Изобретение относится к области медицины, конкретно к фармацевтической технологии, а именно - к способу получения лекарственного средства, обладающего противогрибковым действием, - флуконазола (международное непатентованное наименование, утвержденное Всемирной Организацией Здравоохранения). Фармацевтические препараты, содержащие флуконазол, за время присутствия на рынке зарекомендовали себя как эффективные противогрибковые агенты, мощные селективные ингибиторы синтеза стиролов в клетках грибков.
Препараты флуконазола выпускаются в виде капсул (активное вещество и наполнители), порошка для приготовления суспензий для приема внутрь (активное вещество и фармацевтически приемлемые разбавители), а также в виде растворов для внутривенных инфузий. При очевидных преимуществах флуконазола и препаратов на его основе не до конца остается решенным вопрос о создании высокоочищенной стабильной субстанции, которая с течением времени не подвергалась бы химической деградации в лекарственных формах при их хранении, а в условиях жесткого технологического процесса изготовления готовой лекарственной формы из субстанции не меняла бы своих химико-технологических показателей.
Первоначально фармакологические свойства флуконазола были описаны в патенте США N 4404216, там же описан способ получения. Недостатками этого способа является необходимость сложного синтеза, включающего стадию получения промежуточного продукта - оксирана, который может оставаться в конечном продукте как примесь, а также получение побочного продукта - изофлуконазола.
Известно, что были предприняты попытки получить флуконазол различными способами (патент США N 5484936, патент США N 5508423, заявка WO98/32744). Однако выяснилось, что указанное вещество может существовать в нескольких полиморфных и псевдополиморфных (гидраты) модификациях. Получение кристаллографически чистого флуконазола описано, например, в заявке WO95/07895 (получены полиморфные формы), патенте GB N 2270521 (получен кристаллический моногидрат).
В указанных способах используются токсичные промежуточные продукты, а конечные продукты являются или гидратами, или являются смесью модификаций, что нежелательно для дальнейшей технологической обработки субстанции.
Наиболее близким к предложенному изобретению является способ получения флуконазола путем взаимодействия 2,4-дифтор-2-(1H-1,2,4- триазола-1-ил)-ацетофенона, 1,2,4-триазола и триметилсульфоксония йодида в щелочном водном растворе при нагревании (патент США N 5710280). Недостатком этого способа является тот факт, что конечный продукт не обладает достаточной стабильностью при формировании из него лекарственных форм, также недостаточно стабильна сама субстанции. Необходимость интенсивного неконтролируемого нагревания является нежелательной для конечного продукта и способствует образованию в нем нестабильных высокоэнергетических примесей, что делает продукт кристаллически неоднородным, что в свою очередь затрудняет стандартизацию субстанции. Вышеуказанные недостатки могут помешать использовать продукт, полученный по данному способу, в химико-фармацевтической промышленности достаточно широко.
Физические факторы оказывают влияние на стабильность лекарственных веществ, начиная с момента их получения и до приема больным. Стабильность лекарственной формы в значительной степени зависит от процесса, который применялся при получении исходной субстанции. Важная роль принадлежит исходным продуктам синтеза, выбранным растворителям, очистке промежуточных продуктов.
Стабильность во многом зависит от химической чистоты и физических свойств лекарственного вещества. Известно, что в зависимости от условий кристаллизации могут изменяться размер кристаллов, поверхностная энергия, оформление граней. Эти физические свойства оказывают влияние на гигроскопичность, химическую активность, а следовательно, на стабильность лекарственного препарата. Форма и размер кристаллов находятся в зависимости от природы, степени чистоты растворителя, температурных условий и продолжительности процесса кристаллизации, наличия сопутствующих веществ. Эти факторы непосредственно влияют на образование полиморфных форм субстанций лекарственных веществ.
Процессы получения наиболее стабильных модификаций других соединений описаны, например, в патенте РФ N 2067093; патенте РФ N 2051915; патенте РФ N 2079489. Как следует из этих документов, выбор растворителя для осуществления поставленной задачи и определение условий кристаллизации представляет собой длительный процесс, в котором установка комплекса всех необходимых параметров является следствием экспериментов, причем условия проведения процесса невозможно определить ни из уровня техники, известного на данный момент времени, ни спрогнозировать каким-либо теоретическим путем.
Задачей изобретения является получение кристаллического безводного флуконазола с повышенной стабильностью при одновременном повышении его чистоты.
Поставленная задача достигается подбором комплекса реагентов, растворителей, а также условий проведения процесса взаимодействия полупродуктов, которые позволяют получить флуконазол, пригодный для дальнейшего использования в фармацевтической промышленности.
Для получения флуконазола смешивают 2,4-дифтор-1H-1,2,4 триазола-1-ил- ацетофенон, 1,2,4-триазол и триметилсульфоксония йодид в щелочном водном растворе, смесь настаивают при температуре 4-25oC, нагревают реакционную смесь при температуре 45-50oC в течение 6-28 часов, после чего добавляют метиленхлорид, промывают органический слой водой при pH 8,5-9,5, удаляют метиленхлорид при температуре 45-50oC, промывают толуолом с последующим его удалением при температуре 45-50oC, настаивают в течение 0,5-2 часов, повторно промывают толуолом при температуре 25-30oC и сушат в течение 8-12 часов при температуре 60-65oC.
Полученный продукт имеет высокую степень чистоты, является безводным кристаллическим порошком, по существу, свободным от примеси нестабильных полиморфных модификаций.
Изобретение иллюстрируется следующими примерами.
Пример 1.
В реакционный сосуд помещают 25 мл воды, 2,4-дифтор-2-(1H-1,2,4 триазола-1)-ил-ацетофенона (5 г), 1,2,4-триазола (1,7 г), триметилсульфоксония йодида (5 г), калия гидроксида (3,98 г). Смесь настаивают при температуре 25oC в течение 20 минут при перемешивании, после чего нагревают до температуры 50oC, и по достижении этой температуры продолжают нагрев при перемешивании в течение 24 часов. Реакционную смесь экстрагируют метиленхлоридом (2 раза по 25 мл) при температуре 25oC, при той же температуре промывают органический слой водой (2 раза по 25 мл) до достижения pH 9,5. Удаляют метиленхлорид при пониженном давлении и температуре 50oC. Затем помещают в реакционный сосуд 30 мл толуола при помешивании, после чего толуол удаляют под вакуумом при температуре 50oC. Охлаждают реакционную смесь до температуры 25oC, помешивают и настаивают при той же температуре в течение часа. Промывают на фильтре толуолом (10 мл) при температуре 30oC. Белый порошок, оставшийся на фильтре, сушат 10 часов при температуре 65oC. Получают конечный продукт - 3,25 г безводного флуконазола. Продукт идентифицировали общепринятыми методами, масс-спектрометрией и ЯМР (данные ЯМР приведены в таблице 1).
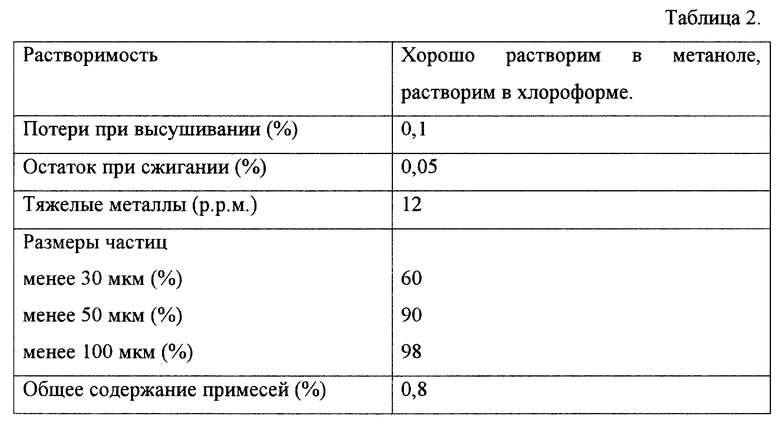
Аналитические показатели субстанции по примеру 1 приведены в таблице 2.
Пример 2.
В реакционный сосуд помещают 50 мл воды, 2,4- дифтор-2(1H-1,2,4 триазола-1-ил)-ацетофенона (44,8 ммоль), 1,2,4- триазола (50,58 ммоль), триметилсульфоксония йодида (44,8 ммоль), калия гидроксида (120 ммоль). Смесь настаивают при температуре 5oC в течение 30 минут при перемешивании, после чего нагревают до температуры 46oC, и по достижении этой температуры продолжают нагрев при перемешивании в течение 12 часов. Реакционную смесь экстрагируют с помощью 70 мл метиленхлорида при температуре 20oC, при той же температуре промывают органический слой водой (3 раза по 50 мл) до достижения pH 8,5. Удаляют метиленхлорид при пониженном давлении и температуре 46oC. Затем помещают в реакционный сосуд 65 мл толуола при помешивании, после чего толуол удаляют под вакуумом при температуре 46oC. Охлаждают реакционную смесь до температуры 20oC, помешивают и настаивают при той же температуре в течение 40 минут. Промывают на фильтре толуолом (25 мл) при температуре 25oC. Белый порошок, оставшийся на фильтре, сушат 11 часов при температуре 61oC. Получают конечный продукт - 6,51 г безводного флуконазола. Продукт идентифицировали аналогично примеру 1.
Температура плавления 138oC. Молекулярная масса 306,2. Конечный продукт - белый кристаллический порошок.
Аналитические показатели субстанции по примеру 2 приведены в таблице 3.
Продукты по примеру 1 и 2, а также продукт, полученный по способу-прототипу, хранили в стандартных условиях, принятых в фармации для субстанций, результаты сравнивали. Данные приведены в таблице 4.
Полученный флуконазол при относительной влажности 95% и температуре 30oC остается безводным:
По примеру 1 (n=6) - в 2,1±0,15 раза дольше сравниваемого образца.
По примеру 2 (n=6) - в 2,4±0,21 раза дольше сравниваемого образца.


Изобретение относится к медицине, конкретно к фармацевтической технологии, а именно к способу получения лекарственного средства, обладающего противогрибковым действием, - флуконазола. Для получения флуконазола смешивают 2,4-дифтор-1H-1,2,4 триазола-1-ил-ацетофенон, 1,2,4-триазол и триметилсульфоксония йодид в щелочном водном растворе, смесь настаивают при температуре 4-25oC, нагревают реакционную смесь при температуре 45-50°С в течение 6-28 ч, после чего добавляют метиленхлорид, промывают органический слой водой при рН 8,5-9,5, удаляют метиленхлорид при температуре 45-50°С, промывают толуолом с последующим его удалением при температуре 45-50°С, настаивают в течение 0,5-2 часов, повторно промывают толуолом при температуре 25-30°С и сушат в течение 8-12 ч при температуре 60-65°С. Предложенный способ позволяет получить флуконазол с более высокой стабильностью и чистотой. 4 табл.
Способ получения флуконазола путем смешения 2,4-дифтор-2-(1H-1,2,4-триазола-1-ил)-ацетофенона, 1,2,4-триазола и триметилсульфоксония йодида в щелочном водном растворе, нагревания реакционной смеси с последующим выделением целевого продукта, отличающийся тем, что перед нагреванием реакционную смесь настаивают при температуре 4 - 25oC, нагревание ведут при температуре 45 - 50oC в течение 6 - 28 ч, после чего добавляют метиленхлорид, промывают органический слой водой до pH - 8,5 - 9,5, удаляют метиленхлорид при температуре 45 - 50oC, промывают толуолом с последующим его удалением при температуре 45 - 50oC, настаивают в течение 0,5 - 2 ч, повторно промывают толуолом при температуре 25 - 30oC и сушат в течение 8 - 12 ч при температуре 60 - 65oC.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 98104064 А1, 27.01.2000 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 5710280 А, 20.01.1998 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 5633386 А, 27.05.1997 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ИВЛЕВА А.Я | |||
| и др | |||
| Медофлюкан (флюконазол) - последнее достижение в лечении микозов | |||
| - Вестник дерматологии и венерологии, 1997, № 3, с | |||
| Способ приготовления сернистого красителя защитного цвета | 1915 |
|
SU63A1 |
