Изобретение относится к новым замещенным в положении 4 1-фенилалкил-1,2,3, 6-тетрагидропиридинам, обладающим нейротрофической и нейрозащитной активностью, способу их получения и содержащим их фармацевтическим композициям.
В европейском патенте 0458696 описывается использование 1-(2-нафтилэтил)-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина для получения лекарственных средств, предназначенных для лечения мозговых и нейронных нарушений.
В международной заявке 93/11107 описываются пиперидины и тетрагидропиридины с защитным действием против расстройств, вызываемых гипоксическими и/или ишемическими состояниями.
В настоящее время найдено, что некоторые фенилалкил-1,2,3,6-тетрагидропиридины, замещенные фенильной или пиридильной группой, оказывают нейротрофическое действие на нервную систему, подобное действию фактора роста нервной ткани (NGF), и могут восстанавливать функцию поврежденных или имеющих аномалию в своих физиологических функциях клеток.
Следовательно, настоящее изобретение, согласно одному из его аспектов, относится к соединениям формулы (I):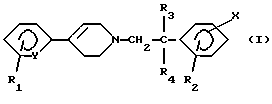
в которой Y означает -СН- или -N-;
R1 означает водород, галоген, трифторметил, (C1-C4)-алкил или (C1-C4)-алкоксил;
R2 означает метил или этил;
каждый из R3 и R4 означает водород или (C1-С3)-алкил;
Х означает:
(а) (C1-С6)-алкил; (C1-C6)-алкоксил, карбокси-(С3-С7)-алкил; (C1-C4)-алкоксикарбонил-(C1-C6)-алкил; карбокси-(С3-С7) -алкоксил; или (C1-C4)-алкоксикарбонил-(C1-C6)-алкоксил;
(б) радикал, выбираемый среди (С3-С7)-циклоалкила, (С3-С7)-циклоалкилоксигруппы, (С3-С7)-циклоалкилметила, (С3-С7)-циклоалкиламиногруппы и циклогексенила, причем указанный радикал может быть замещен галогеном, гидроксилом, (C1-C4)-алкоксилом, карбоксилом, (C1-C4)-алкоксикарбонилом, аминогруппой, моно- или ди[(C1-C4)-алкил]аминогруппой;
или
(в) группу, выбираемую среди фенила, феноксигруппы, фениламиногруппы, N-(C1-С3)-алкилфениламиногруппы, фенилметила, фенилэтила, фенилкарбонила, фенилтиогруппы, фенилсульфонила, фенилсульфинила или стирила, причем указанная группа может быть моно- или полизамещена в фенильной группе галогеном, трифторметилом, (C1-C4)-алкилом, (C1-C4)-алкоксилом, цианогруппой, аминогруппой, моно- или ди [(C1-C4)-алкил]аминогруппой, (C1-C4)-ациламиногруппой, карбоксилом, (C1-C4)-алкоксикарбонилом, аминокарбонилом, моно- или ди[(C1-C4)-алкил]аминокарбонилом, амино-(C1-C4)-алкилом, гидрокси-(C1-C4)-алкилом или галоген-(C1-C4)-алкилом; так же, как к их солям и сольватам и их четвертичным аммониевым солям.
В настоящем описании термин "(C1-С3)-алкил" означает метил, этил, н-пропил и изопропил.
Термин "(C1-C4)-алкил" означает метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор. -бутил и трет. -бутил.
Термин "(C1-С6)-алкил" означает углеводородный радикал с 1-6 атомами углерода, такой как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор. -бутил, трет. -бутил, н-пентил, изопентил, неопентил, трет.-пентил, н-гексил, изогексил, и т.д.
Термин "алкоксил" означает гидроксил, замещенный (C1-С6)-алкилом, предпочтительно (C1-C4)-алкилом, более предпочтительно (C1-С3)-алкилом.
Когда Х означает фенил, номенклатура, применяемая к бифенилу, соответствует правилам ИЮПАК, в частности нумерация положений обоих циклов следующая: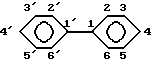
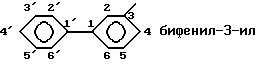
и радикалы, имеющие эту структуру, называют следующим образом: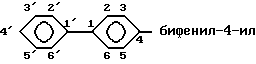
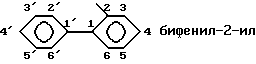
Среди соединений формулы (I), где Х означает группу радикалов (в) , предпочтительными соединениями являются те, в которых фенил замещен 1-3-мя атомами галогена, 1-3-мя трифторметильными группами, 1-3-мя (C1-C4)-алкильными группами, 1-3-мя (C1-C4)-алкоксильными группами, 1-3-мя цианогруппами, 1-3-мя аминогруппами, 1-3-мя моно- или ди[(C1-C4)-алкил] аминогруппами, 1-3-мя (C1-C4)-ациламиногруппами, 1-3-мя карбоксильными группами, 1-3-мя (C1-C4)-алкоксикарбонильными группами, 1-3-мя аминокарбонильными группами, 1-3-мя моно- или ди[(C1-C4)-алкил]аминокарбонильными группами, 1-3-мя амин-(C1-C4)-алкильными группами или 1-3-мя гидрокси-(C1-C4)-алкильными группами, или 1-3-мя галоген-(C1-C4)-алкильными группами. Другую предпочтительную группу образуют соединения формулы (I), где Y означает группу -СН- и R1 означает трифторметил.
Следующую предпочтительную группу составляют соединения формулы (I), где Y означает атом азота и R1 означает атом хлора.
Другую предпочтительную группу образуют соединения формулы (I), где Х означает (C1-C6)-алкил, особенно этил.
Особенно предпочтительными соединениями являются соединения, отвечающие формуле (I'):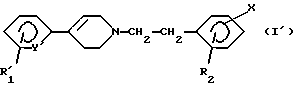
в которой R'1 означает трифторметил и Y' означает CH или R'1 означает хлор и Y' означает азот, причем R2 и Х имеют вышеуказанное значение, и их соли, сольваты и четвертичные аммониевые соли.
Следующую предпочтительную группу образуют соединения формулы (I'), где Х означает (C1-C6)-алкил.
Особенно предпочтительными соединениями согласно настоящему изобретению являются следующие:
1-[2-(3,4-диэтилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин;
1-[2-(3-метил-4-пентилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин;
1-[2-(4-метил-3-пентилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин;
1-[2-(3,4-диэтилфенил)этил] -4-(6-[хлорпирид-2-ил)-1,2,3,6-тетрагидропиридин, и их соли, сольваты и четвертичные аммониевые соли.
Согласно другому из его аспектов настоящее изобретение относится к способу получения соединений формулы (I), их солей или сольватов и их четвертичных аммониевых солей, отличающемуся тем,
что (а) арил-1,2,3,6-тетрагидропиридин формулы (II):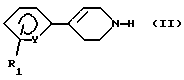
в которой Y и R1 имеют вышеуказанные значения, вводят во взаимодействие с соединением формулы (III):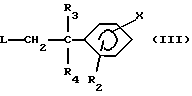
в которой R2, R3, R4 и Х имеют вышеуказанные значения и L означает удаляемую группу, такую как, например, атом хлора, брома, иода; метансульфонилоксигруппа; п-толуолсульфонилоксигруппа; трифторметансульфонилоксигруппа; и
(б) полученное соединение формулы (I) выделяют и в случае необходимости превращают его в одну из его солей или один из его сольватов или в одну из его четвертичных аммониевых солей.
Реакцию проводят в органическом растворителе и при температуре от комнатной до температуры кипения с обратным холодильником используемого растворителя.
В качестве предпочтительного органического растворителя используют алифатический спирт с 1-6 атомами углерода, такой как метанол, этанол, изопропанол, н-бутанол, н-пентанол, однако также могут быть использованы другие растворители, такие как гексан, диметилформамид, диметилсульфоксид, сульфолан, ацетонитрил, пиридин и подобные.
Реакцию предпочтительно проводят в присутствии основного вещества, такого как карбонат щелочного металла или триэтиламин, особенно когда L означает атом галогена.
Температура реакции может изменяться от комнатной (около 20oС) до температуры кипения с обратным холодильником, и в соответствии изменяется время реакции. Обычно реакция заканчивается после кипячения с обратным холодильником в течение 6-12 часов и таким образом полученный целевой продукт может быть выделен обычными способами в виде свободного основания или в виде одной из его солей, причем свободное основание в случае необходимости превращают в одну из его солей путем простого солеобразования в органическом растворителе, таком как спирт, предпочтительно этанол или изопропанол, простой эфир, как 1,2-диметоксиэтан, этилацетат, ацетон или углеводород, как гексан.
Полученное соединение формулы (I) выделяют обычными способами и, в случае необходимости, превращают в одну из его кислотно-аддитивных солей, или, когда имеется кислотная группа, амфотерный характер соединения позволяет осуществлять выделение солей либо с кислотами, либо с основаниями.
Когда получают соли соединения формулы (I) для их введения в качестве лекарственных средств, то нужно, чтобы используемые кислоты или основания были фармацевтическим приемлемыми; если получают соли соединения формулы (I) с другой целью, например для лучшей очистки продукта или для лучшего осуществления аналитических исследований, тогда можно использовать любую кислоту или основание.
Соли с фармацевтическим приемлемыми кислотами представляют собой, например, соли с неорганическими кислотами, такие как гидрохлорид, гидробромид, борат, фосфат, сульфат, гидросульфит, гидрофосфат, дигидрофосфат, и соли с органическими кислотами, такие как цитрат, бензоат, аскорбат, метилсульфат, нафталин-2-сульфонат, пикрат, фумарат, малеат, малонат, оксалат, сукцината, ацетат, тартрат, меэилат, тозилат, изотионат, α-кетоглутарат, α-глицерофосфат, глюкозо-1-фосфат, и т.д.
Соли с фармацевтическим приемлемыми основаниями соединений формулы (I), в которой заместитель Х содержит карбоксил, представляют собой, например, соли с щелочными или щелочноземельными металлами, такими как натрий, калий, кальций, магний, и соли с органическими основаниями, такими как амины, основные аминокислоты (лизин, аргинин, гистидин), трометамол, N-метилглюкамин, и т.д.
Исходные амины формулы (II), где Y означает СН, представляют собой известные соединения, или они могут быть получены способами, аналогичными таковым, используемым для получения известных соединений.
Исходные амины формулы (II), где Y означает азот, могут быть получены путем взаимодействия соответствующего 2-галогенпиридина формулы (р):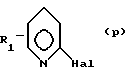
в которой R1 имеет вышеуказанное значение и Hal означает атом галогена, с 1,2,3,6-тетрагидропиридином формулы (q):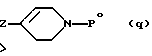
в которой Рo означает защитную группу, такую как, например, бензол, и Z означает заместитель, который позволяет осуществлять нуклеофильное замещение галогена пиридина. Такими заместителями являются, например, триалкилстаннаны, в частности, трибутилстаннан, или соединения Гриньяра.
Затем осуществляют удаление защитных групп от 1,2,3,6-тетрагидропиридина путем отщепления защитной группы в соответствующих условиях.
Соединения формулы (III) могут быть получены
- либо для получения соединения формулы (III), в которой R3=R4=Н, путем взаимодействия соответствующего бензола формулы (г):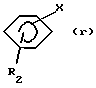
в которой R2 и Х имеют вышеуказанные значения, с ацилгалогенидом формулы L-CH2-CO-Hal, где L и Hal имеют вышеуказанные значения, в присутствии кислоты Льюиса согласно реакции Фриделя-Крафтса и восстановления, таким образом, полученного кетона формулы (s):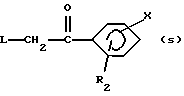
согласно широко описанным в литературе методам;
- либо путем восстановления кислот формулы (V);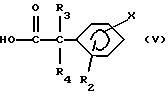
где R2, R3, R4 и X имеют вышеуказанные значения, в спирте, с последующим превращением гидроксила в удаляемую группу.
Кислоты формулы (V) обычно представляют собой описанные в литературе соединения или могут быть получены аналогичным образом.
Примеры получения также приводятся в экспериментальной части.
Активность соединений формулы (I) в отношении нервной системы показана в исследованиях ин витро и ин виво согласно методам, описанным в европейском патенте 0458696, а для оценки нейронной выживаемости использовался тест на выживаемость ин витро, осуществляемый при использовании нейронов, выделенных из рассечений септальной области эмбрионов крыс.
Согласно этому тесту отбирают для анализа септальную область эмбрионов крыс в возрасте 17-18 дней под микроскопом для рассечений в стерильных условиях, затем ее разлагают в среде, состоящей из трипсина и этилендиаминтетрауксусной кислоты. Суспензию клеток помещают в колбу для культивирования в среде, состоящей из модифицированной по способу Дульбекко среды Игла (DME) и питательной смеси Ham's F12 (объем на объем) [R.G. Ham, Proc. Nat. Sci., 53, 288 (1965)], содержащей 5% телячьей сыворотки и 5% лошадиной сыворотки, и выдерживают при температуре 37oС в течение 90 минут. Эта обработка позволяет удалять не нейронные клетки.
Затем проводят посев нейробластов в лунки титрационного планшета по 17•104 клеток на см2, в не содержащей сыворотки культуральной среде, образованной модифицированной по способу Дульбекко средой Игла и питательной смесью Ham's F12, содержащей 30 нмоль селена и 1,25 ммоль трансферрина. Каждую лунку предварительно обрабатывают поли-L-лизином. Планшеты, в которые произведен посев, помещают в термостатируемый инкубатор (37oС; 5% СO2) .
Испытуемые соединения растворяют в диметилсульфоксиде и разбавляют надлежащим образом культуральной средой.
Нейробласты выдерживают в планшетах, содержащих испытуемое соединение или соответствующий растворитель, в течение четырех дней без обмеривания среды.
Спустя 4 дня среду заменяют тетразолиевой солью, растворенной в культуральной среде (0,15 мг/мл). Клетки затем выдерживают в термостате при температуре 37oС в течение четырех часов. Митохондриальные сукцинодегидрогеназы живых клеток восстанавливают тетразолиевую соль до формазана синего цвета, по которому после растворения в диметилсульфоксиде измеряют оптическую плотность при 540 нм. Эта плотность линейно коррелирует с числом живых клеток [Manthorpe и др., Dev. Brain Res., 15, 191-198 (1988)].
Различие между содержащими испытуемые соединения группами и контрольными образцами оценивают путем статистического анализа при использовании двустороннего t-теста Dunnett ("two-tailed Dunnett t-test").
В этом последнем тесте соединения формулы (I) оказались также активными или более активными, чем соединения, описанные в европейском патенте 0458696, причем эффективность некоторых соединений формулы (I) в отношении выживаемости нейронов в два раза больше по отношению к соединению А, описанному в европейском патенте 0458696.
Благодаря этой сильной нейрозащитной активности и их незначительной токсичности, приемлемой для использования в качестве лекарственных средств, соединения формулы (I) так же, как их фармацевтические приемлемые соли, присоединения, их сольваты и их четвертичные аммониевые соли используют для получения фармацевтических композиций, показанных для лечения и/или профилактики всех заболеваний, которые имеют следствием нейронную дегенерацию. В особенности соединения согласно изобретению используют индивидуально, или в совместном приеме, или в ассоциации с другими действующими началами, воздействующими на центральную нервную систему, как, например, ингибиторы ацетилхолинэстеразы, селективные холиномиметические средства M1, антагонисты NMDA, ноотропные средства, такие как пирацетам, особенно при следующих показаниях: нарушения памяти, васкулярная деменция, постэнцефалитические нарушения, постапоплексические нарушения, возникающие вследствие черепно-мозговой травмы, посттравматические синдромы, расстройства, связанные с церебральной аноксией, болезнь Альцгеймера, старческое слабоумие; деменция подкорки головного мозга, такая как хорея Гентингтона и болезнь Паркинсона; провоцируемая СПИДом деменция; нейропатии, возникающие вследствие общей заболеваемости или повреждения симпатических или сенсорных нервов; и заболевания мозга, такие как отек мозга; и спиноцеребеллярные дегенерации, дегенерации мотонейронов, как, например, боковой амиотрофический склероз.
Введение соединений согласно изобретению обычно осуществляют перорально, парентерально, подъязычно или чрескожно. Количество вводимого действующего начала при лечении церебральных и нейронных нарушений согласно способу настоящего изобретения зависит от природы и тяжести излечиваемых болезненных состояний, так же, как массы больных. Однако предпочтительные разовые дозы обычно включают 0,25 -700 мг, предпочтительно 0,5 - 300 мг, особенно предпочтительно 1 - 150 мг, например, от 2 до 50 мг, а именно 2, 5, 10, 15, 20, 25 или 50 мг продукта. Эти разовые дозы обычно вводят один или несколько раз в день, например, 2, 3, 4 или 5 раз в день, предпочтительно 1-3 раза в день, причем общая доза для человека изменяется в пределах от 0,5 до 1400 мг в день, предпочтительно от 1 до 900 мг в день, например, от 2 до 500 мг, более приемлемо 2 - 200 мг в день.
Согласно другому из его аспектов предметом настоящего изобретения является фармацевтическая композиция, содержащая в качестве действующих начал соединение вышеприведенной формулы (I) и соединение, показанное для симптоматического лечения старческого слабоумия типа болезни Альцгеймера (DAT), или их фармацевтически приемлемые соли.
Выражение "соединение, показанное для симптоматического лечения старческого слабоумия типа болезни Альцгеймера (DAT)" означает продукт, который способен улучшать симптоматологическую картину пациентов, пораженных DAT, без воздействия на причины заболевания.
Такими соединениями являются, например, ингибиторы ацетилхолинэстеразы, мускариновые агонисты M1, холиномиметические агонисты, антагонисты рецептора NMDA, ноотропные средства.
Предпочтительными ингибиторами ацетилхолинэстеразы являются донепезил и такрин.
Другими ингибиторами ацетилхолинэстеразы, которые могут быть использованы, являются, например, ривастигмин (SDZ-ENA-713), галантамин, метрифонат, эптастигмин, велнакрин, физостигмин [Drugs, 53(5), 752-768 (1997); Указатель фирмы Мерк, 12-е издание].
Другими ингибиторами ацетилхолинэстеразы являются еще 5,7-дигидро-3-[2-[1-(фенил метил)-4-пиперидинил] этил] -6Н-пирроло[3,2-f]-1,2-бензизоксазол-6-он, называемый также икопезил [J. Med. Chem., 38, 2802-2808 (1995)], MDL-73 745 или зифросилон [Eur. J. Pharmacol., 276, 93-99 (1995)], ТАК-147 [J. Med. Chem., 37, 2292-2299 (1994)].
Другими ингибиторами ацетилхолинэстеразы являются, например, такие, которые описываются в заявке на патент Японии 09-095483; международных заявках 97/13754, 97/21681, 97/19929; заявке на патент Южно-Африканской Республики 96-04565; заявке на патент США 5455245; международной заявке 95-21 822; заявке на европейский патент 637586; заявке на патент США 5401749; заявке на европейский патент 742207; заявке на патент США 5547960; международных заявках 96/20176 и 96/02524; заявке на европейский патент 677516; заявках на патент Японии 07-188177 и 07-133274; заявках на европейский патент 649846 и 648771; заявке на патент Японии 07-048370; заявке на патент США 5391553; международной заявке 94/29272; заявке на европейский патент 627400.
Согласно следующему аспекту, настоящее изобретение относится к фармацевтической композиции, содержащей в качестве действующих начал соединение формулы (I) и агонист рецептора M1, или их фармацевтически приемлемые соли.
Агонистами рецептора M1 являются, например, миламелин, бесипиридин, талсаклидин, ксаномелин, YМ-796 и YM-954 [Eur. J. Pharmacol., 187, 479-486 (1990)] , 3-[N-(2-диэтиламино-2-метилпропил)-6-фенил-5-пропил]пиридазинамин, называемый также SR-46559 (Biorg. Med. Chem. Let., 2, 833-838 (1992)], AF-102, CI-979, L-689 660, LU 25-109, S-9977-2, SB 202 026, тиопилокарпин, WAL 2014 [Pharmacol. Toxicol., 78, 59-68 (1996)].
Согласно другому аспекту изобретение относится к фармацевтической композиции, содержащей в качестве действующих начал соединение формулы (I) и холиномиметический агонист, или их фармацевтически приемлемые соли.
Предпочтительными холиномиметическими агонистами являются, например, МКС-231 [Biorg. Med. Chem. Let., 5 (14), 1495-1500 (1995)], T-588 [Japan J. Pharmacol. , 62, 81-86 (1993)] , ABT-418 (Br. J. Pharmacol., 120, 429-438 (1997)].
Согласно следующему из его аспектов изобретение относится к фармацевтической композиции, содержащей в качестве действующих начал соединение формулы (I) и антагонист рецепторов NMDA, или их фармацевтически приемлемые соли.
Предпочтительным антагонистом рецепторов NMDA является, например, мемантин [Arzneim. Forsch., 41, 773-780 (1991)].
Согласно другому из его аспектов изобретение относится к фармацевтической композиции, содержащей в качестве действующих начал соединение формулы (I) и ноотропное средство, или их фармацевтически приемлемые соли.
Ноотропными средствами, которые могут быть использованы согласно изобретению, являются, например, нетирацетам, небрацетам (Указатель фирмы Мерк, 12-е издание).
Дозы обоих ассоциированных действующих начал обычно выбирают среди доз, которые вводят в случае каждого лекарственного средства при некомбинированном лечении.
Согласно дальнейшему аспекту настоящее изобретение также относится к способу лечения старческого слабоумия типа болезни Альцгеймера, который заключается во введении пораженному этим заболеванием пациенту эффективной дозы соединения формулы (I) или одной из его фармацевтически приемлемых солей и эффективной дозы соединения, показанного для симптоматического лечения DAT, или одной из его фармацевтически приемлемых солей, причем вышеуказанные введения осуществляют одновременно, последовательно или распределено во времени, и эффективные дозы действующих начал могут содержаться в отдельных разовых формах введения или, когда действующие начала вводят одновременно, оба действующих начала предпочтительно содержатся в единой фармацевтической форме.
В фармацевтических композициях согласно настоящему изобретению для перорального, подъязычного, подкожного, внутримышечного, внутривенного, чрескожного или ректального введения действующее начало может быть введено в разовых формах введения либо таким, какое есть, например, в лиофилизированной форме, либо в смеси с классическими фармацевтическими носителями, животным и людям для лечения вышеуказанных заболеваний. Соответствующие разовые формы введения включают формы для введения перорально, такие как таблетки, в случае необходимости расслаиваемые, желатиновые капсулы, порошки, гранулы и оральные растворы или суспензии; формы для введения подъязычно или трансбуккально, формы для подкожного, внутримышечного или внутривенного введения, формы для локального введения и формы для ректального введения.
Когда получают твердую композицию в форме таблеток, главный активный ингредиент смешивают с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные продукты. Таблетки можно покрывать сахарозой или другими соответствующими веществами или еще их можно обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заданное количество действующего начала.
Препарат в виде желатиновых капсул получают путем смешения активного ингредиента с разбавителем и внесения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира может содержать активный ингредиент вместе с подслащивающим, предпочтительно некалорийным средством, метилпарабеном и пропилпарабеном в качестве антисептиков, так же, как с улучшающим вкус веществом и соответствующим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный ингредиент в смеси с диспергаторами или смачивателями, или суспендирующими агентами, как поливинилпирролидон, также, как с подслащивающими веществами или улучшающими вкус веществами.
Для ректального введения используют свечи, которые получают при применении связующих, плавящихся при ректальной температуре, как, например, масло какао или полиэтиленгликоли.
Для парентерального введения используют водные суспензии, солевые растворы и стерильные растворы для инъекций, которые содержат фармакологически приемлемые диспергаторы и/или смачиватели, как, например, пропиленгликоль или бутиленгликоль.
Действующее начало также может быть переведено в лекарственную форму в виде микрокапсул в случае необходимости вместе с одним или несколькими носителями или добавками.
В фармацевтических композициях согласно настоящему изобретению действующее начало также может находиться в виде комплекса-включения в циклодекстринах, их простых или сложных эфирах.
Нижеследующие примеры подробнее иллюстрируют изобретение, однако не ограничивая его объема.
Пример 1
1-[2-(3,4-Диэтилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
1a) 1-Бром-2-(3,4-диэтилфенил)этан
Смесь 4,4 г (0,033 моль) 3,4-диэтилбензола, 50 мл дихлорметана, 8,8 г (0,044 моль) бромацетилбромида охлаждают до температуры 0-5oС и добавляют в нее 5,0 г (0,037 моль) трихлорида алюминия. Перемешивают при температуре 0-5oС в течение одного часа, затем оставляют стоять в течение ночи при комнатной температуре. Выливают в смесь воды со льдом, экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. 2,9 г (0,011 моль) полученного масла смешивают с 6 мл (0,079 моль) трифторуксусной кислоты и 6,7 мл (0,057 моль) триэтилсилана и нагревают при температуре 80oС в течение четырех часов. Затем добавляют водный насыщенный раствор гидрокарбоната натрия вплоть до основного значения рН, экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Таким образом полученное сырое масло очищают путем хроматографической обработки на колонке с силикагелем, элюируя циклогексаном. Получают целевое соединение.
1б) 1-[2-(3,4-Диэтилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
Смесь 2,6 г (0,001 моль) 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина, 60 мл бутанола, 4,1 г (0,025 моль) растертого безводного карбоната калия и 2,6 г (0,00113 моль) полученного в предыдущей стадии продукта кипятят с обратным холодильником в течение 5 часов. Растворитель выпаривают при пониженном давлении, остаток обрабатывают этилацетатом, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Гидрохлорид таким образом полученного масла получают путем обработки насыщенным раствором хлороводорода в изопропаноле. Получают 1,6 г целевого соединения. Т. пл. составляет 220-222oС.
Пример 2
1-[2-(3-Метил-4-пентилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и 1-[2-(4-метил-3-пентилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и их оксалаты
2а) 1-Метил-2-пентилбензол
К раствору 50 мл (0,1 моль) 2 М раствора н-бутилмагнийхлорида в тетрагидрофуране в атмосфере азота прикапывают 4,7 г (0,035 моль) талевого альдегида. Смесь спонтанно нагревается до температуры 40-45oС. Перемешивают при комнатной температуре в течение одного часа, выливают в насыщенный раствор хлорида аммония. Экстрагируют диэтиловым эфиром, экстракт промывают водой, сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Таким образом полученное масло очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексен/этилацетат = 7/3. Выделяют продукт с более высоким показателем Rf. Получают 2,0 г масла. Сырой реакционный продукт растворяют в 25 мл эталона и добавляют туда 1 мл концентрированной серной кислоты и 0,15 г 10%-ного палладия-на-угле. Гидрируют при комнатной температуре в течение 7 часов. Катализатор отфильтровывают, растворитель выпаривают при пониженном давлении и остаток обрабатывают этилацетатом. Промывают водным раствором гидрокарбоната натрия, сушат и растворитель выпаривают при пониженном давлении. Получают 1,35 г целевого продукта.
2б) 1-Бром-2-(3-метил-4-пентилфенил)этан и 1-бром-2-(4-метил-3-пентилфенил)этан
Смесь 1,17 г (0,0054 моль) полученного в предыдущей стадии продукта с 0,62 мл (0,0072 моль) бромацетилбромида охлаждают до температуры 0-5oС и добавляют к ней 0,81 (0,006 моль) трихлорида алюминия. Перемешивают при температуре 0-5oС в течение одного часа и затем при комнатной температуре в течение четырех часов. Выливают на лед, разделяют две фазы, органическую фазу промывают водой, сушат ее и растворитель выпаривают при пониженном давлении. Остаток растворяют в 2,9 мл трифторуксусной кислоты и добавляют туда 3,1 мл (0,0267 моль) триэтилсилана и смесь нагревают при температуре 80oС в течение 5 часов. Выливают в водный раствор гидрокарбоната натрия и экстрагируют диэтиловым эфиром. Экстракт промывают водой, сушат над сульфатом натрия. Получают смесь целевых соединений.
2в) 1-[2-(3-Метил-4-пентилфенил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и 1-[2-(4-метил-3-пентилфенил)этил]-4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин и их оксалаты
Смесь 0,7 г (0,0031 моль) 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина, 16 мл бутанола, 0,9 г (0,0065 моль) растертого безводного карбоната калия и полученного в предыдущей стадии продукта (в теоретически рассчитанном количестве 0,0054 моль) кипятят с обратным холодильником в течение 6 часов. Растворитель выпаривают при пониженном давлении, обрабатывают этил ацетатом, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Таким образом полученное масло очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексен/этилацетат =7/3. Выделяют два продукта с похожими значениями Rf. Продукт с более высоким значением Rf соответствует 1-[2-(3-метил-4-пентилфенил)-этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридину. Оксалат получают в ацетоне. Получают 0,12 г продукта. Т. пл. составляет 140-143oС. Продукт с меньшим значением Rf соответствует изомеру 1-[2-(4-метил-3-пентилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридину. Оксалат получают в ацетоне. Продукт кристаллизуют из ацетона. Получают 0,08 г продукта. Т. пл. составляет 167-169oС.
Пример 3
1-[2-(3,4-Диэтилфенил)этил] -4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидропиридингидрохлорид
3а) (1-Бензил-1,2,3,6-тетрагидропирид-4-ил)трибутилстаннан
При комнатной температуре в течение трех часов перемешивают смесь 15,85 г (0,0837 моль) 1-бензил-4-пиперидона в 140 мл безводного диметоксиэтана и 25 г (0,0837 моль) трисилилгидразина в 140 мл безводного диметоксиэтана. Растворитель выпаривают при пониженном давлении. Остаток обрабатывают с помощью 420 мл безводного гексана и добавляют туда 420 мл безводного тетраметилэтилендиамина. Смесь охлаждают до температуры -78oС и прикапывают к ней 156 мл н-бутиллития (0,25 моль) (1,6 М раствор в гексане). Спустя примерно 30 минут смесь оставляют стоять до повышения температуры вплоть до 0oС и перемешивают в течение 15 минут. Затем к реакционной смеси добавляют 45 мл (0,167 моль) трибутилстаннанхлорида. Спустя 1 час с крайней предосторожностью добавляют смесь воды со льдом. Экстрагируют диэтиловым эфиром, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Получают 70 г сырого продукта, который очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексен/этилацетат = 95/5. Получают целевое соединение в виде масла.
1Н-ЯМР (СDСl3), δ(м.д.): 0,84 (м, 9Н, СH3); 1,19-1,58 (м, 18Н, СН2-цепь); 2,31 (м, 2Н); 2,53 (м, 2Н); 3,02 (м, 2Н); 3,56 (с, 2Н, бензильный метилен); 5,76 (м*, 1Н); 7,18-7,41 (м, 5Н, ароматические).
* полосы-сателлиты 3Jцис(1Н-117Sn) и 3Jцис(1Н-119Sn) .
3б) 1-Бензил-4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидропиридин
18,5 г (0,04 моль) полученного в предыдущей стадии соединения растворяют в атмосфере азота в 200 мл безводного диметилформамида. К раствору добавляют 11,8 г (0,08 моль) 2,6-дихлорпиридина, 0,64 г Pd (II) (Рh3Р)2Сl2, 4,38 г (0,04 моль) тетраметиламмонийхлорида и 2,76 г (0,02 моль) карбоната калия. Нагревают при температуре 110oС в течение 6 часов, затем смесь выливают в 100 мл 5%-ного раствора серной кислоты. Экстрагируют диэтиловым эфиром, в водную фазу добавляют гидроксид аммония до щелочного значения рН и экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Остаток очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексен/этилацетат =1/1.
Получают целевое соединение. Т. пл. составляет 100-102oС.
3в) 4-(6-Хлорпирид-2-ил)-1,2,3,6-тетрагидропиридингидрохлорид
Раствор 7,0 г (0,024 моль) полученного в предыдущей стадии соединения в 110 мл дихлорэтана охлаждают до температуры 0-5oС и добавляют туда 5,8 мл (0,054 моль) хлорэтилхлорформиата. Перемешивают в течение 5 минут, затем кипятят с обратным холодильником в течение полутора часов. Растворитель выпаривают при пониженном давлении, остаток обрабатывают 100 мл метанола и кипятят с обратным холодильником в течение 1 часа. Растворитель выпаривают, остаток обрабатывают изопропанолом и твердое вещество отфильтровывают. Получают целевое соединение, которое кристаллизуют из 90%-ного эталона. Т. пл. составляет 305-307oС.
3г) 1-[2-(3,4-Диэтилфенил)этил] -4-(6-хлорпирид-2-ил)-1,2,3,6-тетрагидропиридингидрохлорид
Следуют методике, описанной в примере 1б), но используя полученный в предыдущей стадии продукт вместо 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина; получают целевое соединение. Т. пл. составляет 234-236oС.
Примеры 4-13
Следуя описанной в примере 2 методике, но используя соответствующий галогенид магния, получают следующие соединения:
1-[2-(3-этил-4-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 4);
1-[2-(4-этил-3-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 5);
1-[2-(3-этил-4-пропилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 6);
1-[2-(4-этил-3-попилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 7);
1-[2-(3-бутил-4-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 8);
1-[2-(4-бутил-3-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 9);
1-[2-(3-изобутил-4-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 10);
1-[2-(4-изобутил-3-метилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 11);
1-[2-(3-изобутил-4-этилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 12);
1-[2-(4-изобутил-3-этилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин (Пример 13).
Пример 14
1-[2-(6-Метил-3-бифенилил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридин
Следуя описанной в примере 2 методике, но используя фениллитий вместо н-бутилмагнийхлорида, получают целевое соединение.
Пример 15
1-[2-(3,4-Диметилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
15а) 1-Бром-2-(3,4-диметилфенил)этан
4,5 г (0,03 моль) 3,4-Диметилацетофенона в 12 мл метанола охлаждают до температуры 0-5oС и прикапывают туда 1,5 мл (0,09 моль) брома. Перемешивают при комнатной температуре в течение 24-х часов, затем оставляют стоять в течение ночи при комнатной температуре. Метанол выпаривают и смесь очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексан/этилацетат = 95/5. 5,3 г (0,013 моль) полученного таким образом продукта смешивают с 12,5 мл (0,162 моль) трифторуксусной кислоты и 18,7 мл (0,011 моль) триэтилсилана и нагревают при температуре 80oС в течение 1 часа. Затем добавляют водный насыщенный раствор гидрокарбоната натрия до щелочного значения рН, экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Получают 5,2 г целевого соединения.
15б) 1-[2-(3,4-Диметилфенил)этил] -4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
Смесь 1,8 г (0,0068 моль) 4-(3-трифторметилфенил)-1,2,3,6-тетрагидропиридина, 25 мл бутанола, 2,4 г (0,017 моль) растертого безводного карбоната калия и 2 г (0,0094 моль) полученного в предыдущей стадии продукта кипятят с обратным холодильником в течение 6 часов. Растворитель выпаривают при пониженном давлении, остаток обрабатывают этилацетатом, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Продукт очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексан/этилацетат = 7/3. Гидрохлорид полученного таким образом масла получают путем обработки насыщенным раствором хлороводорода в изопропаноле. Получают 1,1 г целевого соединения. Т. пл. составляет 270-272oС.
Пример 16
1-[2-(3,4-Диэтилфенил)этил-4-(2-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
16а) 4-Гидрокси-4-(2-трифторметилфенил)пиперидингидрохлорид
3,25 г (0,135 моль) магния смешивают с иодом в количестве на кончике шпателя и прикапывают туда раствор 30,4 г (0,135 моль) 2-бром-1-трифторметилбензола в 125 мл тетрагидрофурана. Перемешивают в течение 1 часа при комнатной температуре и к этой смеси прикапывают 10,1 г (0,041 моль) бензилпиперидона. Перемешивают в течение 1 часа при комнатной температуре и к смеси добавляют насыщенный раствор хлорида аммония. Экстрагируют диэтиловым эфиром, органическую фазу сушат и растворитель выпаривают при пониженном давлении. Продукт очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексан/этилацетат. Получают 6,8 г 1-бензил-4-гидрокси-4-(2-трифторметилфенил)пиперидина, который гидрируют в 75 мл 95%-ного эталола при кислом значении рН за счет добавки соляной кислоты в присутствии 0,7 г 10%-ного палладия-на-угле, нагревая при температуре 60oС в течение 8 часов. Катализатор отфильтровывают и таким образом получают 2,1 г целевого продукта. Т.пл. составляет 247-251oС.
16б) 4-(2-Трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
2,0 г (0,007 моль) полученного в предыдущей стадии продукта растворяют в 12 мл ледяной уксусной кислоты. К полученному раствору прикапывают 3 мл концентрированной серной кислоты и нагревают при температуре 100oС в течение двух часов. Выливают на лед, к смеси добавляют концентрированный раствор гидроксида натрия до щелочного значения рН и экстрагируют дихлорметаном. Органическую фазу сушат и растворитель выпаривают при пониженном давлении. Продукт обрабатывают 15 мл изопропанола и получают 4-(2-трифторметилфенил)-1,2,3,6-тетрагидропиридин. Гидрохлорид получают с помощью раствора хлороводорода в изопропаноле. Получают 0,9 г целевого соединения. Т.пл. составляет 213-215oС.
16в) 1-[2-(3,4-Диэтилфенил)этил] -4-(2-трифторметилфенил)-1,2,3,6-тетрагидропиридингидрохлорид
0,4 г (0,0015 моль) полученного в предыдущей стадии продукта и 0,52 г (0,0037 моль) безводного карбоната калия в 12 мл бутанола кипятят с обратным холодильником в течение 30 минут. Затем туда добавляют 0,41 г (0,0017 моль) полученного в примере 1а) продукта и кипятят с обратным холодильником в течение 6 часов. Растворитель выпаривают, остаток обрабатывают этилацетатом, промывают водой, органическую фазу сушат и растворитель выпаривают при пониженном давлении. Продукт очищают путем хроматографической обработки на колонке с силикагелем, элюируя смесью циклогексан/этилацетат =8/2. Таким образом получают 1-[2-(3,4-диэтилфенил)этил] -4-(2-трифторметилфенил)-1,2,3,6-тетрагидропиридин. Гидрохлорид получают с помощью раствора хлороводорода в изопропаноле. Получают целевое соединение. Т. пл. составляет 184-185oС.
Результаты фармакологического теста на выживаемость нейронов представлены в процентах выживания. Тест проводился по методике, описанной выше. Получены следующие результаты:
Соединение - Выживание нейронов (%)
Пример 1 - 156,8+7,7
Соединение 1 примера 2 - 176,4+18,7
Соединение 2 примера 2 - 183,7+11,5
Эти значения приведены для оптимальной концентрации 250 мкМ и выражены по отношению к контролю, взятому за 100%.
Другие соединения, описанные в заявке, были исследованы в таких же условиях культивирования и показали, что в их присутствии процент выживания нейронов составляет от 164 до 195%. При этом большинство соединений показали выживаемость нейронов, близкую 174,8+8.
Описанный тест является общепринятым тестом на нейрогенез и позволяет охарактеризовать нейротрофическую и нейрозащитную активность.
Заявитель предлагает пример композиции для подтверждения композиции по пункту 11.
Этот пример иллюстрирует состав желатиновой капсулы, содержащей соединение согласно изобретению и соединение, показанное для симптоматического лечения старческого слабоумия типа болезни Альцгеймера (DAT).
Пример 17.
Соединение примера 1 - 2 мг
Донелезил - 10 мг
Коллоидная двуокись кремния - 0,200 мг
Стеарат магния - 0,400 мг
Крахмал - 141,200 мг
Кристаллическая целлюлоза - 26 мгп
Изобретение относится к соединениям формулы (I):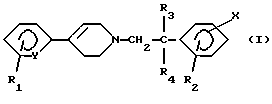
в которой Y означает -СН- или -N-; R1 означает водород, галоген, трифторметил, (С1-С4)-алкил; R2 означает метил или этил; каждый из R3 и R4 означает водород; Х означает (С1-С6)-алкил или фенил. Эти соединения обладают нейротрофической и нейрозащитной активностью и могут быть использованы в медицине. 3 с. и 7 з.п.ф-лы.

в которой Y означает -СН- или -N-;
R1 означает водород, галоген, трифторметил или (С1-С4)алкил;
R2 означает метил или этил;
каждый из R3 и R4 означает водород;
Х означает (C1-C6)-алкил, или фенил, или их соли.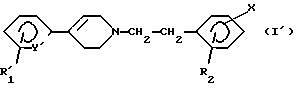
в которой R'1 означает трифторметил и Y' означает СН или R'1 означает хлор и Y' означает азот, a R2 и Х имеют указанные для соединений формулы (I) в п. 1 значения, или его соли.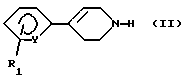
в которой Y и R1 имеют указанные для соединений формулы (I) в п. 1 значения,
вводят во взаимодействие с соединением формулы (III)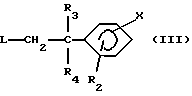
в которой R2, R3, R4 и X имеют указанные для соединений формулы (I) в п. 1 значения и L означает удаляемую группу, и (б) полученное соединение формулы (I) выделяют и, в случае необходимости, превращают его в одну из его солей.
| Аппарат для термообработки мелкодисперсного материала | 1971 |
|
SU458696A1 |
| 1971 |
|
SU412901A1 | |
| СПОСОБ ЗАХОРОНЕНИЯ ТОКСИЧНЫХ ПРОМЫШЛЕННЫХ ОТХОДОВ | 1991 |
|
RU2070102C1 |
| RU 94046310 А1, 27.10.1996. | |||

