Изобретение относится к способу получения кристаллического моногидрата лоракарбефа путем сушки изопропанолата лоракарбефа для получения безводного лоракарбефа и затем гидратирования безводного лоракарбефа для получения моногидрата лоракарбефа.
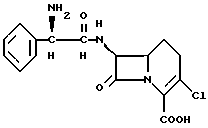
β-Лактамный антибиотик формулы I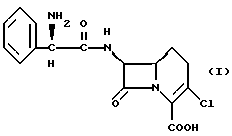
представляет собой сильнодействующий, активный при приеме внутрь антибиотик, известный как лоракарбеф. Антибиотик описан, например, Hashimoto et al., в патенте США N 4335211.
Лоракарбеф выделяют в различных формах, включая кристаллическую моногидратную форму, которая описана в опубликованном Европейском патенте (EPA) N 0311366. Другие известные сольватные формы соединения представлены Eckrich et al. , в патенте США N 4977257. Кристаллическая дигидратная форма лоракарбефа описана в EPA 0369686.
Как указывается в EPA 0369686, кристаллическая моногидратная форма лоракарбефа может быть получена суспендированием сначала дигидрата лоракарбефа в воде и затем получая раствор путем добавления кислоты и последующего регулирования pH основанием или путем добавления основания и затем кислоты. Полученный лоракарбеф можно кристаллизовать и затем выделить фильтрацией. В дальнейшем этот способ будет отнесен к "способу фильтрации". Кристаллический моногидрат лоракарбефа представляет собой тонкие, "похожие на волосы" кристаллы, которые получаются при очень медленной фильтрации.
Кристаллы моногидрата имеют склонность к образованию на фильтре мата, который препятствует завершению фильтрации или значительно замедляет такое завершение, и поэтому кристаллы необходимо промывать водой. Так как моногидрат лоракарбефа умеренно растворим в воле (примерно 10 мг/мл), такая промывка уменьшает выход.
Способ фильтрации считают коммерчески невыгодным способом получения моногидрата лоракарбефа с точки зрения трудностей, с которыми сталкиваются при осуществлении стадии фильтрации. Впоследствии обнаружили, что кристаллический моногидрат лоракарбефа может быть эффективно получен воздействием на кристаллическую изопропанолатную форму лоракарбефа температуры от около 50oC до около 90oC и относительной влажности от около 60% до около 100%. В дальнейшем этот способ будет отнесен к "изопропанолатному способу". Так как этот способ представляет собой превращение твердого изопропанолата лоракарбефа в моногидрат лоракарбефа, его можно осуществлять без фильтрации кристаллического моногидрата, обеспечивая таким образом более эффективный способ.
Однако изопропанолатный способ также считают коммерчески невыгодным, потому что при его осуществлении получают моногидрат лоракарбефа в форме тонкого легкого мелкокристаллического порошка с плотностью примерно 0,2 г/мл. Такая плотность очень затрудняет получение объемного продукта моногидрата лоракарбефа. Поскольку это соединение предназначено для фармацевтического применения, необходимо получать объемный продукт. Для получения объемного продукта желательной плотностью для моногидрата лоракарбефа является плотность более или равная 0,5 г/мл. Таким образом, для получения объемного продукта с такой плотностью, которая является достаточной для того, чтобы продукт можно было использовать в фармацевтике, изопропанолатный способ необходимо модифицировать.
Другой недостаток изопропанолатного способа состоит в том, что полученный моногидрат лоракарбефа может содержать до 5% остаточного изопропанола (% по массе). Так как обычно считается приемлемым, чтобы содержание растворителя в продуктах, предназначенных для использования человеком, было минимальным, большую часть остаточного пропанола из конечного продукта следует удалить. Обычно является желательным уменьшение содержания изопропанола до менее чем 1% (по массе).
Остаточный изопропанол может быть удален из моногидрата лоракарбефа путем сушки кристаллов при пониженном давлении от около 0 мм рт. ст. до около 400 мм рт. ст. при температуре от около 25oC до около 80oC. В результате процесса сушки часть воды, присутствующей в кристаллах моногидрата лоракарбефа, может быть также удалена. Поэтому, если смесь сушат слишком долго, сушка будет оказывать воздействие на химический состав моногидрата лоракарбефа, выражающееся в уменьшении содержания воды, хотя рентгеноструктурный анализ показывает, что кристаллическая структура не изменяется.
Удаление слишком большого количества воды во время процесса сушки создает дополнительные проблемы, так как моногидрат лоракарбефа, имеющий менее чем примерно 5% воды (по массе), будет поглощать влагу из воздуха во время хранения и дальнейшей его обработки. Если произойдет повторная гидратация, эффективность вещества будет изменяться таким образом, что точная доза во время получения препарата не будет получена.
Соответственно настоящее изобретение предусматривает легкое превращение изопропанолата лоракарбефа в моногидрат лоракарбефа, который является коммерчески желательной формой объемного продукта.
Настоящее изобретение предусматривает кристаллическую безводную форму лоракарбефа.
Настоящее изобретение предусматривает способ получения кристаллической безводной формы соединения формулы I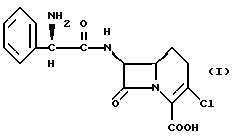
который включает сушку кристаллической изопропанолатной формы соединения формулы I при температуре от около 50oC до около 100oC.
Настоящее изобретение также предусматривает способ получения кристаллической моногидратной формы соединения формулы I, который включает воздействие на кристаллическую безводную форму соединения формулы I относительной влажности от около 90 до около 100%.
Настоящее изобретение также предусматривает способ получения кристаллической моногидратной формы соединения формулы I, имеющего объемный вес более или равный 0,5 г/мл, который включает:
а) воздействие на кристаллическую безводную форму соединения формулы I относительной влажности от около 90 до около 100% для получения кристаллической моногидратной формы соединения формулы I, имеющего содержание воды более или равное 10%; и затем
b) сушку кристаллического моногидрата при температуре от около 55oC до около 75oC при давлении от около 20 мбар до около 50 мбар.
Кроме того, настоящее изобретение предусматривает способ получения кристаллической моногидратной формы соединения формулы I, который включает:
а) сушку кристаллической изопропанолатной формы соединения формулы I при температуре от около 50oC до около 100oC для получения кристаллической безводной формы соединения формулы I; и
b) воздействие на кристаллическую безводную форму соединения формулы I относительной влажности от около 90 до 100%.
И, наконец, настоящее изобретение предусматривает способ получения кристаллической моногидратной формы соединения формулы I, имеющего объемный вес более или равный 0,5 г/мл и остаточное содержание изопропанола менее 1% (по массе), который включает:
а) сушку кристаллической моногидратной формы соединения формулы I при температуре от около 50oC до около 100oC для получения кристаллической безводной формы соединения формулы I;
b) воздействие на кристаллическую безводную форму соединения формулы I относительной влажности от около 90 до около 100% для получения кристаллической моногидратной формы соединения формулы I, имеющего содержание воды более или равное 10%; и затем
с) сушку кристаллического моногидрата при температуре от около 55oC до около 75oC при давлении от около 20 мбар до около 50 мбар.
Термин "содержание воды" относится к количественному измерению содержания воды в конкретном соединении. Содержание воды измеряли в соответствии с методом титрования Карла Фишера.
Настоящее изобретение относится к кристаллическому безводному сольвату соединения формулы I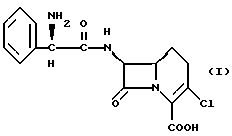
В безводном сольвате соединения формулы I C-2'-асимметрический центр имеет абсолютную R-конфигурацию. Кроме этого, настоящий сольват может включать цвиттерионную форму соединения формулы I.
Предпочтительным воплощением соединения является кристаллический безводный сольват лоракарбефа, имеющий следующую порошковую рентгенограмму:
Ангидрат лоракарбефа
---d--- - --I/I1--
14.8536 - 53.96
10.0551 - 31.90
7.4652 - 100.00
6.7762 - 25.85
5.8288 - 87.78
5.1063 - 32.67
4.9546 - 32.40
4.8040 - 59.97
4.3038 - 34.39
3.9716 - 22.22
3.8515 - 18.12
3.7210 - 20.95
3.5890 - 8.50
3.4888 - 3.49
3.3798 - 46.57
3.2689 - 2.08
3.1825 - 3.59
2.9695 - 12.56
2.8837 - 4.10
2.8135 - 13.75
2.7670 - 41.37
2.6273 - 11.93
d - расстояние между плоскостями, ангстрем;
I/I1 - соотношение интенсивности полос.
Приведенную выше дифрактограмму получили с применением медного источника излучения в охлажденном Si (Li) детекторе твердого состояния Пелтиера. Напряжение на лампе установили равным 50 kv, ток в лампе установили равным 40 мА, апертурная диафрагма имела щель 2 мм, диафрагму рассеянного излучения установили на щель 2 мм, диафрагма детектора имела щель 0,2 мм, скорость развертки прибора ступенчатого сканирования составила 0,02 два тета градуса/ступень для 1,2 сек/ступень и диапазон сканирования составил от 4,0 до 35 два тета градусов. С помощью электронного прибора осуществили вычитание фона, ширину пика установили при значении 0,3 и порог поиска пика был равен 3,0.
Было обнаружено, что, проводя последовательно сушку кристаллического изопропанолата лоракарбефа при температуре от около 50oC до около 100oC для получения кристаллического безводного лоракарбефа и затем воздействие на безводный лоракарбеф относительной влажности от около 90 до 100%, легко превращают твердый изопропанолат лоракарбефа в моногидрат лоракарбефа. Этот способ представляет собой усовершенствованный способ получения моногидрата лоракарбефа, который сводит к минимуму количество изопропанола в конечном продукте без оказания воздействия на химический состав моногидрата лоракарбефа и обеспечивает продукт, имеющий плотность более чем примерно 0,5 г/мл.
Изопропанолатная форма лоракарбефа может быть получена с использованием обычных методов, известных в данной области. Изопропанолат лоракарбефа может быть, например, легко получен путем суспендирования лоракарбефа в изопропаноле или в водном изопропаноле и образования раствора. Раствор обычно получают путем добавления основания или кислоты. Затем подходящий изопропанолат может быть осажден при регулировании pH до значения от 5,8 до 6,2 с использованием соответственно кислоты (например, хлористо-водородной кислоты, бромисто-водородной кислоты или серной кислоты) или основания (например, триэтиламина). Раствор обычно получают при температуре от около 20oC до около 25oC. Изопропанолат может быть выделен известными в данной области методами, например фильтрацией.
Превращение твердого изопропанолата лоракарбефа в безводный лоракарбеф и безводного лоракарбефа в моногидрат лоракарбефа можно осуществить последовательно с тем, чтобы получить желательный результат, т.е. получить моногидрат лоракарбефа с содержанием изопропанола менее 1% и без уменьшения содержания воды. Превращение изопропанолатной формы в безводную форму осуществляют обычно при температуре от около 50oC до около 100oC. Предпочтительный диапазон температуры составляет от около 55oC до около 60oC. Еще один предпочтительный диапазон температур составляет от около 63oC до около 65oC.
Превращение безводной формы в моногидратную форму осуществляют обычно при относительной влажности от около 90 до около 100%. Предпочтительный диапазон влажности составляет от 95 до 100%. Наиболее предпочтительно превращение осуществляют при относительной влажности 100%.
Конкретные аспекты настоящего изобретения дополнительно иллюстрируют последующие примеры.
ПРИМЕР 1
Изопропанолат лоракарбефа.
Изопропиловый спирт (660,0 мл), деионизированную воду (67,0 мл), бис(DMF) сольват лоракарбефа (50,0 г) и хлористо-водородную кислоту (15,6 г) смешивают и перемешивают при комнатной температуре. Если необходимо, для полного растворения можно добавить дополнительное количество хлористо-водородной кислоты. К смеси добавляют деионизированную воду (10,0 мл) и активированный уголь (2 г). Полученную реакционную смесь перемешивают в течение 1 часа и затем для удаления активированного угля смесь фильтруют. К фильтрату для осаждения изопропанолата в течение по меньшей мере 2 часов добавляют аммиак (28%, 12,6 г), и полученную суспензию фильтруют. Отфильтрованный осадок промывают 127,0 мл изопропанола, после этого промывают водой (85,0 мл), и для получения названного продукта влажный осадок сушат в вакууме при температуре 40-45oC.
ПРИМЕР 2
Моногидрат лоракарбефа.
Устанавливают аппаратуру для перегонки Кугельрофа, состоящую из печи Кугельрофа с регулированием температуры пропорционально времени, термопары типа J, 300 мм конденсатора Аллина, присоединенного к бане постоянной температуры, двигателя для перемешивания Кугельрофа, применяемого при перегонке, и автоматического регулятора давления Буши. Пробу изопропанолата лоракарбефа загружают в 300 мм конденсатор Аллина. В 1 л одногорлую круглодонную колбу загружают деионизированную воду (200 г). Колбу помещают в печь Кугельрофа и соединяют с конденсатором. Давление системы уменьшают примерно до 300 мбар. С помощью бани постоянной температуры рубашку конденсатора нагревают до 75oC. Печь Кугельрофа нагревают до температуры 65oC. Затем давление системы уменьшают до 250 мбар и изопропанолат подвергают воздействию относительной влажности 100% при перемешивании примерно в течение времени от 6 до 8 часов. Печь и конденсатор охлаждают до 20-25oC. Систему вентилируют до установления атмосферного давления. Гидратированный продукт удаляют и помещают в вакуумную печь при 40-45oC. Продукт сушат всю ночь в вакууме в слабом токе азота.
ПРИМЕР 3
Изопропанолат лоракарбефа.
Бис (DMF) сольват лоракарбефа (70,50 г), изопропанол (520,0 г) и деионизированную воду (88,8 г) (первоначальная загрузка плюс количество воды из суспензии угля) загружают в 2 л трехгорлую круглодонную колбу с двойными стенками. Затем в суспензию для полного растворения загружают хлористо-водородную кислоту. Растворение завершается при pH 9,0.
В раствор загружают порошок активированного угля (2,0 г). Содержимое колбы перемешивают в течение примерно одного часа при 20-25oC и затем фильтруют через 9 см воронку Бюхнера, предварительно покрытую ускорителем фильтрации, например, Hyflo. Фильтрат возвращают в колбу с двойными стенками и в течение четырех часов с помощью шприцевидного насоса по каплям добавляют аммиак (28%, 12,7 г). Размер кристаллов был большим, что находилось в полном соответствии с ранее полученным изопропанолатным веществом.
Суспензию перемешивают в течение примерно 1 часа при 20-25oC и фильтруют через фильтровальную бумагу Ватмана N 1 (время фильтрации: 2:0.4 мин). Фильтровальный осадок промывают изопропанолом и водой. Промытый материал сушат всю ночь в вакуумной печи при 40-45oC в абсолютном вакууме и в токе азота.
ПРИМЕР 4
Изопропанолат лоракарбефа.
В емкости смешивают изопропанол (440 л), деионизированную воду (25 л), хлористо-водородную кислоту (10 кг) и бис(DMF)сольват лоракарбефа (42,3 кг). Затем стенки смесителя промывают 22 л деионизированной волы. Смесь перемешивают в течение 15 минут и для полного растворения добавляют еще 500 г хлористо-водородной кислоты. Для окончательного завершения растворения в целом добавляют 2 кг хлористо-водородной кислоты, при этом смесь имеет pH 0,7.
В емкость добавляют активированный уголь (1,5 кг), распульпованный в 6 л воды, смесь перемешивают в течение 20 минут и затем фильтруют. Емкость промывают 10 л деионизированной воды. Добавляют аммиак (28%), причем добавление осуществляют до pH раствора 5,8 - 6,2. Полученный кристаллический изопропанолат фильтруют и промывают изопропиловым спиртом. Отфильтрованный осадок сушат в вакууме при температуре от около 42oC до около 48oC.
ПРИМЕР 5
Моногидрат лоракарбефа.
Вещество, полученное в примере 4, превращают в моногидрат лоракарбефа изопропанолатным способом при температуре сушилки между 65oC-75oC, в вакууме 4 фунта/дюйм2 (0,2812 кг/см2) и инжектировании водяного пара в сушилку для поддержания влажности в диапазоне от 95 до 100%. Примерно через один час поддержания таких условий часть содержимого сушилки охлаждают и берут пробу. Вышеприведенные условия для превращения в моногидрат лоракарбефа поддерживают в течение трех часов, при этом полученные в это время показания свидетельствуют о полном завершении превращения в моногидрат лоракарбефа. Затем вещество сушат в абсолютном вакууме (0,7 фунтов/дюйм2) (0,0492 кг/см2) при 45oC в течение примерно 8 часов.
ПРИМЕР 6
Безводный лоракарбеф.
Влажный осадок изопропанолата лоракарбефа (100 г) помещают в аппарат для сушки в кипящем слое и сушат в течение примерно трех часов при температуре от около 63oC до около 65oC потоком воздуха (с помощью воздуходувки) при скорости мешалки 70 оборотов в минуту. По завершении сушки анализ полученного твердого кристаллического вещества показывает, что остаточное содержание изопропанола равно 0,9%.
С использованием рентгеновской кристаллографии кристаллическое твердое вещество идентифицируют как безводный лоракарбеф.
ПРИМЕР 7
Безводный лоракарбеф.
Во вращающуюся вакуумную сушилку (0,85 фут3) (0,0243 м3), снабженную распылительным соплом, помещают влажный осадок изопропанолата лоракарбефа (2,5 кг) и сушат в течение примерно 12,5 часов при давлении около 30 мбар и температуре рециркуляции в рубашке около 70oC. По завершении сушки анализ полученного кристаллического твердого вещества показывает, что остаточное содержание изопропанола равно 0,55%.
ПРИМЕР 8
Моногидрат лоракарбефа.
В этом примере используют вращающуюся вакуумную сушилку, содержащую безводный лоракарбеф, полученный в примере 7. Сначала температуру рециркуляции в рубашке уменьшают до 30oC и затем через распылительное сопло добавляют воду (поток воды: 0,7 мл/мин и поток воздуха в сопло: примерно 15 стандартных фут3/мин (0,425 м3/мин)) в течение примерно 12 часов. Полученное кристаллическое твердое вещество содержит воды 9,4% и с помощью рентгеновской кристаллографии устанавливают, что оно представляет собой моногидрат лоракарбефа.
Затем кристаллическое твердое вещество сушат во вращающейся вакуумной сушилке при 65oC и давлении 30 мбар, при этом получают продукт, имеющий содержание воды 5,3% и объемную плотность 0,21 г/мл.
ПРИМЕР 9
Моногидрат лоракарбефа.
Во вращающуюся вакуумную сушилку (0,85 фут3) (0,0243 м3), снабженную распылительным соплом, помещают влажный осадок изопропанолата лоракарбефа (1,0 кг) и сушат в течение примерно десяти часов при давлении около 40 мбар и температуре рециркуляции в рубашке около 70oC. После завершения сушки анализ полученного кристаллического твердого вещества показывает, что остаточное содержание изопропанола равно 0,70%.
После этого температуру рециркуляции в рубашке уменьшают до 50oC и затем через распылительное сопло добавляют воду (поток воды: 3,0 мл/мин и поток воздуха в сопло: примерно 20 фут3/мин (0,566 м3/мин)) в течение примерно 12 часов. Когда содержание воды в полученном кристаллическом твердом веществе достигает 22% (примерно 385 мл добавленной воды), температуру рециркуляции в рубашке увеличивают до примерно 70oC, давление устанавливают равным примерно 40 мбар, и твердое вещество сушат, при этом получают желательный названный продукт с содержанием воды 5,2% и объемной плотностью 0,43 г/мл. С помощью рентгеновской кристаллографии кристаллическое твердое вещество идентифицируют как моногидрат лоракарбефа.
Изобретение относится к кристаллическому безводному сольвату лоракарбефа и также к способу получения кристаллической моногидратной формы соединения формулы I, который включает сушку кристаллической изопропанолатной формы соединения формулы I при температуре около 50 - 100oC для получения кристаллической безводной формулы соединения формулы I и воздействие на кристаллическую безводную форму соединения формулы I относительной влажности около 90 - 100%. Технический результат - получение новой формы лоракарбефа. 5 с.п. ф-лы.
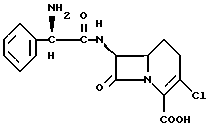
имеющая следующую порошковую рентгенограмму: - I/Iτ
- I/Iτ
14.8536 - 53.96
10.0551 - 31.90
7.4652 - 100.00
6.7762 - 25.85
5.8288 - 87.78
5.1063 - 32.67
4.9546 - 32.40
4.8040 - 59.97
4.3038 - 34.39
3.9716 - 22.22
3.8515 - 18.12
3.7210 - 20.95
3.5890 - 8.50
3.4888 - 3.49
3.3798 - 46.57
3.2689 - 2.08
3.1825 - 3.59
2.9695 - 12.56
2.8837 - 4.10
2.8135 - 13.75
2.7670 - 41.37
2.6273 - 11.93
2. Способ получения кристаллической моногидратной формы соединения формулы I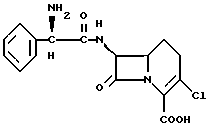
путем обработки кристаллической формы лоракарбефа в условиях относительной влажности около 90 - 100%, отличающийся тем, что в качестве кристаллической формы лоракарбефа используют кристаллическую безводную форму соединения формулы I по п.1.
| US 5352782 A, 20.12.1994 | |||
| Столик микроскопа | 1975 |
|
SU627429A1 |
| Устройство для автоматической зарядки ленточного материала на бобину | 1977 |
|
SU627431A1 |
| Способ получения кристаллической формы сольвата бис(N,N-диметилформамида)-1-карбацефалоспорина | 1989 |
|
RU2002749C1 |
