Изобретение относится к способам получения α- гидроксикислот, которые можно использовать в качестве исходных материалов для синтеза различных фармацевтических средств и агрохимикатов и в качестве добавок к кормам и продуктам.
Общепринятым стандартным методом химического синтеза α- гидроксикислот является гидролиз α- гидроксинитрила минеральной кислотой, такой как серная кислота. В этом случае, однако, образуется более эквивалента соли минеральной кислоты, такой как кислый сульфат. Соль минеральной кислоты должна быть подвергнута переработке, что приводит к проблеме освобождения от большого количества отходов.
Известно также получение свободных α- гидроксикислот путем, по которому соль металла или аммониевую соль α- гидроксикислоты, которую получают биологическим способом, таким как ферментация смеси, содержащей сахар, микроорганизмом или гидролиз α- гидроксинитрила гидролазой, продуцируемой микроорганизмом, подвергают реакции с минеральной кислотой, такой как серная кислота, или обрабатывают ионообменной смолой. В любом способе образуется большое количество солей минеральных кислот, что вызывает ту же проблему, которая указана выше.
Известным способом, идущим без образования солей минеральных кислот, которые становятся отходами, является этерификация аммониевой соли α- гидроксикислоты спиртом (выложенный патент Японии N Hei 7-194387) с последующим гидролизом полученного эфира с кислотным катализатором. Этот способ невыгоден из-за использования спирта и кислотного катализатора как дополнительных добавок. Эти добавки должны быть выделены. Следовательно, не существует выгодного способа получения в промышленном масштабе.
Имеется другой химический синтетический способ: гидролиз α- гидроксинитрила неорганическим основанием, таким как гидроксид натрия. В этом случае для нейтрализации для получения целевой α- гидроксикислоты нужно использовать минеральную кислоту или тому подобный реагент. Когда гидролиз проводят таким образом, образуется эквивалент соли минеральной кислоты, что вызывает ту же проблему освобождения от отходов.
Кроме того, для получения соответствующих свободных кислот из аммониевых солей карбоновых кислот известны следующие способы: небольшое количество воды добавляют к аммониевой соли ненасыщенной жирной кислоты и смесь нагревают при общем кипячении с обратным холодильником при 80oC или выше в органических растворителях для выделения и удаления аммиака, чтобы получить ненасыщенную жирную кислоту (выложенный патент Великобритании N 967352); к 10 ~ 50% водному раствору (мет)акрилата аммония добавляют органический растворитель, который образует азеотропную смесь с водой, и получаемый раствор нагревают до 60 ≈ 100oC для отгонки воды в виде азеотропной смеси и одновременного удаления аммиака, чтобы получить свободную (мет)акриловую кислоту (выложенный патент Японии N Sho 54-115317); 10 ~ 80% водный раствор аммониевой соли кислотной аминокислоты нагревают при добавлении к нему воды для отгонки аммиака и воды и получения кислотной аминокислоты (выложенный патент Японии N Hei 7-330696).
В этих способах, в принципе, аммиак легко удаляется, если карбоновая кислота имеет высокую константу диссоциации. Их недостаток, однако, заключается в том, что степень диссоциации ионов аммония из аммониевых солей карбоновых кислот низка для сильных кислот с pKa < 4, таких как α- гидроксикислоты. Поэтому очень трудно удалить аммоний из сильных кислот. Для удаления большей части аммиака реакцию следует проводить в течение продолжительного периода времени или необходимо добавлять большое количество органических растворителей или большое количество воды. Авторы этого изобретения применяли указанные выше три способа для получения α- гидроксикислот и обнаружили, что они неподходящие в качестве промышленных способов, потому что в каждом из этих способов остается 50% или более аммониевой соли α- гидроксикислоты.
Целью этого изобретения является предоставление способов получения свободных α- гидроксикислот из их аммониевых солей с высоким выходом без создания проблемы освобождения от отходов.
Настоящим изобретением является способ получения α- гидроксикислот. Этот способ состоит из стадии 1: нагревания аммониевой соли α- гидроксикислоты, представленной общей формулой [1] , без растворителя или в органических растворителях и удаления образованных аммиака и воды; и стадии 2: последовательного добавления к остатку воды и нагревания получаемой смеси.
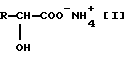 ,
,
где R представляет собой C1-C6-алкил, который может быть замещен C1-C6-алкилтиогруппой, или фенил.
Подробно настоящее изобретение описывается ниже.
α- Гидроксикислоты, которые являются целью этого изобретения, являются соединениями, представленными формулой [1]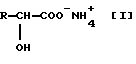 ,
,
где R представляет собой C1-C6-алкил, который может быть замещен C1-C6-алкилтиогруппой, или фенил.
Примерами C1-C6-алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и пентил. Примерами C1-C6-алкилтиогрупп, которыми могут быть замещены C1-C6-алкильные группы, являются метилтио, этилтио, пропилтио, изопропилтио и бутилтио.
Примеры α- гидроксикислот включают молочную кислоту, миндальную кислоту и α- гидрокси-4-метилтиомасляную кислоту.
Аммониевые соли α- гидроксикислот можно получить гидролизом α- гидроксинитрила гидролазой, продуцированной микроорганизмом.
Когда α- гидроксикислоту получают в виде соли металла таким способом, как гидролиз α- гидроксинитрила реакцией с неорганическим основанием или микроорганизмом, соль металла можно превратить в аммониевую соль, например, тем же способом, как способ, описанный в выложенном патенте Японии N Hei 7-194387: NH3 и CO2 добавляют к водному раствору соли металла α- гидроксикислоты для превращения в аммониевую соль α- гидроксикислоты. Полученную аммониевую соль можно использовать в способе этого изобретения.
Этим изобретением является способ, состоящий из следующих двух стадий:
i) Стадия 1
Аммониевую соль α- гидроксикислоты нагревают в органических растворителях для удаления воды и аммиака при превращении ее в поли-α-гидроксикислоты с низкой молекулярной массой.
ii) Стадия 2
После удаления большей части аммиака (после завершения стадии 1) к реакционному раствору добавляют воду для нагревания с целью гидролиза поли α- гидроксикислот с низкой молекулярной массой для получения свободной α- гидроксикислоты.
Стадией 1 является нагревание аммониевой соли α- гидроксикислоты для удаления воды и аммиака при превращении ее в поли-α-гидроксикислоты с низкой молекулярной массой. Чтобы выполнить стадию таким образом, эта реакционная система может быть при пониженном давлении.
В стадии 1 может быть применимо различное перегонное оборудование. Чтобы повысить площадь испарения, особенно выгодно оборудование с мешалкой или оборудование для образования жидкой пленки. Температура реакции обычно находится в диапазоне между 40 и 200oC, особенно предпочтительно в диапазоне между 60 и 170oC. Реакцию проводят при давлении между 0,1 и 760 мм Hg. Конечной точкой реакции является время, когда заканчивается отгонка воды и аммиака. После окончания реакции оставшийся аммиак можно рециркулировать в виде аммониевой соли α- гидроксикислоты. Следовательно, если нужно, реакцию можно останавливать до ее завершения. Когда целевая α- гидроксикислота восприимчива к окислению, воздух можно заменить на инертный газ, такой как азот, аргон или гелий, так чтобы повысить чистоту получаемой α- гидроксикислоты. Когда реакцию проводят при атмосферном давлении, введение инертного газа в реакционный раствор повышает эффективность удаления аммиака.
Органическими растворителями, пригодными в стадии 1, являются растворители, которые не реагируют с α- гидроксикислотами и аммиаком и имеют температуру кипения 40oC или выше. Предпочтительны растворители, образующие азеотропную смесь с водой. Примеры их включают бензол, толуол, ксилол, мезитилен, диэтиловый эфир этиленгликоля, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля, диизобутиловый эфир, ди-н-бутиловый эфир, анизол и декан.
В реакции стадии 1 в качестве побочного продукта может образоваться амид α- гидроксикислоты. Это происходит потому, что растворенный аммиак реагирует с поли-α-гидроксикислотами с низкой молекулярной массой с образованием амида. В этом случае степень образования побочного продукта можно регулировать ниже 2% посредством выбора подходящих органических растворителей, регулирования степени снижения давления или повышения площади испарения. Этот амид α- гидроксикислоты гидролизуют в аммониевую соль α- гидроксикислоты в стадии 2. Следовательно, степень образования побочного продукта далее снижается.
Аммиак, отогнанный в стадии 1, можно рециркулировать в виде газообразного аммиака, он имеет высокую ценность.
Стадией 2 является добавление воды к реакционному раствору для нагревания после окончания реакции стадии 1. Воду обычно добавляют в 0,1-10-кратном, предпочтительно 0,2-3-кратном количестве (частей по массе), от массы остатка после того, как стадия 1 завершилась. Температура реакции составляет 50 ~ 100oC, когда реакцию проводят при атмосферном давлении. Реакцию можно проводить под давлением. При использовании устойчивого к давлению реакционного сосуда можно проводить при 100 ~ 300oC, предпочтительно 120 ~ 170oC, так чтобы время реакции можно было сократить.
В процессе реакции в стадии 2 поли-α-гидроксикислоты с низкой молекулярной массой гидролизуют в α- гидроксикислоту. Амид α- гидроксикислоты, образуемый в стадии 1 в качестве побочного продукта, также частично гидролизуют в аммониевую соль α- гидроксикислоты.
После завершения реакции в стадии 2 большую часть α- гидроксикислоты можно получить в виде свободной кислоты. Часть кислоты может реагировать с аммиаком, оставшимся в стадии 1, и с аммиаком, образуемым путем гидролиза образуемого в качестве побочного продукта амида α- гидроксикислоты, и оставаться в виде аммониевой соли α- гидроксикислоты. Аммониевую соль α- гидроксикислоты, которая образуется вне системы, можно рециркулировать в качестве исходного материала в стадию 1, что не создает особых проблем.
После завершения реакции в стадии 2 из полученного водного раствора α- гидроксикислоты отгоняют воду, получая свободную α- гидроксикислоту с чистотой 80% или выше. Для получения более чистой α- гидроксикислоты раствор предпочтительно экстрагируют подходящими органическими растворителями с последующей отгонкой растворителей. Для экстракции можно использовать, без особых ограничений, органические растворители, нерастворимые в воде и растворяющие свободные α- гидроксикислоты. Примеры их включают толуол, этилацетат, метилизобутилкетон, н-бутанол, диизопропиловый эфир и дихлорэтан. Для экстракции органическими растворителями можно применять непрерывную экстракцию путем противоточного распределения. Эта операция повышает выделение свободной α- гидроксикислоты. Если целевая α- гидроксикислота кристаллическая, свободную кислоту можно осадить в водном растворе после завершения реакции в стадии 2 и выделить фильтрованием вместо экстракции органическими растворителями.
Возможно рециркулирование водных растворов, то есть водных слоев после экстракции органическими растворителями или фильтратов после отделения кристаллов, в качестве исходных материалов стадии 1, когда их концентрируют, после того как α- гидроксикислоту получают вышеуказанным способом.
[Примеры]
Настоящее изобретение далее описывается со ссылкой на следующие примеры. α- Гидроксикислоты, амиды α-гидроксикислот и поли-α-гидроксикислоты анализировали высокоэффективной жидкостной хроматографией. Аммиак определяли методом измерения УФ-поглощения с использованием NADH-глутаминовая кислота-дегидрогеназы (Методы ферментативного анализа, Bergmeyer H.U. 3-rd Edition, Vol.8, pages 454-461).
[Пример 1]
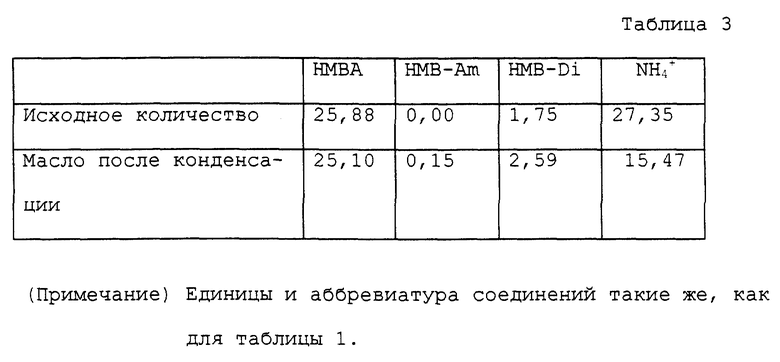
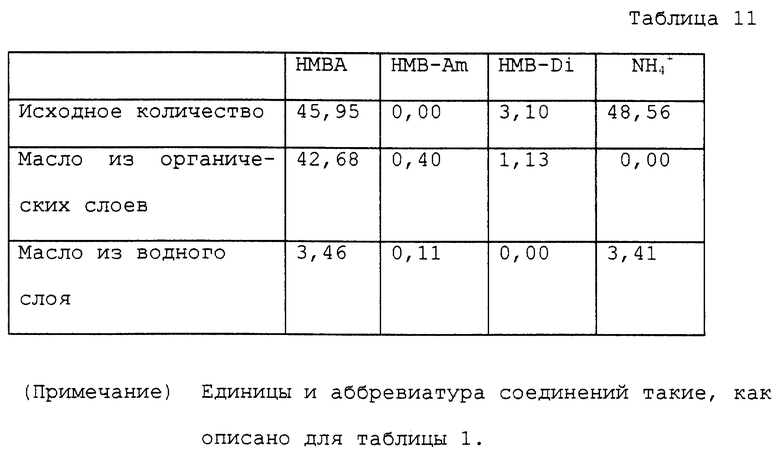
К 100 мл колбе, снабженной мешалкой и термометром, присоединяли ректификационную трубку. Фракционирующую колонку, снабженную термометром и обратным холодильником, соединяли с концом ректификационной трубки. Фракционирующую колонку устанавливали так, чтобы перегнанный органический растворитель был отделен от перегнанной воды и только органический растворитель возвращался в ректификационную трубку для орошения. В эту колбу на 100 мл помещали 14,20 г водного раствора, содержащего 53,40 ммоль α- гидрокси-4-метилтиобутирата аммония и 40 мл ксилола. Колбу помещали на масляную баню с температурой около 150oC для нагревания раствора с перемешиванием. В начале реакции азеотропная смесь ксилола и воды поднималась до верха трубки и температура у верха была 92oC ~ 93oC. С развитием реакции вода отгонялась отдельно. Температура у верха трубки постепенно повышалась и достигала точки кипения ксилола около 140oC. Во время реакции большую часть аммиака, за исключением аммиака, который растворялся в дистиллированной воде, выпускали в виде газа из конца обратного холодильника. После того как раствор нагревали при кипячении с обратным холодильником в течение 4 часов, ксилол перегоняли при пониженном давлении, получая 7,68 г масла. Полученное масло помещали в автоклав с внутренним объемом около 60 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием приблизительно при 150oC в течение 4 часов. Во время нагревания внутреннее давление было приблизительно 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температура и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 9,56 г масла. Аналитические результаты приводятся в таблице 1. В соответствии с результатами оставшаяся часть аммиака составляла 3,4%. Содержание полученного в качестве побочного продукта α- гидрокси-4-метилтиобутирамида и выход свободной α- гидроксиметилтиомасляной кислоты с учетом димеров были 0,9% и 90,4% соответственно.
[Сравнительный пример 1]
Этот пример проводили по способу, описанному в патенте Великобритании N 967352. В колбу на 50 мл, снабженную мешалкой, термометром и обратным холодильником, помещали 3,80 г водного раствора, содержащего 19,50 ммоль α- гидрокси-4-метилтиобутирата аммония и 1,0 мл воды для образования гомогенного раствора. К получаемому раствору далее добавляли 23,0 мл толуола. Колбу помещали на масляную баню с температурой 120oC для нагревания раствора при кипячении с обратным холодильником с перемешиванием. Температура реакционного раствора была 100 ~ 103oC и газообразный аммиак генерировался с верха обратного холодильника. После того как раствор нагревали при кипячении с обратным холодильником в течение 4 часов, толуол и воду отгоняли при пониженном давлении, получая 3,82 г масла. Аналитические результаты приводятся в таблице 2. В соответствии с этими результатами оставшаяся часть аммиака составляла 70,3%. Выход свободной α- гидрокси-4-метилтиомасляной кислоты был 22,1% с учетом димеров,
[Сравнительный пример 2]
Этот пример проводили по способу, описанному в выложенном патенте Японии N Sho 54-115317. В колбу на 200 мл, снабженную мешалкой, термометром, одной дистилляционной колонкой (10 мм внутренний диаметр и 10 см длина) и холодильником, помещали 50% по массе водный раствор, содержащий 25,88 ммоль α-гидрокси-4-метилтиобутирата аммония и 115 мл толуола. Получаемый раствор нагревали при перемешивании, в то время как в паровую фазу вводили небольшое количество высушенного воздуха. Газообразный аммиак был генерирован одновременно, когда была отогнана азеотропная смесь толуола и воды. Перегонку азеотропной смеси останавливали приблизительно через 40 минут после начала перегонки. Реакцию заканчивали приблизительно через 10 минут после того, как был отогнан почти только толуол. Общее количество перегнанного раствора было 64,5 г. Общее количество раствора после реакции было 58,8 г. Раствор после реакции конденсировали при пониженном давлении, получая 5,6 масла. Аналитические результаты приводятся в таблице 3. В соответствии с этими результатами оставшаяся часть аммиака составляла 56,6%. Выход свободной α- гидрокси-4-метилтиомасляной кислоты был 32,8% с учетом димеров.
[Сравнительный пример 3]
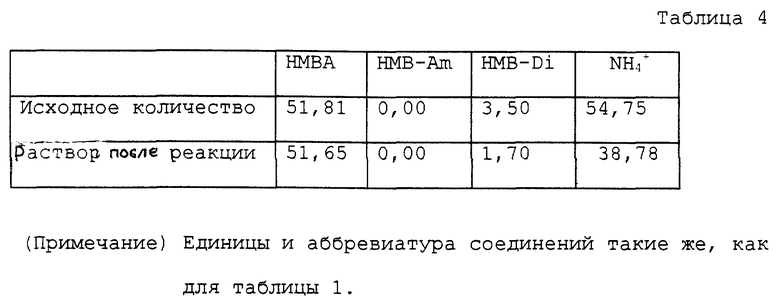
Этот пример проводили по способу, описанному в выложенном патенте Японии Hei 7-330696. В колбу на 50 мл, снабженную мешалкой, термометром, капельной воронкой, одной дистилляционной колонкой (10 мм внутренний диаметр и 10 см длина) и холодильником, помещали 10,35 г водного раствора, содержащего 51,81 ммоль α- гидрокси-4-метилтиобутирата аммония и 12 мл воды. В колбу непрерывно при скорости 20 мл/час подавали воду, предварительно нагретую до 70oC, в то время как из капилляра в колбу при атмосферном давлении продували очень небольшое количество азота. Дно колбы нагревали на масляной бане с температурой 150oC. Водный аммиак перегоняли при температуре перегонки в верхней части 99 ~ 100oC и при скорости перегонки 20 мл/час, тогда как количество оставшегося раствора сохраняли приблизительно постоянным. В течение приблизительно 4 часов получали всего около 80 мл водного аммиака. Общее количество раствора после реакции было 21,9 мл. Аналитические результаты приводятся в таблице 4. В соответствии с этими результатами оставшаяся часть аммиака составляла 70,8%. Выход свободной α- гидрокси-4-метилтиомасляной кислоты был 21,9% с учетом димеров.
[Пример 2]
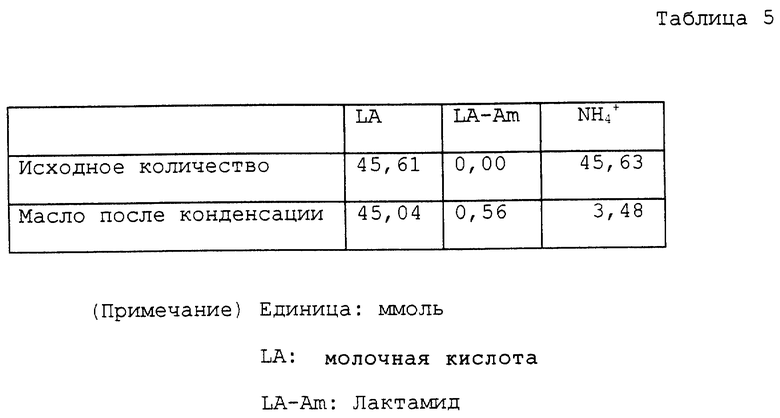
Колбу на 100 мл снабжали теми же устройствами, как устройства, использованные в примере 1. В колбу помещали 5,84 г водного раствора, содержащего 45,61 ммоль лактата аммония и 40 мл ксилола. Колбу помещали на масляную баню с температурой около 150oC для нагревания раствора с перемешиванием. В начале реакции азеотропная смесь ксилола и воды поднималась до верха трубки и температура у верха была 92 ~ 93oC. С развитием реакции вода перегонялась отдельно. Температура у верха трубки постепенно повышалась и достигала точки кипения ксилола около 140oC. В процессе реакции большую часть аммиака, за исключением аммиака, растворенного в дистиллированной воде, удаляли в виде газа из верха обратного холодильника. Раствор нагревали при кипячении с обратным холодильником с перемешиванием в течение всего 4 часов, в то время как реакцию проводили, как описано выше. Затем при пониженном давлении отгоняли ксилол, получая 4,08 г масла. Полученное масло помещали в автоклав с внутренним объемом около 60 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием приблизительно при 150oC в течение 4 часов. Во время нагревания внутреннее давление было приблизительно 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 4,52 г масла. Аналитические результаты приводятся в таблице 5. В соответствии с этими результатами оставшаяся часть аммиака составляла 7,6%. Содержание полученного в качестве побочного продукта лактамида было 1,2%. Выход свободной молочной кислоты был 89,9%.
[Пример 3]
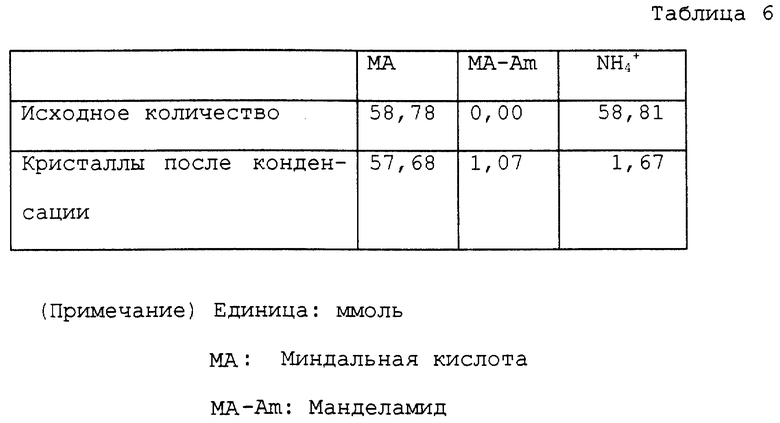
Колбу на 100 мл снабжали теми же устройствами, как устройства, использованные в примере 1. В колбу помещали 10,46 г водного раствора, содержащего 58,78 ммоль манделата аммония и 40 мл ксилола. Колбу помещали на масляную баню с температурой около 150oC для нагревания раствора с перемешиванием. В начале реакции азеотропная смесь ксилола и воды поднималась до верха трубки и температура у верха была 92 ~ 93oC. С развитием реакции вода перегонялась отдельно. Температура у верха трубки постепенно повышалась и достигала точки кипения ксилола около 140oC. В процессе реакции большую часть аммиака удаляли в виде газа из верха обратного холодильника. Раствор нагревали при кипячении с обратным холодильником с перемешиванием в течение всего 4 часов, в то время как реакцию проводили, как описано выше. Затем при пониженном давлении отгоняли ксилол, получая 7,38 г масла. Полученное масло помещали в автоклав с внутренним объемом около 60 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием приблизительно при 150oC в течение 4 часов. Во время нагревания внутреннее давление было приблизительно 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 9,63 г масла. Аналитические результаты приводятся в таблице 6. В соответствии с этими результатами оставшаяся часть аммиака составляла 2,8%. Содержание полученного в качестве побочного продукта манделамида было 1,8%. Выход свободной миндальной кислоты был 93,5%.
[Пример 4]
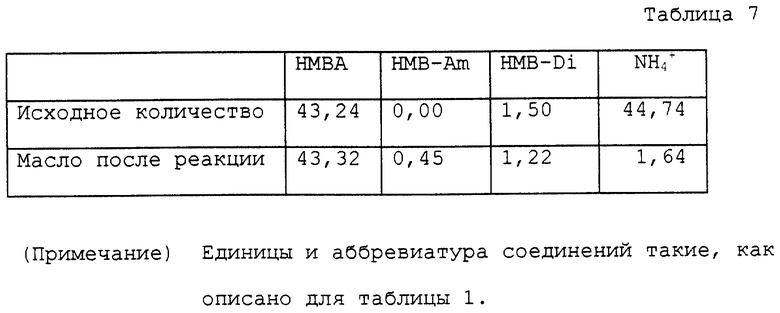
Колбу на 100 мл снабжали теми же устройствами, как устройства, использованные в примере 1. В колбу помещали 10,01 водного раствора, содержащего 43,24 ммоль α- гидрокси-4-метилтиобутирата аммония и 50 мл анизола. Колбу помещали на масляную баню с температурой около 170oC для нагревания раствора с перемешиванием. С развитием реакции вода перегонялась отдельно. Температура у верха трубки постепенно повышалась и достигала точки кипения анизола около 156oC. В процессе реакции большую часть аммиака, за исключением аммиака, растворенного в дистиллированной воде, удаляли в виде газа из верха обратного холодильника. Раствор нагревали при кипячении с обратным холодильником в течение 4 часов. Анизол затем отгоняли при пониженном давлении, получая 6,78 г масла. Полученное масло помещали в автоклав с внутренним объемом около 60 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием приблизительно при 150oC в течение 4 часов. Во время нагревания внутреннее давление было приблизительно 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 7,61 г масла. Аналитические результаты приводятся в таблице 7. В соответствии с этими результатами оставшаяся часть аммиака составляла 3,7%. Содержание полученного в качестве побочного продукта α- -гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты с учетом димеров были 1,0% и 90,1% соответственно.
[Пример 5]
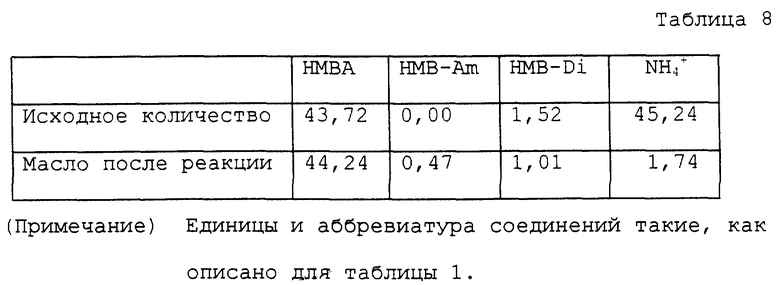
Колбу на 100 мл снабжали теми же устройствами, как устройства, использованные в примере 1. В колбу помещали 10,12 г водного раствора, содержащего 43,72 ммоль α- гидрокси-4-метилтиобутирата аммония и 50 мл диэтилового эфира этиленгликоля. Колбу помещали на масляную баню с температурой около 150oC для нагревания раствора с перемешиванием. С развитием реакции вода перегонялась отдельно. Температура у верха трубки постепенно повышалась и достигала точки кипения диэтилового эфира этиленгликоля около 121oC. В процессе реакции большую часть аммиака, за исключением аммиака, растворенного в дистиллированной воде, удаляли в виде газа из верха обратного холодильника. Раствор нагревали при кипячении с обратным холодильником в течение 4 часов. Диэтиловый эфир этиленгликоля затем отгоняли при пониженном давлении, получая 6,94 г масла. Полученное масло помещали в автоклав с внутренним объемом около 60 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием приблизительно при 150oC в течение 4 часов. Во время нагревания внутреннее давление было приблизительно 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 7,58 г масла. Аналитические результаты приводятся в таблице 8. В соответствии с этими результатами оставшаяся часть аммиака составляла 3,8%. Содержание полученного в качестве побочного продукта α- -гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты при вычислении с учетом димеров были 1,1% и 90,9% соответственно.
[Пример 6]
Прямую трубку присоединяли к колбе на 50 мл, снабженной мешалкой, термометром и трубкой для введения газа. Фракционирующую колонку, снабженную термометром и обратным холодильником, соединяли с концом прямой трубки. В колбу на 50 мл помещали 5,09 г водного раствора, содержащего 20,61 ммоль α- гидрокси-4-метилтиобутирата аммония и 20 мл диметилового эфира диэтиленгликоля. Колбу помещали на масляную баню с температурой около 145oC для нагревания раствора с перемешиванием. При нагревании в реакционный раствор со скоростью 100 мл/мин вводили газообразный азот, чтобы отогнать воду, уже присутствующую в исходных материалах, и воду, образованную во время реакции, в виде азеотропной смеси с диметиловым эфиром диэтиленгликоля за пределы реакционной системы. Большую часть аммиака, образованного во время реакции, за исключением аммиака, растворенного в дистиллированной воде, удаляли в виде газа из верха обратного холодильника. Температура реакционного раствора была приблизительно 115oC в начале реакции из-за присутствия большого количества воды. С развитием реакции температура постепенно повышалась до постоянной около 130oC. После проведения реакции в течение 2 часов диметиловый эфир диэтиленгликоля отгоняли при пониженном давлении, получая 3,65 г масла. Полученное масло переносили в
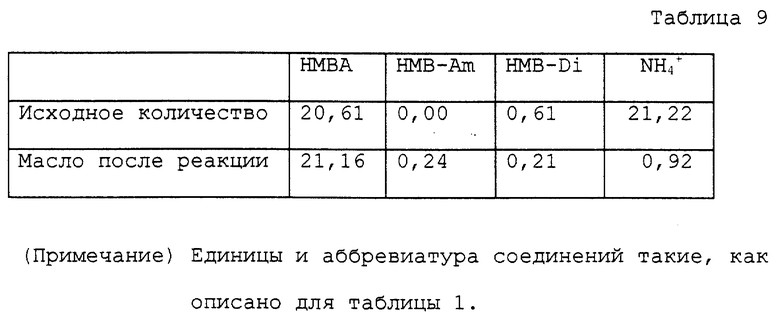
круглодонную колбу на 50 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием при кипячении с обратным холодильником в течение 4 часов при 100oC. Реакционный раствор охлаждали вплоть до комнатной температуры и анализировали. Аналитические результаты приводятся в таблице 9. В соответствии с результатами оставшаяся часть аммиака составляла 4,3%. Содержание полученного в качестве побочного продукта a- гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты с учетом димеров были 1,1% и 92,7% соответственно.
[Пример 7]
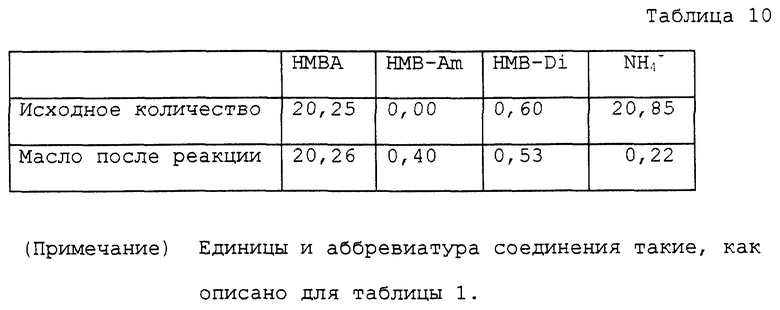
Прямую трубку присоединяли к колбе на 50 мл, снабженной мешалкой и термометром. Фракционирующую колонку, снабженную термометром и дефлегматором, соединяли с концом прямой трубки. Верх дефлегматора соединяли с воздушным отсасывающим устройством, так чтобы внутри реакционной системы было пониженное давление. В колбу на 50 мл помещали 5,00 г водного раствора, содержащего 20,25 ммоль α- гидрокси-4-метилтиобутирата аммония, и 25 мл диметилового эфира диэтиленгликоля. Колбу помещали на масляную баню с температурой около 110oC для нагревания раствора с перемешиванием. При нагревании давление внутри реакционной системы устанавливали до 600 ~ 650 мм Hg посредством отсасывающего устройства, чтобы отогнать воду, уже присутствующую в исходных материалах, и воду, образованную во время реакции, в виде азеотропной смеси с диметиловым эфиром диэтиленгликоля за пределы реакционной системы. Большая часть аммиака, образованного во время реакции, за исключением аммиака, растворенного в дистиллированной воде, была абсорбирована отсасывающим устройством в виде газа. Температура реакционного раствора была приблизительно 90 ~ 93oC в начале реакции. С развитием реакции температура постепенно повышалась до постоянной около 100oC. После проведения реакции в течение 4 часов диметиловый эфир диэтиленгликоля отгоняли при пониженном давлении, получая 4,08 г масла. Полученное масло переносили в круглодонную колбу на 50 мл. К маслу добавляли 20 мл воды для нагревания с перемешиванием при кипячении с обратным холодильником в течение 4 часов при 100oC. Реакционный раствор охлаждали вплоть до комнатной температуры и анализировали. Аналитические результаты приводятся в таблице 10. В соответствии с результатами оставшаяся часть аммиака составляла 1,1%. Содержание полученного в качестве побочного продукта α- гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты с учетом димеров были 1,9 и 93,4% соответственно.
[Пример 8]
9,18 г водного раствора, содержащего 45,95 ммоль α- гидрокси-4-метилтиобутирата аммония, помещали в круглодонную колбу на 50 мл. Этот раствор нагревали при 120 ~ 125oC и 0,8 ~ 1,5 мм Hg в течение 4 часов в роторном испарителе для удаления генерированного аммиака и воды. Общее количество оставшегося реакционного раствора было 7,29 г. Раствор содержал, помимо поли-α-гидрокси-4-метилтиомасляных кислот, 3,06 ммоль α- гидрокси-4-метилтиобутирата аммония и 0,87 ммоль α- -гидрокси-4-метилтиобутирамида. К оставшемуся раствору, полученному после реакции, добавляли 22 мл воды. Получаемый раствор нагревали при кипячении с обратным холодильником при атмосферном давлении в течение 20 часов. Реакционный раствор охлаждали вплоть до комнатной температуры и экстрагировали 25 мл метилизобутилкетона 3 раза. Органические слои объединяли и концентрировали, получая 7,08 г масла. Водный слой после экстракции также концентрировали, получая 0,81 г масла. Аналитические результаты приводятся в таблице 11. В соответствии с результатами оставшаяся часть аммиака составляла 7,0%. Содержание являющегося побочным продуктом α- гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты с учетом димеров были 1,0% и 81,9% соответственно.
[Пример 9]
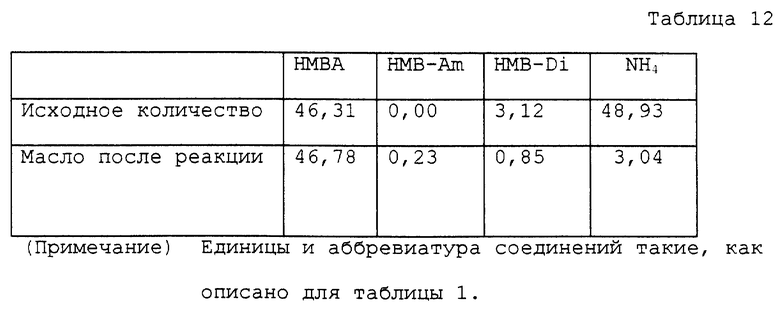
9,25 г водного раствора, содержащего 46,31 ммоль α- гидрокси-4-метилтиобутирата аммония, помещали в круглодонную колбу на 50 мл. Этот раствор нагревали при 135 ~ 140oC и 0,8 ~ 1,5 мм Hg в течение 4 часов в роторном испарителе для удаления генерированного аммиака и воды. Общее количество оставшегося реакционного раствора было 7,20 г. Раствор содержал, помимо поли α- гидрокси-4-метилтиомасляных кислот, 2,54 ммоль α- гидрокси-4-метилтиобутирата аммония и 0,57 ммоль α- гидрокси-4-метилтиобутирамида. Оставшийся раствор, полученный после реакции, переносили в автоклав с внутренним объемом около 60 мл. К раствору добавляли 20 мл воды для нагревания на масляной бане при 170 ~ 175oC в течение 4 часов. Внутреннее давление во время нагревания было 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 7,45 г масла. Аналитические результаты приводятся в таблице 12. В соответствии с результатами оставшаяся часть аммиака составляла 6,2%. Содержание полученного в качестве побочного продукта α- гидрокси-4-метилтиобутирамида и выход свободной α- гидрокси-4-метилтиомасляной кислоты с учетом димеров были 0,4% и 83,2% соответственно.
[Пример 10]
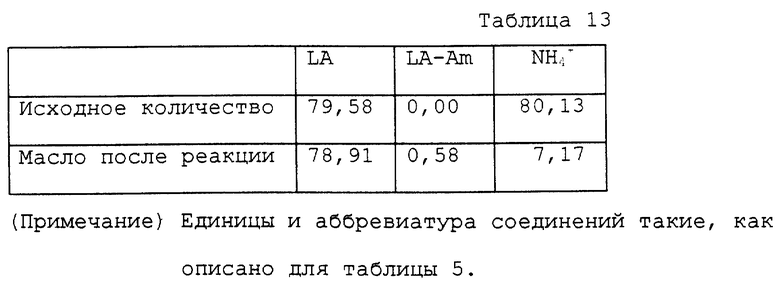
9,04 г водного раствора, содержащего 79,58 ммоль лактата аммония, помещали в круглодонную колбу на 50 мл. Этот раствор нагревали при 118 ~ 120oC и 11 ~ 14 мм Hg в течение 6 часов в роторном испарителе для удаления генерированного аммиака и воды. Общее количество оставшегося реакционного раствора было 6,62 г. Раствор содержал, помимо полимолочных кислот, 6,31 ммоль лактата аммония и 1,04 ммоль лактамида. Оставшийся раствор, полученный после реакции, переносили в автоклав с внутренним объемом около 60 мл. К раствору добавляли 30 мл воды для нагревания на масляной бане при 150 ~ 155oC в течение 3 часов. Внутреннее давление во время нагревания было 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры, переносили в круглодонную колбу на 50 мл и концентрировали при пониженном давлении, так чтобы раствор был приблизительно 80% водным раствором. Получали 9,11 г масла. Аналитические результаты приводятся в таблице 13. В соответствии с результатами оставшаяся часть аммиака составляла 8,9%. Выход свободной молочной кислоты относительно исходного количества лактата аммония был 90,1%.
[Пример 11]
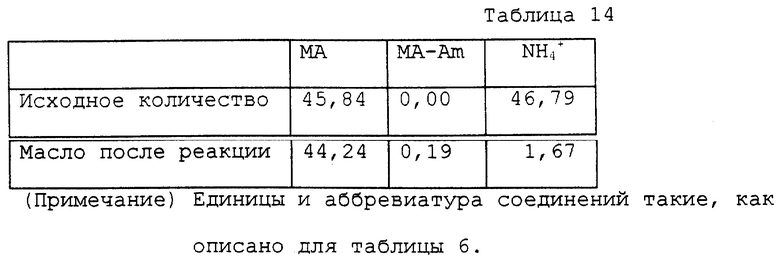
9,06 г водного раствора, содержащего 45,84 ммоль манделата аммония, помещали в круглодонную колбу на 50 мл. Этот раствор нагревали при 118 ~ 120oC и 0,5 ~ 1,0 мм Hg в течение 4 часов в роторном испарителе для удаления генерированного аммиака и воды. Общее количество оставшегося реакционного раствора было 6,09 г. Раствор содержал, помимо полиминдальных кислот, 1,38 ммоль манделата аммония и 0,49 ммоль манделамида. Оставшийся раствор, полученный после реакции, переносили в автоклав с внутренним объемом около 60 мл. К раствору добавляли 20 мл воды для нагревания на масляной бане при 170 ~ 175oC в течение 4 часов. Внутреннее давление во время нагревания было 3 кг/см2. Реакционный раствор охлаждали вплоть до комнатной температуры и переносили в круглодонную колбу на 50 мл. Воду отгоняли при пониженном давлении, получая 6,92 г кристаллов. Аналитические результаты приводятся в таблице 14. В соответствии с результатами оставшаяся часть аммиака составляла 3,6%. Выход свободной миндальной кислоты относительно исходного количества манделата аммония был 92,9%.
Способы этого изобретения являются предпочтительными и выгодными с промышленной точки зрения по следующим причинам:
1. Когда свободную α-гидроксикислоту получают из ее аммониевой соли, аммиак удаляют в виде газообразного аммиака, так что отходы в виде аммониевой соли не образуются.
2. Способы выгодны из-за стоимости продукции благодаря тому, что отсутствует добавление дополнительных веществ, таких как катализаторы или добавки для нейтрализации.
3. Оставшийся аммиак и образующийся в качестве побочного продукта амид α-гидроксикислоты можно отделить от свободной α-гидроксикислоты и рециркулировать в виде их аммониевой соли после проведения гидролитической реакции.
Промышленное применение
Настоящее изобретение является выгодным и эффективным промышленным способом получения свободной α- гидроксикислоты из ее аммониевой соли, имеющим большое значение.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ α-ГИДРОКСИКИСЛОТ С ИСПОЛЬЗОВАНИЕМ НОВОГО МИКРООРГАНИЗМА И НОВЫЙ МИКРООРГАНИЗМ | 1997 |
|
RU2205220C2 |
| ПРОИЗВОДНОЕ БЕНЗОЛА, ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛОМ, И ГЕРБИЦИД | 1997 |
|
RU2162849C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ПОЛУЧЕНИЯ АНАЛОГА МЕТИОНИНА | 2021 |
|
RU2841154C1 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2533708C2 |
| ЦВЕТОПРОЯВЛЯЮЩИЕ КОМПОЗИЦИИ И СОДЕРЖАЩИЙ ИХ РЕГИСТРИРУЮЩИЙ МАТЕРИАЛ | 2009 |
|
RU2456165C2 |
| СПОСОБ ПОЛУЧЕНИЯ СВОБОДНЫХ КИСЛОТ ИЗ ИХ СОЛЕЙ | 2010 |
|
RU2533413C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2137770C1 |
| ЖИДКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СРЕДСТВО НА ЕЕ ОСНОВЕ, КОНТРОЛИРУЮЩЕЕ ЭКТОПАРАЗИТОВ У МЛЕКОПИТАЮЩИХ И ПТИЦ | 2006 |
|
RU2409391C2 |
| ОРТО-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АНИЛИНА И АНТИОКСИДАНТНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2006 |
|
RU2384579C2 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДОКСИМА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО ДЛЯ ЗАЩИТЫ СЕЛЬСКОХОЗЯЙСТВЕННЫХ РАСТЕНИЙ | 1995 |
|
RU2140908C1 |
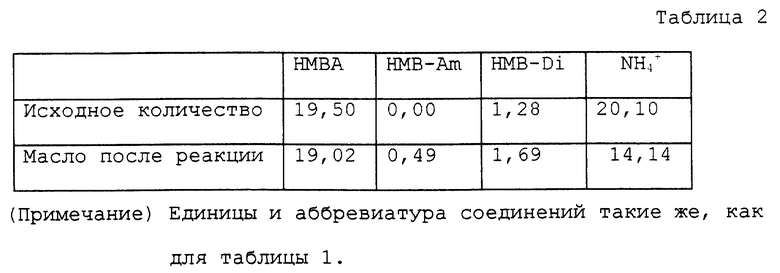
Изобретение относится к области органической химии. Предложен способ получения α-гидроксикислот нагреванием аммониевых солей α-гидроксикислот общей формулы (I)
R-CH(OH)-COO-NH4+,
где R - С1-С6алкил, который может быть замещен тиоалкильной группой, или фенил, возможно содержащие органический растворитель, с удалением образовавшихся аммиака и воды и возможно содержащегося органического растворителя с последующим добавлением воды к остатку и нагреванием полученной смеси. Данный способ позволяет получать свободные α-гидроксикислоты из их аммониевых солей экономичным способом с высоким выходом продукта. 2 с.п. ф-лы, 14 табл.
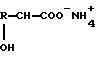
где R - C1 - C6алкил, который может быть замещен C1 - C6 алкилтиогруппой, или фенил,
нагревание проводят с удалением образующихся аммиака и воды и к полученному после удаления аммиака, воды и органического растворителя остатку добавляют воду с получением смеси, которую нагревают.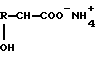
где R - C1 - C6алкил, который может быть замещен C1 - C6 алкилтиогруппой, или фенил,
нагревание водного раствора соответствующей аммониевой соли α-гидроксикислоты с удалением образующихся аммиака и воды проводят при пониженном давлении, добавляют к полученному остатку воду и полученную смесь нагревают.
Приоритет по пунктам:
19.11.1996 по п.1;
26.02.1996 по п.2.
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ аОКСИКМ€ЛвТ- | 0 |
|
SU168284A1 |
| Способ получения алифатических @ -оксикарбоновых кислот | 1988 |
|
SU1512964A1 |
| Способ приготовления сернистого красителя защитного цвета | 1915 |
|
SU63A1 |
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

