Изобретение создано при поддержке правительства США и удостоено премии (grant) под N МН-42705, присвоенной Национальным Институтом Здоровья. Правительство США обладает определенными правами на изобретение.
Область изобретения
Данное изобретение направлено на новые лиганды для допаминовых рецепторов. Более конкретно, данное изобретение относится к необязательно замещенным соединениям тетрагидро-1H-нафт[1,2,3-де]изохинолина и их применению в фармацевтических составах для лечения допаминсвязанной дисфункции центральной и периферической нервной системы.
Обоснование и краткое описание изобретения
Допамин, нейротрансмиттер в центральной нервной системе, вовлечен в многочисленные неврологические расстройства. Например, выдвинута гипотеза, что избыточная стимуляция подгруппы допаминовых рецепторов может привести к шизофрении. Вдобавок, общепризнанно, что либо избыточная, либо недостаточная функциональная допаминергическая активность в центральной и/или периферической нервной системе может вызывать гипертензию, нарколепсию и другие поведенческие, неврологические, физиологические расстройства и нарушения движения, включающие болезнь Паркинсона, хроническое, прогрессирующее заболевание, характеризуемое неспособностью к контролю системы произвольных движений.
Допаминовые рецепторы традиционно подразделяются на два класса (D1 и D2), основанные на фармакологическом и функциональном признаке. D1 рецепторы преимущественно распознают фенилтетрагидробензазепины и приводят к стимуляции ферментативной аденилатциклазы, тогда как D2 рецепторы распознают бутирофеноны и бензамиды и отрицательно (или ничуть не) соединяются с аденилатциклазой. В настоящее время известно, что существует несколько подклассов допаминовых рецепторов и по меньшей мере пять генетических кодов для подклассов допаминовых рецепторов: D1, D2, D3, D4 и D5. Однако, продолжает использоваться и традиционная классификация, по которой D1-подобный класс включает D1 (D1A) и D5 (D1B) рецепторы, тогда как D2-подобный класс состоит из D2, D3 и D4 рецепторов.
Лекарственные средства для центральной нервной системы, обладающие сродством к допаминовым рецепторам, обычно классифицируются не только по их рецепторной селективности, но также по их агонистической (стимулирующей рецептор) или антагонистической (блокирующей рецептор) активности. Хотя физиологические активности, связанные с взаимодействием допамина с различными подклассами рецепторов, полностью не изучены, известно, что лиганды, обладающие селективностью в отношении конкретного подкласса рецепторов, должны давать более или менее прогнозируемые нейрофармацевтические результаты. Изучение пригодности селективных в отношении допаминового рецептора антагонистических и агонистических соединений даст возможность разработки экспериментов, позволяющих лучше понять разнообразные функциональные роли D1 рецепторов, и приведет к новым способам лечения различных расстройств центральной и периферической нервной системы.
Первоначально изучения допаминовых рецепторов были сосредоточены на D2 классе, однако, недавно стала очевидной решающая роль D1 допаминового рецептора в функционировании нервной системы. Первоначально работа по селективным в отношении D1 рецепторов лигандам была главным образом сосредоточена на молекулах одного химического класса, фенилтетрагидробензазепинах, таких как антагонист SCH23390 (1):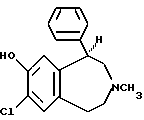
Было найдено, что некоторые из фенилтетрагидробензазепинов служат D1 рецепторными агонистами; однако, у агонистов, являющихся производными от этого класса [включающих, например, SKF38393 (+)-2], обычно полностью отсутствует собственная эффективность. Недавно было показано, что даже SKF 82958, который, как предполагалось, должен быть полным агонистом, не проявляет полной собственной эффективности в препаратах с пониженным рецепторным резервом. Разграничение между агонистами полной и частичной эффективности важно для исследователей в области медицины, ввиду того, что эти соединения оказывают различные воздействия на комплекс событий, опосредованных центральной нервной системой. Например, дигидрексидин и полный агонист, А-77636, обладают необычайным действием против болезни Паркинсона на модели МРТР-обработанной обезьяны, тогда как частичные агонисты не проявляют значительной активности. Недавно полученные результаты наводят на мысль, что полные и частичные агонисты различаются также по их воздействиям на другие комплексные нервные функции. Поэтому, исследователи направили свои усилия на разработку лигандов, являющихся полными агонистами, обладающими полной внутренней (прирожденной) эффективностью. Одним из таких соединений является дигидрексидин (3), гексагидробензо[a]фенантридин формулы: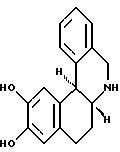
Структура дигидрексидина уникальна в отношении других D1 агонистов, поскольку присоединена дополнительная циклическая система, придающая относительную жесткость молекуле. Исследования по моделированию молекулы дигидрексидина (3) показали, что соединение обладает ограниченным числом низких энергетических конформаций, в которых все ароматические циклы имеют относительное копланарное расположение. Последнее выяснение конфигурации активного энантиомера дигидрексидина (3) совпало с предсказанным на основе этой модели.
В отличие от других близко схожих, с высокой собственной активностью D1 агонистов, таких как 3-замещенные аминометилизохроманы, дигидрексидин (3) обеспечивает полужесткий шаблон для создания модели допаминового лиганда. Существенные особенности этой модели включают присутствие трансоидной β- фенилдопаминовой группы, экваториально ориентированную одну электронную пару на основном азотном атоме и близкую копланарность дополнительного фенильного кольца и кольца пирокатехина. Модель, основанная на дигидрексидине, имеет трансоидный β- фенилдопаминовый радикал, тогда как допаминергические фенилтетрагидробензазепины обладают цисоидной β- фенилдопаминовой конформацией. Модель, основанная на дигидрексидине, служит основой для создания дополнительных D1 рецепторных агонистов. Разработка и синтез D1 рецепторных агонистов, обладающих высокой собственной активностью, представляются важными для медицинского исследования в связи с потенциальной возможностью применения полных агонистов для лечения комплексных явлений, опосредованных центральной нервной системой, а также состояний, в которые вовлечены периферические допаминовые рецепторы. Например, композиции по данному изобретению потенциально применимы в качестве средств для понижения кровяного давления.
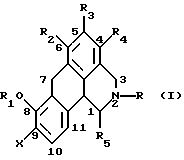
Одним из вариантов воплощения данного изобретения является новый класс допаминовых рецепторных агонистов общей формулы: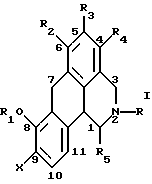
их фармацевтически приемлемые соли и фармацевтические составы таких соединений. Настоящие соединения полезны при лечении пациентов, страдающих допаминсвязанной дисфункцией центральной нервной системы, а также лечении состояний, в которые вовлечены периферические допаминовые рецепторы, что подтверждается наблюдаемыми неврологическим, психологическим, физиологическим или поведенческим расстройствами.
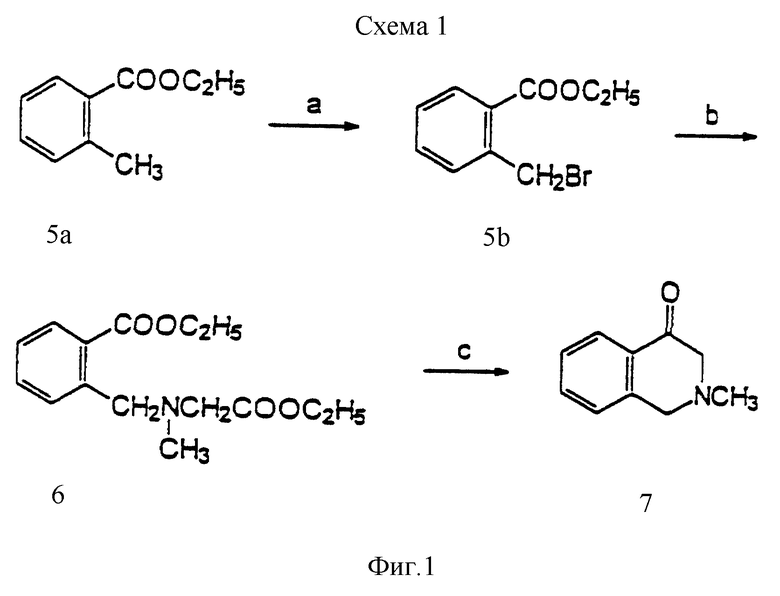
Фиг. 1 иллюстрирует стадии химической реакции по схеме 1, превращение этил-о-толуата в 2-метил-2,3-дигидро-4-(1H)-изохинолон. Реагенты: a) NBS (N-бромсукцинимид), перекись бензоила, CCl4, нагревание до температуры кипения с обратным холодильником; b) этиловый эфир саркозина HCl, K2CO3, ацетон; c) i. NaOEt, EtOH, нагревание до температуры кипения с обратным холодильником, ii. HCl, нагревание до температуры кипения с обратным холодильником.
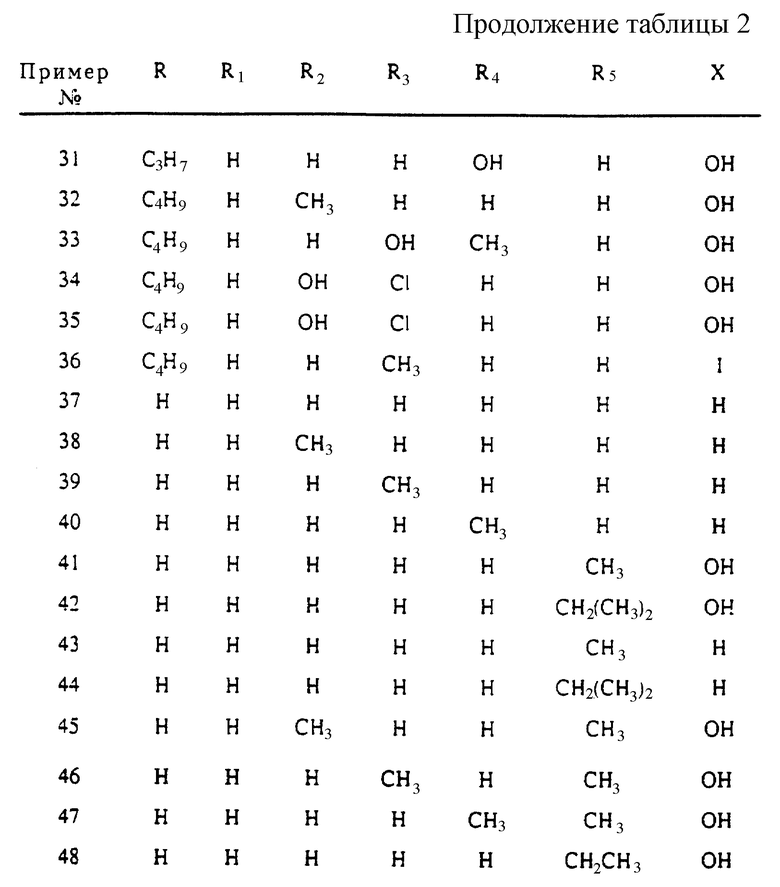
Фиг. 2 иллюстрирует стадии химической реакции по схеме 2, превращения 2,3-диметокси-N, N'-диэтилбензамида в динапсолин. Реагенты: a) i. втор-бутиллитий, TMEDA, Et2O, -78oC, ii. Соединение 7, iii. TsOH, толуол, нагревание до температуры кипения с обратным холодильником; b) i. 1-хлорэтилхлорформиат, (CH2Cl)2, ii. CH3OH; c) TsCl, Et3N; d) H2/Pd-C, HOAc; e) BH3-THF; f) конц. H2SO4, от -40oC до -5oC; g) Na-Hg, CH3OH, Na2HPO4; h) BBr3, CH2Cl2.
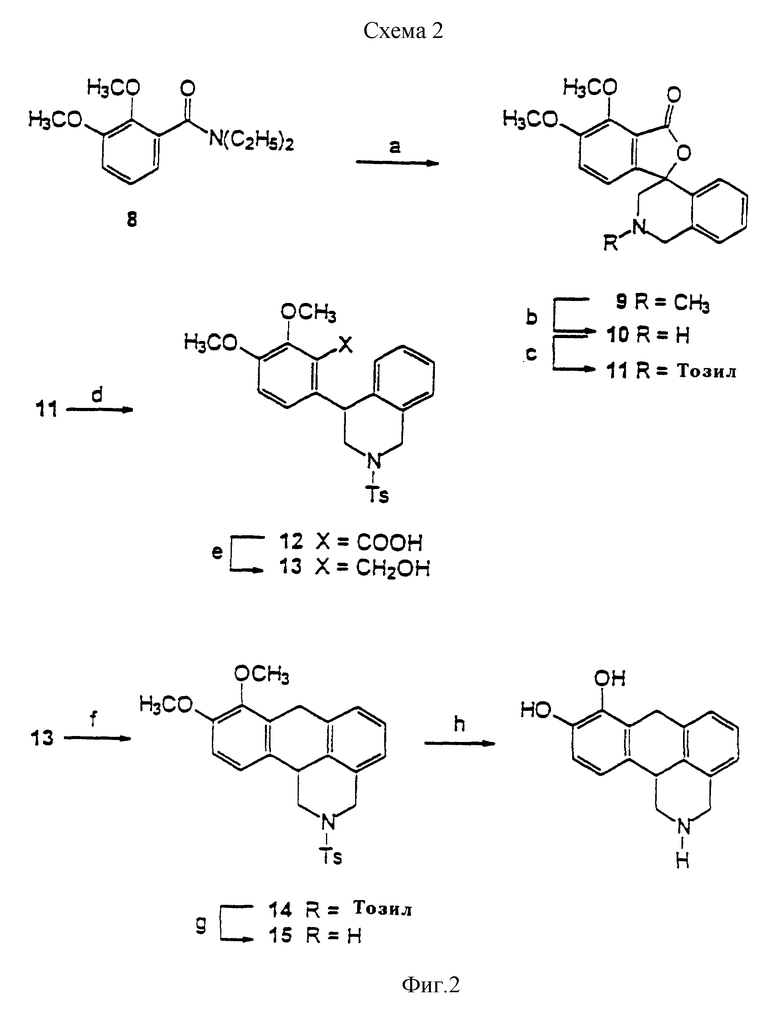
Фиг. 3 выражает графически сродство динапсолина (треугольники), (+)-дигидрексидина (квадраты) и (+)-SCH23390 (закрашенные кружки) к стриальным D1 рецепторам. Стриальные D1 рецепторы крыс метят [3H]SCH23390 (1) и добавляют немеченый динапсолин, (+)-дигидрексидин или (+)-SCH23390 для определения специфического связывания каждого соединения с D1 рецептором.
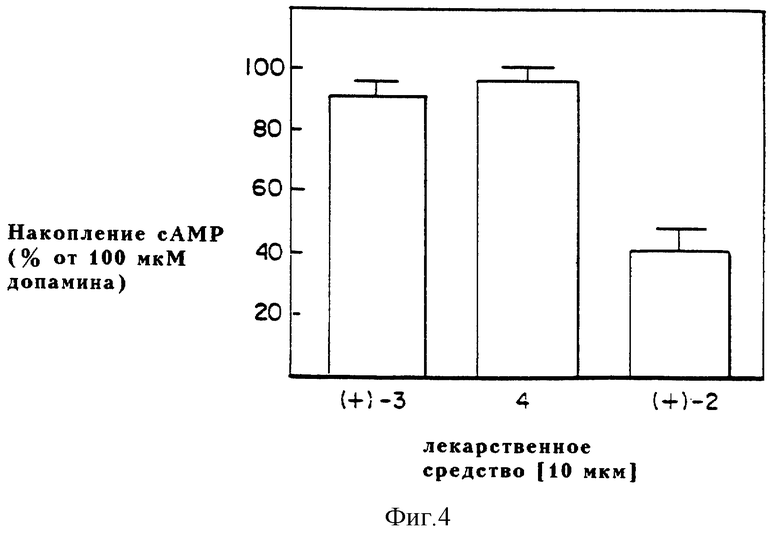
Фиг. 4 выражает графически способность динапсолина (4), (+)-дигидрексидина [(+)-3] и (+)-SKF38393 [(+)-2] к стимуляции аккумуляции cAMP в стерильных гомогенатах крыс относительно допамина.
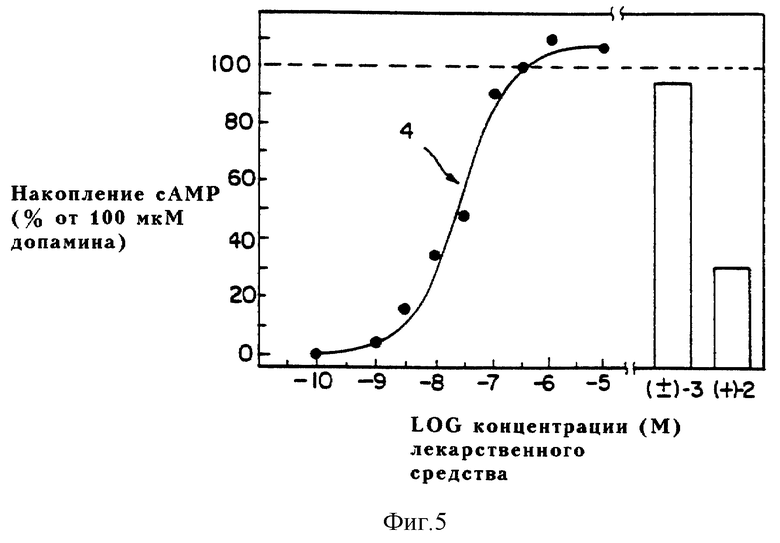
Фиг. 5 выражает графически способность динапсолина (4), (+)-дигидрексидина [(+)-3] и (+)-SKF38393 [(+)-2] к стимуляции аккумуляции cAMP в C-6 клетках глиомы (выражающих D1A рецепторы приматов) относительно допамина.
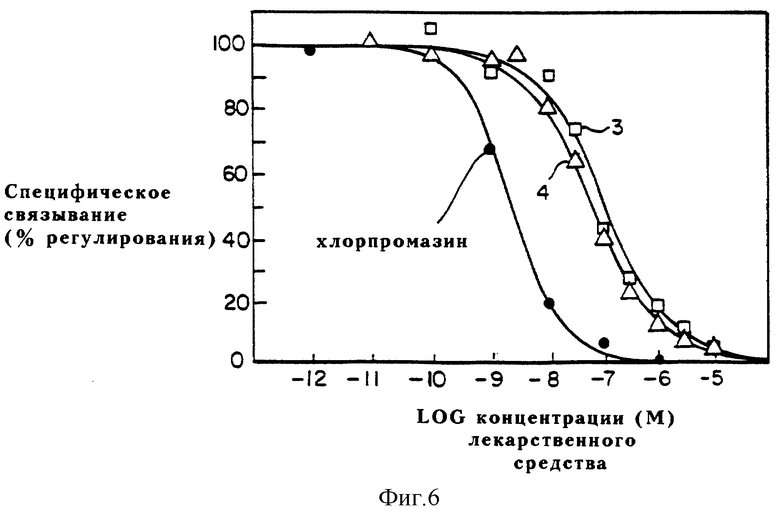
Фиг. 6 выражает графически сродство динапсолина (треугольники), (+)-дигидрексидина (квадраты) и (+)-SCH23390 (закрашенные кружки) к стриальным D2 рецепторам. Стриальные D2 рецепторы крыс метят [3H]SCH23390 и добавляют немеченый динапсолин, (+)дигидрексидин или (+)-SCH23390 для определения специфического связывания каждого соединения с D2 рецептором.
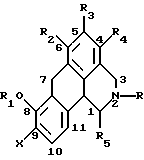
Согласно данному изобретению предложено соединение общей формулы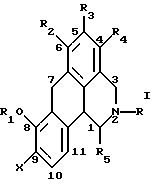
и его фармацевтически приемлемые соли,
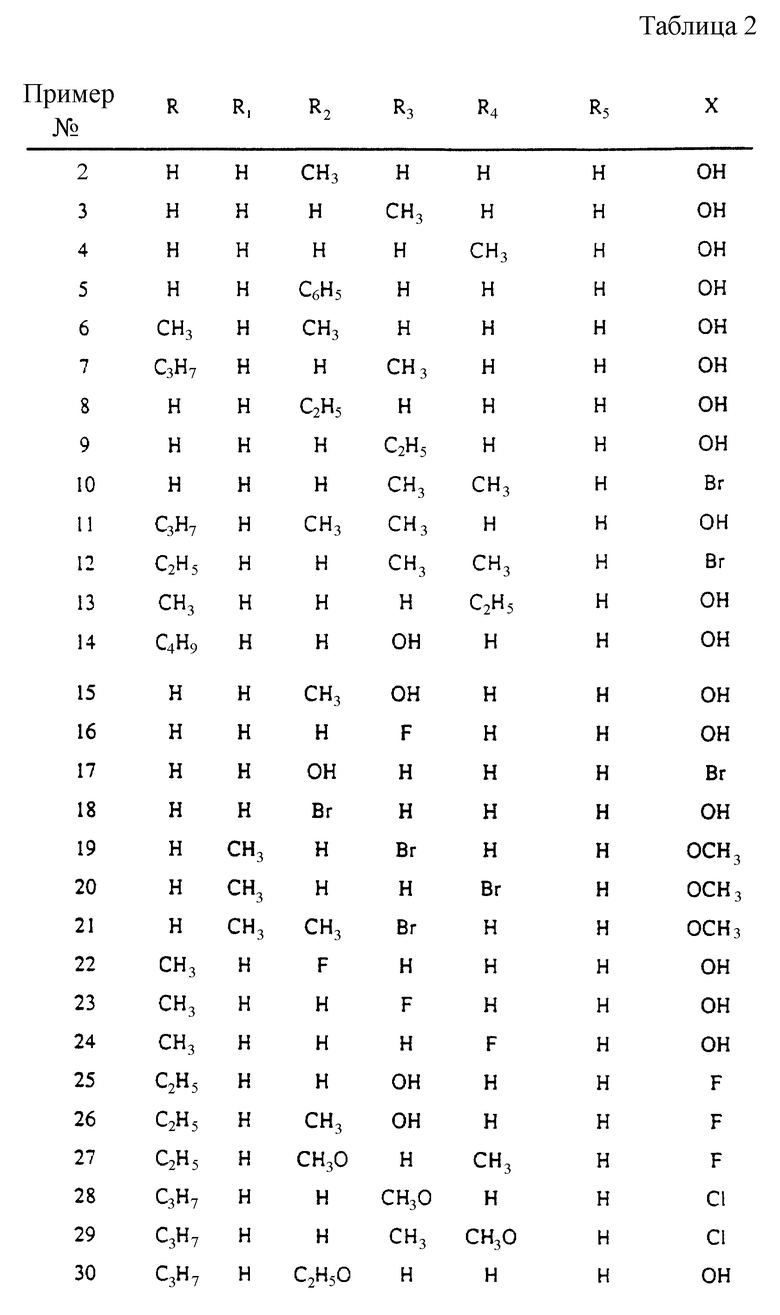
где R и R5 обозначают водород или C1-C4 алкил; R1 обозначает водород, C1-C4 алкил или феноксизащитную группу; X обозначает водород, галоген или группу формулы -OR6, где R6 обозначает водород, C1-C4 алкил или феноксизащитную группу, и R2, R3 и R4 независимо выбирают из группы, включающей водород, C1-C4 алкил, фенил, галоид или группу -OR1, где R1 имеет принятые выше значения, и когда X обозначает группу формулы -OR6, группы R1 и R6 вместе могут образовывать группу формулы -CH2-.
Термин "C1-C4 алкил", как он использован здесь, обозначает разветвленные или линейные алкильные группы с 1-4 углеродными атомами, включающие, но не в порядке ограничения, метил, этил, пропил, изопропил, н-бутил, третбутил и циклопропилметил.
Термин "фармацевтически приемлемые соли" относится к солям, полученным применением органических или неорганических кислот, эти соли пригодны для применения людьми или низшими животными и при этом не вызывают нежелательной токсической, раздражающей, аллергической или тому подобной реакции. Кислоты, пригодные для образования фармацевтически приемлемых солей или биологически активных соединений, имеющих аминофункциональность, хорошо известны в соответствующей области. Соли могут быть получены in situ (на месте) во время окончательного выделения и очистки данных соединений или отдельно, при взаимодействии выделенных соединений в форме свободного основания с подходящей солеобразующей кислотой.
Термин "феноксизащитная группа", как он здесь использован, обозначает заместители на фенольном кислороде, предупреждающие нежелательные реакции и разложение во время синтеза, которые позднее могут быть удалены без воздействия на другие функциональные группы молекулы. Такие функциональные группы и способы их применения и удаления хорошо известны в соответствующей области. Они включают простые эфиры, такие как простые циклопропилметиловые, циклогексиловые, аллиловые эфиры и тому подобные; простые алкоксиалкиловые эфиры, такие как простые метоксиметиловые или метоксиэтоксиметиловые эфиры и тому подобные; простые алкилтиоалкиловые эфиры, такие как простые метилтиометиловые эфиры; простые тетрагидропираниловые эфиры; простые арилалкиловые эфиры, такие как простые бензиловые, 0-нитробензиловые, п-метоксибензиловые, 9-антрилметиловые, 4-пиколиловые эфиры и тому подобные; простые триалкил-силиловые эфиры, такие, как простые триметилсилиловые, триэтилсилиловые, третбутилдиметилсилиловые, трет-бутилдифенилсилиловые эфиры и тому подобные; сложные алкиловые или ариловые эфиры, такие как ацетаты, пропионаты, н-бутираты, изобутираты, триметилацетаты, бензоаты и тому подобные; карбонаты, такие, как метил, этил, 2,2,2-трихлорэтил, 2-триметилсилилэтил, бензил и тому подобные; и карбаматы, такие как метил, изобутил, фенил, бензил, диметил и тому подобные.
Термин "C1-C4 алкокси", как он здесь использован, обозначает разветвленные или линейные алкильные группы из 1-4 углеродных атомов, присоединенные через атом кислорода, включающие, но не в порядке ограничения, метокси, этокси, пропокси и трет-бутокси.
Далее, согласно другому варианту воплощения данного изобретения, настоящие соединения могут быть составлены в общепринятые лекарственные формы для применения в способах лечения пациентов, страдающих допаминсвязанной дисфункцией центральной или периферической нервной системы. Эффективные дозы данных соединений зависят от многих факторов, включающих требующие лечения симптомы, способ введения и общее состояние пациента. Например, для перорального введения установлены эффективные дозы данных соединений приблизительно в интервале от 0,1 до 50 мг/кг, более характерные составляют от 0,5 до 25 мг/кг. Эффективные парентеральные дозы могут составлять порядка от 0,01 до 5 мг/кг веса тела. В основном, лечебные схемы с применением соединений по данному изобретению включают введение около 1 - 500 мг соединений по данному изобретению в день раздельными дозами или единичной дозой.
Жидкие лекарственные формы для перорального применения могут включать фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии и сиропы, содержащие обычно используемые в данной области техники инертные разбавители, такие как вода. Такие композиции могут также включать адъюванты, такие как смачивающие средства, эмульгирующие и суспендирующие средства, подсластители и отдушки. Препараты для инъекций из соединений по данному изобретению могут быть составлены с применением известных в соответствующей области техники продуктов путем диспергирования или растворения эффективных доз соединения в парентерально приемлемом растворителе, таком как вода, или, более предпочтительно, в изотоническом растворе хлористого натрия. Парентеральные составы можно стерилизовать, используя известные в соответствующей области техники приемы микрофильтрации.
Соединения по данному изобретению могут также входить в состав твердых лекарственных форм для перорального введения, таких как капсулы, таблетки, порошки, пилюли и тому подобные. Обычно активное соединение смешивают с инертным разбавителем или носителем, таким как сахар или крахмал, и другими эксципиентами, подходящими для лекарственной формы. Таким образом, таблетируемые составы должны включать приемлемые смазывающие вещества, связывающие и/или разрыхлители. Необязательно порошкообразные композиции, содержащие активное соединение по данному изобретению и, например, носитель в виде сахара или крахмала, могут быть заполнены в желатиновые капсулы для перорального введения. Другие лекарственные формы соединений по данному изобретению могут быть составлены использованием известных в соответствующей области технических приемов в формы, адаптированные к специфическому способу введения.
Одним из полученных согласно данному изобретению соединений является (±)-8,9-дигидрокси-2,3,7,11b-тетрагидро-1H-нафто[1,2,3,-де]изохинолин, называемый ниже "динапсолин". Динапсолин синтезируют из 2-метил-2,3-дигидро-4(1H)-изохинолона по способу, описанному в основном на фиг. 1 и 2. Бромирование боковой цепи этил-о-толуата (5a) с помощью NBS в присутствии перекиси бензоила дает соединение 5b. Алкилирование этилового эфира саркозина соединением 5b дает соединение 6, которое после конденсации Dieckmann'a и последующего декарбоксилирования в ходе кислотного гидролиза дает соединение 7.
Как показано в схеме 2 (фиг. 2), ортонаправленная литизация 2,3-диметокси-N, N'-диэтилбензамида (8) с помощью втор-бутиллития/TMEDA в диэтиловом эфире при -78oC, конденсация содержащих литий групп с соединением 7 и последующее нагревание до температуры кипения с обратным холодильником и с п-толуолсульфокислотой дают спиролактон 9 с умеренным выходом. N-Деметилирование соединения 9 с помощью 1-хлорэтилхлорформиата и последующий метанолиз промежуточного продукта дают соединение 10, которое при обработке п-толуолсульфонилхлоридом и триэтиламином дает соединение 11.
Предыдущие попытки синтеза соединения 11 непосредственной конденсацией 2-п-толуолсульфонил-2,3-дигидро-4(1H)-изохинолона с литиевым соединением 8 в ТГФ или диэтиловом эфире и последующей лактонизацией с кислотой дали только незначительное количество (< 5%) соединения 11. Энолизация 2-п-толуолсульфонил-2,3-дигидро-4(1H)-изохинолона в щелочной среде служит понятным объяснением низкого выхода.
Гидрогенолиз соединения 11 в ледяной уксусной кислоте в присутствии 10% палладия на углероде дает соединение 12, которое при восстановлении дибораном дает промежуточное соединение 13. Циклизация соединения 13 с помощью концентрированной серной кислоты при пониженной температуре приводит к соединению 14. N-Детозилирование соединения 14 с помощью Na/Hg в метаноле, содержащем динатрийгидрофосфатный буфер, дает соединение 15. Наконец, соединение 15 обрабатывают бортрибромидом для расщепления метилового эфира, что приводит к динапсолину (4) в виде его бромистоводородной соли.
Проведено сравнение пространственных структур низкоэнергетических конформаций (+)-транс-10,11 -дигидрокси-5,6,6a,7,8,12b- гексагидробензо[a] фенантридина [(+)-ди-гидрексидин] и 11bR-энантиомера динапсолина, являющегося гомохиральным в отношении (+)-дигидрексидина по 12bS хиральному центру. Легко заметить две основные структурные особенности. Во-первых, стерический объем, обеспечиваемый мостиковой группой C(7)-C(8)этано в дигидрексидине (3), удален. Во-вторых, угол подвешенного фенильного кольца по отношению к плоскости цикла пирокатехина слегка изменен. Это наиболее очевидно при рассмотрении передней плоскости, когда ароматический водород H(1) в дигидрексидине (3) выступает над циклом пирокатехина. Кроме того в динапсолине это положение используют, чтобы присоединить подвешенное фенильное кольцо через метиленовую группу к кольцу пирокатехина: это принуждает подвешенное фенильное кольцо изгибаться в направлении по часовой стрелке относительно дигидрексидина (3), если рассматривать в свете изложенного выше. Аминогруппы занимают аналогичные позиции, обеспечивая степень конформационной гибкости гетероцикла. Кроме того, обе молекулы могут представлять N-H вектор в экваториальной ориентации, что является характерным признаком фармакофора и является важным для D1 рецепторных агонистов. В соответствии с этими наблюдениями фармакологические свойства этих двух молекул аналогичны.
Проведены эксперименты с целью определения связывания динапсолина в отношении D1 рецепторов. Найдено, что динапсолин обладает почти идентичным по сравнению с дигидрексидином (3) сродством (K1 = 5,9 нМ) к стриальным D1 рецепторам крыс. Вдобавок, эксперименты по конкуренции с использованием немеченного SCH23390 (1) в качестве конкурента, показали, что динапсолин проявляет высокое конкурентное сродство, и ему соответствует менее глубокая кривая сравнения, что говорит об агонистических свойствах (см. фиг. 3). Агонистические свойства динапсолина в отношении D1 рецепторов оценивают in vitro, измеряя способность динапсолина увеличивать продуцирование cAMP в полосатом теле крыс и в C-6-mD1-клетках (см. приведенные ниже экспериментальные данные). Как в полосатом теле крыс, так и в C-6-mD1-клетках динапсолин обладает полной агонистической активностью с EC50 около 30 нМ в синтезах стимулирующих cAMP посредством D1 рецепторов.
Таким образом, фармакологические данные приводят к выводу, что динапсолин обладает высоким сродством к допаминовым D1 рецепторам, меченым [3H] SCH23390, что почти идентично сродству (+)-транс-10,11-дигидрокси-5,6,6a, 7,8,12b-гексагидробензо[a] фенантридина (дигидрексидина). Кроме того, (±)-8,9-дигидрокси-2,3,7,11b-тетрагидро-1H-нафто[1,2,3-де] изохинолин (динапсолин), как в стриальных мембранах крыс, так и в клонированных выраженных D1A рецепторах приматов, является полным агонистом по отношению к допамину, подобно дигидрексидину (3), но в отличие от частичного агониста (+)-SKF 38393 (см. фиг. 4 и 5: (+)-SKF 38393 = (+)-2; (±)-транс-10,11-дигидрокси-5,6,6a, 7,8,12b- гексагидробензо[a]фенантридин = (±)-3 и (±)-8,9-дигидрокси-2,3,7,11b-тетрагидро-1H-нафто[1,2,3-де]изохинолин = 4).
На основании общей модели D1 фармакофора следует ожидать, что как сродство, так и собственная активность рацемического динапсолина (и его замещенных аналогов) принадлежит только одному из его энантиомеров - 11bR абсолютной конфигурации (и его гомохиральным аналогам). Можно рассчитывать, что разделение рацемата с применением общепризнанных технических приемов разделения приведет к одному изомеру динапсолина с приблизительно удвоенным относительно рацемата D1 сродством, что сделает его сродство к D1 рецептору аналогичным (+)-транс-10,11- дигидрокси- 5,6,6a,7,8,12b-гексагидробензо [a] фенантридину.
Как показывают фиг. 3 и таблица 1, динапсолин обладает большим сродством к D2-подобным рецепторам, чем дигидрексидин, ожидалось, что он будет в известной степени селективным к D1, а не D2-подобным рецепторам. Однако, определено, что дигидрексидин проявляет только приблизительно десятикратную D1: D2 селективность. Вдобавок, дигидрексидин хотя и обладает ожидаемой допамин-агонистической активностью, обладает также необычным свойством, называемым здесь "функциональной селективностью". В частности, в крысах (in vivo или in vitro) дигидрексидин действует как агонист D2-подобных рецепторов, локализованных постсинаптически, но как антагонист D2-подобных рецепторов, локализованных пре-синаптически. Предполагается, что это обусловлено различиями в лиганд-рецептор-G-протеиновом комплексе, локализованном пост-синаптически иначе, чем пре- синаптически, что определено по наличию специфических G-протеинов в данной клеточной среде.
Показано, что эти D2-свойства дигидрексидина присущи тому же энантиомеру, (т.е., 6aR, 12bS), который служит высоко родственным полным агонистом в отношении D1 рецептора. На этом основании можно было предположить, что как D1, так и D2 свойства динапсолина также сосредоточены в гомохиральном энантиомере. Оптические изомеры динапсолина и соответствующие аналоги представляют собой важные средства для изучения явления "функциональной селективности".
Ранее сообщалось, что дигидрексидин оказывает действие против болезни Паркинсона в МРТР модели болезни Паркинсона, прогнозируется, что и динапсолин должен обладать аналогичным воздействием. Следовательно, динапсолин и его производные имеют потенциальную клиническую применимость при болезни Паркинсона и других состояниях, где стимуляция допаминовых рецепторов может оказывать терапевтическое действие. Вдобавок, сообщается, что соответствующие модификации дигидрексидина дают аналоги, которые могут служить мишенью для специфических групп в популяции класса допаминовых рецепторов. Аналогичные подходы к динапсолину должны привести к соединениям с новой рецепторной селективностью подкласса и/или функциональными профилями.
Что касается следующих описанных экспериментальных способов, температуры плавления определяют прибором для измерения точки плавления Thomas-Hoover'a и не корректируют.
1H-ЯМР-спектры регистрируют с помощью ЯМР-спектрометра Varian VXR 500S (500 мГц) и химические сдвиги приводят в δ величинах (м.д.) относительно ТМС (тетраметилсилана). ИК-спектр регистрируют в таблетках KBr или в жидкой пленке с помощью Percin Elmer 1600 серийного FTIR-спектрометра. Химический ионизационный масс-спектр (CIMS) регистрируют на Finnigan 4000 квадрупольном масс-спектрометре. CI-спектр высокого разрешения регистрируют на Kratos MS50 спектрометре. Данные по элементному анализу получены в микроаналитической лаборатории Purdue University, West Lafayette, IN.
ТГФ перегоняют с натрий-бензофеноном в атмосфере азота непосредственно перед применением; 1,2-дихлорэтан перегоняют с пятиокисью фосфора перед применением.
Пример 1.
Получение 2-метил-2,3-дигидро-4(1H)-изохинолона
Этил 2-бромметилбензоат (5b)
Раствор этил-о-толуата (41,2 г, 0,25 моль) в четыреххлористом углероде (200 мл) добавляют по каплям к перемешиваемой смеси перекиси бензоила (100 мг), четыреххлористого углерода (200 мл) и NBS (44,5 г, 0,25 моль) при 0oC. Смесь нагревают до температуры кипения с обратным холодильником 3,5 часа в атмосфере азота и оставляют охлаждаться до комнатной температуры в течение ночи. Осажденный сукцинимид удаляют фильтрацией и плотный осадок на фильтре промывают четыреххлористым углеродом. Объединенные фильтраты промывают последовательно 2 н. NaOH (100 мл) и водой (2 х 100 мл) и раствор сушат над безводным MgSO4, фильтруют (Целит) и упаривают в вакууме, получая продукт в виде масла. Сушка в глубоком вакууме в течение ночи дает 60,5 г (99%) сырого соединения 5b: 1H-ЯМР продукта показывает наличие около 15% непрореагировавшего исходного вещества. Поскольку смесь нелегко разделить хроматографически или вакуумной перегонкой, ее используют в следующей стадии без дополнительной очистки: 1H-ЯМР (CDCl3) δ 1,43 (т, J = 7 Гц, 3H, CH2CH3), 4,41 (кв, J = 7 Гц, 2H, CH2CH3), 4,96 (с, 1H, CH2Br), 7,24 (м, 1H, ArH), 7,38 (м, 1H, ArH), 7,48 (м, 2H, ArH).
Этиловый эфир N-(2-карбоэтокси)саркозина (6).
К смеси гидрохлорида этилового эфира саркозина (32,2 г, 0,21 моль), карбоната калия (325 меш; 86,9 г, 0,63 моль) и ацетона (800 мл) добавляют раствор соединения 5b (60,7 г, полученное из 0,25 моль этил-0-толуата; 85% превращение в соединение 6; рассчитано 0,21 моль) в ацетоне (100 мл) при комнатной температуре в атмосфере азота. Смесь перемешивают при нагревании до температуры кипения с обратным холодильником 2 часа и затем оставляют при комнатной температуре на 20 часов. Твердый продукт удаляют фильтрацией (Целит) и остаток промывают ацетоном. Фильтраты объединяют и упаривают при пониженном давлении, получая масло. Масло растворяют в 250 мл 3 н. HCl и промывают диэтиловым эфиром. Водный слой подщелачивают водным NaHCO3 и экстрагируют диэтиловым эфиром (3 х 250 мл). Упаривание раствора диэтилового эфира дает масло, которое перегоняют в вакууме, получая 45,33 г (77%) соединения 6: т. кип. 140-142oC (0,5 мм Hg) [т.кип. 182-183oC (10 мм Hg)]; 1H-ЯМР (CDCl3) δ 1,24 (т, 3H, J = 7,1 Гц, CH3), 1,36 (т, 3H, J = 7,1 Гц, CH3), 2,35 (с, 3H, NCH3), 3,27 (с, 2H, CH2Ar), 4,00 (с, 2H, NCH2), 4,14 (кв, 2H, J = 7,1 Гц, CH2CH3), 4,32 (кв, 2H, J = 7,1 Гц, CH2CH3), 7,28 (т, 1H, J = 7,4 Гц, ArH), 7,42 (т, 1H, J = 7,6 Гц, ArH), 7,52 (д, 1H, J = 7,8 Гц, ArH), 7,74 (д, 1H, J = 7,7 Гц, ArH).
2-Метил-2,3-дигидро-4(1H)изохинолон (7). Свеже нарезанный натрий (10,9 г, 0,47 г-атом) добавляют к абсолютному этанолу (110 мл) в атмосфере азота и реакционную смесь нагревают до температуры кипения с обратным холодильником. После исчезновения металлического натрия к реакционной смеси медленно добавляют раствор соединения 6 (35,9 г, 0,128 моль) в сухом толуоле (160 мл). Затем реакционную смесь нагревают до температуры кипения с обратным холодильником и этанол отделяют азеотропно с помощью насадки Дина-Старка. После охлаждения растворитель упаривают при пониженном давлении. Образовавшийся желтый полутвердый остаток растворяют в смеси воды (50 мл), 98% этанола (60 мл) и концентрированной HCl (240 мл) и нагревают до температуры кипения с обратным холодильником 26 часов. После охлаждения смесь концентрируют в вакууме и осторожно подщелачивают твердым NaHCO3. Щелочной раствор экстрагируют эфиром, сушат (MgSO4) и упаривают до масла, которое перегоняют, получая соединение 7 (17,11 г, 83%): т.кип. 130-132oC (5 мм Hg) [т. кип. 81-83oC (0,4 мм Hg); т. пл. (HCl) 250oC]; ИК (неразбавл.) 1694 (C=O) см-1; 1H-ЯМР (CDCl3) δ 2,48 (с, 3H, CH3), 3,31 (с, 2H, CH3), 3,74 (с, 2H, CH2), 7,22 (д, 1H, J = 7,7 Гц, ArH), 7,34 (т, 1H, J = 7,9 Гц, ArH), 7,50 (т, 1H, J = 7,5 Гц, ArH), 8,02 (д, 1H, J = 7,9 Гц, ArH).
Синтез 8,9-дигидрокси-2,3,7,11b-тетрагидро-1H-нафто[1,2,3-де]изохинолина
К раствору 2,3-диметокси-N,N'-диэтилбензамида (соединение 8) (14,94 г, 63 ммоль) в диэтиловом эфире (1400 мл) при -78oC в атмосфере азота добавляют последовательно, по каплям, N,N,N',N'-тетраметилендиамин (TMEDA, 9,45 мл, 63 ммоль) и втор-бутиллитий (53,3 мл, 69 ммоль, 1,3 М раствор в гексане) через каучуковую септу с помощью шприца. Через 1 час к гетерогенной смеси добавляют свеже перегнанное соединение 7 (10,1 г, 62,7 ммоль). Убирают охлаждающую баню и реакционной смеси дают нагреться до комнатной температуры за 9 часов. Затем добавляют насыщенный раствор NH4Cl (400 мл) и смесь перемешивают 15 минут. Эфирный слой отделяют и водный слой экстрагируют дихлорметаном (4 х 100 мл). Органические слои объединяют, сушат (MgSO4) и упаривают до коричневого масла. Масло растворяют в толуоле (500 мл) и нагревают до температуры кипения с обратным холодильником в течение 8 часов с 3,0 г п-толуолсульфокислоты, охлаждают и концентрируют в вакууме. Остаток растворяют в дихлорметане, промывают разбавленным водным NaHCO3, водой и затем сушат (Na2SO4), фильтруют и упаривают, получая смолистый остаток. При растирании с этилацетат-гексаном (50:50) осаждается твердый продукт.
Перекристаллизация из этилацетат-гексана дает 12,75 г (63%) соединения 9 (2',3'-дигидро-4,5-диметокси-2'-метилспиро[изобензофуран- 1(3H)-4'(1'H)изохинолин]-3-он): т. пл. 193-194oC; ИК (KBr) 1752 см-1 (C=O); 1H-ЯМР (CDCl3) δ 2,47 (с, 3H, NCH3), 2,88 (д, 1H, J = 11,6 Гц), 3,02 (д, 1H, J = 11,7 Гц), 3,76 (д, 1H, J = 15,0 Гц), 3,79 (д, 1H, J = 15,1 Гц), 3,90 (с, 3H, OCH3), 4,17 (с, 3H, OCH3), 6,83 (д, 1H, J = 8,4 Гц, ArH), 7,03 (д, 1H, J = 8,2 Гц, ArH), 7,11 (м, 3H, ArH), 7,22 (м, 1H, ArH); МС (Cl) m/z 326 (100); Анал. (C19H19NO4) C, H, N.
2', 3'-Дигидро-4,5- диметокси-спиро [изобензофуран- 1(3H),4'(1'H) изохинолин]-3-он (10).
1-Хлор-этилхлорформиат (5,1 мл, 46,3 ммоль) добавляют по каплям к суспензии соединения 9 (6,21 r, 19,2 ммоль) в 100 мл 1,2-дихлорэтана при 0oC в атмосфере азота. Смесь перемешивают в атмосфере азота в течение 15 минут при 0oC и затем нагревают до температуры кипения с обратным холодильником в течение 8 часов. Смеси дают охладиться и концентрируют при пониженном давлении. К смеси добавляют 75 мл метанола и реакционную смесь нагревают до температуры кипения с обратным холодильником в течение ночи. После охлаждения растворитель упаривают при пониженном давлении, получая хлористоводородную соль соединения 10 с почти количественным выходом. Продукт достаточно чистый для того, чтобы его можно было использовать в следующей стадии без дополнительной очистки:
т.пл. (HCl) 220-222oC; т.пл. (основание) 208-210oC; ИК (CH2Cl2, основание) 1754 см-1 (C=O); 1H-ЯМР (CDCl3, основание) δ 3,18 (д, 1H, J = 13,5 Гц), 3,30 (д, 1H, J = 13,5 Гц), 3,84 (с, 3H, OCH3), 3,96 (с, 3H, OCH3), 4,02 (с, 2H, CH2N), 6,67 (д, 1H, J = 7,5 Гц, ArH), 7,12 (м, 2H, ArH), 7,19 (д, 1H, J = 7,5 Гц, ArH), 7,26 (т, 1H, J = 7,5 Гц, ArH), 7,41 (д, 1H, J = 8,5 Гц, ArH); МС (Cl) m/z 312 (100); HRCIMS рассчитан. для C18H17NO4: 312,1236; Найдено 312,1198; анал. (C18H17NO4) H, N; C: рассчитано 69,44; найдено, 68,01.
2', 3'-Дигидро-4,5- диметокси-2'-п-толуолсульфонилспиро [изобензофуран- 1(3H),4'(1'H)изохинолин] -3-он (11)
7 мл триэтиламина добавляют по каплям к смеси п-толуолсульфонилхлорида (3,6 г, 18,9 ммоль), соединения 10 (в виде HCl-соли, полученной из 19,2 ммоль соединения 9) и хлороформа (100 мл) при 0oC в атмосфере азота. После завершения добавления ледяную баню убирают и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Затем реакционную смесь подкисливают 100 мл охлажденной водной 0,1 н. HCl, экстрагируют дихлорметаном (2 х 100 мл) и органический экстракт сушат (MgSO4), фильтруют и упаривают в вакууме, получая вязкую жидкость, которая при растирании с этилацетат-гексаном при 0oC дает твердый продукт. Перекристаллизация из этилацетат-гексана дает 8,74 г (97%, всего из расчета на соединение 9) соединения 11: т. пл. 208-210oC; ИК (KBr) 1767 см-1 (C=O); 1H-ЯМР (CDCl3) δ 2,43 (с, 1H, CH3), 3,22 (д, 1H, J = 11 Гц), 3,88 (д, 1H, J = 11 Гц), 3,90 (с, 3H, OCH3), 3,96 (д, 1H, J = 15 Гц), 4,17 (с, 3H, OCH3), 4,81 (д, 1H, J = 15 Гц), 6,97 (д, 1H, J = 7,7 Гц, ArH), 7,16 (м, 3H, ArH), 7,26 (м, 1H, ArH), 7,38 (д, 2H, J = 8 Гц, ArH), 7,72 (д, 2H, J = 8 Гц, ArH); МС (Cl) m/z 466 (100); анал. (C25H23NO6S) C, H, N.
3,4-Диметокси-6-[(2-п-толуолсульфонил-1,2,3,4- тетрагидроизохинолин)-4-ил]бензойная кислота (12).
Раствор соединения 11 (2,56 г, 5,51 моль) в ледяной уксусной кислоте (250 мл) с 10% палладием на активированном углероде (6,30 г) встряхивают на гидрогенизаторе Парра при избыточном давлении 50 фунт/кв. дюйм (17,58 кг/см2; см. стр. 18 ориг., строка 4) в течение 48 часов при комнатной температуре. Катализатор удаляют фильтрацией и растворитель упаривают, получая 2,55 г (99%) соединения 12, обладающего чистотой, достаточной для использования в следующей стадии. Аналитический образец перекристаллизовывают из этанол-воды: т. пл. 182-184oC; ИК (KBr) 1717 см-1 (COOH); 1H-ЯМР (ДМСО-d6) δ 2,35 (с, 3H, CH3), 3,12 (м, 1H), 3,51 (дд, 1H, J = 5, 11,5 Гц), 3,71 (с, 6H, OCH3), 4,10 (м, 1H, Ar2CH), 4,23 (с, 2H, ArCH2N), 6,52 (д, 1H, J = 7,5 Гц, ArH), 6,78 (д, 1H, J = 7,5 Гц, ArH), 6,90 (м, 1H, ArH), 7,07 (т, 1H, J = 8 Гц, ArH), 7,14 (т, 1H, J = 6,5 Гц, ArH), 7,20 (д, 1H, J = 7,5 Гц, ArH), 7,38 (д, 2H, J = 8 Гц, ArH), 7,63 (д, 2H, J = 8,5 Гц, ArH), МС (Cl) m/z 468 (16), 450 (63), 296 (100); HRCIMS рассчитан. для C25H25NO6S: 468,1481; найдено 468,1467; анал. (C25H25NO6S) C, H, N.
2-N-п-толуолсульфонил-4-(2-гидроксиметил-3,4-диметоксифенил)-1,2,3,4- тетрагидроизохинолин (13).
К раствору соединения 12 (1,4 г, 2,99 ммоль) в сухом тетрагидрофуране (30 мл) добавляют 1,0 М борантетрагидрофурана (8 мл) при 0oC в атмосфере азота. По завершении добавления смесь перемешивают при нагревании до температуры кипения с обратным холодильником в течение ночи. Добавляют дополнительно диборан (4 мл) и продолжают перемешивание еще 30 минут. После охлаждения и упаривания при пониженном давлении, осторожно добавляют метанол и растворитель удаляют при пониженном давлении. Процедуру повторяют трижды, чтобы гарантировать завершение метанолиза промежуточного боранового комплекса. Упаривание растворителя дает 1,10 г (81%) сырого соединения 13. Аналитические образцы чистят флэш хроматографией (силикагель, EtOAc/гексан) с последующей перекристаллизацией из этилацетат/гексана: т. пл. 162-164oC; 1H-ЯМР (CDCl3) δ 2,38 (с, 3H, CH3), 3,18 (дд, 1H, J = 7,5, 11,9 Гц), 3,67 (дд, 1H, J = 4,5, 11,8 Гц), 3,81 (с, 3H, OCH3), 3,85 (с, 3H, OCH3), 4,27 (д, 1H, J = 15 Гц), 4,40 (д, 1H, J = 15 Гц), 4,57 (т, 1H, J = 6 Гц, CHAr2), 4,71 (с, 2H, CH2OH), 6,58 (д, 1H, J = 8,5 Гц, ArH), 6,74 (д, 1H, J = 8,6 Гц, ArH), 6,84 (д, 1H, J = 7,7 Гц, ArH), 7,08 (т, 2H, J = 7,6 Гц, ArH), 7,14 (т, 1H, J = 6,6 Гц, ArH), 7,27 (д, 2H, J = 8 Гц, ArH), 7,65 (д, 2H, J = 8 Гц, ArH); МС (Cl) m/z 454 (2,57), 436 (100); анал. (C25H27NO5S) C, H, N.
8,9-Диметокси-2-п- толуолсульфонил-2,3,7,11b- тетрагидро-1H- нафт [1,2,3-де]изохинолин (14).
Порошкообразное соединение 13 (427 мг, 0,98 ммоль) добавляют за несколько порций к 50 мл охлажденной концентрированной серной кислоты (50 мл) при -40oC в атмосфере азота при энергичном механическом перемешивании. После добавления реакционную смесь нагревают до -5oC в течение 2 часов и затем выливают на колотый лед (450 г) и оставляют перемешиваться в течение 1 часа. Продукт экстрагируют дихлорметаном (2 х 150 мл), промывают водой (2 х 150 мл), сушат (MgSO4), фильтруют и упаривают, получая масло, которое при растирании с диэтиловым эфиром при 0oC дает соединение 14 (353 мг, 82%) в виде белого твердого продукта, который используют в следующей стадии без дополнительной очистки. Аналитический образец получают центробежной роторной хроматографией, используя 50% этилацетат-гексан в качестве элюента с последующей перекристаллизацией из EtOAc/гексана: т. пл. 204-206oC; 1H-ЯМР (CDCl3) δ 2,40 (с, 3H, CH3), 2,80 (м, 1H, H-1a), 3,50 (дд, 1H, J = 4,5, 17,5 Гц, H-1b), 3,70 (дд, 1H, J = 7,14 Гц, H-3a), 3,828 (с, 3H, OCH3), 3,832 (с, 3H, OCH3), 3,9 (м, 1H, H-11b), 4,31 (д, 1H, J = 17,6 Гц, H-7a), 4,74 (ддд, 1H, J = 1,7, 6,0, 11,2 Гц, H-7b), 4,76 (д, 1H, J = 14,8 Гц, H-3b), 6,77 (д, 1H, J = 8,3 Гц, ArH), 6,87 (д, 1H, J = 8,4 Гц, ArH), 6,94 (д, 1H, J = 7,6 Гц, ArH), 7,13 (т, 1H, J = 7,5 Гц, Ar-H-5), 7,18 (д, 1H, J = 7,2 Гц, ArH), 7,33 (д, 2H, J = 8,1 Гц, ArH), 7,78 (д, 2H, J = 8,2 Гц, ArH); МС (Cl) m/z 436 (55), 198 (86), 157 (100); HRCIMS рассчитан. для C25H25NO4S: 436,1583; найдено 436,1570; анал. (C25H25NO4S) C, H, N.
8,9-Диметокси-2,3,7,11b-тетрагидро- 1H-нафт[1,2,3-де] изохинолин (15).
Смесь соединения 14 (440 мг, 1,01 ммоль), сухого метанола (10 мл), динатрий гидрофосфата (574 мг, 4,04 ммоль) перемешивают в атмосфере азота при комнатной температуре. К этой смеси добавляют тремя порциями 6,20 г 6% Na-Hg и реакционную смесь нагревают до температуры кипения с обратным холодильником в течение 2 часов. После охлаждения добавляют воду (200 мл) и смесь экстрагируют диэтиловым эфиром (3 х 200 мл). Слои диэтилового эфира объединяют, сушат (MgSO4), фильтруют (Целит) и упаривают, получая масло, которое затвердевает в вакууме. После роторной хроматографии получают 142 мг (50%) соединения 15 в виде масла. Масло быстро темнеет на воздухе и его сразу же используют в следующей стадии. Небольшую порцию масла обрабатывают эфирным HCl и хлористоводородную соль соединения 15 перекристаллизовывают из этанол-диэтилового эфира: т. пл. (HCl соль) 190oC (разл.); 1H-ЯМР (CDCl3, основание) δ 3,13 (дд, 1H, J = 10,8, 12 Гц, H-1a), 3,50 (дд, 1H, J = 3,4, 17,4 Гц, H-1b), 3,70 (м, 1H, H-11b), 3,839 (с, 3H, OCH3), 3,842 (с, 3H, OCH3), 4,03 (дд, 1H, J = 6,12 Гц, H-7a), 4,08 (с, 2H, H-3), 4,33 (д, 1H, J = 17,4 Гц, H-7b), 6,78 (д, 1H, J = 8,24 Гц, ArH), 6,92 (м, 3H, ArH), 7,11 (т, 1H, J = 7,5 Гц, ArH), 7,18 (д, 1H, J = 7,5 Гц, ArH); МС (Cl) m/z 282 (100); HRCIMS рассчитан. для C18H19NO2: 282,1494; найдено 282,1497.
8,9-Дигидрокси-2,3,7,11b- тетрагидро-1H-нафт [1,2,3-де] изохинолин (4).
К раствору соединения 15 (25 мг, 0,089 ммоль) в дихлорметане (5 мл) при -78oC добавляют трибромид бора (0,04 мл, 0,106 г, 0,42 ммоль). После перемешивания при -78oC в атмосфере азота в течение 2 часов, охлаждающую баню убирают и реакционную смесь оставляют перемешиваться при комнатной температуре на 5 часов. Затем реакционную смесь охлаждают до -78oC и осторожно добавляют метанол (2 мл). После перемешивания в течение 15 минут при комнатной температуре растворитель упаривают при пониженном давлении. Добавляют дополнительное количество метанола и процедуру повторяют трижды. Образовавшийся серый твердый продукт перекристаллизовывают из этанол-этилацетата, получая в целом 12 мг (41%) бромистоводородной соли соединения 4:
т. пл. 258oC (разл.); 1H-ЯМР (HBr-соль, CD3OD) δ 3,43 (т, 1H, J = 12 Гц, H-1a), 3,48 (дд, 1H, J = 3,5, 18 Гц, H-1b), 4,04 (м, 1H, H-11b), 4,38 (дд, 2H, J = 5,5, 12 Гц, H-7), 4,44 (с, 2H, H-3), 6,58 (д, 1H, J = 8,5 Гц, ArH), 6,71 (д, 1H, J = 8,5 Гц, ArH), 7,11 (д, 1H, J = 7,5 Гц, ArH), 7,25 (т, 1H, J = 7,5 Гц, ArH), 7,32 (д, 1H, J = 7,5 Гц, ArH); МС (Cl) m/z 254 (100); HRCIMS рассчитан. для C16H15NO2: 254,1181; найдено 254,1192.
Фармакология динапсолина
Способы
Взрослых самцов Sprague Dawley крыс (200-250 г) получают из Charles River Breeding лабораторией (Raleigh, NC) или Harlan лабораторий (Indianapolis, IN). Крыс забивают, обезглавливая, извлекают целиком головной мозг и недолго охлаждают в охлажденном льдом растворе соли. Готовят тонкие срезы мозга с помощью рассекающего блока и затем иссекают центральное полосатое тело из двух веночных срезов, содержащих большую часть этой области. Ткань сразу же замораживают с помощью сухого льда и хранят при -70oC до дня испытаний.
Культуры клеток. Клетки С-6 глиомы, выражающие D1A рецептор макак резуса, (C-6-mD1A; Machida et al., 1992) выращивают в DMEM-H-среде, содержащей 4.500 мг/л глюкозы, L-глутамина, 5% сыворотки плода коровы и 600 нг/мл G418 или 2 мг/мл пуромицина. Клетки выдерживают во влажном инкубаторе при 37oC с 5% CO2.
Подготовка мембран. Клетки выращивают в 75 см2 колбах до слияния. Клетки промывают и лизируют с 10 мл охлажденного льдом гипоосмотического буфера (НОВ) (5 мМ Hepes, 2,5 мМ MgCl2, 1 мМ EDTA; pH 7,4) в течение 10 минут при 4oC. Затем клетки выскабливают из колб, используя стерильный клеточный шабер Baxter'a (McGaw Park, IL). Колбы окончательно прополаскивают 5 мл HOB. Конечный объем клеточной суспензии, собранный из каждой колбы, составляет около 14 мл. Затем мембраны, выскобленные из нескольких колб, объединяют. Объединенную суспензию клеток гомогенизируют (10 взмахов), 14 мл за один раз, используя 15 мл стеклянный гомогенизатор. Гомогенаты клеток объединяют и крутят при 43000 об/мин (Sorvall RG-5D/SS-34, Du Pont, Wilmington, DE) при 4oC 20 мин. Удаляют супернатант и осадок в пробирке после центрифугирования ресуспендируют (10 взмахов) в 1 мл охлажденного льдом HOB на каждую исходную колбу гомогенизированных клеток. Этот гомогенат затем снова крутят при 43000 об/мин при 4oC в течение 20 минут. Удаляют супернатант и конечный осадок ресуспендируют (10 взмахов) в охлажденном льдом буфере для хранения (50 мМ Hepes, 6 мМ MgCl2, 1 мМ EDTA; pH 7,4), получая конечную концентрацию около 2,0 мг белка/мл. Аликвоты конечного гомогената хранят в пробирках для микроцентрифугирования при -80oC. Перед применением для испытаний на аденилатциклазу концентрации
белка для каждого мембранного препарата определяют количественно, используя реагент для BCA протеинового испытания (Pierce, Rockford, IL), адаптированный для применения в считывающем устройстве для микропластин (Molecular Devices; Menlo Park, CA).
Испытания на допамин-рецепторное связывание.
Замороженное полосатое тело крыс гомогенизируют несколькими ручными встряхиваниями в Wheaton-Teflon-стеклянном гомогенизаторе в 8 мл охлажденного льдом 50 мМ HEPES буфера с 4,0 мМ MgCl2 (pH 7,4). Ткань центрифугируют при 27000 об/мин в течение 10 минут, супернатант отбрасывают, а осадок в пробирке после центрифугирования гомогенизируют (пять взмахов), ресуспендируют в охлажденном льдом буфере и центрифугируют снова. Конечный осадок в пробирке после центрифугирования ресуспендируют при концентрации 2,0 мг влажного веса/мл. Количество ткани, добавленной в каждую пробирку для испытания, составляет 1,0 мг в конечном объеме для испытания 1,0 мл. D1 рецепторы метят [3H] SCH23390 (0,30 нМ); D2 рецепторы метят [3H]спипероном (0,07 нМ); немеченый кетансерин (50 нМ) добавляют для маскировки связывания по 5-HT2 участкам. Общее связывание определяют как радиолигандное связывание в отсутствии любого конкурирующего лекарственного средства. Неспецифическое связывание оценивают, добавляя немеченый SCH23390 (1 мкМ) или немеченый хлорпромазин (1 мкМ) для испытаний на D1 и D2 рецепторные связывания, соответственно. В качестве внутреннего стандарта в каждое испытание включают кривую сравнения для шести концентраций немеченого SCH23390 (D1 связывание) или хлорпромазина (D2 связывание). Для каждой концентрации лекарственного средства проводят тройное определение. Пробирки для испытания инкубируют при 37oC в течение 15 минут и связывание определяют фильтрацией с охлажденным льдом буфером на 12-ячеечном сборнике клеток Skatron'a (Skatron, Inc., Sterling, VA), используя фильтрующие пластины из стекловолокна (Skatron N 7034). Фильтрам дают высохнуть и добавляют 0,1 мл Optiphase HI-SAF II-сцинтиллирующую жидкость. Радиоактивность определяют на LKB Wallac 1219 RackBeta жидком сцинтилляционном счетчике (Wallac, Gaithersburg, MD). Уровни белка в ткани определяют, используя ВСА реагент для определения белка (Pierce, Rockford, IL).
Данные анализа для радиорецепторных испытаний.
Данные по связыванию из каждого испытания анализируют отдельно. Данные нормализуют, выражая среднее значение dpm для каждой концентрации конкурента в процентах от общего связывания. Затем эти данные подвергают нелинейному регрессивному анализу, используя алгоритм для сигмоидальных кривых в curve-fitting программе InPlot (Graphpad Inc.; San Francisco, CA) или EBDA и LIGAND стандартную программу, адаптированную для IBM-PC by McPherson, для получения К0,5 и коэффициента Hill'a (nн) для каждой кривой. Анализ остаточных вариаций показывает прекрасное соответствие; значения r составляют около 0,99 для всех кривых в данных экспериментах.
Активность аденилатциклазы в полосатом теле крыс. Для измерения активности аденилатциклазы путем отделения cAMP от других меченых нуклеотидов используют способ автоматической ЖХВР Schuiz'a и Mailman'a. Вкратце, стриарную ткань крыс гомогенизируют восемью ручными встряхиваниями в Wheaton-Teflon стеклянном гомогенизаторе в 5 мМ HEPES буфере (pH 7,5), содержащем 2 мМ EGTA (50 мл/г ткани). После добавления и перемешивания 50 мл/г 50 мМ-HEPES буфера (pH 7,5), содержащего 2 мМ EGTA, 20 мкл аликвоту этого гомогената ткани добавляют к полученной реакционной смеси (конечный объем 100 мкл), содержащей 0,5 мМ ATP, 0,5 мМ изобутилметилксантина, [32P]ATP (0,5 мкКюри), 1 мМ cAMP, 2 мМ MgCl2, 100 мМ HEPES буфера, 2 мкМ GTP, 0-100 мкМ допамина, DHX или SKF38393, 10 мМ креатинфосфата и 5 ед. креатинфосфокиназы. Для каждой концентрации лекарственного средства выполняют тройное определение. Реакцию продолжают в течение 15 минут при 30oC и затем обрывают добавлением 100 мкл 3% додецилсульфата натрия (SDS). Белки и большую часть не-циклических нуклеотидов осаждают добавлением по 300 мкл 4,5% ZnSO4 и 10% Ba(OH)2 каждого. Образцы центрифугируют (10000 об/мин в течение 8-9 мин) и супернатанты вкалывают в ЖХВР-систему (водный Z-модуль или RCM 8х10 модуль, снабженный C18, 10-микронным картриджем). Подвижной фазой служит 150 мМ ацетат натрия (pH 5,0) с 23% метанолом. Для количественного определения немеченых cAMP, добавленных к образцам в качестве внутреннего стандарта, используют УФ-детектор (254 нм-детектирование). Радиоактивность каждой фракции определяют радиационным детектором текущего потока (Inus Systems. Tampa, FL), используя счетчик Cerenkov'a. Выделение образца основано на УФ измерении общих площадей пиков, соответствующих немеченым cAMP, рассчитанных с использованием РЕ Nelson (Cupertino, CA) Model 900 data collection modules and TurboChrom стандартной программы. Уровни белка в ткани определяют, используя BCA-реагент для обнаружения белков (Pierce, Rockford, IL).
Анализ на аденилатциклазу в G-6mD1A клетках.
Замороженные мембраны размораживают и добавляют в пробирки для испытаний (10 мг белка/пробирку), содержащие полученную реакционную смесь [100 мМ Hepes, (pH 7,4), 100 мМ NaCl, 4 мМ MgCl2, 2 мМ EDTA, 500 мкМ изобутилметилксантина (IBMX), 0,01% аскорбиновой кислоты, 10 мкМ паргилина, 2 мМ АТР, 5 мкМ GTR, 20 мМ креатинфосфата, 5 единиц креатинфосфокиназы (CPK), 1 мкМ пропранолола] и выбранное лекарственное средство. Конечный реакционный объем составляет 100 мкл.
Базисную cAMP активность определяют инкубацией ткани в реакционной смеси без добавления лекарственного средства. Пробирки для испытаний дублируют и после 15 минут инкубации при 30oC реакцию прекращают добавлением 500 мкл 0,1 н. HCl. Пробирки кратковременно обрабатывают на вихревой воронке и затем в BHG HermLe Z 230 М микроцентрифуге в течение пяти минут при 15000 об/мин для осаждения микрочастиц.
Радиоиммуноанализ (RIA) на cAMP. Концентрацию cAMP в каждом образце определяют с помощью RIA ацетилированного cAMP, модифицированного из ранее описанного. Иодинацию cAMP осуществляют, используя способ, описанный Patel и Linden. Буфером для анализа служит 50 мМ натрий-ацетатный-буфер с 0,1% азида натрия (pH 4,75). Стандартные кривые cAMP получают в буфере при концентрациях 2-500 фмоль/пробирку для испытания. Для повышения чувствительности анализа все образцы и стандарты ацетилируют 10 мкл раствора 2:1 триэтиламин : уксусный ангидрид. Образцы для анализов дублируют. Каждая пробирка для испытания (общий объем 300 мкл) содержит 25 мкл каждого образца, 75 мкл буфера, 100 мкл первичного антитела (овечьи анти-cAMP, 1:100000 разбавление 1% BSA в буфере) и 100 мкл [125I]-cAMP (50000 dpm/100 мкл буфера). Пробирки обрабатывают в вихревой воронке и хранят при 4oC в течение ночи (около 18 часов). Связанную антителами радиоактивность отделяют, добавляя 25 мкл BioMag кролик, anti-goat IgG (Advanced Magnetics, Cambridge MA), после чего подвергают вихревой обработке при 4oC в течение 1 часа. К этим образцам добавляют 1 мл 12% полиэтиленгликоля/50 мМ натрий-ацетатного буфера (pH 6,75) и пробирки центрифугируют при 1700 об/мин в течение 10 мин. Супернатанты отделяют отсасыванием и радиоактивность в осадке после центрифугирования определяют, используя LKB Wallac-гамма счетчик (Gaithersburg, MD).
Данные анализа по исследованию аденилатциклазы.
Данные для каждого образца выражают сначала в пмоль/мг/мин cAMP. Нулевые значения cAMP вычитают из общего количества продуцированного cAMP в случае каждого лекарства. Данные для каждого лекарственного средства выражают в отношении к стимулированию, производимому 100 мкМ DA.
Результаты связывания и функциональных воздействий динапсолина на D1 рецепторы в гомогенатах полосатого тела крыс.
Как показано на фиг. 3, динапсолин конкурирует, проявляя высокое сродство к D1 рецепторам в гомогенатах полосатого тела крыс, обладая сродством, почти идентичным с дигидрексидином, полным D1 агонистом. Как динапсолину, так и дигидрексидину соответствует меньшая крутизна на кривых сравнения, чем SCH 23390 (1), являющемуся прототипом D1 антагониста.
Таблица 1 суммирует сродство (+)-3 и (±)-4 к допаминовым рецепторам в головном мозге крыс. Изучение связывания радиолигандов допаминовыми рецепторами проведено в гомогенатах полосатого тела крыс с использованием 0,3 нМ 3H-SCH23390 (D1 участки) и 0,07 нМ 3H-спиперона в присутствии 50 нМ немеченого кетансерина (D2 участки). Кривые сравнения анализированы по способу нелинейной регрессии с целью определения величин K0,5 и коэффициента крутизны Hill'a (nн). Представлены средние значения величин и стандартная ошибка для трех идентичных испытаний в случае каждого испытуемого соединения.
Способность испытуемых соединений [(+)-SKF 38393 = (+)-2; (±)-транс-10,11-дигидрокси- 5,6,6a, 7,8,12b-гексагидробензо [a]фенантридин = (±)-3 и (±)-8,9-дигидрокси- 2,3,7,11b-тетрагидро- 1H- нафто [1,2,3-де]изохинолин = 4] стимулировать аккумуляцию cAMP исследуют в гомогенатах полосатого тела крыс. Обладающий высоким сродством полный агонист (±)-3 и частичный агонист (+)-2 включены для сравнения. Результаты этих экспериментов приведены на фиг. 4 в виде среднего значения ±SEM (средняя статистическая ошибка) по крайней мере из трех экспериментов. Концентрации насыщения (10 мкМ) как динапсолина, так и дигидрексидина вызывают такую же степень возрастания синтеза cAMP (95,8% ± 4,7 для динапсолина и 91,3% ± 4,6 для дигидрексидина), какую дает максимально эффективная концентрация допамина (100 мкМ). Наоборот, частичный агонист (+)-2 приводит менее чем к 50% стимуляции (40,7 ± 7,0 для SKF 38393). Эти действия блокируются D1 антагонистом SCH23390.
Функциональная эффективность динапсолина исследована также в клонированных D1A рецепторах приматов, выраженных в C-6 клетках глиомы (C-6-mD1 клетки). Как показано на фиг. 5, соединение динапсолин также обладает полной эффективностью в этом препарате, с EC50 около 30 нМ (данные представляют среднее значение из двух экспериментов, проведенных при дублировании). (±)-Транс-10,11-дигидрокси- 5,6,6a, 7,8,12b- гексагидробензо [a]фенантридин (дигидрексидин) также обладает полной эффективностью в этом препарате, тогда как (+)-2 обладает только частичной эффективностью.
Связывание по D2 рецепторам. Исследована способность динапсолина к конкуренции за D2 рецепторы в гомогенатах полосатого тела крыс. Как показано на фиг. 6 и в таблице 1, сродство динапсолина к D2-подобным рецепторам в гомогенатах полосатого тела крыс (K0,5 = 31 нМ) значительно выше, чем сродство дигидрексидина к D2-подобным рецепторам (K0,5 = 50 нМ). Как видно из фиг. 6, крутизна кривых сравнения как для динапсолина, так и для дигидрексидина меньше, чем для прототипа D2 антагониста - хлорпромазина.
Использованием тех же общих способов, что описаны выше в примере 1, синтезированы соединения примеров 2-48, указанные ниже в таблице 2, с использованием исходных соединений, соответствующих соединениям, приведенным в схемах 1 и 2 (фиг. 1 и 2), но замещенных функциональными группами, требующимися для получения замещенных структур, соответствующих конденсированному продукту нафтоизохинолина, показанному для каждого примера. Так, для примера 3, 4 и/или 5 замещенные аналоги соединения 5a (схема 1) дают соответствующие заместители R4, R3 и R2, соответственно, на формуле I. Замещение сложных эфиров N-метилаланина или N-метилвалина на сложные эфиры саркозина на стадии b схемы 1 дает соответствующие соединения формулы I, где R5 обозначает метил и изопропил, соответственно. Применение других 2 и 3 замещенных бензамидов (аналогов соединения 8 в схеме 2) дает соответствующие замещенные структуры при C8 и C9 в формуле I.
Приведенные выше соединения иллюстрируют изобретение, но не ограничивают его описанными соединениями. Подразумевается, что вариации и модификации характерных соединений, которые могут быть получены любым специалистом в соответствующей области, также входят в рамки объема изобретения, как описано в приложенных пунктах.
Пример фармацевтической композиции.
Пример фармацевтической композиции представляет собой раствор динапсолина, растворенного в изотоническом растворе хлорида натрия, а именно изотонический раствор хлорида натрия, содержащий 0,35 мг/мл динапсолина, который получают, например, растворением 350 мг динапсолина при перемешивании в 1 л изотонического раствора хлорида натрия.
Такой раствор используется для парентерального введения динапсолина. Состав и способ получения изотонического раствора хлорида натрия являются широко известными в данной области техники.
Такой раствор динапсолина может быть использован для введения примерно 350 мг динапсолина в день внутривенно. Другие стандартные формы, такая, как стандартная форма для перорального введения, также пригодны для применения согласно настоящему изобретению.

Изобретение относится к области химии и медицины, конкретно к допаминовым рецепторным лигандам формулы (I), фармацевтическим составам таких соединений и к способу применения таких соединений для эффективного лечения пациентов, страдающих допаминсвязанной дисфункцией центральной или периферической нервной системы. 3 с. и 7 з.п. ф-лы, 6 ил., 2 табл.
или их фармацевтически приемлемые соли,
где R и R5 - водород или С1-С4 алкил;
R1 - водород или С1-С4 алкил;
Х - водород, галоген или группа формулы -OR6, где R6 - водород или С1-С4 алкил;
R2, R3 и R4 независимо выбирают из группы, включающей водород, С1-С4 алкил, фенил, галоид или группу -OR1, где R1 имеет принятые выше значения.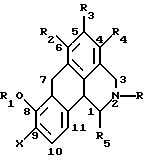
где R и R5 - водород или С1-С4 алкил;
R1 - водород или С1-С4 алкил;
Х - водород, галоген или группа формулы -OR6, где R6 - водород или С1-С4 алкил;
R2, R3 и R4 независимо выбирают из группы, включающей водород, С1-С4 алкил, фенил, галоид или группу -OR1, где R1 имеет принятые выше значения,
или его фармацевтически приемлемой соли в количестве, эффективном для ослабления симптомов указанного нарушения.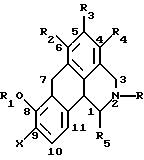
или его фармацевтически приемлемой соли,
где R и R5 - водород или С1-С4 алкил;
R1 - водород или С1-С4 алкил;
Х - водород, галоген или группа формулы -OR6, где R6 - водород или С1-С4 алкил;
R2, R3 и R4 независимо выбирают из группы, включающей водород, С1-С4 алкил, фенил, галоид или группу -OR1, где R1 имеет принятые выше значения,
и фармацевтически приемлемый носитель.
| ПРОИЗВОДНЫЕ БЕНЗО-/С/-ФЕНАНТРИДИНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2073674C1 |
| RU 2063965, C1, 20.07.1996 | |||
| Способ получения производных октагидропиразоло @ 3,4-г @ хинолина или их солей | 1979 |
|
SU1360586A3 |
| Способ получения 9-амино-5,6,6 @ ,7-тетрагидро-4Н-бензо @ , @ тиазоло @ 4,5- @ хинолинов или их кислотно-аддитивных солей в виде рацемата, энантиомеров или смеси энантиомеров | 1987 |
|
SU1480771A3 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
