Изобретение относится к производству полиоксиалкиленгликолей, в частности к способу получения политетрагидрофурана или его сложных моноэфиров монокарбоновых кислот с 1-10 атомами углерода.
Известен способ получения политетрагидрофурана или его сложных моноэфиров монокарбоновых кислот полимеризацией тетрагидрофурана в присутствии катализатора, содержащего каталитически активное количество кислородсодержащего соединения вольфрама и/или молибдена, нанесенного на оксидный носитель, и телогена, в качестве которого можно использовать монокарбоновую кислоту (см. заявку EP N 0503394 Ф2, кл. C 08 G 65/20, 16.09.1992 г.).
Недостатком известного способа является неудовлетворительный выход на единицу объема/времени.
Задачей изобретения является разработка способа, позволяющего получать политетрагидрофуран или его сложного моноэфира с высоким выходом на единицу объема/времени, то есть, с высокой избирательностью при высокой конверсии тетрагидрофурана.
Поставленная задача решается в способе получения политетрагидрофурана или его сложных моноэфиров монокарбоновых кислот с 1-10 атомами углерода полимеризацией тетрагидрофурана в присутствии катализатора, содержащего каталитически активное количество кислородсодержащего соединения вольфрама и/или молибдена, нанесенного на оксидный носитель, и телогена, выбранного из группы, включающей воду, 1,4-бутандиол, политетрагидрофуран с молекулярным весом 200-700 Дальтон, монокарбоновую кислоту с 1-10 атомами углерода, или их смеси, за счет того, что в качестве указанного катализатора используют катализатор, полученный путем нанесения предшественников каталитически активных, кислородсодержащих соединений вольфрама и/или молибдена на гидроксидные предшественники оксидных носителей с последующей сушкой и кальцинированием при температуре 500-1000oC. Предпочтительные признаки предлагаемого способа, изложенные в зависимых пунктах формулы изобретения, будут подробно описаны в следующем.
В качестве оксидных носителей пригодны, например, двуокись циркония, двуокись титана, окись гафния, окись иттрия, окись железа (III), окись алюминия, окись олова (IV), двуокись кремния, окись цинка или смеси данных окисей. Особенно предпочтительными являются двуокись циркония и/или двуокись титана.
Применяемые согласно изобретению катализаторы в общем содержат 0,1 - 50 вес. %, предпочтительно 1 - 30 вес.%, особенно предпочтительно 5 - 20 вес.% каталитически активных, кислородсодержащих соединений молибдена или вольфрама или смеси данных соединений, в пересчете на общий вес катализатора, причем, так как точная химическая структура каталитически активных, кислородсодержащих соединений молибдена и/или вольфрама до сих пор неизвестна, а ее можно лишь догадать, например, на основе данных инфракрасных спектров используемых согласно изобретению катализаторов, в основу расчета берут трехокись молибдена и, соответственно, вольфрама.
Кроме каталитически активных, кислородсодержащих соедниений молибдена и/или вольфрама предлагаемые катализаторы могут содержать еще 0,05 - 10 вес. %, предпочтительно 0,1 - 5 вес.%, в частности 0,25 - 3 вес.% кислородсодержащих соединений серы и/или фосфора, в пересчете на общий вес катализатора. Так как тоже неизвестно, какую химическую структуру имеют данные серо- или фосфорсодержащие соединения в готовом катализаторе, при расчете содержания данных групп в катализаторе исходят из SO4 соответственно PO4.
При получении предлагаемых катализаторов в общем исходят из гидроокисей соответствующих носителей. В том случае, если данные гидроокиси можно приобрести в торговле, их можно использовать в качестве исходных соединений для получения оксидных носителей. Предпочтительно, однако, для получения оксидных носителей используют свежеосажденные гидроокиси, которые после осаждения обычно сушат при 20 - 350oC, предпочтительно при 50 - 150oC, в частности 100 - 120oC, при атмосферном или пониженном давлении.
В качестве исходных соединений для получения данных гидроокисей в общем служат растворимые в воде или поддающиеся гидролизу соли образующих носитель элементов, например, их галогениды, предпочтительно их нитраты или карбоксилаты, в частности, их ацетаты. Пригодными исходными соединениями для осаждения данных гидроокисей являются, например, хлорид или нитрат цирконила или титанила, нитрат или ацетат иттрия или алюминия, нитрат железа (III), галогениды олова (IV), в частности, хлорид олова (IV), нитрат цинка или ацетат цинка. Из растворов данных солей осаждают соответствующие гидроокиси, предпочтительно с использованием водного раствора аммиака. Или же гидроокиси можно получать путем добавления разбавленных или слабых кислот, например, уксусной кислоты, к растворимым в воде гидроксокомплексам соответствующих металлов, до осаждения соответствующей гидроокиси. Другая возможность заключается в получении гидроокисей путем гидролиза органометаллических соединений, например, алкоголятов соответствующих металлов, как, например, тетраэтанолата циркония, тетраизопропилата циркония, тетраметанолата титана, тетраизопропилата титана и т.д.
В общем при осаждении этих гидроокисей образуется гелеобразный осадок, из которого путем сушки получают рентгеноаморфный порошок. Возможно эти рентгеноаморфные осадки состоят, кроме гидроокисей соответствующих металлов, из множества других содержащих гидрокси-группы соединений, например, гидратов окисей, полимерных, не растворимых в воде гидроксокомплексов и т.д. Однако, так как точный химический состав данных осадков нельзя определить, в рамках настоящей заявки во имя простоты исходят из того, что тут речь идет о гидроокисях указанных металлов. Таким образом, в рамках настоящей заявки понятие "гидроокиси" представляет собой общее понятие для всех содержащих гидрокси-группы осадков, полученных в результате вышеупомянутых методов осаждения.
При использовании двуокиси кремния в качестве оксидного носителя для получения используемых согласно изобретению катализаторов предпочтительно берут свежеосажденную кремневую кислоту, которую можно получать, например, путем подкисления раствора жидкого стекла и которую целесообразно сушат перед переработкой описанным выше для гидроокисных осадков методом.
На полученные описанным методом образующие носитель гидроокиси соответственно кремневую кислоту, которые в данной заявке можно также назвать предшественниками носителя, наносят предшественники каталитически активных, кислородсодержащих соединений молибдена и/или вольфрама, предпочтительно путем пропитки носителя водным раствором этих предшественников.
В качестве растворимых в воде предшественников каталитически активных, кислородсодержащих соединений молибдена и/или вольфрама можно назвать, например, растворимые в воде соли вольфрамовой кислоты (H2WO4), образующиеся, например, в результате растворения трехокиси вольфрама в водном аммиаке, то есть, моновольфраматы, и образующиеся из них при подкислении изополивольфраматы, например, паравольфраматы или метавольфраматы, водорастворимые соли молибденовой кислоты (H2MoO4), образующиеся при растворении трехокиси молибдена в водном аммиаке, и образующиеся из них при подкислении изополимолибдаты, в частности, метамолибдаты и парамолибдаты. Предпочтительно в качестве предшественников соединений молибдена и/или вольфрама используют аммониевые соли данных вольфрамовых и молибденовых кислот, которые путем пропитки наносят на служащие в качестве носителей гидроокиси соответственно кремневую кислоту. Насчет номенклатуры, состава и получения молибдатов, изополимолибдатов, вольфраматов и изополивольфраматов указывается на Rompps Chemie Lexikon, 8-е издание, том 4, стр. 2659 - 2660, издательство Франкше Фергалсбуххандлунг, г. Штуттгарт, DE, 1985 г., Rompps Chemie Lexikon, 8-е издание, том 6, стр. 4641 - 4644, г. Штуттгарт, DE, 1988 г., и Comprehensive Inorganic Chemistry, 1-е издание, том 3, стр. 738 - 741 и 766 - 768, издательство Перганон Пресс, Нью-Йорк, 1973 г. Вместо вышеуказанных предшественников каталитически активных соединений молибдена и вольфрама для нанесения молибдена или вольфрама на гидрокисный, то есть, содержащий гидрокси-группы предшественник носителя, можно также использовать гетерополикислоты молибдена или вольфрама, как, например, 12-вольфраматокремневую кислоту (H4[Si{W12O40}]·26H2O) или 12-молибдатокремневую кислоту, или их водорастворимые соли, предпочтительно их аммониевые соли. Пропитанные таким образом гидроокиси, соответственно пропитанную кремневую кислоту сушат в общем при 80 - 350oC, предпочтительно 90 - 150oC, при атмосферном или пониженном давлении.
Также возможно получить катализатор путем тщательного смешивания указанных предшественников каталитически активных, кислородсодержащих соединений молибдена или вольфрама с одной гидроокисью или некоторыми гидроокисями. Кальцинирование предшественника носителя, таким образом снабженного соответствующими соединениями, до используемого согласно изобретению катализатора осуществляют тем же образом, как в случае пропитанных соединениями-предшественниками предшественников носителей. Предпочтительно, однако, для получения используемых согласно изобретению катализаторов используют метод пропитки.
Пропитанные и высушенные таким образом предшественники катализатора преобразуют в готовые катализаторы путем кальцинирования на воздухе при 500 - 1000oC, предпочтительно 550 - 900oC, особенно предпочтительно 600 - 800oC. При кальцинировании гидроокиси носителей соответственно кремневая кислота преобразуется в оксидный носитель, а предшественники каталитически активных, кислородсодержащих соединений молибдена или вольфрама, нанесенных на носитель путем пропитки, преобразуются в указанные каталитически активные соединения. Кальцинирование при указанных высоких температурах является решающим фактором для достижения высокой конверсии и, тем самым, высокого выхода на единицу объема/времени при осуществлении полимеризации тетрагидрофурана. При более низких температурах кальцинирования катализаторы также поощряют полимеризацию тетрагидрофурана, однако, с низкой конверсией. Исходя из инфракрасных исследований получаемых таким образом катализаторов Yinyan и др. (см. Rare Metals 11, 185, 1992 г.), полагают, что в случае нанесенных на носитель катализаторов на основе окиси циркония, дотированных вольфрамом, предшественник каталитически активного, кислородсодержащего соединения вольфрама, путем пропитки нанесенного на гидроокись циркония, при применяемых высоких температурах кальцинирования образует химическое соединение с гидрокси-группами предшественника носителя, причем образуется каталитически активное, кислородсодержащее соединение вольфрама, в отношении химической структуры и химической активности, в частности, что касается каталитических свойств, четко отличается от кислородсодержащих соединений вольфрама, лишь адсорбирванных к служащей в качестве носителя двуокиси циркония. Полагают, что то же самое верно в отношении молибденсодержащих нанесенных на носитель катализаторов, применяемых согласно изобретению.
Как выше указывалось, при осуществлении предлагаемого способа можно предпочтительно применять нанесенные на носитель катализаторы, кроме молибдена и/или вольфрама дополнительно включающие соединения, содержащие серу или фосфор, или серу и фосфор. Данные катализаторы получают путем метода, аналогичного методу, описанному выше в связи с катализаторами, содержащими лишь соединения молибдена и/или вольфрама, причем на служащие в качестве носителя гидроокиси, полученные аналогичным методом, соответственно на кремневую кислоту, путем пропитки дополнительно наносят соединения, содержащие серу и/или фосфор. Нанесение соединений серы или фосфора на носитель можно осуществлять или одновременно с нанесением соединений молибдена и/или вольфрама, или после него. Целесообразно соединения, содержащие серу и/или фосфор, получают путем пропитки служащих в качестве носителей гидроокисей соответственно кремневой кислоты водным раствором соединения, содержащего сульфатные и фосфатные группы, например, серную кислоту или фосфорную кислоту. Кроме того, предпочтительным является использование для пропитки растворов растворимых в воде сульфатов или фосфатов, причем особенно предпочитаются сульфаты, соответственно фосфаты аммония. Дальнейший метод нанесения фосфорсодержащих соединений на предшественник носителя одновременно с соединениями, содержащими молибден или вольфрам, заключается в снабжении служащих в качестве предшественника носителя гидроокиси фосфорсодержащими гетерополикислотами, причем используют вышеописанный метод. В качестве примеров таких гетерополикислот можно назвать 12-вольфраматофосфорную кислоту (H3[P{ W12O4O} ·xH2O) и 12-молибдатофосфорную кислоту (H7(P{Mo2O}O6·28H2O). Для этой цели можно также использовать гетерополикислоты молибдена или вольфрама с органическими кислотами фосфора, например, фосфоновые кислоты. Указанные гетерополикислоты можно также использовать в виде солей, предпочтительно аммониевой соли.
При кальцинировании при вышеуказанных температурах гетерополикислоты разлагаются до каталитически активных, кислородсодержащих соединений молибдена или вольфрама.
Используемые согласно изобретению катализаторы отчасти известны, и их получение описано в заявках JP N 288339/1989 и N 293375/1993, в J. Chem. Soc. Chem. Commun. 1259 (1988 г.), и в Rare Metals 11, 185 (1992 г.). До сих пор данные катализаторы использовали лишь в нефтехимических процессах, например, при алкилировании, изомеризации и крекинге углеводородов, то есть, в способах, не подобных предлагаемому способу.
Используемые согласно изобретению катализаторы могут иметься в виде порошка, например, в случае проведения предлагаемого способа с использованием суспензии, или, целесообразно, в виде формованных изделий, например, в виде цилиндров, шариков, колец, спиралей или гравия, в частности, в случае использования твердого слоя катализатора, что является предпочтительным, например, при использовании снабженного рециркуляционной линией реактора, или при непрерывном осуществлении процесса.
В качестве телогена для получения политетрагидрофурана в виде сложных моноэфиров монокарбоновых кислот в общем служат монокарбоновые кислоты с 1 - 10 атомами углерода, предпочтительно, с 1 - 8 атомами углерода, особенно предпочтительно муравьиная кислота, уксусная кислота, пропионовая кислота, 2-этилгексановая кислота, акриловая кислота и метакриловая кислота.
Особенно неожиданным и особенно выгодным является тот факт, что при использовании воды и/или 1,4-бутандиола в качестве телогенов политетрагидрофуран можно получать согласно изобретению на одной стадии, причем с большим выходом на единицу объема/времени. В случае необходимости низкомолекулярный политетрагидрофуран с отрытой цепью, имеющий молекулярный вес 200 - 700 Дальтон, в качестве телогена можно рециркулировать на стадию полимеризации, где он превращается в политетрагидрофуран с большим молекулярным весом. Так как 1,4-бутандиол и низкомолекулярный политетрагидрофуран имеют две гидроксильные группы, они встроятся не только в качестве телогена на концах цепи политетрагидофурана, а также в качестве мономера в цепь политетрагидрофурана.
Целесообразно телоген подают на полимеризацию в растворенном в тетрагидрофуране виде. Так как телоген приводит к прекращению полимеризации, путем выбора соответствующего количества телогена можно управлять средним молекулярным весом политетрагидрофурана или политетрагидрофурана в виде сложного диэфира. Чем больше содержание телогена в реакционной смеси, тем ниже средний молекулярный вес политетрагидрофурана или его соответствующего производного. В зависимости от содержания телогена в реакционной смеси полимеризации с помощью предлагаемого способа можно целенаправленно получить политетрагидрофуран или его производные со средним молекулярным весом, лежащим между 250 и 10000. Предпочтительно с помощью предлагаемого способа получают политетрагидрофуран, соответственно его производные со средним молекулярным весом 500 - 10000 Дальтон, особенно предпочтительно - 650 - 3000 Дальтон. Для этого телоген добавляют в количестве 0,04 - 17 моль-%, предпочтительно 0,2 - 8 моль-%, особенно предпочтительно 0,4 - 4 моль-% в пересчете на тетрагидрофуран.
Полимеризацию в общем осуществляют при 0 - 80oC, предпочтительно от 25oC до температуры кипения тетрагидрофурана. Обычно давление не является критическим для результата полимеризации, и поэтому реакцию в общем осуществляют при атмосферном давлении или под устанавливающимся при полимеризации давлением.
Для предотвращения образования перекисей эфиров полимеризацию осуществляют предпочтительно в атмосфере инертного газа, например, азота, водорода, двуокиси углерода или благородного газа, причем предпочтительным является азот.
Предлагаемый способ можно осуществлять периодически или непрерывно, причем по причинам экономичности обычно предпочитают непрерывное осуществление.
При периодическом осуществлении предлагаемого способа реагенты, то есть, тетрагидрофуран, соответствующий телоген и катализатор, обычно подвергают взаимодействию при вышеуказанной температуре в снабженном мешалкой котле или в снабженном рециркуляционной линией реакторе до достижения желаемой конверсии тетрагидрофурана. В зависимости от количества катализатора время реакции может составлять 0,5 - 40 ч, предпочтительно 1 - 30 ч. Катализаторы добавляют к стадии полимеризации обычно в количестве 1 - 90%, предпочтительно 4 - 70%, особенно предпочтительно 8 - 60% от веса используемого тетрагидрофурана.
Для переработки продукт реакции в случае периодического осуществления реакции отделяют от суспендированного в нем катализатора, предпочтительно путем фильтрации, декантирования или центрифугирования.
Освобожденный от катализатора продукт полимеризации обычно подвергают перегонке, причем на первой стадии целесообразно отделяют непрореагировавшийся тетрагидрофуран. На второй стадии очистки при желании можно путем перегонки при пониженном давлении отделять низкомолекулярный политетрагидрофуран от конечного продукта полимеризации. Или же летучие олигомеры тетрагидрофурана можно деполимеризовать, например, путем описанного в заявке DE N 3042960 способа, после чего их рециркулируют на реакцию.
Примеры
Получение катализаторов:
Катализатор А получают путем добавления 2600 г гидроокиси циркония к раствору 640 г вольфрамовой кислоты (H2WO4) к 3470 г 25%-ного раствора аммиака. Полученную смесь перемешивают в течение 30 мин, затем сушат при 120oC в течение 2 ч. Пропускают через сито, полученный порошок прессуют в таблетки величиной 3 х 3 мм, которые подвергают кальцинированию при 450oC в течение 2 ч. Содержание вольфрама в полученном катализаторе составляет 20% от его общего веса, рассчитано как трехокись вольфрама.
Катализаторы Б и В получают аналогично вышеописанному методу, причем, однако, катализатор Б подвергают кальцинированию при 600oC, а катализатор В - при 700oC.
Катализатор Г получают путем добавления 1600 г гидроокиси циркония к раствору 425 г вольфрамовой кислоты и 200 г сульфата аммония в 3470 г 25%-ного раствора аммиака. Полученную смесь перемешивают в течение 30 минут, затем сушат при 120oC в течение 2 ч. Пропускают через сито, полученный порошок прессуют в таблетки, которые подвергают кальцинированию при 600oC в течение 2 ч. Содержание вольфрама в полученном катализаторе составляет 18% от его общего веса, рассчитано как трехокись вольфрама, а его содержание серы составляет 7% от общего веса катализатора, рассчитано как SO4.
Катализатор Д получают путем добавления 2600 г гидроокиси циркония к 2260 г 26,5 вес.%-ного раствора трехокиси молибдена в 12%-ном водном аммиаке. Полученную смесь перемешивают в течение получаса, затем сушат при 120oC в течение 16 ч. К высушенной массе добавляют 40 г 75%-ной фосфорной кислоты и 1,4 л воды, и размешивают в течение 30 мин, после чего сушат при 120oC в течение 2 ч. Пропускают через сито, и полученный порошок прессуют в таблетки, которые подвергают кальцинированию при 600oC в течение 2 ч. Полученный катализатор имеет содержание молибдена, рассчитанное как трехокись молибдена, составляющее 20% от общего веса катализатора, и содержание фосфора, рассчитанное как PO4, составляющее 1% от общего веса катализатора.
Катализатор Е получают аналогично катализатору А, с той разницей, что кальцинирование осуществляют при 675oC.
Катализатор Ж получают путем добавления 75 г двуокиси титана (P25, продукт фирмы Дегусса) к раствору 20 г вольфрамовой кислоты (H2WO4) в 100 г 32%-ного водного раствора аммиака. Полученную смесь перемешивают в течение 2 ч, затем сушат при 120oC в течение 12 ч. Измельчают в порошок, который подвергают кальцинированию при 620oC в течение 2 ч. Полученный порошковый катализатор имеет содержание вольфрама, рассчитанного как трехокись вольфрама, составляющее 20% от общего веса катализатора.
Для получения катализатора 3 раствор 16,58 кг TiOCl2 в 43,5 л дистиллированной воды объединяют с 45 кг 10%-ного водного раствора аммиака тем, что оба раствора при 50oC при размешивании и при неизменяющемся pH 6,5, в течение 1 ч добавляют к 30 л дистиллированной воды. После этого дополнительно размешивают в течение часа. Образовавшийся осадок отделяют путем фильтрации, промывают и сушат при 120oC в течение 24 ч. 2,4 кг данной осажденной двуокиси титана добавляют к раствору 638 г вольфрамовой кислоты в 3,65 кг 25%-ного водного раствора аммиака. Полученную смесь размешивают, после чего из нее получают жгуты диаметром 1,5 мм, которые подвергают кальцинированию сперва при 450oC в течение 2 ч, а затем при 610oC в течение дальнейших 2 ч. Содержание вольфрама, рассчитанное как трехокись вольфрама, в полученном катализаторе составляет 18,6% от общего веса катализатора.
Периодическая полимеризация тетрагидрофурана:
10 г катализатора, перед использованием подверженного сушке при 180oC и давлении 0,3 мбар в течение 18 ч для удаления адсорбированной воды, в стеклянной колбе емкостью 100 мл, снабженной приспособлением для охлаждения обратного потока, в атмосфере азота суспендируют в 20 г содержащего телоген тетрагидрофурана, и оставляют стоять при 50oC в течение 24 ч. Затем реакционную смесь разбавляют путем добавления еще 20 г тетрагидрофурана. Катализатор отделяют путем фильтрации и трижды промывают, каждый раз используя 20 г тетрагидрофурана. Фильтраты объединяют, сгущают в ротационном испарителе при температуре 70oC и давлении 20 мбар и взвешивают. Для определения среднего молекулярного веса (Mn) часть полученного политетрагидрофурана подвергают перегонке в трубе с шаровым расширением при 150oC и 0,1 мбар.
Средний молекулярный вес (Mn) полученного таким образом политетрагидрофурана определяют путем гель-проникающей хроматографии. Mn определен следующим уравнением: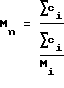
в котором ci - концентрация отдельного типа полимера i в полученной смеси полимеров, а Mi - молекулярный вес отдельного типа полимера i.
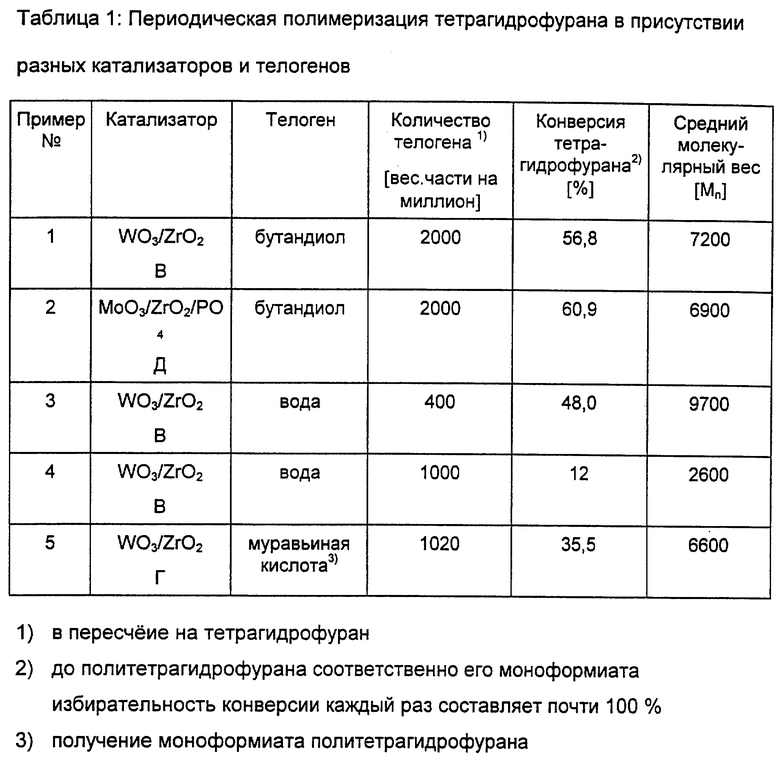
В таблице 1 показан выход политетрагидрофурана при периодической полимеризации в присутствии разных катализаторов и телогенов и их средний молекулярный вес Mn.
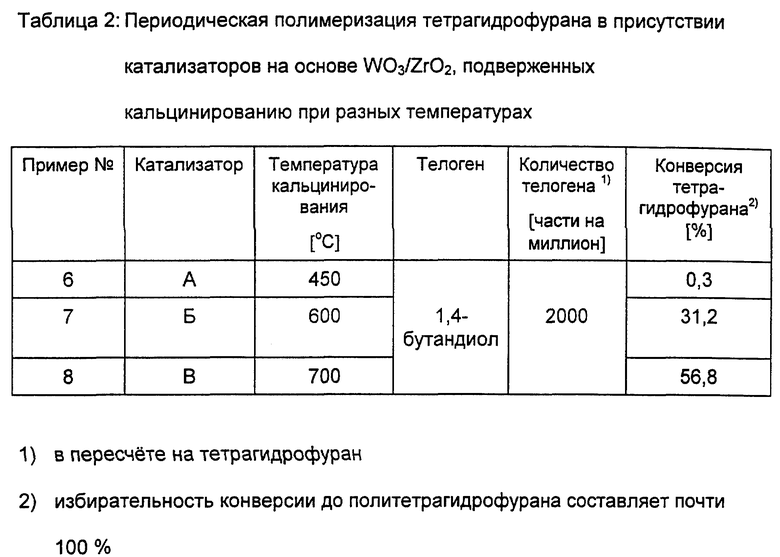
В таблице 2 представлены результаты периодической полимеризации тетрагидрофурана до политетрагидрофурана в присутствии катализаторов, подверженных кальцинированию при разных температурах, при в остальном одинаковых условиях.
Непрерывная полимеризация тетрагидрофурана
Пример 9
В реактор емкостью 250 мл в атмосфере аргона подают 220 мл (372 г) катализатора на основе MoO3/ZrO2/PO43- (катализатора Д), предварительно высушенного при 180oC и 0,3 мбар в течение 20 ч. При подаче в реактор катализатор покрывают тетрагидрофураном, содержащим меньше 0,01 вес.% воды. Для перекачивания реакционной смеси служит циркуляционный насос. После полного наполнения реактора, насоса и линий тетрагидрофураном реакционную смесь при 50oC в течение 24 ч перекачивают по неподвижному слою катализатора, больше не добавляя тетрагидрофурана. После этого в течение 120 ч в установку вводят содержащий 0,2 вес.% 1,4-бутандиола тетрагидрофуран в количестве 8,1 г в час. Соотношение количества циркулируемой смеси к добавляемому реагенту составляет примерно 60. Средний выход тетрагидрофурана за время реакции (120 ч) составляет 48,9%. Средний молекулярный вес Mn полученного политетрагидрофурана согласно данным гель-проникающей хроматографии составляет 2400 Дальтон.
Пример 10
Продолжают описанную в примере 9 непрерывную полимеризацию тетрагидрофурана в присутствии катализатора Д с непрерывным добавлением тетрагидрофурана в одинаковом количестве, однако, без рециркуляции продукта (то есть, насос отключен). Сохраняются остальные условия реакции. После стабилизации выхода политетрагидрофурана продукт реакции собирают в течение 72 ч. После испарения реакционной смеси, как описано для примеров с периодической полимеризацией, получают политетрагидрофуран со средним молекулярным весом 900 Дальтон (согласно данным гель-проникающей хроматографии) при конверсии тетрагидрофурана, составляющей 6,8%.
Периодическая полимеризация тетрагидрофурана
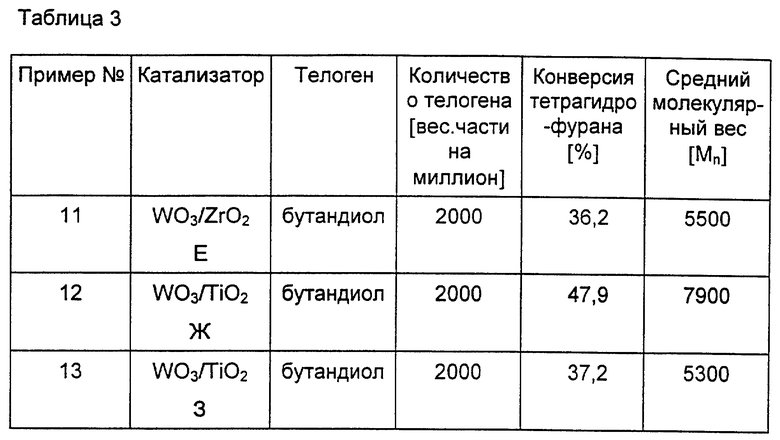
Периодическую полимеризацию в присутствии катализаторов Е - 3 осуществляют описанным выше в примере 1 или 2 методом. Результаты данных примеров приведены в нижеследующей таблице 3.
Непрерывная полимеризация тетрагидрофурана в присутствии бутан-1,4-диола
Пример 14
В реактор емкостью 250 мл в атмосфере аргона подают 332 г (250 мл) катализатора на основе WO3/ZrO2 (катализатора Е), предварительно высушенного при 180oC и при давлении 0,3 мбар в течение 72 ч. Установку полимеризации наполняют тетрагирдфураном, содержащим 0,5 вес.% бутан-1,4-диола. Реакционную смесь при 50oC в течение 24 ч перекачивают по неподвижному слою катализатора. Затем в установку непрерывно вводят 0,166 кг тетрагидрофурана на литр катализатора в час. От получаемого в течение 72 ч продукта полимеризации (3,00 кг) при пониженном давлении отгоняют непрореагировавшийся тетрагидрофуран, а затем продукт полимеризации подвергают перегонке при 150oC и 0,3 мбар. Получают 290 г политетрагидрофурана, средний молекулярный вес Mn которого согласно данным спектра 1H-ЯМР составляет 2100 Дальтон. Выход составляет 9,7%. Достигается конверсия на единицу объема/времени, составляющая 16 г политетрагидрофурана на 2100 л катализатора в час.
Пример 15
Повторяют пример 14 с той лишь разницей, что вместо бутан-1,4-диола полимеризацию проводят в присутствии 2,5 вес.% уксусной кислоты. Получают политетрагидрофуран в виде моноацетата со средним молекулярным весом 2300 Дальтон.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1,4-БУТАНДИОЛА | 1994 |
|
RU2119905C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,4-БУТАНДИОЛА | 1995 |
|
RU2147298C1 |
| СПОСОБ ПОЛУЧЕНИЯ γ-БУТИРОЛАКТОНА | 1996 |
|
RU2138491C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ЛАКТАМОВ | 1994 |
|
RU2120437C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОВ ИЗ ГЛИЦЕРИНА | 2008 |
|
RU2480449C2 |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛИТЕЛЬНОГО АММОНОЛИЗА ЭТИЛЕННЕНАСЫЩЕННЫХ СОЕДИНЕНИЙ НА ОСНОВЕ ОКИСЛОВ МЕТАЛЛОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ОКИСЛИТЕЛЬНОГО АММОНОЛИЗА ЭТИЛЕННЕНАСЫЩЕННЫХ СОЕДИНЕНИЙ | 1994 |
|
RU2136364C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКРИЛОВОЙ КИСЛОТЫ ГЕТЕРОГЕННО КАТАЛИЗИРУЕМЫМ ПАРЦИАЛЬНЫМ ОКИСЛЕНИЕМ ПРОПАНА | 2002 |
|
RU2308446C2 |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ В ГАЗОВОЙ ФАЗЕ С ОПРЕДЕЛЕННЫМ РАСПРЕДЕЛЕНИЕМ ПО РАЗМЕРАМ ЧАСТИЦ ОКСИДА ВАНАДИЯ | 2004 |
|
RU2314867C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ АЛЬФА, ОМЕГА-АМИНОНИТРИЛОВ | 1995 |
|
RU2154630C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ЛАКТАМОВ | 1994 |
|
RU2119912C1 |
Описывается способ получения политетрагидрофурана или его сложных моноэфиров монокарбоновых кислот с 1-10 атомами углерода полимеризацией тетрагидрофурана в присутствии катализатора, содержащего каталитически активное количество кислородсодержащего соединения вольфрама и/или молибдена, нанесенного на оксидный носитель, и телогена, выбранного из группы, включающей воду, 1,4-бутандиол, политетрагидрофуран с молекулярным весом 200-700 Дальтон, монокарбоновую кислоту с 1-10 атомами углерода или их смеси, который заключается в том, что в качестве указанного катализатора используют катализатор, полученный путем нанесения предшественников каталитически активных кислородсодержащих соединений вольфрама и/или молибдена на гидроксидные предшественники оксидных носителей с последующей сушкой и кальцинированием при 500-1000°С. Технический результат - высокая избирательность при высокой конверсии тетрагидрофурана. 9 з.п. ф-лы, 3 табл.
| EP 503394 A2, 16.09.1992 | |||
| US 4658065 A, 14.04.1987 | |||
| Устройство для монтажа длинномерной конструкции | 1989 |
|
SU1754632A1 |
