Данное изобретение принадлежит к области органического химического синтеза и относится к новому способу получения биологически активного 2-[2,4-дифторфенил] -1,3-бис[1H- 1,2,4-триазол-1-ил] -пропан-2-ола, имеющего общее название "флуконазол", который является важным противогрибковым соединением, из монозамещенных гидразинов и s-триазина. Это противогрибковое соединение используется в медицине, ветеринарии и сельском хозяйстве.
Существует необходимость в упрощенном и промышленно применимом способе синтеза флуконазола, который является бистриазольным противогрибковым активным веществом для использования при лечении человека (патент Великобритании GB 2099818, 1982.
Синтез флуконазола, химическое название 2-[2,4-дифторфенил] - 1,3-бис[1H-1,2,4-триазол-1-ил]-пропан-2-ол формулы (I)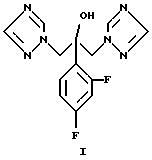
раскрыт в GB 2099818, US 4404216, YU 42770, ES 512882 и ES 520794, ES 618198, WO 95/07895, AT 900961.
В указанных патентах используют два основных пути синтеза, которые схематично показаны на схемах 1 и 2.
Способы синтеза флуконазола осуществляются раскрытием производного оксиэтилена (II) 1,2,4-триазолом (схема 1 ниже) или замещением реакционноспособной группы 1,2,4-триазолом в промежуточном соединении (III), оба способа проводят в сильной щелочной среде. При замещении реакционной группы X (X представляет собой галоген, гидрокси, замещенную гидроксильную группу) в реакции образуется эпоксидное производное, которое, однако, не выделяется в ходе реакции (схема 2 ниже).
Схема 1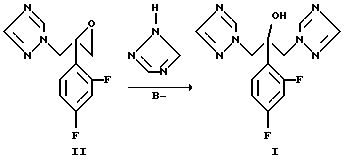
Схема 2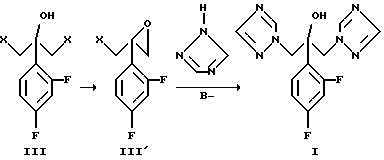
В обоих способах реакцию проводят в щелочной среде, и 1,2,4- триазол является нуклеофилом. 1,2,4-Триазол также частично реагирует по 4 положению и, таким образом, образуются нежелательные изомерные продукты. Такая реакционная способность приводит к низкому выходу реакции и из-за присутствия изомерных продуктов к более низкой чистоте продукта. Если реакцию 1,2,4-триазола проводят с промежуточным соединением (III), где X является хлором, выход составляет до 26%.
Способом данного изобретения, как описано ниже, предотвращают образование нежелательных продуктов, образуемых 1,2,4-триазолом в щелочных условиях, и выход реакции приближают к количественному превращению. Механизм реакции и условия отличаются от тех, которые известны в литературе, и становится возможным избежать образования изомерных продуктов 1,2,4-триазола. Чистота продукта выше.
Объектом данного изобретения является способ получения 2- [2,4-дифторфенил] -1,3-бис[1Н-1,2,4-триазол-1 -ил] -пропан-2-ола (флуконазола) формулы (I)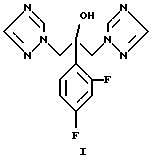
и его фармацевтически приемлемых солей, отличающийся тем, что соединение формулы (IV)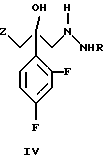
где R является водородом, и Z является радикалом триазола или необязательно в виде соли, взаимодействует с s-триазином формулы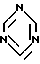
в органическом растворителе в качестве реакционной среды в кислых условиях реакции и при температуре от -60oC до температуры флегмы реакционной смеси.
В качестве органических растворителей могут быть использованы спирты, предпочтительно этанол, метанол, 1- и 2-пропанол, эфиры, такие как диэтиловый эфир, дипропиловый эфир, диизопропиловый эфир, метил трет-бутиловый эфир, ацетонитрил, диметилсульфоксид (ДМСО), диметилформамид (ДМФ), ДМАА (диметилацетамид), N- метилпирролидон.
К растворителю добавляют неорганическую или органическую кислоту, такую как муравьиная кислота, уксусная кислота, трифторуксусная кислота, метансульфокислота, малоновая кислота, малеиновая кислота, яблочная кислота, хлоруксусная кислота, дихлоруксусная кислота, пропановая кислота или паратолуолсульфокислота, соляная кислота, серная кислота и азотная кислота, предпочтительно муравьиная кислота или трифторуксусная кислота.
Также в качестве органического растворителя могут быть использованы сами органические кислоты.
Если используют смесь кислот, например, органических кислот, она может быть составлена при различных соотношениях этих кислот.
Температура реакции находится в интервале от -60oC до температуры кипения используемой реакционной смеси.
Для получения фармацевтически приемлемых солей флуконазола может быть использована муравьиная кислота, уксусная кислота, трифторуксусная кислота, метансульфокислота, малоновая кислота, малеиновая кислота, яблочная кислота, паратолуолсульфокислота, а также соляная кислота, серная кислота и азотная кислота.
Соединение (I) (флуконазол) получают с высоким выходом (80-100%), зависящем от используемой кислоты или растворителя, времени реакции, температуры реакции и pH реакционной среды.
В реакции 2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-3- гидразинпропан-2-ола (IV) с s-триазином в абсолютном спирте и с добавлением соляной кислоты выход составляет 80%. Если в качестве растворителя используют уксусную кислоту, реакция протекает практически количественно (свыше 95%) уже через 45 мин. Если используют муравьиную кислоту, превращение становится количественным уже через 20 мин при температуре в интервале между 0 и 5oC.
Проведенные реакции показывают, что превращение соединений гидразина (IV) реакцией с s-триазином в соединение (1) проходит лучше и быстрее в сильнокислотных условиях реакции, когда соединение (IV) присутствует в виде соли, такой как гидрохлорид, сульфат, ацетат, формиат, метансульфонат и другие. Как отмечено выше, соединение (IV) может быть использовано в виде основания или в виде его соли.
Преимущество данного процесса является очевидным, поскольку в реакции соединения (IV) с s-триазином количественно образуется 2- [2,4-дифторфенил] -1,3-бис[1H-1,2,4-триазол-1-ил]-пропан-2-ол(I) (флуконазол).
Реакцию проводят в кислотных и сильнокислотных условиях реакции (pH равно 1-6). Все известные в данной области техники способы синтеза флуконазола проводят в основных условиях реакции (pH равно 7-14).
Механизм реакции s-триазина с соединением (IV), где R является водородом и Z является радикалом 1,2,4-триазола, представлен на следующей схеме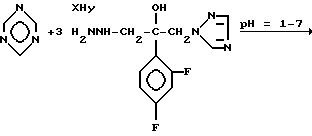
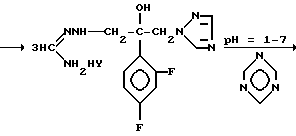
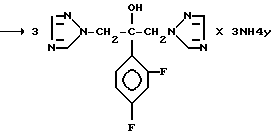
Y=анион кислоты
Соединение формулы (IV) является известным (WO 96/04256). Если его используют в виде соли, оно является солью муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, сульфокислоты, метансульфокислоты, малоновой кислоты, малеиновой кислоты, яблочной кислоты и п-толуолсульфокислоты, а также соляной кислоты, серной кислоты и азотной кислоты.
Соединение формулы (IV), где Z является радикалом триазола, получают по одному из вариантов способа, как описано на схеме 3
Схема 3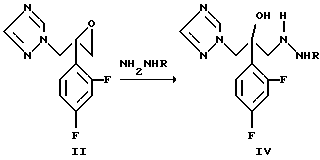
2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)пропан-2,3- эпоксид формулы (II) взаимодействует с соединением гидразина формулы
NH2NHR, (V)
где R такой, как определен выше. Соединение формулы (IV) может быть использовано, когда R является водородом, в виде чистого гидразингидрата, гидразингидрата в воде, гидразингидрата в органических растворителях, таких как ацетонитрил, или в смеси органических растворителей и воды. Соединение формулы (IV), где Z является остатком триазола, получают по другому варианту данного способа, как описано на схеме 4.
Схема 4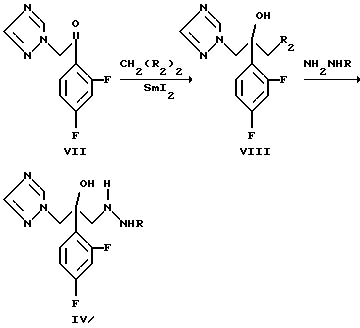
Из 1-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-ил)этан-1-она (VII) галометилированием дигалометаном CH2(R2)2, где R2 является хлором, бромом или йодом, и в присутствии катализатора, который может быть йодидом самария (II) или металлическим самарием, получают 2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-3-галопропан-2-ол формулы (VIII), где R2 такой, как определен выше, и затем в реакции замещения, галоген замещается на соответствующий гидразин формулы (V).
Если R в соединении формулы (V) является водородом, замещение может проводиться с избытком гидразингидрата или с минимальным избытком гидразингидрата в органическом растворителе, предпочтительно ацетонитриле.
Все данные способы получения соединения формулы (IV) проводятся при температуре между -25oC и температурой кипения реакционной смеси. Их проводят в органическом растворителе.
Способ получения соединения формулы (IV), где Z является радикалом триазола, заключается в том, что
a) 2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)пропан- 2,3-эпоксид формулы (II)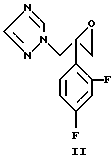
в виде кислотно-аддитивной соли взаимодействует с соединением гидразина формулы
NH2NHR, (V)
где R такой, как определен выше, или
b) 1-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-ил)этан-1-он (VII)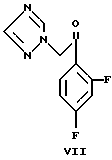
галометилируют дигалометаном CH2(R2)2, где R2 является хлором, бромом или йодом, и в присутствии катализатора, который может быть йодидом самария (II) или металлическим самарием, до 2-(2,4-дифторфенил)-1-(1H-1,2,4- триазол-1-ил)-3-галопропан-2-ола формулы (VIII)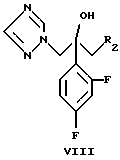
где R2 такой, как определен выше, и затем в реакции замещения галоген замещают на соответствующий гидразин формулы (V).
Исходное соединение формулы (VIII), используемое в варианте b) способа получения соединения формулы (IV), где Z является радикалом триазола, также является новым соединением и представляет собой один из объектов данного изобретения.
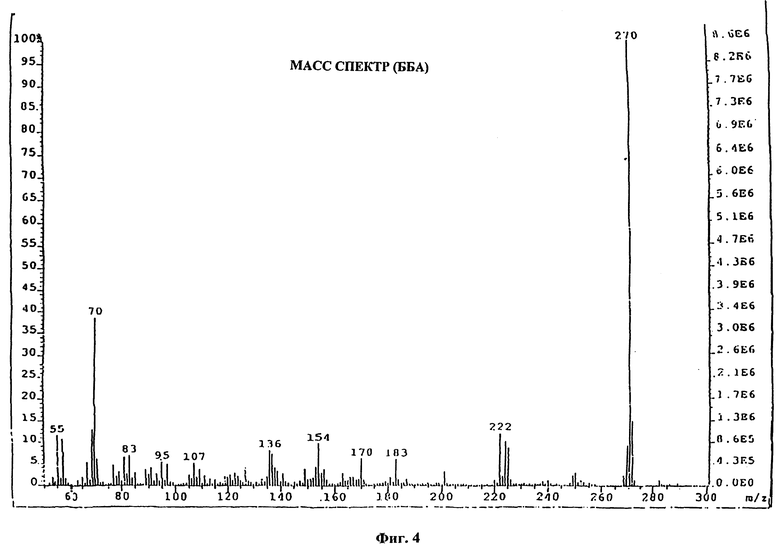
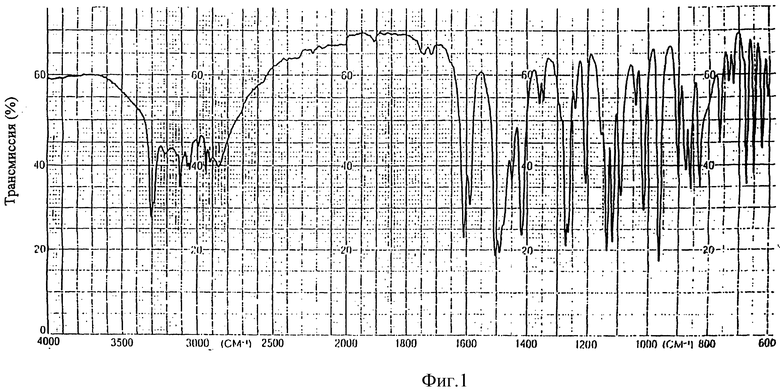
На фиг. 1, 2, 3 и 4 показаны ИК-спектр, ЯМР-спектр, УФ-спектр и масс-спектр (ББА) соединения формулы (IV), где R является водородом и Z является 1,2,4-триазолилом.
Данное изобретение проиллюстрировано, но не ограничено следующими примерами.
Пример 1
Синтез соединения гидразина формулы (IV) (Z является триазолилом, R является водородом) из соединения формулы (II).
К ацетонитрилу (12.5 мл) и эпоксиду формулы (II) в виде метансульфоната (1.67 г, 0.005 моль) добавляют гидразингидрат (0.61 мл, 0.0125 моль; 99%). Получают реакционную смесь, состоящую из трех фаз: две бесцветныe фазы и фаза суспензии с белым твердым веществом. Реакционную смесь нагревают до температуры кипения, и получают только двухфазовую систему из двух бесцветных фаз. Реакционную смесь нагревают при температуре кипения в течение 3 ч, до тех пор пока с помощью тонкослойной хроматографии (хлороформ/метанол 15/2) не установят завершение реакции. Реакционную смесь охлаждают до 0-5oC в течение 10 мин. Полученный белый осадок отфильтровывают и промывают ацетонитрилом. (Получают гидразин метансульфонат (0.64 г).) Фильтрат выпаривают при пониженном давлении на бане, имеющей температуру не выше 40oC. Получают белый продукт (1.35 г), 2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-3-гидразинпропан-2-ол.
Пример 2
Синтез соединения гидразина формулы (IV) (Z является триазолилом, R является водородом) из соединения формулы (II).
Проводят, как описано в примере 1. После завершения реакции реакционную смесь помещают в делительную воронку, добавляют метиленхлорид (25 мл) и насыщенный водный раствор хлорида натрия (15 мл). Смесь встряхивают, отделяют органическую фазу и водную фазу, водную фазу промывают метиленхлоридом (5 мл), органические фазы объединяют, сушат с безводным сульфатом натрия, растворитель выпаривают в вакууме (давление менее 1.33 мбар), и получают целевое соединение (1.28 г), которое кристаллизуют в холодильнике.
Пример 3
Синтез соединения гидразина формулы (IV) (Z является триазолилом, R является этоксикарбонильной группой).
Смесь ацетонитрила (10 мл), эпоксида формулы (II) (0.333 г; 0.001 моль) и этилкарбазата (0.520 г; 0.005 моль) кипятят в течение 4 ч с обратным холодильником. После завершения реакции реакционную смесь охлаждают и добавляют хлороформ (25 мл) и насыщенный водный раствор хлорида натрия (15 мл). Смесь встряхивают в делительной воронке, органическую фазу отделяют от водной фазы и сушат над безводным сульфатом натрия. Растворитель выпаривают и выделяют маслянистый продукт (0.314 г).
ИК (масляная пленка): 3290, 3060, 3020, 2940, 1690, 1600, 1480, 1250, 1130, 945, 835.
Масс-спектр (ББА) м/з: 342 (MH+)
1H ЯМР (CDCl3, 300 МГц) δ (п/м): 1.26 (т, 3H 3J = 7.1 Гц), 2.88 (д, 1H 2J = 4.7 Гц), 2.95 (д, 1H 2J = 4.7 Гц), 3.76 (широкий сигнал, 2H, -NHNH-), 4.16 (кв, 2H 3J = 7.1 Гц), 4.52 (д, 1H 2J = 14.8 Гц), 4.82 (д, 1H 2J = 14.8 Гц), 6.85 (м, 2H), 7.17 (м, 1H), 7.87 (с, 1H), 8.07 (с, 1H).
Пример 4
Синтез соединения гидразина формулы (IV) (Z является триазолилом, R является бутоксикарбонильной группой).
Смесь ацетонитрила (10 мл), эпоксида формулы (II) (0.333 г; 0.001 моль) и трет-бутилкарбазата (0.660 г; 0.005 моль) кипятят в течение 4 ч с обратным холодильником. После завершения реакции реакционную смесь охлаждают и добавляют хлороформ (25 мл) и насыщенный водный раствор хлорида натрия (15 мл). Смесь встряхивают в делительной воронке, органическую фазу отделяют от водной фазы и сушат над безводным сульфатом натрия. Растворитель выпаривают и выделяют маслянистый продукт (0.328 г).
ИК (масляная пленка): 3380, 3070, 3020, 2940, 1680, 1590, 1575, 1470, 1400, 1250, 1115, 945, 850.
Масс-спектр (ББА) м/з: 370 (MH+, 23%), 314 (100%), 289 (15%), 230 (24%), 177 (16%), 89 (12%), 70 (52%), 57 (40%).
1H ЯМР (CDCl3, 300 МГц) δ (п/м): 1.43 (с, 9H), 2.88 (д, 1H 2J = 4.7 Гц), 2.95 (д, 1H 2J = 4.7 Гц), 4.51 (д, 1H 2J = 14.8 Гц), 4.83 (д, 1H 2J = 14.8 Гц), 6.84 (м, 2H), 7.17 (м, 1H), 7.86 (с, 1H), 8.07 (с, 1H).
Пример 5
Синтез 2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-3- йодпропан-2-ола формулы (VIII).
В дважды перегнанный сухой ТГФ (10 мл) помещают дийодметан (2 ммоль; 536 мг) под аргоном и добавляют 0.1 М раствор йодида самария в ТГФ (II) (25 мл). Затем постепенно добавляют раствор 1- (2,4-дифторфенил)-2-(1H-1,2,4-триазоло-1-ил)этан-1-она (VII) в ТГФ (223 мг, 1 ммоль). После добавления раствор перемешивают в течение еще 30 мин при комнатной температуре. Полученный осадок фильтруют отсасыванием, растворитель выпаривают в вакууме, к остатку добавляют метиленхлорид (20 мл), раствор сушат над сульфатом натрия, растворитель выпаривают, и неочищенный продукт кристаллизуют из изопропанола. Получают целевое соединение (335 мг; 82%).
ИК: 3120, 2970, 2960, 1610, 1500, 1420, 1275, 1140, 1105, 970, 855, 680.
1H ЯМР (CDCl3, 300 МГц) δ (п/м): 2.88 (д, 1H 2J = 4.7 Гц), 2.95 (д, 1H 2J = 4.7 Гц), 3.58 (д, 1H 2J = 10.7 Гц), 3.76 (д, 1H 2J = 10.7 Гц), 6.8 (м, 2H), 7.2 (м, 1H), 7.87 (c, 1H), 8.07 (с, 1H).
Пример 6
Синтез гидразина формулы (IV) (Z является триазолилом, R является водородом) из соединения формулы (VIII).
Соединение формулы (VIII) (365 мг; 1 ммоль), полученное по примеру 5, гидразингидрат (125 мг; 2.5 ммоль) и ацетонитрил (10 мл) кипятят в течение 3 ч с обратным холодильником. После завершения реакции добавляют дихлорметан (15 мл) и насыщенный водный раствор хлорида натрия (10 мл), смесь встряхивают в делительной воронке, органическую фазу отделяют от водной фазы, сушат над безводным сульфатом натрия, растворитель выпаривают в вакууме и получают целевое соединение (241 мг; 89.6%).
Пример 7
Синтез флуконазола.
К неочищенному соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют триазин (1.42 г; 0.017 моль; 97%) и уксусную кислоту (10 мл). Желтоватую реакционную смесь кипятят с обратным холодильником (116-118oC) в течение 2 ч 45 мин. Во время этого периода желтая реакционная смесь становится оранжевой. Завершение реакции определяют с помощью тонкослойной хроматографии (хлороформ/метанол 15/2). Уксусную кислоту выпаривают при пониженном давлении и температуре бани 70-75oC. К оранжевому остатку после выпаривания добавляют воду (10 мл) и хлорид натрия (3 г) и добавляют метиленхлорид (25 мл); pH водной фазы 2-3, pH водной фазы доводят до 7 с помощью 33%-ного раствора гидроксида натрия, и затем раствор декантируют. Водную фазу промывают метиленхлоридом (10 мл), органические фазы объединяют и сушат над безводным сульфатом натрия. Их фильтруют, промывают метиленхлоридом, и метиленхлорид выпаривают при температуре 40oC при пониженном давлении. Выделяют розовое твердое вещество (1.49 г; 97.4%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 8
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют триазин (1.42 г; 0.017 моль; 97%) и муравьиную кислоту (10 мл; 98%). Полученную желтоватую реакционную смесь кипятят с обратным холодильником (100-102oC) в течение 40 мин, пока реакционная смесь не станет оранжевой, и завершение реакции определяют с помощью тонкослойной хроматографии. Муравьиную кислоту выпаривают при пониженном давлении и температуре бани 70-71oC. К остатку добавляют воду (10 мл) и хлорид натрия (3 г), добавляют метиленхлорид (25 мл); pH водной фазы 2-3, pH раствора доводят до 7 с помощью 33%-ного раствора гидроксида натрия, и затем раствор декантируют. Органическую фазу промывают метиленхлоридом (10 мл), органические фазы объединяют и сушат над безводным сульфатом натрия. Их фильтруют, промывают метиленхлоридом, и метиленхлорид выпаривают при температуре 40oC при пониженном давлении. Выделяют слегка окрашенный продукт (1.48 г; 96.7%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 9
Синтез флуконазола.
Проводят как в примере 8. Время проведения реакции составляет 70 мин при кипячении с обратным холодильником (100-101oC). Получают слегка окрашенный продукт (1.44 г; 94.1%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 10
Синтез флуконазола.
Проводят как в примере 8. Время проведения реакции составляет 50 мин при температуре 80-85oC. Получают слегка окрашенный продукт (1.50 г; 98%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 11
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют триазин (1.42 г; 0.017 моль; 97%) и муравьиную кислоту (10 мл; 98%). Полученную желтоватую реакционную смесь нагревают до температуры 30oC и затем нагревают в течение 75 мин при 30-35oС. Желтоватая реакционная смесь становится оранжевой, и завершение реакции определяют с помощью тонкослойной хроматографии (хлороформ/метанол 15/2). К реакционной смеси добавляют воду (10 мл) и хлорид натрия (3 г), добавляют метиленхлорид (25 мл); pH водной фазы 1.15, pH раствора доводят до 7 с помощью 33%-ного раствора гидроксида натрия, и декантируют бесцветную водную фазу и желтоватую органическую фазу. Фазы разделяют, водную фазу промывают метиленхлоридом (10 мл), органические фазы объединяют и сушат над безводным сульфатом натрия. Их фильтруют, высушивающее вещество промывают метиленхлоридом, и растворитель выпаривают при температуре 40oC при пониженном давлении. Выделяют белый продукт (1.49 г; 97.4%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 12
Синтез флуконазола.
Проводят как в примере 11. Время проведения реакции составляет 2 ч 35 мин при температуре 20-25oC. Получают слегка окрашенный продукт (1.51 г; 98.7%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 13
Синтез флуконазола.
Проводят как в примере 11. Время проведения реакции составляет 1 ч при температуре 0-5oC. Получают слегка окрашенный продукт (1.50 г; 98%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 14
Синтез флуконазола.
Проводят как в примере 11. Время проведения реакции составляет 90 мин при температуре 0-5oC. Получают белый продукт (1.46 г; 95%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс спектр) соответствуют целевому соединению.
Пример 15
Синтез флуконазола.
Проводят как в примере 11. Время проведения реакции составляет 1 ч при температуре 0-5oC. Используют 97%-ный триазин (0.7 г; 0.009 моль). Получают слегка окрашенный продукт (1.46 г; 95.4%).
Тонкослойная хроматография: хлороформ/метанол 15/2, Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 16
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (20 мл), трифторуксусную кислоту (2 мл; 0.026 моль) и s-триазин (0.8 г; 0.00987 моль). Двухфазную систему нагревают до температуры 37oC и добавляют муравьиную кислоту (4 мл; 98%) до получения желтого раствора. Реакционную смесь выдерживают в течение 1 ч при температуре 35oC. Тонкослойная хроматография показывает завершение реакции. Затем добавляют раствор хлорида натрия (3 г) в воде (10 мл); pH 0.28. Добавляют 33%-ный раствор гидроксида натрия до pH 7.56, и затем раствор декантируют. Водную фазу получают бесцветной, и органическую фазу получают бледно-розовой. Водную фазу промывают метиленхлоридом (10 мл), органические фазы объединяют, сушат над безводным сульфатом натрия, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Выделяют белый продукт (1.39 г; выход 91%).
Тонкослойная хроматография (хлороформ/метанол 15/2) показала, что продукт является очень чистым (один цвет);
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 17
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (10 мл), муравьиную кислоту (4 мл; 98%), трифторуксусную кислоту (1.7 мл; 0.022 моль) и s-триазин (0.6 г; 0.0074 моль). Получают красно-оранжевый раствор. Реакционную смесь выдерживают в течение 1 ч при температуре 25oC. Тонкослойная хроматография показывает завершение реакции. Затем добавляют раствор воды (10 мл) и хлорид натрия (3 г) и метиленхлорид (10 мл); pH 0.05. Добавляют 33%-ный раствор гидроксида натрия до pH 8.14, и затем раствор декантируют. Водную фазу получают бесцветной и органическую фазу получают желтой. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат над безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают бежевый продукт (1.48 г; выход 96.7%).
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 18
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (40 мл), муравьиную кислоту (4 мл; 98%), трифторуксусную кислоту (1.7 мл; 0.022 моль) и s-триазин (0.6 г; 0.0074 моль). Получают оранжевую эмульсию (две фазы), что означает, что реакция проходит в гетерогенной фазе. Реакционную смесь кипятят с обратным холодильником (41oC) и получают те же две фазы. Такую реакционную смесь выдерживают в течение 1 ч при кипячении с обратным холодильником. Тонкослойная хроматография показывает завершение реакции. Затем реакционную смесь охлаждают до комнатной температуры и добавляют раствор хлорида натрия (3 г) в воде (10 мл); pH 0.17. Добавляют 33%-ный раствор гидроксида натрия до pH 8.45, и затем раствор декантируют. Водную фазу получают бесцветной и органическую фазу получают почти бесцветной. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат с безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают белое твердое вещество (1.48 г; выход 96.7%).
Тонкослойная хроматография (хлороформ/метанол 15/2) показала, что продукт является очень чистым (один цвет); Rf 0.51.
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 19
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (20 мл), муравьиную кислоту (4 мл; 98%), трифторуксусную кислоту (1.7 мл; 0.022 моль) и s-триазин (0.6 г; 0.0074 моль). Получают оранжевый раствор (30oC). Реакционную смесь выдерживают в течение 1 ч при температуре (30oC). Тонкослойная хроматография показывает завершение реакции. Добавляют раствор хлорида натрия (3 г) в воде (10 мл); pH 0.27. Добавляют 33%-ный раствор гидроксида натрия до pH 8, и затем раствор декантируют. Водную фазу получают бесцветной, и органическую фазу получают желтой. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат над безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают бежевое твердое вещество (1.51 г; выход 98.7%).
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 20
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (40 мл), муравьиную кислоту (4 мл; 98%) и трифторуксусную кислоту (1.7 мл; 0.022 моль), и реакционную смесь охлаждают до -5oC. Затем добавляют s-триазин (0.6 г; 0.0074 моль), и реакционную смесь выдерживают при температуре от -5 до -10oC в течение 1 ч. Образуются две фазы, что означает, что реакция проходит в гетерогенной фазе. Тонкослойная хроматография показывает завершение реакции. Затем температуру реакционной смеси повышают до 15-20oC и добавляют раствор хлорида натрия (3 г) в воде (10 мл); pH 0.01. Добавляют 33%-ный раствор гидроксида натрия до pH 8.15, и затем раствор декантируют. Водную фазу получают бесцветной, и органическую фазу получают бледно-желтой. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат над безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают твердое вещество (1.49 г; выход 97.4%).
Тонкослойная хроматография (хлороформ/метанол 15/2) показала, что продукт является очень чистым (один цвет).
Данные спектроскопии (ИК, 1H ЯМР, масс-спектр) соответствуют целевому соединению.
Пример 21
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют ацетонитрил (20 мл), муравьиную кислоту (4 мл; 98%) и трифторуксусную кислоту (1.7 мл; 0.022 моль), и реакционную смесь охлаждают до -5oC. Затем добавляют s-триазин (0.6 г; 0.0074 моль) и получают желтый раствор. Систему выдерживают при температуре от -5 до -10oC в течение 1 ч, и затем температуру реакционной смеси повышают до 15-20oC и добавляют метиленхлорид (20 мл) и раствор хлорида натрия (3 г) в воде (10 мл); pH 1.22. Добавляют 33%-ный раствор гидроксида натрия до pH 8.35, и затем раствор декантируют. Водную фазу получают бесцветной, и органическую фазу получают желтой. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат над безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают бежевый продукт (1.53 г; выход 100%).
Тонкослойная хроматография (хлороформ/метанол 15/2) показала, что продукт является очень чистым (одно пятно).
Пример 22
Синтез флуконазола.
К соединению гидразина (IV) из примера 1 (1.35 г; 0.005 моль) добавляют метиленхлорид (40 мл), муравьиную кислоту (4 мл; 98%) и трифторуксусную кислоту (1.7 мл; 0.022 моль), и реакционную смесь охлаждают до -5oC. Получают две фазы. Затем добавляют s-триазин (0.6 г; 0.0074 моль). Получают желтую эмульсию (две фазы), что означает, что реакция проходит в гетерогенной фазе. Реакционную смесь выдерживают при температуре от -5 до -10oC в течение 1 ч, затем температуру реакционной смеси повышают до 15-20oC и добавляют раствор хлорида натрия (3 г) в воде (10 мл); pH 0.57. Добавляют 33%-ный раствор гидроксида натрия до pH 8.29, и затем раствор декантируют. Водную фазу получают бесцветной и органическую фазу получают почти бесцветной. Водную фазу промывают метиленхлоридом (10 мл) и затем снова метиленхлоридом (2х5 мл). Органические фазы объединяют, сушат над безводным сульфатом натрия, фильтруют, промывают метиленхлоридом и выпаривают на роторном испарителе в вакууме (температура бани 40oC). Получают бежевый продукт (1.48 г; выход 96.7%).
Тонкослойная хроматография (хлороформ/метанол 15/2) показала, что продукт является очень чистым (одно пятно).
Пример 23
Синтез флуконазола.
К ацетонитрилу (12.5 мл) и эпоксиду (1.67 г; 0.005 моль) формулы (II) в виде метансульфоната добавляют гидразингидрат (0.61 мл, 0.0125 моль; 99%). Реакционную смесь получают состоящей из трех фаз: две бесцветные фазы и фаза суспензии с белым твердым веществом. Реакционную смесь нагревают до температуры кипения, получают только двухфазную систему из двух бесцветных фаз. Реакционную смесь кипятят с обратным холодильником в течение 3 ч, затем при помощи тонкослойной хроматографии (хлороформ/метанол 15/2) устанавливают завершение реакции.
Реакционную смесь охлаждают до комнатной температуры и затем добавляют метансульфокислоту (0.2 мл), образовавшийся метансульфонат гидразина фильтруют и затем промывают ацетонитрилом (30 мл). Ацетонитрил выпаривают на роторном испарителе, и к неочищенному продукту добавляют 98%-ную муравьиную кислоту (5 мл) и метиленхлорид (5 мл). Систему охлаждают до температуры от 0 до -5oC и добавляют s-триазин (0.6 г; полученный по методике, описанной у W. Kantlehner et al. Synthesis, 1979, 690) с получением желто-оранжевого раствора. Систему выдерживают в течение 1 ч при температуре от 0 до -5oC и затем добавляют раствор хлорида натрия (3 г) в воде (10 мл) а также метиленхлорид (15 мл). Добавляют 33%-ный раствор гидроксида натрия для достижения уровня pH 8.12 (приблизительно 12 мл 33%-ного гидроксида натрия) и декантируют. Водную фазу промывают один раз 10 мл метиленхлорида, затем 5 мл метиленхлорида и еще раз 5 мл метиленхлорида. Органическую фазу сушат над безводным сульфатом натрия, фильтруют, промывают и выпаривают на роторном испарителе в течение 2 ч при температуре бани 80oC. Выход 1.41 г (92%).
Пример 24
Кристаллизация флуконазола.
Вариант 1
Неочищенный флуконазол (2.5 г) смешивают с изопропанолом (20 мл) и получают суспензию. Реакционную среду нагревают до температуры 55oC до растворения нерастворенных частиц, и получают раствор розового цвета. Добавляют активированный уголь (0.5 г), и реакционную смесь нагревают в течение 20 мин при температуре 55-65oC. Твердые частицы отфильтровывают и промывают изопропанолом. Фильтрат концентрируют при пониженном давлении до объема 5 мл и охлаждают в течение 12 ч до температуры 0-5oC. Полученный продукт фильтруют и промывают изопропанолом, охлажденным до -11oC. Получают снежно-белый осадок (1.89 г). Выход 80%.
Температура плавления перекристаллизованного продукта 135-137oC.
Данные спектроскопии (ИК, 1F ЯМР, УФ, масс-спектр) соответствуют целевому соединению.
Маточную жидкость и изопропанол, использованный для промывания, используют для дальнейшей перекристаллизации.
К флуконазолу (2.22 г) добавляют растворитель (20 мл: 7 мл маточной жидкости из предыдущей перекристаллизации и 13 мл изопропанола), и смесь нагревают до 55oC. Добавляют активированный уголь (0.5 г), и реакционную смесь перемешивают в течение 20 мин при температуре 55-60oC, промывают изопропанолом и концентрируют до 5 мл изопропанола. Ее охлаждают до -5oC в течение 12 ч, и флуконазол отфильтровывают. Его промывают охлажденным изопропанолом (3х2.5 мл). Выделяют снежно-белый продукт (1.90 г). Выход 90%.
Температура плавления перекристаллизованного продукта 135- 138oC.
Данные спектроскопии (ИК, 1F ЯМР, УФ, масс-спектр) соответствуют целевому соединению.
К маточной жидкости добавляют воду (30 мл) и метиленхлорид (15 мл). Водную фазу декантируют и экстрагируют метиленхлоридом (2х10 мл). Органические фазы объединяют и сушат над безводным сульфатом натрия, фильтруют, и растворитель выпаривают при пониженном давлении. Выделяют оранжевый продукт (0.64 г). Добавляют изопропанол (0.5 мл), и раствор охлаждают до -5oC. Полученный продукт фильтруют отсасыванием.
Вариант 2
Неочищенный флуконазол (2.5 г) смешивают с этилацетатом (20 мл) и нагревают до температуры кипения, добавляют активированный уголь (0.3 г), и нагревание продолжают с обратным холодильником в течение еще 10 мин. Смесь фильтруют, и фильтрат концентрируют до объема 5 мл. Реакционную смесь охлаждают до температуры 0oC, и полученный продукт фильтруют отсасыванием. Получают белый продукт с температурой плавления 136-138oC (2,25 г; 90%). Маточная жидкость может быть использована для дальнейшей перекристаллизации.
Пример 25
Синтез метансульфоната флуконазола.
Флуконазол (1 г; 0.00327 моль) смешивают с метил-трет-бутиловым эфиром (10 мл), и получают белую суспензию. К реакционной смеси по каплям добавляют метансульфокислоту (0.21 мл; 0.00327 моль). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, фильтруют, и твердый осадок промывают метил-трет-бутиловым эфиром (2х5 мл). Выделяют белый осадок (1.30 г).
Температура плавления 77-80oC.
Данные спектроскопии (ИК, 1H ЯМР, УФ, масс-спектр) соответствуют целевому соединению.
Описывается способ получения 2-[2,4-дифторфенил] -1,3-бис[1Н-1,2,4-триазол-1-ил] -пропан-2-ола (флуконазола) и его фармацевтически приемлемых солей, заключающийся в том, что соединение формулы IV, где R является водородом и Z - триазолом, подвергают взаимодействию с s-триазином. Флуконазол является противогрибковым агентом. Настоящий способ позволяет предотвратить образование нежелательных побочных продуктов и повысить выход. 1 з.п. ф-лы, 4 ил. 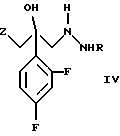
и его фармацевтически приемлемых солей, отличающийся тем, что соединение формулы IV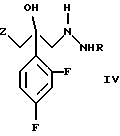
где R является водородом и Z является триазолом, необязательно в виде соли, подвергают взаимодействию с s-триазином формулы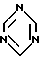
в органическом растворителе в качестве реакционной среды в кислых условиях реакции и при температуре от -60°С до температуры флегмы реакционной смеси.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Способ получения 2-(2,4-дифторфенил)-1,3-бис(1 @ -1,2,4-триазол-1-ил)-пропан-2-ола или его фармацевтически допустимой соли | 1982 |
|
SU1306474A3 |

