Изобретение относится к способу отделения природного нейтрального макролида, особенно трициклического макролида, например рапамицина, 32-дезметилрапамицина, 15-дезоксорапамицина или РК-506, от других природных компонентов, которые получают способами ферментации. Более конкретно настоящее изобретение относится к способу извлечения трициклического макролида рапамицина из концентрата экстракта ферментационного бульона и из концентрата маточного раствора.
Трициклические макролиды, примерами которых являются рапамицин и РК-506, обладают иммунодепрессивной активностью, а также антибиотической и другой фармакологической активностью, они пригодны для лечения отторжения имплантата и трансплантата, воспалительных заболеваний и аутоиммунных заболеваний, например обыкновенной волчанки, ревматического артрита, сахарного диабета и рассеянного склероза.
Трициклические макролиды рапамицина, 32-дезметилрапамицин, 15-дезоксорапамицин и РК-506 получают культивированием различных штаммов Streptomyces в подходящих условиях. Они являются нейтральными соединениями, не содержащими основных аминогрупп, фенольных гидрокси- или карбоксигрупп. Рапамицин получают культивированием S. hygroscopicus NRRL 5491 в водной среде. Мицелин, содержащий трициклический макролид, выделяют из питательной среды и экстрагируют органическим растворителем, например метанолом, для получения смеси, содержащей целевой трициклический макролид, аналогичные соединения, кислотные соединения, например жирные кислоты, соединения основного характера, например алкалоиды и пептиды, и нейтральные липофильные соединения, например жиры. Обычно экстракт ферментационного бульона концентрируют для облегчения транспортировки и/или хранения его до выделения макролида. Выделение трициклических макролидов из экстракта ферментационного бульона и очистку их до настоящего изобретения проводили трудным, дорогим способом с применением различных химических и хроматографических методик для получения очищенного материала [US 5091389, US 3993749, WO 93/11130, Sehgal, J. of Antibiotics 28(10)727(1975)]. Концентрат экстракта ферментационного бульона для рапамицина содержит, например, только 5-15% рапамицина и до 50% кислотных компонентов. Рапамицин нужно отделить от других компонентов. Обычно способы выделения трициклических макролидов из экстрактов ферментационных бульонов включают в себя адсорбцию на активированном угле и десорбцию из него, методы селективного растворения и однократное или многократное проведение дорогого хроматографического разделения с помощью колоночной хроматографии и/или жидкостной хроматографии при высоком давлении. До сих пор избегали условий кислотного и основного характера, поскольку трициклические макролиды, например рапамицин, как полагают, нестабильны в кислотных и основных условиях. Рапамицин в смешиваемом с водой растворе, т.е. метаноле или тетрагидрофуране, подвергается разрушению неорганическими основаниями, например водным раствором едкого натра, органическими основаниями, например 4-диметиламинопиридином (DMAP) или 1,8-диазобицикло[5,4, о] ундец-7-еном (DBU), или водными минеральными кислотами, например соляной кислотой, и кислотами Льюиса, например хлоридом цинка [Steffan et al., Tetrahedron Letters (в печати) D. Yohannes and S. J.Danishefsky, Tetrahedron Letters 33(49), 7469-7472 (1992), Luengo et al., Tetrahedron Letters 34(6), 991-994 (1983) и D.Johannes et al.. Tetrahedron Letters 34(13), 2075-2078 (1993)].
В качестве ближайшего аналога настоящего изобретения можно считать авторское свидетельство N 624578.
Настоящее изобретение предлагает относительно быстрый и эффективный способ извлечения макролида, в частности трициклического макролида, более конкретно рапамацина, из концентратов экстрактов (ферментационного бульона и маточных растворов или их концентратов, полученных из растворов после перекристаллизации, растворителей, применяющихся две растирания макролида, и вод промывания продукта. Способ не включает затратное и дорогое хроматографическое разделение, примеры которого приводятся в US 5091389, US 3993749, WO 93/11130 и Sehgal, J. of Antibiotics 28(10) 727 (1975). Способ предусматривает отделение кислотных и/или основных компонентов от нейтральных компонентов растворением содержащего макролид концентрата в не смешиваемом с водой подходящем растворителе с последующей экстракцией кислотных и/или основных компонентов водным основанием или кислотой соответственно и использует методики селективного растворения или экстракции для отделения нейтрального полярного трициклического макролида от неполярных нейтральных веществ, присутствующих в концентрате. Тогда как в способе, описанном в приведенных ссылках, применяют концентраты экстрактов ферментационных бульонов или маточных растворов, в способе настоящего изобретения можно применять целый экстракционный раствор или маточные растворы, причем растворитель (или смесь растворителей), применяемый для экстракции ферментационного бульона или перекристаллизации, растирания в порошок или промывания, пригоден для настоящего способа и объем растворителя не создает затруднений. Объем растворителя можно уменьшить частичным концентрированием. Любой из растворов, для которого можно применять способ настоящего изобретения, можно именовать содержащим макролид концентратом.
Продукт, полученный настоящим способом, можно очищать до пригодной чистоты обычными методиками, известными в настоящей области науки.
Способ настоящего изобретения изображен на приведенной в конце описания схеме 1. В следующем ниже описании способа изобретения неполярным растворителем является неароматический углеводород, например циклогексан, циклогексен, гексан, гептан, пентам и подобный углеводород. Растворители, которые не смешиваются с неароматическим углеводородным растворителем, включают (но не ограничиваются этими примерами) ацетонитрил и диметилформамид. Термин "экстракция" относится к методике интенсивного перемешивания одного раствора с другим, несмешиваемым раствором, выдерживания несмешиваемых растворов для отделения одного от другого и физического отделения одного слоя или фазы от другой. Термин "промывание для раствора" обозначает процедуру экстракции, а для твердого вещества обозначает промывание этого вещества растворителем, в котором твердое вещество нерастворимо. Термин "маточный раствор" относится к растворам в органических растворителях, полученным из фильтратов кристаллизации, промывочных растворов и обратных экстрактов водных экстрактов и промывочных растворов и продуктов растирания в порошок отобранных твердых веществ. Для макролида применяют растворитель (или смесь растворителей), который растворит макролид и сопровождающие его примеси, например компоненты кислотного или основного характера. Нерастворителем для макролида является растворитель (или смесь растворителей), в которой макролид по существу нерастворим, но в котором растворимы нейтральные компоненты, например жиры. Для кристаллизации макролида применяют растворитель (или смесь растворителей), из которого макролид можно перекристаллизовать или кристаллизовать (из аморфного состояния) при растирании и порошок. Когда способ требует концентрирования раствора, рекомендуется применять растворитель (или смесь растворителей), который имеет достаточную летучесть для отгонки его при температуре и давлении, которые не вызывают разрушение макролида.
В соответствии со способом концентрат, содержащий макролид, независимо от того, является ли он концентратом экстракта ферментационного бульона или концентратом операции перекристаллизации и/или промывания растворителем, растворяют в не смешиваемом с водой растворителе или смеси растворителей, подобранных по способности растворять концентрат и легкости удаления из раствора. Эти растворители включают (но не ограничиваются этими примерами) дихлорметан, трет-бутилметиловый эфир, этилацетат, толуол, 1-бутанол, смесь этилацетат/толуол, смесь гептан/этилацетат или смесь гексан/хлористый метилен. Для экстракции кислотных компонентов пригодны водные растворы оснований, включая едкий натр, бикарбонат натрия, карбонат натрия, гидроксид аммония и подобные основания, предпочтительно раствор едкого натра с концентрацией 0,1-5N, предпочтительно примерно от 0,1 до 1N, более предпочтительно от 0,1 до 0,5N. Органические основания, например триэтиламин, не так эффективны в удалении кислотных компонентов, как более сильные водные неорганические основания. Для экстракции компонентов основного характера пригодны водные растворы минеральных кислот, включая соляную кислоту, первичный кислый фосфат калия, кислый сульфат натрия и подобные соединения, предпочтительно соляной кислоты, с концентрацией 0,1-5N, предпочтительно примерно от 0,1 до 1N, более предпочтительно от 0,1 до 0,5N. Органические кислоты, например трифторуксусная кислота, не так эффективны в удалении компонентов основного характера как минеральные кислоты. Экстракцию кислотных и/или основных компонентов из раствора, содержащего макролид, проводят при - 5-45oC, предпочтительно при -5-30oC и более предпочтительно при -5-10oC. Для избежания возможного разложения макролида под действием кислоты или оснований экстракцию следует проводить без задержки. В зависимости от количества кислотных и/или основных компонентов, присутствующих в растворе, иногда нет необходимости экстрагировать как кислотные, так и основные компоненты. Экстракции только кислотных компонентов или только основных компонентов может быть достаточно для удаления достаточного количества примесей из содержащего макролид раствора, из которого затем можно выделить трициклический макролид.
Выгодно удалять кислотные (или основные) компоненты из раствора, содержащего макролид концентрат, однократной экстракцией. Количество водного основания (или кислоты), необходимое для обеспечения избытка основания (или кислоты) в процессе экстракции, можно определить путем экстракции аликвоты раствора, содержащего макролид концентрата ,водным основанием (или кислотой) и определения объема водного основания (или кислоты), требуемого для получения экстракта аликвоты, имеющего такое значение pH, чтобы присутствовал избыток (относительно cтехиометрического количества) основания (или кислоты). Применяют pH-метр или другое средство для определения pH. Объем водного основания (или кислоты), необходимый для экстракции раствора, из которого взята аликвота, затем определяют на основе соотношения объемов раствора, который будут экстрагировать, и взятой из него аликвоты. Таким образом, экстракцию кислотных (или основных) компонентов можно осуществить в одну операцию вместо того, чтобы проводить многократную экстракцию водным основанием (или кислотой). Очевидно, когда из раствора, содержащего макролид концентрата, нужно удалить как кислотные, так и основные компоненты, требуется провести отдельные экстракции водным основанием и водной кислотой соответственно. Например, при однократной экстракции раствора, содержащего рапамицин концентрата, водным раствором едкого натра достигается pH раствора 12, что достаточно для удаления всего количества кислотных компонентов.
Макролид, извлеченный из раствора указанными выше способами, можно очистить до требуемой степени обычными методиками очистки, известными в данной области науки. Фильтраты и промывочные растворы можно подвергать обработке для извлечения дополнительного количества макролида, если это требуется.
После проведения экстракции для удаления кислотных и/или основных компонентов из содержащего макролид раствора в не смешиваемом с водой растворителе макролид можно отделить от неполярных нейтральных компонентов одним из следующих методов:
(1) Нерастворитель для макролида (или смесь нерастворителей), смешиваемый с содержащим макролид раствором в не смешиваемом с водой растворителе, из которого были удалены кислотные и/или основные компоненты, добавляют в содержащий макролид раствор в количестве, достаточном для того, чтобы сделать макролид (и возможно, аналогичные ему продукты) нерастворимым в образованном растворе и образовать отдельную фазу, масляную или твердую, которую можно затем отделить обычными известными методиками.
(2) Содержащий макролид раствор в не смешиваемом с водой растворителе, из которого экстрагированы кислотные и/или основные компоненты, концентрируют и остаток, содержащий макролид, растворяют в растворяем макролид растворителе, например ацетонитриле или диметилформамиде, и раствор экстрагируют неароматическим углеводородным растворителем, например циклогексаном, гексаном, гептаном или циклогексаном. Слой растворителя, растворяющий макролид, отделяют и концентрируют. Остаток, содержащий макролид, затем растирают с кристаллизующим растворителем для получения макролида, если он твердый, или очищают известными методиками, например хроматографией, если макролид является маслом. В соответствии с другим вариантом растворяющий макролид растворитель после экстракции неароматическим углеводородным растворителем можно обработать смешиваемым с ним не растворяющим макролид растворителем, как указано в методе (1).
(3) Содержащий макролид раствор в не смешиваемом с водой растворителе, из которого экстрагированы кислотные и/или основные компоненты, концентрируют и остаток растирают с кристаллизующим макролид растворителем, например диэтиловым эфиром, диизопропиловым эфиром или трет-бутилметиловым эфиром.
В соответствии с другим вариантом содержащий макролид концентрат можно экстрагировать сначала по методу (1)-(3) для удаления неполярных компонентов и затем остаток, содержащий макролид, растворить в не смешиваемом с водой растворителе, если он уже не находится в этом растворителе, и экстрагировать водным основанием и/или кислотой для удаления кислотных и/или основных компонентов.
Указанный выше способ использует неожиданные данные о том, что макролиды, например рапамицин, не разлагаются в процессе экстракции растворов, содержащих макролид концентратов в не смешиваемом с водой растворителе, водной кислотой или основанием. Ранее на основе наблюдаемого разложения рапамицина в результате обработки его в растворе кислотой или основанием полагали, что будет иметь место разложение рапамицина.
Этот способ экстракции значительно сокращает время, необходимое для извлечения макролидов из этих концентратов, и позволяет исключать времязатратные и дорогие хроматографические методики.
Трициклический макролид рапамицин представляет собой кристаллическое твердое соединение, растворимое в метаноле, ацетоне, диметилформамиде, слабо растворимое в диэтиловом эфире и умеренно растворимое в гексане или нетролейном эфире и нерастворимое в воде. Рапамицин имеет строение, приведенное ниже. Система нумерации атомов та же, которую применяют в химических рефератах.

Следующие примеры только иллюстрируют способ настоящего изобретения для выделения рапамицина из концентратов ферментационных бульонов или маточных растворов. Их не следует рассматривать как ограничение объема этого изобретения. В следующих примерах идентичность выделенного продукта с рапамицином была установлена путем сравнения физических, спектральных и хроматографических свойств с аналогичными свойствами аутентитного рапамицина. Чистоту продукта (неперекристаллизованного) определяли жидкостным хроматографическим анализом при высоком давлении.
ПРИМЕР 1
Раствор концентрированного экстракта ферментационного бульона (157,0 г, содержание рапамицина 10,4%) в хлористом метилене (600 мл) промыли три раза порциями по 150 мл 0,5N раствора NaOH при 0-5oC, промыли водой до достижения нейтральной промывочной воды и затем промыли соляным раствором. Раствор в хлористом метилене концентрировали и остаток (70,5 г) растирали в порошок с применением диэтилового эфира (140 мл). Кристаллический продукт отобрали, промыли диэтиловым эфиром и сушили для получения рапамицина (6,3 г, чистота 91,7%, выход 35,4%). Концентрированием фильтратов (в диэтиловом эфире) получили 63,5 г масла, имеющего содержание рапамицина 13,1%.
ПРИМЕР 2
Раствор концентрированного экстракта ферментационного бульона (206,0 г, содержание рапамицина 11,8% ) в трет-бутилметиловом эфире (800 мл) промыли три раза порциями по 400 мл 0,5N раствора NaOH при 0-5oC и затем промыли водой до достижения нейтральной промывочной воды. Раствор в трет-бутилметиловом эфире концентрировали и остаток (75,0 г) растирали в порошок с применением диэтилового эфира (150 мл). Кристаллический твердый продукт отобрали, промыли диэтиловым эфиром и сушили для получения рапамицина (11,4 г, чистота 92,2%, выход 43,3%). Концентрированием фильтрата (в диэтиловом эфире) получили 58,7 г масла, имеющего содержание рапамицина 11,3%.
ПРИМЕР 3
Раствор концентрированного экстракта ферментационного бульона (10,58 г, содержание рапамицина 10,4%) в ацетонитриле (23 мл) промыли два раза порциями по 23 мл циклогексана и затем концентрировали. Остаток растворили в дихлорметане и промыли последовательно три раза порциями по 23 мл 0,5N раствора NaOH при 0-5oC, два раза порциями по 23 мл 0,5N HCl при 0-5oC и затем водой до достижения нейтральной промывочной воды. Раствор в дихлорметане концентрировали и остаток (4,07 г) растирали в порошок с применением диэтилового эфира. Кристаллический твердый продукт отобрали, промыли диэтиловым эфиром и сушили для получения рапамицина (0,64 г, чистота 89,5%, выход 58,2%). Фильтраты (в диэтиловом эфире) концентрировали для получения 3,36 г масла, имеющего содержание рапамицина 9,8%.
ПРИМЕР 4
Раствор концентрированного экстракта ферментационного бульона (100,0 г, содержание рапамицина 11,8%) в этилацетате (400 мл) промыли один раз порцией 200 мл и два раза порциями по 100 мл 0,5N раствора NaOH при 0-5oC и затем промыли два раза порциями по 200 мл 0,5N раствора соляной кислоты при 0-5oC и в конце водой до достижения нейтральной промывочной воды. Промывочные воды обратно экстрагировали этилацетатом (100 л) и растворы этилацетата объединили. Объединенный раствор в этилацетате концентрировали и остаток (43,2 г) растирали в порошок с применением диизопропилового эфира (45 мл). Кристаллический твердый продукт отобрали, промыли диизопропиловым эфиром и сушили, получив 9,5 г рапамицииа (чистота 85,3%, выход 68,7%). Концентрированием фильтратов (в диизопропиловом эфире) получили масло (31,6 г) с содержанием рапамицина 6,6%.
ПРИМЕР 5
Раствор концентрированного экстракта ферментационного бульона (25,2 г, содержание рапамицина 10,4%) в смеси толуола (120 мл) и этилацетата (25 мл) промыли последовательно три раза порциями по 50 мл 0,5N раствора NaOH при 0-5oC, два раза порциями по 50 мл 0,5N соляной кислоты при 0-5oC и затем водой до достижения нейтральной промывочной воды. Раствор в смеси толуола и этилацетата (128 мл) разделили на две равные части для дальнейшей обработки следующими методами:
Метод А. Раствор в смеси толуола и этилацетата (64 мл) концентрировали и остаток (5,7 г) растирали в порошок с применением диэтилового эфира (11 мл). Кристаллический твердый продукт отобрали, промыли дополнительным количеством диэтилового эфира и сушили для получения 0,64 г рапамицина (чистота 92,7%, выход 64,1%). Концентрированием фильтратов (в диэтиловом эфире) получили 4,3 г масла, имеющего содержание рапамицина 6,8%.
Метод В. Раствор в смеси толуола и этилацетата (64 мл) концентрировали и остаток (9,0 г) растворили в ацетонитриле (50 мл). Раствор в ацетонитриле промыли два раза порциями по 25 мл циклогексана, затем концентрировали. Остаток (4,3 г) растирали в порошок с применением диэтилового эфира (11 мл). Кристаллический твердый продукт отобрали, промыли диэтиловым эфиром и сушили для получения 0,81 г рапамицина (чистота 94,6%, выход 61,8%). Концентрированием фильтратов (в диэтиловом эфире) получили 3,0 г масла с содержанием рапамицина 5,9%.
ПРИМЕР 6
Концентрат маточного раствора (996,0 г, содержание рапамицина 21,6%) растирали с трет-бутилметиловым эфиром (4000 мл). Кристаллический твердый продукт отобрали, промыли трет-бутилметиловым эфиром (500 мл) и сушили для получения 36,2 г рапамицина (чистота 95,1%, выход 13,8%). Фильтраты промыли один раз порцией 2000 мл и два раза порциями по 1000 мл 0,5N раствора едкого натра при 0-5oC. Объединенный экстракт основания промыли один раз трет-бутилметиловым эфиром (500 мл). Фильтрат и экстракт объединили и промыли водой до достижения нейтральной промывочной воды. Водные экстракты объединили и экстрагировали трет-бутилметиловым эфиром (500 мл). Растворы (в трет-бутилметиловом эфире) объединили, концентрировали и остаток (377,1 г) растирали с диизопропиловым эфиром (350 мл). Кристаллический твердый продукт отобрали, промыли диизопропиловым эфиром и сушили для получения 126,7 г рапамицина (чистота 82,4%, выход 58,9%). Таким образом, всего выделено рапамицина из концентрата маточного раствора 162,9 г (72,7%). Концентрированием фильтрата (в диизопропиловом эфире) получили 157,9 г масла, имеющего содержание рапамицина 20,9%.
ПРИМЕР 7
Раствор концентрата маточного раствора (562,1 г, содержание рапамицина 21,6%) в дихлорметане (2000 мл) промыли три раза порциями по 500 мл 0,5N раствора едкого натра при 0-5oC и объединенный водный экстракт основания экстрагировали один раз 200 мл дихлорметана. Органические растворы объединили и промыли два раза порциями по 500 мл 0,5N раствора соляной кислоты при 0-5oC. Объединенный водный экстракт кислот экстрагировали один раз 200 мл дихлорметана. Объединенный органический экстракт промыли водой до достижения нейтральной промывочной воды. Органический раствор концентрировали и остаток (255,0 г) растирали с диизопропиловым эфиром (250 мл). Кристаллический продукт отобрали, промыли диизопропиловым эфиром и сушили для получения 108,6 г рапамицина (чистота 86,6%, выход 77,4%). Концентрированием фильтрата (в диизопропиловом эфире) получили 100,2 г масла с содержанием рапамицина 23,1%.
ПРИМЕР 8
Водный 0,5N раствор едкого натра (400 мл) при 0-5oC добавили в энергично перемешиваемый, охлажденный до 0-5oC раствор концентрированного экстракта ферментационного бульона (198,5 г, содержание рапамицина 8,3%) в 800 мл трет-бутилметилового эфира с такой скоростью, чтобы можно было поддерживать температуру 0-5oC. После энергично перемешивания в течение 6 мин нижний водный слой оснований удалили и хранили при 0-5oC. Органический слой реэкстрагировали два раза порциями по 200 мл 0,5N раствора едкого натра при 0-5oC. Водные экстракты оснований объединили и реэкстрагировали трет-бутилметиловым эфиром (200 мл). Растворы в трет-бутилметиловом эфире объединили и промыли водой до достижения нейтральной промывочной воды (pH 7). Промывные воды объединили и экстрагировали трет-бутилметиловым эфиром (100 мл). Растворы в трет-бутилметиловом эфире объединили, промыли насыщенным водным раствором хлористого натрия и концентрировали в вакууме при 40oC. Остаток растирали с диизопропиловым эфиром (85 мл) при 20-25oC в течение не менее одного часа и смесь охлаждали до 0-5oC в течение ночи. Кристаллический продукт отобрали при помощи воронки Бюхнера из агломерированного стекла и промыли смесью диизопропилового эфира и трет-бутилметилового эфира в соотношении 4: 1 при 20-25oC 5 раз порциями по 20 мл или до получения бесцветного фильтрата. Кристаллический продукт сушили до постоянной массы, получив 12,0 г рапамицина (чистота 91,2%, выход 66,4%). Концентрированием фильтрата (в трет-бутилметиловом эфире) и промывочного раствора получили 63,9 г смолистого продукта с содержанием рапамицина 3,63%.
ПРИМЕР 9
Концентрат маточного раствора (200 г, содержание рапамицина 25%) растворили при перемешивании в трет-бутилметиловом эфире (800 мл) при комнатной температуре. Раствор охладили до 0-5oC и экстрагировали сразу 270 мл 0,65 N раствора едкого натра, предварительно охлажденного до 0-5oC, поддерживая температуру смеси 0-5oC. (Количество водного раствора едкого натра, необходимого для того, чтобы экстракт в конце имел pH 12, определяли по аликвоте концентрированного маточного раствора).
Водный слой основания выдерживали при 0-5oC, в то время как органический слой промыли 5%-ным раствором хлористого натрия, поддерживая температуру смеси 0-5oC. Водный экстракт основания обратно экстрагировали трет-бутилметиловым эфиром (100 мл). Органические слои объединили и промыли три раза порциями по 200 мл 5%-ного раствора хлористого натрия (pH конечного промывочного раствора 7,4). Органический раствор концентрировали при пониженном давлении (80-130 мм рт.ст.) и температуре 25-40oC. К остатку при перемешивании при комнатной температуре медленно, в течение 30 мин добавили циклогексен (80 мл) и перемешивание продолжали до достижения полной кристаллизации (3 ч). Смесь перемешивали при комнатной температуре еще один час, затем охладили до 0-5oC и перемешивали в этих условиях в течение ночи. Белый кристаллический продукт отделили на воронке Бюхнера из агломерированного стекла и промыли пять раз порциями по 40 мл смеси трет-бутилметилового эфира и циклогексена в соотношении 2:3. Продукт сушили до постоянной массы в вакууме при 35-40oC для получения 22,7 г рапамицина (чистота 90,6%, выход 41,4%). Концентрированием фильтратов и промывочных растворов (трет-бутилметиловый эфир и циклогексен) получили 60,1 г масла с содержанием рапамицина 37,5%.
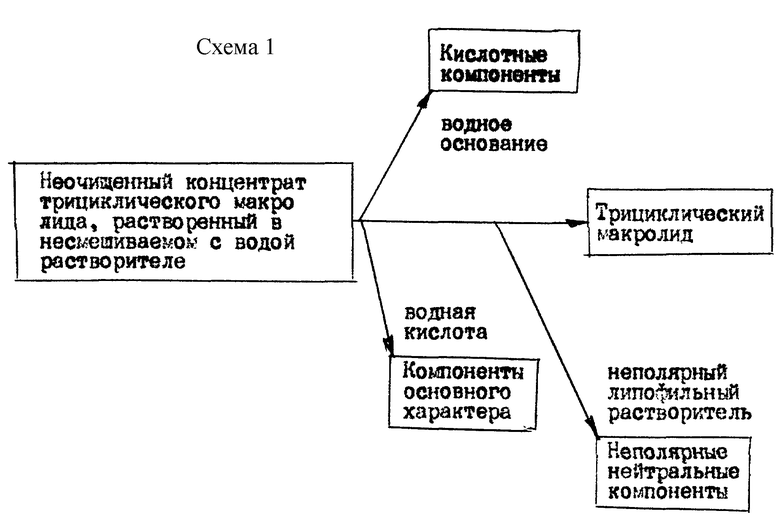
Способ предназначен для извлечения трициклического макролида рапамицина из концентрата экстракта ферментационного бульона и из концентрата маточного раствора. Способ осуществляют в несколько стадий в любом порядке, например экстрагирование водным раствором основания концентрата в не смешиваемом с водой первом растворителе. Далее производят экстрагирование водным раствором щелочи раствора концентрата в не смешиваемом с водой растворителе. И обрабатывают вторым неароматическим углеводородным растворителем раствор концентрата. Изобретение предлагает относительно быстрый и эффективный способ извлечения рапамицина. 6 з.п.ф-лы, 1 ил.
| Способ получения производных полиеновых макролидных антибиотиков | 1972 |
|
SU624578A3 |
| SU 735117 A, 15.08.1981 | |||
| Способ получения антибиотиков | 1967 |
|
SU528883A3 |

