 181:8-12.
Steffan, R. J. and Atlas, R.M. (1988) DNA amplification to enhance detection of genetically engineered bacteria in environmental samples. Appl. Environ. Microbiol. 54: 2185-2191.
Tabor, S. and Richardson, C.C. (1985) A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82:1074-1078.
Talbot, H.W., Johnson, L.M. and Munnecke, D.M. (1984) Glyphosate utilization by Pseudomonas sp. and Alcaligenes sp. isolated from environmental sources. Current Microbiol. 10: 255-260.
Tanaka, J., Kuwano, E. and Eto, M. (1986) Synthesis and pesticidal activities of phosphonate analogs of amino acids. J. Fac.Agr. Kyushu Univ. 30: 209-223.
Timko, M. P., Herdies, L., de Alameida, E., Cashmore, A.R., Leemans, J. and Krebbers, E. (1988) Genetic engineering of nuclear-encoded components of the photosynthetic apparatus of Arabidopsis in The Impact of Chemistry on Biotechnology - A Mutlidisciplinary Discussion. ACS Books, Washington DC pp. 279-295.
Torstensson, L. (1985) Behavior of glyphosate in soils and its degradation, in The Herbicide Glyphosate. eds. E. Grossbard and D.Atkinson. Butterworths. pp. 137-150.
Tsai, Y.-L. and Olson, B.H. (1991) Rapid method for direct isolation of DNA from soil and sediments. Appl. Environ. Microbiol. 57:1070-1074.
Vasil, V., F. Redway and I. Vasil. (1990) Bio/Technology 8:429-434.
Vieira, J. and Messing, J. (1987) Production of single-stranded plasmid DNA. Methods. Enzymol. 153: 3.
Wackett, L.P., Shames, S.L., Venditti, C.P. and Walsh, C.T. (1987a) Bacterial carbon-phosphorus lyase: products, rates, and regulation of phosphonic and phosphinic acid metabolism. J. Bacteriol. 169:710-717.
Wackett, L. P. , Wanner, B.L., Venditti, C.P. and Walsh, C.T. (1987b) Involvement of the phosphate regulon and the psiD locus in the carbon-phosphorus lyase activity of Escherichia coli K-12. J. Bacteriol. 169: 1753-1756.
Weidhase, R. , Albrecht, B., Stock, M., and Weidhase, R.A. (1990) Utilization of glyphosate by Pseudomonas sp. GS. Zentralbl. Mikrobiol. 145:6.
Wong, E.Y., Seetharam, R., Kotts, C.E., Heeren, R.A., Klein, B.K., Braford, S.R., Mathis, K.J., Bishop, B.F., Siegel, N.R., Smith, C.E. and Tacon, W.C. (1988) Expression of excreted insulin-like growth factor-1 in Escherichia coli. Gene 68: 193-203.
Yanisch-Perron, C., Vierra, J. and Messing, J, (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33, 103-119.
Zeleznick, L.D., Meyers, T.C. and Titchener, E.B. (1963) Growth of Escherichia coli on methyl - and ethylphosphonic acids. Biochim. Biophys. Acta. 78: 546-547.



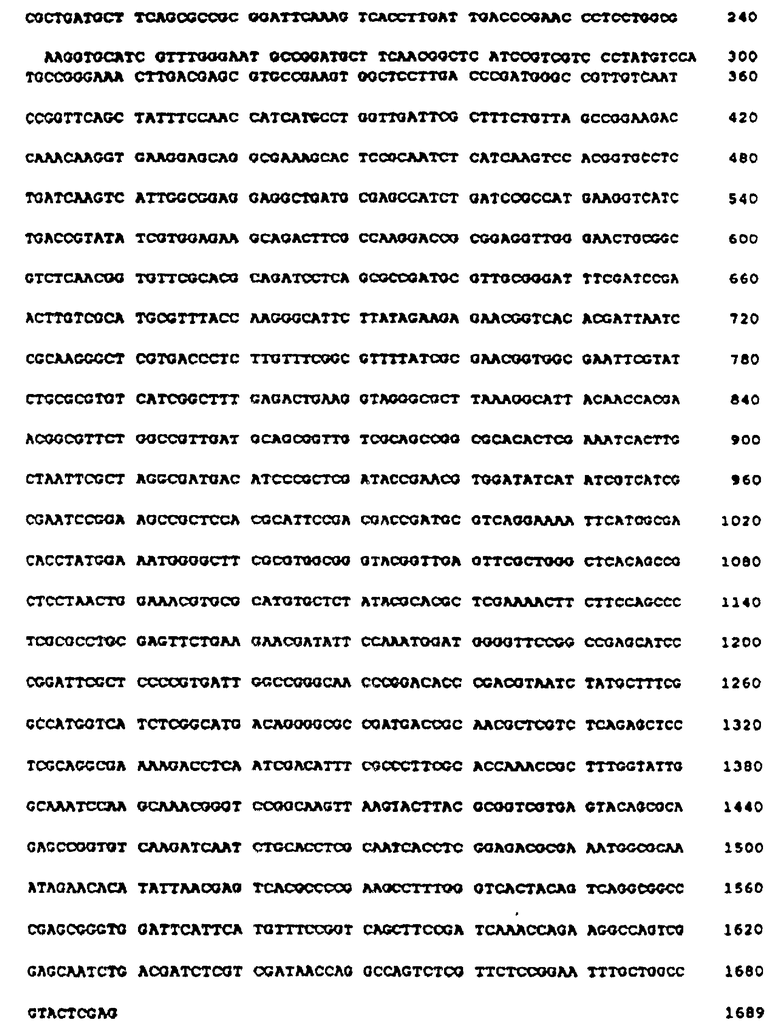




























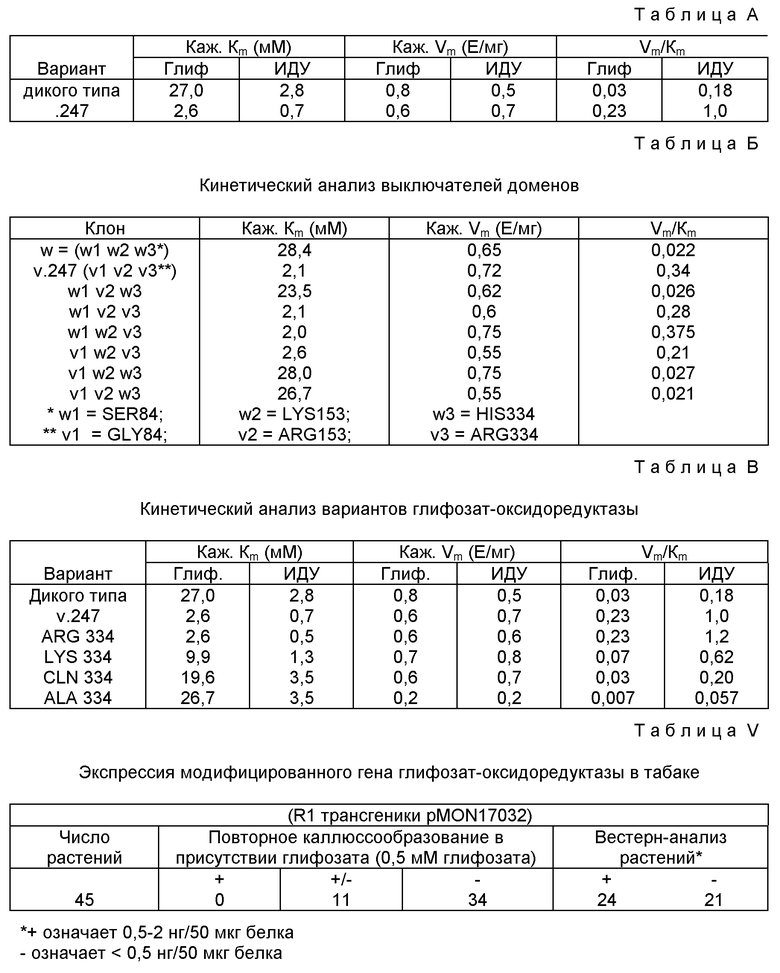
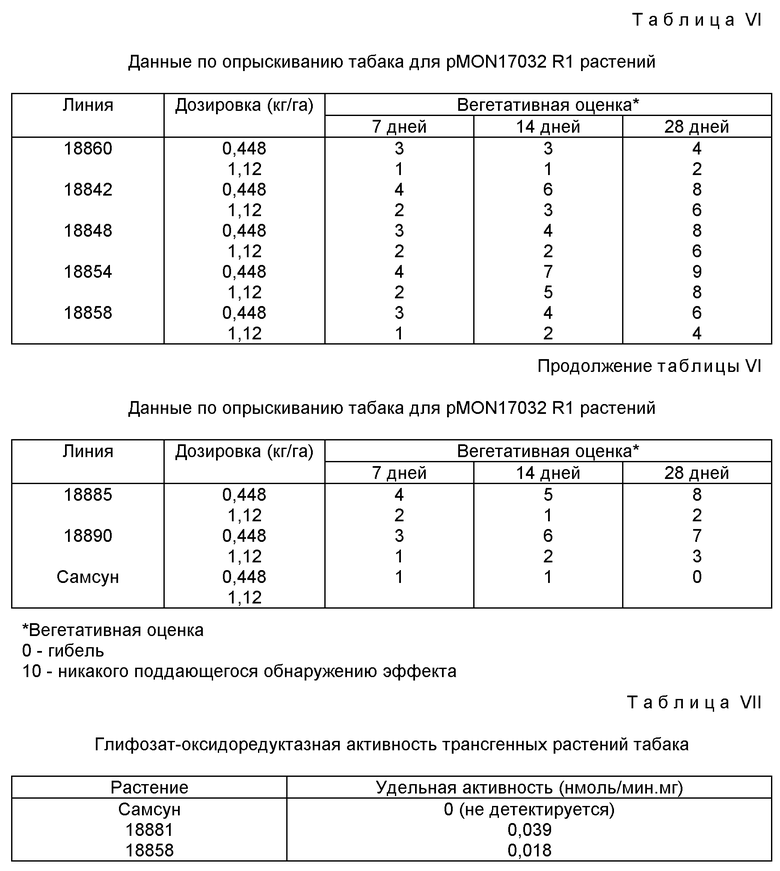
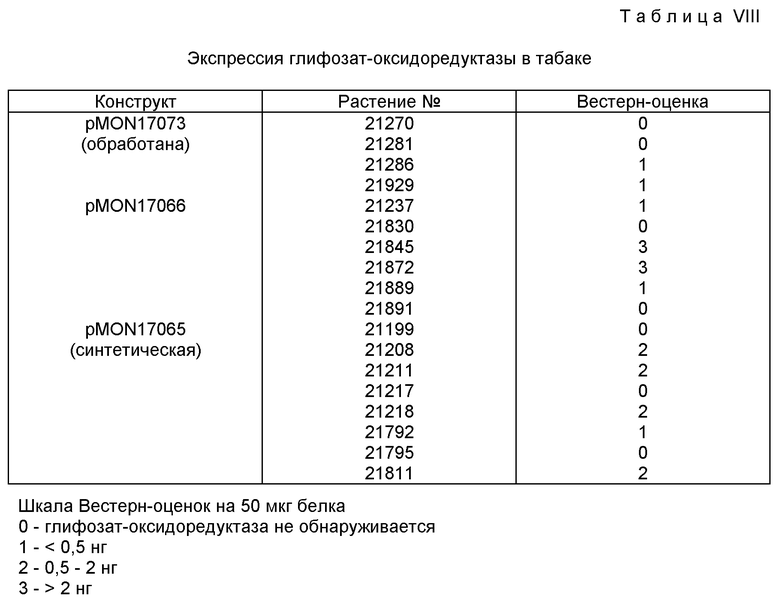
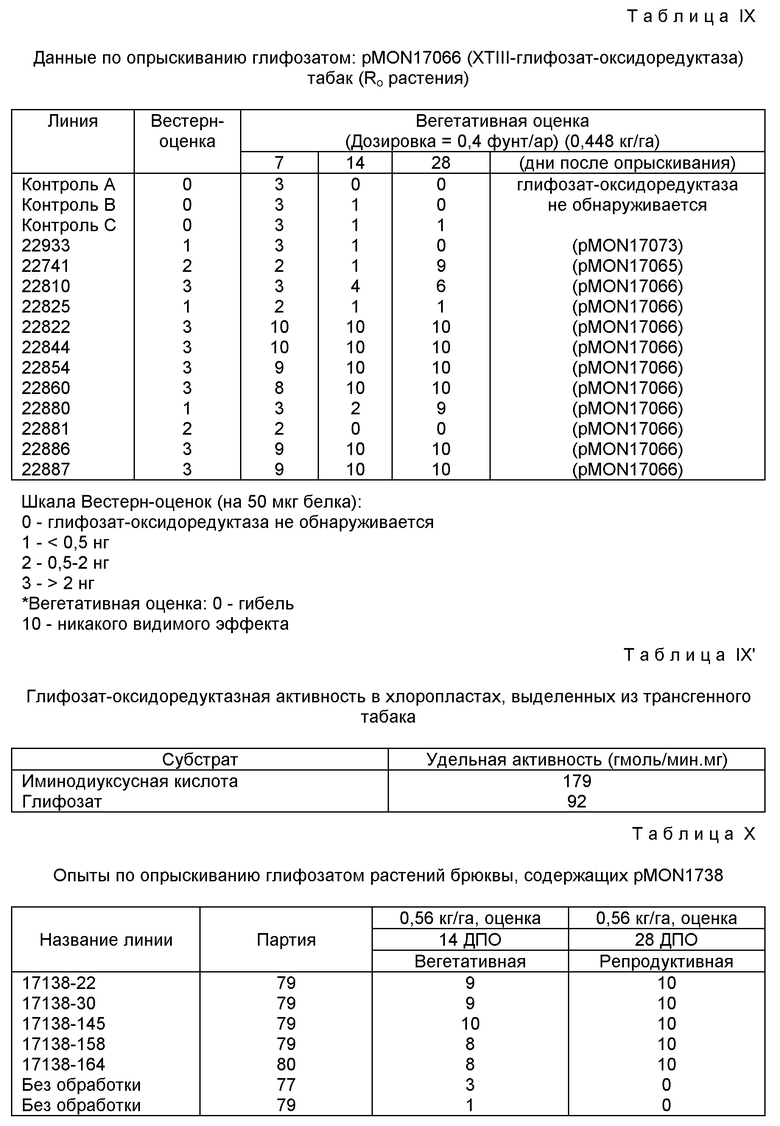
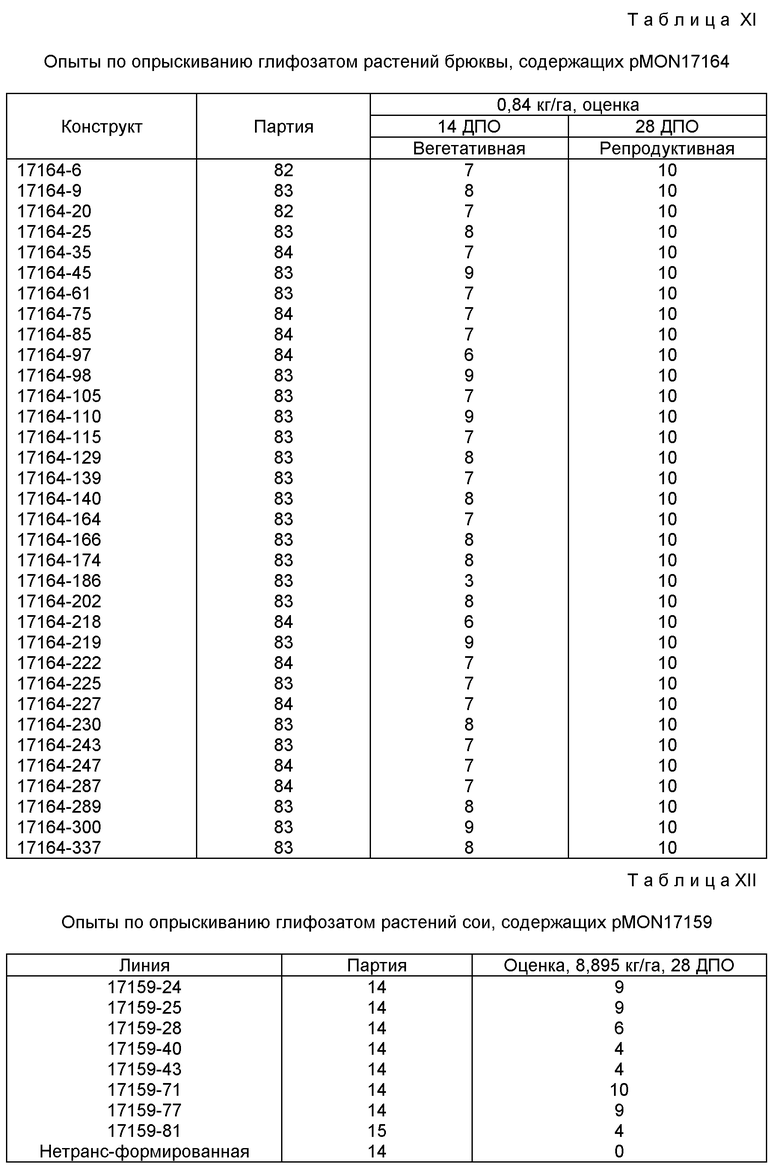
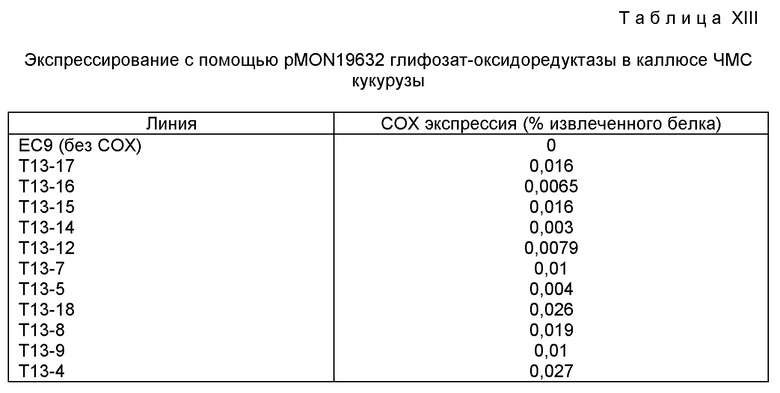







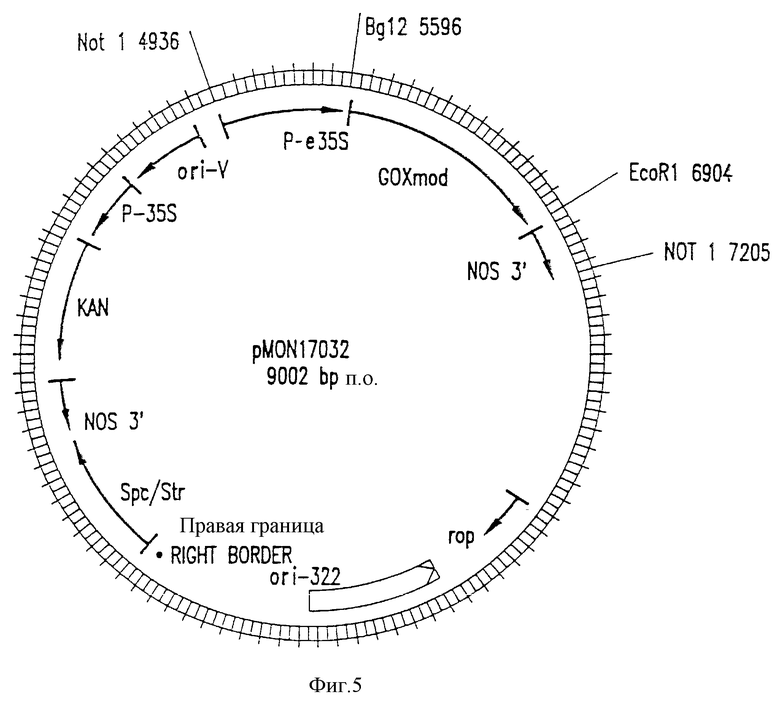

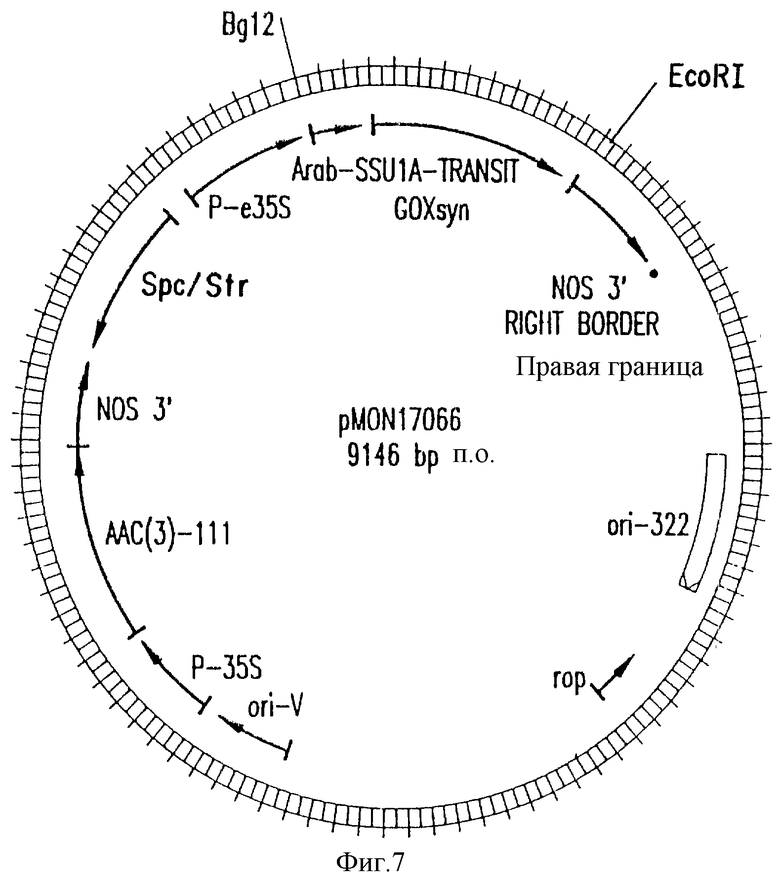
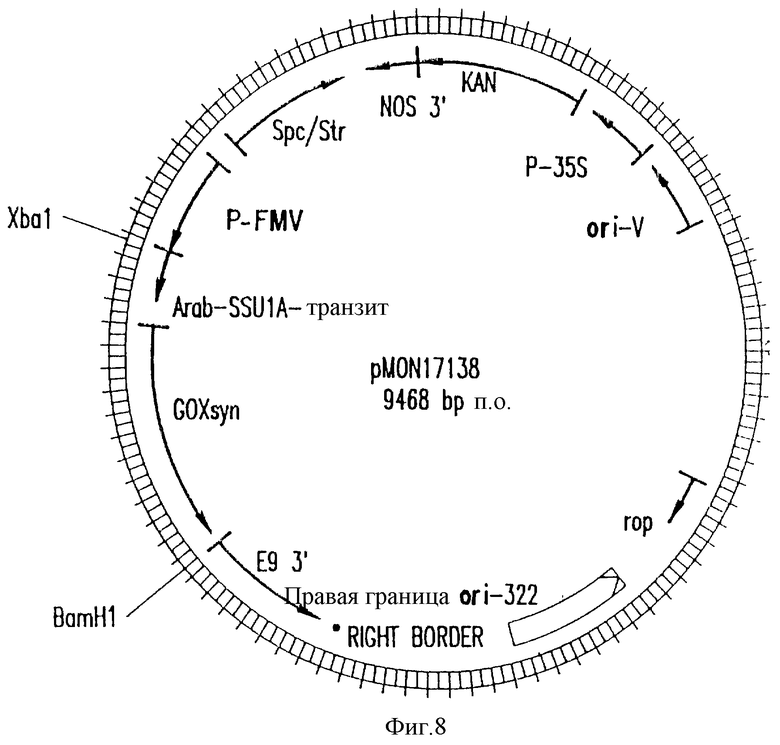
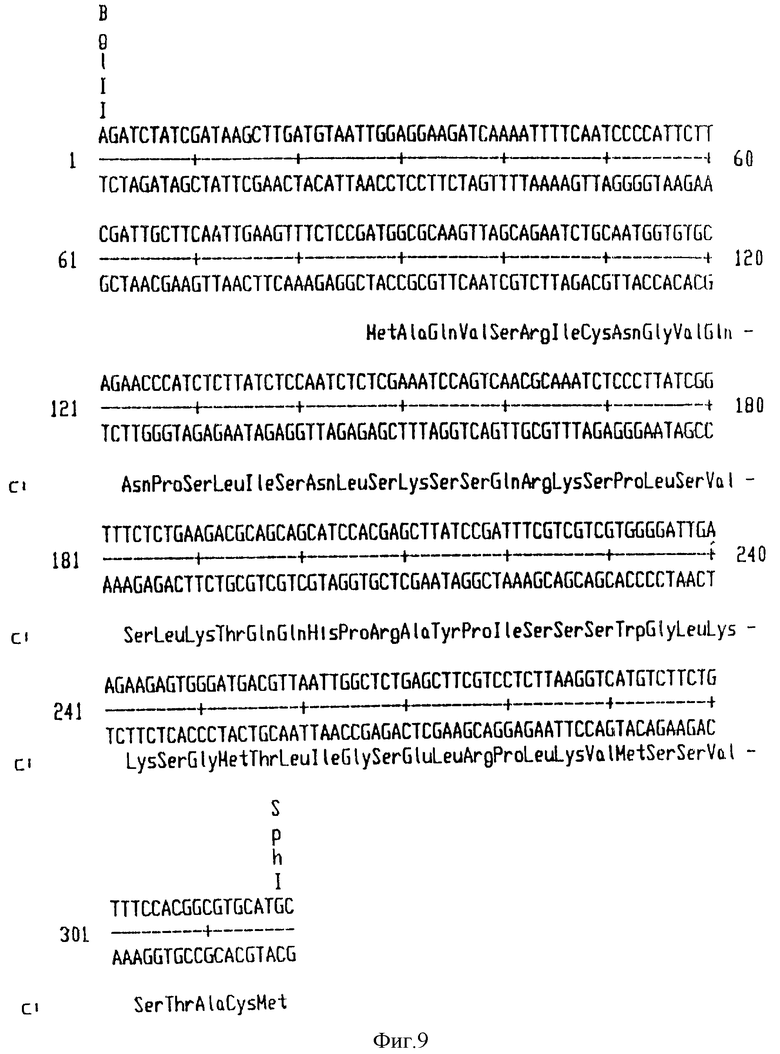
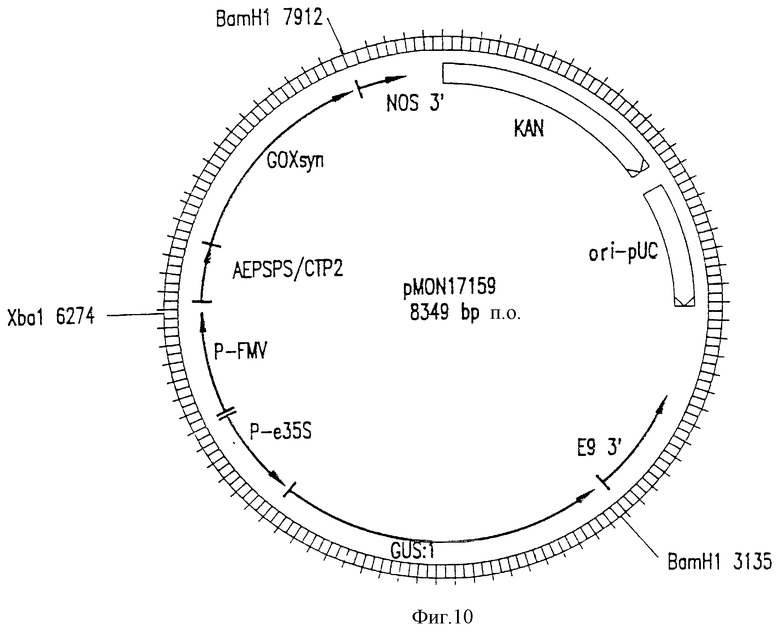
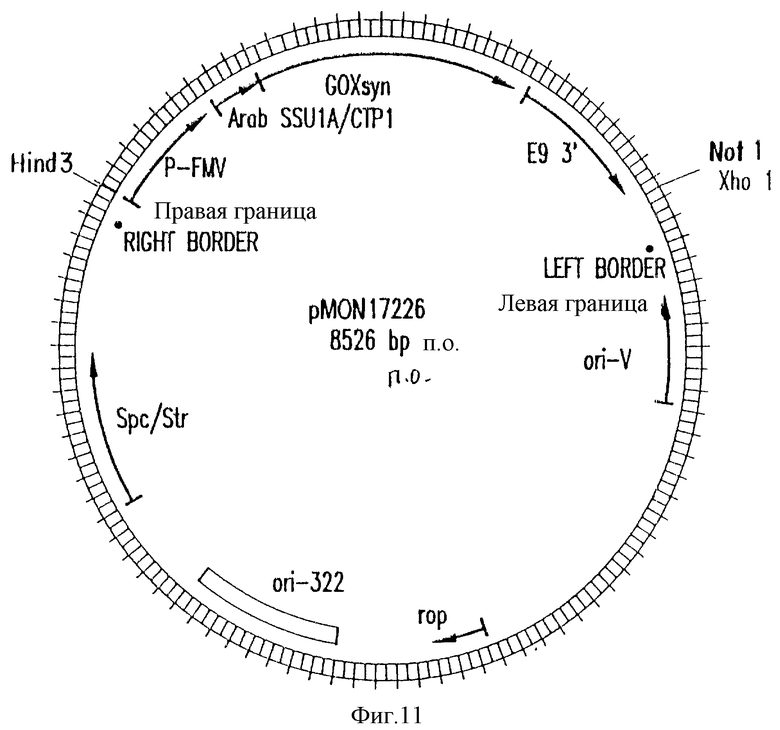
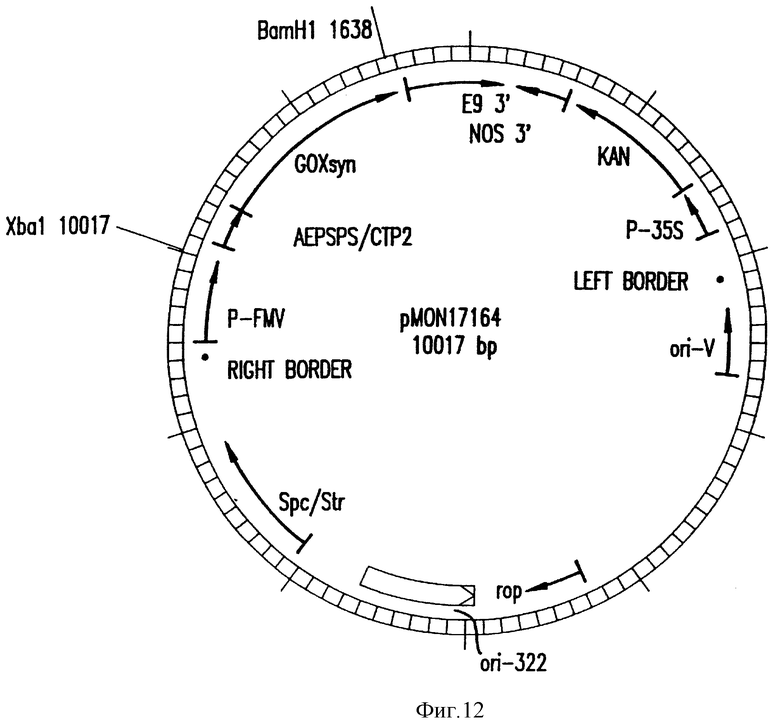

Изобретение может быть использовано в сельском хозяйстве. Трансформирование растений с помощью генов, разрушающих гербицид глифозат, позволяет получить растения с устойчивостью к данному гербициду, применяемому для борьбы с сорняками и др. 3 с. и 6 з.п. ф-лы, 13 ил., 17 табл.
| TIBTECH, vol.8, № 3, 1990 | |||
| TRENDIN GENETICS, vol.4, № 8, 1988 | |||
| APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol.54, № 12, 1988 | |||
| ШМАЛЬЦ Х | |||
| Селекция растений (пер | |||
| с нем.) | |||
| - М.: Колос, 1973. |

