Изобретение относится к новым цефалоспоринам и комплексам карбоцефалоспорин/4-гидроксибензойная кислота, методам их применения и в особенности выделения и очистки антибиотиков, содержащих бета-лактамное кольцо.
Цефаклор, цефалексин, цефрадин и лоракарбеф имеют структуру, представленную ниже: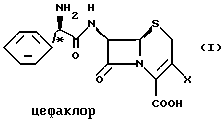
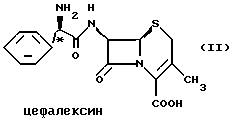
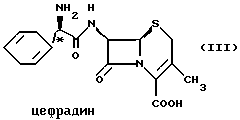
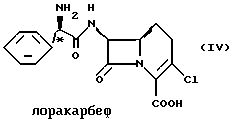
Четыре β-лактамных соединения, в которых асимметрические центры в С-2'-положении имеют абсолютную R-конфигурацию, представляют собой коммерчески важные пероральные антибиотики.
В различных работах рассматриваются процессы выделения и очистки β-лактамных соединений, в частности в Европейской заявке на патент 0341991, опубликованной 15 ноября 1989 года.
В указанной EPO заявке показано использование антрахинон-1,5-дисульфоновой кислоты для получения фармацевтически приемлемой соли, в частности цефалоспорина или карбацефалоспорина, которая позволяет осуществлять регенерацию β-лактама из маточных растворов после кристаллизации.
Поскольку производство этих антибиотиков является дорогостоящим, постоянно ведутся поиски новых более совершенных методов такой регенерации с целью максимизации общего выхода продукта.
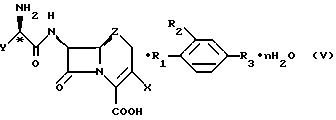
Обнаружено, что коммерчески важные антибиотики цефалексин, цефаклор, цефрадин и лоракарбеф образуют кристаллические комплексы с 4-гидроксибензойной кислотой и родственными соединениями. Настоящее изобретение относится к комплексам со следующей формулой: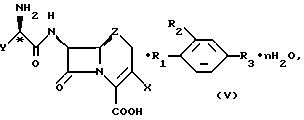
где X - хлор, винил или -CH3;
Z - CH2, S или O;
n = 0 - 5;
Y - фенил или 1,4-циклогексадиен-1-ил;
R1 и R2 - водород или гидроксильная группа при условии, что оба R1 и R2 не являются водородом;
R3 - CO2H, -COO(C1-C4-алкил), или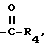
где R4 - C1-С4-алкил.
Далее, изобретение описывает получение указанных выше комплексов для регенерации антибиотика.
Термины "комплекс" и "сокристалл" используются для описания единственной твердой фазы, получающейся в результате комбинации антибиотика и 4-гидроксибензойной кислоты или родственных соединений.
Предпочтительными комплексами настоящего изобретения являются цефрадин • метилпарабен • H2О, цефаклор • метилпарабен • 3H2O, цефаклор • этилпарабен, 2 цефаклор • 4-гидроксибензойную кислоту • 4H2O, цефаклор • метиловый эфир • 3-гидроксибензойной кислоты • (1 или 5) H2O, цефаклор • 4-гидроксиацетофенон, цефаклор • 3- гидроксиацетофенон, цефалексин • метилпарабен • H2O, цефалексин • этилпарабен • 2H2O, цефалексин • метиловый эфир 3-гидроксибензойной кислоты • (0,1 или 5) H2O, цефалексин • 3-гидроксиацетофенон; лоракарбеф • метилпарабен, лоракарбеф • этилпарабен, лоракарбеф • пропилпарабен; лоракарбеф • метиловый эфир 3-гидроксибензойной кислоты • (1 или 5) H2O, лоракарбеф • 4-гидроксиацетофенон или лоракарбеф • 3-гидроксиацетофенон.
Метод получения и выделения комплексов цефалаксина, цефаклора, цефрадина и лоракарбефа с 4-гидроксибензойной кислотой или родственными соединениями достаточно прост. Водные или в основном водные растворы или суспензии β-лактама и антибиотика или родственного соединения смешиваются, и далее компоненты оставляют для образования кристаллического комплекса в диапазоне температур от 0 до 65oC. Предпочтительно добавляется 4-гидроксибензойная кислота или родственное соединение в количестве от 1 до 3 частей на 1 часть используемого β-лактама.
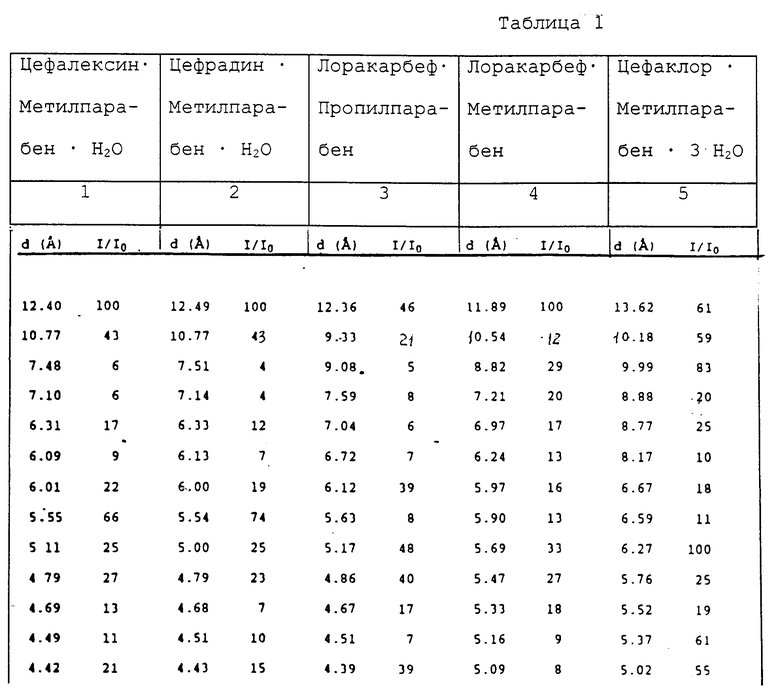
В каждом случае образование комплексов подтверждается характерной для них порошковой рентгенограммой, которая отличается от опубликованных ранее порошковых рентгенограмм цефалексина (L.P. Marelli, Analytical Profiles of Drug Substances, 4, pp. 21-46 (1975)); цефрадина (К. Florey, Analytical Profiles of Drug Substances, 5, pp. 21-59 (1976)); цефаклора (L.S. Lorens, Analytical Profiles of Drug Subctances, 9, pp. 107-123 (1980)); моногидрата лоракарбефа (Pasini, Европейская заявка на патент ЕР 0311366 А1, опубликованная 12 апреля 1989 г.) и дигидрата лоракарбефа (Eckrich et а1. Европейская заявка на патент 0369686 A1, опубликованная 23 мая 1990 г.); а также метил-, этил- и пропилпроизводных 4-гидроксибензойной кислоты, метил-3-оксибензоата и ацетофенонов.
В наших исследованиях ни цефадроксил, ни арилглициновые пенициллины, амоксициллин и ампициллин не образовывали ощутимых количеств сокристаллов с метил- или пропилоксибензойной кислотой.
Кристаллические комплексы β-лактамов с 4-гидроксибензойными кислотами и родственными соединениями могут использоваться в процессах регенерации, выделения и/или очистки β-лактама. Комплексы могут применяться для осаждения β-лактама из разбавленных растворов (например, из маточных растворов или реакционных растворов). Комплексы могут быть выделены фильтрацией. Для регенерации β-лактама комплексы следует растворить в кислом органическом растворителе или в системе, включающей органический растворитель, кислоту или воду.
Подходящими для этой цели кислотами могут быть, к примеру, соляная, серная и бромисто-водородная кислоты.
Пригодные растворители включают этанол, н-бутанол, метилизопропилкетон, диэтиловый эфир, диизопропиловый эфир, этилацетат, хлористый метилен, тетрагидрофуран, диметилформамид, диметилсульфоксид и их водные растворы.
β-лактам выделяется из полученного раствора повышением pH добавлением основания для осаждения β-лактама, тогда как 4-гидроксибензойная кислота и родственные соединения остаются в растворе. Подходящие для этого основания также известны и включают гидроокись аммония, гидроокись натрия и триэтиламин.
Выделение β-лактама может проводиться в температурном интервале от 0 до 50oC. Кроме того, комплексы могут быть растворены в основном органическом растворителе или в системе, содержащей органический растворитель, щелочь и воду, а β-лактам в этом случае осаждается при понижении pH добавлением кислоты.
В любом случае β-лактам может быть отделен в результате фильтрации и далее использоваться в том виде, в каком он получен или может быть превращен в другую более удобную кристаллическую форму.
Экспериментальные данные.
Основные методы.
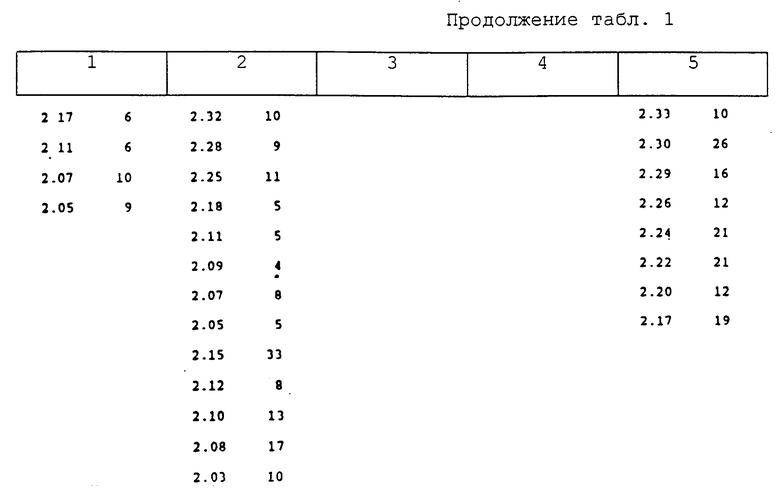
В таблице представлены данные по порошковым рентгенограммам (дебаеграммам) продуктов, приведенных в 1-5 примерах, где d означает межплоскостное расстояние, измеренное в ангстремах, а "I/I0" означает относительную интенсивность.
Пример 1. Цефалексин • метилпарабен • H2O.
Метилпарабен (375 мг, 2,5 ммоль) в 150 мл воды добавляют к моногидрату цефалексина (2,4 г, 6 ммоль) для получения суспензии. После выдерживания суспензии при температуре 5oC в течение 5 дней кристаллы отделяют фильтрованием и промывают водой, при этом образуется 1,06 г комплекса (2,0 ммоль) с температурой плавления 168-169oC.
Рассчитано в % для C24H25N3O7S H2O: С - 55,70; H - 5,26; N - 8,12.
Найдено: С - 55,65; H - 5,28; N - 8,13.
ВЭЖХ (расчет), найдено: (67,1), 67,1% - для цефалексина, (29,4), 28,7% - для метилпарабена.
KF (расчет), найдено: (3,5) 4,0% воды.
Пример 2. Цефрадин • метилпарабен • H2O.
Метилпарабен (250 мг, 1,6 ммоль) в 100 мл воды добавляют к моногидрату цефрадина (1,6 г, 4,4 ммоль) для получения суспензии. После выдерживания суспензии в течение 3 дней при температуре 25oC кристаллы отделяют фильтрованием и промывают водой с получением 0,609 г (1,2 ммоль) комплекса с 1 мол кристаллизационной воды, температура плавления: 167-168oC.
Рассчитано в % для C24H27N3O7S • H2O: С - 55,48; H - 5,63; N - 8,09.
Найдено: С - 55,58; H - 5,57; N - 8,08.
ВЭЖХ (расчет), найдено: (67,2), 67,7% - для цефрадина, (29,3), 28,9% - для метилпарабена.
KF (расчет), найдено: (3,5), 3,7% воды.
Пример 3. Цефаклор • метилпарабен • 3H2O.
Метилпарабен (250 мг, 1,6 ммоль) в 100 мл воды добавляют к моногидрату цефаклора (1,3 г, 3,4 ммоль) для образования суспензии. После выдерживания суспензии в течение 5 дней при температуре 5oC кристаллы отделяют фильтрованием и промывают водой с образованием комплекса, содержащего 3 моля кристаллизационной воды, температура плавления комплекса: 142oC (разлож).
Рассчитано в % для C23H22ClN3O7S • 3H3O: С - 48,13; H - 4,92; N - 7,32.
Найдено: С - 48,01; H - 4,92; N - 6,99.
ВЭЖХ (расчет), найдено: (64,1), 65,0% - для цефаклора, (26,5), 25,4% - для метилпарабена.
KF (расчет), найдено: (9,4), 9,9% воды.
Пример 4. Лоракарбеф • метилпарабен.
Метилпарабен (460 мг, 3,0 ммоль) в 230 мл воды добавляют к моногидрату цефаклора (3,0 г, 8,2 ммоль) для образования суспензии. После выдерживания суспензии при температуре 25oC в течение 25 дней кристаллы отделяют фильтрованием, промывают водой и получают в результате 0,72 г (1,4 ммоль) комплекса, температура плавления 191oC (разложение).
Рассчитано для C24 H24ClN3O7 (в %): С - 57,44; H - 4,82; N - 8,37.
Найдено: С - 57,16; H - 4,92; N - 8,59.
ВЭЖХ (расчет), найдено: (69,7), 70,6% - для лоракарбефа, (30,3), 30,0% - для метилпарабена.
Пример 5. Лоракарбеф • пропилпарабен.
Пропилпарабен (500 мг, 2,8 ммоль) в 500 мл воды добавляют к моногидрату лоракарбефа (5,5 г, 14,9 ммоль) для образования суспензии. После выдерживания суспензии в течение 20 дней при 5oC кристаллы отделяют фильтрованием и промывают водой, при этом получается 1,31 г (2,5 ммоль) комплекса, температура плавления 178oC (разложение).
Рассчитано в % для C26H28ClN3O7: С - 58,9; H - 5,32; N - 7,93.
Найдено; С - 58,69; H - 5,31; N - 7,90.
ВЭЖХ (расчет), найдено: (66,0), 64,31% - для лоракарбефа, (34,0), 33,3% - для пропилпарабена.
Пример 6. Лоракарбеф • этилпарабен.
pH водного маточного раствора моногидрата лоракарбефа (1000 мл, содержащий 9,23 мг/мл моногидрата лоракарбефа) доводят с помощью HCl до 3,6. Этил-п-гидроксибензоат (этилпарабен) (4,52 г) в этаноле (36 мл) добавляют по каплям в течение 15 минут. Примерно через 5 минут начинается процесс осаждения, приводящий в итоге к образованию белых кристаллов. Смесь перемешивают в течение ночи (15 часов) при комнатной температуре, затем фильтруют, промывают водой и высушивают в вакууме при 40oC. Получено 11,72 г продукта, содержащего 73,5% лоракарбефа, теоретический выход лоракарбефа - 8,78 г, выход, таким образом, составляет 98,2%. Ниже приведены характеристики порошковых рентгенограмм комплекса лоракарбеф : этилпарабен:
d - I/I0
12.03778 - 100.0
10.51978 - 2.7
8.87879 - 28.6
7.24748 - 4.0
6.04328 - 1.6
5.77434 - 39.3
5.27012 - 2.9
4.95456 - 3.6
4.80014 - 0.6
4.60833 - 2.7
4.50920 - 2.6
4.03255 - 38.1
3.57845 - 38.9
3.54171 - 4.3
3.35279 - 22.4
3.20988 - 1.4
3.16843 - 0.7
3.02715 - 4.6
2.89099 - 3.9
2.83791 - 0.5
2.80444 - 1.0
2.76760 - 1.9
2.69465 - 1.5
2.62614 - 1.6
Пример 7. Лоракарбеф • метил-3-гидроксибензоат • 5H2O.
Метил-3-гидроксибензоат (496 мг, 3,3 ммоль) в 3 мл в 95% EtOH добавляют к моногидрату лоракарбефа (1,2 г, 3,3 ммоль) в 200 мл воды. После выдерживания смеси при 25oC в течение 19 дней кристаллы отделяют фильтрованием, промывают водой, при этом получают 576 мг (0,97 ммоль) комплекса.
Рассчитано в % для C24 H24ClN3O7 • 5 • H2O: С - 48,69; H - 5,79; N - 7,10.
Найдено: С - 48,84; H - 5,56; N - 7,23.
Пример 8. Лоракарбеф • метилпарабен.
Этилат лоракарбефа (2,55 г, 74,7% содержание лоракарбефа) суспендируют в 100 мл воды. NaOH (1N, 1,08 мл) добавляют в суспензию для повышения pH до 8,9. Смесь перемешивают до полного растворения лоракарбефа. Далее добавляют метил-п-гидроксибензоат (метилпарабен) (0,83 г) в 7 мл этанола, при этом в течение нескольких минут pH падает до значения 8,1.
Смесь становится мутной в связи с началом процесса осаждения. Смесь перемешивают при комнатной температуре в течение 2 часов, фильтруют, промывают водой и высушивают в вакууме при 40oC в течение ночи.
Расчетный выход составляет для комплекса 2,83 г, получено 1,92 г, что составляет 67,8% от теоретически ожидаемого.
Пример 9. Лоракарбеф • бутилпарабен.
Этилат лоракарбефа (2,55 г, 74,6% содержание лоракарбефа) добавляют к 100 мл воды. pH доводится до 8,4 с помощью NaOH (1N, 1,08 мл). После перемешивания в течение 30 минут при комнатной температуре раствор становится практически прозрачным. Бутил-п-гидроксибензоат (бутилпарабен) (1,06 г) в 7 мл этилового спирта добавляют к смеси. Смесь перемешивают в течение нескольких часов при комнатной температуре, фильтруют, промывают водой и высушивают в вакууме при 40oC. Целевой продукт имеет белый цвет.
Расчетный выход комплекса составляет 3,06 г, получено 1,78, что составляет 58,2% от теоретически ожидаемого выхода.
Пример 10. Лоракарбеф • этилпарабен.
Этилат лоракарбефа (2,55 г, 74,7% содержание лоракарбефа) суспендируют в 100 мл воды. В молярном избытке добавляют NaOH для повышения pH до 8,4, при этом смесь становится прозрачной.
Этил-п-гидроксибензоат (0,90 г, 5,44 ммоль) в 7 мл этилового спирта добавляют к смеси, при этом в течение нескольких минут образуется густой осадок. pH составляет 9,95. Смесь перемешивают в течение нескольких часов при комнатной температуре, фильтруют, промывают водой и высушивают в вакууме при 40oC.
Расчетный выход комплекса составляет 2,90 г, получено 2,08 г, выход составляет 71,7%.
Пример 11. Лоракарбеф • пропилпарабен.
Этилат лоракарбефа (2,55 г, 74,7% содержание лоракарбефа) суспендируют в 100 мл воды. Добавляют NaOH (1N, 1,08 мл), и pH становится 7,2. Далее добавляют еще 1N NaOH для повышения pH до 8,2.
Пропил-п-гидроксибензоата (0,90 г, 5,44 ммоль) в 7 мл этилового спирта добавляют к смеси, после чего начинает немедленно выпадать белый твердый осадок. Смесь перемешивают при комнатной температуре в течение нескольких часов. Затем смесь фильтруют, промывают водой и высушивают в вакууме при 40oC.
pH фильтрата опускается до 4,9, но дополнительного образования осадка не наблюдается.
Расчетный выход комплекса составляет 2,98 г, получено 0,90 г, и выход составляет 30,2%.
Пример 12. Лоракарбеф • этилпарабен.
Реакцию ацилирования проводят в соответствии с патентами США 4,312,958, 4,332,896 и 4,335,211. Проводят экстракцию смеси после проведения реакции ацилирования хлористым метиленом, а водную фазу далее фильтруют. Этилпарабен (3,82 г) растворяют в 3 мл этилового спирта и добавляют к водному раствору, содержащему 4,23 г лоракарбефа в форме моногидрата.
В растворе начинается процесс кристаллизации, при этом его pH снижают до 4,3 с помощью соляной кислоты. Смесь перемешивают в течение нескольких часов при комнатной температуре. Смесь охлаждают в течение ночи в холодильнике, фильтруют, промывают водой и высушивают в вакууме при 45oC. Расчетный выход для лоракарбефа составляет 3,99 г, получено 8,37 г продукта с содержанием лоракарбефа 39%, таким образом, выход составляет 81,2%.
Пример 13. Лоракарбеф • этилпарабен.
Этилпарабен растворяют в 30 мл этилового спирта и добавляют к водному раствору моногидрата лоракарбефа (3,88 г, 10,55 мм). pH суспензии снижают до 3,5 с помощью соляной кислоты и оставляют ее для перемешивания в течение нескольких часов при комнатной температуре. Суспензию охлаждают при 5oC в течение нескольких часов, фильтруют и высушивают в вакууме при 55oC.
Расчетный выход составляет 3,69 г, получено 6,35 г продукта с содержанием лоракарбефа, равным 55,3%, при этом выход составляет 95,1%.
Пример 14. Дисольват лоракарбеф • ДМФ.
К смеси лоракарбеф этилпарабен (1,34 г, 65,5% содержание лоракарбефа) в 18 мл ДМФ и 1,8 мл воды добавляют концентрированную соляную кислоту до получения прозрачного раствора. pH раствора медленно повышают до значения 6,9 с помощью триэтиламина, при этом начинается быстрый процесс кристаллизации целевого продукта. Смесь перемешивают в течение 1 часа при комнатной температуре, фильтруют, промывают раствором, содержащим ДМФ: H2O/10:1, и высушивают в вакууме при 40oC.
ВЭЖХ показывает отсутствие этилпарабена.
Расчетный выход в отношении лоракарбефа составляет 0,87 г, получено 1,01 г продукта с содержанием лоракарбефа, равным 73,9%, выход составляет 85,3%.
Пример 15. Дисольват лоракарбеф • ДМФ.
К смеси лоракарбеф • этилпарабен (15,0 г, 49,1% содержание лоракарбефа) в растворителе, содержащем 150 мл ДМФ и 15 мл воды, добавляют по каплям концентрированную соляную кислоту (2,53 мл). Раствор нагревают до 45oC и затем с помощью триэтиламина pH поднимают с 1,84 до 4,0 в течение 45 минут.
После того как начнется процесс кристаллизации, медленно добавляют триэтиламин до повышения pH до 6,7. Смесь перемешивают в течение 1 часа, фильтруют, промывают с помощью ДМФ и этанола и высушивают в вакууме при 40oC.
ВЭЖХ показывает отсутствие этилпарабена. Теоретический выход в отношении лоракарбефа составляет 7,37 г, получено 10,07 г продукта с содержанием лоракарбефа, равным 72%, выход составляет 98,4%.
Пример 16. Лоракарбеф.
Лоракарбеф • этилпарабен (3,0 г, 49,1% содержание лоракарбефа) суспендируют в 30 мл смеси ацетонитрил: вода (1:1). С помощью концентрированной соляной кислоты pH раствора снижают до 1,9, при этом раствор становится прозрачным. Далее, с помощью триэтиламина pH смеси доводят до значения 4,9, при котором быстро образуется белый осадок. Смесь перемешивают при комнатной температуре в течение 1 часа, фильтруют, промывают смесью ацетонитрил: вода/1:1 и высушивают в вакууме при 45oC. Расчетный выход составляет 1,47 г, получено 1,36 г продукта с содержанием лоракарбефа 98,1%, выход составляет 90,3%.
Пример 17. Этилат лоракарбефа.
Лоракарбеф • этилпарабен (11,26 г, 72,9% содержание лоракарбефа) суспендируют в растворе, состоящем из 90 мл этилового спирта и 9 мл воды. Добавляют соляную кислоту (3,5 мл) до получения прозрачного раствора примерно при pH 0,80. К смеси добавляют триэтиламин (5 мл) в 30 мл этанола. Смесь перемешивают в течение 2 часов, фильтруют, промывают этанолом и высушивают в вакууме при 40oC. Расчетный выход составляет 8,21 г, получено 8,22 г продукта с содержанием лоракарбефа, равным 94,8%, выход составляет 95,0%.
Пример 18. Моногидрат лоракарбефа.
Лоракарбеф этилпарабен (30,0 г, 72,7% содержание лоракарбефа) суспендируют в смеси, состоящей из 240 мл этилового спирта и 24 мл H2O. К этой смеси добавляют 9 мл концентрированной соляной кислоты до получения прозрачного раствора. К смеси далее добавляют 15 мл триэтиламина и 84 мл этилового спирта. Смесь перемешивают в течение 1 часа при комнатной температуре, фильтруют, промывают этанолом и высушивают в вакууме при 40oC, в результате чего получается этилат лоракарбефа. Расчетный выход в отношении содержания лоракарбефа составляет 21,81 г, получено 22,59 г этилата лоракарбефа с содержанием лоракарбефа, равным 86,2%, процентный выход составляет 89,3%.
Указанный выше этилат лоракарбефа (5,0 г, 86,2% содержания лоракарбефа) суспендируют в 70 мл H2O и полученную суспензию нагревают до 50oC. Суспензия становится очень густой, что указывает на образование моногидрата. Суспензию перемешивают в течение 1 часа при температуре 50oC, фильтруют, промывают водой и высушивают в вакууме при 40oC. Расчетный выход составляет 4,53 г, получено 3,06 г моногидрата лоракарбефа с содержанием моногидрата лоракарбефа, равным 102,8%, выход составляет 69,3%.
Пример 19. Цефаклор • 4-гидроксиацетофенон.
Раствор 265 мг (1,94 ммоль) 4-гидроксиацетофенола в 1 мл этанола добавляют к 750 мг (1,94 ммоль) моногидрата цефаклора, растворенного в 81 мл воды. Полученный мутный раствор закрывают и выдерживают в течение ночи при комнатной температуре, после чего образовавшиеся очень большие слабо желтые кристаллы отделяют фильтрованием.
Температура плавления 198-203oC (разложение).
Рассчитано CHN в % для C23H22ClN3O6S: С - 54,81; H - 4,36; N - 8,34.
Найдено: С - 54,95; H - 4,51; N - 8,63.
Ниже представлены характеристики порошковых рентгенограмм комплекса цефаклор • 4-гидроксиацетофенон:
d - I/I0
11.4958 - 100.00
10.1960 - 10.80
8.7598 - 16.87
7.3321 - 10.70
6.9139 - 8.61
6.8571 - 6.32
6.4493 - 7.76
5.6711 - 89.55
5.5743 - 30.10
5.1132 - 10.55
4.9524 - 11.19
4.8824 - 13.13
4.7479 - 63.18
4.5850 - 26.57
4.5361 - 45.42
4.3973 - 27.41
4.3076 - 17.56
4.1818 - 5.97
4.1310 - 7.86
4.0050 - 41.00
3.9243 - 30.40
3.8845 - 37.66
3.6794 - 14.88
3.5963 - 22.29
3.5070 - 42.29
3.4101 - 11.74
3.3658 - 14.28
3.3072 - 46.17
3.2420 - 49.50
3.1725 - 13.58
3.1421 - 14.48
3.0758 - 10.95
3.0452 - 11.29
2.9962 - 13.28
2.9223 - 59.60
2.8845 - 15.67
2.8232 - 14.38
2.7527 - 14.63
2.7286 - 7.46
2.6418 - 21.14
2.5815 - 14.73л
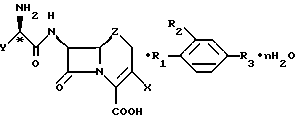
Изобретение относится к новым цефалоспоринам и комплексам карбоцефалоспорин/парааминобензойная кислота, методам их получения и в особенности выделения и очистки антибиотиков, содержащих бета-лактамное кольцо. Описываются комплексы формулы (V), где Х - хлор, водород, винил или -CH3; Z - СН2,S или O; n = 0-5; Y - фенил или 1,4-циклогексадиен-1-ил; R1 и R2 - водород или гидроксигруппа при условии, что оба (R1 и R2) не являются водородом; R3 - -COOН, -СОО(С1-С4-алкил), или
где R4 - С1-С4-алкил. 3 с. и 3 з.п.ф-лы, 1 табл.
где X - хлор, винил или -CH3;
Z - CH2, S или O;
n = 0 - 5;
Y - фенил или 1,4-циклогексадиен-1-ил;
R1 и R2 - водород или гидроксигруппа при условии, что R1 и R2 не являются оба водородом;
R3 - CO2H, -COO(C1 - C4-алкил) или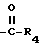
где R4 - C1 - C4-алкил.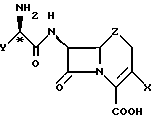
отличающийся тем, что включает стадию растворения комплекса формулы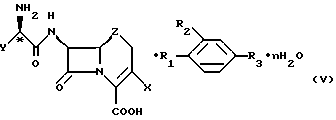
где X - хлор, винил или -CH3;
Z - CH2, S или O;
n = 0 - 5;
Y - фенил или 1,4-циклогексадиен-1-ил;
R1 и R2 - водород или гидроксигруппа при условии, что R1 и R2 не являются оба водородом;
R3 представляет CO2H, -COO(C1 - C4-алкил) или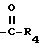
где R4 - C1 - C4-алкил;
в кислом органическом растворителе или в смеси кислого органического растворителя с водой с последующим добавлением достаточного количества основания для осаждения соединения формулы (VI).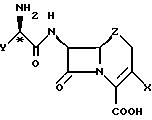
отличающийся тем, что включает стадию растворения комплекса формулы V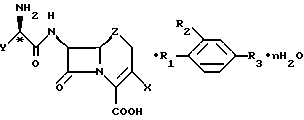
где R1, R2, R3, X, Y, Z и n определены в п.1,
в системе основного органического растворителя или в смеси основного органического растворителя с водой с последующим добавлением достаточного количества кислоты для осаждения соединения формулы (VI).
| Способ получения оптически активных производных цис-7-амино-1-азабицикло-(4,2,0)-окт-2-ен-8-он-2-карбоновой кислоты | 1980 |
|
SU1034607A1 |
| ЗАЖИМ ДЛЯ КРЕПЛЕНИЯ КАНАТА | 0 |
|
SU341991A1 |
| ОДНОВИБРАТОР | 0 |
|
SU369686A1 |
| УСТРОЙСТВО УПРАВЛЕНИЯ ИНВЕРТОРОМ НА ТИРИСТОРАХ С ДВУХСТУПЕНЧАТОЙ КОММУТАЦИЕЙ12 | 0 |
|
SU311366A1 |
| US 5142042 A, 25.08.92. | |||
