Настоящее изобретение относится к способу получения катализатора и к применению катализатора в конверсии синтез-газа по методу Фишера-Тропша, т. е. в получении главным образом жидких углеводородов из смесей CO и H2.
Более подробно, предметом настоящего изобретения является способ получения катализатора, в основном состоящего из Co, Ru и третьего элемента, выбранного из скандия и иттрия, и нанесенного на инертный носитель.
Выбор кобальта обусловлен тем, что кобальт способствует образованию насыщенных продуктов с высокой молекулярной массовой при более низкой температуре по сравнению, например, с системами на основе железа.
Применение катализаторов на основе кобальта возвращает к первым работам Фишера в 1932 г. (Н.H Storch, N.Golumbic, R.B. Anderson, "The Fisher Tropsch and Related Synthesis", John willey & Son, Jnc., New York, 1951), где разработана система Co/Th2/MgO/кизельгур.
Впоследствии развитие таких систем привело к выявлению природы различных промоторов, добавляемых к кобальту для повышения избирательности к углеводородам с высокой молекулярной массой, причем произошло это в основном в течение последних двадцати лет. Фактически повышение цены на нефть в 70-х годах стимулировало исследование других путей производства жидких топлив и химических продуктов.
В патенте US-A-4088671 описан катализатор для синтеза по методу Фишера-Тропша, содержащий в качестве активных компонентов кобальт и рутений, первый из которых присутствует в большем количестве, чем второй.
В патенте US-A-4413064 описан катализатор для реактора с неподвижным слоем, в основном состоящий из кобальта, рутения и тория или оксида лантана, нанесенных на оксид алюминия, и полученный путем пропитки оксида алюминия водным раствором соли кобальта и последующей неводной органической пропитки солью рутения и солью металла, принадлежащего к группе IIIB или IYB. Среди металлов этих групп в патенте US-A-4413064 указаны также скандий и иттрий, но предпочтительными металлами являются торий и лантан. Вышеуказанный катализатор особенно эффективен в конверсии синтез-газа для получения углеводородного продукта с высоким содержанием парафиновых углеводородов, имеющего температуру кипения в области дизельного топлива, т.е. продукта C9-C21. В патенте US-A-4413064 нет никакой информации о способности описанных катализаторов к созданию более тяжелых углеводородов, процесс получения которых является предпочтительным по сравнению с процессом получения углеводородов с точкой кипения в области дизельного топлива.
Был найден способ получения катализатора на инертном носителе, состоящего в основном из большего количества кобальта и меньшего количества рутения и из третьего элемента, выбранного из скандия и иттрия, и особенно эффективного в конверсии синтез-газа в углеводородные продукты, содержащие значительные количества углеводородов с числом углеродных атомов, превышающим или равным 22.
Таким образом, настоящее изобретение касается способа получения катализатора, в основном состоящего из инертного носителя, выбранного из, по крайней мере, одного оксида, по крайней мере, одного элемента, выбранного из Si, Ti, Al, Zr, Zn, Mg, Sn, предпочтительно кремния, и из (в виде элементов или оксидов) большего количества кобальта и меньшего количества рутения и третьего элемента, выбранного из скандия и иттрия, отличающегося тем, что включает в себя, по крайней мере, следующие стадии:
(1) получение первого каталитического предшествующего продукта (А), содержащего кобальт и, по крайней мере, часть инертного носителя, путем осаждения кобальта на инертном носителе с последующими прокаливанием, восстановлением и пассивацией инертного носителя, содержащего кобальт;
(2) получение второго каталитического предшествующего продукта (В), содержащего кобальт, рутений и, по крайней мере, часть инертного носителя, путем осаждения рутения на первом каталитическом предшествующем продукте (А) с последующими прокаливанием, восстановлением и пассивацией инертного носителя, содержащего кобальт и рутений;
(3) получение конечного катализатора путем осаждения элемента, выбранного из скандия и иттрия, на каталитическом предшествующем продукте (В) с последующими прокаливанием, восстановлением и пассивацией инертного носителя, содержащего кобальт, рутений и третий элемент.
Другим предметом настоящего изобретения является катализатор, который может быть получен описанным выше способом.
В способе по настоящему изобретению стадия 1 состоит из первоначального осаждения кобальта на, по крайней мере, части инертного носителя, предпочтительно на всем инертном носителе. Это осаждение, а также осаждение рутения на стадии 2 и третьего элемента на стадии 3 можно осуществлять различными методами, известными специалистам в данной области техники, например путем обмена, пропитки, сухой пропитки (известной также как начальное насыщение влагой), осаждения, желатинирования и механического смешения. В предпочтительном варианте осаждение кобальта на стадии 1 осуществляют методом сухой пропитки. В соответствии с этим методом наносимый материал вводят в контакт с объемом раствора, приблизительно равным по объему пор. На стадии 1 является предпочтительным использовать водные растворы солей кобальта. Могут быть использованы соли кобальта любого типа, например галогениды, нитрат, ацетат, оксалат, сульфат, комплекс, образованный с щавелевой кислотой и оксалатами, комплекс, образованный с молочной кислотой и лактатами, комплекс, образованный с винной кислотой и тартратами, комплекс, образованный другой поликислотой или гидроксикислотой и соответственными солями, комплекс, образованный с ацетилацетонатами.
После осаждения на инертном носителе требуемого количества соли кобальта, предпочтительно нитрата кобальта, следуют стадия прокаливания, затем стадия восстановления и затем стадия пассивации. В соответствии с другим вариантом перед прокаливанием пропитанный носитель подвергают сушке для удаления большей части воды. Эта сушка может быть осуществлена сначала при температурах в пределах между 10 и 30oC и затем при температурах в пределах между 100 и 120oC, предпочтительно при наличии потока газа.
На стадии 1 прокаливание осуществляют при температуре в пределах между 300 и 500oC, предпочтительно в пределах между 350 и 450oC, в воздушной среде, для того чтобы удалить все органические остатки.
Прокаленный указанным образом продукт подвергают затем восстановлению в среде, в основном состоящей из водорода, при температуре в пределах между 300 и 500oC, предпочтительно в пределах между 350 и 450oC. Является предпочтительным постепенно доводить прокаливаемый материал до указанной температуры, например, со скоростью нагрева в пределах между 3 и 20oC в минуту. Обычно операцию восстановления заканчивают при указанной выше температуре в период времени между 10 и 20 часами при расходе H2 в пределах между 1 и 3 литрами в час на грамм катализатора. По окончании восстановления осуществляют операцию пассивации в присутствии кислорода, разбавленного инертным газом (обычно азотом), предпочтительно при температуре в пределах между 10 и 80oC. При использовании, например, азота, содержащего 1-2% O2 (расход 2 литра в час), эта операция может длиться 1-5 часов при 25oC. Очевидно, что по окончании восстановления (и, конечно, перед пассивацией) образец должен быть охлажден.
Вторая стадия способа по настоящему изобретению состоит в осаждении рутения на каталитическом предшествующем продукте (А), полученном после окончания стадии 1.
В отличие от стадии 1 в этом случае является предпочтительным осаждать рутений методом пропитки с использованием органических растворов солей рутения. Например, можно использовать нитрат рутения, растворенный в ацетоне и/или этаноле.
Как и на стадии 1, после осаждения следуют прокаливание, восстановление и затем пассивация. Но в этом случае является предпочтительным осуществлять прокаливание при немного более низкой температуре, чем температура прокаливания на стадии 1, а именно в пределах между 200 и 400oC, предпочтительно между 250 и 350oC. Восстановление же и пассивацию осуществляют при таких же температурных условиях, как и на стадии 1.
По окончании второй стадии получают каталитический предшествующий продукт (В), в основном образованный кобальтом и рутением, осажденными на инертном носителе.
Третья (и последняя) стадия способа по настоящему изобретению состоит в осаждении на предшествующем продукте (В), полученном по окончании второй стадии, третьего элемента, выбранного из иттрия и скандия. В одном из вариантов используют нитрат скандия или иттрия, растворенный в растворителе, выбранном из ацетона, низших спиртов, воды и их смесей. Что касается прокаливания, восстановления и пассивации, то используют те же самые условия, которые описаны для стадии 2. Каталитическая композиция, которая может быть получена способом по настоящему изобретению, содержит Со (в металлической форме или в виде производного), Ru (в металлической форме или в виде производного) и, по крайней мере, третий дополнительный элемент (также в металлической форме или в виде производного), выбранный из Sc и Y, причем все указанные элементы диспергированы на носителе и, если они присутствуют в виде производных, то предпочтительным производным является оксид.
Как описано ранее, указанный носитель состоит из, по крайней мере, оксида, выбранного из, по крайней мере, одного из следующих элементов: Si, Ti, Al, Zr, Zn, Mg. В предпочтительном варианте инертным носителем является диоксид кремния.
Содержание указанных элементов в конечном катализаторе, выраженное для металлов в массовых процентах в сравнении с массой катализатора, изменяется в следующих диапазонах:
Диапазон
Co - 1-50%
Ru - 0,05-5%
Третий - 0,5-5%
Предпочтительные диапазоны
Co - 3-35%
Ru - 0,1-3%
Третий - 0,1-3%
Как уже говорилось, настоящее изобретение касается также способа получения углеводородов из синтез-газа в присутствии вышеописанной каталитической системы.
Что касается синтеза Фишера-Тропша, то его можно рассматривать как способ гидрирования монооксида углерода с целью получения высших углеводородов с, главным образом, линейной цепью. В синтезе Фишера-Тропша избирательность углеводородных продуктов определяется способностью катализатора способствовать реакции развития углеводородной цепи относительно концевого отрезка цепи. Распределение углеводородных продуктов может быть описано через "полимеризационного" типа механизм роста цепи, детально разработанный by Schultz and Flory (P.Biloen, W. MH. Sachtler, Advance in Catalysis, том 30, страницы 169-171, Academic Press, New York, 1981) и приспособленный к синтезу, рассмотренному Андерсоном (R.B.Anderson, Catalysis, vol. IV, P.H.Emmet ed. , Reinhold, New York, 1956). Модель, называемая моделью Андерсона-Шульца-Флори (АШФ, ASF), детально разработана на статистической основе относительно роста цепи, альфа (α), и налагает три условия:
1. Рост цепи должен происходить за счет присоединения промежуточных продуктов с только одним углеродным атомом.
2. Обрыв цепи должен происходить в результате простой десорбции от цепи, например, водородной экстракции.
3. Фактор роста независим от длины цепи.
Математическое представление дано следующей формулой:
Wn = (1-α)2-α(n-1),
где n - число атомов углерода в продукте, Wn - массовая доля продукта и альфа ( α ) - фактор роста, который имеет значения в пределах между 0 и 1.
Из выражения в логарифмической форме:
logWn/n = nlogα+log[(1-α)2/α]
можно получить альфа в виде тангенса угла наклона линейного отношения между log Wn/n и n.
На значения фактора роста альфа влияют как условия реакции, так и состав катализатора. Обычно снижение температуры реакции вызывает повышение избирательности к жидким углеводородам (C5), но неизбежно уменьшает степень конверсии синтез-газа (конв. CO). Существуют, следовательно, ограничения по избирательности и конверсии, вызываемые экономическими соображениями, определяющими точную область целесообразности используемых условий реакции. Но и при низких температурах можно превысить такие пределы, используя каталитические системы, особенно избирательные по отношению к углеводородным фракциям с высокой молекулярной массой (например, C25+).
Настоящее изобретение относится, таким образом, к каталитической композиции, которая позволяет конвертировать смесь CO и H2, известную как синтез-газ, в, по существу, насыщенные неразветвленные углеводороды с процентным содержанием углеводородов C25+ в пределах между 22,5 и 31% по массе и со значениями фактора роста альфа, превышающими 0,90.
Условия эксплуатации таких катализаторов известны в данной области техники как условия для синтеза Фишера-Тропша.
Конверсия синтез-газа в углеводороды происходит при давлении обычно в пределах между 0,1 и 15 МПа, предпочтительно между 1 и 10 МПа, при температуре обычно в пределах между 150 и 350oC, предпочтительно между 170 и 300oC.
Объемная скорость в час обычно находится в пределах между 100 и 20000, предпочтительно между 400 и 5000 (объем синтез-газа на объем катализатора в час); отношение H2/CO в синтез-газе обычно находится в пределах между 1:2 и 5:1, предпочтительно между 1,2:1 и 2,5:1.
Катализатор может быть использован в виде тонкодисперсного порошка (приблизительно 10-700 мкм) или в виде частиц, имеющих эквивалентный диаметр в пределах между 0,7 и 10 мм, соответственно в присутствии жидкой (в эксплуатационных условиях) и газообразной фаз или газообразной фазы. Жидкая фаза может состоять из, по крайней мере, одного углеводорода, имеющего, по крайней мере, 5, предпочтительно, по крайней мере, 10 атомов углерода на молекулу. В предпочтительном варианте жидкая фаза в основном образована тем же самым продуктом реакции.
В качестве примера можно напомнить, что катализаторы по настоящему изобретению могут быть использованы в реакторе с неподвижным слоем катализатора, непрерывно питаемом смесью CO и H2 и работающем при следующих условиях:
- температура реакции 200-215oC;
- давление реакции 20 бар;
- объемная скорость 500 ч-1;
- смесь H2/CO 2/1.
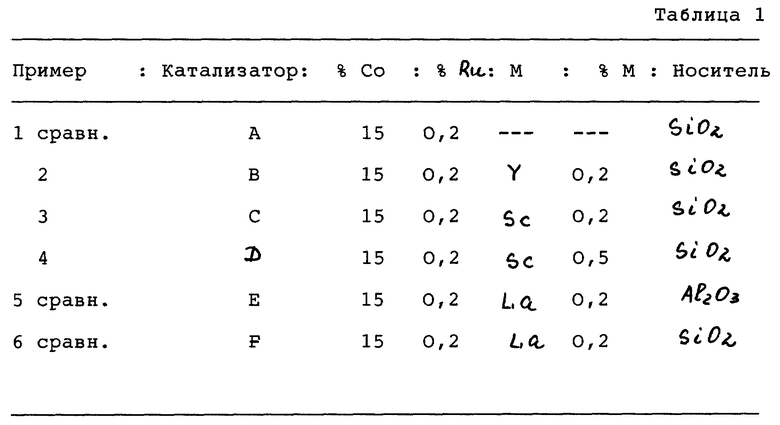
Катализаторы, полученные в примерах 1-6 и имеющие составы, приведенные в таблице 1, оценивали при указанных условиях. Результаты испытаний на химическую активность даны в таблице 2.
Пример 1. Катализатор А (для сравнения)
Используют диоксид кремния с площадью поверхности 300 м2/г, удельным объемом пор 1,3 см3/г, диаметром частиц 20 мкм и удельным весом 0,388 г/см3.
Диоксид кремния подвергают сухой пропитке раствором Co(NO3)2 • 6H2O в азотной кислоте в две стадии, разделенные сушкой при 120oC в течение 16 часов, в таких количествах, чтобы получить процентное содержание Co, равное 15% по массе относительно всего катализатора. Пропитанный указанным образом диоксид кремния прокаливают при 400oC в воздухе в течение 4 часов и затем обрабатывают в потоке H2 с объемной скоростью (GHSV - среднечасовая скорость подачи газа) 1000 ч-1 в трубчатом реакторе при 400oC в течение 16 часов. Восстановленный таким образом образец пассивируют в смеси (1%) O2/(99%)N2 при среднечасовой скорости подачи газа 1000 ч-1 в течение 2 часов при комнатной температуре.
На монометаллический образец Co/SiO2 добавляют 7,5 •10-3 М раствор Ru(NO3)3•H2O, полученный осуществлением следующих операций: осаждение в виде гидроксида с pH 7,2 RuCl3 • H2O, элиминирование хлоридов, повторное растворение в концентрированной HNO3 и разбавление в CH3COCH3 в отношении 1:250 (объем/объем).
Добавляют к образцу раствор рутения в ацетоне в таком количестве, чтобы иметь 0,2% Ru (по массе) по отношению к общей массе. Суспензию оставляют перемешиваться в течение двух часов, после чего сушат под вакуумом при 40oC. Затем следуют фаза прокаливания в воздухе при 300oC в течение 4 часов и восстановление и пассивация, аналогичные тем, что описаны выше.
(Катализатор А: Co/Ru/SiO2, 15% Со, 0,2% Ru)
Пример 2. Катализатор В
Для получения катализатора В к 50 г катализатора А добавляют 10-3 М раствор Y(NO3)3 в ацетоне в таком количестве, чтобы получить конечное массовое процентное содержание иттрия 0,2%.
Полученную при этом суспензию оставляют перемешиваться в течение двух часов и затем сушат под вакуумом при 40oC. Далее образец прокаливают при 300oC в течение 4 часов в воздушной среде, восстанавливают при 400oC в H2 при среднечасовой скорости подачи газа, равной 1000 ч-1, и пассивируют в смеси (1%) O2/(99%) N2 при среднечасовой скорости подачи газа 1000 ч-1 при комнатной температуре.
(Катализатор В: Co/Ru/Y/SiO2, 15% Co, 0,2% Ru, 0,2% Y.)
Пример 3. Катализатор C
Получение катализатора C отличается от описанного в примере 2 использованием 10-3 М раствора Sc(No3)3 в ацетоне в объеме, обеспечивающем получение конечного массового процентного содержания скандия, равного 0,2%.
(Катализатор С: Со/Ru/Sc/SiO2, 15% Co, 0,2% Ru, 0,2% Sc.)
Пример 4. Катализатор D
Получение катализатора D отличается от описанного в примере 3 использованием 10-3 М раствора Sc(NO3)3 в ацетоне в объеме, обеспечивающем получение конечного массового процентного содержания скандия, равного 0,5%.
(Катализатор D: Co/Ru/Sc/SiO2, 15% Со, 0,2% Ru, 0,5% Sc.)
Сравнительный пример 5. Катализатор E
Этот катализатор получают по методике, описанной в US - 4413064, пример 1.
В качестве носителя катализатора используют 100 г гамма-оксида алюминия Харшо (площадь поверхности = 175 м2/г; средний размер пор = 0,5 см3/г; средний диаметр частиц = 40-45 мм; чистота 99%; удельный вес = 0,884 г/см3), который прокаливают при 600oC в течение 2 часов в потоке воздуха.
Затем приготавливают водный раствор нитрата кобальта, растворяя 87,1 г Со(NO3)3 • 6H2O дистиллированной водой до конечного объема 100 см3.
Используя метод начального поглощения влаги, носитель пропитывают этим раствором и после "варки" в течение нескольких часов сушат его в печи в течение 16 часов при 120oC.
Затем приготавливают 0,1 М раствор La(NO3)3 • 6H2O в этаноле и 0,00156 М раствор Ru(NO3)3 в ацетоне. Берут 7,2 см3 первого раствора с разбавлением этанолом до 33,5 см3 и 63,5 см3 второго раствора с доведением объема ацетоном до 66,5 см3. И то, и другое вливают в колбу емкостью 250 см3, где находится 50 г образца Сo/Al2O3, в результате чего получают отношение ацетона к этанолу, приблизительно равное 2, и количество растворителя, равное 2 см3 на грамм носителя. Образец высушивают на роторном испарителе под вакуумом при температуре бани приблизительно 35oC. Для завершения удаления растворителя образец оставляют в печи на 2 часа при 90oC.
Получают продукт, содержащий 15% (м/м) Со, 0,2% (м/м) Ru и 0,2% (м/м) La.
Полученный описанным образом образец загружают в реактор и восстанавливают H2 (35 л/час) в соответствии со следующим температурным профилем:
1. Температуру доводят от 25 до 100oC со скоростью 1oC/мин и держат ее на уровне 100oC в течение 1 часа.
2. С той же самой скоростью температуру повышают до 200oC и на этом уровне держат 2 часа.
3. Затем температуру доводят до 360oC со скоростью 10oC/мин и поддерживают на этом уровне 16 часов.
Далее температуру доводят до 25oC, оставляя катализатор в потоке азота. Затем катализатор пассивируют смесью воздуха (1,2 л/час) и азота (60 л/час) в течение 16 часов.
Сравнительный пример 6. Катализатор F
Этот катализатор получают методом, описанным в патенте US-4413064, пример 1.
В качестве носителя для катализатора используют 25 г диоксида кремния (с теми же самыми характеристиками, которые описаны в примере 1), который прокаливают при 600oC в течение 2 часов в потоке воздуха.
Затем приготавливают водный раствор нитрата кобальта, растворяя 21,77 г Co(NO3)2• 6H2O дистиллированной водой до объема 45 см3.
Используя метод начального насыщения влагой, носитель пропитывают этим раствором и после "варки" в течение нескольких часов сушат 16 часов при 120oC в печи.
Затем приготавливают 0,1 М раствор La(NO3)3 • 6H2O в этаноле и 0,00152 М раствор Ru(NO3)3 в ацетоне. Берут 5,5 см3 первого раствора с разбавлением этанолом до 25 см3 и 50 см3 второго раствора. И то, и другое вливают в колбу емкостью 250 см3, где находится 38 г образца Co/SiO2, в результате чего получают отношение ацетона к этанолу, приблизительно равное 2, и количество растворителя, равное 2 см3 на грамм носителя. Образец высушивают на роторном испарителе под вакуумом при температуре бани приблизительно 35oC. Для завершения удаления растворителя образец оставляют в печи на 2 часа при 90oC.
Состав катализатора: 15% (м/м) Со, 0,2% (м/м) Ru и 0,2% (м/м) La.
Полученный описанным образом образец загружают в реактор и восстанавливают H2 (27 л/час) в соответствии со следующим температурным профилем:
1. Температуру доводят от 25 до 100oC со скоростью 1oC/мин и держат ее на уровне 100oC в течение 1 часа.
2. С той же самой скоростью температуру повышают до 200oC и на этом уровне держат 2 часа.
3. Затем температуру доводят до конечной температуры 360oC со скоростью 10oC/мин и поддерживают на этом уровне 16 часов.
Далее температуру доводят до 25oC, оставляя катализатор в потоке азота. Затем катализатор пассивируют смесью воздуха (0,9 л/час) и азота (45,6 л/час) в течение 16 часов.
Из сравнения катализатора без третьего элемента (сравнительный катализатор А) и катализатора по настоящему изобретению (В, С и D) очевидно, что система с тремя элементами более активна при более низких температурах, причем значения α и избирательности при получении С25+ больше.
Активности катализаторов по настоящему изобретению совершенно одинаковы.
Сравнивая катализатор E (нанесенный на оксид алюминия) и катализатор по настоящему изобретению, можно видеть, что катализатор E дает более низкие значения α и избирательности при получении тяжелых углеводородов.
И наконец, сравнительный катализатор F (нанесенный на диоксид кремния, как и катализатор по настоящему изобретению) показывает намного большее уменьшение значений α и избирательности при C25+ в сравнении с катализатором по настоящему изобретению.

Описывается способ получения катализатора, содержащего инертный носитель, кобальт, рутений и третий элемент, выбранный из скандия и иттрия. Способ включает, по крайней мере, следующие стадии: получение первого каталитического предшествующего продукта (А), содержащего кобальт и инертный носитель, с последующими прокаливанием, восстановлением и пассивацией; получение второго каталитического предшествующего продукта (В), содержащего кобальт, рутений и инертный носитель, путем осаждения рутения на первый каталитический продукт (А) с последующими прокаливанием, восстановлением и пассивацией; получение конечного катализатора путем осаждения элемента, выбранного из скандия и иттрия, на каталитическом предшествующем продукте (В) с последующими прокаливанием, восстановлением и пассивацией. Описывается также композиция для получения катализатора, катализатор и способ синтеза газа. Технический результат - получение катализатора, особенно эффективного в конверсии синтез-газа в углеводородные продукты, содержащие значительные количества углеводородов с числом углеродных атомов, превышающим или равным 22. 4 с. и 5 з.п. ф-лы, 2 табл.
Приоритет по пунктам:
04.08.95 по пп.1,5.6,7,8;
11.04.96 по пп. 2,3,4,9.
| US 4413064 A, 01.11.1983 | |||
| КАТАЛИЗАТОР ДЛЯ КОНВЕРСИИ СИНТЕЗ-ГАЗА В УГЛЕВОДОРОДЫ И СПОСОБ ПОЛУЧЕНИЯ УГЛЕВОДОРОДОВ | 1988 |
|
RU2017517C1 |
| US 4088671 A, 09.05.1978 | |||
| US 5227407 A, 13.07.1993 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| EP 0581619 A1, 02.02.1994. | |||

