Изобретение относится к технологии производства бензилового спирта, в частности к способу непрерывного получения бензилового спирта путем гидролиза бензилхлорида водой при повышенных температурах.
Гидролиз бензилхлорида общеизвестный. Целый ряд литературных источников посвящен этой проблеме с целью выяснения механизма действия и разработки аналитических методов. При этом, как правило, используют гомогенные смеси, состоящие из бензилхлорида, воды и водорастворимого агента растворения, такого, как, например, спирт, ацетон, диоксан или уксусная кислота (см., например, J. Chem. Soc. 1954, стр. 1840 и сл.; Z. Naturf. 1946 (1), стр. 580 - 584; J. Chem. Soc. 1957, стр. 4747 и сл.; Tetrahedron 1957 (1), стр. 129 - 144; Rec. 48, стр. 227 и сл. (1929) и 49, стр. 667 и сл. (1930)). В некоторых случаях гидролиз также проводят в присутствии гидроокисей щелочного металла, карбонатов щелочного металла или карбонатов щелочноземельного металла (см. , например, J. Am. Chem. Soc. 62, стр. 2481 (1940); Rec. 53, стр. 891 и сл. и 869 и сл. (1934)), причем последние из приведенных источников показывают, что гидролиз в присутствии основных соединений ускоряется. Во всех указанных источниках выделение и определение продуктов гидролиза никакой роли не играют.
Имеются, однако, также исследования гидролиза бензилхлорида одной лишь водой, в которых описываются продукты гидролиза. Согласно Ann. 139, стр. 307 и сл. (1866) при гидролизе водой бензилхлорид при температуре 190oC превращается, в частности, в углеводороды и дибензиловый эфир.
В более умеренных условиях (см. Ann. 196, стр. 353 (1879)) при температуре 100 - 110oC и полной конверсии получают выход - правда, очень сильно разбавленного - бензилового спирта, равный 76% теории. Остаток представляет собой высококипящие компоненты, которые не были охарактеризованы.
Тот факт, что реакция конверсии в присутствии щелочи происходит быстрее и успешнее, в промышленности уже рано привело к стремлению связывать образующуюся во время гидролиза бензилхлорида соляную кислоту. Так, например, в патенте Германии N 484662 рекомендуется проведение реакции в присутствии карбоната кальция, а в патенте US N 2221882 реакцию осуществляют сначала в присутствии содового раствора, а потом - натрового щелока. Выход составляет значительно больше 90% теории.
Карбонат натрия очевидно зарекомендовал себя в качестве основания и применялся и впоследствии. Дальнейшие разработки в данной области связаны с техническим выполнением непрерывного осуществления указанного основного гидролиза бензилхлорида (см. заявку DE N 2101810, заявку ЕР N 64486 и патент ГДР N 161067).
Недостатки щелочного гидролиза заключаются, однако, в дополнительном использовании карбоната натрия, образовании хлорида натрия и образовании больших количеств отработанного водного щелока, которые приходится обезвреживать.
Поэтому задачей настоящего изобретения является разработка способа, позволяющего работать без применения карбоната натрия и, таким образом, предотвращающего образование поваренной соли, вызывающей проблемы с обезвреживанием сточных вод. При этом выход бензилового спирта должен был бы хотя бы не хуже уровня техники.
Поставленная задача решается предлагаемым способом непрерывного получения бензилового спирта, который заключается в том, что
а) бензилхлорид и воду в молярном соотношении 1:(10 - 70) подают в реактор с диспергирующими приспособлениями, где имеется температура 80 - 180oC, и смеси дают реагировать до достижения степени конверсии бензилхлорида, равной 30 - 90%,
б) продукт реакции после выхода из реактора разделяют на водную и органическую фазы,
в) органическую фазу в первом перегонном узле разделяют на бензилхлорид и сырой бензиловый спирт,
г) данный бензилхлорид возвращают в реактор,
д) сырой бензиловый спирт во втором перегонном узле рафинируют до высокочистого бензилового спирта, при этом получается содержащий побочные продукты (в частности дибензиловый эфир) кубовый продукт,
е) содержащую разбавленную соляную кислоту водную фазу из сепаратора в экстракторе обрабатывают растворителем, поглощающим еще растворенные в разбавленной соляной кислоте органические компоненты, в частности бензиловый спирт,
ж) экстракт в третьем перегонном узле освобождают от растворителя, возвращающегося в экстрактор, и
з) кубовый продукт третьего перегонного узла подают в первый перегонный узел для получения бензилового спирта.
Пригодным сырьем для предлагаемого способа является бензилхлорид, общедоступный в результате хлорирования в боковую цепь.
Само собой разумеется, что также пригоден бензилхлорид, замещенный галогеном, таким, как хлор, карбоксилом, цианом, метоксилом, метилом и нитрогруппой. Предпочтительным является, однако, незамещенный бензилхлорид.
Пример осуществления способа согласно изобретению представлен на фиг. 1. Температура в реакторе R составляет 80 - 180oC, предпочтительно 100 - 170oC, особенно предпочтительно 110 - 150oC. Реактор может работать в изотермическом режиме за счет того, что теплоту реакции отводят охлаждением, или же преимущественно в адиабатическом режиме за счет того, что исходные соединения E (BCl = бензилхлорид и H2O) по линии 1 подают в смеситель М с температурой, обеспечивающей нагрев реакционной смеси в реакторе до требуемой температуры реакции и выход из реактора по линии 2 с той же самой температурой.
Применяют бензилхлорид BCl и воду H2O в молярном соотношении в диапазоне от 1:10 до 1:70, предпочтительно 1:15 до 1:55, особенно предпочтительно 1:20 до 1:50, в частности 1:25 до 1:50.
Реакцию целесообразно прекращают при достижении степени конверсии бензилхлорида, равной 35 - 90%, предпочтительно 40 - 85%, особенно предпочтительно 45-80%.
В качестве реактора R можно использовать простой котел с мешалкой, который в условиях реакции обеспечивает тонкое диспергирование реакционной смеси.
Более целесообразно, однако, употреблять каскад из котлов с мешалкой, состоящий из 2-7, предпочтительно 3 - 5 котлов.
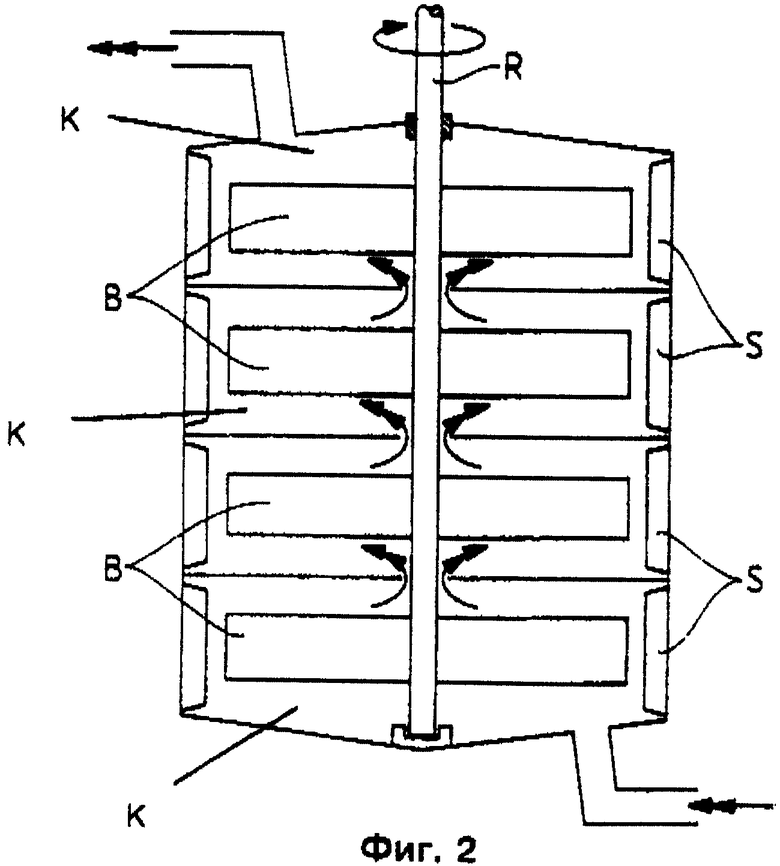
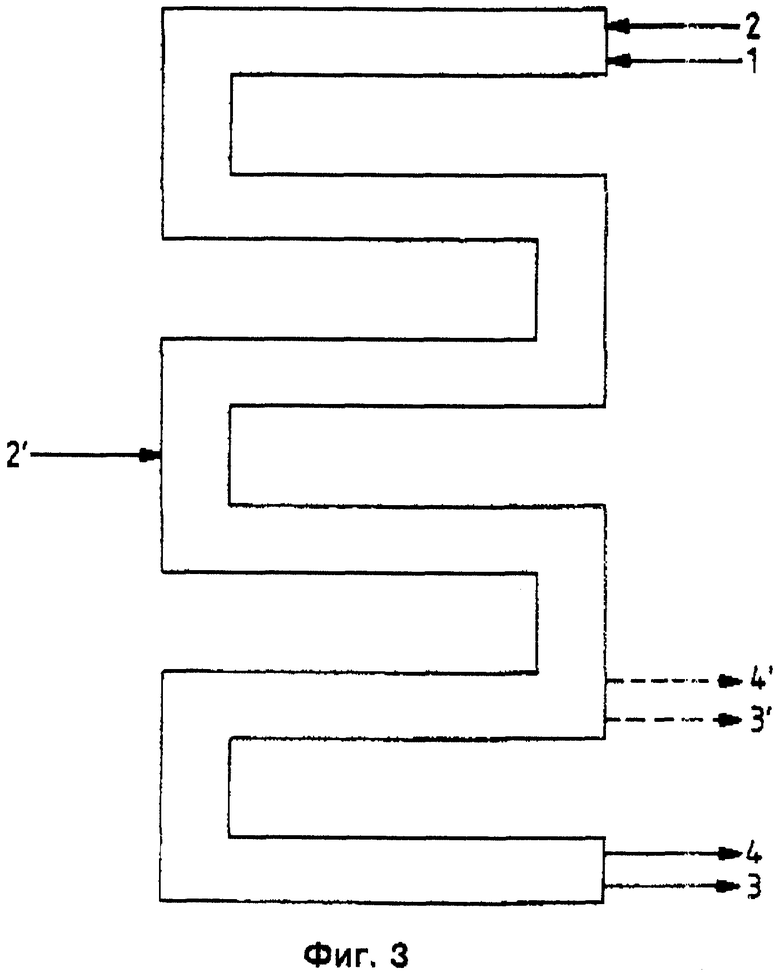
Преимущественными являются и камерные реакторы (см. фиг. 2), в которых перемешивание в отдельных камерах К осуществляется мешалкой R, снабженной 2 - 4 лопатками В по камере. Для улучшения диспергирования каждая камера оснащена двумя или больше турбулизаторами. Камеры непосредственно связаны друг с другом без возможности значительного обратного смешения (например, в случае наличия потока двухфазной смеси) (см. фиг. 2). Реакторы снабжены 2-10, предпочтительно 3 - 8, особенно предпочтительно 3 - 6 камерами и трубчатыми реакторами (фиг. 3), через которые реакционную смесь пробкообразно пропускают с достаточной для возникновения состояния турбулентности скоростью и в которых посредством известных элементов смешивания и диспергирования бензилхлорид как можно более тонко распределяется в воде. При этом реакционную смесь, состоящую из подаваемой по линии 1 органической фазы и подаваемой по линии 2 водной фазы, можно дополнять по линии 2' в любых местах трубчатого реактора. Реакционную смесь бензилхлорида и бензилового спирта, с одной стороны, и разбавленной соляной кислоты, с другой стороны, можно отводить - в зависимости от времени пребывания в трубе - в концевой части реактора по линиям 3 и 4, соответственно, или же раньше по линиям 3' и 4', соответственно. Специалист может вычислять и конструировать подобные трубчатые реакторы по известным методам (см. Perry's Chemical Engineers Handbook, изд-во McGraw Hill Нью-Йорк, 6-ое изд., 1984 г.).
Диаметр капель диспергированной в воде органической фазы может составлять, например, 100 - 400 мкм, предпочтительно 150 - 300 мкм.
Если реакцию проводят при температурах выше 100oC, то необходимо употреблять узлы, в которых можно работать при давлении приблизительно до 50 бар, предпочтительно 30 бар, особенно предпочтительно 20 бар.
Реакционную смесь, выходящую из реактора по линии 2, целесообразно при повышенной температуре, равной 50 - 100oC, предпочтительно 55 - 95oC, в сепараторе Т1 разделяют на органическую фазу и водную разбавленную соляную кислоту, концентрация которой зависит от молярного соотношения реагентов и от достигнутой степени конверсии и составляет, например, 2 - 7 вес.% соляной кислоты. Разделение фаз можно значительно ускорять, в случае необходимости, путем применения сепараторов, например коагуляторов, описанных, например, в источниках Ullmann's Encyclopedia of Ind. Chem. Unit Operations II том В3, стр. 6 - 31, и Chem. Ing. Techn. (1986) 58, стр. 449 и сл.
Если реакцию проводят при температурах выше 100oC, например 130oC, то реакционную смесь перед входом в сепаратор Т1 можно охлаждать путем испарения сбросом давления (2' ---> EV ---> 3'). При этом часть непрореагировавшего бензилхлорида и часть воды из реакционной смеси могут испаряться и возвращаться в реактор по линии 16'. Таким образом можно экономить энергию для нагревания исходных соединений и охлаждающую жидкость для реакционной смеси, а образовавшуюся соляную кислоту можно слегка концентрировать.
Преимущественным является и охлаждение отводимого по линии 2 продукта реакции перед входом в сепаратор Т1 за счет того, что в теплообменнике WT отводят избыточное тепло и с его помощью повторно нагревают выходящую по линии 6 из экстрактора Ех водную фазу либо непосредственным обменом (через 6' ---> 6'') в теплообменнике WT, либо через цикл WK теплоносителя, чтобы путем отгонки в испарителе V по меньшей мере частично по линии 8 отводить остаточный органический материал из водной фазы.
Органическая фаза, отводимая по линии 4 из сепаратора Т1, содержит прежде всего бензиловый спирт и бензилхлорид, дибензиловый эфир в качестве побочного продукта и - еще в растворенном виде - небольшие количества разбавленной соляной кислоты. Соотношение органических веществ определяется достигнутой в реакторе степенью конверсии. Так, содержание бензилового спирта может составлять, например, 40 - 75%, содержание дибензилового эфира - 0,5-6%, а концентрация соляной кислоты - 0,5 - 8%.
Разделение смесей перегонкой D1 не само собой разумеется, так как при повышенной температуре из бензилхлорида и бензилового спирта, а также из бензилового спирта и соляной кислоты могут образоваться побочные продукты, в частности дибензиловый эфир. Согласно источникам литературы эти процессы конденсации осуществляются также во время перегонки подобного продукта гидролиза, как только содержание бензилхлорида составляет выше 1% (см. Chem. Prum. 32 (1982), стр. 586; процитировано в журнале С.А. 98 106890). Для подавления дополнительного образования дибензилового эфира во время перегонки рекомендуется требующее больших расходов взаимодействие остатков бензилхлорида с соединениями азота, например гексаметилентетрамином (см. патент CS N 216042: процитировано в журнале С.А. 102, реф. 45606t). Разделение смеси, содержащей бензилхлорид, бензиловый спирт, дибензиловый эфир и водную соляную кислоту, согласно изобретению осуществляется за счет того, что данную смесь подают по боковому патрубку в непрерывно действующую перегонную колонну с отгонной частью и укрепляющей частью. При этом эксплуатация перегонной колонны происходит при давлении 1 - 950 мбар в верхней части колонны. Из верхней части перегонной колонны отводят смесь, состоящую в основном из бензилхлорида и водной соляной кислоты, а из нижней части перегонной колонны - смесь, состоящую в основном из бензилового спирта и дибензилового эфира.
Поэтому выходящую по линии 4 из сепаратора Т1 смесь сбоку подают в первую перегонную колонну D1. Последняя работает при давлении 1 - 950 мбар, предпочтительно 10 - 500 мбар, особенно предпочтительно 20 - 300 мбар в верхней части колонны. При этом известным специалисту образом в зависимости от установленного давления в верхней части колонны и в зависимости от состава подлежащей разделению смеси в нижней части колонны устанавливается температура, равная 30 - 200oC, предпочтительно 60 - 180oC, особенно предпочтительно 70 - 165oC. Соответствующим образом устанавливается температура в верхней части колонны, составляющая 20 - 175oC, предпочтительно 50 - 155oC, особенно предпочтительно 60 - 140oC. Температура верха колонны при этом всегда ниже температуры нижней части колонны. Боковую подачу подлежащей разделению смеси осуществляют в месте перегонной колонны, в котором температура составляет 25 - 195oC, предпочтительно 55 - 175oC, особенно предпочтительно 65 - 160oC. Установление температурного градиента специалисту известно. Градиент зависит, среди прочего, от состава смеси, а также от ее предварительного нагрева. Перегонную колонну можно эксплуатировать с нагрузкой 0,05 - 1,0 кг органической фазы в подлежащей разделению смеси на литр пустого объема колонны в час. Нагрузка предпочтительно составляет 0,15 - 0,9 кг/л•ч, особенно предпочтительно 0,25 - 0,8 кг/л•ч.
Дополнительное образование дибензилового эфира согласно изобретению практически предотвращается и снижается до незначительных величин, равных меньше 4%, предпочтительно меньше 2%, особенно предпочтительно меньше 0,5%, в пересчете на количество бензилового спирта.
Из верхней части колонны D1 по линии 16 отводится смесь всего непрореагировавшего бензилхлорида и всей еще растворенной соляной кислоты, которая возвращается в реактор R, а из нижней части колонны по линии 17 отводится сырой бензиловый спирт, который практически больше не содержит бензилхлорида, зато все высококипящие компоненты, в частности дибензиловый эфир.
Колонна может быть снабжена известными встроенными элементами, такими, как, например, колпачковые тарелки, ситчатые тарелки и тарелки другой конструкции, и разного рода насадками.
Кубовый продукт колонны D1 по линии 17 или же 17' подают во второй перегонный узел, состоящий из одной колонны D2'' или из двух перегонных колонн D2 + D2'. Если используется лишь одна колонна D2'', то кубовый продукт перегонки бензилхлорида, отводимый из колонны D1 по линии 17, практически свободный от низкокипящих компонентов, в частности от бензилхлорида. Из верха колонны D2'' по линии 21' отводят высокочистый бензиловый спирт ВА с концентрацией свыше 99,99%, а из нижней части по линии 20' отводят смесь высококипящих компонентов, состоящую в основном из дибензилового эфира DB, остаточного бензилового спирта и небольшого количества других высококипящих компонентов, которую можно, в случае необходимости, подвергать дальнейшей периодической обработке.
Если, однако, второй перегонный узел состоит из двух колонн D2 + D2', то в первой колонне D2 отводимый по линии 17 сырой бензиловый спирт можно еще освобождать от отводимых по линии 18 незначительных количеств низкокипящих компонентов, в частности бензилхлорид, а во второй колонне D2' свободный от низкокипящих компонентов и подаваемый по линии 19 сырой бензиловый спирт вышеописанным образом разделяют на высокочистый бензиловый спирт, отводимый по линии 21, и кубовые продукты, отводимые по линии 20.
Не поддающиеся конденсации низкокипящие компоненты и отходящие газы можно отводить из перегонных колонн по линиям 22'- 22IV и подавать на обезвреживание, например, в установке AG сжигания отходящих газов.
Эксплуатация колонн D2 - D2'' осуществляют под вакуумом при давлении 500 - 10 мбар, предпочтительно 250 - 10 мбар, особенно предпочтительно 100 - 10 мбар.
Разбавленную соляную кислоту, отводимую из сепаратора Т1 по линии 5, в экстракторе Ех обрабатывают пригодным растворителем для получения растворенных в водной фазе органических компонентов, прежде всего бензилового спирта, который растворяется - в зависимости от конкретных условий - приблизительно на 2-4% в разбавленной соляной кислоте, подаваемой по линии 5, а также бензилхлорида, который, однако, лишь незначительно растворен. Температура при экстракции Ех составляет 20 - 100oC, предпочтительно 40 -95oC, особенно предпочтительно 50 - 95oC.
Пригодными растворителями являются углеводороды с 4 - 9 атомами углерода, галогенированные углеводороды с 1-8 атомами углерода, предпочтительно ароматические углеводороды, как бензол, толуол, ксилолы, этилбензол и кумол, галогенированные углеводороды как хлористый метилен, хлороформ, дихлорэтан, трихлорэтилен, хлорбензол и бензилхлорид. Особенно предпочитают толуол и бензилхлорид.
Если имеются потери растворителя L, то они могут дополняться по линии 14' на стадии экстракции.
Экстракцию Ех проводят противотоком в непрерывном режиме в известных аппаратах, описанных, например, в источнике Ullmann's Encyclopedia of Ind. Chem. Unit Operations II том В3, стр. 6-14 и сл. В качестве примеров можно назвать насадочные колонны, тарельчатые колонны, пульсационные тарельчатые колонны, пульсационные насадочные колонны, смесительно-отстойные системы аппаратов, колонны с вращающимися встроенными элементами и центробежные экстракторы.
Экстракт, отводимый по линии 13, в третьей перегонной колонне D3 освобождают от экстрагирующего агента и следов разбавленной соляной кислоты, выходящий из верха колонны продукт по линии 14 возвращают в экстрактор Ех и кубовый продукт колонны D3 отводят по линии 15 и вместе с потоком по линии 4 подают в первую перегонную колонну D1, где разделяются бензиловый спирт и бензилхлорид. Третья перегонная колонна D3 работает при давлении 750 - 50 мбар, предпочтительно 500 - 150 мбар, особенно предпочтительно 400 - 200 мбар.
С помощью разбавленной водной соляной кислоты, отводимой по линии 6 из экстрактора Ех, известным образом эксплуатируют установку AS абсорбции соляной кислоты, в которой, например, подаваемый по линии 11 хлористый водород, полученный в результате хлорирования толуола или ароматов, переводят в отводимую по линии 12 высокопроцентную соляную кислоту, которая является очень чистой и пригодной для других видов технического применения или для электролитической переработки до хлора.
Перед подачей отводимой из экстрактора Ех по линии 6 разбавленной соляной кислоты в абсорбционную установку AS целесообразно как можно более полно отделять отгонкой в испарителе V еще растворенные в отводимой из экстрактора Ех по линии 6 соляной кислоте остатки органических соединений, таких, как, например, толуол, бензилхлорид или следы бензилового спирта. Разбавленную соляную кислоту, практически свободную от органических соединений, по линии 7 подают в абсорбционную установку AS. Путем перегонки ее полностью освобождают от следов углеводородных соединений и продукт перегонки по линии 8' подают в сепаратор Т2 и разделяют на воду и органические соединения.
Как уже описано выше, испаритель V снабжается отходящим теплом отводимой по линии 2 реакционной смеси посредством теплообмена (WT непосредственно или через WK). Из испарителя по линии 8 выходит смесь, состоящая главным образом из воды и небольшого количества органических веществ, которую в сепараторе Т2, в случае необходимости вместе с отводимой из абсорбционной установки AS по линии 8' смесью воды и органических остатков, разделяют на водную и органическую фазы. Водную фазу по линии 9 через смеситель М возвращают в реактор R, а органическую фазу можно подавать по линии 10 на хлорирование толуола или же возвращать в третью перегонную колонну D3.
Таким образом осуществляется непрерывный способ получения бензилового спирта из бензилхлорида путем гидролиза водой, который не требует применения оснований, не приводит к образованию ни солей, ни отработанных щелочей, требующих обезвреживания, ни значительных количеств химико-органических отходов, поскольку дибензиловый эфир можно применять либо непосредственно в промышленности, например, в каучуковой промышленности, либо вместе с другими высококипящими компонентами, отводимыми по линиям 20 и 20', и органическими соединениями, отводимыми из сепаратора Т2 по линии 10, на стадии хлорирования толуола для взаимодействия с хлором с получением бензотрихлорида и бензоилхлорида.
Бензиловый спирт представляет собой важный, из-за крайне незначительной токсичности имеющий все возрастающее значение продукт для изготовления духов, косметики, лаков, пластификаторов, а также пользующееся большим спросом вспомогательное средство для каучуковой промышленности, красочной промышленности и текстильной промышленности.
В нижеприведенном примере процентные данные относятся к весу; части представляют собой весовые части.
Пример
Бензилхлорид и воду в молярном соотношении 1:35 при температуре 130oC подвергают реакции, которую прекращают при степени конверсии бензилхлорида, равной 60%, после чего реакционную смесь перерабатывают.
Проведение примера поясняется со ссылкой на фиг. 1. При непрерывном осуществлении способа вещества берут в следующих количествах в час:
14,01 части бензилхлорида BCl в качестве сырья Е с температурой около 35oC, 96,75 части воды в качестве сырья Е с температурой около 150oC, а также 10,51 части подаваемой по линии 16 смеси с температурой около 40oC, являющейся продуктом, выходящим из верха перегонной колонны D1, и состоящей из около 88% бензилхлорида, 4% бензилового спирта, 6% воды и следов соляной кислоты и толуола, и около 18,9 части отводимой из сепаратора Т2 по линии 9 смеси с температурой около 40oC, содержащей больше 99% воды и лишь следы бензилхлорида и бензилового спирта.
Вышеуказанные вещества под давлением около 10 бар насосом подают через смеситель М, а затем через каскад, состоящий из 5 последовательно включенных, снабженных турбулизаторами емкостей с мешалкой, в которых происходит интенсивное перемешивание. При этом температура при хорошем изолировании повышается до 130oC.
После выхода из каскада реакционная смесь содержит около 81% воды, 6,6% бензилхлорида, 8,5% бензилового спирта, 0,27% высококипящих компонентов (прежде всего дибензиловый эфир) и около 2,9% соляной кислоты.
Смесь по линии 2 подают в теплообменник WT, где ее охлаждают до около 90oC. Потом смесь подают в сепаратор Т1 с коагулятором, где органическая фаза отделяется от водной фазы. Последнюю (около 121 части, примерно 94% воды, 3,3% соляной кислоты, 2,6% бензилового спирта и следы бензилхлорида, толуола и дибензилового эфира) в экстракторе Ех, являющемся пульсационной колонной, экстрагируют толуолом. Отводимую по линии 6 водную фазу, содержащую около 96% воды, 0,1% бензилового спирта, 3,4% соляной кислоты, небольшое количество толуола и следы бензилхлорида, подвергают отгонке в испарителе V с помощью отходящего тепла реакционной смеси, получаемого в теплообменнике WT, и далее освобождают от остаточных органических соединений. Потом ее отводят по линии 7 и подают в абсорбционную установку AS соляной кислоты, где последние следы органического материала путем перегонки отводят по линии 8'. Эти дистилляты, отводимые по линии 8 из испарителя V, а по линии 8' - из абсорбционной установки AS, в сепараторе Т2 отделяют от отделившихся перегонкой органических соединений (0,15 - 0,2 части), которые по линии 10 подают в колонну D3 или в экстрактор Ех. Водная фаза (> 99%) из сепаратора Т2 по линии 9 возвращается в смеситель М.
Потери толуола L (0,2 - 0,4 части) на стадии экстракции дополняют из сборника по линии 14'.
Толуольный экстракт из экстрактора Ех по линии 13 подают в выпарную колонну D3, из верхней части которой при 200 - 250 мбар толуол отводится, после чего он по линии 14 возвращается на стадию экстракции. Кубовый продукт с около 94% бензилового спирта, 3,6% высококипящих компонентов и остатками толуола и бензилхлорида фракционируют в колонне D1 вместе с органической фазой, отводимой из сепаратора Т1 по линии 4.
Данная органическая фаза, отводимая по линии 4 из сепаратора Т1 (в то время, как по линии 5 из сепаратора Т1 отводится водная фаза), содержит главное количество органического продукта реакции, а именно примерно 19 частей с около 48% бензилхлорида, 46% бензилового спирта, 1,5% высококипящих компонентов, в частности дибензилового эфира, 3,5% воды, меньше 1% толуола и приблизительно 0,1% соляной кислоты. В перегонной колонне D1 ее подвергают перегонке вместе с кубовым продуктом (около 3 частей), отводимым из колонны D3 по линии 15. При давлении 100-150 мбар из верхней части колонны выходят 10,5 части, которые содержат из больше 88% бензилхлорида, около 4% бензилового спирта, 6% воды, 1% толуола и поданную соляную кислоту, и которые возвращаются через линию 16 в смеситель М, то есть в реакцию.
Из нижней части колонны D1 выходят приблизительно 12,5 части сырого бензилового спирта с концентрацией около 97%, содержащего 3% высококипящих компонентов и около 10 ч/млн бензилхлорида. По линии 17 этот кубовый продукт подают в колонну D2, где при давлении 30 - 40 мбар отделяются последние низкокипящие компоненты, которые по линии 18 возвращаются в колонну D1. Кубовый продукт колонны D2 (около 11,8 части) насосом подают по линии 19 в колонну D2' и при 30 - 50 мбар окончательно перерабатывают до высокочистого спирта (11,2-11,3 части; степень чистоты: больше 99,999%). Кубовый остаток (0,5 - 0,6 части) содержит еще примерно 25% бензилового спирта, который отделяют проведением периодической перегонки под вакуумом и возвращают в колонну D2 или D2'. Таким образом выход повышается до 11,4-11,5 части.
Высококипящие компоненты, отводимые по линии 20, в аппарате хлорирования переводят в бензотрихлорид и бензоилхлорид. Через линии 22' до 22V отходящих газов получается незначительная потеря, состоящая в частности из толуола и воды, которую подают в установку сжигания.
Таким образом, из 14,01 части бензилхлорида в час получают около 11,2 части или же после рекуперации бензилового спирта из нижней части колонны D2' - до 11,5 части бензилового спирта в час.
Это соответствует выходу, равному 94 - 96% теории.
Неутилизируемые побочные продукты практически не получаются.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВОДОСОДЕРЖАЩИЙ ТОЛУИЛЕНДИАМИН, ПРИГОДНЫЙ ДЛЯ ХРАНЕНИЯ ИЛИ ТРАНСПОРТИРОВКИ В ЖИДКОМ ВИДЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ТОЛУИЛЕНДИИЗОЦИАНАТА | 1996 |
|
RU2202537C2 |
| Способ получения полиаминов | 1973 |
|
SU497765A3 |
| Способ получения 1,3-диацетокси-2метиленпропана | 1970 |
|
SU465780A3 |
| Способ получения многоядерных ароматических полиаминов | 1976 |
|
SU602113A3 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ АЦЕСУЛЬФАМА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1993 |
|
RU2106347C1 |
| Способ получения окиси пропилена | 1976 |
|
SU694072A3 |
| Способ получения полиаминов | 1973 |
|
SU494869A3 |
| Способ получения органического раствора надкарбоновой кислоты с числом атомов углерода 1-4 | 1976 |
|
SU638256A3 |
| Способ получения изопрена | 1971 |
|
SU460614A3 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ | 2007 |
|
RU2446151C2 |
Изобретение относится к способу получения бензилового спирта путем гидролиза бензилхлорида водой при повышенных температурах. Бензиловый спирт получают путем гидролиза бензилхлорида водой при температуре 80-180oС. Бензилхлорид и воду в молярном соотношении 1:(10-70) непрерывно подают в реактор с диспергирующими приспособлениями. Реакцию прекращают при достижении величины конверсии 30-90%. Продукт реакции после выхода из реактора разделяют на водную и органическую фазы. Водную фазу, содержащую разбавленную соляную кислоту, экстрагируют растворителем, для поглощения растворенных в разбавленной кислоте органических компонентов, в частности бензилового спирта. Органическую фазу разделяют на бензилхлорид, который возвращают в реактор, и сырой бензиловый спирт, который рафинируют до высокочистого бензилового спирта. Технический результат - упрощение технологии получения бензилового спирта за счет исключения применения карбоната натрия и предотвращения образования поваренной соли. 3 ил.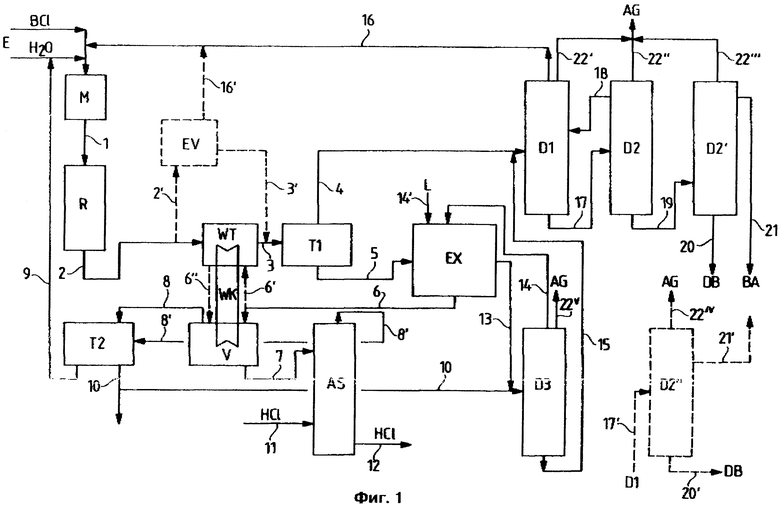
Способ непрерывного получения бензилового спирта путем гидролиза бензилхлорида водой при повышенной температуре, отличающийся тем, что а) бензилхлорид и воду в молярном соотношении 1:(10-70) подают в реактор с диспергирующими приспособлениями, где имеется температура 80-180oС, и смеси дают реагировать до достижения степени конверсии бензилхлорида, равной 30-90%, б) продукт реакции после выхода из реактора разделяют на водную и органическую фазы, в) органическую фазу в первом перегонном узле разделяют на бензилхлорид и сырой бензиловый спирт, г) данный бензилхлорид возвращают в реактор, д) сырой бензиловый спирт во втором перегонном узле рафинируют до высокочистого бензилового спирта, при этом получается содержащий побочные продукты (в частности, дибензиловый эфир) кубовый продукт, е) содержащую разбавленную соляную кислоту водную фазу из сепаратора в экстракторе обрабатывают растворителем, поглощающим еще растворенные в разбавленной соляной кислоте органические компоненты, в частности, бензиловый спирт, ж) экстракт в третьем перегонном узле освобождают от растворителя, возвращающегося в экстрактор, и з) кубовый продукт третьего перегонного узла подают в первый перегонный узел для получения бензилового спирта.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| КОММУТАТОР ДЛЯ ПРЕРЫВАНИЯ ТОКА В ПОСЛЕДОВАТЕЛЬНО СОЕДИНЕННЫХ ПРИЕМНИКАХ ЭЛЕКТРИЧЕСКОГО ТОКА | 1922 |
|
SU550A1 |
| EP 0 064 486 А1, 10.11.1982. | |||

