Данное изобретение относится к способу получения 3-амино-1,2,4-бензотриазин-1,4-диоксида (варианты), далее в описании иногда упоминаемом как тирапазамин, высокой чистоты для системного введения при лечении злокачественных опухолей у млекопитающих.
Тирапазамин является известным соединением, которое как сообщают в патенте США N 3868371, пригодно для использования в качестве противомикробного агента. Кроме того, в патенте США N 3957779 раскрывается использование тирапазамина и его производных в качестве промотора роста для наращивания массы тела животных, например крупного рогатого скота, свиней и домашней птицы.
Тирапазамин также является многообещающим лекарственным средством, которое, как раскрывается в Международной патентной заявке PCT WO 91/047028, пригодно для использования в качестве противоракового агента для лечения млекопитающих, чье описание включено в данную заявку в качестве ссылки. При системном использовании тирапазамина важно, чтобы он имел высокую степень очистки для получения необходимой противораковой активности без побочных эффектов из-за мешающих примесей, которые могут оставаться в нем в процессе его изготовления.
В журнале Angew. Chem. , вып. 84, 1061 (1972) и в патенте США N 3868371 раскрывается способ получения тирапазамина, заключающийся в нижеследующем: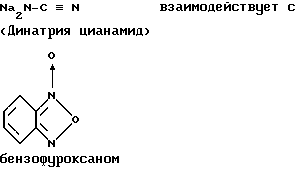
в присутствии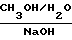
с образованием его натриевой соли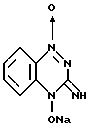
которая при обработке ее уксусной кислотой превращается в 3-амино-1,2,4-бензотриазин-1,4-диоксид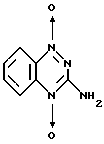
Указанный способ раскрывается подробно далее в данном описании.
К суспензии, содержащей 13,6 г (0,1 моль) бензофуроксана в смеси 40 мл метанола и 40 мл воды, добавляют порциями 17,2 г (0,2 моль) динатрия цианамида при температуре 20oC. Во время его введения температура реакционной массы повышается до 50-60oC, при этом раствор приобретает сине-фиолетовый цвет. Реакционную массу перемешивают в течение еще 40 мин при температуре примерно 60oC, и образующийся преципитат отделяют путем фильтрования из маточного раствора, который затем выдерживают и далее обрабатывают. Преципитат растворяют в воде, раствор фильтруют, фильтрат подкисляют уксусной кислотой. Полученные в результате 12,5 г 3-амино-1,2,4-бензотриазин-1,4-ди-N-оксида (71% от теоретического количества) выделяют в виде красновато-золотистых кристаллов, которые плавятся с разложением при 220oC. После подкисления маточного раствора уксусной кислотой получают еще 3,2 г (18% от теоретического количества) 3-амино-1,2,4-бензотриазин-1,4-диоксида и после перекристаллизации его из диметилформамида указанный продукт плавится при разложении при 220oC. Суммарный выход, как сообщают, составляет 89% от теоретического.
Настоящее изобретение предлагает улучшенный способ получения 3-амино-1,2,4-бензотриазин-1,4 диоксида.
В основу настоящего изобретения положена задача получения 3-амино-1,2,4-бензотриазин-1,4-диоксида такого гранулометрического состава, который подходил бы для использования в твердых лекарственных формах для перорального введения без необходимости после синтеза измельчения его частиц.
Задача решена тем, что заявляемый способ получения 3-амино-1,2,4-бензотриазин-1,4-диоксида согласно изобретению включает стадии, состоящие из:
a) раствор, содержащий бензофуразан-1-оксид в диметилсульфоксиде, при молярном отношении 1: 3 прибавляют к раствору динатрия цианамида в воде при молярном отношении 3: 2, загруженного в реакционную колбу, при температуре от 55 до 65oC с образованием натриевой соли 3-амино-1,2,4-бензотриазин-1,4 диоксида в виде преципитата;
b) полученный преципитат отделяют и суспендируют в воде в объемном соотношении 1: 6;
c) полученную суспензию подкисляют метансульфокислотой, взятой при 3,5 мольной эквивалентной концентрации, с получением раствора;
d) полученный раствор фильтруют для удаления из него загрязняющих нерастворимых остатков кислоты;
e) отфильтрованный раствор загружают в буферный раствор, содержащий избыток ацетата натрия, и дают протекать процессу кристаллизации;
f) полученные кристаллические частицы фильтруют и промывают водой;
g) промытые кристаллические частицы загружают опять в реакционный сосуд и перемешивают примерно пятью объемами воды, и
h) кристаллические частицы отфильтровывают и ополаскивают ацетоном с получением очищенного 3-амино-1,2,4-бензотриазин-1,4 диоксида.
Заявляемый способ может также включать сушку и гомогенизацию очищенного 3-амино-1,2,4-бензотриазин-1,4 диоксида.
Целесообразно указанный раствор бензофуразан-1-оксида в диметилсульфоксиде прибавлять к раствору динатрия цианамида в воде при температуре 60oC.
Получают очищенный продукт (чистота продукта до 98,5%), имеющий размер частиц менее 190 нм, пригодный для использования в твердой лекарственной форме для перорального введения, например, в форме мягкой желатиновой капсулы.
В другом варианте осуществления изобретения способ получения 3-амино-1,2,4-бензотриазин-1,4-диоксида включает стадии, состоящие из:
a) раствор, содержащий бензофуразан-1-оксид в диметилсульфоксиде, при молярном отношении 1: 3 прибавляют к водному раствору цианамида и гидроксида натрия, имеющего молярное отношение 3: 6, при температуре от 55 до 65oC с получением натриевой соли 3-амино-1-2,4-бензотриазин-1,4-диоксида в виде частичной суспензии/раствора;
b) полученную частичную суспензию/раствор разбавляют водой при объемном соотношении примерно 1: 6 с получением в результате раствора, содержащего загрязняющий нерастворимый остаток основания;
c) полученный раствор фильтруют для удаления из него загрязняющего нерастворимого остатка основания;
d) отфильтрованный раствор нейтрализуют небольшим избытком ледяной уксусной кислоты с получением неочищенной кристаллической смеси;
e) кристаллическую смесь охлаждают и собирают твердые частицы фильтрованием в виде мокрой пресс-лепешки;
f) прибавляют при перемешивании смесь ацетона и уксусной кислоты при соотношении примерно 88: 12;
g) образующиеся кристаллические частицы собирают фильтрованием и ополаскивают сначала смесью ацетона и уксусной кислоты, имеющей соотношение 88: 12, а затем водой;
h) к кристаллическим частицам прибавляют примерно пять объемов воды с получением суспензии;
i) полученную суспензию подкисляют метансульфокислотой, взятой при 3,5 мольной эквивалентной концентрации;
j) суспендированные кристаллические частицы собирают фильтрованием, затем загружают в хорошо перемешанный разбавленный раствор ацетата натрия и подвергают их микрокристаллизации с образованием микрокристаллического продукта со средним размером частиц менее 190 нм; и
k) полученные микрокристаллические частицы собирают фильтрованием и ополаскивают сначала водой, а затем ацетоном с получением очищенного 3-амино-1,2,4-бензотриазин-1,4-диоксида.
Указанный способ может также включать сушку и гомогенизацию очищенного 3-амино-1,2,4-бензотриазин-1,4-диоксида.
С целью улучшения технологического процесса целесообразно раствор бензофуразан-1-оксида в диметилсульфоксиде прибавлять к водному раствору цианамида и гидроксида натрия при температуре 60oC.
Тирапазамин является мощным цитотоксическим агентом, который, как свидетельствуют данные, имеет повышенную токсичность и селективность относительно гипоксичных клеток, присутствующих в злокачественных опухолях. Отмечалось, что он эффективен как в препаратах для парентерального введения, так и препаратах в твердой форме для перорального введения.
Для получения высоко чистого тирапазамина, пригодного для использования в таких фармацевтических препаратах, были проведены обширные исследования, раскрываемые ниже в данном описании, с целью улучшения способа получения и качества целевого продукта, которые описаны в журнале Angew. Chem. , вып. 84, 1061 (1972) и патенте США N 3868371.
Условия для протекания гетерогенной реакции
Пример 1
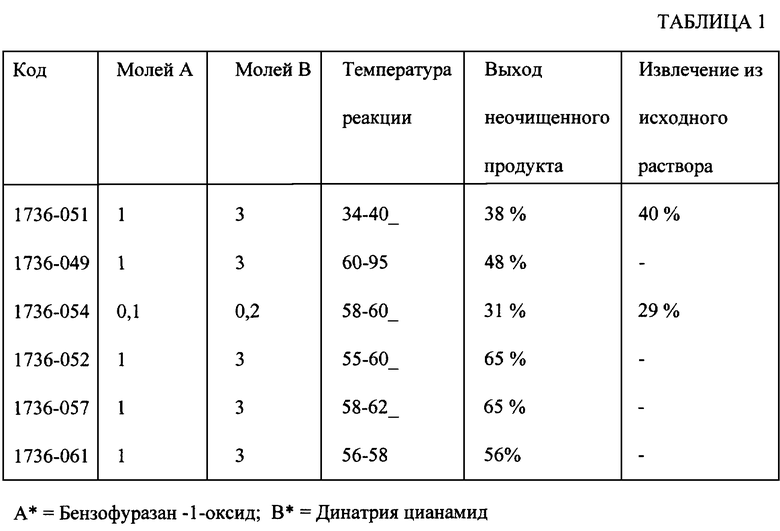
Способ, описанный в вышеуказанных ссылках, осуществляют аналогичным образом, но с модификациями, раскрываемыми в Примерах 1-4. Гетерогенная реакция 1 моля бензофуразан-1-оксида с тремя молями динатрия цианамида в 50% водном растворе метанола сразу же становится экзотермической. В соответствии с противопоставленными ссылками предпочтительная последовательность проведения способа заключается в суспендировании бензофуразан-1-оксида с двумя молями динатрия цианамида в водном растворе метанола. Через несколько минут к реакционной смеси прибавляют еще 1 моль динатрия цианамида. После этого промежуточная натриевая соль осаждается в виде темно-фиолетового маслянистого твердого осадка. Мокрый твердый осадок очень трудно выделить из обычной реакционной колбы, а его фильтрование происходит чрезвычайно медленно и требует больших затрат времени. Неочищенный преципитат растворяют в воде и затем фильтруют для удаления загрязняющего нерастворимого остатка основания. Фильтрат подкисляют уксусной кислотой и продукт выделяют в виде оранжевого твердого осадка. Указанный твердый осадок растворяют примерно в 10 объемах уксусной кислоты при температуре 80oC. Полученный раствор не обеспечивает быстрой кристаллизации, если его не упарить, или, в альтернативном варианте, если его не разбавить менее полярным растворителем.
Пример 2
Используют аналогичный метод, как и в примере 1, за исключением того, что промежуточный продукт, полученный в виде темно-фиолетового маслянистого твердого осадка, растворяют и подкисляют без фильтрации. Продукт извлекают с высоким выходом, но присутствующую в нем загрязняющую примесь, обнаруженную методом тонкослойной хроматографии (ТСХ), очень трудно было удалить из него указанным методом хроматографии. Загрязняющую примесь удаляют флэш-хроматографией и идентифицируют ее как феназина 1,5-ди-N-оксид. Это соединение является наиболее избыточным побочным продуктом реакции, образуемым в результате автоконденсация бензофуразан-1-оксида.
Пример 3
Повторяли методику примера 1 с получением неочищенного продукта, за исключением того, что 2 моля бензофуразан-1-оксида взаимодействовали с 6 молями динатрия цианамида. После введения двух растворов происходила бурная экзотермическая реакция, сопровождаемая выделением газа. Выход продукта после обработки снижался до 22%.
Пример 4
Повторяли методику примера 1 с получением неочищенного продукта, за исключением того, что 1,2 моля бензофуразан-1-оксида взаимодействовали с 3,6 молями динатрия цианамида. После отделения неочищенного преципитата, его затем суспендировали в воде и к полученной суспензии прибавляли 35% гидроксида натрия до получения раствора с проявившейся окраской. Раствор фильтровали и подкисляли уксусной кислотой с образованием кристаллического продукта. Продукт, как установлено, содержит загрязняющую примесь, моно-N-оксид феназина. Кроме того, полученный продукт содержит также хлопьевидный осадок, который можно удалить только путем фильтрования через уксусную кислоту при температуре 85oC. Манипуляции с горячим раствором, содержащим цитотоксичный агент, как установлено, приводят к серьезным проблемам.
Для устранения указанных недостатков и нежелательных характеристик способа, раскрываемого в противопоставленных ссылках, и конечного продукта, полученного таким способом, были проведены обширные исследования для подбора наиболее оптимальных экспериментальных условий и реагентов для использования их в улучшенном способе предлагаемого изобретения.
Условия для гомогенной реакции.
1. Растворитель
Характеристики растворимости реагентов ограничивали выбор растворителя для проведения экспериментов в условия гомогенной реакции. Бензофуразан-1-оксид имеет очень высокую растворимость в неполярных растворителях, например как этилацетат или толуол. Он значительно менее растворим в изопропаноле или метаноле. Однако бензофуразан очень хорошо растворяется в полярных апротонных растворителях, например как диметилформамид, N-метилпирролидон или диметилсульфоксид (ДМСО). Один моль бензофуразан-1-оксида быстро растворяется с поглощением энергии в 3 объемах ДМСО при комнатной температуре.
Динатрия цианамид не растворим в органических растворителях, но растворяется с выделением тепла в воде. Три моля его можно растворить в двух объемах воды с образованием цианамида натрия, который находится в равновесном состоянии с гидроксидом натрия.
Реакционная среда, содержащая крепкий раствор едкой щелочи, является ограничивающим фактором при выборе растворителя для бензофуразан-1-оксида. Предпочтение было отдано диметилсульфоксиду, а не диметилформамиду из-за неоспоримой его стабильности в сильном основании.
2. Метод введения
Под стандартным введением следует понимать как прибавление раствора бензофуразан-1-оксида в ДМСО к раствору, содержащему динатрия цианамида в воде. В одном эксперименте, в котором водный раствор динатрия цианамида добавляли к раствору бензофуразан-1-оксида в метаноле, отмечался низкий выход продукта и главный побочный продукт, образовавшийся в этой реакции, был идентифицирован как феназина моно-N-оксид. При условиях, где присутствовал избыток бензофуразан-1-оксида, например при введении с протеканием обратной реакции, образование родственных феназину побочных соединений имеет благоприятное влияние.
3. Стехиометрия
В соответствии с опубликованными данными (см. Angew. Chem. Int. Ed. , том. 11, 1972, стр. 1009) и на основании наших исследований с использованием условий гетерогенной реакции для получения тирапазамина необходимо 3 эквивалента динатрия цианамида на каждый эквивалент бензофуразан-1-оксида. В одном эксперименте, где один моль-эквивалент взаимодействовал с 2 моль-эквивалентами в условиях гомогенной реакции, получали 26% выход тирапазамина от исходного бензофуразан-1-оксида. Термостабильность водных растворов натриевой соли цианамида хорошо известна специалистам, но при повышенных температурах, как известно, имеет место ее гидролиз до цианата, сопровождаемый выделением аммиака. Известно также, что цианамид полимеризуется с образованием димерных соединений и водонерастворимого тримера, меламина.
Было проведено несколько гомогенных реакций с использованием цианамида и бензофуразан-1-оксида в ДМСО и воде. Образование конечного продукта сопоставимо c данными, полученными в случае динатрия цианамида, но только, если используют три моля цианамида и шесть молей гидроксида натрия.
4. Температура
При стандартных реакционных условиях раствор, содержащий бензофуразан-1-оксид, в ДМСО прибавляли при комнатной температуре к водному раствору динатрия цианамида. Выход продукта был оптимален в случае, когда температуру раствора динатрия цианамида поддерживали во время введения в интервале от 55 до 60oC. Реакция протекала быстро при указанном температурном интервале и не наблюдали никакой экзотермической реакции. В еще одном эксперименте, где температуру поддерживали в интервале от 34 до 40oC, из исходного бензофуразан-1-оксида получали 40% выход целевой продукта.
Подробные данные этого и других проведенных экспериментов, которые продемонстрировали практическую воспроизводимость результатов при оптимальной температуре реакции, приведены в табл. 1.
5. Соотношение растворителя
Большую часть экспериментов проводили в смеси растворителей ДМСО/вода при соотношении примерно 1: 1. В одном эксперименте, в котором использовали смесь ДМСО/вода при соотношении 1: 2, отмечали немного больший выход исходного бензофуразан-1-оксида, а выход тирапазамина при этом был соответственно ниже. Большинство реакций проводили при соотношении 7: 1, об. /вес. , в расчете на бензофуразан-1-оксид. В случае, когда в качестве растворителя вместо ДМСО использовали, например, изопропанол или ТГФ (тетрагидрофуран), то даже при длительном нагреве реакционной массы получали большей частью исходный материал.
6. Кристаллизация
Очистку сырого тирапазамина обеспечивали путем растворения его в 9 объемах уксусной кислоты при температуре 80-90oC. Полученную смесь затем фильтровали для удаления нерастворенных загрязняющих примесей, и фильтрат упаривали до половины от первоначального его объема в условиях неполного вакуума при температуре 60oC. Исключение из реакционного процесса стадии кристаллизации из уксусной кислоты или кислых растворов приводило к проблемам, обусловленным образованием хлопьевидного осадка. Состав хлопьевидного осадка не был идентифицирован. При фильтровании растворов, содержащих флокулянты, на фильтровальной бумаге получали только пятно и недостаточное количество твердого продукта, которое не позволяло определить его характеристики традиционными спектральными методами.
Для исключения стадии дистилляции, более концентрированные растворы можно получить из 6 объемов уксусной кислоты при температуре 106oC. Хотя вышеуказанное имеет практическую реализацию, тем не менее считают, что такая процедура потенциально вредна.
Вначале получали суспензии, содержащие 9 г тирапазамина в 50 мл воды, а затем были выбраны три кислоты (метансульфокислота, серная и соляная кислоты) для изучения их способности к растворению. Раствор метансульфокислоты, как оказалось, наиболее подходит для использования. После нейтрализации реакционного раствора гидроксидом натрия выделили продукт в виде свободного основания. Кристаллы, образующиеся в результате этой процедуры, анализировали на размер частиц, и в результате анализа было выявлено, что они не соответствуют технической спецификации, что 90% состава продукта должно иметь частицы < 190 нм.
Выделение тепла в результате нейтрализации гидроксидом натрия препятствовало быстрому введению реагентов, которое, как полагали, необходимо для образования кристаллов с требуемым размером частиц. Эта проблема, как полагали, обусловлена резким охлаждением кислого раствора в холодном буферном растворе ацетата натрия, который обеспечивал умеренное выделение тепла и очень быстрое введение. Кристаллы, полученные таким образом, отвечали технической спецификации к размеру частиц. Выходы и первоначальный анализ методом ТСХ и ВЭЖХ дали профиль загрязняющих примесей почти такой же, как и в случае уксусной кислоты. Наиболее важным моментом явилось то, что не отмечено выделения какого-либо хлопьевидного осадка из растворов уксусной кислоты, которые выдерживали в течение ночи.
При использовании параметров для протекания гомогенной реакции, полученных в результате исследований, получали тирапазамин по способу, описанному в примере 5.
Пример 5
Раствор, содержащий бензофуразан-1-оксид в диметилсульфоксиде, при молярном отношении 1: 3 прибавляли к водному раствору динатрия цианамида с молярным отношением 3: 2 при температуре примерно 60oC, который был помещен в реакционный сосуд. Продолжительность его введения до 34 мин, и после введения раствора полностью через несколько минут выпадал в осадок продукт в виде натриевой соли. Преципитат отделяли фильтрованием и суспендировали в воде в объемном соотношении 1: 6. Полученная суспензия содержала не растворимое в воде основание и поэтому суспензию фильтровали для его удаления. Суспензию подкисляли метансульфокислотой, взятой при 3,5 мольной эквивалентной концентрации при комнатной температуре. Температуру реакционной смеси повышали до примерно 45oC, а затем теплый раствор фильтровали для удаления нерастворимых загрязняющих кислотных примесей. Фильтрат (отфильтрованный раствор) затем загружали в буферный раствор, содержащий избыток ацетата натрия. Наблюдалась быстрая кристаллизация раствора с образованием 90% кристаллов, имеющих размер частиц менее 190 нм. Кристаллические частицы отфильтровывали и промывали тщательно водой. Кристаллические частицы в виде мокрой пресс-лепешки загружали опять в реакционный сосуд и перемешивали с 5 объемами воды. После промывания полученный твердый осадок фильтровали и ополаскивали ацетоном с получением очищенного тирапазамина. Полученный очищенный продукт сушили в течение 3 дней при температуре примерно от 50 до 60oC. Высушенный твердый продукт гомогенизировали путем пропускания его через сито из нержавеющей стали калибра N 10. Полученный целевой продукт имеет размер частиц менее 190 нм (степень чистоты 98,5%). Выход продукта 63%.
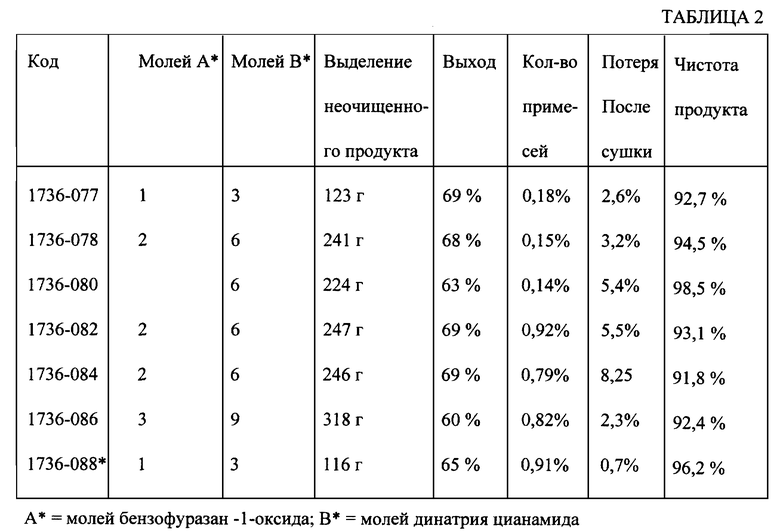
По аналогичной схеме была проведена экспериментальная серия из семи вариантов осуществления способа, результаты которой приведены в табл. 2.
Полученные результаты подтвердили, что заявляемый способ удовлетворяет критерию безопасности и промышленной применимости. Получают в результате продукт в форме кристаллов с размерами частиц менее 190 нм, что является необходимым для использования его в твердой лекарственной форме для перорального введения при лечении злокачественных опухолей.
Пример 6
К водному раствору, содержащему гидроксид натрия при температуре 20-30oC, прибавляли цианамид в молярном соотношении 6: 3. Смесь перемешивали в течение 1 часа при комнатной температуре и затем нагревали до 60oC. Раствор, содержащий бензофуразан-1-оксид в диметилсульфоксиде при молярном отношении 1: 3, прибавляли к водному раствору цианамида и гидроксида натрия в течение примерно 10-20 мин. При завершении введения происходило выделением тепла и реакционный раствор доводили до температуры ниже 75oC путем охлаждения. Смесь затем разбавляли деионизированной водой при объемном соотношении 1: 6, и нерастворимые побочные продукты реакции удаляли фильтрованием при температуре примерно 45oC. Объединенный фильтрат и промывочные воды, полученные при промывке нерастворимых побочных продуктов, подкисляли ледяной уксусной кислотой с получением неочищенной кристаллической смеси. Кристаллическую смесь охлаждали и твердый осадок собирали фильтрованием в виде мокрой пресс-лепешки. Пресс-лепешку ополаскивали деионизированной водой и мокрый твердый осадок переносили в чистый реакционный сосуд. Мокрый твердый осадок разбавляли смесью ацетона и уксусной кислоты в соотношении примерно 88: 12. Смесь перемешивали при комнатной температуре в течение примерно 30 мин. Твердый осадок собирали фильтрованием. Полученную пресс-лепешку также ополаскивали сначала раствором ацетона и уксусной кислоты (88: 12) и затем деионизированной водой. Мокрую лепешку затем переносили в чистый реакционный сосуд.
К мокрому твердому продукту прибавляли деионизированную воду (примерно пять объемов воды), и полученную суспензию подкисляли разбавленной метансульфокислотой, взятой при 3,5 мольной эквивалентной концентрации. Реакционную смесь обрабатывали активированным углем и перемешивали в течение нескольких минут при температуре примерно 45oC. Смесь затем фильтровали в теплом состоянии и повторно очищали активированным углем. После фильтрования теплый фильтрат загружали в хорошо перемешанный охлажденный разбавленный раствор ацетата натрия и подвергали микрокристаллизации с образованием микрокристаллического продукта со средним размером частиц менее 190 нм. Твердый микрокристаллический осадок собирали фильтрованием. Пресс-лепешку ополаскивали деионизированной водой и затем ацетоном. Кристаллический твердый продукт сушили в вакууме примерно при температуре 55oC с получением чистого продукта.
Суммарный выход продукта составляет примерно 40-50% от теоретического. Полученный целевой продукт имеет размер частиц менее 190 нм.
Пример 7
Данный пример иллюстрирует способ крупносерийного производства 3-амино-1,2,4-бензотриазин-1,4-диоксида.
Реактор емкостью 189,266 л (50 гал) (типа К-7) продувают азотом и загружают 17,9 л деионизированной воды и 11,9 кг (297 молей) бусинок гидроксида натрия. Операцию охлаждения использовали для управления экзотермической реакции. Смесь интенсивно перемешивали до получения прозрачного раствора, а затем охлаждали до температуры 15-25oC. В раствор загружали 6,3 кг (150 молей) цианамида и полученную смесь перемешивали в течение 1 часа при температуре 20-30oC. Одновременно, в другой реакционный котел (К-5) загружали 7,4 кг влажного бензофуразан 1-оксида (7,0 кг в расчете на сухой вес) (50 молей) и 25,5 кг диметилсульфоксида. Смесь перемешивали при нагревании до температуры 20-30oC в течение 50 мин.
Раствор, содержащий цианамид (К-7), нагревали до 60oC. Бензофуразан-1-оксид (К-5) пропускали через размещенный на технологической линии патронный фильтр в направлении к раствору цианамида в течение примерно 20 мин. Реакционную смесь перемешивали при охлаждении в течение еще 20 мин, при этом температура не превышала 71oC. К реакционной смеси затем постепенно прибавляли деионизированную воду (50 л). Смесь охлаждали до 44oC и нерастворимые побочные продукты были удалены через действующий патронный фильтр, который выгружал фильтрат в реактор емкостью 189,266 л (К-8). Фильтрат подкисляли введением 19,9 кг (332 моль) уксусной кислоты. Смесь охлаждали до 22oC и перемешивание продолжали в течение еще 1 часа. Твердый осадок собирали фильтрованием. Реактор (К-8) ополаскивали 17,3 л деионизированной воды и промывку сливали на пресс-лепешку. Мокрую лепешку переносили в реактор емкостью 189,266 л (К-8).
Получали раствор, содержащий 34,9 кг ацетона и 6,0 кг уксусной кислоты, и затем добавляли его к неочищенному твердому продукту в реакторе К-8. Смесь перемешивали при температуре 25oC в течение 3 часов, и твердый осадок собирали фильтрованием. Пресс-лепешку ополаскивали раствором, содержащим 10,4 кг ацетона и 1,8 кг уксусной кислоты. В реактор (К-8) загружали 15,0 л деионизированной воды и использовали для промывания пресс-лепешки. Неочищенную мокрую пресс-лепешку использовали на заключительной стадии очистки.
В реактор емкостью 189,266 л (К-8) загружали 11,6 кг (141,5 молей) безводного ацетата натрия и 61,5 л деионизированной воды. Смесь перемешивали и полученный прозрачный раствор охлаждали до температуры 10-20oC.
Одновременно, реактор емкостью 113,560 л (30 гал) (К-11) продували азотом и загружали 35,9 л деионизированной воды и 7,7 кг неочищенного влажного тирапазамина. Полученную суспензию перемешивали в течение 20 мин и добавляли 10,0 кг (102 молей) метансульфокислоты в течение нескольких минут. Реакционную температуру доводили до 40-45oC и к полученному темному раствору добавляли 0,2 кг активированного угля и 0,2 кг солка-флокулянта (solka). Смесь пропускали через размещенный на технологической линии фильтр для отделения твердого осадка. Затем добавляли 2 порцию активированного угля и солка-флокулянта. Фильтрат пропускали через фильтр для окончательной очистки и быстро прибавляли к холодному раствору ацетата натрия (К-8). Смесь перемешивали при охлаждении примерно 1,5 часа и твердый осадок собирали фильтрованием при температуре 10oC. В реактор (К-8) загружали деионизированную воду (34 кг) и использовали ее для промывки пресс-лепешки. Пресс-лепешку промывали последний раз 17,9 кг ацетона и сушили в вакууме при температуре в 55-60oC в течение 43 часов. Вес чистого продукта составлял 3,41 кг (выход 37,2% от теоретического).
Пример 8
Для иллюстрации возможности использования полученного по заявляемому способу 3-амино-1,2,4-бензотриазин-1,4-диоксида в твердых лекарственных формах для перорального применения приводим следующий пример конкретного фармацевтического состава на его основе:
Ингредиент - Мг/капсула
3-амино-1,2,4-бензотриазин-1,4-диоксид - 50 мг
Фракционированное кокосовое масло - 175,9 мг
Сорбитанмонолаурат - 9,26 мг
Гидрированное растительное масло - 37 мг
Желтый воск - 7,4 мг
Указанный состав приготовлен по известной технологии без дополнительного измельчения частиц 3-амино-1,2,4-бензотриазин-1,4-диоксида. Хотя выше описаны предпочтительные варианты осуществления изобретения, специалистам ясно, что изобретение этим не ограничивается и что различные модификации могут иметь место в пределах его сущности.
Описываются улучшенные способы получения 3-амино-1,2,4-бензотриазин-1,4-диоксида путем взаимодействия бензофуразан-1-оксида с динатрия цианамидом в условиях гомогенной реакции - в водном диметилсульфоксиде - при температуре 55-65oC с последующей кристаллизацией в забуференном растворе, содержащем избыток ацетата натрия. В предпочтительном варианте способ включает взаимодействие бензофуразан-1-оксида с динатрия цинамидом в условиях также гомогенной реакции с последующей очисткой в смеси ацетона и уксусной кислоты. 2 с. и 4 з. п. ф-лы, 2 табл.
| US 3868371, 25.02.1975 | |||
| US 5175287, 29.02.1992 | |||
| Способ получения бензо-1,2,4-триазинди- -окисей-(1,4) | 1973 |
|
SU447888A1 |
| Способ получения производных 1,4ди- -окиси 1,2,4-бензтриазина | 1974 |
|
SU583752A3 |

