6-Метил-3,4-дигидро-1,2,3-оксатиазин-4-он-2,2-диоксид (в дальнейшем - Ацесульфам-H) уже некоторое время применяется в форме его калиевой соли (Ацесульфам-К):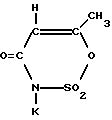
вследствие интенсивного сладкого вкуса в качестве подслащивающего вещества в секторе пищевых продуктов.
Для получения Ацесульфама-К известен ряд различных способов (см., в том числе, Angewandte Chemie, 22 (1973), с. 965-973). Особый интерес в настоящее время представляет собой способ, при котором в растворителе, предпочтительно метиленхлориде, амидосульфокислоту, предпочтительно, однако, растворимую соль амидосульфокислоты, сначала вводят во взаимодействие с дикетеном с образованием ацетоацетамидосоединения. В качестве солей амидосульфокислоты при этом обычно применяют соли щелочных металлов или аммония, предпочтительно триалкиламмониевые соли. При этом при взаимодействии с дикетеном образуется соль ацетоацетамидосульфокислоты формулы (I) соответственно уравнению реакции: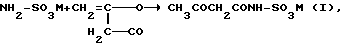
где
M - катион основания, в особенности HN (C1-C6-алкил3).
Растворенное, предпочтительно в метиленхлориде, ацетоацетамидное соединение формулы (I) затем при определенных реакционных условиях вводят во взаимодействие с раствором SO3, предпочтительно в метиленхлориде, и при этом оно циклизуется; SO3 при этом используется предпочтительно в избытке (см. выложенные описания изобретения к неакцептованной заявке на европейский патент 155634; 159516; 217076 и 218076). Таким образом, получается циклический продукт, из которого путем введения во взаимодействие с водой (гидролиз) в гидролизере образуется ацесульфам-H так называемая "сладкая кислота" формулы (II);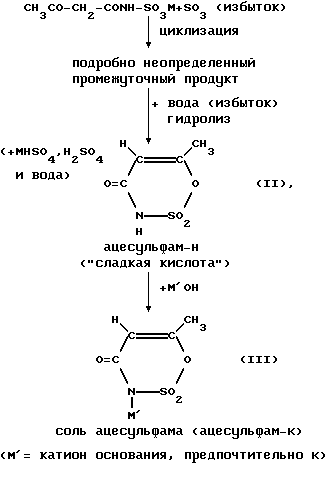
Как циклизация, так и гидролиз представляет собой при этом быстропротекающие, сильноэкзотермические реакции.
Реакционная смесь разделяется на органическую фазу, предпочтительно метиленхлоридную фазу, и водную сернокислую фазу, "Сладкая" кислота формулы (II) находится большей частью (примерно 4/5 в органической фазе, примерно 1/5 ее растворена в сернокислой фазе. Сернокислая фаза содержит почти все количество щелочи или аммония, предпочтительно триалкиламин в виде сульфата, предпочтительно триалкиламмонийгидросульфата. Сернокислую фазу после отделения от еще имеющегося растворителя, предпочтительно метиленхлорида, можно применять далее.
Отделение "сладкой" кислоты в форме желаемой соли формулы (III) осуществляют целесообразно согласно описанному в неакцептованной заявке на европейский патент 218076 методу. При этом сначала с помощью растворителя, предпочтительно метиленхлорида, из сернокислой фазы экстрагируют содержащуюся в ней "сладкую" кислоту в экстракторе. Используемый здесь растворитель объединяют с обычно фазой растворителя, которая содержит большую часть "сладкой" кислоты. Из объединенных фаз растворителей затем с помощью небольшого количества воды в экстракторе экстрагируют растворенный в незначительном количестве в фазе растворителя сульфат и вместе с водой от экстракции возвращают для взаимодействия циклизованного продукта в гидролизере с водой для получения "сладкой" кислоты. Из остающегося не содержащего сульфата раствора "сладкой" кислоты в растворителе извлекают сладкую кислоту в форме целевой соли, предпочтительно калиевой соли, с помощью водного щелочного раствора, предпочтительно раствора гидроксида калия. Водный раствор сладкого вещества формулы (III) можно обрабатывать обычными способами, например путем выпаривания или осаждения с помощью осадителей, с получением чистого кристаллического сладкого вещества формулы (III).
Остающийся растворитель насыщен водой и содержит большую часть образовавшихся во время реакции побочных продуктов, как ацетон, триалкиламин и триалкиламмониевая соль "сладкой" кислоты в случае использования триалкиламмониевой соли амидосульфокислоты, неидентифицированные высококипящие вещества и растворенные смолы.
Таким образом, загрязненный растворитель в этом виде нельзя снова использовать. Так, при смешивании с SO3 в стадии циклизации вместе с примесями образуются окрашенные в темный цвет твердые нерастворимые соединения, которые приводят к засорению трубопроводов. С ацетоном наряду с этим образуются окрашенные продукты, которые проникают в раствор сладкого вещества. Из таких растворов более нельзя выделить бесцветного сладкого вещества. Также при получении ацетоацетамидосоединения формулы (I) из соли амидосульфокислоты и дикетена этот загрязненный растворитель использовать нельзя, так как выход ацетоацетамидного соединения снижается и образуются сильно окрашенные побочные продукты. Они проникают в позднее получаемый раствор сладкого вещества и препятствуют выделению из раствора бесцветной твердой соли ацесульфама формулы (III).
Используемый для смешивания с SO3 и для получения ацетоацетамидного соединения растворитель поэтому должен содержать только незначительные количества загрязнений. Извлечение чистого растворителя из неочищенного растворителя, которое следует за нейтрализацией "сладкой" кислоты в виде ее соли, требует высоких затрат на аппаратуру со значительным потреблением энергии, ибо опри синтезе по причинам выхода и ограниченной растворимости "сладкой" кислоты в растворителе можно работать только с очень разбавленными растворами. Концентрация "сладко" кислоты, например, в метиленхлоридном растворе, который затем нейтрализуют с помощью щелочи, как правило, составляет только примерно 2-3 мас.%. Согласно этому при очистке путем перегонки неочищенного растворителя для получения чистого вновь используемого растворителя при учете необходимого флегмового числа на килограмм получаемого сладкого вещества испаряют вплоть до 100 кг растворителя.
В случае до сих пор обычных способов очистку растворителя осуществляют в более чем двух перегонных колоннах. При этом образующийся в последней колонне зумпфовый продукт, который содержит триалкиламин, более высококипящие компоненты, как ацетон, неидентифицированные высококипящие вещества, твердые остатки, как смола, и незначительные количества соли "сладкой" кислоты, и еще примерно 70% растворителя в качестве агента растворения твердых остатков нужно сжигать. Дальнейшее выпаривание, т.е. продолжающаяся регенерация растворителя, невозможно, так как иначе появляются прилипания твердых веществ, которые приводят к механическим повреждениям.
Учитывая недостатки этого известного способа, задача заключается в разработке способа получения солей ацесульфама, который позволяет осуществлять более простую регенерацию используемого растворителя из неочищенного растворителя путем использования самое большее двух колонн и в котором отсутствует по меньшей мере сжигание образующихся побочных продуктов и остатков.
В настоящее время найден способ, с помощью которого избегают "вышлюзовывания" содержащих растворитель зумпфовых потоков и который дополнительно позволяет осуществлять регенерацию растворителя до чистого растворителя, пригодного для получения ацетоацетамидного соединения формулы (I) и для приготовления смеси с SO3 в одной или двух колоннах для перегонки при ликвидации специальной перегонной колонны для отделения от высококипящих веществ и остатков.
Настоящее изобретение поэтому относится к способу получения солей ацесульфама путем взаимодействия солей амидосульфокислоты с дикетеном с образованием соли ацетоацетамидосульфокислоты формулы (I); циклизации за счет воздействия по меньшей мере примерно эквимолярного количества SO3, причем по меньшей мере реакция циклизации осуществляется в присутствии галогенированного алифатического углеводорода в качестве инертного растворителя; обработки продукта циклизации (органическая фаза) водой и переведения полученного ацесульфама-H формулы (II) в форму нетоксичной соли формулы (III). Сырой инертный растворитель, который образуется при разделении жидкой смеси, перегоняют в одной или двух перегонных колоннах, где после отделения воды и легкокипящих веществ и регенерации достаточно чистого растворителя для повторного использования при получении соединений формулы (I) и/или (II) кубовый остаток, содержащий растворитель, без дополнительной очистки непосредственно вводят в систему после реактора для осуществления циклизации.
Предпочтительно разделение полученной при гидролизе жидкой смеси осуществляют в гидролизе, подключенном после разделения фаз.
Предпочтительно водную фазу из разделителя фаз экстрагируют инертным растворителем в экстракторе, подключенном за разделителем фаз.
Предпочтительно сырой инертный растворитель после разделителя фаз и экстрактора после отделения ацесульфама в виде нетоксичной соли направляют на перегонку.
Предпочтительно в качестве растворителя используют метиленхлорид.
Предпочтительно перегонку осуществляют только в одной перегонной колонне.
Предпочтительно содержащий растворитель кубовый остаток возвращают по меньшей мере в одно место системы, предпочтительно в резервуар для обработки продукта циклизации водой в подключенный после него разделитель фаз и/или следующий за ним экстрактор.
Предпочтительно в качестве соли ацетоацетамидосульфокислоты используют триалкиламмониевые соли ацетоацетамидосульфокислоты, циклизацию осуществляют путем обработки более чем эквимолярным количеством SO3, предпочтительно в метиленхлориде в качестве растворителя, образующийся после циклизации SO3-аддукт гидролизуют в гидролизере до ацесульфама-H, в подключенной емкости для разделения фаз осуществляют разделение на органическую и водную сернокислую фазу. Водную фазу затем в экстракторе экстрагируют метиленхлоридом, и этот метиленхлорид вместе с органической фазой из емкости для разделения фаз подают в следующий экстрактор для промывки водой. Из таким образом очищенной органической фазы получают нетоксичные соли ацесульфама-H путем нейтрализации с помощью основания, и в последующей емкости для разделения фаз осуществляют разделение на водный солевой раствор и метиленхлоридную фазу, и эту метиленхлоридную фазу, в случае необходимости, по меньшей мере через следующую стадию экстракции подают в перегонную колонну.
Предпочтительно сырой растворитель вводят в перегонную колонну, в ее куб или выше, из этой перегонной колонны отводят остаток после перегонки, который наряду с долями растворителя содержит твердые побочные продукты, а также высококипящие вещества, и возвращают в резервуар для обработки продуктов циклизации, в подключенный за ней разделитель фаз и/или в следующий за ним экстрактор для экстракции сернокислой фазы. Из головной части перегонной колонны выводят воду, незначительные количества растворителя и вместе с ним, в случае необходимости, имеющиеся легкокипящие вещества, в то время как основную часть растворителя в виде возврата направляют обратно в перегонную колонну. Из средней части колонны с промежуточной тарелки, которая находится ниже возврата и выше притока вводимого в колонну сырого растворителя, выводят чистый растворитель, и его снова возвращают в систему.
Предпочтительно чистый растворитель возвращают в реактор.
Предпочтительно содержащий растворитель кубовый остаток после перегонки возвращают в гидролизер.
Предпочтительно содержащий растворитель кубовый остаток после перегонки возвращают в находящийся за гидролизером экстрактор для экстракции еще имеющегося в водной фазе ацесульфама-H.
Предпочтительно кубовый остаток после перегонки содержит по меньшей мере еще 10 мас.%, предпочтительно вплоть до 98 мас.% растворителя.
Взаимодействие соли амидосульфокислоты, в особенности триалкиламмониевой соли, с дикетеном осуществляют при температурах преимущественно 0-100oC, предпочтительно при 20-45oC. Температура на стадии циклизации составляет от -20 до 40oC, предпочтительно от -10 до 15oC. На стадии гидролиза SO3-аддукта температура составляет, как правило, 20-50oC, предпочтительно 35-45oC.
В случае используемых в качестве растворителя галогенированных алифатических углеводородов речь идет предпочтительно о таковых с количеством атомов углерода вплоть до четырех, образующих с водой азеотроп, как, например, метиленхлорид, хлороформ, 1,2-дихлор-этан, трихлорэтилен, тетрахлорэтилен, трихлор-фтор-этилен, особенно предпочтителен метиленхлорид.
В предпочтительном варианте осуществления изобретения как для соли ацетоамид-N-сульфокислоты формулы (I), так и для SO3 используют один и тот же растворитель, предпочтительно метиленхлорид.
Молярное соотношение ацетоацетамид-N-сульфоната формулы (I) к SO3 может составлять примерно 1:1, однако предпочтителен вплоть до примерно 20-кратного избыток SO3, предпочтительно примерно 3-10-кратный, в особенности 4-7 кратный, в особенности 4-7-кратный молярный избыток.
В предпочтительном случае использования солей ацетоацетамид-N-сульфокислоты формулы (I) и SO3 в молярном соотношении более чем 1, при реакции циклизации образуется SO3-аддукт, из которого ацесульфам-H формулы (II) нужно выделять путем гидролиза. Гидролиз осуществляют путем добавок воды или льда и/или добавки разбавленной водной серной кислоты по отношению к SO3 примерно в 2-6-кратном молярном избытке. Предпочтительно для гидролиза используют как воду, так и разбавленную водой серную кислоту, которая выходи из подключенного экстрактора с общей концентрацией 2-20 мас.%.
После гидролиза получают двух или (когда уже осажден ацесульфам-H) трехфазную смесь. Ацесульфам-Н формулы (II) находится, по существу, растворенным в органической и в сернокислой фазах. Органическую фазу затем отделяют. Отдельную органическую фазу, соответственно, объединенные органические фазы затем очищают предпочтительно в указанном экстракторе путем экстракции с помощью воды, причем в первую очередь из органической фазы экстрагируется содержащаяся в ней серная кислота.
Объемное отношение органической фазы к водной фазе после экстракции составляет, в общем, примерно (20-5):1. С помощью значительно меньших количеств воды часто можно достигать эффективной очистки.
В простейшем случае экстракцию осуществляют путем перемешивания обеих фаз в круглодонной колбе или котле с мешалкой, в качестве специальных устройств применяют, в принципе, все технологические аппараты для экстракции, как, например, смеситель-отстойник-аппараты, колонны с ситовидным днищем, насадочные колонны, Karr-колонны и т.д. Также можно использовать смесительные элементы, например статическую мешалку для увеличения интенсивности контакта экстракционных фаз.
Доля экстрагируемого одновременно с серной кислотой ацесульфама-H в зависимости от используемого количества воды составляет примерно 2-30% всего содержащегося в органической фазе ацесульфама-H. Для рентабельности способа имеет значение возврат водной фазы снова в процессе гидролиза SO3-аддукта.
Из очищенной органической фазы нетоксичные соли формулы (III) ацесульфама-H получают путем нейтрализации основаниями. В качестве оснований используют основания с нетоксичными катионами. Предпочтительны калиевые основания (растворы KOH, KHCO3 K2CO3 и т.д.), в особенности KOH.
Нейтрализацию ацесульфама-H и получение его нетоксичных солей из содержащей ацесульфам-H органической фазы преимущественно осуществляют путем интенсивного контакта очищенной органической фазы с водным раствором щелочи. Интенсивный контакт осуществляют, в общем, в зависимости от рода и способа экстракции, обычными способами в обычных устройствах, которые описаны выше. Также можно применять смесительные элементы, например статические мешалки.
При нейтрализации добавляют основания до достижения pH водной фазы примерно 5-12, предпочтительно примерно 8-11. Из водной фазы затем обычным образом (путем кристаллизации) получают соль ацесульфама.
Предпочтительно перегонку и регенерацию растворителя (метиленхлорида) согласно изобретению осуществляют таким образом, что неочищенный растворитель вводят в перегонную колонну, в ее нижнюю часть или выше, из этой перегонной колонны выводят остаток после перегонки, который наряду с растворителем содержит твердые побочные продукты, а также высококипящие вещества, и возвращают в систему, предпочтительно в гидролизер, емкость для разделения фаз и/или экстрактор для экстракции сернокислой фазы. Из головной части перегонкой колонны выводят воду, незначительные количества растворителя и вместе с ним имеющиеся в случае необходимости легкокипящие вещества, в то время как основная часть растворителя в виде флегмы возвращается в перегонную колонну. Из средней части колонны с промежуточной тарелки, которая находится ниже возврата и выше притока вводимого в колонну неочищенного растворителя, выводят достаточно очищенный растворитель, и его снова возвращают в систему, предпочтительно в реакторы для получения соединений формулы (I) и/или формулы (II), а также для получения SO3-раствора. Очищенный растворитель, который содержит самое большее 2 мас. %, предпочтительно 1 мас.%, в особенности 0,9-0,1 мас.% примесей, также можно вводить в последующие стадии способа.
Предлагаемый способ обладает неожиданными преимуществами. Значительно уменьшаются затраты на аппаратуру, так как вместо трех колонн необходима только одна колонна или самое большее две колонны. Не получают никаких содержащих растворитель остатков, для утилизации (сжигания) которых необходимы особые устройства. Кроме того, уменьшается расход энергии для очистки перегонкой неочищенного растворителя. Загрязнение раствора калиевой соли ацесульфама, который перерабатывают далее до чистой твердой (калиевой) соли ацесульфама, затрудняет получение ее в чистом виде, если в растворе находятся сильноокрашенные полимеры.
Содержание воды в растворителе, выводимом с промежуточной тарелки, неожиданно значительно меньше, чем в нижней и в головной частях колонны.
Настоящее изобретение относится к устройству для осуществления предлагаемого способа.
Устройство включает реактор для проведения циклизации соли ацетамидосульфокислоты формулы (I):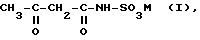 ,
,
где
M - катион основания, в частности NH(C1-C6-алкил)3, путем обработки SO3; подключенный за ним гидролизер для получения ацесульфама-H формулы (II):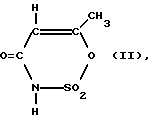 ,
,
связанный с ним разделитель фаз для отделения органической фазы и сернокислой фазы, нейтрализатор для перевода ацесульфама-H в соответствующую нетоксичную соль, емкость для отделения водного раствора этой соли от растворителя и устройство для возврата растворителя. Устройство для возврата растворителя состоит из одной или двух перегонных колонн, зумпфовая зона которых связана трубопроводом с гидролизером, экстрактором и/или разделителем фаз.
Предпочтительно устройство включает одну перегонную колонну, содержащую 15-50 теоретических тарелок.
Предпочтительно устройство дополнительно включает экстрактор для экстракции отделенной сернокислой фазы с помощью растворителя, а также другой экстрактор для экстракции соединенных из разделителя фаз и первого экстрактора органических фаз с помощью воды.
Предпочтительно к перегонной колонне присоединен разделитель фаз, в который переводят конденсированный пар из головной части колонны и в котором осуществляют разделение на водную фазу и фазу растворителя.
Устройство состоит из реактора 3, последовательно подключенного гидролизера 5 для получения кислоты формулы (II); связанной с ним емкости 11 для разделения органической фазы и сернокислой фазы, предпочтительно из одного экстрактора 13 для экстракции отделенной сернокислой фазы с помощью растворителя, предпочтительно дополнительного экстрактора 18 для экстракции соединенных из 11 и 13 органических фаз с помощью воды; нейтрализатора 22 для переведения кислоты формулы (II) в соответствующую соль формулы (III); емкости для разделения фаз 25 для отделения водного раствора соли формулы (III) от растворителя и перегонной колонны 32.
Предпочтительно к перегонной колонне 32 присоединена емкость для разделения фаз 36, в которую поступает конденсат мокрого пара из головной части колонны и здесь разделяется на водную фазу и фазу растворителя. Последняя снова возвращается в колонну. С промежуточной тарелки колонны, которая содержит в общем 15-50, предпочтительно 20-30 теоретических тарелок, выводится чистый растворитель. Из нижней части колонны (зумпф) остаток содержащий растворитель, после дистилляции отводится предпочтительно в гидролизер 5, в емкость для разделения фаз 11 и/или в экстрактор 13. Этот остаток после дистилляции содержит по меньшей мере 10 мас.%, предпочтительно 50-98 мас.%, в особенности 75-95 мас.% растворителя.
Предлагаемый согласно изобретению способ поясняется в качестве примера чертежом.
По трубопроводу 1 раствор SO3 в метиленхлориде подается в реактор (смеситель) 3; по трубопроводу 2 осуществляется подвод раствора ацетамидотриалкиламмониевой соли в метиленхлориде в реактор 3. Через трубопровод 2' в этот раствор дополнительно вводится метиленхлорид для разбавления. Теплота реакции отводится за счет испарения части метиленхлорида, испаренный метиленхлорид и жидкий продукт по трубопроводу 4 вместе поступают в снабженный мешалкой резервуар 5. По трубопроводу 6 в резервуар 5 подается вода; теплота реакции стадии гидролиза также отводится за счет испарения метиленхлорида. Пары метиленхлорида по трубопроводу 7 поступают в конденсатор 8, конденсат по трубопроводу 9 возвращается в резервуар 5. Жидкая смесь из резервуара 5 поступает по трубопроводу 10 в емкость для разделения фаз 11. Нижняя сернокислая фаза из емкости 11 по трубопроводу 12 подается в экстрактор (смеситель-отстойник) 13, в котором эта сернокислая фаза смешивается с метиленхлоридом из трубопровода 14 для экстракции сладкой кислоты. Сернокислая фаза отводится из экстрактора 13 по трубопроводу 15. Содержащий сладкую кислоту метиленхлорид покидает экстрактор 14 по трубопроводу 16. Метиленхлоридная фаза из емкости для разделения фаз 11 выводится по трубопроводу 17 и вместе с метиленхлоридом из трубопровода 16 попадает в экстрактор 13. В экстрактор 18 для экстракции растворенного сульфата снизу по трубопроводу 19 подается вода, которая после выхода из экстрактора 18 по трубопроводу 20 направляется в гидролизер 5. Метиленхлоридная фаза экстрактора 18 по трубопроводу 21 направляется к нейтрализатору с перемешиванием 22, в который по трубопроводу 23 подается водный раствор гидроксида калия, количество которого регулируется через pH-значение в нейтрализаторе с перемешиванием 22. Смесь по трубопроводу 24 вводится в емкость для разделения фаз 25. Верхняя фаза представляет собой водный раствор калиевой соли ацесульфама, который отводится по трубопроводу 26. Нижняя метиленхлоридная фаза по трубопроводу 27 подается в экстрактор 28, в котором она дополнительно промывается небольшим количеством воды, которая подается в экстрактор 28 по трубопроводу 29. Промывная вода по трубопроводу 30 вместе с водным раствором гидроксида калия подается в нейтрализатор 22. Выходящая из экстрактора 28 метиленхлоридная фаза представляет собой неочищенный метиленхлорид, который по трубопроводу 31 направляется в нижнюю часть перегонной колонны 32. Мокрый (соковый) пар из колонны 32 по трубопроводу 33 поступает в конденсатор 34 и двухфазный конденсат по трубопроводу 35 попадает в емкость для разделения фаз 36. Отделившаяся водная фаза в емкости для разделения фаз 36 отводится по трубопроводу 37, метиленхлоридная фаза по трубопроводу 38 в виде возврата отводится в колонну 32. Из нижней части колонны 32 метиленхлорид, содержащий побочные продукты, по трубопроводу 14 отводится в экстрактор 13 для экстракции сладкой кислоты из серной кислоты. С находящейся в колонне промежуточной тарелки по трубопроводу 39 отводится чистый метиленхлорид, который снова используется для смешивания с SO3 и/или для получения раствора триалкиламмониевой соли ацетоацетамидосульфокислоты.
Сравнительный пример 1. Работает только со свежим чистым метиленхлоридом без регенерации. Количественные данные указаны из расчета в час. Раствор 11,0 кг SO3 в 46,0 кг метиленхлорида с температурой +5oC по трубопроводу 1 вводится в реактор 3. По трубопроводу 2 в реактор 3 вводятся 15,0 кг метиленхлоридного раствора при 0oC, который содержит 6,6 кг триэтиламмониевой соли ацетоацетамидосульфокислоты и 1,0 кг полимерных компонентов различного состава, и непрореагировавший исходный материал в небольшом количестве. Через трубопровод 2' этот раствор разбавляется с помощью 12,0 кг метиленхлорида. Приготовление раствора осуществляется в котле с мешалкой из высококачественной стали путем введения 10,0 кг метиленхлорида, добавления 2,5 кг амидосульфокислоты, 2,8 кг триэтиламина, 0,1 кг уксусной кислоты и последующего осторожного добавления 2,2 кг дикетена после того, как предыдущие компоненты полностью растворились. Образовавшаяся в реакторе смесь, которая в виде раствора в метиленхлориде содержит точно не идентифицированный продукт циклизации, образовавшиеся побочные продукты и испаряющийся за счет теплоты реакции метиленхлорид, по трубопроводу 4 подается в так называемый гидролизер 5 (котел с перемешиванием).
По трубопроводу 6 в гидролизер 5 вводят 8,0 кг воды. По трубопроводу 7 20,0 кг испарившегося за счет теплоты реакции метиленхлорида поступают в конденсатор 8, в то время как конденсат по трубопроводу 9 возвращается в гидролизер (котел с перемешиванием 5. Смесь жидкостей из гидролизера 5 , которая поддерживается на постоянном уровне, по трубопроводу 10 вводится в разделитель фаз 11. Нижняя сернокислая фаза по трубопроводу 12 направляется в экстрактор (смеситель-отстойник) 13. В экстрактор 13 по трубопроводу 14 подаются 30,0 кг чистого свежего метиленхлорида. По трубопроводу 15 отводятся 26,0 кг сернокислой фазы. Эта сернокислая фаза содержит 41% серной кислоты, 32% воды, 21% триэтиламмонийгидросульфата, 0,5% уксусной кислоты, 1% ацетона, 0,1% сладкой кислоты, 3% неидентифицированных органических соединений и 1% растворенного метиленхлорида. По трубопроводу 16 метиленхлориднаая фаза из экстрактора 13 поступает в трубопровод 17. Из разделителя фаз 11 по трубопроводу 17 отводятся 100,5 кг метиленхлоридной фазы, которая наряду с другими компонентами содержит 0,2% воды, 2,4% сладкой кислоты и 0,4% сульфата. Объединенные метиленхлоридные потоки из трубопроводов 16 и 17 подаются в экстрактор 18. Снизу в экстрактор по трубопроводу 14 поступают 2,7 кг воды, вода после экстракции по трубопроводу 20 возвращается в гидролизер 5. Метиленхлоридная фаза экстрактора 18 по трубопроводу 21 направляется в нейтрализатор 22 (котел с перемешиванием; охлаждение водой в наружной рубашке); по трубопроводу 23 через регулирование pH-величины в котле 22 при значении, равном 9, в котел 22 поступает 10%-ный раствор гидроксида калия. Образовавшаяся двухфазная смесь по трубопроводу 24 подается в разделитель фаз 25. Из разделителя фаз 25 нижняя метиленхлоридная фаза через трубопровод 27 подводится в экстрактор 28, в котором эта метиленхлоридная фаза промывается поступающей по трубопроводу 29 водой в количестве 6,4 л. Промывная вода по трубопроводу 30 вместе с раствором гидроксида калия поступает в нейтрализатор 22.
По трубопроводу 31 отводятся 99,0 кг метиленхлорида. Метиленхлорид содержит 0,15 воды, 0,1% триэтиламина, 0,4% ацетона и 0,5% полимеров.
В качестве верхней фазы из разделителя фаз 25 по трубопроводу 26 отводятся 17,7 кг раствора ацесульфама калия. Светло-желтый раствор содержит 1% метиленхлорида, 17% ацесульфама калия, 0,3% сульфата калия и 0,5% ацетона, 0,1% триэтиламина и менее 0,05% неизвестных соединений.
При испарении этого раствора сладкого вещества примерно до 30% первоначального объема, охлаждении и фильтрации получают бесцветный ацесульфам калия в виде твердого вещества.
Сравнительный пример 2. Подача неочищенного метиленхлорида в реакцию путем разбавления раствора триэтиламмониевой соли ацетоацетамидосульфокислоты с помощью неочищенного вместо очищенного метиленхлорида.
Порядок проведения опыта и количества такие же, как в сравнительном примере 1, однако для разбавления ацетоацетамидного соединения в трубопроводе 2 вместо чистого метиленхлорида из трубопровода 2' разбавляют поступающим по трубопроводу 31 из сравнительного примера 1 12,0 кг неочищенного метиленхлорида.
При выпаривании отводимого через трубопровод 25 полученного раствора ацесульфама калия, который теперь окрашен в темный цвет, соответственно сравнительному примеру 1, получается окрашенная в желтый цвет калиевая соль ацесульфама в виде твердого вещества, которую можно получать бесцветный только путем нового растворения и дальнейшей обработки раствора с помощью адсорбентов.
Пример 3. Порядок проведения опыта такой же, как в сравнительных примерах. Неочищенный метиленхлорид из экстрактора 28, однако, теперь по трубопроводу 31 попадает в нижнюю часть перегонной колонны 32 для регенерации, через трубопровод 31 - в нижнюю часть колонны 32 (45 ситовых тарелок; промежуточная тарелка для отвода жидкости на уровне тарелки 30).
Неочищенный метиленхлорид по составу соответствует неочищенному метиленхлориду в сравнительном примере 1, также другие индивидуальные потоки соответствуют, прежде всего, индивидуальным потокам сравнительного примера 1.
Нагрев зумпфа колонны 32 регулируется так, чтобы через трубопровод для сокового пара 33 в конденсатор 34 (охлаждение: холодная вода) попадало 110,0 кг дистиллята, которые вводятся по трубопроводу 35 в разделитель фаз 36. По трубопроводу 37 отводятся 0,13 кг водной фазы. Нижняя органическая метиленхлоридная фаза по трубопроводу 38 в виде возврата возвращается в колонну. С промежуточной тарелки по трубопроводу 39 отводятся 66,0 кг чистого метиленхлорида. Этот метиленхлорид содержит в качестве примеси 140 ppm воды, 40 ppm ацетона, менее 30 ppm триэтиламина, его снова используют в качестве растворителя для приготовления ацетоацетамидосоединения и для разбавления раствора до реакции, а также для приготовления SO3-раствора.
Из нижней части колонны 32 по трубопроводу 14 отводятся 29,7 материала, который применяется для экстракции сладкой кислоты из сернокислой фазы в экстракторе 13, и по трубопроводу 16 метиленхлорида фаза снова подается в трубопровод 17.
Зумпфовый материал из колонны 32 в трубопроводе 14 состоит, по существу из метиленхлорида, 300 ppm воды, 1,4% ацетона, 0,3% триэтиламина и 1,7% полимеров.
После многодневной продолжительности опыта, когда все индивидуальные потоки находятся в концентрации равновесия, зумпфовый поток 14 показывает незначительное увеличение концентрации ацетона до 2%, однако нет увеличения концентрации полимеров или других компонентов. Отделенная серная кислота содержит 2% ацетона, 0,5% продуктов конденсации ацетона, 5% полимеров и неидентифицированных органических соединений.
По трубопроводу 25 отводятся 17,6 кг раствора ацесульфама калия; светло-желтый раствор содержит 17% ацесульфама калия, 1% метиленхлорида, 0,3% сульфата калия, 0,6% ацетона, 0,1% триэтиламина, менее 0,5% неизвестных соединений. При выпаривании этого раствора до 30% первоначального объема, охлаждения и фильтрации получают бесцветный ацесульфам калия в виде твердого вещества.
В принципе, такой же результат получают, когда зумпфовый материал колонны 32 вводят в гидролизер 5. В этом случае для экстракции сладкой кислоты из сернокислой фазы в экстракторе 13 можно применять также, например, головной продукт из колонны 32. Разделитель фаз 32 при этом варианте не обязателен.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 6-метил-3,4-дигидро-1,2,3-оксатиазин-4-он-2,2-диоксида | 1986 |
|
SU1535380A3 |
| Способ получения калиевой соли 6-метил-3,4-дигидро-1,2,3-оксатиазин-4-он-2,2-диоксида | 1986 |
|
SU1582988A3 |
| Способ получения 6-метил-3,4-дигидро-1,2,3-оксатиазин-4-он-2,2-диоксида или его калиевой соли и способ получения аммоний ацетоацетамид-N-сульфонатов | 1985 |
|
SU1342418A3 |
| Способ получения многоядерных ароматических полиаминов | 1976 |
|
SU602113A3 |
| Способ получения 2-хлорбутадиена-1,3 | 1972 |
|
SU512694A3 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ИНДЕНОВ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ ДЛЯ ПОЛУЧЕНИЯ МЕТАЛЛОЦЕНОВ И МЕТАЛЛОЦЕНЫ | 1992 |
|
RU2103250C1 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ БЕНЗИЛОВОГО СПИРТА | 1996 |
|
RU2176237C2 |
| ГЛИКОПЕПТИДЫ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ, ОБЛАДАЮЩИЙ АНТИБИОЦИДНЫМ ДЕЙСТВИЕМ | 1992 |
|
RU2099349C1 |
| Способ получения диаминодифенилметанов | 1976 |
|
SU654165A3 |
| Способ получения 1,2-дихлорэтана | 1983 |
|
SU1277887A3 |
Изобретение относится к способу получения солей ацесульфама путем взаимодействия солей амидосульфокислоты с дикетеном с образованием соли ацетоацетамидосульфокислоты формулы (I), циклизации путем воздействия по меньшей мере примерно эквимолярного количества SO3, причем по меньшей мере эту реакцию циклизации осуществляют в присутствии галогенированного алифатического углеводора в качестве инертного растворителя; обработки продукта водой и переведения полученного ацесульфама-Н формулы (II) в форму нетоксичной соли. При перегонке при нормальном давлении образующегося неочищенного растворителя после отделения от воды и легкокипящих веществ и регенерации достаточно чистого для повторного использования при получении соединений формулы (I) и/или (II) растворителя отсаток, содержащий растворитель, после дистилляции без дальнейшей очистки непосредственно возвращается в систему после реактора для осуществления реакции циклизации. Изобретение относится к размещению производнственных единиц для осуществления этого способа. 2 с. и 16 з.п. ф-лы, 1 ил.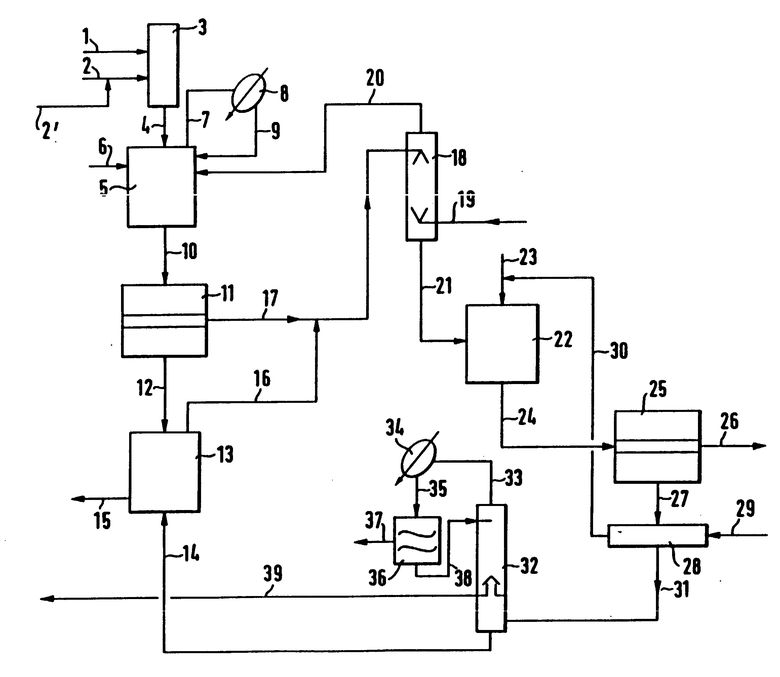
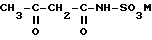
где М - катион основания, в частности HN(алкил-С1-С6)3, которую подвергают циклизации в реакторе путем обработки по меньшей мере эквивалентным количеством SO3 в присутствии галогенированного алифатического углеводорода в качестве инертного растворителя с получением продукта циклизации, который обрабатывают водой в гидролизере с образованием двухфазной жидкой смеси, которую расслаивают в разделителе фаз на водную фазу, содержащую серную кислоту, и органическую фазу, содержащую ацесульфам-Н формулы (II)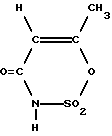
который переводят в форму нетоксичной соли и перерабатывают инертный растворитель перегонкой, отличающийся тем, что сырой инертный растворитель, который образуется при разделении жидкой смеси, перегоняют в одной или двух перегонных колоннах, где после отделения воды и легкокипящих веществ и регенерации достаточно чистого растворителя для повторного использования при получении соединений формулы (I) и/или (II), кубовый остаток, содержащий растворитель, без дальнейшей очистки непосредственно вводят в систему после реактора для осуществления циклизации.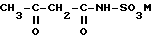
где М - катион основания, в частности NH(С1-С6-алкил)3, путем обработки SO3, подключенный за ним гидролизер для получения ацесульфама-Н формулы (II)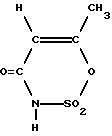
связанный с ним разделитель фаз с отделения органической фазы и сернокислой фазы, нейтрализатор для перевода ацесульфама-Н в соответствующую нетоксичную соль, емкость для отделения водного раствора этой соли от растворителя и устройство для возврата растворителя, отличающееся тем, что устройство для возврата растворителя состоит из одной или двух перегонных колонн, зумпфовая зона которых связана трубопроводом с гидролизером, экстрактором и/или разделителем фаз.
| ЕР, 155634, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ЕР, 159516, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ЕР, 217024, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ЕР, 218076, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
