Изобретение относится к новым соединениям, действующим как агонисты вазопрессина, а также к способам лечения и фармацевтическим композициям, в которых применяются эти соединения.
Предпосылки изобретения
Вазопрессин (антидиуретический гормон, АДГ), который является состоящим из девяти аминокислот пептидным гормоном и нейротрансмиттером, синтезируется в гипоталамусе мозга и транспортируется через расположенный над зрительным трактом гипофизарный тракт к нейрогипофизу (задней доле гипофиза), где он запасается. При восприятии увеличения осмоляльности плазмы осморецепторами мозга или уменьшения объема крови или кровяного давления, регистрируемых барорецепторами и волюморецепторами, вазопрессин высвобождается в кровоток и активирует V1a-рецепторы вазопрессина на сосудах крови, что приводит к сужению сосудов с увеличением кровяного давления, и V2-рецепторы вазопрессина на нефронах почки, чтобы удержать в основном воду и, в меньшей степени, электролиты для увеличения объема крови (Cervoni Р. and Chan P.S., Diuretic Agents, In Kirk-Othiner: Encyclopedia of Chemical Technology, 4th еd., Wiley, Volume 8, 398-432, 1993). О наличии вазопрессина в гипофизе было известно еще в 1895 году (Oliver, Н. and Schaefer, J. Physiol. (London), 18: 277-279, 1895). Определение структуры и полный синтез вазопрессина были осуществлены duVigneaud и сотр. в 1954 году (duVigneaud V., Gish D.T. and Katsoyannis, J. Am. Chem. Soc., 76: 4751-4752, 1954).
V1а-рецепторы вазопрессина медиируются через фосфатидилинозитный путь. Активация V1a-рецепторов вазопрессина вызывает сокращение гладких мышц кровеносных сосудов с повышением кровяного давления. Действия V2-рецепторов вазопрессина опосредуются через активацию аденилатциклазной системы и повышение внутриклеточных уровней содержания цAМФ. Активация V2-рецепторов вазопрессина вазопрессином или вазопрессинподобными (пептидными или непептидными) соединениями увеличивает водопроницаемость собирающих протоков нефрона и делает возможной повторную абсорбцию большого количества свободной воды. Конечным результатом является образование и экскреция концентрированной мочи, с уменьшением объема мочи и увеличением осмоляльности мочи.
Вазопрессин играет жизненно важную роль в сохранении воды за счет концентрирования мочи в месте собирающих протоков почки. Собирающие протоки почки являются относительно непроницаемыми для воды без присутствия вазопрессина при рецепторах, и, следовательно, гипотоническая жидкость, образуемая после фильтрации через гломерулы (почечные клубочки), проходя через проксимальные извитые почечные канальцы, петли Генле и дистальные извитые почечные канальцы, будет экскретироваться в виде разбавленной мочи. Однако во время дегидратации, истощения объема или потери крови вазопрессин высвобождается из мозга и активирует V2-рецепторы вазопрессина в собирающих протоках почки, делая эти протоки очень проницаемыми для воды, и, следовательно, вода повторно абсорбируется и экскретируется концентрированная моча. У пациентов и животных с центральным или нейрогенным несахарным диабетом синтез вазопрессина в мозгу нарушен, и, следовательно, они не вырабатывают вазопрессин или вырабатывают мало вазопрессина, но их рецепторы вазопрессина в почках являются нормальными. Поскольку они не могут концентрировать мочу, они могут производить в 10 раз больший объем мочи в сравнении с их здоровыми аналогами и являются очень чувствительными к действию вазопрессина и агонистов вазопрессина V2. Вазопрессин и десмопрессин, который является пептидным аналогом природного вазопрессина, используются для пациентов с центральным несахарным диабетом. Агонисты вазопрессина V2 применимы также для лечения ночного энуреза, никтурии (ночной полиурии), недержания мочи и способствуют обеспечению способности реципиента к временной задержке мочеиспускания, когда бы это ни было желательно.
Вазопрессин, через активацию его V1а-рецепторов, оказывает сосудосуживающее действие, повышая таким образом кровяное давление. Антагонист V1а-рецептора вазопрессина будет противодействовать этому эффекту. Вазопрессин и агонисты вазопрессина высвобождают фактор VIII и фактор фон Виллебранда, так что они полезны для лечения нарушений, связанных с кровотечением, таких как гемофилия. Вазопрессин и вазопрессинподобные агонисты высвобождают также активатор плазминогена тканевого типа (ТАЛ) в кровоток, так что они полезны в растворении сгустков крови, например, у пациентов с инфарктом миокарда и другими тромбоэмболическими нарушениями (Jackson Е.К., Vasopressin and other agents affecting the renal conservation of water. In: Goodman's and Gilmar's The Pharmacological Basis of Therapeutics, 9th ed., Eds. Hardman, Limbird, Molinoff, Ruddon and Gilman, McGraw-Hill, New York, pp. 715-731, 1996, Lethagen S., Ann. Hematol., 69: 173-180 (1994), Cash J.D. et al. , Brit. J. Haematol, 27:363-364, 1974, David J-L., Regulatory Peptides, 45, 311-317, 1993, и Burggraaf J., et al., Clin. Sci., 86, 497-503 (1994).
Следующие ссылки известного уровня техники описывают пептидные антагонисты вазопрессина: М. Manning et al., J. Med. Chem., 35, 382 (1992); M. Manning et al. , J. Med. Chem., 35, 3895 (1992); H. Gavras and B. Lammek, US Patent 5070187 (1991); M. Manning and W.H. Sawyer, US Patent 5055448 (1991); F. E. Ali, US Patent 4766108 (1988); R.R. Ruffolo et al. Drug News and Perspective, 4(4), 217, (May 1991), P.D. Williarms et аl., сообщали о сильных гексапептидных антагонистах окситоцина [J. Med. Chem., 35, 3905 (1992)], которые также проявляют слабое действие как антагонисты вазопрессина при связывании с V1а- и V2-рецепторами. Недостатком пептидных антагонистов вазопрессина является отсутствие пероральной активности и многие из этих пептидов не являются селективными антагонистами, так как они также проявляют частичное действие как агонисты.
Недавно были описаны непептидные антагонисты вазопрессина. Albright et al. описывают трициклические диазепины в качестве антагонистов вазопрессина и окситоцина в US 5516774 (May 14, 1996); производные тетрагидробензодиазепина в качестве антагонистов вазопрессина описаны в JP 08081460-А (March 26, 1996); Ogawa et al. описывают бензогетероциклические производные в качестве антагонистов вазопрессина и окситоцина и в качестве агонистов вазопрессина в WO 9534540-А; Albright et al. описывают трициклические производные бензазепина в качестве антагонистов вазопрессина в US 5512563 (April 30, 1996) и Venkatesan et al. описывают трициклические производные бензазепина в качестве антагонистов вазопрессина и окситоцина в US 5521173 (May 28, 1996).
Как упоминалось выше, десмопрессин (1-дезамино-8-D-аргинин-вазопрессин) (Huguenin, Boissonnas, Helv. Chim. Acta, 49, 695 (1966)) является агонистом вазопрессина. Это соединение является синтетическим пептидом с вариабельной биологической усваиваемостью. Интраназальный путь является плохо переносимым, а пероральная композиция для ночного энуреза требует в 10-20 раз большей дозы, чем доза, необходимая для интраназального введения.
Соединения этого изобретения являются непептидными и имеют хорошую пероральную усваиваемость. Они являются специфическими агонистами вазопрессина V2 и не имеют агонистического действия в отношении V1а, так что они не повышают кровяное давление. В противоположность этому соединения известного уровня (Ogawa H. et al., WO 9534540-A) являются антагонистами вазопрессина/окситоцина.
Сущность изобретения
Это изобретение относится к новым соединениям, выбранным из соединений общей формулы (I)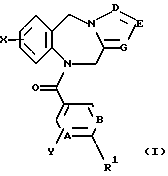
где А, В, Е, G обозначают независимо СН или азот;
D обозначает независимо C-W или азот;
R1 обозначает алканоил из 2-7 атомов углерода, группу, выбранную из CN, COOH, CONH2,
-C≡C-H, -C≡C-R9,
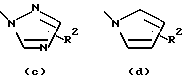
или остаток, выбранный из группы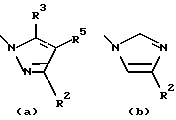





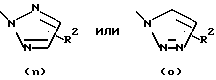
R2, R3 и R5 обозначают независимо водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода, циклоалкил из 3-7 атомов углерода или перфторалкил из 1-6 атомов углерода;
R4 обозначает водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода, циклоалкил из 3-7 атомов углерода, алкоксиалкил из 2-7 атомов углерода или ацильный заместитель, выбранный из группы, состоящей из алканоила из 2-7 атомов углерода, алкеноила из 3-7 атомов углерода, циклоалканоила из 3-7 атомов углерода, ароила или арилалканоила;
Х и Y обозначают независимо водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода, циклоалкил из 3-7 атомов углерода, перфторалкил из 1-6 атомов углерода, алкоксиалкил из 2-7 атомов углерода, галоген (в том числе хлор, бром, фтор и иод), алкокси из 1-6 атомов углерода, гидрокси, СF3 или перфторалкил из 2-6 атомов углерода;
W обозначает водород, галоген (предпочтительно хлор, бром или иод), алкил, алкоксиалкил из 2-7 атомов углерода, гидроксиалкил из 1-6 атомов углерода или CH2NR6R7;
R6 и R7 обозначают независимо водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода или взятые вместе с атомом азота группы CH2NR6R7, R6 и R7 образуют пяти- или шестичленное кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, как, например (но не ограничиваясь указанными), кольца группы


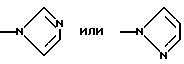
R8 обозначает алкил с прямой цепью из 1-6 атомов углерода;
R9 обозначает независимо водород, триметилсилил или алкил с прямой цепью из 1-6 атомов углерода;
или их фармацевтически приемлемой форме соли, сложного эфира или пролекарства.
Е и G обозначают предпочтительно СН; D обозначает предпочтительно N или C-W, где W обозначает водород, алкил, CH2NR6R7 или галоген, более предпочтительно W обозначает водород, метил, CH2NMe2 или бром.
R2, R3 и R5 обозначают предпочтительно каждый независимо водород, алкил с прямой цепью из 1-6 атомов углерода, циклоалкил из 3-7 атомов углерода или перфторалкил из 1-6 атомов углерода, более предпочтительно водород или алкил с прямой цепью из 1-6 атомов углерода, наиболее предпочтительно водород или метил.
R4 обозначает предпочтительно водород, алкил с прямой цепью из 1-6 атомов углерода или ацильный заместитель, более предпочтительно водород, метил, этил, н-пропил, н-бутил, метоксиметил, ацетил, циклопропилкарбонил, н-пропилкарбонил, 2-тиенилкарбонил, 2-метил, 5-фторфенилкарбонил, 2-метилфенилкарбонил, 2-хлор-4-фторфенилкарбонил, 2,4-дифторфенилкарбонил или 2,4-дифторбензилкарбонил.
X и Y обозначают предпочтительно каждый независимо водород, перфторалкил из 1-6 атомов углерода, галоген, алкокси из 1-6 атомов углерода или гидрокси, более предпочтительно водород, трифторметил, хлор, бром, фтор, метокси или гидрокси. Наиболее предпочтительно по меньшей мере один из X и Y обозначает водород.
R6 и R7 оба обозначают предпочтительно метил.
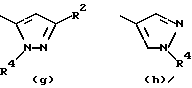
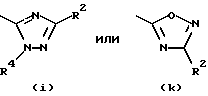
Предпочтительные значения R1 включают CN, CONH2, ацетил или один из следующих остатков:
- остаток а, в котором R2, R3 и R5 обозначают каждый независимо водород или алкил с прямой цепью из 1-6 атомов углерода, при этом более предпочтительно алкил представляет собой метил;
- остаток а, где два из R2, R3 и R5 обозначают водород и третий обозначает циклоалкил из 3-7 атомов углерода или перфторалкил из 1-6 атомов углерода, более предпочтительно, где третий представляет собой циклопропил или трифторметил;
- остаток b, с, d или i, где R2 обозначает водород или алкил с прямой цепью из 1-6 атомов углерода, более предпочтительно, где алкил представляет собой метил;
- остаток f, где R2 обозначает водород и/или R4 обозначает водород, алкил с прямой цепью из 1-6 атомов углерода или ацильный заместитель, выбранный из группы, состоящей из алканоила из 2-7 атомов углерода, алкеноила из 3-7 атомов углерода, циклоалканоила из 3-7 атомов углерода, ароила или арилалканоила, более предпочтительно, где R4 обозначает водород, метил, этил, н-пропил, н-бутил, метоксиметил или ацетил, циклопропилкарбонил, н-пропилкарбонил, 2-тиенилкарбонил, 2-метил, 5-фторфенилкарбонил, 2-метилфенилкарбонил, 2-хлор-4-фторфенилкарбонил, 2,4-дифторфенилкарбонил или 2,4-дифторбензилкарбонил;
- остаток f или g, где R4 обозначает водород и/или R2 обозначает алкил с прямой цепью из 1-6 атомов углерода, более предпочтительно, где этот алкил представляет собой метил;
- остаток k или h, где R2 обозначает метил;
- остаток m, где R2 обозначает водород. Среди более предпочтительных соединений этого изобретения находятся соединения формулы
где А и В обозначают независимо СН или азот;
D обозначает C-W или азот;
R1 обозначает алканоил из 2-7 атомов углерода или группу, выбранную из

R2, R3 и R5 обозначают независимо водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода, циклоалкил из 3-7 атомов углерода или перфторалкил из 1-6 атомов углерода;
R4, X, Y, W, R6, R7 и R8 имеют определенные выше значения,
или их фармацевтически приемлемая соль.
Применяемый в настоящем описании термин алкил в отношении к остатку (части) молекулы или к компоненту остатка (части) молекулы, например алкокси, включает в себя алкильные группы с прямой или разветвленной цепью, например группы метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, пентил, гексил и гептил. Применяемый в настоящем описании термин циклоалкил включает в себя насыщенные и ненасыщенные циклические группы, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопропенил, циклобутены, циклопентены, циклогексены и циклогептены. Предпочтительными являются предельные циклоалкилы.
Для соединений, которые определены выше и на которые ссылаются в настоящем описании, если не указано иначе, ароильные группы включают в себя, например, бензоил, нафтоил, которые могут быть замещены независимо одним или несколькими заместителями из группы, состоящей из водорода, галогена, циано, алкила с прямой цепью из 1-6 атомов углерода, алкила с разветвленной цепью из 3-7 атомов углерода, алкокси из 1-6 атомов углерода, СF3 или фенила (который сам является необязательно замещенным). Гетероароильными группами в данном описании называют карбонил (радикал), непосредственно связанный с атомом углерода пятичленного гетероциклического кольца, имеющего один иди два гетероатома, выбранных из азота, кислорода, серы, например 2-тиеноил. Гетероциклическое кольцо этих гетероароильных групп может также включать в себя, но не ограничивается ими, группы, в которых гетероарильная часть представляет собой группу фуран, пиррол, 2Н-пиррол, имидазол, пиразол, изотиазол, изоксазол, тиофен, пиразолин, имидазолидин или пиразолидин. Гетероарильные группы здесь могут быть замещены независимо одним или несколькими заместителями из группы, состоящей из водорода, галогена, циано, алкила с прямой цепью из 1-6 атомов углерода или алкила с разветвленной цепью из 3-7 атомов углерода.
Арилалканоильными группами в настоящем описании называют карбонильную группу (или радикал), непосредственно связанную с алкильной группой из 1-6 атомов углерода, которая в концевом положении замещена арильной группой, например фенилуксусную кислоту. Арильная группа может быть замещена независимо одним или несколькими заместителями из группы, состоящей из водорода, галогена, циано, алкила с прямой цепью из 1-6 атомов углерода, алкила с разветвленной цепью из 3-7 атомов углерода, алкокси из 1-6 атомов углерода, СF3 или фенила, или замещенного фенила, где заместители выбраны из галогена, циано, алкила с прямой цепью из 1-6 атомов углерода, алкила с разветвленной цепью из 3-7 атомов углерода, алкокси из 1-6 атомов углерода, СF3.
Галогены, упоминаемые здесь, могут быть выбраны из фтора, хлора, брома или иода, если не указано иначе.
Специалистам, работающим в данной области, должно быть понятно, что определение соединений формулы (I), когда R1, R2, R3, R4, R5, R6, R7, X или Y содержат асимметричные атомы углерода, охватывает все возможные стереоизомеры и их смеси, которые обладают обсуждаемой ниже активностью. В частности, оно включает в себя любые оптические изомеры и диастереомеры, а также рацемические и разделенные энантиомерно чистые R и S стереоизомеры, а также другие смеси R и S стереоизомеров и их фармацевтически приемлемые соли, обладающие указанной активностью. Оптические изомеры могут быть получены в чистом виде при помощи стандартных способов разделения. Понятно также, что определение R1, R2, R3, R4, R5, R6, R7, Х или Y соединений формулы (I) включает в себя все возможные региоизомеры и их смеси, которые обладают активностью, обсуждаемой ниже. Такие региоизомеры могут быть получены в чистом виде при помощи стандартных способов разделения, известных специалистам в этой области.
Также среди предпочтительных групп соединений этого изобретения находятся соединения в следующих подгруппах:
а) соединения, имеющие общую формулу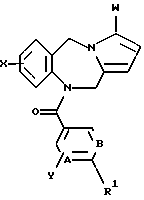
где А, В, W, R1, R2, R3, R4, R5, R6, R7, R8, R9, Х и Y определены, как указано выше;
b) соединения, имеющие общую формулу
где А, В, R1, R2, R3, R4, R5, R9, X и Y определены, как указано выше, и
с) соединения, имеющие общую формулу
где А, В, R1, R2, R3, R4, R5, R9, X и Y определены, как указано выше.
Должно быть понятно, что в составе приведенных выше подгрупп а)-с) можно далее выделить подгруппы, в которых
А и В независимо обозначают СН или азот;
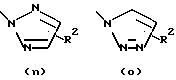
R1 обозначает алканоил из 2-7 атомов углерода или группу, выбранную из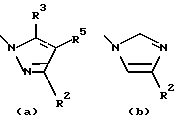



R2, R3 и R5 обозначают независимо водород, алкил с прямой цепью из 1-6 атомов углерода, алкил с разветвленной цепью из 3-7 атомов углерода, циклоалкил из 3-7 атомов углерода или перфторалкил из 1-6 атомов углерода, и
R4, X, Y, W, R6, R7 и R8 определены, как указано выше,
или их фармацевтически приемлемую соль.
Особенно предпочтительными среди соединений группы а), описанной выше, является соединения, в которых W представляет собой Н, А и В, каждый обозначает СН, и R1 обозначает группу алканоил из 2-7 атомов углерода или группу, выбранную из остатков (а), (b), (е), (f), (g), (h), (i) или (k), перечисленных выше.
Фармацевтически приемлемые соли включают в себя соли, произведенные из таких органических и неорганических кислот, как молочная, лимонная, уксусная, винная, янтарная, малеиновая, малоновая, хлористоводородная, бромистоводородная, фосфорная, азотная, серная, метансульфоновая и столь же известные приемлемые кислоты.
В соответствии с данным изобретением предлагается также способ лечения заболеваний, состояний или нарушений, в которых желательным является действие соединения как агониста вазопрессина, причем этот способ предусматривает введение человеку или другому млекопитающему, нуждающемуся в этом, эффективного количества соединения или фармацевтической композиции согласно настоящему изобретению. Представленные способы лечения включают способы лечения заболеваний, состояний или нарушений, при которых является желательным высвобождение фактора VIII и фактора фон Виллебранда в сердечно-сосудистую систему, высвобождение активатора плазминогена тканевого типа (ТАП) в кровоток или воздействие на почечное сохранение воды и концентрирование мочи. Такие способы лечения включают в себя, но не ограничиваются ими, способы лечения несахарного диабета, ночного энуреза, никтурии (ночной полиурии), недержания мочи или нарушений, связанных с кровотечением и свертыванием крови, у человека или других млекопитающих.
Описанные здесь способы включают в себя облегчение для человека или других млекопитающих временной задержки мочеиспускания, которое может быть также описано как регулирование или лечение неспособности временной задержки мочеиспускания, в любой ситуации, когда это является желательным. Предполагается, что этот способ включает в себя процедуры лечения, облегчающие временную задержку мочеиспускания, которые отделены от лечения состояния, известного как ночной энурез, и не включают в себя ночной энурез.
Таким образом, в данном изобретении предлагается фармацевтическая композиция, применимая для лечения вышеупомянутых заболеваний, состояний или расстройств, причем эта фармацевтическая композиция содержит одно или несколько соединений или их фармацевтически приемлемых солей согласно данному изобретению в сочетании или соединении с фармацевтически приемлемым носителем.
Эти композиции предпочтительно приспособлены для перорального введения. Однако они могут быть приспособлены для других способов введения, например парентерального введения для пациента, страдающего от сердечной недостаточности.
Для достижения постоянства введения является предпочтительным, чтобы композиция этого изобретения находилась в форме унифицированной (стандартной) дозы. Подходящие формы унифицированной дозы включают в себя таблетки, капсулы и порошки в мешочках (сашэ) или флаконах. Такие формы унифицированных доз могут содержать от 0,1 до 1000 мг соединения этого изобретения и предпочтительно от 2 до 50 мг. Еще более предпочтительные формы унифицированных доз содержат 5-25 мг соединения данного изобретения. Соединения данного изобретения могут вводиться перорально в диапазоне доз ~0,01-100 мг/кг или предпочтительно в диапазоне доз 0,1-10 мг/кг. Такие композиции могут вводиться от 1 до 6 раз в день, в более типичном случае от 1 до 4 раз в день.
Композиции изобретения могут быть приготовлены с общепринятыми носителями или эксципиентами, такими как наполнители, разрыхлители, связующие вещества, смазывающие вещества, улучшающие вкус и задах агенты и т.п. Их готовят общепринятым способом, например, подобным способу, применяемому для известных гипотензивных агентов, диуретиков и β-блокирующих агентов.
В соответствии с данным изобретением обеспечены также способы получения соединений данного изобретения.
Способ получения согласно изобретению
Соединения данного изобретения могут быть получены в соответствии с одним из общих способов, описанных ниже.
Как показано на Схеме I, трициклический бензодиазепин формулы (1) обрабатывают соответствующим образом замещенным ацетилароил- или (гетероароил)галогенидом, предпочтительно ароил- или (гетероароил)хлоридом формулы (2) в присутствии основания, такого как пиридин или триалкиламин, как, например, триэтиламин, в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, при температурах от -40 до 50oС с получением ацилированного производного формулы (3). Обработка (3) диалкилацеталем диалкиламида формулы (4) в апротонном органическом растворителе, таком как дихлорметан, при температурах в диапазоне от 0oС до температуры кипения растворителя дает енон формулы (5) в соответствии с методикой Lin et al., J. Het. Chem., 14, 345 (1977). Обработка (5) гидроксиламином или замещенным гидразином формулы (6) в уксусной кислоте при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя дает целевые соединения формулы (I), в которых А, В, D, Е, G, Х, Y, R2 и R4 определены, как указано выше, и R1 представляет собой гетероциклический остаток, выбранный из группы, состоящей из гетероциклов (f), (g) или (j), определенных выше.


Предпочтительные замещенные ацетилароил- или (гетероароил)хлориды формулы (2) Схемы I, приведенной в конце описания, легко получают обработкой соответствующих карбоновых кислот тионилхлоридом при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя или оксалилхлоридом в апротонном растворителе, таком как дихлорметан или тетрагидрофуран, в присутствии каталитического количества диметилформамида при температурах в диапазоне от 0 до 40oС. Предпочтительные диалкилацетали диалкиламида либо имеются в продаже, либо известны в литературе или могут быть легко получены в соответствии с методиками, аналогичными мотодикам, описанным в литературе. Kantlehner W. Chem. Ber., 105, 1340 (1972).
Предпочтительными трициклическими бензодиазепинами формулы (1) являются 10,11-дигидро-5Н-пирроло[2,1-c][1,4]бензодиазепин (Albright et al., US Patent No. 5536718, issued July 16, 1996), 10,11-дигидро-5Н-пиразол[5,1-с][1,4] бензодиазепин, Cecchi L. et al., J. Het. Chem., 20, 871 (1983), и 10,11-дигидро-5Н-тетразол[5,1-с] [1,4]бензодиазепин, Klaubert D.H., J. Het. Chem., 22, 333 (1985).
Альтернативный способ получения промежуточных продуктов формулы (3) иллюстрирован на Схеме II, приведенной в конце описания.
Согласно схеме трициклический бензодиазепин формулы (1) обрабатывают подходящим образом замещенным бромароил- (или гетероароил)галогенидом, предпочтительно ароил- (или гетероароил)хлоридом формулы (8) в присутствии органического основания, такого как пиридин или триалкиламин, как, например, триэтиламин, в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, при температурах от -40 до 50oС с получением ацилированного промежуточного продукта формулы (9). Затем промежуточный продукт (9) связывают с ацетиленом, монозамещенным при концевом атоме таким заместителем, как триметилсилил или алкил с прямой цепью из 1-6 атомов углерода, в присутствии пиридина и катализатора, такого как бис(трифенилфосфин)палладий(II)хлорид и иодид меди(I), в органическом основании, таком как триэтиламин, в качестве растворителя, в запаянной пробирке высокого давления при температурах в диапазоне от температуры окружающей среды до 100oС, по существу, в соответствии с методикой Martinez et al., J. Med. Chem., 35, 620 (1992). Затем полученный ацетиленовый промежуточный продукт формулы (10) гидратируют обработкой 1% серной кислотой в апротонном органическом растворителе, таком как тетрагидрофуран, насыщенном сульфатом ртути(II), при температуре окружающей среды, по существу, в соответствии с методикой Reed et al., J. Org. Chem., 52, 3491 1,1987) с получением целевого ацилсоединения формулы (3), где А, В, D, Е, G, Х к Y определены, как указано выше, R9 обозначает водород или алкил с прямой цепью из 1-6 атомов углерода. Альтернативно соединение 9, в котором R9 представляет собой триметилсилил, обрабатывают тетрабутиламмонийфторидом в простом эфире, таком как тетрагидрофуран, в качестве растворителя с получением соединения (10), где R9 представляет собой водород.
Предпочтительные ацилирующие агенты формулы (8) Схемы II легко получают обработкой соответствующим образом замещенной арил(гетероарил)карбоновой кислоты формулы (7) тионилхлоридом при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя или оксалилхлоридом в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, в присутствии каталитического количества диметилформамида при температурах в диапазоне от 0 до 40oС. Защищенные ацетиленовые промежуточные продукты Схемы II либо имеются в продаже, либо известны в данной области или могут быть легко получены при помощи методик, аналогичных процедурам, описанным в данной области.
Как показано на Схеме III, приведенной в конце описания, промежуточные ацетилсоединения (3) Схемы I могут быть получены также реакцией сочетания Штилле (Stille) бромарил (или гетеррарил) соединения формулы (9) Схемы II с (α-этоксивинил)триалкилоловом, предпочтительно (α-этоксивинил)трибутилоловом, в присутствии каталитического количества бис(трифенилфосфин)палладий(II)хлорида в апротонном органическом растворителе, таком как толуол, при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя, по существу, в соответствии с методикой Kosugi et al., Bull. Chem. Soc. Jpn., 60, 767 (1987).
Получение ацетилсоединения (3) может быть также выполнено через катализируемое палладием арилирование винилалкилового простого эфира, такого как винилбутиловый простой эфир, с арилгалогенидным промежуточным продуктом формулы (9) в соответствии с методикой Cabri et al., Tetrahedron Lett., 32, 1753 (1991).
Промежуточные продукты типа (α-алкоксивинил)триалкилолова Схемы III либо доступны коммерчески, либо хорошо известны в данной области или могут быть легко получены посредством методик, аналогичных методикам, описанным в данной области.
В случае, когда R4 на Схеме I представляет собой водород, гетероциклический азот может быть алкилирован или ацилирован в соответствии с реакциями, описанными на схеме IV, приведенной в конце описания.
Согласно схеме IV соединение пиразола формулы (I, R4 представляет собой Н) алкилируют обработкой сильным основанием, таким как гидрид натрия или калия, и алкилирующим агентом, таким как алкилгалогенид, предпочтительно алкилхлорид (бромид или иодид), в апротонном органическом растворителе, таком как диметилформамид или тетрагидрофуран, при температурах в диапазоне от 0 до 80oС с получением соединения (I, R1=(f) или (g)), в котором А, В, D, Е, G, X, Y и R2 определены, как указано выше, a R4 представляет собой алкильную часть молекулы. Альтернативно соединение (I) ацилируют обработкой галогенидом карбоновой кислоты, предпочтительно хлоридом, или ангидридом карбоновой кислоты в присутствии аминного основания, такого как пиридин или триалкиламин, предпочтительно триэтиламин, в апротонном органическом растворителе, таком как дихлорметан иди тетрагидрофуран, без дополнительного растворителя при использовании в качестве основания пиридина, при температурах в диапазоне от -40oС до температуры окружающей среды с получением соединения (I), в котором A, В, D, Е, G, X, Y и R2 определены, как указано выше, а R4 представляет собой ацильный остаток. Алкилирование или ацилирование соединения формулы (I, R4 представляет собой Н) приводит к смеси региоизомеров, где R2 представляет собой водород и R1 представляет собой гетероциклический остаток, выбранный из группы, включающей гетероциклы (f) или (g), определенные выше и иллюстрированные ниже соответственно.

Соединения общей формулы (I) Схемы I, где А и В обозначают углерод, R2 обозначает Н и R1 обозначает гетероциклический остаток, выбранный из определенного выше гетероцикла (g), могут быть получены в соответствии с общим способом, описанным на Схеме V, приведенной в конце описания.
Согласно схеме эфир подходящим образом замещенной галогенарил(или гетероарил)карбоновой кислоты, предпочтительно бром(или иод)метиловый эфир формулы (11) соединяют сочетанием с диалкиламинопропином, предпочтительно 1-диметиламинопропином, в присутствии катализатора, такого как бис(трифенилфосфин)палладий(II)хлорид и иодид меди(I), в органическом основании, таком как триэтиламин, в качестве растворителя и при температурах в диапазоне от температуры окружающей среды до 80oС, по существу, в соответствии с методиками Alami et at., Tetrahedron Lett., 34, 6403 (1993), и Sanogashira et al., Tetrahedron Lett., 4467 (1975), с получением замещенного ацетиленового промежуточного продукта общей формулы (12). Затем промежуточный продукт (12) превращают в его N-оксид обработкой окислителем при помощи любой из ряда стандартных методик окисления (Albini A., Synthesis, 263 (1993), или с использованием диоксирановых реагентов (Murray R.W., Сhеm. Rev., 1187 (1989), в апротонном органическом растворителе, таком как дихлорметан, при температурах ниже температуры окружающей среды. Промежуточный N-оксид не выделяют, а перегруппировывают in situ в енон общей формулы (13) обработкой (предпочтительно с нагреванием) содержащим гидроксил растворителем, в том числе любым растворителем или
комбинацией растворителей, состоящих из воды или содержащих воду, любой C1-8-алкиловый спирт с прямой или разветвленной цепью алкила, этиленгликоль, полиэтиленгликоль, 1,2-пропилендиол, полипропиленгликоль, глицерин, 2-метоксиэтанол, 2-этоксиэтанол, 2,2,2-трифторэтанол, бензиловый спирт, фенол или любой равноценный растворитель, который содержит один или несколько свободных гидроксильных (-ОН) заместителей, известный квалифицированным специалистам в этой области.
Системы растворителей, содержащие один или несколько сорастворителей, вместе с одним или несколькими растворителями, могут быть также использованы для этого процесса перегруппировки N-оксида до целевого енаминона. Упоминаемые здесь сорастворители могут быть определены как разбавители основного растворителя (основных растворителей) и могут быть выбраны из углеводородов, таких как пентан, гексан или гептан; ароматических углеводородов, таких как бензол, толуол или ксилол; простых эфиров, таких как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; хлорированных углеводородов, таких как дихлорметан, хлороформ, дихлорэтан или тетрахлорэтан, или других обычных растворителей, таких как этилацетат, N,N-диметилформамид, N,N-диметилацетамид, ацетонитрил, диметилсульфоксид, ацетон и т.п.
Превращение N-оксида амина в енаминон может быть выполнено введением N-оксида амина в подходящий содержащий гидроксил растворитель, предпочтительно при перемешивании, при комнатной температуре или температуре окружающей среды либо между приблизительно комнатной температурой или температурой окружающей среды и приблизительно температурой кипения растворителя. В других случаях введение N-оксида амина в гидроксильный растворитель, предпочтительно при перемешивании, может быть выполнено в присутствии приемлемого катализатора, такого как содержащий палладий(II) катализатор или содержащий медь(I) катализатор при комнатной температуре или между комнатной температурой и температурой кипения растворителя.
Эта методика обеспечивает новый способ синтеза соединений енаминона из пропаргиламинoв или их N-оксидов в гидроксильных растворителях, которые оказывают влияние на конечный выход этой реакции. Этот новый способ синтеза енаминонов обеспечивает удобную альтернативу существующим способам и дополнительно расширяет диапазон исходных материалов, которые могут быть превращены в енаминоновые продукты.
Хотя точный механизм, посредством которого N-оксид пропаргиламина превращается в енаминоновый продукт, не был строго определен, он, по-видимому, сходен с двумя известными процессами: термической [2,3]-сигматропной перегруппировкой N-оксидов пропаргиламинов (Graig et al., Tetrahedron Lett., 4025, 1979; Hallstrom еt al., Tetrahedron Lett., 667, 1980; Khuthier A-H et al., J. Chem. Soc. Chem. Commun., 9, 1979) и превращением некоторых изоксазолов в енаминоны (Ligouri et al., Tetrahedron, 44, 1255, 1988).
Обработка (13) замещенным гидразином (6) в уксусной кислоте при температурах в диапазоне от окружающей температуры до температуры кипения приводит к смеси региоизомерных соединений общих формул (14) и (15) в переменном соотношении. Основной изомер формулы (14) отделяют при помощи хроматографии и/или кристаллизации и затем гидролизуют до целевой карбоновой кислоты формулы (16).
Затем промежуточный продукт (16) превращают в его ацилирующие варианты, предпочтительно хлорангидрид (бромангидрид или иодангидрид) или смешанный ангидрид формулы (17), посредством методик, аналогичных описанным выше. Затем ацилирующий агент (l7) используют для ацилирования трициклического бензодиазепина формулы (1) посредством любой методики, описанной выше, с получением целевого соединения формулы (I), где А, В представляют собой СН, a D, Е, G, X, Y и R4 определены, как указано выше, R2 представляет собой водород и R1 представляет собой гетероциклический остаток, выбранный из гетероциклической группы (g), иллюстрируемой ниже.

Подобным образом обработка (13) незамещенным гидразином (6, R4 представляет собой Н) в уксусной кислоте при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя дает промежуточный сложный эфир пиразола формулы (18). В этом случае гетероциклический азот может быть aлкилирован или ацилирован, как показано на Схеме VI, приведенной в конце описания, с получением соединений формулы (I), где R2 представляет собой водород и R1 представляет собой гетероциклический остаток, выбранные из гетероциклической группы (f), определенной выше.
Согласно схеме промежуточный сложный эфир формулы (18) алкилируют обработкой сильным основанием, таким как гидрид натрия или калия, и алкилирующим агентом, таким как алкилгалогенид, предпочтительно алкилхлорид (бромид или иодид) в апротонном растворителе, таком как диметилформамид или тетрагидрофуран, при температурах в диапазоне от 0 до 80oС с получением смеси региоизомеров формул (14) и (15) в переменном соотношении. Основной региоизомер формулы (15) отделяют при помощи хроматографии и/или кристаллизации и затем гидролизуют до целевой карбоновой кислоты формулы (19), которую затем превращают в ацилирующий агент, предпочтительно хлорангидрид или смешанный ангидрид, посредством методик, аналогичных методикам, описанным выше. Затем ацилирующие частицы формулы (20) используют для ацилирования трициклического бензодиазепина формулы (1) с получением целевого соединения формулы (I), где А, В, D, Е, G, X, Y и R4 определены, как указано выше, R2 представляет собой водород и R1 представляет собой гетероциклический остаток, выбранный из гетероциклической группы (f), определенной выше.

Соединения общей формулы (I), где R1 обозначает гетероциклический остаток, выбранный из группы (h) в числе гетероциклов R1, определенных выше, могут быть получены, как представлено на Схеме VII, приведенной в конце описания.
Подходящим образом замещенный малоновый диальдегид формулы (21) обрабатывают сначала гидразином в уксусной кислоте при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя и затем промежуточный пиразол окисляют перманганатом калия в основном водном растворе при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя с получением в качестве промежуточного продукта карбоновой кислоты формулы (22). Эту кислоту (22) превращают в ацилирующий агент, предпочтительно хлорангидрид (бромангидрид или иодангидрид) или смешанный ангидрид, посредством методик, аналогичных методикам, описанным выше.
Наконец, этот ацилирующий агент формулы (23) взаимодействует с трициклическим бензодиазепином формулы (1) с образованием соединений общей формулы (I), где А, В, D, Е, G, X, Y и R4 определены, как указано выше, a R1 представляет собой гетероциклический остаток, выбранный из гетероциклической группы (h), определенной выше.

В случае, когда R4 на Схеме VII является водородом, гетероциклический азот может быть алкилирован или ацилирован в соответствии с методиками, описанными выше.
Предпочтительные малоновые диальдегиды формулы (21) и гидразины Схемы VII имеются в продаже или известны в данной области, или могут быть легко получены посредством методик, аналогичныx методикам, описанным в литературе для известных соединений, таких как методики Knorr et al., J. Оrg. Сhеm., 49, 1288 (1984), и Сорроlа et al., J. Het. Сhеm., 11, 51 (1974).
Альтернативное получение промежуточных карбоновых кислот формулы (22) Схемы VII, где Y определен, как указано выше, а R4 является иным, чем водород, представлено на Схеме VIII, приведенной в конце описания.
Оловоорганический реагент формулы (25) взаимодействует в реакции сочетания Штилле (Stille) с соответствующем образом замещенным арил(или гетероарил)галогенидом, предпочтительно бромидом или иодидом формулы (28), в присутствии катализатора, такого как тетракис(трифенилфосфин)палладий(0) и иодид меди(I), в органическом апротонном растворителе, таком как диметилформамид, при температурах в диапазоне от температуры окружающей среды до 150oС, по существу, в соответствии с методиками, аналогичными методикам, описанным в Farina et al., J. Оrg. Chem., 59, 5905 (1994). Щелочной гидролиз полученного сложного эфира формулы (26) гидроксидом натрия или лития в водном спирте или тетрагидрофуране при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя дает целевые карбоновые кислоты формулы (22).
В свою очередь, оловоорганические реагенты формулы (25), где группы R предпочтительно являются алкильными группами, легко получают металлированием 4-бром-N-алкилпиразола формулы (24) триалкилоловогалогенидом, предпочтительно трибутилоловохлоридом (или бромидом), в присутствии металлирующего агента, такого как алкиллитий, например н-бутиллитий, втор-бутиллитий или трет-бутиллитий, в апротонном органическом растворителе, таком как диэтиловый эфир, при температурах в диапазоне от -40oС до температуры окружающей среды в соответствии с методиками, аналогичными методикам, описанным в Martina. et al., Synthesis, 8, 613 (1991).
Предпочтительные N-алкилзамещенные 4-бромпиразолы формулы (24) легко получают из 4-бромпиразола алкилированием алкилгалогенидом, предпочтительно алкилхлоридом (бромидом или иодидом) в присутствии сильного основания, такого как гидрид лития, натрия или калия, в апротонном органическом растворителе, таком как диметилформамид или тетрагидрофуран, при температурах в диапазона от 0 до 80oС. Альтернативно алкилирование 4-бромпиразола можно проводить при помощи алкилирующего агента, упомянутого выше, и сильного щелочного основания, такого как гидроксид лития, натрия или калия, в присутствии катализатора межфазного переноса (Jones R.A. Aldrichimica ACTA, 9(3), 35, 1976), такого как бензилдиметилтетрадециламмонийхлорид или бензилтриметиламмонийхлорид.
Предпочтительные арил(гетероарил)иодиды формулы (28) легко получают диазотированием соответствующих замещенных анилинов формулы (27) с последующим взаимодействием соответствующей соли диазония с иодом и иодидом калия в водной кислой среде, по существу, в соответствии с методиками Street et al., J. Med. Сhеm., 36, 1529 (1993), и Coffen et al., J. Org. Chem., 49, 296 (1984).
Альтернативное получение соединений общей формулы (I) представлено на Схеме IX, приведенной в конце описания.
Трициклический бензодиазепин формулы (1) обрабатывают соответствующим образом замещенным галогенароил(или гетероароил)галогенидом, предпочтительно фторароил- или фтор(или хлор)гетероароилхлоридом формулы (29), в присутствии основания, такого как триэтиламин или диизопропилэтиламин, в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, при температурах от-40oС до температуры кипения растворителя с получением ацилированного производного (30).
Альтернативно ацилирующие частицы могут представлять собой смешанный ангидрид вышеописанной карбоновой кислоты, такой как смешанный ангидрид, полученный реакцией 2,4,6-трихлорбензоилхлорида в растворителе, таком как дихлорметан, в соответствии с методикой Inanaga et al., Bull. Chem. Soc. Jpn, 52, 1989 (1979). Обработка этого смешанного ангидрида общей формулы (29) трициклическим бензодиазепином формулы (I) в растворителе, таком как дихлорметан, и в присутствии органического основания, такого как 4-диметиламинопиридин, при температурах в диапазоне от 0oС до температуры кипения растворителя, дает ацилированное производное (30) в качестве промежуточного продукта Схемы IX.
Затем соединение формулы (30) обрабатывают литиевой, натриевой или калиевой солью подходящим образом замещенного гетероцикла формулы (31) в полярном апротонном органическом растворителе, таком как диметилформамид или тетрагидрофуран, при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя с получением соединения общей формулы (I), где А, В, D, Е, G, X, Y, R2, R3 и R5 определены, как указано выше, а R1 является гетероциклическим остатком, выбранным из группы, состоящей из (а), (b), (с), (d), (l), (n) или (о), определенных выше.



Конденсация промежуточного продукта формулы (30) с промежуточной солью формулы (31) приводит к переменному соотношению региоизомеров общей формулы (I), которые разделяют при помощи хроматографии и/или кристаллизации.
Предпочтительные замещенные фторароил- или фтор(или хлор)гетероароилхлориды формулы (29) имеются в продаже или известны в данной области, или могут быть легко получены при помощи методик, аналогичных описанным в литературе для известных соединений.
Литиевые, натриевые или калиевые соли гетероциклов формулы (31) получают обработкой указанного гетероцикла сильным основанием, таким как гидрид лития, гидрид натрия, гидрид калия или алкоголят металла, при температурах в диапазоне от -40oС до температуры окружающей среды в апротонном органическом растворителе, таком как диметилформамид или тетрагидрофуран.
Альтернативно соединения общей формулы (I), описанные на Схеме IX, могут быть получены в соответствии со способом, представленным на Схеме X, приведенной в конце описания.
Согласно схеме замещенную соответствующим образом фторарил- или фтор(или хлор)гетероарилкарбоновую кислоту формулы (32) этерифицируют при помощи способов, известных в данной области, таких как обработка оксалилхлоридом (или тионилхлоридом) в спиртовом растворителе, таком как метанол, в присутствии каталитического количества диметилформамида, или конденсацией с метанолом в присутствии кислотного катализатора, такого как пара-толуолсульфоновая кислота, при температурах в диапазоне от температуры окружающей среды до температуры кипения.
Полученный сложный эфир формулы (33) взаимодействует с литиевой, натриевой или калиевой солью соответствующим образом замещенного гетероцикла формулы (31) в полярном апротонном органическом растворителе, таком как диметилформамид, при температурах в диапазоне от температуры окружающей среды до 150oС с получением сложного эфира формулы (34) в качестве промежуточного продукта. Конденсация (33) с (31) приводит к переменному соотношению региоизомеров формулы (34), которые разделяют при помощи хроматографии и/или кристаллизации.
Последующий гидролиз промежуточного сложного эфира формулы (34) водным основанием, таким как гидроксид лития, натрия или калия, в метаноле или тетрагидрофуране дает карбоновую кислоту формулы (35).
Затем промежуточную карбоновую кислоту (35) превращают в ацилирующий агент, предпочтительно хлорангидрид или смешанный ангидрид общей формулы (36), при помощи любой из описанных выше методик.
Последующая реакция трициклического бензодиазепина формулы (1) с промежуточным ацилирующим агентом формулы (36) в соответствии с любой из описанных выше методик дает целевые соединения формулы (I) Схемы IX.
Альтернативно замещенные карбоновне кислоты формулы (35), описанные на Схеме X, могут быть получены в соответствии со способом, представленным на Схеме XI, приведенной в конце описания.
Согласно схеме фторарил- или фтор(хлор)гетероарилнитрил формулы (37) взаимодействует с литиевой, натриевой или калиевой солью замещенного гетероцикла формулы (31) в полярном апротонном органическом растворителе, таком как диметилформамид, при температурах в диапазоне от температуры окружающей среды до 150oС с образованием промежуточного продукта общей формулы (38). Реакция (37) с (31) приводит к переменному соотношению региоизомеров формулы (38), которые разделяют при помощи хроматографии и/или кристаллизации. Гидролиз промежуточных нитрилов формулы (38, Y≠СF3) проводят предпочтительно с неорганической кислотой, такой как серная кислота, при температурах в диапазоне от температуры окружающей среды до 60oС.
Альтернативно гидролиз нитрила (38) можно проводить нагреванием в этаноле в присутствии сильного щелочного основания, такого как гидроксид натрия, с катализатором межфазного переноса или без него (Jones R.A. Aldrinchimica Acta, 9(3), 35, 1976), таким как бензилдиметилтетрадециламмонийхлорид.
Затем полученные карбоновые кислоты формулы (35) превращают в целевые соединения формулы (I) Схемы IX с использованием методик, аналогичных методикам, описанным выше.
Альтернативно замещенные карбоновые кислоты формулы (35) Схемы Х могут быть получены в соответствии со способом, представленным на Схеме XII, приведенной в конце описания, последовательной обработкой нитрила формулы (38), где А и В представляют собой СН и где R1 не является алканоилом из 2-7 атомов углерода, алкинилом, (b) или (d), основным пероксидом водорода в диметилсульфоксиде, по существу, в соответствии с методикой Katritzky et al. , Synthesis, 949 (1989) с последующим гидролизом полученного амида формулы (38) предпочтительно посредством обработки разбавленной серной кислотой и нитритом натрия в соответствии с методикой Hanes et al., Tetrahedron, 51, 7403 (1995).
Предпочтительный способ получения промежуточных замещенных карбоновых кислот формулы (35) Схемы X, где R1 является гетероциклическим остатком, выбранным из группы (а), относящийся к гетероциклам R1, описанным выше, представлен на Схеме XIII, приведенной в конце описания.
Диазотирование соответствующим образом замещенного анилина формулы (40) с последующим восстановлением полученной соли диазония формулы (41) хлоридом олова (II) в концентрированной хлористоводородной кислоте в соответствии с процедурой Street et al., J. Med. Chem., 36, 1529 (1993) обеспечивает промежуточную гидрохлоридную соль гидразина формулы (42). Последующая конденсация (42) с производным альдегида формулы (47), где R2 определен, как указано выше, R3 и R5 обозначают Н, а Р является диалкилацеталем, таким как диметилацеталь ацетилацетальдегида, или кетоном формулы (47), где R2, R3 и R5 определены, как указано выше, и Р обозначает =О или (O-алкил)2, в растворителе, таком как водный метанол, при температурах в диапазоне от температуры окружающей среды до 100oС дает после кристаллизации целевой промежуточный сложный эфир формулы (34, R1 обозначает (а) и R5 обозначает Н), который затем превращают в соединения формулы (I), как представлено на Схеме Х выше.

Когда Y обозначает ОСН3, соединения общей формулы (I) Схемы I могут быть легко деметилированы, как представлено на Схеме XIV, приведенной в конце описания.
Согласно схеме реакция соединения (I), где Y обозначает ОСН3, с трибромидом бора в органическом растворителе, таком как дихлорметан, дает соответствующий фенол формулы (I), где Y обозначает ОН и А, В, D, Е, G, X, R2 и R3 определены, как указано выше, и R1 представляет собой гетероциклический остаток, выбранный из группы (а), входящей в число гетероциклов, определенных выше, и иллюстрированной ниже.

Соединения, в которых R1 содержит три гетероатома, получают в соответствии со Схемой XV, приведенной в конце описания.
Согласно схеме трициклический бензодиазепин формулы (1) обрабатывают соответствующим образом замещенным цианоароил(или гетероароил)галогенидом, предпочтительно ароил(или гетероароил)хлоридом формула (43), в присутствии основания в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, при температурах в диапазоне от -40 до 80oС с получением промежуточного нитрила формулы (46, Схема XVI), который, в свою очередь, гидролизуют до промежуточного амида общей формулы (44) с помощью неорганической кислоты, такой как серная кислота, при температуре от температуры окружающей среды до 50oС. Обработка этого амида (44) диалкилацеталем диалкиламида формулы (4) в апротонном органическом растворителе, таком как дихлорметан или тетрагидрофуран, при температурах в диапазоне от 0 до 80oС дает промежуточный продукт формулы (45). Обработка (45) гидроксиламином или гидразином формулы (6) в уксусной кислоте при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя дает целевые соединения формулы (I), где А, В, D, Е, G, X, Y, R2 и R4 определены, как указано выше, и R1 является гетероциклическим остатком, выбранным из групп (е), (i) и (k) из состава определенных выше гетероциклов.


Другой предпочтительный способ получения промежуточного амида формулы (44) (см. Схему XV), где А и В обозначают СН и D не является СН, изображен на Схеме XVI, приведенной в конце описания, и состоит в обработке нитрила формулы (46) основным пероксидом водорода в диметилсульфоксиде, по существу, в соответствии с методикой Katritzky et al., Synthesis, 949 (1989).
Предпочтительный способ получения соединений общей формулы (I), в которых R1 содержит четыре гетероатома и R4 является водородом, изображен на Схеме XVII, приведенной в конце описания.
Обработка промежуточного нитрила формулы (46) Схемы XVI азидом натрия и хлоридом аммония в апротонном органическом растворителе, таком как диметилформамид, при температурах в диапазоне от температуры окружающей среды до температуры кипения растворителя дает целевые соединения формулы (I), где А, В, D, Е, G, Х и Y определены, как указано выше, R4 представляет собой водород и R1 является гетероциклическим остатком, выбранным из группы (m) входящей в число гетероциклов, определенных выше.

Соединения общей формулы (I), где D обозначает CW и W является водородом, могут быть подвергнуты конденсации Манниха, как показано на Схеме XVIII, приведенной в конце описания.
Согласно схеме реакция соединений формулы (I, D обозначает CH) с водным формальдегидом или параформальдегидом, замещенным амином формулы (47) и ледяной уксусной кислотой в спиртовом растворителе, таком как метанол, при температурах в диапазоне от температуры окружающей среды до температуры кипения дает соответствующие основания Манниха общей формулы (I), где А, В, Е, G, X, Y, R2, R3, R5, R6 и R7 определены, как указано выше; D представляет собой CW; W обозначает диалкиламиноалкильный остаток, предпочтительно диметиламинометильный остаток, и R1 обозначает гетероциклический остаток, выбранный из групп (а), (с), (е), (f), (g), (h), (i), (j), (k), (1), (m), (n) и (о), входящих в число гетероциклов, определенных выше.
Подобным же образом соединения общей формулы (I), где D обозначает CH, могут быть подвергнуты галогенированию, как показано на Схеме XIX, приведенной в конце описания.
Согласно схеме реакция (I, D обозначает CH) с N-галогенсукцинимидом, таким как N-хлор(бром или иод)сукцинимид, в полярном апротонном органическом растворителе, таком как дихлорметан, при температурах в диапазоне от -80oС до температуры окружающей среды дает соответствующие галогенированные производные общей формулы (I), где А, В, Е, G, X, R2, R3 и R5 определены, как указано выше, D обозначает CW, W обозначает галоген, такой как хлор (бром или иод), и R1 представляет собой гетероциклический остаток, выбранный из групп (а), (с), (е), (f), (g), (h), (i), (j), (k), (l), (m), (n) и (о), входящих в число гетероциклов, определенных выше.
Рассматриваемые соединения данного изобретения испытывали на биологическую активность в соответствии со следующими методиками.
Действие испытуемых соединений (тест-соединений) в качестве агонистов вазопрессина V2 на нормальных находящихся в сознании крысах, напившихся воды.
Самцам или самкам крыс Sprague-Dawley (Charles River Laboratories, Inc., Kingston, NY), имеющим нормальное кровяное давление, с весом тела 350-500 г предоставляли стандартный пищевой рацион грызунов (Purina Rodent Lab. Chow 5001) и воду ad libitum. В день испытания (теста) крыс помещали по отдельности в метаболические клетки, оборудованные приспособлениями для отделения фекалий от мочи и контейнерами для сбора мочи. Тест-соединение или контрольный препарат давали в виде пероральной дозы 10 мг/кг в объеме 10 мл/кг. Применяемым носителем был 20% диметилсульфоксид (ДМСО) в 2,5% предварительно прокипяченном кукурузном крахмале. Через тридцать минут после введения дозы испытуемого соединения крысам вводили воду в количестве 30 мл/кг в желудок при помощи иглы для кормления. В продолжение всего теста крысам не давали воду или корм. Мочу собирали в течение четырех часов после введения дозы испытуемого соединения. По истечении четырех часов измеряли объем мочи. Осмоляльность мочи определяли при помощи осмометра Fiske One-Ten Osmometer (Fisice Associates, Norwood, MA, 02062) или усовершенствованного осмометра Advanced CRYOMATIC Osmometer, Model 3C2 (Advanced Instruments, Norwood, MA). Определения иона Na+, К+ и Cl- проводили с использованием ионселективных электродов в анализаторе электрической модели системы Весkman synchron EL-ISE Electrolyte System. Осмоляльность мочи должна была пропорционально возрастать. В отборочном тесте-скрининге использовали по две крысы для каждого соединения. Если различие в объеме мочи этих двух крыс составляло более 50%, то использовали третью крысу.
Действие испытуемых соединений (тест-соединений) в качестве агонистов вазопрессина V2 на нормальных находящихся в сознании гомозиготных крысах Brattleboro с центральным несахарным диабетом.
Самцам или самкам гомозиготных крыс Brattleboro (Harlan Sprague-Dawley, Inc. , Indianapolis, IN) с весом тела 250-350 г предоставляли стандартный пищевой рацион грызунов (Purina Rodent Lab. Chow 5001) и воду ad libitum. В день испытания (теста) крыс помещали по отдельности в метаболические клетки, оборудованные приспособлениями для отделения фекалий от мочи и контейнерами для сбора мочи. Испытуемое соединение или контрольный препарат вводили в виде пероральной дозы 1-10 мг/кг в объеме 10 мл/кг. Применяемым носителем был 20% диметилсульфоксид (ДМСО) в 2,5% предварительно прокипяченном кукурузном крахмале. В продолжение всего теста крыс обеспечивали водой ad libitum. Мочу собирали в течение шести часов после введения дозы испытуемого соединения. По истечении шести часов измеряли объем мочи. Осмоляльность мочи определяли при помощи осмометра Fiske One-Ten Osmometer (Fiske Associates, Norwood, MA, 02062) или усовершенствованного осмометра Advanced CRYOMATIC Osmometer, Model 3C2 (Advanced Instruments, Norwood, MA). Определения иона Na+, К+ и Сl- проводили с использованием ионселективных электродов в анализаторе электролитической системы модели Beckman SYNCH-RON EL-ISE Electrolyte System. Эту модель животных использовали в основном для оценки силы и продолжительности действия активных соединений. Результаты этого исследования показаны в таблице .
Нижеследующие примеры представлены для иллюстрации, а не для ограничения объема изобретения.
Пример 1
(4-Фтор-2-трифторметилфенил)-(5Н, 11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Оксалилхлорид (2,0 г) добавляли к суспензии 4-фтор-2-трифторметилбензойной кислоты (2,0 г) в дихлорметане (25 мл). Добавляли две капли диметилформамида и смесь перемешивали в течение 18 часов при комнатной температуре. Полученный раствор выпаривали досуха с получением неочищенного хлорангидрида. Его повторно растворяли в дихлорметане и фильтровали. Выпаривание этого материала давало жидкость, которую затем снова растворяли в гексане, фильтровали и выпаривали с получением хлорангидрида в виде бледно-желтой вязкой жидкости, которую использовали без дополнительной очистки.
Хлорангидрид (2,26 г) в дихлорметане (25 мл) добавляли порциями к смеси 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (1,66 г), дихлорметана (10 мл) и диизопропилэтиламина (1,30 г), охлаждаемый в бане со льдом. После выдерживания при комнатной температуре в продолжение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением 2,57 г указанного в заголовке соединения, т.пл. 154-155oС.
Пример 2
[4-(3-Метилпиразол-1-ил)-2-трифторметилфенил)-(5Н, 11H-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
60% гидрид натрия в масле (0,15 г) промывали гексаном и добавляли безводный диметилформамид (25 мл), а затем 3-метилпиразол (0,25 г). После прекращения выделения водорода добавляли (4-фтор-2-трифторметилфенил)-(5Н,11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (1,0 г). Реакционную смесь нагревали на песочной бане при 110oС в течение 15 часов. Реакционную смесь выливали на лед и добавляли насыщенный солевой раствор. Осадок собирали фильтрованием. Неочищенный продукт реакции растворяли в дихлорметане и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими дополнительными объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана. После охлаждения кристаллы собирали фильтрованием с получением 0,77 г неочищенного продукта. Дальнейшая очистка дополнительным фильтрованием через короткую колонку с водным силикатом натрия-магния с последующим добавлением гексана давала указанное в заголовке соединение в виде кристаллического твердого вещества (0,66 г), т.пл. 194-195oС.
Пример 3
[4-(4-Метилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 2 с использованием (4-фтор-2-трифторметилфенил)-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,8 г), 60% гидрида натрия в масле (0,15 г), 4-метилпиразола (0,20 г) и диметилформамида (25 мл) получали продукт (0,47 г) в виде бесцветното аморфного твердого вещества. MS (масс-спектр), m/z: 437,3 (М+Н)+, 873,2 (2M+H)+.
Пример 4
(4-Пиразол-1-ил-2-трифторметилфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 2 с использованием (4-фтор-2-трифторметилфенил)-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,20 г), пиразола (0,25 г) и диметилформамида (35 мл) получали продукт (0,62 г) в виде бесцветного аморфного твердого вещества. MS, m/z: 423,2 (М+Н)+, 445,2 (M+Na)+, 845,3 (2М+Н)+.
Пример 5
[4-(3-Циклопропилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 2 с использованием (4-фтор-2-трифторметилфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,42 г), 60% гидрида натрия в масле (0,20 г), 3-циклопропилпиразола (0,43 г) и диметилформамида (50 мл) получали продукт (1,22 г) в виде кристаллического твердого вещества, т.пл. 163-164oС.
Пример 6
[4-(4-Метилимидазол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 2 с использованием (4-фтор-2-трифторметилфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,20 г), 4-метилимидазола (0,25 г) и диметилформамида (25 мл) указанное в заголовке соединение (0,66 г) получали в виде аморфного твердого вещества. MS, m/z: 437,2 (М+Н)+, 873,2 (2М+Н)+.
Пример 7
(5Н, 11Н-Пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-(4-[1,2,4]-триазол-1-ил-2-трифторметилфенил)-метанон
Подобно примеру 2 с использованием (4-фтор-2-трифторметилфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,20 г), 1,2,4-триазола (0,20 г) и диметилформамида (25 мл) получали указанное в заголовке соединение (0,36 г) в виде бесцветного аморфного твердого вещества. MS, m/z: 424,2 (М+Н)+, 847,3 (2М+Н)+.
Пример 8
(2-Хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Оксалилхлорид (2,60 г) добавляли к суспензии 2-хлор-4-фторбензойной кислоты (3,44 г) в дихлорметане (50 мл). Добавляли две капли диметилформамида и смесь перемешивали в течение 18 часов при комнатной температуре. Полученный раствор упаривали с получением неочищенного 2-хлор-4-фторбензоилхлорида в виде вязкого масла (3,72 г).
Неочищенный 2-хлор-4-фторбензоилхлорид (3,68 г) в дихлорметане (25 мл) добавляли порциями к перемешиваемому охлаждаемому льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (2,76 г), диизопропилэтиламина (2,47 г) и дихлорметана (50 мл). После 18 часов при комнатной температуре реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (3,85 г), т.пл. 110-112оС.
Пример 9
[2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (изомер А) и [2-хлор-4-(5-метилпиразол-1-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (изомер В)
Способ 1. К 60% гидриду натрия в масле (0,3 г, обезжиренному гексаном) в диметилформамиде (25 мл) добавляли 3-метилпиразол (0,55 г). После прекращения выделения водорода добавляли (2-хлор-4-фторфенил)-(5Н, 11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (1,70 г). Реакционную смесь нагревали в течение 18 часов на песочной бане (внутренняя температура 125oС). Затем реакционную смесь выливали на лед и дополнительно разбавляли насыщенным солевым раствором. Осажденное твердое вещество извлекали фильтрованием. Неочищенный продукт растворяли в дихлорметане, сушили над безводным сульфатом натрия и затем фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенный элюат кипятили с обратным холодильником на горячей пластине с постепенным добавлением гексана до тех пор, пока не наблюдали образование непрозрачного раствора. При охлаждении получали аморфное твердое вещество. При фильтровании этого материала через вторую колонку с водным силикатом натрия-магния и выпаривании растворителя в вакууме получали смесь региоизомеров 9А и 9В в приблизительном соотношении 9:1 в виде аморфного стекла (1,11 г). MS, m/z: 403,2 (М+Н)+.
Способ 2. К предварительно охлажденной перемешиваемой суспензии промытого гексаном 60% гидрида натрия (3,00 г) в безводном диметилформамиде (250 мл) добавляли по каплям в атмосфере азота 3-метилпиразол (5,50 г) при 0oС. Смесь нагревали до комнатной температуры. После прекращения выделения газа добавляли (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон (17,0 г) в виде твердого вещества и смесь нагревали до 130oС в течение одного часа. Реакционную смесь выливали в ледяную воду, осадок собирали фильтрованием и сушили на воздухе. Осадок растворяли в дихлорметане, сушили над безводным сульфатом натрия и фильтровали через короткую колонку с силикагелем, элюируя этилацетатом. Объединенный фильтрат выпаривали в вакууме до остаточной пены (18,5 г). Очистка и разделение региоизомеров колоночной хроматографией низкого давления на силикагеле с элюированием градиентной смесью этилацетат/гексан (10:90-25:75) давали два очищенных региоизомера.
Изомер А, [2-хлор-4-(3-метилпиразол-1-ил)-фенил]-(5Н,11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (13,5 г) в виде бесцветного аморфного твердого вещества. MS (EI), m/z: 402 (М)+. Пробу (0,5 г) кристаллизовали из диэтилового эфира с последующей перекристаллизацией из этанола с получением региоизомера А (0,275 г) в виде бесцветного кристаллического твердого вещества, т.пл. 141-143oС.
Изомер В, [2-хлор-4-(5-мeтилпиpaзoл-l-ил)-фенил]-(5Н,11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (1,93 г) в виде бесцветного аморфного твердого вещества. Пробу кристаллизовали из диэтилового эфира с последующей перекристаллизацией из метанола с получением региоизомера В (1,4 г) в виде бесцветных игольчатых кристаллов, т.пл. 160-163oС. MS (EI), m/z: 402 (М)+. MS (+FAB), m/z: 403 (М+Н)+.
Пример 10
[2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Стадия а): 2-хлор-4-(3-метилпиразол-1-ил)бензонитрил. К охлажденной (0oС) суспензии гидрида натрия (60% в масле, 2,0 г) в диметилформамиде (50 мл) добавляли порциями 3-метилпиразол (3,39 г). После прекращения выделения газообразного водорода добавляли 2-хлор-4-фторбензонитрил (5,17 г) и смесь перемешивали при комнатной температуре в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и полученный осадок собирали фильтрованием. Неочищенный продукт растворяли в дихлорметане, фильтровали через колонку с безводным силикатом натрия-магния и кристаллизовали путем добавления гексана. Перекристаллизация из этанола давала 4,42 г продукта, т.пл. 148-150оС.
Стадия b): 2-хлор-4-(3-метилпиразол-1-ил)бензамид. Суспензию 2-хлор-4-(3-метилпиразол-1-ил)-бензонитрила (4,35 г) со стадии а) в диметилсульфоксиде (20 мл), содержащем карбонат калия (0,40 г) охлаждали на бане со льдом. Добавляли пероксид водорода (30%, 2,4 мл) и смесь нагревали до комнатной температуры на протяжении 1 часа. Полученный осадок извлекали фильтрованием и перекристаллизовывали из этанола с получением 2,44 г продукта в виде тонких игольчатых кристаллов, т.пл. 159-160oС. MS, m/z: 235,9 (М+Н)+.
Стадия с): 2-хлор-4-(3-метилпиразол-1-ил)бензойная кислота. Раствор 2-хлор-4-(3-метилпиразол-1-ил)бензамида (1,09 г) со стадии b) в водной 75% серной кислоте (25 мл) охлаждали в бане со льдом и добавляли нитрит натрия (1,73 г).
Смесь нагревали до комнатной температуры на протяжении 1 часа и выливали на лед. Осадок собирали фильтрованием и непосредственно использовали в следующей реакции.
Стадия d): [2-хлор-4-(3-метилпиразол-1-ил)-фенил]-(5Н-10,11Н-дигидропирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон. Смесь 2-хлор-4-(3-метилпиразол-1-ил)бензойной кислоты (0,69 г), дихлорметана (25 мл) со стадии с), оксалилхлорида (1,0 г) и 1 капли диметилформамида перемешивали при комнатной температуре в течение 18 часов. Смесь упаривали, поглощали дихлорметаном (25 мл) и добавляли к смеси 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепин (0,51 г) в дихлорметане (25 мл), содержащем диизопропилэтиламин (0,76 г). Смесь перемешивали при комнатной температуре в течение 18 часов и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали и полученное вещество кристаллизовали из диэтилового эфира с получением 0,67 г продукта, т.пл. 137-138оС. MS, m/z: 403,2 (М+Н)+, 805,8 (2М+Н)+.
Пример 11
[2-Хлор-4-(4-метилпиразол-1-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,3 г, обезжирен гексаном), 4-метилпиразола (0,48 г) и диметилформамида (25 мл), получали указанное в заголовке соединение (0,74 г) в виде аморфного твердого вещества. MS, m/z: 403,2 (М+Н)+, 425,2 (M+Na)+, 805,3 (2М+Н)+.
Пример 12
[2-Хлор-4-(4-метилимидазол-1-ил)-фенил] -(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,3 г, обезжирен гексаном), 4-метилимидазола (0,48 г) и диметилформамида (25 мл) получали указанное в заголовке соединение (0,38 г) в виде аморфного твердого вещества. MS, m/z: 403,3 (М+Н)+.
Пример 13
[2-Хлор-4-(3-трифторметилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазелин-10-ил)-метанона (0,8 г), 60% гидрида натрия в масле (0,25 г, обезжирен гексаном), 3-трифторметилпиразола (0,61 г) и диметилформамида (25 мл) получали указанное в заголовке соединение в виде аморфного твердого вещества. MS, m/z: 457,2 (М+Н)+.
Пример 14
[2-Хлор-4-(1,2,4-триазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,7 г), 60% гидрида натрия в масле (0,5 г, обезжирен гексаном), 1,2,4-триазола (0,70 г) и диметилформамида (50 мл) получали указанное в заголовке соединение (0,51 г) в виде аморфного твердого вещества. МS, m/z; 390,3 (М+Н)+, 779,3 (2М+Н)+.
Пример 15
(2-Хлор-4-пиррол-1-ил-фенил)-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (1,7 г), 60% гидрида натрия в масле (0,3 г, обезжирен гексаном), пиррола (0,42 г) и диметилформамида (25 мл) получали указанное в заголовке соединение (0,60 г) в виде аморфного твердого вещества. МS, m/z: 388,2 (М+Н)+.
Пример 16
(2-Хлор-4-пиразол-1-ил-фенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,2 г, обезжирен гексаном), пиразола (0,20 г) и диметилформамида (25 мл) получали указанное в заголовке соединение в виде аморфного твердого вещества. MS, m/z: 389,2 (М+Н)+, 777,1 (2M+H)+.
Пример 17
[2-Хлор-4-(1Н-имидазол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-c] [1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-фторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (2,0 г), 60% гидрида, натрия в масле (0,50 г, обезжирен гексаном), имидазола (0,50 г) и диметилформамида (25 мл) получали указанное в заголовке соединение (0,57 г) в виде рыжевато-коричневого аморфного твердого вещества. MS, m/z: 389 (М+Н)+.
Пример 18
[2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(3-метил-5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Стадия а): 1-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-2,2,2-трифторэтанон. К охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (5,62 г) и диизопропилэтиламина (4,0 г) в дихлорметане (75 мл) добавляли по каплям трифторуксусный ангидрид (7,0 г) в дихлорметане. Эту смесь перемешивали при комнатной температуре в течение 18 часов и промывали водой и насыщенным водным раствором бикарбоната натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением 7,70 г продукта в виде тонкик игольчатых кристаллов, т.пл. 134-135oС. MS, m/z: 281 (М+Н)+.
Стадия b): 1-(3-диметиламинометил-5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-2,2,2-трифторэтанон. Смесь 1-(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-2,2,2-трифторэтанона (2,80 г) со стадии а), бисдиметиламинометана (2,04 г), параформальдегида (2,70 г) и уксусной кислоты (1,20 г) в смеси тетрагидрофурана (50 мл) и метанола (50 мл) перемешивали при комнатной температуре в течение 18 часов. Смесь упаривали в вакууме, добавляли воду и водную смесь экстрагировали дихлорметаном. Объединение экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и остаток кристаллизовали из гексана с получением 2,05 г продукта в виде бесцветного твердого вещества, т.пл. 109-110oC. MS, m/z: 338,3 (M+H)+.
Стадия с): триметил-(10-трифторацетил-10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепин-3-ил-метил)аммонийиодид.
Смесь 1-(3-диметиламинометил-5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-2,2,2-трифторэтанона (1,83 г) со стадии b) и иодметана (1,0 г) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 18 часов. Добавляли диэтиловый эфир и полученный осадок собирали фильтрованием с получением 2,54 г продукта в виде бесцветного твердого вещества, т.пл. 140-155oС (разл.).
Стадия d): 10,11-дигидро-3-метил-5Н-пирроло[2,1-с] [1,4]бензодиазепин. Борoгидрид натрия (2,6 г) добавляли в виде двух порций к кипящей с обратным холодильником смеси триметил-(10-трифторацетил-10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепин-3-ил-метил)аммонийиодида (2,60 г) со стадии с) в этаноле. Через 4 часа эту смесь упаривали в вакууме. К остатку добавляли воду и смесь экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и остаток кристаллизовали из гексана с получением 1,14 г продукта, т.пл. 150-151oС. MS, m/z: 199,1 (М+Н)+.
Стадия е): [2-хлор-4-(3-метилпиразол-1-ил)-фенил]-(3-метил-5Н,11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон. Смесь 2-хлор-4-(3-метилпиразол-1-ил) бензойной кислоты (0,18 г) со стадии с) примера 10, оксалилхлорида (0,18 г) и одной капли диметилформамида в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 18 часов.
Смесь упаривали в вакууме и остаток повторно растворяли в дихлорметане и повторно упаривали в вакууме с получением 2-хлор-4-(3-метилпиразол-1-ил)бензоилхлорида. Суспензию хлорангидрида в дихлорметане (25 мл) добавляли по каплям к раствору 10,11-дигидро-3-метил-5Н-пирроло[2,1-с][1,4]бензодиазепина (0,12 г) и диизопропилэтиламина (0,10 г) в дихлорметане (25 мл). Эту смесь перемешивали при комнатной температуре в течение 18 часов и промывали водой и насыщенным водным раствором бикарбоната натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и растирали с диэтиловым эфиром с получением 0,115 г продукта в виде бесцветных кристаллов, т.пл. 178-180oС. MS, m/z: 417,3 (М+Н)+, 833,3 (2М+Н)+.
Пример 19
(2-Хлор-4-трифторметилпиримидин-5-ил)-(5Н, 11Н-пирродо[2,1-с] [1,4] бензодиазепин-10-ил)-метанон
2-Хлор-4-(трифторметил)пиримидин-5-карбонилхлорид (2,57 г) добавляли постепенно к охлаждаемому льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (1,84 г) и диизопропилэтиламина (1,37 г) в дихлорметане (50 мл). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (3,22 г), т.пл. 221-223oС.
Пример 20
[2-(3-Метилпиразол-1-ил)-4-трифторметилпиримидин-5-ил] -(5Н, 11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К 60% гидриду натрия в масле (0,15 г, обезжирен гексаном) в диметилформамиде (25 мл) добавляли 3-метилпиразол (0,25 г). После прекращения выделения водорода добавляли (2-хлор-4-трифторметилпиримидин-5-ил)-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (0,98 г).
Реакционную смесь нагревали в течение 18 часов в песочной бане (внутренняя температура 110oС). Затем смесь выливали на лед и затем разбавляли насыщенным солевым раствором. Осадок фильтровали, повторно растворяли в дихлорметане и сушили над безводным сульфатом натрия. Очистку выполняли фильтрованием через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенный элюат упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения твердое вещество собирали фильтрованием с получением указанного в заголовке соединения (0,54 г) в виде бесцветных кристаллов, т.пл. 202-204oС.
Пример 21
[2-(4-Метилпиразол-1-ил)-4-трифторметилпиримидин-5-ил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно способу 1 примера 9 с использованием (2-хлор-4-трифторметилпиримидин-5-ил)-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанона (0,98 г), 60% гидрида натрия в масле (0,15 г), 4-метилпиразола (0,42 г) и диметилформамида (25 мл) получали указанное а заголовке соединение (0,73 г) в виде кристаллического твердого вещества, т.пл. 214-217oС.
Пример 22
1-[4-(5Н,11Н-Пирроло[2,l-c][1,4]бензодиазепин-10-карбонил)-фенил]-этанон
4-Ацетилбензойную кислоту (5,0 г) и тионилхлорид (10 мл) нагревали на паровой бане в атмосфере аргона в течение 0,75 часа и летучие вещества удаляли при пониженном давлении. Добавляли толуол и снова удаляли летучие компоненты с получением неочищенного хлорангидрида в виде красно-оранжевого масла. Это соединение склонно к отверждению и его использовали как таковое без дополнительных превращений.
Указанный хлорангидрид (4,56 г) в дихлорметане (25 мл) добавляли порциями к охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-c][1,4]бензодиазепина (3,68 г) и диизопропилэтиламина (3,25 г) в дихлорметане (100 мл). После перемешивания при комнатной температурю в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (1,75 г), т.пл. 135-137oС.
Пример 23
3-Диметиламино-1-[4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-фенил]-2-пропен-1-он
Реакционную смесь 1-[4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-фенил] -этанона (1,40 г), трет-бутоксибисдиметиламинометана (5,0 мл) и дихлорметана (10 мл) перемешивали в течение 18 часов. Красно-оранжевый осадок фильтровали с получением указанного в заголовке соединения (1,22 г), т. пл. 203-205oС. Дополнительный продукт (0,18 г) выделяли из реакционной смеси упариванием.
Пример 24
[4-(1Н-Пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Реакционную смесь 3-диметиламино-1-[4-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-фенил] -2-пропен-1-она (1,0 г), безводного гидразина (0,20 г) и ледяной уксусной кислоты (20 мл) нагревали с обратным холодильником в течение 7 часов и упаривали досуха. Неочищенный остаток растворяли в дихлорметане, промывали насыщенным водным раствором бикарбоната натрия и сушили над безводным сульфатом натрия. Раствор фильтровали через короткую колонку с водным силикатом натрия-магния, элюируя несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием. Процедуру с применением колонки повторяли с получением указанного в заголовке соединения (0,65 г), т.пл. 219-221oС.
Пример 25
[4-(1-Метил-1Н-пиразолил-3-ил)-фенил] -(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К смеси 6,0% гидрида натрия в масле (0,35 г, обезжирен гексаном) и диметилформамида (20 мл) добавляли [4-(1Н-пиразол-3-ил)-фенил]-(5Н,11H-пирроло[2,1-с] [1, 4]бензодиазепин-10-ил)-метанон (0,98 г) с последующим добавлением через несколько минут иодметана (0,50 г). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре и затем выливали в воду и экстрагировали дихлорметаном. После высушивания органический слой фильтровали через короткую колонку с водным силикатом натрия-магния, элюируя несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,70 г), т.пл. 194-195oС.
Пример 26
[4-(1-Этил-1Н-пиразол-3-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 25 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,27 г), диметилформамида (25 мл) и этилиодида (0,87 г) получали указанное в заголовке соединение (0,69 г) в виде кристаллического твердого вещества, т.пл. 180-183oС.
Пример 27
[4-(1-Пропил-1Н-пиразол-3-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 25 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,98 г), 60% гидрида натрия в масле (0,30 г), диметилформамида (25 мл) и 1-иодпропана (0,60 г) получали указанное в заголовке соединение (0,32 г) в виде кристаллического твердого вещества, т.пл. 159-161oC.
Пример 28
[4-(1-Бутил-1Н-пиразол-3-ил)-фенил] -(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 25 с использованием [4-(1Н-пиразол-3-ил)-фенил]-(5Н, 11-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,98 г), 60% гидрида натрия в масле (0,30 г), диметилформамида (25 мл) и 1-иодбутана (0,60 г) указанное в заголовке соединение (0,32 г) получали в виде кристаллического твердого вещества, т.пл. 122-123oС.
Пример 29
[4-(1-Метоксиметил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Подобно примеру 25 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (1,0 г), 60% гидрида натрия в масле (0,15 г), диметилформамида (25 мл) и иодметилметилового простого эфира (0,50 г) получали указанное в заголовке соединение (0,26 г) в виде аморфного твердого вещества. MS, m/z: 399,2 (М+Н)+, 797,2 (2М+Н)+.
Пример 30
1-{3-[4-(5Н,11Н-Пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-фенил]-пиразол-1-ил}-этанон
К раствору [4-(1Н-пиразол-3-ил)-фенил]-(5Н,11H-пиролло[2,1-c][1, 4]бензодиазепин-10-ил)-метанона (0,50 г) в безводном пиридине (10 мл) добавляли уксусный ангидрид (0,20 г). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь выливали в воду и осадок собирали фильтрованием. Неочищенный продукт растворяли в дихлорметане и сушили над безводным сульфатом натрия. Этот раствор фильтровали через короткую колонку с водным силикатом натрия-магния, элюируя несколькими объемами дихлорметана. Элюат упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,46 г), т.пл. 192-194oС.
Пример 31
1-{3-[4-(5Н,11H-Пиролло[2,1-с][1,4]бензодиазепин-10-карбонил)-фенил]-пиразол-1-ил)-пропан-1-он
Подобно примеру 30 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1, 4]бензодиазепин-10-ил)-метанона (0,16 г) в безводном пиридине (10 мл) и пропионового ангидрида (0,10 г) получали указанное в заголовке соединение (0,17 г) в виде кристаллического твердого вещества, т.пл. 150-152oС.
Пример 32
[4-(1-Циклопропанкарбонил-1Н-пиразол-3-ил)-фенил]-(5Н,11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил]-метанон
К раствору [4-(1Н-пиразол-3-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанона (1,0 г) в безводном пиридине (10 мл) добавляли циклопропанкарбонилхлорид (0,44 г). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь выливали в воду и осадок собирали фильтрованием. Неочищенный продукт растворяли в дихлорметане и сушили над безводным сульфатом натрия. Этот раствор фильтровали через короткую колонку с водным силикатом натрия-магния, элюируя несколькими объемами дихлорметана. Элюат упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,88 г) в виде кристаллического твердого вещества, т.пл. 197-199oС.
Пример 33
1-{3-[4-(5Н,11Н-Пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-фенил]-пиразол-1-ил}-бутан-1-он
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,71 г) в безводном пиридине (10 мл) и бутирилхлорида (0,32 г) получали указанное в заголовке соединение (0,54 г) в виде твердого вещества, т.пл. 105-110oС. MS, m/z: 424 (М)+.
Пример 34
(5Н, 11Н-Пирроло[2, l-c][1,4]бензодиазепин-10-ил)-{4-[1-(тиофен-2-карбонил)-1Н-пиразол-3-ил]фенил}-метанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11H-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (0,5 г) в безводном пиридине (10 мл) и тиофен-2-карбонилхлорида (0,25 г) получали указанное в заголовке соединение (0,41 г) в виде кристаллического твердого вещества, т. пл. 195-197oС. MS, m/z: 464 (М)+.
Пример 35
{ 4-[1-(5-Фтор-2-метилбензоил)-1H-пиразол-3-ил] -фенил} -5Н, 11Н-пирроло[2,1-c][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (0,35 г) в безводном пиридине (10 мл) и 2-метил-5-фторбензоилхлорида (0,22 г) указанное в заголовке соединение (0,11 г) получали в виде аморфного бледно-желтого твердого вещества. MS, m/z: 490 (М)+.
Пример 36
{ 4-[1-(2-Метилбензоил)-1H-пиразол-3-ил] -фенил} -(5Н,11Н-пирроло[2,1-c] [1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (0,71 г) в безводном пиридине (10 мл) и о-толуилхлорида (0,39 г) получали указанное в заголовке соединение (0,59 г) в виде кристаллического твердого вещества, т.пл. 170-173oС. MS, m/z: 472 (М)+.
Пример 37
{ 4-[1-(2-Хлор-4-фторбензоил)-1H-пиразол-3-ил] -фенил} -(5Н, 11H-пирроло[2,1-c][1,4]бензодиазепин-10-ил)-метанон
2-Хлор-4-фторбензоилхлорид (0,82 г) добавляли порциями к раствору [4-(1H-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,l-c][1,4]бензодиазепин-10-ил)-метанона (1,0 г) и диизопропиламина (0,55 г) в дихлорметане (25 мл), который охлаждали в бане со льдом. Реакционной смеси давали перемешиваться при комнатной температуре в течение ночи. Реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия и сушили над безводным сульфатом натрия. Раствор в дихлорметане пропускали через короткую колонку с водным силикатом натрия-магния, элюируя несколькими объемами дихлорметана. Элюат упаривали досуха с получением 1,06 г продукта в виде твердого вещества, т. пл. 150-157oС. MS, m/z: 510 (М)+.
Пример 38
{4-[1-(2,4-Дихлорбензоил)-1Н-пиразол-3-ил]-фенил}-(5Н,11Н-пирроло[2,l-c] [1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-метанона (0,71 г) в безводном пиридине (20 мл) и 2,4-дихлорбензоилхлорида (0,52 г) получали указанное в заголовке соединение (0,66 г) в виде кристаллического твердого вещества, т. пл. 180-182oС. MS, m/z: 528 (М)+.
Пример 39
2-(2,4-Дихлорфенил)-1-{ 3-[4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-фенил]-пиразол-1-ил}-этанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,71 г) в безводном пиридине (25 мл) и 2,4-дихлорфенилацетилхлорида (0,56 г) получали указанное в заголовке соединение (0,20 г) в виде кристаллического твердого вещества, т.пл. 130-140oС, повторно отверждается, т.пл. 180-182oС.
Пример 40
{ 4-[1-(Бифенил-2-карбонил)-1Н-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 32 с использованием [4-(lH-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанона (0,71 г) в сухом пиридине (20 мл) и 2-бифенилкарбонилхлорида (0,65 г) получали указанное в заголовке соединение (0,49 г) в виде аморфного твердого вещества. MS, m/z: 534 (М)+.
Пример 41
{ 4-[1-(4'-Трифторметилбифенил-2-карбонил)-1H-пиразол-3-ил] -фенил}-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Подобно примеру 32 с использованием [4-(1H-пиразол-3-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанона (0,71 г) в безводном пиридине (20 мл) и 4'-трифторметил-2-бифенилкарбонилхлорида (0,71 г) получали указанное в заголовке соединение (0,59 г) в виде аморфного твердого вещества. MS, m/z: 602 (М)+.
Пример 42
3-Диметиламино-1-[4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-фенил]-2-бутен-1-он
Смесь 1-[4-(5Н,11Н-пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-фенил] -этанона (2,0 г) и диметилацеталя диметилацетамида (15 мл) кипятили с обратным холодильником в инертной атмосфере в течение 15 часов и удаляли летучие компоненты при пониженном давлении. Неочищенное твердое вещество растворяли в дихлорметане и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенный элюат упаривали и постепенно добавляли гексан до тех пор, пока не происходила кристаллизация. Охлажденный раствор фильтровали с извлечением указанного в заголовке соединения (1,03 г) в виде кристаллического твердого вещества, т.пл. 183-185oС.
Пример 43
[4-(5-Метил-1Н-пиразол-3-ил)-фенил]-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Безводный гидразин (0,10 г) добавляли к раствору 3-диметиламино-1-[4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-фенил]-2-бутен-1-она (0,50 г) в ледяной уксусной кислоте (25 мл). Реакционную смесь кипятили с обратным холодильником в течение 18 часов и затем упаривали в вакууме. Твердое вещество экстрагировали дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия, раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на паровой бане при постепенном добавлении гексана с получением непрозрачного раствора. После охлаждения аморфное твердое вещество извлекали фильтрованием с получением продукта (0,33 г). MS, m/z: 368 (М)+.
Пример 44
4-(5Н,11H-Пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-бензонитрил
4-Цианобензойную кислоту (5,0 г) и тионилхлорид (5,0 мл) нагревали на паровой бане в течение одного часа и все летучие компоненты удаляли при пониженном давлении. Добавляли гексан и неочищенный кристаллический хлорангидрид (5,30 г) извлекали фильтрованием и использовали без дополнительной очистки.
К реакционной смеси 10,11-дигидро-5Н-пирроло[2,1-c][1,4]бензодиазепина (3,68 г), дихлорметана (100 мл) и диизопропилэтиламина (2,80 г) добавляли 4-цианобензоилхлорид (2,97 г). После оставления при комнатной температуре на 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (5,05 г), т.пл. 184-186oС.
Пример 45
4-(5Н,11H-Пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-бензамид
4-(5Н,11Н-Пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-бензонитрил (0,5 г) из примера 44 добавляли к концентрированной серной кислоте (5 мл) и эту смесь перемешивали в течение 18 часов при комнатной температуре с получением ярко-желтого раствора. Раствор выливали на лед и подщелачивали добавлением концентрированного гидроксида аммония. Полученное твердое вещество фильтровали, растворяли в дихлорметане и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (5,05 г), т.пл. 226-228oC.
Пример 46
N-(Диметиламинометилен)-4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамид
Смесь 4-(5Н,11Н-пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-бензамида (1,25 г) из примера 45 и диметилацеталя диметилформамида (20 мл) кипятили с обратным холодильником в течение 4 часов и летучие компоненты удаляли в вакууме с получением твердого вещества. Это твердое вещество растворяли в дихлорметане и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на паровой бане с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (1,40 г), т.пл. 232-234oС.
Пример 47
N-(1-Диметиламиноэтилен)-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензамид
Смесь 4-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамида (1,24 г) из примера 45 и диметилацеталя диметилацетамида (5 мл) нагревали на паровой бане в течение 4 часов. При охлаждении в течение 18 часов осаждалось кристаллическое твердое вещество, которое извлекали фильтрованием. Твердое вещество промывали гексаном с получением продукта (1,54 г), т.пл. 210-212oС, MS, m/z: 400 (М)+.
Пример 48
(5Н, 11Н-Пирроло[2,1-с][1,4]бензодиазепин-10-ил)-[4-(2Н-[1,2,4]триазол-3-ил)-фенил]-метанон
Смесь N-(диметиламинометилен)-4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамида (1,0 г) из примера 46, ледяной уксусной кислоты (15 мл) и безводного гидразина (0,16 г) кипятили с обратным холодильником в течение 15 часов и летучие компоненты удаляли в вакууме. Добавляли насыщенный водный раствор бикарбоната натрия и полученное твердое вещество извлекали фильтрованием. Твердое вещество нагревали с обратным холодильником в течение 4 часов и летучие компоненты удаляли в вакууме с получением твердого вещества. Это твердое вещество растворяли в дихлорметане и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на паровой бане с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,39 г), т.пл. 225-227oС. MS, m/z: 355 (М)+.
Пример 49
[4-(2-Метил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н,11Н-пирроло[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Подобно примеру 48 с использованием N-(диметиламинометилен)-4-(5Н,11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-бензамида (1,56 г) из примера 46 в ледяной уксусной кислоте (75 мл) и метилгидразина (0,32 г) получали указанное в заголовке соединение (0,10 г) в виде твердого вещества, т.пл. 155-158oС. MS, m/z: 369 (М)+.
Пример 50
[4-(5-Метил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н,11H-пирроло[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Подобно примеру 48 с использованием N-(1-диметиламиноэтилен)-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензамида (1,00 г) из примера 47 в ледяной уксусной кислоте (75 мл) и безводного гидразина (0,25 г) указанное в заголовке соединение (0,20 г) получали в виде аморфного твердого вещества. MS, m/z: 369 (М)+.
Пример 51
[4-(2,5-Диметил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазедин-10-ил)-метанон
Подобно примеру 48 с использованием N-(1-диметиламиноэтилен)-4-(5Н, 11Н-пирроло[2, l-c] [1,4] бензодиазепин-10-карбонил)-бензамида (1,18 г) из примера 47 в ледяной уксусной кислоте (75 мл) и метилгидразина (0,30 г) указанное в заголовке соединение (0,33 г) получали в виде твердого вещества, т.пл. 193-195oС. MS, m/z: 383 (М)+.
Пример 52
[4-(3-Метил[1,2,4] оксадиазол-5-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Раствор N-(1-диметиламиноэтилен)-4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамида (1,15 г) из примера 47 в ледяной уксусной кислоте (50 мл), содержащий гидрохлорид гидроксиламина (0,40 г) и ацетат калия (1,0 г), кипятили с обратным холодильником в течение 2 часов. Все
летучие компоненты удаляли при пониженном давлении и добавляли насыщенный водный раствор бикарбоната натрия. Эту смесь экстрагировали дихлорметаном и экстракты сушили над безводным сульфатом натрия. Раствор фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу концентрировали на паровой бане с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,38 г), т.пл. 177-179oС. MS, m/z: 371,3 (М)+, 741,3 (2М)+.
Пример 53
1-Метил-4-(4-метилфенил)-lH-пиразол
Смесь 2-(4-метилфенил)-малонового диальдегида (3,05 г), абсолютного этанола (40 мл) и метилгидразина (1,09 г) перемешивали при комнатной температуре в течение 18 часов и летучие компоненты удаляли при комнатной температуре. Добавляли воду и смесь экстрагировали дихлорметаном. После высушивания над безводным сульфатом натрия раствор фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на паровой бане с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (2,91 г), т.пл. 107-108oС.
Пример 54
4-(1-Метил-1Н-пиразол-4-ил)-бензойная кислота
Смесь 1-метил-4-(4-метилфенил)-1H-пиразола (1,70 г), перманганата калия (9,70 г) и 1 н. гидроксида натрия (100 мл) кипятили с обратным холодильником 18 часов. Суспензию фильтровали через диатомовую землю и охлаждали. Водный раствор экстрагировали дихлорметаном, который отбрасывали. Водный раствор подкисляли до рН 5,5. Полученный осадок фильтровался с трудом, и поэтому его экстрагировали дихлорметаном. После выпаривания растворителя полученное твердое вещество перекристаллизовывали из ацетона с получением указанного в заголовке соединения (0,60 г), т.пл. 274-275oС. MS, m/z: 202 (М)+.
Пример 55
[4-(1-Метил-1Н-пиразол-4-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Оксалилхлорид (0,30 г) добавляли к суспензии 4-(1-метил-1Н-пиразол-4-ил)-бензойной кислоты (0,46 г) в дихлорметане (25 мл). Добавляли две капли диметилформамида и смесь перемешивали в течение 16 часов при комнатной температуре. Полученный раствор упаривали досуха с получением неочищенного хлорангидрида (0,57 г), который использовали без дополнительной очистки.
Этот хлорангидрид добавляли к раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепина (0, 37 г) и диизопропилэтиламина (0,58 г) в дихлорметане (50 мл). После 18 часов при комнатной температуре реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,38 г), т.пл. 200-201oС. МS, m/z: 368 (М)+.
Пример 56
6-(1-Метил-1Н-пиразол-4-ил)-пиридин-3-карбоновая кислота
Суспензию 6-(1-формил-2-гидроксивинил)пиридин-3-карбоновой кислоты (1,93 г) (Eastroao Chemicals) в абсолютном этаноле (50 мл) и метилгидразина (0,50 г) перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь фильтровали с получением продукта (1,30 г). Фильтрат упаривали с получением твердого вещества, которое перекристаллизовывали из этилацетата с получением аналитической пробы указанного в заголовке соединения (0,55 г), т. пл. 262-264oС.
Пример 57
[6-(1-Метил-1Н-пиразол-4-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Суспензию 6-(1-метил-1Н-пиразол-4-ил)-пиридин-3-карбоновой кислоты (0,48 г) в тионилхлориде (5,0 мл) перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удаляли при пониженном давлении с получением 6-(1-метил-1Н-пиразол-4-ил)-пиридин-3-карбонилхлорида в виде твердого вещества, которое использовали без дополнительной очистки.
Раствор 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (0,37 г) и диизопропилэтиламина (0,61 г) в дихлорметане (25 мл) охлаждали до 0оС и порциями добавляли раствор 6-(1-метил-1Н-пиразол-4-ил)-пиридин-3-карбонилхлорида в дихлорметане (25 мл). После 18 часов при комнатной температуре реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,31 г), т.пл. 173-175oС. MS, m/z: 370,3 (М+Н)+.
Пример 58
[4-(Пиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К суспензии 4-(пиразол-1-ил)бензойной кислоты (1,56 г) в дихлорметане (25 мл) добавляли оксалилхлорид (1,04 г) и одну каплю диметилформамида. Смесь перемешивали при комнатной температуре в течение 18 часов с получением прозрачного раствора. Летучие вещества удаляли при пониженном давлении с получением 4-(пиразол-1-ил) бензоилхлорида в виде бледно-желтого твердого вещества (1,58 г), которое использовали без дополнительной очистки.
4-(Пиразол-1-ил)бензоилхлорид (0,75 г) добавляли к раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (0,61 г) и диизопропилэтиламина (0,47 г) в дихлорметане (25 мл). После 18 часов при комнатной температуре реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постеленным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,90 г), т.пл. 179-181oС.
Пример 59
[4-(3-Метилпиразол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К суспензии 4-(3-метилпиразол-1-ил) бензойной кислоты (1,84 г) в дихлорметане (25 мл) добавляли оксалилхлорид (1,16 г) и одну каплю диметилформамида. Смесь перемешивали при комнатной температуре в течение 18 часов и летучий материал удаляли при пониженном давлении. Добавляли дихлорметан, раствор фильтровали и растворитель выпаривали при пониженном давлении с получением 4-(3-метилпиразол-1-ил)бензоилхлорида в виде желтого масла (1,76 г), которое использовали без дополнительной очистки.
4-(3-Метилпиразол-1-ил)бензоилхлорид добавляли к охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепина (0,55 г) и диизопропилэтиламина (0,44 г) в дихлорметане (25 мл). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния и затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения получали указанное в заголовке соединение (0,90 г) в виде аморфного твердого вещества. MS, m/z: 369 (М+Н)+.
Пример 60
[4-(4-Метилпиразол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К суспензии 4-(4-метилпиразол-1-ил)бензойной кислоты (0,75 г) в дихлорметане (15 мл) добавляли оксалилхлорид (0,50 г) и одну каплю диметилформамида. Смесь перемешивали при комнатной температуре в течение 18 часов и летучие вещества удаляли при пониженном давлении. Остаток растворяли в гексане и фильтровали через диатомовую землю. Выпаривание растворителя в вакууме давало 4-(4-метилпиразол-1-ил)бензоилхлорид (0,77 г), который использовали без дополнительной очистки.
4-(4-Метилпиразол-1-ил)бензоилхлорид (0,72 г) добавляли к охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (0,60 г) и диизопропилэтиламина (0,48 г) в дихлорметане (25 мл). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,75 г), т.пл. 179-181oС. MS, m/z: 369 (М+Н)+.
Пример 61
[4-(3,5-Диметилпиразол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
К суспензии 4-(3,5-диметилпиразол-1-ил) бензойной кислоты (1,34 г) в дихлорметане (25 мл) добавляли оксалилхлорид (1,0 г) и одну каплю диметилформамида. Смесь перемешивали при комнатной температуре в течение 18 часов и летучие вещества удаляли при пониженном давлении. Остаток растворяли в гексане и фильтровали через диатомовую землю. Выпаривание растворителя в вакууме давало 4-(3,5-диметилпиразол-1-ил)бензоилхлорид (0,80 г), который использовали без дополнительной очистки.
4-(3,5-Диметилпиразол-1-ил)бензоилхлорид (0,75 г) добавляли к охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (0,55 г) и диизопропилэтиламина (0,42 г) в дихлорметане (25 мл). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения указанное в заголовке соединение (0,79 г) получали в виде аморфного твердого вещества. MS, m/z: 383 (M+H)+.
Пример 62
(5Н, 11Н-Пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-[4-(3-трифторметилпиразол-1-ил)-фенил]-метанон
Суспензию 4-(3-трифторметилпиразол-1-ил)бензойной кислоты (1,45 г) в тионилхлориде (5,0 мл) нагревали с обратным холодильником в течение 3 часов. Летучие вещества удаляли при пониженном давлении, остаток растворяли в дихлорметане и фильтровали через диатомовую землю. Выпаривание растворителя в вакууме давало 4-(3-трифторметилпиразол-1-ил)бензоилхлорид (1,45 г), который использовали без дополнительной очистки.
К раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (0,88 г) и диизопропилэтиламина (0,66 г) в дихлорметане (50 мл) добавляли 4-(3-трифторметилпиразол-1-ил)бензоилхлорид (1,40 г). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (1,70 г), т.пл. 166-167oС. MS, m/z: 423,3 (М+Н)+, 845,4 (2М+H)+.
Пример 63
[4-(Имидазол-1-ил)-фенил] -(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Суспензию 4-(имидазол-1-ил)бензойной кислоты (0,90 г) в тионилхлориде (2,0 мл) нагревали на паровой бане в атмосфере аргона в течение одного часа. Выпаривание летучих веществ при пониженном давлении давало остаток, который кристаллизовали при добавлении гексана с получением 4-(имидазол-1-ил)бензоилхлорида в виде гидрохлоридной соли (1,17 г), т.пл. 242-247oС.
К раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (0,75 г), диизопропилэтиламина (1,20 г) и 4-диметиламинопиридина в дихлорметане (50 мл) добавляли гидрохлорид 4-(имидазол-1-ил)бензоилхлорида (1,12 г). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, а затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением указанного в заголовке соединения (0,57 г), т.пл. 171-172oС. MS, m/z: 354 (M+H)+.
Пример 64
[4-(4-Метилимидазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
К суспензии 4-(4-метилимидазол-1-ил)бензойной кислоты (0,80 г) в дихлорметане (25 мл) добавляли оксалилхлорид (0,50 г) и одну каплю диметилформамида. Смесь перемешивали при комнатной температуре в течение 18 часов и летучие вещества удаляли при пониженном давлении с получением 4-(4-метилимидазол-1-ил)бензоилхлорида (1,02 г), который использовали без дополнительной очистки.
4-(4-Метилимидазол-1-ил)бензоилхлорид (0,99 г) добавляли к охлажденному льдом раствору 10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепина (0,64 г) и диизопропилэтиламина (0,60 г) в дихлорметане (25 мл). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь промывали водой и насыщенным водным раствором бикарбоната натрия. Раствор в дихлорметане сушили над безводным сульфатом натрия и фильтровали через короткую колонку с водным силикатом натрия-магния, затем элюировали несколькими объемами дихлорметана. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения получали указанное в заголовке соединение (0,52 г) в виде твердого вещества, т.пл. 140-145oС. MS, m/z: 369 (М+Н)+.
Пример 65
4-Бром-2-хлорбензойная кислота, метиловый эфир
Тионилхлорид (1,64 мл) добавляли по каплям к суспензии 4-бром-2-хлорбензойной кислоты (6,92 г) в метаноле и нагревали до 60oС в течение 2 часов. Растворитель удаляли в вакууме, остаток повторно растворяли в этилацетате и промывали последовательно 0,5 н. гидроксидом натрия (2х), водой и солевым раствором. Органическую фазу сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением указанного в заголовке соединения (7,8 г).
1Н-ЯМР (300 МГц, ДМСО-d6), δ: 3,87 (с, 3Н), 7,68-7,9 (м, 3Н).
Пример 66
2-Хлор-4-(3-диметиламинопропин-1-ил)бензойная кислота, метиловый эфир
К перемешиваемому раствору метилового эфира 4-бром-2-хлорбензойной кислоты (18,69 г) в триэтиламине (110 мл) добавляли 1-диметиламино-2-пропин (12,1 мл), бис(трифенилфосфин)палладий(II) хлорид (1,26 г) и иодид меди(I) (0,136 г).
Эту смесь медленно нагревали до 60oС и указанную температуру поддерживали в течение одного часа. Реакцию охлаждали до комнатной температуры, фильтровали через диатомовую землю и собранное твердое вещество промывали этилацетатом. Растворитель удаляли в вакууме, полученное твердое вещество повторно растворяли в этилацетате и промывали водой (3х). Объединенный органический экстракт сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (225 г), элюируя смесью 40% этилацетат/гексан. После удаления растворителя в вакууме указанное в заголовке соединение получали в виде вязкого масла (17,7 г). MS (+FАB), m/z: 252 (М+Н)+.
Пример 67
2-Хлор-4-(3-диметиламино-2-пропен-1-он-1-ил)-бензойная кислота, метиловый эфир
3-Хлорпероксибензойную кислоту (10,76 г) постепенно добавляли к раствору метилового эфира 2-хлор-4-(3-диметиламинопропин-1-ил)бензойной кислоты (15,07 г) в дихлорметане (40 мл) со скоростью, достаточной для поддержания температуры реакции при -20oС. Смесь перемешивали в течение 10-15 минут. Полученный N-оксид очищали хроматографией на основном оксиде алюминия Activity Grade I (215 г), элюируя смесью 10% метанол/дихлорметан. Растворитель удаляли в вакууме при температуре между 12 и 18oС. Полученный остаток растворяли в метаноле (100 мл) и нагревали при 60-65oС при перемешивании в течение 18 часов. После удаления растворителя в вакууме продукт очищали колоночной хроматографией на силикагеле (190 г), элюируя смесью 70% этилацетат/гексан. Растирание с диэтиловым эфиром, содержащим небольшое количество гексана, давало указанное в заголовке соединение в виде твердого вещества (5,68 г), т.пл. 92-96oС.
Пример 68
2-Хлор-4-(1Н-пиразол-3-ил)-бензойная кислота, метиловый эфир
К суспензии метилового эфира 2-хлор-4-(3-диметиламино-2-пропен-1-он-1-ил)-бензойной кислоты (13,67 г) в этаноле (53 мл) добавляли моногидрохлорид гидразина (7,0 г). Эту смесь нагревали на масляной бане при 75-80oС в течение одного часа. Растворитель удаляли в вакууме. Полученный остаток растворяли в этилацетате, промывали водой и солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением указанного в заголовке соединения в виде неочищенного твердого вещества (12 г). Очищенная проба имела т.пл. 130-131oС.
Пример 69
2-Хлор-4-(1-метил-1Н-пиразол-3-ил)-бензойная кислота, метиловый эфир
К суспензии промытого гексаном гидрида натрия (3,05 г, 60% дисперсия) в диметилформамиде (6 мл) в атмосфере азота добавляли раствор метилового эфира 2-хлор-4-(1H-пиразол-3-ил)-бензойной кислоты (12,0 г) в диметилформамиде (30 мл) на протяжении 15 минут. Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли по каплям иодметан (9,5 мл) на протяжении 15 минут. Смеси давали перемешиваться при комнатной температуре в течение 45 минут. Добавляли дополнительное количество иодметана (5,16 мл) и реакцию перемешивали еще в течение 75 минут. Реакцию разбавляли небольшим количеством воды и упаривали в вакууме. Остаток разбавляли водой (500 мл) и экстрагировали небольшим количеством этилацетата (5х). Объединенную органическую фазу упаривали в вакууме с получением неочищенного продукта (13,48 г). Неочищенный продукт очищали колоночной хроматографией на силикагеле (195 г), элюируя смесью 15% этилацетат/гексан с получением чистого 1-метил-региоизомера (4,29 г) с последующим получением смеси 1-метил- и 2-метил-региоизомеров (4,6 г). Смесь изомеров растирали с гексаном три раза с получением дополнительной пробы чистого 1-метил-региоизомера (2,55 г), т.пл. 66,5-67oС. MS (+FAB), m/z: 251 (М+Н)+.
Пример 70
2-Хлор-4-(1-метил-1Н-пиразол-3-ил)-бензойная кислота.
К раствору метилового эфира 2-хлор-4-(1-метил-1Н-пиразол-3-ил)-бензойной кислоты (6,85 г) в метаноле (32 мл) добавляли 2,5 н. раствор гидроксида натрия (15,3 мл). Реакцию нагревали до 50oС в течение одного часа. Растворитель удаляли в вакууме и остаток растворяли в воде (250 мл), охлаждали в бане со льдом и подкисляли 2 н. хлористоводородной кислотой (24 мл). Полученный осадок отфильтровывали и сушили с получением бесцветного твердого вещества (6,3 г), т.пл. 232-233oС. MS (+FAB), m/z: 236 (М+Н)+.
Пример 71
[2-Хлор-4-(1-метил-1Н-пиразол-3-ил)-фенил] -(5Н,11H-пирроло-[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Хорошо измельченную в порошок 2-хлор-4-(1-метил-1H-пиразол-3-ил)-бензойную кислоту (6,3 г) и диметилформамид (2,16 мл) суспендировали в атмосфере азота в смеси тетрагидрофурана (70 мл) и дихлорметана (15 мл). Добавляли по каплям раствор оксалилхлорида (2,43 мл) в дихлорметане (5 мл) и реакцию перемешивали в течение одного часа. Полученную суспензию 2-хлор-4-(1-метил-1Н-пиразол-3-ил)-бензоилхлорида использовали без дополнительной очистки.
К суспензии 10,11-дигидро-5Н-пирроло[2,1-c][1,4]бензодиазепина (4,93 г) в дихлорметане (15 мл) добавляли диизопропилэтиламин (7 мл). Суспензию свежеприготовленного хлорангидрида постепенно добавляли на протяжении 15 минут под подаваемым насосом током азота. Слегка теплую реакционную смесь перемешивали в атмосфере азота в течение 50 минут. После перемешивания в течение одного часа смесь упаривали в вакууме. Остаток растворяли в дихлорметане, промывали водой, 5% бикарбонатом натрия и водой. Органическую фазу промывали солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением неочищенного продукта (10,95 г). Неочищенный продукт очищали колоночной хроматографией на силикагеле (200 г), нагружая колонку смесью 25% этилацетат/гексан. Менее полярные примеси элюировали смесью 25-30% этилацетат/гексан. Продукт элюировали смесью 30-40% этилацетат/гексан с получением чистой пробы (7,42 г), которую, после внесения затравочных кристаллов, растирали с диэтиловым эфиром, содержащим некоторое количество гексана, в течение 24 часов. Фильтрование давало указанное в заголовке соединение в виде кристаллического твердого вещества (6,88 г), т.пл. 148,5-150oС. MS (EI), m/z: 402 (М)+.
Пример 72
2-Хлор-4-(2-метил-1Н-пиразол-3-ил)-бензойная кислота, метиловый эфир
Указанное в заголовке соединение получали так же, как описано в примере 68, с использованием метил-2-хлор-4-(3-диметиламино-2-пропен-1-он)-бензоата (0,8 г) и метилгидразина (0,319 мл). Основной 2-метил-региоизомер выделяли колоночной хроматографией на силикагеле.
1Н-ЯМР (300 МГц, ДМСО-d6), δ: 3,87 (с, 3Н), 3,89 (с, 3Н), 6,58 (д, 1Н), 7,5 (д, 1Н), 7,62-7,93 (м, 3Н).
Пример 73
2-Хлор-4-(2-метил-1Н-пиразол-3-ил)-бензойная кислота
Указанное в заголовке соединение получали так же, как описано в примере 70, с использованием метилового эфира 2-хлор-4-(2-метил-1Н-пиразол-3-ил)-бензойной кислоты (0,464 г) и 2,5 н. гидроксида натрия (1,04 мл).
1Н-ЯМР (300 МГц, ДМСО-d6), δ: 3,89 (с, 3Н), 6,56 (д, 1Н), 7,49 (д, 1Н), 7,59-7,90 (м, 3Н).
Пример 74
[2-Хлор-4-(2-метил-1Н-пиразол-3-ил)-фенил] -(5Н,11Н-пирроло-[2,1-с][1,4] бензодиазепин-10-ил)-метанон
Указанное в заголовке соединение получали так же, как описано в примере 71, с использованием 2-хлор-4-(2-метил-1Н-пиразол-3-ил)-бензойной кислоты (3,98 г), дающей соответствующий хлорангидрид, и ацилирование 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепином (0,293 г) давало указанное в заголовке соединение в виде пены, т.пл. 78-79oС. MS (EI), m/z: 402 (М)+.
Пример 75
2-Хлор-4-цианобензойная кислота, метиловый эфир
Метиловый эфир 2-хлор-4-аминобензойной кислоты (13,95 г) суспендировали в смеси воды (65 мл) и концентрированной хлористоводородной кислоты (15,7 мл). После перемешивания при комнатной температуре в течение 10 минут суспензию охлаждали до 0oС. Раствор нитрита натрия (5,71 г) в воде (37 мл) постепенно добавляли на протяжении 20 минут, поддерживая температуру реакции 0оС. После перемешивания при 0oС в течение 35 минут реакционную смесь частично нейтрализовали добавлением твердого карбоната натрия (3,16 г) с получением холодного раствора соли диазония.
К предварительно охлажденному раствору цианида меди(I) (8,4 г) и цианида натрия (9,19 г) в воде (112 мл) постепенно добавляли вышеупомянутый раствор соли диазония на протяжении периода 45-50 минут. Температуру раствора соли диазония во время добавления поддерживали при 0oС. Полученную смесь перемешивали в течение 18 часов при комнатной температуре. Осадок фильтровали, сушили на воздухе, растворяли в этилацетате (250 мл) и фильтровали для удаления нерастворимого материала. Органическую фазу сушили над безводным сульфатом магния и растворитель удаляли в вакууме с получением неочищенного продукта в виде коричневого твердого вещества (13,2 г). Неочищенный продукт очищали колоночной хроматографией на силикагеле (250 г), элюируя смесью 5-10% этилацетат/гексан с получением указанного в заголовке соединения (10,9 г) в виде твердого вещества, т.пл. 90-92oС. MS (ЕI), m/z: 195 (М)+.
Пример 76
2-Хлор-4-цианобензойная кислота.
К перемешиваемому раствору метилового эфира 2-хлор-4-цианобензойной кислоты (24,3 г) в метаноле (150 мл) добавляли 2,5 н. гидроксид натрия (54,5 мл). После перемешивания при комнатной температуре в течение 45 минут растворитель удаляли в вакууме. Остаток растворяли в воде, охлаждали в бане со льдом и подкисляли 2 н. хлористоводородной кислотой (14 мл). Полученный осадок фильтровали и сушили в вакууме с получением указанного в заголовке соединения в виде твердого вещества (22,55 г), т.пл. 154-158oС.
Пример 77
3-Хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензонитрил
К охлажденной суспензии 2-хлор-4-цианобензойной кислоты (9,1 г) в смеси дихлорметана (40 мл) и диметилформамида (3,88 мл) добавляли по каплям раствор оксалилхлорида (4,6 мл) в дихлорметане (10 мл) при 0оС. Перемешиваемой реакции давали нагреться до комнатной температуры на протяжении одного часа. Мутный раствор 2-хлор-4-цианобензоилхлорида использовали без дополнительной очистки.
К перемешиваемой суспензии 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (7,32 г) и диизопропилэтиламина (13,6 мл) в дихлорметане (35 мл) добавляли в атмосфере азота мутный раствор 2-хлор-4-цианобензоилхлорида. После одного часа при комнатной температуре смесь разбавляли дихлорметаном и промывали последовательно водой, 5% бикарбонатом натрия и 50% насыщенным солевым раствором. После высушивания над безводным сульфатом натрия растворитель удаляли в вакууме с получением неочищенного продукта (18,0 г). Очистка колоночной хроматографией на силикагеле (250 г) с элюированием смесью 20% этилацетат/гексан с последующим элюированием смесью 25% этилацетат/гексан давала указанное в заголовке соединение (13,56 г) в виде соломенно-желтой пены. MS (EI), m/z: 347 (М)+.
Пример 78
3-Хлор-4-(5Н,11Н-пирроло[2,1-c][1,4]бензодиазепин-10-карбонил)-бензойная кислота
К суспензии 3-хлор-4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензонитрила (90,72 г) в этаноле добавляли 10 н. гидроксид натрия (1,02 мл) и эту смесь нагревали с обратным холодильником в течение двух часов. Растворитель удаляли в вакууме, остаток растворяли в воде и подкисляли 2 н. хлористоводородной кислотой (4,7 мл). Полученный осадок экстрагировали этилацетатом и органическую фазу сушили над безводным сульфатом натрия. После удаления растворителя в вакууме пену растирали с диэтиловым эфиром в течение 18 часов и фильтровали с получением неочищенного продукта (0,69 г). Неочищенный продукт очищали обработкой активированным углем в метаноле. Кристаллизация из смеси метанол/эфир давала указанное в заголовке соединение в виде очищенного твердого вещества (0,29 г), т.пл. 198-199oС. MS (El), m/z: 366 (М)+.
Пример 79
3-Хлор-4-(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамид
Концентрированную серную кислоту (70 мл) добавляли к 3-хлор-4-(5Н,11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-бензонитрилу (12,85 г). Смесь перемешивали при 60oС в течение 3 часов с последующим перемешиванием при комнатной температуре в течение 18 часов. Реакционную смесь выливали на лед и нейтрализовали при 0оС 30% нашатырным спиртом (184 мл). Полученную суспензию экстрагировали этилацетатом. Водную смесь фильтровали и повторно экстрагировали этилацетатом. Объединенную органическую фазу сушили над безводным сульфатом натрия и растворитель удаляли в вакууме. Остаток растирали со смесью диэтилового эфира (50-60 мл) и небольшого количества этилацетата. Фильтрование осадка давало указанное в заголовке соединение в виде кристаллического твердого вещества (10,44 г), т.пл. 211-212oC. MS (EI), m/z: 365 (М)+.
Пример 80
N-(1-Диметиламиноэтилен)-3-хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-бензамид
Суспензию 3-хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-карбонил)-бензамида (5,48 г) и диметилацеталя диметилацетамида (10,97 мл) нагревали при 90oС в течение 20 минут. Избыточный реагент удаляли под пониженным давлением, указанное в заголовке соединение использовали без дополнительной очистки. MS (EI), m/z: 434 (М)+.
Пример 81
[2-Хлор-4-(5-метил-2Н-[1,2,4] триазол-3-ил)фенил]-(5Н,11Н-пирроло[2,1-c] [1,4]бензодиазепин-10-карбонил)-метанон
К раствору N-(1-диметиламиноэтилен)-3-хлор-4-(5Н,11Н-пирроло[2,1-с][1,4] бензодиазепин-10-карбонил)-бензамида (3,01 г) в уксусной кислоте (4 мл) добавляли раствор безводного гидразина (0,435 мл) в уксусной кислоте (4 мл). Реакционную смесь перемешивали при температуре между 85 и 90oС в течение 45 минут. После удаления уксусной кислоты в вакууме реакционную смесь разбавляли водой (35-40 мл), нейтрализовали до рН 7,0 водным бикарбонатом натрия и экстрагировали этилацетатом. Органический экстракт промывали солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением неочищенного продукта (2,68 г). Очистка неочищенного продукта колоночной хроматографией на силикагеле (45 г) с элюированием смесью 70% этилацетат/гексан давала очищенный продукт (2,5 г), который, после растирания с диэтиловым эфиром, давал указанное в заголовке соединение в виде твердого вещества (2 г), т.пл. 211-212oС. MS (EI), m/z: 403 (М)+.
Пример 82
N-(Диметиламинометилен)-3-хлор-4-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамид
Указанное в заголовке соединение получали так же, как описано в примере 80, с использованием 3-хлор-4-(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензамида (1,83 г) и диметилацеталя диметилформамида (5,3 мл). MS (El), m/z: 420 (М)+.
Пример 83
[2-Хлор-4-(2Н-[1,2,4]триазол-3-ил)фенил]-(5Н,11H-пирроло[2,l-c][1,4]бензодиазепин-10-ил)-метанон
Указанное в заголовке соединение получали так же, как описано в примере 81, с использованием N-(диметиламинометилен)-3-хлор-4-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензамида (2,53 г) и гидразина (0,38 мл), т.пл. 174-177oС. MS (EI), m/z: 389 (М)+.
Пример 84
[2-Хлор-4-(2-метил-2Н-[1,2,4] триазол-3-ил)фенил]-(5Н,11Н-пирроло[2,l-c] [1,4]бензодиазепин-10-карбонил)-метанон
Указанное в заголовке соединение получали так же, как описано в примере 48, с использованием N-(диметиламинометилен)-3-хлор-4-(5Н,11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-карбонил)-бензамида (0,572 г) и метилгидразина (0,149 мл), т.пл. 141-143oС. MS (EI), m/z: 403 (М)+.
Пример 85
4-[(2,5-Диметил-2Н-[1,2,4] триазол-3-ил)-2-хлорфенил]-(5Н,11Н-пирроло[2, l-c][1,4]бензодиазепин-10-карбонил)-метанон
Указанное в заголовке соединение получали так же, как описано в примере 48, с использованием N-(1-диметиламиноэтилен)-3-хлор-4-(5Н,11H-пирроло[2, l-c] [1,4] бензодиазепин-10-карбонил)-бензамида (0,51 г) и метилгидразина (0,125 мл), т.пл. 197-199oС. MS (EI), m/z: 417 (М)+.
Пример 86
[2-Хлор-4-(1Н-тетразол-5-ил)-фенил] -(5Н, 11Н-пирроло[2, l-c][1,4]бензодиазепин-10-ил)-метанон
К раствору 3-хлор-4-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-бензонитрила (0,348 г) в диметилформамиде (2 мл) добавляли азид натрия (0,078 г) и хлорид аммония (0,065 г). Смесь нагревали до 100oС в течение 18 часов.
Большую часть диметилформамида удаляли в вакууме. Остаток растворяли в воде (приблизительно 8 мл) и подщелачивали до рН 9,0 2,5 н. гидроксидом натрия (0,6 мл), экстрагировали этилацетатом. Водный экстракт подкисляли 2 н. хлористоводородной кислотой (1,1 мл), повторно экстрагировали этилацетатом, сушили над безводным сульфатом натрия и растворитель удаляли в вакууме с получением неочищенного продукта (0,350 г) в виде масла. Этот маслянистый продукт растирали с диэтиловым эфиром, фильтровали через обработанный кислотой силикагель и элюировали смесью 40% этилацетат/гексан с получением более чистой пробы. Затем ее растирали с диэтиловым эфиром и фильтровали с получением пробы (0,88 г), т.пл. 218-220оС. MS (+FAB): 391 (М+Н)+.
Пример 87
[2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(3-диметиламинометил-5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К перемешиваемому раствору [2-хлор-4-(3-метилпиразол-1-ил)-фенил]-(5Н, 11Н-пирроло[2,1-с] [1, 4]бензодиазепин-10-ил)-метанона (1,61 г), N,N,N',N'-тетраметилдиаминометана (0,82 г) и ледяной уксусной кислоты (0,48 г) в метаноле (25 мл) добавляли раствор 37% водного формальдегида (4 мл). Смесь нагревали до 40oС в течение 10 минут. После перемешивания в течение одного часа при комнатной температуре реакционную смесь упаривали в вакууме, повторно растворяли в дихлорметане и экстрагировали последовательно водным бикарбонатом натрия и водой (4х). Органическую фазу сушили над безводным сульфатом натрия и фильтровали через пробку силикагеля, элюируя этилацетатом. Выпаривание растворителя в вакууме давало масло, которое при растирании с гексаном давало 0,36 г указанного в заголовке соединения в виде бесцветного порошка, т.пл. 100-102oC. MS (+FAB), m/z: 482 (М+Na)+, 460 (М+Н)+.
Пример 88
(3-Бром-5Н, 11Н-пирроло[2,1-c] [1,4] бензодиазепин-10-ил)-[2-хлор-4-(3-метилпиразол-1-ил)-фенил]-метанон
К перемешиваемому предварительно охлажденному раствору [2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанона (1,61 г) в дихлорметане (25 мл) добавляли твердый N-бромсукцинимид (0,712 г) на протяжении 10 минут при -78oС. Реакции давали нагреться до -40оС на протяжении тридцати минут. Смесь разбавляли дихлорметаном и экстрагировали последовательно насыщенным водным раствором бикарбоната натрия (2 х 100 мл) и водой (100 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали через пробку силикагеля и упаривали в вакууме до остатка. Кристаллизация из диэтилового эфира давала 1,47 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 148-149oС (разл.). MS (El), m/z: 480 (М)+.
Пример 89
(4-Бром-2-хлорфенил)-(5Н, 11H-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Диметилформамид (1 каплю) добавляли к раствору 4-бром-2-хлорбензойной кислоты (2,20 г) в безводном тетрагидрофуране (20 мл). Добавляли оксалилхлорид (1,46 г) и смесь нагревали до кипения (с обратным холодильником). Полученный раствор охлаждали до температуры окружающей среды, затем выпаривали досуха с получением неочищенного 4-бром-2-хлорбензоилхлорида в виде вязкой жидкости золотого цвета, которую использовали без дополнительной очистки.
К смеси 10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепина (1,44 г) и триэтиламина (0,95 г) в дихлорметане (40 мл), охлажденной в бане со льдом, добавляли по каплям раствор 4-бром-2-хлорбензоилхлорида (2,42 г) в дихлорметане (20 мл). Охлаждающую баню удаляли и после перемешивания в течение 22 часов реакционную смесь промывали последовательно водой, насыщенным водным раствором бикарбоната натрия, 0,5 н. хлористоводородной кислотой и водой. Раствор в дихлорметане сушили над безводным сульфатом натрия, фильтровали, затем выпаривали в вакууме досуха с получением пены беловатого цвета. Очистка флэш-хроматографией на силикагеле с элюированном смесью гексан/этилацетат (2: 1) приводила к белой пене (3,02 г), т.пл. 77-80oС. MS, m/z: 400 (М)+.
Пример 90
[2-Бром-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н)-пирроло[2,1-с] [1,4] бензодиазепин-10-ил]-метанон
Стадия а): 4-фтор-2-бромбензоилхлорид. Диметилформамид (2 капли) добавляли к раствору 4-фтор-2-бромбензойной кислоты (4,91 г) в безводном тетрагидрофуране (55 мл). Добавляли оксалилхлорид (3,41 г) и смесь нагревали до кипения (с обратным холодильником). Полученный раствор охлаждали до комнатной температуры, выпаривали в вакууме с получением неочищенного хлорангидрида в виде вязкой жидкости золотого цвета, которую использовали без дополнительной очистки.
Стадия b): (4-фтор-2-бромфенил)-(5Н,11H-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон. Раствор 4-фтор-2-бромбензоилхлорида (5,32 г) со стадии а) в дихлорметане (35 мл) добавляли по каплям к раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4]бензодиазепина (3,44 г) и триэтиламина (2,27 г) в дихлорметане (80 мл) и охлаждали в бане со льдом. Охлаждающую баню удаляли и после перемешивания в течение 16 часов реакционную смесь промывали последовательно водой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Раствор в дихлорметане сушили над безводным сульфатом магния, фильтровали и выпаривали в вакууме с получением бледно-лиловой пены. Очистка флэш-хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1: 1) приводила к промежуточному продукту - (4-фтор-2-бромфенил)-(5Н, 11Н-пирроло[2,1-с][1, 4]бензодиазепин-10-ил)-метанону в виде рыжевато-коричневой пены (6,91 г). MS, m/z: 384 (М)+. Это вещество использовали без дополнительной очистки в следующей стадии.
Стадия с): [2-бром-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н)-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Дисперсию 60% гидрида натрия в масле (0,20 г) промывали гексаном и затем суспендировали в диметилформамиде (15 мл). К этой суспензии добавляли 3-метилпиразол (0,41 г). После прекращения выделения газообразного водорода добавляли (4-фтор-2-бромфенил)-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (1,74 г) со стадии b). Реакционную смесь нагревали до 130oС в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры ее выливали в 50% насыщенный водный раствор хлорида натрия и экстрагировали этилацетатом. Раствор в этилацетате сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме с получением коричневого масла. Очистка флэш-хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1: 1) давала бесцветное твердое вещество (0,75 г). Перекристаллизация из метанола давала кристаллическое твердое вещество беловатого цвета (0,53 г), т.пл. 141-14,2,5оС. MS, m/z: 446 (М)+.
Пример 91
(2,4-Дифторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон
Стадия а): 2,4-дифторбензоилхлорид. Суспензию 2,4-дифторбензойной кислоты (3,6 г), содержащую несколько капель диметилформамида, в дихлорметане (40 мл) обрабатывали по каплям в атмосфере азота оксалилхлоридом (2,4 мл). После прекращения выделения газа реакционную смесь кипятили с обратным холодильником еще в течение 15 минут. Раствор упаривали досуха в вакууме и остаток использовали без дополнительной очистки.
Стадия b): (2,4-дифторфенил)-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон. К раствору неочищенного 2,4-дифторбензоилхлорида стадии а) в дихлорметане в атмосфере азота добавляли твердый 10,11-дигидро-5Н-пирроло[2,1-с] [1,4] бензодиазепинамин (2,0 г) и диизопропилэтиламин (3,4 мл). Реакционная смесь становилась желто-оранжевой. После перемешивания при комнатной температуре в течение 10 минут реакционную смесь промывали водой, 1 н. хлористоводородной кислотой, 1 н. гидроксидом натрия и солевым раствором. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха с получением коричневого твердого вещества. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Merck-60) с элюированием смесью 20% этилацетат/гексан с получением 2,9 г указанного в заголовке соединения в виде белой пены. MS (El), m/z: 324 (М)+.
Пример 92
[2-Фтор-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Суспензию промытого гексаном 60% гидрида натрия (0,31 г) в безводном диметилформамиде обрабатывали по каплям 3-метилпиразолом (0,62 мл) в атмосфере азота при комнатной температуре. Перемешивание продолжали до тех пор, пока не прекращалось выделение газа (10 минут). В одной порции добавляли (2,4-дифторфенил)-(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон (2,5 г) со стадии b) примера 91 и перемешивание продолжали до получения прозрачного раствора. Смесь нагревали в предварительно нагретой масляной бане при 130oС в течение одного часа. После охлаждения смесь распределяли между водой и этилацетатом. Органические экстракты сушили над безводным сульфатом натрия и упаривали досуха. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (Merck-60), элюируя смесью 20% этилацетат/гексан с получением 0,82 г указанного в заголовке соединения в виде пены, которое кристаллизовали обработкой ультразвуком из смеси этанол/гексан, т. пл. 192-193oС. MS (EI), m/z: 386 (М)+.
Пример 93
Метил-4-(3-метилпиразол-1-ил)-2-трифторметилбензоат
Стадия а): метил-4-фтор-2-трифторметилбензоат. Суспензию 4-фтор-2-трифторметилбензойной кислоты (25,6 г) и несколько капель диметилформамида в дихлорметане (250 мл) обрабатывали по каплям в атаосфере азота оксалилхлоридом (11,3 мл). После прекращения выделения газа реакционную смесь кипятили с обратным холодильником в течение дополнительных 15 минут. Реакцию охлаждали и добавляли метанол (50 мл). После перемешивания в течение 2 часов реакцию упаривали в вакууме и
остаток распределяли между дихлорметаном и водой. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным сульфатом натрия и упаривали досуха с получением 18,0 г указанного в заголовке соединения в виде масла золотого цвета. MS (EI), m/z: 222 (М)+.
Водный слой подкисляли 2 н. хлористоводородной кислотой с получением бесцветного твердого вещества, которое собирали фильтрованием с получением 7,5 г исходной бензойной кислоты.
Стадия b): метил-4-(3-метилпиразол-1-ил)-2-трифторметилбензоат. Суспензию промытого гексаном 60% гидрида натрия (3,85 г) в безводном диметилформамиде (150 мл) обрабатывали добавлением по каплям раствора 3-метилпиразола (7,75 мл) в диметилформамиде (50 мл) в атмосфере азота при комнатной температуре. Перемешивание продолжали до прекращения выделения газа (10 минут). К прозрачному раствору добавляли по каплям раствор метил-4-фтор-2-трифторметилбензоата (17,8 г) со стадии а) в диметилформамиде (50 мл). После перемешивания в течение 30 минут при комнатной температуре реакцию гасили насыщенным хлоридом аммония и экстрагировали этилацетатом. Органические экстракты (3х) сушили над безводным сульфатом натрия и упаривали досуха. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (Merck-60), элюируя градиентом дихлорметан/гексан (50%-75%) с получением 13,6 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 59-61oС. MS (EI), m/z: 284 (М)+.
Пример 94
4-(3-Метилпиразол-1-ил)-2-трифторметилбензойная кислота
Метил-4-(3-метилпиразол-1-ил)-2-трифторметилбензоат (1,19 г) со стадии b) примера 93 растворяли в метаноле (10 мл) и добавляли раствор 2,5 н. гидроксида натрия (3,3 мл). Реакцию нагревали при кипении с обратным холодильником в течение 90 минут, охлаждали до комнатной температуры и упаривали в вакууме досуха. Остаток распределяли между этилацетатом и 1 н. хлористоводородной кислотой. Объединенные органические экстракты сушили над безводным сульфатом натрия и упаривали в вакууме с получением 1,14 г указанного в заголовке соединения в виде бесцветного твердого вещества. MS (+FAB), m/z: 271 (М+Н)+.
Пример 95
[4-(3-Метилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11H-пиразоло[5,1-с] [1,4]бензодиазепин-10-ил)-метанон
Раствор 4-(3-метилпиразол-1-ил)-2-трифторметилбензойной кислоты (0,26 г) из примера 94 в тетрагидрофуране (5 мл) обрабатывали диметилформамидом (0,020 мл), затем оксалилхлоридом (0,090 мл). Раствор перемешивали при комнатной температуре до прекращения выделения газа и затем раствор нагревали с обратным холодильником до кипения в течение 10 минут. Пробу охлаждали до комнатной температуры, упаривали до твердого вещества и это твердое вещество растворяли в тетрагидрофуране (25 мл). Этот раствор добавляли к раствору (5Н-10,11-дигидропиразоло[5,1-с] [1,4] бензодиазепина (0,143 г) и триэтиламина (0,150 мл) в тетрагидрофуране (20 мл). Раствор перемешивали в течение ночи при комнатной температуре. Образовался осадок. Пробу разбавляли дихлорметаном для растворения осадка и затем эту пробу упаривали в вакууме до приблизительно 1/3 исходного объема. Эту пробу распределяли между дихлорметаном и насыщенным водным хлоридом аммония. Пробу экстрагировали дихлорметаном и органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и упаривали до масла. Масло подвергали флэш-хроматографии на силикагеле с использованием градиента 40% этилацетат/гексаны - 100% этилацетат с получением указанного в заголовке соединения в виде пены (0,30 г). Часть этого материала перекристаллизовывали из смеси ацетон/гексаны с получением тяжелых пластинок, т.пл. 100-102oС. MS, m/z: 437 (М)+.
Пример 96
2-Хлор-4-(3-метил-1Н-пиразол-1-ил)-бензойной кислоты, метиловый эфир и 2-хлор-4-(5-метил-1Н-пиразол-1-ил)-бензойной кислоты, метиловый эфир
Суспензию промытого гексаном гидрида калия (0,424 г) в диметилформамиде (5 мл) обрабатывали одной порцией 3-метилпиразола (0,85 мл) при перемешивании. После прекращения выделения газа к прозрачному раствору добавляли метиловый эфир 2-хлор-4-фторбензойной кислоты (2,0 г) и нагревали при 130oС в течение 15 минут. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом и солевым раствором. Органическую фазу промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаление растворителя в вакууме давало 2,2 г желтого масла (примечание: 20% гидролиз этого эфира определяли при помощи анализа ЯМР-спектра неочищенного продукта). Целевой региоизомер - метиловый эфир 2-хлор-4-(3-метил-1Н-пиразол-1-ил)-бензойной кислоты выделяли из другого изомера (описанного ниже) колоночной флэш-хроматографией на силикагеле (Меrck-60) с элюированием смесью дихлорметан/гексан (2:1), что давало 1,55 г указанного в заголовке соединения в виде бесцветного твердого вещества. MS (ЕI), m/z: 250/252 (М)+.
5-региоизомер, а именно метиловый эфир 2-хлор-4-(5-метил-1Н-пиразол-1-ил)-бензойной кислоты, выделяли при помощи колоночной флэш-хроматографии на силикагеле (Merck-60) дополнительным элюированием смесью дихлорметан/гексан (2: 1), что давало 0,20 г этого продукта в виде бесцветного твердого вещества. MS (EI), m/z: 250/252 (М)+.
Пример 97
2-Хлор-4-(3-метил-1Н-пиразол-1-ил)-бензойная кислота
Раствор метилового эфира 2-хлор-4- (3-метил-1Н-пиразол-1-ил)-бензойной кислоты (1,42 г) из примера 96 и 6 мл 1 М водного гидроксида лития в тетрагидрофуране (20 мл) перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь распределяли между этилацетатом и 1 н. хлористоводородной кислотой. Органический слой промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Выпаривание растворителя в вакууме давало 1,05 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 192-193oC. MS (EI), m/z: 236/238 (М)+.
Пример 98
(2,6-Дихлорпиридин-3-ил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Раствор 2,6-дихлорникотиновой кислоты (3,84 г), оксалилхлорида (2,0 г) и 1 капли диметилформамида в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 18 часов. Раствор упаривали в вакууме с получением 3,50 г 2,6-дихлорникотинилхлорида, который добавляли порциями в дихлорметане (25 мл) к охлажденному на льду раствору 10,11-дигидро-5Н-пирроло[2,1-с][1,4] бензодиазепина (2,15 г) и диизопропилэтиламина (2,03 г) в дихлорметане (50 мл). Смесь перемешивали при комнатной температуре в течение 18 часов и промывали насыщенным водным раствором бикарбоната натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Объединенную органическую фазу упаривали на горячей пластине с постепенным добавлением гексана до тех пор, пока не происходила кристаллизация. После охлаждения кристаллы собирали фильтрованием с получением 2,65 г указанного в заголовке соединения в виде аморфного твердого вещества, т.пл. 115-130oС. MS, m/z: 358,1 (М+Н)+.
Пример 99
(2-Хлор-6-пиразол-1-ил-пиридин-3-ил)-(5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К суспензии 60% гидрида натрия в масле (0,1 г) в диметилформамиде (25 мл) добавляли по каплям пиразол (0,15 г). После прекращения выделения газообразного водорода добавляли (2,6-дихлорпиридин-3-ил)-(5Н,11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (0,67 г) и реакционную смесь нагревали на песочной бане при 110оС в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и растирали с диэтиловым эфиром с получением 0,18 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 133-l35oC. MS, m/z: 390,8 (М+Н)+, 779,1 (2М+Н)+.
Пример 100
[2-Хлор-6-(3-метилпиразол-1-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
К суспензии 60% гидрида натрия в масле (0,1 г) в диметилформамиде (25 мл) добавляли по каплям 3-метилпиразол (0,15 г). После прекращения выделения газообразного водорода добавляли (2,6-дихлорпиридин-3-ил)-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (0,67 г) и реакционную смесь нагревали на песочной бане при 110oС в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Неочищенный продукт очищали препаративной ВЭЖХ (кассета двуокисного кремния Dynamax с60 silica cartridge), элюируя 40% этилацетатом в гексанах с получением 0,21 г бесцветных кристаллов, т.пл. 171-172oC. MS, m/z: 404,2 (М+Н)+, 807,1 (2М+Н)+.
Пример 101
[2-Хлор-6-(4-метилпиразол-1-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
К суспензии 60% гидрида натрия в масле (0,1 г) в диметилформамиде (25 мл) добавляли по каплям 3-метилпиразол (0,45 г). После прекращения выделения газообразного водорода добавляли (2,6-дихлорпиридин-3-ил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (1,79 г) и реакционную смесь нагревали на песочной бане при 110ОС в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Неочищенный продукт очищали препаративной ВЭЖХ (кассета с двуокисью кремния Dynamax c60 silica cartridge) с элюированием 40% этилацетатом в гексанах, что давало 0,26 г бесцветных кристаллов, т. пл, 155-156oС. MS, m/z: 404,2 (М+Н)+, 807,0 (2М+Н)+.
Пример 102
[2-Хлор-4-(3-метил-1,2,4-триазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
К суспензии 60% гидрида натрия в масле (0,3 г) в диметилформамиде (50 мл) добавляли по каплям 3-метил-1,2,4-триазол (0,45 г). После прекращения выделения газообразного водорода добавляли 2-хлор-4-фторфенил-(5Н,11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанол (1,70 г) и реакционную смесь нагревали на песочной бане при 110oС в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и остаток растирали с диэтиловым эфиром с получением 1,25 г указанного в заголовке соединения в виде бесцветных кристаллов, т.пл. 191-193oС. MS, m/z: 404,1 (М+Н)+.
Пример 103
[4-(3-Метил-1,2,4-триазол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
К суспензии 60% гидрида натрия в масле (0,3 г) в диметилформамиде (50 мл) добавляли по каплям 3-метил-1,2,4-триазол (0,45 г). После прекращения выделения газообразного водорода добавляли 4-фтор-2-трифторметилфенил-(5Н, 11Н-пирроло[2,1-с] [1, 4]бензодиазепин-10-ил)-метанон (1,76 г) и реакционную смесь нагревали на песочной бане при 110oС в течение 18 часов. Смесь выливали на лед, разбавляли солевым раствором и экстрагировали дихлорметаном. Объединенные экстракты сушили над безводным сульфатом натрия и фильтровали через короткую колонку с безводным силикатом натрия-магния. Раствор упаривали в вакууме и остаток растирали с диэтиловым эфиром с получением 0,81 г указанного в заголовке соединения в виде бесцветных кристаллов, т.пл. 148-150oС. MS, m/z: 438,2 (М+Н)+, 875,8 (2М+Н)+.
Пример 104
4-Гидразино-2-метоксибензойная кислота, метиловый эфир, гидрохлорид (1: 1), гидрат (2:1)
Перемешиваемую суспензию метилового эфира 4-амино-2-метоксибензойной кислоты (21,74 г) в концентрированной хлористоводородной кислоте (110 мл), которая была охлаждена до -10oС, обрабатывали предварительно охлажденным раствором нитрита натрия (8,5 г) в воде (45 мл) при скорости, необходимой для поддержания температуры реакции ниже 0oС. После завершения добавления реакционную смесь перемешивали при -2oС в течение 10 минут. Полученный мутный оранжевый раствор добавляли по каплям к энергично перемешиваемому предварительно охлажденному раствору дигидрата хлорида олова(II) (101 г) в концентрированной хлористоводородной кислоте (67 мл) при -10oС. Скорость добавления контролировалась таким образом, чтобы поддерживать температуру реакции ниже -5oС. После завершения добавления суспензию кремового цвета нагревали до комнатной температуры и твердое вещество отфильтровывали. Это твердое вещество промывали диэтиловым эфиром и сушили над безводным сульфатом натрия с получением 52 г неочищенного продукта. Неочищенный продукт (20 г) распределяли между водным 2,5 н. гидроксидом натрия и дихлорметаном. Органическую фазу фильтровали через диатомовую землю, промывали солевым раствором и сушили над безводным сульфатом магния. Фильтрование и выпаривание растворителя в вакууме давало твердое вещество кремового цвета (7,1 г), которое при обработке одним эквивалентом раствора безводного хлористого водорода в диэтиловом эфире давало указанное в заголовке соединение в виде моногидрохлоридной соли, т.пл. 76-79oС. MS, m/z: 197 (М+Н)+.
Пример 105
2-Метокси-4-(3-метилпиразол-1-ил)-бензойная кислота, метиловый эфир
К перемешиваемому раствору гидрохлорида метилового эфира 4-гидразино-2-метоксибензойной кислоты (0,88 г) из примера 104 и одной капли концентрированной хлористоводородной кислоты в смеси 1:1 вода/метанол (10 мл) добавляли диметилацеталь ацетилацетальдегида (0,53 г). Реакцию нагревали до 90oС в течение 5 минут, реакцию упаривали в вакууме и распределяли между 1 н. гидроксидом натрия (10 мл) и этилацетатом (50 мл). Органическую фазу удаляли и промывали солевым раствором, сушили над безводным сульфатом магния и фильтровали. Выпаривание растворителя в вакууме давало коричневое масло, которое объединяли с предыдущей партией (0,54 г) и перекристаллизовывали три раза из диизопропилового эфира с получением метилового эфира 2-метокси-4-(3-метилпиразол-1-ил)-бензойной кислоты (0,5 г), т.пл. 167-169oС. MS, m/z: 246 (М)+.
Пример 106
2-Метокси-4-(3-метилпиразол-1-ил)-бензойная кислота
Раствор метилового эфира 2-метокси-4-(3-метилпиразол-1-ил)-бензойной кислоты (0,5 г) из примера 105 в тетрагидрофуране (2,5 мл) обрабатывали 1 н. гидроксидом лития (2,13 мл) при комнатной температуре. Через 14 часов растворитель удаляли в вакууме и указанное в заголовке соединение осаждали доставлением при 0oС 1 н. хлористоводородной кислоты. После высушивания в вакууме 0,42 г указанного в заголовке соединения получали в виде твердого вещества. MS, m/z: 232 (М)+.
Пример 107
[2-Метокси-4-(3-метилпиразол-1-ил)-фенил] (5Н,11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
Оксалилхлорид (0,17 мл) добавляли к перемешиваемому раствору 2-метокси-4-(3-метилпиразол-1-ил)-бензойной кислоты (0,41 г) из примера 106 и диметилформамида (0,004 мл) в безводном тетрагидрофуране (10 мл). Реакцию нагревали при 35oС в течение десяти минут. Полученный раствор выпаривали в вакууме с получением неочищенного карбонилхлорида 2-метокси-4-(3-метилпиразол-1-ил)-бензойной кислоты. После выпаривания совместно с дихлорметаном хлорангидрид растворяли в дихлорметане (10 мл) и добавляли 10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепин (0,31 г). Добавляли диизопропилэтиламин (0,37 мл) и реакцию перемешивали при комнатной температуре в течение 2 часов. Реакцию разбавляли дихлорметаном и промывали водой и затем 1 н. хлористоводородной кислотой. Органическую фазу промывали солевым раствором, сушили над безводным сульфатом магния и упаривали досуха в вакууме. Твердый остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя смесью гексан/этилацетат (2: 1) с получением 0,35 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 92-94oС.
Пример 108
(3-Диметиламинометил-5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)[2-метокси-4-(3-метилпиразол-1-ил)фенил]-метанон
К перемешиваемому раствору [2-метокси-4-(3-метилпиразол-1-ил)-фенил](5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанона из примера 107 (0,57 г) в теплом метаноле (10 мл) добавляли N,N,N',N'-тетраметилдиаминометан (0,392 мл) и уксусную кислоту (0,164 мл). После добавления 37% водного раствора формалина (2,9 мл) реакцию перемешивали в течение пятнадцати минут. Смесь упаривали в вакууме и распределяли между дихлорметаном и гидрокарбонатом натрия. Органическую фазу удаляли, промывали солевым раствором и сушили над безводным сульфатом магния. Раствор фильтровали и растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя смесью хлороформ/метанол (50: 1) с получением твердого вещества. Перекристаллизация этого твердого вещества из ацетона давала указанное в заголовке соединение в виде бесцветного твердого вещества, т.пл. 196-198oС.
Пример 109
[2-Гидрокси-4-(3-метилпиразол-1-ил] (5Н, 11Н-пирроло[2,1-с][1,4]бензодиазепин-10-ил)-метанон
[2-Метокси-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон (0,82 г) из примера 107 растворяли в дихлорметате (20 мл) и охлаждали до -78oС. Добавляли трибромид бора (6,2 мл) и реакцию перемешивали при 0оС в течение пяти минут. Добавляли гидроксид аммония (15 мл) и экстрагировали дихлорметаном. Органическую фазу промывали солевым раствором и сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием и растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя смесью гексан/этилацетат (3:1, затем 2:1) с получением 0,19 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 134-136oС.
Пример 110
2-Хлор-4-иодбензойная кислота, метиловый эфир
Метиловый эфир 4-aминo-2-мeтoкcибeнзойнoй кислоты (22,97 г) охлаждали до внутренней температуры -10oС в концентрированной хлористоводородной кислоте (110 мл) и перемешивали в виде суспензии. К этой смеси добавляли предварительно охлажденный раствор нитрита натрия (98,71 г) в воде (45 мл) при такой скорости, которая позволяла поддерживать температуру реакции ниже 0oС. После перемешивания в течение 25 минут при 0oС реакцию обрабатывали раствором иодида калия (24,44 г) и иода (18,37 г) в воде (50 мл) при такой скорости, которая позволяла поддерживать температуру реакции ниже -4oС. В процессе этого добавления добавляли этилацетат (100 мл) и полученную темную смесь перемешивали при 0oС в течение одного часа. Органический слой разбавляли этилацетатом и хорошо промывали насыщенным водным раствором тиосульфата натрия. Полученный оранжевый раствор промывали солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением масла, которое очищали фильтрованием с отсасыванием через силикагель, элюируя смесью гексан/этилацетат (50:1). Полученное очищенное масло отверждалось при охлаждении с образованием 33,71 г указанного в заголовке соединения. MS, m/z: 296 (М)+.
Пример 111
4-Бром-1-метил-1Н-пиразол
К суспензии предварительно промытого (тетрагидрофураном) 60% гидрида натрия в масле (11,67 г) в тетрагидрофуране (200 мл) добавляли по каплям раствор 4-бромпиразола (39,77 г) в тетрагидрофуране (50 мл). Раствор перемешивали при комнатной температуре в течение двух часов. Добавляли избыток иодметана (33 мл) в тетрагидрофуране (50 мл) при такой скорости, которая была необходима для поддержания небольшого увеличения температуры. Реакцию перемешивали дополнительно в течение двух часов. Растворитель удаляли в вакууме и остаток перемешивали в диэтиловом эфире. Осадок удаляли фильтрованием с отсасыванием и промывали диэтиловым эфиром. Объединенную органическую фазу упаривали в вакууме с получением 42,22 г указанного в заголовке соединения в виде масла. MS, m/z: 160 (М)+.
Пример 112
1-Метил-4-трибутилстаннил-1Н-пиразол
К предварительно охлажденному (внутренняя температура <-10oС) раствору 1,6 М н-бутиллития в гексанах (100 мл) в безводном диэтиловом эфире (100 мл) в атмосфере аргона добавляли раствор 4-бром-1-метил-1Н-пиразола (23,42 г) из примера 111 в диэтиловом эфире (50 мл) при скорости, необходимой для поддержания этой температуры. Реакции давали перемешиваться еще в течение 20 минут перед добавлением хлорида трибутилолова (43,4 мл) в диэтиловом эфире (50 мл). Температуре реакции давали повыситься до 20oС. Реакцию разбавляли диэтиловым эфиром и нерастворимый материал удаляли фильтрованием с отсасыванием. Выпаривание растворителя в вакууме давало 56 г указанного в заголовке соединения в виде масла. MS, m/z: 373 (М+Н)+. Остаточные количества остатков олова удаляли из этого масла отгонкой при помощи аппарата с шаровым холодильником в высоком вакууме при 170oС.
Пример 113
2-Хлор-4-(1-метил-1Н-пиразол-4-ил)-бензойная кислота, метиловый эфир
Дегазированный аргоном раствор в диметилформамиде (70 мл) метилового эфира 3-хлор-4-иодбензойной кислоты (25,4 г) из примера 110, 1-метил-4-трибутилстаннил-1Н-пиразола (31,77 г), тетракис(трифенилфосфин)палладия(0) (1,8 г) и каталитического иодида меди(I) нагревали при 80oС в течение 7 часов. Растворитель удаляли в вакууме и остаток адсорбировали на силикагель. Очистка фильтрованием с отсасыванием через подушку силикагеля с элюированием последовательно гексаном и затем смесью гексан/этилацетат (2:1) давала после выпаривания растворителя твердый остаток, который перекристаллизовывали из диизопропилового эфира с получением 7,82 г указанного в заголовке соединения. MS, m/z: 250 (М)+.
Пример 114
2-Хлор-4-(1-метил-1Н-пиразол-4-ил)-бензойная кислота
К раствору метилового эфира 2-хлор-4-(1-метил-1Н-пиразол-4-ил)-бензойной кислоты (6,25 г) из примера 113 в метаноле (80 мл) добавляли 1 н. гидроксид натрия (30 мл). Реакцию нагревали с обратным холодильником в течение одного часа. Объем растворителя уменьшали в вакууме на три четверти и остаток обрабатывали, 2 н. хлористоводородной кислотой при 0oС. Осадок фильтровали и сушили над безводным сульфатом натрия в вакууме с получением 5,84 г указанного в заголовке соединения. MS, m/z: 237 (М+Н)+.
Пример 115
[2-Хлор-4-(1-метил-1Н-пиразол-4-ил)фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон
Оксалилхлорид (0,49 мл) добавляли к раствору 2-хлор-4-(1-метил-1Н-пиразол-4-ил)-бензойной кислоты (0,41 г) из примера 114 и диметилформамида (0,012 мл) в безводном тетрагидрофуране (20 мл). Реакцию нагревали при 35oС в течение десяти минут. Полученный раствор выпаривали в вакууме досуха с получением неочищенного карбонидхлорида 2-хлор-4-(1-метил-1Н-пиразол-4-ил)-бензойной кислоты. После упаривания совместно с безводным дихлорметаном хлорангидрид растворяли в дихлорметане (20 мл), после чего добавляли 10,11-дигидро-5Н-пирроло[2,1-с] [1,4]бензодиазепин (0,888 г) и диизопропилэтиламин (1,06 мл). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакцию разбавляли дихлорметаном и промывали водой, затем 1 н. хлористоводородной кислотой. Органическую фазу промывали солевым раствором и сушили над безводным сульфатом магния. Раствор в дихлорметане фильтровали и упаривали досуха в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя смесью гексан/этилацетат (2:1) с получением 1,4 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 105-109oС.
Пример 116
[2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(4Н, 10Н-пиразоло[5,1-с] [1,4]-бензодиазепин-5-ил)-метанон
К раствору 2-хлор-4-(3-метилпиразол-1-ил)-бензоилхлорида (0,214 г) стадии е) примера 18 в дихлорметане (10 мл) добавляли 5Н-10,11-дигидропиразоло[5,1-с] [1,4]бензодиазепин (0,153 г) и диизопропилэтиламин (0,173 мл). Реакцию перемешивали при комнатной температуре в течение 2 часов. Реакцию разбавляли дихлорметаном и промывали водой, затем 1 н. хлористоводородной кислотой. Органическую фазу промывали солевым раствором и сушили над безводным сульфатом магния. Раствор в дихлорметане фильтровали и упаривали в вакууме досуха. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя под давлением смесью гексан/этилацетат (1:1), с получением 0,3 г указанного в заголовке соединения в виде бесцветного твердого вещества, т. пл. 187-188oС.
Пример 117
2-Хлор-4-(3-метилпиразол-1-ил)-фенил] -(5,10-дигидро-4Н-тетразоло[5,1-с] [1,4]бензодиазепин-5-ил)-метанон
К раствору 2-хлор-4-(3-метилпиразол-1-ил)-бензоилхлорида (0,18 г) стадии е) примера 18 в дихлорметане (10 мл) добавляли 10,11-дигидро-5Н-тетразол[5,1-с] [1,4] бензодиазепин (0,13 г) и диизопропилэтиламин (0,145 мл). Реакцию перемешивали при комнатной температуре в течение 2 часов. Реакцию разбавляли дихлорметаном и промывали водой, а затем 1 н. хлористоводородной кислотой. Органическую фазу промывали солевым раствором и сушили над безводным сульфатом магния. Раствор в дихлорметане фильтровали и упаривали в вакууме досуха. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя смесью гексан/этилацетат (1:1), с получением 0,14 г указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 110-114oС.
Пример 118
1-[4-(4Н,10Н-Пиразоло[5,1-с][1,4]бензодиазепин-5-карбонил)-фенил]-этанон
Смесь 5,10-дигидpo-4H-пиpазoлo[5,1-c][1,4]бензодиазепина (0,555 г), 4-ацетилбензоилхлорида (0,657 г) и N,N-диизопропилэтиламина (0,464 г) в дихлорметане (15 мл) перемешивали при комнатной температуре в течение 4 часов. Смесь выливали в воду и экстрагировали дихлорметаном. Дихлорметановый экстракт промывали насыщенным гидрокарбонатом натрия, водой и солевым раствором и сушили над безводным сульфатом натрия. Экстракт фильтровали через тонкую подушку водного силиката магния и фильтровальную подушку промывали дихлорметаном. Фильтрат упаривали в вакууме с получением 1,53 г желтого твердого вещества. Растирание этого твердого вещества с этилацетатом давало 0,741 г указанного в заголовке соединения в виде стекловидного продукта, т.пл. 201-210oС. Маточные растворы, оставшиеся после растирания, упаривали и остаток (0,30 г) хроматографировали на пластинках с толстым слоем силикагеля (200 микрон) с использованием смеси гексан/этилацетат (1:1) в качестве растворителя. Твердое вещество растирали с этилацетатом и объединяли с 0,747 г первоначально выделенного продукта. Объединенные твердые вещества осаждали из смеси дихлорметан/гексан с получением 0,73 г продукта в виде стекловидного вещества.
Пример 119
1-[4-(4Н, 10Н-Пиразоло[5,1-c] [1,4] бензодиазепин-5-карбонил)-фенил]-3-(диметиламино)-проп-2-ен-1-он
Смесь 1-[4-(4Н, 10Н-пиразоло[5,1-c][1,4]бензодиазепин-5-карбонил)фенил] -этанона (0,73 г), трет-бутоксибис(диметиламино)метана (0,964 г) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 2 дней. Смесь концентрировали в вакууме и остаток кристаллизовали из смеси дихлорметан/гексан с получением 0,65 г указанного в заголовке соединения в виде желтых кристаллов, т. пл. 225-230oC.
Пример 120
[4-(1-Метил-1Н-пиразол-3-ил)фенил]-(4Н,10Н-пиразоло[5,1-с][1,4]бензодиазепин-5-ил)метанон (изомер А) и [4-(2-метил-1Н-пиразол-3-ил)фенил] -(4Н, 10Н-пиразоло[5,1-с][1,4]бензодиазепин-5-ил)метанон (изомер В)
Смесь 1-[4-(4Н,10Н-пиразоло[5,1-с][1,4]-бензодиазепин-5-карбонил)фенил] -3-(диметиламино)-проп-2-ен-1-она (0,83 г), гидразина (0,198 г) и уксусной кислоты (0,336 г) в 10 мл этанола кипятили с обратным холодильником в течение 4 часов. Летучие компоненты удаляли в вакууме и остаток растворяли в дихлорметане. Раствор промывали водой, 1 н. гидрокарбонатом натрия, водой и солевым раствором и сушили над безводным сульфатом натрия. Раствор фильтровали через тонкую подушку из водного силиката магния и фильтровальную подушку промывали этилацетатом. Фильтрат упаривали в вакууме с получением 0,56 г светло-желтого твердого вещества. Твердое вещество хроматографировали на пластинках с толстым слоем силикагеля (200 микрон) с этилацетатом в качестве растворителя с получением 0,35 г белого твердого вещества в виде смеси А и В (1: 4). Многократные фракционированные (дробные) кристаллизации из этилацетата дают 89 мг кристаллов, т.пл. 155-156oС, в виде смеси А и В (9:1) и 65 мг стекловидного продукта в виде смеси А и В (1:6).
Пример 121
1-[4-(5Н, 11Н-Пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-3-хлорфенил] -этанон
Стадия а). Триэтиламин (8,80 мл) добавляли к раствору (4-бром-2-хлорфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанона (2,37 г) в пиридине (1,80 мл) в трубке Карриуса на 20 мл. Полученный раствор продували азотом в течение 25 минут, затем добавляли (триметилсилил)ацетилен (1,67 мл), бис(трифенилфосфин)палладий(II)хлорид (0,08 г) и иодид меди(I) (0,01 г). Трубку наполняли продутым азотом триэтиламином, запаивали и нагревали на масляной бане при 90oС в течение 80 часов. Раствор охлаждали до комнатной температуры, растворитель удаляли в вакууме и остаток распределяли между дихлорметаном и водой. Дихлорметановый экстракт сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме до коричневой пены. Очистка флэш-хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1: 1) приводила к промежуточному ацетиленовому продукту в виде пены беловатого цвета (2,11 г). MS, m/z: 418 (М)+. Этот продукт использовали без дополнительной очистки в следующей стадии.
Стадия b). Раствор 1% серной кислоты в тетрагидрофуране насыщали сульфатом ртути(II). Промежуточный ацетилен (1,00 г) в тетрагидрофуране (5 мл) перемешивали в течение 50 часов с 30 мл вышеупомянутого раствора сульфата ртути(II) в тетрагидрофуране. Добавляли дополнительное количество сульфата ртути(II) (0,01 г) и воды (0,3 мл). После перемешивания в течение 120 часов реакционную смесь выливали в воду и экстрагировали дихлорметаном. Дихлорметановый раствор промывали последовательно насыщенным водным раствором бикарбоната натрия и водой. Дихлорметановый раствор сушили над безводным сульфатом магния, фильтровали и выпаривали досуха с получением коричневого твердого вещества. Очистка флэш-хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1:1) давала белое твердое вещество (0,30 г), т.пл. 98-100oС. MS, m/z: 364 (М)+.
Пример 122
1-[4-(5Н, 11H-Пирроло[2,1-с][1,4]бензодиазепин-10-карбонил)-3-хлорфенил] -этанон
Трибутил(этоксивинил)олово (1,17 г) добавляли к раствору (4-бром-2-хлорфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанона (1,24 г) в толуоле (10 мл). Полученный раствор продували азотом в течение 10 минут, затем добавляли бис(трифенилфосфин)палладий(II)хлорид (0,11 г). Реакционную смесь нагревали с обратным холодильником до кипения в течение 24 часов. Раствор охлаждали до комнатной температуры и добавляли 5% водную хлористоводородную кислоту (10 мл). После перемешивания в течение одного часа смесь фильтровали через подушку из диатомовой земли. К фильтрату добавляли диэтиловый эфир (5 мл) и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме с получением коричневого стекловидного продукта. Очистка флэш-хроматографией на силикагеле с элюированием смесью гексан/этилацетат (1:1) давала белое твердое вещество (0,30 г). MS, m/z: 364 (М)+.
Пример 123
[2-Хлор-4-(3-метил-4-этинилфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4]бензодиазепин-10-ил)-метанон
Обработка промежуточного ацетилена стадии А примера 121 1 М раствором тетрабутиламмонийфторида в тетрагидрофуране при комнатной температуре обеспечивала при удалении растворителя 84% выход указанного в заголовке соединения в виде оранжево-желтого твердого вещества, т.пл. 84-86oС. MS, m/z: 346 (М)+.




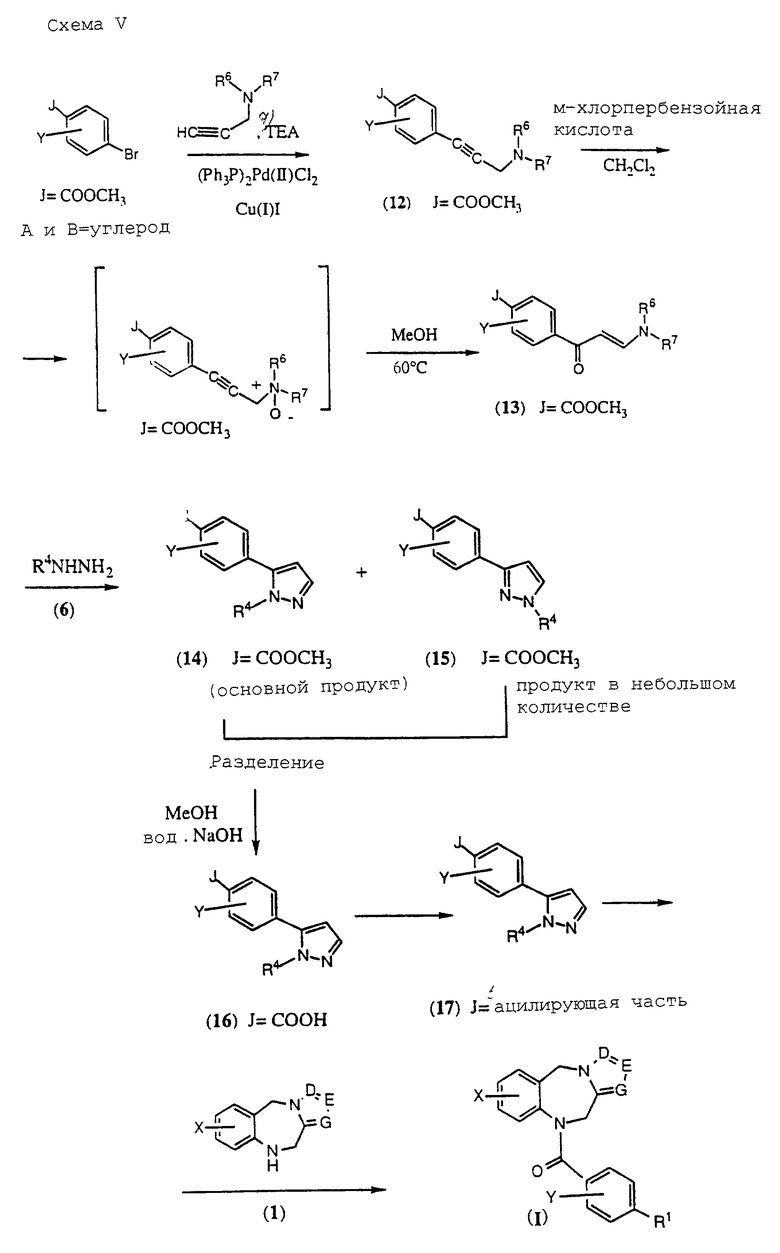

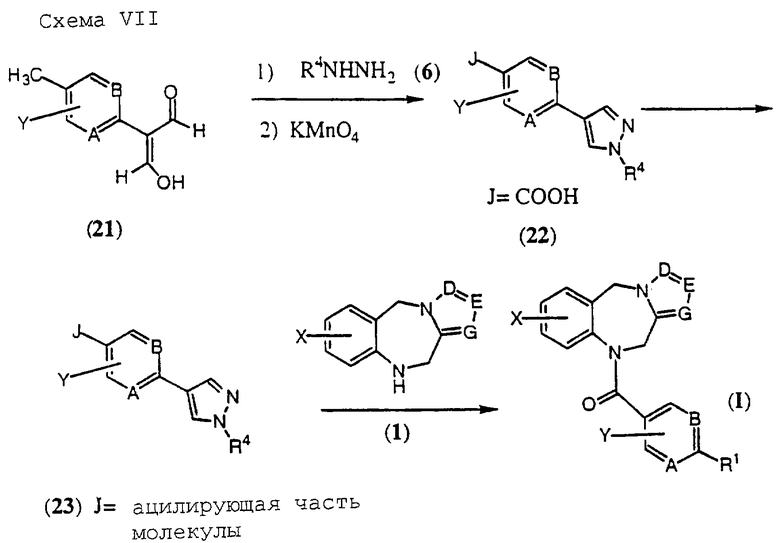




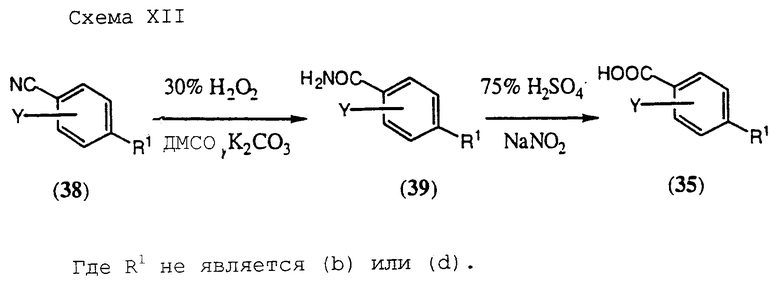





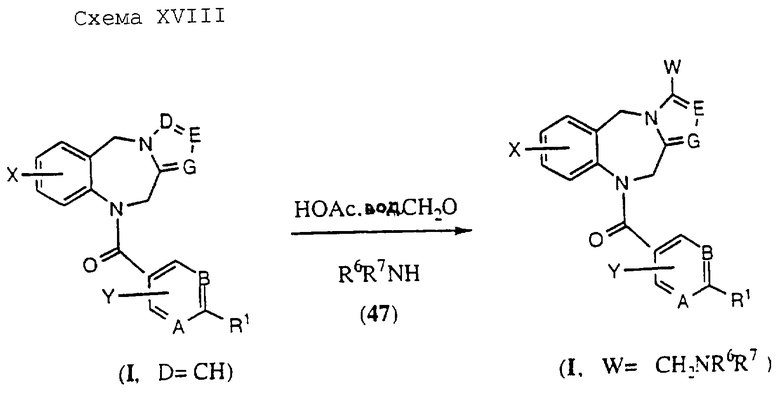



Описываются новые трициклические соединения общей формулы I, где А и В независимо означают СН или азот; D означает С-W или азот; W означает водород, галоген, С1-6-алкил или группу СН2NR6R7, где R6 и R7 независимо означают С1-6-алкил с прямой цепью; Е и G означают одновременно СН или азот; R1 означает алканоил, содержащий 2-7 атомов углерода, группу, выбранную из CN, CONH2, или остаток, выбранный из групп (а), (b), (с), (d), (е), (f), (g), (h), (i), (j), (k) и (m); где R2 - водород или С1-6-алкил с прямой цепью, С3-7-циклоалкил или CF3; R3 и R5 - независимо водород или С1-6-алкил; R4 - водород, С1-6-алкил с прямой цепью, алкоксиалкил, содержащий 2-7 атомов углерода, циклоалканоил, содержащий 3-7 атомов углерода, бензоил, замещенный галогеном, С1-6-алкилом или фенилом, который, в свою очередь, является незамещенным или замещенным CF3, или тиофеналканоил; Х - водород; Y - водород, С1-6-алкил с прямой цепью, С1-6-алкокси, гидрокси, галоген или CF3, или их фармацевтически приемлемые соли, способы их получения, фармацевтическая композиция, обладающая активностью агониста вазопрессина, и способ лечения заболевания или состояния млекопитающего, при котором желательна активность агониста вазопрессина, в частности несахарного диабета. 15 с. и 8 з.п.ф-лы, 1 табл.

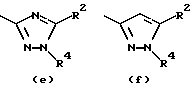
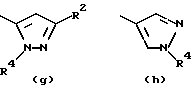
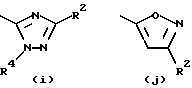


где А и В независимо означают СН или азот;
D означает C-W или азот;
W означает водород, галоген, С1-6-алкил или группу СН2NR6R7;
где R6 и R7 независимо означают С1-6-алкил с прямой цепью;
Е и G означают одновременно СН или азот;
R1 обозначает алканоил, содержащий 2-7 атомов углерода, группу, выбранную из CN, CONH2, или остаток, выбранный из групп (a), (b), (c), (d), (e), (f), (g), (h), (i), (j), (k) и (m)
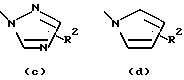




где R2, обозначает водород, С1-6алкил с прямой цепью, С3-7-циклоалкил или CF3;
R3 и R5 означают водород или С1-6алкил с прямой цепью,
R4 обозначает водород, С1-6алкил с прямой цепью, алкоксиалкил, содержащий 2-7 атомов углерода, циклоалканоил, содержащий 3-7 атомов углерода, бензоил, замещенный галогеном, С1-6циклоалкил или фенилом, который, в свою очередь, является незамещенным или замещенным CF3, или тиофеналканоил;
Х обозначает водород;
Y обозначает водород, С1-6алкил с прямой цепью, С1-6алкокси, гидрокси, галоген или СF3,
или их фармацевтически приемлемые соли.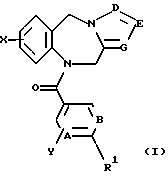
где А и В независимо означают СН или азот;
D означает C-W или азот;
Е и G означают одновременно СН или азот;
R1 означает алканоил, содержащий 2-7 атомов углерода, или остаток, выбранный из групп (а), (b), (e), (f), (g), (h), (i) и (к):
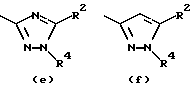
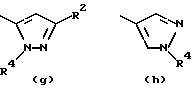

R2, R3, R4, R5, X, Y, W, R6 и R7 определены, как указано в п. 1,
или его фармацевтически приемлемая соль.
где А и В независимо означают СН или N;
D обозначает C-W или N;
R1 означает алканоил, содержащий 2-7 атомов углерода, или остаток, выбранный из групп (а), (b), (e), (f), (g), (h), (i) и (к):
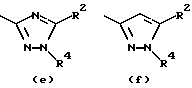
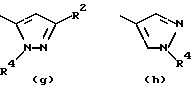
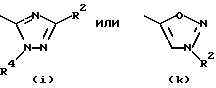
R2, R3, R4, R5, Х, Y, W, R6 R7 определены, как указано в п. 1,
или его фармацевтически приемлемая соль.
где А, В, W, R1, R2, R3, R4, R5, R6, R7, Х и Y определены, как указано в п. 1,
или его фармацевтически приемлемая соль.
где А, В, R1, R2, R3, R4, R5, Х и Y определены, как указано в п. 1,
или его фармацевтически приемлемая соль.
где А, В, R1, R2, R3, R4, R5, Х и Y определены, как указано в п. 1,
или его фармацевтически приемлемая соль.
[4-(3-метилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(4-метилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(4-пиразол-1-ил-2-трифторметилфенил)-(5H, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(3-циклопропилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(4-метилимидазол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-(4-[1,2,4] -триазол-1-ил-2-трифторметилфенил)-метанон,
[2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(5-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(4-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(4-метилимидазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(3-трифторметилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(1,2,4-триазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(2-хлор-4-пиррол-1-илфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(2-хлор-4-пиразол-1-илфенил)-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(1Н-имидазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(3-метил-5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-(3-метилпиразол-1-ил)-4-трифторметилпиримидин-5-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-(4-метилпиразол-1-ил)-4-тpифтopмeтилпиримидин-5-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
1-[4-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)фенил] -этанон,
[4-(1H-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-метил-1H-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-этил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-пропил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-бутил-1H-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-метоксиметил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
1-{ 3-[4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-фенил] -пиразол-1-ил} -этанон,
1-{ 3-[4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-фенил] -пиразол-1-ил} -пропан-1-он,
[4-(1-циклопропанкарбонил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
1-{ 3-[4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-фенил] -пиразол-1-ил} -бутан-1-он,
(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-{ 4-[1-(тиофен-2-карбонил)-1H-пиразол-3-ил] фенил)-метанон,
{ 4-[1-(5-фтор-2-метилбензоил)-1H-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
{ 4-[1-(2-метилбензоил)-1Н-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
{ 4-[1-(2-хлор-4-фторбензоил)-1H-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] -бензодиазепин-10-ил)-метанон,
{ 4-[1-(2,4-дихлорбензоил)-1H-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
2-(2,4-дихлорфенил)-1-{ 3-[4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-фенил] -пиразол-1-ил} -этанон,
{ 4-[1-(бифенил-2-карбонил)-1Н-пиразол-3-ил] фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
{ 4-[1-(4'-трифторметилбифенил-2-карбонил)-1H-пиразол-3-ил] -фенил} -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)метанон,
[4-(5-метил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-[4-(2Н-[1,2,4] триазол-3-ил)-фенил] -метанон,
[4-(2-метил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(5-метил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(2,5-диметил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(3-метил[1,2,4] оксадиазол-5-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(1-метил-1Н-пиразол-4-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[6-(1-метил-1Н-пиразол-4-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(пиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] -бензодиазепин-10-ил)-метанон,
[4-(4-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] -бензодиазепин-10-ил)-метанон,
[4-(3,5-диметилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-[4-(3-трифторметилпиразол-1-ил)-фенил] -метанон,
[4-(имидазол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(4-метилимидазол-1-ил)-фенил] -(5H, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(1-метил-1Н-пиразол-3-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(2-метил-1Н-пиразол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
3-хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензонитрил,
3-хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензойная кислота,
3-хлор-4-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-бензамид,
[2-хлор-4-(5-метил-2Н-[1,2,4] триазол-3-ил)фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-метанон,
[2-хлор-4-(2Н-1,2,4-триазол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бeнзoдиaзeпин-10-ил)-метанон,
[2-хлор-4-(2-метил-2Н-[1,2,4] триазол-3-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-карбонил)-метанон,
4-[(2,5-диметил-2Н-[1,2,4] триазол-3-ил)-2-хлорфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазелин-10-карбонил)-метанон,
[2-хлор-4-(1Н-тетразол-5-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(3-диметиламинометил-5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(3-бром-(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)[2-хлор-4-(3-метилпиразол-1-ил)-фенил] -метанон,
[2-бром-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(2,4-дифторфенил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-фтор-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(3-метилпиразол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пиразоло[5,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(2-хлор-6-пиразол-1-ил-пиридин-3-ил)-(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(2-хлор-6-(3-метилпиразол-1-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-6-(4-метиллиразол-1-ил)-пиридин-3-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(3-метил-1,2,4-триазол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[4-(3-метил-1,2,4-триазол-1-ил)-2-трифторметилфенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-метокси-4-(3-метилпиразол-1-ил)-фенил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
(3-диметиламинометил-5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-[2-метокси-4-(3-метилпиразол-1-ил)-фенил] -метанон,
[2-гидрокси-4-(3-метилпиразол-1-ил] -(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(1-метил-1Н-пиразол-4-ил)фенил] -(5Н, 11H-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон,
[2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(4Н, 10Н-пиразоло[5,1-с] [1,4] бензодиазепин-5-ил)-метанон,
2-хлор-4-(3-метилпиразол-1-ил)-фенил] -(5,10-дигидро-4Н-тетразоло[5,1-с] [1,4] бензодиазелин-5-ил)-метанон,
1-[4-(4Н, 10Н-пиразоло[5,1-с] [1,4] бензодиазепин-5-карбонил)-фенил] -этанон,
[4-(1-метил-1Н-пиразол-3-ил)фенил] -(4Н, 10Н-пиразоло[5,1-с] [1,4] -бензодиазепин-5-ил)-метанон,
[4-(2-метил-1Н-пиразол-3-ил)фенил] -(4Н, 10Н-пиразоло[5,1-с] [1,4] -бензодиазепин-5-ил)-метанон,
1-[4-(5Н, 11Н-пирроло[2,1-с] [1,4)бензодиазепин-10-карбонил)-3-хлорфенил] -этанон,
или[2-хлор-4-(3-мeтил-4-этинилфeнил)(5Н, 11Н-пирроло[2,1-с] [1,4] бензодиазепин-10-ил)-метанон.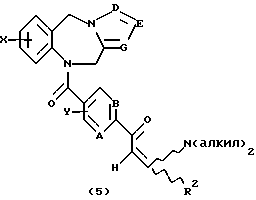
где А, В, D, E, G, X, Y и R2 определен, как указано в п. 1,
с гидроксиламином с получением целевого соединения формулы 1, где R1 представляет собой гетероциклический остаток - группу j, определенную в п. 1.
где каждый из А, В, D, Е, G, X, Y и R2 определен, как указано в п. 1,
с соответствующим образом замещенным гидразином формулы
R4-NHNH2,
где R4 определен, как указано в п. 1,
с получением смеси двух изомеров соединения формулы I, в которых R1 представляет собой один из двух изомерных гетероциклических остатков, определенных в п. 1 как группа f и группа g.
где каждый из D, Е, G и X определен, как указано в п. 1,
с соединением формулы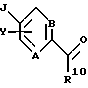
где каждый из А, В и Y определен, как указано в п. 1;
J является ацилирующей группой;
R10 является С1-6-алкильной группой,
с получением соединения формулы I, где каждый из А, В, D, E, G, Х и Y определен, как указано в п. 1;
R1 является С2-7-алканоильной группой.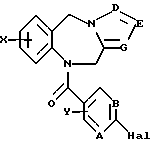
где каждый из А, В, D, Е, G, X и Y определен, как указано в п. 1,
в соответствующее соединение формулы I, где каждый из А, В, D, Е, G, X и Y определен, как указано в п. 1, и R1 является С2-7-алканоильной группой.
где D, Е, G и Х определены, как указано в п. 1,
с ацилирующим агентом формулы 9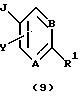
где А и В означают углерод;
J означает ацилирующую группу;
R означает гетероциклический остаток, выбранный из группы g, определенной в п. 1;
R2 обозначает водород,
с получением целевого соединения формулы I, где А и В представляют собой углерод;
R1 представляет собой гетероциклический остаток, выбранный из группы g, определенной в п. 1, и R2 представляет собой водород.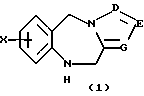
где D, E, G и Х определены, как указано в п. 1,
с ацилирующим агентом формулы 9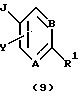
где А и В определены, как указано в п. 1;
J означает ацилирующую группу;
R1 означает гетероциклический остаток, выбранный из группы f, определенной в п. 1;
R2 означает водород,
с получением целевого соединения формулы I, где R1 представляет собой гетероциклический остаток, выбранный из группы f, определенной в п. 1, и R2 представляет собой водород.
где А, В, D, Е, G, Х и Y определен, как указано в п. 1,
с подходящим соединением формулы
R1Н,
где R1 является гетероциклическим остатком, выбранным из групп а, b, с и d, определенных в п. 1,
с получением целевого соединения формулы I, где R1 представляет собой гетероциклический остаток, выбранный из групп а, b, с и d, определенных в п. 1.
где D, Е, G и Х определены, как указано в п. 1,
с ацилирующим агентом формулы 9
где А и В определены, как указано в п. 1;
J означает ацилирующую группу;
R1 означает гетероциклический остаток, выбранный из групп а, b, с и d, определенных в п. 1,
с получением целевого соединения формулы I, где R1 представляет собой гетероциклический остаток, выбранный из групп а, b, с и d, определенных в п. 1.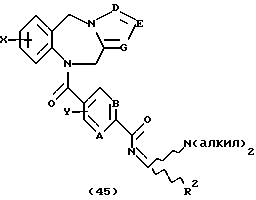
где каждый из А, B, D, Е, G, X, Y и R2 определен, как указано в п. 1,
с гидроксиламином с получением целевого соединения формулы 1, где R1 представляет собой гетероциклический остаток - группу k, определенную в п. 1.
где каждый из А, B, D, Е, G, X, Y и R2 определен, как указано в п. 1,
с соответствующим образом замещенным гидразином формулы
R4-NHNH2,
где R4 определен, как указано в п. 1,
с получением целевого соединения формулы I, в котором R1 представляет собой гетероциклический остаток, выбранный из группы е и группы i, определенных в п. 1.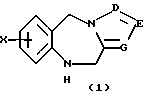
где D, Е, G и Х определены, как указано в п. 1,
с соединением формулы 43
где А, В и Y определены, как указано в п. 1;
J является ацилирующей группой,
с получением целевого соединения формулы 1, где R1 представляет собой CN, и при необходимости, его последующее превращение в соответствии со стандартными методиками в целевое соединение формулы I, где R1 представляет собой СОNH2.
| US 5521173 A, 28.05.1996 | |||
| US 5516774 A, 14.05.1996 | |||
| WO 9722591 A, 26.06.1997 | |||
| RU 94135954 A1, 01.05.1995. |

